
Syndromes Myéloprolifératifs
37
Syndromes Myéloprolifératifs K.Boudjedir Eurocord Hopital Saint louis
description
Syndromes Myéloprolifératifs. K.Boudjedir Eurocord Hopital Saint louis. Définition:. - PowerPoint PPT Presentation
Transcript of Syndromes Myéloprolifératifs

Syndromes Myéloprolifératifs
K.Boudjedir
Eurocord Hopital Saint louis

Définition:
Groupe hétérogène d’hémopathies malignes caractérisée par une prolifération clonale d’un précurseur myéloïde aboutissant à la production de cellules matures en excès (Polynucléaires, GR, Plaquettes)
- Splénomégalie (Séquestration dans la rate). - Le risque initial commun est la thrombose Vx - Le risque évolutif est la transformation en leucémie aigue
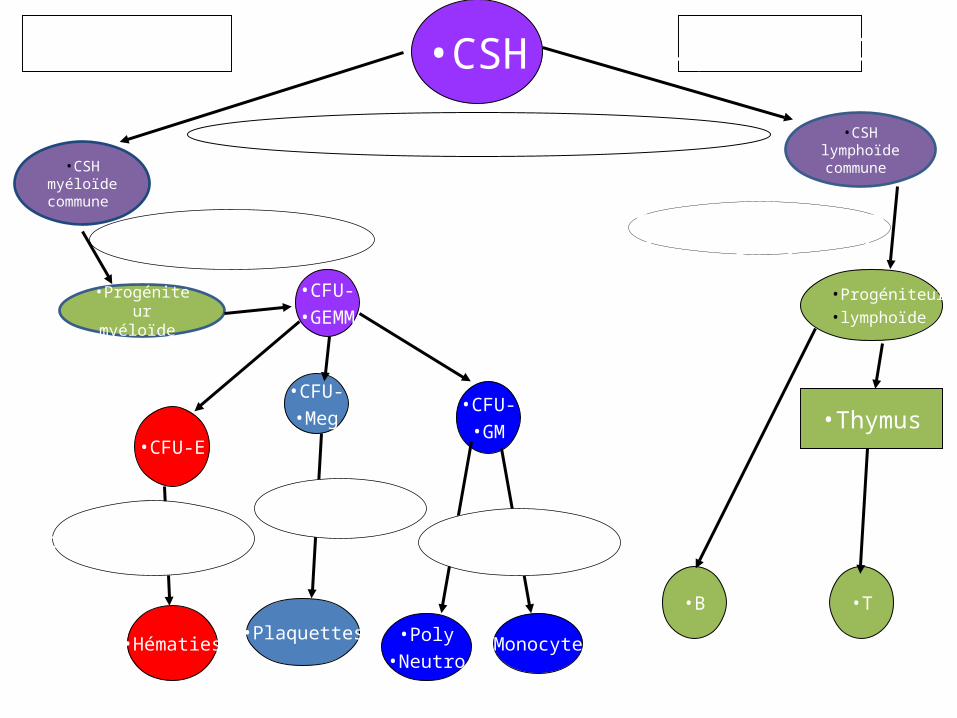
•CSH
•CFU-•GEMM
•Progéniteur•lymphoïde
•CFU-•GM
•CFU-•Meg
•B •T
•Thymus•CFU-E
•Hématies•Plaquettes •Monocytes•Poly
•Neutro
•Myéloïde •Lymphoïde
LA
Sd lymphoPSd myéloP
•CSH myéloïde
commune
•CSH lymphoïde commune
•Progéniteur myéloïde
Polyglobulie LMCTE

CLASSICATION
• Leucémie Myéloïde Chronique Lignée granuleuse
• Maladie de Vaquez ( Polyglobulie I aire) Lignée érythroblastique
• Thrombocytémie essentielle Lignée Mégacaryocytaire
• Myélofibrose primitive (splénomégalie myéloïde) ??
• Syndrome Myéloprolifératif atypique

Leucémie myéloïde chronique
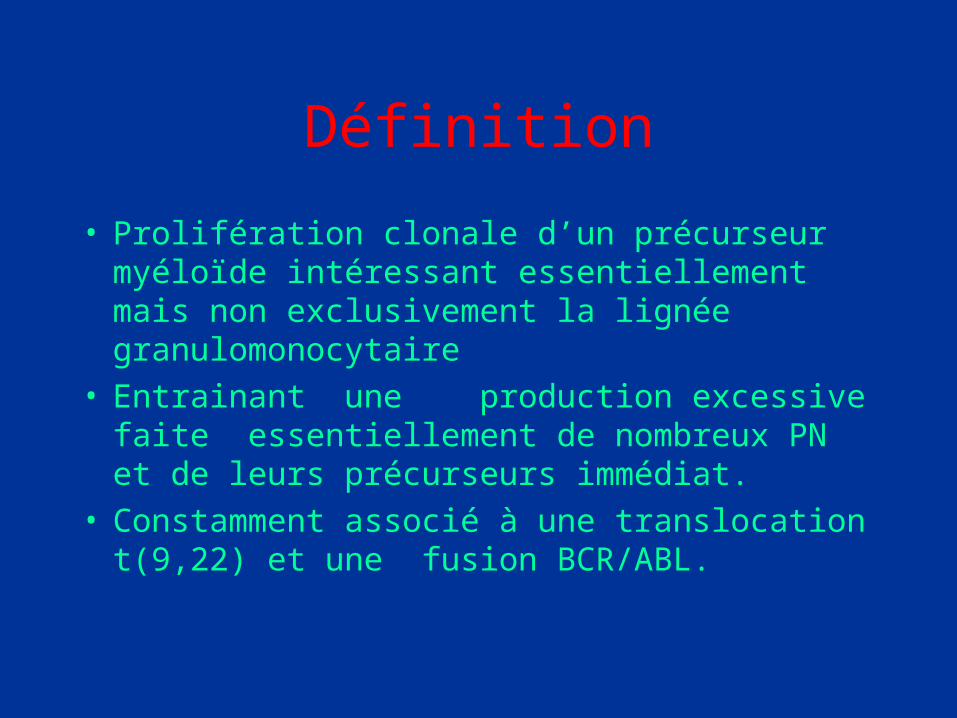
Définition
• Prolifération clonale d’un précurseur myéloïde intéressant essentiellement mais non exclusivement la lignée granulomonocytaire
• Entrainant une production excessive faite essentiellement de nombreux PN et de leurs précurseurs immédiat.
• Constamment associé à une translocation t(9,22) et une fusion BCR/ABL.

LMC: Épidémiologie
• Le plus commun des syndromes myéloprolifératifs.• 15-20% de toutes les leucémies• Incidence: 1,5/100.000• Médiane d’âge: 40 ans• Prédominance masculine• Cause : idiopathique++

Physiopathologie
• Apparition d’une translocation réciproque entre le chromosome 9 et celui du 22 donnant un chromosome pathologique :le chromosome Philadelphia (chromosome 22 raccourcie) dans toutes les cellules myéloïdes.
• Entre le site du gène abl et celui du gène bcr• La proteine codée a une activité tyrosine k

LMC: Signes Cliniques• Asymptomatique: NFS systématique (20-40%)• SF: Asthénie, pesanteur abdominale, perte de
poids, syndrome anémique, sueurs nocturnes• Ex Clinique: Splénomégalie isolée parfois
hépatomégalie• Complications:
– Hyperuricémie (lithiase, crise de goutte)– Thrombose (priapisme…..)– Hémorragies– Infarctus splénique (Douleurs+++)

LMC: Diagnostic * NFS - Hyperleucocytose à PNeutrophile - Myélémie (éléments granuleux immatures médullaires circulants) - Thrombocytose *Caryotype sang et/ou moelle: +++ t(9.22) –Chromosome Ph1 *Biologie Moléculaire:++++ BCR/ABL

Caryotype LMC

LMC: Evolution - Pronostic
• Evolution chronique.• Poussées évolutives: Accélération• Transformation inéluctable vers Leucémie
aiguë (acutisation) en quelques mois voir quelques années .
Toujours (évolution naturelle)

LMC: Evolution - Pronostic
• AcutisationProlifération blastique médullaire sans différenciation =Tableau d’insuffisance médullaire + blastose
2/3 LAM – 1/3 LAL
Pronostic effroyable

LMC: Traitement
• En urgence: Contrôle leucocytose et syndrome tumoral.
• 1/Hydratation 2/Hydroxyurée (Hydrea°) ou le Glivec
• 3/ Tt hypo-uricémiants :Zyloric° (allopurinol) pour éviter les complications liées à l’hyperuricémie)

LMC: Traitement
• Une fois, la phase initiale passée Tt de fond
• Plusieurs possibilités: – Sujet jeune: Allogreffe de CSH : Seule
possibilité curatrice– Immunothérapie: Interféron Alpha en SC
– STI: Nouvelle approche.

•Inhibiteur spécifique de la Tyrosine Kinase du transcrit Bcr-abl•Trt per os 400 mg à 800 mg/j en 2 prises•Effets secondaires :prise de poids, œdèmes périorbitaires, myalgies, atteinte hépatiques….•Excellente réponse
• Hématologique ++•Cytogénétique et Moléculaire•Vers la guérison?
• Excellente tolérance

MALADIE de VAQUEZ

Polyglobulie primitive Définition
• Prolifération clonale d’un précurseur myéloïde intéressant essentiellement mais non exclusivement la lignée érytroblastique
• Entrainant l’augmentation de Hb (>18gr/dl homme, 16 gr/dl femme),l’ augmentation des GR et de HT.
• Augmentation du volume globulaire total• Mis en évidence d’une mutation acquise de la
cellule souche myéloïde :JAK2

Vaquez: Épidémiologie
• Variation géographique à travers le monde.• Incidence en Europe: 8/1.000.000• Prédominance masculine (Sex ratio: 2,5/1)• Pic: 60 ans• Exceptionnel < 20 ans• Idiopathique++

Vaquez: Circonstances de Découverte
• Découverte fortuite à la numération• Érythrose cutanée (visage, mains, conjonctives),
Bouffées congestives, prurit à l’eau.• SF(hyperviscosité): Céphalées, vertiges,
paresthésies, somnolence, tr visuels,.• Splénomégalie• HTA
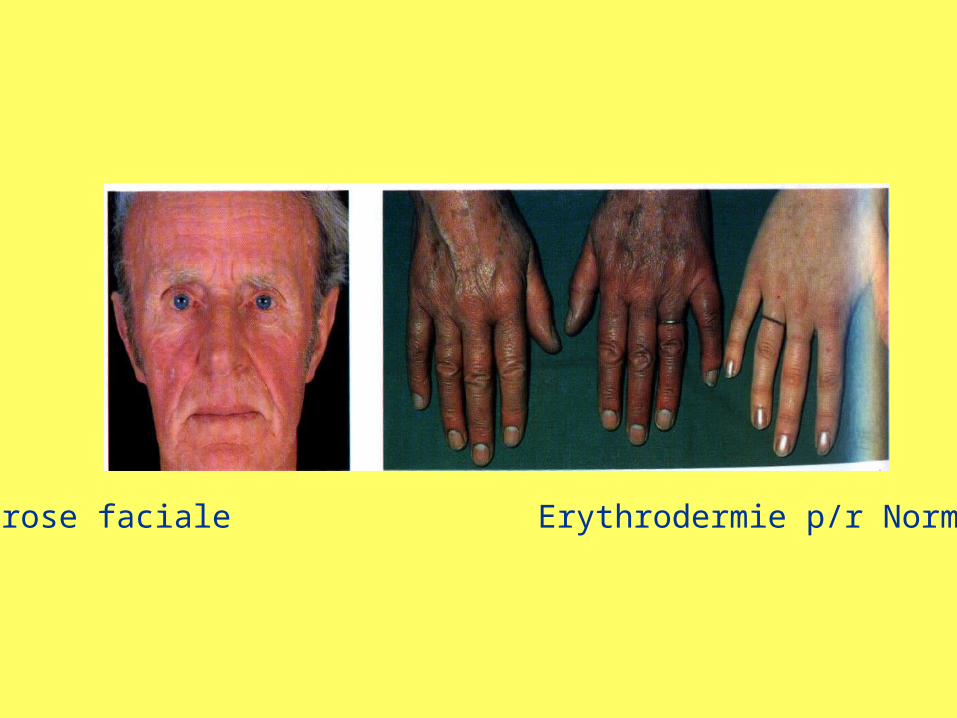
Erythrose faciale Erythrodermie p/r Normal
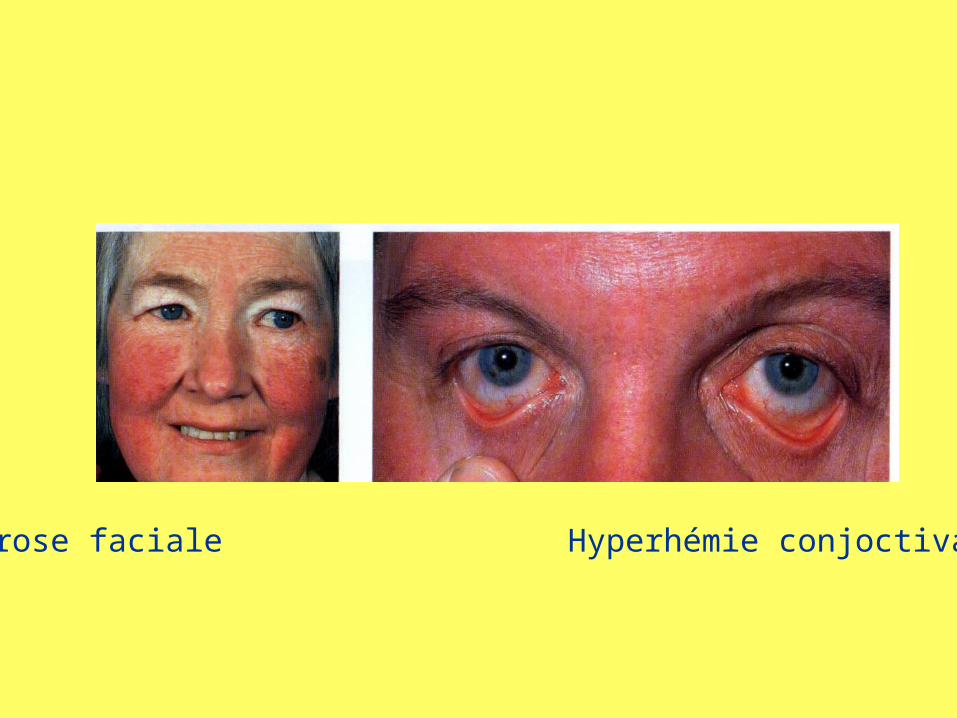
Erythrose faciale Hyperhémie conjoctivale
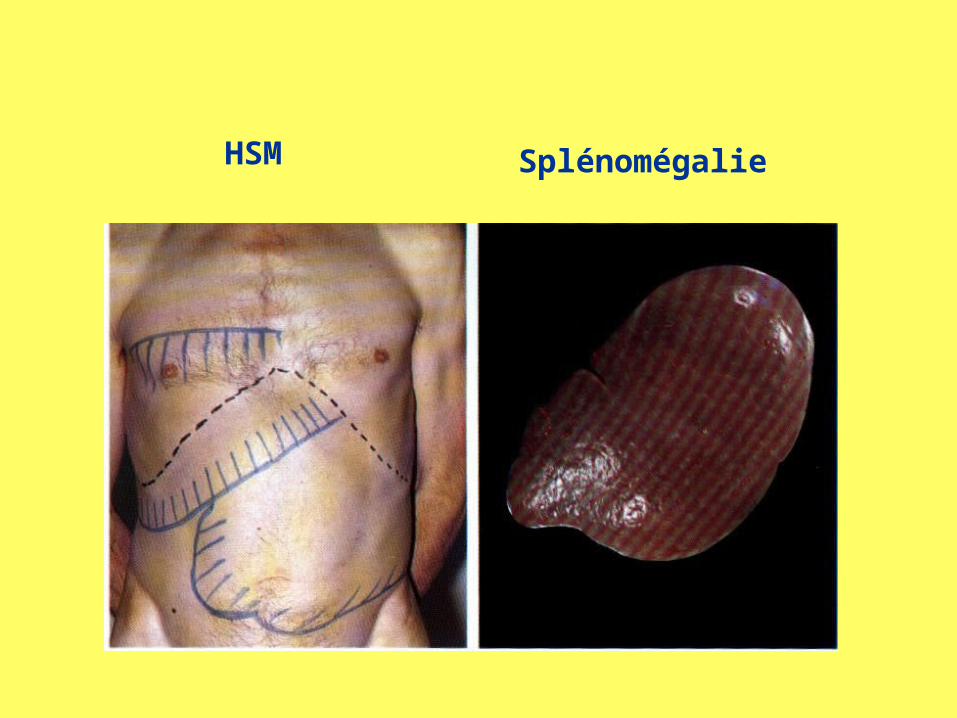
HSM Splénomégalie

Vaquez: Biologie ( 1)
NFS • HB ,GR,HT augmentées• Souvent Thrombocytose• +/- Hyperleucocytose modérée à PN

Biologie (2)
• Biopsie osteomédullaire : montre une hyperplasie myéloïde
• Etude du volume GT: > 36 ml/kg H et > 32 ml /kg ( > 25 % à la valeur théorique
en fonction du poids et de la taille )

Diagnostic
• SPM• Hb, Ht, Gr augmentés • VGT > 36 ml/ kg ou > 32 ml/kg• Eliminer les Polyglobulies
Secondaires(SaO2 > 95%,pas de causes rénales….)
• JAK2 V617F positive (>80%) ++++++

Polyglobulie: Dg différentiel
• Réactionnelle– Adaptée (Erythropoïétine en réaction à un
stimulus hypoxie)/ BPCO – Tabagisme – CO– Inadaptée(Sécrétion anormale Erythropoïétine)/
Tumeurs rénales – Tumeurs Hépatiques – Tumeurs cervelet

Vaquez: Complications
• Thromboses (Risque le +important): veineuses ou artérielles.
• Hémorragies(anomalies des plaq)• Crise de Goutte et lithiases urinaires • Evolution:
– Myelofibrose (30%) Pancytopénie– Acutisation (LA: 10-15%) très péjoratif.

Vaquez: Traitements
• Étiologique:– Saignées (Ht <45%). Modalités ++Risque =
carence en fer et hyperplaquettose. Risque thrombotique
– Hydrea°: Pb = Tt continu. (Attention cytopénie)– Phosphore32( ++ Sujets âgés)
• Symptomatique:– Aspirine– Allopurinol
Thrombocytémie essentielle:Définition-Épidémiologie
• Prolifération clonale d’un précurseur myéloïde intéressant la lignée mégacaryocytaire essentiellement et aboutissant (majoritairement) à la production excessive de plaquettes.
• Incidence: 1 à 2/100.000• Pic: 60 ans• Quelques formes: Femmes 30ans

Thrombocytémie essentielle:Signes cliniques
• Très souvent asymptomatique (NFS systématique)
• Thromboses vasculaires • Hémorragies • Signe Clinique: Splénomégalie

Thrombocytémie essentielle:Signes Biologiques
• Thrombocytose > 600.000/mm3• Biopsie médullaire: hyperplasie myéloïde
avec prédominance mégacaryocytaire
• Attention: LMC atypique!! BCR-ABL de façon systématique

Diagnostics différentiels• Thrombocytose réactionnelle:
– Carence en Fer– Régénération médullaire– Post-splénectomie
• Thrombocytose d’accompagnement (autre syndrome myéloprolifératif)

Evolution-complication
• Risque majeur +++ Thrombose• Risque d’hémorragie++• L’évolution vers la myélofibrose ou une LA

Thrombocytémie essentielle:Thérapeutique
• Limiter les complications thrombotiques:– Anti-agrégants plaquettaires : aspirine 100
mg/j– Hydrea si Plq++++

Autres syndromes myéloprolifératifs

Myélofibrose primitive:
• Fibrose médullaire responsable d’une pancytopénie avec relais par une hématopoïèse splénique.
• Rare. Sujets âgés. Exceptionnel mais existe chez sujets jeunes.
• Risque: Evolution vers Leucémie aiguë.• TRT : sujets jeunes : greffe allogénique• TRT : sujets agés : transfusion + ??


















