
Detyre kursi biologji 1
34
Detyre Kursi nr 1 TEMA : çrregullimet gjenetike autozomale tek njeriu.
-
Upload
danjela-lushaj -
Category
Health & Medicine
-
view
567 -
download
15
Transcript of Detyre kursi biologji 1

Detyre Kursi nr 1
TEMA : çrregullimet gjenetike autozomale
tek njeriu.

Fakulteti I Studimeve Profesionale
Departamenti i Mjekësis
Dega : Ndihmës dentist
Lënda: Biologji
Dorëzoi : Vilson Hoxha
Danjela Lushaj
Eni Veliu
Ardit Osmovi
NenTemat e Grupit : Vilson Hoxha :1) Sindroma Patau
2)Sindroma Edëards
3) Albinizmi
Danjela Lushaj :1) Sindroma Doën
2) Mjaullima maces
3) Talasemia
Eni Veliu : 1) Sindroma Perri
2) Sindroma Marfan
3)Galaktosemia
Ardit Osmovi : 1) Monosomia 2)Poliploidia
Pranoi : Laura Gjyli

OBJEKTIVAT
Të përshkruaj C’janë sëmundjet gjenetike
Të përshkruaj shkaqet e këtyre sëmundjeve
Të përshkruaj crregullimet gjenetike autozomale .
Të përshkruaj crregullimet gjenetike autozomale recesive .
Të përshkruaj Sindromën Patay , shkaqet , simptomat dhe ilustrimet përkatëse .
Të përshkruaj Sindromën e Eduardsit , shkaqet , simptomat dhe ilustrimet përkatëse .
Të përshkruaj Sindromën e Albinizmit , shkaqet , simptomat dhe ilustrimet përkatëse .
Ç’eshte sindroma doën?Karakteristikat e njerezve me
kete semundje

Talasemia-(Njohuri te përgjithshme mbi këtë sëmundje.Nga vjen fjala “Talasemi” Trajtimet e kësaj sëmundje ,3 Llojet e beta-talesemisë,Alfa –Talesemia
Terapia e transfuzionit ,, ulja e përqëndrimit të hekurit në gjak ,Parandalimi
C’është sindroma e mjaullimës se maces? Karakteristikat, foto
Sindroma Marfan, Karakteristikat,Ilustrime
Galaktosemia .Karakteristikat,Ilustrime
Sindroma Perri ,Karakteristikat,Ilustrime
Monosomia,Karakteristikat e kësaj sëmundje ,ilustrime në foto
Poliploidia –Karakteristikat,ilustrimet në foto
C’janë sëmundjet gjenetike ?
Sëmundjet e trashëguara, ose sëmundjet gjenetike, janë sëmundje që transmetohen nga njëri brez në tjetrin. Sëmundjet gjenetike janë aktualisht të pashërueshme, pasi shkenca akoma nuk është në gjendje të korrigjojë defektin,

edhe pse ekzistojnë kura për crregullime të rënda të funksionimit të organeve të njeriut, që bëjnë të pamundur një jetese normale dhe për një kohë të gjatë. Në ndryshim nga sëmundjet e tjera, sëmundjet gjenetike, mbasi shtohen bartësit e fshehtë të këtyre sëmundjeve dhe, për rrjedhojë, rritet mundësia e lindjes së fëmijëve të sëmurë.
Shkaqet : Shkaqet kryesore të sëmundjeve gjenetike janë crregullimet që ndodhin në strukturën e gjeneve, duke sjellë në këtë mënyrë mosfunksionim të tyre. Në bazë të vendndodhjes së alelit me të metë përcaktohet edhe emri i sëmundjes. Mendohet se në shfaqjen e sëmundjeve gjenetike ndikojnë edhe shumë faktorë të tjerë, në këtë rast te jashtëm. Këto faktorë të tjerë mund të jenë rrezatimet ndaj rrezeve të dëmshme, ushqimet, stresi e shumë faktorë të tjerë.
Crregullimet gjenetike autozomale
Janë crregullime të kromosomeve autosomike, janë sëmundje të pakurueshme dhe të lindura ato mund të jenë dhe të trashëgueshme . Shumë prej tyre kanë në bazë një translokacion reciprok e të ekuilibruar,prandaj janë asimptomatike. Vetëm një numër i kufizuar i tyre kanë shfaqje të qarta klinike. Me të shpeshtat janë: -Trizomia 21 (Sindroma e Daën-it) -Trizomia 13 -Trizomia 18
crregullimet gjenetike autozomale recesive .
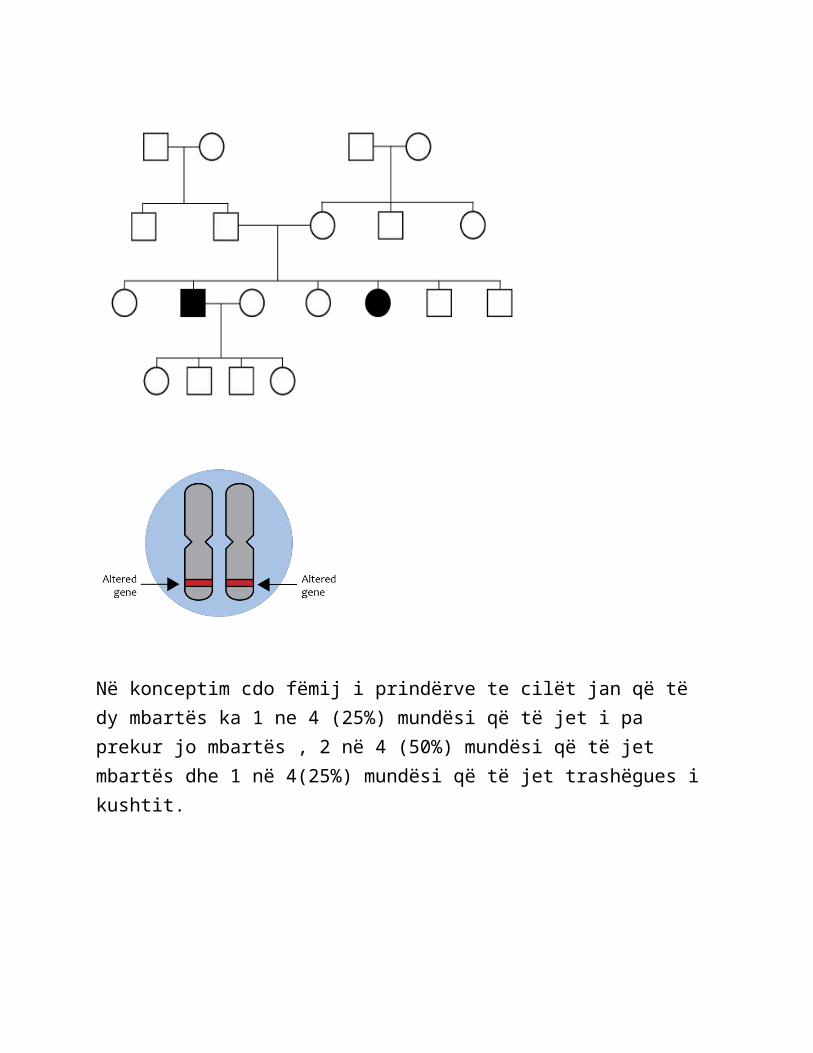
Një crregullim gjenetik autozomal recesive është i shkaktuar nga ndryshime (mutacione) ne një kopie ose cift gjenesh . Të dy gjenet duhet te ken një ndryshim (mutacion) për njeriun që të ket këtë crregullim . Një person i prekur trashëgon një kopje të gjenit me ndryshimin (mutacionin) nga secili prind .Në raste, prindërit e individit të prekur janë bartës të shëndetshëm të një kopjeje të vetme të ndryshuar te genit . Pra kushti është që vetëm në individët që kanë marrë dy kopje të një geni autosomal, një kopje nga secili prind.Geni është në një autosom, një kromozom nonsex. Prindërit janë bartës të cilët kanë vetëm një kopje të genit dhe nuk shfaqin tipar sepse geni është recesiv i genit normal homolog të tij . Në trashëgimin autosomale recesive, njerzit me një kopje të genit të ndryshuar nuk e kan kushtin . Ata mund të thuhet se jan mbartës për kushtin e autozomis recesive.

Në konceptim cdo fëmij i prindërve te cilët jan që të dy mbartës ka 1 ne 4 (25%) mundësi që të jet i pa prekur jo mbartës , 2 në 4 (50%) mundësi që të jet mbartës dhe 1 në 4(25%) mundësi që të jet trashëgues i kushtit.
SINDROMA PATAU
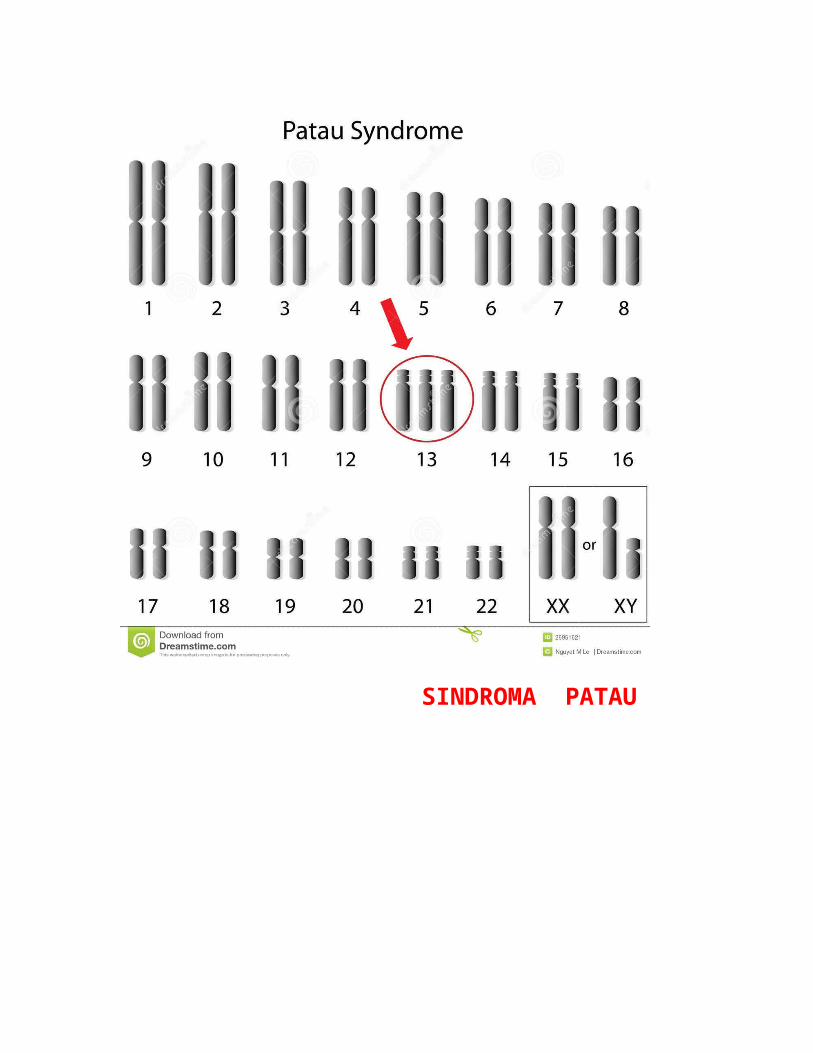
Trisomia 13 (gjithashtu e quajtur sindroma Patau) është një çrregullim gjenetik në të cilin një person ka tre kopje të materialit gjenetik nga kromozomi 13, në vend të dy kopjeve te zakonshme. Rrallë, materiali shtesë mund të jetë bashkëngjitur në një tjetër kromozom (translokacion).
Shkaqet :

Sindroma Patau është rezultat i Trisomis 13, që do të thotë se çdo qelizë në trup ka tre kopje të kromozomit 13 në vend të dy të zakonshmeve. Një përqindje e vogël e rasteve ndodhin kur vetëm disa nga qelizat e trupit kanë një kopje shtesë; raste të tilla janë quajtur mozaiku Patau.
Sindroma Patau gjithashtu mund të ndodhë kur një pjesë e kromozomit 13 bëhet e bashkangjitur në një tjetër kromozom (translokohet) para ose në konceptim në një translokacion Robertsonian. Personat e prekur kanë dy kopje të kromozomit 13, plus material shtesë nga kromozomin 13 bashkangjitur në një tjetër kromozom. Me një translokacion, personi ka një Trisomy të pjesshëm për kromozomin 13 dhe shpesh shenjat fizike të sindromës ndryshojnë nga sindroma tipike Patau.
Shumica e rasteve të sindromës Patau nuk janë të trashëguara, por ndodhin si ngjarje rastësore gjatë formimit të qelizave riprodhuese (vezë dhe spermës). Një gabim në ndarjen qelizore e quajtur non-disjunction ( jo-ndarje) gjat mejozes mund të rezultojë në qelizat riprodhuese me një numër jonormal të kromozomeve. Për shembull, një qelizë vezë ose spermë mund të fitojë një kopje shtesë të kromozomit. Nëse një nga këto qeliza riprodhuese atipike kontribuon në përbërjen gjenetike të fëmijës, fëmija do të ketë një kromozom shtesë 13 në secilin nga qelizat e trupit. Mozaiku i Sindromes Patau gjithashtu nuk është i trashëguar. Ai ndodh si një gabim rastesor gjatë ndarjes qelizore të hershme në zhvillimin e fetusit. Sindroma Patau prek diku midis 1 në 10.000 dhe 1 në 21.700 lindje të gjalla.
Sindroma Patau e shkaktuar nga një translokacion mund të trashëgohet. Një njeri i paprekur mund të mbart një ristrukturim ose rivendosje te materialit gjenetik ndërmjet kromozomit 13 dhe një tjetër kromozomi. Kjo rivendosje është quajtur një translokacion i balancuar sepse nuk ka material shtesë nga kromozomin 13. Edhe pse ata nuk kanë shenja të sindromës Patau, njerëzit të cilët e bartin këtë lloj të translokacionit te balancuar janë në një rrezik në rritje e të pasurit fëmijë me këtë gjendje.
Simptomat :
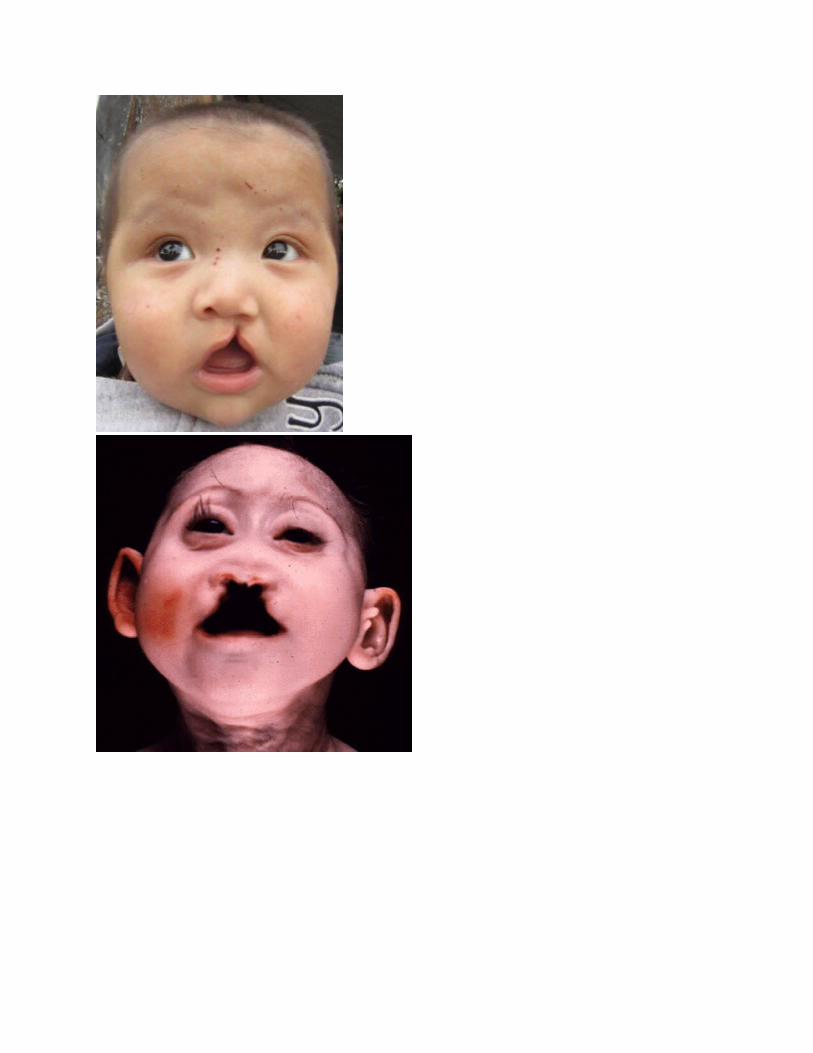
1.Buzë ose qiellëz te ndarë me dysh.
2.Duar të ngjeshura
3.Sy të vendosyr afer - Sytë mund të
bashkohen së bashku në një
4. Me tonin e Ulur te muskujve
5.Gishterinj shtesë te duarve ose
kembeve (polidaktili)
6.Hernie (gjendra): Hernie umbelikale ,
hernie inguinale.
7.Veshë te keqformuar , te vendosur ulet.
8.Neurologjike: Holoprosencephaly, në te
cilen truri nuk është i ndarë plotësisht në gjysma, është i pranishëm dhe është sinjalizuar ne përgjithësi nga prania e defekteve te linjes se mesme te fytyrës.
9.Defekte të kokës (lëkurë e humbur)
10. Nje Rrudhë të vetme te
pëllëmbës .
11. Anomali Skeletore (ne
gjymtyrë) .
12. Sy te vegjël , kok te vogel
(microcephaly).
13. Probleme me sistemin
gastrointestinal.
14. Probleme me zemrën : rreth
80%te individeve .
SINDROMA PATAU


Trisomia 13 u vëzhgua për herë të parë nga Thomas Bartholin në 1657, por natyra kromozonale i sëmundjes është konstatuar nga Dr Klaus Patau në 1960. Sëmundja është emëruar në nder të tij .
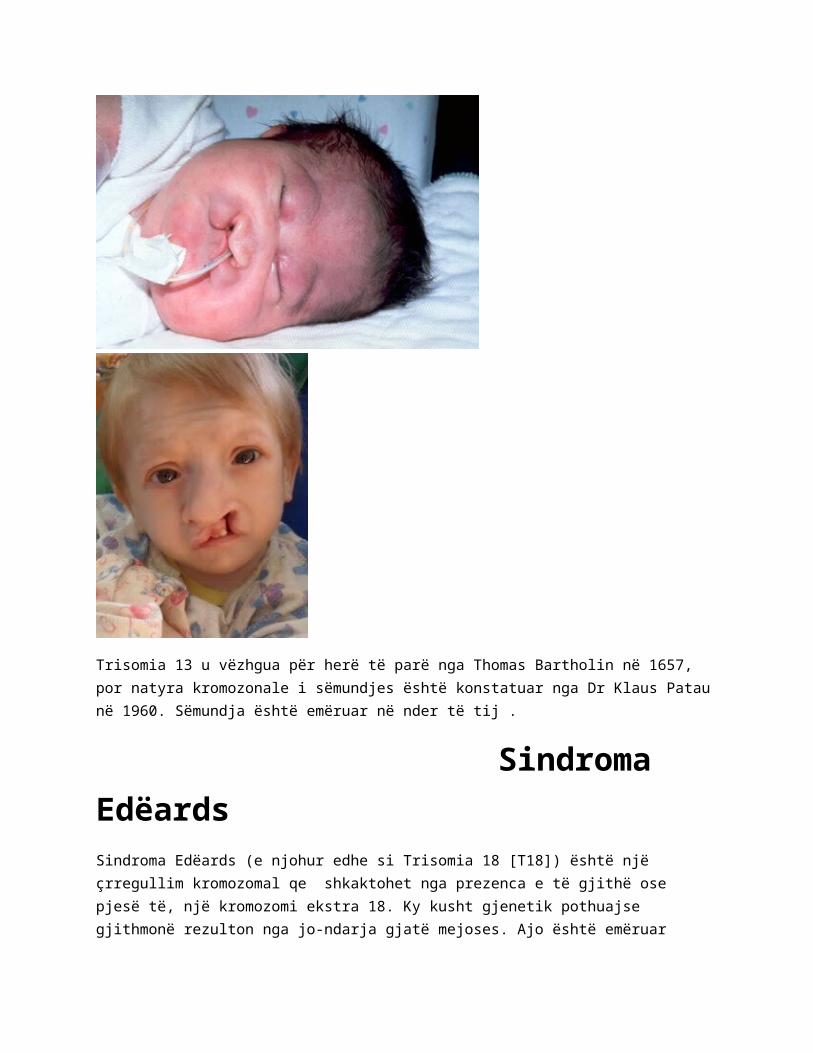
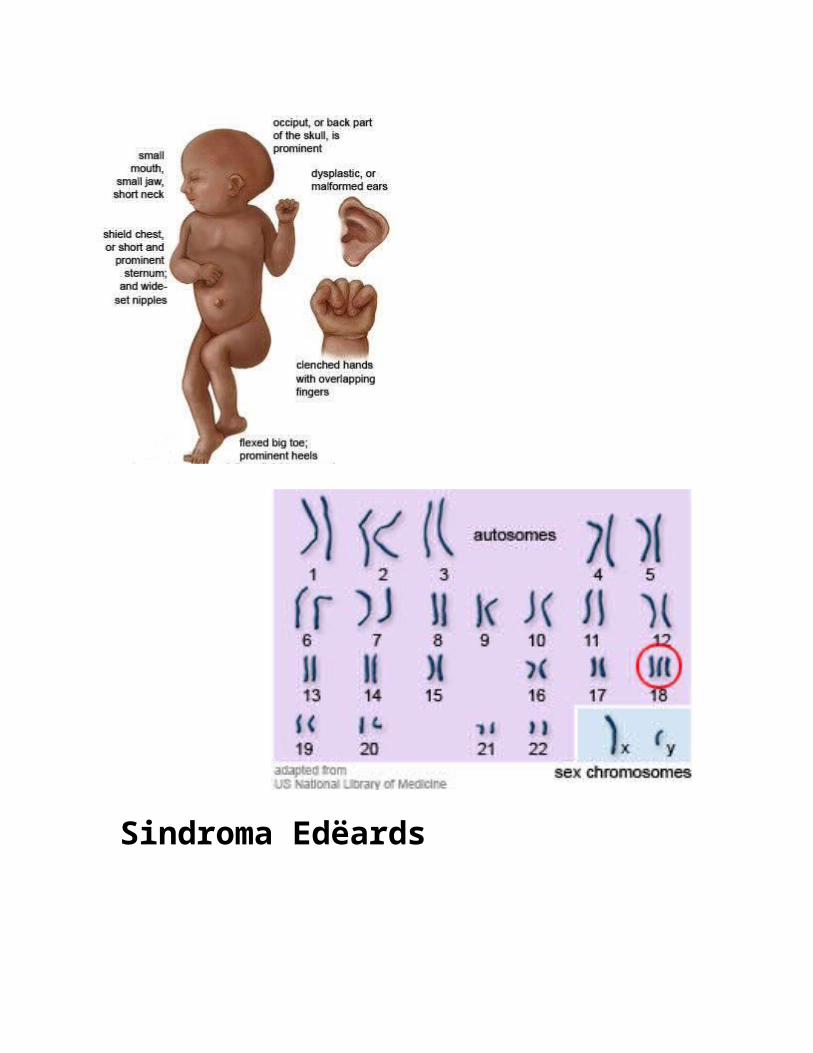
Sindroma EdëardsSindroma Edëards (e njohur edhe si Trisomia 18 [T18]) është një çrregullim kromozomal qe shkaktohet nga prezenca e të gjithë ose pjesë të, një kromozomi ekstra 18. Ky kusht gjenetik pothuajse gjithmonë rezulton nga jo-ndarja gjatë mejoses. Ajo është emëruar sipas John H. Edëards, i cili së pari e përshkroi sindromen në vitin 1960. Është e dyta më e zakonshme nga Trisomit autosomale, pas sindromes Doën .
Shkaqet :
Trisomia 18 (sindroma Edëards) rezulton nga prania e një kopje ekstra të kromozomit 18. Ndonjëherë veza e nënës ose spermatozoidi i babait përmban numrin e gabuar të kromozomeve. Me kombinimin e tyre, ky gabim tejcohet te fëmija.
Nje "Trisomi" do të thotë qe fëmija ka një kromozom shtesë në disa ose të gjitha qelizat e trupit. Në rastin e Trisomis 18, Foshnja ka tre kopje të kromozomit 18. Kjo bën që shumë nga organet e foshnjës të zhvillohen në një mënyrë të parregullt. Bebet me sindromën Edëards 'do të jenë rritur ngadalë në barkun e nënës dhe do të kenë një peshë të ulët, së bashku me një numër të problemeve të tjera serioze mjekësore. Ato që mbijetojnë në lindje, rreth gjysma do të vdesin brenda dy javësh dhe vetëm rreth një në çdo pesë individ do të jetojnë të paktën tre muaj. Rreth një në çdo 12 bebe te lindura me sindromën Edëards mbijetojn përtej një viti, dhe ata do të jetojnë me aftësi të kufizuara të rënda fizike dhe mendore. Disa fëmijë mund të mbijetojë në moshën e rritur te hershme, por kjo është shumë e rrallë. Rreth 1 në çdo 3000 deri ne 6000 foshnje lind me Trisomin 18, dhe shumica janë femra.
Ekzistojnë tri lloje të Trisomy 18:
Trisomia 18 e plote : kromozomi ekstra është në çdo qelizë në trupin e foshnjës. Kjo është lloji më i zakonshëm i Trisomis 18 ,rreth 94% te rasteve.
Trisomia 18 e pjesshme : Fëmija ka vetëm një pjesë të një kromozomi ekstra 18. Kjo pjesë shtesë mund të jenë bashkëngjitur në një tjetër kromozom në vezë apo spermë (i quajtur një Translokacion). Ky lloj i Trisomis 18 është shumë i rrallë.
Trisomia 18 mozaik : Kromozomi ekstra 18 është vetëm në disa nga qelizat e foshnjës. Kjo formë e Trisomis 18 është gjithashtu e rrallë.
Simptomat:
Shenjat fizike të sindromes Edëards 'përfshijnë:
Një kok te vogël në form anormale. Një nofull dhe gojë të vogël . Gishta të gjat të mbivendosur , grushta
te mbledhur te ngjeshur. Vesh te ulet dhe te crregullt.
Këmb të deformuara . Buzen dhe qiellzen te cara . një exomphalos (ku zorrët zhvillohen në
një qeskë jashtë stomakut)
Bebet me sindrom Edëards 'gjithashtu zakonisht kanë:
Probleme të zemrës dhe veshkave . Problemet e të ushqyerit - duke çuar në
rritje te dobët. Probleme të frymëmarrjes . një paaftësi të rëndë të mësuarit , të
lexuarit .
hernie në muret e stomakut të tyre (ku indet e brendshme futen ose shtyjnë nepermjet një dobësie në murin e muskujve) .
infeksionet e shpeshta të mushkërive dhe të sistemit urinar .
Sindroma Edëards
ALBINIZMI

Albinism është një kusht i trashëguar i lindur i karakterizuar ngamungesa ose ulja e një pigment, melaninës, në lëkurë, flokët / qimet dhe në Sytë, ose, thelbesisht , vetëm në sy.
Albinizmi te njerëzit (që nga albus Latinisht, "e bardhë"; i quajtur gjithashtu achromia, achromasia, ose achromatosis) është një çrregullim i lindur karakterizohet nga mungesë e plotë ose e pjesshme e pigmentit në lëkurë, në flokë dhe sy për shkak të mungesës ose defektit të tirosinases, një enzimë që përfshihet në prodhimin e melaninës. Kjo është e kundërta e melanismit .
Albinizmimi rezulton nga trashëgimia e alelit recessiv te gjeneve dhe prek të gjith vertebratët, duke përfshirë edhe njerëzit. Ndërsa një organizëm me mungesë të plotë të melaninës është quajtur një Albino , një organizëm me vetëm një sasi të zvogëluar të melaninës është përshkruar si albinoid.
Te njerëzit, ka dy lloje kryesore të albinizmit: okulokutaneous, që prek sytë, lëkurën dhe flokët, dhe okular që prek vetëm sytë.
Albinizmi Okulokutaneous, OCA , përfshin gjashtë lloje të albinizmit, nga të cilat:katër jo-sindromike (OCA1, OCA2, OCA3, OCA4);dy sindromike (sindroma Hermansky-Pudlak HPS 1-8, dhe Chediak-Higashi CHS)Albinizmi Okular , OA, përfshin vetëm një lloj, OA1.
Shumica e njerëzve me albinism okulokutaneous duken te bardhë apo shumë të zbehtë . Albinizmi Okular rezulton në sy blu te ndritshem, dhe mund të kërkojë testimi gjenetik për tu diagnostikuar.Për shkak se individët me albinizem kan lëkurë që tërësisht i mungon pigmenti i melanines, e cila ndihmon pë të mbrojtur lëkurën nga rrezatimi ultraviolet idiellit, lëkura e tyre mund të digjet më lehtë nga ekspozimi i tepërt ndaj diellit .
Shkaqet:
Simptomat:

Syri i njeriut normalisht prodhon pigment të mjaftueshme për ti dhen irisit ngjyren blu, të gjelbër ose ngjyrë kafe dhe jep opacitet (mbrojtje) në sy. Mungesa e pigment në sy rezulton gjithashtu në probleme me shikimin. Ata që jan të prekur me albinizëm janë përgjithësisht aq të shëndetshëm sa dhe pjesa tjetër e popullsisë , dhe albinism në vetvete nuk shkakton vdekje , edhe pse mungesa e pigmentit që bllokon rrezatimin ultravjolet rrit rrezikun e melanomaseve (kancereve të lëkurës ) dhe probleme të tjera.
Probleme me shikimin :
Lëvizje te shpejta e te pavullnetshme te syve (nystagmus).
Paaftësia e të dy syve për të qëndruar te drejtuar në të njëjtën pikë, ose për të lëvizur në unison (strabismus).
Miopi ose ipermetropi extreme . Ndjeshmëri ndaj dritës (fotofobi).
Lakim jonormal i sipërfaqes se përparme te syrit ose te lentes brenda syrit (astigmatizëm), e cila shkakton shikim te paqartë .
Hipoplazi Optike nervore, moszhvillimi i nervit optik.
Te tjera : Albinizmi sindromike gjithashtu ka probleme me gjakderdhjen , frymëmarrjen, tretjen dhe imunitetin.
Sinteza e Melanines .
ALBINIZMI
Albinizni te kafshet ,i pranishme në të gjitha llojet e kafshëve.
I pranishëm në të gjitha grupet e etnive ( etnia e bardh , etnia e verdh , etnia e zez )

Sindroma Down, Trizomia 21 apo Mongoloizmi quhet një mutacion gjenetik i veçant tek njeriu, tek i cili njeriu posedon kromozomin e 21.Sindroma Daun është një çrreullim kromozonesh që vjen si rezultat i vonesës në zhvillimin fizik, intelektual dhe gjuhësor. Ky është çrregullimi më i njohur i kromozoneve që shoqërohet me vonesë mendore. Njerëzit me sindromën Daun kanë një pamje karakteristike fizike.Sindroma Daun identifikohet si anomali kromozonesh. Me përparimet që janë bërë në fushën e biologjisë molekulare, është zbuluar se njerëzit me Sindromën Daun kanë 47 kromozone në secilën qelizë në vend që të kenë 46. Ky kromozon ekstra, pjesërisht ose plotësisht kromozonin 21, rezulton në karakteristikat që shoqërojnë Sindromën Daun. Dy tipe të tjera anormalitetesh kromozonale shfaqen në Sindromën Daun. Jo ndarje është një nën-ndarje e qelizës të gabuar/me difekt në të cilën ka 3 kromozone me numër 21 në vend që të ketë dy. Kjo ndodh në pothuajse 95% të rasteve të Sindromës Daun. Meqënëse 95% e rasteve të Sindromës Daun ka 3 kopje të kromozonit 21, ky tip referohet si trinomi 21. Nga të gjitha çrregullimet e lidhura me kromozonet, Sindroma Daun është më e përhapur. Zakonisht diagnostikimi i Sindromës Daun dyshohet menjëherë mbas lindjes, në sajë të pamjes fizike të foshnjes, megjithëse këto lloj karakteristikash fizike mund të shihen në popullatën e gjerë. Prandaj, në se dyshohet për Sindromën Daun, një test për kromozonet duhet të bëhet menjëherë mbas lindjes së foshnjes. Mendohet që 1 në 800 deri 1100 lindje të rezultojë një fëmijë me Sindromën Daun. Studimet kanë treguar se më shumë meshkujt se sa femrat lindin me çrregullime kromozonesh. Ose nëna ose babai mund të mbajnë kromozone ekstra. Në 70% - 80% të rasteve, janë nënat ato që mbartin kromozone ekstra. 80% e fëmijëve me Sindromën Daun lindin nga nëna nën moshën 35 vjeç, por shkalla e rastave është më e lartë për gratë mbi moshën 40 vjeç. Në moshën 40 vjeç, shansi për të pasur një fëmijë me Sindromën Daun është afërsisht 1 në 110 lindje, ndërsa në moshën 45 rreziku bëhet më i madh 1 në 35 lindje. Gratë që kanë vetë Sindromën Daun bëjnë fëmijë, por mundësia të kenë fëmijë me Sindromën Daun është 50%. Vdekshmëria e fëmijëve me Sindromën Daun po ulet. Me përparimet e shkencës për trajtimet mjeksore të personave me Sindromën Daun, sot mendohet se mosha e personave me Sindromën Daun ka arritur në 55 vjeç, dhe ka raste edhe më shumë. Shumë individë me Sindromën Daun e shtyjnë krijimin e familjes për më vonë, kur shkalla e ndodhshmërisë është më e lartë. Kështu që numri i rasteve me Sindromën Daun pritet të rritet dhe ka shumë mundësi të dyfishohet brenda 10 vjetëve.Karakteristikat Individët me Sindromën Daun zakonisht kanë një
staturë më të vogël se sa bashkëmoshatarët e tyre pa Sindromën Daun. Jo vetëm zhvillimi i tyre fizik është i ngadaltë por edhe zhvillimi ityre intelektual është i vonuar.Përafërsisht një e treta e fëmijëve të lindur me Sindromën Daun kanë defekte në zemër, shumica e të cilave tashmë mund të korrigjohen me operacione. Të rriturit me Sindromën Daun janë të prirur ndaj arteriosklerozës, e cila mund t’i çojë drejt sëmundjeve të zemrës. Probleme me stomakun janë shumë të përhapura. gjithashtu kanë ndodhshmëri më të lartë se mesatarja të ngushtimit të zorrëve. Edhe problemet e shikimit janë shumë të larta tek personat me Sindromën Daun, si shikimi i vëngër, miopi apo hipermiopi, formim katarakti, etj.Personat me Sindromën Daun janë të prirur edhe ndaj infeksioneve të përsëritura të veshëve, dhe shpesh kanë humbje ose ulje të dëgjimit që shkaktohet nga grumbullimi i lëngut në veshin e brendshëm. Ata shpesh kanë pengesa në të folur për shkak të zmadhimit të gjuhës (si organ). Shumë individë që janë me Sindromën Daun kanë të njëjtat ndryshime në tru ashtu si ata që kanë sëmundjen Alzaimer. Kjo nuk do të thotë që të gjitha rastet me Sindromën Daun shfaqin shenja klinike të sëmundjes Alzaimer. Ka një shans prej 25 % të individëve me Sindromën Daun që janë mbi moshën 35 vjeç të zhvillojnë tipin (dementia) të sëmundjes Alzaimer.Ata gjithashtu, rrezikojnë 5 deri në 20 herë më shumë se sa popullsia tjetër, të zhvillojnë leuçeminë.
Karakteristika: • Probleme të të folurit
• Harmonizim i dobët i muskujve
• Përpëlitje e kapakëve të syrit me copëza lëkure në qoshet e brendshme të syve
• Shenja të bardha në irisin e syrit
• Duar të shkurtra dhe të gjëra me një vizë/të thelluar të vetme në pëllëmbën e njërës dorë ose të të dyja duarve
• Këmbë të gjëra me gishta të shkurtër
• Urë të sheshtë të hundës
• Veshë të shkurtër të ulët
• Qafë të shkurtër dhe kokë të vogël
• Kokën prapa e kanë të sheshtë

• Gojën e kanë të vogël
• Kur janë bebe të qarat i kanë të shkurtra dhe shumë të mprehta, çjerrëse
• Gjuhë të madhe e të zgjatur
• Aftësi të çuditshme ekstremitetet
• Vetëm një nyje përkulëse në gishtin e pestë në vend të dy
Disa njerëz me Sindromën Daun mund të kenë edhe një gjëndje të quajtur instabiliteti atlantoaksikal. Kjo është keq lidhja e dy vertebrave më të larta të qafës. Kjo gjëndje i bën këta individë të jenë më të prirur ndaj vrasjeve, dëmtimeve në se ato marrin pjesë në aktivitete që kërkojnë lëvizjen e qafës.
Talasemia 1. Njohuri të përgjithshme:
a. Për të pasur një molekulë hemoglobine normale duhen 2 vargje nga kromozomi 16, dhe 2 vargje nga kromozomi 11.
b. Çdo njeri normalisht ka 4 gjene për vargjet alfa (2 nga nëna dhe 2 nga babai) në kromozomin 16; në kromozomin 16 gjendet edhe gjeni për vargun zeta, që është homolog i vargut alfa. Çdo njeri ka normalisht 2 gjene beta (1 nga nëna dhe 1 nga babai) në kromozomin 11; në kromozomin 11 gjenden edhe gjenet për vargjet epsilon, gama, dhe delta, që janë homologë të vargut beta.
c. Tek të rriturit, normalisht rreth 96% e hemoglobinës është hemoglobinë A (alfa2beta2); rreth 3% është është hemoglobinë A2 (alfa2delta2), dhe rreth 1% është HbF. Fetusi ka kryesisht hemoglobinë F (alfa2gama2).
2. Fjala "talasemi" e ka prejardhjen nga greqishtja e vjetër dhe do të thotë "sëmundje e detit Mesdhe". Talasemia është sëmundje gjenetike që shkaktohet nga mungesa ose nga pakësimi i prodhimit të vargjeve të hemoglobinës.
3. Në varësi të sasisë të hemoglobinës që prodhohet, talasamia emërohet "majore", "intermedia", ose "minore". a. Talasemia minore është pa simptoma klinike.

b. Talasemia majore bën të domosdoshme dhënien e rregullt të transfuzioneve për të korrigjuar mungesën e theksuar të hemoglobinës.
c. Talasemia intermedia ka simptoma të anemisë të theksuar, por nuk e ka të domosdoshëm transfuzionin.
4. Në varësi të vargut të hemoglobinës që mungon (alfa ose beta), talasemia emërohet "talasemi alfa" ose "talasemi beta".
5. Diagnoza e talasemisë vendoset pas elektroforezës të hemoglobinës.6. Trajtimi:
a. Transfuzion + desferioksaminë për të parandaluar hemokromatozën.
b. Të prekurve u jepet edhe acid folik, dhe mund të bëhet splenektomia.
c. Transplanti i qelizave hemopoetike (transplanti i palcës të kockave) mund ta kurojë sëmundjen për fare (në rreth 80-85% të rasteve).
Ç’eshte talasemia?
Talasemite jane nje grup semundjesh trashgimetare te karakterizuara nga nje defekt gjenetik i sintezes se hemoglobines, qe s’eshte tjeter veçse nje proteine e rruazave te kuqe te gjakut. Nje ose me shume defekte ne hallkat e ketij zinxhiri shkaktojne lloje te ndryshme talasemie, ne menyre te veçante alfa-talasemite dhe beta-talasemite.
Alfa-talasemite gjenden me te perhapura ne Afrike, Azine jugore, Indi, vèndet e lindjes, Kinen jugore dhe ne ndonje rast ne vendet e Mesdheut.
Ekzistojne tre grupe te beta-talasemise:
• beta-talasemia e vogel (minore)• beta-talasemia mesatare (intermedia)• dhe forma me e rende beta-talasemia e madhe (major) ose Morbi i Cooley (Kuli). Ku prodhimi i pamjaftueshem i hemoglobines mund te shkaktoje deme te renda te organeve te brendeshme.

Llogaritet qe personat e semure nga Talasemia Major ne bote jane rreth 3 milion; zona me e goditur eshte ajo e pellgut te Mesdheut. Vetem ne Itali te semuret jane 9000. Mbijetesa e personave te prekur nga semundja varet nga kurimi periodik transufuzional (gati çdo 3 jave), gje qe provokon nje akumulim te hekurit ne organizem. Keshtu qe te semuret duhet ti nenshtrohen nje tjeter terapie kure per te ulur perqendrimin e hekurit ne menyre qe te parandalohen deme serioze ne organe.
Terapia e transfuzionit
Trajtimi per te cilin ka nevoje nje i semure talasemik eshte transfuzioni i rruazave te kuqe, perçuesit e oksigjenit ne organizem. Jane tre arsyet themeltare per te cilen transfuzioni i gjakut eshte i domosdoshem per jeten e te semurit:
• T’ju garantoje indeve dhe organeve nje sasi te mjaftueshme oksigjeni.• Per te mos sforcuar palcen ne prodhimin e rruazave te kuqe ne menyre qe kockat te rriten normalisht.• Te parandaloje ose te pengoje sa te jete e mundur rritjen e dimensioneve te shpretkes dhe sforcimin e organeve te tjera.
Terapia e uljes se perqendrimit te hekurit
Talasemiket akumulojne ne organe nje sasi te konsiderueshme hekuri per shkak te transfuzionit te herepashershem te gjakut. Akumulimi dhe perqendrimi i hekurit demton ne menyre progresive organet e brendshme duke shkaktuar semundje te zemres, diabet, cirrozen e melçise dhe ne kete menyre shkurton jeten e te semureve. Sot ekzistojne menyra te ndryshme per te eliminuar hekurin e tepert ne organizem. Terapia me e perhapur eshte trajtimi me DESFEROXAMIN qe konsiston ne injektimin nen lekure te ketij ilaçi nepermjet nje mikroinjektori per rreth 10-12 ore ne dite. Ky trajtim shkakton shqetesime te renda fizike e psikologjike.
Ne vitet e fundit po perdoren dy ilaçe te reja qe merren nga goja dhe jane

DEFERIPRONE L1 dhe me i funtit i quajtur ICL670A. Perdorimi i ketyre ilaçeve ne te semurit talasemike ben te mundur mbajtjen nen kontroll te sasise se akumulimit te hekurit. Metoda me me pak shqetesime per te semurin dhe me e sigurta per te mos lejuar perqendrimin e hekurit eshte e ashtuquajtura SQUID. Ekzistojne vetem pese aparate SQUID ne bote, njeri prej te cileve gjendet ne Qendren e Mikroçitemise ne Torino, prane Spitalit Regina Margherita/ Sant'Anna.
Parandalimi
Talasemia eshte nje semundje trashegimtare e transmetuar nga bashkimi i dy individeve mikroçitemike ose me mire te themi mbajtes te talasemise pa shprehje te semundjes, ne kodin gjenetik te tyre gjendet nje karakteristike (pjeseza talasemike) qe eshte ne gjendje ta transportoje. Nje individ mikroçitemik i ka rruazat e kuqe me te vogla se normalja e si pasoje me te varfra ne hemoglobine. Personat mikrocitemik kompensojne kete diference me prodhimin e nje numri me te madh rruazash te kuqe dhe keshtu bejne nje jete normale. Ne rast se dy persona mikroçitemik vendosin te bejne nje femije mundesia per te lindur i shendetshem eshte vetem nje e katerta ose 25%. Nga kater femije te lindur tre jane te semur talasemik ose mbajtes te talasemise. Pra dy prinder mikroçitemk bejne qè te linde nje femije talasemik. Ne Itali llogaritet qe mbajtes se semundjes te jene rreth 3 milion.
Talasemia nuk prek rastesisht. Roli Yt ne parandalimin e semundjes eshte themelor dhe eshte e mjaftueshme nje analize gjaku per te ditur se Ti dhe bashkeshorti (bashkeshortja) jeni apo jo transmetues te semundjes.
E ardhmja pa talasemiSherimi nga talasemia sot eshte i mundur nepermjet trapiantimit te palces se shtylles kurrizore, por eshte e domosdoshme te gjendet nje dhurues i te njejtit grup me ate te te semurit talasemik ne menyre qe te evitohet papajtueshmeria. Dhuruesit me te pershtatshem jane vetem vellezerit ose motrat e te semurit, gje qe e ben te mundur trapiantin vetem per pak persona. Ne keto vitet e fundit kerkimi shkencor-mjekesor mbi trapiantet ka

bere te mundur nje tjeter hap perpara: parashikohet trapiantimi i palces edhe midis personave jo familjare.
Teknika mjekesore gjithnje e me te sofistikuara per te bllokuar refuzimin nga organizmi te qelizave embrionale te marra nga kerthiza e te porsalindurve (te pasura me qeliza meme te perberesve te gjakut) po ndryshojne ne menyre te shpejte dhe pozitive kete metode sherimi. Çdo talasemik ne menyre te veçante femijet e te rinjte, presin me shprese te madhe per tu kuruar me me siguri me anen e kesaj teknike qe mund te gjeje zbatim te gjere ne nje dite jo te larget.
Kerkimi mjekesor-shkencorNjohja gjithnje e me e plote e gjenetikes dhe bioteknologjise hapin perspektiven e nje te ardhmeje ne te cilen do te jete e mundur te manipulohen genet. Italia, per fat te keq,investon pak burime ekonomike e shkencore per kerkimin ne kete fushe. Duke munguar financimi shteteror mbetet te presim angazhimin ekonomik te firmave te medha farmaceutike qe natyrisht kerkojne edhe perfitime. Sherbejne laboratore shkencore te specializuara ne shkallen me te larte dhe financime ekonomike te mjaftueshme per te perballuar shpenzimet. Ne qofte se nga njera ane studimet mbi harten gjenetike tregojne se talasemia mund te gjeje zgjidhje ne saje te trajtimit gjenetik, nga ana tjeter vazhdon mungesa e interesit ekonomik per te investuar ne favor te kurimit te semundjeve te rralla.
Gjer me sot keshtu ka qene per ilaçet, keshtu vazhdon te jete e me sa duket do te vazhdoje edhe per kete sektor qe kerkon specializim e teknologji te larte. Me sa duket aty ku duhen me shume investime financiare eshte me i theksuar indiferentizmi. Megjithate mbetet fakt konkret qe eksperimentimi gjenetik eshte nje hipoteze reale sherimi ne nje te ardhshme jo shume te larget, kete e deshmojne edhe trapiantimet e bera kohet e fundit me qeliza staminale.

Dhurimi i gjakut
Mbijetesa e talasemikeve eshte e lidhur me trasfuzimin periodik e te vazhdueshem te gjakut; per fat te keq ky i fundit mund te gjendet vetem nepermjet dhurimit. Nje i semure talasemik ka nevoje per rreth 50 dhurime ne vit, por dhurimet shpesh nuk mbulojne nevojen dhe kerkesen per gjak, gje qe eshte ne dem te te semurit deri ne humbje te jetes se tij. Dhurimi vullnetar i gjakut perben nje gjest real solidariteti sepse dhuron jeten dhe duhet te konsiderohet nje detyrim i çdo individi per te miren e te gjitheve. Perveç kesaj marrja e gjakut eshte edhe nje garanci per shendetin e dhuruesit ne saj te analizave qe i behen gjakut te tij.
Mjaullima Maces


















