
CA LÂM SÀNG - thuchanhthankinh.comthuchanhthankinh.com/userupload-thuchanhthankinh/files/ca lam...
148
CA LÂM SÀNG Ngày 19/1/2015
Transcript of CA LÂM SÀNG - thuchanhthankinh.comthuchanhthankinh.com/userupload-thuchanhthankinh/files/ca lam...

CA LÂM SÀNG
Ngày 19/1/2015

Bệnh án
Bn nữ 75 tuổi nhập viện vì lú lẫn+ giật cơ toàn thân
Bệnh sử:
- 2 tháng nay bệnh nhân biểu hiện hay quên,
- khoảng 1 tháng gần đây quên nhiều, kèm theo mệt, đi lại
chậm, tay chân cứng, cử động khó khăn.
- 10 ngày trước nhập viện bệnh nhân hay la hét, hoảng
sợ, giật tay chân, có đi khám bác sĩ tư được kê toa cho
uống Haloperidol và Artan, cứng đờ toàn thân(rigidity),
kèm theo tiểu không tự chủ, ngưng thuốc điều trị bằng
Olanzapin, bệnh nhân ngủ nhiều hơn, giật cơ và gồng tứ
chi, nhập BV ĐHYD

Tiền căn
1. Bản thân:
- 20 năm trước bệnh nhân có cơn loạn thần la hét sau đó
tự ổn không điều trị gì hết
2. Gia đình:
- Chưa ghi nhận bất thường.

Khám (ngày 3/01/2015)
A/ TỔNG QUÁT:
Tổng trạng:
- Sinh hiệu:
M: 80 lần/phút HA: 125/80 mmHg
T: 37 0 C NT: 16 lần/phút
- Thể trạng gầy. .
- Các cơ quan khác: chưa ghi nhận bất thường

Thần kinh:
1. Hệ thần kinh cao cấp:
Bệnh mở mắt tự nhiên, thực hiện y lệnh lúc đúng lúckhông
2. 12 dây sọ: bt
3. Vận động:
Giật cơ toàn thân, lúc đầu giật bên trái sau đó lan sang phải
Tăng trương lực cơ toàn thân kiểu ngoại tháp
Phản xạ da lòng bàn chân gập 2 bên.

Tóm tắt
BN nữ, 74 tuổi, nhập viện vì lú lẫn, giật cơ
Bệnh khoảng 2 tháng.
Khám ghi nhận:
+ Giảm trí nhớ+ rối loạn tâm thần
+ Giật cơ toàn thân
+ Tăng TLC kiểu ngoại tháp

BÀN LUẬN

Vấn đề của bệnh nhân
Sa sút tâm thần(dementia) tiến triển+loạn thần
Tăng trương lực cơ ngoại tháp
Myoclonus toàn thân

Phân biệt lú lẫn cấp (acute confusional
state) và sa sút tâm thần (dementia)?

Sa sút tâm thần
Sa sút tâm thần là một hội chứng:
1. Tổn thương trí nhớ mãn tính và tiến triển
2. + ít nhất một lãnh vực nhận thức khác(cognitive domain)
- giảm chức năng về kỷ năng
- sự sút kém trong xã hội
- hay chức năng nghề nghiệp

Sa sút tâm thần
Tiêu chuẩn ICD-10 chẩn đoán sa sút tâm thần
Impairment of short- and long-term memory
(more accurately, of anterograde memory)
Ít nhất một trong tiêu chuẩn sau đây(At least one of the
following):
Impairment of abstract thinking(suy nghĩ trừu tương)
Impaired judgment (phán xét)
Other disturbances of higher cortical function(rối loạn chức
năng vỏ não)
Personality change (thay đổi nhân cách)

Sa sút tâm thần
Tiêu chuẩn ICD-10 chẩn đoán sa sút tâm thần
Memory impairment, intellectual impairment causing
significant social and occupational impairment(tổn thương
về mặt xã hội và nghề nghiệp)
Absence of occurrence exclusively during the course of
delirium (không xảy ra được loại trừ trong quá trình mê sảng)
(Cả hai tiêu chuẩn sau đây (Either of the following):
Evidence of an organic factor causing this impaired memory
and intellect (bằng chứng tổn thương thực thể)
Impaired memory and intellect that cannot be accounted for
by any nonorganic mental disorder(không mô tả được rối
loạn tâm thần không thực thực thể)

(Mức độ ý thức)
(Diễn tiến
Gia tăng tktđ
Dự hậu
Tổn thương Không tổn thương
Cấp, bán cấp, dao động mãn
Hồi phục Không hồi phục
có Không có
Phân biệt lú lẫn cấp và SSTT

Phân biệt mê sảng ở bn sa sút tâmthần?(What features distinguish delirium from dementia?)

Không phân biệt chắc chắn, các dấu hiệu giúp phân biệt
1. Khởi phát đột ngột đề nghị delirium, (Thay đổi ý thức, vấn đề rõ ràng, sự tập trung chú ý không cân đối
thiếu hụt khác, hay ảo giác rõ ràng, nhận thức giao động (Khoảng
tình tri giác) tâm thần vận động và/hay hoạt động quá mức tk tự động,
lời nói vỡ tung, ảo giác rõ)
2. Bn sa sút tâm thần mãn tính có thể có delirium thêm vào

Tất cả bn sa sút tâm thần có biểuhiện tâm thần?(Do all patients with dementia develop psychotic features?)

Không, tâm thần có thể tìm thấy trong trong các loại
dementia và không liên quan đến giai đoạn và độ
nặng của dementia
(No. Psychosis is a variable finding in all types of dementia and
is not even clearly related to the stage or severity of dementia0

Các công cụ tầm soát dementia?
(Which screening instruments are commonly used in
diagnosing dementia?)

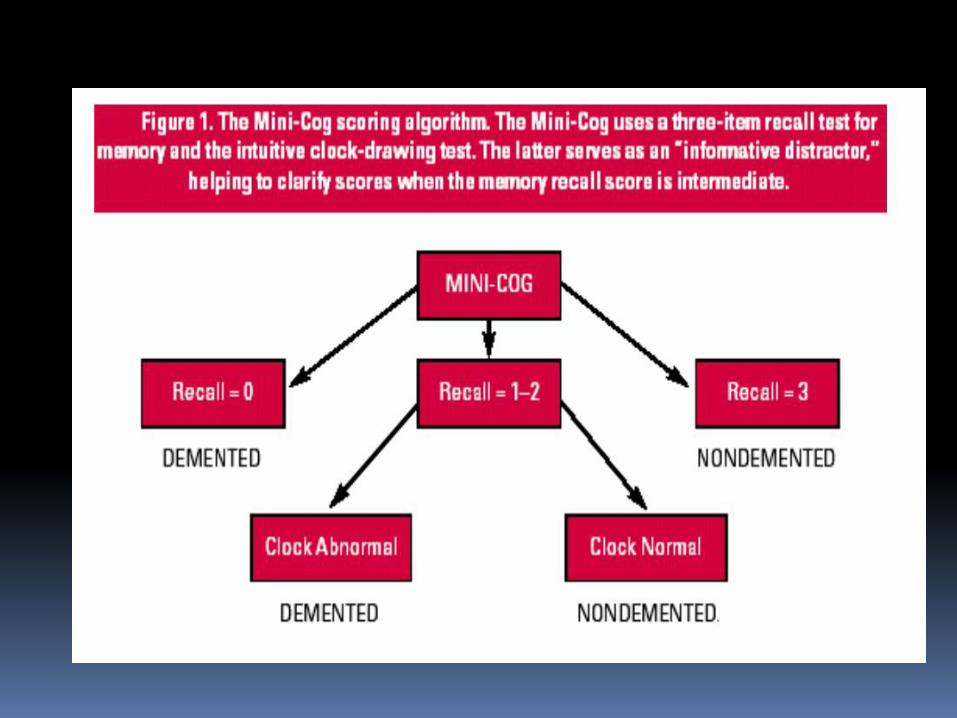
1. The Folstein Mini-Mental Status Examination
(MMSE), minicog test
2. Short Blessed dementia scale,
3. Mattis Dementia Rating Scale
(Thường dùng trong lâm sàng và nghiên cứu
tầm soát dementia và độ nặng của dementia)

Hạn chế MMSE trong đánh giádementia?(What are the limitations of the MMSE in the assessment of dementia?)

1. Dương tính giả bn trầm cảm và âm tính giả trong
dementia sớm(chức năng cao cấp bình thường)
(Besides the fact that it has both false-positive (usually
depression) and false-negative results(usually early dementia in
highly functioning patients),
2 MMSE hạn chế mất tính chất toàn diện
(MMSE also has limitations based on its lack of
comprehensiveness )

Nguyên nhân dementia?
(What are the most common causes of dementia or
conditions resembling dementia?)

1. Alzheimer’s disease là thể thường gặp nhất ở
người lớn (>80%).
(Depression with pseudo dementia is a frequent cause of
cognitive loss and must be ruled out in all patients)
2. Các nguyên nhân quan trọng khác
- multi-infarct or vascular dementia,
- dementia with Lewy bodies,
- frontotemporal dementia,
- dementia-like syndromes (alcohol or chronic use of
certain prescription drugs).

Nguyên nhân ít gặp của dementia?
(What uncommon causes of dementia must be
considered in the differential diagnosis of every
patient with dementia?)

1.Toxins (lead, organic mercury)
2. Vitamin deficiencies (B12,B1, and B6)
3. Endocrine disturbances (hypothyroidism or
hyperthyroidism, hyperparathyroidism,Cushing’s disease,
and Addison’s disease)
4. Chronic metabolic (hyponatremia, hypercalcemia,
chronic hepatic failure, and renal failure)

5. Vasculopathies ảnh hưởng não
6. Bất thường cấu trúc (chronic subdural hematomas,
normal-pressure hydrocephalus, and slow-growing
tumors)
7. Nhiễm hệ thần kinh trung ương (acquired immune
deficiency syndrome [AIDS], Creutzfeldt-Jakob disease,
cryptococcal, tuberculous meningitis….)
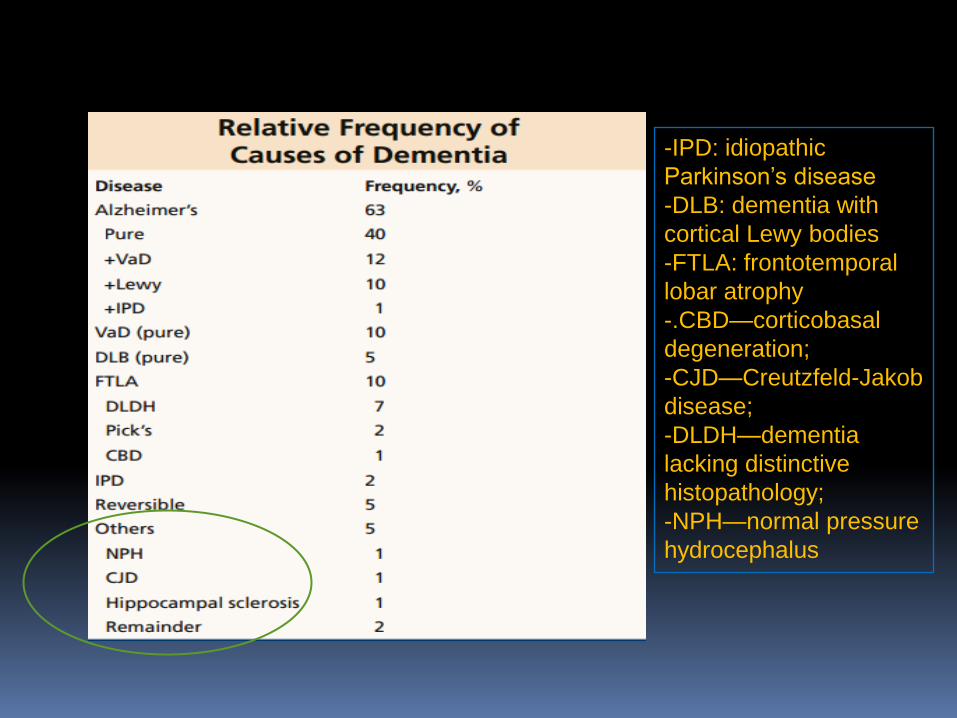
-IPD: idiopathic
Parkinson’s disease
-DLB: dementia with
cortical Lewy bodies
-FTLA: frontotemporal
lobar atrophy
-.CBD—corticobasal
degeneration;
-CJD—Creutzfeld-Jakob
disease;
-DLDH—dementia
lacking distinctive
histopathology;
-NPH—normal pressure
hydrocephalus

Đặc điểm dementias dưới vỏ?
(What are the characteristics of subcortical dementias?)

1. Subcortical dementias lack cortical features, such as
aphasia, apraxia, and acalculia. Recall Memory is
impaired worse than recognition memory. Visuospatial
skills are often impaired.(không có mất ngôn ngữ, thực dụng, tính
toán, tổn thương nhớ lại hơn nhận thức trí nhớ…tổn thương thị giác
không gian)
2. Frontal executive deficits, bradyphrenia, anomia,
personality changes, and psychomotor slowing are
prominent. Dysarthria, abnormal posture and coordination,
and adventitious movements may be present (thiếu hụt thực
hiện thùy trán, suy nghĩ chậm, mất định danh, thay đổi nhân cách và
vận động tình cờ)
‘Brady’ means slower; ‘phrenia’ means thinking—so slowed thinking processes.
Bradyphrenia occurs in some people with Parkinson's disease. It appears to be
related to damage to frontal cortex brain regions -- those regions of the brian
that support thinking, reasoning, and planning.

Phân biệt cortical và subcorticaldementia?
(How do the general features of subcortical dementias
differ from cortical dementias?)

1. Cortical dementias: AD, liên hệ language và
calculations và có thể liên hệ apraxia và cortical
sensory disturbances (e.g., astereognosis,
graphesthesia), liên quan ngôn ngữ, tính toán, thực
dụng….
2. Trong khi subcortical dementias thì không. Cả
recall và recognition memory thường bị tổn thương
trong cortical dementia, recognition memory bình
thường trong subcortical dementia.
Nhớ lại và nhận thức trí nhớ tổn thương trong cortical
dementia….

3. Frontal executive functions mất tương ứng với quá trình
dementia ở vỏ nhưng ưu thế dementia dưới vỏ
dementia.(chức năng thực hiện thùy trán mất, ưu thế dưới vỏ..)
4. Bradykinesia và bradyphrenia, các rối loạn vận động
khác thường không có hay trể trong demtia vỏ, nhưng
sớm dưới vỏ. (giảm động, suy nghĩ chậm ít gặp hay trể ngược
dưới vỏ…)
5. Thay đổi nhân cách (pesonality changes ) trong cả 2 loại,
ưu thế sớm dưới vỏ (thay đổi nhân cách ưu thế sớm dưới vỏ..)

Nguyên nhân subcorticaldementia?
(What disorders or clinical syndromes are typically
associated with subcortical dementia?)

1. Parkinson’s disease
2. Huntington’sdisease
3. Progressive supranuclear palsy
4. Spinocerebellar degeneration
5. Idiopathic basal ganglia calcification
6. Multiple sclerosis
7. Inflammatory conditions involving the basal ganglia
and/or thalamus
8. AIDS
9. Corticobasal degeneration

Myoclonus là gì?

Giật cơ (myoclonus)
Myoclonus: vận động không tự chủ gây co cơ hay
ngăn chận(contractions or inhibitions)
1.Đột ngột (sudden)
2.Ngắn (brief)
3.Shock-like
- Co cơ: "positive myoclonus,“
- Ức chế: "negative myoclonus" or asterixis.
Do nhiều nguyên nhân

Giật cơ (myoclonus)
1. Myoclonus : quick, involuntary muscle jerk.
Thí dụ: hiccups.
(sudden jerks, or "sleep starts," (may experience just before
falling asleep).
2. Hầu hết myoclonus xảy ra:
- nervous system (neurological) disorder,
- epilepsy,
- biến dưỡng(metabolic condition)
- phản ứng thuốc.
(Localized to one part of the body or all over the body
Sometimes severe enough to interfere with eating, speaking or walking)

Giật cơ (myoclonus)
3. Điều trị nguyên nhân kiểm soát triệu chứng myoclonus.
4. Không xác định nguyên nhân điều trị triệu chứng giảm
myoclonus.

Nguyên nhân myoclonus?

1. Physiological myoclonus(sinh lý)
Myoclonus xảy ra ở người bình thường, không cần điều
trị. Bao gồm:
- Hiccups
- Sleep starts
- Run hay co thắc(Shakes or spasms) do anxiety hay
exercise
- Giật cơ trẻ con (Infant muscle twitching) trong sleep
hay sau feeding

2. Essential myoclonus(vô căn)
Essential myoclonus xảy ra tự thân, thường không kèm
triệu chứng khác và không có bệnh khác
Nguyên nhân essential myoclonus:
- unexplained (idiopathic)
- vài trường hợp hereditary.

3. Epileptic myoclonus (dạng động kinh)
Myoclonus xảy ra như một phần của rối loạn động kinh.
Muscle jerks có thể chỉ là dấu hiệu duy nhất hay một
trong nhiều dấu hiệu

4. Symptomatic (secondary) myoclonus
(triệu chứng)
Thể thường gặp của myoclonus. Muscle jerks do bệnh lý
nội khoa bao gồm:
1. Head, spinal cord injury
2. Infection
3. Kidney, liver failure
4. Lipid storage disease
5. Chemical, drug poisoning
6. Prolonged oxygen deprivation
7. Medication reaction

Symptomatic (secondary) myoclonus
8. Autoimmune inflammatory
9. Metabolic disorders
10. Nervous conditions that result in secondary myoclonus:
- Stroke
- Brain tumor
- Huntington's disease
- Creutzfeldt-Jakob disease
- Alzheimer's disease
- Parkinson's disease and Lewy body dementia
- Corticobasal degeneration
- Frontotemporal dementia
- Multiple system atrophy

Phân loại myoclonus

Phân loại myoclonus

Phân loại giải phẫu-sinh lý(anatomic-physiologic categories)
1. Cortical myoclonus:
- posthypoxic myoclonus,
- progressive myoclonus epilepsy
- progressive myoclonus ataxia syndromes, -
- toxic-metabolic myoclonus,
- myoclonus associated neurodegenerative disease,
- myoclonus associated Creutzfeldt-Jakob disease

Phân loại giải phẫu-sinh lý(anatomic-physiologic categories)
2. Cortical-subcortical myoclonus (eg, juvenile myoclonic
epilepsy)
3. Subcortical-nonsegmental myoclonus (eg, myoclonus-
dystonia syndrome)
4. Segmental myoclonus (eg, palatal myoclonus)
5. Peripheral myoclonus (eg, hemifacial spasm)

Điều trị thuốc
Levetiracetam- Trong một báo cáo, levetiracetam hiệu quả trong 3 bệnh
nhân giật cơ tủy (spinal myoclonus )
Levetiracetam : liều đầu tiên 500 - 1000 mg hàng ngày,
chia làm 2 liều.
- Có thể tăng 1000mg mỗi 2 tuần khi cần và có thể lên
3000mg hàng ngày, nếu dung nạp tốt và đáp ứng kém,
liều tối đa hàng ngày 4000mg.
- Điều trị levetiracetam thường dùng liều từ 1000-3000mg
hàng ngày.

Điều trị thuốc
LevetiracetamTác dụng phụ kết hợp với levetiracetam là mệt mỏi
(fatigue), buồn ngủ(somnolence), chóng mặt (dizziness) và
nhiễm trùng(hô hấp trên)
Cắt thuốc đột ngột có thể gây cơn động kinh hay làm nặng myoclonus

Piracetam
1. Liều thấp piracetam (16.8 - 9.6 g daily)
2. Piracetam dùng liều khởi đầu 2.4 g daily, 3 lần/ngày.
3. Liều điều trị piracetam từ 2.4 g- 24 g daily.
4. Tránh cắt đột ngột piracetam , có thể làm nặng hơn
myoclons và/hay cơn động kinh.

Clonazepam
1. Ích lợi của clonazepam trong cortical myoclonus(data from uncontrolled observational studies )
1. Có thể ích lợi trong subcortical-nonsegmental types of myoclonus, including essential myoclonus , myoclonus-dystonia, reticular reflex myoclonus và propriospinalmyoclonus.
2. Clonazepam ích lợi trong essential palatal myoclonus ,3. Clonazepam chọn lựa đầu tiên trong điều trị spinal
segmental myoclonus ,

Clonazepam
1. Khởi đầu 0,5 mg hàng ngày và tăng dần đến 1,5-3mg/ngày, chia 3 liều.
2. Clonazepam liều cao 15 mg/ ngày thường cần thiếtnhưng tăng dần
-The most common side effects: drowsiness, dizziness, fatigue, and sedation. -Abrupt reductions or withdrawals of clonazepam can cause both an exacerbation of myoclonus and withdrawal seizures. - As with other benzodiazepines, clonazepam may produce physical and psychological dependence.- Tolerance with loss of effectiveness may develop over a period of several months in some patients.

Valproic acid
1. Valproic acid tăng dần, khởi đầu 15 mg/kg hàngngày, chia 3 liều. Có thể tăng 5-10mg/ngày hàngtuần khi cần. Liều điều trị 1200-2000mg/ngày
2. Trong thời gian khởi đầu điều trị, bn có thể rốiloạn tiêu hóa thoáng qua, đôi khi nôn ói, ít khiđau bụng, tiêu chảy

Valproic acid
3. Tác dụng phụ khác: weight gain, obesity, hair loss, easy bruising, tremor. (bầm tím)
4. Ít gặp: agranulocytosis, Stevens-Johnson syndrome, aplastic anemia, hepatic failure, dermatitis/rash, serum sickness, and pancreatitis.

Điều trị khác
1. Intrathecal baclofen
2. Botulinum toxin injections

Tóm tắt điều trị myoclonus
1. Tranquilizers: Clonazepam (Klonopin), thuốc
dùng nhiều nhất điều trị triệu chứng myoclonus
tác dụng phụ Clonazepam: chóng mặt, buồn ngủ

Tóm tắt điều trị myoclonus
2. Anticonvulsants: giảm triệu chứng myoclonus
thuốc chống động kinh thường dùng nhất:
levetiracetam (Keppra), valproic acid (Depakene) and
primidone (Mysoline).
- Valproic acid có thể gây : nausea.
- Levetiracetam có thể gây: fatigue vàdizziness.

Tóm tắt điều trị myoclonus
3. Botulinum toxinA (Botox) tiêm có thể điều trị nhiều thể
myoclonus
4. Surgery
Triệu chứng Myoclonus : tumor hay sang thương não,
tủy phẫu thuật có thể chọn lựa. Myoclonus ở mặt hay
tai, phẫu thuật ích lợi.
5. Deep brain stimulation (DBS): myoclonus và các
movement disorders.

Ca lâm sàng:
1. CLS để XĐ CĐ:
- CT scan não -> MRI não.
- Dịch não tủy
2. CLS thường quy: CTM, ĐH, BUN, creatinin,
AST, ALT, Ion đồ, TPTNT, ECG, X quang ngực
thẳng.

- CTM: RBC 4.09 T/L
HB 134 g/L
Hct 37.2 %
WBC 11.94 G/L
Neu 54.5 %, Lym 28.8%
PLT 236 G/L
- PT: 14.4s, INR 1.07, APTT: 30.4s
- VS giờ 1: 6 mm; giờ 2: 14 mm
- CRP: bình thường
- Chức năng gan thận: trong giới hạn bình thường.
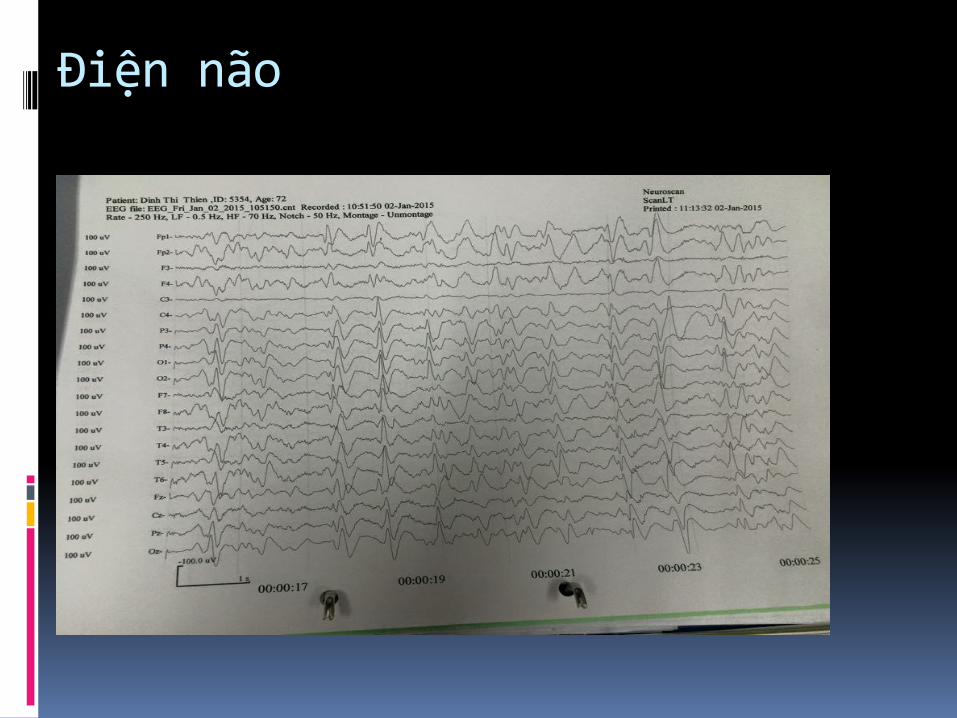
Điện não
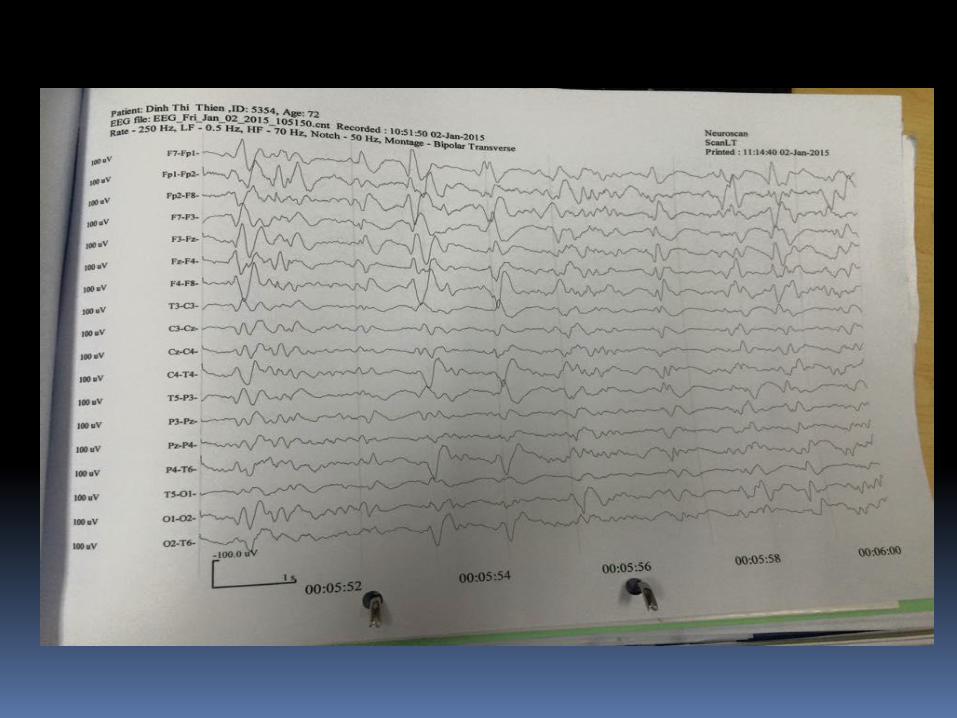

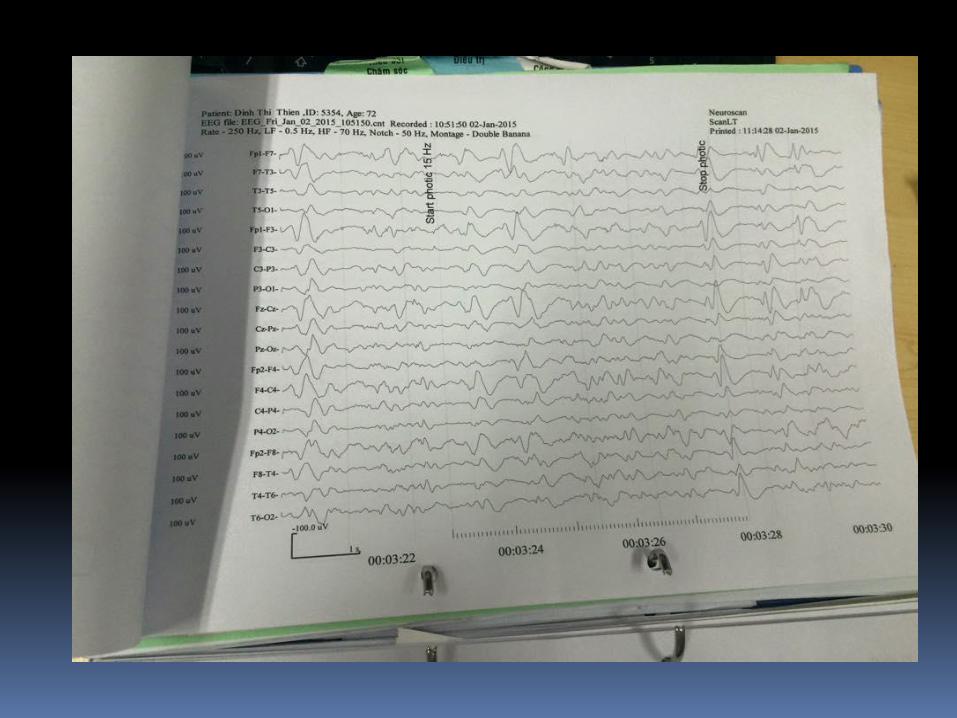


EEG
Thay đổi EEG trong sporadic CJD : periodic bi- and
triphasic waves
(periodic sharp wave complexes, PSWCs).

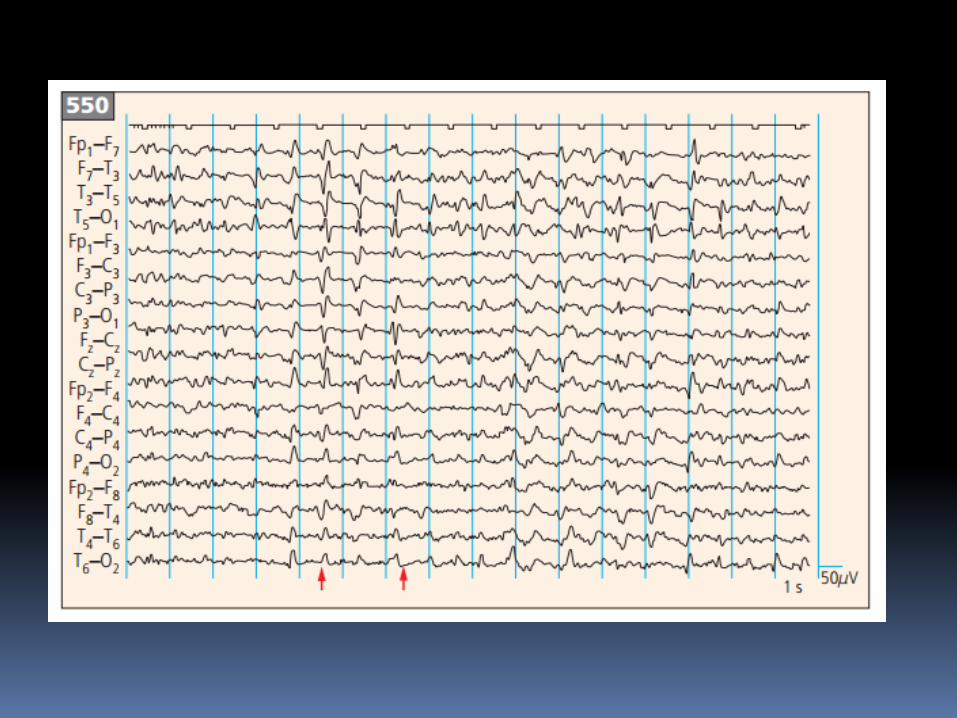
Tiêu chuẩn EEG trong chẩnđoán CJD
- Periodc "sharp-wave" complexes (PSWCs)
- Periodicity (most important criterion)
- Frequency 0.5-2/sec
- Duration between 100 - 600 msec
- Amplitude higher than 150 μV-300 μV
- Generalized activity, with a maximum over the
frontal region, partly also lateralized
- Sometimes followed by burst suppression"
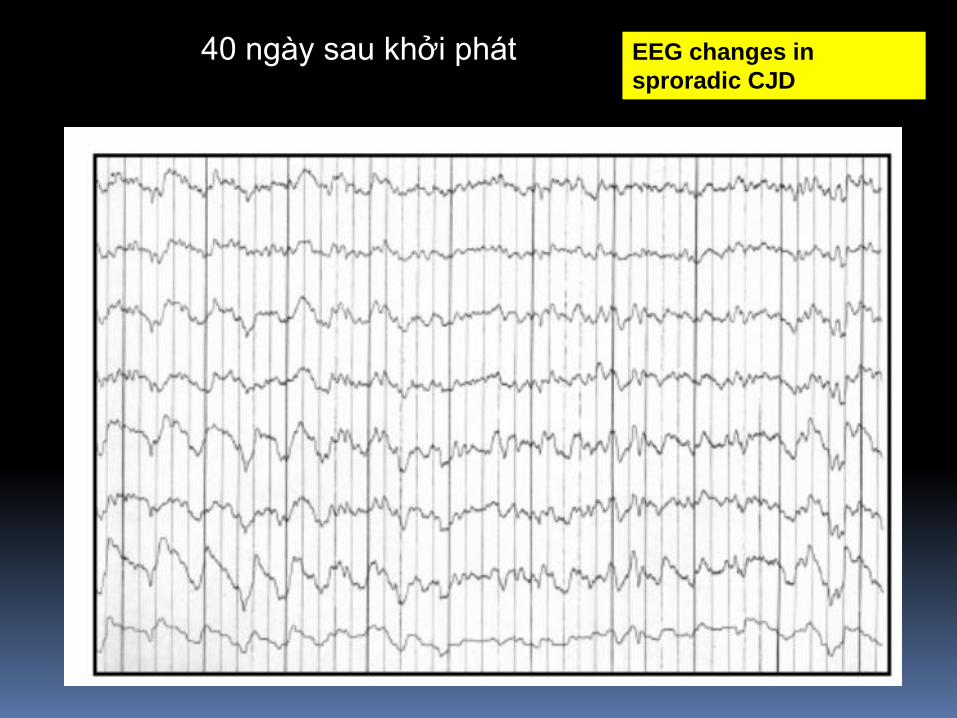
40 ngày sau khởi phát EEG changes in
sproradic CJD
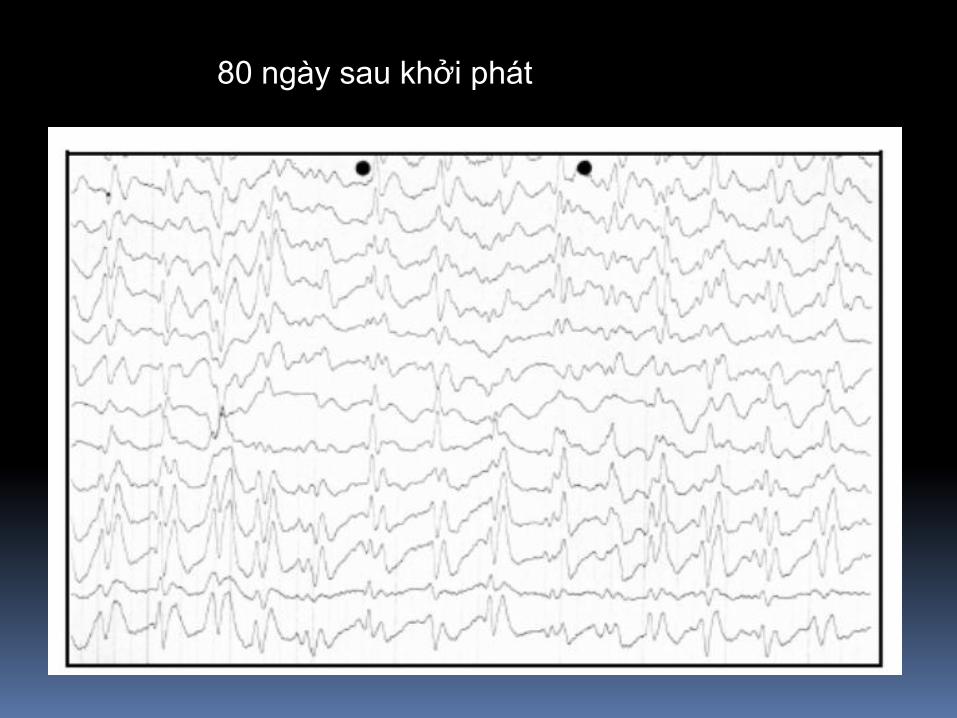
80 ngày sau khởi phát
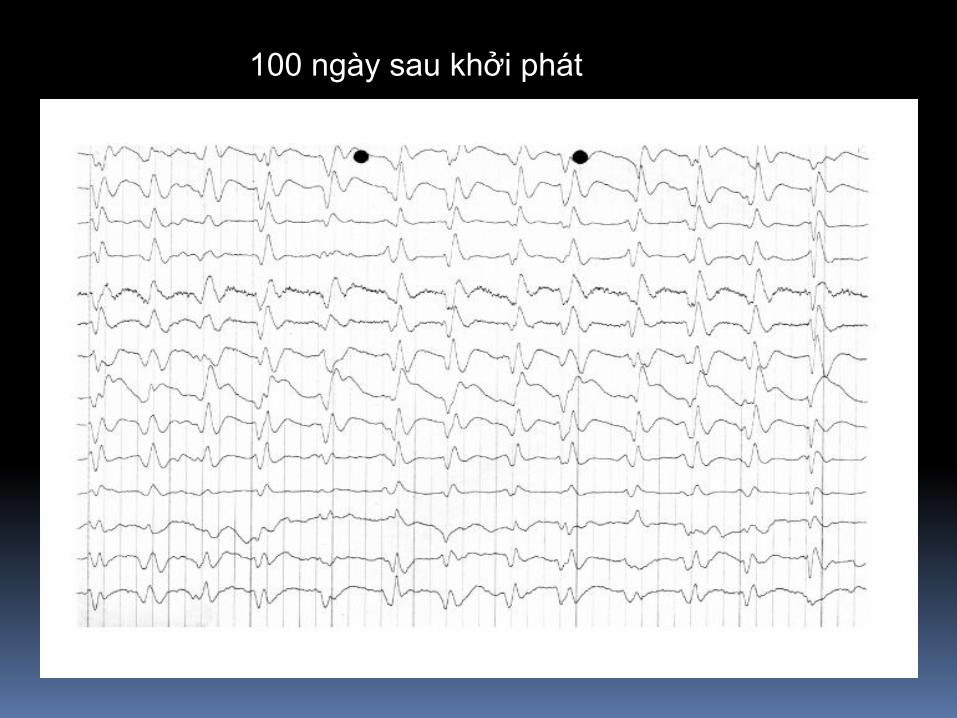
100 ngày sau khởi phát
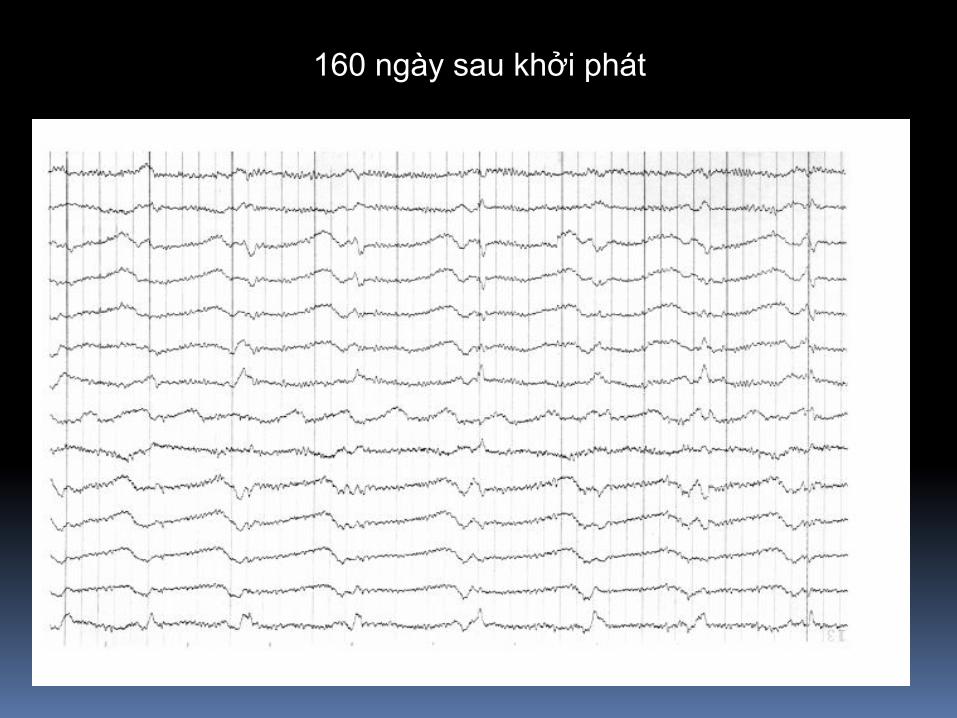
160 ngày sau khởi phát
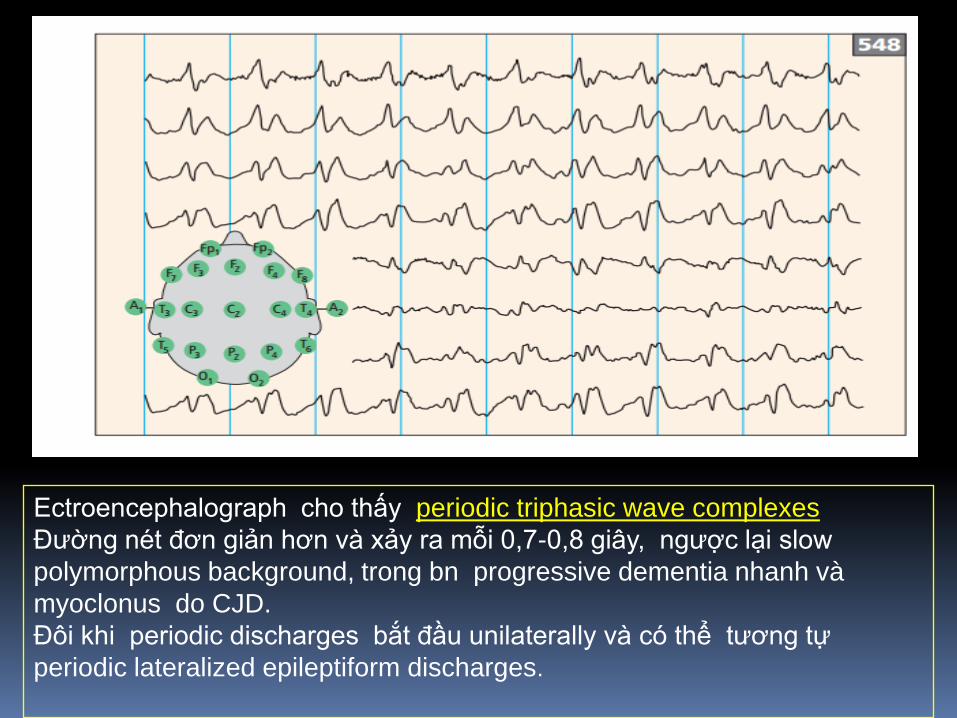
Ectroencephalograph cho thấy periodic triphasic wave complexes
Đường nét đơn giản hơn và xảy ra mỗi 0,7-0,8 giây, ngược lại slow
polymorphous background, trong bn progressive dementia nhanh và
myoclonus do CJD.
Đôi khi periodic discharges bắt đầu unilaterally và có thể tương tự
periodic lateralized epileptiform discharges.
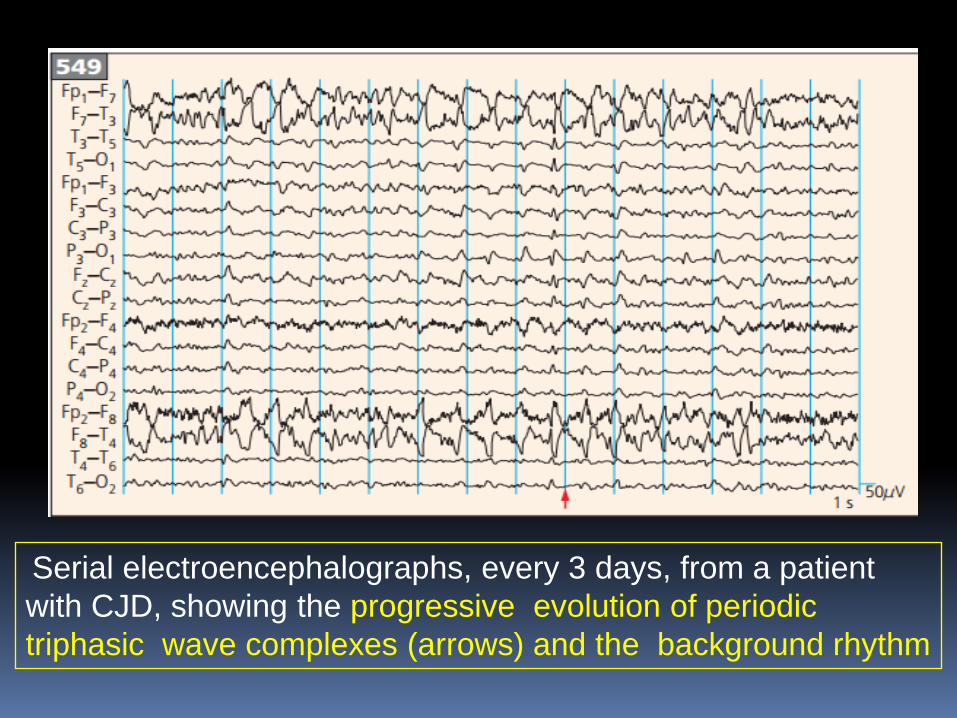
Serial electroencephalographs, every 3 days, from a patient
with CJD, showing the progressive evolution of periodic
triphasic wave complexes (arrows) and the background rhythm
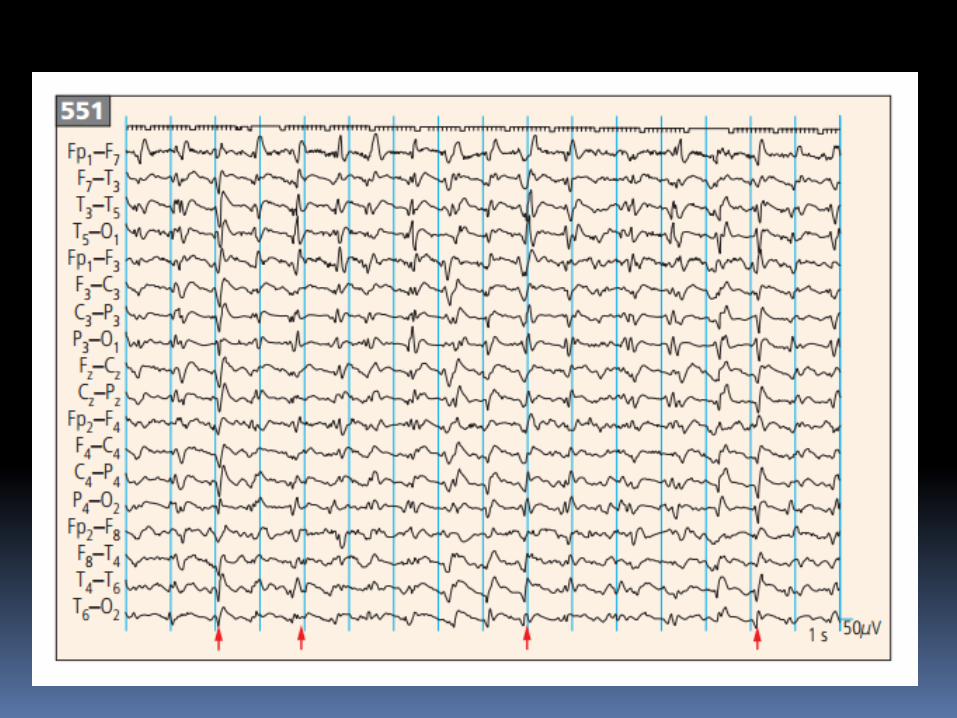


MRI chẩn đoán CJD
Tiêu chuẩn hiện nay của WHO :
1. electroencephalogram (EEG)
2. CSF-analysis for 14-3-3 proteins.
3. Không có tiêu chuẩn MRI chẩn đoán sCJD, mặc dù
có những thay đổi đặc biệt đã được nêu ra

1. Scans are normal (45%) or show cerebral atrophy (30%).
2. DWI MRI commonly reveals high-signal changes in the
basal ganglia or cortical ribbon in cases of CJD
3. Proton density images in vCJD show hyperintensity of
thalamus (especially pulvinar)
4. Fluoro-deoxyglucose PET in FI shows reduction in
thalamic metabolic activity 34
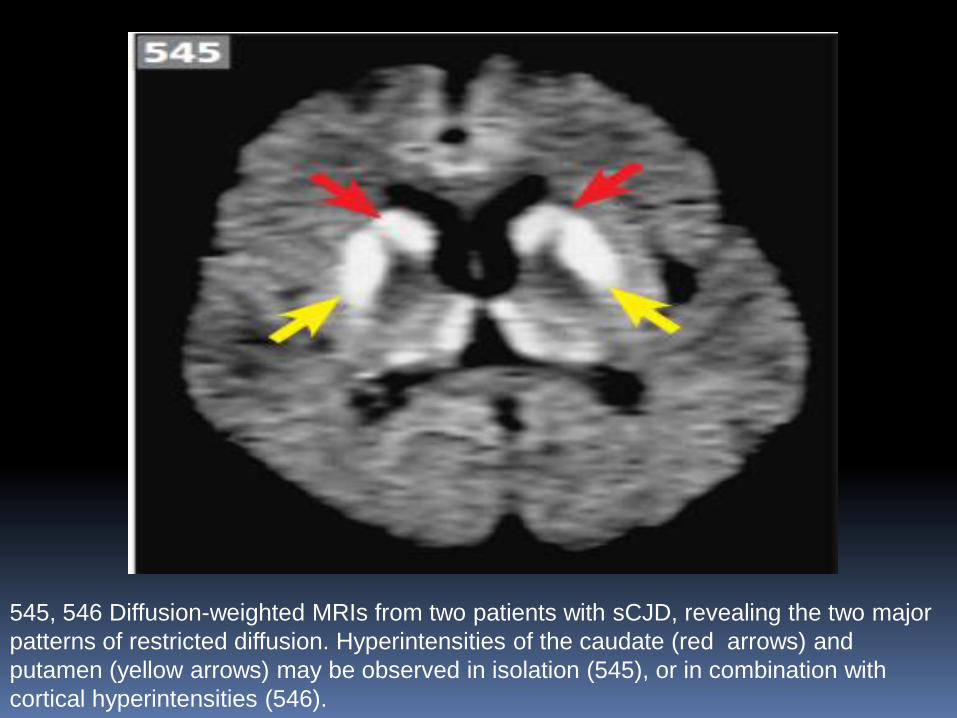
545, 546 Diffusion-weighted MRIs from two patients with sCJD, revealing the two major
patterns of restricted diffusion. Hyperintensities of the caudate (red arrows) and
putamen (yellow arrows) may be observed in isolation (545), or in combination with
cortical hyperintensities (546).
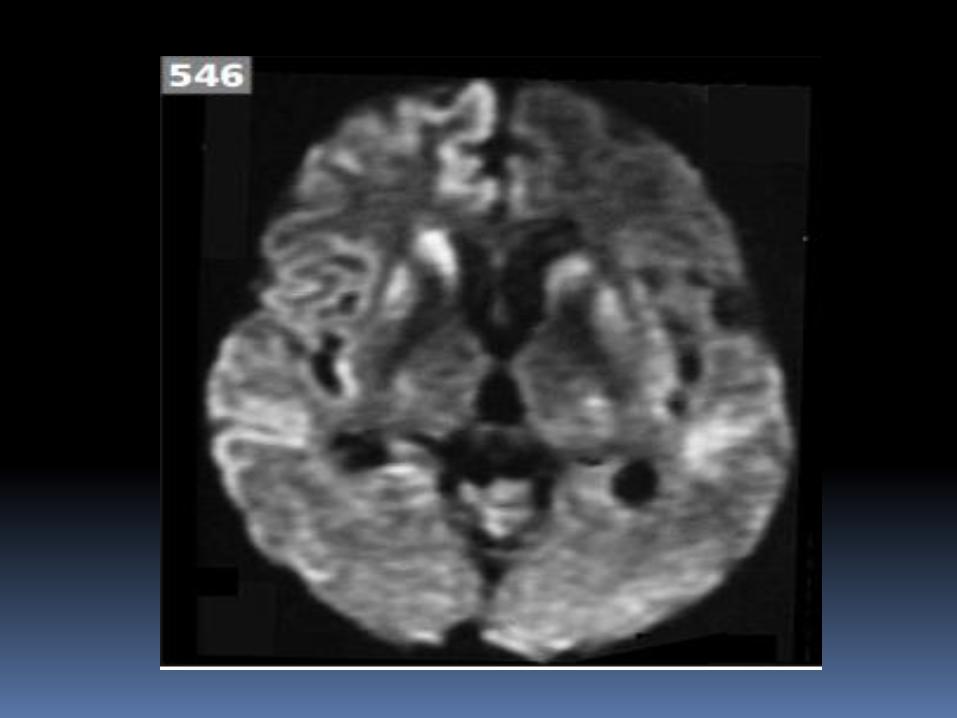

547 Proton-weighted imaging of the brain demonstrates pulvinar hyperintensity in
a patient with vCJD


- X quang ngực: BT.
- SA bụng chưa phát hiện bất thường
- TPTNT: Bình thường
- Dịch não tủy: trong không màu, tế bào 3/uL, chủ
yếu Lympho, Glu 70/168 mg%, Pro 73 mg%,
nấm (-)
- Test nhanh HIV (-)
- CT scan não: Giảm đậm độ lan tỏa chất trắng 2
bán cầu, không ghi nhận u
- MRI não (xem hình):

Tóm tắt
1. Lâm sàng
Sa sút tâm thần(dementia) tiến triển+ rối loạn hành vi
Tăng trương lực cơ ngoại tháp
Myoclonus toàn thân
2. Cận lâm sàng
EEG: sóng alpha mất thay vào sóng chậm ba pha lập lại
CT scan não: Giảm đậm độ lan tỏa chất trắng 2 bán cầu
MRI : tổn thương rải rác vỏ, dưới vỏ
DNT protein tăng nhẹ


Chẩn đoán
Chẩn đoán: sa sút tâm thần tiến triển+ giật cơ
Neurodegenerative disease: classic Creutzfeldt-Jakob
disease?

Bệnh prion?

Prion Diseases(Transmissible Spongiform Encephalopathies,TSEs)
1. Nhóm bệnh thoái hóa thần kinh tiến nặng dần, ít gặp
(family of rare progressive neurodegenerative disorders )
2. Có ở người và động vật
3. Phân biệt thời gian ủ bệnh dài
4. Đặc điểm thay đổi dạng xốp và mất tế bào thần kinh
5. Không gây đáp ứng miễn dịch
6. Thuyết prion được chấp nhận rộng rãi
transmissible degenerative diseases of the central nervous
system caused by novel infectious pathogens designated prions

TSEs có đặc điểm:
Không có đáp ứng miễn dịch
Neuropathology bao gồm:
Thoái hóa não, nhiều vùng trở nên xốp (Spongiform )
Phản ứng gliosis
Mất tế bào thần kinh
Amyloid plaques(mảng tinh bột)
Không phản ứng viêm nhiễm

Nguyên nhận TSEs
1. Tác nhân TSEs: Proteinaceous Infectious particle
(nhiễm mảnh nhỏ có protein)
2. Thuật ngữ "prions" xem như bất thường, tác nhân gây
bệnh lý có thể lan truyền và có thể gây sự gập bất
thường của protein tế bào chuyên biệt bình thường gọi
prion protein rất phong phú trong não. Chức năng của
prion bình thường này vẫn chưa hiểu hết.
3. Sự gập bất thường các prion protein dẫn đến não bị tổn
thương gây triệu chứng bệnh prion, thường tiến triển
nhanh chóng và hầu hết tử vong

Prion là gì ?
Cho đến nay bằng chứng tác nhân nhiễm trong TSEs là protein được khám phá bởi Stanley Prusiner (1996):
1. Người đầu tiên nghiên cứu proteins này.2. Nobel Prize năm 1997. 3. Đặt tên prion proteins (viết tắt PrP) hay prions.
Professor Stanley Prusiner
discovered prions

PrPc
Protein bình thường. Còn gọi PrPc (for cellular). Dễ hòa tan. Dễ tiêu hóa bởi proteases. Mã hóa bở gen PRNP (ở người

PrPC + Cu (Copper)
Antioxidant activity
Resistance to oxidative stress
Prevent neuronal dysfunction
(Brown et al., 2002)
• Antioxidative
Chức năng của Prion protein
• Other functions
?

PrPsc
Bất thường, protein gây bệnh (nhiễm).
Còn gọi PrPsc ( scrapie).
Có chuỗi amino acid giống nhau và số lượng nhưbình thường, cấu trúc nguyên phát được xác địnhnhưng:
- Cấu trúc thứ phát ưu thế beta conformation
- Không hòa tan trong tấc cả dung môi.
- Kháng cao với proteases
- Khi PrPsc đến tiếp xúc PrPc, nó chuyển đổi PrPcthành PrPsc.
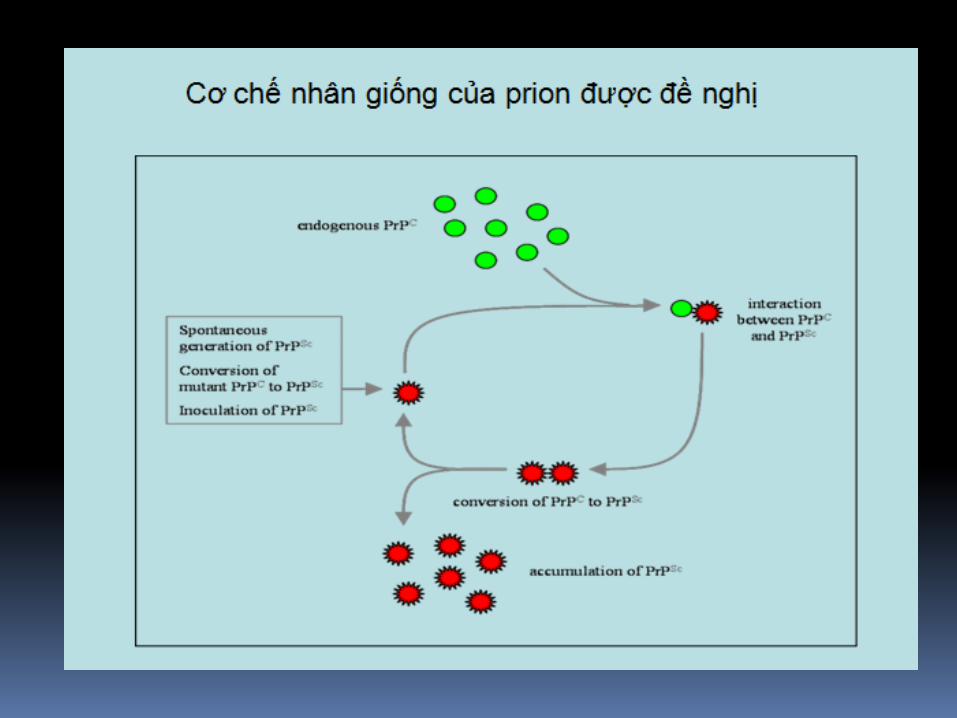
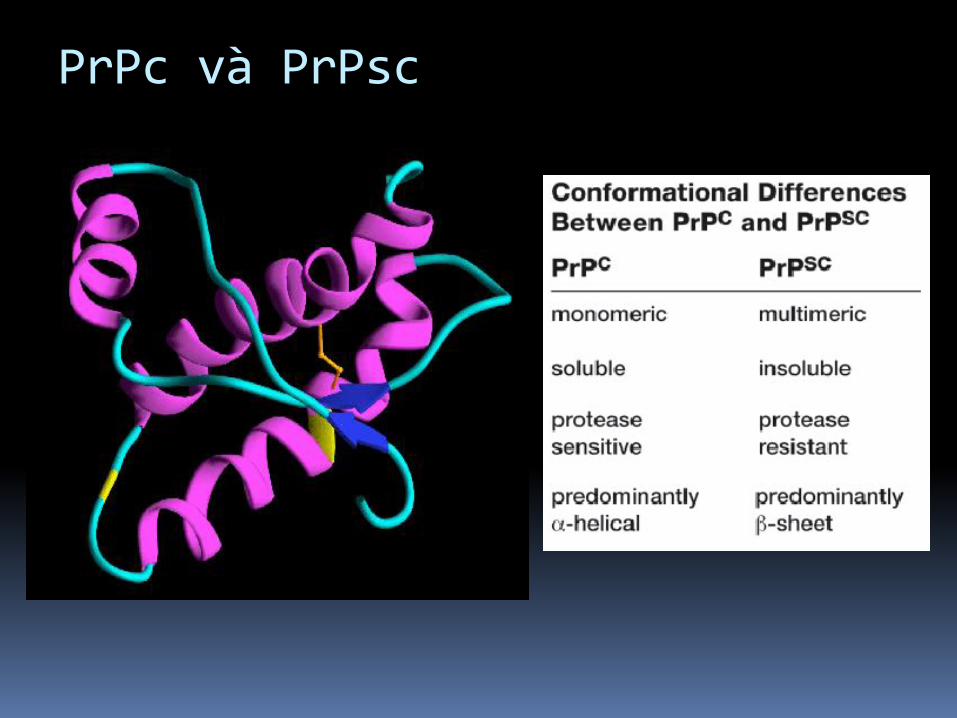
PrPc và PrPsc

Kuru
Fatal Familial Insomnia (FFI)
Creutzfeldt-Jakob disease (CJD)
Scrapie
Bovine Spongiform Encephalopathy (BSE)
Chronic Wasting Disease
(CWD)
Prion Diseases (TSE)
NgườiĐộng vật
Scrapie: bệnh hệ thần kinh dê và cừu, tương tự
CJD và BSE triêu chứng ngứa, không điều khiển
các cơ và thoái hóa hệ thần kinh
Bệnh thoái hóa hệ thần kinh, gây
tử vong ở bộ lạc New Guinea
(Transmissible Spongiform Encephalopathies)
Bò điên
Loại động vật có sừng, móng chẻ nuôi lấy sữa
hoăc ăn thịt và hưu nai trong hoang dã

Human Prion Diseases
Creutzfeldt-Jakob Disease (CJD)
Variant Creutzfeldt-Jakob Disease (vCJD)
Gerstmann-Straussler-Scheinker Syndrome
Fatal Familial Insomnia
Kuru

Kuru (disease)
Kuru : bệnh thoái hóa thần kinh không điều trị được,
bệnh địa phương ở bộ tộc New Guinea. Bệnh gây ra
bởi prion tìm thấy ở người
http://www.biologie.uni-duesseldorf.de
KURU: laughing death -Papua New Guinea (1957)
KURU and prion’s disease: similarities in the symptoms and in
cerebellar ataxia

Fatal familial insomnia
1. Fatal familial insomnia (FFI) rất ít gặp
2. Autosomal dominant inherited prion disease của não
3. Đột biến PrP, có thể tự phát (non-inherited mutation
variant called sporadic fatal insomnia (sFI))
4. FFI tiến triển mất ngủ xấu dần, dẫn đến ảo giác, mê
sảng, lú lẩn giống sa sút tâm thần
5. Trung bình sống 18 tháng khi khởi phát

Animal Prion Diseases
Bovine Spongiform Encephalopathy (BSE)
Chronic Wasting Disease (CWD)
Scrapie(dê và cừu)
Transmissible mink encephalopathy(chồn)
Feline spongiform encephalopathy(họ mèo)
Ungulate spongiform encephalopathy(động vật
4 chân có móng vuốt)

- Chronic wasting disease (CWD): North American cervids (động vật
hưu, nai) .
- CWD was first identified as a fatal wasting syndrome in captive mule
deer(loài hưu lớn bắc mỹ) in Colorado in the late 1960s and in the wild in
1981.
- It was recognized as a spongiform encephalopathy in 1978.
- To date, no strong evidence of CWD transmission to humans has been
reported.
CWD= Chronic wasting disease

Bệnh xốp não ở bò(bệnh bò điên)?(What is bovine spongiform encephalopathy?or Mad Cow Disease)

Bovine spongiform encephalopathy (BSE)
Còn gọi bệnh bò điên” mad cow disease”
1. Bệnh gây tử vong thoái hóa hệ thần kinh ở động
vật có sừng móng chẻ
2. Gây thoái hóa dạng xốp não và tủy
3. BSE thời gian ủ bệnh từ 2,5-8 năm, thường súc
vật trưởng thành 4-5 năm

Bovine spongiform encephalopathy (BSE):
1. Các triệu chứng thần kinh tiến triển ở động vật có sừng
móng chẻ
2. Nhiễm tác nhân truyền bệnh không quy ước prion
(unconventional transmissible).
3. Hiện nay hầu hết chấp nhận giả thuyết tác nhân gây
bệnh là sự sửa đổi protein bình thường có tên prion
protein.
(More than 184,000 cases of BSE have been confirmed in the United
Kingdom.)
Cow affected by Bovine Spongiform Encephalitis
www.jonbarron.org

1. Bằng chứng dịch tễ học và ở phòng thí nghiệm;
liên quan nguyên nhân bệnh prion mới ở người:
variant Creutzfeldt-Jakob disease (vCJD) được báo
cáo đầu tiên ở Anh năm 1996 và sự bùng phát BSE
2. Thực phẩm nhiễm BSE(1984-1986) và sự khởi phát
ca vCJD đầu tiên (1994-1996) thì trùng hợp thời gian ủ
bệnh cho thể bệnh prion ở người

Creutzfeldt-Jakob’s Disease
(Mad cow disease or Prion disease)

CJD (Creutzfeldt-Jakob Disease, Classic)
1.Classic CJD là prion disease.ở người
2.Neurodegenerative disorder.
3.Tiến triển nhanh và luôn luôn tử vong.
4.Những bệnh này thường chết trong 1 năm
khởi phát.
(Important Note: Classic CJD is not related to "mad cow"
disease. Classic CJD also is distinct from "variant CJD",
another prion disease that is related to BSE).
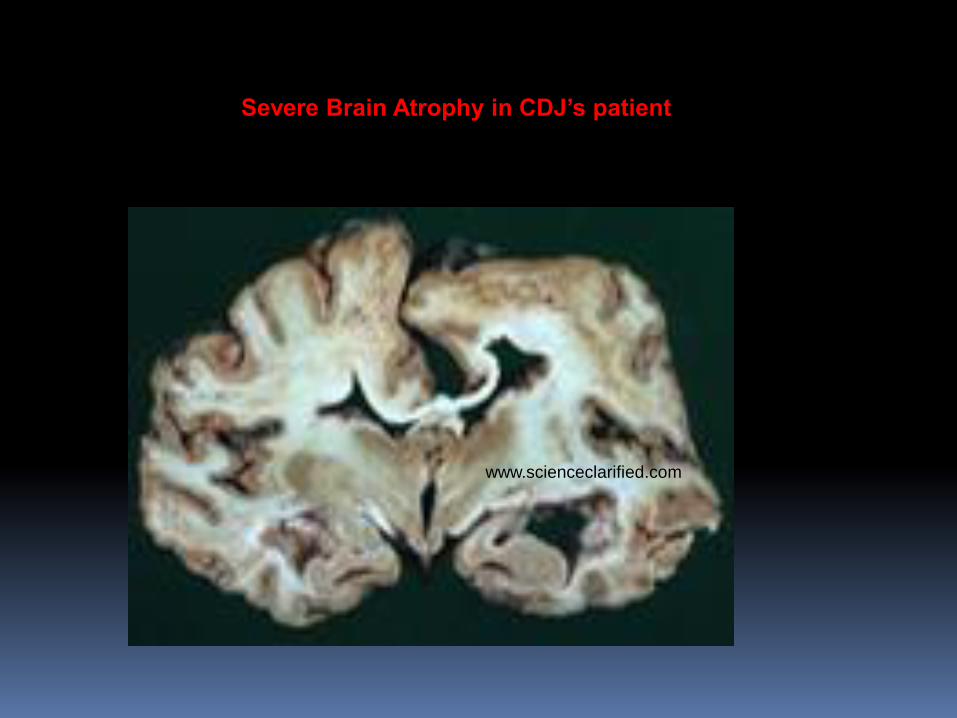
Severe Brain Atrophy in CDJ’s patient
www.scienceclarified.com

Classic CJD
Classic CJD: phát hiện sớm từ năm 1920.
Hầu hết classic CJD xảy ra rải rác
Nguyên nhân: spontaneous transformation of normal prion proteins into abnormal prions.
Sporadic disease xảy ra trên toàn thế giới, Mỹ 1-2 ca/1triệu dân/năm

Ai nguy cơ?
1. CJD xảy ra toàn thế giới
2. Mỗi năm 1/1triệu người ở Mỹ
( Nguy cơ CDJ gia tăng theo tuổi, Hầu hết khoảng
60. ít xảy ra dưới 40 tuổi.)

Lan rộng như thế nào?(How is it spread?)
1. Không có bằng chứng CJD truyền qua không khí, thức
ăn hay sữa hay trực tiếp person-to-person.
2. 85% trường hợp, CJD xảy ra lác đác (sporadically), ở
người không biết yếu tố nguy cơ hay đột biến gen (no
known risk factors or inherited genetic mutations). Được
biết như sporadic CJD.
3. Một số ít trường hợp (5 đến15%) là di truyền và xảy ra
trong gia đình có tiền sử CJD (familial CJD).

Lan rộng như thế nào?(How is it spread?)
4. Một số ca báo cáo:
- CJD lây truyền cho bệnh nhận ghép giác mạc, màng
cứng, kích thích tố tuyến yên của những bn mắc CJD
(iatrogenic CJD).

Làm thế nào chẩn đoán
1. Chỉ có brain biopsy hay khám nghiệm mô não sau
chết có thể xác định chẩn đoán CJD
2. Autopsy khuyến cáo xác định chẩn đoán.
3. Clinical symptoms, a neurological exam, diagnostic
tests có thể giúp chẩn đoán nhưng không xác định
CJD.

Trị liệu thế nào?
(How is it treated?)
Hiện nay chưa biết điều trị CJD, tuy nhiên điều trị giảm
triệu chứng, nâng đở cho bệnh nhân

Lâm sàng CJD
Khởi phát âm thầm .
1. Bất thường hành vi sớm (10% ca):
- Personality change, withdrawal, apathy, depression,
sleep disturbance.
- Agitation, fear, paranoia
2. Dementia tiến triển nhanh và nặng: forgetful, confused,
visual distortions , hallucinations.
3. Myoclonus, thường stimulus-sensitive (80% bn CJD).

Lâm sàng CJD
Bất thường vận động:
- Cerebellar ataxia.
- Extrapyramidal signs: tremor, rigidity, bradykinesia,
dystonic posturing; choreoathetosis.
- Pyramidal signs: weakness, spasticity, hyperreflexia,
và Babinski signs.
• Generalized seizures có thể xảy ra

Variant Creutzfeldt-Jakob Disease (variant CJD)
1. Variant CJD mô tả đầu tiên ở Anh năm1996
2. Khác biệt lâm sàng và bệnh học với classic CJD
3. Mỗi bệnh có particular genetic profile of the prion protein
gene.
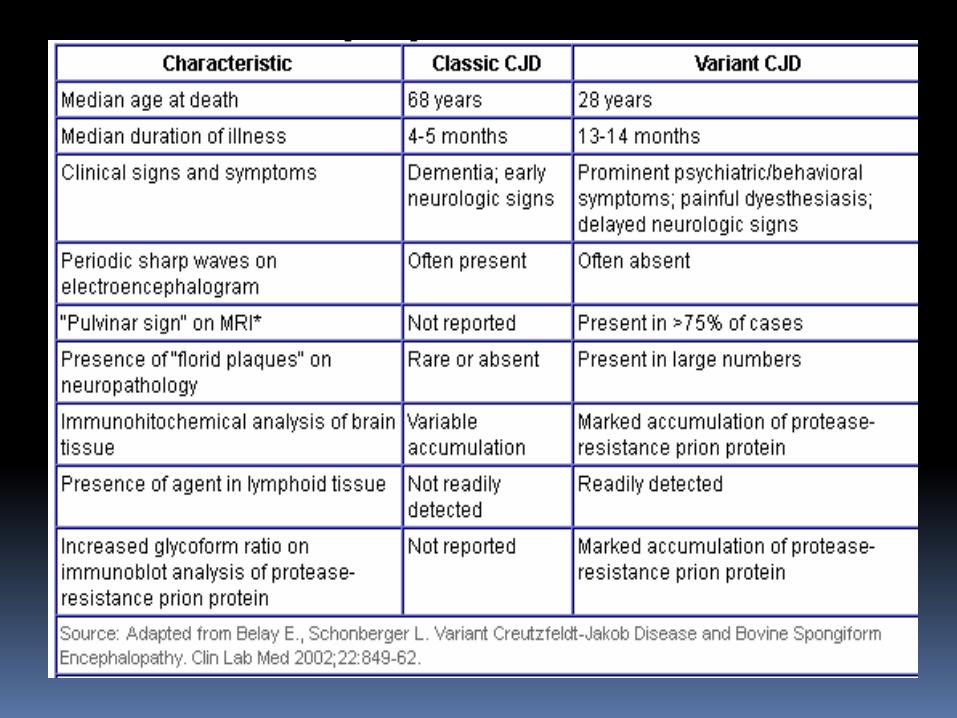
4. Tuổi trung bình chết vCJD: 28 tuổi, CJD classic: 68 tuổi
5. Thời gian bệnh trung bình vCJD 14 tháng, so với 5
tháng classic CJD.

Bằng chứng sự liên hệ BSE (Mad Cow Disease)
1. 1996, bằng chứng sự bùng phát giữa BSE và vCJD.
2. Có bằng chứng khoa học mạnh tác nhân gây bùng phát
bệnh prion disease ở bò, tương tự tác nhân bùng phát
vCJD ở người

Bằng chứng sự liên hệ BSE (Mad Cow Disease)
3. Cả 2 rối loạn là bệnh lý não tử vong và không thường có
thời gian ủ bệnh dài tính bằng năm và là tác nhân lan
truyền không quy ước
4. Tuy nhiên bằng chứng thấp vCJD ngay cả ăn thịt nhiễm
bệnh.
5. 1996, bởi vì tăng vCJD ở Anh, gia tăng kiểm soát ở Mỹ

Tuổi trung bình chết 68 years (Classical)
28 years (Variant)
Thời gian trung bình của bệnh 4-5 months (Classical)
13-14 months (Variant)
Triệu chứng lâm sàng Dementia; triệu chứng thần kinh sớm (Classical)
Ưu thế psychiatric/behavioral symptoms; neurologic signs trể (Variant)
Lâm sàng và đặc điểm bệnh học của CJD
và biến thể CJD

Cách thức có thể lan truyền(Possible Modes of Transmission)
Sporadic(rời rạc) Trường hợp không biết yếu tố nguy cơ,
Infection qua mắc phải
Inherited(di truyền) It is an autosomal and dominant trait.
Iatrogenic(gây ra do khám hay điều trị) Acquired infection Diet
Medical procedures Surgery
Growth hormone
Corneal transplants

Tiêu chuẩn chẩn đoán vCJD ở Mỹ?
(Diagnostic Criteria for Variant
Creutzfeldt-Jakob Disease in the United States)

Chẩn đoán xác định Variant CJD
Neuropathologic: mô não để xác định chẩn đoán
vCJD.
1.Một số lan rộng tấm amyloid kuru-type amyloid
plaques xung quanh là các vacuoles trong cả
tiễu não và não

Chẩn đoán nghi ngờ Variant CJD
a. Tuổi hiện thời hay lúc chết <55 tuổi (a brain autopsy is
recommended, however, for all physician-diagnosed
CJD cases).
b. Triệu chứng tâm thần lúc khởi phát bệnh và/hay hằng
định cảm giác đau (frank pain and/or dysesthesia).
c. Dementia, và phát triển ≥4 tháng sau khởi phát bệnh ít
nhất 2 trong 5 triệu chứng thần kinh sau đây: poor
coordination, myoclonus, chorea, hyperreflexia, or visual
signs.
(If persistent painful sensory symptoms exist, ≥4 months delay in the
development of the neurologic signs is not required).

Chẩn đoán nghi ngờ Variant CJD
d. Normal hay abnormal EEG, nhưng không chẩn đoán
thay đổi EEG thường gặp trong classic CJD.
e. Thời gian bệnh trên 6 tháng.
f. Không tiền sử nhận cadaveric human pituitary growth
hormone hay dura mater graft.
g. Không tiền sử liên hệ bệnh nhân CJD (first degree
relative) hay bn đột biến gen PrP

GHI CHÚ:
Nếu bn có tăng tín hiệu đặc biệt 2 bên pulvinar high trên
MRI scan,
- Chẩn đoán nghi ngờ variant CJD đòi hỏi sự hiện diện :
progressive neuropsychiatric disorder, và các tiêu chuẩn d,
e, f và g.
- Và 5 tiêu chuẩn sau đây:
1) early psychiatric symptoms (anxiety, apathy, delusions,
depression, withdrawal);
2) persistent painful sensory symptoms (frank pain and/or
dysesthesia);
3) ataxia;
4) myoclonus or chorea or dystonia;
5) dementia.

GHI CHÚ :
2. Tiền sử tiếp xúc bovine spongiform encephalopathy (BSE):
- residence
- travel to a BSE-affected country after 1980 increases the
index of suspicion for a variant CJD diagnosis.

BSE có ở Việt Nam?

Preventing vCJD
1. Giảm nguy cơ mắc phải vCJD từ thực phẩm, du lịch châu
Âu hay vùng có BSE nói chung có thể xem xét tránh cả
thịt bò và phó sản của bò hay chọn phần thịt cơ cứng
(selecting beef or beef products, such as solid pieces of muscle meat
rather than brains or beef products like burgers and sausages)
2. Milk và phó sản từ bò không tin chắc bất cứ nguy cơ
truyền tác nhân BSE.

Treatment
Điều trị nâng đở
Không có điều trị đặc hiệu giảm tiến triển bệnh

Chẩn đoán Creutzfeld-Jacob Disease
Trước tiên loại trừ dementia có thể điều trị như
encephalitis hay chronic meningitis.
Test chẩn đoán cơ bản bao gồm :
DNT (14-3-3 protein)
Electroencephalogram (EEG)
Computerized tomography
Magnetic resonance imaging (MRI)
Chỉ chẩn đoán xác định CJD qua:
brain biopsy hay autopsy.

Lâm sàng và đặc điểm bệnh học của CJD
và biến thể
Periodic sharp waves on electroencephalogram Often present (Classical)
Often absent (Variant)
Phân tích miễn dịch nhu mô não Variable accumulation (Classical)
Marked accumulation of protease-resistance prionprotein (Variant) (sự tích tụ of protease-resistance prion protein)
Presence in lymphoid tissue Not readily detected (Classical)
Readily detected (Variant)

Diagnosis Sporadic CJD
Definite:
•Neuropathologically confirmed, and/or
•Immunohistochemically confirmed proteinase-
resistant PrP in tissue sections or Western blot

Diagnosis Sporadic CJD
Probable:
•Progressive dementia.
•Typical periodic EEG, positive MRI, or positive
CSF study, and:
• At least two of the following clinical features:
•Myoclonus.
•Visual or cerebellar disturbance.
•Pyramidal/extrapyramidal dysfunction.
•Akinetic mutism..

Diagnosis Sporadic CJD
Possible:
•Progressive dementia.
•Two of the clinical features listed above.
•Either a negative diagnostic test (EEG, MRI, or
CSF) or they are unavailable.
•Duration 2 years.

Iatrogenic CJD
•Progressive cerebellar syndrome in a pituitary
hormone recipient.
•Sporadic CJD with a recognized exposure risk
(e.g. dura mater transplant).

Familial CJD
•Definite or probable CJD plus definite or
probable CJD in a first-degree relative.
•Neuropsychiatric disorder plus disease-specific
PRNP mutation.

vCJD
1a: Progressive neuropsychiatric disorder.
1b: Duration of illness 6 months.
1c: Routine investigations do not suggest an
alternative diagnosis.
1d: No history of potential iatrogenic exposure.

vCJD
2a: Early psychiatric symptoms (especially apathy
and depression).
2b: Persistent painful sensory symptoms.
2c: Ataxia.
2d: Myoclonus or chorea or dystonia.
2e: Dementia.

vCJD
3a: EEG does not show the (typical) PSWCs of
CJD (or no EEG performed).
3b: Posterior thalamic (pulvinar) high signal on
DWI or proton density MRI brain scan.

vCJD
Definite vCJD: 1a and neuropathologic
confirmation of vCJD.
Probable: 1 and four-fifths of 2, and 3a and 3b.
Possible: 1 and four-fifths of 2.
.

Câu hỏi ?


















