







Languages
Pages
Legal





Bibliografische Informationen der Deutschen Bibliothek
Die Deutsche Bibliothek verzeichnet diese Publikation in der Deutschen
Nationalbibliografie;
Detaillierte bibliografische Daten sind im Internet über http://dnb.ddb.de abrufbar.
1. Auflage 2012
© 2012 by Verlag: Deutsche Veterinärmedizinische Gesellschaft Service GmbH,
Gießen
Printed in Germany
ISBN 978-3-86345-0
Verlag: DVG Service GmbH
Friedrichstraße 17
35392 Gießen
0641/24466
www.dvg.net
70-0

Institut für Pharmakologie, Toxikologie und Pharmazie Tierärztliche Hochschule Hannover
Prokonvulsiva als pharmakologische Strategie zur Epilepsieprävention
THESE
zur Erlangung des Grades eines DOCTOR OF PHILOSOPHY
– Ph.D. –
im Fachgebiet Pharmakologie
durch die Tierärztliche Hochschule Hannover und das Zentrum für Systemische
Neurowissenschaften (ZSN) Hannover
vorgelegt von
Marta Małgorzata Rattka
Bielsko-Biała
Hannover 2012

Supervisor: Prof. Dr. W. Löscher
Wissenschaftliche Betreuung:
Prof. Dr. W. Löscher
Prof. Dr. G. Bicker
Prof. Dr. E. Ponimaskin
1. Gutachten:
Prof. Dr. W. Löscher (Institut für Pharmakologie, Toxikologie und
Pharmazie der Stiftung Tierärztliche Hochschule Hannover)
Prof. Dr. G. Bicker (Institut für Tierökologie und Zellbiologie der Stiftung Tierärztliche
Hochschule Hannover)
Prof. Dr. E. Ponimaskin (Institut für Neurophysiologie der Medizinischen Hochschule
Hannover)
2. Gutachten: Prof. Dr. R. Köhling (Institut für Physiologie der Universität Rostock)
Tag der mündlichen Prüfung: 30.03.2012
Gefördert durch ein Stipendium der Friedrich-Ebert-Stiftung. Diese Arbeit ist ein
Teilprojekt der DFG-Forschergruppe Neurodegeneration und -regeneration bei ZSN
-Erkrankungen des Hundes (Forschergruppe 1103).

Zuzannie Gąsior


Inhaltsverzeichnis
Inhaltsverzeichnis
1. Einleitung ....................................................................................................................... 1 2. Literaturübersicht ........................................................................................................... 3
2.1 Epilepsie .......................................................................................................................3
2.2 Temporallappenepilepsie ..............................................................................................4
2.3 Epileptogenese und Epilepsieprävention ......................................................................6
2.4. Antikonvulsiva in der Epileptogeneseforschung ...........................................................8
2.4.1 Untersuchungen im Tiermodell ............................................................................. 8 2.4.2 Klinische Studien .................................................................................................10
2.5 Prokonvulsiva in der Epileptogeneseforschung ........................................................... 11
2.6 GABA und Epilepsie ................................................................................................... 12
2.6.1 Theorie der exzitatorischen GABA .......................................................................13 2.6.2 Synchronisations-Theorie ....................................................................................14
2.7 Tiermodelle für Temporallappenepilepsie ................................................................... 15
2.7.1 Das Lithium-Pilokarpin-Modell .............................................................................17 2.7.2 Das fokale Kainsäure-Modell ...............................................................................18
2.8 Pentylenterazol (PTZ) ................................................................................................. 19
3. Zielsetzung und Arbeitshypothesen ..............................................................................21 4. Material und Methoden .................................................................................................23
4.1 Tiere ........................................................................................................................... 23
4.2 Stereotaktische Operation .......................................................................................... 24
4.3.1 SE-Induktion im Pilokarpin-Modell .......................................................................28 4.3.2 SE-Induktion im fokalen Kainsäure-Modell ...........................................................29
4.5 Studie 1: Etablierung des fokalen Kainsäure-Modells ................................................. 33
4.5.1 Studiendesign ......................................................................................................33 4.5.2 Implantation von Führungsrohren und Ableitelektroden .......................................33 4.5.3 SE-Induktion ........................................................................................................34 4.5.4 EEG- und Videoüberwachung ..............................................................................34 4.5.5 Verhaltenstests ....................................................................................................35
4.5.5.1 Der Hyperexzitabilitätstest (HET) ..................................................................36 4.5.5.2 Der Elevated-Plus Maze-Test (EPM) ............................................................37 4.5.5.3 Der Open Field-Test (OF) .............................................................................39 4.5.5.4 Der Morris Water Maze-Test (MWM) ............................................................39
4.5.6 Phenobarbital-Selektion .......................................................................................41 4.5.7 Tötung der Tiere und Histologie ...........................................................................43
4.5.7.1 Transkardiale Perfusion ................................................................................43 4.5.7.2 Dekapitation .................................................................................................44
4.5.7.2.1 Histologie nach Dekapitation ..................................................................44 4.6 Studie 2: PTZ-Schwellenversuche im Pilokarpin- und fokalen Kainsäure-Modell ........ 45
4.6.1 Studiendesign ......................................................................................................45 4.6.2 Implantation von Führungsrohren und Ableitelektroden .......................................46 4.6.3 SE-Induktion ........................................................................................................47
4.6.3.1 SE-Induktion im Pilokarpin-Modell .................................................................47 4.6.3.2 SE-Induktion im fokalen Kainsäure-Modell ....................................................48
4.6.4 PTZ-Schwellenbestimmung .................................................................................48 4.6.5 Bestimmung der Latenzzeit im Pilokarpin-Modell .................................................49 4.6.6 Diazepam-Vorversuch im Pilokarpin-Modell .........................................................50

Inhaltsverzeichnis
4.6.7 Bestimmung des ovariellen Zyklusstandes ..........................................................50 4.7 Studie 3: Infusionsversuche mit PTZ im Pilokarpin- und fokalen Kainsäure-Modell .... 50
4.7.1 Pharmakokinetische Vorversuche ........................................................................51 4.7.1.1 PTZ-Halbwertszeitbestimmung .....................................................................51 4.7.1.2 Etablierung des Infusionsprotokolls ...............................................................51
4.7.2 Studiendesign PTZ-Behandlung ..........................................................................52 4.7.3 Implantation von Ableitelektroden und Führungsrohren ...................................53
4.7.4 Implantation von Kathetern in die Vena jugularis externa .....................................54 4.7.5 SE-Induktion ........................................................................................................55
4.7.5.1 SE-Induktion im Pilokarpin-Modell .................................................................55 4.7.5.2 SE-Induktion im fokalen Kainsäure-Modell ....................................................56
4.7.6 PTZ-Infusion ........................................................................................................56 4.7.7 EEG- und Videoüberwachung ..............................................................................58
4.8 Statistik ....................................................................................................................... 58
5. Ergebnisse ...................................................................................................................59 5.1 Studie 1: Etablierung des fokalen Kainsäure-Modells ................................................. 59
5.1.1 SE-Induktion im fokalen Kainsäure-Modell ...........................................................59 5.1.2 Entwicklung von spontanen, wiederkehrenden, epileptische Anfällen im fokalen Kainsäure-Modell ..........................................................................................................61 5.1.3 Verhaltensänderungen im fokalen Kainsäure-Modell ...........................................66
5.1.3.1 Der Hyperexcitabilitätstest (HET) ..................................................................66 5.1.3.2 Der Elevated-Plus Maze-Test (EPM) ............................................................69 5.1.3.4 Der Morris Water Maze-Test (MWM) ............................................................75
5.1.4 Die Phenobarbital-Selektion .................................................................................77 5.1.5 Histologie .............................................................................................................80
5.1.5.1 Histologie der 1. Gruppe ...............................................................................80 5.1.5.2 Histologie der 2. Gruppe ...............................................................................80
5.2 Studie 2: Schwellenversuche im Pilokarpin- und fokalen Kainsäure-Modell ................ 83
5.2.1 Die SE-Induktion ..................................................................................................83 5.2.1.1 Die SE-Induktion und Latenzzeit im Pilokarpin-Modell ..................................83 5.2.1.2 Die SE-Induktion im fokalen Kainsäure-Modell ..............................................85
5.2.2 Der PTZ-Schwellentest ........................................................................................87 5.2.2.1 Schwellenveränderungen nach SE im Pilokarpin-Modell ...............................89 5.2.2.2 Schwellenveränderungen nach SE im fokalen Kainsäure-Modell ..................90
5.2.3 Vergleich beider SE-Modelle ................................................................................91 5.2.4 Einfluss des ovariellen Zyklusstandes auf die PTZ-Schwelle ...............................92
5.3 Studie 3: Infusionsversuche mit PTZ im Pilokarpin- und fokalen Kainsäure-Modell .... 98
5.3.1 Pharmakokinetische Vorversuche ........................................................................98 5.3.1.1 Bestimmung der Plasmahalbwertszeit von PTZ ............................................98 5.3.1.2 Korrelation zwischen konvulsiver PTZ-Dosis und dem Plasmaspiegel ........ 100 5.3.1.3 Etablierung des Infusionsprotokolls ............................................................. 101
5.3.2 Infusionsversuche mit PTZ im Pilokarpin-Modell (60 Minuten SE-Gruppe) ........ 102 5.3.2.1 SE-Induktion ............................................................................................... 102 5.3.2.2 PTZ-Behandlung ......................................................................................... 102 5.3.2.3 Entwicklung von spontanen Anfällen nach PTZ-Behandlung ....................... 102
5.3.3 Infusionsversuche mit PTZ im Pilokarpin-Modell (90 Minuten SE-Gruppe) ........ 103 5.3.3.1 SE-Induktion ............................................................................................... 103 5.3.3.2 PTZ-Behandlung ......................................................................................... 103 5.3.3.3 Entwicklung von spontanen Anfällen nach PTZ-Behandlung ....................... 104
5.3.4 Infusionsversuche im fokalen Kainsäure-Modell (13-16 Stunden-Gruppe) ......... 105 5.3.4.1 SE-Induktion ............................................................................................... 105 5.3.4.2 PTZ-Behandlung ......................................................................................... 105

Inhaltsverzeichnis
5.3.4.3 Entwicklung von spontanen Anfällen nach PTZ-Behandlung ....................... 105 5.3.5 Infusionsversuche im fokalen Kainsäure-Modell (7 Tage-Gruppe) ..................... 106
5.3.5.1 SE-Induktion ............................................................................................... 106 5.3.5.2 PTZ-Behandlung ......................................................................................... 107 5.3.5.3 Entwicklung von spontanen Anfällen nach PTZ-Behandlung ....................... 107 5.3.5.4 Gemeinsame Auswertung der Kainsäure-Gruppen ..................................... 109
6. Diskussion .................................................................................................................. 110 6.1 Einleitung .................................................................................................................. 110
6.2 Studie 1: Etablierung des fokalen Kainsäure-Modells. .............................................. 110
6.2.1 SE-Induktion ...................................................................................................... 110 6.2.2 Anfallsausprägung nach SE ............................................................................... 112 6.2.3 Verhaltensänderungen ....................................................................................... 112 6.2.4 Pharmakologische Untersuchungen .................................................................. 115 6.2.5 Neurodegeneration ............................................................................................ 117 6.2.6 Zusammenfassung ............................................................................................ 118
6.3 Studie 2: Schwellenversuche im Pilokarpin- und im fokalen Kainsäure-Modell ......... 118
6.3.1 Einfluss von Diazepam auf die individuelle Krampfschwelle ............................... 118 6.3.2 Latenzzeit im Pilokarpin-Modell ......................................................................... 119 6.3.3 Veränderungen der individuellen Krampfschwelle bei Kontrollen ....................... 120 6.3.4 Veränderungen der Krampfschwelle nach SE .................................................... 120 6.3.5 Einfluss des Zyklusstandes auf die Krampfschwelle .......................................... 122
6.4 Studie 3: PTZ-Behandlung zur Epilepsieprävention .................................................. 123
6.4.1 Etablierung des Behandlungsprotokolls ............................................................. 123 6.4.2 PTZ-Behandlung ................................................................................................ 124
6.5 PTZ zur Epilepsieprävention: Ergebnisse und Ausblick ............................................ 125
7. Zusammenfassung ..................................................................................................... 129 8. Summary .................................................................................................................... 131 9. Literatur ...................................................................................................................... 133 10. Anhang ....................................................................................................................... 142 Publikationen ............................................................................................................... 151 Erklärung ..................................................................................................................... 152 Danksagung ................................................................................................................ 153

Abbildungsverzeichnis
Abbildungsverzeichnis:
Abb. 1: Epilepsieentstehung und –progression und Möglichkeiten der Intervention. ...............8 Abb. 2: Aufsicht auf einen Rattenschädel mit eingezeichneten Kreuzungen der Knochennähte ................................................... 26 Abb. 3: Schematische Darstellung der Lokalisation der Ableitelektrode . ................. 27 Abb. 4: Schematische Darstellung der Lokalisation des Führungsrohres ............. 27 Abb. 5 :Schematische Darstellung der Lokalisation der Injektionsnadel ............................... 30 Abb. 6 :Intrahippokampale Mikroinjektion von Kainsäure ..................................................... 30 Abb. 7: Schematische Darstellung des zeitlichen Ablaufs der Studie 1 ................................ 33 Abb. 8: Schematischer Aufbau des Elevated Plus Maze. ..................................................... 38 Abb. 9: Schematischer Aufbau des Open field. .................................................................... 39 Abb. 10: Schematische Darstellung des Morris Water Maze. ............................................... 40 Abb. 11: Schematische Darstellung des zeitlichen Ablaufs der Studie 2 ............................... 46 Abb. 12 (A-C): EEG-Veränderungen vor und nach Kainsäure-Mikroinjektion ...................... 60 Abb. 13 (A-C): Spontane epileptische Anfälle 8 Wochen nach SE-Induktion ....................... 64 Abb. 14: Tägliche Anfallsfrequenz in der 2. Tiergruppe ........................................................ 65 Abb. 15 (A-H): Ergebnisse des HET ................................................................................... 68 Abb. 16 (A-J): Ergebnisse des EPM. ................................................................................... 70 Abb. 17 (A-F): Ergebnisse des OF I ..................................................................................... 72 Abb. 18 (A-F): Ergebnisse des OF II .................................................................................... 73 Abb. 19 (A-F): Ergebnisse OF III ......................................................................................... 74 Abb. 20 : Ergebnisse des MWM. .......................................................................................... 76 Abb. 21 (A-B): MWM: Mittlere Schwimmgeschwindigkeit und Plattformkreuzungen ............ 76 Abb. 22: Tägliche Anfallsfrequenz während der PB-Selektion. ............................................ 79 Abb. 23 (A-D): Neurodegeneration im Hippokampus 40 Wochen nach Kainsäure . 82 Abb. 24: Typisches EEG-Muster eines spontanen, epileptischen Anfalls ............................ 84 Abb. 25: Kein Enfluss von Diazepam (3x10 mg/kg) auf die PTZ-Schwelle............................ 86 Abb. 26 (A-E): EEG-Ableitung während einer PTZ-Schwellenbestimmung. ........................ 88 Abb. 27 (A-C): PTZ-Schwellenveränderungen im Pilokarpin-Modell .................................... 93 Abb. 28 (A-F): Zusammengefasste Verhaltensänderungen nach PTZ-Infusion .................. 94 Abb. 29 (A-C): PTZ-Schwellenveränderungen im fokalen Kainsäure-Modell ....................... 95 Abb. 30 (A-C): Weitere Endpunkte nach PTZ-Schwellentest ............................................... 96 Abb. 31 (A-C): Verhaltensänderungen nach Ende des PTZ-Schwellentests ......................... 97 Abb. 32 :Vergleich der PTZ-Schwelle ................................................................................... 98 Abb. 33: PTZ-Plasmaspiegel ................................................................................................ 99 Abb. 34: Darstellung der konvulsiven PTZ-Dosis ................................................................ 100 Abb. 35: Wöchentliche Anfallsfrequenz im Pilokarpin-Modell ........................... . 104 Abb. 36: Wöchentliche Anfallsfrequenz im fokalen Kainsäure-Modell I. ................... 106 Abb. 37 :Wöchentliche Anfallsfrequenz im fokalen Kainsäure-Modell II. ................. 108 Abb. 38 :Wöchentliche Anfallsfrequenz nach PTZ-Behandlung kombinierte Daten ............ 109

Tabellenverzeichnis
Tabellenverzeichnis Tabelle 1: Stereotaktische Koordinaten für die Implantation von DG-Elektroden und Führungsrohren. .................................................................................................. 25 Tabelle 2: Modifizierte Anfallsskala nach Racine (1972) ....................................................... 32 Tabelle 3: Tabellarische Darstellung des zeitlichen Ablaufs der Verhaltensbatterie ............ 35 Tabelle 4: Verwendeter Hypolokomotion-, Hyperlokomotion- und Ataxie-Score. ................. 43 Tabelle 5: Übersicht über die Tiergruppen in Studie 2. ........................................................ 46 Tabelle 6: Score-Systeme zur Beurteilung der Verhaltensänderungen ................................ 49 Tabelle 7: Übersicht über die Behandlungsgruppen in Studie 3 ............................................ 53 Tabelle 8: Übersicht über die Zeitpunkte der Katheter-Implantation Studie 3 ........................ 54 Tabelle 9: Übersicht über die zum SE-Abbruch verwendeten Substanzen ........................... 56 Tabelle 10: Übersicht über die Überwachungszeitpunkte in den einzelnen Tiergruppen . ..... 58 Tabelle 11: Durchschnittliche tägliche Anfallsfrequenz ........................................................ 63 Tabelle 12: Phenobarbital-Plasmakonzentration .................................................................. 77 Tabelle 13: Mittlerer Neurodegenerationsscore .................................................................... 81 Tabelle 14: Latenzzeit und Anfälle in den ersten 3 Tagen nach SE ..................................... 84 Tabelle 15: PTZ-Halbwertszeit der einzelnen Tiere nach 12 mg/kg PTZ Injektion ............... 99 Tabelle 16: PTZ-Plasmakonzentrationen 24 h und 48 h (1. Infusionsprotokoll) ................. 101 Tabelle 17: PTZ-Plasmakonzentrationen 24 h und 48 h (2. Infusionsprotokoll) .................. 101 Tabelle 18: Anfälle während der 48 h PTZ-Behandlung...................................................... 107

Abkürzungen
Abkürzungen
Aqua dest. destilliertes Wasser
bzw. beziehungsweise
CA1 Cornu Ammonis Region 1
CA2 Cornu Ammonis Region 2
CA3a Cornu Ammonis Region 3a
CA3c Cornu Ammonis Region 3c
cm Zentimeter
DG Gyrus Dentatus
ECoG Elektrokortikogramm
EDTA Ethylendiamintetraacetat
EEG Elektroenzephalogramm
EPM Elevated-Plus Maze
g Gramm
GABA Gamma-Amino-Buttersäure
GABAA-Rezeptor Gamma-Amino-Buttersäure-Rezeptor Typ A
h Stunde
HPLC Hochleistungsflüssigkeitschromatographie
(High Performance Liquid Chromatography)
Hz Herz
ILAE Internationale Liga gegen Epilepsie
(International League Against Epilepsy)
i. p. intraperitoneal
i. v. intravenös
KCC2 Kalium-Chlorid-Cotransporter Subtyp 2
kg Kilogramm
M Mol
Mg Milligramm
min Minute
mmol Millimol
mm Millimeter
mV Millivolt
MWM Morris Water Maze

Abkürzungen
NaCl Natriumchlorid
NKCC1 Natrium-Kalium-Chlorid-Cotransporter
Subtyp 1
NMDA N-Methyl-D-Aspartat
OF Open Field
p.a. pro analysi
PTZ Pentylentetrazol
RNA Ribonukleinsäure
s Sekunde
s. c. subkutan
SE Status epileptikus
SEM gemittelter Standartdfehler (Standard Error of
Mean)
µl Mikroliter
WHO Weltgesundheitsorganisation (World Health
Organisation)
z. B. zum Beispiel


Einleitung
1
1. Einleitung
Epilepsie ist die häufigste chronische neurologische Erkrankung des Menschen. Sie
ist charakterisiert durch das Auftreten von spontanen, wiederkehrenden epileptischen
Anfällen (CHANG und LOWENSTEIN 2003). Sie kann einhergehen mit psychischen
Begleiterkrankungen wie Depressionen, Angststörungen oder kognitiven
Dysfunktionen (JACOBS et al. 2009, JONES et al. 2010). Die häufigste
Epilepsieform bei Erwachsenen ist die Temporallappenepilepsie. Bei
symptomatischen Epilepsieformen, wie der Temporallappenepilepsie, ist ein
Hirninsult (z. B. Schädel-Hirn-Trauma, Status epileptikus) der primäre Auslöser der
Erkrankung. Der Prozess zwischen Hirninsult und Epilepsieentstehung wird als
Epileptogenese bezeichnet (LÖSCHER und BRANDT 2010 a, PITKÄNEN und
LUKASIUK 2011). Das Risiko einer posttraumatischen Epilepsie liegt je nach Insult
durchschnittlich bei 20%, das bedeutet, dass 20% der Patienten nach Insult an
Epilepsie erkranken werden. Bei schwersten, penetrierenden Schädel-Hirn-
Traumata, z. B. nach Schussverletzungen, steigt das Risiko sogar auf 50% (TEMKIN
et al. 1995, AARABI et al. 2000, Richtlinien der American Association for the Trauma
Surgery 2001, EFTEKHAR et al. 2009). Die Erkrankung tritt in den meisten Fällen in
den ersten zwei Jahren nach Hirninsult auf (ENGLANDER et al. 2003). Die derzeitige
Therapie der Epilepsie ist auf eine symptomatische Anfallsunterdrückung beschränkt.
Obwohl Patienten aus der Risikogruppe leicht identifiziert werden können, gibt es bis
heute keine Möglichkeit, diese Menschen präventiv zu behandeln (DICHTER 2009
PITKÄNEN und LUKASIUK 2011).
Aus diesem Grund ist es dringend notwendig, Strategien zu entwickeln, die die
Epileptogenese verhindern oder zumindest modulieren könnten (DICHTER 2009
LÖSCHER und BRANDT 2010 a, PITKÄNEN und LUKASIUK 2011). Der Versuch,
Patienten präventiv mit Medikamenten zu behandeln, die zur symptomatischen
Anfallsunterdrückung verabreicht werden, war nicht erfolgreich, da sich bis heute
keiner dieser Stoffe als antiepileptogen erwies (TEMKIN et al. 1990 und 1999,
TEMKIN 2001 und 2009). Während der Epileptogenese kommt es zu einer Reihe
von Veränderungen im Gehirn.

Einleitung
2
Zu ihnen zählen der Verlust von Neuronen, Entzündungsprozesse und die
Entstehung von neuronaler Übererregbarkeit (STABLES et al. 2002). Zu rationalen
Strategien der Antiepileptogenese zählen die Neuromodulation, die
Entzündungshemmung, die Neuroprotektion und die Immunmodulation (LÖSCHER
und BRANDT 2010 a, PITKÄNEN 2010, PITKÄNEN und LUKASIUK 2011). Die
Neuromodulation ist eine Strategie der Antiepileptogenese, die das Entstehen der
neuronalen Übererregbarkeit nach einem Insult verhindern soll (LÖSCHER und
BRANDT 2010 a). In dieser Arbeit soll die Neuromodulation nach Insult durch
Behandlung mit einem GABAA-Rezeptor-Antagonisten erfolgen. Unter
physiologischen Bedingungen prokonvulsiv wirkende GABAA-Rezeptor-Antagonisten
konnten in experimentellen Untersuchungen antikonvulsive Effekte in epileptischen
Netzwerken entfalten (KHAZIPOV und HOLMES 2003, KLAASSEN et al. 2006). Der
Einfluss solcher Mittel auf die Epileptogenese wurde bislang nicht untersucht.
Ziel dieser Arbeit war deshalb die Behandlung mit dem GABAA-Rezeptor-
Antagonisten Pentylentetrazol (PTZ) als pharmakologische Strategie der
Epilepsieprävention. Das antiepileptogene Potential von PTZ wurde in zwei
Rattenmodellen für Temporallappenepilepsie untersucht. Der Behandlungserfolg
wurde anhand des Auftretens von spontanen Anfällen nach Behandlung beurteilt.

Literaturübersicht
3
2. Literaturübersicht
2.1 Epilepsie
Epilepsien sind die häufigsten neurologischen Erkrankungen des Menschen. Etwa 50
Millionen Menschen sind weltweit von Epilepsien betroffen, das entspricht etwa 1%
der Weltbevölkerung (WHO 2009). Es handelt sich bei Epilepsien um eine seit
langem bekannte Gruppe von Erkrankungen, die bereits im Altertum beschrieben
wurden. Epilepsien sind charakterisiert durch spontane wiederkehrende Anfälle, die
infolge von pathologischen, synchronen Neuronenentladungen auftreten (FISHER et
al. 2005). Nach Nomenklatur der Internationalen Liga gegen Epilepsie (International
League Againist Epilepsy ILAE) lassen sich sowohl Epilepsien, als auch epileptische
Anfälle verschiedenen Gruppen zuordnen.
Epilepsien kann man je nach Ätiologie in idiopathisch, symptomatisch und kryptogen
einteilen (SHORVON 2011). Bei idiopathischen Epilepsien handelt es sich um
Erkrankungen genetischen Ursprungs. Symptomatische Epilepsien haben einen
primären Hirninsult, wie z. B. ein Schädel-Hirn-Trauma als Auslöser. Kryptogene
Epilepsien sind unbekannten Ursprungs, vermutlich symptomatisch. Durch immer
genauer werdende diagnostische Verfahren werden heutzutage auch kleinste
Hirnveränderungen besser erkannt, so dass eine Vielzahl von früher kryptogen
eingestuften Epilepsien nun als symptomatisch klassifiziert wird. Aus diesem Grund
wird vermutet, dass die Mehrzahl kryptogener Epilepsien eigentlich symptomatischer
Natur ist (SHORVON 2011).
Epileptische Anfälle können ebenfalls klassifiziert werden. Je nachdem, ob während
eines Anfalls beide Hirnhemisphären betroffen sind oder nicht, werden Anfälle als
fokal oder generalisiert bezeichnet. Bei fokalen Anfällen entsteht der Anfall im
epileptischen Fokus, meist in den Strukturen des limbischen Systems im
Temporallappen und bleibt auf den Fokus begrenzt. Fokale Anfälle können bei
erhaltenem Bewusstsein auftreten (einfache fokale Anfälle), oder mit
Bewusstseinsverlust einhergehen (komplex fokale Anfälle).

Literaturübersicht
4
Wenn sich ein fokaler Anfall über den Fokus hinaus auf beide Hirnhemisphären
ausbreitet, sprechen wir von sekundär generalisierten Anfällen. Primär generalisierte
Anfälle können mit tonisch-klonischen Krämpfen einhergehen (konvulsiver Anfall).
Es kann aber auch zu einer kurzen Bewusstseinstrübung ohne Krämpfe kommen
(Absence-Anfall) (SHORVON 2011) .
Epilepsien werden ganzheitlich als Epilepsie-Syndrom zusammengefasst (FISHER et
al. 2005). Neben epileptischen Anfällen kann die Krankheit mit einer Reihe von
psychiatrischen Begleiterkrankungen einhergehen, die bedeutend für die
Lebensqualität der Betroffenen sind. Zu den häufigsten psychiatrischen
Komorbiditäten zählen Depressionen, Angststörungen, Psychosen und kognitive
Dysfunktionen (KANNER 2011). Ein weiterer Faktor, der für die Lebensqualität der
Patienten von Bedeutung ist, ist die immer noch große Stigmatisierung der
Erkrankung (GARCIA-MORALES et al. 2008, JONES et al. 2010).
2.2 Temporallappenepilepsie
Temporallappenepilepsie ist die am häufigsten auftretende Epilepsieform bei
Erwachsenen. Sie ist außerdem die Epilepsieform mit der höchsten Anzahl der
therapieresistenten Fälle und somit von besonderer Bedeutung für die medizinische
Forschung (ENGEL 1996 a). Auf dem Markt vorhandene Medikamente können vielen
Patienten helfen, die Anfälle zu kontrollieren, jedoch wird weniger als die Hälfte der
Patienten dauerhaft anfallsfrei (SPENCER 2002).
Bei dieser Erkrankung ist der Epilepsieherd in den Strukturen des Temporallappens
lokalisiert (vorwiegend im Hippokampus). Die Temporallappenepilepsie ist
gekennzeichnet durch wiederkehrende, spontane Anfälle (komplex-fokal und
sekundär generalisiert), neuronale Degeneration und psychiatrische
Begleiterkrankungen wie Depressionen und Lern- und Gedächtnisstörungen
(MAJORES et al. 2004, JONES et al. 2010). Ein möglicher Grund für die
Verknüpfung psychiatrischer Auffälligkeiten mit Temporallappenepilepsien ist die
Tatsache, dass die Anfälle im Temporallappen entstehen und dieser für emotionales
Verhalten eine besondere Rolle spielt (SCHACHER et al. 2006).

Literaturübersicht
5
Die Neurodegeneration, auch hippokampale Sklerose genannt, betrifft bestimmte
Regionen des Hippokampus (Hiluszellen des Gyrus Dentatus,
Pyramidalzellschichten CA1 und CA3c) (MARGERISON und CORSELLIS 1966,
SLOVITER 1994, KÄLVIÄINEN et al. 1998, MAJORES et al. 2004).
Die Temporallappenepilepsie ist eine symptomatische Epilepsieform. Der
Erkrankung geht ein primärer Hirninsult voraus. Eine Vielzahl von Ereignissen kann
das Entstehen einer Temporallappenepilepsie begünstigen. Zu den häufigsten
Hirninsulten zählen Schädel-Hirn-Traumata, Hirntumore, ein lang anhaltender
epileptischer Anfall, welcher als Status epileptikus (SE) bezeichnet wird,
Fieberkrämpfe, Schlaganfälle oder Infektionen des zentralen Nervensystems
(STABLES et al. 2002). Der Prozess zwischen Hirninsult und ersten spontanen
epileptischen Anfällen wird als Epileptogenese bezeichnet. Während der
Epileptogenese kommt es zu einer Reihe von Veränderungen im Gehirn, unter
anderem zu neuronaler Übererregbarkeit, Entzündung und Neurodegeneration.
Welcher dieser Prozesse ursächlich für die Epilepsieentstehung ist, ist zur Zeit nicht
bekannt (LÖSCHER und BRANDT 2010 a).
Da es bislang nicht möglich ist, der Epilepsieentstehung bei Risikopatienten durch
prophylaktische Behandlung nach einem Insult entgegenzuwirken, ist die
gegenwärtige Therapie auf eine symptomatische beschränkt (DICHTER 2009 Alle
auf dem Markt erhältlichen Antiepileptika sind antikonvulsive Stoffe, welche
symptomatisch die Anfälle unterdrücken, ohne zu einer Heilung zu führen. Die
Einnahme von Antiepileptika ist mit Nebenwirkungen verbunden, welche einen
Einfluss auf die Lebensqualität der Betroffenen haben können. Das größte Problem
der gegenwärtigen Therapie mit Antiepileptika ist jedoch die Tatsache, dass bis zu
75% der Patienten mit Temporallappenepilepsie pharmakoresistent sind (SPENCER
2002). Das bedeutet, dass sie trotz Therapie mit zwei verschiedenen Antiepileptika
(kombiniert oder als Monotherapie) nicht dauerhaft anfallsfrei werden (KWAN et al.
2010). Für pharmakoresistente Patienten ist eine chirurgische Resektion des
epileptischen Fokus oftmals die einzige Alternative. Auch nach einem solchen
Eingriff werden nicht alle Patienten anfallsfrei. Viele sind trotz Resektion auf eine
Therapie mit Antiepileptika angewiesen (FOLDVARY et al. 2001, LÖSCHER und
SCHMIDT 2006). Aus diesen Gründen ist die Antiepileptogeneseforschung von
größter Bedeutung für Patienten aus der Risikogruppe.

Literaturübersicht
6
2.3 Epileptogenese und Epilepsieprävention Als Epileptogenese werden die Prozesse bezeichnet, die im Gehirn zur
Epilepsieentstehung nach einem initialem Insult führen. Die Latenzzeit zwischen
primärem Hirninsult und der Epilepsieentstehung bietet ein Zeitfenster für eine
präventive Behandlung (STABLES et al. 2002, STAFSTROM und SUTULA 2005).
Leider ist nicht bekannt, welcher Zeitpunkt nach Insult optimal für eine präventive
Behandlung ist. Es gibt Hinweise aus tierexperimentellen Arbeiten, dass eine solche
Behandlung schnellstmöglich erfolgen sollte (CHEN et al. 2007, ECHEGOYEN et al.
2009). In einer Studie von PITKÄNEN et al. (2004) konnte jedoch selbst eine
Behandlung sieben Tage nach Hirninsult noch zu krankheitsmodifizierenden Effekten
führen. Die Latenzzeit kann bei Menschen viele Jahre andauern (FRENCH et al.
1993, SILLANPÄÄ und SCHMIDT 2006). In Nagermodellen, die zur
Epilepsieforschung etabliert wurden, ist sie auf einige Tage bis Wochen beschränkt.
Während der Epileptogenese kommt es zu verschiedenen Veränderungen im Gehirn.
Insultassoziierter Neuronenverlust, Entzündung, synaptische Reorganisation und
neuronale Übererregbarkeit sind einige von ihnen (STABLES et al. 2002). Da bis
heute nicht geklärt ist, welche dieser Prozesse ursächlich an der Epilepsieentstehung
beteiligt sind, und welche eine Konsequenz der Epilepsie darstellen, ist ein rationaler
antiepileptogener Ansatz schwierig. Als rationale Strategien der Antiepileptogenese
sind die antiinflammatorische, die immunmodulatorische, die neuromodulatorische
sowie die neuroprotektive Strategie zu nennen (PITKÄNEN et al. 2004, VEZZANI
und GRANATA 2005, BRANDT et al. 2006, JUNG et al. 2006, ANDRE et al. 2007,
ZENG et al. 2009, BRANDT et al. 2010, POLASCHECK et al. 2010, STARK und
BAZAN 2011). Neuroprotektion wurde lange Zeit als Strategie zur
Epilepsieprävention verfolgt. Obwohl neuroprotektive Stoffe identifiziert werden
konnten, blieb die Neuroprotektion ohne Auswirkungen auf die Anfallsentwicklung
(ANDRE et al. 2001, BRANDT et al. 2006, ANDRE et al. 2007, POLASCHECK et al.
2010, LANGER et al. 2011). Leider konnte bis heute kein Mittel identifiziert werden,
welches einen wahren antiepileptogenen Effekt hervorruft, das heißt, die
Epilepsieentstehung nach einem Insult verhindern kann (LÖSCHER und BRANDT
2010 a).

Literaturübersicht
7
Der Prozess der Epilepsieentstehung und Progression (Epileptogenese) bietet
mehrere Möglichkeiten der therapeutischen Intervention.
Am Anfang des Prozesses steht der initiale Insult. Das sind meist Schädel-Hirn-
Traumata, Fieberkrämpfe im Kleinkindalter, ein SE, ein Schlaganfall oder auch eine
Infektion des zentralen Nervensystems. Wenn es nicht gelingt, den durch den Insult
entstandenen Schaden zu reparieren, kann es durch begünstigende Faktoren wie
„second hit“, Empfindlichkeitsgene oder Komorbiditäten zum Auftreten von
spontanen epileptischen Anfällen kommen (WALKER et al. 2002). Der
Krankheitsverlauf kann progressiv sein, es können psychiatrische Komorbiditäten
auftreten. Oftmals ist die Epilepsie pharmakoresistent, das bedeutet, dass es zu
keiner vollständigen Anfallsunterdrückung durch Medikamente kommt (KWAN et al.
2010). Die Zeit zwischen dem initialem Insult und der klinischen Manifestation der
Epilepsie, dem Auftreten von spontanen Anfällen, wird als Latenzzeit bezeichnet. Die
Latenzzeit bietet mehrere Möglichkeiten der therapeutischen Intervention (LÖSCHER
et al. 2008). Das Ziel aller Antiepileptogenese-Studien ist es, eine wirklich
antiepileptogene Substanz zu finden, welche das Auftreten von epileptischen
Anfällen nach Hirninsult verhindern könnte. Patienten aus der Risikogruppe könnten
mit einer solchen Substanz prophylaktisch nach Insult behandelt werden. Leider ist
es bis heute nicht gelungen eine solche Substanz zu identifizieren (DICHTER 2009
Es gibt verschiedene Probleme bei therapeutischen Ansätzen. Zum einen die Wahl
des geeigneten Behandlungszeitpunktes und der Behandlungsdauer mit einer
antiepileptogenen Substanz. Zum anderen die Abgrenzung einer wirklich
antiepileptogenen Wirkung von einer Insultmodifizierung und Krankheitsmodifizierung
(LÖSCHER und BRANDT 2010 a, PITKÄNEN und LUKASIUK 2011).
Die Antiepileptogenese ist nur während der Latenzzeit, vor dem ersten Auftreten von
epileptischen Anfällen möglich. Eine andere Möglichkeit der therapeutischen
Intervention ist die Insultmodifizierung. Eine Insultmodifizierung ist z. B. ein
schnellstmöglicher SE-Abbruch oder Fiebersenkung im Falle von Fieberkrämpfen.
Durch Insultmodifizierung soll das Risiko von spontanen Anfällen gesenkt werden,
sie kann aber auch einen krankheitsmodulierenden Effekt zufolge haben z. B. einen
milderen, nichtprogressiven Krankheitsverlauf. Eine Insultmodifizierung ist nur
zeitnah zum Insult möglich. Ein krankheitsmodulierender Effekt wäre auch nach
klinischer Manifestation der Epilepsie möglich. Eine symptomatische

Literaturübersicht
8
Anfallsunterdrückung mit Antikonvulsiva ist derzeit die einzige Therapie für
Epilepsiepatienten (LÖSCHER und BRANDT 2010 a, PITKÄNEN 2010, MANI et al.
2011). Aus diesem Grund ist die Suche nach prophylaktischen
Behandlungsmaßnahmen von größter Bedeutung. Die Epilepsieentstehung und
Progression ist schematisch in Abbildung 1 dargestellt.
2.4. Antikonvulsiva in der Epileptogeneseforschung Eine Reihe von Antiepileptika, die zur symptomatischen Anfallsunterdrückung
verwendet werden, wurde auf ihr antiepileptogenes Potential hin untersucht.
Solche Untersuchungen fanden in Tiermodellen, aber auch in klinischen Studien an
Patienten statt.
2.4.1 Untersuchungen im Tiermodell
Im Kindling-Modell konnte ein antiepileptogener Effekt für Phenobarbital, Valproat
und Levetiracetam gezeigt werden (TURNER et al. 1977, SILVER et al. 1991,
LÖSCHER et al. 1998, STRATTON et al. 2003). Ein Nachteil des Kindling-Modells ist
Primärer Hirninsult
Intervention
Insult-Modifizierung
Reparatur
Keine Reparatur Keine Konsequenz
Spontane Anfälle
Chronische Epilepsie
Progression der Epilepsie
Keine Progression
Epileptogenesez. B. durch „second hit“
Antiepileptogenese
Anfallsunterdrückung
Krankheitsmodifizierung
Abb. 1: Epilepsieentstehung und –progression und Möglichkeiten der Intervention. Modifiziert nach LÖSCHER et al. (2008).

Literaturübersicht
9
jedoch die Tatsache, dass die Tiere in diesem Modell nicht chronisch epileptisch
sind, sondern nur akut induzierte Anfälle haben.
Da sie jedoch im Laufe des Kindling-Prozesses immer empfindlicher auf die
induzierten Anfälle reagieren und es somit zu chronischen Veränderungen der
individuellen Krampfschwelle kommt, wird das Kindling-Modell als Epileptogenese-
Modell eingestuft. Weder Phenobarbital, noch Valproat zeigten antiepileptogene
Effekte in klinischen Studien (TEMKIN 2001). In post SE-Epilepsiemodellen wurden
ebenfalls verschiedene Antiepileptika untersucht. Solche Modelle unterscheiden sich
vom Kindling-Modell dadurch, dass die Tiere eine chronische Epilepsie nach einem
SE entwickeln (in diesen Modellen ist ein SE der initiale Insult). Bis auf Topiramat
konnte für keinen dieser Stoffe ein antiepileptogener Effekt gezeigt werden (BRANDT
et al. 2006, SUCHOMELOVA et al. 2006, BRANDT et al. 2007, JESSBERGER et al.
2007, BRANDT et al. 2010, FRANCOIS et al. 2011). Die Behandlung mit den
Antiepileptika Valproat, Carisbamat und Topiramat führte zur
Krankheitsmodifizierung in post SE-Epilepsiemodellen (BRANDT et al. 2006,
SUCHOMELOVA et al. 2006, JESSBERGER et al. 2007, FRANCOIS et al. 2011) .
Eine Behandlung mit dem Antiepileptikum Valproat war in Rattenmodellen für
Temporallappenepilepsie neuroprotektiv, ein antiepileptogener Effekt konnte jedoch
nicht erzielt werden (BRANDT et al. 2006, JESSBERGER et al. 2007). Die einzige
Studie, in der ein antiepileptogener Effekt einer Valproat-Behandlung gezeigt werden
konnte wurde von BOLANOS et al. (1998) durchgeführt. Da in dieser Arbeit die
Anfallsausprägung während der Phase ds Valproatausschleichens untersucht wurde,
ist nicht sicher, ob der beobachtete Effekt einen wahren antiepileptogenen Effekt
darstellt oder nur auf die antikonvulsive Wirkung des Valproats zurückzuführen ist.
Für Carisbamat konnte im Tiermodell ebenfalls ein starker neuroprotektiver Effekt
gezeigt werden. Zusätzlich hatte Carisbamat durch eine Insultmodifikation einen
krankheitsmodifizierenden Effekt bei der Ratte, wenn es direkt nach pilokarpin-
induziertem SE verabreicht wurde (FRANCOIS et al. 2011). Für Topiramat konnte
ebenfalls ein neuroprotektiver Effekt gezeigt werden (RIGOULOT et al. 2004). In
einer Studie von SUCHOMELOVA et al. (2006) war eine Topiramat-Behandlung
krankheitsmodifizierend. In einer Arbeit von (2002) konnte ein antiepileptogener
Effekt mit einer Topiramat-Behandlung erzielt werden, die Studie wurde 2002 als
Abstract, jedoch bislang nicht als Manuskript veröffentlicht. Eine klinische Studie zum

Literaturübersicht
10
Einsatz von Topiramat nach Schädel-Hirn-Trauma wird derzeit durchgeführt, die
Ergebnisse sind noch nicht bekannt (Quelle: clinicaltrials Datenbank).
Insgesamt ist es gelungen neuroprotektive Substanzen zu identifizieren, die
Neuroprotektion hatte jedoch keinen Einfluss auf die Epilepsieentstehung. Somit ist
es nicht gelungen einen antiepileptogenen Effekt über Neuroprotektion zu erreichen.
2.4.2 Klinische Studien
Es wurden mehrere klinische Studien mit verschiedenen Antiepileptika durchgeführt
(MANI et al. 2011). In einer verblindeten, placebokontrollierten Studie von TEMKIN et
al. (1990) wurde das Antiepileptikum Phenytoin bei Patienten mit Schädel-Hirn-
Trauma untersucht. Die prophylaktische Behandlung startete 24 h nach Insult und
wurde ein Jahr lang fortgeführt. Sie hatte keinen Effekt auf die spätere
Anfallsentwicklung. Es gab keinen signifikanten Unterschied in der Anfallsentstehung
zwischen behandelter Gruppe und Placebo-Gruppe. In einem weiteren Versuch
dieser Arbeitsgruppe (TEMKIN et al. 1999) wurden die Antiepileptika Phenytoin und
Valproat bei Patienten mit Schädel-Hirn-Trauma untersucht. Drei verschiedene
Studiendesigns wurden gewählt: 1. Phenytoin-Behandlung über eine Woche, 2.
Valproat-Behandlung über eine Woche, 3. Valproat-Behandlung über sechs Monate.
Auch in dieser Studie konnte kein antiepileptogener Behandlungseffekt gezeigt
werden. In einer Metaanalyse verschiedener Antiepileptogenesestudien konnte für
keinen der getesteten Stoffe ein antiepileptogener Effekt gezeigt werden (TEMKIN
2001). Untersucht wurden Diazepam und Phenobarbital bei Patienten mit
Fieberkrämpfen, Phenytoin und Valproat bei Hirntumorpatienten und Carbamazepin
und Phenobarbital bei Schädel-Hirn-Trauma Patienten (TEMKIN 2001). In einer
weiteren Studie von TEMKIN (2009) wurde der Einsatz von Phenobarbital,
Carbamazepin, Phenytoin, Valproat und Magnesium bei Patienten nach Schädel-
Hirn-Trauma untersucht. Mit keinem dieser Stoffe konnte ein antiepileptogener Effekt
erzielt werden. Keines der getesteten Antiepileptika erwies sich bei Risikopatienten
zur prophylaktischen Behandlung geeignet. Diese Befunde lassen darauf schliessen,
dass Prozesse, die während der Epileptogenese zur Epilepsieentstehung führen,
sich von dem Prozess der akuten Anfallsentstehung, welcher durch Antiepileptika

Literaturübersicht
11
unterdrückt wird, unterscheiden (WEAVER 2003). Aus diesem Grund werden
zunehmend andere Strategien der Antiepileptogenese verfolgt.
2.5 Prokonvulsiva in der Epileptogeneseforschung Für verschiedene Stoffe, die bei naiven Tieren prokonvulsiv wirken (HALONEN et al.
1995, WALLACE et al. 2003, CHROSCINSKA-KRAWCZYK et al. 2011), konnte
experimentell ein antikonvulsiver, neuromodulierender oder
krankheitsmodifizierender Effekt gezeigt werden (ARMSTRONG et al. 2009).
Untersucht wurden der Adenosin-Rezeptor-Agonist Koffein, der α2-Rezeptor-
Antagonist Atipamezol und der Typ 1 Cannabinoid-Rezeptor-Antagonist Rimonabant.
In einer Studie von RIGOULOT et al. (2003) wurde der Einfluss einer Koffein-
Behandlung im Lithium-Pilokarpin-Modell untersucht. Ratten wurden hierfür 15 Tage
vor SE und 7 Tage danach oral mit Koffein behandelt. Die Behandlung hatte keinen
antiepileptogenen Effekt, wirkte jedoch neuroprotektiv. Die Ergebnisse dieser Studie
sind insofern kritisch zu sehen, da die Behandlung bereits vor Insult (SE) startete,
was zu einer Modifikation des Insults führen kann und für eine Behandlung von
Risikopatienten nicht umsetzbar ist. Das Prokonvulsivum Atipamezol wurde in einer
Studie von PITKÄNEN et al. (2004) in einem Rattenmodell für
Temporallappenepilepsie (elektrisches post SE-Epilepsiemodell) untersucht. Das
Atipamezol wurde verwendet, da es bereits im Rattenmodell für Schlaganfall zum
Einsatz kam und dort zu positiven Effekten führte (PUURUNEN et al. 2001). Eine
systemische Behandlung mit Atipamezol konnte die Epilepsieentstehung zwar nicht
verhindern, die Krankheit hatte jedoch einen milderen, nichtprogressiven Verlauf
(PITKÄNEN et al. 2004). Zudem hatte die Behandlung einen neuroprotektiven Effekt.
Die Behandlung führte insgesamt zu einer Krankheitsmodifizierung.
Der Typ 1 Cannabinoid-Rezeptor-Antagonist Rimonabant wurde in mehreren Studien
auf sein antiepileptogenes Potential hin untersucht (CHEN et al. 2007, ECHEGOYEN
et al. 2009, DUDEK et al. 2010). Der Einsatz von Cannabinoid-Rezeptor-
Antagonisten sollte über Neuromodulation zur Verhinderung der neuronalen
Übererregbarkeit führen. Es gibt Hinweise, dass Veränderungen der Cannabinoid-
Rezeptoren, die nach einem Insult erfolgen, die Entstehung von neuronaler
Übererregbarkeit begünstigen können. Eine Applikation von Cannabinoid-Rezeptor-

Literaturübersicht
12
Antagonisten sollte die Entstehung der neuronalen Übererregbarkeit durch eine
Blockade der Plastizität der Cannabinoid-Rezeptoren nach einem Hirninsult
verhindern. In der Studie von CHEN et al. (2007) wurde eine Rimonabant-Applikation
2 min nach Induktion von Fieberkrämpfen bei Ratten vorgenommen.
Sechs Wochen nach Behandlung wurde die neuronale Erregbarkeit anhand einer
Kainsäure-Krampfschwelle ermittelt. Die Behandlung mit Rimonabant konnte das
Entstehen einer neuronalen Übererregbarkeit sechs Wochen nach Behandlung
verhindern. Das Auftreten von spontanen Anfällen wurde in dieser Studie jedoch
nicht untersucht. In der Studie von ECHEGOYEN et al. (2009) wurde ein ähnliches
Studiendesign in einem Schädel-Hirn-Trauma-Modell bei Ratten angewendet. Auch
in diesem Modell gelang es durch eine einmalige Rimonabant-Gabe direkt nach
Insult, die neuronale Übererregbarkeit sechs Wochen später zu verhindern. Die
neuronale Erregbarkeit wurde auch hier mittels Kainsäure-Krampfschwelle ermittelt.
Da diese Studien vielversprechende Ergebnisse lieferten, wurde Rimonabant auch in
einer Antiepileptogenese-Studie untersucht. In der Arbeit von DUDEK et al. (2010)
wurde Rimonabant im systemischen Kainsäure-Modell (chemisches post SE-
Epilepsiemodell) bei der Ratte untersucht. Die Rimonabant-Applikation erfolgte
einmalig nach SE-Induktion. Der Behandlungserfolg wurde mittels EEG- und
Videoüberwachung bis zwei Wochen nach SE überprüft. Die Rimonabant-
Behandlung hatte in dieser Untersuchung keinen Einfluss auf die Anfallsentwicklung.
Es konnte somit kein antiepileptogener Effekt erzielt werden.
Prokonvulsive GABAA-Rezeptor-Antagonisten wurden ebenfalls in der
Epilepsieforschung verwendet. Sie werden im nächsten Kapitel näher beschrieben.
2.6 GABA und Epilepsie GABA ist als wichtigster inhibitorischer Neurotransmitter von besonderem Interesse
für die Epilepsieforschung. Lange wurde Epilepsie als Ungleichgewicht zwischen
Inhibition und Exzitation gesehen und die Anfallsentstehung wurde als Konsequenz
von Veränderungen des GABAergen Systems beschrieben (ENGEL 1996 b, DALBY
und MODY 2001). Eine Reihe von antikonvulsiven Stoffen (Phenobarbital,
Diazepam, Vigabatrin, Valproat) wirkt zumindest teilweise über eine Verstärkung des
GABAergen Sytems. Neuere Hypothesen belegen jedoch, dass GABA auch

Literaturübersicht
13
proepileptogen wirken kann (COHEN et al. 2002, KHAZIPOV und HOLMES 2003,
KLAASSEN et al. 2006).
2.6.1 Theorie der exzitatorischen GABA
Es konnte gezeigt werden, dass GABA nach Insult depolarisierend und somit
exzitatorisch wirken kann. Bei Epilepsiepatienten kann es zu einem GABA-switch
von inhibitorisch zu exzitatorisch kommen. Dieser beruht auf einer veränderten
Exprimierung von Chlorid-Cotransportern in bestimmten Hirnregionen (Subiculum).
Eine Hochregulierung von NKCC1 (Subtyp 1 Natrium-Kalium-Chlorid-Cotransporter)
bei gleichzeitig gesenkter Exprimierung von KCC2 (Sybtyp 2 Kalium-Chlorid-
Cotransporter) an Neuronen führt zu Veränderungen des Chloridhaushaltes. Der
intrazelluläre Chloridgehalt steigt an. Das hat zufolge, dass GABA depolarisiered
wirkt (COHEN et al. 2002, PALMA et al. 2006). Dieser Zustand ist neonatal
physiologisch (BEN-ARI et al. 1989). Es wird vermutet, dass das Gehirn nach einem
Insult auf neonatale Funktionsweisen zurückgreift. Somit wird während der
Epileptogenese die Ontogenese wiederholt. Dieser ursächlich als
Reparaturmechanismus bestehende Prozess ist in diesem Fall kontraproduktiv
(KÖHLING 2002, BEN-ARI und HOLMES 2005). In reseziertem humanen
Temporallappengewebe von chronisch epileptischen Patienten konnte eine
depolarisiernde Wirkung von GABA im Subiculum gezeigt werden (COHEN et al.
2002). In einer Studie von PALMA et al. (2006) konnte die unphysiologische
Exprimierung von NKKC1 und KCC2 in Temporallappengewebe von chronisch
epileptischen Patienten auf RNA-Ebene gezeigt werden. Ein GABA-switch nach
Hirninsult konnte auch tierexperimentell gezeigt werden (PATHAK et al. 2007). In
der Studie von PATHAK et al. (2007) konnte bereits 24 h nach pilokarpin-induziertem
SE ein positiver EGABA-Shift (GABA Umkehr-Potential) im Hippokampus festgestellt
werden. Das lässt auf eine depolarisiernde Wirkung von GABA schließen. Zwei
weitere Arbeiten geben indirekt Hinweise auf den GABA-switch (LI et al. 2008,
BRANDT et al. 2010). In der Studie von LI et al. (2008) wurde im Maus Pilokarpin-
Modell (chemisches post SE-Epilepsiemodell) eine NKCC1-Expression im
Hippokampus bereits einen Tag nach SE auf RNA- und Protein-Ebene gezeigt. In
der Arbeit von BRANDT et al. (2010) konnte die NKCC1-Expression 24 h sowie vier

Literaturübersicht
14
Tage nach pilokarpin-induziertem SE bei Ratten immunhistochemisch gezeigt
werden. All das lässt vermuten, dass es nach einem Insult durch eine Veränderte
Exprimierung von Chlorid-Cotransportern zu einem GABA-switch von inhibitorisch zu
exzitatorisch kommt.
Da eine depolarisiernde Wirkung von GABA neonatal physiologisch ist, lassen sich
Anfälle bei Kleinkindern nur schwer mit traditionellen Antikonvulsiva unterdrücken. In
verschiedenen tierexperimentellen in vivo und in vitro Studien konnte eine
pharmakologische Blockade des NKCC1-Transporters mit dem selektiven NKCC1-
Inhibitor Bumetanid Anfälle bei Neonaten unterdrücken (DZHALA et al. 2005 und
2008, GLYKYS et al. 2009). Es wurden ebenfalls Epileptogenesestudien mit
Bumetanid vorgenommen, sowohl im modifizierten Kindlig-Modell, als auch in einem
post SE-Epilepsiemodell (MAZARATI et al. 2009, BRANDT et al. 2010). In der
Kindling-Studie von MAZARATI et al. (2009) gab es Hinweise auf einen
antiepileptogenen Effekt einer Bumetanid-Behandlung bei neonatalen Ratten. In der
Studie von BRANDT et al. (2010) konnte im Pilokarpin-Modell kein antiepileptogener
Effekt mit einer Bumetanid-Behandlung erzielt werden. Dies könnte jedoch an
methodischen Schwierigkeiten liegen (kurze Plasmahalbwertszeit und schlechte
Penetration der Blut-Hirn-Schranke durch Bumetanid).
Der Einsatz von GABAA-Rezeptor-Antagonisten wäre in Hinsicht auf diese Theorie
ebenfalls eine Möglichkeit, um durch antagonistische Wirkung am GABAA-Rezeptor
depolarisierende GABA-Aktionen zu verhindern.
2.6.2 Synchronisations-Theorie
GABA kann auch proepileptogen wirken, ohne direkte exzitatorische Eigenwirkung
zu entfalten. Nach einem Insult kommt es initial zu einem kompensatorischen
Anstieg der GABAergen Inhibition (GREEN 1986). Dadurch werden erregende
glutamaterge Neurone zunächst gehemmt. Die gesteigerte GABAerge Inhibition kann
jedoch nicht lange aufrechterhalten werden. Nach ihrem Kollaps werden viele
erregende glutamaterge Neurone gleichzeitig aus ihrer Hemmung entlassen und
können synchron feuern (MANN und MODY 2008). Diese Netzwerk-
Synchronisierung führt zu einer Übererregbarkeit in der betroffen Hirnregion, was die
Entstehung von epileptischen Anfällen begünstigen kann (MANN und MODY 2008).

Literaturübersicht
15
Der Einsatz von GABAA-Rezeptor-Antagonisten wäre in Hinsicht auf diese Theorie
sinnvoll, um den initialen Anstieg der GABAergen Inhibition zu verhindern. Das
würde in Konsequenz die Netzwerk-Synchronisierung und die daraus resultierende
neuronale Übererregbarkeit verhindern.
Es wurden bereits Studien zum Einsatz von GABAA-Rezeptor-Antagonisten in der
Epilepsieforschung durchgeführt. In einer Studie von KHAZIPOV und HOLMES
(2003) wurden elektrophysiologische Untersuchungen des Hippokampus in vivo an
anästhesierten Ratten durchgeführt. Epileptiforme Aktivität wurde mittels Kainsäure
erzeugt. Eine Zugabe des GABAA-Rezeptor-Antagonisten Bicucullin konnte die
epileptiforme Aktivität unterbinden. Es wurde postuliert, dass GABA zu einer
Netzwerk-Synchronisierung führte, die durch Bicucullin-Gabe unterbrochen wurde.
Somit hatte in dieser Untersuchung die Behandlung mit einem GABAA-Rezeptor-
Antagonisten einen antikonvulsiven Effekt. In einer Studie von KLAASSEN et al.
(2006) wurden Untersuchungen in einem genetischen Mausmodell für humane
autosomal dominante nokturnale Frontallappenepilepsie durchgeführt. In dieser
Studie konnte eine subkonvulsive Dosis des GABAA-Rezeptor-Antagonisten
Picrotoxin die klinische und elektrographische Anfallsausprägung unterdrücken. In
diesen Studien konnte in zwei verschiedenen Modellen und Untersuchungsdesigns
gezeigt werden, dass GABAA-Rezeptor-Antagonisten antikonvulsiv wirken können.
Der antikonvulsive Effekt wird auf eine Verhinderung der Netzwerk-Synchronisierung
zurückgeführt. Es wurden jedoch bislang keine Antiepileptogenesestudien mit
GABAA-Rezeptor-Antagonisten durchgeführt, obwohl es nach einem Insult
wahrscheinlich zu einer Übererregbarkeit infolge von Netzwerk-Synchronisierung
kommt. Aus diesem Grund haben wir entschieden eine Behandlung mit dem GABAA-
Rezeptor-Antagonisten Pentylentetrazol (PTZ) als Strategie der Epilepsieprävention
durchzuführen.
2.7 Tiermodelle für Temporallappenepilepsie Um Substanzen auf ihr antiepileptogenes Potential hin zu untersuchen, sind
geeignete Tiermodelle notwendig. Ein ideales Tiermodell der
Temporallappenepilepsie sollte die Konditionen der Temporallappenepilepsie beim
Menschen widerspiegeln, d.h. die Epilepsie sollte infolge eines Hirninsults und nach

Literaturübersicht
16
einer gewissen Latenzzeit auftreten, die Tiere sollten komplex fokale und sekundär
generalisierte Anfälle haben, mit Ursprung im Temporallappen, und es sollten
pathologische Veränderungen im Hippokampus auftreten (hippokampale Sklerose)
(CURIA et al. 2008). Ferner sollten die Tiere Verhaltensauffälligkeiten zeigen, welche
den psychiatrischen Komorbiditäten der Humanpatienten ähnlich sind.
Ein geeignetes Modell sollte Prädiktivität hinsichtlich der klinischen Wirksamkeit der
getesteten Arzneimittel aufweisen. Es gibt kein „ideales“ Epilepsiemodell, welches
allen Anforderungen genügt und zusätzlich praktikabel ist, aus diesem Grund kann
es sinnvoll sein, verschiedene Modelle ergänzend zu nutzen, um falsch-positive und
falsch-negative Ergebnisse zu vermeiden (LÖSCHER und BRANDT 2010 a). Es gibt
eine Vielzahl an Epilepsie- und Anfallsmodellen, nicht alle sind jedoch gleichermaßen
für Antiepileptogenestudien geeignet. Schädel-Hirn-Trauma-Modelle spiegeln die
Ätiologie der humanen Epilepsie gut wider. Sie sind jedoch aufgrund von
Praktikabilitätsgründen (lange Latenzzeit und die geringe Anzahl an epileptischen
Tieren nach Insult) nur bedingt für Antiepileptogenesestudien geeignet (BOLKVADZE
und PITKÄNEN 2011, PITKÄNEN et al. 2011 ). Die NIH/NINDS (National Institutes of
Health/National Institute of Neurological Disorders and Stroke) empfehlen das
Kindling-Modell und post SE-Epilepsiemodelle zur Erforschung von
antiepileptogenen Therapien (STABLES et al. 2002). Im Kindling-Modell sind die
Tiere nicht chronisch epileptisch, sie haben keine spontanen Anfälle, sondern nur
akut induzierte. Trotzdem kommt es bei den Tieren zu chronischen Veränderungen
(Senkung der Krampfschwelle). In post SE-Epilepsiemodellen sind die Tiere
chronisch epileptisch und haben spontane epileptische Anfälle. Da in dieser Arbeit
ausschließlich post SE-Epilepsiemodelle an der Ratte verwendet werden, wird auf
diese im Weiteren näher eingegangen.
In post SE-Epilepsiemodellen ist ein SE der initiale Hirninsult. Dieser kann elektrisch,
durch Stimulation bestimmter Hirnareale oder chemisch, durch Krampfgifte ausgelöst
werden. Diese können entweder systemisch oder fokal also direkt in Zielareale des
Gehirns verabreicht werden. Nach einer modellabhängig unterschiedlich langen
Latenzzeit kommt es bei den Tieren zu spontanen, wiederkehrenden epileptischen
Anfällen. Die Epilepsie bleibt meist lebenslänglich bestehen (LÖSCHER 2002).

Literaturübersicht
17
2.7.1 Das Lithium-Pilokarpin-Modell
Pilokarpin ist ein Parasypathomimetikum welches die Blut-Hirn-Schranke passieren
kann und dadurch sowohl periphere, als auch zentrale Wirkungen entfaltet. Das
systemische Pilokarpin-Modell, als Rattenmodell für Temporallappenepilepsie, wurde
erstmals von TURSKI et al. (1983) beschrieben.
Die Applikation von Pilokarpin führt über eine Aktivierung von muskarinergen M1-
Rezeptoren zu Krämpfen, die in einen SE übergehen (HAMILTON et al. 1997). Der
pilokarpin-induzierte SE wird nicht ausschließlich über eine Aktivierung von
muskarinergen Rezeptoren vermittelt, eine Aktivierung der NMDA-Rezeptoren ist für
die Aufrechterhaltung des SE verantwortlich (NAGAO et al. 1996, SMOLDERS et al.
1997). Die Pilokarpin-Applikation kann entweder als Bolus (400 mg/kg) oder
fraktioniert (10 mg/kg in 30 min Abständen) erfolgen (TURSKI et al. 1983, GLIEN et
al. 2001). Die fraktionierte Pilokarpin-Gabe ermöglicht es, durch eine individuelle
Dosisanpassung die ansonsten hohe Mortalitätsrate zu senken (GLIEN et al. 2001).
Eine weitere Möglichkeit der Dosissenkung besteht bei einer Vorbehandlung der
Tiere mit Lithium. Lithium, das in der Humanmedizin als Antipsychotikum
Verwendung findet, führt über eine periphere Entzündungsreaktion zu Schädigungen
der Blut-Hirn-Schranke. Es kommt zu einer Ausschüttung von Interleukin 1β und
Monozyten, die die Blut-Hirn-Schranke schädigen. Aus diesem Grund kann 12-16 h
vor Pilokarpin-Applikation eine Behandlung mit Lithium erfolgen, um durch die
Vorschädigung der Blut-Hirn-Schranke die Pilokarpin-Dosis zu senken (MARCHI et
al. 2009). Der SE wird für gewöhnlich nach 60-120 min pharmakologisch (meist
durch Diazepam) unterbrochen, da die Tiere sonst sterben würden. Nach einer
Latenzzeit von Tagen bis Wochen kommt es bei den Tieren zum Auftreten von
spontanen, wiederkehrenden epileptischen Anfällen, die lebenslang bestehen
bleiben (LEITE et al. 1990 a). Die epileptischen Tiere zeigen hyperexzitables
Verhalten sowie kognitive Störungen (RICE et al. 1998, LEITE et al. 1990 b). Bei den
Tieren kommt es außerdem zu histopathologischen Veränderungen im Gehirn, die
dem Bild der hippokampalen Sklerose des Menschen entsprechen. Die
Neuronenverluste sind jedoch stärker ausgeprägt und sind nicht ausschliesslich auf
Strukturen des Temporallappens beschränkt (SLOVITER 2005). Zusammenfassend
kann man sagen, dass das Lithium-Pilokarpin-Modell ein gutes, durch die
systemische Applikation einfaches und praktikables Modell der

Literaturübersicht
18
Temporallappenepilepsie darstellt, welches viele der Konditionen der humanen
Temporallappenepilepsie widerspiegelt (CURIA et al. 2008). Wie jedes Modell hat es
auch Nachteile, das sind vor allem das Ausmaß und die Ausprägung der
histologischen Veränderungen und Verhaltensänderungen.
Es wird auch vermutet, dass der pilokarpin-induzierte SE einen derart schweren
Insult darstellt, dass es nur bedingt möglich ist, in diesem Modell antiepileptogene
Stoffe zu entdecken (SLOVITER 2005). Aus diesem Grund haben wir beschlossen,
unsere Arbeit vergleichend an zwei Rattenmodellen für Temporallappenepilepsie
durchzuführen.
2.7.2 Das fokale Kainsäure-Modell
Kainsäure ist ein aus der Alge Dignea simplex gewonnenes Glutamat-Analogon,
welches, wie Glutamat selbst, erregend wirkt. Es entfaltet seine Wirkung über
ionotrope Glutamat-Rezeptoren (Kainat-Rezeptoren) (OLNEY et al. 1974, LODGE et
al. 1979, BEN-ARI und COSSART 2000). Eine fokale intracerebrale Kainsäure-
Applikation führt bei den Tieren zu epileptischen Anfällen (komplex fokal und
sekundär generalisiert), die in einen limbischen SE übergehen (BEN-ARI 1985). Der
SE wird in diesem Modell nicht unterbrochen und kann mehrere Stunden andauern.
Trotzdem ist die Mortalitätsrate sehr gering (RAEDT et al. 2009). Die meisten
Arbeiten zu diesem Modell wurden in den achtziger Jahren des 20. Jahrhunderts
durchgeführt. Neuere Arbeiten wurden von Bragin et al. (1999, 2004, 2005 und 2007)
und RAEDT et al. durchgeführt. Die Kainsäure-Applikation kann entweder unter
Narkose oder an wachen Ratten erfolgen (CAVALHEIRO et al. 1982, BRAGIN et al.
1999, RAEDT et al. 2009). Eine Applikation unter Narkose ist zwar einfacher
durchzuführen, die Narkose kann aber einen insultmodifizierenden Effekt haben und
dadurch die spätere Entstehung von epileptischen Anfällen beeinflussen. In einer
Studie von CAVALHEIRO et al. (1982) konnte ein kainsäure-induzierter SE zwar
spontane epileptische Anfälle auslösen, diese hörten jedoch nach 30 Tagen auf
(spontane Remission). Vermutlich ist dies auf einen insultmodifizierenden Effekt der
Narkose zurückzuführen. Aus diesem Grund haben wir uns für eine SE-Induktion an
wachen Ratten entschieden. Auch in diesem Modell kommt es zur Ausprägung von
epileptischen Anfällen nach einer Latenzzeit von Tagen bis Wochen (BABB et al.

Literaturübersicht
19
1995, RAEDT et al. 2009). In der Studie von RAEDT et al. (2009) konnte eine
Progression der Krankheit gezeigt werden. Die Tiere zeigen Verhaltensänderungen
wie gesteigerte Aggressivität und kognitive Dysfunktionen (HANDELMANN und
OLTON 1981, CAVALHEIRO et al. 1982).
Eine einseitige Kainsäure-Injektion in die CA3-Region des Hippokampus führt primär
zu Schäden der ipsilateralen Injektionsseite. Betroffen sind vor allem die CA3a- und
CA3c-Region, sowie die Hilusneurone des Gyrus Dentatus. Die CA1-Region und die
Körnerzellschicht des Gyrus Dentatus werden relativ verschont (RAEDT et al. 2009).
Die Veränderungen treten dosisabhängig auf (HANDELMANN und OLTON 1981,
CAVALHEIRO et al. 1982) . Die histopathologischen Veränderungen spiegeln das
Bild der hippokampalen Sklerose bei Humanpatienten gut wider (BABB et al. 1995,
RAEDT et al. 2009). Dadurch, dass der SE in diesem Modell nicht abgebrochen wird,
variiert die SE-Länge von Tier zu Tier, was einen Nachteil des Modells darstellt.
Durch die fokale Applikation der Kainsäure sind die Neurodegeneration und die
Verhaltensänderungen moderater als in systemischen post SE-Epilepsiemodellen.
Aus diesem Grund wollten wir dieses Modell für unser Labor etablieren.
2.8 Pentylenterazol (PTZ) PTZ ist ein GABAA-Rezeptor-Antagonist, der seine Wirkung durch Bindung an der
Picrotoxin-Bindungsstelle im Inneren des Chloridionophors entfaltet
(RAMANJANEYULU und TICKU 1984, HUANG et al. 2001). Durch seine
antagonistische Wirkung am GABAA-Rezeptor ist PTZ prokonvulsiv (MACDONALD
und BARKER 1977). PTZ wurde erstmalig in den dreißiger Jahren des 20.
Jahrhunderts zur Krampftherapie bei Behandlung von Patienten mit Depression
eingesetzt (FINK 1972 und 1984). Diese Form der Behandlung wurde durch die
elektrokonvulsive Therapie ersetzt. PTZ wurde außerdem als zentrales Analeptikum
bei Narkosezwischenfällen verwendet. Durch seine geringe therapeutische Breite ist
es für diese Indikation jedoch obsolet (COPER und HERRMANN 1988). PTZ wird
experimentell als anxiogener (JUNG et al. 2002) und aufmerksamkeitssteigernder
Stoff verwendet. In einem Maus-Modell des Down-Syndroms konnte eine PTZ-
Behandlung die Langzeitpotenzierung und dadurch die Kognition verbessern
(FERNANDEZ et al. 2007).

Literaturübersicht
20
In der experimentellen Epilepsieforschung wird PTZ zur Anfallsinduktion verwendet.
Die akute Anfallsinduktion wird auch im PTZ-Schwellentest genutzt. Mittels des PTZ-
Schwellentests kann die individuelle Krampfschwelle bestimmt werden. Der Test
kann durch subkutane, intraperitoneale oder intravenöse Applikation durchgeführt
werden.
Die intravenöse Applikation erhöht die Sensitivität des Tests durch individuelle
Dosierung. Während des Tests wird PTZ intravenös bis zum Auftreten eines
Krampfes verabreicht. Anhand der Zeit bis zum Auftreten des Anfalls, der PTZ-
Konzentration und -Infusionsrate und des Gewichtes des Tieres, kann die individuelle
Krampfschwelle ermittelt werden. Diese wird ausgedrückt in der PTZ-Dosis [mg/kg],
die nötig war, um den Anfall auszulösen. Der Test kann angewendet werden, um
Medikamente auf eventuelle antikonvulsive oder prokonvulsive Wirkung zu
untersuchen und findet breite Verwendung in der pharmazeutischen Industrie
(ORLOFF et al. 1949, LÖSCHER 2009). Der PTZ-Schwellentest kann bei einem Tier
mehrmals wiederholt werden und ermöglicht es, Änderungen der individuellen
Krampfschwelle zu erfassen, z. B. nach Prämedikation mit verschieden
Medikamenten. Dadurch, dass PTZ seine Wirkung am GABAA-Rezeptor entfaltet, ist
es vorwiegend prädiktiv für Medikamente mit GABA-potenzierendem
Wirkungsmechanismus und könnte Hinweise auf Veränderungen des GABAergen
Systems geben (LÖSCHER 2009 und2011).
Es gibt eine Reihe von Untersuchungen, die den PTZ-Metabolismus bei Ratten
beschreiben. Die PTZ-Plasmahalbwertszeit liegt bei 2-3h (ESPLIN und WOODBURY
1956, VOHLAND und ZUFELDE 1976, RAMZAN und LEVY 1985). PTZ wird bei der
Ratte in der Leber metabolisiert und renal ausgeschieden. Es penetriert die Blut-Hirn-
Schranke und breitet sich im Gehirn aus (ESPLIN und WOODBURY 1956). Die
prokonvulsive Wirkung wird durch den Hippokampus und parahippokampale
Regionen (GOLARAI et al. 1992, STRINGER 1994, WALSH et al. 1999, QIAN et al.
2011) aber auch durch den mamillothalamischen Trakt vermittelt (MIRSKI und
FERRENDELLI 1986 und 1987). Es gibt bislang keine Studien über den Einsatz
einer PTZ-Behandlung zur Epilepsieprävention.

Zielsetzung und Arbeitshypothesen
21
3. Zielsetzung und Arbeitshypothesen
Es ist bislang nicht möglich, die Epilepsieentstehung nach einem Hirninsult zu
verhindern. In zahlreichen Studien wird versucht, die Epileptogenese durch
unterschiedliche Strategien zu verhindern. Während der Epileptogenese kommt es
zu einer Reihe von Veränderungen im Gehirn, unter anderem zur neuronalen
Übererregbarkeit, zur Entzündung und zu Neuronenverlusten. In dieser Arbeit haben
wir uns auf die neuronale Übererregbarkeit fokussiert. Mit einer pharmakologischen
Strategie wollen wir die neuronale Übererregbarkeit nach einem Insult verhindern,
um in Konsequenz die Epileptogenese zu verhindern. Da es bis heute keine
Möglichkeit gibt, Patienten aus klar definierbaren Risikogruppen nach einem Insult
präventiv zu behandeln, ist eine Forschung auf diesem Gebiet von größter
Bedeutung. Da konservative Strategien einer Behandlung mit antikonvulsiven Mitteln
nach Insult bis heute nicht erfolgreich waren, wird der Einsatz von prokonvulsiven
Mitteln in der Antiepileptogeneseforschung immer häufiger diskutiert. Grund dafür ist
die Annahme, dass bei der Epileptogenese andere Mechanismen eine Rolle spielen
als bei der akuten Anfallsentstehung und deshalb der Einsatz von Antikonvulsiva
ohne Erfolg blieb. Es gibt Hinweise, dass der Einsatz von Stoffen, die eigentlich
prokonvulsiv wirken, von Vorteil sein könnte. In diesem Projekt soll der prokonvulsive
GABAA-Rezeptor-Antagonist PTZ zur Verhinderung der neuronalen Übererregbarkeit
verwendet werden. Die Wahl eines GABAA-Rezeptor-Antagonisten hat folgende
Gründe: Trotz der früheren Annahmen, dass Epilepsie durch einen Mangel an
GABAerger Inhibition ausgelöst wird, gibt es zunehmend Hinweise, dass der
inhibitorische Transmitter GABA auch proepileptogen wirken kann (KHAZIPOV und
HOLMES 2003, KLAASSEN et al. 2006). Zum einen konnte gezeigt werden, dass
eine nach Hirninsult kompensatorisch gesteigerte GABAerge Inhibition nach ihrem
Kollaps erregende Netzwerke synchronisieren kann (MANN und MODY 2008). Zum
anderen wurde ein GABA-switch von inhibitorisch zu exzitatorisch gezeigt (COHEN
et al. 2002, PATHAK et al. 2007). Dieser beruht auf einer Veränderung in der
Expression von Chlorid-Cotransportern nach einem Insult.

Zielsetzung und Arbeitshypothesen
22
Ziel dieser Arbeit war es, durch Applikation von PTZ in subkonvulsiver Dosierung die
initiale GABA-Hochregulierung nach einem Insult zu verhindern und somit der
Netzwerk-Synchronisierung entgegenzuwirken. Eine Behandlung mit PTZ wäre
ebenfalls bei einem GABA-switch sinnvoll, da PTZ die depolarisiernde Wirkung von
GABA antagonisieren könnte. Für andere GABAA-Rezeptor-Antagonisten (Picrotoxin,
Bicucullin) konnte sowohl in vivo, als auch in vitro, ein antikonvulsiver Effekt auf
epileptische Netzwerke gezeigt werden (KHAZIPOV und HOLMES 2003,
KLAASSEN et al. 2006). Der Einfluss eines solchen Stoffes auf die Epileptogenese
wurde jedoch noch nicht untersucht. Unsere Arbeitshypothese lautete:
Eine präventive Behandlung mit subkonvulsiven PTZ-Dosen nach Insult wirkt
antiepileptogen.
Diese Arbeitshypothese wollten wir in zwei Rattenmodellen für
Temporallappenepilepsie testen, im systemischen Pilokarpin-Modell und im fokalen
Kainsäure-Modell. Da wir das fokale Kainsäure-Modell für unser Institut erst
etablieren mussten, ergab sich für die Modelletablierung folgende Arbeitshypothese:
Das fokale Kainsäure-Modell ist ein geeignetes Rattenmodell für
Temporallappenepilepsie, welches ein moderateres Modell darstellt als das
systemische Pilokarpin-Modell.
Um Veränderungen des GABAergen Systems nach Insult zu beschreiben, haben wir
Veränderungen der individuellen Krampfschwelle nach SE untersucht. Das sollte uns
helfen, den am besten geeigneten Behandlungszeitpunkt nach SE herauszufinden.
Diese Untersuchungen wurden vergleichend in beiden Epilepsiemodellen
durchgeführt. Unsere Arbeitshypothese lautete hier:
Ein SE führt als initialer Insult zu Veränderungen des GABAergen Systems,
welche sich modellunabhängig im PTZ-Schwellentest als Veränderungen der
individuellen Krampfschwelle erfassen lassen.
Das Hauptziel dieser Arbeit war die Untersuchung des antiepileptogenen Potentials
von PTZ. Die Schwellenversuche waren Vorrausetzung zur Festlegung eines
Behandlungszeitfensters. Darüber hinaus sollten sie zusätzliche Informationen über
den Prozess der Epileptogenese und die zeitlichen Abläufe während dieses
Prozesses geben. Die Etablierung des fokalen Kainsäure-Modells sollte es
ermöglichen, dieses chemische SE-Modell bei der Ratte besser zu charakterisieren.

Material und Mehtoden
23
4. Material und Methoden
Im Anhang befindet sich eine Auflistung der verwendeten Substanzen,
Verbrauchsmaterialien und Geräte sowie die Herstellungsprotokolle verwendeter
Lösungen und die Färbeprotokolle.
Im Rahmen dieser Arbeit wurden drei Studien durchgeführt.
Studie 1: Etablierung des fokalen Kainsäure-Modells.
Studie 2: PTZ-Schwellenversuche im Pilokarpin- und fokalen Kainsäure-Modell.
Studie 3: Infusionsversuche mit PTZ im Pilokarpin- und fokalen Kainsäure-Modell.
In allen Studien wurden die Methoden der stereotaktischen Operation, der Status
epileptikus-Induktion (SE-Induktion) und der EEG- und Videoüberwachung
angewendet, aus diesem Grund werden sie einleitend beschrieben. Die
studienspezifischen Details sind dann bei der entsprechenden Studie eingehend
erklärt.
4.1 Tiere Für die Versuche dieser Studien wurden weibliche Sprague Dawley-Ratten von
Harlan (Horst, Niederlande) und Charles River (Sulzfeld, Deutschland) verwendet.
Alle Ratten wogen bei ihrer Ankunft 200-220 g (Alter ~ 9 Wochen). Die Tiere mit
EEG-Aufsätzen wurden einzeln in durchsichtigen Makrolonkäfigen Typ III (Ebeco,
Caustrop-Rauxel) auf Weichholzgranulat gehalten. Tiere ohne EEG-Aufsätze wurden
in Gruppen von 5 Ratten in Makrolonkäfigen Typ IV (Ebeco, Caustrop-Rauxel)
gehalten. Sie erhielten Leitungswasser und Standardnagerdiät (Altromin, Lage) ad
libitum. Das Umsetzen in saubere Käfige erfolgte einmal pro Woche. Das Futter
wurde einmal pro Woche aufgefüllt, das Wasser zweimal pro Woche erneuert. Die
weiblichen Ratten wurden ohne männliche Tiere gehalten, um einen synchronen
Zyklusstand zu vermeiden. Die Raumtemperatur betrug 22-24° C, die Luftfeuchtigkeit
lag bei 50-60%. Die Tiere wurden in einem 12 Stunden Hell-Dunkel-Zyklus gehalten
(helle Phase von 6-18 Uhr, mitteleuropäische Zeit).

Material und Mehtoden
24
Nach Ankunft hatten die Tiere mindestens eine Woche, um sich an die neue
Umgebung zu gewöhnen, in dieser Zeit wurden sie täglich an den Umgang mit
Menschen und die späteren Eingriffe (Fixieren etc.) gewöhnt.
4.2 Stereotaktische Operation Nach der Eingewöhnungsphase erfolgte die stereotaktische Operation. Dieser
Eingriff diente der Implantation von EEG-Ableitelektroden und bei Tieren aus dem
fokalen Kainsäure-Modell auch von Führungsrohren zur Kainsäure-Mikroinjektion.
Die bipolaren Ableitelektroden wurden in unserem Institut aus teflonisoliertem,
rostfreien Stahldraht (Science Products, Hofheim) hergestellt. Die Führungsrohre
wurden, ebenfalls in unserem Institut, aus rostfreiem Edelstahldraht (Schoeller
Werke, Hellenthal) hergestellt, waren 17 mm lang und hatten einen
Innendurchmesser von 0,52 mm.
Die Methode der stereotaktischen Operation ermöglicht es, bestimmte
Gehirnregionen anhand ihrer Lage relativ zu sichtbaren Kreuzungen von
Knochennähten an der Schädeloberfläche (z. B. Bregma, Lambda) zu lokalisieren.
Mithilfe von ermittelten Koordinaten aus dem Rattenhirnatlas von PAXINOS und
WATSON (2007) kann die Lage von spezifischen Gehirnregionen bestimmt werden.
Diese Technik kann angewendet werden, um Ableit-, Stimulationselektroden, oder
auch Führungsrohre zu implantieren oder auch um Substanzen im Verfahren der
Mikroinjektion in bestimmte Hirnareale zu injizieren.
Die Ableitelektrode wurde in den rechten Gyrus Dentatus (DG) des Hippokampus
implantiert (siehe Abbildung 3). Die Koordinaten waren bereits in unserem Labor
etabliert. Das Führungsrohr wurde analog zu BRAGIN et al. (2004) in den rechten,
posterioren Hippokampus, 1 mm über der Zielregion (CA3-Region) implantiert (siehe
Abbildung 4). Die Koordinaten wurden in Vorversuchen ermittelt und waren anterior-
posterior für Ratten von Harlan und Charles River unterschiedlich. Die
stereotaktischen Koordinaten für Elektrode und Führungsrohr sind in nachfolgender
Tabelle dargestellt
.

Material und Mehtoden
25
Lage relativ zu
Bregma EEG-Elektrode Führungsrohr
Harlan-Ratten Charles-River-Ratten
Anterior-posterior
(AP) -3,9 mm -5,8 mm -5,6 mm
Lateral (L) -1,7 mm -4,5 mm
Ventral (V) -3,5 mm -4,3 mm
Tabelle 1: Stereotaktische Koordinaten für die Implantation von DG-Elektroden und Führungsrohren.
Der Zahnbalken des Stereotakten war auf -3,9 mm eingestellt, damit Bregma
(rostraler Kreuzungspunkt der Knochennähte) und Lambda (kaudaler
Kreuzungspunkt der Knochennähte) auf einer Höhe lagen.
Die Tiere wurden mit Chloralhydrat (400 mg/kg i.p., Konzentration: 36 mg/ml,
Injektionsvolumen: 10 ml/kg) oder Isofluran (als Inhalationsnarkose, 3% zur
Narkoseeinleitung, 1,5% zur Aufrechterhaltung der Narkose) narkotisiert. Die Wahl
der Narkose wird bei den einzelnen Studien eingehend beschrieben. Die
Narkosetiefe wurde anhand des Zwischenzehenreflexes beurteilt. Im Falle einer zu
flachen Chloralhydrat-Narkose wurde ein Viertel der Ausgangsdosis nachinjiziert.
Anschließend wurde die Kopfhaut rasiert. Die Tiere wurden in den Stereotakten
eingespannt, indem die Ohrbalken in den knöchernen Gehöhrgang geschoben
wurden. Nach gerader Positionierung wurde die Kopfhaut mit 70%igem Ethanol
desinfiziert. Die desinfizierte Kopfhaut wurde nun mit Lokalanästhetikum (Tetracain
2%, Einwirkzeit 5 Minuten) benetzt. Nach Einwirken der Anästhesie wurde ein 2-3 cm
langer Schnitt in der Medianen vorgenommen, und die Haut wurde mit
Bulldogklemmen zu den Seiten hin aufgeklappt. Die Schädeldecke wurde mit
Lokalanästhetikum benetzt (Bupivacain 0,2%, Einwirkzeit 5 Minuten). Nach
Einwirken der Anästhesie wurde das Periost mit einem Periostschaber entfernt und
die Schädeldecke wurde mit Wasserstoffperoxid (35%) gereinigt, um die
Knochennähte darzustellen. Die Lage von Bregma wurde nun bestimmt und markiert.
Anhand der Koordinaten relativ zu Bregma wurden die Bohrlöcher markiert und
gebohrt. Die Bohrlöcher wurden mit einem Dentalbohrer (Dremel, Leinfelden-
Echterdingen) gebohrt.

Material und Mehtoden
26
Insgesamt wurden 4 Bohrlöcher bei der Implantation einer Ableitelektrode gebohrt
(eins für die Elektrode, 3 für Halteschrauben). Bei der Implantation von
Ableitelektrode und Führungsrohr wurden 6 Bohrlöcher benötigt (entsprechend 2 für
Elektrode und Führungsrohr und 4 für Halteschrauben). Die Elektrode und das
Führungsrohr wurden anhand der ermittelten Koordinaten in den Bohrlöchern
implantiert. Zusätzlich zur Elektrode und dem Führungsrohr wurden Halteschrauben
(Hummer & Rieß, Nürnberg) am Schädel angebracht. An der Halteschraube rechts
neben der Elektrode, wurde die indifferente Erdungselektrode angebracht (siehe
Abbildung 2). Das implantierte Führungsrohr wurde durch einen Mandrin
verschlossen.
Die Befestigung am Schädel erfolgte mit kaltaushärtendem Zahnzement (Paladur,
Heraeus Kulzer, Hanau). Die unterste Schicht war mit Marbofloxacin-Pulver versetzt,
um postoperativen Infektionen vorzubeugen. Aus dem Zahnzement entstand ein
Aufsatz, welcher eine für den Stecker des EEG-Kabels kompatible Endung hatte.
Nach Aushärtung des Zahnzements wurden die Wundränder um den Aufsatz herum
adaptiert und mit resorbierbarem Nahtmaterial (Surgicryl PGA, Hünningen, Belgien),
durch Knopfhefte verschlossen. Die Tiere erhielten acht Tage lang Antibiose,
beginnend zwei Tage vor dem Eingriff (Marbofloxacin ca. 3 mg/kg, 0,1 ml/Tier
zweimal täglich s.c.). Nach dem Eingriff hatten die Tiere eine zwei- bis dreiwöchige
Erholungsphase. In dieser Zeit wurden sie zweimal pro Woche an Umgang mit
Menschen und die späteren Eingriffe gewöhnt.
Elektrode
Erde
Führungsrohr
Halteschraube
Abb. 2: Aufsicht auf einen Rattenschädel mit eingezeichneten Kreuzungen der Knochennähte (Bregma, Lambda) und Lokalisation von Elektrode, Erde und Führungsrohr. Modifiziert nach PAXINOS und WATSON (2007)

Material und Mehtoden
27
Abb. 3: Schematische Darstellung der Lokalisation der Ableitelektrode . Modifiziert nach PAXINOS und WATSON (2007).
Abb. 4: Schematische Darstellung der Lokalisation des Führungsrohres . Modifiziert nach PAXINOS und WATSON (2007).

Material und Mehtoden
28
4.3 SE-Induktion In den Studien dieser Arbeit wurden zwei SE-Modelle angewendet, das systemische
Pilokarpin-Modell und das fokale Kainsäure-Modell. Da es im Pilokarpin-Modell zu
massiver Neurodegeneration und Verhaltsänderungen kommt, wollten wir zusätzlich
ein moderateres Modell für unsere Untersuchungen nutzen. Aus diesem Grund
haben wir uns entschlossen, das fokale Kainsäure-Modell in unserem Institut zu
etablieren und die weiteren Studien in beiden Modellen vergleichend durchzuführen.
Modellabhängig erfolgte die SE-Induktion also durch systemische Pilokarpin-Gabe
oder durch fokale Kainsäure-Mikroinjektion. Beide Methoden der SE-Induktion sollen
im Weiteren näher beschrieben werden.
4.3.1 SE-Induktion im Pilokarpin-Modell
Die SE-Induktion im Pilokarpin-Modell erfolgte fraktioniert, analog eines an unserem
Institut etablierten Protokolls von GLIEN et al. (2001). Zwölf bis sechzehn Stunden
vor Pilokarpin-Gabe erhielten die Ratten Lithiumchlorid (127 mg/kg in 3 ml/kg aqua
dest.) oral. Aufgrund der Lithiumchlorid-Vorbehandlung ist es möglich, die Pilokarpin-
Dosis zu reduzieren. Die periphere Wirkung des Pilokarpins wurde durch
intraperitoneale (i.p.) Verabreichung von Methylscopolamin (1 mg/kg, gelöst in 1
ml/kg aqua dest.) 30 min vor Pilokarpin-Gabe antagonisiert. Das Pilokarpin wurde
ebenfalls intraperitoneal verabreicht. Das fraktionierte Pilokarpin-Protokoll ermöglicht
eine individuelle Dosierung und senkt so die Sterblichkeitsrate.
Die Tiere erhielten 10 mg/kg Pilokarpin (gelöst in 10 ml aqua dest.) in
dreißigminütigen Abständen bis zum Auftreten eines generalisierten Anfalls. Wenn
die generalisierten Anfälle nicht mehr aufhörten oder das Tier das Bewusstsein
zwischen den Anfällen nicht wiedererlangte, wurde dieser Zustand als Eintritt des SE
beschrieben. Die Zahl der Injektionen pro Tier war auf 5 beschränkt.
Erfahrungsgemäß führen weitere Injektionen nur noch bei wenigen Tieren zum SE,
sie steigern die Mortalitätsrate jedoch deutlich.
Der SE wurde, versuchsabhängig nach 60 oder 90 min (im Detail bei den einzelnen
Versuchen beschrieben), pharmakologisch unterbrochen. Der SE-Abbruch erfolgte
entweder durch Diazepam oder durch eine kombinierte Behandlung aus Diazepam,
Phenobarbital und Scopolamin (im Detail bei den einzelnen Versuchen beschrieben).

Material und Mehtoden
29
Der SE-Abbruch war notwendig, um zum einen die Mortalitätsrate zu senken, und
zum anderen, um die Schwere des Insults (SE) für alle Tiere gleich zu halten.
Die Kontrollen erhielten die gleiche Behandlung, lediglich das Pilokarpin wurde durch
isotone Natriumchloridlösung ersetzt (sham SE). Tiere, die trotz fünfmaliger
Pilokarpin-Gabe keinen SE hatten, wurden versuchsabhängig entweder als eigene
Untergruppe zusammengefasst oder vom Versuch ausgeschlossen (im Detail bei
den einzelnen Versuchen beschrieben).
Bei den Ratten, bei denen eine Ableitelektrode implantiert wurde, wurde die SE-
Induktion unter EEG-Kontrolle durchgeführt. Die Tiere wurden je nach
Gewichtszunahme und Allgemeinbefinden unterschiedlich lange (3 bis 5 Tage) mit
Babynahrung zwangsernährt. Im Falle der Dehydrierung erhielten die Ratten
zusätzlich einmal täglich isotone Natriumchloridlösung als Injektion (4 ml pro Ratte
i.p.).
4.3.2 SE-Induktion im fokalen Kainsäure-Modell
Die SE-Induktion erfolgte in diesem Modell durch fokale Kainsäure-Mikroinjektion in
den Hippokampus (siehe Abbildung 5). Die Mikroinjektion wurde nach einem an
unserem Institut etablierten Protokoll von GERNERT und LÖSCHER (2001) an frei
beweglichen Ratten durchgeführt (siehe Abbildung 6). Für die Mikroinjektion wurde
Kainsäure von Sigma-Aldrich verwendet. Die Kainsäure-Konzentration betrug 0,4 µg
in einem Injektionsvolumen von 0,2 µl isotoner Natriumchloridlösung, analog
BRAGIN et al. (2005). Das Mikroinjektionssystem war wie folgt aufgebaut: Eine
stählerne Injektionskanüle (1 mm länger als das Führungsrohr, um die CA3-Region
des Hippokampus zu erreichen) war durch einen 20-30 cm langen, flexiblen
Polyethylenschlauch mit einer 0,5 µl Hamiltonspritze verbunden. Nach Entfernen des
Mandrins wurde die Injektionskanüle durch das Führungsrohr eingeführt. Die
Injektionskanüle verblieb erst 1 Minute im Gehirn, um Druckreaktionen des
Gewebes abzuwarten. Nach einer Minute begann die eigentliche Mikroinjektion. Die
Kainsäure wurde über 4 Minuten injiziert (0,05 µl/Minute). Das Injektionsvolumen
wurde anhand der Vorwärtsbewegung eines Markerfarbstoffes (Thionin) kontrolliert,
welcher durch Luftblasen und destilliertes Wasser von der eigentlichen Substanz
getrennt war.

Material und Mehtoden
30
Nach Ende der Mikroinjektion wurde die Kanüle für eine weitere Minute im Gehirn
belassen, um der injizierten Substanz Zeit zur Diffusion zu geben. Während der
gesamten Prozedur wurden die Tiere beobachtet. Auftretende Anfälle oder
Stereotypien wurden notiert. Die Tiere wurden mindestens 10 Minuten vor der
Mikroinjektion EEG-überwacht, um ein Basis-EEG zu registrieren. Direkt nach der
Mikroinjektion wurden die Tiere ebenfalls an das EEG angeschlossen. Die EEG-
Überwachung dauerte bis zum nächsten Morgen (mindestens 20 h). Der SE wurde
nicht abgebrochen, er hörte bei allen Tieren spontan auf (nach etwa 11 h). Die
Ratten erholten sich sehr schnell und mussten im Gegensatz zum Pilokarpin-Modell
nicht mit Babynahrung gefüttert werden.
Abb. 5 : Schematische Darstellung der Lokalisation der Injektionsnadel zur Mikroinjektion. Modifiziert nach PAXINOS und WATSON (2007).
Abb. 6 : Intrahippokampale Mikroinjektion von Kainsäure bei einer wachen, freibeweglichen Ratte.

Material und Mehtoden
31
4.4 EEG- und Videoüberwachung
EEG-Überwachungssystem
Um epileptische Anfälle registrieren zu können, wurden die Tiere mittels EEG und
Video überwacht. Das EEG-Überwachungssystem bestand aus 16 Ein-Kanal-
Verstärkern (BioAmp ADInstruments, Spechbach) und einem 16-Kanal Analog-
Digital-Wandler (Power Lab ADInstruments, Spechbach). Die Daten wurden mit der
LabChart 6 oder LabChart 7 Windows-Software (ADInstruments, Spechbach)
aufgenommen und analysiert. Die Frequenz der EEG-Aufzeichnung lag bei 200 Hz.
Der Aufnahmebereich lag zwischen 0,1 s (Highpass-Filter) und 60 Hz (Lowpass-
Filter). Ein Notch-Filter oder Mains-Filter (Software-abhängig) filterten Frequenzen
von 50 Hz (Netzstrom) aus. Die EEG-Überwachung war mit einem
Videoüberwachungssystem kombiniert.
Das Videoüberwachungssystem verwendete zuerst digitale Webcams (Logitech,
Morges, Schweiz) mit Rotlichtlampen als Lichtquelle. Da die Bildqualitität
insbesondere nachts suboptimal war, wurde das System auf sensitivere Infrarot-
Kameras umgestellt (NYCTO Vision, CaS Services, Wunstorf) mit Infrarotlampen als
Lichtquelle. Die Kameras erlaubten eine gleichzeitige Aufnahme von 8 Tieren. Das
digitalisierte Videosignal wurde auf dem EEG-aufzeichnenden PC aufgenommen.
Die Chart-Software ermöglichte eine simultane Aufnahme von EEG- und digitaler
Video-Datei. Die über den gesamten Zeitraum eingeschalteten Infrarot-, bzw.
Rotlicht-Lampen, die über den Käfigen angebracht waren, dienten nachts als
Lichtquelle.
Die Ratten waren während der Überwachung in speziellen Einzelkäfigen aus
Plexiglas untergebracht. Zweiadrige, ummantelte Kabel mit Abschirmung dienten als
Ableitkabel und waren über einen Binder-Stecker mit den an den Köpfen der Ratten
befindlichen Aufsätzen verbunden. Die Kabel wurden anfangs in unserem Institut
hergestellt (mit Materialen von Menzel Electronic, Hannover), später bezogen wir sie
von der Firma Hopa Kabelkonfektion (Hopa Kabelkonfektion GmbH, Langenhagen).
Um den Tieren Bewegungsfreiheit zu gewährleisten, waren die Ableitkabel in
beweglichen Telefonentzwirlern an der Decke der Käfige befestigt. Die Kabel hatten
eine Aluminiumummantelung als Beißschutz.

Material und Mehtoden
32
Datenanalyse
Die Analyse der so gesammelten Daten erfolgte visuell. Wenn ein Anfall im EEG
gefunden wurde, wurde das entsprechende Video analysiert, um den Anfallstyp zu
bestimmen. Die Anfallstypen wurden nach einer modifizierten Anfallsskala von
RACINE (1972) klassifiziert. Wenn im EEG eindeutige Anfälle erkannt wurden, die im
Video nicht eindeutig sichtbar waren, z. B. wenn die Ratte mit dem Rücken zur
Kamera saß, oder die Aufnahme zu dunkel war, wurden diese als nicht klassifizierbar
eingestuft. Fokale Anfälle wurden im Video oft als unklassifiziert eingestuft, weil sie
schwer zu erkennen sind. Die verwendete, modifizierte Skala für Anfallstypen nach
RACINE (1972) ist in der nachfolgenden Tabelle dargestellt.
Anfallstyp Verhalten des Tieres
Typ I Stereotypes Schnüffeln,
Blinzeln, schwache
Kaubewegungen Fokale Anfälle
Typ II Kauen, Kopfnicken
Typ III Unilateraler
Vorderbeinklonus
Typ IV Bilateraler
Vorderbeinklonus mit
Aufrichten
Generalisierte Anfälle
Typ (V) Typ V ohne Umfallen, da
die Ratte durch einen
Hinterbeinklonus nach
vorne oder zur Seite fällt
Typ V Bilateraler
Vorderbeinklonus mit
Aufrichten und Umfallen
Tabelle 2: Zur Datenanalyse verwendete, modifizierte Anfallsskala nach RACINE (1972)

Material und Mehtoden
33
4.5 Studie 1: Etablierung des fokalen Kainsäure-Modells
4.5.1 Studiendesign
In dieser Studie sollte das fokale Kainsäure-Modell als moderates (im Vergleich zum
Pilokarpin-Modell) Temporallappenepilepsie-Modell etabliert werden. Die Studie
wurde an zwei Tiergruppen durchgeführt. In der ersten Gruppe wollten wir Tiere von
zwei verschiedenen Züchtern (Harlan und Charles River) vergleichen. Da es in
dieser Gruppe entzündungsbedingt zu vielen Verlusten der EEG-Aufsätze kam,
wurde eine zweite Tiergruppe unter optimierten Bedingungen mit Kainsäure
behandelt. Dadurch, dass wir in unserem Labor nur in wenigen Versuchen Tiere von
Charles River verwenden und keine Unterschiede in den Versuchen der ersten
Gruppe aufgefallen waren, wurden in der zweiten Gruppe ausschließlich Tiere von
Harlan verwendet. Beide Gruppen wurden zusammen ausgewertet.
1. Gruppe: Tiere von Charles River (n=10) und Harlan (n=10).
2. Gruppe: Tiere von Harlan (n=16).
Der zeitliche Ablauf der Studie ist in nachfolgender Abbildung gezeigt.
4.5.2 Implantation von Führungsrohren und Ableitelektroden
Die Implantation von Führungsrohren und Ableitelektroden erfolgte mittels
stereotaktischer Operation, wie in Kapitel 4.2 beschrieben. Alle Tiere dieser Studie
(n=36) wurden mit Chloralhydrat narkotisiert, da das Isofluran-System zu dieser Zeit
in unserem Labor noch nicht etabliert war.
Ankunft
der
Tiere
Stereotaktische
OP
SE-Induktion EEG- und
Videoüberwachung
Verhaltenstests PB-Selektion Histologie
4, 6 und 8 Wochen nach SE für 1-2,5 Wochen 24 h/Tag
1 Wo 2 Wo
13-30 Wo
nach SE
33 Wo
nach SE
40 Wo
nach SEEpileptogenese
Stereotaktische
OP
SE-Induktion EEG- und
Videoüberwachung
Verhaltenstests PB-Selektion Histologie
4, 6 und 8 Wochen nach SE für 1-2,5 Wochen 24 h/Tag
1 Wo 2 Wo
13-30 Wo
nach SE
33 Wo
nach SE
40 Wo
nach SEEpileptogenese
Abb. 7: Schematische Darstellung des zeitlichen Ablaufs der Studie 1 (PB - Phenobarbital).

Material und Mehtoden
34
Die Tiere erhielten auch in dieser Studie acht Tage lang Antibiose, beginnend zwei
Tage vor dem Engriff. Als Unterschied ist zu verzeichnen, dass die erste Tiergruppe
zuerst eine Kombination aus Gentamicin (Genta 5%, 50 mg/ml, 0,1 ml/Tier einmal
täglich s.c.) und Chloramphenicolsuccinat (112,5 mg verdünnt in 1 ml 0,9% NaCl-
Lösung, 0,1 ml/Tier, zweimal täglich i.m.) als Antibiose erhielten. Nach vermehrt
auftretenden Entzündungen und Verlusten der Aufsätze wurde ein Resistenztest
durchgeführt und die Antibiose wurde auf Marbofloxacin umgestellt. Während der
zweiwöchigen Erhohlungsphase nach der Operation wurden die Ratten zweimal pro
Woche an den Umgang mit Menschen und die späteren Eingriffe gewöhnt. Eine
Ratte starb in Narkose, acht weitere verloren die Aufsätze, so dass nur 27 Tiere in
weiteren Versuchen dieser Studie verwendet wurden.
4.5.3 SE-Induktion
Zwei Wochen nach der stereotaktischen Operation wurde bei den Tieren (n=27) ein
SE durch fokale Kainsäure-Mikroinjektion induziert. Die SE-Induktion erfolgte wie in
Kapitel 4.3.2 beschrieben. Der SE wurde nicht abgebrochen, er hörte bei allen Tieren
spontan auf. Die Ratten erholten sich sehr schnell und mussten nicht mit
Babynahrung gefüttert werden. Eine Ratte starb 2 Wochen nach SE, 4 andere
verloren ihre Aufsätze nach der Mikroinjektion, so dass 22 Ratten in weiteren
Versuchen dieser Studie verwendet wurden.
4.5.4 EEG- und Videoüberwachung
Die EEG- und Videoüberwachung erfolgte wie in Kapitel 4.4 beschrieben. Die erste
Tiergruppe (n=12) wurde 4, 6 und 8 Wochen nach SE-Induktion für jeweils 7 Tage
(24 h/d) überwacht. Die zweite Tiergruppe (n=10) wurde 8 Wochen nach SE-
Induktion zwischen 11 und 18 Tagen (24 h/d) überwacht. Die Unterschiede in der
Überwachungsdauer ergaben sich daraus, dass die erste Gruppe im Falle eines
Aufsatzverlustes weiterhin videoüberwacht wurde. Die zweite Tiergruppe sollte
ursprünglich 18 Tage überwacht werden, im Falle eines Aufsatzverlustes wurde die
Überwachung jedoch abgebrochen, da in diesem Modell oft fokale Anfälle auftreten,
welche im Video leicht übersehen werden können.

Material und Mehtoden
35
Die Ratten aus der zweiten Gruppe wurden zusätzlich nach 33 Wochen im Rahmen
eines Substanzversuches videoüberwacht (siehe Kapitel 4.5.6).
4.5.5 Verhaltenstests
Um die epilepsieassoziierten Verhaltensänderungen zu beurteilen, wurden bei den
Tieren diverse Verhaltenstests durchgeführt. Der Open Field- und Elevated-Plus
Maze-Test zur Beurteilung von explorations- und angstassoziiertem Verhalten, der
Hyperexzitabilitätstest zur Beurteilung der taktilen und akustischen Übererregbarkeit
und der Morris Water Maze-Test zur Beurteilung der räumlichen Orientierung und
des Lernverhaltens. Als Kontrolltiere dienten nichtepileptische Ratten (n=7). Die
Verhaltenstests wurden bei den zwei Versuchsgruppen zu unterschiedlichen
Zeitpunkten durchgeführt, da sie nicht von Anfang an in diesem Umfang geplant
waren. Der zeitliche Ablauf ist in nachfolgender Tabelle dargestellt. Aufgrund
baulicher Umbaumaßnahmen, die im Verhaltenslabor stattfanden, war es leider nicht
möglich, die zweite Tiergruppe im Morris Water Maze-Test zu testen.
Test Zeitpunkt der
Durchführung 1. Gruppe
Zeitpunkt der
Durchführung 2. Gruppe
HET: Hyperexzitabilitätstest 13 Wochen nach SE 15 Wochen nach SE
EPM: Elevated-Plus Maze-Test 26 Wochen nach SE 17 Wochen nach SE
OF: Open Field-Test 27 Wochen nach SE 18 Wochen nach SE
MWM: Morris Water Maze-Test 30 Wochen nach SE Nicht durchgeführt
Tabelle 3: Tabellarische Darstellung des zeitlichen Ablaufs der Verhaltensbatterie in beiden Tiergruppen.
Alle Verhaltensuntersuchungen wurden in einem speziellen Verhaltenslabor
durchgeführt, in dem standardisierte Bedingungen herrschten. Das Labor bestand
aus zwei voneinander getrennten Räumen. In einem Raum wurden die eigentlichen
Versuche durchgeführt, im anderen wurden die Tiere vor dem Versuch beobachtet
und konnten zur Ruhe kommen. Dadurch konnte gewährleistet werden, dass die
Ratten während des Versuchs nicht vom Experimentator beeinflusst wurden. Alle
Ratten wurden eine Stunde vor Versuchsbeginn in den Vorbereitungsraum gebracht,
um eine stressfreie Versuchsdurchführung zu gewährleisten.

Material und Mehtoden
36
Während dieser Zeit wurden sie beobachtet, um das Auftreten von epileptischen
Anfällen auszuschließen. Ein Tier wurde nur dann im Verhaltenstest untersucht,
wenn es mindestens eine Stunde vor Versuchsbeginn anfallsfrei war. Bei Anfällen,
die während des Versuchs auftraten, wurde das Tier in den Käfig zurückgesetzt und
nach einer Stunde erneut getestet. Bei einem zweiten epileptischen Anfall wurde das
Tier vom Versuch ausgeschlossen. Um den Einfluss des Experimentators auf das
Tier zu begrenzen, befanden sich die Experimentatoren während des Versuchs im
Vorbereitungsraum. Die Datenerhebung erfolgte über das Video-Tracking-System
EthoVision (Noldus, Wageningen, Niederlande). Die einzige Ausnahme hierzu war
der Hyperexzitabilitätstest, in welchem die Ratten direkt von den Experimentatoren
beurteilt wurden.
4.5.5.1 Der Hyperexzitabilitätstest (HET)
Der HET ermöglicht es, taktile sowie akustische Übererregbarkeit von epileptischen
Ratten zu beschreiben (RICE et al. 1998). Er besteht aus vier verschiedenen, einfach
durchzuführenden Tests. Vier verschiedene Experimentatoren, die über die
Behandlung der Ratten verblindet sind, führen nacheinander alle vier Tests durch.
Die Ratten werden in ihrem Heimatkäfig getestet. Das Verhalten der Tiere wird nach
einem Score-System bewertet. Dieser Test wurde ohne Kontrollratten durchgeführt.
Die Ergebnisse wurden mit Tieren aus anderen post SE-Epilepsiemodellen und
naiven Tieren aus anderen Versuchen unseres Labors verglichen.
Score-System:
Approach-response-Test
Ein Stift wird vertikal langsam zur Schnauze der Ratte hingeführt
Bewertung:
1: keine Reaktion
2: die Ratte schnüffelt am Stift
3: die Ratte bewegt sich vom Stift weg
4: die Ratte erstarrt
5: die Ratte springt vom Stift weg
6: die Ratte attackiert den Stift

Material und Mehtoden
37
Touch-response-Test
Die Ratte wird am Rumpf vorsichtig mit dem Stift angestupst.
Beurteilung:
1: keine Reaktion
2: die Ratte dreht sich der Berührungsstelle langsam zu
3: die Ratte läuft vorwärts, von der Berührung weg
4: die Ratte erstarrt
5: die Ratte dreht sich schnell/ruckartig zur Berührungsstelle hin
6: die Ratte dreht sich zur nicht berührten Körperseite
7: die Ratte springt mit oder ohne Vokalisation weg
Finger-snap-Test
Ein Klicker wird zügig von hinten über den Rücken der Ratte geführt (mit wenigen
Zentimetern Abstand) und geklickert.
Beurteilung:
1: keine Reaktion
2: normale Reaktion (die Ratte zuckt zusammen, wackelt mit den Ohren, springt
ein wenig, erstarrt)
3: die Ratte springt dramatisch
Pick-up-Test
Es wird versucht, die Ratte anzuheben, indem man sie um den Rumpf fasst.
Beurteilung:
1: sehr einfach
2: einfach, aber mit Vokalisation
3: etwas schwierig; die Ratte richtet sich auf und dreht sich zur Hand
4: die Ratte meidet die Hand und versucht zu entkommen
5: die Ratte springt aus dem Käfig
6: die Ratte attackiert die Hand
4.5.5.2 Der Elevated-Plus Maze-Test (EPM)
Im EPM werden explorations- und angstassoziiertes Verhalten untersucht. Das
verwendete EPM hatte eine Höhe von 86 cm und bestand aus vier plusförmig
angeordneten, schwarzen Armen (siehe Abbildung 8).

Material und Mehtoden
38
Zwei Arme waren offen (Länge 50 cm, Breite 14 cm), zwei geschlossen (Länge 50
cm, Breite 14 cm, Höhe 31 cm). Die offenen Arme hatten eine 0,5 cm hohe
Plexiglaskante, die das Abrutschen der Tiere verhindern sollte. An den Kreuzungen
der Arme befand sich das Zentrum (Länge 14 cm, Breite 14 cm). Der
Versuchsdurchlauf dauerte 5 Minuten pro Tier. Die Ratten wurden mit dem Kopf
immer in Richtung desselben geschlossenen Arms im Zentrum eingesetzt. Bewertet
wurden: Zahl der Eintritte in die einzelnen Kompartimente (offene Arme,
geschlossene Arme, Zentrum), Aufenthaltsdauer in den einzelnen Kompartimenten
und zurückgelegte Strecke. Diese Parameter wurden durch das Video Tracking-
System von EthoVision erfasst und quantifiziert. Aufrichten, Putzen und Headdips
(das Herauslehnen über den Rand der offenen Arme) wurden manuell gewertet,
indem eine Strichliste dieser Verhaltensweisen vom Experimentator erstellt wurde.
Diese Verhaltensweisen konnten nicht durch das Video Tracking-System erfasst
werden, es sind jedoch wichtige Verhaltensparameter. Bei epileptischen Ratten kam
es oft zu Absprüngen vom EPM, in solchen Fällen wurden die Tiere schnellstmöglich
auf das EPM zurückgesetzt. Bei zwei Absprüngen wurde das Tier vom Versuch
ausgeschlossen. Nach jedem Versuchsdurchlauf wurde das EPM mit 0,1%iger
Essigsäure gereinigt, um zu vermeiden, dass die Tiere durch Geruchsspuren der
davor getesteten Ratte abgelenkt wurden.
Abb. 8: Schematischer Aufbau des Elevated Plus Maze (modifiziert nach Verhaltens-SOP des Instituts).

Material und Mehtoden
39
4.5.5.3 Der Open Field-Test (OF)
Der OF-Test ist, ähnlich wie der EPM-Test, ein Test, in welchem explorations- und
angstassoziiertes Verhalten untersucht werden. Das verwendete OF hatte eine runde
Form, einen Randhöhe von 25 cm bei einem Innendurchmesser von 80 cm und war
beige. Es wurde in drei Zonen mit einem Radius von jeweils 13-13,5 cm unterteilt.
Die drei Zonen waren entsprechend von innen nach außen: Zentrum, innerer Ring
und äußerer Ring (siehe Abbildung 9). Der Versuchsdurchlauf dauerte 5 Minuten pro
Tier. Die Ratten wurden im Zentrum eingesetzt. Bewertet wurden: Zahl der Eintritte in
die einzelnen Kompartimente (innerer Ring, äußerer Ring, Zentrum),
Aufenthaltsdauer in den einzelnen Kompartimenten und zurückgelegte Strecke.
Diese Parameter wurden mittels EthoVision erfasst. Aufrichten und Putzen wurden
manuell gewertet. Diese Verhaltensweisen konnten nicht durch das Video Tracking-
System erfasst werden, es sind jedoch wichtige Verhaltensparameter. Nach jedem
Versuchsdurchlauf wurde das OF mit 0,1%iger Essigsäure gereinigt, um zu
vermeiden, dass die Tiere durch Geruchsspuren der davor getesteten Ratte
abgelenkt wurden.
4.5.5.4 Der Morris Water Maze-Test (MWM)
Im MWM werden räumliche Orientierung und Lernverhalten untersucht (MORRIS
1984). In diesem Test lernen die Versuchstiere, eine unter der Wasseroberfläche
versteckte Plattform anhand von visuellen Anhaltspunkten am Beckenrand zu finden.
Das verwendete MWM war ein schwarzes, rundes Wasserbecken mit einer Höhe
von 60 cm und einem Durchmesser von 150 cm. Die Plattform war ebenfalls
schwarz, hatte Abmessungen von 10 cm x 10 cm und befand sich 1,5 cm unter der
Wasseroberfläche.
Äusserer Ring
Innerer Ring
Zentrum
Abb. 9: Schematischer Aufbau des Open field.

Material und Mehtoden
40
Das MWM war in 4 Quadranten unterteilt, die mit den Himmelsrichtungen
übereinstimmten: Nord-Ost, Nord-West, Süd-Ost und Süd-West. Norden (N), Süden
(S), Osten (O) und Westen (W) waren als Einsatzpunkte der Versuchstiere markiert.
Am Beckenrand war ein Spielzeuggummihuhn als visueller Orientierungspunkt
angebracht (siehe Abbildung 10). Das Becken wurde bis auf eine Höhe von 27 cm
mit Wasser befüllt, die Wassertemperatur betrug 19 ± 1 °C. Die Wassertemperatur
wurde bewusst so niedrig gewählt, um die Ratten zum Auffinden der Plattform zu
motivieren. Durch die niedrige Wassertemperatur ist in diesem Test das Auffinden
der Plattform eine Belohnung. Durch das Auffinden der Plattform wird ein negativer,
als unangenehm empfundener Reiz (kaltes Wasser) weggenommen. Der Versuch
war in 5 Aquisitionstage gegliedert, denen ein Habituationstag voranging. Am
Habituationstag wurde jede Ratte für 60 s in das Becken (ohne Plattform) gesetzt,
um Tiere mit Schwimmdefiziten oder deutlicher Quadrantenpräferenz auszusortieren.
Während der Aquisitionstage hatte jede Ratte 4 Schwimmproben je 60 s. Zwischen
den Proben hatten die Tiere 60 s Ruhepause. Die Lage der Plattform und des
visuellen Anhaltspunktes (Huhn) war konstant, die Ratte wurde bei jeder
Schwimmprobe an unterschiedlichen Stellen (N, W, S, O) ins Wasser gesetzt. Wenn
die Ratte die Plattform gefunden hatte, musste sie 3 s darauf verbleiben. Im Falle
des Nichtauffindens wurde die Ratte nach 60 s vom Experimentator zur Plattform
hingeleitet. Die letzte Schwimmprobe des letzten Aquisitionstages (Probe trial) wurde
ohne Plattform durchgeführt und diente der Überprüfung des Kurzzeitgedächtnisses
und der Abfrage des Erlernten. Tiere, die gelernt hatten die Plattform zu finden,
schwammen in diesem Testdurchlauf wiederholt über der ehemaligen
Plattformposition. Bewertet wurden: Zeit zum Auffinden der Plattform und Zahl der
Kreuzungen über der Plattform (bei dem Probe trial).
Abb. 10: Schematische Darstellung des Morris Water Maze (modifiziert nach Verhaltens-SOP des Instituts).

Material und Mehtoden
41
4.5.6 Phenobarbital-Selektion
Dreiunddreißig Wochen nach SE-Induktion wurde bei 8 Ratten ein Phenobarbital-
Behandlungsversuch gestartet. Diese Untersuchung wurde vorgenommen, um das
Auftreten von Phenobarbital-Respondern und -Nonrespondern in diesem
Epilepsiemodell zu untersuchen. Responder sind Ratten, die auf eine Behandlung
mit einem Antiepileptikum mit einer Reduktion der Anfallsfrequenz um mindestens
50% reagieren. Nonresponder sind solche Tiere, bei denen es unter Behandlung zu
keiner 50%igen Reduktion der Anfallsfrequenz kommt. Für das basolaterale
Amygdala Stimulations-Modell (elektrisches post SE-Epilepsiemodell) konnte in
unserem Labor bereits das Auftreten von Phenobarbital-Respondern und
-Nonrespondern gezeigt werden (BRANDT et al. 2004).
Phenobarbital ist ein klinisch zugelassenes, früher häufig verwendetes
Antikonvulsivum. Es kann auch bei Ratten angewendet werden. Die
Plasmahalbwertszeit beträgt bei weiblichen Sprague Dawley-Ratten 16.9 ± 1,43
Stunden (BRANDT et al. 2004).
Das Dosierungsprotokoll wurde ebenfalls in unserem Labor etabliert (BRANDT et al.
2004). Die therapeutische Plasmakonzentration von Phenobarbital liegt zwischen 10-
40 µg/ml (BRANDT et al. 2004, LÖSCHER 2007). Um diese zu erreichen, wurde den
Ratten erst ein Bolus von 25 mg/kg i.p. verabreicht, 10 Stunden später erhielten sie
15 mg/kg i.p.. Danach wurde Phenobarbital in einer Dosis von 15 mg/kg zweimal
täglich im 10-14 h Interwall i.p. verabreicht. Das Phenobarbital wurde in destilliertem
Wasser gelöst, das Injektionsvolumen betrug 3 ml/kg. Der Versuchsablauf war
folgender:
4 Tage Kontrollphase: Das Auftreten von spontanen Anfällen wird bei den Tieren
untersucht
4 Tage Behandlungsphase: die Tiere werden, wie oben beschrieben, behandelt
4 Tage Auswaschphase: die Tiere werden nicht behandelt, das Auftreten von
spontanen Anfällen wird untersucht

Material und Mehtoden
42
Während des gesamten Versuchs wurden die Tiere kontinuierlich (24 h/d)
videoüberwacht, um das Auftreten von spontanen Anfällen zu detektieren. Eine
gleichzeitige EEG-Überwachung war nicht möglich, da die Tiere keine EEG-Aufsätze
mehr hatten. Die Videos wurden visuell ausgewertet.
Um die Konzentration von Phenobarbital im Plasma zu bestimmen, wurde 14 h nach
der letzten Injektion eine retrobulbäre Blutentnahme vorgenommen (unter 2%iger
Tetracain-Lokalanästhesie). Die Blutprobe (~0,5 ml/Ratte) wurde in einem
Eppendorfgefäß (mit 10 µl Ethylendiamintetraacetat-Lösung 5 mmol/ml Vollblut zur
Gerinnungshemmung) aufgefangen. Die Proben wurden anschließend zur
Plasmagewinnung 2,5 min bei 12000 Umdrehungen/Minute zentrifugiert.
Die Plasmaproben wurden mittels Hochleistungsflüßigkeitschromatographie (high
performance liquid chromatography, HPLC), analog POTSCHKA und LÖSCHER
(2001), analysiert. Dieses Verfahren ermöglicht die Quantifizierung der analisierten
Substanzen anhand eines bekannten Standards.
Um die phenobarbital-assozierten Nebenwirkungen zu erfassen, wurde während der
Kontrollphase und während der Behandlungsphase (60 min nach Behandlung) ein
verblindetes Ataxie-Scoring vorgenommen. Dafür wurden die Ratten nacheinander
von einem Experimentator im Open field zwei Minuten lang beobachtet. Die
Beurteilung war wie folgt:
Hypolokomotion/Sedation
Score Verhalten
1 Tendenz zu verminderter Lokomotion
2 Reduzierte Lokomotion mit häufigen Ruhepausen
3 Keine Vorwärtsbewegung
Hyperlokomotion
Score Verhalten
1 Tendenz zu erhöhter Lokomotion
2 Deutlich erhöhte Lokomotion
3 Stark erhöhte Lokomotion

Material und Mehtoden
43
Ataxie
Score Verhalten
1 Leichte Ataxie
2 Deutliche Ataxie
3 Dauerhafter Verlust der Stellreflexe, mit Versuch der Vorwärtsbewegung
Tabelle 4: Verwendeter Hypolokomotion-, Hyperlokomotion- und Ataxie-Score. Ein Score=0 bedeutete keine Veränderung des Verhaltens.
4.5.7 Tötung der Tiere und Histologie
Vierzig Wochen nach SE-Induktion wurden die Tiere getötet und ihre Gehirne
entnommen und histologisch untersucht.
4.5.7.1 Transkardiale Perfusion
Die Ratten aus der ersten Gruppe wurden durch transkardiale Perfusion getötet.
Zuerst wurden sie mit Chloralhydrat tief narkotisiert (bis zum Aussetzen der Atmung,
nach Wirkung dosiert). Anschließend wurden die Bauchdecke mithilfe einer Schere
eröffnet. Die Brusthöhle wurde ebenfalls eröffnet und das Zwerchfell durchtrennt.
Das Herz wurde freigelegt und eine Kanüle, die über einen Schlauch mit der
Perfusionspumpe verbunden war, wurde durch die Herzspitze in die Aorta
geschoben. Der rechte Vorhof wurde aufgeschnitten, um den Blutabfluss zu
ermöglichen. Zuerst wurde das Blut mit phosphatgepufferter Saline aus dem Gewebe
gespült (80-100 ml/Ratte), danach wurde das Gewebe fixiert. Als Fixans diente
4%iges Paraformaldehyd (250 ml/Ratte). Nach Fixierung wurden die Gehirne
entnommen, in 10%ige Saccharoselösung überführt und für 24 Stunden bei 7°C
gelagert. Nach Ablauf von 24 Stunden wurden die Gehirne in 30%ige
Saccharoselösung überführt und verblieben bis zur weiteren Aufarbeitung bei 7°C.
Die Lagerung in Saccharoselösung diente als Gefrierschutz vor dem Schneiden an
einem Gefriermikrotom.

Material und Mehtoden
44
4.5.7.1.1 Histologie nach transkardialer Perfusion
Die entnommenen Gehirne wurden am Gefriermikrotom, im Bereich von 2 mm bis 7
mm posterior zu Bregma, in 40 µm dicke Schnitte geschnitten. Von jedem Gehirn
wurden 4 Schnittserien angefertigt, 3 davon wurden im flüssigen Gefriermedium bei
-20°C eingefroren. Eine Serie wurde zur Elektroden- und Führungsrohrlokalisation
mit Thionin-Färbung angefärbt.
4.5.7.2 Dekapitation
Die Tiere der zweiten Gruppe wurden dekapitiert. Dafür wurden die Ratten mittels
CO2 (5%) tief narkotisiert und mit einer Nagerguillotine dekapitiert. Anschließend
wurden die Gehirne entnommen und in Behälter aus Aluminiumfolie überführt. Die
Gehirne wurden mit Gefrierschutzmedium bedeckt. Die Aluminiumbehälter wurden
für einige Minuten in einen Behälter mit 2-Methyl-Butan gestellt, das 2-Methyl-Butan
wiederum wurde durch flüssigen Stickstoff auf -80°C gekühlt. Die so eingefrorenen
Gehirne wurden bis zur weiteren Aufarbeitung bei -20°C gelagert.
4.5.7.2.1 Histologie nach Dekapitation
Die Gehirne der dekapitierten Ratten wurden am Kryostaten in 14 µm dicke Schnitte
geschnitten. Die Schnittebenen waren 2 mm, 3 mm, 4 mm, 5 mm und 6 mm posterior
zu Bregma. Pro Schnittebene wurden ca. 10 Schnitte angefertigt. Eine Serie wurde
zur Beurteilung der Neurodegeneration mittels Thionin-Färbung angefärbt. Die
Neurodegeneration wurde in den Ebenen 3 mm und 4 mm posterior zu Bregma
ausgewertet. Die späteren Sektionen waren ungeeignet zur
Neurodegenerationsauswertung, da die Einstichkanäle von Elektrode und
Führungsrohr zu Artefakten führten. Das Ausmaß der Neurodegeneration wurde mit
einem Score-System bewertet. Das Score-System ist nachfolgend beschrieben.
0: keine Neurodegeneration
1: Veränderungen der Struktur, ohne sichtbaren Neuronenverlust
2: 20% bis 50% Neuronenverlust
3: über 50% Neuronenverlust

Material und Mehtoden
45
Im Hilus wurde Neurodegeneration nur als vorhanden oder nichtvorhanden
beschrieben, da es aufgrund der Schnittdicke schwierig war, das Score-System in
dieser Region anzuwenden. Die Score-Daten wurden für beide Schnittebenen als
Mittelwert zusammengefasst.
4.6 Studie 2: PTZ-Schwellenversuche im Pilokarpin- und fokalen Kainsäure-Modell
4.6.1 Studiendesign
Die Versuche dieser Studie wurden an zwei Rattenmodellen für
Temporallappenepilepsie durchgeführt, dem systemischen Pilokarpin-Modell und
dem fokalen Kainsäure-Modell. Die Studie wurde für das jeweilige Modell separat
ausgewertet. Im Pilokarpin-Modell wurde der Versuch in drei Tiergruppen
durchgeführt, diese wurden in der Auswertung zusammengefasst. In der Pilokarpin-
Studie wurde zusätzlich zu den eigentlichen Schwellenversuchen noch die Latenzzeit
zwischen SE und dem Auftreten von ersten spontanen Anfällen bestimmt. Dafür
wurden die Tiere direkt nach SE mittels EEG und Video bis zum Auftreten des ersten
Anfalls überwacht. Für diese Studie wurden 7 Tiere ohne PTZ-Schwellenbestimmung
als Kontrollen und die SE-Tiere aus der zweiten und dritten PTZ-Gruppe verwendet.
Da der SE bei den Pilokarpin-Tieren mittels Diazepam unterbrochen wurde, wurde in
einem separaten Vorversuch der Einfluss von Diazepam auf die PTZ-Schwelle
untersucht. Der zeitliche Ablauf der Studie ist in Abbildung 11 gezeigt. Eine Übersicht
über die einzelnen Tiergruppen der Studie 2 ist in Tabelle 5 aufgeführt.
Es ergaben sich folgende Behandlungsgruppen:
SE-Tiere - Tiere mit modellabhängig induziertem SE (Kainsäure oder Pilokarpin)
Kontrollen - Tiere mit sham SE
Tiere ohne SE – Tiere bei denen es trotz SE-Induktion nich zum SE-Eintritt kam

Material und Mehtoden
46
Studie Tierzahl Ableitelektroden PTZ-Schwelle
PTZ-Schwelle im
Pilokarpin-Modell
1. Gruppe
SE-Tiere n=11
Kontrollen n=10
Tiere ohne SE n=6
nein 2, 4, 6, 9, 14, 22 Tage
post SE
PTZ-Schwelle im
Pilokarpin-Modell
2. Gruppe
SE-Tiere n=2
Kontrollen n=3
Tiere ohne SE n=2
ja 2, 6, 13-14 Tage post
SE
PTZ-Schwelle im
Pilokarpin-Modell
3. Gruppe
SE-Tiere n=2
Kontrollen n=6
Tiere ohne SE n=2
ja 2, 6, 14 Tage post SE
Latenzzeitbestimmung
nach Pilokarpin-
induziertem SE
SE-Tiere n=7#
SE+PTZ-Tiere n=4* ja
2, 6, 14 Tage post SE
(nur Tiere der
SE+PTZ-Gruppe)
Diazepam-Vorversuch Tiere n=8 nein
2 Tage nach
Diazepam-Gabe
PTZ-Schwelle im
fokalen Kainsäure-
Modell
SE-Tiere n=20
Kontrollen n=15
Tiere ohne SE n=1
ja 2, 4, 6, 9, 14, 22 Tage
post SE
Tabelle 5: Übersicht über die Tiergruppen in Studie 2. *SE-Tiere aus dem 2. und 3. PTZ-Schwellenversuch #
nur sechs Tiere im Versuch verwendet (1 Ratte ohne EEG-Ableitung)
4.6.2 Implantation von Führungsrohren und Ableitelektroden
Wie in Tabelle 5 dargestellt, erfolgte bei allen Tieren mit Ausnahme der ersten
Pilokarpin-Gruppe eine Implantation von Ableitelektroden (Pilokarpin-Gruppe) oder
von Ableitelektroden und Führungsrohren (Kainsäure-Gruppe). Die stereotaktische
Operation verlief wie im Kapitel 4.2 beschrieben.
Tage nach
SE
Ankunft
der Tiere
SE-Induktion-5 +2
PTZ-Schwellenbestimmung
+4 +6 +9 +14 +22
Abb. 11: Schematische Darstellung des zeitlichen Ablaufs der Studie 2.

Material und Mehtoden
47
Als Änderung sind zwei zusätzliche Halteschrauben, die bei Tieren der Kainsäure-
Gruppe am Schädel angebracht wurden, zu vermerken. Die Halteschrauben sollten
helfen, entzündungsbedingte Verluste der EEG-Aufsätze zu vermeiden, da diese
trotz Antibioseumstellung häufig auftraten. Des Weiteren wurden in der dritten
Pilokarpin-Gruppe zusätzlich zu einer EEG-Ableitelektrode im Gyrus Dentatus
Kortexschrauben zur Elektrocorticogramm-Ableitung (ECoG-Ableitung) implantiert
(Koordinaten AP, -2,2 mm und L, +/- 4,8 mm relativ zu Bregma). So konnte während
der Versuche entweder hippokampales EEG oder kortikales ECoG abgeleitet
werden. Eine gleichzeitige Ableitung war technisch nicht möglich. Da jedoch in den
ersten Versuchen mit EEG ein PTZ-induzierter Myoklonus nicht sichtbar war, sollte
die kortikale Ableitung in dieser Hinsicht untersucht werden. Auch in dieser Studie
erhielten die Tiere Marbofloxacin zur postoperativen Antibiose und hatten zwei bis
drei Wochen Zeit, um sich von dem operativen Eingriff zu erholen. Während dieser
Phase wurden die Tiere mindestens zweimal wöchentlich an den Umgang mit
Menschen und spätere Eingriffe gewöhnt.
4.6.3 SE-Induktion
Zwei bis drei Wochen nach stereotaktischer Operation wurde bei den Tieren ein SE
induziert. Modellabhängig erfolgte das entweder durch systemische Pilokarpin-Gabe
oder durch fokale Kainsäure-Gabe.
4.6.3.1 SE-Induktion im Pilokarpin-Modell
Die SE-Induktion im Pilokarpin-Modell erfolgte wie im Kapitel 4.3.1 beschrieben. Die
SE-Dauer betrug 90 Minuten. Der SE wurde pharmakologisch mittels Diazepam
(Faustan, 10 mg/kg in 2ml/kg i.p.) unterbrochen. Im Falle von weiteren Krämpfen
konnte die Diazepam-Injektion bis zu zweimal im zehnminütigen Abstand wiederholt
werden. Die Kontrollen erhielten die gleiche Behandlung, lediglich das Pilokarpin
wurde durch isotone Natriumchloridlösung ersetzt (sham SE).
Der Versuch wurde an 41 Ratten durchgeführt. Nur eine Ratte starb während der SE-
Induktion. Tiere, die trotz fünfmaliger Pilokarpin-Gabe keinen SE hatten, wurden vom
Versuch ausgeschlossen. Bei den Ratten, bei denen eine Ableitelektrode implantiert
wurde, wurde der Versuch unter EEG-Kontrolle durchgeführt

Material und Mehtoden
48
4.6.3.2 SE-Induktion im fokalen Kainsäure-Modell
Die SE-Induktion durch fokale Kainsäure-Gabe erfolgte wie im Kapitel 4.3.2
beschrieben. Die Kontrollen erhielten isotone Natriumchloridlösung anstelle von
Kainsäure (sham SE). Um einen Einfluss des ovariellen Zyklustandes auf die SE-
Induktion ausschließen zu können, wurde bei allen Tieren direkt vor Mikroinjektion
ein Vaginalabstrich zur Zyklusbestimmung durchgeführt (siehe Kapitel 4.6.7).
4.6.4 PTZ-Schwellenbestimmung
Die Bestimmung der individuellen Krampfschwelle erfolgte im intravenösen PTZ-
Schwellentest, beschrieben bei LÖSCHER (2009 und 2011). Den Ratten wurde eine
Kanüle in die laterale Schwanzvene eingeführt und mittels Klebeband fixiert. Die
Kanüle war über einen Polyethylenschlauch mit einer Infusionspumpe verbunden.
Während des ganzen Versuchs waren die Ratten frei beweglich. Die PTZ-Infusion
(PTZ-Konzentration 0,8%, gelöst in isotoner Natriumchloridlösung) erfolgte mit einer
konstanten Flussrate von 1 ml/min Die Infusion wurde beim Auftreten des ersten
Myoklonus gestoppt. Die individuelle PTZ-Krampfschwelle wurde anhand der PTZ-
Dosis, die zum Auslösen des ersten Myoklonus nötig war, des Körpergewichts der
Ratte und der PTZ-Konzentration und -Flussrate berechnet und in mg/kg
ausgedrückt. Die Schwellenbestimmung erfolgte bei allen Tieren 5 Tage vor SE-
Induktion (Kontrollschwelle), sowie 2, 4, 6, 9, 14 und 22 Tage danach. Aufgrund der
Ergebnisse aus dem ersten Versuch, wurden in der zweiten und dritten Pilokarpin-
Gruppe keine Schwellenbestimmmungen nach 4, 9 und 22 Tagen durchgeführt. Drei
Tiere starben infolge des PTZ-Schwellentests. Die Daten aller Gruppen waren sehr
konsistent und wurden kombiniert. In der Kainsäure-Gruppe wurden alle 7
Schwellenbestimmungen durchgeführt, um beide Modelle zu vergleichen. Alle
Versuche wurden zur gleichen Tageszeit (8:00-14:00 Uhr) durchgeführt. Die Tiere
wurden eine Stunde vor Versuchsbeginn in den Versuchraum gebracht und
beobachtet, um das eventuelle Auftreten von spontanen Anfällen zu detektieren. Im
Falle von spontanen Anfällen wurde die Schwellenbestimmung erst eine Stunde
nach dem Anfall durchgeführt. Bei allen Tieren, die EEG-Ableitelektroden hatten,
wurde der Versuch unter EEG-Kontrolle durchgeführt.

Material und Mehtoden
49
Bei den Tieren aus der dritten Pilokarpin-Gruppe, die sowohl hippokampale, als auch
kortikale Ableitelektroden hatten, wurde der Versuch mit kortikaler ECoG-Ableitung
durchgeführt. Obwohl die Infusion nach dem ersten Myoklonus gestoppt wurde,
konnten weitere Endpunkte beobachtet werden. Weitere Endpunkte der Infusion
waren: Klonus, Klonus mit Verlust der Stellreflexe und Vorderbeintonus. Das
Auftreten von weiteren Endpunkten wurde mit einem Score-System bewertet und
notiert. Da epileptische Ratten nach Ende der Infusion noch Verhaltensänderungen
zeigten, wurden diese ebenfalls mit einem Score-System bewertet. Die beiden
Score-Systeme sind in nachfolgender Tabelle dargestellt.
Score I „Weitere Endpunkte nach PTZ-Infusion“
0 Keine weiteren Endpunkte
1 Klonus
2 Klonus mit Verlust der Stellreflexe
3 Klonus mit Verlust der Stellreflexe, Running and bouncing
4 Klonus mit Verlust der Stellreflexe, Running and bouncing, Vorderbeintonus
Score II „Verhalten nach PTZ-Schwellenbestimmung“
0 Keine Veränderungen
1 Myoklonien, verminderte Ansprechbarkeit, Stereotypien mit einer Dauer < 2 min
2 Myoklonien, verminderte Ansprechbarkeit, Stereotypien mit einer Dauer > 2 min
3 Generalisierte Anfälle (Typ IV oder V)
4 Myoklonien und generalisierte Anfälle
Tabelle 6: Score-Systeme zur Beurteilung der Verhaltensänderungen nach PTZ-Schwellentest.
4.6.5 Bestimmung der Latenzzeit im Pilokarpin-Modell
Um die Latenzzeit (Zeit zwischen SE und dem ersten spontanen Anfall) im
fraktionierten Pilokarpin-Modell zu ermitteln, wurden 6 SE-Tiere ohne PTZ-
Schwellenbestimmung und 4 SE+PTZ-Tiere (aus dem 2. und 3. PTZ-
Schwellenversuch) verwendet. Die Tiere ohne PTZ-Schwellenbestimmung waren
notwendig, um den Einfluss von PTZ auf die Latenzzeit auszuschliessen. Das
Studiendesign war wie folgt: Die Tiere wurden ab der SE-Induktion kontinuierlich bis
zu 4 Wochen EEG- und Video-überwacht, um das Auftreten von spontanen Anfällen
zu registrieren. Die SE-Induktion verlief wie im Kapitel 4.3.1 beschrieben.

Material und Mehtoden
50
Die Überwachung erfolgte wie in Kapitel 4.4 beschrieben. Die Auswertung der Daten
erfolgte visuell. Die Latenzzeit wurde in Tagen bis zum Auftreten des ersten
spontanen Anfalls ausgedrückt.
4.6.6 Diazepam-Vorversuch im Pilokarpin-Modell
Dieser Vorversuch wurde durchgeführt, um zu untersuchen, ob der SE-Abbruch
mittels Diazepam einen Einfluss auf die PTZ-Schwelle 48 h nach SE hat. Zu diesem
Zweck wurde bei 8 Ratten eine PTZ-Kontrollschwelle bestimmt. Drei Tage später
erhielten die Ratten Diazepam in der bei einem SE-Abbruch üblichen Dosierung (3 x
10 mg/kg im Abstand von 10 Minuten). Die PTZ-Schwelle wurde erneut nach 48 h
bestimmt und mittels Student`s t-Test mit der Kontrollschwelle verglichen.
4.6.7 Bestimmung des ovariellen Zyklusstandes
Um den Einfluss des Zyklusstandes auf die individuelle PTZ-Krampfschwelle
beschreiben zu können, wurden in der Kainsäure-Gruppe Vaginalabstriche zur
Zyklusbestimmmung entnommen. Zu diesem Zweck wurde bei jedem Tier
unmittelbar vor der Schwellenbestimmung ein Vaginalabstrich aus dem dorsalen
Scheidendach mittels Impföse entnommen. Dieser wurde auf einem Objektträger in
isotoner Natriumchloridlösung ausgestrichen und getrocknet. Die Abstriche wurden
mit der Lösung nach SHORR (1940) gefärbt. Die Auswertung erfolgte am
Lichtmikroskop. Anhand des Zellbildes im Vaginalausstrich konnten folgende
Zyklusstadien und -zwischenstadien unterschieden werden: Proöstrus, Proöstrus-
Östrus, Östrus, Östrus-Metöstrus, Metöstrus, Metöstrus-Diöstrus, Diöstrus, Diöstrus-
Proöstrus.
4.7 Studie 3: Infusionsversuche mit PTZ im Pilokarpin- und fokalen Kainsäure-Modell
Ziel der Studie 3 war die Behandlung von Ratten nach SE mit subkonvulsiven PTZ-
Dosen. Die Tiere sollten nach Insult (SE) präventiv behandelt werden. Da PTZ ein
prokonvulsiver Stoff ist, musste zuerst ein geeignetes Behandlungsprotokoll erstellt
werden, um die PTZ-Dosis im subkonvulsiven Bereich zu halten.

Material und Mehtoden
51
4.7.1 Pharmakokinetische Vorversuche
Die Vorversuche setzten sich aus der Halbwertszeitbestimmung von PTZ und der
Ermittlung von konvulsiven PTZ-Plasmakonzentrationen zusammen. Anhand der
Daten aus diesen Versuchen konnte ein Behandlungsprotokoll erstellt werden.
Obwohl PTZ laut Literaturangaben nicht im Gewebe kumuliert, bestand doch die
Gefahr, dass es bei einer chronischen Behandlung über 48 h zu einer Kumulation
von PTZ kommt, was wiederum in Krämpfen bei den behandelten Tieren resultieren
würde.
4.7.1.1 PTZ-Halbwertszeitbestimmung
Die PTZ-Halbwertszeit beträgt laut Literaturangaben 2-3 Stunden (ESPLIN und
WOODBURY 1956 VOHLAND und ZUFELDE 1976 RAMZAN und LEVY 1985). Da
es keine Angaben zur Halbwertszeit bei weiblichen Sprague Dawley-Ratten gibt,
haben wir diese bestimmt. Die PTZ-Halbwertszeitbestimmung wurde an einer
Gruppe von 4 Ratten durchgeführt. Es wurden 2 naive Tiere und 2 Tiere 24 h nach
pilokarpin-induziertem SE verwendet. Die Tiere erhielten eine einmalige PTZ-
Injektion (12 mg/kg gelöst in isotoner Natriumchloridlösung) über die laterale
Schwanzvene. Zur PTZ-Plasmakonzentrationsbestimmung wurde den Tieren 15 min,
1 h, 2 h und 4 h nach Applikation retrobulbär Blut entnommen (siehe Kapitel 4.5.6).
Die PTZ-Konzentration wurde mittels HPLC bestimmt. Die Halbwertszeiten wurden
mit dem PK Solutions 2.0 Programm (Summit Research Services, Montrose, CO,
USA) berechnet.
4.7.1.2 Etablierung des Infusionsprotokolls
Nach Bestimmung der Halbwertszeit sollte die konvulsive PTZ-Plasmakonzentration
bestimmt werden. Zu diesem Zweck wurde bei 2 naiven Ratten und bei 2 Ratten 24 h
nach pilokarpin-induziertem SE direkt nach der PTZ-Schwellenbestimmung (siehe
Kapitel 4.6.4) retrobulbär Blut entnommen. Die PTZ-Plasmakonzentration wurde
mittels HPLC gemessen.
Basierend auf den Ergebnissen dieser Vorversuche haben wir eine subkonvulsive
Zielkonzentration von 10 µl/ml festgelegt und ein Behandlungsprotokoll erstellt.

Material und Mehtoden
52
Das erste Behandlungsprotokoll erstellt zur Erhaltung der 10 µg/ml
Plasmakonzentration, sah einen Bolus von 10 mg/kg und eine Infusion von 15
µg/kg/min zur Aufrechterhaltung vor. Es wurde mithilfe des PK Solution Programms
erstellt. Das Behandlungsprotokoll wurde an naiven Tieren getestet. Die verabreichte
PTZ-Dosis blieb im subkonvulsiven Bereich, sie reichte jedoch nicht aus, um den
Zielplasmaspiegel von 10 µg/ml zu erreichen. Anhand der Plasmawerte der
behandelten Tiere wurde ein zweites Behandlungsprotokoll erstellt. Das zweite
Behandlungsprotokoll zur Erhaltung der 10 µg/ml Plasmakonzentration sah einen
Bolus von 10 mg/kg und eine Infusion von 60 µg/kg/min zur Aufrechterhaltung vor.
Es wurde ebenfalls mithilfe des PK Solution Programms erstellt. Auch dieses
Behandlungsprotokoll wurde zuerst an naiven Tieren getestet.
4.7.2 Studiendesign PTZ-Behandlung
Die Infusionsversuche dieser Studie wurden, wie bereits in Studie 2 beschrieben, in
zwei Modellen durchgeführt, in dem systemischen Pilokarpin-Modell und in dem
fokalen Kainsäure-Modell. Es gab folgende Behandlungsgruppen:
Saline + Saline: sham SE-Induktion mit isotoner Natriumchloridlösung + Behandlung
mit isotoner Natriumchloridlösung
SE + Saline: SE-Induktion mit Pilokarpin oder Kainsäure + Behandlung mit isotoner
Natriumchloridlösung
SE + PTZ: SE-Induktion mit Pilokarpin oder Kainsäure + Behandlung mit PTZ
Die teilweise sehr kleinen Gruppengrößen haben mehrere Ursachen. In einem
Infusionsversuch konnten maximal 8 Ratten gleichzeitig behandelt werden. Diese 8
Tiere wurden auf zwei oder drei Behandlungsgruppen aufgeteilt. Die
Infusionsversuche sollten erste Hinweise über den Einfluss einer PTZ-Behandlung
auf die Epileptogenese liefern. Bei vielversprechenden Ergebnisssen wären die
Gruppen aufgefüllt worden.

Material und Mehtoden
53
Da diese Versuche jedoch eine große Belastung für die Versuchstiere darstellten,
war eine Auffüllung der Gruppen bei den gegenwärtigen Ergebnissen aus
Tierschutzgründen abzulehnen.
Die nachfolgende Tabelle zeigt die Behandlungsgruppen in den einzelnen
Versuchen.
Studie
Behandlungsgruppen Dauer der
PTZ-
Behandlung
Start der
PTZ-
Behandlung Saline+Saline SE+Saline SE+PTZ
1. Gruppe:
PTZ-Infusion im
Pilokarpin-
Modell
- - n=6 48 h Direkt nach SE-
Abbruch
2. Gruppe:
PTZ-Infusion
im Pilokarpin-
Modell
- n=4 n=4 48 h Direkt nach SE-
Abbruch
1. Gruppe:
PTZ-Infusion
im fokalen
Kainsäure-
Modell
n=2 n=2 n=5 48 h 13-16 h nach
SE-Induktion
2. Gruppe:
PTZ-Infusion
im fokalen
Kainsäure-
Modell
n=2 n=2 n=4 48 h 7 d nach SE-
Induktion
Tabelle 7: Übersicht über die Behandlungsgruppen in Studie 3.
4.7.3 Implantation von Ableitelektroden und Führungsrohren
Bei allen Tieren dieser Studie wurden EEG-Ableitelektroden implantiert. Die Tiere
aus dem fokalen Kainsäure-Modell hatten zusätzlich Führungsrohre zur Kainsäure-
Mikroinjektion. Der genaue Ablauf der stereotaktischen Operation ist in Kapitel 4.2
beschrieben. Die Tiere dieser Studie hatten zusätzliche Halteschrauben, die seitlich
am Schädel befestigt waren, um Verluste der EEG-Aufsätze zu vermeiden.

Material und Mehtoden
54
Ein Teil der Tiere wurde wie in Kapitel 4.2 beschrieben mit Chloralhydrat narkotisiert,
ein Teil mit dem Inhalationsnarkotikum Isofluran. Die Isofluran-Narkose wurde neu in
unserem Labor etabliert. Die Vorteile einer Inhalationsnarkose sind die bessere
Steuerbarkeit einer solchen und daraus resultierend eine tiefere Narkose bei
gleichzeitig weniger Narkosezwischenfällen. Da wir jedoch nur über einen Isofluran-
Verdampfer verfügen, wurde ein Teil der Tiere weiterhin mit Chloralhydrat
narkotisiert. Isofluran wurde in einer Konzentration von 3% zur Narkoseeinleitung
und 1,5% zur Narkoseaufrechterhaltung verabreicht. Auch in dieser Studie erhielten
die Tiere Marbofloxacin zur postoperativen Antibiose und hatten zwei bis drei
Wochen Zeit, um sich von dem operativen Eingriff zu erholen. Während dieser Phase
wurden sie mindestens zweimal wöchentlich an den Umgang mit Menschen und die
späteren Eingriffe gewöhnt.
4.7.4 Implantation von Kathetern in die Vena jugularis externa
Da die PTZ-Behandlung intravenös erfolgte, wurden den Ratten Dauerkatheter in die
Vena jugularis implantiert. In den verschiedenen Behandlungsgruppen erfolgte die
Implantation zu unterschiedlichen Zeitpunkten. Eine Übersicht dazu ist in
nachfolgender Tabelle dargestellt.
Studie Zeitpunkt Katheter-Operation
1. Gruppe PTZ-Infusion im Pilokarpin-Modell 2 Tage vor SE-Induktion
2. Gruppe PTZ-Infusion im Pilokarpin-Modell 2 Tage vor SE-Induktion
1. Gruppe PTZ-Infusion im fokalen Kainsäure-
Modell 2 Tage vor SE-Induktion
2. Gruppe PTZ-Infusion im fokalen Kainsäure-
Modell 5 Tage nach SE-Induktion
Tabelle 8: Übersicht über die Zeitpunkte der Katheter-Implantation in den einzelnen Tiergruppen der Studie 3.
Für die Operation wurden 50 cm lange Katheter aus Polyethylenschlauch verwendet.
Der Katheter sollte auf 3 cm in die Vene implantiert werden, so dass er im rechten
Vorhof lag. Diese Länge (3 cm) war auf dem Katheter markiert. Die Ratten wurden
mit Chloralhydrat oder Isofluran narkotisiert, je nach Verfügbarkeit des Isofluran-
Verdampfers, da die Isofluran-Narkose bevorzugt wurde.

Material und Mehtoden
55
Die Operationsfläche wurde anschließend rasiert und mit 70%igem Ethanol gereinigt.
Der Katheter wurde in die rechte V. jugularis externa implantiert und mit Garn
befestigt. Der implantierte Katheter wurde unter der Haut in den Nacken geführt, wo
er zwischen den Schulterblättern wieder austrat. Die Wunde wurde durch Knopfhefte
mit resorbierbarem Nahtmaterial verschlossen. Der Katheter wurde durch eine
Spiralfeder gefädelt, um die Ratten am Durchbeißen des Katheters zu hindern. Die
Feder war mit einem Geschirr verbunden. In diesem Geschirr verblieben die Ratten
für die Dauer der Infusion.
Die Tiere erhielten sieben Tage lang Antibiose, beginnend zwei Tage vor dem Engriff
(Marbofloxacin 3 mg/kg, 0,1 ml/Tier zweimal täglich erst s.c. nach
Katheterimplantation i.v.). Nach der Operation wurden die Ratten einzeln in
durchsichtigen Plexiglaskäfigen gehalten. Die Katheter waren über einen
beweglichen Swivel und eine spezielle Aufhängung am Käfig befestigt, was den
Ratten eine gewisse Bewegungsfreiheit gewährleistete. Die Tiere waren an eine
Infusionspumpe angeschlossen, die es ermöglichte, 8 Ratten gleichzeitig zu
infundieren. Während der Erholungsphase (2-3 Tage) vor der eigentlichen
Substanzbehandlung erhielten die Ratten heparinisierte isotone
Natriumchloridlösung (25 IE Heparin/ml) mit einer Infusionsgeschwindigkeit von 50
µl/h. Nach Ende der Dauerinfusion wurden die Katheter kurz abgeschnitten und mit
einem Stück Draht verschlossen, so dass die Tiere zurück in ihre Heimatkäfige
konnten.
4.7.5 SE-Induktion
Zwei Tage nach beziehungsweise fünf Tage vor Katheterimplantation (siehe Tabelle
8) wurde bei den Tieren ein SE induziert. Modellabhängig erfolgte das entweder
durch systemische Pilokarpin-Gabe, oder durch fokale Kainsäure-Gabe.
4.7.5.1 SE-Induktion im Pilokarpin-Modell
Die SE-Induktion erfolgte ähnlich wie im Kapitel 4.3.1 beschrieben. Die Ratten
wurden mit Lithiumchlorid und Methylscopolamin vorbehandelt. Um die Zahl der
Tiere, die nach Pilokarpin-Gabe einen SE entwickeln zu steigern, wurde das
fraktionierte Pilokarpin-Protokoll modifiziert.
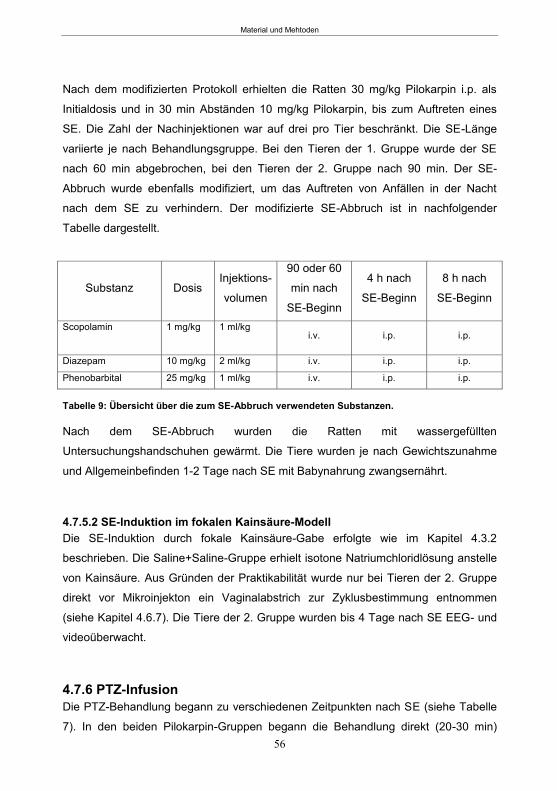
Material und Mehtoden
56
Nach dem modifizierten Protokoll erhielten die Ratten 30 mg/kg Pilokarpin i.p. als
Initialdosis und in 30 min Abständen 10 mg/kg Pilokarpin, bis zum Auftreten eines
SE. Die Zahl der Nachinjektionen war auf drei pro Tier beschränkt. Die SE-Länge
variierte je nach Behandlungsgruppe. Bei den Tieren der 1. Gruppe wurde der SE
nach 60 min abgebrochen, bei den Tieren der 2. Gruppe nach 90 min. Der SE-
Abbruch wurde ebenfalls modifiziert, um das Auftreten von Anfällen in der Nacht
nach dem SE zu verhindern. Der modifizierte SE-Abbruch ist in nachfolgender
Tabelle dargestellt.
Substanz Dosis Injektions-
volumen
90 oder 60
min nach
SE-Beginn
4 h nach
SE-Beginn
8 h nach
SE-Beginn
Scopolamin 1 mg/kg 1 ml/kg i.v. i.p. i.p.
Diazepam 10 mg/kg 2 ml/kg i.v. i.p. i.p.
Phenobarbital 25 mg/kg 1 ml/kg i.v. i.p. i.p.
Tabelle 9: Übersicht über die zum SE-Abbruch verwendeten Substanzen.
Nach dem SE-Abbruch wurden die Ratten mit wassergefüllten
Untersuchungshandschuhen gewärmt. Die Tiere wurden je nach Gewichtszunahme
und Allgemeinbefinden 1-2 Tage nach SE mit Babynahrung zwangsernährt.
4.7.5.2 SE-Induktion im fokalen Kainsäure-Modell
Die SE-Induktion durch fokale Kainsäure-Gabe erfolgte wie im Kapitel 4.3.2
beschrieben. Die Saline+Saline-Gruppe erhielt isotone Natriumchloridlösung anstelle
von Kainsäure. Aus Gründen der Praktikabilität wurde nur bei Tieren der 2. Gruppe
direkt vor Mikroinjekton ein Vaginalabstrich zur Zyklusbestimmung entnommen
(siehe Kapitel 4.6.7). Die Tiere der 2. Gruppe wurden bis 4 Tage nach SE EEG- und
videoüberwacht.
4.7.6 PTZ-Infusion
Die PTZ-Behandlung begann zu verschiedenen Zeitpunkten nach SE (siehe Tabelle
7). In den beiden Pilokarpin-Gruppen begann die Behandlung direkt (20-30 min)

Material und Mehtoden
57
nach SE-Abbruch. In der 1. Kainsäure-Gruppe begann die Behandlung 13-16 h nach
SE-Induktion, in der 2. Gruppe 7 Tage nach SE-Induktion. Eigentlich sollte die PTZ-
Behandlung in der 1. Kainsäure-Gruppe 12 h nach SE-Induktion beginnen, weil zu
diesem Zeitpunkt der kainsäure-induzierte SE bei den meisten Tieren spontan
aufhört. Der PTZ-Bolus wurde jedoch versehentlich zu spät appliziert, so dass die
Behandlung, von Tier zu Tier unterschiedlich, entweder 13 h oder 16 h nach SE-
Induktion erfolgte. Das PTZ wurde über 48 Stunden infundiert, die SE+Saline-Gruppe
erhielt isotone Natriumchloridlösung statt PTZ. Um die Zielkonzentration von 10
µg/ml Plasma zu erreichen, wurde den Ratten ein Bolus von 10 mg/kg PTZ (gelöst in
2 ml isotoner Natriumchloridlösung) i.v. verabreicht. Zur Aufrechterhaltung dieser
Plasmakonzentration bekamen die Tiere PTZ in einer Dosierung von 60 µg/kg/min
(gelöst in isotoner Natriumchloridlösung). Das PTZ wurde über eine Infusionspumpe
verabreicht. Die Infusionsrate lag bei 1 µl/min/g Ratte (da die Körpergewichte
zwischen den Tieren nicht stark schwankten, wurde das gemittelte Gewicht aller
angeschlossenen Tiere verwendet). Während der gesamten Infusionsdauer (48 h)
wurden die Tiere beobachtet, um bei eventuellen PTZ-Nebenwirkungen (Krämpfe),
die Infusion zu stoppen. Wenn die Infusion zu einzelnen Anfällen (fokal oder
generalisiert) führte, wurde sie nicht gestoppt, nur im Falle von wiederholten Anfällen,
welche nicht spontan endeten. Um eine kontinuierliche Beobachtung der Ratten über
48 Stunden zu gewährleisten, haben sich immer 2 Experimentatoren tags und nachts
abgewechselt. Nach Ende der Behandlung wurden bei den Tieren retrobulbär
Blutproben entnommen, um die PTZ-Plasmakonzentration zu bestimmen (siehe
Kapitel 4.5.6). Bei Tieren der 1. Pilokarpin-Gruppe wurden Blutproben 24 h und 48 h
nach Infusionsbeginn entnommen, bei den Tieren der weiteren Behandlungsgruppen
nur nach 48 h. Die PTZ-Konzentration wurde mittels HPLC gemessen.

Material und Mehtoden
58
4.7.7 EEG- und Videoüberwachung
Um das Auftreten von spontanen Anfällen zu detektieren, wurden die Tiere EEG- und
videoüberwacht. Die einzelnen Gruppen wurden zu unterschiedlichen Zeitpunkten
nach Behandlung überwacht (siehe Tabelle 10). Die Überwachungsanlage ist in
Kapitel 4.4 beschrieben.
Studie Zeitpunkt der Überwachung
1. Gruppe: PTZ-Infusion im Pilokarpin-Modell 4 und 7 Wochen nach SE
2. Gruppe: PTZ-Infusion im Pilokarpin-Modell 8 Wochen nach SE
1. Gruppe: PTZ-Infusion im fokalen Kainsäure-
Modell
8 und 12 Wochen nach SE
2. Gruppe: PTZ-Infusion im fokalen Kainsäure-
Modell
8 und 16 Wochen nach SE
Tabelle 10: Übersicht über die Überwachungszeitpunkte in den einzelnen Tiergruppen der Studie 3.
Die Auswertung der Überwachungsdateien erfolgte visuell.
4.8 Statistik Die statistische Auswertung der Studien erfolgte mit der GraphPad Prism Software
5.0 (La Jolla, CA, USA). Alle Werte sind, sofern nicht anders angegeben, als
Mittelwertsfehler angegeben (Mean + SEM standard error of mean). Bei einem
Vergleich von zwei Gruppen wurde entweder der Student`s t-Test (bei
parametrischen Daten), oder der Mann-Whitney-U-Test (bei nichtparametrischen
Daten) verwendet. Bei einem Vergleich von drei, oder mehr Gruppen, von denen
parametrische Daten erhoben wurden, wurde eine einfaktorielle Varianzanalyse
(ANOVA) mit Bonferroni oder Student`s t-Test als post hoc Test durchgeführt. Bei
nichtparametrischen Daten wurde entsprechend der Kruskal-Wallis-Test mit Dunn`s
oder Mann-Whitney-U-Test als post hoc Test durchgeführt. Alle statistischen Tests
wurden zweiseitig durchgeführt. Ein Singnifikanzniveau von p<0,05 wurde festgelegt.

Ergebnisse
59
5. Ergebnisse
5.1 Studie 1: Etablierung des fokalen Kainsäure-Modells
5.1.1 SE-Induktion im fokalen Kainsäure-Modell
Bei allen 27 Ratten konnte durch Kainsäure-Mikroinjektion ein SE induziert werden.
Dieser war klinisch charakterisiert durch verminderte Ansprechbarkeit, „Straub tail“
(senkrecht hochgestellter Schwanz), „Wet dog shakes“ (charakteristisches Schütteln
des Rumpfes), Verharren und Stereotypien wie Schnüffeln, Kauen und Kopfnicken.
Diese Verhaltensänderungen waren von generalisierten tonisch-klonischen Anfällen
begleitet. Da Kainsäure einen limbischen SE induziert, bei welchem es nicht
durchgehend zu generalisierter tonisch-klonischer Krampfaktivität kommt, wurde der
SE anhand des EEG-Bildes beurteilt. Vor der SE-Induktion wurde ein Basis-EEG
aufgenommen (siehe Abbildung 12 A). Nach der SE-Induktion konnten
charakteristische EEG-Veränderungen (zuerst erhöhte Frequenz und Amplitude der
Basislinie, paroxysmale Veränderungen, später hochamplitudige Veränderungen der
Basislinie (siehe Abbildung 12 B, C) beobachtet werden. Die SE-Länge und -
Schwere wurde anhand der EEG-Veränderungen (EEG-Anfälle, siehe Abbildung 12
B) bestimmt. Die ersten paroxysmalen EEG-Veränderungen traten nach etwa 10 min
auf (12,78 ± 1,19 min). Die Zahl der auszählbaren EEG-Anfälle während der 20 h
Überwachung nach SE lag im Mittel bei etwa 65 (66,8 ± 7,8). Die Dauer der
paroxysmalen EEG-Veränderungen variierte von Tier zu Tier und lag im Schnitt bei
11,2 h (3,7 - 20,5 h). Die Veränderungen der Basislinie (erhöhte Frequenz und
Amplitude, Spikes), hielten länger an. Der SE endete spontan bei allen Tieren. Die
beschriebenen EEG-Veränderungen sind in nachfolgender Abbildung dargestellt.
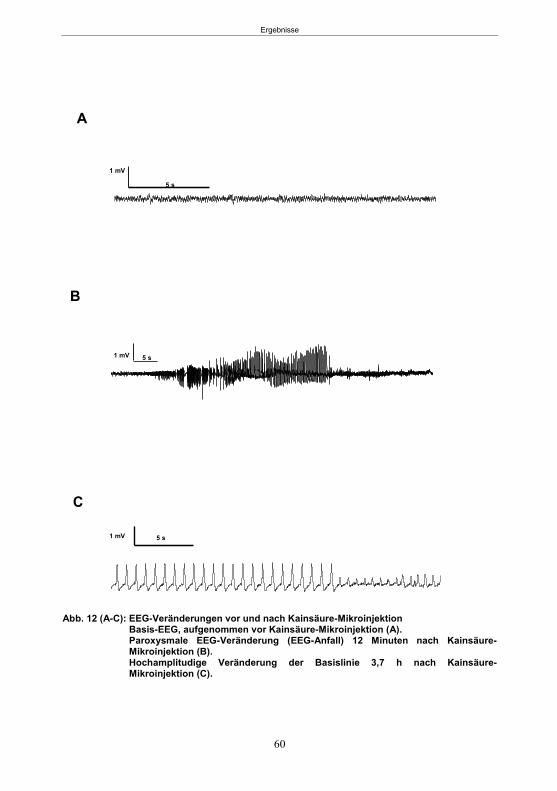
Ergebnisse
60
5 s
1 mV
5 s1 mV
5 s1 mV
A
B
C
Abb. 12 (A-C): EEG-Veränderungen vor und nach Kainsäure-Mikroinjektion Basis-EEG, aufgenommen vor Kainsäure-Mikroinjektion (A). Paroxysmale EEG-Veränderung (EEG-Anfall) 12 Minuten nach Kainsäure- Mikroinjektion (B). Hochamplitudige Veränderung der Basislinie 3,7 h nach Kainsäure- Mikroinjektion (C).

Ergebnisse
61
5.1.2 Entwicklung von spontanen, wiederkehrenden, epileptische Anfällen im fokalen Kainsäure-Modell
Die Studie wurde an zwei Tiergruppen durchgeführt. Die 1. Gruppe setzt sich aus
Tieren von Harlan (n=4) und Tieren von Charles River (n=8) zusammen. In der 2.
Gruppe wurden ausschließlich Tiere von Harlan (n=10) verwendet. Da es keinen
Unterschied in der SE-Induktion zwischen den Ratten beider Züchter gab, wurden
die Tiere in der Auswertung zusammengefasst. Die Tiere der ersten Gruppe wurden
nach 4, 6 und 8 Wochen, für einen Zeitraum von einer Woche überwacht. Die Tiere
der zweiten Gruppe wurden nach 8 Wochen für einen Zeitraum von 11-18 Tagen
überwacht. Acht Ratten dieser Gruppe wurden zusätzlich nach 33 Wochen im
Rahmen des Phenobarbital-Selektionsversuchs videoüberwacht (siehe Kapitel 4.5.6
und 5.1.4). Die Anfallsentwicklung war wie folgt: Sieben von 12 Tieren entwickelten
Anfälle während der ersten Überwachungsphase (4 Wochen nach SE), zwei weitere
Tiere hatten Anfälle während der zweiten Überwachungsphase (6 Wochen nach SE)
und ein weiteres Tier während der dritten Überwachungsphase (8 Wochen nach SE).
Insgesamt entwickelten 10 von 12 Ratten der ersten Gruppe epileptische Anfälle
(83%). In der zweiten Tiergruppe hatten 9 von 10 Tieren Anfälle während der ersten
Überwachungsphase 8 Wochen nach SE (90%). Bei einer der Ratten, die keine
Anfälle während der Überwachungsphase entwickelte, konnten später Anfälle
beobachtet werden, so dass insgesamt 20 von 22 Ratten dieser Studie, als
epileptisch bezeichnet werden können (91%).
Die meisten Anfälle waren sekundär generalisiert (Typ IV oder V, siehe Abbildung 13
A), es traten aber auch fokale Anfälle auf (Typ I-III, siehe Abbildung 13 B). Der
Anfallstyp (fokal oder generalisiert) konnte nicht im EEG, sondern nur mithilfe der
Videodateien bestimmt werden. Fokale Anfälle unterscheiden sich im EEG nicht von
generalisierten Anfällen, die paroxysmalen Veränderungen sind unabhängig vom
Anfallstyp. Bei den Tieren der ersten Gruppe und bei der späten Überwachung (33
Wochen nach SE) der zweiten Gruppe konnten aufgrund der hohen Verlustrate der
EEG-Aufsätze meist nur generalisierte Anfälle erfasst werden. Obwohl sich fokale
Anfälle gut von generalisierten Anfällen im Video unterscheiden lassen, ist es bei
alleiniger Videoüberwachung oft schwierig, fokale Anfälle zu erkennen.
Die mittlere Anfallsfrequenz lag während der Überwachungsphase 8 Wochen nach
SE bei 6,65 Anfällen pro Woche.

Ergebnisse
62
Bei Tieren, die sowohl 8 als auch 33 Wochen nach SE überwacht wurden, stieg die
mittlere Anfallsfrequenz von 9,26 auf 52,4 Anfälle pro Woche, was für eine
Progression der Krankheit bei diesen Tieren spricht. Einige Ratten hatten während
der Überwachung EEG-Auffälligkeiten (siehe Abbildung 13 C), welche nicht eindeutig
als Anfall klassifiziert werden konnten, aber immer mit Verhaltensänderungen
einhergingen (Verharren, seitliche Kopfbewegungen). Möglicherweise könnten diese
EEG-Ereignisse eine Form von fokalen, nichtkonvulsiven Anfällen darstellen.
Die wöchentliche Anfallsfrequenz (Zahl der Anfälle während der gesamten
Überwachungsdauer geteilt durch sieben Tage) in den einzelnen
Überwachungsphasen ist in nachfolgender Tabelle dargestellt. Die niedrigere
Anfallsfrequenz der 1. Gruppe könnte daher kommen, dass bei vielen Tieren ohne
EEG-Aufsätze nur generalisierte Anfälle detektiert werden konnten. Die tägliche
Anfallsfrequenz während der Überwachung ist für jedes Tier der 2. Gruppe individuell
dargestellt (siehe Abbildung 14).

Ergebnisse
63
Tier Züchter 4 Wochen
nach SE
6 Wochen
nach SE
8 Wochen
nach SE
33 Wochen
nach SE
MR 57 Charles River 1 1 0 -
MR 58 Charles River 2 1 0 -
MR 60 Charles River 1 0 0 -
MR 61 Charles River 1 3 2 -
MR 63 Charles River 0 0 1 -
MR 64 Charles River 0 0 0 -
MR 65 Charles River 0 0 0 -
MR 66 Charles River 3 8 5 -
MR 67 Harlan 2 3 0 -
MR 73 Harlan 1 4 3 -
MR 74 Harlan 0 4 2 -
MR 76 Harlan 0 4 6 -
MR 122 Harlan - - 4,28 16
MR 123 Harlan - - 0 -
MR 124 Harlan - - 0,54 106
MR 126 Harlan - - 0,78 12,3
MR 127 Harlan - - 5,25 56
MR 129 Harlan - - 6,61 -
MR 131 Harlan - - 26,6 80,5
MR 132 Harlan - - 10,18 26
MR 133 Harlan - - 21,78 53,4
MR 134 Harlan - - 4,67 69
Tabelle 11: Durchschnittliche wöchentliche Anfallsfrequenz (Zahl der Anfälle während der gesamten Überwachungsdauer geteilt durch sieben Tage) in den einzelnen Überwachungsphasen.

Ergebnisse
64
Abb. 13 (A-C): Spontane epileptische Anfälle 8 Wochen nach SE-Induktion. Spontaner generalisierter Anfall Typ IV (A). Spontaner fokaler Anfall Typ III (B). Spontaner nichtkonvulsiver Anfall (C).
5 s1 mV
5 s1 mV
1 mV5 s
A
B
C
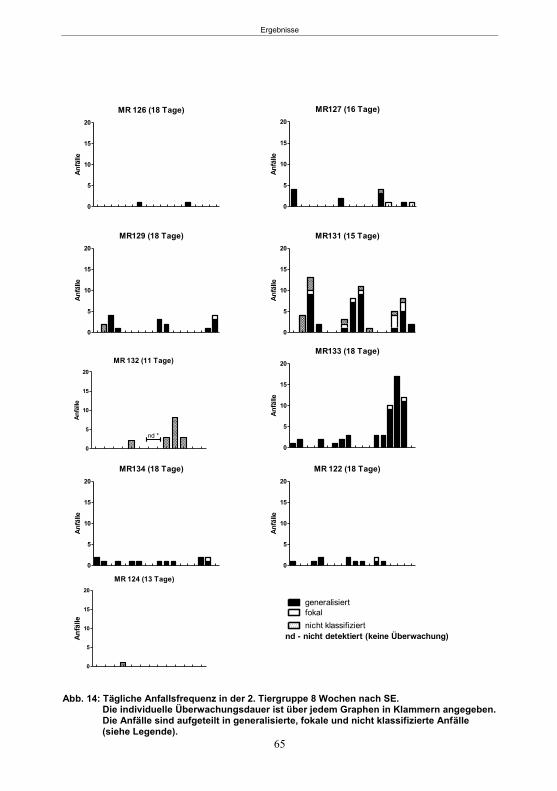
Ergebnisse
65
MR 126 (18 Tage)
0
5
10
15
20
An
fälle
MR127 (16 Tage)
0
5
10
15
20
An
fälle
MR129 (18 Tage)
0
5
10
15
20
An
fälle
MR131 (15 Tage)
0
5
10
15
20
An
fälle
MR 132 (11 Tage)
0
5
10
15
20
nd *
An
fälle
MR133 (18 Tage)
0
5
10
15
20
An
fälle
MR134 (18 Tage)
0
5
10
15
20
An
fälle
MR 122 (18 Tage)
0
5
10
15
20
An
fälle
MR 124 (13 Tage)
0
5
10
15
20
An
fälle
generalisiert
fokal
nicht klassifiziert
nd - nicht detektiert (keine Überwachung)
Abb. 14: Tägliche Anfallsfrequenz in der 2. Tiergruppe 8 Wochen nach SE. Die individuelle Überwachungsdauer ist über jedem Graphen in Klammern angegeben. Die Anfälle sind aufgeteilt in generalisierte, fokale und nicht klassifizierte Anfälle (siehe Legende).

Ergebnisse
66
5.1.3 Verhaltensänderungen im fokalen Kainsäure-Modell
Im Vergleich zu Ratten aus dem systemischen Pilokarpin-Modell waren die
Kainsäure-Ratten in den ersten Wochen nach SE weniger aggressiv und einfacher
zu handhaben. Um die bei epileptischen Ratten typischen Verhaltensänderungen zu
beschreiben, haben wir eine Verhaltensbatterie durchgeführt. In den Endergebnissen
sind alle Tiere (von Harlan und von Charles River) zusammengefasst. Die
Ergebnisse im Züchtervergleich werden ebenfalls dargestellt, sie unterscheiden sich
aber grundsätzlich nicht von den zusammengefassten Daten.
5.1.3.1 Der Hyperexcitabilitätstest (HET)
Der HET ermöglicht es, taktile sowie akustische Übererregbarkeit von epileptischen
Ratten zu beschreiben und wurde in dieser Studie 13 (1. Gruppe), bzw. 15 (2.
Gruppe) Wochen nach SE durchgeführt (siehe Kapitel 4.5.5.1). Die Ratten dieser
Studie wurden mit Kontrollratten aus einem anderen Projekt verglichen
(POLASCHECK et al. 2010). Die Ergebnisse werden durch ein Score-System
beschrieben (siehe Kapitel 4.5.5.1). Die epileptischen Ratten unterscheiden sich im
Touch-Response- und Finger-Snap-Test von Kontrollen (siehe Abbildung 15 C, E),
nicht jedoch im Pick-up-Test und Approach-Response-Test (siehe Abbildung 15 G,
A). Im Züchtervergleich unterscheiden sich im Touch-Response-Test nur die
epileptischen Tiere von Harlan von den Kontrollen (siehe Abbildung 15 D). Im Finger-
Snap-Test gibt es keinen Unterschied zwischen Züchtervergleich und
Gesamtergebnissen (siehe Abbildung 15 E, F). Die Ergebnisse des Finger-Snap-
Tests sowie des Touch-Response-Tests sind ein Hinweis auf eine akustische und
taktile Übererregbarkeit der epileptischen Ratten. Im Pick-up-Test unterscheiden sich
die Kainsäure-Ratten nicht von Kontrollen, was übereinstimmt mit der besseren
Handhabbarkeit und geringeren Aggressivität der Kainsäure-Ratten im Vergleich zu
Pilokarpin-Ratten. Es lässt sich jedoch eine deutliche Teilung der Gruppe erkennen.
Ein Teil der epileptischen Tiere liegt mit den Score-Werten auf Kontrollniveau, ein
Teil liegt deutlich oberhalb des Kontrollniveaus. Die Ursachen dieser Spaltung
innerhalb der Gruppe sind nicht geklärt, da die hohen Score-Werte weder mit einer
längeren SE-Dauer, noch mit einer höheren Anfallsfrequenz korrelierten. Diese
Ergebnisse zeigen die unterschiedliche Ausprägung der epilepsieassozierten

Ergebnisse
67
Verhaltensänderungen in verschiedenen Nagermodellen für
Temporallappenepilepsie.
Kontr
ollen
KA-T
iere
0
1
2
3
4
5
6
7
*
p=0,0033
Sc
ore
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
1
2
3
4
5
6
7 *
p=0,002
Sc
ore
Kontr
ollen
KA-T
iere
0
2
4
6
p=0,2802
Sc
ore
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
2
4
6
p=0,5263
Sc
ore
Approach-Response-Test
Touch-Response-Test
A
C D
B
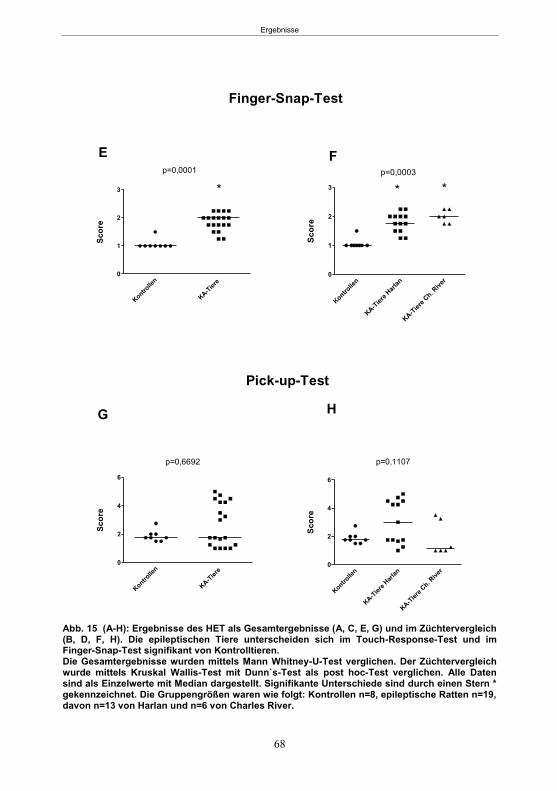
Ergebnisse
68
Abb. 15 (A-H): Ergebnisse des HET als Gesamtergebnisse (A, C, E, G) und im Züchtervergleich (B, D, F, H). Die epileptischen Tiere unterscheiden sich im Touch-Response-Test und im Finger-Snap-Test signifikant von Kontrolltieren. Die Gesamtergebnisse wurden mittels Mann Whitney-U-Test verglichen. Der Züchtervergleich wurde mittels Kruskal Wallis-Test mit Dunn`s-Test als post hoc-Test verglichen. Alle Daten sind als Einzelwerte mit Median dargestellt. Signifikante Unterschiede sind durch einen Stern * gekennzeichnet. Die Gruppengrößen waren wie folgt: Kontrollen n=8, epileptische Ratten n=19, davon n=13 von Harlan und n=6 von Charles River.
Kontr
ollen
KA-T
iere
0
1
2
3 *
p=0,0001
Sc
ore
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
1
2
3 * *
p=0,0003
Sc
ore
Kontr
ollen
KA-T
iere
0
2
4
6
p=0,6692
Sc
ore
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
2
4
6
p=0,1107
Sc
ore
Finger-Snap-Test
Pick-up-Test
E F
G H

Ergebnisse
69
5.1.3.2 Der Elevated-Plus Maze-Test (EPM)
Das EPM ist ein Test zur Untersuchung von explorations- und angstassoziiertem
Verhalten. In dieser Studie wurde er 27 (1. Gruppe), beziehungsweise 17 (2. Gruppe)
Wochen nach SE durchgeführt (siehe Kapitel 4.5.5.2). In diesem Test wird die
Aufenthaltsdauer in den verschiedenen Kompartimenten des EPM bewertet. Naive
Ratten empfinden die offenen Arme des EPM als aversiv und halten sich vermehrt in
den geschlossenen Armen auf. Eine längere Aufenthaltsdauer in den offenen Armen
deutet auf eine Explorationsfreudigkeit und reduziertes Angstverhalten hin.
Haeddips, also das Herauslehnen über den Rand der offenen Arme, und Absprünge
vom EPM sind relativ selten. Die Ergebnisse sind in Abbildung 16 dargestellt.
Die Frequenz der Eintritte in die einzelnen Kompartimente war in beiden Gruppen
gleich (siehe Abbildung 16 A, C, E). Die Aufenthaltsdauer in den verschiedenen
Kompartimenten war ebenfalls nicht signifikant unterschiedlich zwischen den beiden
Gruppen (siehe Abbildung 16 B, D, F). Bei den epileptischen Ratten lies sich jedoch
ein Trend zu einer verlängerten Aufenthaltsdauer in den offenen Armen beobachten
(p=0,0855), bei gleichzeitig tendenziell verkürzter Aufenthaltsdauer in den
geschlossenen Armen (p=0,058), (siehe Abbildung 16 D, F). Beide Gruppen
unterschieden sich nicht in der zurückgelegten Gesamtstrecke und der Frequenz des
Aufrichtens im EPM (siehe Abbildung 16 G, I). Die nichtepileptischen Kontrollen
putzten sich signifikant häufiger im Vergleich zu den epileptischen Ratten (siehe
Abbildung 16 J). In der epileptischen Gruppe konnte man einen Trend zu vermehrten
Headdips beobachten (p=0,0614), (siehe Abbildung 16 H). Die Ergebnisse im
Züchtervergleich unterscheiden sich nicht von den Gesamtergebnissen und sind
daher nicht abgebildet. Absprünge vom EPM konnte man nur bei den epileptischen
Ratten beobachten (nicht abgebildet). Acht von 18 epileptischen Ratten sind vom
EPM abgesprungen, ein Tier wurde nach 2 Absprüngen vom Versuch
ausgeschlossen. Abschließend kann man sagen, dass die epileptischen Tiere
tendenziell gesteigertes Explorationsverhalten und weniger angstassoziiertes
Verhalten im Vergleich zu nichtepileptischen Kontrollen zeigten (verlängerte
Aufenthaltsdauer in den offenen Armen, Headdips, Absprünge). Der Unterschied war
jedoch nicht sehr stark ausgeprägt.

Ergebnisse
70
Abb. 16 (A-J): Ergebnisse des EPM. Dargestellt sind die Frequenz der Eintritte (A, C, E) und die Aufenthaltsdauer (B, D, F) in den einzelnen Kompartimenten sowie die zurückgelegte Strecke (G) und beurteilte Verhaltensweisen (H, I, J) im EPM. Das Putzverhalten ist signifikant nidriger bei den epileptischen Ratten im Vergleich zu Kontrollen (J). Es gibt eien Trend zu vermehrten Headdips und einer längeren Aufenthaltsdauer in den offenen Armen (H, D). Alle Daten sind als Mittelwert mit SEM dargestellt und wurden mittels Student`s t-Test verglichen. Signifikante Unterschiede sind durch einen Stern * gekennzeichnet. Gruppengrößen: Kontrollen n=7, Kainsäure-Tiere n=17.
EPM Frequenz der Eintritte ins Zentrump=0,5534
Kontrollen KA-Tiere0
5
10
15
Fre
qu
en
z
EPM Frequenz der Eintritte in die offenen Armep=0,1215
Kontrollen KA-Tiere0
2
4
6
8
Fre
qu
en
z
EPM Frequenz der Eintritte in die geschlossenen Armep=0,8007
Kontrollen KA-Tiere0
2
4
6
8
10
Fre
qu
en
z
EPM Aufenthaltsdauer in den offenen Armenp=0,0855
Kontrollen KA-Tiere0
10
20
30
40
50
Au
fen
tha
lts
da
ue
r [s
]
EPM Aufenthaltsdauer im Zentrump=0,321
Kontrollen KA-Tiere0
10
20
30
40
Au
fen
tha
lts
da
ue
r [s
]EPM Aufenthaltsdauer in den geschlossenen Armen
p=0,0582
Kontrollen KA-Tiere0
100
200
300
Au
fen
tha
lts
da
ue
r [s
]
EPM zurückgelegte Streckep=0,4123
Kontrollen KA-Tiere0
500
1000
1500
2000
zu
rüc
kg
ele
gte
Str
ec
ke
[c
m]
EPM headdipsp=0,0614
Kontrollen KA-Tiere0
2
4
6
8
10
Fre
qu
en
z
EPM Aufrichtverhalten0,976
Kontrollen KA-Tiere0
5
10
15
20
Fre
qu
en
z
EPM Putzverhaltenp=0,0014
Kontrollen KA-Tiere0.0
0.5
1.0
1.5
2.0
2.5
*
Fre
qu
en
z
A B
C D
E F
G H
I J

Ergebnisse
71
5.1.3.3 Der Open Field-Test (OF)
Der OF-Test untersucht, ähnlich wie der EPM-Test, explorations- und
angstassoziiertes Verhalten. In dieser Studie wurde er 26 (1. Gruppe)
beziehungsweise 18 (2. Gruppe) Wochen nach SE durchgeführt (siehe Kapitel
4.5.5.3). Auch hier wird die Aufenthaltsdauer in den einzelnen Kompartimenten
gewertet. Naive Ratten halten sich bevorzugt im äußeren Ring auf, der innere Ring
und das Zentrum werden als aversiv empfunden. In dieser Studie unterscheiden sich
die epileptischen Ratten von den nichtepileptischen Kontrollen in der Frequenz der
Eintritte ins Zentrum (siehe Abbildung 17 A) und in den inneren Ring (siehe
Abbildung 17 C). Wenn man den Versuch nach Züchtern getrennt auswertet,
unterscheiden sich die epileptischen Tiere in der Frequenz der Eintritte ins Zentrum
nicht von den Kontrollen (siehe Abbildung 17 B), was vermutlich an den zu kleinen
Tierzahlen liegt. Bei den epileptischen Tieren von Harlan ist die Frequenz der
Eintritte in den inneren Ring höher im Vergleich zu den Kontrollen, was den
Gesamtergebnissen entspricht (siehe Abbildung 17 D). Die Frequenz der Eintritte in
den äußeren Ring war insgesamt nicht verändert im Vergleich zur Kontrollgruppe
(siehe Abbildung 17 E, F). Was die Aufenthaltsdauer in den einzelnen
Kompartimenten betrifft, so gab es keine Unterschiede zwischen den Gruppen in der
Aufenthaltsdauer im Zentrum (siehe Abbildung 18 A, B). Im Züchtervergleich
betrachtet, hatten die Tiere von Harlan eine verlängerte Aufenthaltsdauer im inneren
Ring, was in einer entsprechend verkürzten Aufenthaltsdauer im äußeren Ring
resultierte (siehe Abbildung D, F). Diese Ergebnisse sah man als Trend (p=0,0564)
der epileptischen Ratten zu einer verlängerten Aufenthaltsdauer im inneren Ring, in
den Gesamtergebnissen (siehe Abbildung 18 C). Die Aufenthaltsdauer im äußeren
Ring war in beiden Gruppen gleich, mit einem Trend der epileptischen Tiere zur
verkürzten Aufenthaltsdauer (siehe Abbildung 18 E). Die Gruppen unterscheiden sich
weder in der zurückgelegten Strecke, noch im Putzverhalten oder im
Aufrichtverhalten (siehe Abbildung 19). Im Züchtervergleich sind die zurückgelegte
Strecke und das Aufrichtverhalten signifikant höher bei den Tieren von Charles River
(siehe Abbildung 19 B, D). Zusammenfassend kann man sagen, dass die
epileptischen Ratten eine Reihe von Verhaltensänderungen im OF-Test zeigten, wie
weniger angstassoziiertes Verhalten und vermehrtes Explorationsverhalten.
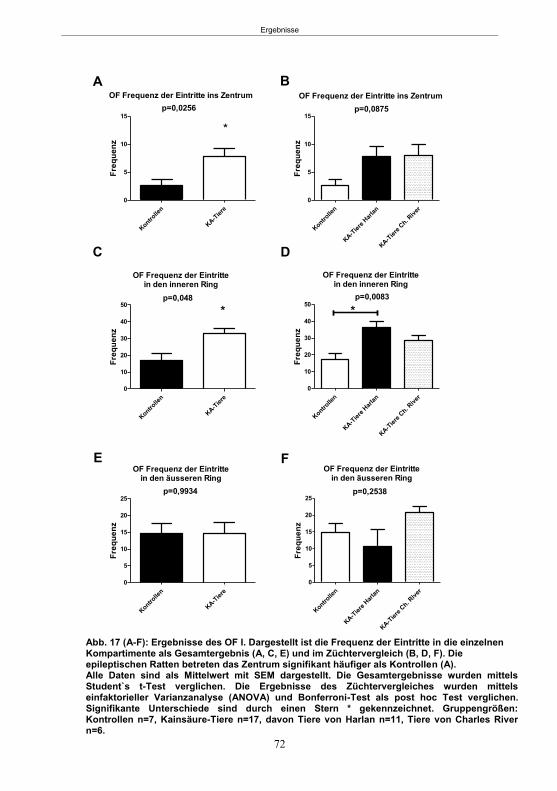
Ergebnisse
72
OF Frequenz der Eintritte ins Zentrum
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
5
10
15
Fre
qu
en
z
OF Frequenz der Eintrittein den inneren Ring
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
10
20
30
40
50
Fre
qu
en
z
OF Frequenz der Eintrittein den äusseren Ring
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
5
10
15
20
25
Fre
qu
en
z
OF Frequenz der Eintritte ins Zentrum
Kontr
ollen
KA-T
iere
0
5
10
15
*
Fre
qu
en
z
OF Frequenz der Eintrittein den inneren Ring
Kontr
ollen
KA-T
iere
0
10
20
30
40
50
Fre
qu
en
z
OF Frequenz der Eintrittein den äusseren Ring
Kontr
ollen
KA-T
iere
0
5
10
15
20
25
Fre
qu
en
z
A B
C D
E F
p=0,0256 p=0,0875
p=0,048
p=0,9934 p=0,2538
p=0,0083
* *
Abb. 17 (A-F): Ergebnisse des OF I. Dargestellt ist die Frequenz der Eintritte in die einzelnen Kompartimente als Gesamtergebnis (A, C, E) und im Züchtervergleich (B, D, F). Die epileptischen Ratten betreten das Zentrum signifikant häufiger als Kontrollen (A). Alle Daten sind als Mittelwert mit SEM dargestellt. Die Gesamtergebnisse wurden mittels Student`s t-Test verglichen. Die Ergebnisse des Züchtervergleiches wurden mittels einfaktorieller Varianzanalyse (ANOVA) und Bonferroni-Test als post hoc Test verglichen. Signifikante Unterschiede sind durch einen Stern * gekennzeichnet. Gruppengrößen: Kontrollen n=7, Kainsäure-Tiere n=17, davon Tiere von Harlan n=11, Tiere von Charles River n=6.

Ergebnisse
73
OF Aufenthaltsdauer im Zentrum
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
5
10
15
20
Au
fen
tha
lts
da
ue
r [s
]
OF Aufenthaltsdauer im inneren Ring
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
50
100
150 *A
ufe
nth
alt
sd
au
er
[s]
OF Aufenthaltsdauer im äusseren Ring
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
100
200
300 *
Au
fen
tha
lts
da
ue
r [s
]
OF Aufenthaltsdauer im Zentrum
Kontr
ollen
KA-T
iere
0
5
10
15
20
Au
fen
tha
lts
da
ue
r [s
]
OF Aufenthaltsdauer im inneren Ring
Kontr
ollen
KA-T
iere
0
50
100
150
Au
fen
tha
lts
da
ue
r [s
]
OF Aufenthaltsdauer im äusseren Ring
Kontr
ollen
KA-T
iere
0
100
200
300
Au
fen
tha
lts
da
ue
r [s
]
A B
C D
E F
p=0,1583 p=0,3491
p=0,0564 p=0,0185
p=0,0548 p=0,0304
Abb. 18 (A-F): Ergebnisse des OF II. Dargestellt ist die Aufenthaltsdauer in den einzelnen Kompartimenten als Gesamtergebnis (A, C, E) und im Züchtervergleich (B, D, F). Die epileptischen Ratten von Harlan halten sich länger in inneren Ring und kürzer im äusseren Ring auf im Vergleich zu Kontrolltieren (D, F). Alle Daten sind als Mittelwert mit SEM dargestellt. Die Gesamtergebnisse wurden mittels Student`s t-Test verglichen. Die Ergebnisse des Züchtervergleiches wurden mittels einfaktorieller Varianzanalyse (ANOVA) und Bonferroni-Test als post hoc Test verglichen. Signifikante Unterschiede sind durch einen Stern * gekennzeichnet. Gruppengrößen: Kontrollen n=7, Kainsäure-Tiere n=17, davon Tiere von Harlan n=11, Tiere von Charles River n=6.

Ergebnisse
74
Zurückgelegte Strecke
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
1000
2000
3000 *
zu
rüc
kg
ele
gte
Str
ec
ke
[c
m]
Aufrichtverhalten
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0
10
20
30
40
*F
req
ue
nz
Putzverhalten
Kontr
ollen
KA-T
iere
Har
lan
KA-T
iere
Ch. R
iver
0.0
0.5
1.0
1.5
2.0
2.5
Fre
qu
en
z
Zurückgelegte Strecke
Kontr
ollen
KA-T
iere
0
1000
2000
3000zu
rüc
kg
ele
gte
Str
ec
ke
[c
m]
A B
C D
E F
Aufrichtverhalten
Kontr
ollen
KA-T
iere
0
10
20
30
40
Fre
qu
en
z
Putzverhalten
Kontr
ollen
KA-T
iere
0.0
0.5
1.0
1.5
2.0
2.5
Fre
qu
en
zp=0,7316 p=0,0296
p=0,1298 p=0,0488
p=0,3539 p=0,4988
Abb. 19 (A-F): Ergebnisse OF III. Dargestellt sind die zurückgelegte Strecke, das Aufrichtverhalten und das Putzverhalten als Gesamtergebnis (A, C, E) und im Züchtervergleich (B, D, F). Die epileptischen Tiere von Charles River sind aktiver im Vergleich zu Kontrolltieren und epileptischen Ratten von Harlan (B, D). Alle Daten sind als Mittelwert mit SEM dargestellt. Die Gesamtergebnisse wurden mittels Student`s t-Test verglichen. Die Ergebnisse des Züchtervergleiches wurden mittels einfaktorieller Varianzanalyse (ANOVA) und Bonferroni-Test als post hoc Test verglichen. Signifikante Unterschiede sind durch einen Stern * gekennzeichnet. Gruppengrößen: Kontrollen n=7, Kainsäure-Tiere n=17, davon Tiere von Harlan n=11, Tiere von Charles River n=6.

Ergebnisse
75
5.1.3.4 Der Morris Water Maze-Test (MWM)
Im MWM werden räumliche Orientierung und Lernverhalten untersucht. Dieser Test
konnte nur in der ersten Gruppe durchgeführt werden. Der Versuch wurde 30
Wochen nach SE durchgeführt. Der Test wurde an 5 aufeinanderfolgenden Tagen
durchgeführt (siehe Kapitel 4.5.5.4). Ein Lernerfolg, gemessen in verkürzter
Latenzzeit [s] bis zum Auffinden der Plattform im Vergleich zum Vortag konnte bei
den nichtepileptischen Kontrollen ab dem zweiten Tag festgestellt werden. Im
Gegensatz dazu konnte man bei den epileptischen Ratten erst ab dem dritten Tag
einen Lernerfolg verzeichnen (siehe Abbildung 20). Am letzten Tag sah man keinen
Unterschied mehr zwischen den beiden Gruppen. Am dritten und vierten Tag war die
Zeit zum Auffinden der Plattform bei den epileptischen Ratten signifikant erhöht im
Vergleich zu den Kontrollen. Die epileptischen Ratten lernten langsamer als
Kontrollen. Diese Ergebnisse lassen auf Schwierigkeiten im konditionierten Lernen
und bei der räumlichen Orientierung sowie auf Gedächtnisdefizite der epileptischen
Ratten schließen. Im letzten Tetslauf des fünften Tages wurde ein Probe trial
durchgeführt, in welchem die Plattform aus dem Becken entfernt und die Zahl der
Kreuzungen der ehemaligen Plattformposition detektiert wurde. In diesem Test
unterscheiden sich die epileptischen Ratten deutlich von den Kontrollen (siehe
Abbildung 21 B). Diese Unterschiede konnten nicht auf Schwimmdefizite der
epileptischen Ratten zurückgeführt werden, da die mittlere Schwimmgeschwindigkeit
in beiden Gruppen gleich war (siehe Abbildung 21 A). Das Schwimmverhalten beider
Gruppen zeigte ebenfalls erhebliche Unterschiede. Die epileptischen Ratten zeigten
häufig ein zielloses Schwimmverhalten, schwammen oft am Beckenrand
(Thigmotaxis) ohne zu versuchen, die Plattform zu finden. Oft blieben sie auch nicht
auf der Plattform sitzen, wenn sie sie einmal gefunden hatten. Die Thigmotaxis kann
als angstassoziiertes Verhalten gewertet werden. All das lässt nicht nur auf Defizite
im räumlichen Gedächtnis schließen. Durch das Verhalten auf der Plattform ist es
auch sehr wahrscheinlich, dass für die epileptischen Ratten konditioniertes Lernen
ein Problem darstellte, da sie nicht imstande waren, die Zusammenhänge im MWM-
Test zu verknüpfen. Züchtervergleich: Da in diesem Versuch nur 2 Tiere von Harlan
untersucht wurden, kann über die Harlan-Tiere keine Aussage getroffen werden.
Wenn ausschließlich die Charles River Tiere gegen die Kontrollen getestet werden,

Ergebnisse
76
gibt es keinen Unterschied im Vergleich zu den kombinierten Ergebnissen (nicht
abgebildet).
Kontrollen KA-Tiere0
10
20
30
40
A Mittlere Schwimmgeschwindigkeit p=0,0665
Gesch
win
dig
keit
[cm
/ s
]
Kontrollen KA-Tiere0
2
4
6
*
B Kreuzungen der Plattform p=0,0096
Fre
qu
en
z
Abb. 21 (A-B): Die Ratten unterscheiden sich nicht in der mittleren Schwimmgesvchwindigkeit (A). Die Zahl der Plattformkreuzungen beim Probe Trial ist niedriger bei epileptischen Ratten (B). Alle Daten sind als Mittelwert mit SEM dargestellt. Die Unterschiede zwischen den Gruppen wurden mittels Student`s t-Test untersucht und sind durch einen Stern * angezeigt. Die Gruppengrößen: Kontrollen n=7, Kainsäure-Tiere n=9.
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
0
20
40
60Kontrollen
KA-Tiere
* * *
** * *
# #
Zeit bis zum Auffinden der Plattform
Zeit
[s]
Abb. 20 : Ergebnisse MWM. Beide Gruppen zeigen über die Zeit einen Lernerfolg. Die epileptischen Ratten lernen signifikant langsamer als Kontrolltiere. Alle Daten sind als Mittelwert mit SEM dargestellt. Die Daten innerhalb einer Gruppe wurden mittels einseitiger Varianzanalyse mit Bonferroni-Test als post hoc-Test untersucht. Signifikante Unterschiede werden innerhalb der Gruppe durch einen Stern * angezeigt. Die Unterschiede zwischen den Gruppen wurden mittels Student`s t-Test untersucht und werden durch eine Raute # angezeigt. Die Gruppengrößen: Kontrollen n=6, Kainsäure-Tiere n=8.

Ergebnisse
77
5.1.4 Die Phenobarbital-Selektion
Dreiunddreißig Wochen nach SE wurden 8 Tiere mit Phenobarbital behandelt. Ziel
der Behandlung war die Selektion von Phenobarbital-Respondern und
Phenobarbital-Nonrespondern im fokalen Kainsäure-Modell. Phenobarbital-
Responder sind Tiere, die auf eine Behandlung ansprechen (mit einer
Anfallsreduktion um mindestens 50%). Bei Phenobarbital-Nonrespondern resultiert
die Behandlung nicht in einer Anfallsreduktion in diesem Ausmaß (siehe Kapitel
4.5.6). Der Versuch dauerte 12 Tage. Während der gesamten Zeit wurden die Tiere
kontinuierlich videoüberwacht. Der Versuch war unterteilt in 4 Tage Kontrollphase, 4
Tage Behandlungsphase und 4 Tage Auswaschphase (siehe Kapitel 4.5.6). Bei allen
Tieren lag die Phenobarbitalplasmakonzentration im therapeutischen Bereich
zwischen 10-40 µg/ml (siehe Tabelle 12).
Tier Phenobarbital im Plasma [µg/ml]
MR 124 19,05
MR 126 49,66
MR 131 18,94
MR 133 41,20
MR 122 26,63
MR 127 32,63
MR 132 25,84
MR 134 11,78
Tabelle 12: Phenobarbital-Plasmakonzentration [µg/ml] während der Behadlungsphase, bei den einzelnen Tieren. Der Phenobarbital-Gehalt im Plasma wurde 14 h nach letzer Behandlung bestimmt.
In dem viertägigen Behandlungszeitraum konnte bei allen Ratten die Anfallsfrequenz
um mindestens 50% reduziert werden (siehe Abbildung 22), somit konnten keine
Phenobarbital-Nonresponder identifiziert werden. Wir können nicht ausschließen,
dass Grund hierfür die kurze (4 Tage) Behandlungsdauer war. Die Tiere hatten keine
EEG-Aufsätze mehr und konnten ausschließlich videoüberwacht werden. Aus
diesem Grund wurde der Versuch auf 12 Tage beschränkt, da die Auswertung der
Videoüberwachung extrem zeitaufwändig ist. Es wäre sinnvoll, diesen Versuch über
einen längeren Zeitraum zu wiederholen. Phenobarbital hatte initial eine gute
Wirkung bei allen Tieren, in einem Folgeversuch mit längerer Behandlung müsste die
eventuelle Toleranzentwicklung untersucht werden.

Ergebnisse
78
Die Tiere wurden während des Versuchs einem Ataxie-, Hyperlokomotion-,
Hypolokomotion-Scoring unterzogen und zeigten unter Phenobarbital-Behandlung
leichte bis deutliche Ataxie. Alle Tiere hatten Anfälle während der Kontrollphase.
Zwei Ratten (MR 122 und MR 132) konnten nur bedingt mit in die Endauswertung
aufgenommen werden, da sie nur wenige Anfälle während der Kontrollphase zeigten.
Die durchschnittliche Anfallsfrequenz während der 4 Tage lag bei 20,4 ± 6,32.
Während der Behandlungsphase wurden 4 Ratten anfallsfrei. Die Zahl der Anfälle lag
in diesem Zeitraum im Mittel bei 1,75 ± 0,96, und war damit signifikant gesunken
(p=0,0147). In der Auswaschphase kam es zu einem signifikanten Anstieg der
Anfallsfrequenz auf 36,5 ± 9,8 (p=0,007). Die Zahl der Anfälle pro Tag im Verlauf des
Versuchs ist für jedes Tier separat in Abbildung 22 dargestellt.

Ergebnisse
79
Abb. 22: Tägliche Anfallsfrequenz während der PB-Selektion. Dargestellt ist die Zahl der Anfälle pro Tag individuell für jede Ratte in den jeweiligen Versuchsphasen (PB – Phenobarbital).
MR 124
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
Tag 6
Tag 7
Tag 8
Tag 9
Tag 1
0
Tag 1
1
Tag 1
2
0
10
20
30
40Kontrollphase PB-Behandlung Auswaschphase
An
fälle
MR 126
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
Tag 6
Tag 7
Tag 8
Tag 9
Tag 1
0
Tag 1
1
Tag 1
2
0
10
20
30
40Kontrollphase PB-Behandlung Auswaschphase
An
fälle
MR 131
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
Tag 6
Tag 7
Tag 8
Tag 9
Tag 1
0
Tag 1
1
Tag 1
2
0
10
20
30
40Kontrollphase PB-Behandlung Auswaschphase
An
fälle
MR 133
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
Tag 6
Tag 7
Tag 8
Tag 9
Tag 1
0
Tag 1
1
Tag 1
2
0
10
20
30
40Kontrollphase PB-Behandlung Auswaschphase
An
fälle
MR 122
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
Tag 6
Tag 7
Tag 8
Tag 9
Tag 1
0
Tag 1
1
0
10
20
30
40Kontrollphase PB-Behandlung Auswaschphase
An
fälle
MR 127
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
Tag 6
Tag 7
Tag 8
Tag 9
Tag 1
0
Tag 1
1
0
10
20
30
40Kontrollphase PB-Behandlung Auswaschphase
An
fälle
MR 132
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
Tag 6
Tag 7
Tag 8
Tag 9
Tag 1
0
Tag 1
1
0
10
20
30
40Kontrollphase PB-Behandlung Auswaschphase
An
fälle
MR 134
Tag 1
Tag 2
Tag 3
Tag 4
Tag 5
Tag 6
Tag 7
Tag 8
Tag 9
Tag 1
0
Tag 1
1
0
10
20
30
40Kontrollphase PB-Behandlung Auswaschphase
An
fälle

Ergebnisse
80
5.1.5 Histologie
5.1.5.1 Histologie der 1. Gruppe
Die Tiere der ersten Gruppe wurden transkardial perfundiert und die Gehirne mittels
Gefriermikrotom geschnitten. Eine Serie der Hirnschnitte (Schnittdicke 40 µm) wurde
mittels Thionin-Färbung angefärbt. Die Lage der Elektroden und Führungsrohre sollte
lichtmikroskopisch beurteilt werden. Da die Tiere erst spät nach dem Verlust der
EEG-Aufsätze getötet wurden, konnten die Schnitte nicht ausgewertet werden, denn
das Gewebe war durch die Verluste der Aufsätze und damit einhergehende
entzündliche Prozesse stark verändert. Der Einstichkanal war durch Gliose stark
vernarbt und deshalb nicht gut erkennbar.
5.1.5.2 Histologie der 2. Gruppe
Die Tiere der zweiten Gruppe wurden dekapitiert und die Gehirne mittels Kryostat
geschnitten. Eine Serie der Hirnschnitte (Schnittdicke 14 µm) wurde mittels Thionin-
Färbung angefärbt. Die Neurodegeneration wurde in den Ebenen 3 mm und 4 mm
posterior zu Bregma, mittels Score-System bewertet (siehe Kapitel 4.5.7.2.1).
Ausgewertet wurden folgende Regionen des Hippokampus: Pyramidenzellschichten
CA1, CA3a, CA3c, Körnerzellschicht des Gyrus Dentatus (DG) und Hilus des Gyrus
Dentatus. Die mittleren Score-Werte für die jeweilige Region sind in der
nachfolgenden Tabelle dargestellt.
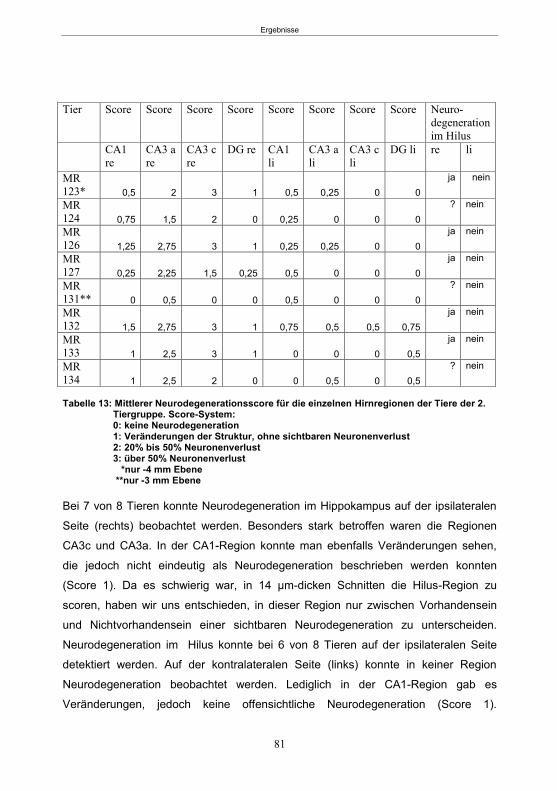
Ergebnisse
81
Tier Score Score Score Score Score Score Score Score Neuro-
degeneration
im Hilus
CA1
re
CA3 a
re
CA3 c
re
DG re CA1
li
CA3 a
li
CA3 c
li
DG li re li
MR
123* 0,5 2 3 1 0,5 0,25 0 0
ja nein
MR
124 0,75 1,5 2 0 0,25 0 0 0
? nein
MR
126 1,25 2,75 3 1 0,25 0,25 0 0
ja nein
MR
127 0,25 2,25 1,5 0,25 0,5 0 0 0
ja nein
MR
131** 0 0,5 0 0 0,5 0 0 0
? nein
MR
132 1,5 2,75 3 1 0,75 0,5 0,5 0,75
ja nein
MR
133 1 2,5 3 1 0 0 0 0,5
ja nein
MR
134 1 2,5 2 0 0 0,5 0 0,5
? nein
Tabelle 13: Mittlerer Neurodegenerationsscore für die einzelnen Hirnregionen der Tiere der 2. Tiergruppe. Score-System: 0: keine Neurodegeneration 1: Veränderungen der Struktur, ohne sichtbaren Neuronenverlust 2: 20% bis 50% Neuronenverlust 3: über 50% Neuronenverlust *nur -4 mm Ebene **nur -3 mm Ebene
Bei 7 von 8 Tieren konnte Neurodegeneration im Hippokampus auf der ipsilateralen
Seite (rechts) beobachtet werden. Besonders stark betroffen waren die Regionen
CA3c und CA3a. In der CA1-Region konnte man ebenfalls Veränderungen sehen,
die jedoch nicht eindeutig als Neurodegeneration beschrieben werden konnten
(Score 1). Da es schwierig war, in 14 µm-dicken Schnitten die Hilus-Region zu
scoren, haben wir uns entschieden, in dieser Region nur zwischen Vorhandensein
und Nichtvorhandensein einer sichtbaren Neurodegeneration zu unterscheiden.
Neurodegeneration im Hilus konnte bei 6 von 8 Tieren auf der ipsilateralen Seite
detektiert werden. Auf der kontralateralen Seite (links) konnte in keiner Region
Neurodegeneration beobachtet werden. Lediglich in der CA1-Region gab es
Veränderungen, jedoch keine offensichtliche Neurodegeneration (Score 1).
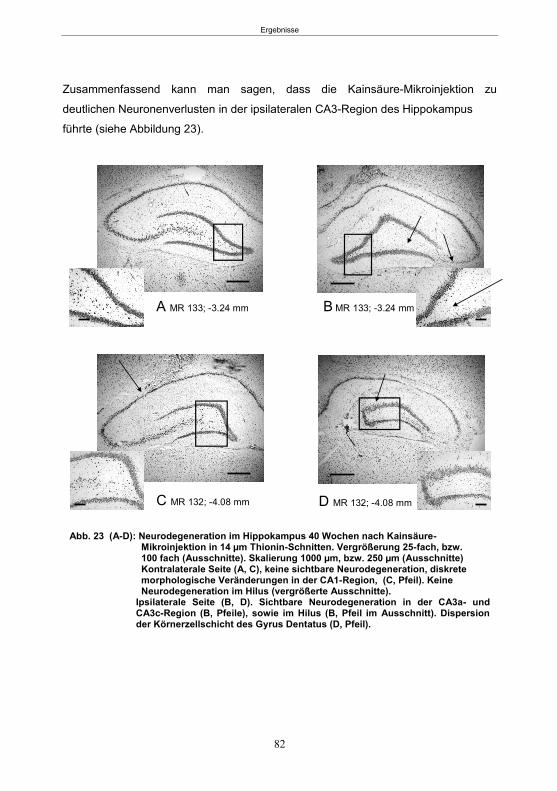
Ergebnisse
82
Zusammenfassend kann man sagen, dass die Kainsäure-Mikroinjektion zu
deutlichen Neuronenverlusten in der ipsilateralen CA3-Region des Hippokampus
führte (siehe Abbildung 23).
A MR 133; -3.24 mm B MR 133; -3.24 mm
C MR 132; -4.08 mm D MR 132; -4.08 mm
Abb. 23 (A-D): Neurodegeneration im Hippokampus 40 Wochen nach Kainsäure- Mikroinjektion in 14 µm Thionin-Schnitten. Vergrößerung 25-fach, bzw. 100 fach (Ausschnitte). Skalierung 1000 µm, bzw. 250 µm (Ausschnitte) Kontralaterale Seite (A, C), keine sichtbare Neurodegeneration, diskrete morphologische Veränderungen in der CA1-Region, (C, Pfeil). Keine Neurodegeneration im Hilus (vergrößerte Ausschnitte). Ipsilaterale Seite (B, D). Sichtbare Neurodegeneration in der CA3a- und CA3c-Region (B, Pfeile), sowie im Hilus (B, Pfeil im Ausschnitt). Dispersion der Körnerzellschicht des Gyrus Dentatus (D, Pfeil).

Ergebnisse
83
5.2 Studie 2: Schwellenversuche im Pilokarpin- und fokalen Kainsäure-Modell
5.2.1 Die SE-Induktion
Die Ergebnisse der SE-Induktion werden im Folgenden für beide SE-Modelle
getrennt beschrieben.
5.2.1.1 Die SE-Induktion und Latenzzeit im Pilokarpin-Modell
Die SE-Induktion
Für die SE-Induktion wurden insgesamt 41 Ratten verwendet. Bei 23 Tieren (56%)
konnte ein SE ausgelöst werden. Die mittlere Pilokarpin-Dosis lag bei 37 mg/kg, die
mittlere Zeit zum SE-Eintritt betrug 103 Minuten. Eine Ratte starb im SE. Während
des SE kam es zu limbischer Krampfaktivität, die sich im Verhalten durch
Stereotypien (Kaubewegungen, Blinzeln, Speicheln, verminderte Ansprechbarkeit
etc.) äußerte. Die limbische Krampfaktivität wurde von sehr häufigen generalisierten
Anfällen begleitet. Im Vergleich zu einem durch fokale Kainsäure-Mikroinjektion
induzierten SE zeigten die Tiere deutlich mehr generalisierte tonisch-klonische
Krampfaktivität. Es war somit möglich, den SE nur am Verhalten des Tieres, ohne
EEG-Ableitung festzumachen, wohingegen der SE nach Kainsäure-Mikroinjektion
hauptsächlich am charakteristischen EEG-Bild bestimmt wurde. Die Tiere der
zweiten und dritten Pilokarpin-Gruppe, sowie die Pilokarpin-Latenzzeit-Tiere, wurden
während der SE-Induktion EEG- und videoüberwacht. Im EEG konnten
hochfrequente paroxysmale Veränderungen beobachtet werden. Der SE wurde nach
90 min mittels Diazepam abgebrochen (siehe Kapitel 4.3.1). Der Abbruch mittels
Diazepam konnte den SE nicht zuverlässig unterbrechen. In der Nacht nach SE kam
es bei den Tieren zu erneuter Krampfaktivität.
Die Latenzzeit im Pilokarpin-Modell
In diesem Versuch wurden 6 SE-Ratten ohne PTZ-Schwellenbestimmung und 4 SE-
Ratten mit wiederholter PTZ-Schwellenbestimmung verwendet.

Ergebnisse
84
Die Tiere wurden bis zu 4 Wochen nach SE kontinuierlich überwacht. Anfälle, die
während der ersten 3 Tage nach SE auftraten, wurden als insultassoziiert betrachtet
und gingen nicht in die Latenzzeit-Bestimmung mit ein. Zwei Ratten hatten
insultassoziierte Anfälle. Die Latenzzeit zum Auftreten der ersten spontanen Anfälle
(siehe Abbildung 24 und Tabelle 14) lag bei den SE-Tieren durchschnittlich bei 7,3 ±
0,62 Tagen und reichte von 6 bis 10 Tagen. Bei den PTZ-Tieren lag die Latenzzeit
bei zwei Tieren entsprechend bei 6 und 9 Tagen, die anderen beiden Ratten hatten
keine Anfälle während der Überwachungszeit (nicht abgebildet). Ob die verlängerte
Latenzzeit eine normale Variation darstellt, oder als Folge der PTZ-
Schwellenversuche zu sehen ist, müsste an einer repräsentativeren Tierzahl
untersucht werden. Der erste Anfall konnte sowohl fokal als auch generalisiert (Typ
IV oder V) sein. Die individuelle Latenzzeit ist für die einzelnen Tiere in
nachfolgender Tabelle abgebildet.
Tier Dauer der Überwachung nach SE Anfälle in den ersten drei Tagen nach SE
(insultassoziiert)
Erster spontaner Anfall
MR 106 22 Tage Nein 6 Tage nach SE
MR 109 13 Tage Nein 6 Tage nach SE
MR 110 15 Tage Ja 10 Tage nach SE
MR 112 29 Tage Nein 8 Tage nach SE
MR 113 17 Tage Ja 7 Tage nach SE
MR 119 29 Tage Nein 7 Tage nach SE
Tabelle 14: : Latenzzeit und Anfälle in den ersten 3 Tagen nach SE (insultassoziierte Anfälle) bei den Kontrolltieren der Latenz-Studie im Pilokarpin-Modell.
1 mV
5 s
Abb. 24: Typisches EEG-Muster eines spontanen, epileptischen Anfalls einer Pilokarpin-Ratte.

Ergebnisse
85
5.2.1.2 Die SE-Induktion im fokalen Kainsäure-Modell
Für die SE-Induktion wurden insgesamt 21 Ratten verwendet. Bei 20 Tieren (95%)
konnte durch Kainsäure-Mikroinjektion ein SE induziert werden. Dieser war
charakterisiert durch fokale Anfälle, stereotypes Schnüffeln, Kopfnicken, „Straub tail“,
verminderte Ansprechbarkeit, „Wet dog shakes“, Verharren. Diese Stereotypien und
Verhaltensänderungen waren von generalisierten tonisch-klonischen Anfällen
begleitet. Die EEG-Veränderungen waren wie in Kapitel 5.1.1 beschrieben. Die Zahl
der auszählbaren EEG-Anfälle während der Überwachung nach SE lag im Mittel bei
etwa 90 (92,05 ±8,8). Die Dauer der paroxysmalen EEG-Veränderungen variierte von
Tier zu Tier und lag im Schnitt bei 11,8 h (5,8-16,5 h). Die Veränderungen der
Basislinie (erhöhte Amplitude, Spikes), hielten länger an. Der SE endete spontan bei
allen Tieren. Eine Ratte, die während des SE extrem viele generalisierte Anfälle
hatte, starb 48 Stunden nach SE.
Der Diazepam-Vorversuch im Pilokarpin-Modell
In diesem Versuch sollte der Einfluss von Diazepam auf die PTZ-Krampfschwelle
untersucht werden. Da der SE im Pilokarpin-Modell durch Diazepam abgebrochen
wurde, war es wichtig, den Einfluss von Diazepam auf die individuelle
Krampfschwelle zu untersuchen. Dieser Versuch war nur für das Pilokarpin-Modell
wichtig, da im fokalen Kainsäure-Modell der SE nicht abgebrochen wurde. Zu diesem
Zeck wurde bei 8 naiven Ratten die PTZ-Krampfschwelle 48 h nach Diazepam-Gabe,
mittels PTZ-Infusionstest bestimmt. Die Diazepam-Dosis entsprach der Dosis, die
zum SE-Abbruch verwendet wurde (3 x 10 mg/kg). Die Krampfschwelle 48 h nach
Diazepam wurde mit der zuvor ermittelten Kontrollschwelle verglichen. Es gab keinen
signifikanten Unterschied zwischen Kontrollschwelle und post Diazepam-Schwelle,
p=0,2429 (siehe Abbildung 25). Aufgrund dieser Ergebnisse haben wir entschieden,
in weiteren Versuchen Diazepam zum SE-Abbruch zu verwenden, da dies keinen
Einfluss auf die PTZ-Schwelle 48 h nach SE hatte.

Ergebnisse
86
Abb. 25: Kein Enfluss von Diazepam (3x10 mg/kg) auf die PTZ-Schwelle bei naiven Tieren. Die Daten sind als Mittelwert mit SEM dargestellt und wurden mittels gepaartem Student`s t-Test verglichen, Gruppengröße n=8.
Kontrollschwelle Nach Diazepam0
10
20
30
Kontrollschwelle versus Schwelle 48 h nach Diazepam p=0,2429
PT
Z [
mg
/kg
]

Ergebnisse
87
5.2.2 Der PTZ-Schwellentest
In diesem Test sollten die Veränderungen der Hirnerregbarkeit nach einem SE,
ermittelt als Veränderungen der individuellen PTZ-Krampfschwelle, untersucht
werden. Fünf Tage vor SE-Induktion wurde bei allen Tieren eine Kontrollschwelle
mittels PTZ-Infusionstest bestimmt (siehe Kapitel 4.6.4). Die Kontrollschwelle lag bei
den Tieren bei etwa 20 mg/kg. In der Pilokarpin-Gruppe lag die Kontrollschwelle bei
19,6 ± 0,49 mg/kg, in der Kainsäure-Gruppe bei 18,8 ± 0,43 mg/kg. Drei Tiere aus
der Pilokarpin-Gruppe starben nach der Kontrollschwellenbestimmung. Die Infusion
von PTZ wurde bei dem Auftreten des ersten Myoklonus gestoppt. Bei den meisten
Ratten kam es danach noch zu einem Klonus. Weitere Endpunkte nach dem
Myoklonus wurden durch ein Score-System erfasst (siehe Kapitel 4.6.4 Score I). Für
gewöhnlich zeigten die Tiere 3 bis 5 Minuten nach Schwellenbestimmung wieder
normales Verhalten. Verhaltensänderungen nach Ende der PTZ-Infusion wurden
durch ein Score-System erfasst (siehe Kapitel 4.6.4 Score II). Bei Tieren mit einer
EEG-Ableitelektrode wurden die Schwellenversuche unter EEG-Kontrolle
durchgeführt (siehe Abbildung 26). Der Myoklonus wurde von keinen spezifischen
EEG-Ereignissen begleitet und war weder bei der hippokampalen noch bei der
kortikalen Ableitung im EEG bzw. ECoG sichtbar (siehe Abbildung 26 A-E). Der auf
den Myoklonus folgende Klonus ging mit paroxysmalen EEG-Veränderungen einher
(siehe Abbildung 26). Das EEG-Bild zeigte ableitungsabhängig (kortikal versus
hippokampal) keine Unterschiede. Es gab auch keine Unterschiede im EEG-Bild
zwischen den SE-Tieren und Kontrollen (siehe Abbildung 26). Lediglich die Dauer
der EEG-Veränderungen war bei den SE-Tieren verlängert (siehe Abbildung 26).
Dies entspricht dem durch das Score-System II beschriebenen klinischen Bild.

Ergebnisse
88
1 mV
M K/V RBPTZ
10 s
1 mV
KM K/V M (für 17 min)
PTZ
10 s
RB
PTZ M K
VT
1 mV10 s
1 mV 10 s
PTZ M K
PTZ M K K/VRB
1 mV 10 s
A
B
C
D
E
Abb. 26 (A-E): EEG-Ableitung während einer PTZ-Schwellenbestimmung. Der Pfeil zeigt den Anfang der PTZ-Infusion. Abkürzungen: M – Myoklonus, K – Klonus, K/V – Klonus mit Verlust der Stellreflexe, VT – Vorderbeintonus, RB – Running and bouncing. Paroxysmale Veränderungen sind im EEG ab dem klonischen Anfall sichtbar. Dargestellt sind PTZ-Schwellenbestimmungen im Pilokarpin-Modell (A, B, C) und im fokalen Kainsäure-Modell (D, E). Kortikale Ableitung 5 Tage vor sham SE (A). Hippokampale Ableitung 4 Tage nach sham SE (B). Hippokampale Ableitung 2 Tage nach pilokarpin-induziertem SE (C). Hippokampale Ableitung 2 Tage nach sham SE (D). Hippokampale Ableitung 2 Tage nach kainsäure-induziertem SE (E).

Ergebnisse
89
5.2.2.1 Schwellenveränderungen nach SE im Pilokarpin-Modell
Der PTZ-Schwellentest wurde in dieser Studie an 3 Tiergruppen durchgeführt. Die
Ergebnisse aller Gruppen wurden kombiniert, um eine Gruppengröße zu erreichen,
die eine statistische Aussage zulässt. Es gab Unterschiede in der Zahl der
Schwellenbestimmungen in den einzelnen Tiergruppen. Die Schwellenbestimmung
erfolgte 2, 4, 6, 9, 14 und 22 Tage nach SE in der 1. Gruppe und 2, 6 und 14 Tage
nach SE in den beiden anderen Gruppen (siehe Kapiltel 4.6.1 Tabelle 5).
Veränderungen der Krampfschwelle in der Kontrollgruppe
Bei den Kontrollen mit sham SE (n=18) stieg die individuelle Krampfschwelle nach
wiederholter Durchführung. Die dritte und sechste Schwelle war signifikant erhöht im
Vergleich zur Kontrollschwelle (siehe Abbildung 27 A). Auch weitere Endpunkte
während der Infusion (siehe Score I) und das Verhalten nach PTZ-Infusion (siehe
Score II) änderten sich nach wiederholter Krampfschwellenbestimmung (siehe
Abbildung 28). Die Ratten zeigten schwerere Anfälle (mehr weitere Endpunkte) im
Anschluss an die PTZ-Infusion, die nach dem ersten Myoklonus gestoppt wurde. Für
gewöhnlich folgte auf den Myoklonus ein Klonus. Nach der fünften und sechsten
Schwellenbestimmung traten vermehrt Anfälle mit Verlust der Stellreflexe, „Running
and bouncing“ und Vorderbeintonus auf (siehe Abbildung 28 A). Es traten auch
Veränderungen im Verhalten nach Ende der PTZ-Infusion auf. Ab der vierten
Schwellenbestimmung zeigten die Ratten Myoklonien nach Ende der PTZ-Infusion,
dieser Unterschied wurde nach der fünften Schwellenbestimmung signifikant (siehe
Abbildung 28 D). Diese Veränderungen sind womöglich eine Reaktion auf die
wiederholte PTZ-Infusion in kurzen Zeitabständen. Eine Kontrollratte starb nach der
zweiten Krampfschwellenbestimmung.
Veränderungen der Krampfschwelle in der SE-Gruppe
In der SE-Gruppe (n=15) gab es keine Veränderungen der Krampfschwelle, wenn die
Ergebnisse mit einer Varianzanalyse untersucht wurden. In der Varianzanalyse gab
es einen Trend (p=0,95). Daraufhin haben wir einen direkten Vergleich der Schwelle
2 Tage nach SE mit der Kontrollschwelle mittels Student`s t-Test durchgeführt. Wenn
man die 2-Tage-Schwelle gegen die Kontrollschwelle testete, so war sie signifikant

Ergebnisse
90
niedriger (p=0,0183) (siehe Abbildung 27 B). Die gesteigerte Empfindlichkeit
gegenüber PTZ drückte sich nicht nur in einer gesenkten Krampfschwelle aus, die
SE-Tiere zeigten nach der PTZ-Infusion deutliche Verhaltensänderungen. Während
bei den Kontrolltieren erst nach wiederholter PTZ-Infusion Verhaltensänderungen
auftraten, hatten die Pilokarpin-Ratten ab der ersten Schwellenbestimmung nach SE
(nach 2 Tagen) deutliche Verhaltensänderungen (Myoklonien, generalisierte Anfälle,
Stereotypien etc.), die bis zu 1 Stunde nach Infusion anhielten (siehe Abbildung 28
E). Die Schwere der Anfälle im Anschluss an die PTZ-Infusion (weitere Endpunkte
Score I) änderte sich im Gegensatz zu den Kontrolltieren nicht (siehe Abbildung 28
B). Wenn man die Pilokarpin-Tiere mit den Kontrollen vergleicht, so ist die
Krampfschwelle bei den post SE-Tieren zu allen Zeitpunkten signifikant niedriger als
bei den Kontrolltieren, obwohl es keinen Unterschied in der Kontrollschwelle (fünf
Tage vor SE) zwischen den beiden Gruppen gab (siehe Abbildung 27 C). Da PTZ
seinen Wirkungsort am GABAA-Rezeptor hat, lassen die Ergebnisse auf
Veränderungen des GABAergen Systems nach einem pilokarpin-induzierten SE
schließen. Die Ratten, die trotz Pilokarpin-Gabe keinen SE hatten, wurden vom
Versuch ausgeschlossen.
5.2.2.2 Schwellenveränderungen nach SE im fokalen Kainsäure-Modell
Der PTZ-Schwellentest wurde in dieser Studie an einer Tiergruppe durchgeführt. Die
Schwellenbestimmung erfolgte 2, 4, 6, 9, 14 und 22 Tage nach SE. Keine der Ratten
starb während oder nach einem PTZ-Schwellentest.
Veränderungen der Krampfschwelle in der Kontrollgruppe
Die individuelle Krampfschwelle blieb bei den Kontrollratten (n=15) über den
gesamten Zeitraum unverändert im Vergleich zur Kontrollschwelle (siehe Abbildung
29 A).
Was die Schwere der Anfälle nach PTZ-Infusion betrifft (weitere Endpunkte Score I),
so hatten die Tiere nach der letzten Schwellenbestimmung tendenziell häufiger
Klonus mit Verlust der Stellreflexe, „Running and bouncing“ und Vorderbeintonus im
Vergleich zur Kontrollschwelle, was jedoch durch die große Streuung der Daten nicht
signifikant war (siehe Abbildung 30 A). Nach sieben Schwellenbestimmungen kam es

Ergebnisse
91
bei den Kontrolltieren zu Verhaltensänderungen (Myoklonien, siehe Score II), die
nach dem letzten PTZ-Schwellentest signifikant wurden (siehe Abbildung 31 A).
Veränderungen der Krampfschwelle in der SE-Gruppe
Die individuelle Krampfschwelle war bei den SE-Tieren (n=19) an Tag 2, 9 und 22
post SE signifikant niedriger im Vergleich zur Kontrollschwelle (siehe Abbildung 29
B). Wenn man die SE-Tiere direkt mit den Kontrollen vergleicht, so haben die SE-
Tiere zu fast allen Zeitpunkten eine signifikant niedrigere Krampfschwelle. Die
einzige Ausnahme ist die Schwelle 6 Tage nach SE (siehe Abbildung 29 C). Die
Schwere der Anfälle nach PTZ-Infusion war am Tag 4 nach SE signifikant niedriger
im Vergleich zur Kontrollschwelle (siehe Abbildung 30 B). Ansonsten variierte sie
nicht signifikant von Schwellenbestimmung zu Schwellenbestimmung. Im direkten
Vergleich beider Gruppen sieht man ebenfalls, ausser bei der Schwelle 4 Tage nach
SE, keine Unterschiede (siehe Abbildung 30 C). Im Gegensatz zu den Kontrolltieren,
die für gewöhnlich 3 bis 5 Minuten nach Versuchsende wieder Normalverhalten
zeigten, kam es in der SE-Gruppe zu Verhaltensänderungen nach Infusion. Das
Verhalten nach Ende der PTZ-Infusion (Score II) war bereits 2 Tage nach SE stark
verändert. Die Tiere zeigten Myoklonien, verminderte Ansprechbarkeit und
Stereotypien, die bei einigen Tieren, ähnlich wie in der Pilokarpin-Gruppe, bis zu 1
Stunde nach Versuchsende anhielten (siehe Abbildung 31 B). Im direkten Vergleich
zeigen die SE-Tiere 2 und 6 Tage nach SE signifikant stärkere
Verhaltensänderungen im Vergleich zu Kontrollen (siehe Abbildung 31 C).
5.2.3 Vergleich beider SE-Modelle
Sowohl im Pilokarpin-Modell als auch im fokalen Kainsäure-Modell sank die PTZ-
Schwelle 2 Tage nach SE. Dieser Effekt war bei den Pilokarpin-Tieren nur im
direkten Vergleich zwischen der Kontrollschwelle und der 2-Tages Schwelle sichtbar,
was vermutlich daran lag, dass diese Gruppe sich aus drei kleineren
zusammensetzte, was die Streuung innerhalb der Gruppe erhöhte. Dass dieser
Effekt jedoch robust war, zeigte sich im Kainsäure-Modell, in dem es ebenfalls zu
einer Senkung der PTZ-Schwelle 2 Tage nach SE kam. Im direkten Vergleich beider
Gruppen gab es nur an Tag 4 nach SE signifikante Unterschiede zwischen den

Ergebnisse
92
Gruppen. Zu diesem Zeitpunkt war die PTZ-Schwelle deutlich niedriger bei den
Kainsäure-Ratten im Vergleich zur Pilokarpin-Gruppe (siehe Abbildung 32).
5.2.4 Einfluss des ovariellen Zyklusstandes auf die PTZ-Schwelle
Der Zyklusstand wurde mittels Vaginalabstrichen anhand des Zellbildes
lichtmikroskopisch bestimmt. Diese Untersuchungen wurden ausschließlich in der
fokalen Kainsäure-Gruppe durchgeführt. Die weiblichen Ratten wurden ohne
männliche Ratten gehalten, was dazu führte, dass ihr Zyklus nicht synchron war.
Viele Tiere (26 von 31) hatten keinen regelmäßigen Zyklus. Es konnte kein
Zusammenhang zwischen Zyklusstand und Höhe der PTZ-Schwelle festgestellt
werden, so dass ein hormoneller Einfluss auf die PTZ-Schwelle nicht anzunehmen
ist.

Ergebnisse
93
-5 +2 +4 +6 +9 +14 +220
10
20
30
* *
PTZ-Schwelle Kontrollen
PTZ [
mg/k
g]
-5 +2 +4 +6 +9 +14 +220
10
20
30
*
PTZ-Schwelle Pilokarpin-Tiere
PTZ [
mg/k
g]
-5 +2 +4 +6 +9 +14 +220
10
20
30
#
# ## #
#
PTZ-SchwelleKontrollen versus Pilokarpin-Tiere
PTZ [
mg/k
g]
A
B
C
Kontrollen
Pilokarpin-Tiere
Abb. 27 (A-C): PTZ-Schwellenveränderungen, ausgedrückt in mg/kg, im Pilokarpin-Modell im Zeitverlauf. Dargestellt sind Schwellenveränderungen bei Kontrolltieren (A) und Pilokarpin-Tieren (B) in Relation zur Kontrollschwelle, sowie ein Vergleich beider Gruppen (C). Signifikante Unterschiede in Bezug auf die Kontrollschwelle (-5) werden durch einen Stern * angezeigt. Signifikante Unterschiede zwischen den Gruppen werden durch eine Raute # angezeigt. Alle Daten sind als Mittelwert mit SEM dargestellt. Die Ergebnisse der Kontrolltiere wurden mithilfe einer einfaktoriellen Varianzanalyse (ANOVA) und dem Bonferroni-Test als post hoc-Test verglichen. Die Ergebnisse der Pilokarpin-Tiere wurden mittels Student`s t-Test verglichen (Kontrollschwelle mit 2-Tages Schwelle). Der Vergleich beider Gruppen erfolgte mittels Student`s t-Test. Gruppengrößen: Kontrollen n=18, Pilokarpin-Tiere n=15.
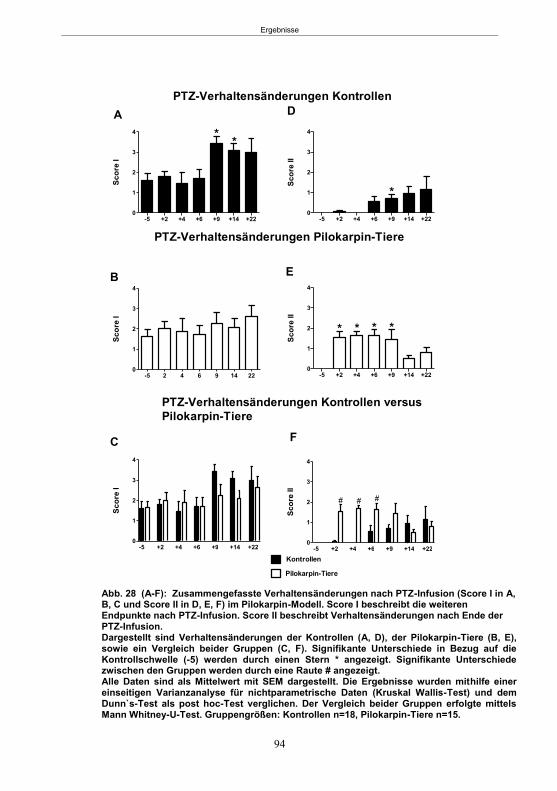
Ergebnisse
94
-5 +2 +4 +6 +9 +14 +220
1
2
3
4 **
Sc
ore
I
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
*
Sc
ore
II
-5 2 4 6 9 14 220
1
2
3
4
Sc
ore
I
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
* * * *
Sc
ore
II
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
Sc
ore
I
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
# # #
Sc
ore
II
A
B
C
D
E
F
PTZ-Verhaltensänderungen Kontrollen
PTZ-Verhaltensänderungen Pilokarpin-Tiere
PTZ-Verhaltensänderungen Kontrollen versus
Pilokarpin-Tiere
Kontrollen
Pilokarpin-Tiere
Abb. 28 (A-F): Zusammengefasste Verhaltensänderungen nach PTZ-Infusion (Score I in A, B, C und Score II in D, E, F) im Pilokarpin-Modell. Score I beschreibt die weiteren Endpunkte nach PTZ-Infusion. Score II beschreibt Verhaltensänderungen nach Ende der PTZ-Infusion. Dargestellt sind Verhaltensänderungen der Kontrollen (A, D), der Pilokarpin-Tiere (B, E), sowie ein Vergleich beider Gruppen (C, F). Signifikante Unterschiede in Bezug auf die Kontrollschwelle (-5) werden durch einen Stern * angezeigt. Signifikante Unterschiede zwischen den Gruppen werden durch eine Raute # angezeigt. Alle Daten sind als Mittelwert mit SEM dargestellt. Die Ergebnisse wurden mithilfe einer einseitigen Varianzanalyse für nichtparametrische Daten (Kruskal Wallis-Test) und dem Dunn`s-Test als post hoc-Test verglichen. Der Vergleich beider Gruppen erfolgte mittels Mann Whitney-U-Test. Gruppengrößen: Kontrollen n=18, Pilokarpin-Tiere n=15.
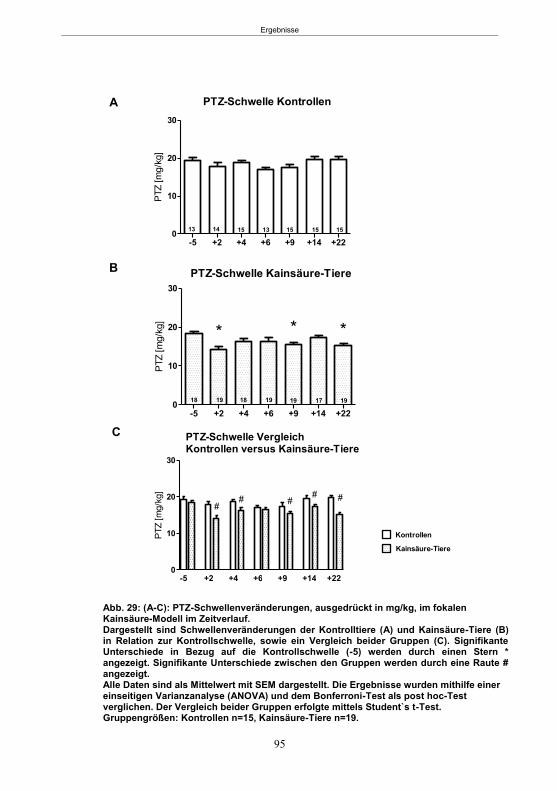
Ergebnisse
95
-5 +2 +4 +6 +9 +14 +220
10
20
30
13 14 15 13 15 15 15
PTZ-Schwelle Kontrollen
PTZ [
mg/k
g]
-5 +2 +4 +6 +9 +14 +220
10
20
30
* * *
18 19 18 1719 19 19
PTZ-Schwelle Kainsäure-Tiere
PTZ [
mg/k
g]
-5 +2 +4 +6 +9 +14 +220
10
20
30
## #
# #
PTZ-Schwelle VergleichKontrollen versus Kainsäure-Tiere
PTZ [
mg/k
g]
A
B
C
Kontrollen
Kainsäure-Tiere
Abb. 29: (A-C): PTZ-Schwellenveränderungen, ausgedrückt in mg/kg, im fokalen Kainsäure-Modell im Zeitverlauf. Dargestellt sind Schwellenveränderungen der Kontrolltiere (A) und Kainsäure-Tiere (B) in Relation zur Kontrollschwelle, sowie ein Vergleich beider Gruppen (C). Signifikante Unterschiede in Bezug auf die Kontrollschwelle (-5) werden durch einen Stern * angezeigt. Signifikante Unterschiede zwischen den Gruppen werden durch eine Raute # angezeigt. Alle Daten sind als Mittelwert mit SEM dargestellt. Die Ergebnisse wurden mithilfe einer einseitigen Varianzanalyse (ANOVA) und dem Bonferroni-Test als post hoc-Test verglichen. Der Vergleich beider Gruppen erfolgte mittels Student`s t-Test. Gruppengrößen: Kontrollen n=15, Kainsäure-Tiere n=19.

Ergebnisse
96
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
PTZ-Verhaltensänderungen Kontrollen
Sc
ore
I
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
*
PTZ-Verhaltensänderungen Kainsäure-Tiere
Sc
ore
I
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
#
PTZ-VerhaltensänderungenKontrollen versus Kainsäure-Tiere
Sc
ore
I
A
B
C
Kontrollen
Kainsäure-Tiere
Abb. 30 (A--C): Weitere Endpunkte nach PTZ-Schwellentest, ausgedrückt durch ein Score-System (Score I) im fokalen Kainsäure-Modell im Zeitverlauf. Dargestellt sind weitere Endpunkte der Kontrolltiere (A) und der Kainsäure-Tiere (B), sowie ein Vergleich beider Gruppen (C). Signifikante Unterschiede in Bezug auf die Kontrollschwelle (-5) werden durch einen Stern * angezeigt. Signifikante Unterschiede zwischen den Gruppen werden durch eine Raute # angezeigt. Alle Daten sind als Mittelwert mit SEM dargestellt. Die Ergebnisse wurden mithilfe einer einseitigen Varianzanalyse für nichtparametrische Daten (Kruskal Wallis-Test) und dem Dunn`s-Test als post hoc-Test verglichen. Der Vergleich beider Gruppen erfolgte mittels Mann Whitney-U-Test. Gruppengrößen: Kontrollen n=15, Kainsäure-Tiere n=19.
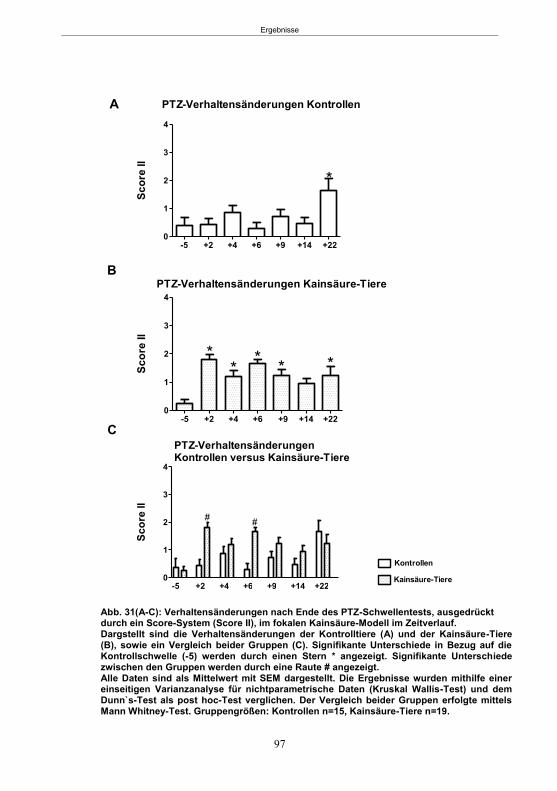
Ergebnisse
97
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
*
PTZ-Verhaltensänderungen Kontrollen
Sc
ore
II
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
**
** *
PTZ-Verhaltensänderungen Kainsäure-Tiere
Sc
ore
II
-5 +2 +4 +6 +9 +14 +220
1
2
3
4
##
PTZ-VerhaltensänderungenKontrollen versus Kainsäure-Tiere
Sc
ore
II
A
B
C
Kontrollen
Kainsäure-Tiere
Abb. 31(A-C): Verhaltensänderungen nach Ende des PTZ-Schwellentests, ausgedrückt durch ein Score-System (Score II), im fokalen Kainsäure-Modell im Zeitverlauf. Dargstellt sind die Verhaltensänderungen der Kontrolltiere (A) und der Kainsäure-Tiere (B), sowie ein Vergleich beider Gruppen (C). Signifikante Unterschiede in Bezug auf die Kontrollschwelle (-5) werden durch einen Stern * angezeigt. Signifikante Unterschiede zwischen den Gruppen werden durch eine Raute # angezeigt. Alle Daten sind als Mittelwert mit SEM dargestellt. Die Ergebnisse wurden mithilfe einer einseitigen Varianzanalyse für nichtparametrische Daten (Kruskal Wallis-Test) und dem Dunn`s-Test als post hoc-Test verglichen. Der Vergleich beider Gruppen erfolgte mittels Mann Whitney-Test. Gruppengrößen: Kontrollen n=15, Kainsäure-Tiere n=19.

Ergebnisse
98
5.3 Studie 3: Infusionsversuche mit PTZ im Pilokarpin- und fokalen Kainsäure-Modell
5.3.1 Pharmakokinetische Vorversuche
Um ein Infusionsprotokoll für PTZ zu etablieren, wurden pharmakokinetische
Versuche durchgeführt. Wichtig am Infusionsprotokoll war die Aufrechterhaltung
einer konstanten PTZ-Konzentration im Plasma, die jedoch über die gesamte
Behandlungsdauer im subkonvulsiven Bereich bleiben sollte. Zum einen wurde die
PTZ-Plasmahalbwertszeit bestimmt. Zum anderen sollte der Zusammenhang
zwischen individueller PTZ-Krampfschwelle [mg/kg] und dazugehöriger PTZ-
Plasmakonzentration [µg/ml] untersucht werden. Des Weiteren wurden die beiden
errechneten Infusionsdosen von 15 und 60 µg/kg/min auf Nebenwirkungen und
Erreichen der Zielplasmakonzentration untersucht.
5.3.1.1 Bestimmung der Plasmahalbwertszeit von PTZ
Die Bestimmung der PTZ-Plasmahalbwertszeit wurde an 4 Tieren durchgeführt, an 2
naiven Ratten und an 2 Ratten 24 h nach pilokarpin-induziertem SE.
-5 +2 +4 +6 +9 +14 +220
10
20
30
# Pilokarpin-Tiere
Kainsäure-Tiere
PTZ-Schwelle ModellvergleichP
TZ [
mg/k
g]
Abb. 32 : : Vergleich der PTZ-Schwelle, ausgedrückt in mg/kg, in beiden SE-Modellen im Zeitverlauf (siehe Legende). Signifikante Unterschiede werden durch eine Raute # angezeigt. Die Daten sind als Mittelwert mit SEM dargestellt und wurden mittels Student`s t-Test verglichen. Gruppengrößen: Kainsäure-Tiere n=19, Pilokarpin-Tiere n=15.

Ergebnisse
99
Alle Ratten erhielten eine intravenöse Applikation von 12 mg/kg PTZ. Nach 15 min, 1
h, 2 h und 4 h wurde bei den Tieren retrobulbär Blut entnommen und die PTZ-
Plasmakonzentration wurde mittels HPLC bestimmt. Die Ergebnisse sind in
nachfolgender Tabelle und Abbildung 33 dargestellt. Bei einem Tier (MR 144) war
der PTZ-Plasmaspiegel von Anfang an sehr niedrig, was vermutlich auf eine
paravenöse Injektion zurückzuführen ist. Die mittels PK Solutions Software
errechnete mittlere PTZ-Halbwertszeit lag bei 3,3 h (Daten aller Tiere als Mittelwert).
0 1 2 3 40
5
10
15
naive Tiere n=2
SE-Tiere n=2
PTZ-Plasmahalbwertszeit
Zeit [h]
PT
Z [
µg
/m
l]
Abb. 33: PTZ-Plasmaspiegel [µg/ml] im Zeitverlauf, als Rechtecke dargestellt naive Tiere, als Kreise dargestellt post SE-Tiere (siehe Legende).
Tier PTZ [µg/ml]
0,25 h
PTZ [µg/ml]
1 h
PTZ [µg/ml]
2 h
PTZ [µg/ml]
4 h
Halbwertszeit
[h]
MR 105 11,41 10,57 5.94 4,45 2,59
MR 108 13,03 12,14 9,14 3,07 1,73
MR 151 post SE-Tier
11,48 11,22 10,13 7,4 5,69
MR 144 post SE-Tier
7,02 3,89 3,03 2,78 3,22
Tabelle 15: PTZ-Halbwertszeit der einzelnen Tiere nach 12 mg/kg PTZ Injektion i.v.. Die ermittelten Plasmaspiegel sind in µg/ml Plasma angegeben.

Ergebnisse
100
5.3.1.2 Korrelation zwischen konvulsiver PTZ-Dosis und dem Plasmaspiegel
Um den Zusammenhang zwischen konvulsiver PTZ-Dosis und dem dazugehörigen
Plasmaspiegel zu erfassen, haben wir den PTZ-Schwellentest durchgeführt und
direkt nach dem Krampf (Myoklonus) PTZ-Plasmaspiegel bestimmt. Der PTZ-
Schwellentest wurde wie in Kapitel 4.6.4 beschrieben durchgeführt. Direkt nach dem
Myoklonus wurde bei den Tieren retrobulbär Blut entnommen (unter Tetracain-
Lokalanästhesie). Der PTZ-Gehalt der Plasmaproben wurde mittels HPLC analysiert.
Der Versuch wurde an 2 naiven Ratten und 2 Ratten 24 h nach pilokarpin-
induziertem SE durchgeführt. Die Plasmaspiegel [µg/ml] waren hochsignifikant mit
der errechneten PTZ-Schwelle [mg/kg] korreliert, Korrelationskoeffizient r=0,9831
(p=0,0085). Es gab keinen Unterschied zwischen den naiven- und den post SE-
Tieren. Da die niedrigste konvulsive Dosis bei 14 mg/kg lag und diese mit einem
Plasmaspiegel von 18 µg/ml korrelierte, haben wir uns entschlossen in den
Infunsionsversuchen eine Plasmakonzentration von 10 µg PTZ pro ml Plasma
anzustreben. Das Ziel war, einen Plasmaspiegel aufrechtzuerhalten, der im
subkonvulsiven Bereich liegt.
Abb. 34: Darstellung der konvulsiven PTZ-Dosis [mg/kg] in Verhältnis zur PTZ- Plasmakonzentration [µg/ml]. Dargestellt die Tiere 24 h nach pilokarpin-induziertem SE, und naive Tiere (siehe Legende). Die Korrelation wurde mittels Pearson`s–Korrelationstest ermitelt.
0 10 20 30 400
10
20
30
40
PTZ-Schwelle versus Plasmaspiegel
SE-Tiere
naive Tiere
PTZ im Plasma [µg/ml]
PT
Z [
mg
/kg
]

Ergebnisse
101
5.3.1.3 Etablierung des Infusionsprotokolls
Als subkonvulsive PTZ-Plasmakonzentration haben wir anhand der PTZ-
Schwellenversuche und der pharmakokinetischen Vorversuche PTZ-Plasmaspiegel
von 10 µg/ml festgelegt. Um diese Plasmakonzentration aufrechtzuerhalten, haben
die Ratten (n=3) der ersten Behandlungsgruppe einen Bolus von 10 mg/kg und zur
Aufrechterhaltung eine Infusion von 15 µg/kg/min erhalten. Vierundzwanzig und 48
Stunden nach Infusionsbeginn wurde bei den Tieren retrobulbär Blut entnommen und
der PTZ-Plasmaspiegel mittels HPLC analysiert. Die PTZ-Plasmaspiegel sind in
nachfolgender Tabelle dargestellt. Da die Zielkonzentration von PTZ im Plasma nicht
erreicht werden konnte, wurde in der nächsten Tiergruppe (n=2) das
Infusionsprotokoll auf 60 µg/kg/min umgestellt. Bei beiden Tieren lag die ermittelte
Plasmakonzentration im Bereich der Zielplasmakonzentration (siehe Tabelle 17).
Tier PTZ [µg/ml] nach 24 h PTZ [µg/ml] nach 48 h
MR 138 2,48 -
MR 139 1,75 3,11
BUM 36 1,63 1,15
Tabelle 16: PTZ-Plasmakonzentrationen 24 h und 48 h nach Infusion von 15 µg/kg/min PTZ. Ein leeres Feld bedeutet, dass keine Blutprobe entnommen werden konnte.
Tier PTZ [µg/ml] nach 24 h PTZ [µg/ml] nach 48 h
MR 156 8,38 9,53
MR 157 7,18 10,23
Tabelle 17: PTZ-Plasmakonzentrationen 24 h und 48 h nach Infusion von 60 µg/kg/min PTZ.
Weder in der 15 µg-Gruppe, noch in der 60 µg-Gruppe kam es bei den naiven Ratten
zu PTZ-induzierten Nebenwirkungen. Aus diesem Grund haben wir uns
entschlossen, das zweite Infusionsprotokoll in allen weiteren Versuchen zu
verwenden.

Ergebnisse
102
5.3.2 Infusionsversuche mit PTZ im Pilokarpin-Modell (60 Minuten SE-Gruppe)
5.3.2.1 SE-Induktion
In diesem Versuch konnte der SE bei 6 von 6 Ratten induziert werden. Wie bereits in
Kapitel 5.2.1.1 beschrieben hatten die Tiere während des SE generalisierte Anfälle,
die mit stereotypem Verhalten einhergingen. Die mittlere Pilokarpin-Dosis lag bei
46,7 mg/kg, die mittlere Zeit zum SE-Eintritt betrug 73,8 Minuten. Eine Ratte starb an
Atemdepression nach SE-Abbruch, so dass nur 5 Tiere in den weiteren Versuchen
dieser Studie verwendet wurden. Durch den modifizierten SE-Abbruch gelang es, die
motorische Krampfaktivität und paroxysmale EEG-Veränderungen effektiv zu
unterdrücken (siehe Kapitel 4.7.5.1).
5.3.2.2 PTZ-Behandlung
Die PTZ-Behandlung erfolgte durch intravenöse Infusion. Bei den Tieren konnten
während der gesamten Behandlungsdauer keine generalisierten Anfälle beobachtet
werden. Zwei der 5 Tiere hatten Myoklonien (über ~ 10 min) 20 bzw. 25 h nach
Beginn der Infusion. Die Zielplasmakonzentration konnte bei allen Tieren erreicht
werden.
5.3.2.3 Entwicklung von spontanen Anfällen nach PTZ-Behandlung
Insgesamt wurden 7 Tiere mit PTZ behandelt, zwei naive Tiere (MR 156 und MR
157 aus den Kinetik-Versuchen) ohne SE und 5 SE-Tiere (MR 154, MR 158, MR
159, MR 161, MR 162). Von den 5 SE-Tieren wurden 3 Tiere (MR 159, MR 161, MR
162) 4 und 7 Wochen nach der Behandlung EEG- und videoüberwacht. Die anderen
2 SE-Tiere (MR 154 und MR 158) und die 2 naiven Tiere (MR 156 und MR 157 aus
den Kinetik-Versuchen) wurden 8 Wochen nach Behandlung überwacht. Die
unterschiedlichen Überwachungszeitpunkte kommen infolge der Verfügbarkeit der
Überwachungsplätze aus organisatorischen Gründen zustande. Die
Überwachungsdauer betrug jeweils 1 Woche. Die naiven Ratten (n=2) hatten keine
Anfälle. Das lässt darauf schließen, dass die alleinige Behandlung mit PTZ nicht
proepileptogen wirkte.

Ergebnisse
103
Von den 5 Ratten mit SE hatte nur eine (MR 161) Anfälle während der Überwachung
(3 Anfälle pro Woche, 4 Wochen nach Behandlung und 11 Anfälle pro Woche, 7
Wochen nach Behandlung). Alle anderen Tiere blieben anfallsfrei. Dieser Effekt ist
jedoch nicht auf die PTZ-Behandlung zurückzuführen, da Tiere aus anderen
Projekten, die nach dem 60 min SE-Protokoll behandelt wurden, ebenfalls keine
Epilepsie entwickelten (1 von 10 Tieren hatte 40 Wochen nach SE fokale Anfälle).
Aus diesem Grund war anzunehmen, dass ein SE von 60 Minuten kein ausreichend
starker Insult war, um spontane Anfälle auszulösen. Nach diesen Experimenten
haben wir uns entschlossen, die SE-Dauer auf 90 Minuten zu erhöhen und die
Versuche mit einem modifizierten Protokoll zu wiederholen.
5.3.3 Infusionsversuche mit PTZ im Pilokarpin-Modell (90 Minuten SE-Gruppe)
5.3.3.1 SE-Induktion
In diesem Versuch konnte der SE bei 8 von 9 Ratten induziert werden (88,9%). Wie
bereits in Kapitel 4.3.1 beschrieben, hatten die Tiere während des SE generalisierte
Anfälle, die mit stereotypem Verhalten einhergingen. Die mittlere Pilokarpin-Dosis lag
bei 43,75 mg/kg, die mittlere Zeit zum SE-Eintritt betrug 61,75 Minuten. Eine Ratte
starb nach SE-Abbruch, so dass nur 7 Tiere in den weiteren Versuchen dieser Studie
verwendet wurden.
5.3.3.2 PTZ-Behandlung
Die PTZ-Behandlung erfolgte auch hier durch intravenöse Infusion. Pro
Versuchsdurchlauf konnten maximal 8 Ratten gleichzeitig behandelt werden. Die
Tiere wurden in zwei Behandlungsgruppen eingeteilt: SE+PTZ (n=4) und SE+Saline
(n=3). Es traten keine generalisierten Anfälle während der Infusion auf. Ein Tier hatte
einen generalisierten Anfall (Typ IV) während der Blutentnahme nach Ende der
Behandlung. Die Zielplasmakonzentration konnte bei 3 von 4 PTZ-Ratten (eine Ratte
zog sich den Katheter vor Ende der Behandlung) erreicht werden. Eine der
Kontrollratten hatte PTZ im Plasma, was vermutlich auf einer Verwechslung der
Infusionssysteme beruhte. Beide Tiere wurden von der Auswertung ausgeschlossen.
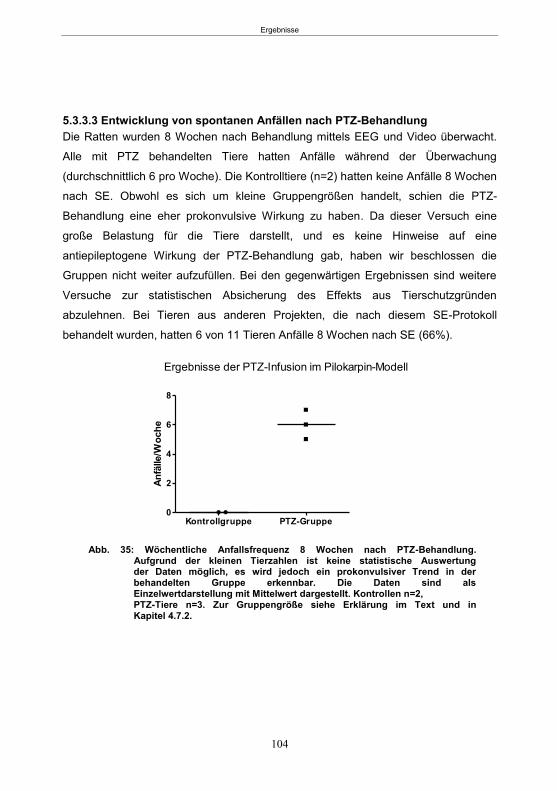
Ergebnisse
104
5.3.3.3 Entwicklung von spontanen Anfällen nach PTZ-Behandlung
Die Ratten wurden 8 Wochen nach Behandlung mittels EEG und Video überwacht.
Alle mit PTZ behandelten Tiere hatten Anfälle während der Überwachung
(durchschnittlich 6 pro Woche). Die Kontrolltiere (n=2) hatten keine Anfälle 8 Wochen
nach SE. Obwohl es sich um kleine Gruppengrößen handelt, schien die PTZ-
Behandlung eine eher prokonvulsive Wirkung zu haben. Da dieser Versuch eine
große Belastung für die Tiere darstellt, und es keine Hinweise auf eine
antiepileptogene Wirkung der PTZ-Behandlung gab, haben wir beschlossen die
Gruppen nicht weiter aufzufüllen. Bei den gegenwärtigen Ergebnissen sind weitere
Versuche zur statistischen Absicherung des Effekts aus Tierschutzgründen
abzulehnen. Bei Tieren aus anderen Projekten, die nach diesem SE-Protokoll
behandelt wurden, hatten 6 von 11 Tieren Anfälle 8 Wochen nach SE (66%).
Kontrollgruppe PTZ-Gruppe0
2
4
6
8
An
fälle/W
och
e
Ergebnisse der PTZ-Infusion im Pilokarpin-Modell
Abb. 35: Wöchentliche Anfallsfrequenz 8 Wochen nach PTZ-Behandlung. Aufgrund der kleinen Tierzahlen ist keine statistische Auswertung der Daten möglich, es wird jedoch ein prokonvulsiver Trend in der behandelten Gruppe erkennbar. Die Daten sind als Einzelwertdarstellung mit Mittelwert dargestellt. Kontrollen n=2, PTZ-Tiere n=3. Zur Gruppengröße siehe Erklärung im Text und in Kapitel 4.7.2.

Ergebnisse
105
5.3.4 Infusionsversuche im fokalen Kainsäure-Modell (13-16 Stunden-Gruppe)
5.3.4.1 SE-Induktion
In diesem Versuch konnte der SE bei 6 von 6 Ratten induziert werden. Wie bereits
beschrieben zeigten die Tiere Stereotypien, welche von generalisierten Anfällen
begleitet wurden und mit paroxysmalen EEG-Veränderungen einhergingen. Die Zahl
und Dauer der auszählbaren EEG-Anfälle konnte bei diesen Tieren nicht bestimmt
werden, da zu vorgegebenen Zeitpunkten nach SE die PTZ-Behandlung erfolgte.
5.3.4.2 PTZ-Behandlung
Die PTZ-Behandlung startete 13 h (n=1) oder 16 h (n=3) nach Kainsäure-
Mikroinjektion. Die PTZ-Behandlung erfolgte auch hier durch intravenöse Infusion.
Pro Versuchsdurchlauf konnten maximal 8 Ratten gleichzeitig behandelt werden. Alle
Tiere hatten Anfälle unter der PTZ-Infusion, 3 von 4 direkt nach Bolus-Applikation.
Ein Tier (MR 177) wurde nur 5 Stunden lang infundiert, weil danach der Katheter
beschädigt wurde. Da auch die beiden Kontrolltiere während des
Behandlungszeitraums Anfälle zeigten, haben wir beschlossen den Zeitpunkt der
Behandlung zu wechseln. Da Kontrollratten in diesem Zeitraum nach SE Anfälle
hatten, ist es nicht auszuschließen, dass ein Teil der beobachteten Anfälle SE-
induziert war, und PTZ-unabhängig auftrat. Eine Behandlung 13-16 h nach
Kainsäure-Mikroinjektion konnte bei einigen Tieren in die SE-Dauer fallen und diesen
verstärken. Da der SE nicht abgebrochen wurde und die SE-Dauer von Tier zu Tier
variierte, war es schwierig einen SE-nahen Behandlungszeitpunkt festzulegen. Aus
diesem Grund wurde im nächsten Versuch eine Behandlung 7 Tage nach SE
vorgenommen.
5.3.4.3 Entwicklung von spontanen Anfällen nach PTZ-Behandlung
Alle Tiere wurden 8 Wochen nach PTZ-Behandlung EEG- und videoüberwacht. Die
Tiere, die keine Anfälle 8 Wochen nach Behandlung zeigten, wurden nach 12
Wochen erneut überwacht. Die Dauer der Überwachung betrug jeweils eine Woche.
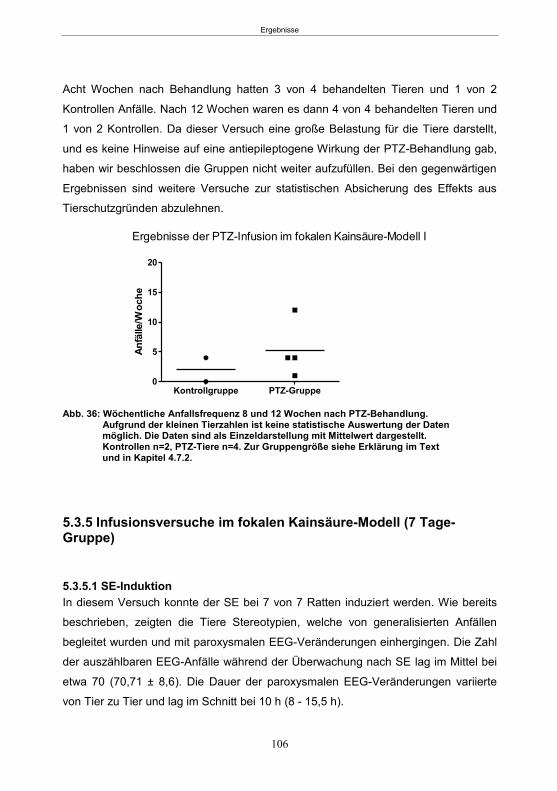
Ergebnisse
106
Acht Wochen nach Behandlung hatten 3 von 4 behandelten Tieren und 1 von 2
Kontrollen Anfälle. Nach 12 Wochen waren es dann 4 von 4 behandelten Tieren und
1 von 2 Kontrollen. Da dieser Versuch eine große Belastung für die Tiere darstellt,
und es keine Hinweise auf eine antiepileptogene Wirkung der PTZ-Behandlung gab,
haben wir beschlossen die Gruppen nicht weiter aufzufüllen. Bei den gegenwärtigen
Ergebnissen sind weitere Versuche zur statistischen Absicherung des Effekts aus
Tierschutzgründen abzulehnen.
5.3.5 Infusionsversuche im fokalen Kainsäure-Modell (7 Tage-Gruppe)
5.3.5.1 SE-Induktion
In diesem Versuch konnte der SE bei 7 von 7 Ratten induziert werden. Wie bereits
beschrieben, zeigten die Tiere Stereotypien, welche von generalisierten Anfällen
begleitet wurden und mit paroxysmalen EEG-Veränderungen einhergingen. Die Zahl
der auszählbaren EEG-Anfälle während der Überwachung nach SE lag im Mittel bei
etwa 70 (70,71 ± 8,6). Die Dauer der paroxysmalen EEG-Veränderungen variierte
von Tier zu Tier und lag im Schnitt bei 10 h (8 - 15,5 h).
Kontrollgruppe PTZ-Gruppe0
5
10
15
20
Ergebnisse der PTZ-Infusion im fokalen Kainsäure-Modell I
An
fälle/W
och
e
Abb. 36: Wöchentliche Anfallsfrequenz 8 und 12 Wochen nach PTZ-Behandlung. Aufgrund der kleinen Tierzahlen ist keine statistische Auswertung der Daten möglich. Die Daten sind als Einzeldarstellung mit Mittelwert dargestellt. Kontrollen n=2, PTZ-Tiere n=4. Zur Gruppengröße siehe Erklärung im Text und in Kapitel 4.7.2.

Ergebnisse
107
Da die Tiere erst 7 Tage nach SE-Induktion behandelt werden sollten, wurden sie 4
Tage nach SE, EEG- und videoüberwacht. Drei von 7 Tieren entwickelten Anfälle in
den ersten 2 Tagen nach SE (siehe Tabelle 18). Diese Anfälle waren möglicherweise
insultassoziiert und auf die SE-Induktion zurückzuführen (siehe Kapitel 5.2.1.1).
5.3.5.2 PTZ-Behandlung
Die PTZ-Behandlung startete in dieser Gruppe 7 Tage nach SE-Induktion. Die PTZ-
Behandlung erfolgte auch hier durch intravenöse Infusion. Pro Versuchsdurchlauf
konnten maximal 8 Ratten gleichzeitig behandelt werden.Vier von 5 behandelten
Tieren hatten Anfälle während der Infusion, 2 von ihnen unter Bolus-Gabe. Im
Vergleich dazu hatte 1 von 2 Kontrollratten (mit isotoner Natriumchloridlösung
infundiert) Anfälle in diesem Zeitraum (siehe Tabelle 18). Bei allen Tieren lag der
PTZ-Plasmaspiegel im angestrebten Bereich.
SE+PTZ-Tiere Anfälle während der PTZ-
Infusion
Anfälle in den ersten
48 h nach SE
(insultassoziiert)
MR 185 ja nein
MR 186 ja nein
MR 187 nein nein
MR 189 ja ja
MR 191 ja ja
SE+Saline-Tiere Anfälle während der NaCl-Infusion
Anfälle in den ersten 48 h nach SE
(insultassoziiert) MR 190 ja nein
MR 194 nein ja
Tabelle 18: Anfälle während der 48 h PTZ-Behandlung und Anfälle in den ersten 48 h nach SE (insultassoziierte Anfälle).
5.3.5.3 Entwicklung von spontanen Anfällen nach PTZ-Behandlung
Alle Tiere wurden 8 Wochen nach PTZ-Behandlung EEG- und videoüberwacht. Die
Tiere, die keine Anfälle 8 Wochen nach Behandlung zeigten, wurden nach 16
Wochen erneut überwacht. Die Dauer der Überwachung betrug jeweils eine Woche.
Acht Wochen nach Behandlung hatten 3 von 5 behandelten Tieren und 1 von 2
Kontrollen Anfälle.

Ergebnisse
108
Nach 16 Wochen waren es dann 4 von 5 behandelten Tieren und 2 von 2 Kontrollen.
Da dieser Versuch eine große Belastung für die Tiere darstellt, und es keine
Hinweise auf eine antiepileptogene Wirkung der PTZ-Behandlung gab, haben wir
beschlossen die Gruppen nicht weiter aufzufüllen. Bei den gegenwärtigen
Ergebnissen sind weitere Versuche zur statistischen Absicherung des Effekts aus
Tierschutzgründen abzulehnen.
Kontrollgruppe PTZ-Gruppe0
5
10
15
20
Ergebnisse der PTZ-Infusion im fokalen Kainsäure-Modell II
An
fälle/W
och
e
Abb. 37 : Wöchentliche Anfallsfrequenz 8 und 16 Wochen nach PTZ-Behandlung. Aufgrund der kleinen Tierzahlen ist keine statistische Auswertung der Daten möglich. Die Daten sind als Einzelwertdarstellung mit Mittelwert dargestellt. Kontrollen n=2, PTZ-Tiere n=5. Zur Gruppengröße siehe Erklärung im Text und in Kapitel 4.7.2.
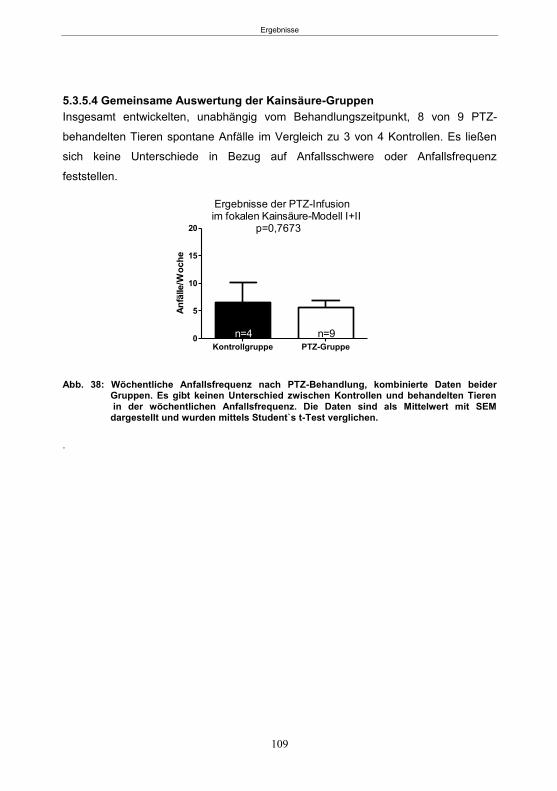
Ergebnisse
109
5.3.5.4 Gemeinsame Auswertung der Kainsäure-Gruppen
Insgesamt entwickelten, unabhängig vom Behandlungszeitpunkt, 8 von 9 PTZ-
behandelten Tieren spontane Anfälle im Vergleich zu 3 von 4 Kontrollen. Es ließen
sich keine Unterschiede in Bezug auf Anfallsschwere oder Anfallsfrequenz
feststellen.
Abb. 38: Wöchentliche Anfallsfrequenz nach PTZ-Behandlung, kombinierte Daten beider Gruppen. Es gibt keinen Unterschied zwischen Kontrollen und behandelten Tieren in der wöchentlichen Anfallsfrequenz. Die Daten sind als Mittelwert mit SEM dargestellt und wurden mittels Student`s t-Test verglichen.
.
Kontrollgruppe PTZ-Gruppe0
5
10
15
20
n=4 n=9
Ergebnisse der PTZ-Infusionim fokalen Kainsäure-Modell I+II p=0,7673
An
fälle/W
och
e

6. Diskussion
6.1 Einleitung Im Rahmen dieser Arbeit sollte der Einsatz des prokonvulsiven GABAA-Rezeptor-
Antagonisten PTZ als Strategie zur Epilepsieprävention untersucht werden. Da es bis
heute keine Möglichkeit gibt, Risikopatienten präventiv zu behandeln, werden
dringend antiepileptogene Behandlungsmöglichkeiten gesucht. Die naheliegendste
Strategie wäre eine Behandlung mit antikonvulsiven Mitteln, es konnte jedoch für
eine Reihe dieser Stoffe gezeigt werden, dass sie kein antiepileptogenes Potential
haben (TEMKIN 2001 und 2009). Der Einsatz von prokonvulsiven Mitteln als
neuromodulatorische Strategie der Epilepsieprävention wird aufgrund von neuen
Studien immer häufiger diskutiert (ARMSTRONG et al. 2009). Aus diesem Grund
sollte auch in dieser Studie ein prokonvulsiver Stoff (PTZ) auf sein antiepileptogenes
Potential hin untersucht werden. Um das zu erreichen, wurden drei verschiedene
Studien durchgeführt. Da die Untersuchungen vergleichend in zwei
Epilepsiemodellen durchgeführt werden sollten, wurde zuerst das fokale Kainsäure-
Modell für unser Labor etabliert (Studie 1). Um Veränderungen der neuronalen
Erregbarkeit nach Insult zu untersuchen, wurden in der zweiten Studie
Veränderungen der individuellen Krampfschwelle nach SE untersucht (Studie 2). Als
letztes wurden pharmakokinetische Versuche vorgenommen, und es wurde ein
prophylaktischer Behandlungsversuch mit PTZ unternommen (Studie 3). Die
Ergebnisse werden im Folgenden einzeln für jede Studie diskutiert.
6.2 Studie 1: Etablierung des fokalen Kainsäure-Modells.
6.2.1 SE-Induktion
In dieser Studie gelang es uns, durch Kainsäure-Mikroinjektion an wachen,
freibeweglichen Ratten ein neues Modell für Temporallappenepilepsie für unser
Labor zu etablieren.

Diskussion
111
Die Kainsäure-Mikroinjektion führte bei allen Tieren zum Eintritt eines SE, was
übereinstimmt mit der Arbeit zum fokalen Kainsäure-Modell von RAEDT et al. (2009)
und einen Vorteil darstellt gegenüber dem systemischen Pilokarpin-Modell und
anderen Modellen (GLIEN et al. 2001, NISSINEN et al. 2000 NISSINEN und
PITKÄNEN 2007 FURTADO et al. 2011), in denen nicht bei allen behandelten Tieren
ein SE auslösbar ist. Wie bei RAEDT et al. (2009) korrelierte auch bei uns weder die
SE-Dauer noch die im EEG quantifizierten EEG-Veränderungen mit dem späteren
Auftreten von Anfällen oder der Anfallsfrequenz. Dadurch, dass es nur eine bipolare
Ableitelektrode im Hippokampus gab, so dass der Unterschied zwischen fokalen und
generalisierten Anfällen nicht bestimmt werden konnte, war die Messung von EEG-
Veränderungen auch nur begrenzt möglich. Aus diesem Grund kann man nicht
ausschliessen, dass es einen Zusammenhang gibt zwischen der Schwere des SE
und der späteren Anfallsausprägung, vielmehr ist anzunehmen, dass die SE-
Schwere nicht ausschließlich am ermittelten EEG-Bild festgemacht werden kann. In
einer Arbeit von BRANDT et al. (2003) konnte für das basolaterale Amygdala
Stimulations-Modell (elektrisches post SE-Epilepsiemodell) gezeigt werden, dass die
SE-Schwere von großer Bedeutung für die spätere Anfallsentwicklung ist. Die SE-
Schwere wurde anhand der auftretenden klinischen Anfälle bestimmt. Tiere mit
schwerstem SE, bei denen kontinuierlich generalisierte tonisch-klonische Anfälle
auftraten, entwickelten fast alle (> 90%) Anfälle. Im Vergleich dazu hatten nur etwa
30% der Tiere mit fokalen Anfällen während des SE auch später epileptische Anfälle.
Da in unserer Arbeit der SE nur im EEG aufgrund einer bipolaren Ableitelektrode im
Hippokampus und nicht im Verhalten beurteilt wurde, ist eine Korrelation zwischen
ausgewerteten SE-Parametern und späterer Anfallsausprägung nicht zu erwarten.
Ein weiterer Vorteil dieses Modells war die geringe Mortalität nach SE (insgesamt
starb in allen Versuchen nur eins von 42 Tieren nach SE) und die Tatsache, dass die
Ratten sich sehr schnell vom SE erholten und schon am nächsten Tag normale
Futteraufnahme zeigten. Das ist ein Unterschied zum systemischen Pilokarpin-
Modell sowohl in der hochdosierten Variante als auch im fraktionierten Modell
(GLIEN et al. 2001). Wie bei GLIEN et al. (2001) beschrieben, ist das hochdosierte
Pilokarpin-Modell durch eine hohe Mortalitätsrate charakterisiert. Diese lässt sich
durch die fraktionierte Pilokarpin-Gabe senken. Flüssigkeitssubstitution und
Zwangsernährung über mehrere Tage nach SE lassen sich jedoch nicht vermeiden.

Diskussion
112
Aus diesem Grund stellt das fokale Kainsäure-Modell eine vorteilhaftere Möglichkeit
der SE-Induktion dar, was das Wohlbefinden der Tiere nach SE betrifft. Analog zu
den Studien von BRAGIN et al. (1999, 2004 und 2007) und RAEDT et al. (2009)
wurde der SE in diesem Modell nicht pharmakologisch abgebrochen. Aufgrund der
vorhandenen Literaturdaten wussten wir nicht welche SE-Länge notwendig ist, damit
die Tiere später eine chronische Epilepsie entwickeln. Dies war ein Nachteil dieses
Modells, da die SE-Länge von Tier zu Tier variierte, und somit der Insult nicht bei
allen Tieren von gleicher Dauer war. Aufgrund der ermittelten durchschnittlichen SE-
Länge von etwa 11 h könnte man in weiterführenden Untersuchungen versuchen,
den SE nach 8 h oder 10 h abzubrechen.
6.2.2 Anfallsausprägung nach SE
Die Ratten wurden in diesem Versuch vier, sechs und acht Wochen nach SE
überwacht. In diesem Zeitraum entwickelten 86% der Tiere spontane Anfälle. Bei
RAEDT et al. (2009) entwickelten entsprechend 87% der Tiere spontane Anfälle in
den ersten acht Wochen nach SE, bei BRAGIN et al. (2004) waren es 73%. Acht
Ratten wurden 33 Wochen nach SE erneut überwacht. Alle Tiere entwickelten in
diesem Zeitraum Anfälle. Die mittlere wöchentliche Anfallsfrequenz stieg signifikant,
was für eine Progression der Krankheit spricht. Die Progression der durch Kainsäure-
Mikroinjektion induzierten Epilepsie wurde ebenfalls von Raedt et al. (2009)
beschrieben. Im Gegensatz dazu beschrieb CAVALHEIRO et al. (1982), dass Anfälle
nur etwa 30 Tage nach der SE-Induktion durch Kainsäure-Mikroinjektion auftreten.
Da in der Studie von CAVALHEIRO et al. (1982) die SE-Induktion an anästhesierten
Ratten durchgeführt wurde, kann man annehmen, dass die Anästhesie einen
insultmodifiziereden Effekt hatte. Dieser konnte bei einer Mikroinjektion, durchgeführt
an wachen Ratten, eliminiert werden.
6.2.3 Verhaltensänderungen
Da psychiatrische Komorbiditäten eine große Rolle bei Patienten mit
Temporallappenepilepsie spielen (HERMANN et al. 2008), ist es wichtig, Tiermodelle
zu entwickeln, die diese widerspiegeln.

Diskussion
113
In der vorliegenden Arbeit wurden Verhaltensversuche in diesem Modell (Kainsäure-
Mikroinjektion an wachen Ratten) erstmals durchgeführt. Wir nahmen an, dass die
Verhaltensänderungen durch den moderaten Insult weniger schwer sind als in
systemischen Modellen. Es ließ sich eine Reihe von Verhaltensauffälligkeiten bei den
epileptischen Tieren beobachten.
In dem Hyperexzitabilitätstest nach RICE et al. (1998) zeigten die epileptischen
Ratten im fokalen Kainsäure-Modell Anzeichen von Hyperexzitabilität im Touch-
Response- und Finger-Snap-Test, nicht aber im Approach-Response- und Pick-up-
Test. Im Gegensatz dazu äußert sich die Hyperexzitabilität im systemischen
Pilokarpin-Modell durch Auffälligkeiten im Touch-Response- und Pick-up-Test (RICE
et al. 1998). Die Tatsache, dass die Kainsäure-Ratten keine Auffälligkeiten im Pick-
up-Test zeigten, stimmt mit Beobachtungen während des Umgangs mit den Tieren
überein. Die Tiere waren in den ersten Monaten nach SE einfach zu handhaben und
zeigten Artgenossen gegenüber soziales Verhalten. Sie zeigten keine Aggressivität
gegenüber den Experimentatoren. Dies änderte sich zwar zunehmend mit der
Progression der Krankheit, die epilepsieassoziierte Hyperexzitabilität war jedoch
insgesamt nicht so stark ausgeprägt wie es bei Ratten aus dem systemischen
Piokarpin-Modell der Fall sein kann (RICE et al. 1998, POLASCHECK et al. 2010).
Somit stellt das fokale Kainsäure-Modell eine Möglichkeit dar, moderate
Hyperexzitabilität bei Ratten zu erzeugen.
Die Ratten des fokalen Kainsäure-Modells zeigten tendenziell weniger
angstassoziiertes- und mehr Explorationsverhalten im EPM und OF im Vergleich zu
Kontrollen. Dies stimmt überein mit der Arbeit von MUNOZ und GROSSMAN 1980,
in welcher Ratten nach einseitiger Hippokampus-Läsion durch Kainsäure eine
Hyperlokomotion im OF im Vergleich zu Kontrollen zeigten, diese jedoch erst bei
beidseitig lädierten Tieren signifikant wurde. Insgesamt treten bei epileptischen
Tieren oft stark ausgeprägte Verhaltensabnormalitäten, wie verändertes
angstassoziiertes Verhalten und Hyper- oder Hypolokomotion auf (HELFER et al.
1996, DETOUR et al. 2005 BRANDT et al. 2006 SUN et al. 2009 POLASCHECK et
al. 2010). In Studien im Amygdala-Kindling-Modell konnte bei Ratten oft
Hypolokomotion und vermehrtes angstassoziiertes Verhalten beobachtet werden
(ADAMEC 1990, HELFER et al. 1996). In Studien mit systemischer SE-Induktion gibt
es kontroverse Ergebnisse.

Diskussion
114
In der Arbeit von SUN et al. (2009) zeigten Ratten nach systemischer SE-Induktion
mit Kainsäure Hyperlokomotion und gesteigertes angstassoziiertes Verhalten. Im
Gegensatz dazu konnte in mehreren Studien zum systemischen Pilokarpin-Modell
eine Hyperlokomotion mit vermindertem angstassoziierten Verhalten beobachtet
werden (DETOUR et al. 2005, BRANDT et al. 2006, POLASCHECK et al. 2010).
Diese Verhaltensänderungen der epileptischen Ratten sind nicht konform mit der
klinischen Situation, da Angststörungen eine häufige epilepsieassoziierte
Komorbidität darstellen (HERMANN et al. 2008, KANNER 2011). Sie lassen sich
jedoch durch pilokarpin-verursachte großflächige Läsionen des limbischen Systems
erklären (von besonderer Bedeutung ist die neuronale Verschaltung zwischen
Hippokampus und Amygdala), welches wichtig für Emotionen und emotionales
Verhalten ist. Das könnte bei den betroffenen Tieren dazu führen, dass aversive
Situationen fehlinterpretiert werden (DETOUR et al. 2005). Im Hinblick auf diese
Theorie erscheint es logisch, dass Tiere mit fokalen Läsionen weniger von solchen
Verhaltensabnormalitäten betroffen sind als Tiere nach systemischer SE-Induktion
mit Pilokarpin.
Im MWM nach MORRIS (1984), in dem räumliche Orientierung und Gedächtnis
untersucht werden, lernten die epileptischen Ratten im fokalen Kainsäure-Modell
langsamer als die Kontrollen, die Plattformposition zu finden. Das spricht für
Störungen der hippokampalen Integrität, welche für räumliche Orientierung und
Gedächtnis verantwortlich ist (D'HOOGE und DE DEYN 2001). Auch im Probe trial,
in welchem die Beibehaltung und Abfrage des Erlernten getestet wird (BOLDING und
RUDY 2006), schnitten die epileptischen Tiere deutlich schlechter ab als Kontrollen.
Dies deutet darauf hin, dass es den Tieren nicht nur schwerer fiel, neue Aufgaben zu
erlernen, auch die Abfrage des einmal Erlernten war gestört. Diese Daten stimmen
überein mit Untersuchungen im fokalen Kainsäure-Modell bei Mäusen von
GRÖTICKE et al. (2008), in denen die epileptischen Mäuse im MWM langsamer
lernten im Vergleich zu Kontrollen. Gedächtnisprobleme bei den Ratten sprechen für
das Modell, da solche Probleme auch bei humanen Epilepsiepatienten eine große
Rolle spielen (HOPPE et al. 2007).

Diskussion
115
Eine weitere Erklärung der Schwierigkeiten von epileptischen Ratten im MWM
könnte die Tatsache sein, dass die Tiere von der Komplexizität der Aufgabe
überfordert waren, da einige Tiere niemals auf der Plattform sitzen blieben und das
Auffinden der Plattform nicht als Ausweg aus der unangenehmen Situation begriffen.
Da diese Aufgabe nicht ausschließlich räumliche Orientierung sondern auch
Fähigkeit zu konditioniertem Lernverhalten voraussetzt, ist es wahrscheinlich, dass
beides für epileptische Tiere ein Problem darstellt. Auch bei epileptischen Patienten
werden kognitive Dysfunktionen als häufige Komorbidität beschrieben (JENSEN
2011).
6.2.4 Pharmakologische Untersuchungen
Ein wichtiger Bestandteil der Studie im fokalen Kainsäure-Modell war die Behandlung
der epileptischen Ratten mit Phenobarbital. Hier sollte beurteilt werden, wie die Tiere
auf eine Behandlung mit einem Standard-Antiepileptikum ansprechen. Da
Pharmakoresistenz ein großes Problem in der Behandlung der
Temporallappenepilepsie darstellt, sollte ein Rattenmodell idealerweise auch diese
Kondition widerspiegeln. Aus diesem Grund wurde eine Behandlung zur Selektion
von Phenobarbital-Respondern und Phenobarbital-Nonrespondern vorgenommen.
Die Untersuchung wurde an acht Ratten durchgeführt, die 33 Wochen nach SE für
vier Tage behandelt wurden. Der Behandlung ging eine viertägige Kontrollphase
voraus. Auf die Behandlung folgte eine viertägige Auswaschphase. Bei allen Tieren
konnte eine Phenobarbital-Plasmakonzentration im wirksamen Bereich erzielt
werden. Während der Behandlungsphase kam es bei allen Tieren zu einer
signifikanten Anfallsreduktion. Vier Tiere wurden anfallsfrei. Alle Tiere sprachen
somit gut auf die Behandlung an. Dieses Ergebnis war insofern unerwartet, da die
Behandlung zu einem späten Zeitpunkt (33 Wochen) nach SE-Induktion erfolgte, und
eine lang unbehandelte Epilepsie klinisch ein Prädiktor für Pharmakoresistenz ist
(LÖSCHER und BRANDT 2010 b). In verschiedenen Untersuchungen sowohl
unserer (GLIEN et al. 2002, BRANDT et al. 2004, BETHMANN et al. 2007) als auch
anderer Arbeitsgruppen (NISSINEN und PITKÄNEN 2007) konnte gezeigt werden,
dass epileptische Ratten aus verschiedenen Epilepsiemodellen sich in ihrer
individuellen Reaktion auf eine Therapie mit Antiepileptika unterscheiden.

Diskussion
116
So reagieren Ratten, die nach einem SE durch elektrische Stimulation der
basolateralen Amygdala epileptisch werden im Schnitt zu etwa 30% nicht mit einer
mindestens 50%igen Anfallsreduktion auf die Behandlung (BRANDT et al. 2004).
Diese Tiere werden als Nonresponder bezeichnet. Nonresponder sind oft Tiere mit
einer hohen Anfallsfrequenz und starken epilepsieassoziierten
Verhaltensänderungen (GASTENS et al. 2008, LÖSCHER und BRANDT 2010 b).
Rattenmodelle für Temporallappenepilepsie, in denen Responder und Nonresponder
identifiziert werden können, bieten eine Grundlage zur Erforschung von
Mechanismen der Pharmakoresistenz bei Epilepsiepatienten. In unserer Studie im
fokalen Kainsäure-Modell konnten keine Nonresponder identifiziert werden, selbst
Tiere mit einer hohen Anfallsfrequenz sprachen gut auf die Behandlung mit
Phenobarbital an. Dieses Ergebnis war umso überraschender, da in einer Studie von
RIBAN et al. (2002) im fokalen Kainsäure-Modell bei der Maus gezeigt werden
konnte, dass paroxsysmale EEG-Veränderungen durch akute Diazepam-Gabe
unterdrückt wurden, nicht jedoch durch Carbamazepin, Phenytoin oder Valproat.
Diazepam verlor seine Wirksamkeit bei chronischer Behandlung über mehrere Tage.
Aus diesem Grund wurde in dieser Studie das fokale Kainsäure-Modell bei der Maus
als gutes Modell zur Untersuchung von Pharmakoresistenz beim Menschen
beschrieben. Die Untersuchungen wurden drei bis sechs Wochen nach SE
durchgeführt, und der Behandlungserfolg wurde ausschließlich anhand des EEG-
Bildes beurteilt. In unserer Studie hingegen wurde der Behandlungserfolg anhand
der spontanen Anfälle im Verhalten beurteilt, und die Behandlung erfolgte über
mehrere Tage, was die Unterschiede zwischen beiden Studien erklären kann.
Natürlich können auch Speziesunterschiede in diesem Fall eine Rolle spielen.
Wahrscheinlich ist jedoch die Dauer der Behandlung maßgeblich für das Ergebnis
verantwortlich, da eine Toleranz und die damit verbundene schlechtere
Ansprechbarkeit auf die Behandlung erst später im Laufe der Behandlung auftritt,
diese in unserer Studie aber auf vier Tage limitiert war. In einer Studie von GLIEN et
al. (2002) im Pilokarpin-Modell konnte gezeigt werden, dass während einer
zweiwöchigen Behandlungsphase mit dem Antiepileptikum Levetiracetam einige
Ratten in der zweiten Behandlungswoche schlechter auf die Behandlung
ansprachen, was eine Toleranzentstehung vermuten lässt.

Diskussion
117
Zusammenfassend kann man sagen, dass epileptische Tiere aus dem fokalen
Kainsäure-Modell gut auf eine Behandlung mit einem Standard-Antiepileptikum
ansprechen. Ob während einer längeren Behandlung bei einigen Tieren Toleranz
gegenüber Phenobarbital eintreten würde, welche eine Selektion von Respondern
und Nonrespondern ermöglichte, müsste in weiterführenden Untersuchungen
ermittelt werden. Die Pharmakologie von anderen Antiepileptika in diesem Modell
sollte ebenfalls in weiterführenden Studien untersucht werden.
6.2.5 Neurodegeneration
In dieser Studie zeigten sieben von acht Tieren des fokalen Kainsäure-Modells
Neurodegeneration im Hippokampus ipsilateral zur Kainsäure-Injektion. Betroffen
waren die Regionen CA3a und CA3c, sowie der Hilus des Gyrus Dentatus. In der
Körnerzellschicht des Gyrus Dentatus war eine Dispersion der Zellen sichtbar. Die
CA1-Region war weniger stark betroffen. Es gab morphologische Auffälligkeiten, die
jedoch nicht eindeutig als Neurodegeneration eingestuft werden konnten.
Kontralateral traten nur vereinzelt Auffälligkeiten in der CA1-Region auf. Ein
ähnliches Bild zeigte sich in der Studie von GRÖTICKE et al. (2008), in welcher
epileptische Mäuse im fokalen Kainsäure-Modell untersucht wurden. Auch hier war
der Neuronenverlust ipsilateral am deutlichsten. Mittels spezifischer Färbung von
Neuronen konnten aber auch diskrete Veränderungen im Hilus und Gyrus Dentatus
kontralateral gefunden werden. In der Arbeit von HANDELMANN und OLTON (1981)
konnten Veränderungen auch außerhalb des Hippokampus, in Amygdala und
Claustrum gefunden werden. In unserer Arbeit wurden nur Veränderungen des
Hippokampus mittels Score-System beurteil, aus diesem Grund können wir keine
Aussage zur Neurodegeneration in anderen Hirnregionen treffen. In der Arbeit von
RAEDT et al. (2009) konnte Neurodegeneration sowohl in der CA3- als auch in der
CA1-Region ipsilateral beschrieben werden. Die Tatsache, dass Kainsäure-
Mikroinjektion zu moderatem, unilateralem Neuronenverlust führt, stellt einen Vorteil
gegenüber Modellen mit systemischer SE-Induktion dar, in denen es zu einer
ausgeprägten, bilateralen Neurodegeneration im Hippokampus, aber auch in
anderen Hirnregionen kommt (TURSKI et al. 1987, SLOVITER 2005, CURIA et al.
2008).

Diskussion
118
Die moderaten Veränderungen, vorrangig auf den epileptischen Fokus begrenzt,
kommen dem Bild der hippokampalen Sklerose des Menschen näher als die massive
Neurodegeneration, welche nach systemischer SE-Induktion auftritt.
6.2.6 Zusammenfassung
Zusammenfassend kann man sagen, dass es uns in Studie 1 gelungen ist, ein
moderates Epilepsiemodell für unser Labor zu etablieren. Das fokale Kainsäure-
Modell zeichnet sich durch eine moderate Neurodegeneration im Hippokampus
sowie moderate Verhaltensänderungen bei den Tieren aus. Die moderaten
Verhaltensänderungen waren von Vorteil, da die epileptischen Tiere einfacher zu
handhaben waren und weniger aggressives Verhalten gegenüber den
Experimentatoren zeigten. Durch SE-Induktion an wachen Ratten konnten wir den
insultmodifizierenden Effekt der Anästhesie umgehen. Es kam dadurch zu einer
zuverlässigen Anfallsentwicklung und einem progressiven Krankheitsverlauf bei den
Tieren. Die spontanen Anfälle ließen sich leicht durch Phenobarbital unterdrücken,
was zunächst keine Selektion von Respondern und Nonrespondern erlaubt. Eine
weitere pharmakologische Charakterisierung dieses Modells ist jedoch notwendig.
Ein Nachteil des Modells ist die SE-Länge. Dadurch, dass der SE nicht abgebrochen
wurde, variierte die SE-Länge von Tier zu Tier, was eine unterschiedliche Insult-
Stärke zur Folge hatte. Ein nächster Schritt wäre es, einen SE-Abbruch in diesem
Modell einzuführen. Da das fokale Kainsäure-Modell die Konditionen von Patienten
mit Temporallappenepilepsie gut widerspiegelt, haben wir es in unseren Studien zur
Epileptogenese verwenden können.
6.3 Studie 2: Schwellenversuche im Pilokarpin- und im fokalen Kainsäure-Modell Den eigentlichen Schwellenversuchen gingen im Pilokarpin-Modell zwei Versuche
voraus, diese sollen im Weiteren zu Anfang besprochen werden.
6.3.1 Einfluss von Diazepam auf die individuelle Krampfschwelle
Es konnte gezeigt werden, dass 48 h nach Diazepam-Applikation im Pilokarpin-
Modell in einer Dosierung von 30 mg/kg keine Auswirkungen auf die individuelle

Diskussion
119
Krampfschwelle vorhanden waren. Das war zu erwarten, da die Halbwertszeit von
Diazepam bei Ratten bei 1,4 h liegt (LÖSCHER 2007).
6.3.2 Latenzzeit im Pilokarpin-Modell
Die in dieser Arbeit ermittelte Latenzzeit nach Pilokarpin-induziertem SE beträgt
durchschnittlich 7,3 Tage. Diese Zeitspanne stimmt überein mit Daten für männliche
Wistar-Ratten aus der Arbeit von GOFFIN et al. Die Latenzzeit lag in dieser Arbeit
durchschnittlich bei 7,2 Tagen. Die Ermittlung der Latenzzeit war für uns wichtig, um
das Zeitfenster der Behandlung festzulegen. Das Konzept der Latenzzeit in
chemischen SE-Modellen wird aufgrund einiger Studien kontrovers diskutiert
(SLOVITER 2008). Manche interpretieren die anfallsfreie Zeit als verlängerte Periode
einer subklinischen Abnormalität und nicht als präepileptischen Zustand
(BUMANGLAG und SLOVITER 2008). In der Arbeit von BUMANGLAG und
SLOVITER (2008) wurde die Latenzzeit bei Tieren nach pilokarpin-induziertem und
elektrisch-induziertem (Stimulation des Tractus perforans) SE ermittelt. Nahezu alle
Tiere entwickelten spontane Anfälle. Die Zeit zum Auftreten von ersten spontanen
Anfällen lag im Pilokarpin-Modell zwischen zwei und neun Tagen (durchschnittlich
4,9 Tage) und im elektrischen Modell zwischen einem und acht Tagen
(durchschnittlich 2,6 Tage). Diese Ergebnisse werden in der Arbeit als Argument
gegen das Konzept einer Latenzzeit nach Insult interpretiert, da die Anfälle in beiden
Modellen bei einigen Tieren direkt (ein bis zwei Tage) nach SE auftraten. Solche
Anfälle können aber auch als insultassoziiert betrachtet werden, wenn sie direkt in
den ersten Tagen nach SE auftreten. Auch bei Menschen können unmittelbar nach
Insult epileptische Anfälle auftreten, die eigentliche Epilepsie kann sich unter
Umständen erst Jahre nach Insult entwickeln, z. B. im Falle von Fieberkrämpfen
(DELANTY et al. 1998, LHATOO et al. 2001). In dieser Arbeit konnten zwar bei
einigen Ratten insultassoziierte Anfälle beobachtet werden, bei allen Tieren kam es
nichtsdestotrotz zu einer anfallsfreien Periode nach initialem Insult, so dass man in
diesem Fall von einer Latenzzeit zwischen Insult und Epilepsieentstehung sprechen
kann.

Diskussion
120
6.3.3 Veränderungen der individuellen Krampfschwelle bei Kontrollen
Die PTZ-Schwellenversuche wurden durchgeführt, um Veränderungen der
Krampfschwelle nach SE zu erfassen, denen wahrscheinlich Veränderungen des
GABAergen Systems und der neuronalen Erregbarkeit zugrunde liegen. Bei beiden
Kontrollgruppen kam es nach wiederholter Schwellenbestimmung zu einer erhöhten
Empfindlichkeit gegenüber PTZ, welche sich in tendenziell zunehmender
Anfallsschwere während der PTZ-Infusion und Verhaltensänderungen nach Ende der
PTZ-Infusion äußerte (Anfälle und Stereotypien). Dies war deutlicher ausgeprägt bei
den Kontrollen aus dem Pilokarpin-Versuch, bei denen das Verhalten an Tag 4 und
Tag 14 nach sham-SE signifikant verändert war. Bei den Kontrollen aus der fokalen
Kainsäure-Gruppe kam es erst bei der letzten Schwellenbestimmung an Tag 22 nach
sham SE zu signifikanten Verhaltensänderungen. Die erhöhte Empfindlichkeit
gegenüber PTZ kann als Kindling-ähnlicher Effekt erklärt werden (GODDARD et al.
1969). Gleichzeitig kam es bei den Kontrollen aus der Pilokarpin-Gruppe zu einer
Erhöhung der Krampfschwelle bei wiederholter Auslösung. Diese kann dadurch
erklärt werden, dass nach einen Anfall gegenläufige Prozesse im Gehirn ablaufen,
zum einen solche, die die Empfindlichkeit neuen Insulten gegenüber steigern (z. B.
Kindling-ähnliche Effekte), zum anderen jedoch solche, die protektiv und
anfallslimitierend wirken (z. B. verstärkte Inhibition nach Insult), (LÖSCHER und
KÖHLING 2010). In einer Arbeit von GREEN (1986) wurden Veränderungen des
GABAergen Systems nach elektrisch-induzierten Anfällen beschrieben. Es kam
einerseits zu einem Anstieg der funktionellen GABA-Wirkung, was in einer
gesteigerten Krampfschwelle bis 3 h nach Anfall resultierte. Andererseits kam es zu
einer fast vollständigen Hemmung der GABA-Synthese und -Ausschüttung. Diese
Mechanismen können eine Erklärung für die scheinbar paradoxen Veränderungen
der PTZ-Empfindlichkeit in der Pilokarpin-Kontrollgruppe sein.
6.3.4 Veränderungen der Krampfschwelle nach SE
Sowohl im Pilokarpin-Modell als auch im fokalen Kainsäure-Modell kam es zu
Veränderungen der PTZ-Empfindlichkeit nach SE. Die Tiere reagierten nach SE mit
einer höheren Empfindlichkeit auf PTZ.

Diskussion
121
Das äußerte sich nicht nur in einem Abfall der individuellen Krampfschwelle, auch
das Verhalten nach Ende der PTZ-Infusion war verändert (Anfälle und Stereotypien).
In beiden Gruppen kam es zu einer Senkung der Krampfschwelle zwei Tage nach
SE. Dies war deutlicher im fokalen Kainsäure-Modell, was durch die einheitliche
Gruppenzusammensetzung und die daraus resultierende geringere Streuung erklärt
werden kann. Dass dieser Effekt jedoch modellunabhängig auftritt, spricht dafür,
dass er robust ist. Im fokalen Kainsäure-Modell war die Krampfschwelle auch an Tag
9 und Tag 22 nach SE gesenkt. In beiden Gruppen waren die Krampfschwellen nach
SE, mit Ausnahme der Schwelle 4 Tage nach SE in der Kainsäure-Gruppe, zu allen
Zeitpunkten niedriger als bei Kontrollen. Verhaltensänderungen nach PTZ-Infusion
traten ebenfalls in beiden Gruppen schon ab dem ersten Test nach SE auf. Das
deutet auf permanente Veränderungen der neuronalen Erregbarkeit nach SE hin. Da
der PTZ-Test durch die Wirkungsweise von PTZ Veränderungen des GABAergen
Systems erfasst, gehen wir davon aus, dass die gesteigerte Empfindlichkeit
gegenüber PTZ Veränderungen des GABAergen Systems widerspiegelt. Diverse in
vivo (GREEN 1986, AUVIN et al. 2009) und in vitro (LEROY et al. 2004,
BARMASHENKO et al. 2011, PAVLOV et al. 2011) Studien beschreiben eine
gesteigerte neuronale Erregbarkeit und Veränderungen des GABAergen Systems
nach Hirninsult.
Die Veränderung des GABAergen Systems und die daraus resultierende neuronale
Übererregbarkeit, welche direkt nach SE, also noch in der Latenzphase auftrat, kann
verschiedene Ursachen haben. Zum einen kann eine verstärkte GABAerge Inhibition
nach ihrem Kollaps zur Netzwerk-Synchronisierung führen und somit neuronale
Übererregbarkeit erzeugen (MANN und MODY 2008). Zum anderen kann ein GABA-
switch von inhibitorisch zu exzitatorisch stattfinden, was ebenfalls zu einer
Übererregbarkeit führen würde und in Konsequenz in einer erregenden Wirkung von
GABA in der betroffenen Hirnregion resultieren würde (PATHAK et al. 2007,
BARMASHENKO et al. 2011). Auch Veränderungen in der GABAA-Rezeptor-
Pharmakologie (COULTER 2001) und Verluste von GABAergen Interneuronen
(MORIMOTO et al. 2004), beides Prozesse, die nach SE stattfinden, können für
diese neuronale Übererregbarkeit verantwortlich sein. Eine systemische PTZ-
Applikation ermöglicht keine Aussage darüber, welcher dieser Prozesse und in
welchem Ausmaß für die Ergebnisse dieser Studie ursächlich verantwortlich war.

Diskussion
122
Elektrophysiologische Untersuchungen könnten darüber Aufschluss geben, indem
man die Funktionalität des GABAergen Systems nach Insult in Hippokampus-
Schnittpräparaten untersuchen würde.
Da in beiden Modellen zwei Tage nach SE die markanteste Veränderung der
Krampfschwelle auftrat, könnte dies darauf hinweisen, dass zu diesem Zeitpunkt der
Kollaps der GABAergen Inhibition stattfindet. Der initiale Anstieg der GABAergen
Inhibition, welchen wir mit der PTZ-Behandlung verhindern wollten, müsste
demzufolge vorher, also sehr zeitnah zum SE auftreten. Dies wird auch gestützt
durch die Arbeit von GREEN (1986), in welcher die individuelle Krampfschwelle bis
zu 3 h nach Anfall erhöht war. Aufgrund dieser Annahmen und der Tatsache, dass
zwei Tage nach SE die Krampfschwelle erniedrigt war, haben wir zuerst
beschlossen, die PTZ-Behandlung direkt nach SE zu starten und auf 48 h zu
beschränken. Die Anfälle, die unter PTZ-Behandlung bei den Kainsäure-Tieren
auftraten, veranlassten uns später zu einer 48 h Behandlung 7 Tage nach SE,
analog der Studie von PITKÄNEN et al. (2004).
6.3.5 Einfluss des Zyklusstandes auf die Krampfschwelle
In unseren Untersuchungen konnte kein Einfluss des ovariellen Zyklusstandes auf
die individuelle PTZ-Schwelle gefunden werden. Die Tiere hatten keinen synchronen
Zyklus, da sie ohne Kontakt zu männlichen Ratten gehalten wurden. Das konnte
auch für andere Studien unserer Arbeitsgruppe gezeigt werden (KÜCKER et al.
2010). Obwohl einige Studien, unter anderem die von SCHARFMAN und
MACLUSKY (2006) dem Zyklusstand einen hohen Einfluss auf die neuronale
Erregbarkeit zuschreiben, scheint dieser doch substanzabhängig zu sein. In der
Arbeit von FINN und GEE (1994) wurde gezeigt, dass der ovarielle Zyklusstand
keinen Einfluss auf die individuelle PTZ-Krampfschwelle hatte. Aufgrund dieser
Ergebnisse und der Studien von KÜCKER et al. (2010) und FINN und GEE (1994)
können wir davon ausgehen, dass die Tatsache, dass in dieser Arbeit weibliche
Ratten verwendet wurden, keinen Einfluss auf die erzielten Ergebnisse hatte.

Diskussion
123
6.4 Studie 3: PTZ-Behandlung zur Epilepsieprävention
6.4.1 Etablierung des Behandlungsprotokolls
Eine Behandlung mit dem GABAA-Rezeptor-Antagonisten PTZ zur
Epilepsieprävention wurde bislang nicht durchgeführt. Aus diesem Grund gab es in
der Literatur keine Angaben über die Auswirkungen einer chronischen PTZ-
Applikation auf die Epileptogenese. In dieser Studie wurde PTZ in subkonvulsiven
Dosen verabreicht und der PTZ-Gehalt im Plasma sollte über die gesamte
Behandlungsdauer, in diesem Fall über 48 h, im subkonvulsiven Bereich liegen. Da
PTZ eine Substanz ist, die schon seit langem in der Medizin verwendet wird, gab es
diverse Arbeiten zum Metabolismus von PTZ bei der Ratte (ESPLIN und
WOODBURY 1956, VOHLAND und ZUFELDE 1976, RAMZAN und LEVY 1985). Um
ein geeignetes Behandlungsprotokoll zu etablieren, haben wir einige
Untersuchungen bei weiblichen Sprague Dawley-Ratten wiederholt. Die
Plasmahalbwertszeit liegt laut Literarurangaben bei 2-3 h (ESPLIN und WOODBURY
1956, VOHLAND und ZUFELDE 1976, RAMZAN und LEVY 1985). Die von uns
ermittelte Halbwertszeit von 3,3 h lag im Rahmen der Literaturdaten. In Arbeiten von
RAMZAN und LEVY (1985) und MARCUCCI et al. (1971) wird beschrieben, dass
PTZ-Gehalte in Serum und Gehirn weitgehend gleich sind, so dass man annehmen
konnte, dass der PTZ-Plasmagehalt den Gehalt im Gehirn widerspiegelt. Laut
Literaturangaben wird PTZ bei Ratten über die Leber metabolisiert und renal
ausgeschieden. Die Plasmaproteinbindung ist vernachlässigbar gering und es gibt
keine Hinweise auf Kumulation (ESPLIN und WOODBURY 1956). Ein wichtiges
Ergebnis der pharmakokinetischen Versuche war die hohe Korrelation (r=0,9831) der
Krampfschwelle mit dem PTZ-Plasmaspiegel. Die mittlere Krampfschwelle lag bei
unseren Tieren bei ~ 20 mg/kg und die niedrigste Krampfschwelle 24 h nach SE lag
bei 14 mg/kg, was einem Plasmaspiegel von 18 µg/ml entspricht. Aus diesem Grund
haben wir uns entschlossen, eine Zielplasmakonzentration von 10 µg/ml
anzustreben. In vitro-Daten zeigten, dass die halbe inhibitorische Konzentration
(IC50) von PTZ auf den GABAA-Rezeptor bei 1 mmol/l, liegt, während unsere
Zielplasmakonzentration bei 72 µmol/l lag (BLOMS-FUNKE et al. 1996).

Diskussion
124
Die Zielplasmakonzentration war jedoch ein Kompromiss zwischen höchstmöglicher-
und verträglicher Dosis, die jedoch stets im subkonvulsiven Bereich bleiben sollte.
Mit dem aufgrund der Ergebnisse etablierten Behandlungsprotokoll gelang es uns,
die Zielplasmakonzentration über die gesamte Behandlungsdauer (48 h)
aufrechtzuerhalten. Bei den PTZ-behandelten Tieren ohne SE traten keine
Nebenwirkungen unter der Behandlung auf. Somit war es uns bei naiven Ratten
gelungen, ein Behandlungsprotokoll für eine PTZ-Dauerbehandlung zu etablieren.
6.4.2 PTZ-Behandlung
Die PTZ-Infusion hatte modellabhängig verschiedene Auswirkungen. Im Pilokarpin-
Modell, in dem die Behandlung direkt nach SE-Abbruch begann, hatten die Tiere
keine Anfälle während der Bolus-Gabe. Auch im weiteren Verlauf der Infusion kam
es nur in vereinzelten Fällen zu Myoklonien oder fokalen Anfällen. Das lässt sich
dadurch erklären, dass Diazepam und Phenobarbital zur SE-Unterbrechung
verabreicht worden sind. Da beides GABAA-Rezeptor-Agonisten sind, können sie
den prokonvulsiven Wirkungsmechanismus von PTZ aufheben.
Anders war die Situation im fokalen Kainsäure-Modell. Im ersten Versuch, in
welchem die Infusion 13 h oder 16 h nach SE-Beginn startete, hatten alle Tiere
Anfälle während der PTZ-Behandlung. Da der SE in diesem Modell nicht
abgebrochen wurde, und auch die Kontrolltiere in diesem Zeitraum Anfälle hatten,
kann es eine normale Variation der SE-Länge oder insultassoziierte Anfälle
darstellen. Zum anderen kann es bedeuten, dass PTZ die Anfälle im SE verstärkt
hat. Dafür spricht die Tatsache, dass 3 von 4 Tieren Anfälle direkt unter Bolus-Gabe
entwickelten und somit empfindlicher auf die PTZ-Dosis reagierten. Für die zweite
Hypothese spricht auch die Tatsache, dass Tiere nach PTZ-Behandlung längere
Anfälle hatten als Kontrollen. Durch die geringen Gruppengrößen ist jedoch keine
eindeutige Aussage zu den Ursachen möglich.
Da der SE in diesem Modell nicht abgebrochen wurde, war es schwierig einen
Behandlungszeitpunkt zu wählen, der SE-nah ist, ohne gleichzeitig den SE durch die
Behandlung zu verstärken. Aufgrund der lang anhaltenden Krampfaktivität im fokalen
Kainsäure-Modell (durchnittliche SE-Länge von etwa 11 h) können epileptogene
Veränderungen möglicherweise später auftreten als im Pilokarpin-Modell.

Diskussion
125
Deshalb könnte eine Behandlung zu einem späteren Zeitpunkt nach SE sinnvoll sein.
In einer Studie von PITKÄNEN et al. (2004) konnte gezeigt werden, dass eine
Behandlung mit dem prokonvulsiven α2-Rezeptor-Antagonisten Atipamezol 7 Tage
nach einem elektrisch induziertem SE einen insultmodifizierenden Effekt hatte. In
Anlehnung an diese Studie und aufgrund der Ergebnisse der ersten
Behandlungsgruppe haben wir uns entschlossen einen späteren Zeitpunkt der
Behandlung (7 Tage nach SE) zu wählen. Auch in dieser Behandlungsgruppe hatten
4 von 5 Tieren Anfälle unter der Behandlung. Das spricht dafür, dass PTZ trotz
etabliertem Behandlungsschema prokonvulsiv wirkte. Eine Erklärung dafür wäre die
gesteigerte Empfindlichkeit gegenüber PTZ im fokalen Kainsäure-Modell. Das
bestätigen die Schwellenversuche, in welchen die Kainsäure-Tiere eine
Schwellenabsenkung 2, 9 und 22 Tage nach SE zeigten. Da auch eines von 2
Kontrolltieren Anfälle in diesem Zeitraum zeigte, ist es auch möglich, dass erste
spontane Anfälle in diesem Modell früher auftreten als angenommen. Eine sinnvolle
Lösung wäre zum einen ein SE-Abbruch, um den Insult für alle Tiere gleich zu
halten. Zum anderen sollte aufgrund der erhöhten Empfindlichkeit das etablierte
Behandlungsprotokoll für das fokale Kainsäure-Modell überarbeitet werden.
6.5 PTZ zur Epilepsieprävention: Ergebnisse und Ausblick Ziel der Arbeit war die Untersuchung des antiepileptogenen Potentials einer PTZ-
Behandlung nach Hirninsult. Der Einsatz von Prokonvulsiva zur Epilepsieprävention
wird in letzter Zeit häufig diskutiert (ARMSTRONG et al. 2009, LÖSCHER und
BRANDT 2010 a, PITKÄNEN 2010, PITKÄNEN und LUKASIUK 2011). Wichtig für
unsere Untersuchungen waren jedoch hauptsächlich zwei Theorien über die Rolle
von GABA in der Epilepsieentstehung, da wir konkret einen GABAA-Rezeptor-
Antagonisten einsetzen wollten. Es ist seit langem bekannt, dass während der
Epileptogenese Veränderungen des GABAergen Systems auftreten. Es gibt
zunehmend Hinweise, dass GABA auch proepileptogen wirken kann. Das kann zum
einen über den GABA-switch von inhibitorisch zu exzitatorisch erfolgen (PATHAK et
al. 2007). Zum anderen kann eine kompensatorische Hochregulierung von GABA,
welche nach Insult auftritt (NUTT et al. 1981, GREEN 1986), nach ihrem Kollaps eine
Netzwerk-Synchronisierung hervorrufen (MANN und MODY 2008).

Diskussion
126
Das war die Grundlage, welche uns zum Einsatz eines GABAA-Rezeptor-
Antagonisten veranlasste. Im Sinne der Synchronisationstheorie würde eine
prophylaktische Behandlung mit subkonvulsiven PTZ-Dosen nach Insult die
Netzwerk-Synchronisierung verhindern, indem die initiale GABA-Hochregulierung
verhindert wird. Auch bei einer exzitatorischen GABA-Wirkung wäre eine Behandlung
mit PTZ sinnvoll, um der exzitatorischen Wirkung von GABA in
Neuronensubpopulationen im Hippokampus entgegenzuwirken.
Trotz der Annahme, dass eine PTZ-Behandlung die Netzwerk-Synchronisierung
nach Insult verhindern und dadurch antiepileptogen wirken könnte, ist es uns nicht
gelungen, mit dem verwendeten Behandlungschema das erwartete Resultat zu
erzielen. Zehn von elf Tieren entwickelten, unabhängig von Epilepsiemodell und
Behandlungszeitpunkt, spontane Anfälle 8 bis 16 Wochen nach PTZ-Behandlung. In
der Pilokarpin-Gruppe schien die PTZ-Behandlung eher proepileptogen zu wirken, da
8 Wochen nach Behandlung alle behandelten Tiere epileptisch waren, während bei
den Kontrollen zu diesem Zeitpunkt noch keine Anfälle detektiert werden konnten.
Allerdings ist die Tierzahl der Gruppe zu gering, um einen zufälligen Effekt
ausschliessen zu können. Es gab auch keine Unterschiede in der Anfallsfrequenz
zwischen epileptischen PTZ-behandelten Tieren und epileptischen Kontrollen. Diese
Ergebnisse können mehrere Ursachen haben. Die Dauer der Behandlung, sowie das
gewählte Behandlungszeitfenster könnten inadäquat gewesen sein. Die Wahl der
geeigneten PTZ-Dosis und die Art der Behandlung (systemisch versus fokal) war
eine weitere Schwierigkeit dieser Studie. Da die zeitlichen Abläufe der
Veränderungen des GABAergen Systems nach Insult nur unvollständig geklärt sind,
war diese Arbeit ein Versuch, Behandlungszeitpunkt und -dauer anhand der
Schwellenversuche zu ermitteln. Wir haben versucht, zwei Behandlungszeitfenster
zu wählen, ein SE-nahes und ein SE-fernes, es ist jedoch trotzdem möglich, dass die
gewählten Behandlungszeitpunkte nicht die richtigen waren. Die Dauer der
Behandlung könnte ebenfalls für die Ergebnisse verantwortlich sein, falls sie zu kurz
oder zu lang gewählt worden war. Die Wahl der PTZ-Dosis erfolgte aufgrund von
Ergebnissen der Schwellenversuche, sowie der pharmakokinetischen Versuche. Die
Dosis sollte einen Kompromiss darstellen zwischen höchstmöglicher aber systemisch
verträglicher Dosis.

Diskussion
127
Dies ist nur teilweise gelungen, da die gewählte Behandlungsdosis bei den
Kainsäure-Ratten zu Anfällen führte, also möglicherweise zu hoch war. Ein weiterer
Punkt ist die systemische Behandlung. Möglicherweise laufen die Prozesse, welche
wir mit unserer Behandlung beeinflussen wollen, nicht gleichermaßen im gesamten
Gehirn ab, sondern nur im epileptischen Fokus. PTZ entfaltet seine Wirkung über
den Hippokampus (GOLARAI et al. 1992, STRINGER 1994, WALSH et al. 1999,
QIAN et al. 2011) aber auch über den mammillothalamischen Trakt (MIRSKI und
FERRENDELLI 1986 und 1987), möglicherweise wäre also eine fokale Behandlung
sinnvoll. Aus diesem Grund ist eine systemische Behandlung mit PTZ
möglicherweise nicht der richtige Weg, um diese Prozesse zu beeinflussen.
Verschiedene Studien zeigen paradoxe Effekte einer Behandlung mit prokonvulsiven
Stoffen. In einer Studie von KLAASSEN et al. (2006) konnte gezeigt werden, dass
der GABAA-Antagonist Picrotoxin in subkonvulsiver Dosierung einen antikonvulsiven
Effekt in einem genetischen Mausmodell für Epilepsie hatte. In einer Studie von
KHAZIPOV und HOLMES (2003) konnte der GABAA-Antagonist Bicucullin
epileptische Aktivität im Rattenhippokampus unterdrücken. Der Effekt einer solchen
Behandlung auf die Epileptogenese wurde jedoch nicht untersucht. Auch mit anderen
prokonvulsiven Stoffen wurde die Strategie der Neuromodulation zur
Epilepsieprävention verfolgt. Cannabinoid-Rezeptor-Antagonisten sollten die
Veränderungen des Cannabinoid-Rezeptor Systems nach Insult verhindern, die zu
einer neuronalen Übererregbarkeit führen kann. Atipamezol und Koffein wurden
aufgrund ihrer neuroprotektiven und krankheitsmodifizierenden Effekte in anderen
Krankheitsmodellen ausgewählt. Der α2-Rezeptor-Antagonist Atipamezol hatte
keinen Effekt auf die Anfallsausprägung in einem Rattenmodell für Epilepsie, der
Krankheitsverlauf konnte jedoch gemildert werden (PITKÄNEN et al. 2004). Weitere
Studien befassen sich mit dem Typ 1 Cannabinoid Rezeptor-Antagonisten
Rimonabant. In der Arbeit von CHEN et al. (2007) hat die einmalige Applikation von
Rimonabant in einem Rattenmodell für Fieberkrämpfe eine spätere neuronale
Übererregbarkeit verhindert. Dasselbe war der Fall in einem Rattenmodell für
Schädel-Hirn-Trauma, in welchem die einmalige Applikation von Rimonabant eine
spätere neuronale Übererregbarkeit verhinderte (ECHEGOYEN et al. 2009).

Diskussion
128
Eine Behandlung mit dem Cannabinoid Rezeptor-Antagonisten Rimonabant hatte
jedoch keinen Einfluss auf die Epileptogenese in einem Rattenmodell für Epilepsie
(DUDEK et al. 2010). Obwohl also prokonvulsive Stoffe und insbesondere GABAA-
Antagonisten antikonvulsiv oder neuromodulierend wirken können, ist es bis heute
nicht gelungen, eine wirklich antiepileptogene Substanz zu entdecken.
Die Behandlung mit PTZ hatte zwar keinen Einfluss auf die Epileptogenese, wir
können aber nicht ausschließen, dass eine lokale Behandlung direkt im epileptischen
Fokus erfolgreich wäre. Da es bis heute keine Möglichkeit gibt, Patienten zu helfen,
die ein erhöhtes Risiko haben an Epilepsie zu erkranken, ist es wichtig, nach neuen
Strategien zu suchen. Eine lokale Applikation von PTZ zur Epilepsieprävention in
Rattenmodellen wäre ein weiterer logischer Schritt.

Zusammenfassung
129
7. Zusammenfassung Marta Rattka
Prokonvulsiva als pharmakologische Strategie zur Epilepsieprävention
Epilepsie ist die häufigste chronische neurologische Erkrankung des Menschen.
Symptomatische Epilepsieformen, wie die Temporallappenepilepsie, entwickeln sich
nach einem Hirninsult, wie z. B. Schädel-Hirn-Trauma oder Status epileptikus (SE).
Es ist somit eine Erkrankung mit klar definierbarer Risikogruppe. Trotzdem gibt es bis
heute keine Möglichkeit, solche Patienten präventiv zu behandeln. Die einzige
Behandlungsmöglichkeit ist die symptomatische Anfallsunterdrückung mit
Antiepileptika. Ein weiteres Problem der Temporallappenepilepsie ist eine hohe
Anzahl an pharmakoresistenten Patienten. Das bedeutet, dass diese Patienten trotz
Behandlung nicht anfallsfrei werden. Aus diesem Grund ist es dringend notwendig,
Strategien zur Epilepsieprävention zu entwickeln, um Patienten aus der Risikogruppe
zu helfen. Es konnte gezeigt werden, dass paradoxerweise Mittel, die eigentlich
Krämpfe auslösen, also prokonvulsiv wirken, in subkonvulsiven Dosen verabreicht
einen akut antikonvulsiven Effekt haben können oder neuronale Übererregbarkeit
verhindern. Aus diesem Grund wird der Einsatz solcher prokonvulsiver Stoffe in der
Antiepileptogenese-Forschung immer häufiger diskutiert. In der Gruppe der
Prokonvulsiva waren GABAA-Rezeptor-Antagonisten für uns von besonderem
Interesse, da es zunehmend Hinweise gibt, dass der inhibitorische Transmitter GABA
die Entstehung einen neuronalen Übererregbarkeit nach Hirninsult begünstigen
kann. Zum einen kann eine nach Insult kompensatorisch gesteigerte GABAerge
Inhibition nach ihrem Kollaps zur Synchronisierung von erregenden Netzwerken
führen. Zum anderen kann, durch veränderte Exprimierung von Chlorid-
Cotransportern, ein GABA-switch von inhibitorisch zu exzitatorisch stattfinden. Das
war für uns der Grund den GABAA-Rezeptor-Antagonisten Pentylentetrazol (PTZ) in
unserer Studie zur Epilepsieprävention einzusetzen. Ziel dieser Arbeit war eine
Untersuchung der Auswirkungen einer prophylaktischen PTZ-Behandlung nach Insult
auf die Epileptogenese. Wir gingen davon aus, dass eine solche Behandlung
antiepileptogen sein würde. Unsere Arbeitshypothese sollte an zwei Rattenmodellen

Zusammenfassung
130
für Epilepsie, dem systemischen Pilokapin-Modell und dem fokalen Kainsäure-
Modell, untersucht werden.
Das fokale Kainsäure-Modell musste zunächst an unserem Institut etabliert werden.
Im zweiten Schritt haben wir dann in beiden Modellen zeitliche Veränderungen der
neuronalen Erregbarkeit nach Hirninsult, in unserem Fall nach SE, mittels
Bestimmung der PTZ-Krampfschwelle untersucht, um ein Behandlungszeitfenster
festlegen zu können. Zuletzt haben wir ein Behandlungsprotokoll für eine
subkonvulsive PTZ-Dosierung erstellt und eine prophylaktische PTZ-Behandlung
nach einem Insult (in unseren Modellen nach SE) durchgeführt. Der
Behandlungserfolg wurde 8 bis 16 Wochen nach Behandlung anhand des Auftretens
von spontanen epileptischen Anfällen beurteilt. Es ist uns nicht gelungen, mit einer
systemischen PTZ-Behandlung eine Epilepsieentstehung zu verhindern, weder im
systemischen Pilokarpin-Modell, noch im fokalen Kainsäure-Modell. Wir konnten
durch unsere Behandlung weder die Anfallsausprägung, noch die Anfallsfrequenz
beeinflussen. Im Pilokarpin-Modell schien die PTZ-Behandlung eher proepileptogen
zu wirken. Eine systemische Behandlung mit dem GABAA-Rezeptor-Antagonisten
PTZ, hatte, so wie wir sie durchgeführt haben, also keinen antiepileptogenen Effekt.
Es ist uns jedoch gelungen, neue Erkenntnisse über das fokale Kainsäure-Modell zu
gewinnen, insbesondere über Verhaltensänderungen und Pharmakologie in diesem
Modell. Des Weiteren haben wir Veränderungen der neuronalen Erregbarkeit nach
SE im Zeitverlauf mittels individueller Krampfschwellenbestimmung erfasst. Diese
Ergebnisse zeigen Veränderungen des GABAergen Systems nach SE und leisten
somit einen Beitrag zum besseren Verständnis der Epileptogenese. Die Ursachen
dieser Veränderungen sollten in weiterführenden Studien untersucht werden, um das
Verständnis der epileptogenen Mechanismen weiterhin zu verbessern. Obwohl eine
systemische PTZ-Behandlung nicht antiepileptogen war, kann man jedoch nicht
ausschließen, dass eine lokale Behandlung direkt im epileptischen Fokus erfolgreich
wäre. Der nächste Schritt könnte eine lokale PTZ-Behandlung sein, da Prozesse, die
wir beeinflussen wollten, möglicherweise nur im epileptischen Fokus ablaufen, was
die unwirksame systemische Behandlung erklären könnte.

Summary
131
8. Summary Marta Rattka
Proconvulsants as pharmacological strategy for epilepsy prevention
Epilepsy is the most common chronic neurological disorder in humans. Symptomatic
epilepsies are often a consequence of a brain insult, for example traumatic brain
injury or SE. The patients at risk can be easily identified, but currently we have no
possibility to prevent the disease. The only treatment for patients with epilepsy is a
symptomatic suppression of the seizures. Patients with an onset of symptomatic
epilepsy are often pharmacoresistent, which means that a treatment with common
antiepileptic drugs does not lead to seizure control. To help patients at risk, the
development of antiepileptogenic strategies is an urgent need. Experimental studies
have shown that paradoxically proconvulsant agents, at a subconvulsive dose, can
exert anticonvulsive effects, or prevent the development of neuronal hyperexcitability
after brain insult, a hallmark of epileptogenesis. The use of proconvulsant drugs for
studies on antiepileptogenesis is a new strategy. The GABAA-receptor-antagonists
play an important role among the group of proconvulsive drugs. New hypotheses
propose that the inhibitory neurotransmitter GABA can play a crucial role in the
establishment of neuronal hyperexcitability after brain insult. A compensatory up-
regulation of GABA after a brain insult can lead, after its collapse, to network
synchronisation. Another explanation is a switch of GABA from an inhibitory to an
excitatory action, due to a change in expression pattern of chloride co-transporters.
This prompted us to perform our experiments on epilepsy prevention with the
GABAA-receptor-antagonist pentylenetetrazole (PTZ). The goal of our study was the
prevention of epileptogenesis by a prophylactic PTZ-treatment after brain insult. We
tested this working hypothesis in two rat models of temporal lobe epilepsy, in the
systemic pilocarpine model and in the intrahippocampal kainic acid model. The
intrahipocampal kainic acid model was not established in our institute, so this was
our first step. Then we assessed excitability changes after brain insult in the two rat
models by determining the PTZ seizure threshold. After this we developed a
subconvulsive treatment protocol for PTZ and started with the prophylactic treatment

Summary
132
after SE. Eight to 16 weeks after the treatment we assessed the occurrence of
spontaneous seizures in our rats.
The systemic treatment with PTZ did not prevent the development of epilepsy,
neither in the pilocarpine model, nor in the kainic acid model. The treatment was not
able to affect the occurrence of spontaneous seizures or the seizure frequency. In
the pilocarpine model our intervention seems to have rather proepileptogenic
consequences. Thus the treatment with PTZ had no antiepileptogenic effect.
Nevertheless our studies provide new insights into the intrahippocampal kainc acid
model, especially with respect to behavioural alterations and the pharmacological
response of the spontaneous seizures. We also described changes in the neuronal
excitability at several time points after SE, by assessing changes in individual seizure
threshold. This results leads to a better understanding of the process of
epileptogenesis due to describing changes of the GABAergic system after SE. Our
systemic treatment failed, but we can not exclude the possibility of a successful local
treatment with PTZ. The next step should be a local treatment, because it is possible
that the processes we want to influence occur only in the epileptic focus, so that a
systemic treatment is ineffective.

Literatur
133
9. Literatur
AARABI, B., M. TAGHIPOUR, A. HAGHNEGAHDAR, M. FAROKHI und L. MOBLEY (2000). "Prognostic factors in the occurrence of posttraumatic epilepsy after penetrating head injury suffered during military service." Neurosurg Focus 8(1).
ADAMEC, R. E. (1990). "Amygdala kindling and anxiety in the rat." Neuroreport 1(3-4): 255-258. ANDRE, V., C. DUBE, J. FRANCOIS, C. LEROY, M. A. RIGOULOT, C. ROCH, I. J. NAMER und A.
NEHLIG (2007). "Pathogenesis and pharmacology of epilepsy in the lithium-pilocarpine model." Epilepsia 5: 41-47.
ANDRE, V., A. FERRANDON, C. MARESCAUX und A. NEHLIG (2001). "Vigabatrin protects against hippocampal damage but is not antiepileptogenic in the lithium-pilocarpine model of temporal lobe epilepsy." Epilepsy Res 47(1-2): 99-117.
ARMSTRONG, C., R. J. MORGAN und I. SOLTESZ (2009). "Pursuing paradoxical proconvulsant prophylaxis for epileptogenesis." Epilepsia 50(7): 1657-1669.
AUVIN, S., N. PORTA, A. NEHLIG, C. LECOINTE, L. VALLEE und R. BORDET (2009). "Inflammation in rat pups subjected to short hyperthermic seizures enhances brain long-term excitability." Epilepsy Res 86(2-3): 124-130.
BABB, T. L., J. PEREIRA-LEITE, G. W. MATHERN und J. K. PRETORIUS (1995). "Kainic acid induced hippocampal seizures in rats: comparisons of acute and chronic seizures using intrahippocampal versus systemic injections." Ital J Neurol Sci 16(1-2): 39-44.
BARMASHENKO, G., S. HEFFT, A. AERTSEN, T. KIRSCHSTEIN und R. KOHLING (2011). "Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus." Epilepsia 52(9): 1570-1578.
BEN-ARI, Y. (1985). "Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy." Neuroscience 14(2): 375-403.
BEN-ARI, Y., E. CHERUBINI, R. CORRADETTI und J. L. GAIARSA (1989). "Giant synaptic potentials in immature rat CA3 hippocampal neurones." J Physiol 416: 303-325.
BEN-ARI, Y. und R. COSSART (2000). "Kainate, a double agent that generates seizures: two decades of progress." Trends Neurosci 23(11): 580-587.
BEN-ARI, Y. und G. L. HOLMES (2005). "The multiple facets of gamma-aminobutyric acid dysfunction in epilepsy." Curr Opin Neurol 18(2): 141-145.
BETHMANN, K., C. BRANDT und W. LÖSCHER (2007). "Resistance to phenobarbital extends to phenytoin in a rat model of temporal lobe epilepsy." Epilepsia 48(4): 816-826.
BLOMS-FUNKE, P., M. MADEJA, U. MUSSHOFF und E. J. SPECKMANN (1996). "Effects of pentylenetetrazol on GABA receptors expressed in oocytes of Xenopus laevis: extra- and intracellular sites of action." Neurosci Lett 205(2): 115-118.
BOLANOS, A. R., M. SARKISIAN, Y. YANG, A. HORI, S. L. HELMERS, M. MIKATI, P. TANDON, C. E. STAFSTROM und G. L. HOLMES (1998). "Comparison of valproate and phenobarbital treatment after status epilepticus in rats." Neurology 51(1): 41-48.
BOLDING, K. und J. W. RUDY (2006). "Place learning in the Morris water task: making the memory stick." Learn Mem 13(3): 278-286.
BOLKVADZE, T. und A. PITKÄNEN (2011). "Development of Post-Traumatic Epilepsy after Controlled Cortical Impact and Lateral Fluid-Percussion Induced Brain Injury in the Mouse." J Neurotrauma 24: 24.
BRAGIN, A., A. AZIZYAN, J. ALMAJANO, C. L. WILSON und J. ENGEL, JR. (2005). "Analysis of chronic seizure onsets after intrahippocampal kainic acid injection in freely moving rats." Epilepsia 46(10): 1592-1598.
BRAGIN, A., J. ENGEL, JR., C. L. WILSON, E. VIZENTIN und G. W. MATHERN (1999). "Electrophysiologic analysis of a chronic seizure model after unilateral hippocampal KA injection." Epilepsia 40(9): 1210-1221.
BRAGIN, A., C. L. WILSON, J. ALMAJANO, I. MODY und J. ENGEL, JR. (2004). "High-frequency oscillations after status epilepticus: epileptogenesis and seizure genesis." Epilepsia 45(9): 1017-1023.

Literatur
134
BRAGIN, A., C. L. WILSON und J. ENGEL, JR. (2007). "Voltage depth profiles of high-frequency oscillations after kainic acid-induced status epilepticus." Epilepsia 5: 35-40.
BRANDT, C., A. M. GASTENS, M. SUN, M. HAUSKNECHT und W. LÖSCHER (2006). "Treatment with valproate after status epilepticus: effect on neuronal damage, epileptogenesis, and behavioral alterations in rats." Neuropharmacology 51(4): 789-804.
BRANDT, C., M. GLIEN, A. M. GASTENS, M. FEDROWITZ, K. BETHMANN, H. A. VOLK, H. POTSCHKA und W. LÖSCHER (2007). "Prophylactic treatment with levetiracetam after status epilepticus: lack of effect on epileptogenesis, neuronal damage, and behavioral alterations in rats." Neuropharmacology 53(2): 207-221.
BRANDT, C., M. GLIEN, H. POTSCHKA, H. VOLK und W. LOSCHER (2003). "Epileptogenesis and neuropathology after different types of status epilepticus induced by prolonged electrical stimulation of the basolateral amygdala in rats." Epilepsy Res 55(1-2): 83-103.
BRANDT, C., M. NOZADZE, N. HEUCHERT, M. RATTKA und W. LÖSCHER (2010). "Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy." J Neurosci 30(25): 8602-8612.
BRANDT, C., H. A. VOLK und W. LÖSCHER (2004). "Striking differences in individual anticonvulsant response to phenobarbital in rats with spontaneous seizures after status epilepticus." Epilepsia 45(12): 1488-1497.
BUMANGLAG, A. V. und R. S. SLOVITER (2008). "Minimal latency to hippocampal epileptogenesis and clinical epilepsy after perforant pathway stimulation-induced status epilepticus in awake rats." J Comp Neurol 510(6): 561-580.
CAVALHEIRO, E. A., D. A. RICHE und G. LE GAL LA SALLE (1982). "Long-term effects of intrahippocampal kainic acid injection in rats: a method for inducing spontaneous recurrent seizures." Electroencephalogr Clin Neurophysiol 53(6): 581-589.
CHANG, B. S. und D. H. LOWENSTEIN (2003). "Epilepsy." N Engl J Med 349(13): 1257-1266. CHEN, K., A. NEU, A. L. HOWARD, C. FOLDY, J. ECHEGOYEN, L. HILGENBERG, M. SMITH, K.
MACKIE und I. SOLTESZ (2007). "Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures." J Neurosci 27(1): 46-58.
CHROSCINSKA-KRAWCZYK, M., M. JARGIELLO-BASZAK, M. WALEK, B. TYLUS und S. J. CZUCZWAR (2011). "Caffeine and the anticonvulsant potency of antiepileptic drugs: experimental and clinical data." Pharmacol Rep 63(1): 12-18.
COHEN, I., V. NAVARRO, S. CLEMENCEAU, M. BAULAC und R. MILES (2002). "On the origin of interictal activity in human temporal lobe epilepsy in vitro." Science 298(5597): 1418-1421.
COPER, H. und W. M. HERRMANN (1988). "Psychostimulants, analeptics, nootropics: an attempt to differentiate and assess drugs designed for the treatment of impaired brain functions." Pharmacopsychiatry 21(5): 211-217.
COULTER, D. A. (2001). "Epilepsy-associated plasticity in gamma-aminobutyric acid receptor expression, function, and inhibitory synaptic properties." Int Rev Neurobiol 45: 237-252.
CURIA, G., D. LONGO, G. BIAGINI, R. S. JONES und M. AVOLI (2008). "The pilocarpine model of temporal lobe epilepsy." J Neurosci Methods 172(2): 143-157.
D'HOOGE, R. und P. P. DE DEYN (2001). "Applications of the Morris water maze in the study of learning and memory." Brain Res Brain Res Rev 36(1): 60-90.
DALBY, N. O. und I. MODY (2001). "The process of epileptogenesis: a pathophysiological approach." Curr Opin Neurol 14(2): 187-192.
DELANTY, N., C. J. VAUGHAN und J. A. FRENCH (1998). "Medical causes of seizures." Lancet 352(9125): 383-390.
DELORENZO, R., MORRIS TA, BLAIR RE, WALLACE M und R. B (2002). "Topiramate is both neuroprotective and antiepileptogenic in the pilocarpine model of status epilepticus." Epilepsia 43(Suppl 7): 15.
DETOUR, J., H. SCHROEDER, D. DESOR und A. NEHLIG (2005). "A 5-month period of epilepsy impairs spatial memory, decreases anxiety, but spares object recognition in the lithium-pilocarpine model in adult rats." Epilepsia 46(4): 499-508.
DICHTER, M. A. (2009 ). "Emerging concepts in the pathogenesis of epilepsy and epileptogenesis." Arch Neurol 66(4): 443-447.
DUDEK, F. E., W. A. POULIOT, C. A. ROSSI und K. J. STALEY (2010). "The effect of the cannabinoid-receptor antagonist, SR141716, on the early stage of kainate-induced epileptogenesis in the adult rat." Epilepsia 3: 126-130.

Literatur
135
DZHALA, V. I., A. C. BRUMBACK und K. J. STALEY (2008). "Bumetanide enhances phenobarbital efficacy in a neonatal seizure model." Ann Neurol 63(2): 222-235.
DZHALA, V. I., D. M. TALOS, D. A. SDRULLA, A. C. BRUMBACK, G. C. MATHEWS, T. A. BENKE, E. DELPIRE, F. E. JENSEN und K. J. STALEY (2005). "NKCC1 transporter facilitates seizures in the developing brain." Nat Med 11(11): 1205-1213.
ECHEGOYEN, J., C. ARMSTRONG, R. J. MORGAN und I. SOLTESZ (2009). "Single application of a CB1 receptor antagonist rapidly following head injury prevents long-term hyperexcitability in a rat model." Epilepsy Res 85(1): 123-127.
EFTEKHAR, B., M. A. SAHRAIAN, B. NOURALISHAHI, A. KHAJI, Z. VAHABI, M. GHODSI, H. ARAGHIZADEH, M. R. SOROUSH, S. K. ESMAEILI und M. MASOUMI (2009). "Prognostic factors in the persistence of posttraumatic epilepsy after penetrating head injuries sustained in war." J Neurosurg 110(2): 319-326.
ENGEL, J., JR. (1996 a). "Introduction to temporal lobe epilepsy." Epilepsy Res 26(1): 141-150. ENGEL, J., JR. (1996 b). "Excitation and inhibition in epilepsy." Can J Neurol Sci 23(3): 167-174. ENGLANDER, J., T. BUSHNIK, T. T. DUONG, D. X. CIFU, R. ZAFONTE, J. WRIGHT, R. HUGHES
und W. BERGMAN (2003). "Analyzing risk factors for late posttraumatic seizures: a prospective, multicenter investigation." Arch Phys Med Rehabil 84(3): 365-373.
ESPLIN, D. W. und D. M. WOODBURY (1956). "The fate and excretion of C14-labeled pentylenetetrazol in the rat, with comments on analytical methods for pentylenetetrazol." J Pharmacol Exp Ther 118(2): 129-138.
FERNANDEZ, F., W. MORISHITA, E. ZUNIGA, J. NGUYEN, M. BLANK, R. C. MALENKA und C. C. GARNER (2007). "Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome." Nat Neurosci 10(4): 411-413.
FINK, M. (1972). "CNS effects of convulsive therapy: significance for a theory of depressive psychosis." Proc Annu Meet Am Psychopathol Assoc 60: 93-115.
FINK, M. (1984). "Meduna and the origins of convulsive therapy." Am J Psychiatry 141(9): 1034-1041. FINN, D. A. und K. W. GEE (1994). "The estrus cycle, sensitivity to convulsants and the
anticonvulsant effect of a neuroactive steroid." J Pharmacol Exp Ther 271(1): 164-170. FISHER, R. S., W. VAN EMDE BOAS, W. BLUME, C. ELGER, P. GENTON, P. LEE und J. ENGEL,
JR. (2005). "Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE)." Epilepsia 46(4): 470-472.
FOLDVARY, N., W. E. BINGAMAN und E. WYLLIE (2001). "Surgical treatment of epilepsy." Neurol Clin 19(2): 491-515.
FRANCOIS, J., K. GERME, A. FERRANDON, E. KONING und A. NEHLIG (2011). "Carisbamate has powerful disease-modifying effects in the lithium-pilocarpine model of temporal lobe epilepsy." Neuropharmacology 61(1-2): 313-328.
FRENCH, J. A., P. D. WILLIAMSON, V. M. THADANI, T. M. DARCEY, R. H. MATTSON, S. S. SPENCER und D. D. SPENCER (1993). "Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination." Ann Neurol 34(6): 774-780.
FURTADO, M. A., O. W. CASTRO, F. DEL VECCHIO, J. A. DE OLIVEIRA und N. GARCIA-CAIRASCO (2011). "Study of spontaneous recurrent seizures and morphological alterations after status epilepticus induced by intrahippocampal injection of pilocarpine." Epilepsy Behav 20(2): 257-266.
GARCIA-MORALES, I., P. DE LA PENA MAYOR und A. M. KANNER (2008). "Psychiatric comorbidities in epilepsy: identification and treatment." Neurologist 14(6 Suppl 1): S15-25.
GASTENS, A. M., C. BRANDT, J. P. BANKSTAHL und W. LÖSCHER (2008). "Predictors of pharmacoresistant epilepsy: pharmacoresistant rats differ from pharmacoresponsive rats in behavioral and cognitive abnormalities associated with experimentally induced epilepsy." Epilepsia 49(10): 1759-1776.
GERNERT, M. und W. LÖSCHER (2001). "Lack of robust anticonvulsant effects of muscimol microinfusions in the anterior substantia nigra of kindled rats." Eur J Pharmacol 432(1): 35-41.
GLIEN, M., C. BRANDT, H. POTSCHKA und W. LÖSCHER (2002). "Effects of the novel antiepileptic drug levetiracetam on spontaneous recurrent seizures in the rat pilocarpine model of temporal lobe epilepsy." Epilepsia 43(4): 350-357.
GLIEN, M., C. BRANDT, H. POTSCHKA, H. VOIGT, U. EBERT und W. LÖSCHER (2001). "Repeated low-dose treatment of rats with pilocarpine: low mortality but high proportion of rats developing epilepsy." Epilepsy Res 46(2): 111-119.

Literatur
136
GLYKYS, J., V. I. DZHALA, K. V. KUCHIBHOTLA, G. FENG, T. KUNER, G. AUGUSTINE, B. J. BACSKAI und K. J. STALEY (2009). "Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures." Neuron 63(5): 657-672.
GODDARD, G. V., D. C. MCINTYRE und C. K. LEECH (1969). "A permanent change in brain function resulting from daily electrical stimulation." Exp Neurol 25(3): 295-330.
GOFFIN, K., J. NISSINEN, K. VAN LAERE und A. PITKANEN (2007). "Cyclicity of spontaneous recurrent seizures in pilocarpine model of temporal lobe epilepsy in rat." Exp Neurol 205(2): 501-505.
GOLARAI, G., J. E. CAVAZOS und T. P. SUTULA (1992). "Activation of the dentate gyrus by pentylenetetrazol evoked seizures induces mossy fiber synaptic reorganization." Brain Res 593(2): 257-264.
GREEN, A. R. (1986). "Changes in gamma-aminobutyric acid biochemistry and seizure threshold." Ann N Y Acad Sci 462: 105-119.
GRÖTICKE, I., K. HOFFMANN und W. LÖSCHER (2008). "Behavioral alterations in a mouse model of temporal lobe epilepsy induced by intrahippocampal injection of kainate." Exp Neurol 213(1): 71-83.
HALONEN, T., T. KOTTI, J. TUUNANEN, A. TOPPINEN, R. MIETTINEN und P. J. RIEKKINEN (1995). "Alpha 2-adrenoceptor agonist, dexmedetomidine, protects against kainic acid-induced convulsions and neuronal damage." Brain Res 693(1-2): 217-224.
HAMILTON, S. E., M. D. LOOSE, M. QI, A. I. LEVEY, B. HILLE, G. S. MCKNIGHT, R. L. IDZERDA und N. M. NATHANSON (1997). "Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice." Proc Natl Acad Sci U S A 94(24): 13311-13316.
HANDELMANN, G. E. und D. S. OLTON (1981). "Spatial memory following damage to hippocampal CA3 pyramidal cells with kainic acid: impairment and recovery with preoperative training." Brain Res 217(1): 41-58.
HELFER, V., C. DERANSART, C. MARESCAUX und A. DEPAULIS (1996). "Amygdala kindling in the rat: anxiogenic-like consequences." Neuroscience 73(4): 971-978.
HERMANN, B., M. SEIDENBERG und J. JONES (2008). "The neurobehavioural comorbidities of epilepsy: can a natural history be developed?" Lancet Neurol 7(2): 151-160.
HOPPE, C., C. E. ELGER und C. HELMSTAEDTER (2007). "Long-term memory impairment in patients with focal epilepsy." Epilepsia 9: 26-29.
HUANG, R. Q., C. L. BELL-HORNER, M. I. DIBAS, D. F. COVEY, J. A. DREWE und G. H. DILLON (2001). "Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: mechanism and site of action." J Pharmacol Exp Ther 298(3): 986-995.
JACOBS, M. P., G. G. LEBLANC, A. BROOKS-KAYAL, F. E. JENSEN, D. H. LOWENSTEIN, J. L. NOEBELS, D. D. SPENCER und J. W. SWANN (2009). "Curing epilepsy: progress and future directions." Epilepsy Behav 14(3): 438-445.
JENSEN, F. E. (2011). "Epilepsy as a spectrum disorder: Implications from novel clinical and basic neuroscience." Epilepsia 1: 1-6.
JESSBERGER, S., K. NAKASHIMA, G. D. CLEMENSON, JR., E. MEJIA, E. MATHEWS, K. URE, S. OGAWA, C. M. SINTON, F. H. GAGE und J. HSIEH (2007). "Epigenetic modulation of seizure-induced neurogenesis and cognitive decline." J Neurosci 27(22): 5967-5975.
JONES, R., H. RICKARDS und A. E. CAVANNA (2010). "The prevalence of psychiatric disorders in epilepsy: a critical review of the evidence." Funct Neurol 25(4): 191-194.
JUNG, K. H., K. CHU, S. T. LEE, J. KIM, D. I. SINN, J. M. KIM, D. K. PARK, J. J. LEE, S. U. KIM, M. KIM, S. K. LEE und J. K. ROH (2006). "Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus." Neurobiol Dis 23(2): 237-246.
JUNG, M. E., H. LAL und M. B. GATCH (2002). "The discriminative stimulus effects of pentylenetetrazol as a model of anxiety: recent developments." Neurosci Biobehav Rev 26(4): 429-439.
KÄLVIÄINEN, R., T. SALMENPERA, K. PARTANEN, P. VAINIO, P. RIEKKINEN und A. PITKÄNEN (1998). "Recurrent seizures may cause hippocampal damage in temporal lobe epilepsy." Neurology 50(5): 1377-1382.
KANNER, A. M. (2011). "Anxiety disorders in epilepsy: the forgotten psychiatric comorbidity." Epilepsy Curr 11(3): 90-91.

Literatur
137
KHAZIPOV, R. und G. L. HOLMES (2003). "Synchronization of kainate-induced epileptic activity via GABAergic inhibition in the superfused rat hippocampus in vivo." J Neurosci 23(12): 5337-5341.
KLAASSEN, A., J. GLYKYS, J. MAGUIRE, C. LABARCA, I. MODY und J. BOULTER (2006). "Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy." Proc Natl Acad Sci U S A 103(50): 19152-19157.
KÖHLING, R. (2002). "Neuroscience. GABA becomes exciting." Science 298(5597): 1350-1351. KÜCKER, S., K. TÖLLNER, M. PIECHOTTA und M. GERNERT (2010). "Kindling as a model of
temporal lobe epilepsy induces bilateral changes in spontaneous striatal activity." Neurobiol Dis 37(3): 661-672.
KWAN, P., A. ARZIMANOGLOU, A. T. BERG, M. J. BRODIE, W. ALLEN HAUSER, G. MATHERN, S. L. MOSHE, E. PERUCCA, S. WIEBE und J. FRENCH (2010). "Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies." Epilepsia 51(6): 1069-1077.
LANGER, M., C. BRANDT, C. ZELLINGER und W. LÖSCHER (2011). "Therapeutic window of opportunity for the neuroprotective effect of valproate versus the competitive AMPA receptor antagonist NS1209 following status epilepticus in rats." Neuropharmacology 61(5-6): 1033-1047.
LEITE, J. P., Z. A. BORTOLOTTO und E. A. CAVALHEIRO (1990 a). "Spontaneous recurrent seizures in rats: an experimental model of partial epilepsy." Neurosci Biobehav Rev 14(4): 511-517.
LEITE, J. P., E. M. NAKAMURA, T. LEMOS, J. MASUR und E. A. CAVALHEIRO (1990 b). "Learning impairment in chronic epileptic rats following pilocarpine-induced status epilepticus." Braz J Med Biol Res 23(8): 681-683.
LEROY, C., P. POISBEAU, A. F. KELLER und A. NEHLIG (2004). "Pharmacological plasticity of GABA(A) receptors at dentate gyrus synapses in a rat model of temporal lobe epilepsy." J Physiol 557(Pt 2): 473-487.
LHATOO, S. D., J. W. SANDER und D. FISH (2001). "Temporal lobe epilepsy following febrile seizures: unusually prolonged latent periods." Eur Neurol 46(3): 165-166.
LI, X., J. ZHOU, Z. CHEN, S. CHEN, F. ZHU und L. ZHOU (2008). "Long-term expressional changes of Na+ -K+ -Cl- co-transporter 1 (NKCC1) and K+ -Cl- co-transporter 2 (KCC2) in CA1 region of hippocampus following lithium-pilocarpine induced status epilepticus (PISE)." Brain Res 24: 141-146.
LODGE, D., G. A. JOHNSTON, D. R. CURTIS und J. C. BORNSTEIN (1979). "Kainate neurotoxicity and glutamate inactivation." Neurosci Lett 14(2-3): 343-348.
LÖSCHER, W. (2002). "Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy." Epilepsy Res 50(1-2): 105-123.
LÖSCHER, W. (2007). "The pharmacokinetics of antiepileptic drugs in rats: consequences for maintaining effective drug levels during prolonged drug administration in rat models of epilepsy." Epilepsia 48(7): 1245-1258.
LÖSCHER, W. (2009). "Preclinical assessment of proconvulsant drug activity and its relevance for predicting adverse events in humans." Eur J Pharmacol 610(1-3): 1-11.
LÖSCHER, W. (2011). "Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs." Seizure 20(5): 359-368.
LÖSCHER, W. und C. BRANDT (2010 a). "Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research." Pharmacol Rev 62(4): 668-700.
LÖSCHER, W. und C. BRANDT (2010 b). "High seizure frequency prior to antiepileptic treatment is a predictor of pharmacoresistant epilepsy in a rat model of temporal lobe epilepsy." Epilepsia 51(1): 89-97.
LÖSCHER, W., M. GERNERT und U. HEINEMANN (2008). "Cell and gene therapies in epilepsy--promising avenues or blind alleys?" Trends Neurosci 31(2): 62-73.
LÖSCHER, W., D. HÖNACK und C. RUNDFELDT (1998). "Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy." J Pharmacol Exp Ther 284(2): 474-479.
LÖSCHER, W. und R. KÖHLING (2010). "Functional, metabolic, and synaptic changes after seizures as potential targets for antiepileptic therapy." Epilepsy Behav 19(2): 105-113.

Literatur
138
LÖSCHER, W. und D. SCHMIDT (2006). New Horizons in the development of antiepileptic drugs: Innovative strategies, Epilepsy Res. 2006 Jun;69(3):183-272.
MACDONALD, R. L. und J. L. BARKER (1977). "Pentylenetetrazol and penicillin are selective antagonists of GABA-mediated post-synaptic inhibition in cultured mammalian neurones." Nature 267(5613): 720-721.
MAJORES, M., J. EILS, O. D. WIESTLER und A. J. BECKER (2004). "Molecular profiling of temporal lobe epilepsy: comparison of data from human tissue samples and animal models." Epilepsy Res 60(2-3): 173-178.
MANI, R., J. POLLARD und M. A. DICHTER (2011). "Human clinical trails in antiepileptogenesis." Neurosci Lett 497(3): 251-256.
MANN, E. O. und I. MODY (2008). "The multifaceted role of inhibition in epilepsy: seizure-genesis through excessive GABAergic inhibition in autosomal dominant nocturnal frontal lobe epilepsy." Curr Opin Neurol 21(2): 155-160.
MARCHI, N., Q. FAN, C. GHOSH, V. FAZIO, F. BERTOLINI, G. BETTO, A. BATRA, E. CARLTON, I. NAJM, T. GRANATA und D. JANIGRO (2009). "Antagonism of peripheral inflammation reduces the severity of status epilepticus." Neurobiol Dis 33(2): 171-181.
MARCUCCI, F., M. L. AIROLDI, E. MUSSINI und S. GARATTINI (1971). "Brain levels of metrazol determined with a new gas chromatographic procedure." Eur J Pharmacol 16(2): 219-221.
MARGERISON, J. H. und J. A. CORSELLIS (1966). "Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes." Brain 89(3): 499-530.
MAZARATI, A., D. SHIN und R. SANKAR (2009). "Bumetanide inhibits rapid kindling in neonatal rats." Epilepsia 50(9): 2117-2122.
MIRSKI, M. A. und J. A. FERRENDELLI (1986). "Anterior thalamic mediation of generalized pentylenetetrazol seizures." Brain Res 399(2): 212-223.
MIRSKI, M. A. und J. A. FERRENDELLI (1987). "Interruption of the connections of the mammillary bodies protects against generalized pentylenetetrazol seizures in guinea pigs." J Neurosci 7(3): 662-670.
MORIMOTO, K., M. FAHNESTOCK und R. J. RACINE (2004). "Kindling and status epilepticus models of epilepsy: rewiring the brain." Prog Neurobiol 73(1): 1-60.
MORRIS, R. (1984). "Developments of a water-maze procedure for studying spatial learning in the rat." J Neurosci Methods 11(1): 47-60.
MUNOZ, C. und S. P. GROSSMAN (1980). "Some behavioral effects of selective neuronal depletion by kainic acid in the dorsal hippocampus of rats." Physiol Behav 25(4): 581-587.
NAGAO, T., A. ALONSO und M. AVOLI (1996). "Epileptiform activity induced by pilocarpine in the rat hippocampal-entorhinal slice preparation." Neuroscience 72(2): 399-408.
NISSINEN, J., T. HALONEN, E. KOIVISTO und A. PITKANEN (2000). "A new model of chronic temporal lobe epilepsy induced by electrical stimulation of the amygdala in rat." Epilepsy Res 38(2-3): 177-205.
NISSINEN, J. und A. PITKÄNEN (2007). "Effect of antiepileptic drugs on spontaneous seizures in epileptic rats." Epilepsy Res 73(2): 181-191.
NUTT, D. J., P. J. COWEN und A. R. GREEN (1981). "Studies on the post-ictal rise in seizure threshold." Eur J Pharmacol 71(2-3): 287-295.
OLNEY, J. W., V. RHEE und O. L. HO (1974). "Kainic acid: a powerful neurotoxic analogue of glutamate." Brain Res 77(3): 507-512.
ORLOFF, M. J., H. L. WILLIAMS und C. C. PFEIFFER (1949). "Timed intravenous infusion of metrazol and strychnine for testing anticonvulsant drugs." Proc Soc Exp Biol Med 70(2): 254-257.
PALMA, E., M. AMICI, F. SOBRERO, G. SPINELLI, S. DI ANGELANTONIO, D. RAGOZZINO, A. MASCIA, C. SCOPPETTA, V. ESPOSITO, R. MILEDI und F. EUSEBI (2006). "Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory." Proc Natl Acad Sci U S A 103(22): 8465-8468.
PATHAK, H. R., F. WEISSINGER, M. TERUNUMA, G. C. CARLSON, F. C. HSU, S. J. MOSS und D. A. COULTER (2007). "Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy." J Neurosci 27(51): 14012-14022.
PAVLOV, I., N. HUUSKO, M. DREXEL, E. KIRCHMAIR, G. SPERK, A. PITKANEN und M. C. WALKER (2011). "Progressive loss of phasic, but not tonic, GABA(A) receptor-mediated inhibition in dentate granule cells in a model of post-traumatic epilepsy in rats." Neuroscience 194: 208-219.

Literatur
139
PAXINOS, G. und C. WATSON, Eds. (2007). The Rat Brain in Stereotactic Coordinates. Amsterdam. PITKÄNEN, A. (2010). "Therapeutic approaches to epileptogenesis--hope on the horizon." Epilepsia 3:
2-17. PITKÄNEN, A., T. BOLKVADZE und R. IMMONEN (2011 ). "Anti-epileptogenesis in rodent post-
traumatic epilepsy models." Neurosci Lett 497(3): 163-171. PITKÄNEN, A. und K. LUKASIUK (2011). "Mechanisms of epileptogenesis and potential treatment
targets." Lancet Neurol 10(2): 173-186. PITKÄNEN, A., S. NARKILAHTI, Z. BEZVENYUK, A. HAAPALINNA und J. NISSINEN (2004).
"Atipamezole, an alpha(2)-adrenoceptor antagonist, has disease modifying effects on epileptogenesis in rats." Epilepsy Res 61(1-3): 119-140.
POLASCHECK, N., M. BANKSTAHL und W. LÖSCHER (2010). "The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy." Exp Neurol 224(1): 219-233.
POTSCHKA, H. und W. LÖSCHER (2001). "In vivo evidence for P-glycoprotein-mediated transport of phenytoin at the blood-brain barrier of rats." Epilepsia 42(10): 1231-1240.
PUURUNEN, K., J. JOLKKONEN, J. SIRVIO, A. HAAPALINNA und J. SIVENIUS (2001). "An alpha(2)-adrenergic antagonist, atipamezole, facilitates behavioral recovery after focal cerebral ischemia in rats." Neuropharmacology 40(4): 597-606.
QIAN, B., Y. SUN, Z. WU, L. WAN, L. CHEN, S. KONG, B. ZHANG, F. ZHANG, Z. Y. WANG und Y. WANG (2011). "Epileptiform response of CA1 neurones to convulsant stimulation by cyclothiazide, kainic acid and pentylenetetrazol in anaesthetized rats." Seizure 20(4): 312-319.
RACINE, R. J. (1972). "Modification of seizure activity by electrical stimulation. II. Motor seizure." Electroencephalogr Clin Neurophysiol 32(3): 281-294.
RAEDT, R., A. VAN DYCKE, D. VAN MELKEBEKE, T. DE SMEDT, P. CLAEYS, T. WYCKHUYS, K. VONCK, W. WADMAN und P. BOON (2009). "Seizures in the intrahippocampal kainic acid epilepsy model: characterization using long-term video-EEG monitoring in the rat." Acta Neurol Scand 119(5): 293-303.
RAMANJANEYULU, R. und M. K. TICKU (1984). "Interactions of pentamethylenetetrazole and tetrazole analogues with the picrotoxinin site of the benzodiazepine-GABA receptor-ionophore complex." Eur J Pharmacol 98(3-4): 337-345.
RAMZAN, I. M. und G. LEVY (1985). "Kinetics of drug action in disease states. XIV. Effect of infusion rate on pentylenetetrazol concentrations in serum, brain and cerebrospinal fluid of rats at onset of convulsions." J Pharmacol Exp Ther 234(3): 624-628.
RIBAN, V., V. BOUILLERET, B. T. PHAM-LÊ, J. M. FRITSCHY, C. MARESCAUX und A. DEPAULIS (2002). "Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy." Neuroscience 112(1): 101-111.
RICE, A. C., C. L. FLOYD, B. G. LYETH, R. J. HAMM und R. J. DELORENZO (1998). "Status epilepticus causes long-term NMDA receptor-dependent behavioral changes and cognitive deficits." Epilepsia 39(11): 1148-1157.
RICHTLINIEN DER AMERICAN ASSOCIATION FOR THE TRAUMA SURGERY (2001). "Antiseizure prophylaxis for penetrating brain injury." J Trauma 51(2 Suppl): S41-43.
RIGOULOT, M. A., E. KONING, A. FERRANDON und A. NEHLIG (2004). "Neuroprotective properties of topiramate in the lithium-pilocarpine model of epilepsy." J Pharmacol Exp Ther 308(2): 787-795.
RIGOULOT, M. A., C. LEROY, E. KONING, A. FERRANDON und A. NEHLIG (2003). "Prolonged low-dose caffeine exposure protects against hippocampal damage but not against the occurrence of epilepsy in the lithium-pilocarpine model in the rat." Epilepsia 44(4): 529-535.
SCHACHER, M., R. WINKLER, T. GRUNWALD, G. KRAEMER, M. KURTHEN, V. REED und H. JOKEIT (2006). "Mesial temporal lobe epilepsy impairs advanced social cognition." Epilepsia 47(12): 2141-2146.
SCHARFMAN, H. E. und N. J. MACLUSKY (2006). "The influence of gonadal hormones on neuronal excitability, seizures, and epilepsy in the female." Epilepsia 47(9): 1423-1440.
SHORR, E. (1940). "A New Technic for Staining Vaginal Smears." Science 91(2361): 321-322. SHORVON, S. D. (2011). "The etiologic classification of epilepsy." Epilepsia 52(6): 1052-1057. SILLANPÄÄ, M. und D. SCHMIDT (2006). "Natural history of treated childhood-onset epilepsy:
prospective, long-term population-based study." Brain 129(Pt 3): 617-624. SILVER, J. M., C. SHIN und J. O. MCNAMARA (1991). "Antiepileptogenic effects of conventional
anticonvulsants in the kindling model of epilespy." Ann Neurol 29(4): 356-363.

Literatur
140
SLOVITER, R. S. (1994). "The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy." Ann Neurol 35(6): 640-654.
SLOVITER, R. S. (2005). "The neurobiology of temporal lobe epilepsy: too much information, not enough knowledge." C R Biol 328(2): 143-153.
SLOVITER, R. S. (2008). "Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis: the importance of the "latent period" and other concepts." Epilepsia 9: 85-92.
SMOLDERS, I., G. M. KHAN, J. MANIL, G. EBINGER und Y. MICHOTTE (1997). "NMDA receptor-mediated pilocarpine-induced seizures: characterization in freely moving rats by microdialysis." Br J Pharmacol 121(6): 1171-1179.
SPENCER, S. S. (2002). "When should temporal-lobe epilepsy be treated surgically?" Lancet Neurol 1(6): 375-382.
STABLES, J. P., E. H. BERTRAM, H. S. WHITE, D. A. COULTER, M. A. DICHTER, M. P. JACOBS, W. LOSCHER, D. H. LOWENSTEIN, S. L. MOSHE, J. L. NOEBELS und M. DAVIS (2002). "Models for epilepsy and epileptogenesis: report from the NIH workshop, Bethesda, Maryland." Epilepsia 43(11): 1410-1420.
STAFSTROM, C. E. und T. P. SUTULA (2005). "Models of epilepsy in the developing and adult brain: implications for neuroprotection." Epilepsy Behav 7(3): 19.
STARK, D. T. und N. G. BAZAN (2011). "Synaptic and extrasynaptic NMDA receptors differentially modulate neuronal cyclooxygenase-2 function, lipid peroxidation, and neuroprotection." J Neurosci 31(39): 13710-13721.
STRATTON, S. C., C. H. LARGE, B. COX, G. DAVIES und R. M. HAGAN (2003). "Effects of lamotrigine and levetiracetam on seizure development in a rat amygdala kindling model." Epilepsy Res 53(1-2): 95-106.
STRINGER, J. L. (1994). "Pentylenetetrazol elicits epileptiform activity in the dentate gyrus of the urethane anesthetized rat by activation of the entorhinal cortex." Brain Res 636(2): 221-226.
SUCHOMELOVA, L., R. A. BALDWIN, H. KUBOVA, K. W. THOMPSON, R. SANKAR und C. G. WASTERLAIN (2006). "Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects." Pediatr Res 59(2): 237-243.
SUN, Q. J., R. S. DUAN, A. H. WANG, W. SHANG, T. ZHANG, X. Q. ZHANG und Z. F. CHI (2009). "Alterations of NR2B and PSD-95 expression in hippocampus of kainic acid-exposed rats with behavioural deficits." Behav Brain Res 201(2): 292-299.
TEMKIN, N. R. (2001). "Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials." Epilepsia 42(4): 515-524.
TEMKIN, N. R. (2009). "Preventing and treating posttraumatic seizures: the human experience." Epilepsia 2: 10-13.
TEMKIN, N. R., S. S. DIKMEN, G. D. ANDERSON, A. J. WILENSKY, M. D. HOLMES, W. COHEN, D. W. NEWELL, P. NELSON, A. AWAN und H. R. WINN (1999). "Valproate therapy for prevention of posttraumatic seizures: a randomized trial." J Neurosurg 91(4): 593-600.
TEMKIN, N. R., S. S. DIKMEN, A. J. WILENSKY, J. KEIHM, S. CHABAL und H. R. WINN (1990). "A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures." N Engl J Med 323(8): 497-502.
TEMKIN, N. R., M. M. HAGLUND und H. R. WINN (1995). "Causes, prevention, and treatment of post-traumatic epilepsy." New Horiz 3(3): 518-522.
TURNER, I. M., S. M. NEWMAN, S. LOUIS und H. KUTT (1977). "Pharmacological prophylaxis against the development of kindled amygdaloid seizures." Ann Neurol 2(3): 221-224.
TURSKI, L., E. A. CAVALHEIRO, S. J. CZUCZWAR, W. A. TURSKI und Z. KLEINROK (1987). "The seizures induced by pilocarpine: behavioral, electroencephalographic and neuropathological studies in rodents." Pol J Pharmacol Pharm 39(5): 545-555.
TURSKI, W. A., E. A. CAVALHEIRO, M. SCHWARZ, S. J. CZUCZWAR, Z. KLEINROK und L. TURSKI (1983). "Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study." Behav Brain Res 9(3): 315-335.
VEZZANI, A. und T. GRANATA (2005). "Brain inflammation in epilepsy: experimental and clinical evidence." Epilepsia 46(11): 1724-1743.
VOHLAND, H. W. und H. ZUFELDE (1976). "Factors responsible for reduced pharmacological activity in rats of pentetrazol administered orally." Naunyn Schmiedebergs Arch Pharmacol 293(3): 277-283.

Literatur
141
WALKER, M. C., H. S. WHITE und J. W. SANDER (2002). "Disease modification in partial epilepsy." Brain 125(Pt 9): 1937-1950.
WALLACE, M. J., R. E. BLAIR, K. W. FALENSKI, B. R. MARTIN und R. J. DELORENZO (2003). "The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy." J Pharmacol Exp Ther 307(1): 129-137.
WALSH, L. A., M. LI, T. J. ZHAO, T. H. CHIU und H. C. ROSENBERG (1999). "Acute pentylenetetrazol injection reduces rat GABAA receptor mRNA levels and GABA stimulation of benzodiazepine binding with No effect on benzodiazepine binding site density." J Pharmacol Exp Ther 289(3): 1626-1633.
WEAVER, D. F. (2003). "Epileptogenesis, ictogenesis and the design of future antiepileptic drugs." Can J Neurol Sci 30(1): 4-7.
ZENG, L. H., N. R. RENSING und M. WONG (2009). "The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy." J Neurosci 29(21): 6964-6972.
Anhang
142
10. Anhang
Tierhaltung und -versorgung
Verbrauchsmaterial Bezugsquelle
Einstreu (Weichholzgranulat Altromin) Altromin, Lage
Makrolonkäfig (Typ III und Typ IV) Ebeco, Caustrop-Rauxel
Milchbrei (Früchte oder Banane) Milupa GmbH, Friedrichsdorf
Tierfutter (Standardnagerdiät Atromin
1324)
Altromin, Lage
Stereotaktische Operation
Gerät Bezugsquelle
Bohrer (0,13/ 0,11 mm) Hager & Meisinger GmbH, Neuss
Dentalbohrer (Mod. 732 T1) Dremel Leinfelden-Echterdingen
Stereotakt I David Kopf Tujunga, USA
Stereotakt II Stölting Wood Dale, USA
Verbrauchsmaterial Bezugsquelle
Bepanthen Augensalbe Bayer Vital, Leverkusen
Bupivacainhydrochlorid (Carbostesin) Astra Zeneca GmbH, Wedel
Chloralhydrat Roth, Karlsruhe
Chloramphenicol-Succinat Roth, Karlsruhe
Dentalzement (Paladur®) Heraeus Kulzer, Hanau
Draht für Elektroden und Erden (SS-
8T/SA)
Science Products, Hofheim
Draht für Führungsrohre Schoeller Werker, Hellenthal
Gentamicinsulfat Caelo, Hilden
Isofluran Cp Pharma, Burgdorf
Isotone Natriumchloridlösung (NaCl
0,9%)
Braun, Tuttlingen
Marbofloxacin (Marbocyl® FD1 %) WDT,Garbsen

Anhang
143
Nahtmaterial (Surgicryl® PGA
polyglycolic acid)
SMI, Hünningen, Belgien
Sauerstoff Linde AG, Pullach
Schrauben (aus nicht rostendem Stahl) Hummer & Rieß, Nürnberg
Tetracainhydrochlorid Caesar&Loretz GmbH, Hilden
Wasserstoffperoxid (35%) AppliChem GmbH, Darmstadt
SE-Induktion Pilokarpin
Substanz Bezugsquelle
Diazepam (Faustan®) Temmler Pharma GmbH & Co. KG,
Marburg
Isotone Natriumchloridlösung (NaCl
0,9%)
Braun, Tuttlingen
Lithiumchlorid Sigma Aldrich, Steinheim
Methylscopolamin Sigma Aldrich, Steinheim
Phenobarbital-Natrium Serva, Heidelberg
Pilocarpin Sigma Aldrich, Steinheim
Scopolamin Sigma Aldrich, Steinheim
SE-Induktion Kainsäure
Substanz Bezugsquelle
Destilliertes Wasser Eigenherstellung
Isotone Natriumchloridlösung (NaCl
0,9%)
Braun, Tuttlingen
Kainsäure Sigma Aldrich, Steinheim
Thionin (Acetatsalz) Sigma Aldrich, München
Anhang
144
Gerät/Verbrauchsmaterial Bezugsquelle
Feinwaage „CPA225D“ Sartorius AG, Göttingen
Hamilton-Spritze (5 µl, 24 G) Hamilton Bonaduz AG, Bonaduz,
Schweiz
Draht für Injektionsnadel 18 mm Schoeller Werke, Hellenthal
Mikroinjektionsschlauch CMA Microdialysis, Schweden
EEG- und Videoüberwachung
Gerät Bezugsquelle
Ableitkabel Hopa Kabelkonfektion, Langenhagen
Ableitkabel Eigenproduktion Material von Radio Menzel, Hannover
Digitalkamera (Logitech, 2MPC903) Conrad Elektronic, Hannover
Infrarotkamera (Nycto Vision) CaS Buisness Servicess, Wunstorf
Infrarotlampen und Generator (Nycto
Vision)
CaS Buisness Servicess, Wunstorf
Rotlichtlampen und Generator Conrad Elektronic, Hannover
16-Ein-Kanal-Verstärker (BioAmp) ADInstuments, Spechbach
16-Kanal-Analog-Digitalwandler
(Powerlab/16SP)
ADInstuments, Spechbach
Verhaltensversuche
Gerät/Substanz Bezugsquelle
Essigsäure Riedel de Häen, Hannover
Etho Vision Software Noldus, Wageningen, Niederlande
Kamera Panasonic
Klimaanlage Fujitsu General Euro GmbH, Düsseldorf
Open field Arena Firma Zimmermann, Hannover
PC Dell
Waage, Sartorius Universal Omnilab, Göttingen
White Noise Generator 60 dB Eigenbau
Anhang
145
Histologie
Verbrauchsmaterial/Gerät Bezugsquelle
Chloralhydrat AppliChem GmbH, Darmstadt
CO2 Kohlensäurewerk, Räthen
Eideckmedium (Entellan) Merck, Darmstadt
Gefriermedium Jung, Nussloch
Gefriermikrotom, Frigomobil CM1325 Leica, Wetzlar
Kryostat HM560M Microm, Walldorf
Lichtmikroskop Leica, Wetzlar
Lichtmikroskop Zeiss, Göttingen
N-Methylbutan Fulka Chemie GmbH, Buchs, Schweiz
Objektträger und Deckgläschen Roth, Karlsruhe
Paraformaldehyd Roth, Karlsruhe
Perfusionspumpe ISM897A Ismatech Laboratoriums GmbH,
Wertheim-Modfeld
Stickstoff Westfalen Gas, Münster
Thionin Sigma Aldrich, München
Xylol Roth, Karlsruhe
Xylol Ersatzmedium (XEM) Roth, Karlsruhe
PTZ-Schwellentest
Verbrauchsmaterial/Gerät Bezugsquelle
Infusionspumpe (PHD 2000) Harvard Appartus, Holliston, USA
Injektionskanüle 24 G Terumo, Leuven, Belgien
Isotone Natriumchloridlösung (NaCl
0,9%)
Braun, Tuttlingen
Pentylentetrazol Caesar & Loretz, Hilden
Polyethylenschlauch Kleinfeld Labortechnik, Gehrden

Anhang
146
PTZ-Infusion
Verbrauchsmaterial/Gerät Bezugsquelle
Ein-Kanal-Swivel Instech Laboratories, Plymouth Meeting,
USA
Geschirr mit Federspirale Instech Laboratories, Plymouth Meeting,
USA
Infusionspumpe (PHD 2000) Harvard Appartus, Holliston, USA
Isotone Natriumchloridlösung (NaCl
0,9%)
Braun, Tuttlingen
Kanülen 21 G, stumpf B. Braun, Melsungen
Pentylentetrazol Caesar & Loretz, Hilden
Polyethylenschlauch Katheter Kleinfeld Labortechnik, Gehrden
Tubing Adapter (C3409500) Axel Semrau, Sprockhövel
Tygonschlauch (S-54-HL) Kleinfeld Labortechnik, Gehrden
PTZ-Analytik
Gerät Bezugsquelle
Entgaser (ERC - 3512) Erma-Elektronic, Immendingen
Filter (Millipore Kit; 0,22 μm) Millipore, Billerica, MA, USA
Hauptsäule (HPLC column EC 250/4.6; Nuceosil 120-5 C18, 250 x 4,6 mm)
Macherey-Nagel, Düren
Integrator (C - R4A) Shimadzu, Duisburg
Pumpen (LC - 6A) Shimadzu, Duisburg
Pumpencontroler (SCL-6A; Fluß: 1 ml/min 35°C)
Shimadzu, Duisburg
Säulenofen (Knauer Column Oven,
35°C)
Knauer, Berlin
UV-Detektor (SPD – 6A; Wellenlänge:
210 nm)
Shimadzu, Duisburg
Vorsäule (Nucleosil 120-5 C 18, 60 x 4
mm)
Knauer, Berlin
Zentrifuge (5415 D) Eppendorf, Hamburg

Anhang
147
Verbrauchsmaterial Bezugsquelle
Acetonnitril p.a. (HPLC FarUV) Lab-Scan, Gliwice, Polen
Calciumchlorid-Dihydrat p.a. Merck, Darmstadt
Kaliumchlorid p.a. Merck, Darmstadt
Kaliumdihydrogenphosphat p.a. Merck, Darmstadt
Magnesiumsulfat-Heptahydat p.a. Merck, Darmstadt
Methanol p.a. Lab-Scan, Gliwice, Polen
Natriumchlorid p.a. Roth, Karlsruhe
Natriumdihydrogencarbonat p.a. Merck, Darmstadt
Natriumdihydrogenphosphat p.a. Sigma-Aldrich, München
Natriumhydroxid p.a. Sigma-Aldrich, München
Pentylentetrazol Caesar&Loretz, Hilden
Salzsäure p.a. (37%ig) Merck, Darmstadt
Tetracainhydrochlorid Caesar & Loretz, Hilden
Titriplex III p.a. Merck, Darmstadt
Puffer und Lösungen
0,4 M Phosphatpuffer (Stammlösung)
800 ml Aqua dest.
57 g Di-Natriumhydrogenphosphat-Dihydrat
12,48 g Natriumdihydrogenphosphat-Dihydrat
Mit 1 M Natronlauge auf pH 7,4 einstellen
Ad 1.000 ml Aqua dest.
0,5 M Tris-gepufferte Saline (TBS)
9 g Natriumchlorid
6,057 g Tris
800 ml Aqua dest.
Mit 37%iger Salzsäure auf pH 7,6 einstellen
Ad 1.000 ml Aqua dest.

Anhang
148
8% Paraformaldehydlösung
800 ml Aqua dest. auf 60-70°C erhitzen
80 g Paraformaldehyd-Pulver unter Rühren hinzugeben
Mit 1 M Natronlauge bis zur Klärung titrieren
Auf Raumtemperatur abkühlen lassen und dann filtrieren
Ad 1.000 ml Aqua dest.
4% Paraformaldehydlösung
8%ige Paraformaldehydlösung mit 0,2 M PBS auf eine 4%ige Lösung verdünnen
10%ige Saccharoselösung (pro Rattengehirn)
3 g Zucker
Ad 30 ml 0,1M PBS
30%ige Saccharoselösung (pro Rattengehirn)
9 g Zucker
Ad 30 ml 0,1M PBS
Chromgelantine
7 g Gelantine in 1.000 ml Aqua dest. auf 60°C erwärmen
Etwas Thymol (wenige mg) zugeben
0,7 g Chrom-(III) Kaliumsulfat-Dodecanhydrat nach dem Erwärmen hinzugeben
Auf Eis auf 20°C abkühlen
filtrieren

Anhang
149
Gefriermedium
85,6g Glucose-Monohydrat
1,4g Magnesium-Chlorid-Hexahydrat
In 500 ml 0,1 M PBS lösen
Mit 87%igem Glyzerin auf 1.000 ml auffüllen
Thionin-Lösung
100 ml 1 M Essigsäure
36 ml 1 M Natronlauge
Ad 1.000 ml Aqua dest.
Auf 60-70°C erhitzen
1,25 g Thionin darin lösen
1 Stunde bei 60-70°C rühren und dann heiß filtrieren
Bei 60°C lagern (bis zu drei Monate haltbar)
Färbeprotokolle
Thioninfärbung
3 min in 100% Ethanol inkubieren
3 min in 95% Ethanol inkubieren
3 min in 70% Ethanol inkubieren
3 min in 50% Ethanol inkubieren
3 min in Aqua dest. inkubieren
75-90 sek. in Thionin-Färbelösung (42- 48°C) inkubieren
3 min in 50% Ethanol inkubieren

Anhang
150
3 min in 70% Ethanol inkubieren
3 min in 95% Ethanol inkubieren
3 min in 100% Ethanol inkubieren
3 min in Terpineol/Xylol-Ersatzmedium 1:1 inkubieren
3 min in Xylol-Ersatzmedium inkubieren
3 min in frischem Xylol-Ersatzmedium inkubieren
Sofortiges Eindecken der Schnitte mit Entellan® und Trocknen an der Luft
Färbung nach Shorr
10-mal in 70% Ethanol eintauchen
10-mal in Aqua dest. Wässern
2 min in Meyers Hämalaun inkubieren
10-mal in Aqua dest. Wässern
1 min in Lithium-Carbonat (gesättigte Lösung 1:1 mit Aqua dest. verdünnt) inkubieren
10-mal in Aqua dest. Wässern
10-mal in 70% Ethanol inkubieren
6 min in Shorr´scher Färbelösung inkubieren
10-mal in 95% Ethanol eintauchen
30 sek. In 95% Ethanol inkubieren
Lufttrocknen lassen

151
Publikationen
Originalarbeiten
C. BRANDT, M. NOZADZE, N. HEUCHERT, M. RATTKA and W. LÖSCHER (2010)
"Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in
the pilocarpine model of temporal lobe epilepsy."
J Neurosci 30(25): 8602-8612.
M. RATTKA, C. BRANDT, M. BANKSTAHL, S. BRÖER and W. LÖSCHER (2011)
"Enhanced susceptibility to the GABA antagonist pentylenetetrazole during the latent
period following a pilocarpine-induced status epilepticus in rats."
Neuropharmacology 60(2-3): 505-512.
Poster
RATTKA, M., C. BRANDT, M. BANKSTAHL, S. BRÖER and W. LÖSCHER (2010)
“Alterations in pentylenetetrazole seizure threshold after pilocarpine-induced status
epilepticus”
FENS Abstr., vol.5, 106.36
M. RATTKA, C. BRANDT, M. BANKSTAHL, S. BRÖER and W. LÖSCHER (2010)
„Alterations in seizure threshold to the GABAA receptor antagonist pentylenetetrazole
after pilocarpine-induced status epilepticus.”
40 th Annual Meeting Society for Neuroscience, San Diego, 350.2/K14
M. RATTKA, C: BRANDT and W: LÖSCHER (2011)
“Effects of subconvulsive pentylenetetrazole infusion on epileptogenesis in a status
epilepticus model in Sprague-Dawley rats.”
Epilepsia 52, Issue Supplement s6, 159

152
Erklärung Hiermit erkläre ich, dass ich die Dissertation „Prokonvulsiva als pharmakologische
Strategie zur Epilepsieprävention“ selbstständig verfasst habe.
Bei der Anfertigung wurden folgende Hilfen Dritter in Anspruch genommen:
Frau M. Gramer und Frau M. Hausknecht (Technische Angestellte am
Institut für Pharmakologie, Toxikologie und Pharmazie) führten für mich die HPLC-
Analyse der PTZ-Plasmakonzentrationen durch. Herr Michael Weißing (Technischer
Angestellter am Institut für Pharmakologie, Toxikologie und Pharmazie) fertigte die
Kryostat-Schnitte der Gehirne an. Frau Nicole Ernst, Frau J. Hinz, Herr S. Obili
(Technische Angestellte am Institut für Pharmakologie, Toxikologie und Pharmazie)
und Herr Phillip Demmer (Biologie-Diplomand) haben mich bei der Beobachtung der
Tiere während der PTZ-Infusion unterstützt.
Ich habe keine entgeltliche Hilfe von Vermittlungs- bzw. Beratungsdiensten
(Promotionsberater oder andere Personen) in Anspruch genommen. Niemand hat
von mir unmittelbar oder mittelbar entgeltliche Leistungen für Arbeiten erhalten, die
im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen.
Ich habe die Dissertation an folgenden Institution angefertigt:
Institut für Pharmakologie, Toxikologie und Pharmazie der Tierärztlichen Hochschule
Hannover. Die Dissertation wurde bisher nicht für eine Prüfung oder Promotion oder
für einen ähnlichen Zweck zur Beurteilung eingereicht. Ich versichere, dass ich die
vorstehenden Angaben nach bestem Wissen vollständig und der Wahrheit
entsprechend gemacht habe.
Unterschrift

153
Danksagung
Mein größter Dank gilt Herrn Professor Doktor W. Löscher für die Überlassung des
Themas, die wissenschaftliche Anleitung bei der Durchführung der Versuche und der
Abfassung dieser Dissertation sowie die jederzeit gewährte freundliche
Unterstützung.
Herrn Professor Doktor G. Bicker und Herrn Professor Doktor E. Ponimaskin danke
ich für die kompetente und freundliche Betreuung während des PhD-Studiums.
Frau Claudia Brandt, PhD möchte ich für die freundliche Betreuung, die Hilfe bei der
Durchführung der Versuche und die geduldigen Korrekturen meiner hoffnungslos
unformatierten Präsentationen danken. Ich danke ihr auch für die sehr nette
Arbeitsatmosphäre und die geduldige Beantwortung alle Fragen, sowie für die
schöne Zeit, die ich mit Marlow verbringen durfte.
Ich danke allen Mitarbeitern des Instituts für die nette Arbeitsatmosphäre und stetige
Hilfsbereitschaft. Insbesondere möchte ich Nicole Ernst, Julia Hinz, Philip Demmer
und Stefan Obili für ihre Hilfe bei allen Versuchen danken. Martina Gramer und Maria
Hausknecht möchte ich für die Durchführung der Analytik danken. Michael Weißing
möchte ich für seine Hilfe bei der Histologie danken. Bianca Backofen-Wehrhahn
möchte ich für ihre Hilfe bei der Formatierung dieser Arbeit danken.
Allen Doktoranden, Diplomanden, Auszubildenden und Praktikanten danke ich für die
Hilfe bei den Versuchen und für die schöne Zeit, die wir zusammen verbracht haben.
Meiner Oma Zuzanna Gąsior möchte ich für Ihre Liebe und Unterstützung danken.
Ich danke meinem Mann Grzegorz Żuk für seine Liebe und Geduld.
Ich danke dem Redakteur Jerzy Urban für seine Feuilletons in der „Täglichen
Wochenzeitung NIE“, die mich während der gesamten PhD-Arbeit begleitet haben.

154
Ich danke der Friedrich-Ebert-Stiftung für die Gewährung eines
Promotionsstipendiums, welches mir das PhD-Studium ermöglicht hat. Ferner
möchte ich der Stiftung für die Möglichkeit der Teilnahme an den interessanten
studienbegleitenden Seminaren danken.
Diese Arbeit ist ein Teilprojekt der DFG-Forschergruppe Neurodegeneration und –
regeneration bei ZSN-Erkrankungen des Hundes (Forschergruppe 1103).


Top Related