
Magazyn TÜV Rheinland Polska 4/2016 ISSN 2299-6249 · ryzyka w biznesie. Jacek Sadowski, Kierownik...
32
Magazyn TÜV Rheinland Polska 4/2016 ISSN 2299-6249 Jakosc Zarządzanie ryzykiem w biznesie Temat Numeru: Czy zarządzanie ryzykiem jest trudne? Technologie i biznes: 06 Proces zarządzania ryzykiem w branży wyrobów medycznych Bezpieczenstwo: 14 Rola ludzi w systemach zarządzania – dyskusja praktyków Wiedza i praktyka: 16
Transcript of Magazyn TÜV Rheinland Polska 4/2016 ISSN 2299-6249 · ryzyka w biznesie. Jacek Sadowski, Kierownik...

Magazyn TÜV Rheinland Polska 4/2016ISSN 2299-6249
Jakosc
Zarządzanie ryzykiem
w biznesie
Temat Numeru:
Czy zarządzanie ryzykiem jest trudne?
Technologie i biznes:
06 Proces zarządzania ryzykiemw branży wyrobów medycznych
Bezpieczenstwo:
14 Rola ludzi w systemach zarządzania – dyskusja praktyków
Wiedza i praktyka:
16

„Statek w porcie jest bezpieczny, ale nie po to buduje się statki” to
zdanie Johna A. Shed’a pochodzi z książki wydanej w 1928 roku.
Trzeba przyznać, że przez lata nie straciło na aktualności. Ta metafora
wydaje się szczególnie trafna w kontekście prowadzenia działalności
biznesowej. Stając za sterem firmy, trzeba mieć świadomość
możliwości wystąpienia sztormów i mielizn. Innymi słowy, zanim
wyruszy się w rejs, trzeba oszacować ryzyko.
Im bardziej skomplikowany lub nietypowy wyrób
lub im większe oczekiwania i potrzeby użytkowników
czy klientów, tym większe ryzyko pojawia się
w procesie cyklu życia takiego produktu. W artykule
odpowiadamy na pytanie, jak zminimalizować ryzyko
w projektowaniu, produkcji i sprzedaży wyrobów.
Dzielenie się wiedzą i doświadczeniami z codziennej
pracy związanej z systemami zarządzania było
głównym celem konferencji zorganizowanej tej
jesieni w Krakowie. Dyskusja panelowa prelegentów
przerodziła się w prawdziwe forum wymiany
pozytywnych i negatywnych doświadczeń
na temat roli ludzi w systemach zarządzania.
6
20
Słowo wstępne
WYDAWCA: TÜV Rheinland Polska Sp. z o.o.
ul. Wolności 327, 41-800 Zabrze
tel. 32 271 64 89 w.105 fax. 32 271 64 88
REDAKCJA: Agata Tynka, e-mail: [email protected]
Na okładce: Jacek Sadowski, Kierownik Sekcji Certyfikacji Systemów
Zarządzania, TÜV Rheinland Polska., Fot. Tomasz Grajek
Źródła zdjęć: Shutterstock, Picjumbo, Thinkstock, źródła własne.
SKŁAD I GRAFIKA:
Tomasz Grajek
TÜV Rheinland Polska
KOREKTA:
Joanna Owczarczyk,
Anna Banach
TÜV Rheinland Polska
W tym numerze Jakości sporo miejsca poświęcamy tematowi
ryzyka w biznesie. Jacek Sadowski, Kierownik Sekcji Certyfikacji
Systemów Zarządzania TÜV Rheinland Polska, odnosząc się do
norm ISO, odpowiada na pytania redakcji dotyczące podejścia
do ryzyka w różnych branżach.
Poza rozwiązaniami systemowymi warto także rozważyć metody
minimalizowania ryzyka poprzez badania i certyfikację wyrobów.
Pisze o tym Milena Zielińska, Kierownik ds. Rozwoju Certyfikacji
Wyrobów TÜV Rheinland Polska, zwracając uwagę na grupy
wyrobów o podwyższonym ryzyku. Taką szczególną grupą są
wyroby medyczne, regulacjom na tym rynku przygląda się
nasz ekspert Maciej Ściera, Kierownik Produktu INMETRO.
Kwestie dotyczące zarządzania ryzykiem to poza procesami
i wymaganiami prawnymi także ludzie. O ich szczególnej roli
rozmawiali praktycy systemów zarządzania w trakcie jesiennego
Forum wymiany doświadczeń. Dla osób, które nie miały okazji
przysłuchiwać się dyskusji panelowej, zamieszczamy obszerną
relację z debaty.
Korzystając z okazji, pragnę życzyć Państwu realizacji
noworocznych postanowień i bezproblemowej drogi
do ich osiągnięcia.
Zapraszam i życzę miłej lektury
Janusz Grabka
Prezes Zarządu TÜV Rheinland Polska

4 Molex Sulęcin – 10 lat ciągłych zmian dzięki Lean Six Sigma
6 Czy zarządzanie ryzykiem jest trudne?
14 Proces zarządzania ryzykiem w branży wyrobów medycznych
20 Jak zminimalizować ryzyko w projektowaniu, produkcji i sprzedaży wyrobów?
10 Norma ISO 9001:2015 oczami audytora
16 Rola ludzi w systemach zarządzania – dyskusja praktyków
22 Zmiany w prawie odnośnie ochrony danych osobowych – obowiązki przedsiębiorców
25 Z pamiętnika audytora:Ryzyko w biznesie
26 Zmienia się ISO/TS – standard zarządzania jakością w przemyśle motoryzacyjnym
Technologie i biznes
Bezpieczeństwo
Wiedza i praktyka
Nowe rozporządzenie UE nr 2016/679 z 27 kwietnia 2016
roku (RODO UE ) w sprawie ochrony danych osobowych
reguluje m.in. zasady ochrony przetwarzanych w firmach
zbiorów tych danych. W Polsce trwają prace legislacyjne
nad ujednoliceniem rozwiązań instytucjonalnych.
W praktyce do zmiany jest około 800 aktów prawnych.
Jak podejść do tematu świadomego zarządzania ryzykiem
według nowych wytycznych norm ISO 9001 i 14001 i czy
słusznie temat budzi obawy. Do rozmowy zaprosiliśmy
Jacka Sadowskiego, Kierownika Sekcji Certyfikacji Systemów
Zarządzania TÜV Rheinland Polska.
24
20
News
24 Jak pomóc klientowi dokonać właściwego wyboru produktu?
24 TÜV Rheinland Polska laureatem programu Inwestor w Kapitał Ludzki 2016
!! !

Technologie i biznes
4
Molex Sulęcin – 10 lat ciągłych zmian dzięki Lean Six SigmaMolex jest jednym z największych światowych producentów złączy i akcesoriów połączeniowych. Innowacyjne rozwiązania elektroniczne oferowane przez firmę znajdują zastosowanie w wielu branżach, w tym w elektronice użytkowej, przemysłowej, motoryzacyjnej, samochodowej i medycznej. O tym, jak wyglądają procesy produkcyjne w polskim oddziale Molex rozmawiamy z Piotrem Goryńskim, Continuous Improvement Leader Black Belt w Molex Sulęcin.
Proszę krótko opowiedzieć, czym zajmuje się polski oddział firmy Molex. Jak duży jest zakład w Sulęcinie?
Zajmujemy się produkcją wiązek kablowych, głównie dla branży automotive i przemysłowej. Poziom zatrudnienia to obecnie około 1000 pracowników.
W trakcie listopadowego Forum wymiany doświadczeń, zorganizowanego przez TÜV Rheinland Polska opowiedział Pan o doświadczeniach związanych z prowadzeniem projektów Lean Six Sigma. To już blisko 10 lat ciągłych zmian, jak podsumowałby Pan ten czas?
Był to czas zdobywania doświadczenia
oraz wiedzy i jej implementacji przez cały zespół. Było i jest wciąż sporo wyzwań, jednakże radzimy sobie z nimi coraz lepiej. Cały proces Global Lean Six Sigma ma się bardzo dobrze. Przez niespełna 10 lat usprawniliśmy nasze procesy produkcyjne i administracyjne, zarówno pod względem poprawy jakości, jak i produktywności. Oszczędności sięgnęły powyżej 10

Technologie i biznes
5
milionów USD. Najważniejsza jest jednak dla nas świadomość i wiedza pracowników dotycząca 5S i metod rozwiązywania problemów. Co za tym idzie – wszelkie pomysły na usprawnienia wychodzą od nich samych. Ważne jest, aby stworzyć takie środowisko i kulturę, które będą angażować do proponowania zmian na lepsze. Nie jest to proces, który można wykreować w jeden miesiąc, czy rok – potrzeba na to zdecydowanie dłuższego czasu.
Na czym polega sekret sukcesu projektów GLSS w Molex Sulęcin?
Nie uważam, że GLSS w Molex Sulęcin ma jakiś sekret – jest to raczej ciężka praca całego zespołu pracowników oraz wsparcie kierownictwa. Wsparcie kierownictwa dla procesu GLSS ma fundamenty w wiedzy. Każdy z kierowników jest Green Belt’em lub Lean Belt’em. Także każdy jest przeszkolonym Championem. Także zrozumienie Lean Six Sigma na każdym poziomie jest jego esencją. Warto równierz dodać , że liderzy produkcji, magazynu oraz działu utrzymania ruchu oraz pracownicy administracji są przeszkoleni z zakresu
podstawowych narzędzi służących do rozwiązywania problemów (Yellow Belt).
Jakie bariery trzeba było pokonać, aby ten sukces był możliwy? Z czym muszą się liczyć firmy, które planują wdrożenie w swoich zakładach projektów Lean?
Na początku myślałem, że same szkolenia i pozyskana wiedza wystarczą. Jednak z perspektywy czasu wiem, że głównym czynnikiem sukcesu jest respekt dla wszystkich pracowników oraz zaangażowanie głównego kierownictwa. Dodając do tego wiedzę Lean oraz Six Sigma, mamy prawie gotową receptę na sukces. Prawie, bo dochodzą jeszcze chęci do przeprowadzania usprawnień i gotowość na krytykę stanu zastanego i po samych usprawnieniach, bo to już jest stan zastany. Pytamy ponownie (PDCA), jak możemy to zrobić lepiej i szybciej.
W firmie poza Leanem funkcjonują certyfikowane systemy zarządzania ISO 9001, ISO 14001, ISO TS 16949. Jak w praktyce wygląda równoległe funkcjonowanie tych rozwiązań?
W naszej firmie rozwiązania te się uzupełniają, można powiedzieć, że żyją w symbiozie. Mamy jedną wspólną księgę jakości, wspólne procedury. Audyty systemu i procesu są przeprowadzane wspólnie z GLSS. Każdy audytor jest Green/Lean Belt’em i w pełni uczestniczy w analizie przyczyn niezgodności. Opracowują oni także działania zapobiegawcze i korygujące. Cele oraz projekty, które wspomagają osiągniecie tych celów, są projektami Lean Six Sigma, np. projekty jakościowe, redukcji złomu, a także zużycia energii, sprężonego powietrza i innych zasobów, co powiązane jest z celami środowiskowymi. Wyniki audytów procesów są źródłem inspiracji, jak i gotowymi tematami dla kolejnych projektów Lean Six Sigma. Mapowanie procesów także przebiega zgodnie z VDA, TS. Warto nadmienić również, że spełnianie celów ma wymierny wpływ na oszczędności naszej organizacji. Wyniki projektów Lean Six Sigma mają wpływ nie tylko na dany proces, ale są wdrażane na inne jako dobra praktyka.

Technologie i biznes
6
Czy zarządzanie ryzykiem jest trudne?Większość osób związanych zawodowo z normami ISO słyszała o zmianach w standardach ISO 9001 oraz ISO 14001. Stare wersje tych norm stracą ważność 15 września 2018 roku, już teraz pojawiają się jednak pytania, jak podejść do tematu świadomego zarządzania ryzykiem według nowych wytycznych. Do rozmowy na ten temat zaprosiliśmy Jacka Sadowskiego, Kierownika Sekcji Certyfikacji Systemów Zarządzania TÜV Rheinland Polska.
Dwie najpopularniejsze normy ISO 9001 oraz ISO 14001 w najnowszych wersjach, przetłumaczonych na język polski kilka miesięcy temu, nakładają na organizacje obowiązek szacowania zagrożeń dla zaplanowanych procesów biznesowych. Teoretycznie to wymaganie nie jest niczym nowym, czy firmy są na nie gotowe? Czy świadome zarządzanie ryzykiem jest trudne?
Położyłbym tutaj nacisk na „świadome” zarządzanie ryzykiem, konieczność rozważenia zagrożeń, aby ograniczyć możliwy wpływ „nieoczekiwanego”. Każdy menedżer prowadzący jakąkolwiek działalność w jakiś sposób bierze pod uwagę zagrożenia, które mogą zaburzyć funkcjonowanie jego biznesu. U niektórych proces ten odbywa się gdzieś z tyłu głowy.
Ryzyko definiowane jest jako wpływ niepewności na cele. Kluczowe zatem jest rozważenie, co i w jaki sposób organizacja chce osiągnąć prowadząc biznes, w jakim otoczeniu funkcjonuje, co wpływa na jej działalność i jak ona oddziałuje na świat wokół niej. Wszystko to nazywa się kontekstem organizacyjnym. Właściwe przemyślenie tych kwestii pozwala na określenie większości zagrożeń, z którymi firma może mieć do czynienia, pomaga wybrać te najistotniejsze (najczęściej z punktu widzenia prawdopodobieństwa zdarzenia i potencjalnych skutków), dla których powinny zostać przewidziane działania prewencyjne.
Zapobieganie zidentyfikowanym ryzykom buduje stabilną sytuację organizacji na rynku i zwiększa zaufanie do niej. Powoduje, że zarządzający potrafią przewidywać trendy, zamiast podążać za innymi.
Świadome zarządzanie ryzykiem nie jest więc trudne, ale powinno być prowadzone
systemowo, aby jego wyniki były jak najlepiej dostosowane do działalności organizacji.
Wspomniane zmiany w normach ISO wprowadzają jednakowe definicje i układ dla wszystkich systemów zarządzania. Czy można się zatem spodziewać, że każda nowelizowana norma ISO będzie w podobny sposób regulowała kwestie ryzyka? Jak to wygląda obecnie, np. w przypadku ISO 27001 czy ISO 50001?
Analizowanie ryzyk, zarządzanie nimi oraz podejmowanie działań w celu minimalizacji najważniejszych z nich jest nieodłącznym elementem świadomego zarządzania. Trudno więc się dziwić, że elementy te pojawiają się również w standardach opisujących wymagania dla poszczególnych systemów zarządzania.
W niektórych z nich elementy te widoczne były od początku. Tak było w przypadku systemu zarządzania bezpieczeństwem i higieną pracy, w którym opisywane były wymagania dla zarządzania ryzykiem związanym z wykonywaną pracą. Podobnie w systemie zarządzania bezpieczeństwem informacji rozważane są ryzyka i zabezpieczenia związane z aktywami informacyjnymi. W systemie zarządzania bezpieczeństwem żywności natomiast jest mowa o identyfikacji zagrożeń jakości zdrowotnej żywności.
Wymagania dotyczące analizowania ryzyka i zapobiegania mu pojawiły się również w nowych wydaniach norm opisujących systemy zarządzania jakością i środowiskiem (działania związane z ryzykiem zastąpiły wcześniej opisywane działania zapobiegawcze). Należy się spodziewać, że wymagania dotyczące analizowania ryzyka i działań ukierunkowanych na nie będą pojawiały się również w kolejnych nowelizowanych standardach.

Technologie i biznes
7

Technologie i biznes
8
Eksperci jednostki TÜV Rheinland Polska mają styczność z przedsiębiorstwami z różnych branż, od przemysłu spożywczego przez producentów chemii gospodarczej, po przemysł ciężki. Szczególnie w przypadku wyrobów konsumenckich trzeba brać pod uwagę zagrożenie dla zdrowia i życia użytkowników. Proszę powiedzieć, jak ten temat jest regulowany przez standardy branżowe i jak te rozwiązania mają się do wytycznych nowej wersji ISO 9001.
Norma ISO 9001 mówi o „myśleniu opartym na ryzyku”. O tym, że należy zastanowić się nad zagrożeniami, które mogą wystąpić w zaprojektowanych procesach i w uzasadnionych przypadkach podjąć działania, aby im przeciwdziałać. ISO 9001 jest standardem ogólnym – może ją wdrożyć każda organizacja niezależnie od działalności, jaką prowadzi. Stąd wynika również ogólność rozwiązań w niej opisanych.
W wielu dziedzinach mamy jednak normy bardziej szczegółowe (często oparte na wymaganiach ISO 9001), dostosowane do specyfiki poszczególnych sektorów. Takimi normami są np. norma dla producentów wyrobów medycznych ISO 13485, czy normy w zakresie bezpieczeństwa żywności. W standardach tych często obowiązują bardziej szczegółowe wymagania dotyczące zarządzania ryzykiem i działań podejmowanych na podstawie analizy ryzyka.
Jak podejście do zarządzania ryzykiem różni się w zależności od specyfiki organizacji? Czy, co do zasady, zarządzanie ryzykiem w służbie zdrowia różni się od zarządzania ryzykiem w przedsiębiorstwie produkcyjnym? Pytam o podejście systemowe i metodologię wdrożenia.
Wszystko zależy od kontekstu organizacji. Co i dla kogo robi, w jakim otoczeniu funkcjonuje, jakie wymagania prawne i inne regulują jej działalność. Rozważenie tych kwestii pozwala na zaprojektowanie odpowiednich procesów tak, aby dostarczane usługi czy wyroby odpowiadały wymaganiom klientów. Pozwala to również na zidentyfikowanie zagrożeń, które mogą te procesy zaburzyć i spowodować, że w efekcie powstanie wyrób nie odpowiadający wymaganiom czy usługa, która nie spełnia przepisów prawnych. To jest ta część zarządzania ryzykiem, która musi być specyficzna dla każdej organizacji, bo każda działa w innym, specyficznym dla niej kontekście.

Technologie i biznes
9
Jacek Sadowski – Kierownik Sekcji Certyfikacji Systemów Zarządzania TÜV Rheinland Polska. Absolwent Wydziału Mechanicznego Politechniki Lubelskiej na kierunku Mechanika i Budowa Maszyn. Audytor wiodący systemów zarządzania jakością, środowiskowego, BHP i bezpieczeństwa informacji. Od 2005 roku pracownik TÜV Rheinland Polska odpowiedzialny za usługi certyfikacji systemów zarządzania.
Inne będą uwarunkowania działalności szpitala, który świadczy usługi medyczne w sposób sztywno określony wymaganiami przepisów, zależny każdorazowo od indywidualnego badania i diagnozy pacjenta. W wypadku szpitala działalność podlega kontrolom wielu instytucji. Inaczej będzie działał producent odkuwek, dla którego wymaganiami będą przede wszystkim specyfikacje klienta i normy techniczne.
Również kontekst dwóch organizacji wytwarzających to samo będzie się najczęściej różnił, ponieważ zwykle działają na różnym rynku lokalnych klientów i konkurentów, zatrudniają pracowników z lokalnego (różniącego się) rynku pracy, inne mogą być także miejscowe uregulowania prawne.
Kiedy każdy określi już na własne potrzeby odpowiednie procesy i zdarzenia, które mogą je zaburzyć, w następnym kroku należy określić wartości tych ryzyk. Najczęściej robi się to na podstawie prawdopodobieństwa wystąpienia potencjalnych zagrożeń i ich konsekwencji (tutaj należy zastanowić się np. nad możliwymi karami za niedotrzymanie wymagań, spowodowane zaburzeniem procesu, utratą korzyści, utratą zaufania). Następnie należy ocenić, które z tych ryzyk organizacja jest w stanie zaakceptować, a skutki których są zbyt wysokie, aby się z nimi pogodzić. Dla ryzyk zbyt wysokich trzeba zastanowić się nad działaniami, które pomogą te ryzyka zmniejszyć. Ta część zarządzania ryzykiem (jeżeli chodzi o sposób postępowania) jest w zasadzie niezależna od rodzaju organizacji.
Oczywiście sposoby zmniejszania ryzyka muszą być specyficzne – dostosowane do kontekstu organizacji i jej procesów. Ryzyko można minimalizować na wiele sposobów – m.in. przebudowując procesy (np. zwiększając kontrolę), podnosząc kwalifikacje pracowników, szukając innych dostawców czy wreszcie ubezpieczając się od konkretnych ryzyk. Ponieważ otoczenie zmienia się nieustannie, określone wcześniej procesy i zagrożenia
powinny być regularnie przeglądane, aby zidentyfikować pojawiające się ryzyka i przypisane im wartości.
Od roku 2009 istnieje standard ISO 31000, poświęcony procesom zarządzania ryzykiem. Czy może Pan ocenić jego popularność wśród polskich firm i organizacji? Jak Pan ocenia jego przydatność?
Norma ISO 31000 opisuje ogólne zasady i wytyczne zarządzania ryzykiem, postępowanie według schematu:1. Ustanowienie kontekstu zarządzania
ryzykiem2. Identyfikacja ryzyka3. Analiza ryzyka4. Wartościowanie ryzyka5. Opracowanie sposobów postępowania
z ryzykiem
Opisuje również ustanowienie struktury zarządzania ryzykiem, sposób komunikacji w zarządzaniu ryzykiem itp. Nie omawia natomiast, z uwagi na swoją ogólność, szczegółowych metodologii, jak ryzyko wartościować. A metodologii tych funkcjonuje wiele – jakościowych i ilościowych – głównie opartych na prawdopodobieństwie wystąpienia niepożądanych zdarzeń (materializacji zagrożeń) i ciężkości ich następstw. Metodologię każdy musi wybrać odpowiednią dla siebie, dostosowaną do kontekstu organizacji.
Norma ISO 31000 jest normą ogólną – tak, jak w przypadku systemu zarządzania jakością norma ISO 9001 – skierowaną do wszystkich organizacji. Jest w związku z tym tylko jednym z narzędzi zarządzania ryzykiem. W poszczególnych dziedzinach funkcjonują często inne narzędzia, dostosowane do specyfiki sektora. Zasady opisane w normie ISO 31000 należałoby potraktować jako zdroworozsądkową listę – jeżeli w zarządzaniu ryzykiem będziemy zgodnie z nimi postępować – wyniki analizy ryzyka powinny być wiarygodne, a zarządzanie ryzykiem powinno przynieść oczekiwane rezultaty.

Wiedza i praktyka
10
Norma ISO 9001:2015 oczami audytoraMinął już ponad rok od publikacji normy ISO 9001:2015, a od 6 lipca 2016 roku dostępne jest oficjalne polskie wydanie oznaczone jako PN-EN ISO 9001:2015-10. Póki co niewiele firm poddało się certyfikacji na nową normę. Może to wskazywać na problemy z interpretacją wymagań i wykorzystaniem ich w praktyce. A jak sprawa wygląda z perspektywy audytora wiodącego tej normy?
Zadziwiający był fakt bardzo dużej aktywności informacyjno-szkoleniowej na temat zmian w najnowszym wydaniu normy ISO 9001:2015 w okresie, kiedy jeszcze trwały prace nad jej projektami – wstępnym ISO/DIS 9001:2014 oraz ostatecznym, ale nadal tylko projektem, ISO/FDIS 9001:2015. Ta aktywność nie przełożyła się dotychczas na etap następny, czyli praktyczne dostosowanie obecnych systemów zarządzania jakością do wymagań normy ISO 9001:2015.
Problemy z interpretacjąOd momentu publikacji oficjalnego polskiego wydania ww. standardu na
palcach jednej ręki można policzyć firmy, które już poddały się certyfikacji na nową normę. Powyższa sytuacja wskazuje na to, że ostatnie wydanie jest najtrudniejsze do zrozumienia i wykorzystania w praktyce. Jeśli interpretacja nowych wymagań przez pełnomocników ds. jakości, konsultantów, trenerów i audytorów okaże się zgodna z założeniami ich autorów, a zawarte tam wymagania zostaną prawidłowo wdrożone i audytowane, wtedy system zarządzania jakością ma szansę stać się bardzo przydatnym firmie narzędziem. System ten będzie najsilniej powiązanym z prowadzoną działalnością gospodarczą, wtedy stwierdzenie, iż „system zarządzania
jakością jest wartościowym narzędziem w rękach najwyższego kierownictwa, które wesprze jego członków w rozwoju firmy”, przestanie być tylko pustym frazesem. Skoro założenia są tak dobre, to skąd się bierze ten duży opór przed zmianą?
Czy to już koniec ery pełnomocników?Jednym z powodów, dla których pełnomocnicy ds. jakości nie spieszą się z rozpoczęciem prac dostosowawczych może być obawa, że wdrożenie zmian będzie oznaczało koniec ich przygody z nadzorowaniem i doskonaleniem systemu w firmie. W nowej normie ISO 9001 nie ma już bowiem wymagania pt.

Wiedza i praktyka
11
„Przedstawiciel kierownictwa”. Nigel Croft – przewodniczący Komitetu Technicznego ISO/TC176 SC2 odpowiedzialnego za normę ISO 9001, podczas swojego wykładu na konferencji 10th National Quality Conclave, która odbyła się w Delhi w Indiach w dniach 7-8 sierpnia 2015 roku, wyjaśnia podjętą decyzję o usunięciu tego wymagania: „Zdaję sobie sprawę z tego, że w praktyce przedstawicielami kierownictwa często nie są osoby będące członkami kadry zarządzającej najwyższego szczebla, nie
mają dostępu do strategii firmy i jej planowanych kierunków rozwoju. Jeżeli system zarządzania jakością ma wynikać z kontekstu organizacji, tj. określenia nie tylko obecnych, ale i przyszłych klientów firmy wynikających ze strategicznych kierunków jej rozwoju oraz poznania ich wymagań, wyboru stron zainteresowanych, czynników zewnętrznych, a nie tylko wewnętrznych, to tylko najwyższe kierownictwo może ustalić, kto będzie odpowiedzialny za spełnienie wymagań z rozdziału 4 „Kontekst organizacji” i podejścia opartego na ryzyku. Dlatego uznaliśmy, że lepiej będzie usunąć z normy ISO
9001:2015 wymaganie odnoszące się do przedstawiciela kierownictwa i całą odpowiedzialność za system zarządzania jakością przekazać członkom kadry zarządzającej. Oni następnie będą musieli podjąć świadomą decyzję, czy całą odpowiedzialność za system zostawią w swoich rękach, czy przekażą ją dotychczasowym pełnomocnikom ds. jakości lub jeszcze innym osobom”. Jest o tym wzmianka w p. 5.1.1 j„…wspieranie innych właściwych członków kierownictwa w wykazywaniu
przywództwa w obszarach ich odpowiedzialności”.Dotychczasowe wymagania dla przedstawiciela kierownictwa nie zniknęły zupełnie. Zmieniły tylko swoje miejsce. Teraz są zamieszczone w dwóch wymaganiach: 5.3 „Role, odpowiedzialność i uprawnienia w organizacji” oraz p. 5.1 „Przywództwo i zaangażowanie” i 5.1.1 „Postanowienia ogólne”.
Zamieszczono tam wymagania odnoszące się stricte do najwyższego kierownictwa („zapewnienie dostępności zasobów potrzebnych w systemie zarządzania
jakością”) i pełnomocnika ds. jakości („komunikowanie znaczenia skutecznego zarządzania jakością i zgodności z wymaganiami systemu zarządzania jakością”), ale również do obu wyżej wymienionych stron („zapewnienie zintegrowania wymagań systemu zarządzania jakością z procesami biznesowymi organizacji”, czy „zapewnienie ustanowienia polityki jakości i celów jakościowych dla systemu zarządzania jakością i ich zgodności z kontekstem oraz strategicznym kierunkiem organizacji”).
Dobrze będzie ustalić, kto ma być odpowiedzialny w firmie za spełnianie poszczególnych podpunktów powyżej wspomnianych wymagań. Potrzebna będzie rozmowa dotychczasowego pełnomocnika ds. jakości z członkami kadry zarządzającej firmą na temat podziału odpowiedzialności za system i ustalenie, w jaki sposób poszczególne wymagania zawarte w p. 5.1.1 i 5.3 mają być spełniane. Bez tego kroku nie będzie można określić zakresu obowiązków i odpowiedzialności dla pełnomocnika.
To, czy zakres zostanie bez zmian, czy ulegnie modyfikacji, będzie zależało od dotychczasowego dostępu do informacji o celach biznesowych i strategii. Wspomniane spotkanie okaże się cenne również dlatego, że członkowie najwyższego kierownictwa mogą uświadomić sobie, iż nieprzekazywanie pełnomocnikowi ds. jakości tych informacji może spowodować poważne konsekwencje dla wyników certyfikacji. Audytor będzie mógł bowiem stwierdzić niezgodność z wymaganiem 4.1 „Zrozumienie organizacji i jej kontekstu”. Takie działanie ma na celu zapobieganie powtórzeniu się sytuacji, gdy system zarządzania jakością był oderwany od biznesowej rzeczywistości i żył swoim własnym, raczej nikomu niepotrzebnym, życiem.
Pełnomocnicy ds. jakości stoją teraz przed dużą szansą na nowe, ciekawsze wyzwania, zostania partnerami dla kadry zarządzającej i osobami współodpowiedzialnymi za rozwój firm, dla których pracują.
Może pojawić się pokusa, by system zarządzania jakością dostosować pod wymagania audytora. Takie założenie jest dużym błędem. Może spowodować, że osoby odpowiedzialne za system zarządzania jakością utracą z pola widzenia to, co najważniejsze, czyli klientów
Wiedza i praktyka
12
firmy i strategiczne kierunki jej rozwoju. Właśnie spełnianiu ich wymagań i celów podporządkowany ma być cały system zarządzania jakością, każdy proces, każda wykonywana czynność, każdy dokument i zapis.
Klient i jego wymagania Główne przesłanie normy ISO 9001 nie zmieniło się od jej pierwszego wydania w 1987 roku, tj. poprzez system zarządzania jakością spełniać wymagania klientów obecnych i przyszłych. W wersji z 2015 roku wprowadzono dodatkowe wzmocnienia w tych elementach normy ISO 9001, które dotychczas, szczególnie od wydania z 2000 roku, nie były spełniane w firmach w taki sposób, jakiego oczekiwali autorzy tej normy. Praktyka pokazała, że od roku 2000 wspomniana istota normy ISO 9001 gdzieś się zagubiła.
Wprowadzenie podejścia procesowego tylko połowicznie spełniło swoje zadanie. Umożliwiło firmom handlowym i tym świadczącym usługi łatwiejsze korzystanie z tego standardu. Jednak w wielu firmach tak bardzo skoncentrowano się na procesach, które funkcjonują w ich wnętrzu, że stracono z pola widzenia powód projektowania tych procesów, tj. spełnianie wymagań klientów.
W 2008 roku wprowadzono do normy ISO 9001 bardzo ważną zmianę – wszędzie tam, gdzie dotychczas w tekście wymagań znajdowało się słowo „jakość”, zamieniono je na sformułowanie „zgodność z wymaganiami dotyczącymi wyrobu”. Wykorzystano do tego definicję „jakości” Philipa Crosby’ego – „jakość to zgodność
z wymaganiami”. Było to „światełko w tunelu”, które jednak do tej pory z tego tunelu nie wyszło. Konieczne zatem okazało się wprowadzenie większych zmian, aby skierować uwagę użytkowników tej normy na właściwe tory.
Najważniejsze wymagania nowej normy Normę ISO 9001 warto więc traktować jako zbiór „dobrych praktyk”, które pomogą firmie rozwijać się w sposób zrównoważony. W jej trzech ostatnich wydaniach znajdziemy niezmiennie te same fragmenty, które odnoszą się do roli klienta firmy i spełniania jego wymagań, poprzez sprzedawane mu wyroby i usługi oraz miejsce wymagań normy ISO 9001 w takim zestawieniu. W ISO 9001:2015 są to następujące wymagania: p. 1 „Zakres normy” i p. 0.1 „Wprowadzenie”.
Kontekst organizacjiCzytając wymagania normy ISO 9001:2015, od rozdziału 4 trudno znaleźć fragment tekstu, który brzmiałby znajomo. Pewnie dlatego jedyne zmiany, które wielu zauważa to brak wymagania dla przedstawiciela kierownictwa, brak osobnego wymagania dla księgi jakości, nadzoru nad dokumentami i nadzoru nad zapisami.
Kolejną trudnością, na jaką można się natknąć w kontakcie z normą ISO 9001:2015, jest pojawienie się całego nowego rozdziału 4. „Kontekst organizacji”, a szczególnie pierwszego zdania z p. 4.1 „Zrozumienie organizacji i jej kontekstu”.
Być może dlatego część pełnomocników
ds. jakości, chcąc ułatwić sobie zadanie, chce tylko wiedzieć: „Co musimy zmienić w swojej dokumentacji SZJ, żeby dostosować SZJ do normy ISO 9001:2015?”.
Na co wskazuje tak postawione pytanie w odniesieniu do dostosowania systemu zarządzania jakością do wymagań normy ISO 9001:2015? Osoby, które je zadają nie mają jeszcze wyrobionego biznesowego nastawienia do systemu zarządzania jakością. Na tym etapie są jeszcze bardziej nastawione na procesy wewnątrz firmy i odnoszącą się do nich dokumentację systemową. Powyższe pytanie oczywiście w pewnym momencie padnie. Tylko zanim to nastąpi, trzeba będzie wykonać kilka kroków.
Krokiem, od którego należy zacząć proces dostosowawczy jest właściwe zrozumienie pierwszego zdania w p. 4.1. Mając w pamięci, że celem systemu zarządzania jakością jest spełnianie wymagań klienta, to zadanie staje się znacznie prostsze do zrozumienia. Zadania do wykonania są następujące:1. Na podstawie celu istnienia
organizacji określamy obecnych klientów firmy.
2. Na podstawie strategicznych kierunków rozwoju określamy potencjalnych klientów firmy.
3. Poznajemy wymagania, potrzeby i oczekiwania klientów obecnych i potencjalnych.
Wymagania klientów stanowią solidną podstawę do tworzenia systemu zarządzania jakością, w tym do projektowania lub modyfikacji procesów. Jeżeli pełnomocnik ds. jakości nie jest członkiem najwyższego kierownictwa, to może nie znać strategii firmy. Zna za to cel istnienia organizacji. Chcąc spełniać wymagania z rozdziału 4, może wybrać jedną z dwóch opcji:a) bazuje na celu istnienia organizacji,
tj. na obecnych klientach; po określeniu czynników wewnętrznych i zewnętrznych, które mogą wpływać na spełnienie przez firmę wymagań tych klientów oraz zidentyfikowaniu ryzyk i szans, prosi członków kadry zarządzającej o strategiczne kierunki rozwoju; następnie w oparciu o nie przeprowadza drugi raz analizy dla przyszłych klientów,
b) od razu przeprowadza analizy dlaobecnych i przyszłych klientów firmy.Wybór opcji zależy od sytuacji w danej firmie. Zalecana jest opcja a).
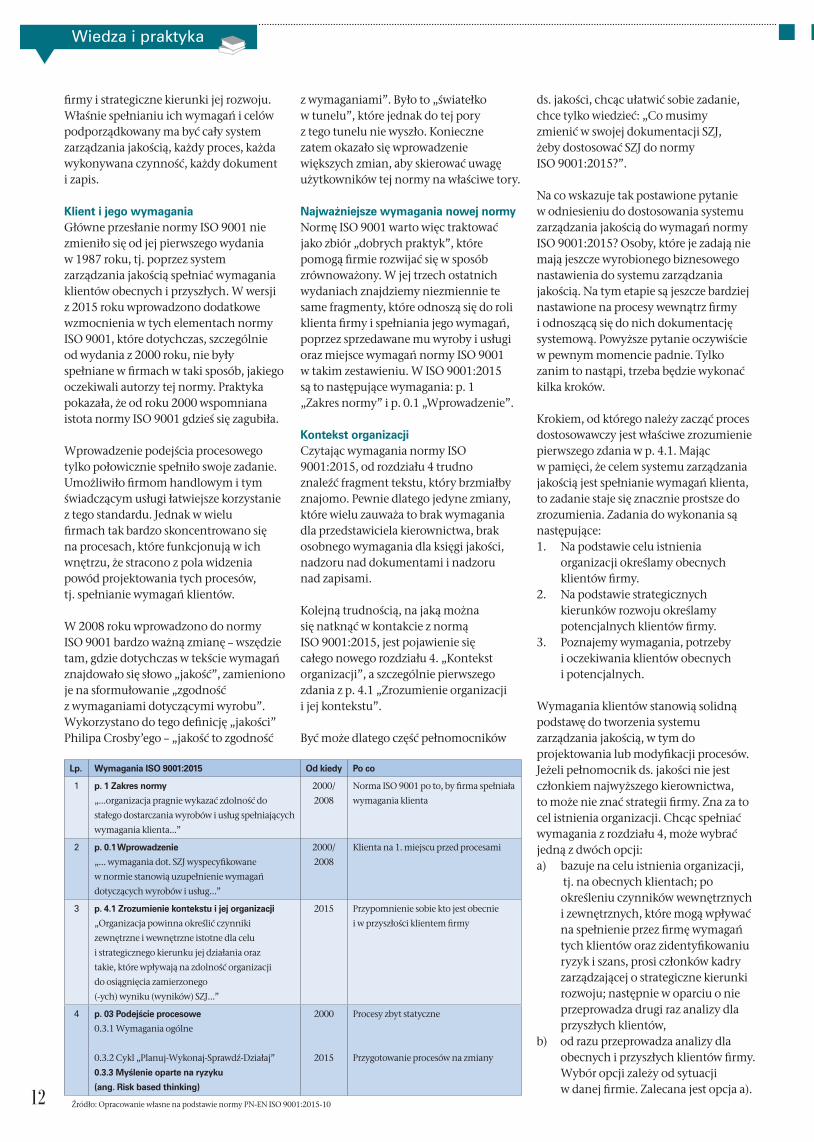
Lp. Wymagania ISO 9001:2015 Od kiedy Po co
1 p. 1 Zakres normy
„...organizacja pragnie wykazać zdolność do
stałego dostarczania wyrobów i usług spełniających
wymagania klienta...”
2000/
2008
Norma ISO 9001 po to, by firma spełniała
wymagania klienta
2 p. 0.1 Wprowadzenie
„... wymagania dot. SZJ wyspecyfikowane
w normie stanowią uzupełnienie wymagań
dotyczących wyrobów i usług...”
2000/
2008
Klienta na 1. miejscu przed procesami
3 p. 4.1 Zrozumienie kontekstu i jej organizacji
„Organizacja powinna określić czynniki
zewnętrzne i wewnętrzne istotne dla celu
i strategicznego kierunku jej działania oraz
takie, które wpływają na zdolność organizacji
do osiągnięcia zamierzonego
(-ych) wyniku (wyników) SZJ...”
2015 Przypomnienie sobie kto jest obecnie
i w przyszłości klientem firmy
4 p. 03 Podejście procesowe
0.3.1 Wymagania ogólne
0.3.2 Cykl „Planuj-Wykonaj-Sprawdź-Działaj”
0.3.3 Myślenie oparte na ryzyku
(ang. Risk based thinking)
2000
2015
Procesy zbyt statyczne
Przygotowanie procesów na zmiany
Źródło: Opracowanie własne na podstawie normy PN-EN ISO 9001:2015-10

Wiedza i praktyka
13
– jednego z guru jakości, wiele lat temu, mają być odtąd zarządzane zarówno poszczególne procesy, jak i interakcje pomiędzy nimi jako system. Z kolei według koncepcji „podejścia opartego na ryzyku”, jak wyjaśnia Nigel Croft, nie wszystkie realizowane procesy mają dokładnie taki sam wpływ na zdolność organizacji do dostarczania na rynek takich wyrobów i usług, które na pewno będą zgodne z odnoszącymi się do nich wymaganiami. Należy spośród wszystkich procesów wybrać te, które mają największy wpływ na tę zdolność. Kryterium wyboru stanowią zidentyfikowane ryzyka. Procesom uznanym za istotne trzeba będzie poświęcić więcej uwagi, by zapobiegać występowaniu problemów, które mogą wpłynąć na jakość wyrobu gotowego i usługę. Więcej uwagi może oznaczać więcej punktów kontroli, więcej środków nadzoru, więcej zasobów, w tym informacji i dokumentów czy zapisów. Cyklowi PDCA została podporządkowana również struktura normy ISO 9001:2015, jak przedstawiono na poniższym rysunku.
Rysunek 4 Struktura normy ISO 9001:2015 w cyklu PDCA
Bibliografia:
1. Norma PN-EN ISO 9001:2015-10 „Systemy zarządzania
jakością – Wymagania”
2. ISO: Discover the new ISO 9001:2015!, dostęp online
[https://www.youtube.com/watch?v=Lp6xP-We5yY]
3. Croft N.: wykład podczas konferencji 10th NATIONAL
QUALITY CONCLAVE, Delhi, Indie, 7-8. Sierpnia
2015, dostęp on line [https://www.youtube.com/
watch?v=Ar5BdhpwG_w]
4. Schmidt B.: Trendy – Zadowolenie klienta jest ważniejsze
od samego produktu; Raport specjalny Harward Business
Review Polska, listopad 2016
Aact
Pplan
Ccheck
Ddo
Doskonalenie
Kontekst organizacji PrzywództwoPlanowanie
Wsparcie
Działania operacyjneOcena efektów działania
Źródło: Opracowanie własne na podstawie normy PN-EN ISO 9001:2015-10
Beata Łuczak – Audytor wiodący ISO 9001. Od 14 lat pomaga firmom wdrażać i doskonalić systemy zarządzania jakością. Jest aktywnym uczestnikiem europejskiego programu „Uczenie się przez całe życie”. Autorka ponad 40 artykułów w czasopismach specjalistycznych. Wykładowca w licznych szkoleniach, seminariach i konferencjach krajowych i międzynarodowych z zakresu systemów zarządzania jakością i audytowania, m.in. podczas 20th ASQ Audit Division Conference w Reno, USA w 2011 roku. Absolwentka Wydziału Biologii specjalność Biologia molekularna na UAM w Poznaniu oraz Studium Podyplomowego z Marketingu i Zarządzania Jakością w Wyższej Szkole Bankowej w Poznaniu.
Ta zmiana nie dotyczy wyłącznie normy ISO 9001 i jej ostatniej rewizji. Wynika z opracowanej przez niezależną grupę roboczą w Międzynarodowej Organizacji Normalizacyjnej ISO, struktury wyższego rzędu nazwanej Anex SL. Ma ona poprawić stopień zgodności norm ISO dotyczących różnych systemów zarządzania.
Traf sprawił, że ta sama koncepcja, tj. cykl PDCA w normie ISO 9001:2015 występuje podwójnie – w odniesieniu do całego systemu zarządzania jakością i do jego poszczególnych procesów. Jednak ten traf może przynieść przyszłym systemom zarządzania jakością i firmom, w których będą funkcjonować wiele dobrego.
Dalsze postępowanie, aż do ustalenia koniecznych zmian w dokumentacji systemu zarządzania jakością, wynika z poszczególnych wymagań normy ISO 9001:2015. Obrazuje poniższy rysunek.
Trzy koncepcje, na których opiera się norma ISO 9001:2015Praktyka w ostatnich latach pokazała, że zarządzanie procesowe okazało się trafionym posunięciem. Jednak często procesy są zbyt statyczne niż wskazują na to zmiany zachodzące w ich otoczeniu zewnętrznym i wewnętrznym. Za rzadko wprowadzane są do nich modyfikacje. Dlatego ten element systemu zarządzania jakością został wzmocniony w normie ISO 9001:2015. W p. 0.3 „Podejście procesowe” dodano dwie koncepcje: cykl „Planuj – Wykonaj – Sprawdź – Działaj” oraz podejście oparte na ryzyku (ang. Risk-based thinking).
Zgodnie z cyklem PDCA, spopularyzowanym przez W.E. Deminga
Cel działania organizacji i strategiczne kierunki rozwoju:
obecni i przyszli klienci i ich wymagania (p. 4.1)
Czynniki zewnętrzne i wewnętrzne (p. 4.1)
Istotne strony zainteresowane i ich wymagania (p. 4.2)
Identyfikacja ryzyk i szans zewnętrznych i wewnętrznych
Działania minimalizujące ryzyko (p. 6.1)
Zmiany w procesach (p. 4.4)
Zmiany w dokumentach
Źródło: Opracowanie własne na podstawie normy PN-EN ISO 9001:2015-10

14
Bezpieczeństwo
Proces zarządzania ryzykiem w branży wyrobów medycznychRynek wyrobów medycznych jest szczególnym obszarem, gdzie produkowane wyroby mają wpływ na zdrowie i życie ludzi. Wytwórcy wyrobów medycznych, poza oceną ryzyka podejmowanych decyzji biznesowych, muszą również zarządzać ryzykiem związanym z samymi wyrobami medycznymi, wprowadzanymi przez nich na rynek.
Zarządzanie ryzykiem związanym z wyrobami medycznymi jest obowiązkiem wytwórcy. Taki obowiązek nakładają na niego Dyrektywa dla wyrobów medycznych 93/42/EWG, Dyrektywa dla wyrobów medycznych do diagnostyki in vitro 98/79/WE i Dyrektywa dla aktywnych implantowanych wyrobów medycznych 90/385/EWG.
Regulacje prawneNa polskim rynku wymagania dotyczące zarządzania ryzykiem zostały wprowadzone do prawa krajowego przez Ustawę o wyrobach medycznych z dnia 20 maja 2010 (z późniejszymi zmianami) oraz rozporządzenia w sprawie wymagań zasadniczych oraz procedur oceny zgodności dla wyrobów medycznych z dnia 12 stycznia 2011 (Dz.U.2011.16.74, Dz.U.2011.16.75, Dz.U.2011.16.76).
To, w jaki sposób zarządzać ryzykiem związanym z wyrobami, zostało szczegółowo opisane w normie PN-EN ISO 14971 „Wyroby medyczne – Zastosowanie zarządzania ryzykiem do wyrobów medycznych”. Norma ta jest zharmonizowana ze wspomnianymi wcześniej dyrektywami. Oznacza to, że zastosowanie jej pozwala domniemywać, iż proces zarządzania ryzykiem prowadzony według tej normy jest zgodny z wymaganiami dyrektyw.
Zarządzanie ryzykiem to proces ciągłyZarządzanie ryzykiem nie jest pojedynczym aktem analizy zidentyfikowanych zagrożeń związanych z stosowaniem wyrobu medycznego.
Zarządzanie ryzykiem jest procesem obejmującym cały cykl życia wyrobu medycznego.
Proces ten rozpoczyna się na etapie formułowania założeń dla projektowanego wyrobu medycznego. Na tym etapie powstają pierwsze informacje – do czego wyrób ma służyć, czyli jakie jest przeznaczenie danego wyrobu, ogólna zasada działania tego wyrobu oraz parametry. Niejednokrotnie dane te nie powstają „od zera”, często są zaczerpnięte z informacji o podobnych wyrobach dostępnych na rynku lub wcześniejszych projektach opracowanych przez firmę. Na tym etapie powstaje wstępna analiza ryzyka wyrobu. Na podstawie tych założeń można definiować pierwsze zagrożenia związane z charakterem wyrobu.
Załącznik E normy PN-EN ISO 14971, podpowiada wytwórcy przykłady zagrożeń związanych z zastosowanymi źródłami energii, cechami biologicznymi i chemicznymi wyrobów, oraz przykłady zagrożeń wynikających z niewłaściwego użycia wyrobu lub niedostatecznej informacji dołączonej do niego.
Dodatkowym źródłem informacji o potencjalnych zagrożeniach, jakie należy rozważyć w procesie zarządzania ryzykiem, są normy zharmonizowane mające zastosowanie do danej rodziny wyrobów medycznych, wśród nich na przykład norma PN-EN 62366-1 „Wyroby medyczne – Część 1: Zastosowanie inżynierii użyteczności do wyrobów medycznych”, norma PN-EN 62304 „Oprogramowanie wyrobów medycznych
– Procesy cyklu życia oprogramowania”, czy norma PN-EN 60601-1 „Medyczne urządzenia elektryczne – Część 1: Wymagania ogólne dotyczące bezpieczeństwa podstawowego oraz funkcjonowania zasadniczego” wraz z zestawem norm szczegółowych. O ile norma PN-EN ISO 14971 w sposób ogólny odnosi się do identyfikacji potencjalnych zagrożeń, o tyle we wspomnianych normach szczegółowych można znaleźć informacje, jakie zagadnienia należy ująć w zarządzaniu ryzykiem.
Ocena zagrożeń Wszystkie zagrożenia, które zostały uznane przez wytwórcę za prawdopodobne, muszą zostać ocenione. Zagrożenia można oceniać na wiele różnych sposobów. Należy tu wymienić kilka popularnych metod, wśród nich wstępną analizę zagrożeń PHA (Preliminary Hazard Analysis), analizę drzewa błędów FTA (Fault Tree Analysis), analizę rodzajów i skutków uszkodzeń FMEA (Fault Mode and Effect Analysis), analizę zagrożeń i zdolności do działania HAZOP (HAZard and OPerability Study) oraz analizę zagrożeń i krytyczne punkty kontroli HACCP (Hazard Analysis and Critical Control Points).
Najczęściej wykorzystywanymi przez wytwórców wyrobów medycznych są metody PHA oraz FMEA. Podczas oceny ryzyka, każdemu ryzyku przypisywana jest wartość, najczęściej oparta na dwóch wskaźnikach – prawdopodobieństwie wystąpienia awarii (probability) i stopniu szkody, jakie może spowodować awaria (severity). Wartości te pozwalają ocenić, jak wysokie jest ryzyko. Norma PN-EN ISO 14971 nie podaje, jaki poziom ryzyka jest akceptowalny, określenie kryteriów akceptowalności pozostawiono do zdefiniowania wytwórcom wyrobów medycznych.
Minimalizowanie zagrożeńAnalizując wymagania zasadnicze dyrektyw dotyczących wyrobów medycznych, można znaleźć zapis, że każde ryzyko należy sprowadzić do najniższego możliwego poziomu („As Low As Possible”) oraz, że każde ryzyko należy porównać z korzyściami wynikającymi ze stosowania danego rozwiązania lub wyrobu medycznego. Jak ocenić, czy ryzyko zostało sprowadzone do poziomu ALAP? Dobrym odniesieniem jest aktualny stan wiedzy i techniki („state of the art”).
Wyniki wstępnej analizy ryzyka mają posłużyć do rozwoju bezpiecznego wyrobu medycznego. Dla każdego zagrożenia,

15
Bezpieczeństwo
które nie zostało zaakceptowane, wytwórca ma obowiązek zastosować środki sterowania ryzykiem, których celem jest zredukowanie zagrożenia do poziomu tak niskiego, jak to możliwe. Środki te powinny być stosowane według ściśle określonego trzystopniowego algorytmu.
W pierwszej kolejności należy zapewnić inherentne bezpieczeństwo wyrobu medycznego, poprzez wyeliminowanie możliwych zagrożeń, bądź zmniejszenie prawdopodobieństwa wystąpienia, bądź
szkody w przypadku awarii. W tym celu możliwe jest wykorzystanie elementów o wyższej niezawodności lub na przykład zmniejszenie napięć stosowanych w urządzeniu medycznym do SELV (Safety Extra Low Voltage).
Kolejnym krokiem jest zastosowanie elementów zabezpieczających takich, jak osłony lub systemy alarmowe, które mają na celu zmniejszenie prawdopodobieństwa wystąpienia zagrożenia. Ostatnim środkiem sterowania ryzykiem jest poinformowanie użytkownika o ryzyku resztkowym, którego nie dało się wyeliminować. W tym celu na wyrobach medycznych umieszczane są różnego rodzaju graficzne symbole ostrzegawcze, natomiast w instrukcjach obsługi liczne ostrzeżenia.
Stosując kolejne środki sterowania ryzykiem, nie należy zapomnieć, że
wprowadzenie tych rozwiązań może prowadzić do powstania nowych zagrożeń. Takie zagrożenia również należy w procesie zarządzania ryzykiem zidentyfikować i ocenić.
Zakończeniem procesu projektowania jest zwolnienie wyrobu medycznego do produkcji. Aby mogło to nastąpić, wszystkie zidentyfikowane zagrożenia oraz ogólne ryzyko resztkowe musi być sprowadzone do poziomu akceptowalnego. Koniec etapu
projektowania nie wiąże się z zakończeniem procesu zarządzania ryzykiem. Jak już wspomniano, proces zarządzania ryzykiem obejmuje cały cykl życia wyrobu medycznego.
Nadzór po wprowadzeniu do obrotuWytwórca wyrobów medycznych ma obowiązek zbierać i analizować informacje o swoich wyrobach medycznych również po ich wyprodukowaniu i sprzedaży. Szczególnie istotne z punktu widzenia
zarządzania ryzykiem są informacje o awariach wyrobu medycznego (reklamacjach) oraz informacje o incydentach medycznych z udziałem wyrobu medycznego. Każdą taką informację, a w szczególności informacje o incydentach medycznych, należy rozważyć pod kątem potencjalnych nowych zagrożeń. W zakresie działań zapobiegawczych, wytwórca powinien wprowadzić zmiany w wyrobie lub jego dokumentacji towarzyszącej, mające na celu zapobiegnięcie powtórzeniu się
takiej awarii lub incydentu. Działania te sprowadzają się do analizy nowych zagrożeń i wprowadzeniu stosownych środków sterowania ryzykiem.
Wyniki i doświadczenia z zarządzania ryzykiem wyrobu medycznego nie są wykorzystywane tylko do tego jednego wyrobu. Dane te są cenną wskazówką podczas opracowywania kolejnych projektów, dzięki czemu każdy kolejny wyrób medyczny jest bardziej bezpieczny.
Maciej Ściera – Kierownik Produktu INMETRO TÜV Rheinland Polska. Przez sześć lat pracował jako konstruktor w Żywieckiej Fabryce Sprzętu Szpitalnego FAMED S.A. Z TÜV Rheinland Polska związany od 2009 roku. Członek Komitetu Technicznego 67 ds. Elektrycznej Aparatury Medycznej przy Polskim Komitecie Normalizacyjnym. Audytor wiodący, ekspert certyfikujący i audytor senior systemu zarządzania jakością wyrobów medycznych wg EN ISO 13845. Od 2014 roku zajmuje się koordynacją certyfikacji wyrobów medycznych wg brazylijskich wymagań INMETRO w Europie.

16
Wiedza i praktyka
Dzielenie się wiedzą i doświadczeniami z codziennej pracy związanej z systemami zarządzania, było głównym celem konferencji zorganizowanej tej jesieni w Krakowie przez TÜV Rheinland Polska. Do aktywnego udziału, poza ekspertami jednostki, zaproszone zostały osoby zajmujące się innowacjami, zarządzaniem według lean six sigma oraz pełnomocnicy systemów zarządzania według ISO. W pierwszym dniu konferencji zaplanowana została dyskusja panelowa prelegentów, która w efekcie przerodziła się w prawdziwe forum wymiany pozytywnych i negatywnych doświadczeń praktyków.
Andrzej Szastok: Pierwsze pytanie pozwolę sobie skierować do Grzegorza Grabki. Bazując na blisko dwudziestoletnim doświadczeniu jednostki w certyfikacji polskich firm, jak oceniacie wykorzystanie przez organizacje systemów zarządzania jakością do ich rozwoju i przystosowania do, wciąż zmieniającej się, rzeczywistości?
Grzegorz Grabka: Faktycznie, nasze doświadczenie to tysiące przeprowadzonych audytów, co pozwala wyciągnąć już konkretne wnioski. Wśród audytowanych firm, jak w każdej populacji, można wydzielić kilkanaście procent orłów, około 60% rzetelnych, około 15% tych z tyłu. Dla tych ostatnich certyfikaty są zwykle wymaganiem klienta. Do orłów zaliczam te organizacje, gdzie decyzja o wdrażaniu systemu była dobrze przemyślana przez kadrę zarządzającą. To są te firmy, w których dyrektor zarządzający postanawia, że wprowadza system zarządzania po to, po co został wymyślony, a więc do realizacji celów. Tutaj nie ma znaczenia, czy to narzędzie będzie oparte o lean czy o ISO 9001, jeśli zarządzający potrafi jasno określić cel. Jest też taka grupa organizacji, które zrobią wszystko, żeby spełnić wymagania wynikające ze standardu. Dla nich nie jest istotne zrozumienie przydatności tych wymagań. Istotne jest to, że skoro spełniają wymagania to dostają certyfikat.
Andrzej Szastok: Czyli, jeśli dobrze zrozumiałem, sytuacja wygląda tak, że jeśli system zarządzania, niezależnie czy według normy ISO czy lean managment, będzie środkiem do osiągnięcia celu to będzie ok, jeśli będzie to cel sam w sobie to…
Grzegorz Grabka: Nie będzie to najskuteczniejsze narzędzie.
Andrzej Szastok: No więc właśnie, mam podobne doświadczenie. Dostałem kiedyś zadanie uporządkowania pewnego zespołu: czterdzieści kobiet, ja sam. Zauważyłem, że w pewnej części tego zespołu panie wykonywały prace podwójnie tzn. najpierw ewidencjonowały pewne dane w systemie komputerowym, a następnie wyciągały kartoteki w formacie A3, wkręcały w maszynę i tam wprowadzały te same dane. Zapytałem, dlaczego tak robią. One spojrzały na mnie zdziwione i odpowiedziały, że zawsze tak było. No i właśnie tutaj pojawia się pytanie, co można zrobić i jak radzić sobie z takim sposobem myślenia. Jak przygotować pracowników na zmiany, które wiążą się z wdrożeniem systemu zarządzania. Jakie są wasze pomysły?
Beata Łuczak: Moim zdaniem, żeby osiągnąć zmianę to trzeba samemu do tej zmiany być przekonanym. Zdarza mi się, że słyszę w trakcie szkoleń pytanie:
Rola ludzi w systemach zarządzania –dyskusja praktyków

17
Wiedza i praktyka
jakie dowody mam przedstawić, żeby być przygotowanym na przyjazd audytora? Jeśli będziemy mieć takie przekonanie to nie osiągniemy efektu, o jaki chodzi. W takiej sytuacji system będzie przygotowany pod oczekiwania audytora, a nie pod to, żeby firma się faktycznie rozwijała. Natomiast jeśli osoba odpowiedzialna za wdrożenie i utrzymanie systemu będzie sama przekonana to będzie potrafiła zmotywować innych.
Rafał Lewandowski: To, o czym mówimy, jest bardzo ważne na samym początku budowania zaangażowania pracowników i odwracania ich nastawienia. Można to zrobić w formie zabaw i gier. Ważne, żeby pokazać ludziom na prostych przykładach, że oni sami decydują, co robią. Trzeba pokazać, że mimo ograniczeń norm i przepisów prawnych to oni mogą mieć wpływ na procesy w organizacji, bo to oni za nie odpowiadają. Podam przykład mojej firmy, gdzie wdrożyliśmy grę „leanową”. Ludzie przychodzą bardzo sceptyczni, z laptopami, komórkami, obstawiają się szklankami i kubkami. Mówię im wtedy, że nawet nie zdążą włączyć tych komputerów. Po grze przyznają, że poznali coś fajnego. Ja wychodzę z takiego założenie, że przez zaangażowanie i pokazanie, że coś można zrobić, ludzie łapią tego „bakcyla” i potem ciągną to dalej.
Jacek Sadowski: Myślę, że kluczem jest pokazanie ludziom, że uczestniczą w tych procesach i że doskonalenie procesów na ich własnym poziomie jest po prostu opłacalne. Bo jeśli przemyślimy te procesy dobrze to znikną wspomniane szafy z dokumentami, z których trzeba wyjmować kartoteki i coś tam robić jeszcze raz. Jeżeli przemyślimy zagrożenia, które w tych procesach mogą się pojawić to doprowadzimy do tego, że będą one bezpieczniejsze. W ten sposób poprawi się też bezpieczeństwo firmy oraz bezpieczeństwo pracownika.
Andrzej Szastok: To może ja włożę kij w mrowisko. Znam taką firmę, gdzie pracownicy mają obowiązek wymyślania „kaizenów” i tak się dzieje. Oni te „kaizeny” wymyślają, wdrażają, ale okazuje się, że – z perspektywy dwóch lat – sami kręcą na siebie bicz, bo np. na linii produkcyjnej skraca się czas o kilka sekund, co w związku z tym wymaga od nich przyśpieszenia pracy.
Piotr Goryński: I to jest właśnie ten trick, nie chodzi o to, żeby człowiek dostawał mniej czasu na wykonanie zadań, ale żeby

Wiedza i praktyka
18
wprowadzić takie usprawnienia, żeby ta praca mogła być wykonywana szybciej.
Renata Ruman-Dzido, Dyrektor Szpitala Wojewódzkiego w Opolu (głos z sali): Jeśli można się wtrącić, chciałabym lekko odejść od tematów produkcyjnych. Od szesnastu lat zarządzam szpitalem i mam za sobą wyprowadzenia organizacji z problemów finansowych. Powiem tak, żeby myśleć o jakości to najpierw pewną bazę podstawowych potrzeb trzeba mieć zabezpieczoną. Kiedy trzeba walczyć z problemami finansowymi to nie jest najlepszy moment, żeby przekonywać ludzi. Dlatego w 2006 roku zdecydowałam, że wdrażamy systemy zarządzania jednocześnie z wdrażaniem standardów akredytacji Centrum Monitorowania Jakości w Ochronie Zdrowia.
Moim zdaniem, absolutnie kluczowe jest to, czy prowadzący firmę jest do tego przekonany. Mówię ze swojego doświadczenia, żeby system funkcjonował, konieczny jest przywódca, który jest po prostu entuzjastą. Uważam, że w początkowej fazie pewne rzeczy powinny być wpisane w zakres obowiązków. Osobiście obserwowałam, jak ludzie z czasem przekonywali się do tych rozwiązań i stawali się ich zwolennikami. Nie wolno odpuścić na początku tylko dlatego, że jest więcej przeciwników.
Andrzej Szastok: Proszę nam odpowiedzieć na pytanie, jak przekonała Pani lekarzy?
Renata Ruman-Dzido: Tym, którzy byli mniej przekonani wysyłałam pozaplanowe audyty. Później na przeglądach kierownictwa byłam bezwzględna i pokazywałam dobre, ale i złe praktyki.
Teresa Osadczuk-Witkowska, Pełnomocnik Szpitala Wojewódzkiego w Opolu (głos z sali): Tu nie chodzi o wykorzystanie siły. Nasi lekarze z czasem zauważyli poprawę. Jeżeli my stawiamy na lepsze zapisy w dokumentacji to dla nich jest wartość, bo te zapisy ich chronią. To uporządkowało wiele spraw. W przypadku niezgodności wystawiam kartę i lekarz musi się sprawą zająć, bo potem oceniam skuteczność podjętego działania naprawczego. Nawet ci, którzy początkowo byli sceptyczni, uważają teraz, że te procedury są potrzebne, bo wpływają na bezpieczeństwo pacjentów.
Jacek Sadowski: Czyli podsumowując, od sprawnego procesu, przez bezpieczeństwo organizacji, do bezpieczeństwa osobistego.
Ilona Turczyn, Wojewódzki Ośrodek Medycyny Pracy w Lublinie (głos z sali): Pozwolę sobie nie zgodzić się z moją przedmówczynią, panią dyrektor szpitala. Ja też jestem entuzjastką, ale reprezentuję specjalistykę ambulatoryjną. Tutaj rzeczywistość wygląda trochę inaczej, lekarz przyjmuje dwie godziny np. popołudniu. W takim układzie ciężko jest nawet o bezpośrednią komunikację, a co dopiero o przekonanie go, żeby się angażował.
Andrzej Szastok: No właśnie i tutaj pojawia się pytanie, co zrobić z takimi pracownikami, którzy wpadają na 1/8 etatu i nie czują więzi, jak ze swoim podstawowym pracodawcą?
Grzegorz Grabka: Z mojego doświadczenia wynika, że jest jedna rzecz, do której pracownika nie można zmusić. Mianowicie nie da się zmusić podwładnego, żeby polubił własną pracę. Wszystko inne da się zrobić. Mogę zmusić pracownika, żeby przychodził do pracy codziennie z akwarium ze złotą rybką, jestem w stanie to zrobić. Czy on się będzie z tym zgadzał czy nie to jest już zupełnie inna kwestia. Natomiast jeśli mówimy o motywowaniu to ja osobiście nie wierzę w motywowanie zewnętrzne. Bo działa ono tak długo, jak długo stoi nadzorca z batem. Co więc można zrobić? Była już dzisiaj o tym mowa, podstawą jest szacunek do pracownika. Jedną z podstawowych potrzeb człowieka jest bycie docenianym, bycie ważnym. Jeżeli menadżer potrafi swoim ludziom pokazać, że oni są ważni, potrafi delegować, potrafi ich nauczyć poprzez dawanie przykładu to wtedy osiąga swój cel. Tak, jak mówiła tutaj pani dyrektor szpitala, podstawą powinno być pokazanie własnego zaangażowania.
Rafał Lewandowski: Powiedźmy sobie szczerze, że tu nigdzie nie padło stwierdzenie, że te wszystkie narzędzia: lean, six sigma, ISO są proste. Każda organizacja ma swoją specyfikę, ma swoje branże, ma ludzi. Przede wszystkim właśnie składa się z ludzi, którzy w jednym dniu są pozytywnie nastawieni, w drugim mogą tacy nie być, bo coś im się w życiu wydarzy. Podstawą jest to, żeby menadżer wiedział o tym i żeby to nie wpłynęło na prace innych osób. To ludzie generują atmosferę w organizacji.
Beata Łuczak: Zauważmy, że nasza dyskusja koncentruje się na tym, co wewnątrz organizacji, na menedżerach, pracownikach, na tym, czego oni oczekują.

Wiedza i praktyka
19
Mam wrażenie, że zapomnieliśmy trochę o systemach i o tym, po co tu jesteśmy. Odniosę się do przykładów podawanych przez panie, mianowicie do korzyści, jakie szpital czy przychodnia odniosły z wprowadzenia systemów zarządzania jakością. Wspomniałyście panie, że uporządkowanie, że doposażenie stanowisk pracy. Ale pani z Lublina wspomniała też o tym, czego tak naprawdę potrzebuje pacjent. I tutaj trzeba zadać sobie pytanie, kto jest dla nas ważniejszy? Czy NFZ, bo on jest płatnikiem, czy pacjent czyli końcowy użytkownik?
Jeśli lekarz pracuje w szpitalu na etacie i tam wykonuje zabiegi, i dojeżdża na dwie godziny do ZOZu, gdzie się regularnie spóźnia, to można się domyślić, czego potrzebują pacjenci. Przecież jeśli my idziemy do lekarza to chcemy być przyjęci o tej godzinie, na którą zostaliśmy zapisani, a nie czekać dlatego, że lekarz nie poinformował o tym, że się spóźni, bo przedłużył się zabieg w szpitalu. Wymagania pacjentów czy klientów to nie są jakieś bardzo wyimaginowane rzeczy. To jest proste do zidentyfikowania, a nie zawsze na nie jest zwracana uwaga.
Renata Ruman-Dzido: Dla mnie to kwestia oczywista, że jakość w jednostkach medycznych jest skoncentrowana na pacjencie. Wszyscy kluczowi pracownicy muszą być zaangażowani. Dlaczego w szpitalu tak ważne jest, żebyśmy zadbali o pacjenta, o jego bezpieczeństwo? Ano dlatego, że z tego się bierze bezpieczeństwo firmy, bezpieczeństwo indywidualne pracowników medycznych. My też badamy oczekiwania pacjentów i to nie różni się bardzo od badania oczekiwania klientów. W naszej branży nie ma mowy o reklamacjach, bo odpowiadamy za zdrowie i życie ludzi.
Andrzej Szastok: I tu niestety muszę postawić kropkę, gdyż skończył się nam czas przewidziany na tę debatę. Dziękuję Państwu bardzo i zapraszam do dalszej dyskusji w kuluarach.
Dyskusję prowadził Andrzej Szastok, doświadczony trener biznesu.
W panelu wzięli udział: • Grzegorz Grabka, Dyrektor Działu Certyfikacji Systemów
TÜV Rheinland Polska• Beata Łuczak, Audytor wiodący ISO 9001 • Rafał Lewandowski, trener, certyfikowany Six Sigma Black Belt • Piotr Goryński, Continuous Improvement Leader Black Belt
Molex Sp. z o.o. • Jacek Sadowski, Kierownik Sekcji Certyfikacji Systemów
Zarządzania TÜV Rheinland Polska

Bezpieczeństwo
20
Jak zminimalizować ryzyko w projektowaniu, produkcji i sprzedaży wyrobów?Ryzyko związane z wyrobem może być rozpatrywane w różnych kategoriach. Pierwszy aspekt związany jest z legalnością i bezpieczeństwem wyrobów. Realizacja wymagań bezpieczeństwa dóbr obecnych w obrocie handlowym jest absolutnym minimum, koniecznym do zagwarantowania przez producentów lub wprowadzających wyrób do obrotu. Z drugiej strony istnieje również ryzyko powiązane z oczekiwaniami i potrzebami przyszłych użytkowników czy klientów, dlatego oprócz niezbędnego bezpieczeństwa produktów istnieje cała gama kryteriów warunkujących powodzenie czy sukces biznesowy.
Im bardziej skomplikowany lub nietypowy wyrób lub im większe oczekiwania i potrzeby użytkowników czy klientów, tym większe ryzyko pojawia się w procesie cyklu życia takiego produktu. To z kolei wiąże się z koniecznością wkalkulowania tego ryzyka w sam biznes wytwórcy.
Ryzyko porażki rynkowejDla przykładu wystarczy wyobrazić sobie sytuację, w której produkty np. kaski motocyklowe, spełniające wszelkie kryteria bezpieczeństwa, wytworzone z użyciem najnowocześniejszej technologii, przy wykorzystaniu całej dostępnej na dany moment wiedzy eksperckiej i technicznej – nie znajdują zainteresowania na rynku, gdyż zwyczajnie nie odpowiadają potrzebom i oczekiwaniom użytkowników (np. nie pasują do głowy użytkownika: są za małe, za ciasne, za ciężkie itp.). Innym przykładem mogą być zaawansowane, lecz zbyt skomplikowane rozwiązania high-tech lub proste w obsłudze, lecz funkcjonalnie archaiczne systemy, które nie uzyskają niezbędnej aprobaty użytkowników. Produkty, dla których nie dokonano analizy wszelkich możliwych ryzyk w biznesie, mogą stać się bezużytecznym gadżetem.
Konieczna analiza W celu stworzenia konkretnych wymagań stawianych różnego rodzaju wyrobom, konieczne jest przeprowadzenie analizy ryzyka, uwzględniającej cechy deterministyczne, potrzeby i oczekiwania przyszłych użytkowników, parametry techniczne, funkcjonalność czy planowane zastosowanie wyrobów. Analizę taką można przeprowadzić w oparciu o uznane, międzynarodowe narzędzia i techniki diagnostyczne, takie jak Diagram Ishikawy czy FMEA (ang. Failure Mode and Effects Analysis). Analiza ryzyka wyrobu poszerzona o zastosowanie innych metod
związanych z kontrolą procesu cyklu życia produktu poprzez audyty miejsca produkcji, badania laboratoryjne czy inspekcje produktów gotowych, może być skutecznym sposobem ograniczającym ryzyko w biznesie przedsiębiorcy.
Wiedza o zagrożeniach na każdym etapie cyklu życia wyrobuIlość zastosowanych metod weryfikacji w całym cyklu projektowania i życia wyrobu jest wprost proporcjonalna do prawdopodobieństwa zminimalizowania ryzyka. Nie ma możliwości 100% wyeliminowania ryzyka, ale świadome analizowanie i kontrolowanie procesu życia wyrobu może je skutecznie redukować.
Na załączonym wykresie zostały przedstawione propozycje działań minimalizujących ryzyko powstawania nieprawidłowych produktów. Działania te stanowią podstawę zapewnienia bezpieczeństwa i jakości wyrobów. Kompleksowy charakter oraz holistyczne ujęcie procesowe oparte o metodologię optymalizacji procesów i wyrobów DMAIC (Define, Measure, Act, Improve, Check) dają gwarancję pełnej wiedzy o zagrożeniach na każdym etapie cyklu życia wyrobu, od procesu projektowania począwszy, przez
produkcję seryjną, po wyrób gotowy, na obsłudze posprzedażowej kończąc.
Stosowanie metodyki weryfikacji i kontroli towaru ogranicza również ryzyko wyprodukowania wyrobów niezgodnych lub niebezpiecznych, a co za tym idzie zagrożenie poniesienia wyższych kosztów, które rośnie wraz z kolejnym krokiem cyklu życia wyrobu. Wybór towaru przez użytkownika jest determinowany przez szereg czynników takich jak: cena, bezpieczeństwo, design (moda), marka, jakość wykonania, funkcjonalność, ergonomia, użyteczność i inne. Spełnienie wszystkich kryteriów niesie ze sobą wielkie wyzwanie i odpowiedzialność zarówno dla producenta, wprowadzającego wyrób do obrotu, jak również jednostek kontrolujących. Odpowiedzią może być pełne zadowolenie i polecanie wyrobu innym, bądź reklamacja i niezadowolenie klienta, a nawet utrata wizerunku marki.
Rola niezależnej jednostkiW procesie projektowania, wytwarzania i funkcjonowania w obrocie wyrobu uczestniczy wielu interesariuszy/stron. Na poniższym wykresie zależność pomiędzy stronami została przedstawiona w ujęciu projektowym, gdyż każdy wyrób
Define Measure
Analyze
ImproveControl
Przepływ procesu i działania minimalizujące ryzyko w biznesie
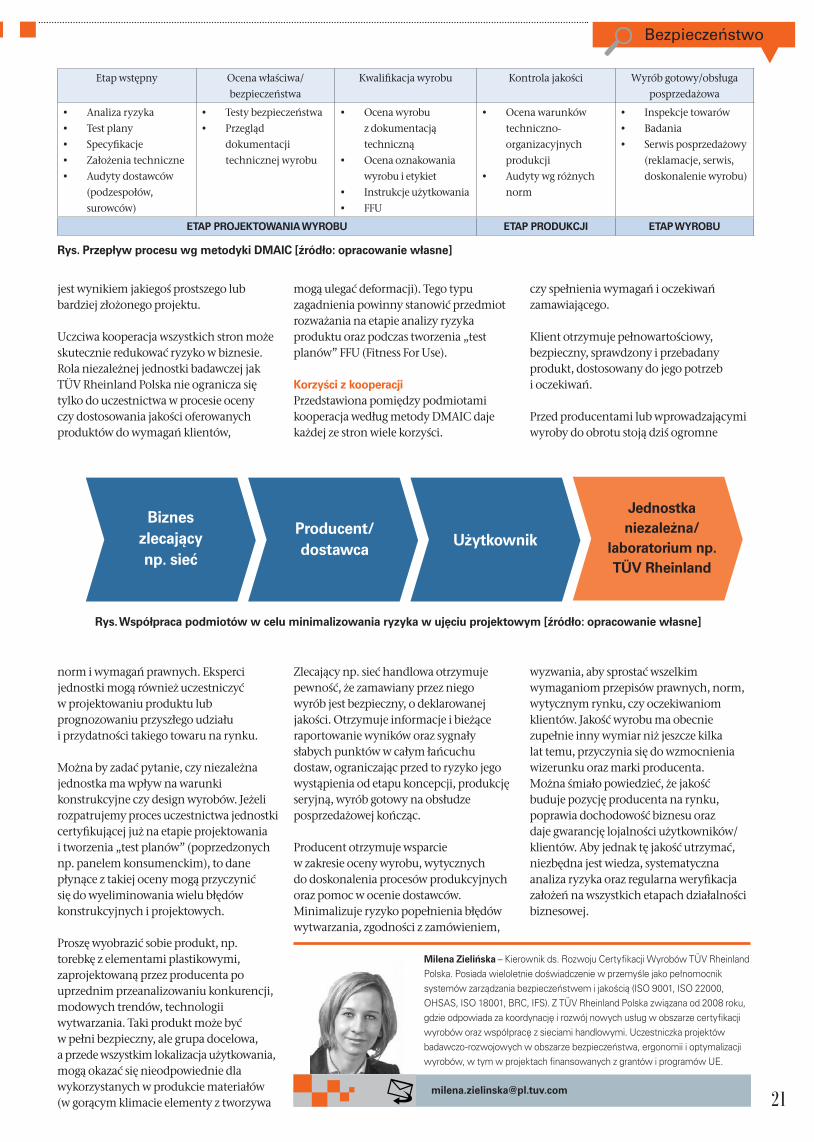
Bezpieczeństwo
21
Milena Zielińska – Kierownik ds. Rozwoju Certyfikacji Wyrobów TÜV Rheinland Polska. Posiada wieloletnie doświadczenie w przemyśle jako pełnomocnik systemów zarządzania bezpieczeństwem i jakością (ISO 9001, ISO 22000, OHSAS, ISO 18001, BRC, IFS). Z TÜV Rheinland Polska związana od 2008 roku, gdzie odpowiada za koordynację i rozwój nowych usług w obszarze certyfikacji wyrobów oraz współpracę z sieciami handlowymi. Uczestniczka projektów badawczo-rozwojowych w obszarze bezpieczeństwa, ergonomii i optymalizacji wyrobów, w tym w projektach finansowanych z grantów i programów UE.
jest wynikiem jakiegoś prostszego lub bardziej złożonego projektu.
Uczciwa kooperacja wszystkich stron może skutecznie redukować ryzyko w biznesie. Rola niezależnej jednostki badawczej jak TÜV Rheinland Polska nie ogranicza się tylko do uczestnictwa w procesie oceny czy dostosowania jakości oferowanych produktów do wymagań klientów,
mogą ulegać deformacji). Tego typu zagadnienia powinny stanowić przedmiot rozważania na etapie analizy ryzyka produktu oraz podczas tworzenia „test planów” FFU (Fitness For Use).
Korzyści z kooperacjiPrzedstawiona pomiędzy podmiotami kooperacja według metody DMAIC daje każdej ze stron wiele korzyści.
czy spełnienia wymagań i oczekiwań zamawiającego.
Klient otrzymuje pełnowartościowy, bezpieczny, sprawdzony i przebadany produkt, dostosowany do jego potrzeb i oczekiwań.
Przed producentami lub wprowadzającymi wyroby do obrotu stoją dziś ogromne
Etap wstępny Ocena właściwa/
bezpieczeństwa
Kwalifikacja wyrobu Kontrola jakości Wyrób gotowy/obsługa
posprzedażowa
• Analiza ryzyka
• Test plany
• Specyfikacje
• Założenia techniczne
• Audyty dostawców
(podzespołów,
surowców)
• Testy bezpieczeństwa
• Przegląd
dokumentacji
technicznej wyrobu
• Ocena wyrobu
z dokumentacją
techniczną
• Ocena oznakowania
wyrobu i etykiet
• Instrukcje użytkowania
• FFU
• Ocena warunków
techniczno-
organizacyjnych
produkcji
• Audyty wg różnych
norm
• Inspekcje towarów
• Badania
• Serwis posprzedażowy
(reklamacje, serwis,
doskonalenie wyrobu)
ETAP PROJEKTOWANIA WYROBU ETAP PRODUKCJI ETAP WYROBU
Rys. Przepływ procesu wg metodyki DMAIC [źródło: opracowanie własne]
Bizneszlecający np. sieć
Producent/dostawca
Użytkownik
Jednostka niezależna/
laboratorium np.TÜV Rheinland
Rys. Współpraca podmiotów w celu minimalizowania ryzyka w ujęciu projektowym [źródło: opracowanie własne]
norm i wymagań prawnych. Eksperci jednostki mogą również uczestniczyć w projektowaniu produktu lub prognozowaniu przyszłego udziału i przydatności takiego towaru na rynku.
Można by zadać pytanie, czy niezależna jednostka ma wpływ na warunki konstrukcyjne czy design wyrobów. Jeżeli rozpatrujemy proces uczestnictwa jednostki certyfikującej już na etapie projektowania i tworzenia „test planów” (poprzedzonych np. panelem konsumenckim), to dane płynące z takiej oceny mogą przyczynić się do wyeliminowania wielu błędów konstrukcyjnych i projektowych.
Proszę wyobrazić sobie produkt, np. torebkę z elementami plastikowymi, zaprojektowaną przez producenta po uprzednim przeanalizowaniu konkurencji, modowych trendów, technologii wytwarzania. Taki produkt może być w pełni bezpieczny, ale grupa docelowa, a przede wszystkim lokalizacja użytkowania, mogą okazać się nieodpowiednie dla wykorzystanych w produkcie materiałów (w gorącym klimacie elementy z tworzywa
Zlecający np. sieć handlowa otrzymuje pewność, że zamawiany przez niego wyrób jest bezpieczny, o deklarowanej jakości. Otrzymuje informacje i bieżące raportowanie wyników oraz sygnały słabych punktów w całym łańcuchu dostaw, ograniczając przed to ryzyko jego wystąpienia od etapu koncepcji, produkcję seryjną, wyrób gotowy na obsłudze posprzedażowej kończąc.
Producent otrzymuje wsparcie w zakresie oceny wyrobu, wytycznych do doskonalenia procesów produkcyjnych oraz pomoc w ocenie dostawców. Minimalizuje ryzyko popełnienia błędów wytwarzania, zgodności z zamówieniem,
wyzwania, aby sprostać wszelkim wymaganiom przepisów prawnych, norm, wytycznym rynku, czy oczekiwaniom klientów. Jakość wyrobu ma obecnie zupełnie inny wymiar niż jeszcze kilka lat temu, przyczynia się do wzmocnienia wizerunku oraz marki producenta. Można śmiało powiedzieć, że jakość buduje pozycję producenta na rynku, poprawia dochodowość biznesu oraz daje gwarancję lojalności użytkowników/ klientów. Aby jednak tę jakość utrzymać, niezbędna jest wiedza, systematyczna analiza ryzyka oraz regularna weryfikacja założeń na wszystkich etapach działalności biznesowej.

Wiedza i praktyka
22
Zmiany w prawie odnośnie ochrony danych osobowych – obowiązki przedsiębiorców
Nowe rozporządzenie UE nr 2016/679 z 27 kwietnia 2016 roku (RODO UE ) w sprawie ochrony danych osobowych reguluje m.in. zasady ochrony przetwarzanych w firmie zbiorów danych osobowych. W Polsce trwają prace legislacyjne nad ujednoliceniem rozwiązań instytucjonalnych. Rozwiązania powinny być wypracowane w ramach aktów wykonawczych, niezbędnych dla wdrożenia RODO UE w okresie jego vacatio legis tzn. do 24 maja 2018 roku.
Ochrona danych osobowych to problem złożony, a kolejne zmiany w prawie budzą niepokój wielu przedsiębiorców, którzy zadają sobie pytanie o obowiązki, jakie ich czekają i prawidłowość dotychczasowych działań.
Czym jest RODO UE?Wprowadzane RODO UE jest dokumentem ustawowym ujednolicającym zasady ochrony danych osobowych w całym Europejskim Obszarze Gospodarczym (EOG). Komisja Europejska zwróciła uwagę na istniejące zagrożenia, dlatego też w obszernej treści rozporządzenia (łącznie 99 art.) znalazły się istotne dla przedsiębiorców zapisy, mające zapewnić spójny stopień ochrony osób fizycznych w Unii. Regulacje te mają także zapobiegać rozbieżnościom hamującym swobodny przepływ danych osobowych na rynku wewnętrznym, gwarantując podmiotom gospodarczym pewność prawa, jego przejrzystość, a osobom fizycznym we wszystkich państwach członkowskich ten sam poziom prawnie egzekwowalnych praw oraz obowiązków i zadań administratorów/podmiotów przetwarzających. RODO UE pozwala spójnie monitorować przetwarzanie danych osobowych, a także zapewnia równoważne kary we wszystkich państwach członkowskich oraz skuteczną współpracę organów nadzorczych z tych krajów.
Co to oznacza dla przedsiębiorcy?Przyjęte założenia z zakresu różnicowania rozwiązań w dopasowaniu do wielkości
podmiotów oparte są na zasadzie technologicznej neutralności – pozostawiają dobór środków ochronnych po stronie Administratora Danych Osobowych (ADO). Działania takie ułatwia wprowadzone w RODO UE nowe ujęcie pojęć – dane osobowe, przetwarzanie, zbiór danych. Istotne nowości to enumeratywny katalog uprawnień co do czynności przetwarzania oraz wprowadzone definicje profilowania i pseudonimizacji danych osobowych (w tym medycznych – o stanie zdrowia, genetyczne, biometryczne).
Należy pamiętać, że art. 42 RODO UE zachęca do dobrowolnej certyfikacji wprowadzonych systemów bezpieczeństwa w ramach audytu ochrony danych przez krajowe akredytowane (art. 43 ust.1, pkt. b) jednostki certyfikujące, co ma potwierdzić wzmocnienie kontroli podmiotu nad własnymi danymi oraz promować wdrażanie mechanizmów zwiększających ochronę prywatności (tzn. privacy by design – na etapie projektowania; oraz privacy by default – jako ustawienia domyślne).
Podsumowując, przedsiębiorca (ADO) nie tylko powinien zapoznać się szczegółowo z treścią RODO UE, ale także wprowadzać zmiany wynikające z aktów wykonawczych. Te będą się niebawem pojawiały, w związku z aktualizacją polskiej ustawy (UODO). W praktyce profilaktyka bezpieczeństwa danych osobowych wg RODO UE, wymaga dla swej poprawnej realizacji zmiany w Polsce ok. 800 aktów prawnych.
Marek Blim – magister inżynier elektronik, doktor nauk wojskowych. Wykładowca: ASG, WAT, UMCS, PW, WWSI, AGH. Od 2001 roku audytor PCBC/BSI (ISO 9001/BS-7799). Były biegły sądowy (2002-2007) z zakresu OIN oraz menedżer TQM EQO (2004-2012). Aktualnie: rzeczoznawca STZOiMoZB Stowarzyszenia POLALARM (od 2005); ekspert ISO 9001 (od 2006); audytor ISO 27001 (od 2010). Uhonorowany za działalność naukowo-techniczną Srebrną (2008) i Złotą (2014) Odznaką NOT oraz medalem pamiątkowym FSNT NOT im. Feliksa Kucharzewskiego (2012).

Wiedza i praktyka
23
Uproszczony opis zapisów rozporządzenia Gdzie
Zachowując rozumienie danych osobowych, rozszerzono znacznie zakres zdefiniowanych
pojęć (m.in.: zgoda i ograniczenie przetwarzania, profilowanie, pseudonimizacja, dane
genetyczne, dane biometryczne, dane dotyczące zdrowia, przetwarzanie trans graniczne,
usługa społeczeństwa informacyjnego)
Art.4
ust.1 –
ust.26
Rozszerzono dotychczasowe zasady przetwarzania danych oraz ich zgodność z prawem
(rozliczalność administratora danych, ograniczenie przechowania, integralność
i poufność)
Art.5
Art.6
Wymóg zachowania formy zgody na przetwarzanie danych osobowych (oświadczenie
lub wyraźne działanie potwierdzające) szczególnie gdy przetwarzamy dane dziecka
(13 lat < wiek dziecka < 16 lat) w celu bezpośredniego oferowania mu usług – udzielenie
lub zaaprobowanie zgody przez osobę sprawującą władzę rodzicielską nad dzieckiem
Art.7
Art.8
Możliwe jest przetwarzanie danych bez identyfikacji osoby której dotyczą, jeżeli cele
przetwarzania jej nie wymagają lub już nie wymagają – istotne dla ponoszonych kosztów
Art.11
Istotnie doprecyzowano wymóg zachowania obowiązku informacyjnego o przetwarzaniu
danych, wskazując jego sposób i formułę
Art.13
Art.14
Wprowadzono wymóg zachowania prawa dostępu do przetwarzanych danych oraz
ich kontroli i sprostowania przez właściciela danych
Art.15
Art.16
Szczególne znaczenie ma dla przedsiębiorcy realizacja przez klienta „prawa do bycia
zapomnianym”, zwłaszcza gdy jako ADO musi on wykazać uzasadnioną konieczność
wyłączenia możliwości skorzystania z tego prawa przez osobę wnioskującą (np.
przetwarzanie musi być niezbędne do ustalenia, dochodzenia lub obrony roszczeń),
aby mógł zachować prawo do ograniczenia przetwarzania
Art.17
Art.18
Podtrzymana jest zasada zabraniająca przetwarzania danych szczególnych kategorii
(dane wrażliwe – patrz: definicje rozszerzone zapisane w Art.4)
Art.27
Przedsiębiorca, jako ADO planując swój system przetwarzania danych osobowych, ma
obowiązek uwzględniania ochrony danych w fazie projektowania oraz zachowania
domyślnej ochrony poprzez przeprowadzenie procesu analizy zagrożeń i oceny ryzyka
dla bezpieczeństwa procesów przetwarzania
Art.25
Art.29
Przedsiębiorca ma obowiązek zapewnienia bezpieczeństwa przetwarzanych danych, a dla
przypadku jego naruszenia – obowiązek informowania organu nadzoru (DPA/GIODO)
Art.32
Art.33
Przedsiębiorca, jako ADO ma obowiązek uwzględnienia/ocenienia prawdopodobieństwa
wystąpienia i wagi zagrożeń dla praw i wolności osoby, której przetwarzane dane dotyczą
Art.35
Art.36
Działania powyższe przedsiębiorca powinien przeprowadzić (ADO – gestor) w przypadku
wyrażania zgody na przetwarzanie danych dla procesora oraz dla ew. subprocesora
Art.28
Art.31
Przedsiębiorca zgodnie z zapisem warunków działania zawartych w ust.1 art. 37 musi
wyznaczyć inspektora ochrony danych (DPO – data protection official) z uprawnieniami
określającymi jego pozycję oraz gwarantującymi jego status i niezależność
Art.37
Art.38
Przedsiębiorca zapewnia dla powołanego DPO komplet warunków dla poprawnego
realizowania zadań własnych oraz dla asystowania przy postępowaniach organu nadzoru
Art.39
Przedsiębiorca powinien zadbać o właściwe stosowanie treści rozporządzenia – wykorzystać
kodeksy postępowania i mechanizmy certyfikacji stosowanych zabezpieczeń
Art.40
Art.42
Przedsiębiorca musi mieć świadomość międzynarodowej współpracy w zakresie ochrony
danych osobowych oraz niezależności statutowej organów nadzorczych (EROD)
Art.50
Art.52
Przedsiębiorca musi mieć świadomość zakresu zadań i uprawnień posiadanych przez
organy nadzorcze – w tym prawa do skutecznego stosowania środka ochrony prawnej
przed sądem i rzetelnego procesu, zgodnie z Kartą praw podstawowych UE
Art.57
Art.58
Przedsiębiorca musi mieć świadomość, że najwyższym organem w zakresie rozstrzygania
sporów jest Europejska Rada Ochrony Danych (EROD)
Art.65
Art.68
Przedsiębiorca musi mieć świadomość, że każda osoba której dane dotyczą, ma prawo
do wniesienia skargi do organu nadzorczego oraz prawo do skutecznego środka ochrony
prawnej przed sądem przeciwko administratorowi lub podmiotowi przetwarzającemu
Art.77
Art.78
Art.79
Przedsiębiorca musi mieć świadomość, że każda osoba, która poniosła szkodę majątkową
lub niemajątkową w wyniku naruszenia niniejszego rozporządzenia, ma prawo uzyskać
od administratora lub podmiotu przetwarzającego odszkodowanie za poniesioną szkodę
Art.82
Art.83
Art.84
Zestawienie głównych wskazań i zaleceń dla przedsiębiorców w RODO UE

24
News !!!Jak pomóc klientowi dokonać właściwego wyboru produktu?W kolejnym odcinku JakośćTV odpowiadamy, co kryje się za znakami na produktach użytkowych i wyjaśniamy, czym są badania Fitness For Use (FFU).
- Kupując dowolny sprzęt, dajmy na to AGD, klient staje przed wyborem jednego spośród kilku bądź kilkunastu podobnych produktów. Aby dokonać właściwego wyboru, warto zwrócić uwagę na znaki umieszczone na opakowaniu produktu – przekonuje Jakub Dubowik, Koordynator Obszaru Klient Sieciowy TÜV Rheinland Polska.
Rozpiętość tematyczna kanału JakośćTV odpowiada różnorodności usług realizo-wanych przez ekspertów TÜV Rheinland Polska. W poprzednich odcinkach jest więc mowa zarówno o popularnych nor-mach ISO 9001 czy ISO 14001, ale także zmianach w dyrektywie ciśnieniowej, oznakowaniu CE, alergenach w żywności czy rolnictwie ekologicznym.
Oglądaj JakośćTV na tuv.pl/jakosctv
TÜV Rheinland Polska laureatem programu Inwestor w Kapitał Ludzki 2016Uroczysta gala, na której wręczono godła Inwestor w Kapitał Ludzki odbyła się 17 listopada 2016 roku w Warszawie. Fundacja Obserwatorium Zarządzania uhonorowała w ten sposób dziewięciu pracodawców. Wśród wyróżnionych firm znalazła się także jednostka TÜV Rheinland Polska.
Godło Inwestor w Kapitał Ludzki mogą otrzymać podmioty, które zgłoszą się do programu i przejdą szczegółowy, niezależny proces badawczy i spełnią określone w nim standardy. Podstawą do zdobycia nagrody są tutaj oceny dokonywane przez pracowników podczas niezależnego, anonimowego badania.
- Nasz udział w programie był podyktowany chęcią porównania pozycji TÜV Rheinland Polska jako pracodawcy z innymi firmami wiodącymi na rynku. Badania satysfakcji pracowników są prowadzone w naszej firmie regularnie, co dwa lata. Osoby zatrudnione we
wszystkich oddziałach koncernu mają możliwość udziału w globalnej, anonimowej ankiecie” – mówi Grażyna Bohdziewicz-Szpor, Kierownik Personalny TÜV Rheinland Polska. – Cieszy nas, że opinie naszych pracowników pozwoliły na zdobycie tego wyróżnienia. Godło Inwestor w Kapitał Ludzki to znak dla przyszłych pracowników, że zostaliśmy z pozytywnym wynikiem ocenieni przez niezależną instytucję – dodaje.
Więcej informacji na temat programu można znaleźć na stronie http://inwestorwkapitalludzki.pl/

25
Wiedza i praktyka
Z pamiętnika audytora:Ryzyko w biznesie
W roku 1996 firma, w której pracowałem, podjęła decyzję o wdrożeniu systemu zapewnienia jakości według modelu ISO 9001. Było to poważne wyzwanie, ponieważ firma zatrudniała około tysiąca osób, posiadała kilka lokalizacji oraz miała bardzo zróżnicowany profil produkcji. Budując procedurę przeglądu umowy, bardzo dużą uwagę poświęciliśmy procesowi opiniowania projektów umów, aby redukować ryzyka związane z niekorzystnymi dla firmy zapisami w umowach.
W działania związane z opiniowaniem zaangażowane były różne komórki organizacyjne, które wydawały opinie pod kątem własnego merytorycznego zakresu. Przekrój opiniodawców był bardzo szeroki – od produkcji poprzez głównego mechanika, kontrolę jakości, zakupy, po księgowość i opinie prawne. Proces ten był w rzeczywistości nieco czasochłonny, ale znacznie redukował ryzyka biznesowe. Przez kilka lat proces przeglądu umów funkcjonował prawidłowo. Dzięki temu firma ustrzegła się wielu „wpadek”, które zdążyła w międzyczasie zaliczyć konkurencja.
Około roku dwutysięcznego dział marketingu, który od początku krytykował procedurę przeglądu umowy, doprowadził do jej uproszczenia, tłumacząc to faktem, iż jest ona nadmiernie biurokratyczna i czasochłonna. Pomimo mojego sprzeciwu – a byłem wówczas pełnomocnikiem do spraw SZJ,
zarząd zgodził się na uproszczenia. Niestety na efekty nie trzeba było długo czekać. Okazało się, że pewna umowa, o bardzo dużej wartości, obejmowała błędne założenia w zakresie AKPiA. Chodziło o ściśle dedykowane do realizacji kontraktu urządzenia z zakresu automatyki. W konsekwencji, firma zamiast zarobić na kontrakcie, musiała do niego dopłacić. Myślicie pewnie Państwo, że ktoś poszedł po rozum do głowy i przywrócił pierwotne postępowanie związane z opiniowaniem umów? Nic bardziej mylnego. Żeby było ciekawiej, za taki stan rzeczy winą obarczono ISO – oczywiście nieoficjalnie. „Korytarz” mówił o tym, że jest to struktura biurokratyczna i przeszkadzająca w działaniu firmy. Sytuacja powtórzyła się kilkakrotnie, w różnych konfiguracjach. Dodatkowo na to nałożył się kryzys w branży i firma wpadła w poważne kłopoty. Ale to już było zmartwienie następnych zarządów. Wielu zarządów.
FELIETON
Jarosław Nowicki Audytor wiodący TÜV Rheinland Polska. Od 20 lat w branży związanej z normami ISO. Rzeczoznawca Stowarzyszenia Inżynierów i Techników Mechaników Polskich.
Zastanawiali się Państwo nad tym, jak to jest po drugiej stronie na audycie?
O blaskach i cieniach tej pracy w luźnej formie felietonu pisze Jarosław Nowicki, audytor wiodący TÜV Rheinland Polska.

26
Wiedza i praktyka
Zmienia się ISO/TS – standard zarządzania jakością w przemyśle motoryzacyjnymStworzona przez IATF norma ISO/TS 16949, najbardziej rozpowszechniony standard motoryzacyjny został zastąpiony dokumentem IATF 16949:2016, opublikowanym w październiku 2016 roku. Oznacza to, że dostawców w branży motoryzacyjnej czekają spore zmiany.Organizacje certyfikowane zgodnie z ISO/TS 16949:2009 powinny przejść na IATF 16949 poprzez audyt przejścia zgodnie z bieżącym cyklem audytów ISO/TS 16949:2009 (tzn. w regularnie zaplanowanym audycie wznowienia certyfikacji albo audycie podtrzymania certyfikacji).
Kiedy trzeba być gotowym na zmianę?TÜV Rheinland będzie oferować wszystkim klientom przejście na nowy IATF 16949 od dnia 1 kwietnia 2017. Wszystkie organizacje ubiegające się o audyt certyfikacyjny mogą być certyfikowane zgodnie z ISO/TS 16949:2009 do 1 października 2017, jednak certyfikat ISO/TS 16949:2009 będzie ważny tylko do 14 września 2018. Po 1 października 2017 nie będą już przeprowadzane żadne audyty (certyfikacyjne, podtrzymania certyfikacji, wznowienia certyfikacji oraz transferu)
zgodnie z ISO/TS 16949:2009.
Jak będzie wyglądał audyt?Audyt przejścia powinien odpowiadać zakresem i czasem trwania audytowi wznowienia certyfikacji. Przegląd dokumentacji, poza zakładem, powinien być przeprowadzony przed audytem przejścia.
Przegląd powinien zawierać, jako minimum, przegląd dokumentacji systemu zarządzania jakością klienta (tzn. Księgę jakości i procedury), w tym dowody potwierdzające zgodność z wymaganiami IATF 16949 (zwłaszcza nowe wymagania motoryzacyjne jak SPICE i TPM).
Jeżeli organizacja nie dostarczy wymaganych informacji, plan audytu powinien zawierać dodatkowo minimum pół dnia audytowego na miejscu, w celu zebrania oraz przeglądu brakujących
informacji.
Wszystkie funkcje wspomagające na miejscu albo zamiejscowe powinny być uwzględnione w procesie przejścia zgodnie z bieżącym cyklem audytu ISO/TS 16949:2009 i powinny być uwzględnione w audycie przejścia.
Organizacje certyfikowane według ISO/TS 16949:2009 nie mogą przejść na nowy IATF 16949 poprzez audyt transferu do nowej, uznanej przez IATF jednostki certyfikującej, nie mogą tego zrobić także w audycie specjalnym.
Kompetencje audytorówAudyt przejścia powinien być przeprowadzony wyłącznie przez wykwalifikowanych audytorów trzeciej strony, którzy wykazali swoje kompetencje do audytowania według
Harmonogram przejścia z ISO/TS 16949 na IATF 16949
1 kwietnia2017
1 października2017
17 maja2018
Możliwy początek
przejścia na
IATF 16949
Brak dalszych
audytów zgodnie
z ISO/TS 16949:2009
Koniec fazy
przejścia do
IATF 16949:2009
Zalecany okres przejściowy120 dni

27
Wiedza i praktyka
IATF 16949.
Jednostka certyfikująca może wyznaczyć więcej niż jednego audytora z poprzedniego cyklu audytu do udziału w audycie przejścia (czyli tzw. „audytorów z przenoszeniem”). Jeden z członków zespołu przeprowadzający audyt przejścia, będzie mógł uczestniczyć również w następnych audytach. Jednostka certyfikująca może wyznaczyć jednego audytora w przypadku, gdy audyt przekracza pięć dni, ale nie więcej niż siedem dni.
Konsekwencje niezgodnościGdy jednostka certyfikująca zidentyfikuje niezgodności w audycie przejścia, klient i jednostka certyfikująca powinni stosować wszystkie wymogi dla procesu zarządzania niezgodnościami, jak określono w Zasadach IATF. Jeżeli są
zidentyfikowane niezgodności krytyczne w audycie przejścia, decyzja zawieszenia certyfikatu powinna być zgodna z Zasadami IATF.
Zasady IATFJednostka certyfikująca powinna spełniać wszystkie wymagania procesu decyzyjnego certyfikacji jak określono w Zasadach IATF, z następującymi poniższymi wymaganiami: • Jednostka certyfikująca może podjąć
pozytywną decyzję certyfikacyjną po
upływie ważności certyfikatu ISO/TS 16949:2009, pod warunkiem, że decyzję certyfikacyjną podjęto w ciągu maksymalnie 120 dni kalendarzowych od ostatniego dnia audytu przejścia.
• Po pozytywnej decyzji certyfikacyjnej jednostka certyfikująca powinna wydać organizacji certyfikat IATF 16949. Certyfikat powinien zawierać datę wydania (data pozytywnej decyzji certyfikacyjnej) i datę ważności (data wydania plus maksymalnie trzy lata minus jeden dzień). Nowy certyfikat powinien zawierać nowy numer IATF.
IATF 16949 a ISO 9001Po 1 października 2017 roku organizacja może być tylko audytowana i certyfikowana zgodnie z normą IATF 16949. Przy określaniu liczby dni audytu dla organizacji ubiegających się o audyt certyfikacyjny
IATF 16949, redukcje mogą być udzielone w określonych sytuacjach/organizacjach.
Podczas podniesienia do IATF 16949 z istniejącego ISO 9001:2015, dni drugiego etapu audytu certyfikacyjnego nie powinny być redukowane o więcej niż 30%, przy czym jednostka certyfikująca dla certyfikacji IATF 16949 powinna być tą samą jednostką jak dla istniejącej certyfikacji ISO 9001:2015. W sytuacjach, kiedy klient transferuje certyfikowane ISO 9001:2015 do nowej jednostki certyfikującej, przynajmniej jeden audyt podtrzymania certyfikacji, powinien być przeprowadzony zgodnie z ISO 9001:2015 przez nową jednostkę certyfikującą, zanim zostanie przeprowadzony audyt podniesienia na IATF 16949.
Jeżeli jest rozszerzony zakres w audycie certyfikacyjnym, powinno być stosowane uaktualnienie bez redukcji. Powinno być stosowane wymagane 100% wymaganej liczby dni drugiego etapu certyfikacyjnego. Jeżeli organizacja ma ważny istniejący certyfikat ISO 9001:2008, powinno być stosowane uaktualnienie bez żadnych redukcji. Powinno być stosowane 100% wymaganej liczby dni drugiego etapu certyfikacyjnego.
KonkluzjeDość to skomplikowane i wymaga nie lada skupienia, aby spełnić powyższe wymagania. Od momentu wydania pierwszego komunikatu przez IATF padło wiele pytań ze strony organizacji, ale także samych jednostek certyfikujących. Wątpliwości budziły kwestie związane z terminarzem poszczególnych działań, jakie dowody potwierdzające zgodność z wymaganiami IATF 16949 należy przygotować, co wreszcie z wykazaniem trendów z ostatnich 12 miesięcy.
Jak widać temat przejścia na nowy standard jest dość trudny i wymaga dogłębnego zapoznania się z wymaganiami IATF. Dlatego warto śledzić stronę internetową IATF oraz pozostać w kontakcie z jednostką certyfikującą. Można także zapoznać się z nowymi wymaganiami specyfikacji IATF 16949:2016, która jest już dostępna w Polsce, choć na razie tylko w języku angielskim.
Damian Popielarz – Kierownik Produktu ISO/TS TÜV Rheinland Polska.Absolwent Akademii Techniczno-Rolniczej w Bydgoszczy, gdzie ukończył studia na kierunku Mechanika i Budowa Maszyn. Szeroko pojętą problematyką systemów zarządzania jakością, w tym systemów dedykowanych przemysłowi motoryzacyjnemu zajmuje się już od 1999 roku. Audytor wiodący systemów zarządzania jakością ISO 9001, ISO/TS 16949 oraz ISO14001.
14 września2018
11 stycznia2019
Utrata ważności wszystkich
certyfikatów ISO/TS 16949
Ostatnia możliwa data
wydania certyfikatu
po przeprowadzeniu
audytu przejścia
Nie ma możliwości audytu przejścia, wymagany jest teraz
audyt certyfikacyjny
Ten czas pozwala zakończyć proces zamknięcia niezgodności
120 dni

Wiedza i praktyka
28
Sposób na sukces wedługBriana Tracy’egoBrian Tracy to amerykański mentor ludzi sukcesu, znany i ceniony na całym świecie. W podtytule książki „Cele” składa czytelnikowi obietnicę: „Zdobędziesz wszystko czego pragniesz, szybciej niż myślisz”. Jak doszedł do takiego przekonania i na czym polega sekret jego metody, wyjaśnia na 270 stronach swojej książki, wydanej niedawno przez Wydawnictwo Helion.
Szybki rzut oka na spis treści pozwala odpowiedzieć na to pytanie. Dwadzieścia jeden rozdziałów, z których każdy zawiera konkretne instrukcje, np. zastanów się nad swoimi przekonaniami, zostań ekspertem w swojej dziedzinie, otaczaj się odpowiednimi ludźmi, mądrze organizuj swój czas. Wygląda to jak spis przykazań dla przyszłego człowieka sukcesu. Różnica polega na tym, że autor nie tylko daje czytelnikom listę zaleceń, ale w kolejnych rozdziałach odwołuje się do przykładów z własnego życia i opisuje, jak poradzić sobie z wątpliwościami.
Strategia Bazując na wieloletnim doświadczeniu trenerskim, Tracy pisze, żeby nie szukać wymówek i nie skupiać się na negatywnych emocjach: „Antidotum na wszelkiego rodzaju negatywne emocje to wziąć na siebie pełną odpowiedzialność za bieżącą sytuację”. Chcąc kierować własnym życiem, trzeba stać się swoim szefem. Idąc tym tropem, należy wyznaczyć sobie strategie działania i wziąć na siebie obowiązki podobne do obowiązków prezesa firmy. „Skoro jesteś prezesem, sprawujesz również pełną kontrolę nad swoim osobistym działem badań i rozwoju, nad szkoleniem samego siebie oraz nad nieustannym rozwijaniem własnych umiejętności i poszerzaniem wiedzy. (…) Przykra prawda jest taka, że nikt nie przejmuje się Tobą bardziej niż Ty sam.”
WartościTracy zwraca uwagę na sprawy
fundamentalne, jak wartości, na których chce się budować życiowy sukces. Podpowiada także, że aby je zdefiniować, każdy powinien przyjrzeć się swojemu postępowaniu w sytuacjach kryzysowych. Istotne jest tutaj też poczucie własnej wartości jako źródło odczuwalnego szczęścia. Co kilka stron powtarza: „Pamiętaj, że stajesz się tym, o czym myślisz przez większość czasu”.
PrzyszłośćW kontekście planów autor używa sformułowania „orientacja przyszłościowa” i podkreśla, żeby nie zapominać o fantazjach i marzeniach. Radzi zadawać sobie pytania dotyczące idealnego życia zawodowego, rodzinnego, kondycji fizycznej i zaangażowania społecznego. Po wykonaniu takiego ćwiczenia Tracy sugeruje ułożenie kalendarza, który będzie zawierał działania ujęte we wcześniejszych odpowiedziach.
Cele – metoda dwunastu krokówZgodnie z powszechnie znaną definicją celów biznesowych autor podpowiada, żeby posłużyć się jasnymi zasadami ich wyznaczania. Cel musi być zatem jasny, konkretny, szczegółowy i zapisany. Musi być także wymierny i obiektywny, ograniczony czasowo, ambitny, zgodny z wyznawanymi wartościami. Ostatnia zasada wymieniona przez Tracy’ego to zalecenie: „Twoje życie musi mieć jakiś jeden główny sens”. Oczywiście poza tymi podstawowymi zasadami autor podaje swoją autorską metodę dwunastu kroków, którą przekazał już ponad milionowi ludzi.
Pragnij czegoś – czego tak naprawdę chcesz?
Uwierz, że Twój cel jest osiągalny
Zapisz swój cel
Określ swój punkt wyjścia
Określ, dlaczego tego chcesz
Wyznacz termin
Zidentyfikuj przeszkody
Określ, jakich dodatkowych umiejętności i jakiej wiedzy
potrzebujesz
Wskaż ludzi, których pomocy będziesz potrzebował
Połącz wszystko w jeden plan
Nieustannie wizualizuj sobie swój cel
Nigdy się nie poddawaj
Metoda12 kroków
1.
2.
3.
4.
5.
6
7.
8.
9.
10.
11.
12.

Wiedza i praktyka
Agata Tynka - Specjalista ds. Public Relations, Redaktor naczelna magazynu Jakość. Absolwentka Uniwersytetu Jagiellońskiego, na kierunku Dziennikarstwo i komunikacja społeczna ze specjalizacją public relations. Karierę zawodową rozpoczęła w 2004 roku pracą w agencji PR. Kolejne doświadczenia, w tym praca w wydawnictwie książkowym, pozwoliły jej zdobyć kompetencje potrzebne do prowadzenia redakcji firmowego magazynu. W TÜV Rheinland Polska odpowiada za tworzenie i realizację strategii komunikacji zewnętrznej, działania content marketingowe i copywriting.
29
Doskonalenie Brian Tracy twierdzi, że ludzie sukcesu w pewnym momencie swojej kariery podejmują decyzję o koncentracji na doskonałości. Właśnie obranie takiej drogi opisuje w rozdziale na temat stania się ekspertem w swojej dziedzinie. Sugeruje oczywiście, że najlepiej jest skupić się na tym, w czym jest się dobrym. Autor opisuje aż osiem sposobów identyfikacji talentów. Na pierwszym miejscu jest tutaj prosta prawda: „Zawsze będziesz najlepszy i najszczęśliwszy, robiąc to, co uwielbiasz robić. Gdyby było Cię na to stać, zajmowałbyś się tym za darmo.”
Życie rodzinne i zdrowieAutor przenosi metodę dwunastu kroków także na życie rodzinne i kwestie związane ze zdrowiem. W kolejnych rozdziałach książki podaje dowody na uniwersalność swojej metody osiągania celów w każdej sferze życia. Istotne jest oczywiście, aby te cele się zazębiały. Do pełnego sukcesu potrzebne jest poczucie szczęścia, a to jest możliwe tylko w wypadku osiągnięcia harmonii. To oczywiście upragniony „work life balance”.
Monitoring postępówTracy skupia się także na kontroli i weryfikacji celów. To metody zaczerpnięte z zarządzania biznesem. Przytacza przy tym zabawne pytanie: „Jak zjeść słonia?” i odpowiedź „Po kawałku”. Radzi, aby tak właśnie traktować cele, dzieląc je na te mniejsze, które doprowadzą do realizacji celu głównego.
Konsekwencja i wytrwałośćOstatni rozdział książki to swoisty apel do czytelnika: „Nie poddawaj się, dopóki nie osiągniesz sukcesu”. Tutaj autor skupia się na sposobach zneutralizowania lęków oraz na odwadze koniecznej do podjęcia ryzyka. Najwięcej miejsca poświęca jednak udowodnieniu czytelnikom, że najcenniejszym zasobem ludzi sukcesu jest wytrwałość.
PodsumowanieSceptykom książka ta wyda się pewnie kolejnym poradnikiem przeznaczonym dla osób, które przed lustrem trenują rolę zwycięzcy. Tym osobom radziłabym zrobienie rachunku sumienia, bo mimo kliku oczywistych prawd zawartych w tej książce, jest ona niezłym przewodnikiem dla tych, którzy szukają swojej drogi.

Do wygrania:• 3 książki „Cele” autorstwa
Briana Tracy’ego ufundowane przez wydawnictwo Helion SA (www.onepress.pl)
• 3 kubki ceramiczne z hasłem „Zasmakuj w Jakości” i znakiem certyfikacji TÜV Rheinland Polska
• 3 zestawy figurek „Eksperci TÜV Rheinland”
Litery z pól oznaczonych kolorem tworzą hasło krzyżówki. Hasło prosimy przesłać mailem na adres [email protected] do 28 lutego 2017 roku.
Dane osobowe uczestników konkursu będą przetwarzane przez organizatora konkursu tj. TÜV Rheinland Polska Sp. z o.o., w celu realizacji umowy przystąpienia do konkursu i jego prawidłowego przeprowadzenia lub marketingu własnych produktów organizatora konkursu. Gwarantujemy prawo do wglądu do swoich danych osobowych oraz ich poprawiania, jak również żądania zaprzestania przetwarzania danych. Dane osobowe nie będą udostępniane innym podmiotom. Podanie danych jest dobrowolne.
4. jego wymagania powinny być w centrum zainteresowania firmy
5. taki m.in. musi być cel8. takie ma być bezpieczeństwo
wyrobu medycznego9. w procesie PDCA występuje po
etapie „działaj”11. może je otrzymać producent od
jednostki oceniającej13. zagrożenia muszą zostać…16. przemysł, którego dotyczy
standard ISO/TS17. najcenniejszy zasób ludzi sukcesu18. dotyczą ich zmiany w prawie odnośnie
ochrony danych osobowych
Poziomo1. ryzyko dla wyrobów można…
dzięki badaniom2. przypisywana jest każdemu
ryzyku podczas jego oceny3. tylko tacy audytorzy mogą
prowadzić audyty przejścia na nową wersję standardu
6. … z udziałem wyrobów medycznych muszą byćanalizowane
7. do niej dążą osoby zdeterminowane do osiągnięcia sukcesu
10. okres od 1.04.2017 do 1.10.2017 dla firm z ISO/TS
12. wpływ niepewności na cele14. jest potrzebny, aby system
sprawnie funkcjonował15. trzeba go określić dla
organizacji
Pionowo
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
30
Wpisz hasło

Zapisz się na bezpłatną prenumeratę drukowanej Jakości
Jakosc
Zapisz się na newsletter z Jakością
Adres email
Imię
Nazwisko
Firma
Stanowisko
Adres do wysyłki
Skan lub zdjęcie wypełnionego formularza z tej strony prosimy przesłać na adres [email protected] lub faksem na numer +48 32 271 64 88.
Skan lub zdjęcie wypełnionego formularza prosimy przesłać na adres [email protected] lub faksem na numer +48 32 271 64 88. Na podany wyżej adres email prześlemy wiadomość z linkiem potwierdzającym chęć zapisu na elektroniczną wersję Jakości.
2016/04

Pracując w TÜV Rheinland Polska stale uczysz się czegoś nowego. Niezależnie od rodzaju
wykonywanej pracy posiadasz dostęp do wiedzy oraz technologii, które pozwalają lepiej rozumieć
otaczającą nas rzeczywistość. Jako niezależna strona trzecia, TÜV Rheinland testuje techniczne
innowacje, produkty, usługi i systemy pod kątem bezpiecznego i odpowiedzialnego użytkowania.
Dzięki różnorodności świadczonych usług i branż z jakimi współpracujemy, a także dzięki
atmosferze sprzyjającej wymianie doświadczeń Ty także zyskasz szansę stania się ekspertem.
www.tuv.pl/praca
Poznawaj świat oczami ekspertów.Razem wiemy więcej
2016







![Dnevni avaz [broj 6249, 12.1.2013]](https://static.fdocument.pub/doc/165x107/553f93a74a7959e10e8b47e8/dnevni-avaz-broj-6249-1212013.jpg)










