
童途有你 幼兒家長講座系列三 - Education Bureau · •對成長最有利的管教方式 ... •每個人的情緒是獨特的,不同的人對同一件事 可以有不同的情緒反應

快速定位基因的方法:
Mapping by sequencing
By 陈秭如

做MBS需要的知识储备
Mapping by sequencing (MBS)← 生物信息学 + 遗传学
生物信息学:
1. 熟悉Linux/unix操作
2. 会至少1门编程语言:R / python / perl
遗传学:
• 三大遗传学定律
• 图位克隆

学习生信:推荐的课程和读物
生物信息入门:
• István Albert的一个文字版在线课程:
http://www.personal.psu.edu/iua1/courses/code-repository-2014.html
• 三本书:

学习生信:推荐几个微信公众号
生信媛 生信技能树
生信分析
宏基因组
宏基因组分析
Biobabble
R语言

从一篇cell封面文章说起
Sebastian Soyk . et al. Bypassing Negative Epistasis on Yield in Tomato Imposed by a Domestication Gene. Cell 169, 1142–1155 (2017).
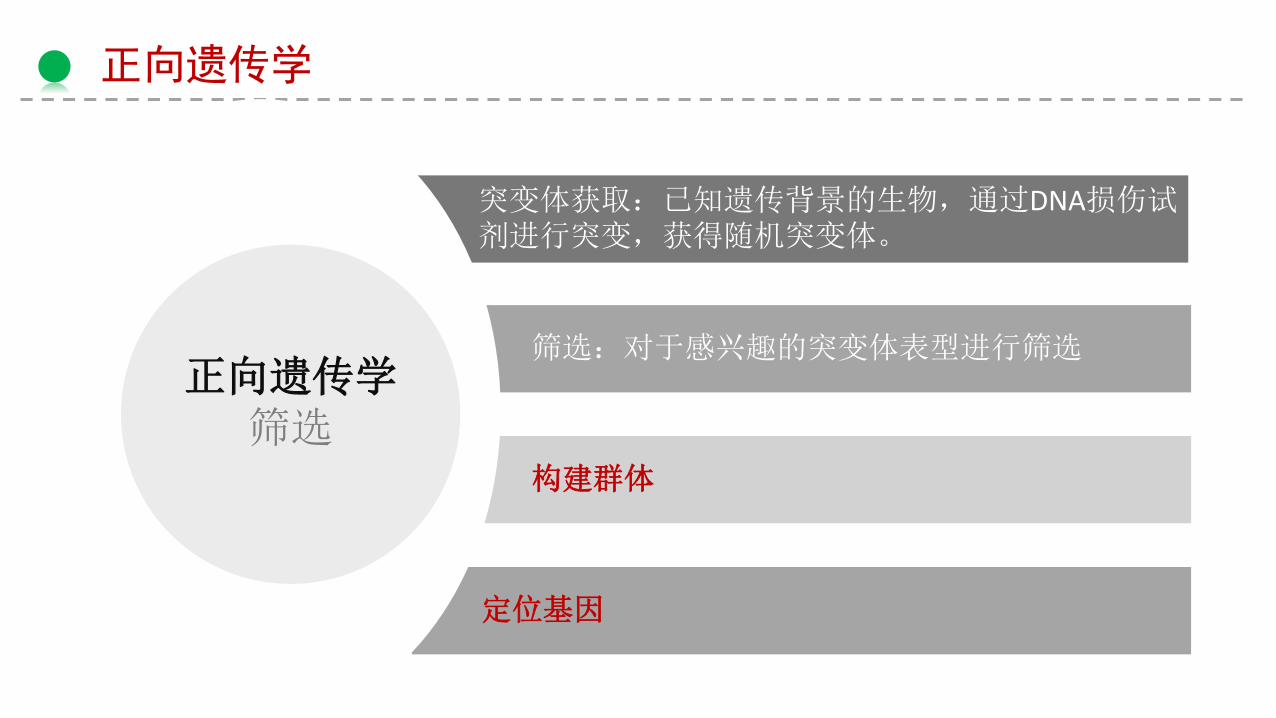
突变体获取:已知遗传背景的生物,通过DNA损伤试剂进行突变,获得随机突变体。
筛选:对于感兴趣的突变体表型进行筛选
构建群体
定位基因
正向遗传学
正向遗传学筛选

群体的类型
• 暂时性分离群体:F2、 F3、 BC(backcross 回交)等
• 永久性分离群体:RIL (Recombinant Inbred Lines 重组自交系)、DH( doublehaploid 双单倍体)等
[1] Cheng Zou. et al. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnology Journal 14, 1941–1955(2016).

基因定位原理·染色体交换
1:……ATC TAGGG CCT……2:……ATC - - - - - CCT……
1 2

基因定位的方式·检测每个个体的marker
• 对群体的每个个体进行marker检测:定位QTL常用
• 对极端个体进行混池[1,2]:BSA(Bulk segregant analysis):常用来定位质量性状
[1] Michelmore, R.W., Paran, I. and Kesseli, R.V. (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl Acad. Sci. USA, 88, 9828–9832.[2] Giovannoni, J.J., Wing, R.A., Ganal, M.W. and Tanksley, S.D. (1991) Isolation of molecular markers from specific chromosomal intervals using DNA pools from existing mapping populations. Nucleic Acids Res. 19, 6553–6568.
亲本1
亲本2×
……
果实大
果实小
亲本1-M4:……ATC TAGGG CCT……亲本2-M4:……ATC - - - - CCT……
以M4为例:
亲本1 亲本2 1 2 3 4 5
凝胶电泳图(以InDel marker为例)
个体
作图软件:R/qtlMapMaker……
不同的单株
LOD
5
4
3
2
1
0

基因定位·BSA
Sandal N. et al. (2005) Mapping and map-based cloning. In: Márquez A.J. (eds) Lotus japonicus Handbook. Springer, Dordrecht
亲本1 亲本2 Pool1(大) pool(小)
BSA-凝胶电泳图(以InDel marker为例)
亲本1-M4:……ATC TAGGG CCT……亲本2-M4:……ATC - - - - CCT……
以M4为例:
不同的单株
亲本1
亲本2×
……
分离群体
果实大
果实小
传统的基因定位的缺点:• 需要设计marker• 需要借助凝胶电泳等方式来“看”marker• 多步骤,时间长(几个月-几年)
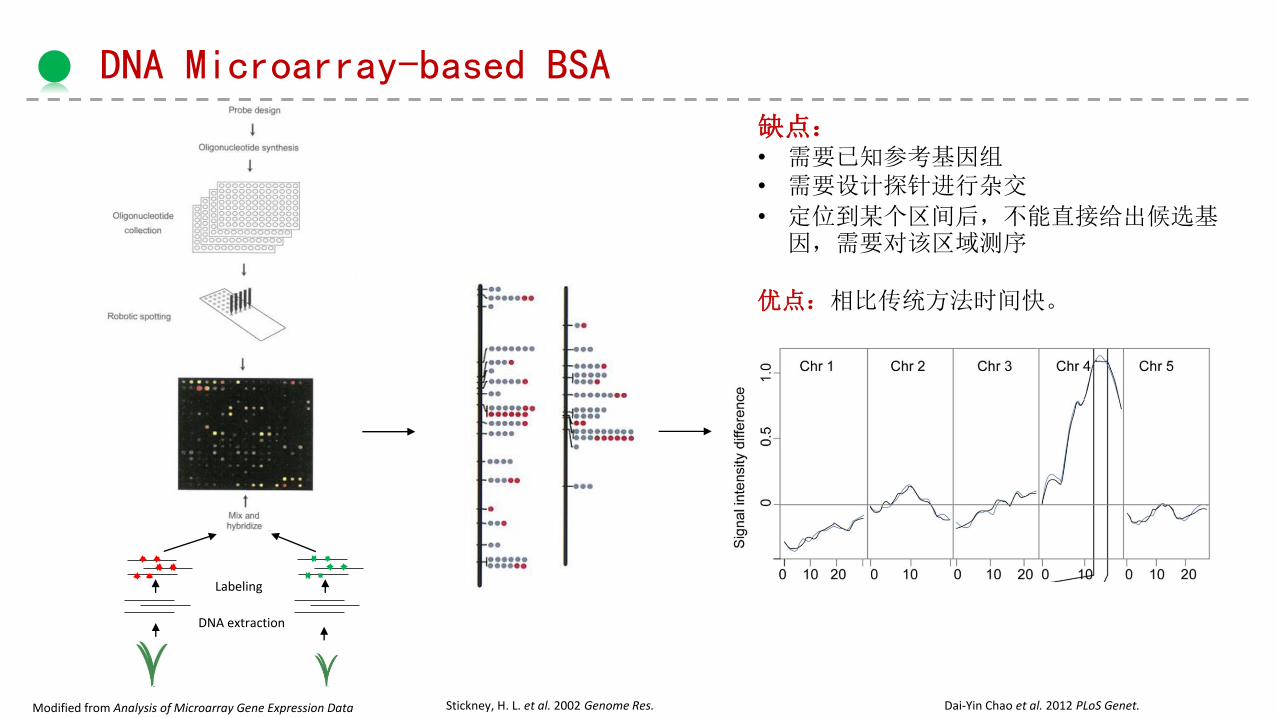
DNA Microarray-based BSA
Modified from Analysis of Microarray Gene Expression Data Stickney, H. L. et al. 2002 Genome Res. Dai-Yin Chao et al. 2012 PLoS Genet.
DNA extraction
Labeling
缺点:• 需要已知参考基因组• 需要设计探针进行杂交
• 定位到某个区间后,不能直接给出候选基因,需要对该区域测序
优点:相比传统方法时间快。

Mapping by Sequencing
Genome sequencing reveals agronomically important loci in rice using MutMap
MBS = BSA + 二代测序
优势:• 分子标记更密集
• 分辨率高:如果混池足够大,可以直接将目的基因作图到比较近的分子标记之间
• 测序reads在目的区间覆盖深度足够时,可以直接找到目的基因
• 时间快速,只需要几天的时间
MBS的适用范围:一般用于突变体的基因(质量性状)定位或者主效QTL的定位。

MBS的基本原理和分析流程
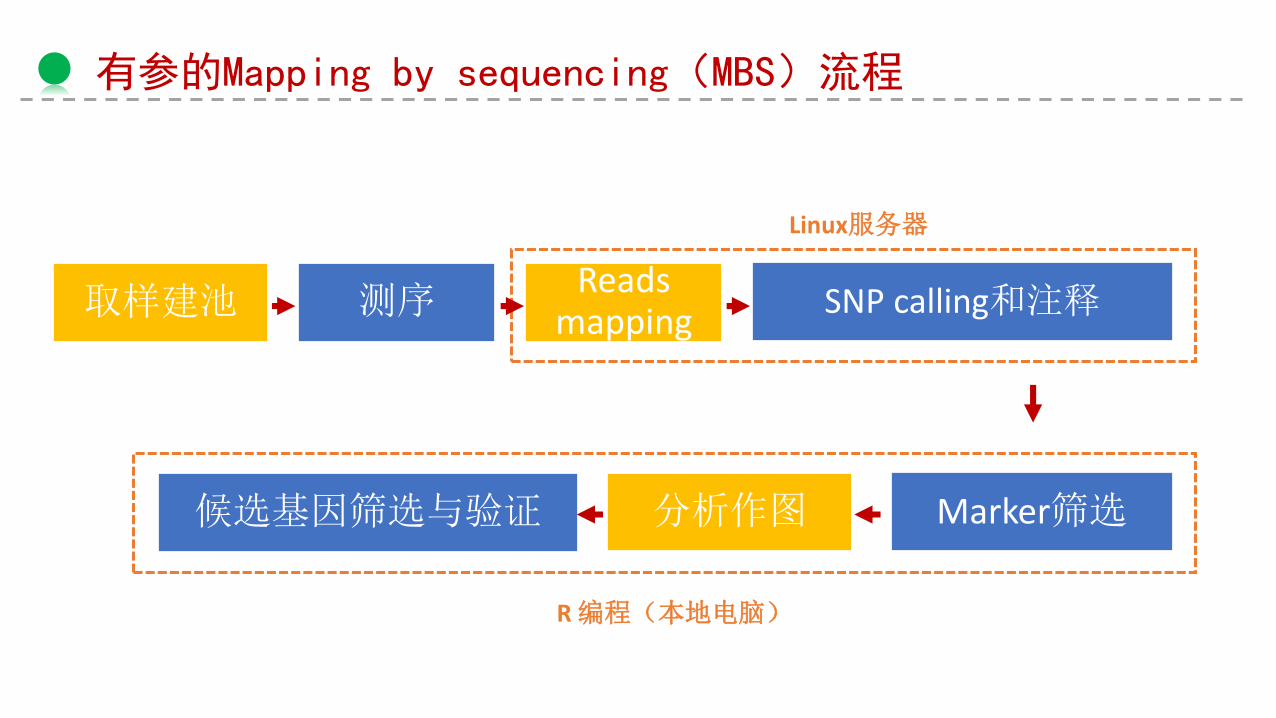
有参的Mapping by sequencing(MBS)流程
取样建池 测序Reads
mappingSNP calling和注释
Marker筛选分析作图
Linux服务器
R 编程(本地电脑)
候选基因筛选与验证

illumina 二代测序
http://www.sohu.com/a/139848890_653813
fastq文件
一个完整的reads一般包含4行:第1、3行:序列名称(为了节省存储空间有时会省略第三行“+”后面的序列名称第2行:reads的序列第4行:reads的每个碱基对应的质量
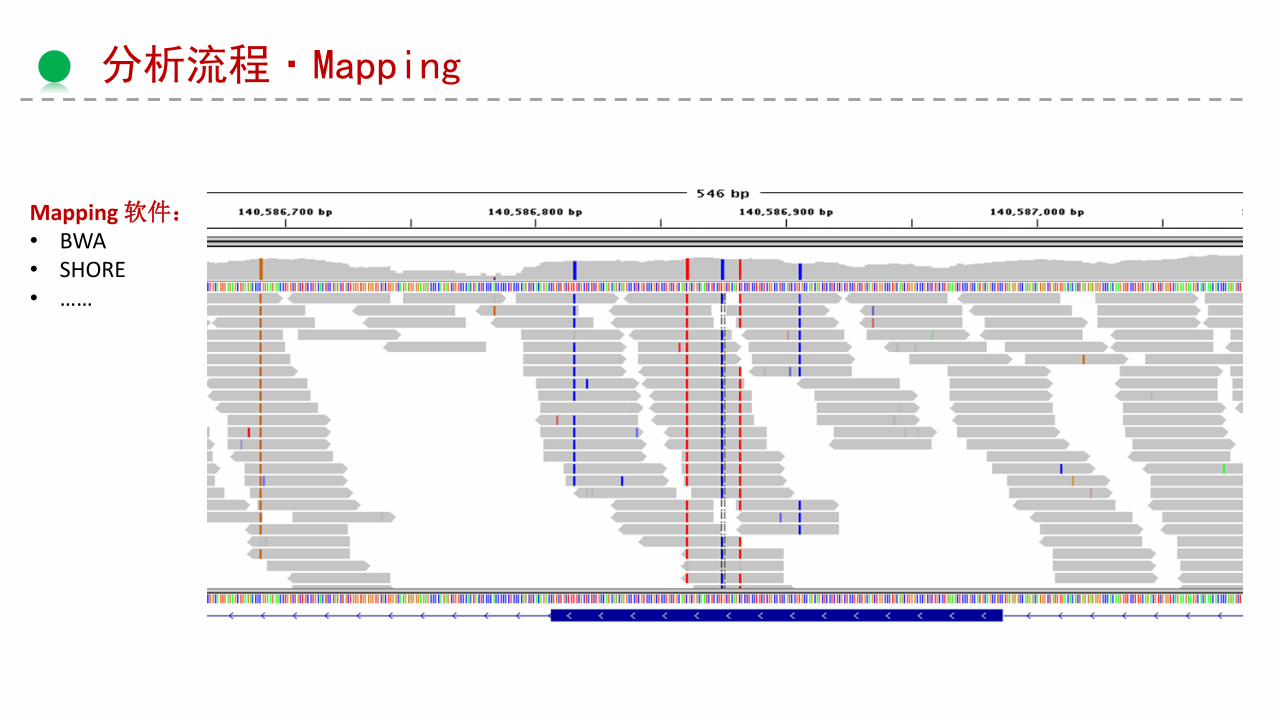
分析流程·Mapping
Mapping 软件:• BWA• SHORE• ……
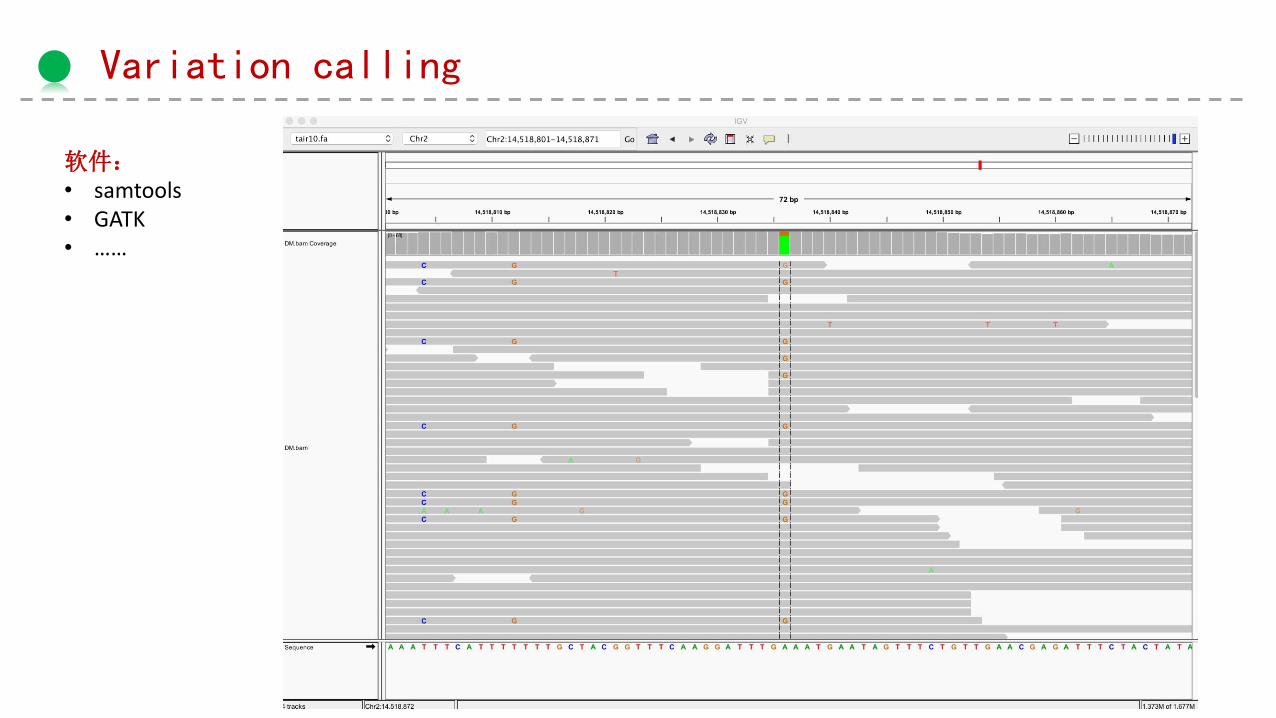
Variation calling
软件:• samtools• GATK• ……

Variation callingVCF文件
Ref Alt
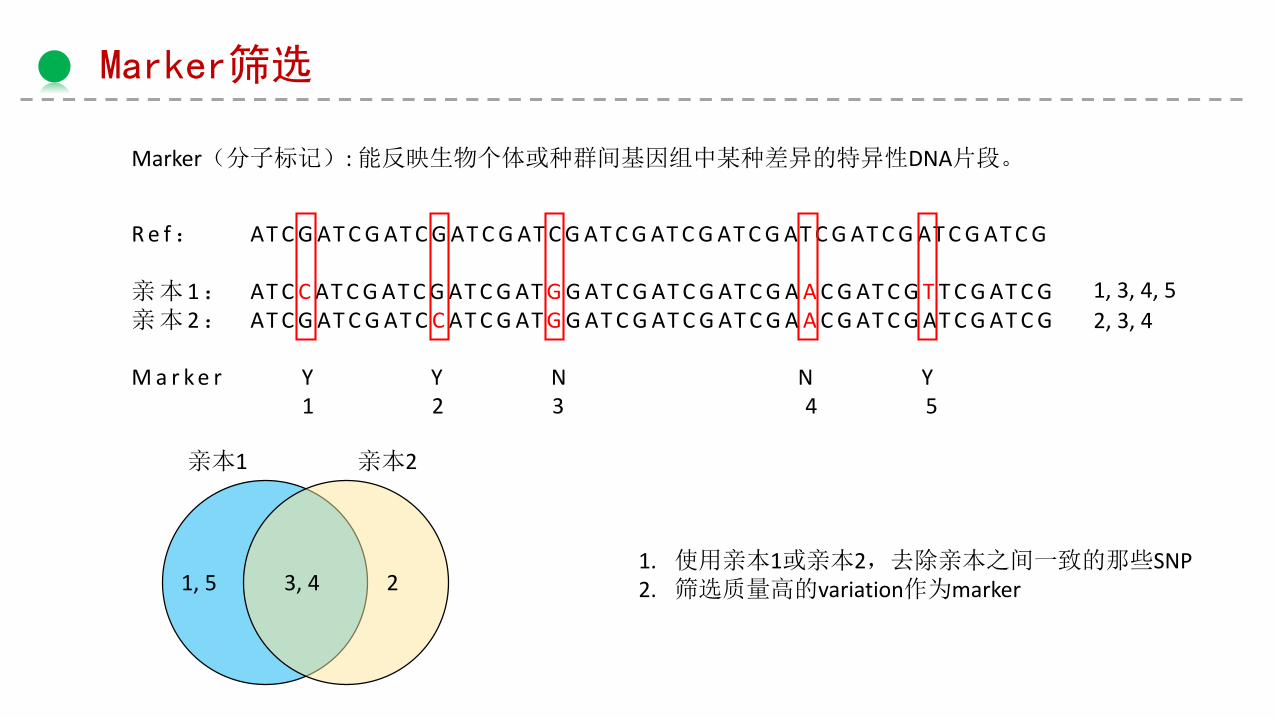
Marker筛选
R e f: AT C G AT C G AT C G AT C G AT C G AT C G AT C G AT C G AT C G AT C G AT C G AT C G
亲本 1: AT C C AT C G AT C G AT C G AT G G AT C G AT C G AT C G A A C G AT C G T T C G AT C G亲本 2: AT C G AT C G AT C C AT C G AT G G AT C G AT C G AT C G A A C G AT C G AT C G AT C G
M a r k e r Y Y N N Y1 2 3 4 5
亲本1 亲本2
1, 3, 4, 5
2, 3, 4
1, 5 3, 4 2
Marker(分子标记): 能反映生物个体或种群间基因组中某种差异的特异性DNA片段。
1. 使用亲本1或亲本2,去除亲本之间一致的那些SNP2. 筛选质量高的variation作为marker

分析作图:利用Allele Frequency(AF)来定位候选基因
Takagi H, et al. (2013) , Plant J 74(1):174-183.
隐性质量性状的突变体在causal基因的位点:突变体表型池:aa
含100% 的a,0%的A野生型表型池:1(AA)+2(Aa)=4A+2a
约含33.3%的a,66.7%的A
与causal基因不连锁的位点:随机,A和a各含50%
滑窗口求平均:相邻点的allele frequency数值是剧烈变化的
Korbinian Schneeberger.Nature Review Genetics.15, 662-76(2014)
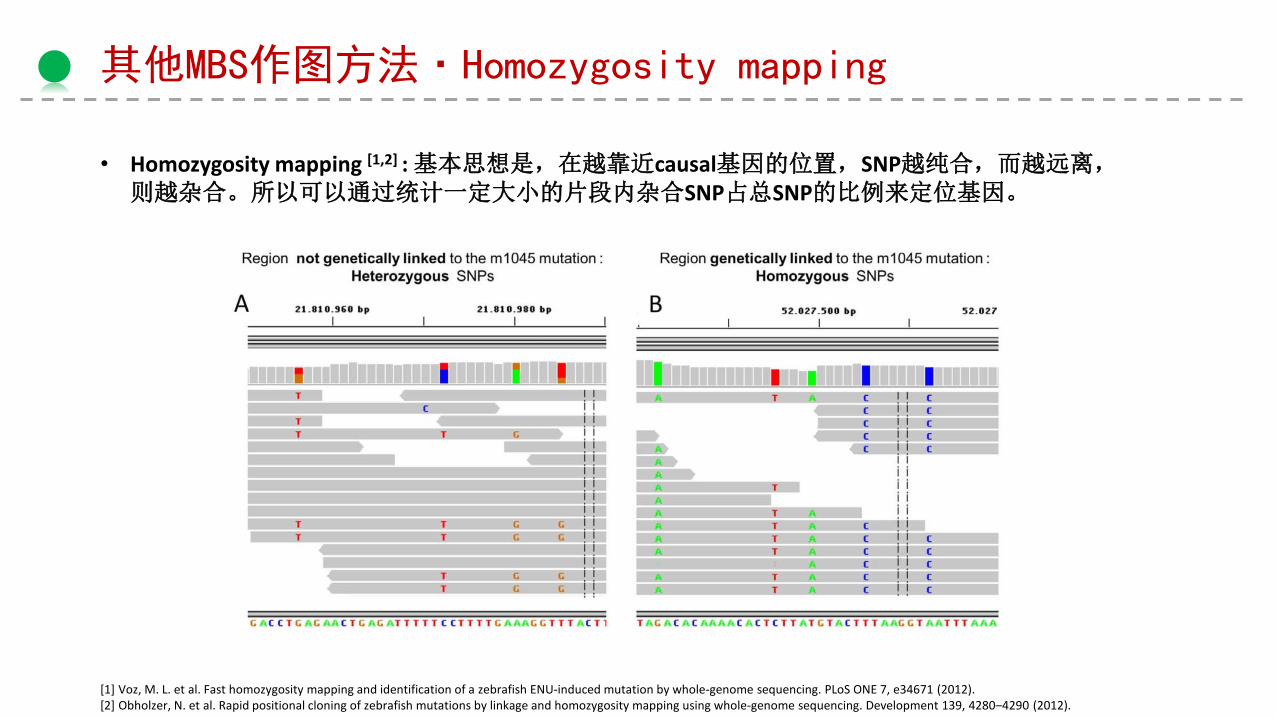
其他MBS作图方法·Homozygosity mapping
• Homozygosity mapping [1,2] : 基本思想是,在越靠近causal基因的位置,SNP越纯合,而越远离,则越杂合。所以可以通过统计一定大小的片段内杂合SNP占总SNP的比例来定位基因。
[1] Voz, M. L. et al. Fast homozygosity mapping and identification of a zebrafish ENU-induced mutation by whole-genome sequencing. PLoS ONE 7, e34671 (2012). [2] Obholzer, N. et al. Rapid positional cloning of zebrafish mutations by linkage and homozygosity mapping using whole-genome sequencing. Development 139, 4280–4290 (2012).

其他MBS作图方法· Allelic distance
• Allelic distance [1]
[1] Hill, J. T. et al. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 23, 687–697 (2013).
Each letter (A, C, G, T) corresponds to the frequency of its corresponding DNA nucleotide.
In practice, SNP loci with more than two variants are extremely rare, so two of the terms will be zero.

候选基因筛选
得到候选区间
分析候选突变软件:snpEff等
找到候选基因 基因功能验证
需要注意的问题:
• 获得的候选区间测序深度是否足够?(不足有可能导致causal的变异不能被软件识别出来)
• 区间是否存在gap( causal的变异有可能在gap里)
• 目标表型是由于基因突变导致还是染色体畸变导致?(比如染色体倒位)

无参考基因组的MBS
参考基因组的作用:
• 作为测序比对的模板• 作为物理图谱
无参考基因组的MBS策略:
• 基于近源物种的共线性区域[1]
• 基因组直接比较[2] - NIKS (needle inthe k-stack):无需分离群体、遗传图谱和参考基因组
无参考基因组的基因定位更建议用RAD-seq等方法进行基因定位。
[1] Galvao, V. C . et al. Synteny-based mapping-by-sequencing enabled by targeted enrichment. Plant J. 71, 517-526 (2012).[2] Nordstrom, K. J . et al. Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers. Nature Biotech. 31, 325–330 (2013).

MBS现有软件和应用
•1、SHOREmap : outcross / backcross; mutant or QTL;sliding-window based analysis of
allele frequencies
•2、MutMap系列: analysis of allele frequencies
· MutMap :
· MutMap+:发育早期纯合致死的突变体
· MutMap-Gap:重测序品系的Reads部分无法比对到参考序列上,结合了MutMap和de novo组装
•3、 QTL-Seq:性状存在差异的一对亲本之间杂交得到F2群体或者近等基因系(RILs)
•4、复杂基因组:全外显子组捕获测序(whole exome capture)BSA和BSR-Seq(Bulked
Segregant RNA-Seq)

Korbinian Schneeberger.Nature Review Genetics.Using next-generation sequencing to isolate mutantgenes from forward genetic screens.(2014)

常见问题
1. 选用什么样的群体?

常用的群体构建策略·outcross
Takagi, H. et al. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183 (2013).
优点:• 分子多态性更多,理论上有助于候选区域的缩小。• 受测序深度的影响小(由于marker密度大,对AF
的准确性要求没那么高)
缺点:• 如果是检测突变体,可能引入新的QTL,导致分池
可能会有污染
• 在突变体的基础上进行诱变的情况下,需要分离纯化原来的突变体,比较麻烦耗时。
• 由于背景复杂,需要的群体会比回交的大• 确定causal变异的时候难度比较大,因为混杂了另
一个亲本的变异。
建议:• 杂交群体用于QTL mapping而不用于突变体的基因
定位。
总的变异 = 亲本1的变异(比如EMS产生的SNP) + 亲本2的变异

常用的群体构建策略·backcross
Abe, A. et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nature Biotech. 30, 174–178 (2012).
优点:• 背景简单,获得候选区间后能更容易找到causal的
变异。• 需要的群体更小
缺点:• 可用于作为定位的marker少(诱变产生的变异)• 受测序深度的影响大(由于marker密度小,对AF
的准确性要求会更高)
建议:• 定位突变体的causal基因时,使用回交群体。
总的变异 = 亲本1的变异(比如EMS产生的SNP)
回交群体的marker只能使用测序的方式获得,而不同用Microarray的方式,因为这些marker来源是突变产生的。

常见问题
2. 需要多大的群体,混池多少个体?测序深度多少?
群体越大、正确混池的个体越多、测序深度越深则越有利于提高mapping的分辨率。但是考虑到
时间和经济问题,是否有性价比最高的选择?很可惜,不同的物种不同的实验设计,有不同的
突变密度和不同的重组率,并没有一个可以广泛适用的标准。
这里有一些物种的推荐个体数和测序深度:
• 拟南芥:User guide for mapping- by-sequencing in Arabidopsis.
• 斑马鱼:Perspectives for identification of mutations in the zebrafish: making use of next-
generation sequencing technologies for forward genetic approaches.
• 秀丽隐杆线虫:Deep sequencing strategies for mapping and identifying mutations from genetic
screens.

代码实践
• Rapid identification of causal mutations in tomato EMS populations via mapping-by-
sequencing. (Nature Protocols )
• 我就是Super Star——基因定位之BSA(by 李广伟)
http://mp.weixin.qq.com/s/cE-hccTyW1l6t584B_iPEw



















