
Nghiên cứu lý thuyết cấu trúc, một số tính ấ ại...
14
Nghiên cứu lý thuyết cấu trúc, một số tính chất của các clusters kim loại và lưỡng kim loại của Rhodi Trần Diệu Hằng Trường Đại học Khoa học tự nhiên Luận văn ThS. ngành: Hóa lý thuyết và hóa lý; Mã số: 60 44 31 Người hướng dẫn: PGS.TS. Nguyễn Thị Minh Huệ Năm bảo vệ: 2013 Abstract. Tìm hiểu cơ sở hóa học lượng tử, các phương pháptính toán và các phần mềm tính toán được sử dụng trong hóa học lượng tử. Sưu tầm và đọc các bài báo, các tài liệu về các cluster kim loại và lưỡng kim loại của các nguyên tố, đặc biệt là các kim loại chuyển tiếp. Lựa chọn phương pháp tính toán tốt nhất để khảo sát hệ nghiên cứu. Sử dụng phương pháp đã chọn để tối ưu hóa cấu trúc, tính năng lượng điểm đơn, năng lượng điểm không...để tìm ra cấu trúc bền nhất của các cluster kim loại và lưỡng kim loại của rhodi. Từ các thông số thu được về cấu trúc và năng lượng của các cluster lưỡng kim loại, so sánh các kết quả để tìm ra quy luật và sự biến đổi về bán kính, cấu trúc, năng lượng của các cluster rhodi trước và sau khi pha tạp nguyên tố khác. Từ các kết quả thu được, nghiên cứu một số tính chất của các cluster kim loại và lưỡng kim loại Rhodi. Keywords. Hóa lý học; Kim loại; Lưỡng kim loại; Hóa học lượng tử; Lý thuyết cấu trúc Content MỞ ĐẦU 1. Lí do chọn đề tài Cùng với sự phát triển không ngừng của kinh tế, hiện nay, khoa học công nghệ cũng đãvà đang đạt được những thành tựu vượt bậc đánh dấu những bước tiến quan trọng trong sự phát triển của toàn nhân loại. Trong đó, đáng chú ý nhất chính là sự ra đời và phát triển nhanh chóng của một lĩnh vực mới được gọi là khoa học nano.Lĩnh vực này mở rộng sang vật lý, hóa học, kỹ thuật và các lĩnh vực khác, từ khoa học cơ bản cho đến một loạt các ứng dụng công nghệ, vì thế nó được gọi là công nghệ nano.Những năm gần đây, công nghệ nano ra đời không những tạo nên bước nhảy đột phá trong ngành hóa học vật liệu, điện tử, tin học, y sinh học mà còn được ứng dụng rộng rãi trong đời sống như gạc chữa bỏng được phủ nano bạc, nước rửa rau sống, chất diệt khuẩn khử mùi trong máy lạnh…Công nghệ nano làm thay đổi cuộc sống của chúng ta nhờ vào khả năng can thiệp của con người tại kích thước nanomet(nm). Tại quy mô đó, vật liệu nano thể hiện những tính chất đặc biệt và lý thú khác hẳn với tính chất của chúng ở các kích thước lớn hơn.
Transcript of Nghiên cứu lý thuyết cấu trúc, một số tính ấ ại...

Nghiên cứu lý thuyết cấu trúc, một số tính
chất của các clusters kim loại và lưỡng kim
loại của Rhodi
Trần Diệu Hằng
Trường Đại học Khoa học tự nhiên
Luận văn ThS. ngành: Hóa lý thuyết và hóa lý; Mã số: 60 44 31
Người hướng dẫn: PGS.TS. Nguyễn Thị Minh Huệ
Năm bảo vệ: 2013
Abstract. Tìm hiểu cơ sở hóa học lượng tử, các phương pháptính toán và các phần
mềm tính toán được sử dụng trong hóa học lượng tử. Sưu tầm và đọc các bài báo,
các tài liệu về các cluster kim loại và lưỡng kim loại của các nguyên tố, đặc biệt là
các kim loại chuyển tiếp. Lựa chọn phương pháp tính toán tốt nhất để khảo sát hệ
nghiên cứu. Sử dụng phương pháp đã chọn để tối ưu hóa cấu trúc, tính năng lượng
điểm đơn, năng lượng điểm không...để tìm ra cấu trúc bền nhất của các cluster kim
loại và lưỡng kim loại của rhodi. Từ các thông số thu được về cấu trúc và năng
lượng của các cluster lưỡng kim loại, so sánh các kết quả để tìm ra quy luật và sự
biến đổi về bán kính, cấu trúc, năng lượng của các cluster rhodi trước và sau khi pha
tạp nguyên tố khác. Từ các kết quả thu được, nghiên cứu một số tính chất của các
cluster kim loại và lưỡng kim loại Rhodi.
Keywords. Hóa lý học; Kim loại; Lưỡng kim loại; Hóa học lượng tử; Lý thuyết cấu
trúc
Content
MỞ ĐẦU
1. Lí do chọn đề tài
Cùng với sự phát triển không ngừng của kinh tế, hiện nay, khoa học công nghệ cũng
đãvà đang đạt được những thành tựu vượt bậc đánh dấu những bước tiến quan trọng trong sự
phát triển của toàn nhân loại. Trong đó, đáng chú ý nhất chính là sự ra đời và phát triển nhanh
chóng của một lĩnh vực mới được gọi là khoa học nano.Lĩnh vực này mở rộng sang vật lý,
hóa học, kỹ thuật và các lĩnh vực khác, từ khoa học cơ bản cho đến một loạt các ứng dụng
công nghệ, vì thế nó được gọi là công nghệ nano.Những năm gần đây, công nghệ nano ra đời
không những tạo nên bước nhảy đột phá trong ngành hóa học vật liệu, điện tử, tin học, y sinh
học mà còn được ứng dụng rộng rãi trong đời sống như gạc chữa bỏng được phủ nano bạc,
nước rửa rau sống, chất diệt khuẩn khử mùi trong máy lạnh…Công nghệ nano làm thay đổi
cuộc sống của chúng ta nhờ vào khả năng can thiệp của con người tại kích thước
nanomet(nm). Tại quy mô đó, vật liệu nano thể hiện những tính chất đặc biệt và lý thú khác
hẳn với tính chất của chúng ở các kích thước lớn hơn.

Trong số các vật liệu có kích thước nano, các cluster chiếm một vị trí rất quan trọng vì
chúng là các khối xây dựng nên khoa học nano. Các cluster được định nghĩa là một tập hợp
có từ một vài đến hàng ngàn nguyên tử ở kích cỡ nm hoặc nhỏ hơn.Chính vì ở kích thước nm
nên nó có những tính chất vật lý và hóa học khác biệt với khi ở dạng khối.Có lẽminh chứng
rõ ràng nhất cho hiện tượng này đó chính là việc khám phá ra các cluster kim loại vàng, một
vật liệu được biết đến với sự thụ động hóa học của nó khi ở dạng khối, nhưng lại hoạt động
hóa học mạnh và trở thành vật liệu xúc tác tuyệt vời cho nhiều phản ứng như oxy hóa CO,
khử NO [7, 36, 39]... Walter Knight và các cộng sự [55] đã mở ra một kỉ nguyên mới trong
lĩnh vực nghiên cứu cluster khi điều chế và phát hiện ra các cluster kim loại kiềm có đến 100
nguyên tửbằng cách cho bay hơi kim loại natri và dẫn hơi kim loại qua ống phun siêu âm.
Các nghiên cứu được mở rộng với những cluster kim loại có kích thước lớn hơn, nhưng có
lớp vỏ electron giống với các cluster kim loại kiềm đó là các cluster thuộc nhóm kim loại
quý: Cu, Ag, Au…và các kim loại chuyển tiếp có phân lớp d chưa bão hòa. Nhữngelectron ở
orbitan dchưa bão hòa đóng vai trò quan trọng trong quá trình hình thành liên kết hóa học và
vì thế nó được dự đoán sẽ tạo ra những đặc tính khác biệt đối với các cluster. Hơn nữa, các
cluster kim loại khi tương tác với ánh sáng, chúng sẽ trải qua các chuyển mức năng lượng, hệ
quả là xảy ra các hiện tượng hấp thụ, phát xạ ánh sáng và hiện tượng cộng hưởng bề mặt.
Tính chất quang học của các hạt có kích thước nm của các kim loại đã được nghiên cứu trong
những năm gần đây và được đưa vào ứng dụng có hiệu quả cao trong công nghiệp hiện đại
được ứng dụng trong nghiên cứu về y học, vật liệu bán dẫn….Tuy nhiên, vẫn chưa có lí
thuyết đầy đủ để giúp chúng ta dự đoán các cấu trúc bền của các cluster kim loại ở trong các
phân tử và các chất rắn. Hơn nữa, chúng ta vẫn tương đối ít biết về mối quan hệ phức tạp và
tinh vi giữa cấu trúc, electron và nguyên tử với độ bền và khả năng phản ứng của hợp chất.
Do đó, việc nghiên cứu các tính chất độc đáo, khác biệt và khả năng ứng dụng rộng rãi của
các cluster của nhiều kim loại chuyển tiếp đang là mối quan tâm của rất nhiều nhà khoa học
trên thề giới. Trong đó, rhodi là một trong những kim loại chuyển tiếp đang được quan tâm
và nghiên cứu hiện nay vì những ứng dụng to lớn của kim loại này trong các lĩnh vực khác
nhau như là sản xuất sợi thủy tinh, làm điện cực cho bugi của tàu bay, làm vật liệu chế tạo
tiếp điểm do điện trở thấp…và đặc biệt là chất xúc tác hữu ích của nhiều quy trình công
nghiệp, đáng chú ý là nó được sử dụng trong hệ thống xúc tác của các bộ chuyển đổi xúc
tác trong ô tô và để cacbonyl hóa có xúc tác của metanol nhằm sản xuất axít axetic theo quy
trình Monsanto [8, 20, 21]. Đã có nhiều công trình nghiên cứu về cấu trúc của cluster kim
loại rhodi và các cluster của rhodi với các nguyên tố khác như rutheni, hidro…hay phân tử
khác như CO…nhưng chưa có nghiên cứu nào về cấu trúc cũng như tính chất của các cluster
lưỡng kim loại của rhodi với Ni, Co...
Hóa học lượng tử là một ngành khoa học ứng dụng cơ học lượng tử vào giải quyết các
vấn đề của hóa học. Cụ thể nó cho phép tiến hành các nghiên cứu lí thuyết về cấu trúc phân
tử và khả năng phản ứng, giúp tiên đoán nhiều thông số của phản ứng trước khi tiến hành thí
nghiệm. Hơn thế nữa,cùng với sự tiến bộ của công nghệ số trong thời đại ngày nay, máy tính
có thể tính toán một cách nhanh chóng những phép tính phức tạp, giúp cho việc phát triển
cácphương pháp và phần mềm tính toán hóa học lượng tử. Áp dụng các phương pháp và phần
mềm này để tính toán không những cho biết các tham số về cấu trúc, về các loại năng lượng,
bề mặt thế năng, cơ chế phản ứng, các thông số nhiệt động lực học… mà còn cho chúng ta
biết các thông tin về phổ hồng ngoại, phổ khối lượng, phổ UV-VIS... Nhờ vậy các phương
pháp tính hóa học lượng tử và các phần mềm tính toán trở thành công cụ đắc lực trong việc
nghiên cứu, khảo sát các cấu trúc phân tử, cơ chế của rất nhiều phản ứng hóa học trong các
điều kiện khác nhau mà đôi khi thực nghiệm rất khó thực hiện hoặc không thể thực hiện
được. Điều đó cho thấy tầm quan trọng của việc nghiên cứu lí thuyết bằng cách sử dụng các
phần mềm tính toán hóa học lượng tử hiện đại. Với tất cả những lí do trên, chúng tôi chọn đề

tài: “ Nghiên cứu lý thuyết cấu trúc, một số tính chất của các clusters kim loại và lưỡng
kim loại của Rhodi”.
Chƣơng 1. CƠ SỞ LÍ THUYẾT VỀ HÓA HỌC LƢỢNG TỬ
1.1.Phƣơng trình Schrödinger
1.2. Toán tử Hamilton
1.3. Hàm sóng của hệ nhiều eletron
1.4. Cấu hình eletron và bộ hàm cơ sở
1.4.1. Cấu hình electron
1.4.2. Bộ hàm cơ sở
1.4.2.2. Một số khái niệm về bộ hàm cơ sở
1.4.2.3. Phân loại bộ hàm cơ sở .
1.5. Các phƣơng pháp gần đúng hóa học lƣợng tử
1.5.1. Phƣơng pháp Hartree-Fock
1.5.2. Phƣơng pháp nhiễu loạn Møller-Plesset (MPn)
1.5.2.1. Lý thuyết nhiễu loạn cho bài toán không suy biến
1.5.2.2. Lý thuyết nhiễu loạn cho bài toán suy biến
1.5.3. Phƣơng pháp tƣơng tác cấu hình (CI)
1.5.4. Phƣơng pháp tƣơng tác chùm (CC)
1.6. Phƣơng pháp phiếm hàm mật độ (DFT - Density Functional Theory)
1.6.1. Mô hình Thomas - Fermi
1.6.2. Các định lý Hohenberg-Kohn
1.6.3. Các phƣơng trình Hohenberg-Kohn
1.6.4. Một số phiếm hàm trao đổi
1.6.5. Một số phiếm hàm tƣơng quan
1.6.6. Các phiếm hàm hỗn hợp
1.6.7. Một số phƣơng pháp DFT thƣờng dùng
Chƣơng 2. TỔNG QUAN VỀ HỆ CHẤT NGHIÊN CỨU
2.1. Hệ chất nghiên cứu
2.1.1. Cluster kim loại
2.1.2. Cluster lƣỡng kim loại
2.1.3. Kim loại Rhodi
2.2. Phƣơng pháp nghiên cứu
2.2.1. Phần mềm tính toán
Để nghiên cứu các cluster kim loại và lưỡng kim loại Rhodi (Rhn và Rhn-1M với M =
Fe, Co, Ni) bằng phương pháp hoá học lượng tử, chúng tôi đã sử dụng hai phần mềm chính là
Gaussian 03 và Gaussview.
2.2.2. Phƣơng pháp nghiên cứu
Khảo sát các phương pháp thuộc nhóm phương pháp phiếm hàm mật độ(DFT) như
B3LYP, BP86, B3PW91, BPE1…để lựa chọn phương pháp phù hợp với chất nghiên cứu.
Tối ưu hoá cấu trúc đồng thời tính năng lượng điểm đơn, năng lượng điểm không và
các thông số nhiệt động học của các phân tử theo phương pháp đã chọn ở trên với bộ hàm cơ sở
tương ứng là LANL2DZ, Aug-cc-pvdz-pp, Aug-cc-pvtz-pp …
Sau khi tìm được các cấu trúc bền nhất của các cluster rhodi, chúng tôi thay các
nguyên tử rhodi bằng các kim loại khác như Fe, Co, Ni. Tiến hành tối ưu hóa để tìm ra các
cấu trúc có năng lương cực tiểu. Từ các cấu trúc bền thu được của các cluster kim loại và
lưỡng kim loại rhodi khảo sát một số tình chất của các cluster này như năng lượng ion hóa,

năng lượng liên kết, năng lượng liên kết trung bình, mức chêch lệch năng lượng LUMO-
HOMO…
Sử dụng phương pháp phiếm hàm mật độ phụ thuộc thời gian (TD-DFT) để xác định phổ
UV-VIS của một số cluster kim loại và lưỡng kim loại của rhodi.
Chƣơng 3. KẾT QUẢ VÀ THẢO LUẬN
3.1. KHẢO SÁT PHƢƠNG PHÁP TÍNH TOÁN
Chúng tôi chọn một số phương pháp phiếm hàm mật độ DFT thường được sử dụng
như: B3LYP, B3PW91, PB86, PBE1…để xác định cấu trúc và tính chất của cluster Rh2, từ
kết quả thu được so sánh với số liệu thực nghiệm. Phương pháp phù hợp tốt nhất sẽ được lựa
chọn để khảo sát các cluster Rhn và Rhn-1M với M = Fe, Co, Ni trong khuôn khổ luận văn
này. Từ các tính toán trên ta thu được các giá trị sau:
Bảng 3.1. Giá trị độ dài liên kết Rh-Rh (Å) và năng lượng liên kết trung bình (eV) ELKTB của
cluster Rh2
Rh2 B3LYP B3PW91 BP86 Thực nghiệm
Độ bội spin 5 5 5 5
Độ dài liên kết Rh-Rh (Å) 2,320 2,303 2,257 2,28
ELKTB (eV) 0,744 0,751 1,488 1,46
Phân tích các kết quả trên chúng tôi nhận thấy phương pháp BP86 cho giá trị năng
lượng thấp nhất bên cạnh đó các giá trị về độ dài liên kết và năng lượng liên kết trung bình
phù hợp tốt nhất với các số liệu thực nghiệm (bảng 3.1). Chúng tôi cũng tính toán năng lượng
ion hóa thứ nhất của nguyên tử kim loại rhodi theo các phương pháp khác nhau và kết quả
cho thấy phương pháp BP86 với giá trị thế ion hóa thứ nhất là 7,84 eV là phương pháp cho
giá trị gần với thực nghiệm nhất 7,46 eV. Như vậy phương pháp BP86 là phương pháp phù
hợp nhất trong khuôn khổ các phương pháp khảo sát. Vì vậy để tối ưu hóa cấu trúc hình học,
xác định năng lượng và một số tính chất của cluster Rhn và Rhn-1M chúng tôi sử dụng phương
pháp này cùng với các bộ hàm cơ sở thích hợp.
3.2. CẤU TRÚC VÀ TÍNH CHẤT CỦA CLUSTER Rhn
3.2.1. Khảo sát dạng bền của các cluster Rhn
Sử dụng phương pháp BP86 với bộ hàm LANL2DZ, chúng tôi khảo sát một số
cấu trúc có thể có của các cluster Rhn.Bảng 3.3 chỉ ra các cấu trúc hình học có năng lượng
thấp và các dạng đồng phân của các cluster Rhn với n = 2-13. Chúng tôi sử dụng dấu chấm (.)
ngăn cách giữa hàng đơn vị và hàng thập phân trong số liệu độ dài liên kết trong tất cả các
cấu trúc.
Từ những tính toán bước đầu ta đã thu được một số kết quả về giá trị năng lượng
tương ứng với các đồng phân trên của cluster Rhn, từ các giá trị này cấu trúc bền nhất
của mỗi dạng cluster là cấu trúc có năng lượng thấp nhất.
3.2.2 Tính chất của các cluster Rhn bền
Các dạng bền của các cluster Rhn được biểu diễn trong bảng 3.5. Các dạng bền của các
Rhn tương ứng đều có cấu trúc khá đối xứng trong nhóm các đồng phân có năng lượng thấp.
Bảng 3.5. Cấu trúc bền của các cluster Rhn
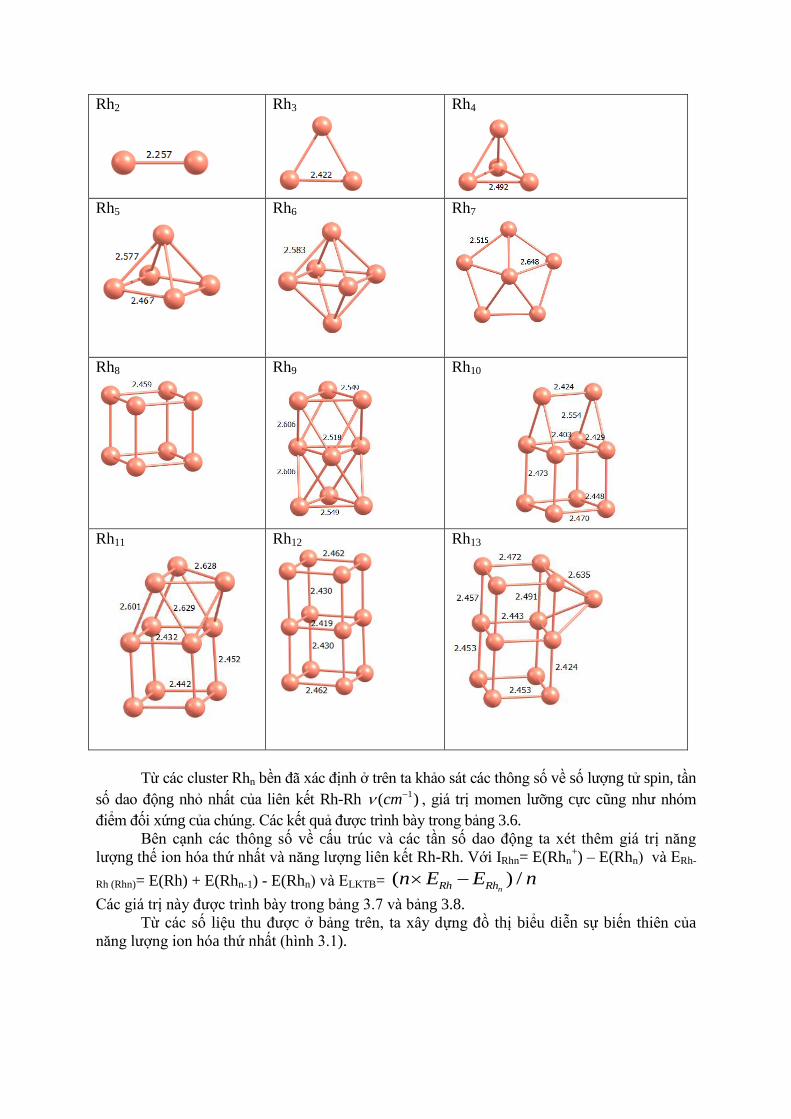
Rh2
Rh3
Rh4
Rh5
Rh6
Rh7
Rh8
Rh9
Rh10
Rh11
Rh12
Rh13
Từ các cluster Rhn bền đã xác định ở trên ta khảo sát các thông số về số lượng tử spin, tần
số dao động nhỏ nhất của liên kết Rh-Rh 1( )cm , giá trị momen lưỡng cực cũng như nhóm
điểm đối xứng của chúng. Các kết quả được trình bày trong bảng 3.6.
Bên cạnh các thông số về cấu trúc và các tần số dao động ta xét thêm giá trị năng
lượng thế ion hóa thứ nhất và năng lượng liên kết Rh-Rh. Với IRhn= E(Rhn+) – E(Rhn) và ERh-
Rh (Rhn)= E(Rh) + E(Rhn-1) - E(Rhn) và ELKTB= ( ) /nRh Rhn E E n
Các giá trị này được trình bày trong bảng 3.7 và bảng 3.8.
Từ các số liệu thu được ở bảng trên, ta xây dựng đồ thị biểu diễn sự biến thiên của
năng lượng ion hóa thứ nhất (hình 3.1).
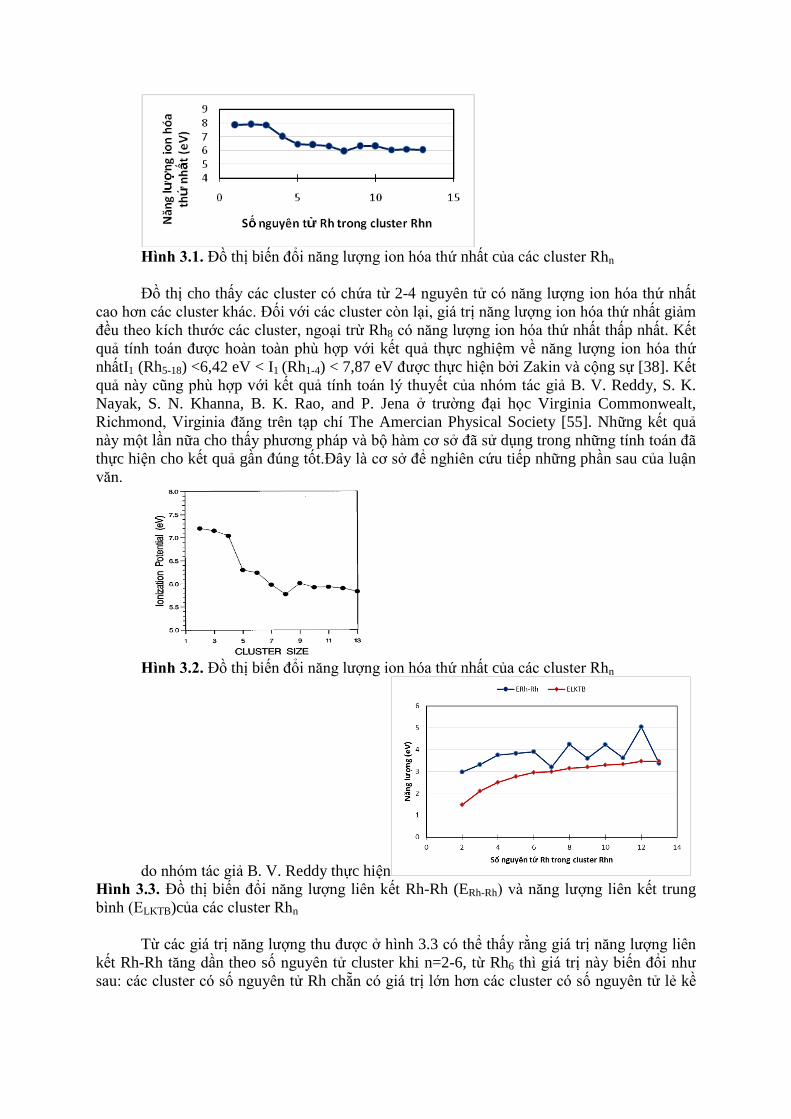
Hình 3.1. Đồ thị biến đổi năng lượng ion hóa thứ nhất của các cluster Rhn
Đồ thị cho thấy các cluster có chứa từ 2-4 nguyên tử có năng lượng ion hóa thứ nhất
cao hơn các cluster khác. Đối với các cluster còn lại, giá trị năng lượng ion hóa thứ nhất giảm
đều theo kích thước các cluster, ngoại trừ Rh8 có năng lượng ion hóa thứ nhất thấp nhất. Kết
quả tính toán được hoàn toàn phù hợp với kết quả thực nghiệm về năng lượng ion hóa thứ
nhấtI1 (Rh5-18) <6,42 eV < I1 (Rh1-4) < 7,87 eV được thực hiện bởi Zakin và cộng sự [38]. Kết
quả này cũng phù hợp với kết quả tính toán lý thuyết của nhóm tác giả B. V. Reddy, S. K.
Nayak, S. N. Khanna, B. K. Rao, and P. Jena ở trường đại học Virginia Commonwealt,
Richmond, Virginia đăng trên tạp chí The Amercian Physical Society [55]. Những kết quả
này một lần nữa cho thấy phương pháp và bộ hàm cơ sở đã sử dụng trong những tính toán đã
thực hiện cho kết quả gần đúng tốt.Đây là cơ sở để nghiên cứu tiếp những phần sau của luận
văn.
Hình 3.2. Đồ thị biến đổi năng lượng ion hóa thứ nhất của các cluster Rhn
do nhóm tác giả B. V. Reddy thực hiện
Hình 3.3. Đồ thị biến đổi năng lượng liên kết Rh-Rh (ERh-Rh) và năng lượng liên kết trung
bình (ELKTB)của các cluster Rhn
Từ các giá trị năng lượng thu được ở hình 3.3 có thể thấy rằng giá trị năng lượng liên
kết Rh-Rh tăng dần theo số nguyên tử cluster khi n=2-6, từ Rh6 thì giá trị này biến đổi như
sau: các cluster có số nguyên tử Rh chẵn có giá trị lớn hơn các cluster có số nguyên tử lẻ kề

nó. Tuy nhiên giá trị năng lượng liên kết trung bình lại tăng đều mặc dù tỉ lệ tăng lại giảm khi
số nguyên tử Rh trong cluster tăng dần.
Một ứng dụng quan trọng của cluster kim loại nói chung cũng như Rhodi nói riêng là
tính chất quang được ứng dụng rộng rãi trong điện tử đặc biệt là vật liệu bán dẫn. Tính chất
ảnh hưởng trực tiếp ứng dụng này là khoảng năng lượng thuộc vùng cấm của các vật
liệu.Năng lượng vùng cấm được tính bằng giá trị chênh lệch năng lượng của LUMO và
HOMO. Ta thu được các kết quả năng lượng ở bảng 3.9.
Hình 3.4. Đồ thị biến đổi EHOMO (eV), ELUMO(eV) và ELUMO-HOMO (eV) của các cluster Rhn
Phân tích đồ thị trên ta thấy mức chênh lệch năng lượng của LUMO – HOMO biến
đổi không đều, giá trị cao nhất là cluster Rh 3,733 eV và thấp nhất là cluster Rh5 0,056 eV.
Ngoài ra, cluster Rh11 cũng có giá trị ELUMO-HOMO tương đối thấp. Từ Rh7 các giá trị này
biến đổi ít hơn so với các cluster trước đó. So sánh với mức năng lượng LUMO-HOMO của
một số vật liệu bán dẫn được sử dụng phổ biến hiện nay (bảng 3.10) có thể dự đoán rằng các
cluster Rhn sẽ trở thành vật liệu đầy tiềm năng trong công nghệ bán dẫn đặc biệt là các cluster
Rh5 và Rh11.
Một số hình ảnh LUMO, HOMO của cluster Rh, Rh5 và Rh12được biểu diễn trong bảng 3.11
của luận văn.
3.2.3. Phổ UV-VIS của một số cluster Rhn
Để xác định phổ UV-VIS của một số cluster Rhn ta sử dụng phương pháp phiếm hàm
mật độ phụ thuộc vào thời gian (TD-DFT) (time-dependent density functional). Kết quả các
pic đặc trưng và hình phổ UV-VIS được trình bày trong bảng 3.12. Kết quả này được sử dụng
làm tài liệu tham khảo cho các nghiên cứu thực nghiệm về sau.
Bảng 3.12. Kết quả phổ UV-VIS của một số cluster Rhn
Rhn Pic đặc trưng (nm)
Rh3 456
341
Rh4 274
359
630
Rh5 420
589
Rh6 833
Rh7 757
Rh9 960
Rh10 1604
Rh11 1496
Rh12 960

3.3. Cấu trúc và tính chất của các cluster lƣỡng kim loại Rhn-1M
Để có thể xác định dạng bền của các lưỡng kin loại Rhn-1M với M = Fe, Co, Ni, ta sử
dụng các cấu trúc cluster Rhn đã xác định được từ những tính toán ở trên và một cách tương
tự, chúng tôi chọn phương pháp BP86 cùng với bộ hàm cơ sở LANL2DZ để khảo sát các
đồng phân có thể có của cluster lưỡng kim loại RhnM. Sau đó, dựa vào năng lượng điểm đơn,
năng lượng dao động điểm không và tính đối xứng của các cấu trúc để lựa chọn cấu trúc bền
tương ứng với trạng thái ổn định nhất cho các cluster lưỡng kim loại này. Từ đó, khảo sát một
số tính chất đặc trưng và các quy luật biến đổi của chúng, kết quả được trình bày ở những
phần sau.
3.3.1 RhM
Đối với cluster dạng RhM, sau khi tối ưu hóa chúng tôi thu được các cấu trúc có cực
tiểu năng lượng của các cluster RhM như trong bảng 3.13.
Một trong những ứng dụng quan trọng của các cluster kim loại nói chung và rhodi nói
riêng đó chính là tính chất quang được ứng dụng trong vật liệu bán dẫn, điều này có liên quan
mật thiết đến giá trị năng lượng HUMO, LUMO đặc biệt là sự chênh lệch năng lượng
LUMO-HOMO (∆ELUMO-HOMO), đây chính là khoảng cách giừa vùng hóa trị và vùng cấm
(LUMO-HOMO band gap). Vì thế, khi thay thế các nguyên tử Rh bằng các kim loại khác với
hi vọng tìm kiếm những vật liệu cho giá trị ELUMO-HOMO bé. Kết quả tính toán được cho thấy
giá trị này giảm dần khi lần lượt thay thế một nguyên tử Rh bằng Ni, Co, Fe trong cluster
RhM, trong đó cluster Rh-Fe cho giá trị ELUMO-HOMO nhỏ nhất, giá trị cụ thể được trình bày
trong bảng 3.13. Các kết quả này sẽ góp phần định hướng cho các nhà nghiên cứu thực
nghiệm trong việc tìm kiếm các vật liệu bán dẫn.
3.3.2 Rh2M
Các kết quả đối với cluster Rh2M được tóm tắt trong bảng 3.14. Như dự đoán, các cấu
trúc cluster Rh2M thu được đều có dạng đối xứng C2v. Giá trị độ dài liên kết M-Rh tăng dần
từ Fe đến Ni, tuy nhiên có một điều thú vị đó là độ dài liên kết Rh-Rh trong các cluster này
giảm dần, cụ thể như sau dRh-Rh(Rh2Ni) < dRh-Rh(Rh2Co) < dRh-Rh(Rh3) < dRh-Rh(Rh2Fe). Cũng
như cluster RhFe, cluster Rh2Fe có giá trị năng lượng liên kết trung bình lớn nhất trong tất cả
các cluster, thậm chí lớn hơn cả cluster Rh3.
Các kết quả về mức chênh lệch năng lượng LUMO-HOMO cũng cho thấy sự giảm
đáng kể khi thay thế một nguyên tử Rh bằng các kim loại Fe, Co, Ni, trong đó cluster Rh2Co
cho giá trị ∆ELUMO-HOMO bé nhất.
3.3.3.Rh3M
Tất cả các cluster thu được đều có dạng bền là dạng tứ diện, ngoài Rh3Fe có dạng đối
xứng là C1, hai cluster còn lại đều có dạng đối xứng là C3v. Các giá trị về độ dài liên kết biến
đổi không đều, tuy nhiên nhìn chung quy luật biến đổi cũng có nhiều nét tương đồng với
cluster lưỡng kim loại Rh2M, cụ thể độ dài liên kết của Rh-Ni lớn hơn đội dài liên kết Rh-Co
và Rh-Fe, nhưng liên kết Rh-Rh trong cluster Rh3Ni lại cho giá trị nhỏ nhất, nhỏ hơn độ dài
của Rh-Rh trong Rh4. Trong khi đó độ dài liên kết Rh-Rh trong hai cluster Rh3Fe và Rh3Co
cho độ dài lớn hơn Rh-Rh trong cluster Rh4. Các kết quả tính toán được trình bày trong bảng
3.15.
Giá trị ∆ELUMO-HOMO của các cluster lưỡng kim loại Rh3M đều nhỏ hơn so với
∆ELUMO-HOMO của cluster Rh4, trong đó cluster Rh3Ni cho kết quả bé nhất, và có giá trị nhỏ
hơn Rh4 0,403eV.
3.3.4. Rh4M
Các cấu trúc cũng như năng lượng điểm đơn và năng lượng dao động điểm không của các
đồng phân của cluster lưỡng kim loại Rh4M được trình bày lần lượt trong bảng 3.16 và
3.17.Từ các giá trị năng lượng này, ta xác định được các cấu trúc bền đối với các cluster
lưỡng kim loại Rh4M. Từ các cấu trúc này ta khảo sát một số tính chất như: năng lượng liên

kết trung bình, giá trị năng lượng HOMO, LUMO, mức chênh lệch năng lượng LUMO-
HUMO…Các kết quả này được trình bày trong bảng 3.18.
Cấu trúc của các cluster Rh4M vẫn là cấu trúc chóp tứ giác, tuy nhiên vị trí thay thế
Rh của các nguyên tử kim loại là khác nhau. Fe và Ni thay thế cho một nguyên tử Rh ở đáy
của hình vuông và phá vỡ cấu trúc tháp vuông, hình thành nên cấu trúc có đối xứng Cs. Điều
này có thể được giải thích là do sự tương đồng về số electron độc thân của Fe và Ni, Fe có
cấu hình 3d64s
2, do đó ở lớp ngoài cùng có 4 electron độc thân và Ni (3d
84s
2) ở trạng thái
kích thích cũng có 4 electron độc thân.
Trong khi đó Co và Rh cùng thuộc 1 phân nhóm trong bảng tuần hoàn do đó có cùng
số electron lớp ngoài cùng nên đều kiểu cấu trúc đối xứng cao đối với cluster Rh4M là C4v.
Sự biến đổi bán kính cũng khá phức tạp, tuy nhiên nhìn chung đối với Rh4Fe và Rh4Ni sự
biến đổi cũng khá giống với sự biến đổi bán kính ở các cluster Rh3Fe, Rh3Ni và Rh3Fe,
Rh2Ni. Cluster Rh4Co có độ dài liên kết Rh-Co nhỏ hơn Rh-Rh trong Rh5, vì bán kính rCo <
rRh. Điều này cho thấy sự ảnh hưởng rất lớn của độ bội spin đến cấu trúc và bán kính của các
cluster lưỡng kim loại.
Quy luật biến đổi về mức chênh lệch LUMO-HUMO dường như không còn đúng với
Rh4M. Cluster Rh5 cho giá trị ∆ELUMO-HOMO nhỏ nhất không chỉ trong tất cả các cluster kim
loại Rhn được tính mà còn thấp hơn các cluster lưỡng kim loại Rh4M (M = Fe, Co, Ni). Kết
quả về giá trị năng lượng liên kết trung bình vẫn cho thấy Rh4Fe là cluster có năng lượng lớn
nhất.
3.3.5. Rh5M
Các cấu trúc bền đối với các cluster Rh5Fe và Rh5Co là cấu trúc có dạng lưỡng tháp
ngũ giác và đều có đối xứng cao C4v còn đối với Rh5Ni cấu trúc bền là dạng C1 bị biến dạng
khỏi cấu trúc lưỡng tháp ngũ giác. Sự biến đổi cấu trúc đối với cluster Rh5Ni có thể được giải
thích là do độ bội spin, lúc này độ bội spin của Rh5Ni là 8 khác với độ bội spin 6 trong
Rh5Fe. Độ dài các liên kết cũng biến đổi khá phức tạp, phụ thuộc vào độ bội spin và tương
quan giữa các nguyên tố với nhau.Quy luật đối với năng lượng liên kết khá ổn định, cluster
Rh5Fe vẫn là cluster cho năng lượng liên kết trung bình lớn nhất.
Đối với mức chênh lệch năng lượng LUMO-HOMO, ngoại trừ sự bất thường đối với
cluster Rh4M, thì các cluster lưỡng kim loại Rh5M vẫn cho các giá trị ∆ELUMO-HOMO nhỏ hơn
so với cluster Rh6 giống như các cluster lưỡng kim loại trước đó. Trong đó, Rh5Ni là cluster
có mức năng lượng này thấp nhất
3.3.6. Rh6M
Các cấu trúc bền của các cluster Rh6M đều là dạng thay thế một nguyên tử Rh trên
đỉnh của cấu trúc lưỡng chóp ngũ giác đều D5h của Rh7, trong đó Rh6Fe, Rh6Co có đối xứng
C5v, còn cluster Rh6Ni cấu trúc bị biến dạng khỏi dạng C5v và chuyển thành C1. Cluster Rh6Fe
có giá trị năng lượng liên kết trung bình lớn nhất, điều này cho thấy sự phù hợp quy luật về
sự biến đổi năng lượng liên kết từ RhM đến Rh6M.Đối với cluster Rh6M, lại thấy sự lặp lại về
sự biến đổi mức năng lượng LUMO-HOMO khi Rh7 lại cho giá trị ∆ELUMO-HOMO nhỏ hơn các
cluster lưỡng kim loại.
3.3.7. Rh7M
Tối ưu hóa các đồng phân có thể có của cluster lưỡng kim loại Rh7M, từ đó tính giá trị
năng lượng điểm đơn, năng lượng dao động điểm không, năng lượng tổng và năng lượng
tương đối của chúng để xác định cấu trúc bền của các cluster lưỡng kim loại này.
Dựa vào các kết quả thu được ta xác định được các cấu trúc bền nhất đối với các
cluster Rh7M, sau đó khảo sát một số tính chất quan trọng đối với các cấu trúc thu được này.
Các kết quả được trình bày trong bảng 3.25.
Các cluster Rh7Fe và Rh7Co vần giữ cấu trúc giống với cấu trúc của Rh8, trong đó
Rh7Fe có dạng đối xứng cao hơn là C3v còn Rh7Co bị biến dạng về C1. Cấu trúc Rh7Ni
chuyển về dạng cấu trúc mới và có đối xứng là Cs.

Cluster Rh7Fe vẫn là cấu trúc cho năng lượng liên kết trung bình lớn nhất. Mặc dù
Rh7Co có mức chênh lệch LUMO-HOMO là thấp nhất, nhưng giá trị này của Rh8 vẫn nhỏ
hơn của Rh7Fe và Rh7Co.
3.4. Chênh lệch mức năng lƣợng giữa LUMO-HOMO của các cluster lƣỡng kim loại
Rhn-1M
Các kết quả về mức chênh lệch năng lượng LUMO-HOMO của các clusterkim loại
Rhn và cluster lưỡng kim loại Rhn-1Fe, Rhn-1Co, Rhn-1Ni với n = 2 – 8 được trình bày trong
bảng 3.26.
Từ các giá trị trong bảng 3.26 nhìn chung với n=2-6 thì khi thay thế một nguyên tử Rh
bằng các nguyên tố Fe, Co, Ni thì các giá trị ∆ELUMO-HOMOcủa các cluster lưỡng kim loại Rhn-
1M nhỏ hơn ∆ELUMO-HOMO của các Rhn tương ứng, ngoại trừ cluster Rh5 có ∆ELUMO-HOMO
>∆ELUMO-HOMO (Rh4M) , đến Rh6M thì xu hướng này có dường như không còn phù hợp. Như
vậy khi có sự mở rộng nghiên cứu, chúng tôi sẽ tiếp tục khảo sát thêm các cấu trúc với n>8
để tìm ra quy luật biến đổi giá trị ∆ELUMO-HOMO.So sánh các giá trị ∆ELUMO-HOMO của các
cluster lưỡng kim loại Rhn-1M với các vật liệu bán dẫn được sử dụng phổ biến hiện nay (bảng
3.10) cho thấy các cluster lưỡng kim loại này có giá trị tương đối thấp và được dự đoán là vật
liệu bán dẫn đầy tiềm năng trong nhiều ngành công nghiệp khác nhau.
Biễu diễn một số hình ảnh HOMO, LUMO của một số cluster lưỡng kim loại trong bảng 3.27
3.5. Năng lƣợng liên kết trung bình của các cluster lƣỡng kim loại Rhn-1M
Từ các giá trị trong bảng 3.28 và hình 3.5 có thể thấy rằng năng lượng liên kết trung
bình của các cluster Rhn và Rhn-1M tăng dần khi số nguyên tử Rh tăng dần. Nhìn chung, năng
lượng liên kết trung bình giảm dần theo chiều Rhn-1Fe > Rhn> Rhn-1Co > Rhn-1Ni ngoại trừ
Rh4Ni và Rh6Ni có năng lượng liên kết trung bình lớn hơn Rh4Co và Rh6Co.
Hình 3.5. Đồ thị biểu diễn năng lượng liên kết trung bình của các cluster Rhn và Rhn-1M
3.6. Phổ UV-VIS của một số cluster lƣỡng kim loại
Để xác định phổ UV-VIS của một số cluster lưỡng kim loại Rhn-1M chúng tôi sử dụng
phương pháp phiếm hàm mật độ phụ thuộc vào thời gian (TD-DFT) (time-dependent desity
functional).So sánh với phổ UV-VIS của các cluster kim loại Rhn.Kết quả các pic đặc trưng
và hình phổ UV-VIS được trình bày trong bảng 3.29 và 3.30.Kết quả này được sử dụng làm
tài liệu tham khảo cho các nghiên cứu thực nghiệm về sau.
3.6.1. Rh3M
3.6.2. Rh6M

KẾT LUẬN
Từ các kết quả nghiên cứu của đề tài, một số kết luận được rút ra như sau:
1. Đã tối ưu hóa hơn 50 cấu trúc bền của các cluster kim loại Rhn (n=2-13) và cluster
lưỡng kim loại của rhodi Rhn-1M (M = Fe, Co, Ni) với n=2-8 ở nhiều trạng thái spin khác
nhau bằng phương pháp phiếm hàm mật độ BP86/LANL2DZ. Tính được năng lượng điểm
đơn và tần số dao động của mỗi cấu trúc, từ đó xác định được cấu trúc bền nhất của mỗi dạng
cluster tương ứng. Đồng thời cũng xác ddingj được các tham số cấu trúc như : độ dài liên kết,
góc liên kết và góc nhị diện, nhóm
điểm đối xứng…
2. Từ các cấu trúc bền thu được tiếp tục khảo sát một số tính chất đặc trưng của chúng,
cụ thể là:
- Đã tính được giá trị năng lượng ion hóa thứ nhất của các cluster Rhn. So sánh các kết
quả thu được với thực nghiệm và với kết quả tính toán lí thuyết cho thấy có sự phù hợp cao.
Điều này một lần nữa khẳng định tính đúng đắn của phương pháp sử dụng.
-Trong mỗi dạng cluster thu được, chúng tôi cũng đã tính được các giá trị năng lượng
liên kết Rh-Rh và Rh-M và năng lượng liên kết trung bình của chúng. So sánh các kết quả thu
được để thấy sự biến đổi các giá trị của các cluster Rhn trước và sau khi thêm các nguyên tố
kim loại Fe, Co, Ni.
- Sử dụng phương pháp phiếm hàm mật độ phụ thuộc thời gian (TD-DFT), chúng tôi đã
tính toán phổ UV-VIS của một số cluster Rhn và cluster Rhn-1M để làm số liệu tham khảo cho
các nghiên cứu thực nghiệm sau này.
- Đã so sánh sự thay đổi về cấu trúc, bán kính, năng lượng liên kết trung bình của các
cluster Rhn trước và sau khi thay thế các nguyên tố kim loại Fe, Co, Ni, bước đầu nghiên cứu
sự ảnh hưởng của độ bội spin đến cấu trúc, bán kính và năng lượng liên kết.
- Đã tính được mức chênh lệch năng lượng LUMO – HUMO của các cluster kim loại
và lưỡng kim loại của rhodi. So sánh với ∆ELUMO – HUMO của một số vật liệu bán dẫn được sử
dụng phổ biến hiện nay. Kết quả thu được cho thấy các cluster kim loại Rhn nói chung và
cluster lưỡng kim loại Rhn-1M nói riêng là những vật liệu bán dẫn đầy tiềm năng cho công
nghiệp khác nhau, đặc biệt là các cluster Rh5 (0,056 eV), Rh11 (0,081 eV), Rh7Co (0,086 eV).
Chúng tôi hi vọng những kết quả nghiên cứu ở trên sẽ trở thành tài liệu tham khảo hữu
ích cho các nhà hóa học thực nghiệm hoặc làm cơ sở cho các nghiên cứu tiếp theo.
Hƣớng phát triển của đề tài:
1. Mở rộng nghiên cứu cấu trúc và tính chất của dạng bền đối với các cluster có kích
thước lớn (n>13) và xác định quy luật biến đổi của chúng.
2. Mở rộng nghiên cứu cấu trúc và tính chất của các cluster lưỡng kim loại Rhn-mMm
khi số nguyên tử kim loại M thay thế tăng lên (m > 1) và mở rộng đối với các nguyên tố M
khác.
3. Mở rộng nghiên cứu tính chất của các cluster kim loại và lướng kim loại rhodi để
định hướng làm xúc tác cho các phản ứng hóa học khác nhau.
4. Mở rộng nghiên cứu đối với các kim loại chuyển tiếp khác như : Pd, Pt, Ir...
References
TIẾNG VIỆT
1. Eyring H,, Walter J,, Kimball G, E, (1976), Hóa học lượng tử (bản dịch tiếng việt), Nhà
xuất bản Khoa học và Kỹ thuật Hà Nội,

2. Nguyễn Đình Huề, Nguyễn Đức Chuy (2003), Thuyết lượng tử về nguyên tử và phân tử
(Tái bản lần thứ nhất), Tập (1, 2) , Nhà xuất bản Giáo dục,
3. Trần Thành Huế (2003), Hóa học đại cương, Tập 1, Nhà xuất bản Giáo dục,
4. Lâm Ngọc Thiềm (2007), Nhập môn hóa học lượng tử, Nhà xuất bản Đại học Quốc gia
Hà Nội,
5. Lâm Ngọc Thiềm (Chủ biên), Phạm Văn Nhiêu, Lê Kim Long (2007), Cơ sở hóa học
lượng tử, Nhà xuất bản Khoa học và Kỹ thuật Hà Nội,
6. Đào Đình Thức (1980), Cấu tạo nguyên tử và liên kết hóa học, Tập 2, Nhà xuất bản Đại
học và Trung học chuyên nghiệp,
TIẾNG ANH
7. A, Sanchez, S, Abbet, U, Heiz, W,D, Schneider, H, Haekkinen, R,N, Barnett and U,
Landman, J, Phys, Chem, A, 1999, 103, 9573,
8. Amatayakul, W (2001), "Life cycle assessment of a catalytic converter for passenger
cars", Journal of Cleaner Production 9 (5): 395
9. Becke A,D, (1988), “Density-functional exchange-energy approximation with correct
asymptotic behaviour”, Phys, Rev,A38, pp, 3098-3100,
10. B, V, Reddy, S, K, Nayak, S, N, Khanna, B, K, Rao, and P, Jena (1999), “electronic
structure and magnetism of Rhn (n=2 – 13) clusters”, The American Physical Society,
59(7), 5214-5222
11. C, Lee, W, Yang, and R,G, Parr (1988), “Development of the Colle-Salvetti correlation-
energy formula into a functional of the electron density”, Phys, Rev,,B37, pp, 785-789,
12. Chattaraj P, K, (2009), Chemical Reactivity Theory: A Density Functional View, Taylor &
Francis Group, USA,
13. D, M, P, Mingos and D, J Wales, Introduction to cluster chemistry, Prentice Hall, 1990,
ISBN 0-13-479049-9
14. Dunning, T, H, (1989), “Gaussian Basis Sets for Use in Correlated Molecular
Calculations, I, The Atoms BoronThrough Neon and Hydrogen”, J, Chem, Phys,, 90, pp,
1007-1023
15. Eschrig H, (1996),TheFundamentals of Density Functionals Density, B, G, Teubner
Verlagsgesellschaft Stuttgart – Leipzig, Germany,
16. F, Baletto and R, Ferrando (2005), Rev, Mod, Phys,, 77, 371,
17. Feller D, (1992), “Application of Systematic Sequences of Wave Functions to the Water
Dimer”, J, Chem, Phys,, 96, pp, 6104-6114,
18. Foresman J, B,, Frish E, (1990), Exploring Chemistry with Electronic Structure Methods
(Second Edition), Gaussian, Inc,, Pittsburgh, PA,
19. Frish M, J,, Frish A, E,, Foresman J, B, (1995), Gaussian 98 User’s Reference, Gaussian,
Inc,, Pittsburgh, PA,
20. Heck, R (2001), "Automobile exhaust catalysts", Applied Catalysis A: General, 221, 443,
21. Heck, R (2001), "The application of monoliths for gas phase catalytic
reactions",Chemical Engineering Journal, 82, 149,
22. G, Schmid, Adv, Eng, Mater,, 2001, 3, 737,
23. I, Katakuse, T, Ichihara, Y, Fujita, T, Matsuo, T, Sakurai and H, Matsuda (1985),Int, J,
Mass Spectrom, Ion Processes, 67, 229,
24. I, Katakuse, T, Ichihara, Y, Fujita, T, Matsuo, T, Sakurai and H, Matsuda (1986), Int, J,
Mass Spectrom, Ion Processes, 74, 33,
25. I, Sinfelt, J, H, (1977), Act, Chem, Res,10, 15
26. J, A, Alonso (2000), Chem, Rev,, , 100, 637,
27. J, A, Alonso and N, H, March (1989), Electrons in Metals and Alloys; Academic:
London, 1989,

28. J,M, Thomas, W,J, Thomas (1997), Principles and Practice of Heterogeneous Catalysis,
VCH Verlagsgessellschaft mbH, Weinheim, New York,
29. James E, Huheey, Inorganic Chemistry Huheey, 3rd ed, Harper and Row, New York
30. K, A, Gingerich and D, L, Cocke (1972), J, Chem, Soc, Chem, Commun,1, 536
31. Koch W,, Holthausen M, C, (2001), A Chemist’s Guide to Density Functional Theory
(Second Edition), Villey-VCH, Germany,
32. [16]Kohn W,, Sham L, J, (1965), “Self-Consistent Equations Including Exchange and
Correlation Effects", Phys, Rev,, 140, pp, 1133-1138,
33. Levine I, N, (2000), Quantum Chemistry (Fifth Edition), Prentice-Hall, Inc,, New Jersey,
USA,
34. Lowe J, P,, Peterson K, A, (2006),Quantum Chemistry (Third Edition), Elsevier
Acadamic Press, USA,
35. Lu P,, Liu G, Q,, Li J, C, (2005), “Existing Problems in Theoretical Determination of
Red- Shifted or Blue-Shifted Hydrogen”, J, Mol, Struct,(THEOCHEM), 723, pp, 95-100,
36. M, Haruta (1997), Catal, Today, 36, 153
37. M,A, Hayat (1991), Colloidal Gold: Principles, Methods, and Applications, Academic
Press, San Diego,
38. M,R, Zakin, D,M, Cox and A, Kaldor (1988), J, Chem, Phys,, 89, 1201,
39. M, Valden, X, Lai and D, W, Goodman (1998), Science, 281, 1647,
40. Moller C,, Plesset M, S, (1934), “Note on an Approximation Treatment for Many-
Electron Systems”, Phys, Rev,, 46, pp, 618-622,
41. Nesbet R, K, (2004), Variational Principles and Methods in Theoretical Physics and
Chemistry, Cambridge University Press, New York,
42. P, Pyykkö (1988), Chem, Rev,, 88, 563,
43. P, Schwerdtfeger, M, Dolg, W, H, E, Schwarz, G, A, Bowmaker and P, D, W, Boyd, J,
Chem, Phys,, 1989, 91, 1762,
44. Parr G,, Yang W, (1989), Density Functional Theory of Atoms and Molecules, Oxford
University Press, Oxford,
45. Perdew J,P,, Chevary J,A,, Vosko S,H,, Jackson K,A,, Pederson M,R,, Fiolhais C, (1992)
“Atoms, molecules, solids and surfaces: Applications of the generalized gradient
approximation for exchange and correlation”, Phys, Rev,, B46, pp, 6671-6687,
46. Perdew J,P,, Wang Y, (1992), “Accurate and simple analytic representation of the
electron-gas correlation energy”, Phys, Rev,, B45, pp, 13244-13249,
47. S, Loth, S, Baumann, C, P, Lutz, D, M, Eigler and A, J, Heinrich (2012), Science, 335,
196,
48. Szabo A,, Ostlund N, S, (1989), Modern Quantum Chemistry: Introduction to Advanced
Structure Theory, Dover Publications, Inc,, Mineola, New York,
49. Slater, J, (1974), The Self-Consisternt Field for Molecules and Solids: Quantum Theory
of Molecules and Solids, Vol, 4, McGraw-Hill: New York,,
50. Vosko S,H,, Wilk L,, Nusair M, (1980), “Accurate spin-dependent electron liquid
correlation energies for local spin density calculations: a critical analysis”, Can, J, Phys,
58, pp, 1200-1211,
51. Y, Kawazoe, T, Kondow and K, Ohno (2002), Clusters and nanomaterials: Theory and
experiment, Spinger, Berlin,,
52. Yensen F, (2007), Introduction to Computaional Chemistry (Second Edition), John Wiley
& Sons, Ltd, England,
53. W, A, de Heer (1993), Rev, Mod, Phys,, 65, 611,
54. W, A,de Heer, W, D, Knight, M, Y, Chou and M, L, Cohen (1987), Solid State Phys,,
40, 93,

55. W, D, Knight, , K, Clemenger, W, A, de Heer, W, A, Saunders, M, Y, Chou and M, L,
Cohen (1984), Phys, Rev, Lett,, 52, 2141,
56. Wollaston, W, H, (1804), "On a New Metal, Found in Crude Platina",Philosophical
Transactions of the Royal Society of London 94: 419–430.





![ể ấ ị trường] 21/09/2021](https://static.fdocument.pub/doc/165x107/618a32cea214f416a77d0d39/-trng-21092021.jpg)












