
Genetica acondroplasia
20
ACONDROPLASIA NICOLE BARRERA Y AMBAR OÑATE
-
Upload
ambar-macias -
Category
Documents
-
view
184 -
download
0
Transcript of Genetica acondroplasia

ACONDROPLASIA
NICOLE BARRERA Y AMBAR OÑATE

ESQUEMA GENERAL
La acondroplasia es un trastorno genético del crecimiento óseo que es evidente desde el nacimiento. Se presenta en aproximadamente uno de cada 15,000 a uno de cada 40,000 bebés y ocurre en todas las razas y en ambos sexos.
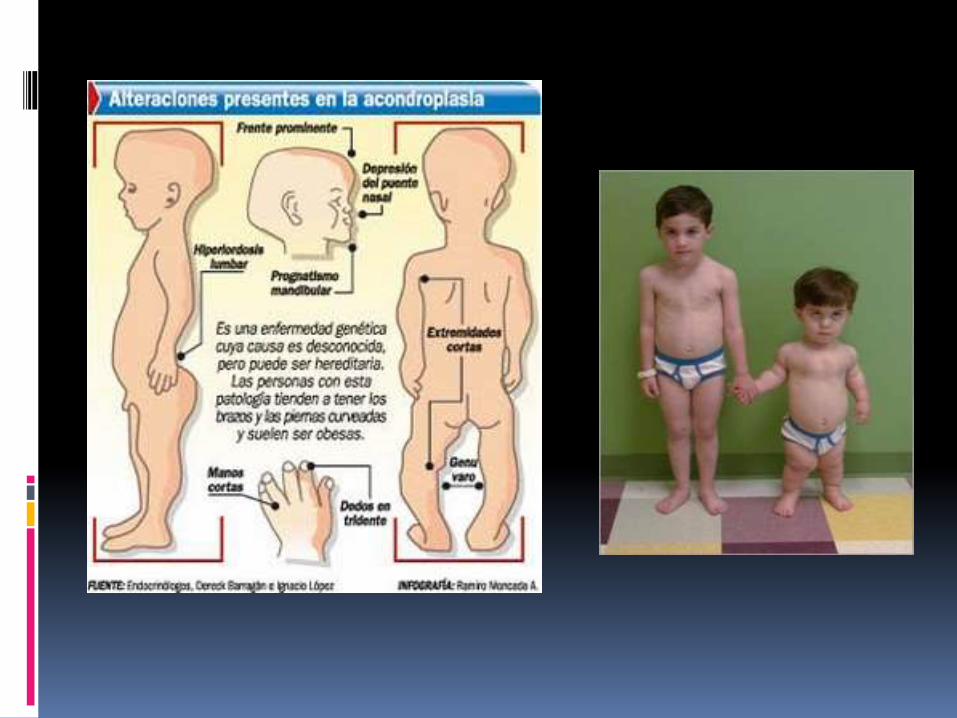
La acondroplasia es el más común de un grupo de defectos de crecimiento que se caracterizan por anormalidad en las proporciones del cuerpo. Los individuos afectados tienen brazos y piernas muy cortos, mientras que el torso tiene un tamaño casi normal.
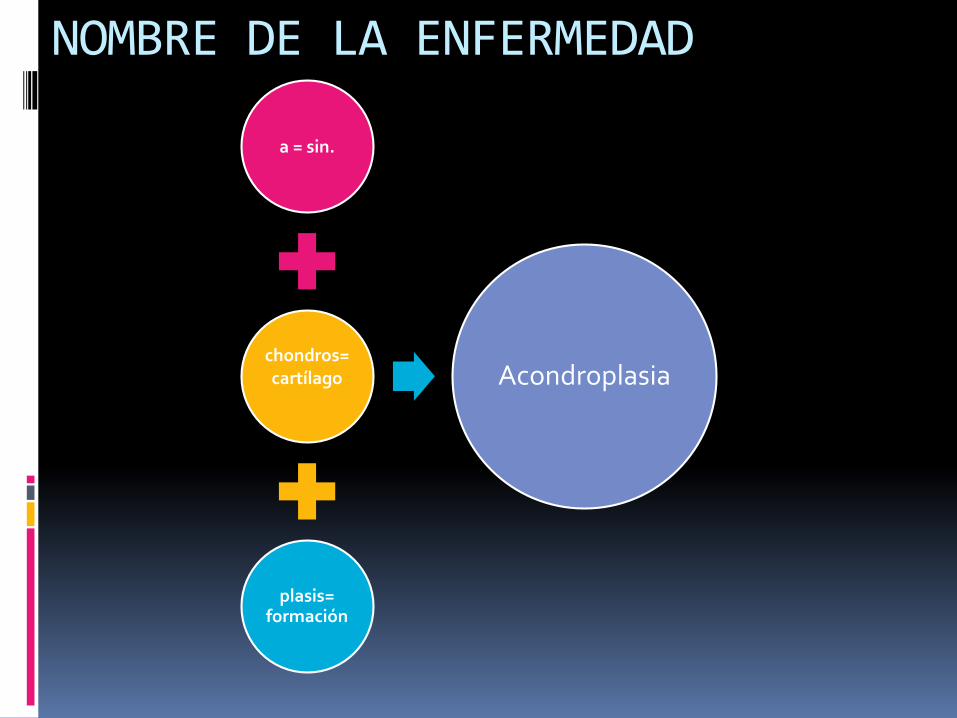
NOMBRE DE LA ENFERMEDAD
a = sin.
chondros= cartílago
plasis= formación
Acondroplasia

SINONIMIAS
Condrodistrofia Fetal (Kauffmann)
Enfermedad de Parrot
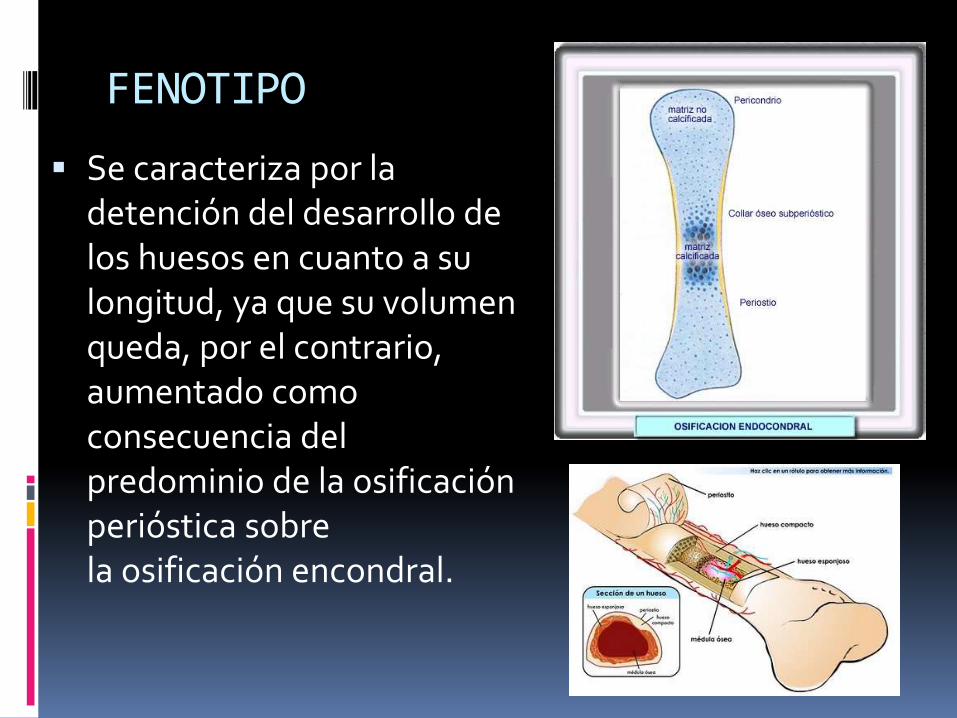
FENOTIPO
Se caracteriza por la detención del desarrollo de los huesos en cuanto a su longitud, ya que su volumen queda, por el contrario, aumentado como consecuencia del predominio de la osificación perióstica sobre la osificación encondral.

MANIFESTACIONES CLINICAS
Clínicamente se manifiesta por un enanismo que afecta únicamente a los miembros (especialmente a los segmentos proximales: micromeliarizomélica de P. Marie); la cabeza es voluminosa, mientras que el tronco es poco más o menos normal

INCIDENCIA
La incidencia de este trastorno varía entre uno de cada 25.000 a uno de cada 40.000 bebés nacidos.
La Acondroplasia no tiene preferencia por ningún sexo. Es decir se da por igual en hombres que en mujeres.

FRECUENCIA EN POBLACIONES
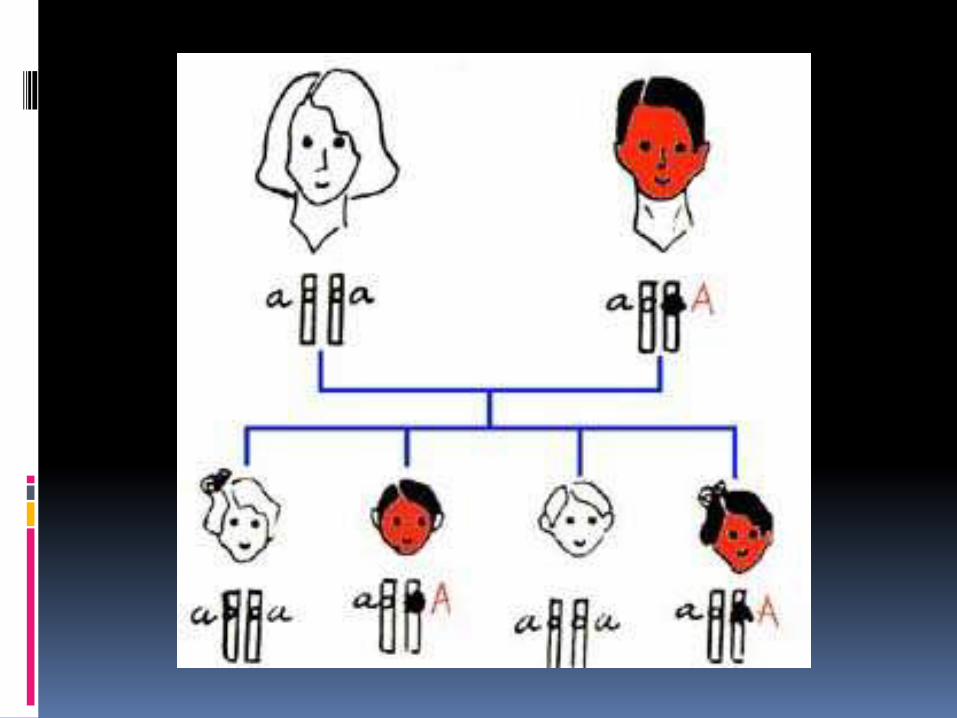
La acondroplasia es un desorden autosomaldominante, pero entre el 75 y 90% de los casos son debidos a nuevas mutaciones de un gen dominante. La tasa de mutación es relativamente elevada y se estima estar entre 1.72 y 5.57x10-5 por gameto y generación.
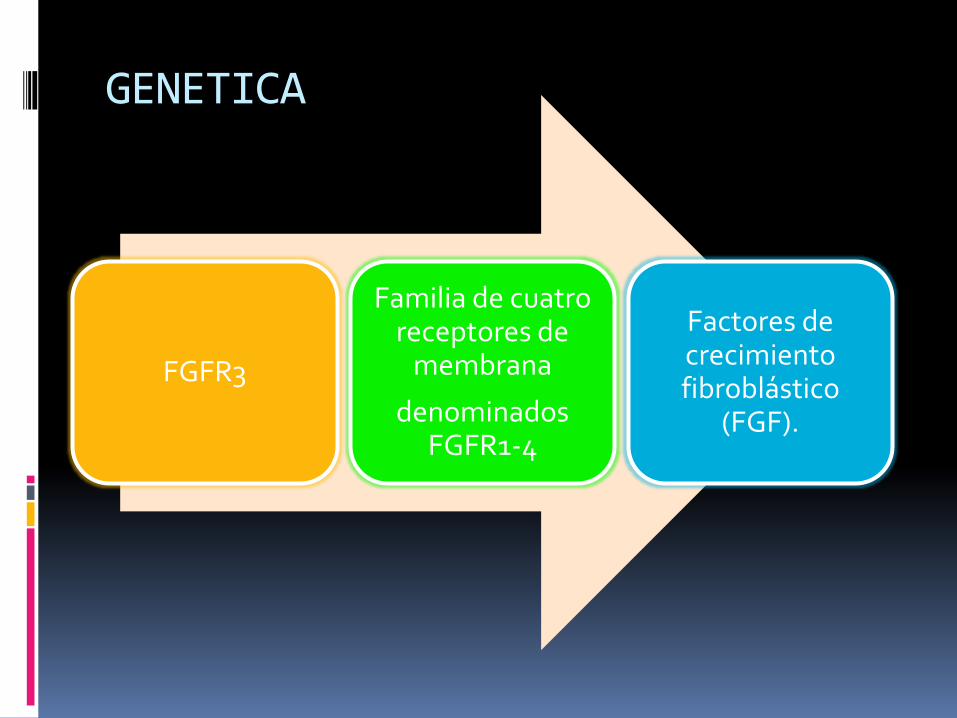
GENETICA
FGFR3
Familia de cuatro receptores de
membrana
denominados FGFR1-4
Factores de crecimiento fibroblástico
(FGF).
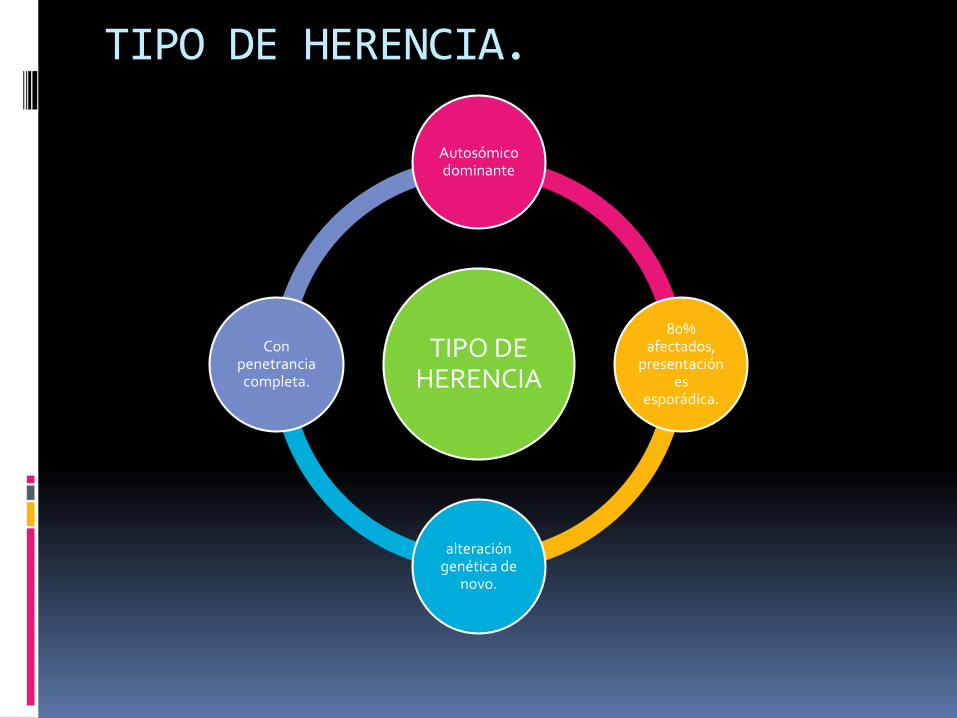
TIPO DE HERENCIA.
TIPO DE HERENCIA
Autosómicodominante
80% afectados,
presentación es
esporádica.
alteración genética de
novo.
Con penetranciacompleta.
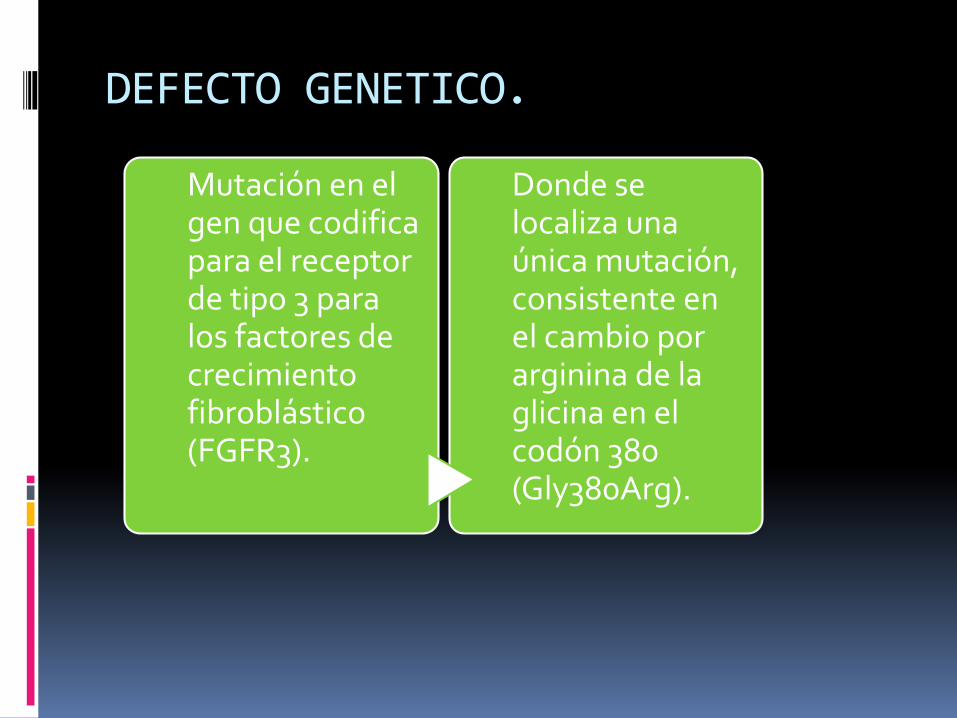
DEFECTO GENETICO.
Mutación en el gen que codifica para el receptor de tipo 3 para los factores de crecimiento fibroblástico(FGFR3).
Donde se localiza una única mutación, consistente en el cambio por arginina de la glicina en el codón 380 (Gly380Arg).
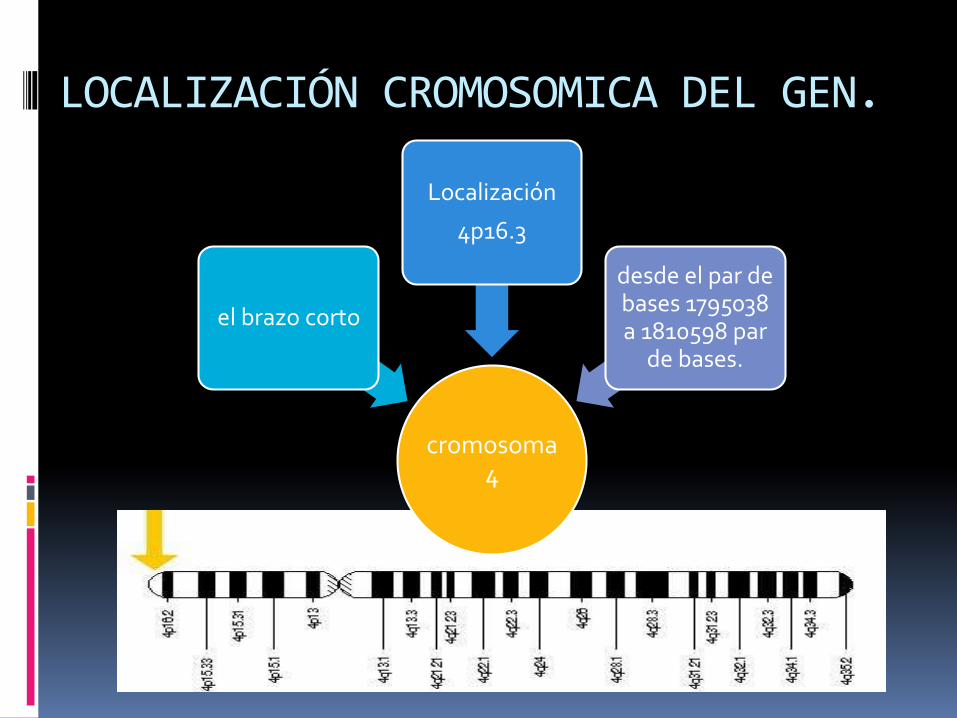
LOCALIZACIÓN CROMOSOMICA DEL GEN.
cromosoma 4
el brazo corto
Localización
4p16.3
desde el par de bases 1795038 a 1810598 par
de bases.
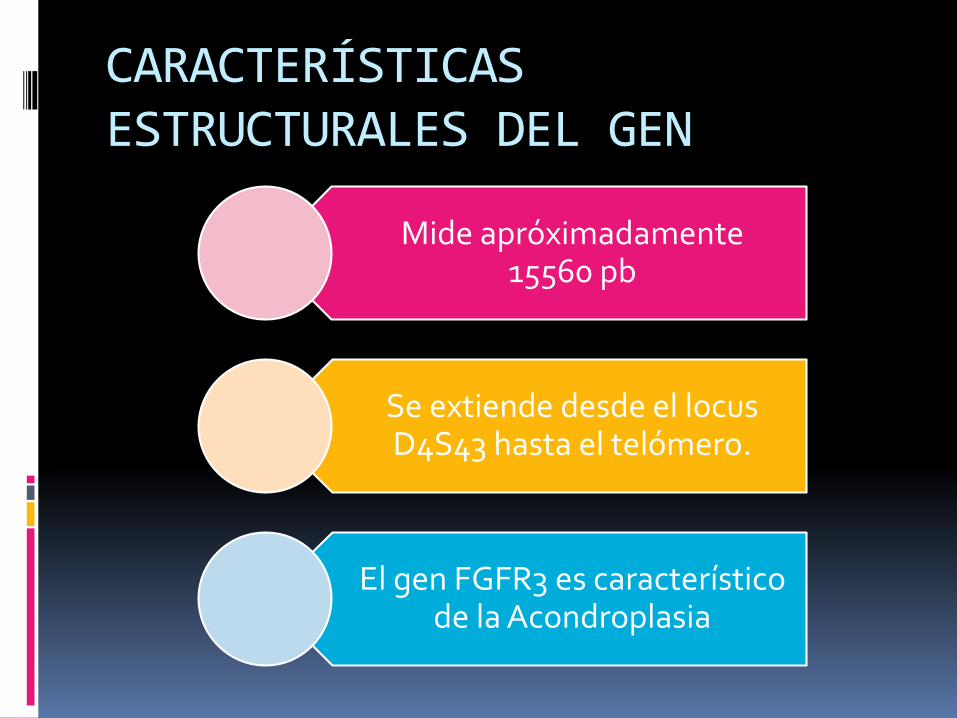
CARACTERÍSTICAS ESTRUCTURALES DEL GEN
Mide apróximadamente15560 pb
Se extiende desde el locus D4S43 hasta el telómero.
El gen FGFR3 es característico de la Acondroplasia

• Dos mutaciones posibles en el exón 10 que afectan a este gen: G1138A y G1138C. Ambas son puntuales, donde dos pares de bases complementarias del ADN se intercambian:
• Mutación G1138A: en el nucleótido número 1138, la guanina es sustituida por adenina. En el 98% de los casos de Acondroplasia, se sufre esta mutación.
• Mutación G1138C: tiene lugar el cambio de guanina por citosina, también en el nucleótido 1138. La frecuencia de esta alteración es mucho menor, apenas en el 2% de los casos.
• En ambos casos se da el cambio del aminoácido arginina por una glicina, dicha mutación puede darse de dos formas distintas: por herencia autosómica dominante, cuando hay antecedentes familiares de enfermedad (alrededor del 10% de los casos) y por una mutación de novo, con padres sanos (es la causa más frecuente, hasta en el 90% de los pacientes).
TIPOS DE MUTACIONES

Acondroplásicos tienen alto grado de homogeneidad
genética, pues se encuentra una de esas dos mutaciones en la
mayoría de los casos.
Sin embargo, Superti-Furga y colaboradores, en 1995,
describieron el caso de un recién nacido acondroplásico
que no tenía ninguna de las dos mutaciones del nucleótido 1138.
Pero que presentaba una sustitución de glicina por
cisteína en el codón 375 del gen del FGFR3.
Lo que indica heterogeneidad alélica para este gen y, además, reafirma el papel etiológico del FGFR3 en la patogénesis de la
Acondroplasia.
HETEROGENEIDAD ALELICA, NO ALELICA O AMBAS
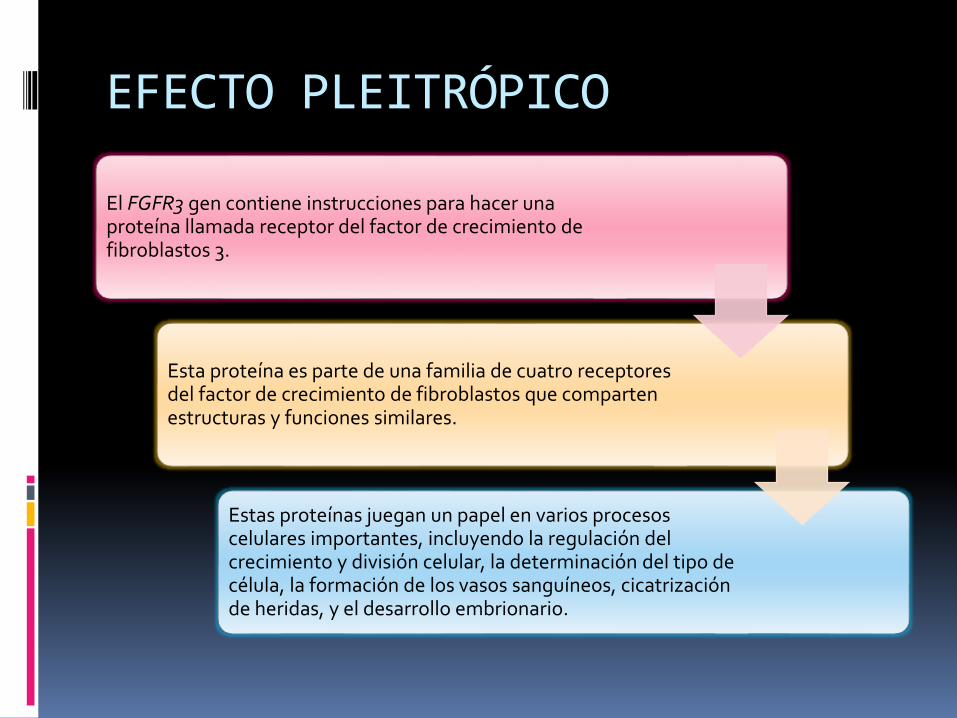
El FGFR3 gen contiene instrucciones para hacer una proteína llamada receptor del factor de crecimiento de fibroblastos 3.
Esta proteína es parte de una familia de cuatro receptores del factor de crecimiento de fibroblastos que comparten estructuras y funciones similares.
Estas proteínas juegan un papel en varios procesos celulares importantes, incluyendo la regulación del crecimiento y división celular, la determinación del tipo de célula, la formación de los vasos sanguíneos, cicatrización de heridas, y el desarrollo embrionario.
EFECTO PLEITRÓPICO
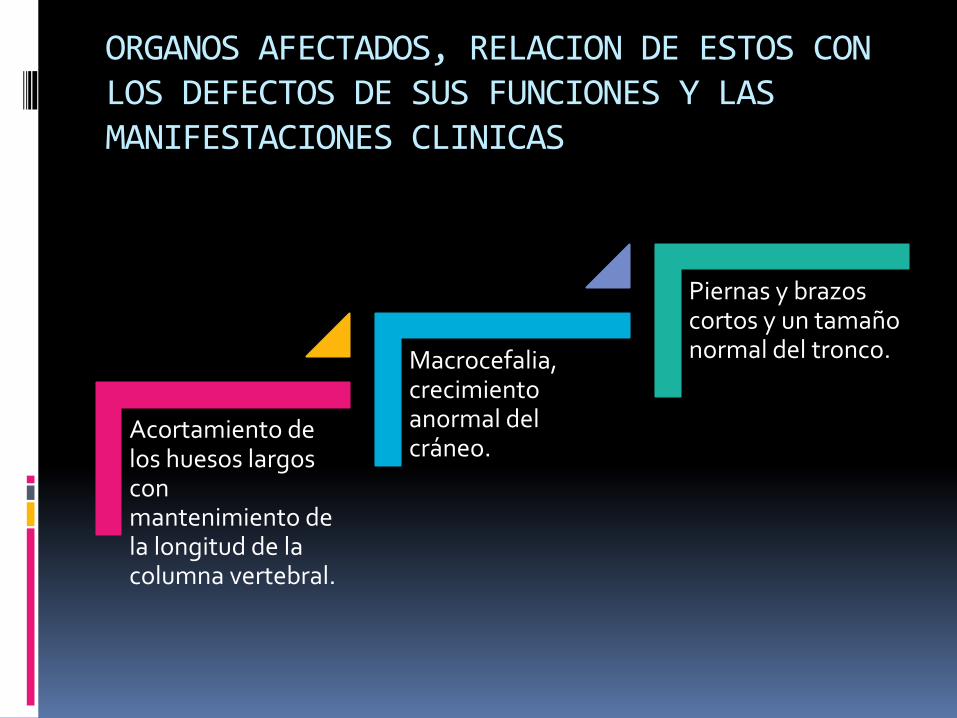
Acortamiento de los huesos largos con mantenimiento de la longitud de la columna vertebral.
Macrocefalia, crecimiento anormal del cráneo.
Piernas y brazos cortos y un tamaño normal del tronco.
ORGANOS AFECTADOS, RELACION DE ESTOS CON LOS DEFECTOS DE SUS FUNCIONES Y LAS MANIFESTACIONES CLINICAS

Bibliografía Shiang R, Thompson LM, Zhu YZ, Church DM,Fielder TJ, Bocian M et al. Mutations in the transmembrane domain in FGFR3
cause the most common genetic form of dwarfism, achondroplasia. Cell 1994; 78: 335-342.
Rousseau F, Bonaventure J, Legeal-Mallet L, Pelet A, Rozet JM, Maroteaux P et al. Mutations in the gene encoding fibroblastgrowth factor receptor-3 in achondroplasia. Nature (Lond) 1994;371: 252-254.
Entrada 100800. En: McKusick VA, editor. Online Mendelian inheritance in man. Johns Hopkins University, 1996. [online] http://www.acondroplasiauruguay.org/documentos/biblioteca/Acondroplasiaestudiomolecularde28pacientesenMadrid.pdf
Murdoch JL, Walker BA, Hall JG, Abbey H, Smith KK, Mckusick VA. Achondroplasia: a genetic and statistical survey. Ann Hum Genet 1970;33:227-4.
Gadner RJM. A new estimate of the achondroplasia mutation rate. Clin Genet 1977;11:31-8.
Oberklaid F, Danks DM, Jensen F, Stace L, Rosshandler S. Achondroplasia and hypoachondroplasia: comments on frequency, mutation rate and radiological features in skull and spine. J Med Genet 1979;16:140-6.
Stoll C, Dott B, Roth MP, Alembik Y. Birth prevalence rates of skeletal dysplasias. Clin Genet 1989;35:88-92.
Gorlin RJ, Cotten M, Levin LS. Syndromes of the Head and Neck. 3rd ed. New York: Oxford University Press; 1990.p.171-9.
Mckusick VA. Mendelian inheritance in man. Catalogues of autosomal dominant, autosomal recessive and X-linkedphenotypes. The Johns Hopkins University Press. OMIN; 1996.
Nicoletti B, Kopits SE, Ascani E, Mckusick VA, editors. Human achondroplasia. A multidisciplinary approach. Proceedings of the First Symposium on Human Achondroplasia; 1986 Nov 19-21; Rome, Italy.New York: Plenum Press; 1988.
Francomano CA, Ortiz de Luna RI, Hefferon TW, Bellus GA, Turner CE, Taylor E, et al. Localization of the achondroplasiagene to distal 2,5 Mb of human chromosome 4p. Hum Mol Genet 1994;3:787-92.
Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, et al. Mutations in transmembrane domain of R3FCF cause the most common genetic form of dwarfism, achondroplasia. Cell 1994;78:335-42.
Velinov M, Slaugenhaupt SA, Stoilov I, Scott CI, Gusella JF, Tsipouras P. The gene for achondroplasia maps to the telomericregion of chromosome 4p. Nat Genet 1994;6:312-7.
Rousseau F, Bonaventure J, Legeall-Mallet L, Pelet A, Rozet JM, Maroteaux P, et al. Mutations in the gene encodingfibroblast growth factor receptor in achondroplasia. Nature 1994;371: 252-4.


















