
Termodinamica: concetti di base - INFN · 1 Termodinamica: concetti di base Sistema termodinamico:...
27
1 Termodinamica: concetti di base Sistema termodinamico: porzione di universo separata da tutto il resto del mondo sistema Ambiente esterno confini del sistema Stato del sistema: definito dal valore delle variabili termodinamiche, nel caso di un gas P,V,T ( in uno stato di equilibrio) P V T 3 T 2 T 1 PV=nRT
Transcript of Termodinamica: concetti di base - INFN · 1 Termodinamica: concetti di base Sistema termodinamico:...

1
Termodinamica: concetti di base
Sistema termodinamico: porzione di universo separata da tutto il resto del mondo
sistema
Ambiente esterno confini del sistema
Stato del sistema: definito dal valore delle variabili termodinamiche, nel caso di un gas P,V,T ( in uno stato di equilibrio)
P
V
T3 T2 T1
PV=nRT

2
Trasformazione: passaggio da uno stato ad un altro per intervento dell’ambiente esterno (il sistema in uno stato di equilibrio non cambia spontaneamente)
Trasformazioni reali o irreversibili:il percorso della trasformazione ?? (solo stato di equilibrio di partenza stato di equilibrio di arrivo) e non può essere percorsa in senso inverso
A
B
P
V Trasformazioni ideali o reversibili:inesistenti in natura
• successione continua di stati di equilibrio
• compiute in un senso e in senso contrario non lasciano tracce nell’ambiente
Trasformazioni quasi statiche: avvengono lentamente da poter essere considerate come sequenza di stati di equilibrio. Perché possano essere considerate reversibili occorre ridurre il più possibile fenomeni dissipativi e che gli scambi di calore avvengano tra corpi con una differenza piccola di temperatura (limite ⇒0)
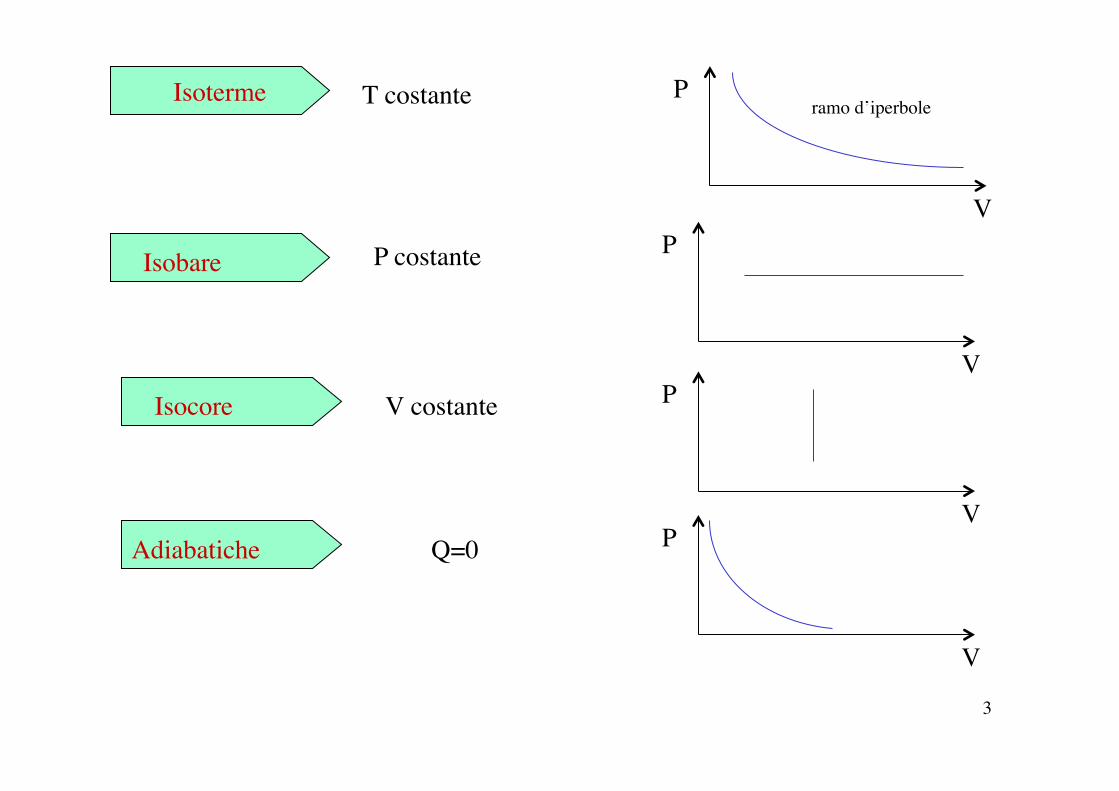
3
Isoterme T costante
V
P ramo d’iperbole
Isobare P costante
V
P
Isocore V costante
V
P
Adiabatiche Q=0
V
P

4
V
P cicliche A ad A A
Espansione libera Trasformazione irreversibile NON ha una rappresentazione grafica
Funzioni di stato Se la grandezza G è una funzione di stato, allora ad ogni stato del sistema corrisponde un ben determinato valore della grandezza G;
G=f(stato)
• La sua variazione dipende solo dallo stato iniziale e finale e non dalla trasformazione seguita (ad esempio nel caso di un gas la pressione P)
• Viceversa, se una certa quantità è indipendente dalla trasformazione, ma dipende solo dallo stato iniziale e finale, allora quella quantità può essere interpretata come la variazione di una funzione di stato
Sono funzioni di stato l’energia interna e l’entropia di un sistema

5
Scambi energetici tra sistema e ambiente esterno Lavoro e/o calore
modi di fare lavoro⇔forze che possono essere esercitate su o da un sistema
Lavoro ⇒variazione di volume
nel caso di un gas si ha lavoro in corrispondenza di un cambiamento di volume
h
F
Espansione L>0
h F
Compressione L<0
Nel caso di un lavoro positivo si ha diminuzione di energia interna del sistema, se la trasformazione è adiabatica il sistema si raffredda
L=FΔx=PAΔx=PΔV

6
P
V V1 V2
P1=P2
P
V V1 V2
P1
P2
P
V
P1
P2
Isoterma
L=nRT lnV2/V1
Isobara
L=P(V2-V1)
Isocora
L=0
Nel caso di un gas:L=area sottesa dalla trasformazione nel piano P-V
Per una trasformazione ciclica il lavoro è dato dall’area racchiusa dal ciclo

7
si ha scambio di calore tutte le volte che tra sistema ed ambiente esterno esiste una differenza di temperatura
Q>0 quando è assorbito dal sistema
Q<0 quando è ceduto dal sistema
Calore
Energia interna
Qual’è l’energia di un bicchiere di acqua? Su scala macroscopica il bicchiere d’acqua sembra non possedere alcuna energia
Il sistema possiede una data energia interna dovuta all’energia cinetica e potenziale (associata alle forze attrattive) delle molecole

8
il Io P. della T. ⇒Conservazione dell’energia totale
Relazione tra Energia Interna_Calore scambiato_Lavoro
Q-L=ΔU Il calore assorbito - il lavoro fatto dal sistema sull’ambiente
• U “energia interna” è una funzione di stato del sistema (ΔU, al contrario di Q ed L, non dipende dal tipo di trasformazione termodinamica ma solo dal valore che le variabili termodinamiche assumono allo stato iniziale e finale)
• Si misura in J e si possono misurare solo ΔU
• In un gas perfetto U dipende solo dalla temperatura
• ΔU quantità di energia scambiata tra il sistema e l’ambiente ⇒ conservazione dell’energia
Nel caso di un sistema isolato (Q=0 e L=0) l’energia interna si deve conservare U=costante
Il Primo Principio della Termodinamica

9
Dal Io P. segue:
• L e Q devono essere trattati nello stesso modo
• Si ottengono ΔU con Q o L o loro combinazioni e sono indipendenti dal modo con cui si ottengono
ΔU=f(Pi,Vi,Ti;Pf,Vf,Tf)
Calore
ΔU=Q-W Q=cmΔT
scambi
Q/t=kAΔT/d
lavo
ro
1cal=4.1868J

10
Applicazioni del primo principio • Trasformazione isoterma : in un gas perfetto U è la somma delle energie cinetiche delle molecole
Ecin=3/2KBT
U=N 3/2KBT
E quindi se T non varia anche U non varia
ΔU=0 ossia Q-L=0 Q=L
• Trasformazione adiabatica :(Q=0)
ΔU=-L
• Trasformazione isocora: (V=cost)
L=0
ΔU=Q=cVmΔt
• Trasformazione isobara: (P=cost)
ΔU=cPmΔt-PΔV
• Trasformazione ciclica:
ΔU=0 Q=L
In una trasformazione qualsiasi di un gas perfetto
ΔU=nCV(T2-T1)
Cv calore molare a volume costante

11
Il secondo principio della Termodinamica
Il I P. NON permette di prevedere il rapporto tra il lavoro fatto e il calore fornito
Il II P. limita le possibilita` di scambio di calore e del suo impiego come energia meccanica (lavoro). Permette di calcolare il rendimento di una macchina termica ideale.
Una macchina termica deve compire un ciclo e tornare al proprio stato termodinamico iniziale (ΔU=0) dopo aver scambiato calore e lavoro con l’ambiente esterno
€
η =LQ1
=Q1 − Q2
Q1
=1−Q2
Q1
ηrev =1− T2
T1
T.di Carnot
T1=cost >T2
T2=costante
L Q1
Q2

12
I due enunciati del II P.
• Kelvin-Planck: è impossibile realizzare una macchina che lavorando ciclicamente, trasformi in lavoro meccanico il calore scambiato con un’unica sorgente (non è possibile come unico risultato estrarre calore da un’oggetto e convertirlo interamente in lavoro)
• Clausius: è impossibile realizzare una macchina che lavorando ciclicamente abbia come unico risultato il passaggio di una certa quantita` di calore dalla sorgente fredda a quella calda (il calore non fluisce MAI spontaneamente da una sorgente fredda ad una piu` calda)

13
€
Teorema di Carnot
ηrev =1− T2
T1
Il T. di Carnot stabilisce che date due sorgenti di calore alle temperature T1 e T2 < T1, tutte le macchine termiche reversibili funzionanti con le due sorgenti hanno lo stesso rendimento ηrev mentre ogni altra macchina irreversibile che funzioni con le stesse sorgenti ha un rendimento η< ηrev

14
Interpretazione del II P.
-microscopica: i sistemi tendono ad evolvere da configurazioni caratterizzate da un grande ordine verso configurazioni più disordinate e più probabili
-macroscopica: una certa grandezza (funzione di stato) detta ENTROPIA (S) tende ad assumere il massimo valore possibile
€
dS =dQrev
TΔS ≥ 0
dQ è la quantita` di calore scambiata con il sistema a temperatura T Entropia totale (del sistema e dell’ambiente) non può mai diminuire ; ΔS=0 nel caso di trasformazioni reversibili e ΔS>0 nel caso di trasformazioni irreversibili
Si può dimostrare l’equivalenza con i precedenti enunciati del II P.
ΔS si misura in J/oK

15
€
ΔS = SB − SA =dQrev
T>
A
B
∫ dQTA
B
∫Dalla definizione di variazione di entropia
Segue che se la trasformazione è adiabatica ossia calore scambiato=0 ΔS=0 per una trasformazione adiabatica reversibile ΔS>0 per una trasformazione adiabatica irreversibile
Poiche ` si puo` considerare l’universo in cui viviamo un sistema isolato (non c’è nulla al di fuori con cui scambiare calore) possiamo formulare il IIo pricipio della T. dicendo che per ogni trasformazione termodinamica deve valere la relazione ΔSu≥0 dove vale l’ugualianza solo per trasformazioni reversibili Le trasformazioni che avvengono in natura sono sempre irreversibili quindi sono sempre accompagnate da un aumento di entropia dell’universo

16
NB: -Gli unici cambiamenti possibili in un sistema isolato (come l’Universo) sono quelli per cui ΔS ≥ 0 -In natura le trasformazioni sono sempre irreversibili e quindi producono sempre un aumento di entropia -L’entropia dell’universo non puo` mai diminuire, questo non implica che l’entropia di un singolo elemento dell’universo non possa diminuire, cio` puo` avvenire purche` per qualche altro componenete dell’universo abbia un aumento di entropia maggiore

17
Calcolo della variazione di Entropia • individuare una trasformazione reversibile che colleghi il punto iniziale A e finale B
• nel caso di Gas perfetti si dimostra che :
• per una trasformazione reversibile isoterma T=cost dove Qrev è tutto il calore scambiato
ad esempio nel caso di cambiamento di stato
€
ΔS = SB − SA =dQrev
TA
B
∫
€
dQ = dU + dL dL = PdV dU = ncV dT
ΔS = SB − SA = ncV ln TBTA
+ nR lnVB
VA
€
ΔS = SB − SA =Qrev
T
€
ΔS = SB − SA =λmT

18
• Nel caso di un corpo di massa m e calore specifico c inizialmente alla temperatura TA che scambia calore con l’esterno e subisce una variazione di temperatura fino a TB la variazione di entropia è data dall’espressione:
• Per calcolare la variazione di entropia dell’Universo occorre sommare la variazione di entropia dei singoli sistemi che partecipano allo scambio di calore €
ΔS = SB − SA =dQrev
TA
B
∫ =cmdTTA
B
∫ = cm lnTBTA

19
I Potenziali Termodinamici
Funzioni dette ”potenziali termodinamici" utili per descrivere l’evoluzione di reazioni chimiche e processi non ciclici sono: - l’energia interna U - L’entalpia H - L’energia libera di Helmholtz F - L’energia libera di Gibbs G
ENERGIA INTERNA U
l’energia interna U rappresenta il calore scambiato dal sistema in una reazione a Volume costante
Dal Io principio della T. segue che: ΔU=Q - PΔV se V=costante ΔU=Q

20
H = U + PV ENTALPIA
E` utile per descrivere le trasformazioni che avvengono a pressione P=costante (ad esempio reazioni chimiche) ΔH = ΔU + PΔV=ΔU+L=Q la variazione di entalpia è pari al calore scambiato (assorbito o ceduto) nella trasformazione a pressione costante.
Q ceduto Q<0 ΔH<0 l’entalpia diminuisce Q assorbito Q>0 ΔH>0 l’entalpia aumenta
H o meglio ΔH rappresenta l’energia termica che il sistema deve scambiare per compiere una determinata trasformazione a pressione costante.

21
ENERGIA LIBERA (o potenziale ) DI HELMOLTZ
Utile per descrivere le trasformazioni che avvengono a V e T costanti. La variazione:
Pone un limite superiore al lavoro che il sistema puo` compiere portandosi dallo stato A allo stato B. Nel caso il sistema non compia lavoro (V=costante) L=0 ΔF≤0 FB≤FA indica che il sistema (la reazione) evolve verso stati di energia libera F decrescente e l’equilibrio si raggiunge per il valore minimo di F
€
SB − SA ≥dQTA
B
∫ =1T
dQA
B
∫ =QT
Q ≤ T(SB − SA )
T=costante
Per il Io Principio
€
ΔU =Q− LL =Q−ΔU ≤ T(SB − SA ) − (UB −UA )
€
ΔU =Q− LL =Q−ΔU ≤ T(SB − SA ) − (UB −UA )L ≤ −ΔF

22
ENERGIA LIBERA DI GIBBS
Molto spesso reazioni chimiche avvengono a temperatura e pressione costante cosi` pure trasformazioni che implicano una transizione di fase. L’energia libera di Gibbs risulta molto utile per descrivere queste trasformazioni chimiche e fisiche e gli stati di equilibrio dei sistemi ad esse soggetti

23
ENERGIA LIBERA DI GIBBS G = U + PV-TS
È utile per descrivere le trasformazioni che avvengono a T e P costante, condizione in cui avvengono tutte le reazioni chimiche in una cellula. A P e T costante la variazione ΔG:
In termini di calore scambiato ed usando la relazione tra entropia e calore Qrev
Applicando il IIo P. della Termodinamica
Risulta il segno di ugualianza vale solo se la trasformazione è reversibile. Nel caso di reazioni chimiche irreversibili il processo avverra` nella direzione che riduce G fino a raggiungere, all’equilibrio, il valore minimo.
€
ΔG = ΔU + PΔV −TΔS = ΔH −TΔS
€
ΔG =Q−Qrev
€
Qrev
T≥QT
€
ΔG ≤ 0

24
Per un sistema termodinamico a contatto termico con un ambiente a temperatura T che si trasformi a pressione costante G non puo` aumentare e gli stati in cui G è minima rappresentano stati di equilibrio. La spontaneita` della reazione è legata al segno relativo di ΔH e ΔS ed al valore di di T. (vedi tabella i casi in cui ΔG<0)
ΔH ΔS ΔH<0 ΔS>0 Reazione spontanea esotermica ΔH<0 ΔS<0 Reazione spontanea esotermica a T basse ΔH>0 ΔS>0 Reazione spontanea endotermica a T alte ΔH>0 ΔS<0 Reazione non spontanea
€
L ≤ TΔS −ΔUΔG = ΔU + PΔV −TΔS = ΔH −TΔS ≤ 0

25

26
Alcune formule per il Gas Perfetto
€
Equazione di stato per il gas perfetto PV = nRT
R = 8.31 Jmole ⋅K
; n = numero di moli
relazione di Mayer per il calore specificocp - cv = R
cp calore specifico a pressione costante (cp =52R per gas monoatomici, cp =
72R per gas biatomici)
cv calore specifico a volume costante (cv =32R per gas monoatomici, cv =
52R per gas biatomici)
γ =cp cv
(γ =53
per gas monoatomici, γ =75
per gas biatomici)
differenza di energia interna tra due stati A e BΔU = ncv (TB - TA )differenza di entropia tra due stati A e B
ΔS = ncvlnTBTA
+nRln VB
VA

27
Alcune trasformazioni del Gas Perfetto
€
Isocora V = costante L = 0 Q = ΔU = ncv (TB - TA )PATA
=PBTB
Isobara P = costante L = P(VB -VA ) Q = ncp (TB - TA )VA
TA=VB
TB
Isoterma T = costante L = nRTlnVB
VA
Q = L
PAVA = PBVB
Adiabatica L = -ΔU = −ncv (TB - TA ) Q = 0PAVA
γ = PBVB
γ
TAVA
γ −1 = TBVB
γ −1


















