







Languages
Pages
Legal


BAB IPENDAHULUAN
1.1 Latar Belakang
Penyakit Hirschsprung’s disease merupakan gangguan kompleks yang
terjadi karena tidak adanya sel ganglion di pleksus submukosa dan myenterik pada
dinding usus yang menimbulkan obstruksi fungsional dan dilatasi usus mulai dari
proksimal ke segmen yang terkena (Monajemzadeh et al, 2011). Hal ini dianggap
sebagai akibat dari pemberhentian dini dari migrasi kraniokaudal pada sel kres
neuron vagal pada dinding usus antara minggu ke-5 sampai minggu ke-12 usia
kehamilan untuk membentuk sistem saraf enterik (Haricharan dan Georgeson,
2008). Penyakit ini merupakan kelainan congenital yang sering dijumpai pada
kasus bedah anak (Iqbal et al, 2010).
Angka kejadian Hirschsprung’s disease diperkirakan sekitar 1:5000
kelahiran. Namun, kejadian bervariasi sesuai dengan kelompok-kelompok suku,
seperti pada Hispanic 1:10000 kelahiran, Amerika-Kaukasian 1,5:10000, Afro-
Amerika 2,1:10000, dan Asia 2,8:10000. Laki-laki lebih sering terkena
dibandingkan perempuan dengan rasio 4:1 (Amiel et al, 2008). Di Indonesia
diperkirakan lahir 1200 bayi dengan Hirschsprung’s disease setiap tahun. Di
Rumah Sakit Cipto Mangunkusumo Jakarta tercatat datang berobat 20-40 pasien
setiap tahun (Kartono, 1993). Selain itu, jumlah pasien Hirschsprung’s disease di
Banda Aceh pada tahun 2011 didapatkan pasien sebanyak 76 orang dengan
persentase laki-laki sekitar 73,08% dan perempuan 26,92% (Nasrizarni dan
Muntadhar, 2012).
Gejala umum Hirschsprung’s disease ini terdiri dari pengeluaran mekonium
yang terlambat, distensi abdomen, muntah bercampur empedu dan intoleransi
makanan (Haricharan dan Georgeson, 2008). Hirschsprung’s disease ini dicurigai
pada bayi baru lahir dengan konstipasi dan/atau gagal mengeluarkan mekonium
dalam 48 jam setelah lahir. Gejala ini bisa tidak ditemukan pada 6%-42% pasien.
Gejala-gejala lain selain konstipasi bisa dengan tanda obstruksi usus bagian
bawah seperti distensi abdomen dan muntah yang bercampur empedu. Setelah

periode bayi baru lahir, temuan yang umum adalah konstipasi, distensi abdomen
diiringi dengan gagal tumbuh (Schulten dan Travassos, 2011).
Penelitian yang dilakukan di Irlandia, mayoritas pasien mengalami
komplikasi enterokolitis berlanjut pada ganggguan fungsi usus beberapa tahun
kemudian (Menezes dan Puri, 2006). Selain itu, pada tahun 2010 di kota yang
sama, pasien memiliki fungsi usus yang normal pasca tindakan operasi pull-
through (Doodnath dan Puri, 2010). Penelitian yang dilakukan di Rumah Sakit
Universitas Zagazig, outcome jangka pendek dan panjang pascaoperasi transanal
pull-through terbilang baik. Namun, follow-up jangka panjang dibutuhkan untuk
mengetahui dan mengobati komplikasi dan disfungsi usus. Enterokolitis yang
didapatkan sebelum operasi meningkatkan insiden pascaoperasi seperti diare,
enterokolitis dan ekskoriasi perineum (Saleh et al., 2009).

BAB IITINJAUAN PUSTAKA
2.1 Definisi
Hirschsprung’s disease merupakan kelainan obstruksi usus yang sering
terjadi pada bayi baru lahir. Penyakit ini ditandai dengan tidak adanya sel
ganglion di bagian distal usus yang dimulai dari sfingter ani interna sampai ke
bagian proksimal usus dengan panjang yang bervariasi, selalu melibatkan anus
dan setidaknya melibatkan rektum. Bagian-bagian usus yang terlibat meliputi
bagian rektosigmoid, kolon asenden dan transversum dan aganglionosis kolon
total (Puri dan Montedonico, 2008).
Hirschsprung’s disease’s adalah kelainan kongenital yang ditandai dengan
tidak adanya sel ganglion pada saluran pencernaan bawah. Aganglionosis ditandai
dengan adanya kelainan pada enteric nervous system (ENS) dimana sel ganglion
gagal membentuk persarafan pada saluran gastrointestinal bagian bawah selama
masa embrionik (Garcia-Barcelo et al, 2007).
Obstruksi fungsional dan dilatasi usus pada penyakit ini pertama kali
diperkenalkan pada 1888 oleh Harald Hirschprung. Dia menyebutkan penyakit ini
merupakan kelainan kompleks yang diakibatkan oleh tidak adanya sel ganglion
pada dinding usus yang menyebabkan obstruksi fungsional dan dilatasi usus
dimulai dari distal menuju ke proksimal pada segmen yang aganglion. Kelainan
ini dapat muncul pada masa neonatus sebagai obstruksi usus. Umumnya kelainan
ini terjadi pada anak-anak, namun dapat juga terjadi pada anak yang lebih tua
dengan konstipasi kronik, dimana pengobatan konvensional tidak berespons
(Monajemzadeh et al, 2011).
Konstipasi merupakan masalah umum yang terjadi pada anak-anak dan
hanya sebagian kecil pasien yang mengalami konstipasi yang disebabkan oleh
penyebab organik, untuk itu perlu dibedakan antara dua kondisi. Konstipasi pada
Hirschprung’s disesase didefinisikan sebagai kegagalan neonatus dalam
mengeluarkan mekonium dalam 48 jam setelah dia lahir dan pada anak yang lebih
tua tidak teraturnya pengeluaran feses karena peningkatan konsistensi. Hanya

sebagian kecil anak-anak mengalami konstipasi dengan Hirschsprung’s disease
(Monajemzadeh et al, 2011).
2.2 Epidemiologi
Insiden Hirschsprung’s disease diperkirakan terjadi pada 1 dari 5000
kelahiran. Survei yang dilakukan oleh California Birth Defects Monitoring
Program dari tahun 1983-1997 diperoleh hasil bahwa penyakit ini terjadi pada 2,8
dari 10.000 kelahiran di Asia, 2,1 dari 10.000 kelahiran di Afrika-Amerika, 1,5
dari 10.000 kelahiran pada orang Kulit Putih dan 1 dari 10.000 kelahiran pada
orang Hispanic. Rasio antara laki-laki dan perempuan pada bagian rektosigmoid
adalah 4:1, sedangkan pada segmen panjang 1:1 sampai 2:1 (Haricharan dan
Georgeson, 2008).
Sekitar 20% kasus penyakit ini merupakan keturunan. Sekitar 30% pasien
menunjukkan hubungan dengan kelainan lain seperti abnormalitas kromosomal
atau neurokristopati yang berbeda dan dengan variasi anomali yang lain. Pasien
dengan sindrom Down memiliki risiko tinggi terhadap Hirschsprung’s disease’s
dan rasio laki-laki dengan perempuan seimbang (Lantieri et al, 2008).
2.3 Etiologi
Pada dasarnya, Hirschsprung’s disease berawal di tingkat molekuler dari
gangguan sinyal pada tahap perkembangan. Akhirnya, sinyal yang
mengendalikan perpindahan sel kres neuron memberikan hasil yang kacau
sehingga menyebabkan aganglionosis usus distal. Kelainan bersifat kompleks,
seperti yang terlihat pada jumlah gen yang terlibat dalam patogenesisnya
(Haricharan dan Georgeson, 2008).
Gangguan seluler dan molekuler selama perkembangan dari sistem saraf
usus dan perpindahan sel kres neural ke dalam usus yang sedang berkembang
merupakan etiologi primer dari Hirschsprung’s disease. Pertama kali, neuroblast
yang berasal dari sel kres neural muncul dalam perkembangan esofagus pada usia
gestasi minggu ke-5 pada fetus. Sel-sel ini bermigrasi dalam bentuk kraniokaudal
menuju ke sisa pembentukan usus mulai dari minggu ke-5 sampai ke-12 usia
gestasi. Faktor lain yang berpengaruh seperti perubahan komponen matriks
ekstraseluler, abnormalitas pada faktor neurotropik, dan daya lekat molekul sel

neuron, juga ditunjukkan berperan terhadap terjadinya Hirschsprung’s disease
(Haricharan dan Georgeson, 2008).
Hirschsprung’s disease adalah malformasi kongenital usus yang ditandai
dengan tidak adanya sel ganglion intrinsik parasimpatis pada pleksus myenterikus
dan submukosa. Hal ini terjadi akibat tertahannya migrasi kraniokaudal pada sel
kres neural vagal pada usus antara minggu ke-5 dan minggu ke-12 masa
kehamilan dalam membentuk enteric nervous system (ENS). Sfingter ani interna
merupakan batas akhir dari kelainan ini. Kelainan ini disebut short-segment
Hirschsprung’s disease (80% kasus) jika kelainan hanya sebatas sfingter ani
interna sampai dengan sigmoid, sedangkan disebut long-segment Hirschsprung’s
disease (20% kasus) jika kelainan dimulai dari sigmoid ke proksimal (Amiel et al,
2008).
2.4 Patofisiologi
Ciri khas patofisiologi dasar Hirschsprung’s disease adalah obstruksi
fungsional yang disebabkan oleh segmen kolon distal yang aganglionik sehingga
menghambat pergerakan peristaltik usus. Pada usia neonatal, usus bisa saja dalam
keadaan normal, namun pada usia anak, usus bagian proksimal mengalami
hipertrofi dan menjadi semakin tebal dan panjang dari normal. Lapisan otot
longitudinal terlihat mengelilingi kolon sepenuhnya. Selama ini disadari bahwa
gejala obstruktif Hirschsprung’s disease merupakan motilitas yang abnormal dari
segmen distal usus, namun masih ada penjelasan yang kurang jelas untuk proses
terjadinya dinding usus bagian distal yang mengecil pada penyakit ini. Penyakit
ini ditandai dengan tidak adanya sel ganglion di bagian pleksus myenterik dan
mukosa. Aganglionosis ini meluas ke bagian rektosigmoid pada hampir 80%
pasien. Aganglionosis ini juga berlanjut sampai ke bagian proksimal zona transisi.
Panjang zona ini bisa bervariasi dan memanjang hingga beberapa sentimeter dan
dicirikan dengan hipoganglionosis. Beberapa kelainan yang lain telah
digambarkan berhubungan dengan Hirschsprung’s disease sehingga mungkin
berperan dalam patofisiologinya dan mungkin bisa menjelaskan ketidaksesuaian
antara panjang kolon yang nonfungsional dan derajat obstruksinya (Puri dan
Montedonico, 2008).

Pada aganglionosis terdapat peningkatan aktivitas saraf kolinergik di zona
intermuskular dan submukosa pada segmen aganglionik. Serabut ini muncul
sebagai saraf yang tebal dan sesuai dengan saraf parasimpatis preganglionik
ekstrinsik. Asetilkolin secara terus-menerus dilepaskan dari akson saraf
parasimpatis ini menghasilkan akumulasi yang berlebihan pada enzim
asetilkolinesterase yang ditemukan pada mukosa lamina propria, mukosa otot dan
otot sirkular dengan menggunakan teknik pewarnaan histokimia. Konsentrasi
asetilkolinesterase ditemukan meningkat dalam serum dan eritrosit pada anak
yang mengalami Hirschsprung’s disease. Hiperplasia nervus kolinergik dianggap
sebagai penyebab dari spastisitas segmen aganglionik karena asetilkolinesterase
merupakan neurotransmitter eksitatori utama (Puri dan Montedonico, 2008).
Penelitian dengan menggunakan fluorescent-histochemical menunjukkan
bahwa terjadi peningkatan jumlah dari persarafan adrenergik pada kolon yang
aganglionik pada Hirschsprung’s disease dan memiliki persebaran yang tidak
merata. Peningkatan itu juga ditemukan pada lapisan otot sirkular dan
longitudinal, sedangkan pada normalnya hal tersebut hampir tidak ada. Secara
normal persarafan adrenergik bekerja untuk merelaksasikan usus, tidak mungkin
hiperaktivitas adrenergik bertanggung jawab terhadap peningkatan tonus pada
kolon aganglionik (Puri dan Montedonico, 2008).
2.5 Manifestasi Klinis
Hirschsprung’s disease ini dicurigai pada bayi baru lahir dengan konstipasi
dan/atau gagal mengeluarkan mekonium dalam 48 jam setelah lahir. Gejala-gejala
lain selain konstipasi bisa dengan tanda obstruksi usus bagian bawah seperti
distensi abdomen dan muntah yang bercampur empedu. Penyakit ini juga bisa
disertai dengan enterokolitis dan sepsis, terutama pada bentuk yang lebih luas.
Setelah periode bayi baru lahir, temuan yang umum adalah konstipasi, distensi
abdomen diiringi dengan gagal tumbuh (Schulten dan Travassos, 2011).
Sekitar 60%-90% pasien dengan Hirschsprung’s disease gagal
mengeluarkan meconium, sedangkan pasien yang mengalami distensi abdomen
sekitar 63%-91% dan yang muntah bercampur empedu sekitar 19%-37%. Pasien
Hirschsprung’s disease yang enterokolitis berkisar antara 5%-44%. Keadaan diare
yang berbau busuk secara terus-menerus, demam dan distensi abdomen

mengindikasikan pasien Hirschsprung’s disease dengan enterokolitis, dimana
ketika tidak disadari dapat memperburuk keadaan megakolon pada pasien
(Haricharan dan Georgeson, 2008).
Terkadang, diagnosis penyakit ini harus dipertimbangkan adanya perforasi
yang tidak diketahui pada sekum dan appendiks, meskipun hal ini merupakan
temuan yang jarang. Beberapa anak tidak mengalami obstruksi pada periode
neonatus dan terjadi kemudian pada periode infant atau dewasa dengan keadaan
konstipasi yang berat, distensi abdomen kronik dan gagal tumbuh. Ini merupakan
hal yang umum terjadi di antara bayi yang masih menyusui yang memiliki
kemungkinan terjadinya konstipasi pada saat menyusui. Namun, para peneliti
berpikir bahwa temuan ini tidak dapat dipercaya. Setelah anamnesis dan
pemeriksaan fisik, tahap diagnostik berikutnya termasuk pemeriksaan radiologi,
manometri anorektal dan biopsi rektum (Puri dan Montedonico, 2008).
Pada banyak kasus, diagnosis Hirschsprung’s disease pada anak-anak
apabila terjadi obstruksi usus dengan manifestasi klinis seperti: (1) pengeluaran
mekonium terlambat (24 jam setelah kelahiran), (2) distensi abdomen, (3) muntah,
dan (4) enterokolitis neonatus. Beberapa pasien terlambat terdiagnosis pada masa
bayi atau masa dewasa dengan konstipasi berat, distensi abdomen kronik, muntah,
dan gagal tumbuh. Pada foto polos abdomen terdapat usus kecil dan kolon
proksimal yang distensi. Gambaran klasik adalah kolon proksimal yang distensi
dengan corong aganglionik terletak ke distal usus. Pada barium enema, rektum
dapat terlihat kontraksi yang tidak terkoordinasi. Zona transisi merepresentasikan
daerah dimana usus yang aganglionik bersatu dengan usus yang ganglionic
(Amiel et al, 2008).

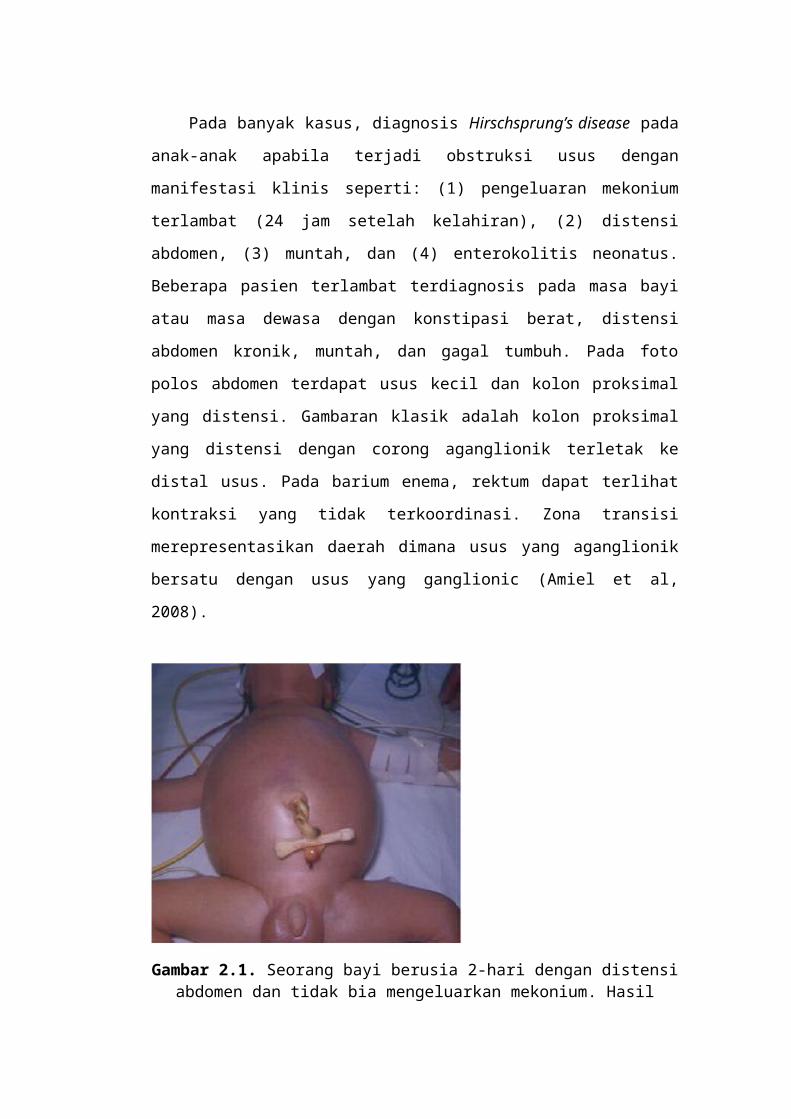
Gambar 2.1. Seorang bayi berusia 2-hari dengan distensi
abdomen dan tidak bia mengeluarkan mekonium. Hasil pemeriksaan biopsi isap rektum mengkonfirmasi adanya
penyakit Hirschsprung.4
Berdasarkan penelitian yang dilakukan oleh Izadi et al (2007), kebanyakan
pasien mempunyai distensi abdomen dan konstipasi yang kronis tanpa
membedakan perbedaan tipe aganglionik yang terlibat, usia pasien dan temuan
klinisnya. Luas daerah yang dipengaruhi tidak berefek pada onset dan tipe gejala
penyakit tersebut.
2.6 Letak Kelainan
Letak kelainan Hirschsprung’s disease dapat diklasifikasikan menjadi
rektosigmoid, segmen panjang dan aganglionosis total. Aganglionosis intestinal
total dan segmen pendek juga terkadang dideskripsikan. Letak kelainan yang
paling berat dan paling jarang ditemukan adalah aganglionosis intestinal total
yang dimulai dari duodenum sampai rektum. Selain itu, letak kelainan yang paling
sering ditemukan adalah rektosigmoid (Haricharan dan Georgeson, 2008).
Selain klasifikasi di atas, Hirschsprung’s disease juga dapat diklasifikasikan
segmen pendek dimana segmen yang aganglionosis tidak meluas di atas sigmoid,
segmen panjang dimana segmen aganglionosis memanjang ke fleksus splenikus
atau kolon transversum dan aganglionosis kolon total jika aganglionosis mencapai
kolon atau ujung ileum (Puri dan Montedonico, 2008).

2.7 Penyakit Penyerta
Hirschsprung’s disease ini bisa berhubungan dengan kecacatan kongenital
yang lain, termasuk sindrom Down, Smith-Lemli-Opitz, Waarden burg, hipoplasia
kartilago rambut, hipoventilasi kongetinal dan abnormalitas sistem urogenital dan
kardiovaskular. Selain itu, penyakit ini berhubungan dengan mikrosefali, retardasi
mental dan fasies yang abnormal dengan autisme atau dengan bibir sumbing,
hidrosefalus, dan mikrognathia. Kelainan genetik telah diketahui dalam gen
multipel yang mengkode protein yang menandai RET (Rearranged during
transfection factor) dan yang terlibat dalam jalur reseptor endothelin (EDN) tipe
B (Wyllie, 2007).
2.8 Diagnosis
Hirschsprung’s disease dicurigai pada neonatus sesuai dengan manifestasi
klinis yang telah disebutkan sebelumnya. Pemeriksaan yang dapat dilakukan
untuk mendiagnosis penyakit ini seperti contrast enema (CE), anorectal
manometry (ARM), biopsi rektal tebal , dan rectal suction biopsy (RSB).
Pemeriksaan pertama untuk mendiagnosis Hirschsprung’s disease ini berbeda
sesuai dengan ketersediaan pemeriksaan dan keahlian di masing-masing rumah
sakit. Pemeriksaan dengan radiologi CE memiliki sensitivitas 65%-80% dan
spesifitas 66%-100%, ARM dengan sensitivitas 75%-100% dan spesifitas 85%-
97% dan RSB dengan sensitivitas 91%-100% dan spesifitas 97%-100%
(Haricharan dan Georgeson, 2008).
2.9 Pemeriksaan Penunjang
a. Foto Polos Abdomen
Foto polos abdomen pada neonatus dengan Hirschsprung akan menunjukkan loop dilatasi pada usus dengan fluid level and airless pelvis. Kadang-kadang, mungkin saja dapat terlihat sejumlah kecil udara dalam rektum yang tidak distensi dan dilatasi kolon di atasnya yang meningkatkan kecurigaan. Radiografi polos abdomen yang diperoleh dari pasien dengan aganglionosis kolon total (TCA) mungkin menunjukkan tanda-

tanda karakteristik obstruksi ileum dengan air-fluid level atau distensi gas sederhana pada loop usus halus.4
Pada pasien dengan enterokolitis akibat Hirschsprung, radiografi polos abdomen dapat menunjukkan penebalan dinding usus dengan ketidakteraturan dari mukosa usus atau loop terlalu melebar, yang menunjukkan megakolon toksik. Pneumoperitoneum dapat ditemukan pada pasien dengan perforasi. Perforasi spontan dari saluran usus telah dilaporkan terjadi pada 3% pasien Hirschsprung.4
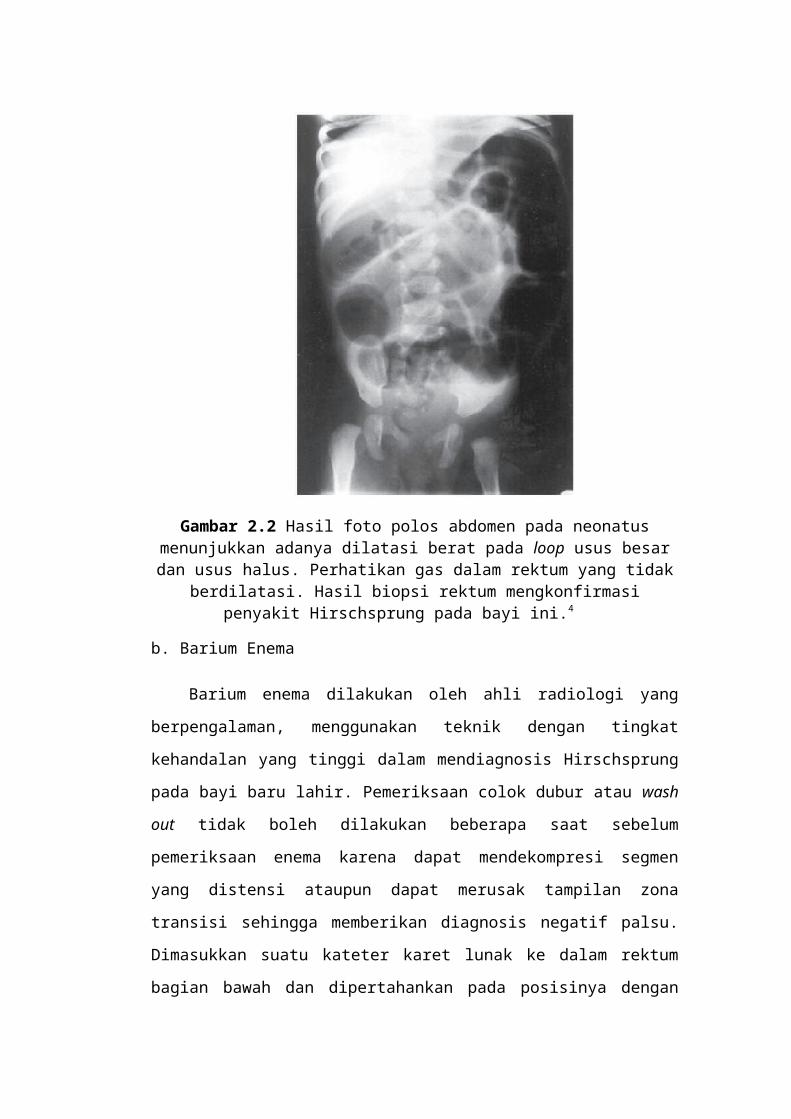
Gambar 2.2 Hasil foto polos abdomen pada neonatus menunjukkan adanya dilatasi berat pada loop usus besar dan
usus halus. Perhatikan gas dalam rektum yang tidak berdilatasi. Hasil biopsi rektum mengkonfirmasi penyakit Hirschsprung pada
bayi ini.4
b. Barium Enema
Barium enema dilakukan oleh ahli radiologi yang berpengalaman, menggunakan teknik dengan tingkat kehandalan yang tinggi dalam mendiagnosis Hirschsprung pada bayi baru lahir. Pemeriksaan colok dubur atau wash out tidak boleh dilakukan beberapa saat sebelum pemeriksaan enema karena dapat mendekompresi segmen yang distensi ataupun dapat merusak tampilan zona transisi sehingga memberikan diagnosis negatif palsu. Dimasukkan suatu kateter karet lunak ke dalam rektum bagian bawah dan dipertahankan pada posisinya dengan bokong diplester ketat. Tidak boleh menggunakan balon kateter karena resiko terjadi perforasi dan kemungkinan distorsi zona transisi. Barium harus disuntikkan secara perlahan dalam jumlah kecil di bawah kontrol fluoroskopik dengan bayi dalam posisi lateral. Suatu kasus khas Hirschsprung akan menunjukkan aliran barium dari rektum yang tidak berdilatasi melalui zona transisi bentuk kerucut menjadi dilatasi kolon. Beberapa kasus dapat menunjukkan transisi segera antara kolon proksimal yang berdilatasi dan segmen aganglionik distal, menjadikan sedikit keraguan pada diagnosis.4
Apabila pada foto barium enema tidak terlihat tanda khas penyakit
hirschsprung dan juga pada anak yang lebih besar, maka dapat dilanjutkan dengan
foto retensi barium setelah 24-48 jam barium dibiarkan membaur dengan feses.
Gambaran khasnya adalah terlihatnya barium yang membaur dengan feses ke arah
proksimal kolon. Sedangkan pada penderita yang bukan hirschsprung namun

disertai dengan obstipasi kronis, barium terlihat menggumpal di daerah rektum
dan sigmoid.3, 12
Gambar 2.3 (a) Gambaran distensi abdomen pada penderita hirschprung(b) Gambaran barium enema pada bayi tersebut1
c. Manometri anorektal
Meskipun manometri anorektal jarang digunakan pada neonatus untuk
menegakkan diagnosis, teknik ini berguna pada anak-anak yang berusia di
atasnya. Temuan klasiknya adalah tidak adanya refleks penghambatan rectoanal
ketika rektum distensi. Pada anak normal, distensi rektum menyebabkan
peningkatan sementara tekanan anus dengan penurunan tekanan sfingter ani
internal. Anak-anak dengan Hirschsprung mengalami kekurangan refleks
penghambatan ini dan relaksasi sfingter internal.2
Pada inervasi usus normal, distensi rektum menghasilkan relaksasi sfingter internal berupa refleks rectosphincteric. Pada orang normal, setelah distensi balon rektal dengan udara, rektum segera merespon dengan kenaikan sementara tekanan yang berlangsung selama 15-20 detik, pada saat yang sama

aktivitas ritmik sfingter internal menurun atau hilang dan tekanannya turun sebesar 15-20 cm, durasi relaksasi bertepatan dengan gelombang rektal.4
Pada pasien dengan penyakit Hirschsprung, rektum sering menunjukkan gelombang spontan dari berbagai amplitudo dan frekuensi dalam fase istirahat. Aktivitas ritmik sfingter internal akan lebih berat. Pada distensi rektum, dengan adanya kenaikan udara, terjadi relaksasi sfingter internal tidak lengkap. 4
Gambar 2.4 Manometri Anorektal (a) refleks rectosphincteric normal pada inflasi balon rektum. (b) Tidak adanya refleks rectosphincteric dan aktivitas ditandai ritmik sfingter internal berat pada pasien dengan penyakit Hirschsprung.4
d. Biopsi Rektum
Diagnosis penyakit Hirschsprung dapat dikonfirmasi dengan pemeriksaan spesimen biopsi rektum. Pengenalan teknik pewarnaan histokimia untuk deteksi aktivitas acetylcholinesterase (AChE) pada suction rectal biopsy telah menghasilkan metode yang sederhana dan dapat diandalkan untuk diagnosis Hirschsprung. Biopsi rektal dengan ketebalan penuh (full thickness rectal biopsy) jarang diindikasikan untuk diagnosis Hirschsprung kecuali aganglionosis kolon total. Pada orang normal, aktivitas acetylcholinesterase yang dapat

dideteksi diamati dalam lamina propria dan muskularis mukosa, dan pewarnaan sel-sel ganglion submukosa dapat jelas dengan acetylcholinesterase. Pada Hirschsprung, terjadi peningkatan yang signifikan pada aktivitas acetylcholinesterase dalam lamina propria dan muskularis yang jelas sebagai serabut saraf kolinergik kasar berwarna cokelat sampai hitam.4
Nakao dkk melaporkan sensitivitas sebesar 91% penggunaan pewarnaan
acetylcholinesterase untuk diagnosis Hirschsprung dan tingkat false-negatif
sebesar 8%. Biopsi hisap rektum jelas lebih sensitif dibandingkan manometri
anorectal maupun enema kontras untuk diagnosis Hirschsprung. Namun, biopsi
harus cukup dalam untuk menunjukkan tidak adanya sel-sel ganglion dan adanya
serabut saraf hipertrofik. Menariknya, anak-anak dengan Hirschsprung kolon total
dapat mengalami peningkatan aktivitas acetylcholinesterase dalam rektum dan
kolon kiri tetapi mungkin menunjukkan tingkat aktivitas proksimal
acetylcholinesterase mendekati normal ke fleksura lienalis.3
Gambar 2.5 (A) Spesimen biopsi pada ganglionik usus normal ini telah diwarnai dengan hematoxylin dan eosin. Sebuah sel ganglion (panah) terlihat pada submukosa. (B)
Spesimen biopsi rektal pada neonatus dengan penyakit Hirschsprung ini telah diwarnai dengan hematoxylin dan eosin. Sel-sel ganglion tidak ditemukan pada dinding rektum.
Batang saraf submukosa (tanda panah) juga terlihat diameternya lebih besar dari 40 pM, yang sangat berhubungan dengan aganglionosis. (C) Spesimen biopsi rektal pada
neonatus dengan penyakit Hirschsprung ini telah diwarnai dengan acetylcholinesterase.
Pewarnaan yang meningkat pada mukosa dan submukosa (tanda panah) merupakan diagnostik penyakit Hirschsprung.
2.10 Penatalaksanaan
Penatalaksanaan penyakit ini adalah dengan pembedahan. Setelah
penanganan preoperasi yang hati-hati, prinsip yang mendasar adalah dengan
menempatkan usus yang normal ke anus untuk menimbulkan kontraksi pada
sfingter ani interna. Semenjak munculnya teknik Swenson pada tahun 1948,
beberapa pendekatan operasi mulai banyak dikembangkan seperti teknik Soave
dan Duhamel. Teknik operasi satu tahap mungkin jika diagnosis ditegakkan
dengan segera, sebelum terjadi dilatasi kolon, pada kelainan short segment. Untuk
kelainan long segment dan aganglionosis kolon total, enterostomi sering
dilakukan pada tahap pertama sebelum dilakukan operasi definitif. Laparoskopi
dan transanal pull-through sudah mulai dikenal untuk penanganan penyakit ini
(Amiel et al, 2008).
Tindakan Bedah Definitif
Telah dijelaskan beberapa pendekatan operasi untuk pengelolaan penyakit
Hirschsprung. Semua operasi ini sesuai dengan konsep aslinya untuk mengoreksi
penyakit Hirschsprung yang didukung oleh Swenson. Prinsip-prinsip untuk
pengobatan bedah secara efektif ini meliputi reseksi bagian aganglionik dari usus
dan identifikasi gangliona usus proksimal yang normal dengan anastomosis
coloanal atau enteroanal. Selain itu, operasi tersebut harus memperhatikan
kontinensia fekal dan urin dan fungsi seksual normal. Operasi yang paling umum
digunakan untuk penyakit Hirschsprung telah dikembangkan oleh Swenson,
Duhamel, dan Soave.3
Teknik Swenson menggunakan proktektomy yang hampir total dengan diseksi
dekat dengan bagian luar dinding otot rektum. Swenson percaya bahwa reseksi
diagonal pada rektum distal menghasilkan striktur yang sedikit pada garis
anastomotik.3

Gambar 2.6 Prinsip-prinsip prosedur pull-through Swenson dapat dilihat pada gambar-gambar berikut. (A) Usus ganglionated proksimal di-grasp melalui insisi di stump
rectosigmoid yang prolaps. (B) Usus ganglionated kemudian dijahit ke anus.3
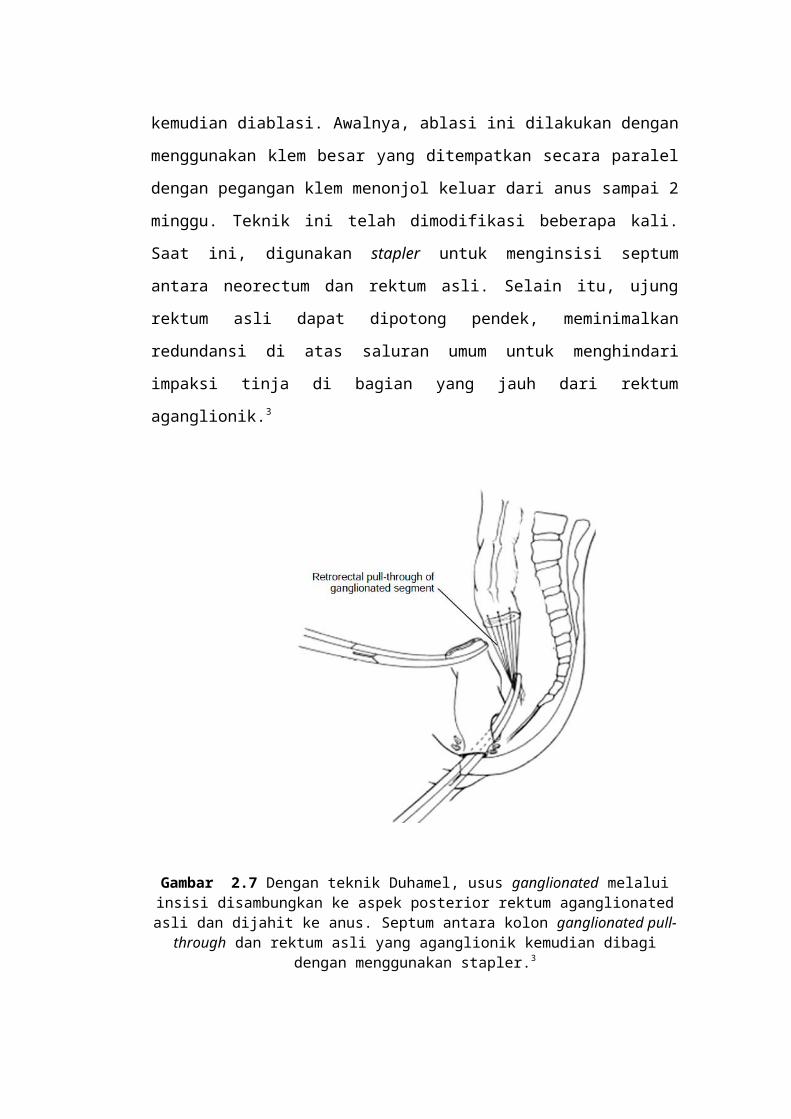
Teknik Duhamel menggunakan cara diseksi di belakang rektum dan
meninggalkan rektum aganglionik di tempatnya. Kemudian rektum dan usus
proksimal aganglionik direseksi. Usus ganglionated dibawa di belakang rektum,
menciptakan dua silinder sejajar pada lumen rektum asli, kira-kira 2 cm di atas
garis dentate. Septum antara dua silinder usus tersebut kemudian diablasi.
Awalnya, ablasi ini dilakukan dengan menggunakan klem besar yang ditempatkan
secara paralel dengan pegangan klem menonjol keluar dari anus sampai 2 minggu.
Teknik ini telah dimodifikasi beberapa kali. Saat ini, digunakan stapler untuk
menginsisi septum antara neorectum dan rektum asli. Selain itu, ujung rektum asli
dapat dipotong pendek, meminimalkan redundansi di atas saluran umum untuk
menghindari impaksi tinja di bagian yang jauh dari rektum aganglionik.3

Gambar 2.7 Dengan teknik Duhamel, usus ganglionated melalui insisi disambungkan ke aspek posterior rektum aganglionated asli dan dijahit ke anus. Septum antara kolon ganglionated pull-through dan rektum asli yang aganglionik kemudian dibagi dengan
menggunakan stapler.3
Teknik Soave menggunakan cara mukosektomi rektum, meninggalkan
suatu cuff muskuler. Soave awalnya menjelaskan eksteriorisasi neorektum keluar
anus dan meninggalkannya sebagai suatu tunggul (stump) yang menonjol. Stump
tersebut dipotong 2 sampai 3 minggu kemudian. Boley menjelaskan pendekatan
yang sama dengan anastomosis primer. Semua prosedur ini menggunakan suatu
laparoskopi atau transanal counterpart.3

Gambar 2.8 (A) Untuk operasi Soave, dilakukan diseksi extramucosal rektum setelah insisi melingkar pada mukosa dubur. (B) Kolon ganglioik ditarik melalui manset rektum
aganglionik, dan dilakukan anastomosis coloanal.3
Pada tahun 1980, So dkk memperkenalkan konsep pull-through primer
untuk penyakit Hirschsprung. Suatu prosedur satu tahap menghindari komplikasi
yang terkait dengan kolostomi dan tidak membutuhkan untuk dilakukan operasi
lain guna menutup kolostomi tersebut. Suatu pull-through satu tahap primer
sesuai untuk sebagian besar bayi dan anak-anak yang didiagnosis dengan
Hirschsprung.3
Ketika diindikasikan, dilakukan kolostomi akhir hanya bagian proksimal
ke zona transisi biopsi yang didapatkan. Teknik Brooke digunakan untuk
mematangkan kolostomi tersebut. Kolon harus hati-hati diamankan ke dinding
perut bagian dalam pada jarak minimal 4 cm untuk menghindari prolaps
kolostomi karena kolon proksimal ukurannya sering menyusut setelah
pembentukan kolostomi. Kolon distal tersisa sebagai kantong Hartman kecuali
zona transisi berada pada atau di atas fleksura lienalis.3

Pull-through satu tahap untuk Hirschsprung menjadi lebih populer dengan
pengenalan laparoscopicassisted transanal endorectal pull-through (LATEP).
LATEP memungkinkan dilakukan pull-through tanpa insisi yang besar pada
abdomen. Selanjutnya, de la Torre menjelaskan tentang transanal pull-through
(TAP) tanpa eksplorasi atau biopsi perut. LATEP dan TAP telah menjadi dua
metode yang paling populer untuk prosedur perbaikan utama Hirschsprung pada
bayi dan anak-anak. Di Rumah Sakit Anak Alabama, LATEP lebih banyak
dilakukan pada neonatus dan anak-anak yang menjalani operasi utama untuk dua
alasan. Pertama, biopsi laparoskopi memungkinkan dikonfirmasinya sejauh mana
aganglionosis sebelum terjadi pembagian mesenterium kolon atau ablasi rektum.
Hal ini terutama menguntungkan pada neonatus di mana enema kontras agak
tidak bisa diandalkan dalam memprediksi tingkat zona transisi. Jika diidentifikasi
aganglionosis segmen panjang, penundaan operasi definitif sampai tersedia hasil
histopatologis dari bagian permanen dapat mencegah reseksi segmen panjang usus
yang tidak perlu akibat kesalahan dalam analisis bagian beku. Kedua, penggunaan
laparoskopi membantu dalam melakukan anastomosis coloanal tension-free dengan melepaskan ligamen yang menahan ke usus desenden dan memastikan
bahwa tidak ada pemutaran dari pull-through usus selama anastomosis. Di Rumah
Sakit Anak Alabama, neonatus dengan aganglionosis kolon total ditangani dengan
meratakan enterostomi usus kecil awal, diikuti 6 sampai 12 bulan kemudian
dengan prosedur Duhamel dengan bantuan laparoscopik. Teknik Duhamel
menawarkan keuntungan membuat reservoir anus yang lebih besar pada pasien
dengan Hirschsprung kolon subtotal atau Hirschsprung kolon total.3
Melakukan ileostomi awal dan menunda prosedur hingga 6 sampai 12 bulan
juga mengurangi kejadian ruam popok yang parah pasca operasi terkait dengan
pull-through usus kecil pada bayi baru lahir. Pada neonatus, kecuali ada
kontraindikasi, LATEP dilakukan segera setelah diagnosis dikonfirmasi dengan
biopsi isap rektum. Kontraindikasi untuk dilakukan pull-through primer pada
neonatus meliputi:

Enterocolitis berat
Dilatasi proksimal masif
Tidak mampu untuk ditentukan zona transisi
Komorbiditas yang mengancam hidup
Dekompresi preoperatif usus dilakukan dengan kombinasi pelebaran rektum
dengan jari dan irigasi kolon dengan saline melalui tabung rektal dengan ujung
tabung diposisikan di atas zona transisi. Neonatus diberikan cairan secara
intravena. Diberikan dua dosis rejimen antibiotik oral yang terdiri dari eritromisin
dan neomycin dalam waktu 8 dan 4 jam sebelum operasi dijadwalkan. Segera
diberikan antibiotik spektrum luas intravena sebelum pasien dibawa ke ruang
operasi.3
Pull-Through Laparoskopi
Bayi diposisikan melintang di dekat ujung meja operasi untuk
memungkinkan akses ke perut dan perineum. Penetapan posisi berjalan baik jika
kepala pasien diputar ke sisi kanan dan menghadap dokter anestesi. Dengan
pengaturan pobsisi ini, ahli bedah laparoskopi memiliki akses yang lebih baik ke
bagian sigmoid pasien dan kolon sebelah kiri. Kaki yang dibungkus dalam bidang
operasi setelah prepping. Pneumoperitoneum diperoleh dengan menggunakan
teknik cut-down terbuka melalui umbilikus. Tekanan dari 10 sampai 12 mmHg
ditoleransi dengan baik pada semua kelompok umur. Hiperventilasi sederhana ini
berguna untuk mencegah hiperkarbia pada pasien ini. Digunakan teleskop 4 mm,
30-derajat pada bayi, dan 5 mm, lingkup 30 derajat sangat membantu pada pasien
yang lebih besar.3

Gambar 2.9. Foto menunjukkan port penempatan untuk operasi ini. Biasanya diperlukan tiga atau empat port. Port umbilikal dimasukkan
menggunakan teknik terbuka, dan port lainnya diperkenalkan di bawah visualisasi langsung. Teleskop (panah putus-putus) ditempatkan
melalui port 5-mm di perut kanan bagian atas. Dua port kerja utama dokter bedah adalah port umbilikal untuk tangan kiri dan port perut
bagian bawah kanan untuk tangan kanan. Sebuah instrumen retraksi (panah padat) sering membantu dan dapat dimasukkan melalui insisi tusukan di perut bagian atas sebelah kiri bayi. Dimasukkan kateter
kemih untuk membantu dekompresi kandung kemih.3
2.11 KomplikasiKomplikasi yang dapat terjadi mungkin termasuk
peningkatan insiden enterokolitis pasca operasi dengan prosedur Swenson, konstipasi setelah perbaikan dengan teknik Duhamel, serta diare dan inkontinensia dengan pull-through menggunakan prosedur Soave (Lee SL., 2012).
Secara umum, komplikasi kebocoran anastomosis dan pembentukan striktur adalah sebesar (5-15 %), obstruksi usus (5%), abses pelvis (5%), infeksi luka (10%), dan dehiscence luka dan reseksi tidak lengkap yang membutuhkan operasi ulang (5%). Pasien dengan operasi dua tahap juga dapat mengembangkan komplikasi stomal, seperti prolaps atau striktur (Lee SL., 2012).
Komplikasi yang terjadi kemudian terkait dengan pengelolaan bedah penyakit Hirschsprung’s disease termasuk enterocolitis, gejala obstruktif berlanjut, inkontinensia, konstipasi

kronis (6-10%), dan kematian, kebanyakan mempengaruhi pasien dengan penyakit segmen panjang (1-5%). Fistula rectovesical juga telah dilaporkan dalam literatur pernah terjadi (Lee SL., 2012).
2.12 Prognosis
Hasil jangka panjang setelah perbaikan definitif penyakit Hirschsprung’s disease sulit untuk ditentukan karena dari laporan penelitian dan literatur hasilnya saling bertentangan. Beberapa peneliti melaporkan tingkat kepuasan yang tinggi, sementara yang lain melaporkan kejadian kontipasi dan inkontinensia yang signifikan (Lee SL., 2012).
Sayangnya, sekitar 1% dari pasien dengan penyakit Hirschsprung’s disease membutuhkan kolostomi permanen untuk memperbaiki inkontinensia. Pasien dengan terkait trisomi 21 cenderung memiliki hasil klinis yang lebih buruk (Lee SL., 2012).
Secara umum, lebih dari 90% pasien dengan penyakit Hirschsprung’s disease memiliki hasil yang memuaskan, meskipun banyak pasien mungkin mengalami gangguan fungsi usus selama beberapa tahun sebelum mengembangkan kontinensia yang normal (Lee SL., 2012).
BAB IIIKESIMPULAN
1. Hirschsprung’s disease’s disease merupakan kelainan obstruksi usus yang
sering terjadi pada bayi baru lahir. Penyakit ini ditandai dengan tidak adanya
sel ganglion di bagian distal usus yang dimulai dari sfingter ani interna sampai
ke bagian proksimal usus dengan panjang yang bervariasi, selalu melibatkan
anus dan setidaknya melibatkan rektum.
2. Insidensi penyakit ini adalah sekitar 1 per 5000 kelahiran hidup. Anak-anak
Asia tampaknya memiliki insiden tertinggi yang hampir 3 per 5000 kelahiran
hidup. Rasio laki-laki dan perempuan adalah sekitar 4:1.
3. Pada umumnya, diagnosis Hirschsprung’s disease bayi baru lahir gambaran
klinisnya yaitu, pengeluaran mekonium yang terlambat (>24 jam setelah lahir),
distensi abdomen, dan muntah hijau.
4. Hirschsprung’s disease dicurigai pada neonatus sesuai dengan manifestasi
klinis yang telah disebutkan sebelumnya. Pemeriksaan yang dapat dilakukan
untuk mendiagnosis penyakit ini seperti contrast enema (CE), anorectal
manometry (ARM), biopsi rektal tebal , dan rectal suction biopsy (RSB).
5. Penatalaksanaan penyakit ini adalah dengan pembedahan. Setelah penanganan
preoperasi yang hati-hati, prinsip yang mendasar adalah dengan menempatkan
usus yang normal ke anus untuk menimbulkan kontraksi pada sfingter ani
interna.

6. komplikasi dapat berupa kebocoran anastomosis dan pembentukan striktur, obstruksi usus, abses pelvis, infeksi luka, dan dehiscence luka dan reseksi tidak lengkap yang membutuhkan operasi ulang, inkontinensia, dan enterokolitis.
7. Secara umum, sebagian besar pasien dengan penyakit Hirschsprung’s disease memperoleh hasil yang memuaskan, meskipun banyak pasien mungkin mengalami gangguan fungsi usus selama beberapa tahun sebelum mengembangkan kontinensia yang normal.
DAFTAR PUSTAKA
1. Monajemzadeh, M. et al. 2011. Hirschsprung's Disease: A Clinical and
Pathologic Study in Iranian Constipated Children. Iran J Pediatr, 21(3),
pp.362-66.
2. Haricharan, R.N. dan Georgeson, K.E. 2008. Hirschsprung Disease. In
Seminars in Pediatric Surgery.
3. Iqbal, M.Z. et al. 2010. Hirschsprung’s Disease; Modified Duhamel
(Martin Modification), A Procedure of Choice (A study at Sheikh Zayed
Hospital Rahim Yar Khan). Professional Med J, 17(2), pp.223-31.
4. Amiel, J. et al. 2008. HIrschsprung Disease, Associated Syndromes and
Genetics: A Review. J Med Genet, (45), pp.1-14.
5. Kartono, D. 1993. Penyakit Hirschsprung: Perbandingan Prosedur
Swenson dan Prosedur Duhamel Modifikasi. Disertasi. Universitas
Indonesia,
6. Nasrizarni dan Muntadhar. 2012. Profil Penderita Penyakit Hirschsprung
di Rumah Sakit Dr. Zainoel Abidin dan Rumah Sakit Ibu dan Anak Banda
Aceh. Skripsi. Universitas Syiah Kuala.
7. Schulten, D. dan Travassos, V. 2011. Clinical and Surgical Aspects of
Hirschsprung’s Disease. Utrecht: Coloplast Peristeen.

8. Menezes, M. dan Puri, P. 2006. Long-term outcome of patients with
enterocolitis complicating. Pediatr Surg Int, 22, pp.316-18.
9. Doodnath, R. dan Puri, P. 2010. A systematic review and meta-analysis of
Hirschsprung’s. Pediatr Surg Int, 26, pp.1107-10.
10. Saleh, A.M. et al. 2009. Hirschsprung's Disease: Early and Late Outcome
after Correction by Transanal. Annals of Pediatric Surgery, 5(1), pp.27-30.
11. Puri, P. dan Montedonico, S. 2008. Swenson’s Procedure. Dalam A.M.
Holschneider dan P. Puri, eds. Hirschsprung´s Disease and Allied
Disorders. New York: Springer Berlin Heidelberg.
12. Garcia-Barcelo et al. 2007. Correlation Between Genetic Variations in
Hox Clusters and Hirschsprung’s Disease. Annals of Human Genetics, 71,
pp. 526–536.
13. Izadi, M. et al. 2009. Clinical Manifestations of Hirschsprung’s Disease: A
Six Year Course Review of Admitted Patients in Gilan, Northern Iran.
Middle East Journal of Digestive Diseases, 1(2).
14. Lantieri, F. et al. 2008. The Molecular Genetics of Hirschsprung’s
Disease. Dalam A.M. Holschneider dan P. Puri, eds. Hirschsprung's
Disease and Allied Disorders. New York: Springer Berlin Heidelberg.
15. Wyllie, R. 2007. Motility disorders and Hirschsprung disease. Dalam R.M.
Kliegman, R.E. Behrman, H.B. Jenson dan B.F. Stanton, eds. Nelson
Textbook of Pediatrics. Philadelphia: Saunders Elsevier.
16. Lee SL. 2012. Hirschsprung Disease. Medscape Reference. Accessed from http//:emedicine.medscape.com/article/178493. Accessed on March 20th, 2014.
17. Holschneider A. M., Puri P. (Eds.). 2008. Hirschsprung´s Disease and Allied Disorders, Third Edition. New York: Springer-Verlag.
18. Kartono, Darmawan. 2004. Penyakit Hirschsprung. Jakarta:
Sagung Seto

19. Georgeson KE. 2010. Hirschsprung’s Disease. In: Ashcraft KW, Holcomb GW, Murphy JP (editor). Ashcraft’s Pediatric Surgery,
5th ed. Philadelphia: Sauders Elsevier; 456-467.
20. Kelleher J, Blake N. 2008. Diagnosis of Hirschsprung’s Disease and
Allied Disorders. Hirschsprung’s Disease and Allied Disorders. 3rd
Edition. New York: Springer; P. 145-151.