
Enfermedad de Huntington
25
El enigma de la enfermedad de Huntington Cattaneo, E., Rigamonti D., y Zucatto C. (2003). El enigma de la enfermedad de Huntington. Scientific American México, 1 (8) : 32-37. Revisión de un artículo científico
-
Upload
christian-arzaluz -
Category
Health & Medicine
-
view
263 -
download
2
Transcript of Enfermedad de Huntington

El enigma de la enfermedad de
Huntington
Cattaneo, E., Rigamonti D., y Zucatto C. (2003). El enigma de la enfermedad de Huntington. Scientific American México, 1
(8) : 32-37.
Revisión de un artículo científico

Panorama de la enfermedad de
Huntington
• Es una enfermedad que incapacita y causa demencia a la mitad de la vida de las personas que la contraen.
• Esta enfermedad resulta de una mutación en el gen huntingtino localizado en el cromosoma 4 (4p16.3)
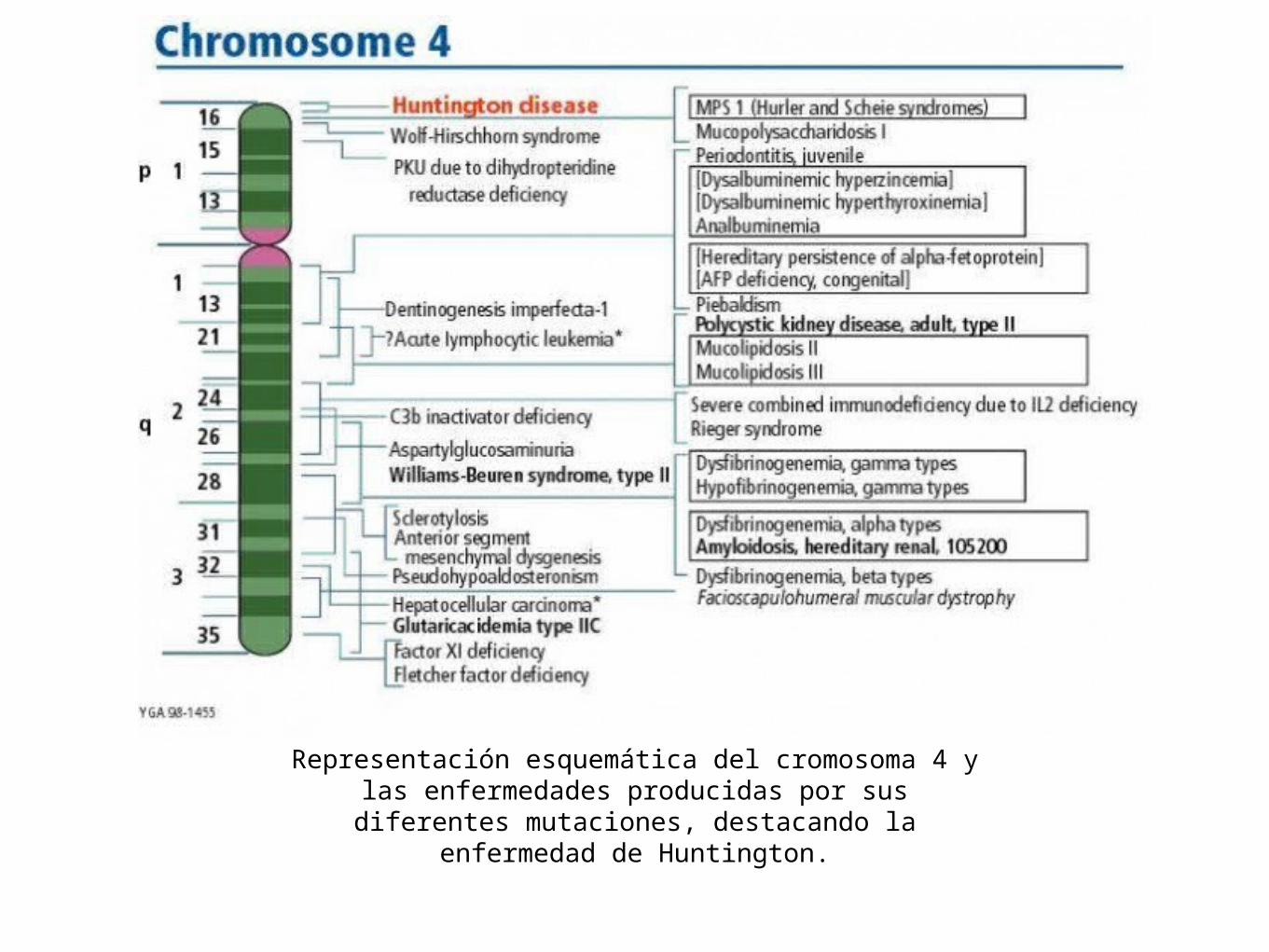
Representación esquemática del cromosoma 4 y las enfermedades producidas por sus diferentes mutaciones,
destacando la enfermedad de Huntington.
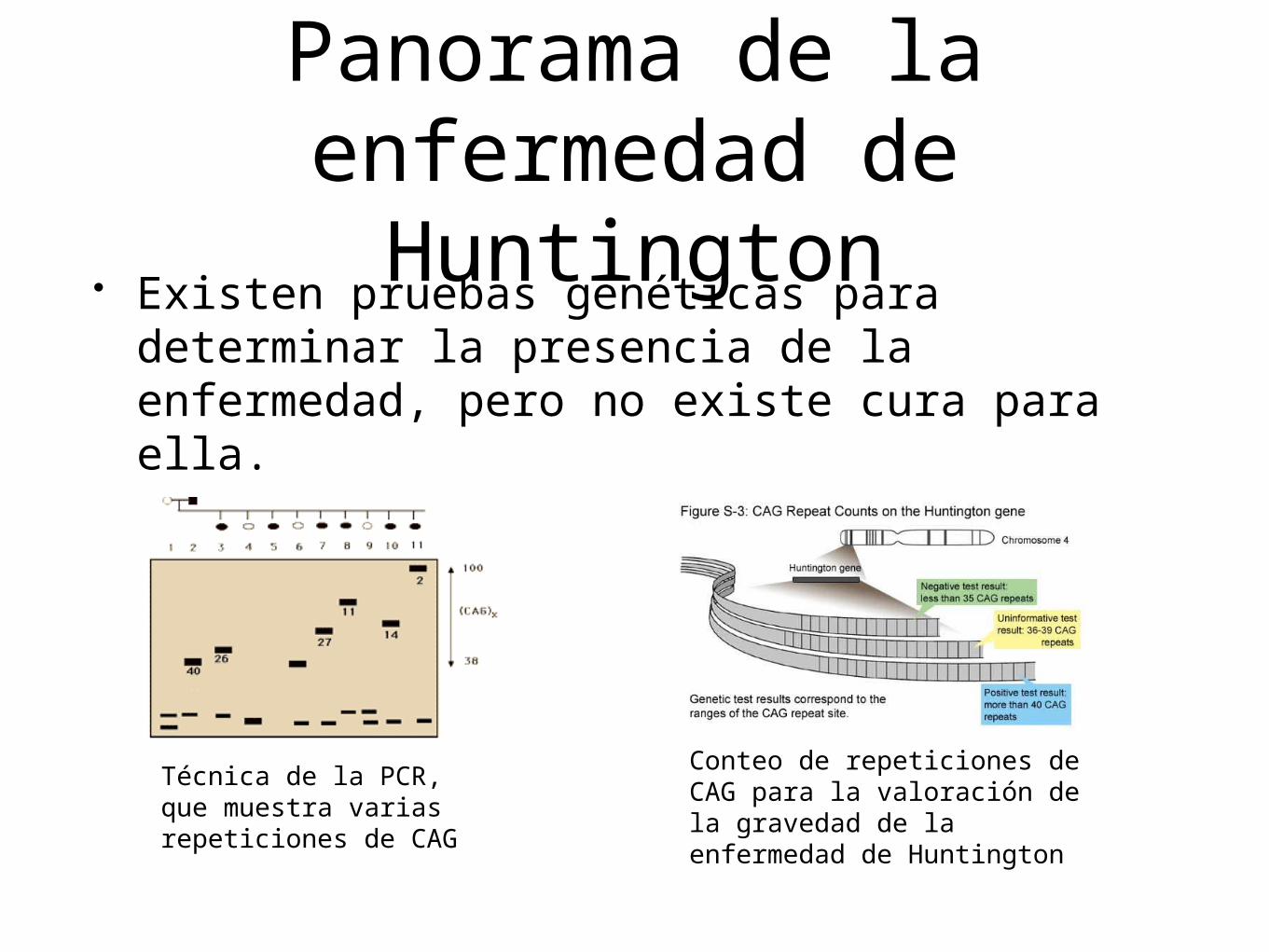
Panorama de la enfermedad de
Huntington• Existen pruebas genéticas para determinar la
presencia de la enfermedad, pero no existe cura para ella.
Técnica de la PCR,que muestra varias repeticiones de CAG
Conteo de repeticiones de CAG para la valoración de la gravedad de la enfermedad de Huntington

Panorama de la enfermedad de
Huntington• Las personas destinadas a adquirir la enfermedad, presentan un tartamudeo en el gen huntingtino, que hace que se produzca la proteína huntingtina mal conformada, en la cual se repite el mismo aminoácido glutamina docenas de veces.
Esquema comparativo entre el gen huntingtino normal y la proteína huntingtina normal; y el gen huntingtino mutado, con la proteína huntigntina mal conformada con 37-80 repeticiones de glutamina.

Panorama de la enfermedad de
Huntington• La proteína huntingtina mutante parece ser
tóxica para las células nerviosas clave en el cerebro; además de ser incapaz de activar el gen de un importante factor de crecimiento (FNDC)
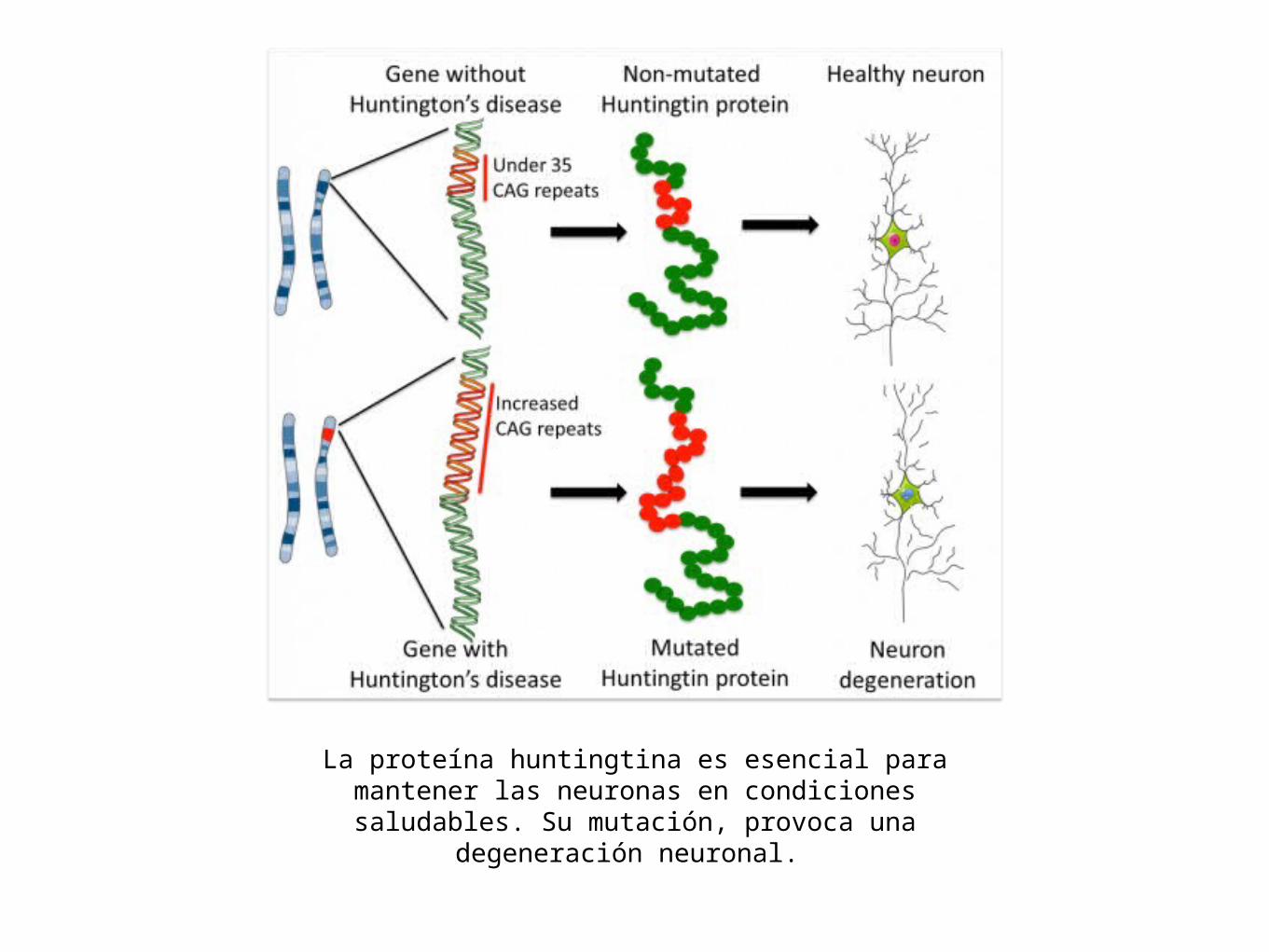
La proteína huntingtina es esencial para mantener las neuronas en condiciones saludables. Su mutación, provoca
una degeneración neuronal.
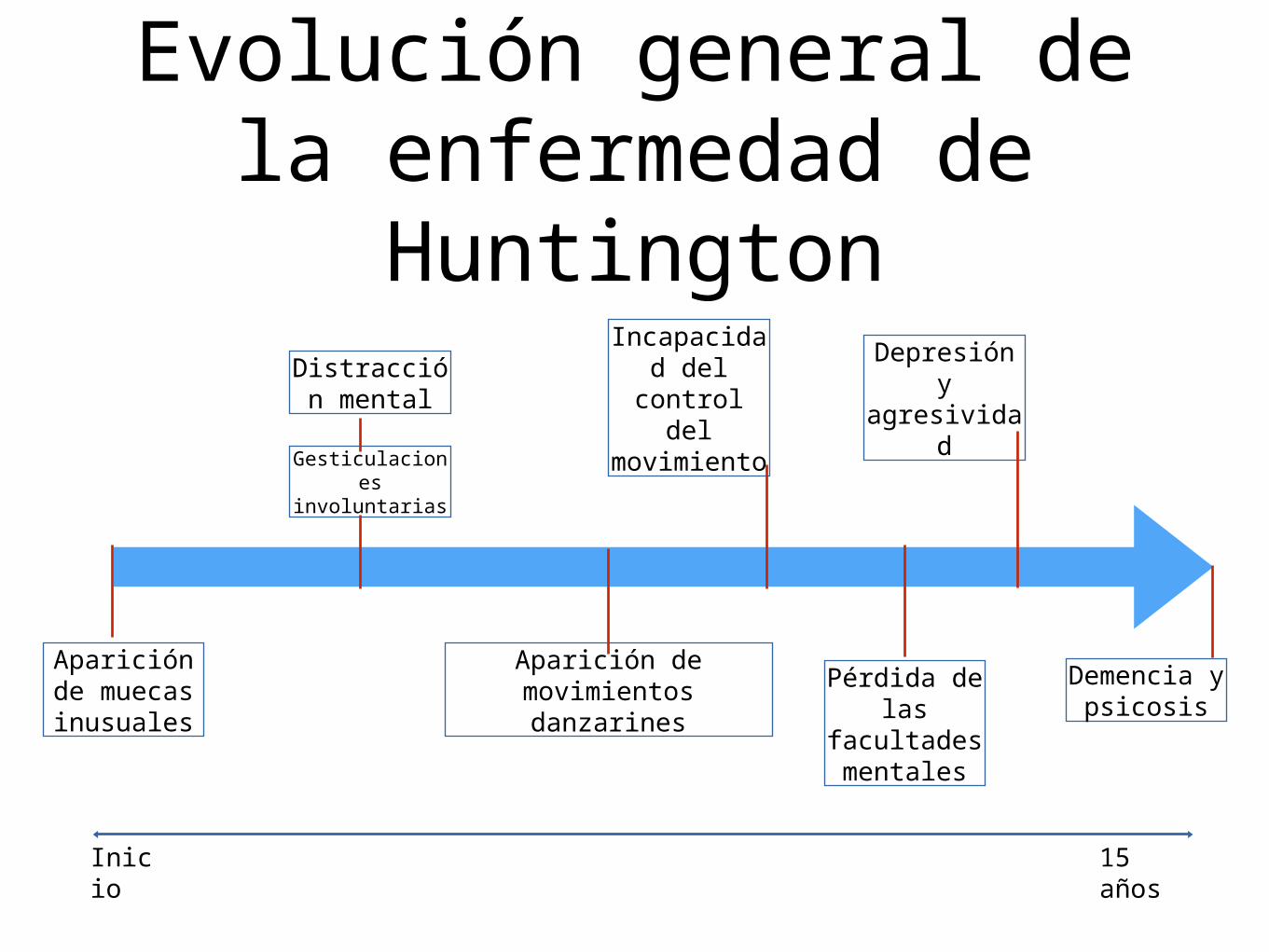
Evolución general de la enfermedad de
Huntington
Aparición de muecasinusuales
Distracción mental
Gesticulaciones involuntarias
Aparición de movimientosdanzarines
Incapacidad del control
del movimiento
Pérdida de las
facultades mentales
Depresión y agresividad
Demencia y psicosis
Inicio
15 años
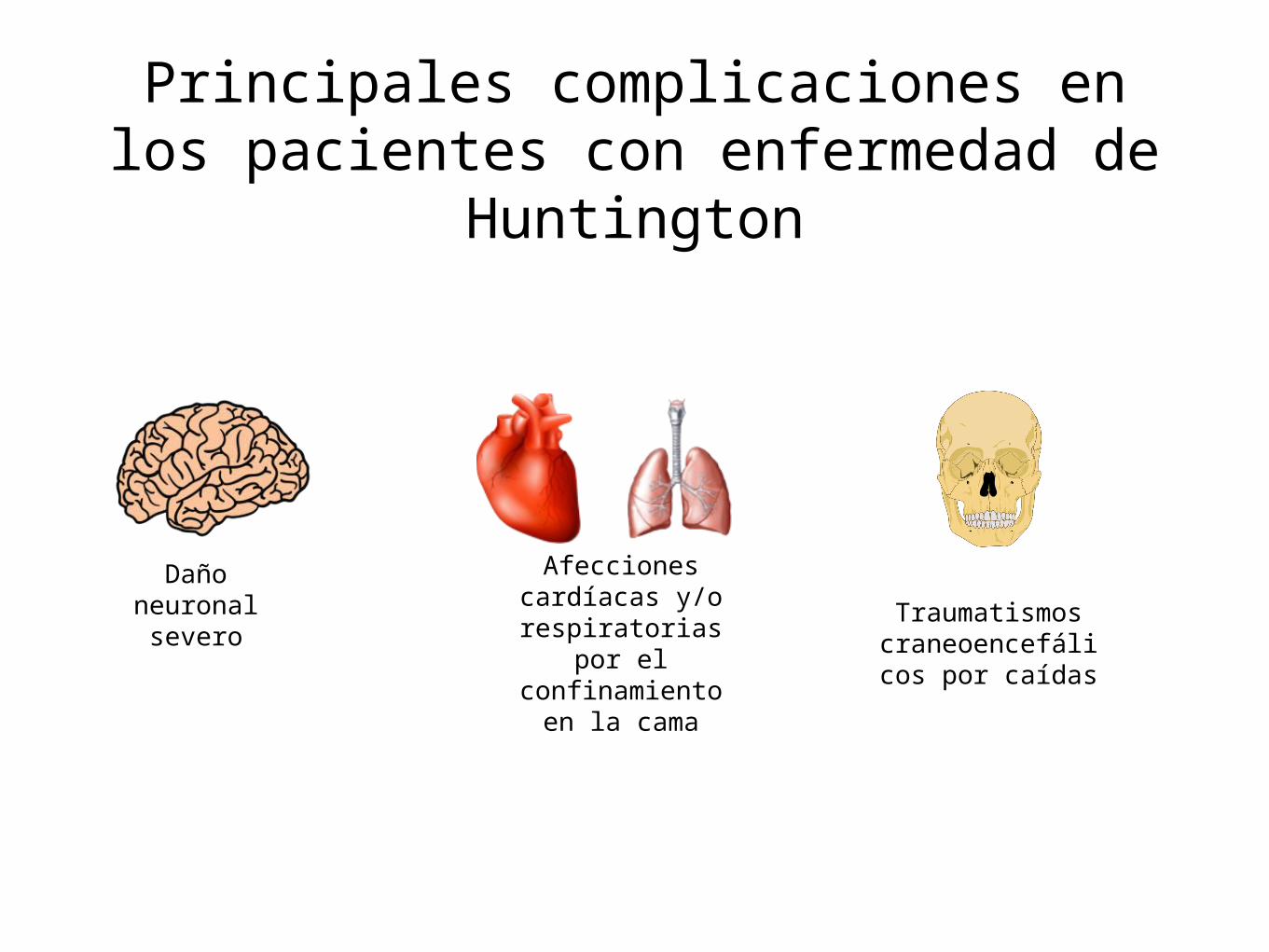
Principales complicaciones en los pacientes con enfermedad de
Huntington
Daño neuronal severo
Afecciones cardíacas y/o
respiratorias por el confinamiento
en la cama
Traumatismos craneoencefálico
s por caídas
Fisiopatogenia de la enfermedad de Huntington
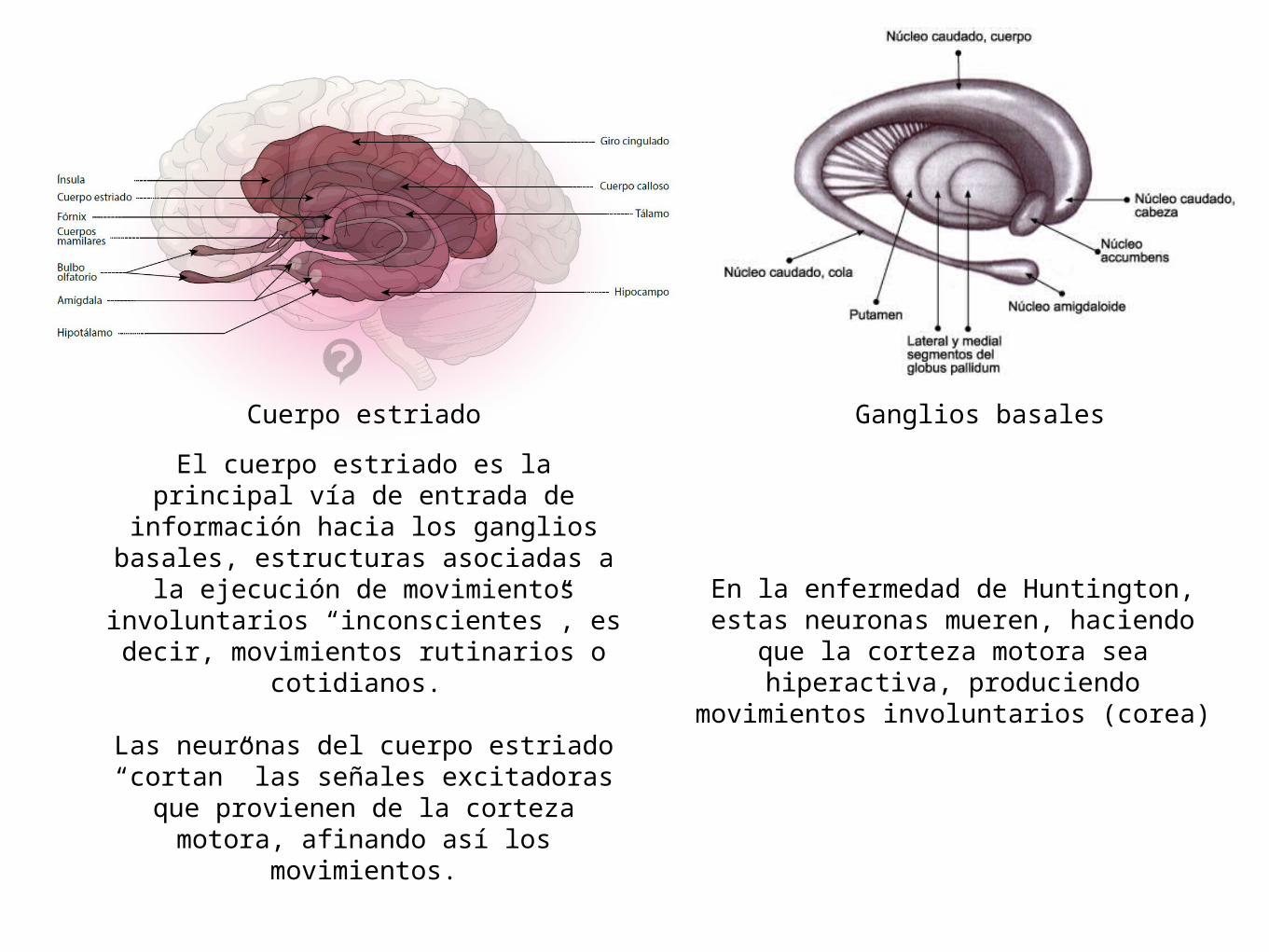
El cuerpo estriado es la principal vía de entrada de información hacia los
ganglios basales, estructuras asociadas a la ejecución de movimientos
involuntarios “inconscientes”, es decir, movimientos rutinarios o cotidianos.
Las neuronas del cuerpo estriado “cortan” las señales excitadoras que
provienen de la corteza motora, afinando así los movimientos.
Cuerpo estriado Ganglios basales
En la enfermedad de Huntington, estas neuronas mueren, haciendo que la
corteza motora sea hiperactiva, produciendo movimientos involuntarios
(corea)
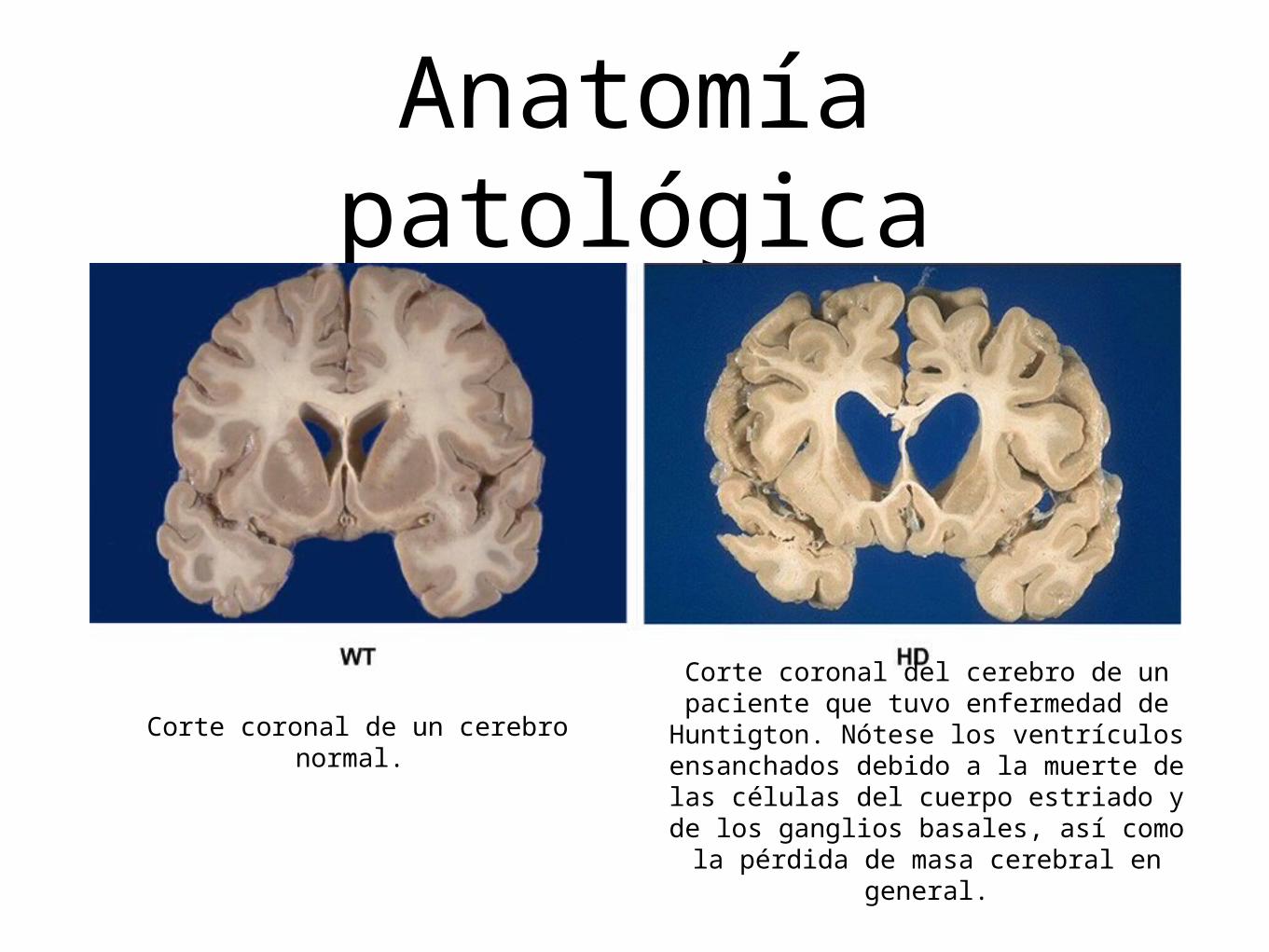
Anatomía patológica
Corte coronal de un cerebro normal.
Corte coronal del cerebro de un paciente que tuvo enfermedad de Huntigton. Nótese los ventrículos
ensanchados debido a la muerte de las células del cuerpo estriado y de los
ganglios basales, así como la pérdida de masa cerebral en general.
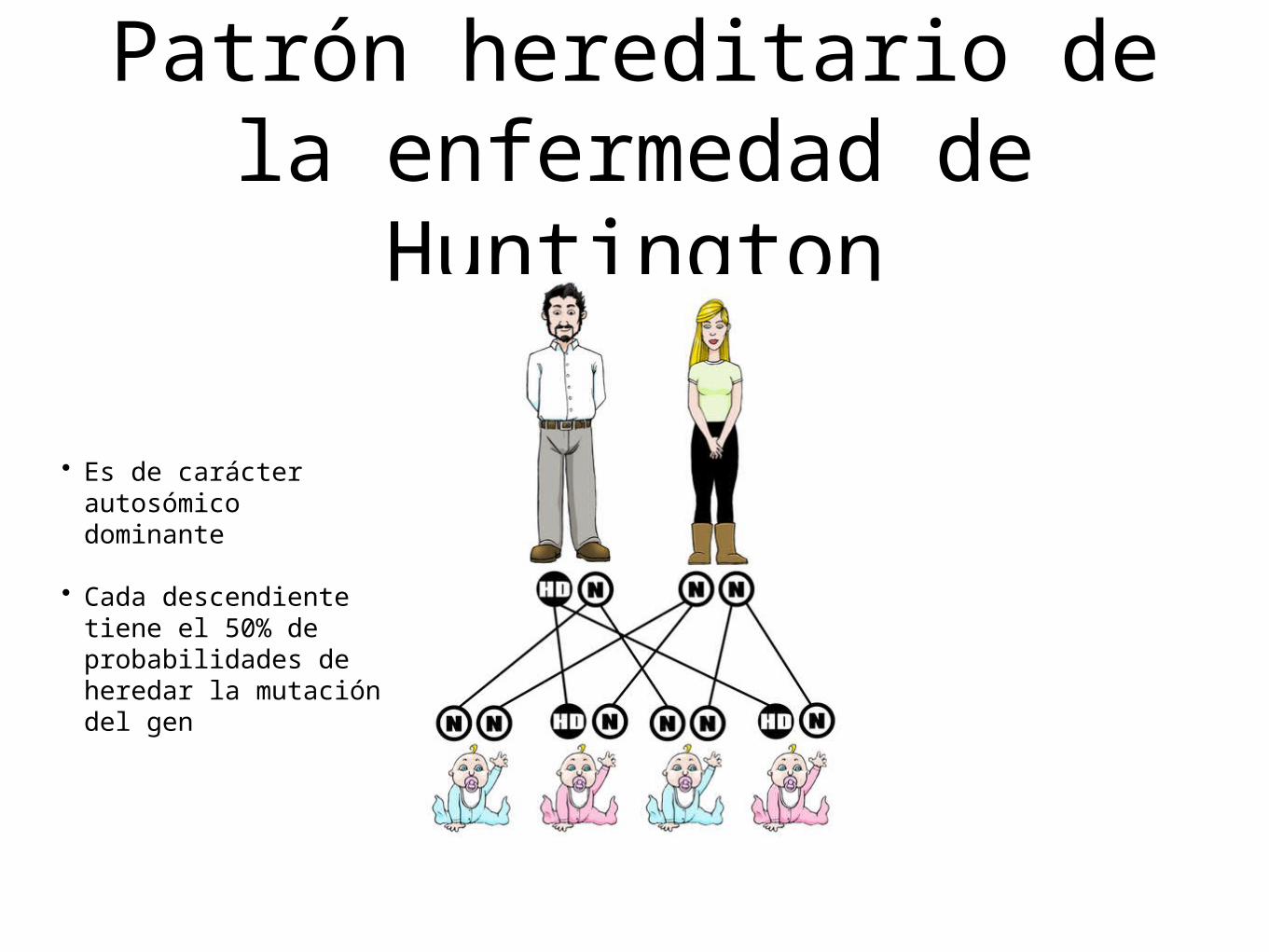
Patrón hereditario de la enfermedad de
Huntington
• Es de carácter autosómico dominante
• Cada descendiente tiene el 50% de probabilidades de heredar la mutación del gen
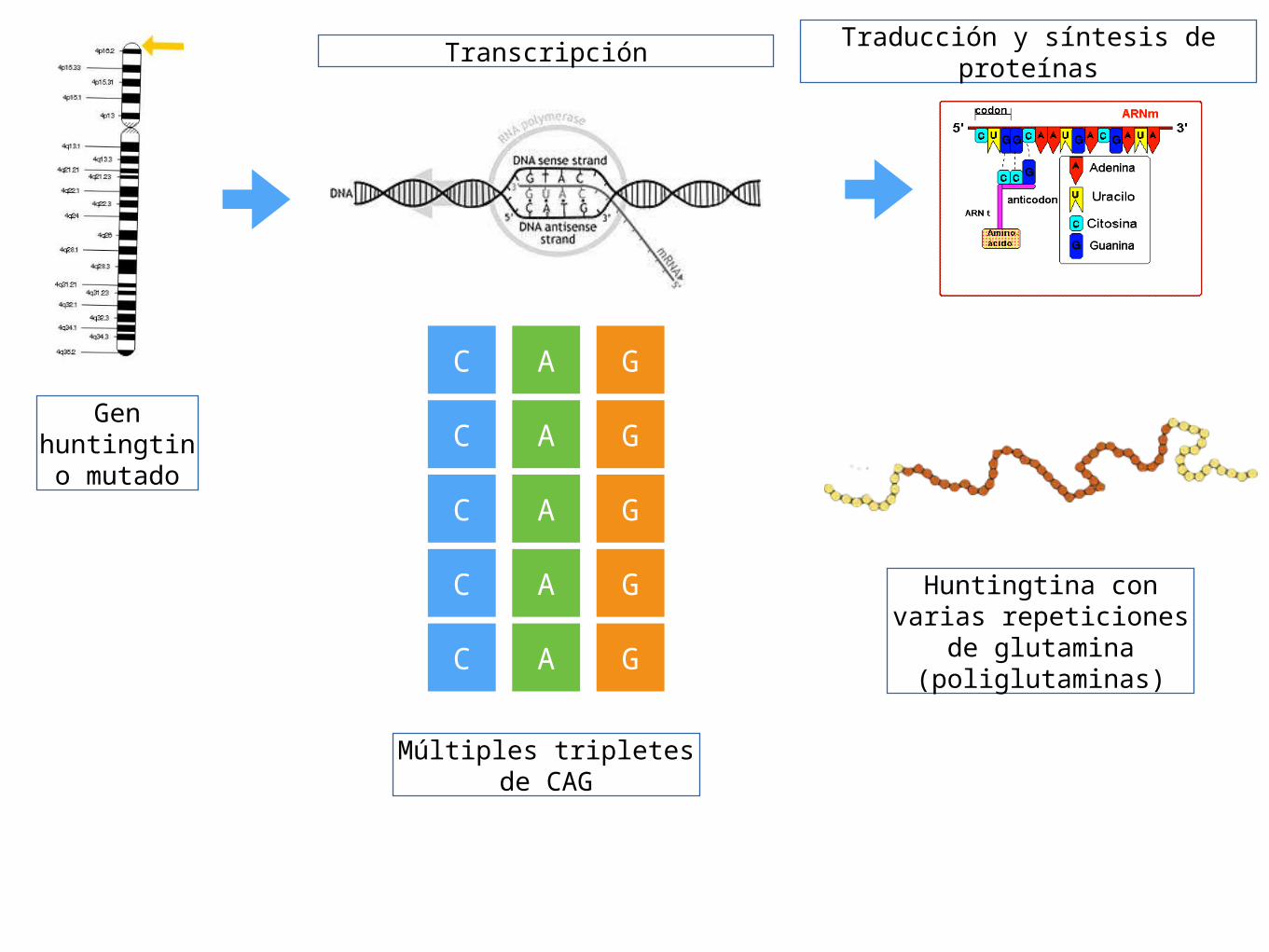
Gen huntingtino
mutado
Transcripción
C A G
C A G
C A G
C A G
C A G
Múltiples tripletes de CAG
Traducción y síntesis de proteínas
Huntingtina con varias repeticiones de
glutamina (poliglutaminas)

Normalmente, hay cierto número de repeticiones de CAG (10-35). Cuando estas repeticiones aumentan de 36-39, hay un riesgo pregenético para el desarrollo de la enfermedad.
Cuando estas repeticiones sobrepasan las 40, es muy probable que se desarrolle la enfermedad.
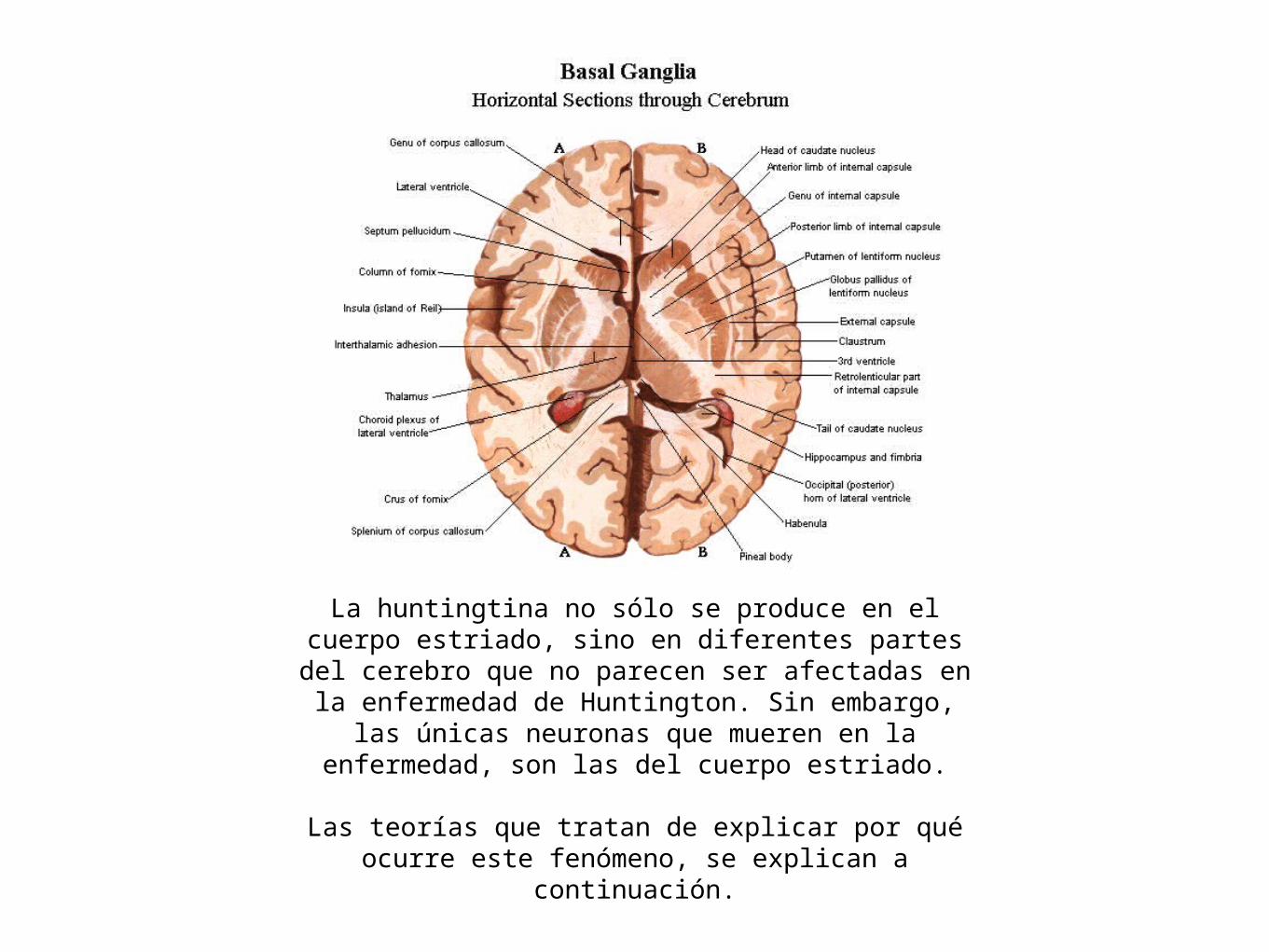
La huntingtina no sólo se produce en el cuerpo estriado, sino en diferentes partes del cerebro que no parecen ser afectadas en la enfermedad de Huntington. Sin embargo,
las únicas neuronas que mueren en la enfermedad, son las del cuerpo estriado.
Las teorías que tratan de explicar por qué ocurre este fenómeno, se explican a continuación.

Teoría de la pérdida de función
Cuando se altera la estructura de una proteína, también se altera su función.
Sin embargo, esto no explica por qué la huntingtina mal conformada no afecta las demás zonas del cerebro, ni tampoco el por qué si se tiene un alelo normal y
uno afectado, se sigue produciendo el daño neuronal.

Teoría de los agregados de huntingtina
La huntingtina mal conformada, forma agregados con otras proteínas,
especialmente la huntingtina con estructura normal, lo cual explica el
carácter dominante de la enfermedad.
La huntingtina mal conformada forma láminas beta con otras proteínas, actuando como un “pegamento”. Estos agregados deterioran la función normal
de la huntingtina; sin embargo, esta teoría no explica por qué los agregados proteicos provocan el daño celular, ni por qué no son eliminados por los
proteosomas.
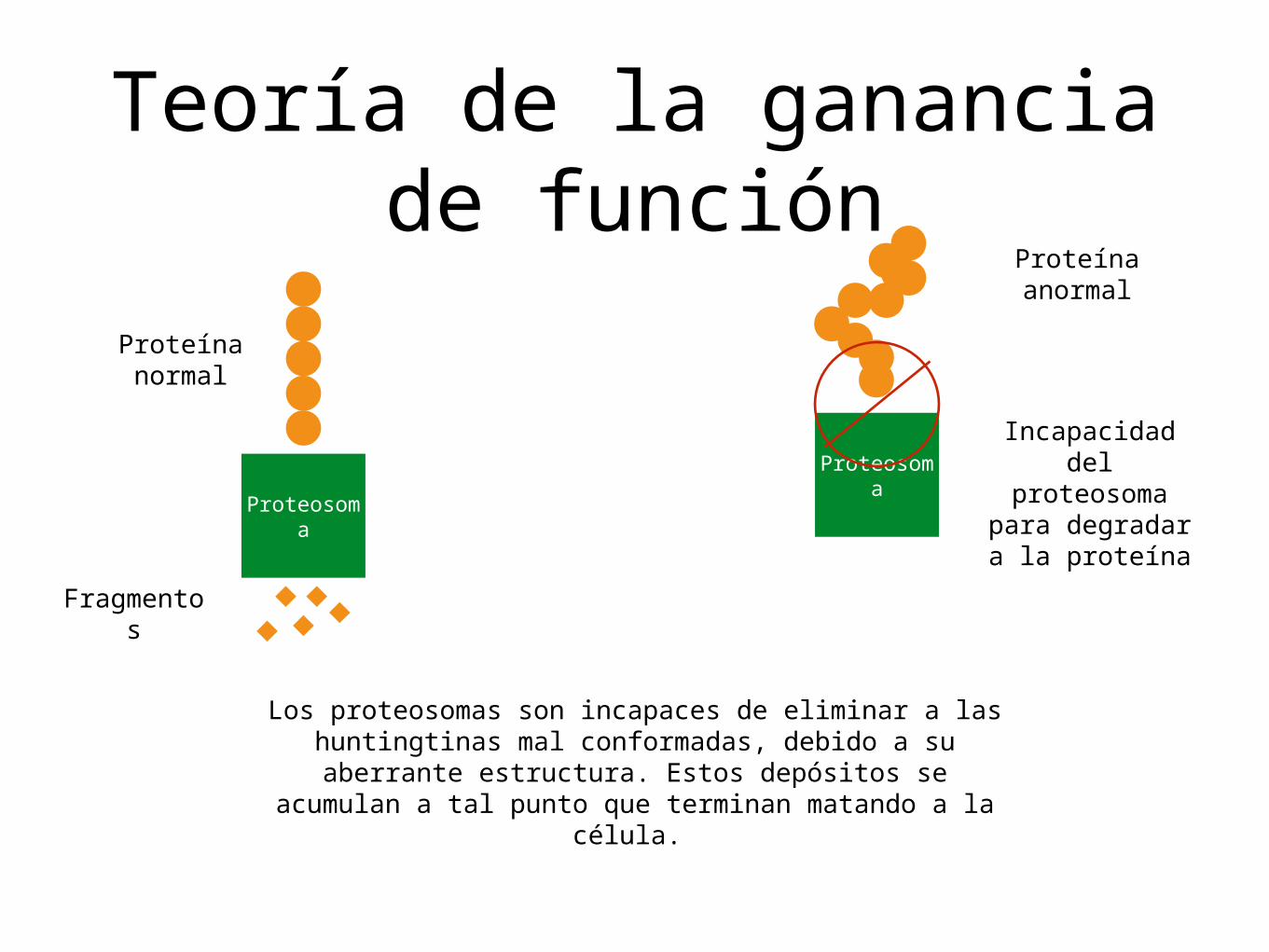
Teoría de la ganancia de función
Los proteosomas son incapaces de eliminar a las huntingtinas mal conformadas, debido a su aberrante
estructura. Estos depósitos se acumulan a tal punto que terminan matando a la célula.
Proteosoma
Proteína normal
Fragmentos
Proteosoma
Proteína anormal
Incapacidad del proteosoma
para degradar a la proteína
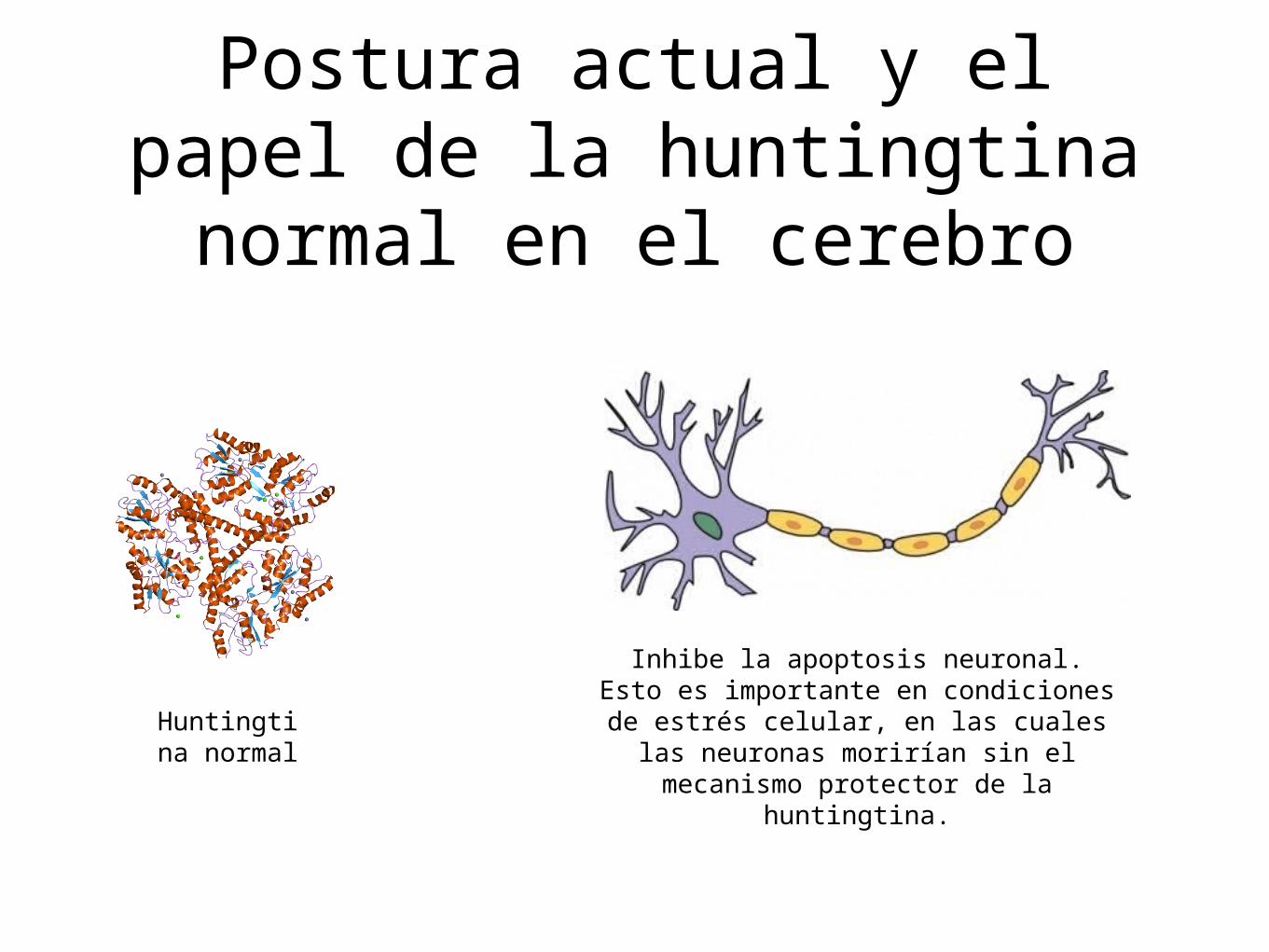
Postura actual y el papel de la huntingtina normal en el
cerebro
Huntingtina normal
Inhibe la apoptosis neuronal. Esto es importante en condiciones de estrés celular, en las cuales las neuronas
morirían sin el mecanismo protector de la huntingtina.
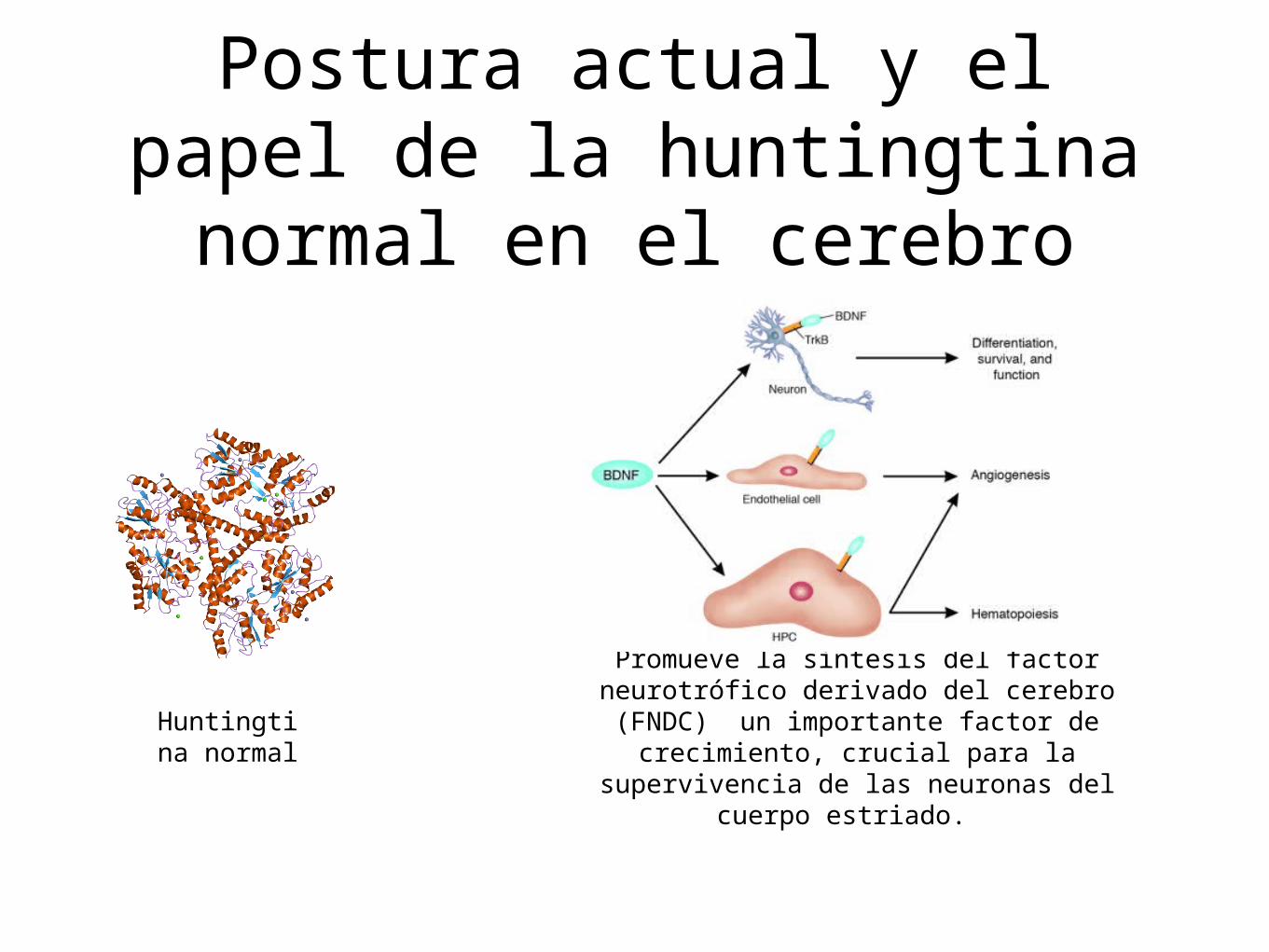
Postura actual y el papel de la huntingtina normal en el
cerebro
Huntingtina normal
Promueve la síntesis del factor neurotrófico derivado del cerebro (FNDC) un importante factor de
crecimiento, crucial para la supervivencia de las neuronas del
cuerpo estriado.

Postura actual y el papel de la huntingtina normal en el
cerebroLa postura actual de la fisiopatología de la enfermedad de Huntington, se define como la combinación de los siguientes factores :
1. La huntingtina mutada inhibe a la huntingtina normal (del alelo sano), por lo cual su función protectora de la apoptosis neuronal queda inhibida.
2. La huntingtina mutada es incapaz de producir el FNDC, haciendo propensas a las células del cuerpo estriado a morir en situaciones de estrés celular.
3.La destrucción progresiva del cuerpo estriado, provoca los signos y síntomas clínicos observados en la enfermedad.

Tratamiento de la enfermedad de
HuntingtonNo existe cura para la enfermedad de Huntington, por lo cual el tratamiento se enfoca en disminuir los síntomas. Se pueden utilizar los siguientes medicamentos, aunque su efectividad varía de persona a persona :
• Sedantes : Disminuyen la corea, aunque disminuye la dopamina, acentuando la depresión en el paciente
• Antidepresivos : Combaten la depresión, aunque pueden exacerbar la corea.
• Neurolépticos : Controlan la psicosis, pero pueden aumentar los movimientos espásticos.
También se ha intentado realizar transplantes celulares y la inducción de factores de crecimiento neuronales, la mayoría obtenidos de fetos abortados, por lo cual este tratamiento es de controversia en muchos países.

Un tratamiento en estudio, consiste en la administración de factores de crecimiento, especialmente neuronales. Sin embargo, se tiene la problemática que si son ingeridos por vía oral, son degradados en el estómago; y si son inyectados, no pueden atravesar fácilmente la barrera hematoencefálica. Actualmente se estudian unas cápsulas que se insertan en los ventrículos , que contienen células genéticamente modificadas que producen factores de crecimiento.
Se espera que en un futuro, la terapia génica y farmacológica pueda abordar correctamente la enfermedad de Huntington.


















