
ALLEGATO I RIASSUNTO DELLE CARATTERISTICHE DEL...
22
1 ALLEGATO I RIASSUNTO DELLE CARATTERISTICHE DEL PRODOTTO
Transcript of ALLEGATO I RIASSUNTO DELLE CARATTERISTICHE DEL...

1
ALLEGATO I
RIASSUNTO DELLE CARATTERISTICHE DEL PRODOTTO

2
1. DENOMINAZIONE DEL MEDICINALE FIRDAPSE 10 mg compresse 2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA Ogni compressa contiene amifampridina fosfato equivalente a 10 mg di amifampridina.
Per l’elenco completo degli eccipienti, vedere paragrafo 6.1.
3. FORMA FARMACEUTICA Compresse. Compresse bianche, rotonde, piatte, incise su un lato. La compressa può essere divisa in due metà uguali. 4. INFORMAZIONI CLINICHE 4.1 Indicazioni terapeutiche Trattamento sintomatico della sindrome miastenica di Lambert-Eaton (LEMS) negli adulti. 4.2 Posologia e modo di somministrazione Il trattamento deve iniziare sotto il controllo di un medico con esperienza nel trattamento della malattia. FIRDAPSE deve essere somministrato in dosi separate, tre o quattro volte al giorno. La dose iniziale raccomandata è di 15 mg al giorno, e può essere aumentata di 5 mg alla volta ogni 4 o 5 giorni, fino a un massimo di 60 mg al giorno. La dose singola non deve mai superare i 20 mg. La compressa deve essere assunta insieme al cibo. Solo per uso orale. Pazienti pediatrici e adolescenti L’uso di FIRDAPSE non è raccomandato nei pazienti di età inferiore ai 18 anni, a causa della mancanza di dati sulla sicurezza e l’efficacia (vedere paragrafo 5.2). Pazienti con insufficienza renale o epatica FIRDAPSE deve essere usato con cautela nei pazienti con insufficienza renale o epatica. Nei pazienti che presentano un’insufficienza moderata o grave della funzione renale o epatica si raccomanda una dose iniziale di 5 mg di FIRDAPSE. Per i pazienti con insufficienza lieve della funzione renale o epatica si raccomanda una dose iniziale di 10 mg di FIRDAPSE. Per tali pazienti si raccomanda una titolazione più lenta, rispetto a quelli che non soffrono di insufficienza renale o epatica: le dosi si devono aumentare di 5 mg per volta ogni 7 giorni. Se si verificano reazioni avverse, è necessario interrompere la titolazione verso l’alto della dose (vedere paragrafi 4.4 e 5.2).

3
4.3 Controindicazioni • Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti • Epilessia • Asma incontrollata • Uso concomitante con sultopride (vedere paragrafi 4.5 e 5.1) • Uso concomitante con medicinali a basso indice terapeutico (vedere paragrafo 4.5) • Uso concomitante con medicinali di cui sia noto il potenziale di causare prolungamento dell’intervallo
QTc • Sindrome congenita del QT (vedere paragrafo 4.4) 4.4 Avvertenze speciali e precauzioni di impiego Non sono stati effettuati studi su pazienti affetti da insufficienza renale o epatica. A causa del rischio di un significativo aumento dell’esposizione al medicinale, i pazienti affetti da insufficienza renale o epatica devono essere sottoposti ad un attento controllo. Per i pazienti affetti da insufficienza renale o epatica, la titolazione della dose di amifampridina deve essere più lenta rispetto ai pazienti la cui funzione renale ed epatica è normale. Se si verificano reazioni avverse, è necessario interrompere la titolazione verso l’alto della dose (vedere paragrafo 4.2). L’esposizione all’amifampridina si associa a un aumento del rischio di crisi epilettiche. Il rischio di crisi è dose-dipendente ed è maggiore nei pazienti che presentano fattori di rischio che abbassano la soglia epilettica; è compreso anche l’uso in associazione con altri medicinali di cui è noto l’effetto di abbassare la soglia epilettica (vedere paragrafo 4.5). In caso di crisi, il trattamento deve essere interrotto. L’amifampridina non è stata completamente testata nei modelli di cancerogenicità, e il rischio di cancerogenicità associato al trattamento non è stato determinato. L’uso di amifampridina in pazienti affetti dalla forma non paraneoplastica di LEMS deve iniziare solo dopo un’accurata valutazione del rapporto rischio/beneficio per il paziente. Il controllo clinico e il monitoraggio elettrocardiografico (ECG) sono indicati all’inizio del trattamento e in seguito a cadenza annuale. In caso di segni e sintomi che indichino aritmie cardiache, l’ECG deve essere effettuato immediatamente. I pazienti devono essere istruiti ad informare i medici dell’assunzione del farmaco; può infatti rendersi necessario un attento monitoraggio di una patologia concomitante (in particolare l’asma). 4.5 Interazioni con altri medicinali ed altre forme di interazione Non sono stati effettuati studi di interazione in vitro o in vivo. Inoltre, non è noto in che modo l’amifampridina venga eliminata. Interazioni farmacocinetiche Medicinali eliminati tramite metabolismo o secrezione attiva Non vi sono dati che indichino quali enzimi siano coinvolti nel metabolismo dell’amifampridina, né quali siano gli effetti dell’amifampridina sul metabolismo o la secrezione attiva di altri medicinali. Occorre quindi seguire con cura particolare i pazienti sottoposti a un trattamento concomitante con medicinali eliminati tramite metabolismo o secrezione attiva. Se possibile, il monitoraggio è consigliato. Se necessario, si dovrà adeguare la dose del medicinale somministrato in associazione. È controindicato l’uso concomitante di medicinali a basso indice terapeutico (vedere paragrafo 4.3).

4
Sostanze che sono potenti inibitori degli enzimi che metabolizzano i medicinali (vedere paragrafo 5.2) Il modo in cui l’amifampridina viene eliminata non è noto. Potenti inibitori di enzimi come per esempio cimetidina o ketoconazolo possono inibire il metabolismo dell’amifampridina, provocando un aumento dell’esposizione all’amifampridina. Quando si inizia il trattamento con un potente inibitore di enzimi, occorre controllare attentamente il manifestarsi nei pazienti di possibili reazioni avverse. Se il trattamento con un potente inibitore viene interrotto, è necessario controllare i pazienti per verificare l’efficacia, poiché può rendersi necessario un aumento dell’amifampridina. Sostanze che sono potenti induttori di enzimi che metabolizzano i medicinali (vedere paragrafo 5.2) Potenti induttori di enzimi che metabolizzano i medicinali, come per esempio i barbiturici, la carbamazepina, le rifamicine, possono aumentare l’eliminazione dell’amifampridina e provocare un’esposizione subterapeutica all’amifampridina. Nel corso delle prime settimane dopo l’inizio o la fine di tale trattamento può rendersi necessario un adeguamento della dose. Attualmente non esistono informazioni sull’interazione tra amifampridina e citocromo P450. Interazioni farmacodinamiche Sulla base delle proprietà farmacodinamiche di FIRDAPSE, occorre tener conto delle seguenti considerazioni: L’uso concomitante con il sultopride è controindicato, poiché tale associazione può aumentare il rischio di tachicardia ventricolare, e in particolare di torsione di punta (vedere paragrafi 4.3 e 5.1). Associazioni che richiedono precauzioni di impiego Medicinali di cui è noto l’effetto di abbassare la soglia epilettica L’uso concomitante di FIRDAPSE e di sostanze di cui sia noto l’effetto di abbassare la soglia epilettica aumenta il rischio di crisi. La decisione di somministrare sostanze pro-convulsive o tali da abbassare la soglia epilettica deve essere valutata con estrema attenzione, data la gravità dei rischi associati. Tali sostanze comprendono gran parte degli antidepressivi (antidepressivi triciclici, inibitori selettivi della captazione della serotonina), neurolettici (fenotiazine e butirrofenoni), mefloquina, bupropione e tramadolo (vedere paragrafi 4.4 e 5.1). Associazioni da valutare Medicinali con effetti atropinici L’uso concomitante di FIRDAPSE e di medicinali con effetti atropinici può ridurre l’effetto di entrambi i principi attivi e quindi deve essere valutato. I medicinali con effetti atropinici comprendono gli antidepressivi triciclici, gran parte degli antistaminici atropinici H1, gli anticolinergici, i medicinali anti-Parkinson, gli antispasmodici atropinici, la disopiramide, i neurolettici fenotiazinici e la clozapina. Medicinali con effetti colinergici L’uso concomitante di FIRDAPSE e di medicinali con effetti colinergici (per esempio inibitori diretti e indiretti della colinesterasi) può aumentare l’effetto di entrambi i principi attivi e quindi deve essere valutato. Medicinali con effetti miorilassanti non depolarizzanti L’uso concomitante di FIRDAPSE e di medicinali con effetti miorilassanti non depolarizzanti (per esempio mivacurium e pipercurium) può ridurre l’effetto di entrambi i principi attivi e quindi deve essere valutato. Medicinali con effetti miorilassanti depolarizzanti L’uso concomitante di FIRDAPSE e di medicinali con effetti miorilassanti depolarizzanti (per esempio sussametonio) può ridurre l’effetto di entrambi i principi attivi e quindi deve essere valutato.

5
4.6 Gravidanza e allattamento Non sono disponibili dati clinici adeguati sull’esposizione all’amifampridina in gravidanza. Non sono disponibili dati preclinici di sicurezza in merito agli effetti dell’amifampridina sulla funzione riproduttiva (vedere paragrafo 5.3). FIRDAPSE non deve essere usato durante la gravidanza. Uomini e donne potenzialmente fertili devono utilizzare metodi contraccettivi efficaci durante il trattamento con FIRDAPSE. Non è noto se l’amifampridina venga escreta nel latte materno umano. L’escrezione di amifampridina nel latte non è stata studiata negli animali. FIRDAPSE non deve essere usato durante l’allattamento. 4.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchinari Non sono stati effettuati studi sulla capacità di guidare veicoli e di usare macchinari. Tuttavia, a causa di reazioni avverse come sonnolenza, capogiri, convulsioni e visione offuscata, FIRDAPSE può alterare lievemente o moderatamente la capacità di guidare veicoli o di usare macchinari (vedere paragrafo 4.8). 4.8 Effetti indesiderati La sindrome miastenica di Lambert-Eaton è una patologia genetica estremamente rara. Di conseguenza, esistono scarsissime informazioni sulle reazioni avverse al trattamento con amifampridina, a causa del ridotto numero di pazienti interessati. Le reazioni avverse segnalate più frequentemente dalla letteratura pubblicata sono le parestesie (tra cui le parestesie periferiche e peribuccali) e le patologie gastrointestinali (tra cui epigastralgia, diarrea, nausea e dolori addominali). Intensità e incidenza di gran parte delle reazioni avverse sono dose-dipendenti. Sono state segnalate anche le seguenti reazioni avverse: • Disturbi psichiatrici: Disturbi del sonno. • Patologie del sistema nervoso: Convulsioni, ansia, sonnolenza, vertigini, senso di debolezza, spossatezza,
cefalea, corea, mioclonia. • Patologie dell’occhio: Visione offuscata. • Patologie cardiache: Disturbi del ritmo cardiaco, palpitazioni. • Patologie vascolari: Sindrome di Raynaud, estremità fredde. • Patologie respiratorie, toraciche e mediastiniche: Tosse, ipersecrezione bronchiale, attacchi asmatici in
pazienti asmatici o pazienti con un’anamnesi di asma. • Patologie epatobiliari: Livelli elevati di enzimi epatici (transaminasi). In considerazione dei limitatissimi dati disponibili, non è possibile stimare la frequenza delle singole reazioni avverse. 4.9 Sovradosaggio L’esperienza relativa al sovradosaggio è limitata. Dal momento che gli effetti sono dose-dipendenti, è prevedibile che il sovradosaggio acuto si manifesti con debolezza generale associata a parestesie diffuse, nausea, vomito, convulsioni e disturbi del ritmo cardiaco. In caso di sovradosaggio, il paziente deve interrompere il trattamento. Non sono noti antidoti specifici. Una terapia integrativa deve essere somministrata secondo le indicazioni cliniche, che comprendono un attento controllo dei segni vitali e delle condizioni cardiache del paziente.

6
5. PROPRIETÀ FARMACOLOGICHE 5.1 Proprietà farmacodinamiche Categoria farmacoterapeutica: altri medicinali per il sistema nervoso, codice ATC: N07XX05. L’amifampridina blocca i canali voltaggio-dipendenti del potassio, e in tal modo prolunga la depolarizzazione della membrana presinaptica cellulare. Il prolungamento del potenziale d’azione favorisce il trasporto del calcio nelle terminazioni nervose. Il conseguente incremento delle concentrazioni di calcio intracellulare agevola l’exocitosi delle vescicole contenenti acetilcolina, e ciò a sua volta favorisce la trasmissione neuromuscolare. Tutto questo migliora la forza muscolare e l’ampiezza della risposta muscolare (CMAP) a riposo di una media ponderata differenziale complessiva di 1,69 mV (95% CI da 0,60 a 2,77). Il profilo farmacodinamico dell’amifampridina è stato studiato su una gamma di dosi. Uno studio prospettivo randomizzato, controllato verso placebo, su 26 pazienti affetti da sindrome miastenica di Lambert-Eaton (LEMS) ha segnalato l’efficacia clinica dell’amifampridina alla dose standard massima raccomandata di 60 mg al giorno (Sanders et al 2000). Due studi ulteriori, su un totale di 57 pazienti affetti da LEMS, hanno riferito dati per dosi più elevate di amifampridina. McEvoy et al 1989 riporta i dati di uno studio a breve termine su 12 pazienti affetti da LEMS, il quale ha dimostrato che la somministrazione di amifampridina a dosi fino a 100 mg al giorno per un periodo di tre giorni è risultata efficace per il trattamento dei sintomi autonomici e motori della LEMS. Sanders et al 1998 offre dati sull’efficacia e la sicurezza del trattamento con amifampridina a dosi fino a 100 mg al giorno per 45 pazienti affetti da LEMS, sottoposti al trattamento per un periodo medio di 31 mesi. Perciò, in circostanze eccezionali dosi più elevate, fino a un massimo di 80 mg al giorno, possono avere effetti positivi se somministrate con un adeguato controllo di sicurezza. Si raccomanda di effettuare la titolazione della dose da 60 a 80 mg al giorno con aumenti di 5 mg ogni 7 giorni. La titolazione della dose verso l’alto deve essere interrotta se si riscontrano eventi avversi o anomalie nell’ECG. Questo medicinale è stato autorizzato in “circostanze eccezionali”. Ciò significa che data la rarità della malattia non è stato possibile ottenere informazioni complete su questo medicinale. L’Agenzia Europea dei Medicinali (EMA) esaminerà annualmente qualsiasi nuova informazione che si renderà disponibile sul medicinale e questo Riassunto delle Caratteristiche del Prodotto (RCP) verrà aggiornato, se necessario. 5.2 Proprietà farmacocinetiche Assorbimento: L’amifampridina viene assorbita rapidamente, e le concentrazioni di picco del plasma (Tmax) vengono raggiunte da 20 minuti a 1 ora dopo l’assunzione. L’effetto di una concomitante assunzione di cibo sull’assorbimento dell’amifampridina non è stato ancora studiato. Distribuzione: Non sono stati effettuati studi. Biotrasformazione: Non esistono dati sui metaboliti dell’amifampridina, e gli enzimi metabolizzanti non sono stati identificati. Eliminazione: La modalità di eliminazione dell’amifampridina non è nota. È possibile che l’amifampridina venga eliminata principalmente attraverso i reni. L’emivita di eliminazione è di circa 2 ore. 24 ore dopo la somministrazione l’amifampridina non è più rilevabile nel siero.

7
Popolazioni speciali: Non esistono dati sulla farmacocinetica dell’amifampridina nei pazienti pediatrici e nei pazienti affetti da insufficienza renale o epatica (vedere paragrafi 4.2 e 4.4). L’effetto dell’età sulla farmacocinetica dell’amifampridina non è stato studiato. 5.3 Dati preclinici di sicurezza I dati preclinici disponibili per l’amifampridina sono limitati. Negli studi di farmacologia della sicurezza sui topi, non si sono osservati effetti correlati al sistema nervoso centrale fino a 40 mg/kg. In uno studio di tossicità a dosi ripetute su topi e cani, si sono osservati affetti sul sistema nervoso centrale, un aumento del peso del fegato e dei reni, nonché effetti cardiaci (blocco atrioventricolare di secondo grado). Negli studi sugli animali non si sono ottenuti margini di sicurezza per l’esposizione umana, a causa della sensibilità dei modelli animali utilizzati. Non sono stati effettuati studi di tossicità a lungo termine di durata superiore alle 4 settimane. L’amifampridina non è risultata genotossica in una batteria standard di test in vitro e in vivo, ma non sono disponibili i risultati di studi di cancerogenicità completi. Non sono stati effettuati studi di cancerogenicità o tossicità in gravidanza. 6. INFORMAZIONI FARMACEUTICHE 6.1 Elenco degli eccipienti Cellulosa microcristallina Silice colloidale anidro Calcio stearato 6.2 Incompatibilità Non pertinente. 6.3 Periodo di validità 3 anni. 6.4 Precauzioni particolari per la conservazione Non conservare a temperatura superore ai 30°C. Conservare nella confezione originale per proteggere il medicinale dalla luce e dall’umidità. 6.5 Natura e contenuto del contenitore Blister termoformati perforati monodose (con copertura termoformata di alluminio-PVC/PVDC) contenenti 10 compresse. Una scatola contiene 100 compresse, suddivise in 10 strip da 10 compresse ciascuna.

8
6.6 Precauzioni particolari per lo smaltimento Il medicinale non utilizzato ed i rifiuti derivati da tale medicinale devono essere smaltiti in conformità alla normativa locale vigente. 7. TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO BioMarin Europe Limited, 164 Shaftesbury Avenue London, WC2H 8HL Regno Unito 8. NUMERO(I) DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO EU/1/09/601/001 9. DATA DELLA PRIMA AUTORIZZAZIONE/RINNOVO DELL’AUTORIZZAZIONE 23 Dicembre 2009 10. DATA DI REVISIONE DEL TESTO Informazioni più dettagliate su questo medicinale sono disponibili sul sito web della Agenzia Europea dei Medicinali (EMA): http://www.ema.europa.eu

9
ALLEGATO II
A. TITOLARE DELL’AUTORIZZAZIONE ALLA PRODUZIONE RESPONSABILE DEL RILASCIO DEI LOTTI
B. CONDIZIONI DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN
COMMERCIO
C. OBBLIGHI SPECIFICI PER IL TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO

10
A. TITOLARE DELL’AUTORIZZAZIONE ALLA PRODUZIONE RESPONSABILE DEL RILASCIO DEI LOTTI
Nome ed indirizzo del produttore responsabile del rilascio dei lotti AGEPS (Agence générale des équipements et produits de santé) 7, rue du Fer à Moulin F-75005 Paris Francia B. CONDIZIONI DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO • CONDIZIONI O LIMITAZIONI DI FORNITURA E DI UTILIZZAZIONE IMPOSTE AL
TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO Medicinale soggetto a prescrizione medica limitativa (Vedere Allegato I: Riassunto delle Caratteristiche del Prodotto, paragrafo 4.2). • CONDIZIONI O RESTRIZIONI PER QUANTO RIGUARDA L’USO SICURO ED
EFFICACE DEL MEDICINALE Non pertinente • ALTRE CONDIZIONI Sistema di farmacovigilanza Il titolare dell’autorizzazione all’immissione in commercio deve assicurare che il sistema di farmacovigilanza, come descritto nella versione 1.4 di data 15 ottobre 2009 presentata nel Modulo 1.8.1. della domanda di autorizzazione all’immissione in commercio, esista e sia operativo prima e durante la commercializzazione del medicinale. Piano di Gestione del Rischio (Risk Management Plan, RMP) Il titolare dell’autorizzazione all’immissione in commercio si impegna ad effettuare gli studi e le ulteriori attività di farmacovigilanza descritti nel piano di farmacovigilanza, come concordato nella versione 0.2 di data 20 aprile 2009 del RMP incluso nel Modulo 1.8.2. della domanda di autorizzazione all’immissione in commercio, e qualsiasi aggiornamento del RMP approvato dal Comitato per i Medicinali per Uso Umano (Committee for Medicinal Products for Human Use, CHMP). In accordo con la linea guida del CHMP sui sistemi di gestione del rischio per i medicinali per uso umano, il RMP aggiornato deve essere presentato insieme al successivo Rapporto periodico di aggiornamento sulla sicurezza (Periodic Safety Update Report, PSUR). Inoltre, il RMP aggiornato deve essere presentato:
• quando si ricevono nuove informazioni che possano avere impatto sulle specifiche di sicurezza, sul piano di farmacovigilanza o sulle attività di minimizzazione del rischio in vigore
• entro 60 giorni dal raggiungimento di un importante obiettivo (di farmacovigilanza o di minimizzazione del rischio)
• su richiesta dell’EMA
11
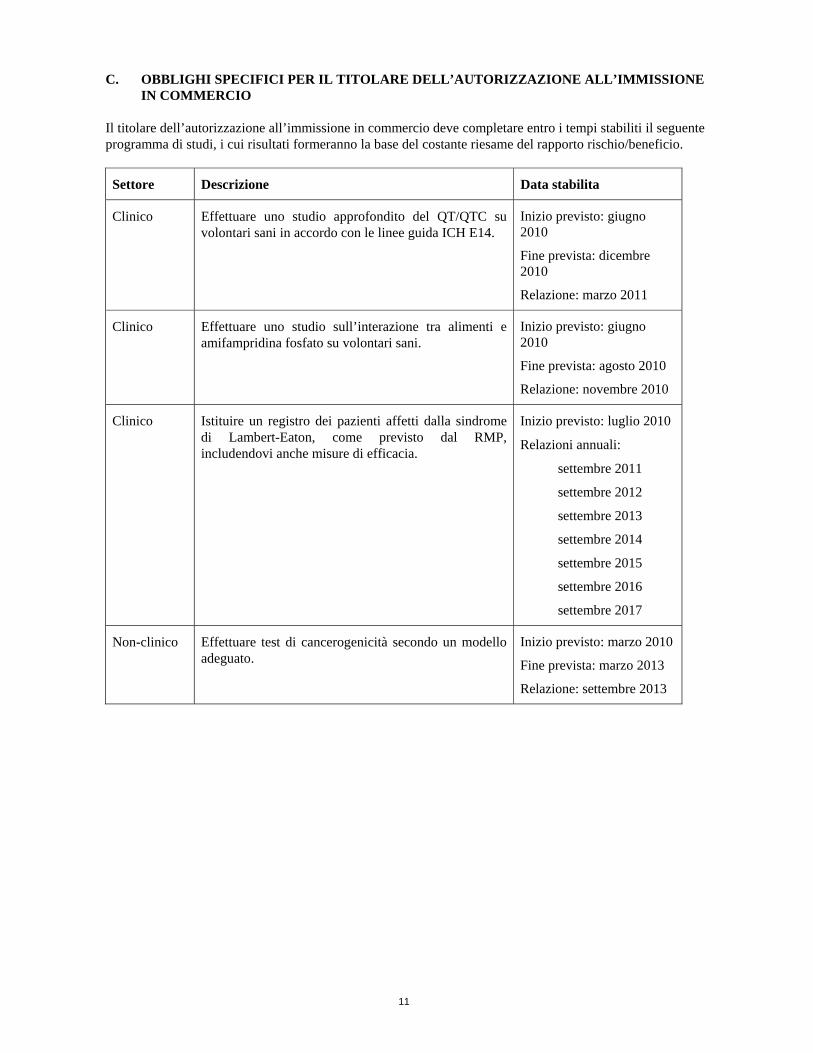
C. OBBLIGHI SPECIFICI PER IL TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
Il titolare dell’autorizzazione all’immissione in commercio deve completare entro i tempi stabiliti il seguente programma di studi, i cui risultati formeranno la base del costante riesame del rapporto rischio/beneficio.
Settore Descrizione Data stabilita
Clinico Effettuare uno studio approfondito del QT/QTC su volontari sani in accordo con le linee guida ICH E14.
Inizio previsto: giugno 2010
Fine prevista: dicembre 2010
Relazione: marzo 2011
Clinico Effettuare uno studio sull’interazione tra alimenti e amifampridina fosfato su volontari sani.
Inizio previsto: giugno 2010
Fine prevista: agosto 2010
Relazione: novembre 2010
Clinico Istituire un registro dei pazienti affetti dalla sindrome di Lambert-Eaton, come previsto dal RMP, includendovi anche misure di efficacia.
Inizio previsto: luglio 2010
Relazioni annuali:
settembre 2011
settembre 2012
settembre 2013
settembre 2014
settembre 2015
settembre 2016
settembre 2017
Non-clinico Effettuare test di cancerogenicità secondo un modello adeguato.
Inizio previsto: marzo 2010
Fine prevista: marzo 2013
Relazione: settembre 2013

12
ALLEGATO III
ETICHETTATURA E FOGLIO ILLUSTRATIVO

13
A: ETICHETTATURA

14
INFORMAZIONI DA APPORRE SUL CONFEZIONAMENTO SECONDARIO CARTONE 1. DENOMINAZIONE DEL MEDICINALE FIRDAPSE 10 mg compresse amifampridina 2. COMPOSIZIONE QUALITATIVA E QUANTITATIVA Ogni compressa contiene amifampridina fosfato equivalente a 10 mg di amifampridina. 3. ELENCO DEGLI ECCIPIENTI 4. FORMA FARMACEUTICA E CONTENUTO 100 compresse 5. MODO E VIA(E) DI SOMMINISTRAZIONE Uso orale Leggere il foglio illustrativo prima dell’uso. 6. AVVERTENZA PARTICOLARE CHE PRESCRIVA DI TENERE IL MEDICINALE FUORI
DALLA PORTATA E DALLA VISTA DEI BAMBINI Tenere fuori dalla portata e dalla vista dei bambini. 7. ALTRA(E) AVVERTENZA(E) PARTICOLARE(I) SE NECESSARIO 8. DATA DI SCADENZA Scad. {MM/AAAA} 9. PRECAUZIONI PARTICOLARI PER LA CONSERVAZIONE Non conservare a temperatura superiore ai 30ºC. Conservare nella confezione originale per proteggere il medicinale dalla luce e dall’umidità.

15
10. PRECAUZIONI PARTICOLARI PER LO SMALTIMENTO DEL MEDICINALE NON UTILIZZATO O DEI RIFIUTI DERIVATI DA TALE MEDICINALE, SE NECESSARIO 11. NOME E INDIRIZZO DEL TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO BioMarin Europe Limited, 164 Shaftesbury Avenue London, WC2H 8HL Regno Unito 12. NUMERO(I) DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO EU/1/09/601/001 13. NUMERO DI LOTTO Lotto 14. CONDIZIONE GENERALE DI FORNITURA Medicinale soggetto a prescrizione medica. 15. ISTRUZIONI PER L’USO 16. INFORMAZIONI IN BRAILLE FIRDAPSE

16
INFORMAZIONI MINIME DA APPORRE SU BLISTER O STRIP Blister termoformati perforati monodose 1. DENOMINAZIONE DEL MEDICINALE FIRDAPSE 10 mg amifampridine 2. NOME DEL TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO BioMarin Europe Limited 3. DATA DI SCADENZA EXP 4. NUMERO DI LOTTO Lot 5. ALTRO

17
B: FOGLIO ILLUSTRATIVO

18
FOGLIO ILLUSTRATIVO: INFORMAZIONI PER L’UTILIZZATORE
FIRDAPSE 10 mg compresse amifampridina
Legga attentamente questo foglio prima di prendere questo medicinale. • Conservi questo foglio. Potrebbe aver bisogno di leggerlo di nuovo. • Se ha qualsiasi dubbio, si rivolga al medico o al farmacista. • Questo medicinale è stato prescritto per lei. Non lo dia ad altre persone, anche se i loro sintomi sono
uguali ai suoi, perché potrebbe essere pericoloso. • Se uno qualsiasi degli effetti indesiderati peggiora, o se nota la comparsa di un qualsiasi effetto
indesiderato non elencato in questo foglio, consulti il medico o il farmacista. Contenuto di questo foglio: 1. Che cos’è FIRDAPSE e a che cosa serve 2. Prima di prendere FIRDAPSE 3. Come prendere FIRDAPSE 4. Possibili effetti indesiderati 5. Come conservare FIRDAPSE 6. Altre informazioni 1. CHE COS’È FIRDAPSE E A CHE COSA SERVE FIRDAPSE viene usato per trattare negli adulti i sintomi di una patologia neuromuscolare chiamata sindrome miastenica di Lambert-Eaton (LEMS). Tale patologia interessa la trasmissione degli impulsi nervosi ai muscoli, provocando debolezza muscolare. Essa può associarsi ad alcuni tipi di tumore (forma paraneoplastica di LEMS) o all’assenza di tali tumori (forma non paraneoplastica di LEMS). Nei pazienti affetti da questa patologia, una sostanza chimica denominata acetilcolina, che trasmette gli impulsi nervosi ai muscoli, non viene rilasciata normalmente e il muscolo non riceve, in tutto o in parte, i segnali nervosi. FIRDAPSE aumenta il rilascio dell’acetilcolina e favorisce la recezione dei segnali nervosi da parte dei muscoli.

19
2. PRIMA DI PRENDERE FIRDAPSE Non prenda FIRDAPSE • Se è allergico (ipersensibile) all’amifampridina, o ad uno qualsiasi degli eccipienti di FIRDAPSE, • Se è affetto da asma incontrollata, • Se è epilettico, • In associazione al sultopride (un medicinale prescritto per il trattamento di alcuni disturbi
comportamentali negli adulti). • In associazione a medicinali che possano alterare l’attività elettrica del cuore (prolungamento
dell’intervallo QT – individuabile nell’elettrocardiogramma) • In associazione a medicinali che hanno la dose terapeutica vicina alla dose massima di sicurezza • Se ha problemi cardiaci dalla nascita (sindrome congenita del QT). In caso di dubbi, si rivolga al medico o al farmacista. Faccia particolare attenzione con FIRDAPSE Informi il medico se ha • Asma • Un’anamnesi di convulsioni • Problemi renali • Problemi al fegato Il medico controllerà attentamente gli effetti di FIRDAPSE su di lei, e potrà modificare la dose dei medicinali che lei assume. Egli inoltre le effettuerà una vista cardiologica all’inizio del trattamento e in seguito annualmente. Se lei è affetto da LEMS ma non da tumori, prima di cominciare il trattamento il medico effettuerà un’accurata valutazione dei potenziali rischi di tumore associati a FIRDAPSE. Informi tutti i medici che consulta del fatto che sta usando FIRDAPSE. Interrompa il trattamento e consulti immediatamente il medico in caso di: • Convulsioni • Asma. Assunzione di FIRDAPSE con altri medicinali Potrebbe essere necessario prendere precauzioni particolari o modificare il dosaggio di FIRDAPSE, se sta assumendo FIRDAPSE insieme ad altri medicinali. È particolarmente importante informare il medico se sta assumendo uno dei seguenti medicinali: • Antimalarici (per esempio alofantrina e mefloquina) • Disopiramide (un farmaco antiaritmico) • Tramadol (un antidolorifico) • Antidepressivi – antidepressivi triciclici (per esempio clomipramina, amoxapina), inibitori selettivi della
ricaptazione della serotonina (per esempio citalopram, dapoxetina) e antidepressivi atipici (per esempio buproprione)
• Medicinali per disturbi mentali (per esempio aloperidolo, carbamazapina, cloropromazina, clozapina) • Medicinali anti-Parkinson - anticolinergici (per esempio triesilfenidile, mesylato), inibitori MAO-B (per
esempio selegilina, deprenyl), inibitori COMT (per esempio entacapone) • Antiallergici - antistaminici (per esempio terfenadina, astemizolo, cimetidina) • Medicinali per il trattamento di disturbi della digestione (per esempio cisapride, domperidone) • Medicinali per il trattamento delle infezioni - antibiotici (per esempio rifampicina) e antifungini (per
esempio ketoconazolo) • Miorilassanti - (per esempio mivacurium, pipercurium, sussametonio) • Sedativi (per esempio barbiturici)

20
Informi il medico se sta assumendo o ha recentemente assunto qualsiasi altro medicinale, compresi quelli senza prescrizione medica. Gravidanza e allattamento FIRDAPSE non deve essere usato durante la gravidanza. Deve fare uso di un contraccettivo efficace durante il trattamento. Se la paziente scopre di essere incinta durante il trattamento, dovrà informare immediatamente il medico. Durante il trattamento con FIRDAPSE non si deve allattare. Chieda consiglio al medico o al farmacista prima di prendere qualsiasi medicinale. Guida di veicoli e utilizzo di macchinari Questo medicinale può provocare sonnolenza, CAPOGIRI, convulsioni e visione offuscata e quindi può alterare la capacità di guidare veicoli o usare macchinari. Non guidi e non usi macchinari se riscontra tali effetti indesiderati. 3. COME PRENDERE FIRDAPSE Prenda sempre FIRDAPSE seguendo esattamente le istruzioni del medico. Se ha dubbi consulti il medico o il farmacista. La dose da assumere viene stabilita dal medico in base all’intensità dei sintomi. Tale dose è adatta solo a lei. La dose iniziale è di 5 mg (mezza compressa) tre volte al giorno (cioè 15 mg al giorno). Il medico può aumentare gradualmente tale dosaggio, dapprima a 5 mg (mezza compressa) quattro volte al giorno (cioè 20 mg al giorno). In seguito il medico può continuare ad aumentare la dose giornaliera complessiva aggiungendo 5 mg (mezza compressa) al giorno, ogni 4 o 5 giorni. La dose massima raccomandata è di 60 mg al giorno (cioè un totale di sei compresse da prendere a intervalli durante la giornata). Le dosi giornaliere complessive superiori a 20 mg devono essere divise in dosi separate (da due a quattro). La dose singola non deve mai superare i 20 mg (due compresse). Le compresse hanno una linea di incisione che consente di dividerle a metà. Assumere le compresse con un po’ d’acqua ingerendo anche del cibo. Pazienti affetti da patologie renali e del fegato: I pazienti affetti da patologie renali e del fegato devono usare FIRDAPSE con cautela. Si raccomanda una dose iniziale di 5 mg di FIRDAPSE ai pazienti affetti da insufficienza moderata o grave della funzione renale o del fegato. Ai pazienti affetti da insufficienza lieve della funzione renale o del fegato si raccomanda una dose iniziale di 10 mg di FIRDAPSE. In questi pazienti, la dose di FIRDAPSE deve essere aumentata più lentamente, rispetto ai pazienti che non lamentano disturbi renali o del fegato: aumentare la dose di 5 mg ogni 7 giorni. In caso di eventi avversi, si prega di consultare il medico, in quanto potrebbe essere necessario interrompere l’aumento della dose. Se prende più FIRDAPSE di quanto deve Se prende più FIRDAPSE di quanto deve, potrà avvertire nausea e debolezza, e un lieve formicolio o intorpidimento in alcune parti del corpo. A seconda della quantità di FIRDAPSE assunta, potrebbe anche accusare convulsioni, vomito o problemi cardiaci (disturbi del ritmo cardiaco). Se avverte uno qualsiasi di questi sintomi, consulti immediatamente il medico o il farmacista.

21
Se dimentica di prendere FIRDAPSE Se dimentica di prendere FIRDAPSE, non prenda una dose doppia per compensare la dose dimenticata, ma continui il trattamento secondo le prescrizioni del medico. Se interrompe il trattamento con FIRDAPSE Se interrompe il trattamento, potrebbe avvertire sintomi come stanchezza, riflessi lenti e stipsi. Non interrompa il trattamento senza consultare il medico. Se ha qualsiasi dubbio sull’uso di FIRDAPSE, si rivolga al medico o al farmacista. 4. POSSIBILI EFFETTI INDESIDERATI Come tutti i medicinali, FIRDAPSE può causare effetti indesiderati, sebbene non tutte le persone li manifestino. Interrompa il trattamento e consulti immediatamente il medico in caso di: • Convulsioni • Asma Gli effetti indesiderati segnalati più frequentemente sono: • Formicolio e intorpidimento intorno alla bocca e alle estremità (mani e piedi), • Mal di stomaco, diarrea, nausea e dolori addominali. Atri effetti indesiderati: L’intensità e l’incidenza di gran parte degli effetti indesiderati dipende dalla dose che sta assumendo. Sono stati segnalati, tra gli altri, i seguenti effetti indesiderati (la frequenza non può essere definita sulla base dei dati disponibili): • Convulsioni, • Tosse, muco eccessivo o viscoso nelle vie aeree, attacchi asmatici nei pazienti asmatici o in quelli che
hanno avuto episodi di asma in passato, • Sindrome di Raynaud (disturbo circolatorio che interessa le dita delle mani e dei piedi), mani e piedi
freddi, • Visione offuscata, • Disturbi del ritmo cardiaco, battito cardiaco accelerato o irregolare (palpitazioni), • Debolezza, stanchezza, cefalea, • Ansia, capogiri, disturbi del sonno, sonnolenza, • Corea (disturbo motorio), mioclonia (spasmo o contrazione muscolare), • Aumento di alcuni enzimi del fegato (transaminasi) rivelato dalle analisi del sangue. Se uno qualsiasi degli effetti indesiderati peggiora, o se nota la comparsa di un qualsiasi effetto indesiderato non elencato in questo foglio illustrativo, informi il medico o il farmacista. 5. COME CONSERVARE FIRDAPSE Tenere FIRDAPSE fuori dalla portata e dalla vista dei bambini. Non usi FIRDAPSE dopo la data di scadenza che è riportata sulla confezione dopo Scad. La data di scadenza si riferisce all’ultimo giorno del mese. Non conservare a temperatura superiore ai 30°C. Conservare nella confezione originale, per proteggere il medicinale dalla luce e dall’umidità. I medicinali non devono essere gettati nell’acqua di scarico e nei rifiuti domestici. Chieda al farmacista come eliminare i medicinali che non utilizza più. Questo aiuterà a proteggere l’ambiente.

22
6. ALTRE INFORMAZIONI Cosa contiene FIRDAPSE • Il principio attivo è l’amifampridina. Ogni compressa contiene amifampridina fosfato equivalente a
10 mg di amifampridina. • Gli eccipienti sono cellulosa microcristallina, silice colloidale anidro e calcio stearato. Descrizione dell’aspetto di FIRDAPSE e contenuto della confezione Compressa bianca, rotonda, piatta, incisa su un lato. La compressa può essere divisa in due metà uguali. Blister termoformati perforati monodose (con copertura termoformata di alluminio-PVC/PVDC) contenenti 10 compresse. Una scatola contiene 100 compresse, suddivise in 10 strip da 10 compresse ciascuna. Titolare dell’autorizzazione all’immissione in commercio e produttore Titolare dell’autorizzazione all’immissione in commercio: BioMarin Europe Limited, 164 Shaftesbury Avenue London, WC2H 8HL Regno Unito Produttore: AGEPS-EPHP (Agence Générale des Equipements et Produits de Santé – Etablissement pharmaceutique des hôpitaux de Paris) 7, rue du Fer à Moulin – BP 09 F-75221 Paris Cedex 05 FRANCIA Questo foglio illustrativo è stato approvato l’ultima volta nel {MM/AAAA} A questo medicinale è stata rilasciata una autorizzazione in “circostanze eccezionali”. Ciò significa che data la rarità della sua malattia non è stato possibile ottenere informazioni complete su questo medicinale. L’Agenzia Europea dei Medicinali (EMA) esaminerà annualmente qualsiasi nuova informazione sul medicinale e questo foglio illustrativo verrà aggiornato, se necessario. Informazioni più dettagliate su questo medicinale sono disponibili sul sito web della Agenzia Europea dei Medicinali (EMA): http://www.ema.europa.eu


















