
DESENVOLVIMENTO DE METODOLOGIA EXPERIMENTAL...
113
DESENVOLVIMENTO DE METODOLOGIA EXPERIMENTAL PARA DETERMINAÇÃO DO TDS DE AMOSTRAS DE SALMOURAS CARACTERÍSTICAS DO PRÉ-SAL BRASILEIRO Lucas Rego Barros Rebello Thauara Siepman Projeto de Graduação apresentado ao Curso de Engenharia de Petróleo da Escola Politécnica, Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Engenheiro de Petróleo. Orientador: Prof. Santiago Gabriel Drexler, M. Sc. Coorientador: Prof. Paulo Couto, Dr. Eng. Rio de Janeiro Setembro de 2018
Transcript of DESENVOLVIMENTO DE METODOLOGIA EXPERIMENTAL...

DESENVOLVIMENTO DE METODOLOGIA EXPERIMENTAL
PARA DETERMINAÇÃO DO TDS DE AMOSTRAS DE
SALMOURAS CARACTERÍSTICAS DO PRÉ-SAL BRASILEIRO
Lucas Rego Barros Rebello
Thauara Siepman
Projeto de Graduação apresentado ao Curso de
Engenharia de Petróleo da Escola Politécnica,
Universidade Federal do Rio de Janeiro, como
parte dos requisitos necessários à obtenção do
título de Engenheiro de Petróleo.
Orientador: Prof. Santiago Gabriel Drexler, M. Sc.
Coorientador: Prof. Paulo Couto, Dr. Eng.
Rio de Janeiro
Setembro de 2018


iii
Rego Barros Rebello, Lucas
Siepman, Thauara
Desenvolvimento de Metodologia Experimental para
Determinação do TDS de Amostras de Salmouras
Características do Pré-Sal Brasileiro/Lucas Rego Barros
Rebello, Thauara Siepman. – Rio de Janeiro: UFRJ/Escola
Politécnica, 2018.
XXI, 92p.: il.; 29,7 cm.
Orientador: Santiago Gabriel Drexler
Coorientador: Paulo Couto
Projeto de Graduação – UFRJ / Escola Politécnica /
Engenharia do Petróleo, 2018.
Referências Bibliográficas: p. 89-92.
1. TDS. 2. Condutividade Elétrica. 3. Salmoura. 4.
Caracterização de Fluidos. 5. Software OLI. 6. Pré-Sal
Brasileiro. I. Drexler, Santiago Gabriel & Couto, Paulo. II.
Universidade Federal do Rio de Janeiro, Escola
Politécnica, Curso de Engenharia de Petróleo. III. Título.

iv
Agradecimentos
Gostaríamos de agradecer primeiramente a Deus, que nos guia constantemente
não só em nossas trajetórias acadêmicas como em todos os passos de nossas vidas. Nosso
profundo deslumbramento por Sua criação serve como uma inesgotável fonte de
inspiração para tentar compreendê-la.
Eu, Lucas Rebello, agradeço aos meus pais, Alvaro e Marta, pelo amor e apoio
incondicionais a mim dedicados e pelo contínuo investimento na minha educação,
acreditando fortemente em seu valor inestimável como forma de engrandecimento
pessoal e profissional, e também como um dos mais poderosos instrumentos de
transformação da realidade. Nesse sentido, agradeço também aos meus demais familiares
por compartilharem dos mesmos princípios e, em especial, à minha tia Andréa, à minha
avó Nereida e à minha avó Maria Joanna (in memoriam). Dedico ainda um agradecimento
a Dilson Losso e Ruy Carvalho, os quais considero parte da minha família e que também
muito me apoiaram.
Eu, Lucas Rebello, agradeço à minha namorada, Amanda Vilhena, com quem
aprendo diariamente a ser uma pessoa melhor. Seu apoio, companheirismo e
aconselhamentos foram fundamentais ao longo da execução deste trabalho e durante toda
a minha graduação.
Eu, Lucas Rebello, agradeço aos vários amigos que fiz ao entrar na universidade
e com quem pude dividir as inúmeras alegrias e dificuldades inerentes à árdua caminhada
de me tornar um Engenheiro pela UFRJ. Em especial, agradeço ao meu amigo Daniel
Teixeira, pelo companheirismo imprescindível e por me provar o valor de uma grande
amizade.
Eu, Lucas Rebello, quero agradecer ainda à minha estimada amiga e também
autora deste projeto, Thauara Siepman, por aceitar o desafio de escrever este trabalho
comigo e pela amizade durante os anos de graduação.
Eu, Thauara Siepman, agradeço aos meus pais, Erico e Silvia, pelo amor
incondicional, por apoiarem a minha escolha de estudar em outro estado e por me
ensinarem a nunca desistir dos meus sonhos, por mais difíceis que eles pareçam ser.
Agradeço também à minha irmã, Rafaella, pelo amor e amizade, e por sempre me motivar

v
a ser uma pessoa melhor. Dedico ainda um agradecimento aos meus tios Marisa e
Henrique por todo o apoio, e aos meus avós maternos e paternos, que me ensinaram a ser
forte e persistente.
Eu, Thauara Siepman, agradeço ao meu namorado, Lucas Sathler, pelo
companheirismo, amor, confiança e paciência para suportar a distância e meu eventual
mau humor durante a execução deste trabalho.
Eu, Thauara Siepman, agradeço também às minhas irmãs de coração, Jéssica e
Aninha, que dividiram apartamento comigo durante os cinco anos de graduação, e aos
meus amigos, Felipe Relvas, Lucas Pestana, Henrique Lemos, Gabriel Magalhães, Thiago
Gabetto e Renata Zimbres. Vocês tornaram a experiência de morar longe de casa mais
fácil e prazerosa.
Eu, Thauara Siepman, quero agradecer ainda ao meu estimado amigo e também
autor deste projeto, Lucas Rebello, pela amizade e paciência, tornando a realização deste
trabalho divertida (e gramaticalmente correta).
Agradecemos ao nosso orientador, Prof. M. Sc. Santiago Drexler, pelo atencioso
acompanhamento e direcionamento deste projeto e também pelos valiosos conhecimentos
ministrados ao longo das disciplinas do curso de Engenharia de Petróleo. Sua didática
notável e seu vasto saber muito contribuíram para o desenvolvimento do nosso fascínio
pela indústria de petróleo.
Agradecemos ao nosso coorientador, Prof. Dr. Paulo Couto, igualmente pelos
relevantes conhecimentos transmitidos no decorrer das disciplinas do curso e também
pelo primoroso trabalho desempenhado como coordenador do curso de Engenharia de
Petróleo na Escola Politécnica da UFRJ. Acreditamos que nossos orientadores espelham
o que há de melhor em termos de excelência acadêmica dentro do corpo docente da UFRJ.
Agradecemos ao Laboratório de Recuperação Avançada de Petróleo (LRAP) da
COPPE/UFRJ por proporcionar a infraestrutura e os recursos financeiros necessários para
a realização dos experimentos descritos neste trabalho. Agradecemos também a todos os
alunos e técnicos que trabalham no LRAP pelo auxílio que nos foi prestado na execução
dos experimentos.
Agradecemos à pesquisadora Merlin Bandeira do Laboratório de Ensaios Não
Destrutivos, Corrosão e Soldagem (LNDC) da COPPE/UFRJ por nos fornecer o acesso

vi
ao programa OLI, que utilizamos para realizar as simulações computacionais presentes
neste trabalho. Somos muito gratos a ela pela gentileza e enorme presteza com que nos
recebeu e nos auxiliou a operar o programa.
Por fim, gostaríamos de agradecer à Escola Politécnica e à UFRJ por permitir
direta ou indiretamente que tudo isso fosse possível, de modo que agora carregamos
conosco com grande responsabilidade a exímia tradição e o notável prestígio desta ilustre
instituição.

vii
“Humanity’s deepest desire for knowledge is
justification enough for our continuing quest.”
Stephen Hawking

viii
Resumo do Projeto de Graduação apresentado à Escola Politécnica/UFRJ como parte
dos requisitos necessários para a obtenção do grau de Engenheiro de Petróleo.
DESENVOLVIMENTO DE METODOLOGIA EXPERIMENTAL
PARA DETERMINAÇÃO DO TDS DE AMOSTRAS DE
SALMOURAS CARACTERÍSTICAS DO PRÉ-SAL BRASILEIRO
Lucas Rego Barros Rebello
Thauara Siepman
Setembro de 2018
Orientador: Prof. Santiago Gabriel Drexler, M. Sc.
Coorientador: Prof. Paulo Couto, Dr. Eng.
Curso: Engenharia de Petróleo
O Total de Sólidos Dissolvidos, ou Total Dissolved Solids – TDS, é uma propriedade
importante na caracterização de fluidos provenientes de campos petrolíferos. A
determinação do TDS de salmouras que são produzidas juntamente com óleo e gás de
poços de petróleo permite avaliar, de forma conjunta com outras condições, a
possibilidade de ocorrência de corrosão e de formação de incrustações de sais inorgânicos
em ambientes que abrangem desde as rochas reservatório até a planta de processamento,
incluindo todos os dutos de escoamento ao longo do trajeto. Além disso, o conhecimento
do TDS também é fundamental na garantia da estabilidade química de fluidos injetados
nos poços a fim de se aumentar o volume de óleo recuperado. Nesse sentido, este projeto
tem como objetivo encontrar uma metodologia experimental para se determinar o TDS a
partir de correlações com a condutividade elétrica de amostras de salmouras
características do Pré-sal brasileiro. Para isso, também foi analisada a variação da
condutividade com a concentração e a temperatura das amostras. Por fim, para verificar
a validade dos resultados obtidos, foi utilizado um programa computacional apropriado
em que foram simulados todos os experimentos realizados e, com isso, possibilitou-se
elaborar interpretações úteis.
Palavras-chave: TDS, Condutividade elétrica, salmoura, caracterização de fluidos,
software OLI, Pré-sal brasileiro.

ix
Abstract of Undergraduate Project presented to POLI/UFRJ as a partial fulfillment of
the requirements for the degree of Petroleum Engineer.
DEVELOPMENT OF EXPERIMENTAL METHODOLOGY FOR TDS
DETERMINATION OF BRINE SAMPLES CHARACTERISTIC OF THE
BRAZILIAN PRE-SALT
Lucas Rego Barros Rebello
Thauara Siepman
September/2018
Advisor: Prof. Santiago Gabriel Drexler, M. Sc.
Co-advisor: Prof. Paulo Couto, Dr. Eng.
Course: Petroleum Engineering
Total Dissolved Solids (TDS) is an important property in the characterization of fluids
from oilfields. Determination of the TDS of brines that are produced together with oil and
gas from oil wells allows to evaluate, jointly with other conditions, the possibility of
corrosion occurrence and formation of inorganic salts scale in environments that cover
from the reservoir rock to the processing plant, including all flow ducts along the fluids
path. In addition, knowledge of TDS is also critical to ensuring the chemical stability of
injected fluids in wells in order to increase the volume of recovered oil. In this sense, this
project aims to find an experimental methodology to determine the TDS from correlations
with the electrical conductivity of brine samples characteristic of the Brazilian Pre-salt.
For this purpose, the variation of the conductivity with the concentration and the
temperature of the samples was also analyzed. Finally, to verify the validity of the results
obtained, an appropriate software was used in which all experiments were simulated and,
thus, it was possible to elaborate useful interpretations.
Keywords: TDS, Electrical conductivity, brine, fluid characterization, OLI software,
Brazilian Pre-salt.

x
SUMÁRIO
LISTA DE FIGURAS ................................................................................................ xiii
LISTA DE TABELAS ............................................................................................... xvi
LISTA DE SÍMBOLOS ............................................................................................. xix
1. INTRODUÇÃO .................................................................................................... 1
1.1. Motivação e Objetivos ................................................................................... 1
1.2. Estruturação do Trabalho ............................................................................... 3
2. PRINCIPAIS APLICAÇÕES PARA O TDS ......................................................... 5
2.1. Caracterização de Fluidos do Reservatório..................................................... 5
2.2. Incrustações de Sais Inorgânicos .................................................................... 7
2.2.1. Solubilidade de Sais Inorgânicos .............................................................. 7
2.2.1.1. Índice de Saturação ......................................................................... 10
2.2.2. Condições para Formação de Incrustações Inorgânicas .......................... 10
2.2.3. Tipos Mais Comuns de Incrustação ........................................................ 13
2.2.4. Problemas Operacionais Devido a Incrustações...................................... 13
2.2.5. Prevenção e Remediação........................................................................ 15
2.3. Corrosão por Sais Inorgânicos ..................................................................... 17
2.3.1. Mecanismos dos Processos Corrosivos e Formas de Ocorrência ............. 17
2.3.2. Corrosão pela Água de Formação .......................................................... 21
2.3.3. Métodos para Inibição da Corrosão ........................................................ 22
2.4. Estabilidade Química de Fluidos Injetados................................................... 26
2.4.1. Polímeros............................................................................................... 26
2.4.1.1. Tipos de Polímeros ......................................................................... 27
2.4.1.2. Influência da Composição da Água ................................................. 27
2.4.2. Surfactantes ........................................................................................... 29
2.4.2.1. Tipos de Surfactantes ...................................................................... 29

xi
2.4.2.2. Efeito da Salinidade ........................................................................ 30
3. FUNDAMENTAÇÃO TEÓRICA ....................................................................... 33
3.1. Condutometria Direta .................................................................................. 33
3.1.1. Condutividade Elétrica das Soluções Iônicas .......................................... 34
3.1.2. Fatores que influenciam a Condutividade Elétrica das Soluções Iônicas . 35
3.1.2.1. Efeito da Geometria da Célula de Condutividade ............................ 35
3.1.2.2. Efeito da Temperatura .................................................................... 37
3.1.2.3. Efeito da Concentração e da Natureza dos Eletrólitos ...................... 38
3.2. Gravimetria ................................................................................................. 44
3.2.1. Gravimetria por Precipitação.................................................................. 44
3.2.1.1. Agente Precipitante ......................................................................... 45
3.2.2. Gravimetria de Volatilização.................................................................. 45
3.2.3. Eletrogravimetria ................................................................................... 46
3.2.4. Metodologia para Determinação do TDS por Gravimetria ...................... 47
3.2.4.1. Problemas do Método ..................................................................... 47
3.3. Método de Correlação do TDS com a Condutividade Életrica ...................... 48
3.3.1. Características do Método ...................................................................... 48
3.3.2. Fator TDS .............................................................................................. 49
4. METODOLOGIA EXPERIMENTAL E DE SIMULAÇÕES COMPUTACIONAIS
51
4.1. Materiais Utilizados ..................................................................................... 51
4.1.1. Água Deionizada ................................................................................... 51
4.1.2. Sais Inorgânicos ..................................................................................... 51
4.2. Equipamentos .............................................................................................. 52
4.3. Descrição da Metodologia Empregada ......................................................... 55
4.3.1. Procedimento Experimental ................................................................... 55
4.3.1.1. Soluções de Cloreto de Sódio.......................................................... 55

xii
4.3.1.2. Salmoura Característica do Pré-Sal Brasileiro ................................. 57
4.3.2. Programa OLI ........................................................................................ 59
4.3.3. Testes para Verificação da Correlação Experimental .............................. 60
5. RESULTADOS E DISCUSSÕES ....................................................................... 62
5.1. Sistema Simplificado: Solução de NaCl ....................................................... 62
5.2. Salmoura Característica do Pré-Sal Brasileiro .............................................. 69
5.3. Simulações Computacionais no Programa OLI ............................................ 78
5.4. Análises Comparativas ................................................................................ 81
5.5. Testes para Verificação da Correlação Experimental.................................... 84
6. CONCLUSÕES .................................................................................................. 85
6.1. Propostas para Trabalhos Futuros ................................................................ 86
REFERÊNCIAS BIBLIOGRÁFICAS ......................................................................... 89

xiii
LISTA DE FIGURAS
Figura 1 - Etapas da formação da incrustação. FONTE: Adaptado de SANTANA e
MANZELA (2016). .................................................................................................... 11
Figura 2 - Duto com incrustação de BaSO4. Fonte: SANTANA e MANZELA (2016). 16
Figura 3 - Duto depois da remoção. Fonte: SANTANA e MANZELA (2016). ............ 16
Figura 4 - Exemplo de processo de corrosão com mecanismo químico. Fonte:
FRAUCHES-SANTOS et al. (2014). .......................................................................... 18
Figura 5 - Mecanismo eletroquímico da corrosão em diferentes meios. Fonte:
FRAUCHES-SANTOS et al. (2014). .......................................................................... 19
Figura 6 - Representação esquemática de algumas formas de corrosão. Fonte: FREIRE e
FLORIDO (2016). ...................................................................................................... 21
Figura 7 - Representação esquemática da proteção anódica. Fonte: FRAUCHES-
SANTOS et al. (2014). ................................................................................................ 24
Figura 8 - Representação esquemática da proteção catódica. Fonte: FRAUCHES-
SANTOS et al. (2014). ................................................................................................ 24
Figura 9 - Ação da solução polimérica na eficiência de varrido areal. Fonte: Adaptado de
RIOS (2014). .............................................................................................................. 27
Figura 10 - Efeito das forças iônicas na forma de um polieletrólito de cadeia flexível,
como HPAM: (a) Em baixa salinidade e (b) Em alta salinidade. Fonte: RIOS (2014). . 28
Figura 11 - Representação esquemática dos tipos de surfactantes. Fonte: SALAS (2014).
................................................................................................................................... 29
Figura 12 - Diagrama de fases para o sistema Windsor I. Fonte: SANABRIA (2013). . 31
Figura 13 - Diagrama de fases para o sistema Windsor II. Fonte: SANABRIA (2013). 31
Figura 14 - Diagrama de fases para o sistema Windsor III. Fonte: SANABRIA (2013).
................................................................................................................................... 32
Figura 15 - Constantes de célula de condutividade apropriadas para cada faixa de
condutividade típica das amostras. Fonte: SARTORELLI (2015). ............................... 36
Figura 16 - Perfil típico da condutividade da solução em função da sua concentração.
Fonte: MARTÍNEZ (2017). ........................................................................................ 38
Figura 17 - Perfil típico da condutividade molar da solução em função da sua
concentração. Fonte: MARTÍNEZ (2017). .................................................................. 39

xiv
Figura 18 - Gráficos da condutividade em função da concentração para um eletrólito forte
em (a) e para um eletrólito fraco em (b). Fonte: Adaptado de PILLING (s.d.). ............ 40
Figura 19 - Gráfico da condutividade molar do KCl versus a raiz quadrada da
concentração. Fonte: Adaptado de PILLING (s.d.). ..................................................... 41
Figura 20 - Gráfico do inverso da condutividade molar (1/𝛬𝑚) do ácido acético versus o
produto da condutividade molar com a concentração (𝛬𝑚𝐶). Fonte: Adaptado de
PILLING (s.d.)............................................................................................................ 43
Figura 21 - Etapas da análise gravimétrica por precipitação. Fonte: MURITO e FINETE
(2009). ........................................................................................................................ 44
Figura 22 - (a) Arranjo para eletrólise com potencial não controlado; (b) Arranjo para
eletrólise com potencial controlado. Fonte: SKOOG et al. (2005). ............................... 46
Figura 23 - Balança analítica de precisão NewClassic MS da marca METLLER
TOLEDO. ................................................................................................................... 53
Figura 24 - Agitador magnético C-MAG HS7 da marca IKA®. .................................. 53
Figura 25 - Medidor de condutividade SevenExcellence™ da marca METTLER
TOLEDO. ................................................................................................................... 54
Figura 26 - Purificador de água OS10LXE da marca GEHAKA. ................................. 54
Figura 27 - Bomba de vácuo nXDS IFT 20 da marca Edwards. ................................... 55
Figura 28 - Representação esquemática de todas as configurações de medição. ........... 56
Figura 29 - Representação esquemática de todas as configurações de medição. ........... 57
Figura 30 - Gráfico da condutividade versus a temperatura para amostras com diferentes
concentrações.............................................................................................................. 64
Figura 31 - Gráfico da condutividade versus a concentração para amostras medidas em
diferentes temperaturas. .............................................................................................. 65
Figura 32 - Gráfico dos Fatores TDS experimentais versus a concentração (ppm) para
diferentes temperaturas. .............................................................................................. 69
Figura 33 - Gráfico da condutividade versus a temperatura para amostras com diferentes
concentrações.............................................................................................................. 72
Figura 34 - Gráfico da condutividade versus a concentração para amostras medidas em
diferentes temperaturas. .............................................................................................. 73
Figura 35 - Gráfico do TDS versus a concentração para amostras medidas em diferentes
temperaturas. .............................................................................................................. 74
Figura 36 - Gráfico dos Fatores TDS experimentais versus a concentração (ppm) para
diferentes temperaturas. .............................................................................................. 77

xv
Figura 37 - Gráfico da condutividade versus a temperatura para a salmoura característica
do Pré-sal brasileiro simulada para diferentes concentrações. ...................................... 78
Figura 38 - Gráfico do TDS versus as condutividades simuladas para diferentes
temperaturas para a salmoura característica do Pré-sal brasileiro. ................................ 79

xvi
LISTA DE TABELAS
Tabela 1 - Valores de condutividade elétrica a 25 °C e fator TDS para vários tipos de
água. Fonte: Adaptado de RUSYDI (2018). ................................................................ 50
Tabela 2 - Sais utilizados para preparação das amostras com respectivas marcas e níveis
de pureza..................................................................................................................... 51
Tabela 3 - Equipamentos utilzados na realização dos experimentos. ............................ 52
Tabela 4 - Composições planejadas e experimentais para as salmouras características do
Pré-sal brasileiro. ........................................................................................................ 58
Tabela 5 - Dados de entrada do programa: concentração dos íons (ppm) que compõem as
amostras de 226363, 215000 e 200000 ppm. ............................................................... 60
Tabela 6 - Dados de entrada do programa: concentração dos íons (ppm) que compõem as
amostras de 150000, 100000 e 50000 ppm. ................................................................. 60
Tabela 7 - Composições experimentais das amostras de teste. ..................................... 61
Tabela 8 - Resultados experimentais das condutividades das três amostras das soluções
de NaCl para a concentração de 230000 ppm medidas nas temperaturas de 25, 40, 55 e
70 °C. ......................................................................................................................... 62
Tabela 9 - Resultados experimentais das condutividades das três amostras das soluções
de NaCl para a concentração de 215000 ppm medidas nas temperaturas de 25, 40, 55 e
70 °C. ......................................................................................................................... 62
Tabela 10 - Resultados experimentais das condutividades das três amostras das soluções
de NaCl para a concentração de 200000 ppm medidas nas temperaturas de 25, 40, 55 e
70 °C. ......................................................................................................................... 63
Tabela 11 - Resultados experimentais das condutividades das três amostras das soluções
de NaCl para a concentração de 150000 ppm medidas nas temperaturas de 25, 40, 55 e
70 °C. ......................................................................................................................... 63
Tabela 12 - Resultados experimentais das condutividades das três amostras das soluções
de NaCl para a concentração de 100000 ppm medidas nas temperaturas de 25, 40, 55 e
70 °C. ......................................................................................................................... 63
Tabela 13 - Resultados experimentais das condutividades das três amostras das soluções
de NaCl para a concentração de 50000 ppm medidas nas temperaturas de 25, 40, 55 e 70
°C. .............................................................................................................................. 64

xvii
Tabela 14 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 31.
................................................................................................................................... 65
Tabela 15 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 31.
................................................................................................................................... 66
Tabela 16 - Valores do coeficiente 𝛼𝑐𝑜𝑛𝑐. 𝑖 para cada concentração. .......................... 66
Tabela 17 - Fatores TDS experimentais para diferentes concentrações e temperaturas das
amostras de soluções de NaCl. .................................................................................... 68
Tabela 18 - Fatores TDS calculados para 40, 55 e 70°C utilizando-se αmédio para corrigir
os valores de condutividade medidos a 25°C das amostras de soluções de NaCl. ......... 68
Tabela 19 - Erros relativos (%) entre os fatores TDS experimentais e os calculados
exibidos nas Tabelas 17 e 18, respectivamente. ........................................................... 68
Tabela 20 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 226363 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C. ............................................................................. 70
Tabela 21 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 215000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C. ............................................................................. 70
Tabela 22 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 200000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C. ............................................................................. 70
Tabela 23 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 150000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C. ............................................................................. 71
Tabela 24 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 100000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C. ............................................................................. 71
Tabela 25 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 50000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C. ............................................................................. 71
Tabela 26 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 33.
................................................................................................................................... 73
Tabela 27 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 34.
................................................................................................................................... 74

xviii
Tabela 28 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 35.
................................................................................................................................... 75
Tabela 29 - Valores do coeficiente 𝛼𝑐𝑜𝑛𝑐. 𝑖 para cada concentração. .......................... 75
Tabela 30 - Fatores TDS experimentais para diferentes concentrações e temperaturas das
amostras de salmouras características do Pré-sal brasileiro. ......................................... 76
Tabela 31 - Fatores TDS calculados para 40, 55 e 70°C utilizando-se αmédio para corrigir
os valores de condutividade medidos a 25°C das amostras de salmouras características do
Pré-sal brasileiro. ........................................................................................................ 76
Tabela 32 - Erros relativos (%) entre os fatores TDS experimentais e os calculados
exibidos nas Tabelas 30 e 31, respectivamente. ........................................................... 77
Tabela 33 - Resultados simulados das condutividades para a salmoura característica do
Pré-sal brasileiro para as mesmas configurações de medição experimentais. ............... 78
Tabela 34 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 37.
................................................................................................................................... 79
Tabela 35 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 38.
................................................................................................................................... 80
Tabela 36 - Valores do coeficiente 𝛼𝑐𝑜𝑛𝑐. 𝑖 para cada concentração. .......................... 80
Tabela 37 - Erro relativo (%) entre as condutividades experimentais das soluções de NaCl
e da salmoura característica do Pré-sal brasileiro. ........................................................ 82
Tabela 38 - Erro relativo (%) entre as condutividades experimentais e as simuladas para
a salmoura característica do Pré-sal brasileiro. ............................................................ 83
Tabela 39 - Resultados dos testes experimentais para verificação da correlação
experimental. .............................................................................................................. 84

xix
LISTA DE SÍMBOLOS
NOMENCLATURA
a𝐴 - Atividade dos cátion A
a𝐵 - Atividade dos cátion B
A - Área da seção transversal uniforme
[An] - Concentração dos ânions
C - Concentração da solução
Ci - Concentração molar do íon i
Cv - Coeficiente de variação da população
E - Erro da faixa de operação do equipamento
E - Força eletromotriz
Ed - Potencial de decomposição
𝑓 - Fator TDS
𝑓𝑇𝑐𝑜𝑛𝑐.𝑖 - Fator TDS para uma temperatura 𝑇 e concentração i
G - Condutância de material ou solução
Gref - Condutância de uma solução de referência
𝐾𝑑 - Constante de equilíbrio da equação de dissociação
𝐾ps - Constante do produto de solubilidade
𝐿 - Comprimento
M - Massa
n - Número de elementos da população
[Me] - Concentração dos cátions dos metais
P - Pressão
𝑖 - Corrente elétrica
I - Força iônica
R - Resistência
Rref - Resistência da solução de referência
Rs - Razão de solubilidade
R2 - Coeficiente de determinação
T - Temperatura

xx
LETRAS GREGAS
α - Coeficiente de temperatura
αconc.i - Coeficiente de temperatura na concentração i
𝛼𝑑 - Grau de dissociação
𝛼𝑚é𝑑𝑖𝑜 - Coeficiente de temperatura médio
γ - Coeficiente de atividade
𝛾𝑖 - Coeficiente de atividade do íon i
𝛿 - Constante de célula de condutividade
Δ𝑇 - Variação de temperatura
𝜅 - Condutividade de material ou solução
𝜅25 °𝐶 - Condutividade medida a 25 °C
𝜅𝑟𝑒𝑓 - Condutividade de uma solução de referência
𝜅𝑇𝑟𝑒𝑓 - Condutividade da solução medida a uma temperatura de referência
𝜅𝑇 - Condutividade da solução medida a uma temperatura 𝑇
𝜅25 °𝐶𝑐𝑜𝑛𝑐,𝑖
- Condutividade medida a 25 °C para uma concentração i
𝜆+ - Condutividade iônica limite por mol de eletrólito do cátion
𝜆𝐾+ - Condutividade iônica limite por mol de eletrólito de 𝐾+
𝜆− - Condutividade iônica limite por mol de eletrólito do ânion
𝜆𝐶𝑙− - Condutividade iônica limite por mol de eletrólito de 𝐶𝑙−
𝜈+ - Coeficiente estequiométrico do cátion
𝜈− - Coeficiente estequiométrico do ânion
ξ - Constante experimental da lei de Kohlrausch
𝜌 - Resistividade de material ou solução
𝜎𝑚𝑜 - Tensão da interface óleo-microemulsão
𝜎𝑚𝑤 - Tensão da interface água-microemulsão
Tref - Temperatura de referência
Tmax - Temperatura máxima de operação de equipamento
V - Tensão elétrica
V - Volume
𝑧𝑖 - Carga do íon i

xxi
𝜎𝛼25 °𝐶 - Desvio-padrão dos coeficientes de temperatura medidos a 25 °C
Λ𝑚 - Condutividade molar
Λ𝑚𝑜 - Condutividade molar à diluição infinita
(Λ𝑚𝑜 )𝐾𝐶𝑙 - Condutividade molar à diluição infinita do 𝐾𝐶𝑙
ABREVIAÇÕES
EOR - Enhanced Oil Recovery
TS - Total de Sólidos ou Total Solids
TDS - Total de Sólidos Dissolvidos ou Total Dissolved Solids
TSS - Total de Sólidos em Suspensão ou Total Suspended Solids
IS - Índice de Saturação
PAM - Poliacrilamida
HPAM - Poliacrilamida Hidrolizada
SCC - Stress Corrosion Cracking

1
1. INTRODUÇÃO
1.1. Motivação e Objetivos
No cenário internacional, o petróleo é o principal produto comercializado,
satisfazendo a uma grande parcela das demandas energéticas globais. Com isso, no atual
padrão tecnológico, em que a energia é essencial tanto na produção quanto na
comercialização de qualquer produto, o petróleo é um dos elementos-chave da economia
mundial. No caso da indústria brasileira de petróleo e gás natural, mudanças primordiais
foram introduzidas pela Lei do Petróleo (Lei nº 9.478, de 1997), que flexibilizou o
monopólio da Petrobras na exploração, produção, importação e no transporte de petróleo
e gás natural. Com isso, a abertura da exploração e produção no Brasil culminou em uma
nova fase para a indústria petrolífera nacional (FERREIRA, 2014).
O ambiente de produção de petróleo, assim como toda a cadeia envolvida nessas
atividades, foi substancialmente transformado pela recentes descobertas de grandes
reservas nas camadas de Pré-sal. Os volumes existentes demandam e justificam a
concepção de novos métodos e tecnologias para sua extração, bem como criam
oportunidades para instalação de novas indústrias de bens e serviços no Brasil. Do ponto
de vista da rede logística relacionada à exploração e produção desse petróleo, deve-se
considerar sua localização em regiões mais distantes da costa, as maiores profundidades
em que ele se encontra e, ainda, a falta de infraestrutura implantada nesses locais. Dessa
maneira, desafios inéditos se colocam: restrições mais estreitas, necessidade de abordar o
problema em variados níveis hierárquicos e em diferentes tempos de implantação e
obrigatoriedade da consideração dos riscos e incertezas no tratamento dos problemas e na
tomada de decisão (FERREIRA, 2014).
A produção de água está sempre vinculada às atividades de exploração e produção
de petróleo, uma vez que ela cresce ao longo da vida produtiva do campo petrolífero
paralelamente ao declínio da produção de óleo em razão do consumo da energia natural
contida no reservatório e também da eventual aplicação de injeção de água nos poços.
Em campos maduros, a fração de água nos líquidos produzidos, ou water cut, pode atingir
valores acima de 90% (TÁVORA, 2007). Sendo assim, a análise química da água
produzida apresenta grande relevância nesse contexto e, em especial, o conhecimento do

2
Total de Sólidos Dissolvidos, ou, no termo em inglês, Total Dissolved Solids – TDS,
como é mais conhecido.
O TDS pode ser definido como a medida da concentração total de matéria
dissolvida em uma amostra de água. Desse modo, ele contabiliza todos os íons orgânicos
e inorgânicos dissociados bem como as substâncias dissolvidas não dissociadas
(HUBERT e WOLKERSDORFER, 2014). Neste momento, faz-se necessário diferenciar
do TDS os seguintes termos: Sólidos Totais, ou Total Solids – TS e Total de Sólidos em
Suspensão, ou Total Suspended Solids – TSS. O TS é o termo aplicado ao resíduo material
que resta em um vaso após a evaporação de uma amostra de água e subsequente secagem
do resíduo. Assim, o TS envolve o TSS, que, por sua vez, é a porção de sólidos totais em
uma amostra que pode ser retida por um filtro, enquanto que o TDS é a porção capaz de
passar por um filtro (CARLSON, 2005).
Dessa forma, o TDS de águas de formação é uma propriedade importante na
caracterização de fluidos provenientes de campos petrolíferos, podendo auxiliar, por
exemplo, na interpretação de perfis elétricos dos poços e na identificação da fonte de água
intrusiva. Além disso, o TDS é fundamental para a avaliação do potencial de formação
de incrustações de sais inorgânicos e de ocorrência de corrosão, que podem ser
ocasionados por uma incompatibilidade da água de injeção com a água da formação
quando elas se misturam, bem como por oscilações de pH, agitação e mudanças nas
condições termodinâmicas do meio. Outra aplicação relevante do TDS consiste em
determinar a influência dos sais dissolvidos na água produzida na estabilidade química
de fluidos injetados nos poços a fim de se aumentar o volume de óleo recuperado, como
polímeros e surfactantes.
O método padrão para determinar o TDS é a gravimetria, no entanto, ela é
dispendiosa e pouco prática, já que suas etapas precisam ser inteiramente repetidas para
cada medição, o que a torna demorada e, assim, inadequada em situações que exigem
uma rápida tomada de decisão. Nesse contexto, o objetivo deste trabalho é encontrar uma
metodologia experimental para determinar o TDS a partir de correlações com a
condutividade elétrica de amostras de uma salmoura característica do Pré-sal brasileiro.
Para tanto, também foram analisadas como as variações de temperatura e concentração
podem afetar a condutividade e, consequentemente, o TDS.

3
Diante da relevância das reservas descobertas recentemente nas camadas do Pré-
sal brasileiro, justifica-se a necessidade da constante elaboração de novos estudos para a
caracterização dos fluidos típicos dessa localidade. Além disso, conforme será melhor
explicado na Seção 3.3, a condutividade elétrica de soluções apresenta, dentre outros
fatores, forte dependência da composição química dos íons presentes nas águas de
formação produzidas juntamente com o óleo e o gás em poços de petróleo. Tal carência
de especificidade inerente ao resultado da condutividade total de uma solução com
múltiplos eletrólitos impossibilita a proposição de uma relação geral entre o TDS e a
condutividade para qualquer tipo de salmoura. Desse modo, este método obriga que seja
realizada uma correta representação do sistema aquoso que se deseja caracterizar a fim
de se evitarem erros elevados na determinação de seu TDS. Portanto, neste trabalho, a
designação de amostras de uma salmoura do Pré-sal brasileiro com uma composição
específica como objeto de estudo para o desenvolvimento de correlações visa possibilitar
que se desenvolva uma metodologia repetível para outros sistemas com uma composição
bem definida.
1.2. Estruturação do Trabalho
O presente trabalho foi estruturado visando tornar a compreensão dos assuntos a
mais clara e natural possível. Dessa maneira, ele foi dividido em seis capítulos de modo
que a fundamentação teórica dos conceitos necessários e a metodologia empregada
fossem abordadas antes da apresentação dos resultados e da elaboração de interpretações
para os mesmos.
O Capítulo 1 introduz o assunto central deste trabalho e expõe as motivações e os
objetivos gerais, destacando a utilidade do tema para a indústria de petróleo.
O Capítulo 2, por sua vez, revisa os conceitos teóricos essenciais para a
assimilação da metodologia descrita no Capítulo 4 e para a análise dos resultados dos
experimentos e das simulações computacionais discutida no Capítulo 5. Para tanto,
incluíram-se a explicação dos fundamentos da Condutometria Direta e dos métodos
existentes para a determinação do TDS de salmouras.
No Capítulo 3 são detalhadas as principais aplicações do TDS para a indústria de
petróleo, dentre as quais se incluem a caracterização de fluidos do reservatório, o

4
potencial de ocorrência de corrosão e de formação de incrustações de sais inorgânicos e,
também, a garantia da estabilidade química de fluidos injetados em poços petrolíferos.
No Capítulo 4 é descrita a metodologia empregada neste estudo, desde os
equipamentos e materiais utilizados até a explicação de como foram realizadas cada etapa
de execução do procedimento experimental. Além disso, também serão introduzidos os
aspectos relevantes do programa computacional utilizado, bem como a descrição das
simulações realizadas.
Já o Capítulo 5 contém a discussão dos dados obtidos nos experimentos e nas
simulações computacionais que foram descritos no capítulo anterior. Para isso, serão
exibidos todos os gráficos gerados para um modelo simplificado de uma solução de
cloreto de sódio e para uma salmoura característica do Pré-sal brasileiro. Dessa forma, a
análise conjunta dos resultados experimentais e das simulações computacionais permitiu
a elaboração de interpretações úteis.
Por fim, o Capítulo 6, que finaliza este trabalho, faz uma revisão dos principais
pontos abordados ao longo de todo o projeto para se chegar às conclusões pertinentes,
assim como fornece propostas para trabalhos futuros em função do que foi tratado nos
capítulos anteriores.

5
2. PRINCIPAIS APLICAÇÕES PARA O TDS
2.1. Caracterização de Fluidos do Reservatório
WEBB e KUHN (2004) afirmam que a água de formação pode ser descrita em
termos de elemento, íon, composição molecular e isotópica, bem como por propriedades
físicas, exibindo variações significativas em relação a todos esses parâmetros.
▪ Íons maiores: Na, Ca, K, Sr, Mg, Ba, Cl−, SO42−, HCO3
−;
▪ Íons menores: Li, Fe, Zn, Si, B, Mn, Pb, F−, etc;
▪ Gases dissolvidos: CO2, H2S, CH4, C2H6, etc;
▪ Compostos orgânicos: ácidos graxos, hidrocarbonetos aromáticos policíclicos,
benzeno, tolueno, etil benzeno, xileno, naftenatos, etc;
▪ Propriedades físicas normalmente determinadas: resistividade, TDS, densidade,
viscosidade, etc.
▪ Razões isotópicas: Sr087 / Sr0
86 , O018 / O0
16 , S034 / S0
32 , H02 / H0
1 , etc.
As análises químicas de águas produzidas juntamente com óleo em campos
petrolíferos são de grande utilidade na produção de petróleo, permitindo identificar a
fonte de água intrusiva, planejar projetos de injeção de água e de descarte de água salgada,
bem como tratar a água produzida a fim de prevenir problemas de corrosão na
recuperação primária e secundária. A determinação da fonte de água intrusiva em um
poço de petróleo, por exemplo, visa permitir que o revestimento possa ser assentado e
cimentado de modo a impedir que essa água alastre os horizontes de óleo ou de gás. Em
alguns poços, um vazamento pode se desenvolver no revestimento ou no cimento e, nesse
caso, as análises da água são empregadas para delinear o horizonte da água para que a
área de vazamento possa ser reparada. Com a atual preocupação mais acentuada na
prevenção da poluição da água, torna-se fundamental localizar a fonte de uma salmoura
poluente, a fim de que uma ação corretiva possa ser tomada (COLLINS, 1975).
A interpretação de perfis elétricos demanda um conhecimento do TDS e da
composição da água intersticial. Essa informação também possibilita a correlação de
unidades estratigráficas e dos aquíferos dentro dessas unidades, assim como em estudos
de movimentação de águas de subsuperfície. Portanto, a compreensão dos processos que

6
levam à acumulação de petróleo ou de outros minerais exige um conhecimento da
natureza dessas águas de formação (COLLINS, 1975).
A composição dos sólidos dissolvidos presentes em águas de formação
provenientes de campos petrolíferos sofre influência de diversos fatores, como
(PETROWIKI, 2016):
▪ Composição da água de formação no ambiente deposicional da rocha sedimentar;
▪ Alterações subsequentes por interação da água com a rocha durante a
compactação de sedimentos;
▪ Alterações por interação da água com a rocha durante a migração de água (caso a
migração ocorra);
▪ Modificações resultantes da mistura com outras águas, incluindo a infiltração de
águas mais recentes, como as águas meteóricas.
Eventualmente, algumas águas associadas ao óleo sofrem uma diluição quando
óleo passa a ser produzido. Essa redução nas concentrações pode ser decorrente da
movimentação de águas diluídas oriundas da compactação de jazidas de argilas
encontradas na proximidade do reservatório de petróleo quando a pressão diminui em
razão da retirada contínua de óleo e salmoura. As alterações de composição dessas águas
podem proporcionar informações úteis para a compreensão dos processos de migração do
petróleo. Interpretando tais variações do ponto de vista da geologia e da história geológica
da área, pode-se, em aguns casos, identificar as rotas prováveis de fluxo e a direção da
água conata. Os fatores que a influenciam também podem ter atuado na migração do óleo
no passado de forma que a água e o óleo podem ter fluído através das mesmas vias e na
mesma direção. Dessa maneira, as informações obtidas da interpretação regional da
composição da água permitem auxiliar na limitação dos modelos de migração do óleo e
contribuir para a localização da prospecção (TEIXEIRA, 2007).
As variações na composição da água refletem a falta de tempo para se procesar a
homogeneização da mistura por advecção e/ou difusão. Essas alterações podem ter sido
resultantes de recentes ou atuais reações da água com a rocha ou influxo de fluidos,
preservando essas modificações. Outros possíveis fatores são as pequenas variações de
pressão que reduzem o fluxo advectivo, a baixa permeabilidade e uma região contendo
apenas óleo ou uma rocha que possui grande volume e porosidade, abrigando grande
volume de fluido que necessita ser misturado. Dentro das possibilidades de explicação

7
para as modificações nas concentrações, uma delas consiste na variação na
permeabilidade do reservatório (TEIXEIRA, 2007).
O conhecimento da composição química da água conata também é essencial para
a determinação da saturação inicial da água no reservatório e para a sua caracterização,
uma vez que alterações de composição podem apontar a extensão do reservatório ou a
região associada a um aquífero sob um campo. Sendo assim, a posse de informações
acerca das modificações nas concentrações da água pode auxiliar na identificação da zona
de produção de água de um determinado poço, assim como no reconhecimento de
misturas de águas de formação e também na determinação da contribuição de diferentes
zonas no poço para o total da água produzida. Além disso, tais informações colaboram
no sentido da avaliação do potencial de incrustação dos poços e na tomada de ações para
sua remediação (TEIXEIRA, 2007).
2.2. Incrustações de Sais Inorgânicos
Um dos problemas mais significativos no campo da Garantia de Escoamento, que
envolve até mesmo os sistemas de injeção de água, é a formação de depósitos de sais
inorgânicos, conhecido na indústria de petróleo como incrustação ou scale. Pode-se
definir incrustações como componentes inorgânicos de baixa solubilidade em água, que,
quando precipitam, podem se aglomerar em diferentes pontos do sistema de produção.
Além disso, podem limitar ou bloquear o escoamento do óleo ao obstruir a matriz da
rocha, bem como fraturas e canhoneados (ARAL e DUARTE, 2010).
A grande incidência de incrustações encontradas nos campos produtores de óleo
se deve à precipitação direta na água que é produzida naturalmente nas rochas
reservatório, ou como um resultado da água produzida que se torna saturada com
componentes incrustantes que se encontram poço abaixo (VIDAL, 2015).
2.2.1. Solubilidade de Sais Inorgânicos
Para se determinar o potencial de incrustação, é de suma importância saber a
solubilidade do sal, uma vez que a deposição de sais inorgânicos só acontecerá em
salmouras em que a solubilidade do mesmo for baixa. Alguns sais são pouco solúveis em

8
água, como é o caso dos sulfatos de bário e estrôncio. A solubilidade do carbonato de
cálcio, por sua vez, depende fortemente das variações de pressão. Podemos dividir o
processo de solubilização de um sal em água em duas fases:
i. Separação dos íons do retículo cristalino;
ii. Solvatação dos íons separados.
Ao analisarmos a energia global do sistema, observamos o fornecimento de
energia para remoção dos íons do retículo e, em seguida, a liberação de energia no
processo de hidratação. A partir desse balanço, verificamos a maior ou menor
solubilidade do sal em água. A relação entre a energia do retículo cristalino e as variáveis
termoquímicas foi desenvolvida em 1917 pelos cientistas alemães Max Born e Fritz
Haber, para explicar o fenômeno da solubilidade (ARAL e DUARTE, 2010).
Para quantificar o fenômeno, pode-se aplicar a lei de Hess. De acordo com essa
lei, a variação total de energia de um processo depende somente das energias dos estados
inicial e final, e não do caminho seguido (RODRIGUES, s.d.). Portanto, a baixa
solubilidade de um sal pode ser entendida como um resultado de uma baixa energia
liberada no processo de solvatação e uma alta energia reticular para a quebra de ligações
(ARAL e DUARTE, 2010).
Segundo HUNTER (1993), a razao de saturação Rs é definida como a razão do
produto das atividades dos íons pelo produto de solubilidade 𝐾ps:
Rs =[Me]. [An]
𝐾ps
(1)
onde [Me] é a concentração dos cátions dos metais, [An] é a concentração dos ânions e
𝐾ps é obtido pela expressão:
𝐾ps = [a𝐴]. [a𝐵] (2)
sendo a𝐴 e a𝐵 as atividades dos cátions A e B, respectivamente.
A atividade representa a concentração efetiva, ou seja, o quanto as interações entre
as moléculas em uma mistura se desviam da idealidade. A razão entre a concentração real

9
e a prevista no preparo de uma solução é o coeficiente de atividade γ, que é função da
temperatura, da pressão e da força iônica (NETZ e ORTEGA, 2002).
Em geral, a atividade é menor que a sua correspondente concentração analítica,
consequentemente, o coeficiente de atividade é, em geral, menor que 1. Ou seja, quanto
maior a concentração analítica, menor é a atividade correspondente e menor é o valor do
coeficiente de atividade. No caso de soluções extremamente diluídas, a atividade se iguala
à sua concentração analítica, por conseguinte, o coeficiente de atividade é igual a 1
(ANDRADE, 2016).
Para determinar o coeficiente de atividade, um método bastante utilizado foi
proposto por Debye e Hückel em 1923, no qual são utilizados os três passos descritos a
seguir (ARAL e DUARTE, 2010):
a) Calcula-se a força iônica I por meio da seguinte expressão:
I =
1
2∑ Ci. 𝑧i
2 (3)
onde Ci é a concentração molar do íon i presente na solução e 𝑧𝑖 é a sua carga.
b) Utiliza-se a seguinte formula para a obtenção dos coeficientes de atividade 𝛾𝑖 de
cada íon i:
log 𝛾𝑖 =−0,51. 𝑧i
2√I
1 + √I (4)
c) Calcula-se a atividade de um íon i de carga 𝑧𝑖 pela expressão:
𝑎𝑖 = 𝛾𝑖 . C𝑖 (5)
sendo 𝛾𝑖 o coeficiente de atividade do íon i e Ci a sua concentração.
Através da Equação (3) é possível observar que, quanto maior a concentração de
íons na solução, maior é o valor da força iônica, por se tratar de um somatório. Além
disso, pelas Equações (3), (4) e (5), percebe-se que quanto maior a força iônica, menores

10
serão as atividades dos íons, e consequentemente, maior a solubilidade dos sais, em razão
da diminuição do denominador (Kps) da Equação (1) para um numerador constante
(ARAL e DUARTE, 2010).
2.2.1.1. Índice de Saturação
O índice de saturação é um termo mais utilizado na indústria de petróleo no lugar
da razão de saturação mencionada anterioriormente. De acordo com SANTANA e
MANZELA (2016), o Índice de Saturação de uma solução (IS) é a medida da relação
entre a quantidade de sal dissolvida na água e a concentração máxima de equilíbrio. O IS
depende da temperatura, da pressão, do pH e da composição da água, ou seja, da presença
de outros sais dissolvidos. Pode-se expressar o IS como log(Rs):
IS = log(Rs) = log ([Me]. [An]
Kps) (6)
IS = log([Me]. [An]) + p. Kps (7)
Segundo ARAL e DUARTE (2010), os possíveis estados de uma solução salina
são:
i. Se IS > 1, então a solução está supersaturada e há risco de precipitação;
ii. Se IS = 1, então a solução está saturada, em equilíbrio e não há precipitação;
iii. Se IS < 1, então a solução está subsaturada e não há precipitação.
2.2.2. Condições para Formação de Incrustações Inorgânicas
Antes de um poço ser perfurado e completado, os fluidos in situ se encontram em
equilíbrio naquele ambiente. Quando esses fluidos escoam para o poço produtor, ocorrem
oscilações de pressão, variações de temperatura e turbulência. Com essas alterações, há
um deslocamento do equilíbrio, visto que as condições em que os fluidos se encontram já
não são mais as mesmas que as iniciais, e processos como o aparecimento de incrustação
inorgânica acontecem (SANTANA e MANZELA, 2016). Nesse sentido, admite-se que,

11
para ocorrer a cristalização de um composto presente na água, algumas condições devem
ocorrer simultaneamente (AFFONSO e ANDRADE, 2017):
i. Supersaturação;
ii. Nucleação;
iii. Tempo de contato adequado para o crescimento de cristais.
Figura 1 - Etapas da formação da incrustação. FONTE: Adaptado de SANTANA e MANZELA
(2016).
a) Supersaturação
Classificamos como solução saturada uma mistura constituída pela dissolução da
quantidade máxima de soluto pelo solvente em uma determinada temperatura. Quando
uma solução contém concentrações mais elevadas de componentes dissolvidos que a
concentração de equilíbrio, ocorre a supersaturação. Algumas razões que podem causar
supersaturação são (AFFONSO e ANDRADE, 2017):
i. Alterações de temperatura, visto que o aumento ou a diminuição da temperatura
pode desestabilizar a água e desencadear a formação de incrustações;
ii. Mudanças no pH, uma vez que, por exemplo, a elevação do pH da água gasosa
contendo íon ferro irá desestabilizar o carbonato de cálcio e o óxido de ferro;
iii. Alterações na pressão, pois uma redução da pressão de CO2, por exemplo,
ocasiona a desestabilização do carbonato de cálcio, que forma a incrustação;
iv. Agitação;

12
v. Mistura de diferentes tipos de águas, como a água de formação com a água de
injeção, por exemplo.
b) Nucleação
Por definição, a nucleação é o início de um processo de precipitação. Existem dois
tipos de nucleação possíveis, com mecanismos distintos (ARAL e DUARTE, 2010):
i. Nucleação Homogênea: esse mecanismo não necessita da presença de uma
substância que não seja própria do local específico para iniciar o processo de
precipitação e pode ocorrer em qualquer ponto do ambiente. Todavia, é muito
improvável na produção de petróleo e gás, já que é comum a presença de tais
substâncias (VIDAL, 2015).
ii. Nucleação Heterogênea: esse mecanismo requer a presença de uma substância
estranha ao local específico para acionar a nucleação, podendo ser, por exemplo,
produtos de corrosão, pontos de solda ou pontos de corrosão sobre superfícies
metálicas, arranhões em superfícies de metal, sólidos em suspensão e até
microrganismos (VIDAL, 2015).
Em uma solução supersaturada, os íons individuais estão em constante movimento
e se movem para dentro e para fora da zona de influência de outros íons. Esses íons, que
são eletricamente carregados e são atraídos pelas cargas opostas, formam continuamente
aglomerados de íons que, em seguida, se quebram. Uma vez que esses aglomerados se
tornam suficientemente grandes e estáveis, continuam a crescer e, ao invés de se
quebrarem, produzem cristais. A formação de cristais a partir desses aglomerados é
conhecida como processo de nucleação. Quando a nucleação alcançar o estágio de cristal,
irá continuar a produzir mais cristais, reduzindo a supersaturação do sistema (AFFONSO
e ANDRADE, 2017).
c) Tempo de Contato
Ainda que a nucleação ocorra em soluções supersaturadas, não há garantia de que
a incrustação aderente irá se formar. De modo geral, é necessário que haja um tempo de
contato suficiente para que o processo de crescimento do cristal prossiga e possa formar
a incrustação. O tempo requerido depende do grau de supersaturação (quanto maior o

13
grau, menor o tempo de contato), do tipo de mineral (quanto menos solúvel, menor o
tempo de conato), da temperatura, da pressão e da agitação (VIDAL, 2015).
2.2.3. Tipos Mais Comuns de Incrustação
De acordo com ARAL e DUARTE (2010), os tipos mais comuns de incrustações
encontrados na indústria de petróleo e gás são:
i. Misturas de sulfatos de bário (barita), estrôncio (celestita), cálcio anidro (anidrita)
e cálcio di-hidratado (gesso), e de rádio, que ocorrem pela incompatibilidade entre
a água do mar e a água de formação rica em cátions de metais alcalino terrosos;
ii. Cristais de ferro, na forma de carbonato (siderita) e sulfeto (pirita);
iii. Misturas de carbonatos de metais alcalino terrosos (precipitam antes do
breakthrough da água do mar) são gerados a partir da liberação de CO2 da água
da formação, o que gera a variação do pH em pressões mais baixas que a pressão
de bolha. A precipitação dos carbonatos é favorecida pelo aumento da temperatura
e pela redução da pressão;
iv. Cristais de haletos que ocorrem principalmente na forma de NaCl (halita)
precipitado, em face à redução da temperatura de evaporação de parte da água de
formação;
v. Silica precipitada (calcedônio ou opala amorfa), em um processo normalmente
associado à lixiviação térmica (injeção de vapor) ou alcalina da rocha
reservatório;
vi. Incrustaçoes com níveis altos de radioatividade em um processo no qual uma parte
do metal alcalino terroso, rádio, lixiviado da rocha reservatorio pela água de
injeção, co-precipita como sulfato à semelhança dos outros metais alcalinos
terrosos, enquanto a outra parte pode permanecer soluvel na água produzida.
2.2.4. Problemas Operacionais Devido a Incrustações
Os problemas causados por incrustações de sais inorgânicos relacionados à
Garantia de Escoamento não estão ligados exclusivamente a uma etapa em particular da
cadeia produtiva do petróleo, ou de um equipamento desta. Alguns casos, no entanto, são
considerados mais graves que outros devido à menor facilidade de remoção e ao custo de

14
remediação. Nas subseções a seguir serão apresentados alguns dos casos mais relevantes,
como explicado por FERREIRA e CARDOSO (2011).
a) Perfuração e Completação de Poços
Os problemas relacionados à formação de incrustações são oriundos da
incompatibilidade entre o fluido de perfuração e/ou salmoura de completação com a água
da formação. Esse cenário é ainda mais crítico quando se trata de um poço pioneiro em
um novo prospecto, com a utilização de um fluido de perfuração com densidade maior do
que a necessária, o que resulta na invasão do fluido para dentro da formação, danificando
os poros da mesma pela geração de incrustações.
b) Injeção de Água
Quando novos poços de injeção são perfurados há o risco de surgimento de
problemas operacionais com incrustações caso a água de injeção não seja compatível com
a água da formação. Essa incompatibilidade pode ocorrer se a água injetada se tornar
instável durante a elevação ou o tratamento. Já no caso de a água ser estável, porém
incompatível com o aquífero, ainda poderão serem geradas incrustações na formação.
c) Produção de Água
Conforme já foi mencionado, quando o poço começa a produzir água, há o risco
da formação de incrustações. A gravidade do problema é função da composição química
da água, do drawdown, da pressão e da temperatura. No caso do drawdown, a precipitação
ocorrerá ainda dentro da formação. Quando ocorre o breakthrough da água injetada no
poço produtor, podem ser desencadeados graves problemas de incrustações, uma vez que,
para certas condições, a água da formação e de injeção, provenientes do mar, fluem juntas
em direção ao poço, de modo que, no caso de incompatibilidade, resulta na precipitação
dos sais na forma de incrustações no local em que ocorre a mistura. Os danos nesse caso,
serão notados no revestimento e, em seguida, na formação.

15
d) Instalações de Produção
Para elevados valores de watercut, equipamentos e tubulações da planta de
processamento estão sujeitos a sofrer com a precipitação de sais por supersaturação
causada por alterações nas condições de temperatura e pressão da água, ou pela mistura
com águas incompatíveis. O processo de precipitação de incrustações é desencadeado
pela mistura de águas provenientes de diferentes poços de produção ou pela mistura da
água do mar com a água da formação.
2.2.5. Prevenção e Remediação
Existem duas formas de solucionar os problemas operacionais decorrentes das
incrustações de sais inorgânicos: os métodos corretivos e os métodos preventivos. Como
as operações de workover (intervenção) para reparar linhas e equipamentos que
apresentem obstruções relacionadas a incrustações são demoradas e, portanto,
dispendiosas, as técnicas de prevenção ganham especial relevância nesse contexto.
a) Métodos Corretivos
Os metódos corretivos são necessários quando já há a presença de incrustações no
sistema de produção e são divididos em remoção mecânica e química:
i. Remoção química: é considerado o primeiro método a ser executado em função
de seu menor custo, especialmente quando a incrustação não é facilmente
acessível ou quando os métodos mecânicos não são efetivos ou caros demais para
serem utilizados (FERREIRA e CARDOSO, 2011).
ii. Remoção mêcanica: pode ser realizada por meio de diversas ferramentas e
técnicas aplicáveis na coluna de produção e nos canhoneados. Um dos métodos
mais antigos é o uso de explosivos, que geram uma grande quantidade de energia
de impacto, removendo a inscrustação. Esse método, no entanto, pode danificar a
coluna e o cimento. Com o intuito de diminuir os problemas causados pelos
explosivos, foram desenvolvidos métodos com explosivos mais fracos. Quando a
camada é espessa, recomendam-se as técnicas de perfuração de rochas ou
trituradores de aço. Brocas de impacto e tecnologia de trituração foram

16
desenvolvidas para serem usadas em flexitubos dentro da configuração da coluna
(SANTANA e MANZELA, 2016). Embora a remoção mecânica tenha um custo
alto quando comprado a outros tratamentos, ela é necessária nos casos em que a
gravidade da incrustação é elevada, de forma que não é possível retirá-la apenas
com tratamentos químicos (FERREIRA e CARDOSO, 2011).
Figura 2 - Duto com incrustação de BaSO4. Fonte: SANTANA e MANZELA (2016).
Figura 3 - Duto depois da remoção. Fonte: SANTANA e MANZELA (2016).
b) Métodos Preventivos
Com a finalidade de reduzir e até mesmo evitar a incrustação inorgânica. A
indústria petrolífera vem desenvolvendo substâncias orgânicas e inorgânicas que possam
atuar nesse processo. Os principais tipos são descritos a seguir, de acordo com os estudos
realizados por SANTANA e MANZELA (2016) e FERREIRA e CARDOSO (2011):

17
i. Inibidores químicos: são produtos químicos que atrasam ou previnem a formação
de hidratos quando adicionados em baixas concentrações à água com propensão
a formar incrustações. Seu princípio de funcionamento se dá pela adsorção à
superfície dos cristais, impedindo seu crescimento ou apenas diminuindo a
aderências dos cristais em superfícies sólidas.
ii. Diluição: é o método mais simples utilizado para a prevenção de incrustação de
sais na coluna de produção por meio do controle da precipitação de halitas em
poços de alta salinidade. A diluição diminui a saturação no poço por estar sempre
enviando água a todo o sistema de produção, inclusive para dentro da formação.
iii. Inibidores de superfície: os inibidores do tipo modificadores de superfície
interagem quimicamente com os sítios de nucleação dos cristais, diminuindo
significativamente suas taxas de crescimento.
iv. Inibidores quelantes: possuem agentes que bloqueiam a precipitação ou
crescimento da incrustação, mas apenas até certo ponto de supersaturação. Como
os agentes quelantes consomem os íons de incrustação em proporções
estequiométricas, a eficiência e o custo-benefício são baixos.
v. Filtração da água injetada: para facilitar a extração do óleo, as companhias
petrolíferas injetam água nos poços para forçar a saída do óleo, e essa água é
extraída do próprio mar, tratada e retornada ao poço. Para tratá-la, são utilizadas
técnicas de ultrafiltração e nanofiltração através de uma membrana seletiva que
consegue retirar somente algumas impurezas da água e manter outras que deverão
permanecer na água ou que não prejudiquem o processo.
2.3. Corrosão por Sais Inorgânicos
2.3.1. Mecanismos dos Processos Corrosivos e Formas de Ocorrência
Segundo GENTIL (2007), a seleção do material mais apropriado para ser utilizado
em determinados equipamentos ou instalações que estarão sujeitas à corrosão requer
sempre uma análise conjunta das variáveis dependentes do material metálico, do meio
corrosivo e das condições operacionais, como descritas a seguir:

18
▪ Material metálico: composição química, presença de impurezas, processo de
obtenção, tratamentos térmicos e mecânicos, estado da superfície, forma, união
de materiais (solda, rebites, etc.), contato com outros metais;
▪ Meio corrosivo: composição química, concentração, impurezas, pH, temperatura,
teor de oxigênio, pressão, sólidos suspensos;
▪ Condições operacionais: solicitações mecânicas, movimento relativo entre
material metálico e meio, condições de imersão no meio (total ou parcial), meios
de proteção contra a corrosão, operação contínua ou intermitente.
Os principais mecanismos dos processos corrosivos são o mecanismo químico e
o eletroquímico. No mecanismo químico, um agente químico ataca diretamente o
material, que pode ser metálico ou não, havendo reações químicas que resultam na
formação de um produto de corrosão sobre a sua superfície, porém sem a transferência de
cargas ou elétrons e, assim, não é gerada corrente elétrica. Esse mecanismo geralmente
se processa em temperaturas elevadas, como em fornos, caldeiras e unidades de processo.
A Figura 4 exemplifica uma placa de ferro reagindo com o sulfeto de hidrogênio na
ausência de umidade, em que, inicialmente, ocorre a adsorção do gás H2S na superfície
do ferro e, em seguida, o ataque, gerando uma película de sulfeto ferroso. Com isso, o
processo corrosivo pode ser inibido ou impedido em razão da formação dessa película
sobre a superfície do metal. Além disso, outros metais como cádmio, cobre, prata e zinco
também são suscetíveis a esse mesmo mecanismo (FRAUCHES-SANTOS et al., 2014).
Figura 4 - Exemplo de processo de corrosão com mecanismo químico. Fonte: FRAUCHES-
SANTOS et al. (2014).
Já a corrosão eletroquímica se caracteriza por ser um processo espontâneo, capaz
de ocorrer quando o metal ou liga está em contato com um eletrólito, em que se
processam, concomitantemente, as reações anódicas (oxidação) e catódicas (redução),
acarretando assim, a deterioração do metal. Tais eletrólitos podem ser a água do mar, a

19
água de formação, o ar atmosférico com umidade, o solo, entre outros. A transferência
dos elétrons da região anódica para a catódica é efetuada através de um condutor metálico,
e a difusão de ânions e cátions na solução fecha o circuito elétrico. O número de cargas
de íons que se descarregam no catodo ou o número de elétrons que migram do anodo para
o catodo permitem avaliar a intensidade do processo corrosivo, como apontam os
mecanismos exibidos na Figura 5 (FRAUCHES-SANTOS et al., 2014).
Figura 5 - Mecanismo eletroquímico da corrosão em diferentes meios. Fonte: FRAUCHES-
SANTOS et al. (2014).
Quanto às formas de corrosão caracterizadas a seguir e ilustradas na Figura 6, elas
fornecem informações úteis quanto à elucidação do mecanismo envolvido no processo
corrosivo e na aplicação de medidas apropriadas de proteção (GENTIL, 2007):
▪ Uniforme: a corrosão se apresenta em toda a extensão da superfície, havendo
perda uniforme de espessura;
▪ Por placas: apenas determinadas regiões da superfície metálica exibem a corrosão,
formando placas com escavações;
▪ Alveolar: a corrosão ocasiona sulcos ou escavações na superfície metálica
semelhante a alvéolos, com fundo arredondado e profundidade geralmente
inferior ao seu diâmetro;
▪ Puntiforme ou por pite: a corrosão se localiza em pontos ou em pequenas áreas da
superfície metálica gerando pites, que são cavidades que exibem o fundo em
forma angulosa e profundidade geralmente superior ao seu diâmetro;
▪ Intergranular (ou intercristalina): a corrosão se processa entre os grãos da rede
cristalina do metal, o que compromete suas propriedades mecânicas a ponto de
poder fraturar dependendo da solicitação por esforços mecânicos, tendo-se assim
a corrosão sob tensão fraturante (Stress Corrosion Cracking – SCC);
▪ Intragranular (ou transgranular ou transcristalina): a corrosão ocorre nos grãos da
rede cristalina do metal, levando à perda de suas propriedades mecânicas de modo
que pode fraturar mediante a menor solicitação mecânica, havendo-se também
corrosão sob tensão fraturante;

20
▪ Filiforme: a corrosão se apresenta sob a forma de filamentos finos e pouco
profundos que se propagam em diferentes direções sem se ultrapassarem. A razão
para isso é que o produto da corrosão, em estado coloidal, possui carga positiva,
gerando repulsão. Este tipo de corrosão é encontrado geralmente em superfícies
metálicas revestidas com tintas ou com metais, provocando o deslocamento do
revestimento. Além disso, é mais frequentemente observada para circunstâncias
em que a umidade relativa do ar é superior a 85% e também em revestimentos
mais permeáveis à penetração de oxigênio e água ou exibindo falhas, como riscos,
ou em regiões de arestas;
▪ Por esfoliação: a corrosão se processa de forma paralela à superfície metálica, se
apresentando em chapas ou componentes extrudados nos quais seus grãos foram
alongados e achatados, de modo a fornecer as condições para que inclusões ou
segregações presentes no material sejam convertidas em plaquetas alongadas em
função do trabalho mecânico. No caso de uma superfície de ligas de alumínio com
essas características na qual se inicie um processo corrosivo, o ataque pode atingir
as inclusões ou segregações alongadas de forma que a corrosão se manifestará
através de planos paralelos à superfície metálica e, mais comumente, em frestas.
O produto de corrosão, volumoso, leva à separação das camadas que se encontram
entre as regiões que sofrem ação corrosiva e, por conseguinte, o material
desintegra-se na forma de placas paralelas à superfície.
▪ Grafítica: a corrosão ocorre no ferro fundido cinzento em temperatura ambiente e
o ferro metálico é transformado em produtos de corrosão, restando a grafite
intacta. A área corroída adquire um aspecto escuro, próprio do grafite, e sua
retirada pode ser realizada facilmente com uma espátula.
▪ Dezincificação: é a corrosão que se apresenta em ligas de cobre-zinco (latões), em
que se observa o surgimento de regiões com coloração avermelhada, que
contrastam com a coloração amarela típica dos latões. Considera-se que se
processa uma corrosão preferencial do zinco, restando o cobre com sua
característica cor avermelhada. A dezincificação e a corrosão grafítica são
exemplos de corrosão seletiva em razão da corrosão preferencial de zinco e ferro,
respectivamente.
▪ Empolamento pelo hidrogênio: o hidrogênio atômico penetra no metal e difunde-
se rapidamente em função de seu pequeno volume atômico. Com isso, ele se

21
transforma em hidrogênio molecular em regiões com descontinuidades, como
inclusões e vazios, exercendo pressão e provocando a formação de bolhas.
▪ Em torno do cordão de solda: é a forma de corrosão observada em torno do cordão
de solda, de modo que se processa intergranularmente em aços inoxidáveis não
estabilizados ou com teores de carbono maiores que 0,03%.
Figura 6 - Representação esquemática de algumas formas de corrosão. Fonte: FREIRE e FLORIDO (2016).
2.3.2. Corrosão pela Água de Formação
A corrosão que mais acomete a indústria de petróleo é a eletroquímica, em razão,
principalmente, da influência dos constituintes do fluido de perfuração e da água de
formação, entre outros fatores (FRAUCHES-SANTOS et al., 2014). A corrosividade da
água de formação é influenciada de forma considerável pelas concentrações das espécies
dissolvidas, como gases e sais dissolvidos, além de sólidos suspensos e microorganismos;
bem como pelas variáveis do processo, como pH, temperatura e velocidade de circulação
do fluido (VASCONCELOS e BROSEGUINI, 2013).

22
De um modo geral, a condutividade elétrica reflete a quantidade de íons
dissolvidos na água, de forma que essa propriedade exibe valores maiores quanto maior
o número de íons em solução. Sendo assim, a condutividade e o TDS, que está
diretamente relacionado, permitem prever o potencial corrosivo da água de formação a
fim de que métodos de inibição possam ser aplicados no âmbito da produção de petróleo
(OGDEN, 2008).
Os sais dissolvidos apresentam efeitos muitos importantes no processo corrosivo
da água, de modo que podem causar uma aceleração ou um retardamento da velocidade
do processo. Cloretos, sulfatos, sais hidrolisáveis, sais oxidantes e bicarbonatos de cálcio,
magnésio e ferro estão entre os íons e sais que influenciam os processos de corrosão com
maior frequência, podendo causar diversos efeitos, tanto individualmente quanto por
meio de suas interações. Além disso, no caso de formação de incrustações de sais
inorgânicos, também se observa um aumento da corrosão (VASCONCELOS e
BROSEGUINI, 2013).
Os íons responsáveis pela dureza (Ca2+ e Mg2+) e os íons bicarbonato (HCO3-) são
inibidores do processo corrosivo, acarretando a sua desaceleração; por outro lado, os íons
cloreto (Cl-) e sulfato (SO4−) geralmente aceleram o ataque corrosivo. Além disso, no caso
da presença de íons sulfato, deve-se levar em consideração também a possibilidade de
ocorrência de corrosão microbiológica provocada por bactérias redutoras de sulfato, o
que acelera o processo corrosivo. Já o cloreto de sódio, por ser um eletrólito forte, tem
seu efeito caracterizado pela elevação de condutividade, que é essencial no mecanismo
eletroquímico de corrosão. No caso do processo corrosivo de ferro em água saturada de
ar em temperatura ambiente, percebe-se que a taxa de corrosão inicialmente aumenta com
a concentração de NaCl e em seguida diminui. A solubilidade do oxigênio em água
decresce continuamente com o incremento da concentração de NaCl, o que justifica a
redução da taxa de corrosão para altas concentrações de NaCl (VASCONCELOS e
BROSEGUINI, 2013).
2.3.3. Métodos para Inibição da Corrosão
Os métodos para inibição da corrosão que serão descritos a seguir foram extraídos
de FRAUCHES-SANTOS et al. (2014).

23
a) Revestimentos
Os revestimentos anticorrosivos podem ter suas ações protetoras justificadas em
virtude da formação de películas de óxidos, hidróxidos e outros compostos pela reação
de metais, como alumínio, cromo, níquel e zinco, com os oxidantes do meio corrosivo.
Os revestimentos podem ser metálicos, não metálicos inorgânicos, compósitos ou não
metálicos orgânicos (tintas).
As principais exigências para um revestimento são: baixa permeabilidade,
resistência química ao meio de operação, dilatação térmica compatível com o substrato,
propriedades físicas apropriadas às solicitações e agressões que experimentará por
abrasão, tráfego, impacto, flexão, entre outras. Além disso, espera-se do revestimento que
seja monolítico (sem emendas), praticamente incapaz de apresentar trincas ou fissuras,
não admita infiltrações, tenha uma aplicação fácil e rápida, permita reparos localizados,
utilize equipamentos de suporte simples e seja de baixo custo.
b) Proteção Catódica e Anódica
Os materiais metálicos podem ser protegidos mediante a aplicação de corrente
anódica ou impedindo a difusão de oxigênio por meio de processo catódico. Os protetores
anódicos são aqueles que interferem nas reações anódicas, de forma que migram para a
superfície anódica provocando passivação em presença de oxigênio dissolvido. Para isso,
este tipo de protetor reage com o produto de corrosão inicialmente formado e origina um
filme aderente em sua superfície, que a protege por ser extremamente insolúvel. A
proteção anódica é adotada com êxito apenas para os metais e ligas que geram películas
protetoras, como o titânio, o cromo, ligas de ferro-cromo e ligas de ferro-cromo-níquel.
A sua aplicação encontra maior interesse para eletrólitos de elevada agressividade
(eletrólitos fortes), como, por exemplo, um tanque metálico destinado a armazenar ácidos.
A proteção anódica não só permite a formação da película protetora, como também,
principalmente, garante a sua estabilidade. A Figura 7 a seguir ilustra o processo de
proteção anódica.

24
Figura 7 - Representação esquemática da proteção anódica. Fonte: FRAUCHES-SANTOS et al.
(2014).
Já a proteção catódica, apesar de ser empregada com eficiência para proteger
estruturas metálicas sem qualquer revestimento, sua utilização torna-se extremamente
econômica e mais simples quando as superfícies são previamente revestidas. Nesses
casos, sua função consiste em complementar a proteção fornecida pelos revestimentos, já
que eles sempre apresentam poros e falhas, além de se tornarem deficientes com o tempo.
Os protetores catódicos neutralizam o processo corrosivo por meio do deslocamento do
potencial de corrosão para valores negativos, elevando o pH do meio e reduzindo a
solubilidade do íon ferroso. Além disso, a proteção catódica é aplicada para estruturas
enterradas ou submersas, não podendo ser empregada em estruturas aéreas em razão da
necessidade de um eletrólito contínuo, o que não se consegue na atmosfera.
Os protetores catódicos são espécies químicas que exibem íons metálicos capazes
de reagir com a alcalinidade catódica, gerando, com isso, compostos insolúveis. Esses
compostos cobrem toda a superfície catódica, impedindo a difusão do oxigênio e dos
elétrons e, por conseguinte, inibem o processo catódico. Em comparação aos protetores
anódicos, a eficiência dos protetores catódicos no concreto é razoavelmente baixa. Tal
fato pode ser explicado por alguns estudos que demonstram que inibidores catódicos não
aderem à superfície metálica como os anódicos, tornando-os menos efetivos. A Figura 8
abaixo ilustra o processo de proteção catódica.
Figura 8 - Representação esquemática da proteção catódica. Fonte: FRAUCHES-SANTOS et
al. (2014).

25
c) Técnicas de Modificação do Meio Corrosivo
Outra forma de se inibir a corrosão consiste na modificação do meio corrosivo por
meio do controle do pH e da desaeração. O controle de pH tem o objetivo de favorecer a
passivação dos metais, que necessita de um pH levemente básico. Nesse sentido, devem
ser tomadas precauções especiais em relação aos metais anfóteros, já que eles perdem a
resistência à corrosão em meios muito básicos, bem como com a precipitação de
compostos de cálcio e magnésio que se tornam insolúveis em pH alto, podendo acarretar
problemas de incrustação.
Já a desaeração consiste na remoção de oxigênio do meio, uma vez que a sua
retirada contribui para a polarização catódica com a decorrente redução da intensidade do
processo corrosivo. Os processos de remoção de oxigênio podem ser químicos ou
mecânicos. No processo químico, a remoção é efetuada pelos agentes sequestradores de
oxigênio, enquanto que, no processo mecânico, ela é realizada por meio da desaeração
por arraste do oxigênio por outro gás, comumente vapor de água, ou, em câmara de vácuo,
na qual a descompressão favorece a saída de gases.
d) Inibidores de Corrosão
O uso de inibidores de corrosão específicos é um dos principais métodos
empregados pela indústria a fim de prevenir ou minimizar o processo corrosivo. Tais
inibidores são compostos orgânicos ou inorgânicos que, quando inseridos no meio
corrosivo, evitam ou reduzem o desenvolvimento das reações de corrosão. Normalmente
esses compostos são adsorvidos, formando um filme muito fino e persistente, que leva a
uma redução na taxa de corrosão em função da atenuação das reações anódicas, catódicas
ou ambas. Dessa forma, os inibidores de corrosão podem atuar impedindo reações
anódicas (inibidores anódicos), reações catódicas (inibidores catódicos) ou ambas
(inibidores mistos). Vale destacar que esses compostos podem agir tanto no período
inicial do tratamento como também no período de propagação, diminuindo a taxa de
corrosão em ambas as circunstâncias.
Os compostos químicos empregados para esse propósito devem satisfazer alguns
requisitos relativos à estrutura e ao comportamento químico. Os compostos inorgânicos,
por exemplo, devem ser capazes de oxidar o metal, gerando uma camada passiva sobre a

26
sua superfície. Já os inibidores orgânicos são compostos orgânicos que apresentam
insaturações e/ou grupamentos fortemente polares em sua estrutura e contendo átomos de
nitrogênio, oxigênio ou enxofre. Em geral, esse tipo de inibidores é adequado para
proteger os materiais metálicos em meio ácido e se denominam inibidores de adsorção,
que adsorvem nas superfícies metálicas catódicas e/ou anódicas, protegendo-as.
2.4. Estabilidade Química de Fluidos Injetados
Atualmente, a produção mundial de petróleo é composta por inúmeros campos
maduros que já se encontram em declínio. Concomitantemente, as incertezas em relação
à descoberta de novas reservas têm suscitado o estudo e o desenvolvimento de técnicas
avançadas de recuperação de óleo, conhecidas também como métodos de EOR (Enhanced
Oil Recovery), que visam incrementar o fator de recuperação de óleo após uma etapa de
recuperação primária, caracterizada pela utilização da energia natural do reservatório, ou
secundária, representada pela injeção de água ou gás. Entre as técnicas utilizadas para a
recuperação avançada de óleo encontram-se os métodos químicos, em que há a aplicação
de compostos químicos, tais como polímeros, surfactantes e algumas sustâncias alcalinas
que visam modificar as condições do deslocamento de óleo ao longo do meio poroso e,
consequentemente, aumentar o volume de óleo recuperado durante o processo
(SANABRIA, 2013).
Nas subseções a seguir, será explicado como a presença de sais inorgânicos pode
afetar a estabilidade dos compostos químicos injetados e, portanto, prejudicar a eficiência
dos métodos EOR.
2.4.1. Polímeros
A injeção de polímeros como método de EOR tem como objetivo principal reduzir
a mobilidade da água no meio poroso. Como a água injetada normalmente possui uma
viscosidade menor que a do óleo, ela se desloca com maior facilidade, criando caminhos
preferenciais entre os poços injetores e os produtores. Com a adição de polímeros à água
de injeção, aumenta-se a sua viscosidade e, consequentemete, reduz-se a razão de
mobilidade, dificultando a formação dos caminhos preferenciais e aumentando a região
varrida pelo fluido injetado (FERREIRA, 2016). A Figura 9 ilustra a ação da solução

27
polimérica na eficiência de varrido areal, em que (a) representa uma recuperação em
condição de alta razão de mobilidade (desfavorável) e (b) com razão de mobilidade baixa
(favorável).
Figura 9 - Ação da solução polimérica na eficiência de varrido areal. Fonte: Adaptado de RIOS
(2014).
2.4.1.1. Tipos de Polímeros
Usualmente, os dois polímeros mais aplicados para recuperação do óleo são os
sintéticos, como a poliacrilamida (PAM) e os biopolímeros, como a goma xantana. Como
as poliacrilamidas são fortemente adsorvidas nas superfícies minerais, elas precisam ser
parcialmente hidrolisadas para diminuir os níveis de adsorção, gerando as poliacrilamidas
hidrolisadas (HPAM) (SHENG, 2010).
Em muitas condições, as soluções com HPAM apresentam uma viscoelasticidade
ligeiramente maior que aquelas com goma xantana e, por isso, são as mais utilizadas em
aplicações de EOR. A goma xantana, por sua vez, apresenta uma boa resistência à
degradação mecânica e não é tão sensível à salinidade (FERREIRA, 2016).
2.4.1.2. Influência da Composição da Água
Tanto a goma xantana quanto a HPAM são polieletrólitos e interagem fortemente
com a presença de íons em solução. Todavia, como a cadeia da poliacrilamida é flexível,
a HPAM apresenta uma resposta mais intensa às forças iônicas, tornando suas

28
propriedades mais sensíveis à salinidade/dureza do que uma solução com goma xantana
nas mesmas condições. Os efeitos da salinidade e dureza são descritos a seguir:
a) Salinidade
A presença de íons monovalentes de sais inorgânicos como Na+ e K+ pode
influenciar bastante as propriedades da solução polimérica, como a adsorção, a
mobilidade e a redução de permeabilidade dependendo do tipo de polímero utilizado,
sendo mais visível no caso da HPAM que da goma xantana. Quando a HPAM é dissolvida
em água, os grupos carboxila (−COO-) da cadeia molecular se repelem, fazendo com que
sua estrutura permaneça estendida, o que proporciona um volume hidrodinâmico maior à
molécula, resultando no aumento da viscosidade da solução polimérica. Contudo, com a
adição de um sal monovalente à solução, como NaCl ou KCl, ocorre a neutralização das
cadeias laterais negativas da HPAM pelos cátions positivos dos sais, reduzindo a repulsão
entre trechos da cadeia que não permanecem tão esticados, o que resulta na diminuição
da viscosidade. A Figura 10 ilustra o encolhimento molecular devido à alta salinidade
(RIOS, 2014).
Figura 10 - Efeito das forças iônicas na forma de um polieletrólito de cadeia flexível, como HPAM: (a) Em baixa salinidade e (b) Em alta salinidade. Fonte: RIOS (2014).
b) Dureza
O nível de dureza da água pode ser caracterizado pela concentração dos íons Ca2+
e Mg2+ diluídos. A presença desses íons divalentes na solução polimérica tem um efeito
mais acentuado em suas propriedades que em espécies de íons monovalentes, como citado
anteriormente. As HPAM interagem fortemente com os cátions divalentes, o que resulta

29
na redução da viscosidade, formação de gel e até a precipitação do polímero (RIOS,
2014).
De acordo com um estudo realizado por WARD (1981) sobre a viscosidade da
HPAM na presença de Ca2+ e Mg2+, percebeu-se que a viscosidade da solução sofria uma
redução mais acentuada para os íons de cálcio que para os íons de magnésio. Ele também
mostrou que a redução de viscosidade causada por cátions divalentes é mais forte quanto
mais concentrada for a solução polimérica e quanto maior for seu grau de hidrólise.
2.4.2. Surfactantes
Os surfactantes representam uma classe de moléculas anfifílicas especiais
conhecidas como agentes de superfície ativa ou tensoativos, de caráter ambivalente, em
que uma parte da molécula é formada por um grupo polar ou iônico, com afinidade pela
água, e a outra parte por grupos apolares, com afinidade por compostos orgânicos, como
o óleo. Por meio da aplicação dos surfactantes, é possível modificar o comportamento
molecular na interface de um sistema de dois fluidos imiscíveis pela redução da tensão
interfacial, como é o caso das misturas de óleo e água presentes nos poros da rochas
reservatório (SANABRIA, 2013).
2.4.2.1. Tipos de Surfactantes
Os surfactantes podem ser divididos em dois grupos de acordo com a natureza da
porção hidrofílica: iônicos e não iônicos (SALAS, 2014). A Figura 11 mostra a
representação esquemática dos tipos de surfactantes.
Figura 11 - Representação esquemática dos tipos de surfactantes. Fonte: SALAS (2014).

30
i. Surfactantes iônicos: são classificados de acordo com a estrutura do grupo
hidrofílico de íons que se dissocia em que o meio aquoso. As cargas dos
surfactantes iônicos tendem a se repelir na superfície e constituem um
mecanismo de estabilidade em sistemas dispersos, podendo ser
classificados em: surfactantes aniônicos, catiônicos e anfóteros.
ii. Surfactantes não iônicos: não possuem carga em suas moléculas, mas
apresentam uma parte lipofílica e outra hidrofílica que se solubiliza em
água, ligando-se através de pontes de hidrogênio. Eles possuem algumas
características particulares que são compatíveis quimicamente com a
maioria dos outros surfactantes e suas propriedades são pouco afetadas
pelo pH (SALAS, 2014).
2.4.2.2. Efeito da Salinidade
Durante a aplicação do método de recuperação avançada de óleo, os surfactantes
devem manter o sistema quimicamente estável, gerar a redução da tensão interfacial e
manter uma baixa taxa de adsorção à rocha. A instabilidade gerada na mistura é o
resultado da redução da solubilidade do surfactante na salmoura pela alta concentração
de eletrólitos. Para avaliar o comportamento dos sistemas de fases surfactante-óleo-água
são utilizados os diagramas ternários de pseudo-componentes, em que o surfactante é
representado no topo, o óleo no vértice direito e a salmoura no vértice esquerdo. Esse tipo
de diagramas é muito relevante, uma vez que o efeito da concentração de eletrólitos na
tensão interfacial é especifico para cada sistema óleo-salmoura-surfactante formado.
a) Windsor I (ou Windsor II(-))
Nos casos em que a salinidade é baixa, a solubilidade do surfactante é maior na
fase aquosa, formando um sistema chamado de Windsor I (ou Windsor II(-)). Esse tipo
de ambiente caracteriza-se pela presença de no máximo duas fases e por linhas de
amarração (tie lines) com derivada negativa, como mostra a Figura 12. As fases formadas
são óleo puro em excesso e uma microemulsão que contém salmoura, surfactante e uma
pequena quantidade de óleo solubilizado (SANABRIA, 2013).

31
Figura 12 - Diagrama de fases para o sistema Windsor I. Fonte: SANABRIA (2013).
b) Windsor II (ou Windsor II(+))
Para sistemas de alta salinidade, chamados de Windsor II (ou Windsor II (+)),
também são formadas no máximo duas fases, no entanto, as linhas de amarração
caracterizam-se pela derivada positiva. Nesse tipo de ambiente formam-se uma salmoura
de alta salinidade e uma microemulsão que contém óleo, surfactante e uma pequena
quantidade de salmoura, como ilustra a Figura 13 (SANABRIA, 2013).
Figura 13 - Diagrama de fases para o sistema Windsor II. Fonte: SANABRIA (2013).

32
c) Windsor III
Nos sistemas de salinidade intermediária, chamados de Windsor III, podem existir
três fases em equilíbrio, como mostra a Figura 14. Estes sistemas caracterizam-se pela
geração de tensões interfaciais ultrabaixas, considerado como a condição ótima para o
processo de injeção de surfactantes. Os diagramas ternários para os pseudocomponentes
evidenciam uma região de uma fase localizada na parte superior, duas regiões bifásicas e
uma região trifásica na parte inferior (região de baixas concentrações de surfactante)
formando um triângulo (SANABRIA, 2013).
Figura 14 - Diagrama de fases para o sistema Windsor III. Fonte: SANABRIA (2013).
Ao se considerar a existência de três possíveis fases no sistema, geram-se dois
tipos de interfaces: a interface entre a fase aquosa e a fase da microemulsão, e a interface
entre a fase oleosa e a fase da microemulsão. A tensão da interface óleo-microemulsão
(𝜎𝑚𝑜) decresce de forma considerável em função da salinidade, devido ao aumento da
solubilidade do surfactante na fase oleosa nessa direção. Consequentemente, o efeito
contrário ocorre com a tensão da interface água-microemulsão (𝜎𝑚𝑤), ou seja, a
solubilidade dos surfactantes será maior quando a concentração de eletrólitos dissolvidos
for menor, promovendo uma menor tensão interfacial (SANABRIA, 2013).

33
3. FUNDAMENTAÇÃO TEÓRICA
3.1. Condutometria Direta
A Condutometria é um ramo da Química Analítica que se baseia em medições de
condutância elétrica de soluções iônicas. De modo geral, a condução da eletricidade
através das soluções iônicas ocorre mediante a migração de íons positivos e negativos
com a aplicação de um campo eletrostático. A condutância elétrica desse tipo de soluções
depende do número de íons presentes, bem como de suas das cargas e suas mobilidades.
A condutometria envolve duas técnicas analíticas: a condutometria direta e a titulação
condutométrica. As medidas de condutância também são utilizadas para outros fins, como
a determinação de constantes de ionização, produtos de solubilidade, condutâncias-
equivalentes, formação de complexos e efeitos de solventes (OHLWEILLER, 1981).
A condutometria direta consiste em medições de condutância ou condutividade
com a finalidade de se determinar a concentração de um eletrólito em solução e também
pode ser aplicada em situações nas quais há o interesse apenas pela condutividade total
da solução. No entanto, como a condutância de uma solução é o somatório das
condutâncias individuais de todas as espécies iônicas presentes, deve-se atentar ao fato
de que o resultado da medição dessa propriedade carece de especificidade. Na titulação
condutométrica, por sua vez, analisa-se o aumento ou o decréscimo de condutânica ou
condutividade da solução a fim de relacioná-los às variações de concentração das espécies
iônicas que participam de uma determinada reação. Para isso, são realizadas diversas
medidas da condutividade, antes e depois do ponto de equivalência, de modo que o ponto
final da titulação se caracteriza como uma descontinuidade na variação da condutividade
(OHLWEILLER, 1981).
Em razão do objetivo principal deste trabalho ser encontrar uma correlação
experimental entre o TDS de amostras de uma salmoura característica de um campo
petrolífero e suas respectivas condutividades, serão abordados nesta seção apenas os
conceitos fundamentais relativos à condutometria direta. Nesse sentido, será tratada a
fundamentação teórica a respeito da condutividade elétrica das soluções iônicas e os
fatores que exercem influência significativa sobre a mesma.

34
3.1.1. Condutividade Elétrica das Soluções Iônicas
As soluções iônicas, assim como os materiais, de modo geral, apresentam a
característica de resistir ao fluxo de carga elétrica. Essa propriedade física é conhecida
como resistência (R) e para qualquer material com uma área da seção transversal uniforme
A e comprimento 𝐿, tem-se que
R = 𝜌𝐿
A (8)
onde a constante de proporcionalidade 𝜌 é conhecida como resistividade do material e
tem unidade de ohms-metro (ohm.m) no Sistema Internacional (SI). Por outro lado, a
habilidade de uma solução iônica ou de um material em conduzir corrente elétrica é
conhecida como condutância (G), que é o inverso da resistência, e tem unidade de ohm-1
ou siemens (S), permitindo-se escrever:
G = 𝜅A
𝐿 (9)
onde a constante de proporcionalidade 𝜅 é conhecida como condutividade do material e
tem unidade de ohm-1.m-1 ou S. m-1 (ALEXANDER e SADIKU, 2013).
O físico alemão Georg Simon Ohm (1787-1854) propôs a lei de Ohm, que
estabelece a relação entre corrente (i) e tensão (v) para um resistor (ALEXANDER e
SADIKU, 2013). Matematicamente, é dada por:
v = Ri (10)
Aplicando-se uma diferença de potencial, os íons em uma solução são quase
instantaneamente acelerados em direção ao eletrodo de carga oposta, porém a velocidade
de migração dos íons é limitada pela resistência do solvente ao movimento das partículas.
A velocidade de migração dos íons varia linearmente com a diferença de potencial
aplicada; as soluções de eletrólitos seguem à lei de Ohm, ou seja, a corrente i é
diretamente proporcional à força eletromotriz E e inversamente proporcional à resistência

35
R da solução. Nas condições em que se faz necessária a aplicação de um potencial Ed
(potencial de decomposição) para vencer os efeitos de polarização dos eletrodos, a forma
aplicável da lei de Ohm é (OHLWEILLER, 1981):
i =(E − Ed)
R (11)
3.1.2. Fatores que influenciam a Condutividade Elétrica das Soluções
Iônicas
3.1.2.1. Efeito da Geometria da Célula de Condutividade
Pode-se reescrever a Equação (9) de modo a isolar a variável da condutividade,
na seguinte forma:
κ = G (𝐿
A) (12)
onde o parâmetro 𝐿/𝐴 na equação acima é chamado de constante de célula de
condutividade e, para uma célula padrão, essa constante é 1,0 cm-1. No entanto, para
minimizar o erro nas medições de amostras de soluções iônicas com baixa condutividade
devem ser utilizadas células de condutividade com valores mais baixos para essa
constante, na ordem de 0,1 cm-1. Inversamente, para a medição de amostras de soluções
com alta condutividade devem ser utilizadas células de condutividade com valores mais
altos para essa constante, na ordem de 10,0 cm-1 (SARTORELLI, 2015). A Figura 15 a
seguir mostra os valores apropriados para essa constante conforme as faixas de
condutividade típicas das amostras que se planeja realizar uma medição, bem como exibe
as curvas dos erros associados à escolha de cada uma dessas constantes.

36
Figura 15 - Constantes de célula de condutividade apropriadas para cada faixa de condutividade
típica das amostras. Fonte: SARTORELLI (2015).
Na prática, as células eletrolíticas raramente são geometricamente simples a ponto
de que a constante de célula de condutividade possa ser determinada diretamente. Em
particular, a superfície A do eletrodo é influenciada por sua rugosidade em nível
microscópico, que geralmente não pode ser determinada. Dessa forma, a dependência
com a geometria da célula deve ser determinada experimentalmente. Sendo assim,
conhecida a condutividade de uma solução de referência, 𝜅𝑟𝑒𝑓, a dependência da
condutividade da célula com a geometria é representada por um único parâmetro, δ, que
é obtido pela medição da resistência da solução de referência, 𝑅𝑟𝑒𝑓, à passagem de
corrente elétrica por:
δ = 𝑅𝑟𝑒𝑓𝜅𝑟𝑒𝑓 (13)
de modo que a condutividade de outras soluções pode ser determinada a partir de
(MARTÍNEZ, 2017).
κ =δ
R (14)

37
Usualmente, o aparelho de medição reporta diretamente o valor da condutância.
Nesse caso, determina-se a constante de célula de condutividade, δ, por meio da medição
da condutância de uma solução de referência, 𝐺𝑟𝑒𝑓, cuja condutividade 𝜅𝑟𝑒𝑓 é conhecida,
de acordo com:
δ =κref
Gref (15)
Com isso, possibilita-se que todas os valores de condutância medidos sejam convertidos
em condutividade por meio da constante de célula de condutividade (MARTÍNEZ, 2017).
3.1.2.2. Efeito da Temperatura
Nos metais a corrente elétrica é transportada por elétrons, enquanto que nas
soluções é transportada por íons (RADIOMETER ANALYTICAL, 2009). A resistência
e a condutância sofrem uma influência significativa da temperatura. Na condução
eletrônica em metais, a condutividade geralmente decresce com o aumento da
temperatura. Já na condução iônica, ocorre o inverso, de forma que 𝑑𝜅/𝑑𝑇 é da ordem de
+1 a +2% por grau. Em geral, as condutividades dos eletrólitos em solução são muito
inferiores às dos metais (OHLWEILLER, 1981).
A dependência da condutividade da solução com a temperatura é representada
pelo coeficiente de temperatura 𝛼. Como o coeficiente 𝛼 varia com a composição da
solução e também com a sua concentração, esse parâmetro deve ser obtido
experimentalmente para uma determinada solução e uma determinada concentração da
mesma (SARTORELLI, 2015). Uma maneira de se fazer isso é medir a condutividade de
uma amostra de uma certa solução em estudo em uma concentração fixa para várias
temperaturas. Em seguida, constrói-se um gráfico da condutividade da amostra versus a
temperatura e se faz um ajuste linear dos dados experimentais. Com isso, caso os dados
apresentem um baixo desvio em relação à linearidade, o coeficiente 𝛼, com unidade de
%/°𝐶, para uma dada concentração i da solução pode ser calculado pela seguinte
equação:
αconc.i =coeficiente angular
κTref
. 100 (16)

38
onde no numerador substitui-se o coefiente angular da reta obtida no ajuste linear dos
dados experimentais e 𝜅𝑇𝑟𝑒𝑓 é a condutividade da solução medida a uma temperatura de
referência (𝑇𝑟𝑒𝑓), em geral, 25 °C. Pode-se repetir esse procedimento experimental
preparando-se amostras com várias concentrações e, assim, analisar como o coeficiente
𝛼 varia com a concentração da solução em estudo.
3.1.2.3. Efeito da Concentração e da Natureza dos Eletrólitos
A condutividade de uma solução eletrolítica depende fortemente de sua
concentração. Uma solução que não possua nenhum íon é incapaz de conduzir
eletricidade. A água pura, por exemplo, possui íons e, assim, apresenta uma certa
condutividade, ainda que baixa. Quando eletrólitos são adicionados, a condutividade
naturalmente aumenta. No entanto, a condutividade não aumenta sempre na mesma
proporção da concentração em função das interações que ocorrem entre os íons na
solução. Por exemplo, o fluxo de íons de carga positiva na direção do eletrodo negativo
tende a aumentar a concentração de íons na proximidade do eletrodo. Esse aumento repele
os próprios íons, reduzindo o fluxo e, portanto, a corrente elétrica. Dessa forma, a variação
da condutividade com a concentração geralmente tem um perfil semelhante ao exibido na
Figura 16 (MARTÍNEZ, 2017).
Figura 16 - Perfil típico da condutividade da solução em função da sua concentração. Fonte:
MARTÍNEZ (2017).
Para facilitar a comparação das condutividades de soluções de diferentes
eletrólitos, foi introduzido o conceito de condutividade molar (OHLWEILLER, 1981),

39
ou seja, a condutividade por íon na solução. Esta condutividade, por unidade de
concentração do eletrólito, reflete a capacidade dos íons, em cada solução, de se mover
em direção aos eletrodos da célula de condutividade, de modo que depende das interações
dos íons entre si e das interações com o solvente. Sendo assim, define-se a condutividade
molar Λ𝑚 , como (MARTÍNEZ, 2017):
Λm =κ
C (17)
Com isso, pode-se repensar a variação da condutividade molar com a
concentração. Em concentrações muito baixas, os íons não interagem entre si e a
condutividade é resultante da velocidade com que eles são arrastados no solvente pela
diferença de potencial. Em concentrações maiores, as interações entre íons são mais
significativas e dificultam o arraste, logo, a condutividade molar diminui. O perfil da
condutividade molar Λ𝑚 em função da concentração torna-se similar ao exibido na Figura
17 a seguir (MARTÍNEZ, 2017).
Figura 17 - Perfil típico da condutividade molar da solução em função da sua concentração. Fonte: MARTÍNEZ (2017).
Segundo PILLING (s.d.), eletrólito é toda a substância que, dissociada ou
ionizada, origina íons positivos (cátions) e íons negativos (ânions), pela adição de um
solvente ou aquecimento e, desse modo, torna-se um condutor de eletricidade. Nesse
sentido, a natureza do eletrólito, que pode ser forte ou fraco, causa uma influência
considerável no comportamento de sua condutividade em função da concentração.

40
Para um eletrólito forte, o valor da condutividade cresce marcadamente com a
concentração. Nas soluções diluídas, o valor de 𝜅 aumenta quase linearmente com a
concentração. Por outro lado, as condutividades dos eletrólitos fracos aumentam de forma
bastante gradual com a concentração (OHLWEILLER, 1981). Tais comportamentos
podem ser observados na Figura 18 a seguir, que mostra em (a) o caso do cloreto de
potássio, um eletrólito forte, e em (b) o caso do ácido acético, um eletrólito fraco.
Figura 18 - Gráficos da condutividade em função da concentração para um eletrólito forte em
(a) e para um eletrólito fraco em (b). Fonte: Adaptado de PILLING (s.d.).
Em ambos os casos, a elevação da condutividade é devida ao incremento do
número de íons por unidade de volume da solução. No caso dos eletrólitos fortes, o
número de íons por unidade de volume aumenta na proporção da concentração; nas
soluções mais concentradas, a não linearidade no aumento de 𝜅 com a concentração é
decorrente das interações iônicas. Por outro lado, a variação gradual de 𝜅 nas soluções de
eletrólitos fracos se relaciona com a ionização parcial do soluto e a redução do grau de
ionização com a elevação da concentração (OHLWEILLER, 1981).
a) Eletrólitos Fortes
Para eletrólitos fortes, a variação da condutividade molar com a concentração foi
definida pela lei de Kohlrausch, formulada inicialmente em 1874:
Λm = Λmo − ξ√C (18)

41
onde Λ𝑚𝑜 é a condutividade molar à diluição infinita, que é o valor limite da condutividade
molar quando a concentração é virtualmente nula, e 𝜉 é uma constante experimental com
unidade de S. (cm7. mol3)0,5 (PILLING, s.d.).
Segundo essa lei, que é válida para altas diluições, pode-se, por exemplo, construir
um gráfico da condutividade molar do eletrólito versus a raiz quadrada da concentração
e, em seguida, ajustar uma reta aos dados experimentais relacionados às soluções com
grande diluição. Dessa maneira, é possível determinar o valor de Λ𝑚𝑜 a partir do
coeficiente linear e o valor de 𝜉 a partir do coeficiente angular da reta ajustada, como se
observa na Figura 19 para um exemplo com cloreto de potássio (PILLING, s.d.).
Figura 19 - Gráfico da condutividade molar do KCl versus a raiz quadrada da concentração. Fonte: Adaptado de PILLING (s.d.).
A lei de Kohlrausch também estabelece que, em diluição infinita, a dissociação
do eletrólito é completa e, assim, cada íon liberado contribui para a condutividade do
eletrólito. Sendo assim, a condutividade do eletrólito em diluição infinita é a soma
algébrica da condutividade à diluição infinita dos seus constituintes iônicos (𝜆+, 𝜆−).
Λ𝑚𝑜 = 𝜈+𝜆+ + 𝜈−𝜆− (19)
onde 𝜆+ e 𝜆− são as condutividades iônicas limite por mol de eletrólito do cátion e do
ânion, respectivamente e, 𝜈+ e 𝜈− são os coeficientes estequiométricos do cátion e do
ânion, respectivamente, no eletrólito. Por exemplo, para o KCl, 𝜈+ = 1 e 𝜈− = 1, de

42
modo que (Λ𝑚𝑜 )𝐾𝐶𝑙 = 𝜆𝐾+
+ 𝜆𝐶𝑙−. Com o incremento da concentração da solução, a lei
de Kohlrausch torna-se inaplicável em razão do aumento das interações entre os íons,
bem como outros motivos (PILLING, s.d.).
b) Eletrólitos Fracos
Os eletrólitos fracos não se dissociam completamente e apresentam condutividade
inferior à dos eletrólitos fortes. Com o incremento da concentração, o equilíbrio de
dissociação é deslocado na direção das moléculas não dissociadas (PILLING, s.d.). Nesse
caso, o grau de dissociação de eletrólitos fracos é determinante. Para um eletrólito fraco
monoprótico que se envolve em um equilíbrio de dissociação do tipo
HA(aq) ⇋ H(aq)+ + A(aq)
− (20)
define-se o grau de dissociação 𝛼𝑑 por
αd =[A−]
[HA] + [A−] (21)
onde [𝐻𝐴] + [𝐴−] = 𝐶𝐴 é a concentração total do eletrólito. Para soluções infinitamente
diluídas, o grau de dissociação de eletrólitos fracos também pode ser dado pela razão
entre a condutividade molar Λ𝑚 e a condutividade molar à diluição infinita Λ𝑚𝑜
(MARTÍNEZ, 2017):
αd =Λm
Λmo (22)
Como já foi dito, o grau de dissociação decresce com o aumento da concentração.
Além disso, utilizando-se a seguinte equação usual para a constante de equilíbrio de
dissociação (𝐾𝑑):
Kd =[H+][A−]
[HA] (23)

43
pode-se mostrar que (MARTÍNEZ, 2017):
Kd =𝛼𝑑
2C
1 − 𝛼𝑑=
Λm2 C
(Λmo − Λm)Λm
o (24)
Como o valor limite da condutividade molar à diluição infinita Λ𝑚𝑜 de eletrólitos
fracos é alcançada apenas para concentrações extremamente baixas, na prática torna-se
impossível fazer medições exatas nessas concentrações. Consequentemente, o valor de
Λ𝑚𝑜 não pode ser encontrado extrapolando-se as curvas obtidas a partir dos gráficos de
Λ𝑚 versus √𝐶 para eletrólitos fracos. Dessa forma, a partir da Equação (24) obtém-se
uma expressão linear entre o inverso da condutividade molar e o produto da condutividade
molar com a concentração de eletrólitos fracos (PILLING, s.d.):
1
Λm=
1
Λmo +
ΛmC
(Λmo )2Kd
(25)
A Equação (25) acima é conhecida como a lei de diluição de Ostwald. A partir de
tal expressão, pode-se construir um gráfico de 1/Λ𝑚 versus Λ𝑚𝐶 em que a condutividade
molar à diluição infinita Λ𝑚𝑜 pode ser obtida igualando-se o valor do coeficiente linear da
reta a 1/Λ𝑚𝑜 . Em seguida, pode-se determinar a constante de equilíbrio de dissociação 𝐾𝑑
igualando-se o valor do coeficiente angular da reta a 1/[(Λ𝑚𝑜 )2𝐾𝑑]. A Figura 20 exibe
um gráfico desse tipo para o ácido acético (PILLING, s.d.).
Figura 20 - Gráfico do inverso da condutividade molar (1/𝛬𝑚) do ácido acético versus o produto
da condutividade molar com a concentração (𝛬𝑚𝐶). Fonte: Adaptado de PILLING (s.d.).

44
3.2. Gravimetria
A análise gravimétrica ou gravimetria é a determinação da concentração de um ou
mais analitos, de composição química conhecida, presentes na amostra original, por meio
de medidas de massa feitas com uma balança analítica (MURITO e FINETE, 2009).
Antes de ser pesada, a substância a ser analisada deve ser separada da amostra e, para
isso, existem diversos tipos de análise gravimétrica, os principais são gravimetria por
precipitação, gravimetria de volatilização e eletrogravimetria (FERNANDES, 2014).
3.2.1. Gravimetria por Precipitação
Neste método, é adicionado um agente precipitante à amostra, de forma que o
analito seja convertido em um precipitado pouco solúvel que se deposita no fundo do
recipiente. Em seguida, esse precipitado é filtrado, lavado para remoção de possíveis
impurezas e, se necessário, convertido a outro composto, geralmente por tratamento
térmico adequado ou, em alguns casos, por desidratação e pesado, como ilustra a Figura
21. A massa do sólido pode ser usada juntamente com equações estequiométricas para se
calcular a concentração de compostos iônicos de uma solução (SKOOG et al., 2005).
Figura 21 - Etapas da análise gravimétrica por precipitação. Fonte: MURITO e FINETE (2009).

45
3.2.1.1. Agente Precipitante
De acordo com SKOOG et al. (2005), o agente precipitante precisa reagir
especificamente ou, pelo menos, seletivamente com o analito. Os reagentes específicos
são mais raros e reagem com apenas uma espécie química. Por outro lado, os reagentes
seletivos, que são mais comuns, reagem com um número limitado de espécies. Além
disso, o reagente precipitante ideal deve provocar uma reação com o analito para formar
um produto que seja:
a) Facilmente filtrado e lavado para remoção de contaminantes;
b) Possuir solubilidade suficientemente baixa para que não haja perda significativa
do analito durante a filtração e a lavagem;
c) Ser não reativo com os constituintes da atmosfera;
d) Ter a composição química conhecida após sua secagem ou, se necessário,
calcinação.
3.2.2. Gravimetria de Volatilização
Os dois métodos gravimétricos mais comuns baseados na volatilização são
aqueles para a determinação de água. A gravimetria de volatilização utiliza a volatilidade
da água por meio da aplicação de energia química ou térmica, de modo que seu vapor
entre em contato direto com um absorvedor de massa previamente conhecida. Assim,
após toda a volatilização, a massa da água é determinada pela subtração da massa final e
inicial do absorvedor. Este método também é conhecido como volatilização direta
(FERNANDES, 2014). O método indireto, no qual a quantidade de água é estabelecida
pela perda de massa da amostra durante o aquecimento, é menos satisfatório porque
precisa-se considerar que a água é o único componente volatilizado, o que é
frequentemente indevido, pois o aquecimento de muitas substâncias resulta em sua
decomposição e consequente variação na massa, não podendo ser relacionada com a
presença da água (SKOOG et al., 2005).

46
3.2.3. Eletrogravimetria
O método da eletrogravimetria é bastante utilizado em determinação metálica e
porcentagem de sais em água. Na maioria das aplicações, o metal é depositado em um
cátodo de platina previamente pesado e o aumento da massa é determinado, porém alguns
métodos empregam a deposição anódica. Existem dois tipos gerais de métodos
eletrogravimétricos. Em um deles, não há nenhum controle no potencial do eletrodo de
trabalho e o potencial de célula aplicado é mantido em um nível mais ou menos constante,
o que fornece uma corrente suficientemente alta para completar a eletrólise em um
intervalo de tempo razoável. O segundo tipo é um método de potencial controlado, ou
método potenciostático, uma ferramenta potente para a separação e determinação de
espécies metálicas que tenham potenciais padrão que diferem por apenas alguns décimos
de volt (SKOOG et al., 2005).
A Figura 22 (a) a seguir apresenta o equipamento para a eletrodeposição de metais
sem controle do potencial do cátodo. Note que esta é uma célula com dois eletrodos. Já
na Figura 22 (b), tem-se o arranjo para eletrólise com potencial controlado. O voltímetro
digital monitora o potencial entre o eletrodo de trabalho e o eletrodo de referência. A
tensão aplicada entre o eletrodo de trabalho e o contra-eletrodo varia pelo ajuste do
contato do potenciômetro mostrado em C, para manter o eletrodo de trabalho (neste caso
o cátodo) sob um potencial constante contra o eletrodo de referência. A corrente no
eletrodo de referência permanece essencialmente igual a zero durante todo o tempo
(SKOOG et al., 2005).
Figura 22 - (a) Arranjo para eletrólise com potencial não controlado; (b) Arranjo para eletrólise
com potencial controlado. Fonte: SKOOG et al. (2005).

47
3.2.4. Metodologia para Determinação do TDS por Gravimetria
A metodologia padrão para a determinação do TDS é a gravimetria, em que o
volume é medido com precisão na amostra filtrada e evaporado a seco a uma dada
temperatura em um cadinho de platina. Em seguida, ocorre o resfriamento em um
dessecador e a determinação do peso do resíduo (HUBERT e WOLKERSDORFER,
2014).
3.2.4.1. Problemas do Método
O estudo realizado por WALTON (1988) apontou alguns dos problemas
observados nesse método:
a) Após a precipitação dos sais dissolvidos durante a evaporação, um pouco de água
de cristalização pode ser incorporada na estrutura cristalina, que será então pesada
como material sólido.
b) Devido ao elevado custo da platina, é comum a substituição por porcelana ou
outro prato/cadinho. Isto pode resultar em erros devido à precipitação de sais
dentro dos poros da porcelana que se tornam difíceis de remover, fornecendo erros
de pesagem.
c) A amostra de água pode espirrar durante o aquecimento, causando perda de sais.
d) Especialmente em pH baixo, algum conteúdo aniônico, como cloreto e nitrito,
pode ser perdido pela volatilização de ácidos.
e) Ganho de massa também pode acontecer devido à oxidação ou transformação em
hidróxidos.
f) Alguns parâmetros podem causar um tempo de secagem mais longo. Tais como o
elevado conteúdo mineral, alta concentração de bicarbonato ou grandes
concentrações de cálcio, magnésio, cloreto e sulfato.
g) Para amostras de baixo TDS, a falta de precisão na pesagem, mesmo com uma
boa balança eletrônica, levará a erros significativos.

48
3.3. Método de Correlação do TDS com a Condutividade
Életrica
3.3.1. Características do Método
Conforme foi discutido na Subseção 3.2.4, o método gravimétrico para
determinação do TDS apresenta diversos problemas que podem ocorrer durante a sua
execução, acarretando, em muitos casos, uma perda significativa de precisão. Além disso,
o fato de esse método ser demorado e dispendioso acaba desencorajando a sua utilização
em certas circunstâncias. Sendo assim, para águas de formação, que em geral são
hipersalinas, um método adequado, mais prático e menos custoso é o desenvolvimento de
uma correlação entre o TDS e a condutividade elétrica.
Como já foi dito no Capítulo 1, o TDS inclui todos os íons orgânicos e inorgânicos
dissociados bem como as substâncias dissolvidas não dissociadas (HUBERT e
WOLKERSDORFER, 2014). Nesse sentido, embora apenas os componentes iônicos ou
eletrolíticos possam contribuir efetivamente para a condução de corrente elétrica, este
método de correlação do TDS com a condutividade encontra ampla aplicação para
salmouras oriundas de reservatórios de petróleo, visto que a composição das mesmas se
caracteriza pela presença majoritária de sais inorgânicos.
A relação entre o TDS e a condutividade é comumente descrita na literatura pela
seguinte correlação:
TDS = 𝑓. κ (26)
onde o TDS tem unidade de 𝑚𝑔/ℓ ou 𝑝𝑝𝑚, a condutividade 𝜅 tem unidade de 𝜇𝑆/𝑐𝑚 e
𝑓 é uma constante chamada de fator TDS, que, em geral, pertence ao intervalo de 0,55 a
0,9. Tal correlação é uma aproximação, uma vez que, como já mencionado, espécies não
iônicas não contribuem para a condutividade e, além disso, as espécies iônicas individuais
possuem massas molares distintas. O valor apropriado do fator TDS depende da atividade
dos íons dissolvidos específicos e da atividade média de todos os íons presentes na
amostra, que, por suas vezes, são influenciados pela temperatura, pela quantidade relativa

49
de cada íon e pela concentração total de sólidos dissolvidos na amostra (THIRUMALINI
e JOSEPH, 2009).
Portanto, a relação entre o TDS e a condutividade é complexa e, em muitos casos,
não linear, já que a influência de diferentes eletrólitos na condutividade não é a mesma,
o que compromete a capacidade dessa propriedade de ser um indicador fidedigno do valor
do TDS. Embora a aplicabilidade de eletrodos seletivos de íons tenha crescido
ultimamente como uma das formas de solucionar essa questão, é improvável, por motivos
técnicos e financeiros, que essa tecnologia esteja extensamente disponível para todos os
principais componentes químicos de águas de formação em um futuro próximo
(MARANDI et al., 2013).
No entanto, existem muitas circunstâncias nas quais a composição química de
salmouras é razoavelmente constante em uma dada região ou local de estudo e, assim, a
relação entre o TDS e a condutividade pode ser estabelecida em laboratório com uma
precisão satisfatória para uma ampla faixa de concentrações e temperaturas. Nesses casos,
uma vez encontrada uma correlação para um sistema bem definido, as medições de
condutividade são relativamente rápidas e pouco dispendiosas, de modo que oferecem
uma vantagem considerável em relação ao emprego da gravimetria (SIOSEMARDE et
al., 2010).
3.3.2. Fator TDS
A relação entre a condutividade e a concentração para soluções com um único
eletrólito é linear para as concentrações mais baixas e achatada para as mais elevadas,
visto que a mobilidade iônica diminui com concentração crescente em razão de
interferências e interações entre os íons. No entanto, a inclinação do segmento linear e o
o grau de achatamento nas concentrações mais altas diferem para diferentes eletrólitos
dissolvidos. Como as salmouras provenientes da produção de poços de petróleo não são
soluções simples e contêm várias espécies iónicas e não dissociadas com quantidades e
proporções bastante variáveis, a proposição de uma relação mais genérica entre o TDS e
a condutividade torna-se complicada (HUBERT e WOLKERSDORFER, 2014).
Frequentemente são utilizados valores predefinidos para o fator TDS que não
tenham sido validados para um local específico (HUBERT e WOLKERSDORFER,

50
2014). RUSYDY (2018) classificou valores de condutividade elétrica e fator TDS para
vários tipos de água, como se observa na Tabela 1.
Tabela 1 - Valores de condutividade elétrica a 25 °C e fator TDS para vários tipos de água.
Fonte: Adaptado de RUSYDI (2018).
Tipos de água Condutividade elétrica a 25 °C Fator TDS
Água de formação 500 − 3000 𝜇𝑆/𝑐𝑚 0,55 − 0,75
Água destilada 1 − 10 𝜇𝑆/𝑐𝑚 0,5
Água doce 300 − 800 𝜇𝑆/𝑐𝑚 0,55
Água do mar 45000 − 60000 𝜇𝑆/𝑐𝑚 0,7
Salmoura 65000 − 85000 𝜇𝑆/𝑐𝑚 0,75
O TDS também foi classificado segundo TODD e MAYS (2005) em quatro tipos:
o tipo I é água doce com TDS inferior a 1000 𝑚𝑔/ℓ; o tipo II é água salobra com TDS
entre 1000 e 10000 𝑚𝑔/ℓ; o tipo III é água salina com TDS de 10000 até 100000 𝑚𝑔/ℓ;
e o tipo IV é salmoura com TDS superior a 100000 𝑚𝑔/ℓ. Além disso, RHOADES,
KANDIAH e MASHALI (1992) também classificaram as águas em seis tipos de acordo
com suas condutividades: o tipo I é não salino com condutividade inferior 700 𝜇𝑆/𝑐𝑚; o
tipo II é ligeiramente salino com condutividade de 700 a 2000 𝜇𝑆/𝑐𝑚; o tipo III é
moderadamente salino com condutividade entre 2000 e 10000 𝜇𝑆/𝑐𝑚; o tipo IV é
altamente salino com condutividade de 10000 a 25000 𝜇𝑆/𝑐𝑚; o tipo V é muito altamente
salino com condutividade entre 25000 e 45000 𝜇𝑆/𝑐𝑚; e o tipo IV é salmoura com
condutividade superior a 45000 𝜇𝑆/𝑐𝑚.
Contudo, conforme já foi mencionado, os reais valores do fator TDS podem variar
em uma extensão significativa em função dos principais íons dominantes, dos locais de
amostragem e, eventualmente, do momento no tempo. Sendo assim, o cálculo do TDS
por um fator generalizado é uma estimativa que, em muitos casos, carece de precisão
satisfatória e, por isso, faz-se necessário determinar fatores específicos para uma região
em particular, bem como para concentrações e temperaturas fixas (HUBERT e
WOLKERSDORFER, 2014).

51
4. METODOLOGIA EXPERIMENTAL E DE
SIMULAÇÕES COMPUTACIONAIS
Neste capítulo serão descritos os materiais e os equipamentos utilizados no
procedimento experimental. Em seguida, será explicada a metodologia empregada tanto
para os experimentos quanto para as simulações computacionais.
4.1. Materiais Utilizados
4.1.1. Água Deionizada
A fim de se evitar que os íons e demais partículas presentes na água do sistema de
distribuição urbano influenciem as medições de condutividade, foi utilizada água
deionizada no preparo das salmouras. A deionização foi realizada por um purificador de
água OS10LXE da marca GEHAKA.
4.1.2. Sais Inorgânicos
A Tabela 2 a seguir lista os sais inorgânicos utilizados na preparação das amostras
com suas respectivas marcas e níveis de pureza.
Tabela 2 - Sais utilizados para preparação das amostras com respectivas marcas e níveis de
pureza.
Sais Marca Pureza
Cloreto de sódio (NaCl) ISOFAR ≥ 99,0%
Cloreto de cálcio di-hidratado (CaCl2.2H2O) ISOFAR ≥ 99,0%
Cloreto de magnésio hexa-hidratado (MgCl2.6H2O) Sigma-Aldrich ≥ 99,0%
Cloreto de potássio (KCl) ISOFAR ≥ 99,0%
Cloreto de bário di-hidratado (BaCl2.2H2O) Sigma-Aldrich ≥ 99,0%
Cloreto de estrôncio hexa-hidratado (SrCl2.6H2O) ISOFAR ≥ 99,0%
Sulfato de sódio (Na2SO4) Sigma-Aldrich ≥ 99,0%

52
4.2. Equipamentos
A Tabela 3 descreve os equipamentos utilizados para a realização dos
experimentos e as Figuras de 23 a 27 mostram fotografias dos mesmos. Na Subseção
4.3.1, que virá a seguir, serão explicadas as circunstâncias nas quais esses equipamentos
foram empregados durante o procedimento experimental para a preparação e a medição
de amostras.
Tabela 3 - Equipamentos utilzados na realização dos experimentos.
Equipamento Modelo Marca Limites de
operação Aplicação
Balança
analítica de
precisão
NewClassic MS METTLER
TOLEDO
Máx = 220g Medição da
massa de sais Mín = 0,01g
e = ± 0,001g
Agitador
magnético C-MAG HS7 IKA®
Tmáx = 550 °C Solubilização de
amostras e = ± 5 °C
Medidor de
Condutividade SevenExcellence™
METTLER
TOLEDO
Máx = 2000 mS/cm Medição da
condutividade
de amostras
Mín = 0.001 μS/cm
e = ± 0.5%
Purificador de
água OS10LXE GEHAKA
𝜅 < 1,0 μS/cm
a 25 °C
Deionização da
água por osmose
reversa
Pressão de entrada:
0,2 a 6 kgf/cm2
Vazão de serviço e
de rejeito: 10 a 15
ℓ/h
Bomba de
vácuo nXDS IFT 20 Edwards Máx = 0,5 bar
Desaeração de
amostras de
salmouras

53
Figura 23 - Balança analítica de precisão NewClassic MS da marca METLLER TOLEDO.
Figura 24 - Agitador magnético C-MAG HS7 da marca IKA®.

54
Figura 25 - Medidor de condutividade SevenExcellence™ da marca METTLER TOLEDO.
Figura 26 - Purificador de água OS10LXE da marca GEHAKA.

55
Figura 27 - Bomba de vácuo nXDS IFT 20 da marca Edwards.
4.3. Descrição da Metodologia Empregada
4.3.1. Procedimento Experimental
Foram realizados experimentos com dois tipos de sistemas aquosos. O primeiro
tipo consiste de soluções contendo apenas cloreto de sódio dissolvido em água
deionizada, enquanto o segundo é constituído de salmouras com uma composição
característica das encontradas no Pré-sal brasileiro. A preparação de amostras dessas duas
variedades de solução visou comparar a resposta da condutividade entre uma solução com
apenas um eletrólito dissolvido e uma salmoura complexa com múltiplos eletrólitos
dissolvidos. Além disso, também se objetivou avaliar a influência das variações de
concentração e temperatura na condutividade das amostras de ambos os tipos de soluções.
4.3.1.1. Soluções de Cloreto de Sódio
Neste modelo simplicado de soluções contendo apenas cloreto de sódio, planejou-
se medir a condutividade de amostras preparadas para seis concentrações (230000,
215000, 200000, 150000, 100000 e 50000 ppm), nas quais, em cada uma delas, foram
realizadas medições em quatro temperaturas (25, 40, 55 e 70 °C), totalizando 24

56
configurações de medição distintas. Além disso, no intuito de minimizar os possíveis
erros experimentais, foram preparadas três amostras para cada uma dessas configurações
de medição. Dessa forma, o resultado da condutividade para cada concentração e
temperatura foi obtido pela média aritmética dos valores medidos de cada grupo dessas
três amostras. A Figura 28 a seguir ilustra todas as 24 configurações utilizadas, em que
cada grupo de três quadrados pretos (numerados de 1 a 3) representam as amostras
medidas em condições idênticas.
Figura 28 - Representação esquemática de todas as configurações de medição.
Para o preparo de amostras de 100 mℓ de solução, inicialmente foi calculado a
massa de NaCl necessária para produzir a primeira amostra, de maior concentração
(230000 ppm). Para isso, foi utilizada a definição de concentração volumétrica:
𝐶 ≡𝑚
𝑉 (27)
Em seguida, a massa calculada foi pesada em uma balança analítica de precisão e,
posteriormente, dissolvida em 100 mℓ de água deionizada com o auxílio do agitador
magnético. Havendo ocorrida a completa homogeneização da solução, foi possível
realizar as medições com o medidor de condutividade nas temperaturas de 25, 40, 55 e
70 °C, que eram alcançadas por meio do aquecimento gerado pelo agitador magnético.
Além disso, foram utilizados vidros de relógio para cobrir o topo dos béqueres a fim de

57
se minimizar as perdas por evaporação durante o aquecimento, visto que a água evaporada
condensava no vidro e escorria de volta para a solução em constante agitação.
Para a preparação e a medição das amostras com as concentrações subsequentes,
foi repetido o mesmo procedimento experimental, recalculando-se a massa de sal
necessário a partir da Equação (27).
4.3.1.2. Salmoura Característica do Pré-Sal Brasileiro
Neste modelo de uma salmoura característica do Pré-sal brasileiro contendo
diversos sais dissolvidos, também planejou-se medir a condutividade em amostras
preparadas para as mesmas configurações de medição descritas na subseção anterior,
salvo uma pequena alteração no valor da maior concentração (226363 ppm), conforme se
observa na Figura 29 abaixo.
Figura 29 - Representação esquemática de todas as configurações de medição.
Inicialmente, foram preparadas três salmouras de 1 ℓ visando-se alcançar a
composição planejada que está mostrada na Tabela 4. Para tanto, para cada amostra, foi
pesada a massa necessária de cada um dos sais com a balança analítica de precisão e, em
seguida, esses sais foram dissolvidos em 800 mℓ de água deionizada com o auxílio do
agitador magnético até ser observada a completa homogeneização. Posteriormente, para
remover o ar incorporado no processo de agitação, cada kitasato com uma amostra de
salmoura foi conectado a uma bomba de vácuo, que foi deixada em funcionamento por

58
uma hora. Após esse tempo, as amostras foram transferidas para um balão volumétrico,
no qual foram avolumadas para 1 ℓ.
Tabela 4 - Composições planejadas e experimentais para as salmouras características do Pré-sal
brasileiro.
Sais
Composição
planejada
(g/𝓵)
Composição
experimental
da 1ª amostra
(g/𝓵)
Composição
experimental
da 2ª amostra
(g/𝓵)
Composição
experimental
da 3ª amostra
(g/𝓵)
NaCl 146,31 146,3107 146,3100 146,3100
CaCl2.2H2O 88,81 88,8128 88,8100 88,8100
MgCl2.6H2O 17,73 17,7306 17,7317 17,7310
KCl 2,29 2,2911 2,2910 2,2909
BaCl2.2H2O 0,04 0,0405 0,0408 0,0409
SrCl2.6H2O 3,83 3,8303 3,8305 3,8301
Na2SO4 0,08 0,0800 0,0801 0,0807
Com as três salmouras prontas, de TDS = 226363 ppm, foi retirada de cada uma
delas uma amostra de 50 mℓ. Com isso, foi possível realizar as medições com o medidor
de condutividade nas temperaturas de 25, 40, 55 e 70 °C, que eram alcançadas por meio
do aquecimento gerado pelo agitador magnético. Assim como nas amostras de soluções
de NaCl, também foram utilizados vidros de relógio para cobrir o topo dos béqueres a fim
de se minimizar as perdas por evaporação durante o aquecimento.
Para a preparação e medição da condutividade das amostras com as concentrações
subsequentes, planejou-se preparar amostras, também de 50 mℓ, provenientes de
diluições de alíquotas retiradas das salmouras (com TDS = 226363 ppm). Dessa forma,
foi utilizada a Equação (28) abaixo para se calcular o volume da alíquota a ser retirada a
fim de se atingir a concentração estabelecida para cada amostra e, com isso, saber a
quantidade de água deionizada a ser adicionada para se alcançar um volume final de 50
mℓ por amostra.
𝐶1𝑉1 = 𝐶2𝑉2 (28)

59
Na Equação (28), 𝐶1 é a concentração de cada salmoura (226363 ppm), 𝑉1 é o
volume da alíquota retirada da mesma, 𝐶2 é a concentração que se deseja atingir e 𝑉2 é o
volume final da amostra diluída (50 mℓ), de modo que o volume de água deionizada que
deve ser adicionado em cada diluição é dado por 𝑉2 − 𝑉1.
4.3.2. Programa OLI
É fundamental que cientistas e engenheiros compreendam a influência da química
aquosa em seus processos, o que inclui os efeitos de componentes de rastreamento, pH,
temperatura e outros fatores em seus sistemas de processo. Nas últimas três décadas, a
OLI refinou um programa que modela com precisão soluções aquosas multifásicas e
multicomponentes para praticamente qualquer mistura de espécies químicas, além de
apresentar muitas outras funcionalidades. A base para o programa da OLI é o OLI Engine,
que é composto por Solvers e Databanks (OLI Systems, Inc., 2011).
Dentro do contexto de simulações aquosas, o OLI Databank contém coeficientes
próprios para a previsão das propriedades termodinâmicas, de transporte e físicas de 80
elementos inorgânicos da tabela periódica, e suas espécies aquosas associadas, assim
como mais de 8000 espécies orgânicas. Assim, a maioria das misturas de espécies
químicas pode ser modelada, desde que o solvente da solução seja a água. Além disso, a
pedido do usuário, o modelo aquoso pode incorporar química redox, co-precipitação e
cinética de reação. Também estão disponíveis fenômenos de superfície, como troca
iônica, complexação superficial e adsorção molecular, bem como propriedades de
transporte, como condutividade elétrica, viscosidade e difusividade (OLI Systems, Inc.,
2011).
Nesse sentido, o programa OLI Analyser Studio foi útil para simular as
condutividades da salmoura característica do Pré-sal brasileiro a fim se de comparar os
resultados simulados com os obtidos experimentalmente. Para tanto, foi necessário inserir
como dados de entrada no programa as concentrações dos íons da salmoura em estudo,
bem como suas temperaturas e pressão atmosférica. Vale destacar que as condições de
simulação foram as mesmas das descritas nos experimentos das Subseções 4.3.1.1 e
4.3.1.2, de modo que as concentrações dos íons inseridas em cada simulação são
proporcionais aos valores estabelecidos para as concentrações totais de cada amostra,
como mostram as Tabelas 5 e 6.

60
Tabela 5 - Dados de entrada do programa: concentração dos íons (ppm) que compõem as
amostras de 226363, 215000 e 200000 ppm.
Íon
Concentração dos íons
(ppm) na amostra com
TDS = 226363 ppm
Concentração dos íons
(ppm) na amostra com
TDS = 215000 ppm
Concentração dos íons
(ppm) na amostra com
TDS = 200000 ppm
Na+ 57584 54693 50878
Ca2+ 24218 23002 21397
Mg2+ 2121 2015 1874
K+ 1202 1142 1062
Ba2+ 23 22 20
Sr2+ 1259 1196 1112
SO42- 54 51 48
Cl- 139904 132881 123610
Tabela 6 - Dados de entrada do programa: concentração dos íons (ppm) que compõem as
amostras de 150000, 100000 e 50000 ppm.
Íon
Concentração dos íons
(ppm) na amostra com
TDS = 150000 ppm
Concentração dos íons
(ppm) na amostra com
TDS = 100000 ppm
Concentração dos íons
(ppm) na amostra com
TDS = 50000 ppm
Na+ 38158 25439 12719
Ca2+ 16048 10699 5349
Mg2+ 1405 937 468
K+ 797 531 266
Ba2+ 15 10 5
Sr2+ 834 556 278
SO42- 36 24 12
Cl- 92708 61805 30903
4.3.3. Testes para Verificação da Correlação Experimental
Após realizadas as simulações no programa OLI, foram comparados os resultados
experimentais com os simulados a fim de validar uma ou mais correlações específicas.
Em seguida, foram preparadas duas amostras de teste a fim de se verificar se essas
correlações validadas pelo programa conseguem prever adequadamente o valor do TDS
dessas amostras. Para isso, a primeira amostra de teste foi preparada com TDS = 208000
ppm e a segunda com TDS = 185008 ppm. Vale salientar que ambas apresentam os
mesmos íons e a mesma proporção da composição iônica da salmoura característica do
Pré-sal brasileiro, como se observa na Tabela 7 a seguir.

61
Tabela 7 - Composições experimentais das amostras de teste.
Sais
Composição
experimental da amostra
com TDS = 208000 ppm
(g/𝓵)
Composição
experimental da amostra
com TDS = 185008 ppm
(g/𝓵)
NaCl 134,4417 120,6032
CaCl2.2H2O 81,6081 71,1829
MgCl2.6H2O 16,2922 14,5197
KCl 2,1052 1,8812
BaCl2.2H2O 0,0372 0,0415
SrCl2.6H2O 3,5196 3,1200
Na2SO4 0,0735 0,0803

62
5. RESULTADOS E DISCUSSÕES
5.1. Sistema Simplificado: Solução de NaCl
As Tabelas de 8 a 13 apresentam os resultados de condutividade obtidos
experimentalmente para cada grupo de três amostras de soluções de NaCl variando-se a
concentração, de 50000 a 230000 ppm, e para as temperaturas de 25, 40, 55 e 70 °C. Ao
final de cada tabela também foram exibidas as médias aritméticas e os desvios-padrão
dos resultados referentes a cada grupo de três amostras com a mesma configuração de
medição. Esses valores médios são os que serão utilizados para as análises subsequentes,
a fim de se minimizarem os erros experimentais de cada amostra individualmente.
Tabela 8 - Resultados experimentais das condutividades das três amostras das soluções de NaCl
para a concentração de 230000 ppm medidas nas temperaturas de 25, 40, 55 e 70 °C.
Condutividades das soluções NaCl (µS/cm) a 230000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 211283 219058 220186 223917
Amostra 2 207172 219440 221344 226564
Amostra 3 211703 219482 219612 226984
Média 210052,67 219326,67 220380,67 225821,67
Desvio-padrão 2044,14 190,75 720,36 1357,67
Tabela 9 - Resultados experimentais das condutividades das três amostras das soluções de NaCl
para a concentração de 215000 ppm medidas nas temperaturas de 25, 40, 55 e 70 °C.
Condutividades das soluções NaCl (µS/cm) a 215000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 201488 210746 213990 218524
Amostra 2 203546 209436 215256 217964
Amostra 3 205938 209419 216254 218155
Média 203657,33 209867,00 215166,67 218214,33
Desvio-padrão 1818,41 621,59 926,43 232,44
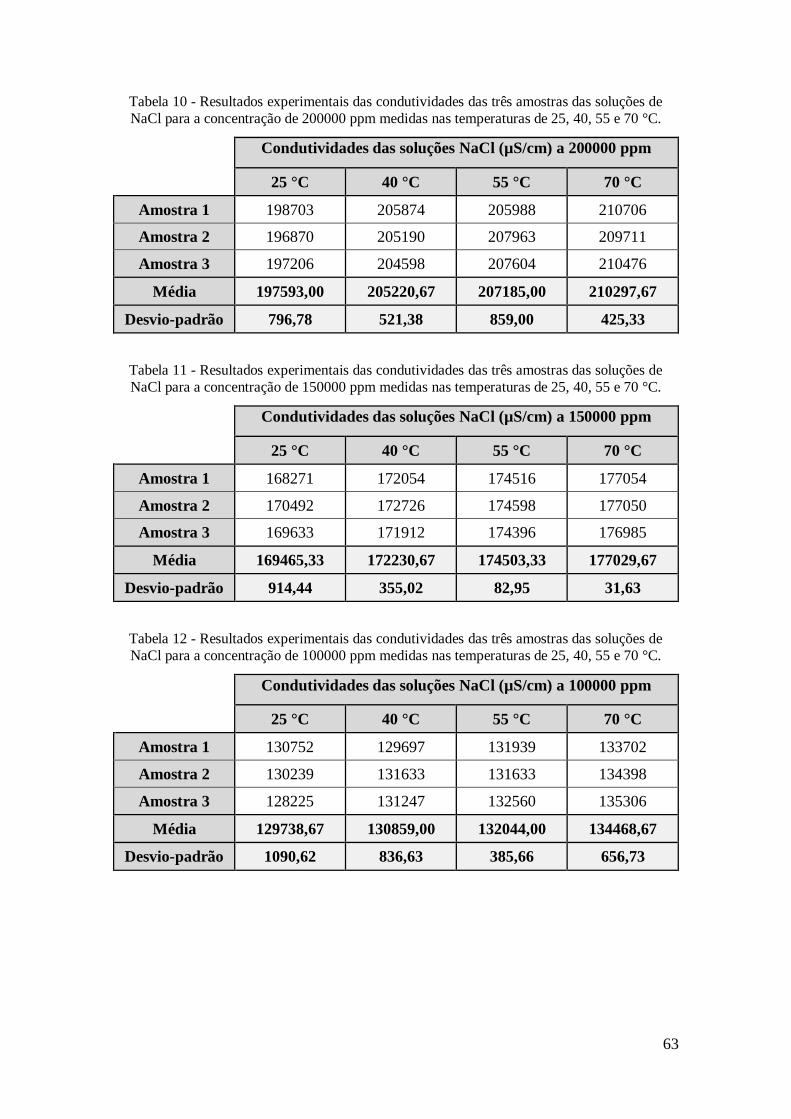
63
Tabela 10 - Resultados experimentais das condutividades das três amostras das soluções de
NaCl para a concentração de 200000 ppm medidas nas temperaturas de 25, 40, 55 e 70 °C.
Condutividades das soluções NaCl (µS/cm) a 200000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 198703 205874 205988 210706
Amostra 2 196870 205190 207963 209711
Amostra 3 197206 204598 207604 210476
Média 197593,00 205220,67 207185,00 210297,67
Desvio-padrão 796,78 521,38 859,00 425,33
Tabela 11 - Resultados experimentais das condutividades das três amostras das soluções de
NaCl para a concentração de 150000 ppm medidas nas temperaturas de 25, 40, 55 e 70 °C.
Condutividades das soluções NaCl (µS/cm) a 150000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 168271 172054 174516 177054
Amostra 2 170492 172726 174598 177050
Amostra 3 169633 171912 174396 176985
Média 169465,33 172230,67 174503,33 177029,67
Desvio-padrão 914,44 355,02 82,95 31,63
Tabela 12 - Resultados experimentais das condutividades das três amostras das soluções de
NaCl para a concentração de 100000 ppm medidas nas temperaturas de 25, 40, 55 e 70 °C.
Condutividades das soluções NaCl (µS/cm) a 100000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 130752 129697 131939 133702
Amostra 2 130239 131633 131633 134398
Amostra 3 128225 131247 132560 135306
Média 129738,67 130859,00 132044,00 134468,67
Desvio-padrão 1090,62 836,63 385,66 656,73

64
Tabela 13 - Resultados experimentais das condutividades das três amostras das soluções de
NaCl para a concentração de 50000 ppm medidas nas temperaturas de 25, 40, 55 e 70 °C.
Condutividades das soluções NaCl (µS/cm) a 50000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 74325,8 76721,1 76971,9 79242,2
Amostra 2 74047,5 76259,1 77131,6 78398,4
Amostra 3 75756,7 76367,8 78690,9 79068,6
Média 74710,00 76449,33 77598,13 78903,07
Desvio-padrão 748,80 197,23 775,45 363,82
Analisando-se as tabelas mostradas acima, observa-se que os valores calculados
dos desvios-padrão de cada grupo de três amostras com a mesma configuração de
medição são inferiores às diferenças das médias de condutividade entre duas temperaturas
de medição consecutivas. Dessa forma, constata-se que as variações de condutividade
com a temperatura são maiores do que os erros experimentais previstos na preparação e
na medição das amostras, o que garante a repetitividade deste método para fins de
caracterização do comportamento de fluidos.
A Figura 30 exibe graficamente, a partir dos dados das tabelas acima, como as
condutividades médias das amostras com diferentes concentrações variam com a
temperatura.
Figura 30 - Gráfico da condutividade versus a temperatura para amostras com diferentes
concentrações.
50000,00
70000,00
90000,00
110000,00
130000,00
150000,00
170000,00
190000,00
210000,00
230000,00
250000,00
15 20 25 30 35 40 45 50 55 60 65 70 75 80 85
Co
nd
uti
vid
ade
(µS/
cm)
Temperatura (°C)
Condutividade (µS/cm) vs. temperatura (°C)
230000 ppm
215000 ppm
200000 ppm
150000 ppm
100000 ppm
50000 ppm

65
Com isso, observa-se que a dependência da condutividade com a temperatura para
diferentes concentrações pode ser ajustada por retas com um baixo desvio em relação à
linearidade, uma vez que o menor valor de 𝑅2 obtido foi de 0,9096, conforme mostra a
Tabela 14.
Tabela 14 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 31.
Concentração das
amostras (ppm)
Correlações obtidas pelos
ajustes lineares
𝑹𝟐 dos ajustes
lineares
230000 𝜅 = 322,41𝑇 + 203581 (29) 𝑅2 = 0,9096
215000 𝜅 = 326,47𝑇 + 196219 (30) 𝑅2 = 0,9789
200000 𝜅 = 267,19𝑇 + 192383 (31) 𝑅2 = 0,9155
150000 𝜅 = 166,44𝑇 + 165401 (32) 𝑅2 = 0,9986
100000 𝜅 = 102,5𝑇 + 126909 (33) 𝑅2 = 0,9599
50000 𝜅 = 91,52𝑇 + 72568 (34) 𝑅2 = 0,9921
A Figura 31 apresenta graficamente, de forma complementar à Figura 31, como
as condutividades médias das amostras medidas em diferentes temperaturas variam com
a concentração.
Figura 31 - Gráfico da condutividade versus a concentração para amostras medidas em
diferentes temperaturas.
0,00
50000,00
100000,00
150000,00
200000,00
250000,00
300000,00
0 50000 100000 150000 200000 250000
Co
nd
uti
vid
ade
(µS/
cm)
Concentração (ppm)
Condutividade (µS/cm) vs. concentração (ppm)
25 °C
40 °C
55 °C
70 °C

66
Nota-se também que, por sua vez, a dependência da condutividade com a
concentração para diferentes temperaturas pode, similarmente, ser muito bem ajustada
por retas, já que o menor valor de 𝑅2 obtido foi de 0,9747, conforme mostra a Tabela 15.
Tabela 15 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 31.
Temperaturas das
amostras (°C)
Correlações obtidas pelos
ajustes lineares
𝑹𝟐 dos ajustes
lineares
25 𝜅 = 0,7345𝐶 + 48514 (35) 𝑅2 = 0,9747
40 𝜅 = 0,7773𝐶 + 46567 (36) 𝑅2 = 0,9828
55 𝜅 = 0,7874𝐶 + 47127 (37) 𝑅2 = 0,9829
70 𝜅 = 0,8034𝐶 + 47591 (38) 𝑅2 = 0,9848
A partir dos coeficientes angulares obtidos nos ajustes lineares das equações de
(29) a (34) da Tabela 14 e das condutividades médias medidas a 25 °C apresentadas nas
Tabelas de 8 a 13, foi possível, por meio da Equação (16), calcular os coeficientes de
temperatura 𝛼𝑐𝑜𝑛𝑐.𝑖 para as amostras com diferentes concentrações. A Tabela 16 abaixo
exibe os resultados desses cálculos.
Tabela 16 - Valores do coeficiente 𝛼𝑐𝑜𝑛𝑐.𝑖 para cada concentração.
Concentração (ppm) Coeficiente 𝜶𝒄𝒐𝒏𝒄.𝒊 (%/°C)
230000 0,1535
215000 0,1603
200000 0,1352
150000 0,0982
100000 0,0790
50000 0,1225
𝜶𝒎é𝒅𝒊𝒐 0,1248
Para verificar se o valor médio do coeficiente de temperatura (𝛼𝑚é𝑑𝑖𝑜) pode
representar adequadamente os demais, foi calculado inicialmente o desvio-padrão σ𝛼𝑐𝑜𝑛𝑐.𝑖
da população de valores de 𝛼𝑐𝑜𝑛𝑐.𝑖 expostos na Tabela 16 por meio da Equação (39) a
seguir:

67
σ𝛼𝑐𝑜𝑛𝑐.𝑖= √∑
(𝛼𝑐𝑜𝑛𝑐.𝑖 − 𝛼𝑚é𝑑𝑖𝑜)2
𝑛
𝑛
𝑖=1
(39)
onde 𝛼𝑐𝑜𝑛𝑐.𝑖 é o coeficente de temperatura calculado para a concentração i e 𝑛 é o número
total de dados da população. Em seguida, foi calculado o coeficiente de variação 𝐶𝑣 da
população a partir da razão entre o desvio-padrão e a média dos dados, como mostra a
Equação (40) abaixo:
𝐶𝑣 =σ𝛼𝑐𝑜𝑛𝑐.𝑖
𝛼𝑚é𝑑𝑖𝑜 (40)
O valor calculado para 𝐶𝑣 foi de 23,14%, no entanto, para examinar com mais
profundidade a capacidade do coeficiente de temperatura médio em representar
apropriadamente os demais 𝛼𝑐𝑜𝑛𝑐.𝑖, foram calculados os fatores TDS por meio de 𝛼𝑚é𝑑𝑖𝑜
a fim de aferir os erros relativos por ele fornecidos em relação aos valores obtidos
experimentalmente. Para tanto, a Tabela 17 apresenta os valores experimentais para os
fatores TDS calculados pela Equação (26), enquanto que a Tabela 18 fornece os valores
dos fatores TDS calculados para as temperaturas de 40, 55 e 70 °C utilizando-se 𝛼𝑚é𝑑𝑖𝑜
para corrigir os valores de condutividade medidos a 25 °C, de acordo com a seguinte
equação:
𝑓𝑇𝑐𝑜𝑛𝑐.𝑖 =
TDS
𝜅25 °𝐶𝑐𝑜𝑛𝑐.𝑖(1 + Δ𝑇
𝛼𝑚é𝑑𝑖𝑜
100 ) (41)
onde 𝑓𝑇𝑐𝑜𝑛𝑐.𝑖 é o fator TDS para uma temperatura 𝑇 e concentração i, 𝜅25 °𝐶
𝑐𝑜𝑛𝑐.𝑖 é a
condutividade medida a 25 °C para a amostra de concentração i e Δ𝑇 é diferença entre a
temperatura de referência, 25 °C, e a temperatura que se deseja corrigir a condutividade
para a amostra de concentração i.

68
Tabela 17 - Fatores TDS experimentais para diferentes concentrações e temperaturas das
amostras de soluções de NaCl.
Fatores TDS experimentais
TDS (ppm) 25 °C 40 °C 55 °C 70 °C
230000 1,0950 1,0487 1,0436 1,0185
215000 1,0557 1,0245 0,9992 0,9853
200000 1,0122 0,9746 0,9653 0,9510
150000 0,8851 0,8709 0,8596 0,8473
100000 0,7708 0,7642 0,7573 0,7437
50000 0,6693 0,6540 0,6443 0,6337
Tabela 18 - Fatores TDS calculados para 40, 55 e 70°C utilizando-se αmédio para corrigir os
valores de condutividade medidos a 25°C das amostras de soluções de NaCl.
Fatores TDS calculados utilizando-se 𝛂𝐦é𝐝𝐢𝐨 = 𝟎, 𝟏𝟐𝟒𝟖 %/°𝐂
TDS (ppm) 25 °C 40 °C 55 °C 70 °C
230000 - 1,0748 1,0555 1,0367
215000 - 1,0363 1,0176 0,9996
200000 - 0,9936 0,9757 0,9584
150000 - 0,8689 0,8532 0,8381
100000 - 0,7566 0,7430 0,7298
50000 - 0,6570 0,6451 0,6337
A Tabela 19 a seguir expõe os erros relativos (%) obtidos entre os fatores TDS
experimentais e os calculados por meio de 𝛼𝑚é𝑑𝑖𝑜 a partir dos valores de condutividade
medidos a 25 °C, em que se observa que o maior erro relativo encontrado foi de 2,50% e
o erro relativo médio total foi de 1,17%. Portanto, é possível assumir um único valor
médio para o coeficiente de temperatura a fim de representar os demais 𝛼𝑐𝑜𝑛𝑐.𝑖, visto que
há uma fraca dependência de 𝛼 com a concentração.
Tabela 19 - Erros relativos (%) entre os fatores TDS experimentais e os calculados exibidos nas
Tabelas 17 e 18, respectivamente.
Erros relativos (%) entre os fatores TDS experimentais e os
calculados
TDS (ppm) 25 °C 40 °C 55 °C 70 °C
230000 - 2,50 1,13 1,79
215000 - 1,16 1,84 1,45
200000 - 1,95 1,07 0,77
150000 - 0,24 0,74 1,09

69
100000 - 0,99 1,90 1,87
50000 - 0,45 0,12 0,00
Erro relativo médio total = 1,17%
A Figura 32 abaixo exibe graficamente os resultados experimentais para os fatores
TDS mostrados na Tabela 17, em que a proximidade das retas ajustadas é compatível com
a adoção de único valor médio para o coeficiente de temperatura com a finalidade de
representar satisfatoriamente os demais.
Figura 32 - Gráfico dos Fatores TDS experimentais versus a concentração (ppm) para diferentes
temperaturas.
5.2. Salmoura Característica do Pré-Sal Brasileiro
As Tabelas de 20 a 25 exibem os resultados de condutividade obtidos
experimentalmente para cada grupo de três amostras de salmouras características do Pré-
sal brasileiro variando-se a concentração, de 50000 a 226363 ppm, e para as temperaturas
de 25, 40, 55 e 70 °C. Também foram apresentadas ao final de cada tabela as médias
aritméticas e os desvios-padrão dos resultados referentes a cada grupo de três amostras
com a mesma configuração de medição. Analogamente ao que foi feito para as amostras
de soluções de NaCl, esses valores médios são os que serão utilizados para as análises
subsequentes, a fim de se minimizarem os erros experimentais de cada amostra
individualmente.
0,5000
0,6000
0,7000
0,8000
0,9000
1,0000
1,1000
1,2000
0 50000 100000 150000 200000 250000
Fato
r TD
S
Concentração/TDS (ppm)
Fatores TDS experimentais
25 °C
40 °C
55 °C
70 °C

70
Tabela 20 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 226363 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C.
Condutividades das salmouras características do Pré-sal
brasileiro (µS/cm) a 226363 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 230318 256533 268531 281707
Amostra 2 231051 259764 279852 306569
Amostra 3 228979 264376 275168 303268
Média 230116,00 260224,33 274517,00 297181,33
Desvio-padrão 857,87 3218,39 4644,65 11024,68
Tabela 21 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 215000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C.
Condutividades das salmouras características do Pré-sal
brasileiro (µS/cm) a 215000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 210956 255768 259893 276656
Amostra 2 222558 248698 267050 291769
Amostra 3 223111 253598 270483 294450
Média 218875,00 252688,00 265808,67 287625,00
Desvio-padrão 5604,13 2957,17 4411,55 7833,10
Tabela 22 - Resultados experimentais das condutividades das três amostras das salmouras características do Pré-sal brasileiro para a concentração de 200000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C.
Condutividades das salmouras características do Pré-sal
brasileiro (µS/cm) a 200000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 205960 242184 253916 268456
Amostra 2 208016 243094 262079 275157
Amostra 3 204717 243821 263661 271124
Média 206231,00 243033,00 259885,33 271579,00
Desvio-padrão 1360,38 669,69 4270,08 2754,53

71
Tabela 23 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 150000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C.
Condutividades das salmouras características do Pré-sal
brasileiro (µS/cm) a 150000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 179348 207890 230627 253754
Amostra 2 176675 209132 235344 254741
Amostra 3 177660 208184 221899 250538
Média 177894,33 208402,00 229290,00 253011,00
Desvio-padrão 1103,76 529,96 5569,72 1794,50
Tabela 24 - Resultados experimentais das condutividades das três amostras das salmouras
características do Pré-sal brasileiro para a concentração de 100000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C.
Condutividades das salmouras características do Pré-sal
brasileiro (µS/cm) a 100000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 132045 147077 159546 169990
Amostra 2 127045 149748 162946 174725
Amostra 3 134112 151190 161564 177381
Média 131067,33 149338,33 161352,00 174032,00
Desvio-padrão 2966,76 1703,93 1396,12 3056,89
Tabela 25 - Resultados experimentais das condutividades das três amostras das salmouras características do Pré-sal brasileiro para a concentração de 50000 ppm medidas nas
temperaturas de 25, 40, 55 e 70 °C.
Condutividades das salmouras características do Pré-sal
brasileiro (µS/cm) a 50000 ppm
25 °C 40 °C 55 °C 70 °C
Amostra 1 75471,1 86674,6 92888,7 102155,0
Amostra 2 69270,5 83280,6 85540,4 89601,4
Amostra 3 73205,8 85597,6 90101,7 92346
Média 72649,13 85184,27 89510,27 94700,80
Desvio-padrão 2561,81 1416,08 3028,94 5388,69

72
Analisando-se as tabelas mostradas acima, percebe-se, similarmente ao caso das
amostras de soluções de NaCl, que os valores calculados dos desvios-padrão de cada
grupo de três amostras com a mesma configuração de medição são inferiores às diferenças
das médias de condutividade entre duas temperaturas de medição consecutivas. Sendo
assim, também se constata neste caso que as variações de condutividade com a
temperatura são maiores do que os erros experimentais previstos na preparação e na
medição das amostras, o que garante, novamente, a repetitividade deste método para fins
de caracterização do comportamento de fluidos.
A Figura 33 apresenta graficamente, a partir dos dados das tabelas acima, como
as condutividades médias das amostras com diferentes concentrações variam com a
temperatura.
Figura 33 - Gráfico da condutividade versus a temperatura para amostras com diferentes
concentrações.
Nota-se, com isso, que a dependência da condutividade com a temperatura para
diferentes concentrações pode ser ajustada por retas com um baixo desvio em relação à
linearidade, dado que o menor valor de 𝑅2 obtido foi de 0,9308, como mostra a Tabela
26.
50000,00
100000,00
150000,00
200000,00
250000,00
300000,00
350000,00
15 20 25 30 35 40 45 50 55 60 65 70 75 80 85
Co
nd
uti
vid
ade
(µS/
cm)
Temperatura (°C)
Condutividade (µS/cm) vs. temperatura (°C)
226363 ppm
215000 ppm
200000 ppm
150000 ppm
100000 ppm
50000 ppm

73
Tabela 26 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 33.
Concentração das
amostras (ppm)
Correlações obtidas pelos
ajustes lineares
𝑹𝟐 dos ajustes
lineares
226363 𝜅 = 1436,6𝑇 + 197272 (42) 𝑅2 = 0,9818
215000 𝜅 = 1462,5𝑇 + 186782 (43) 𝑅2 = 0,9681
200000 𝜅 = 1419,3𝑇 + 177765 (44) 𝑅2 = 0,9308
150000 𝜅 = 1641,6𝑇 + 139174 (45) 𝑅2 = 0,9937
100000 𝜅 = 939,38𝑇 + 109327 (46) 𝑅2 = 0,9898
50000 𝜅 = 469,87𝑇 + 63192 (47) 𝑅2 = 0,9338
A Figura 34, de forma complementar à Figura 33, exibe graficamente como as
condutividades médias das amostras com diferentes temperaturas variam com a
concentração.
Figura 34 - Gráfico da condutividade versus a concentração para amostras medidas em
diferentes temperaturas.
Constata-se também que, por sua vez, a dependência da condutividade com a
concentração para diferentes temperaturas pode, analogamente, ser muito bem ajustada
por retas, já que o menor valor de 𝑅2 obtido foi de 0,9540, conforme mostra a Tabela 27.
0
50000
100000
150000
200000
250000
300000
350000
0 50000 100000 150000 200000 250000
Co
nd
uti
vid
ade
(µS/
cm)
Concentração (ppm)
Condutividade (µS/cm) vs. concentração (ppm)
25 °C
40 °C
55 °C
70 °C

74
Tabela 27 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 34.
Temperaturas das
amostras (°C)
Correlações obtidas pelos
ajustes lineares
𝑹𝟐 dos ajustes
lineares
25 𝜅 = 0,8536𝐶 + 38880 (48) 𝑅2 = 0,9850
40 𝜅 = 0,9769𝐶 + 46550 (49) 𝑅2 = 0,9819
55 𝜅 = 1,0250𝐶 + 52583 (50) 𝑅2 = 0,9655
70 𝜅 = 1,1019𝐶 + 56803 (51) 𝑅2 = 0,9540
No gráfico da Figura 35 a seguir inverteram-se os eixos do gráfico da Figura 34,
já que o objetivo deste trabalho é obter uma correlação para o TDS em função da
condutividade das amostras de salmouras. Além disso, como as salmouras analisadas não
possuem sólidos em suspensão (TSS = 0), o TS (Total Solids) resume-se ao TDS e, assim,
pode-se considerar o eixo das ordenadas como sendo diretamente o TDS, em vez da
concentração.
Figura 35 - Gráfico do TDS versus a concentração para amostras medidas em diferentes temperaturas.
Naturalmente, pode-se isolar a concentração (ou o TDS) das Equações (48) a (51),
obtendo-se as Equações (52) a (55) expostas na Tabela 28.
0
50000
100000
150000
200000
250000
0 50000 100000 150000 200000 250000 300000 350000
TDS
(pp
m)
Condutividade (µS/cm)
TDS (ppm) vs. condutividade (µS/cm)
25 °C
40 °C
55 °C
70 °C

75
Tabela 28 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 35.
Temperaturas das
amostras (°C)
Correlações obtidas pelos ajustes
lineares 𝑹𝟐 dos ajustes lineares
25 𝑇𝐷𝑆 = 1,1540𝜅 − 42519 (52) 𝑅2 = 0,9850
40 𝑇𝐷𝑆 = 1,0051𝜅 − 43941 (53) 𝑅2 = 0,9819
55 𝑇𝐷𝑆 = 0,9420𝜅 − 44122 (54) 𝑅2 = 0,9655
70 𝑇𝐷𝑆 = 0,8657𝜅 − 41950 (55) 𝑅2 = 0,9540
Com base nos coeficientes angulares obtidos nos ajustes lineares das equações de
(42) a (47) da Tabela 26 e das condutividades médias medidas a 25 °C apresentadas nas
Tabelas de 20 a 25, foi possível, por meio da Equação (16), calcular os coeficientes de
temperatura 𝛼𝑐𝑜𝑛𝑐.𝑖 para as amostras com diferentes concentrações. A Tabela 29 a seguir
exibe os resultados desses cálculos.
Tabela 29 - Valores do coeficiente 𝛼𝑐𝑜𝑛𝑐.𝑖 para cada concentração.
Concentração (ppm) Coeficiente 𝜶𝒄𝒐𝒏𝒄.𝒊 (%/°C)
230000 0,6243
215000 0,6682
200000 0,6881
150000 0,9225
100000 0,7167
50000 0,6468
𝜶𝒎é𝒅𝒊𝒐 0,7111
Similarmente ao procedimento descrito no caso das amostras de soluções de NaCl,
calculou-se o coeficiente de variação 𝐶𝑣 da população de valores de 𝛼𝑐𝑜𝑛𝑐.𝑖 por meio das
Equações (39) e (40) a fim de verificar se o valor médio do coeficiente de temperatura
(𝛼𝑚é𝑑𝑖𝑜) pode representar adequadamente os demais.
Nesse caso, o valor encontrado para 𝐶𝑣 foi de 13,92%, todavia, para analisar com
mais profundidade a capacidade do coeficiente de temperatura médio em representar
apropriadamente os demais 𝛼𝑐𝑜𝑛𝑐.𝑖, foram calculados os fatores TDS por meio de 𝛼𝑚é𝑑𝑖𝑜
com o propósito de verificar os erros relativos por ele fornecidos em relação aos valores
obtidos experimentalmente. Para tanto, a Tabela 30 apresenta os valores experimentais

76
para os fatores TDS calculados pela Equação (26), enquanto que a Tabela 31 fornece os
valores dos fatores TDS calculados para as temperaturas de 40, 55 e 70 °C utilizando-se
𝛼𝑚é𝑑𝑖𝑜 para corrigir os valores de condutividade medidos a 25 °C, de acordo com a
Equação (41).
Tabela 30 - Fatores TDS experimentais para diferentes concentrações e temperaturas das
amostras de salmouras características do Pré-sal brasileiro.
Fatores TDS experimentais
TDS (ppm) 25 °C 40 °C 55 °C 70 °C
226363 0,9837 0,8699 0,8246 0,7617
215000 0,9823 0,8509 0,8089 0,7475
200000 0,9698 0,8229 0,7696 0,7364
150000 0,8432 0,7198 0,6542 0,5929
100000 0,7630 0,6696 0,6198 0,5746
50000 0,6882 0,5870 0,5586 0,5280
Tabela 31 - Fatores TDS calculados para 40, 55 e 70°C utilizando-se αmédio para corrigir os valores de condutividade medidos a 25°C das amostras de salmouras características do Pré-sal
brasileiro.
Fatores TDS calculados utilizando-se 𝛂𝐦é𝐝𝐢𝐨
TDS (ppm) 25 °C 40 °C 55 °C 70 °C
226363 - 0,8889 0,8107 0,7452
215000 - 0,8876 0,8096 0,7442
200000 - 0,8763 0,7993 0,7347
150000 - 0,7619 0,6949 0,6388
100000 - 0,6894 0,6288 0,5780
50000 - 0,6219 0,5672 0,5214
A Tabela 32 a seguir expõe os erros relativos (%) obtidos entre os fatores TDS
experimentais e os calculados por meio de 𝛼𝑚é𝑑𝑖𝑜 a partir dos valores de condutividade
medidos a 25 °C, em que se percebe que o maior erro relativo obtido foi de 7,75% e o
erro relativo médio total foi de 3,06%. Sendo assim, é possível adotar um único valor
médio para o coeficiente de temperatura a fim de representar os demais 𝛼𝑐𝑜𝑛𝑐.𝑖, uma vez
que há uma fraca dependência de 𝛼 com a concentração (ou o TDS).

77
Tabela 32 - Erros relativos (%) entre os fatores TDS experimentais e os calculados exibidos nas
Tabelas 30 e 31, respectivamente.
Erros relativos (%) entre os fatores TDS experimentais e os
calculados
TDS (ppm) 25 °C 40 °C 55 °C 70 °C
230000 - 2,18 1,68 2,16
215000 - 4,32 0,09 0,45
200000 - 6,49 3,86 0,24
150000 - 5,86 6,23 7,75
100000 - 2,96 1,46 0,59
50000 - 5,95 1,55 1,25
Erro relativo médio total = 3,06%
A Figura 36 a seguir apresenta graficamente os resultados experimentais para os
fatores TDS mostrados na Tabela 30, em que a proximidade das retas ajustadas é coerente
com a adoção de único valor médio para o coeficiente de temperatura com a finalidade
de representar satisfatoriamente os demais.
Figura 36 - Gráfico dos Fatores TDS experimentais versus a concentração (ppm) para diferentes temperaturas.
0,4000
0,5000
0,6000
0,7000
0,8000
0,9000
1,0000
1,1000
1,2000
0 50000 100000 150000 200000 250000
TDS
fact
or
Concentração/TDS (ppm)
Fatores TDS experimentais
25 °C
40 °C
55 °C
70 °C

78
5.3. Simulações Computacionais no Programa OLI
A Tabela 33 a seguir expõe os resultados das simulações de condutividade no
programa OLI da salmoura característica do Pré-sal brasileiro para as mesmas
configurações de medição experimentais.
Tabela 33 - Resultados simulados das condutividades para a salmoura característica do Pré-sal
brasileiro para as mesmas configurações de medição experimentais.
Condutividades simuladas (µS/cm) por concentração e temperatura da
salmoura característica do Pré-sal brasileiro
T (°C) 226363
ppm
215000
ppm
200000
ppm
150000
ppm
100000
ppm
50000
ppm
25 222894,00 217603,00 210752,00 179103,00 134179,00 75835,40
40 288126,00 281730,00 272813,00 231772,00 173691,00 98299,70
55 358552,00 351007,00 339615,00 287866,00 215377,00 121876,00
70 431564,00 422343,00 408178,00 344909,00 257484,00 145588,00
A Figura 37 exibe graficamente, a partir dos dados da Tabela 33, como as
condutividades simuladas em diferentes concentrações variam com a temperatura.
Figura 37 - Gráfico da condutividade versus a temperatura para a salmoura característica do Pré-
sal brasileiro simulada para diferentes concentrações.
0,00
50000,00
100000,00
150000,00
200000,00
250000,00
300000,00
350000,00
400000,00
450000,00
500000,00
15 25 35 45 55 65 75 85
Co
nd
uti
vid
ade
(µS/
cm)
Temperatura (°C)
Condutividade (µS/cm) vs. temperatura (°C)
226363 ppm
215000 ppm
200000 ppm
150000 ppm
100000 ppm
50000 ppm

79
Similarmente ao que foi realizado para os resultados experimentais, a dependência
da condutividade com a temperatura para diferentes concentrações foi ajustada por retas
com baixíssimo desvio em relação à linearidade, uma vez que o menor valor de 𝑅2
encontrado foi de 0,9994, como mostra a Tabela 34.
Tabela 34 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 37.
Concentração das
amostras (ppm)
Correlações obtidas pelos
ajustes lineares
𝑹𝟐 dos ajustes
lineares
226363 𝜅 = 4642,9𝑇 + 104746 (56) 𝑅2 = 0,9994
215000 𝜅 = 4556,6𝑇 + 101730 (57) 𝑅2 = 0,9994
200000 𝜅 = 4393,9𝑇 + 99131 (58) 𝑅2 = 0,9995
150000 𝜅 = 3690,1𝑇 + 85634 (59) 𝑅2 = 0,9997
100000 𝜅 = 2744,0𝑇 + 64842 (60) 𝑅2 = 0,9998
50000 𝜅 = 1552,2𝑇 + 36669 (61) 𝑅2 = 0,9998
A Figura 38 também apresenta graficamente os dados da Tabela 33, porém, desta
vez, evidenciando como o TDS varia com as condutividades simuladas em diferentes
temperaturas.
Figura 38 - Gráfico do TDS versus as condutividades simuladas para diferentes temperaturas
para a salmoura característica do Pré-sal brasileiro.
0
50000
100000
150000
200000
250000
0 50000 100000 150000 200000 250000 300000 350000 400000 450000 500000
TDS
(pp
m)
Condutividade (µS/cm)
TDS (ppm) vs. condutividade (µS/cm)
25 °C
40 °C
55 °C
70 °C

80
As correlações do TDS com as condutividades simuladas para diferentes
temperaturas também foram obtidas por meio de ajustes lineares dos dados da Figura 38
com alta precisão, já que o menor valor de 𝑅2 encontrado foi de 0,9789, como mostra a
Tabela 35.
Tabela 35 - Correlações obtidas pelos ajustes lineares dos dados do gráfico da Figura 38.
Temperaturas das
amostras (°C)
Correlações obtidas pelos ajustes
lineares 𝑹𝟐 dos ajustes lineares
25 𝑇𝐷𝑆 = 1,1983𝜅 − 50892 (62) 𝑅2 = 0,9790
40 𝑇𝐷𝑆 = 0,9270𝜅 − 51130 (63) 𝑅2 = 0,9789
55 𝑇𝐷𝑆 = 0,7426𝜅 − 50338 (64) 𝑅2 = 0,9797
70 𝑇𝐷𝑆 = 0,6148𝜅 − 49070 (65) 𝑅2 = 0,9810
A partir dos coeficientes angulares obtidos nas retas ajustadas das equações de
(56) a (61) da Tabela 34 e das condutividades simuladas a 25 °C mostradas na Tabela 33,
foi possível, por meio da Equação (16), calcular os coeficientes de temperatura 𝛼𝑐𝑜𝑛𝑐.𝑖
para para as amostras com diferentes concentrações. A Tabela 36 abaixo apresenta os
resultados desses cálculos, em que os valores encontrados são próximos de 2%, o que
corresponde ao valor esperado para 𝛼 conforme foi descrito na Subseção 3.1.2.2.
Tabela 36 - Valores do coeficiente 𝛼𝑐𝑜𝑛𝑐.𝑖 para cada concentração.
Concentração (ppm) Coeficiente 𝜶𝒄𝒐𝒏𝒄.𝒊 (%/°C)
230000 2,0830
215000 2,0940
200000 2,0849
150000 2,0603
100000 2,0450
50000 2,0468
𝜶𝒎é𝒅𝒊𝒐 2,0690
Analogamente ao procedimento descrito para os resultados experimentais,
calculou-se o coeficiente de variação 𝐶𝑣 da população de valores de 𝛼𝑐𝑜𝑛𝑐.𝑖 por meio das
Equações (39) e (40) com o objetivo de verificar se o valor médio do coeficiente de

81
temperatura (𝛼𝑚é𝑑𝑖𝑜) pode representar adequadamente os demais. Nesse caso, o valor
encontrado para 𝐶𝑣 foi de 0,93%, que, por ser um valor muito baixo, garante a validade
de 𝛼𝑚é𝑑𝑖𝑜 para descrever a variação de temperatura em todas as concentrações.
5.4. Análises Comparativas
Conforme foi mencionado na teoria de condutometria direta explicada na
Subseção 3.1.2.3, a elevação da condutividade com a concentração é decorrente do
incremento do número de íons por unidade de volume da solução. No caso dos eletrólitos
fortes, o número de íons por unidade de volume aumenta na proporção da concentração,
contudo, nas soluções mais concentradas, a não linearidade geralmente observada no
aumento da condutividade com a concentração é oriunda das interações iônicas. No
entanto, tanto para as amostras das soluções de NaCl quanto para aquelas das salmouras
características do Pré-sal brasileiro, que são sistemas compostos majoritariamente por
eletrólitos fortes, predominou uma forte tendência linear dos dados experimentais do TDS
com a concentração, havendo apenas um desvio da linearidade levemente mais
pronunciado para as concentrações mais altas.
Vale destacar ainda que, no primeiro sistema existia apenas um tipo de eletrólito
enquanto que no segundo havia diversos tipos. Embora essa diferença de composição não
tenha afetado a tendência linear observada no comportamento da condutividade, foram
medidos valores maiores para essa propriedade no caso das amostras de salmouras com
mais de um tipo de sal em comparação às amostras de soluções contendo apenas NaCl,
com erros relativos de até 30% entre esses sistemas aquosos, ainda que ambos tivessem
sido avaliados nas mesmas concentrações1 e temperaturas, como se pode verificar na
Tabela 37. Conforme foi discutido na Seção 3.3, tal fato comprova a importância de serem
desenvolvidas correlações para o TDS específicas para cada solução em estudo com uma
composição em particular, a fim de se evitarem resultados imprecisos.
1 Rigorosamente, houve uma pequena diferença no valor da maior concentração de comparação, que foi de
230000 ppm para as soluções de NaCl e 226363 ppm para as salmouras características do Pré-sal brasileiro.
No entanto, essa diferença não resultou em um prejuízo significativo para a elaboração das interpretações
dos dados. As demais concentrações de comparação foram precisamente iguais.

82
Tabela 37 - Erro relativo (%) entre as condutividades experimentais das soluções de NaCl e da
salmoura característica do Pré-sal brasileiro.
Erro relativo (%) entre as condutividades experimentais das soluções
de NaCl e das salmouras características do Pré-sal brasileiro
T (°C) 226363
ppm
215000
ppm
200000
ppm
150000
ppm
100000
ppm
50000
ppm
25 8,72 6,95 4,19 4,74 1,01 2,84
40 15,72 16,95 15,56 17,36 12,37 10,25
55 19,72 19,05 20,28 23,89 18,16 13,31
70 24,01 24,13 22,56 30,03 22,73 16,68
Quanto à resposta da condutividade em relação à temperatura, as amostras de
soluções de NaCl apresentaram um coeficiente 𝛼𝑚é𝑑𝑖𝑜 de 0,1248 %/°C, enquanto que as
amostras das salmouras características do Pré-sal brasileiro resultaram em um valor
médio de 0,7111 %/°C. Essa diferença resulta do fato de que os coeficientes angulares
das retas ajustadas nos gráficos de condutividade versus temperatura são maiores no caso
da salmoura do Pré-sal em comparação às soluções de NaCl, como se observa nos gráficos
das Figuras 30 e 33, respectivamente. Entretanto, ambos esses sistemas exibiram uma
dependência da condutividade com a temperatura inferior ao intervalo esperado de 1 a 2
%/°C mencionado na Subseção 3.1.2.2. Diante disso, em função do maior valor de 𝛼𝑚é𝑑𝑖𝑜
encontrado para as amostras das salmouras características do Pré-sal brasileiro, conclui-
se que a resposta da condutividade dessa salmoura é mais sensível à variação de
temperatura do que a da solução de NaCl. Esses resultados também reforçam a relevânica
de se representar adequadamente o sistema aquoso que se deseja caracterizar, visto que a
dependência da condutividade com a temperatura varia de acordo com a composição
química do sistema.
Na Tabela 38, foram calculados os erros relativos entre os valores médios das
condutividades experimentais e as simuladas no programa OLI para a salmoura
característica do Pré-sal. Nessa tabela, percebe-se que o erro relativo entre as
condutividades medidas e as simuladas para 25 °C não excede 5%, no entanto, observam-
se erros crescentes bastante sistemáticos ao longo de todas as concentrações conforme a
temperatura aumenta. Além dos usuais erros experimentais, tal fato pode ser justificado
majoritariamente pela dificuldade prática em se evitar a evaporação de água durante as

83
medições da condutividade em maiores temperaturas, apesar da utilização do vidro de
relógio no topo dos béqueres que continham as amostras.
Tabela 38 - Erro relativo (%) entre as condutividades experimentais e as simuladas para a
salmoura característica do Pré-sal brasileiro.
Erro relativo (%) entre as condutividades experimentais e as simuladas
para a salmoura característica do Pré-sal brasileiro
T (°C) 226363
ppm
215000
ppm
200000
ppm
150000
ppm
100000
ppm
50000
ppm
25 3,14 0,58 2,19 0,68 2,37 4,39
40 10,72 11,49 12,25 11,21 16,31 15,40
55 30,61 32,05 30,68 25,55 33,48 36,16
70 45,22 46,84 50,30 36,32 47,95 53,73
Portanto, em função do baixo erro relativo encontrado entre as medições e as
simulações para 25 °C, a Equação (52), proveniente de um ajuste linear e exibida
novamente abaixo, provou-se uma correlação experimental válida para a salmoura
característica do Pré-sal brasileiro avaliada a 25°C:
𝑇𝐷𝑆 = 1,1540𝜅25 °𝐶 − 42519 (66)
Além disso, em função dos erros sistemáticos evidenciados pelas simulações na
medição experimental da condutividade para as temperaturas mais elevadas, como
mostrado na Tabela 38, sugere-se o acoplamento do coeficiente de temperatura médio
obtido nas simulações computacionais (𝛼𝑚é𝑑𝑖𝑜 = 2,0690 %/°C) na Equação (66) acima a
fim de se poder corrigir para 25°C o valor da condutividade medida em outras
temperaturas, resultando na correlação expressa pela Equação (67) a seguir:
𝑇𝐷𝑆 = 1,1540𝜅𝑇
1 + 0,02069(T − 25)− 42519 (67)
onde 𝜅𝑇 é a condutividade da amostra medida a uma temperatura T.

84
5.5. Testes para Verificação da Correlação Experimental
Na Tabela 39 a seguir estão expostos os resultados dos testes experimentais
descritos na Subseção 4.3.3 para verificação da correlação dada pela Equação (66) para a
salmoura característica do Pré-sal brasileiro. Nessa tabela, observam-se que os erros
relativos entre o TDS experimental e o calculado foram baixos, o que fornece uma
comprovação adicional para a correlação encontrada.
Tabela 39 - Resultados dos testes experimentais para verificação da correlação experimental.
Amostras de
salmouras
TDS
experimental
(ppm)
Condutividade
(µS/cm) medida
a 25 °C
TDS calculado
pela correlação
(ppm)
Erro relativo
(%) entre o
TDS
experimental
e o calculado
Amostra de
teste 1 208000 207128 196506,71 5,53
Amostra de
teste 2 185008 199857 188115,98 1,68

85
6. CONCLUSÕES
O TDS é uma propriedade importante na caracterização de fluidos, bem como
possui diversas aplicações. A determinação do TDS de salmouras na indústria de petróleo
permite avaliar, de forma conjunta com outras condições, a possibilidade de formação de
incrustações de sais inorgânicos e de ocorrência de corrosão ao longo de todos os
ambientes que entram em contato com as salmouras, como, por exemplo, todos os dutos
de escoamento de petróleo. Outra importante aplicação é voltada para a análise da
estabilidade química de fluidos injetados, como polímeros e surfactantes utilizados como
métodos especiais de recuperação (EOR).
O método de determinação do TDS a partir de correlações com a condutividade
elétrica busca explorar o fato de que os íons em soluções eletrolíticas apresentam a
capacidade de conduzir corrente elétrica. No entanto, como cada tipo de íon gera uma
contribuição diferente para a condutividade total da solução, este método carece de
especificidade, de modo que a condutividade pode resultar em valores diferentes para
amostras de mesmas concentrações e temperaturas, mas com composições de íons
distintas. Dessa forma, tal conclusão impede a proposição de uma relação geral entre o
TDS e a condutividade para qualquer tipo de salmoura. Contudo, conforme foi
mencionado na Seção 3.3, alguns autores defendem, como forma de superar essa
limitação, que se desenvolvam correlações específicas para determinadas regiões de
composição razoavelmente constante e durante um certo momento no tempo que preserve
essa condição.
Com base nessa carência de especificidade inerente ao resultado da condutividade
total de uma solução com múltiplos eletrólitos mencionada na literatura, este trabalho
objetivou avaliar a influência da diferença de composição química entre uma solução
contendo apenas cloreto de sódio e uma salmoura mais complexa, característica do Pré-
sal brasileiro, na resposta de suas condutividades em face às variações de concentração e
temperatura. Primeiramente, concluiu-se que a diferença na quantidade de eletrólitos
distintos dissolvidos nas amostras de soluções de NaCl e de salmouras características do
Pré-sal brasileiro não provocou alteração na tendência linear encontrada no
comportamento da condutividade. No entanto, foram medidos valores maiores para essa
propriedade no caso das amostras de salmouras com mais de um tipo de sal dissolvido

86
em comparação às amostras de soluções contendo NaCl apenas, ainda que ambas
tivessem sido avaliadas nas mesmas concentrações e temperaturas. Os erros relativos
entre esses dois tipos de soluções foram de até 30%, como mostra a Tabela 37, o que
representa a primeira evidência no sentido de reforçar a importância de se representar
corretamente o sistema aquoso que se deseja caracterizar a fim de se evitarem erros
elevados na determinação de seu TDS.
Nesse sentido, outra importante evidência foi encontrada ao se analisar a
dependência da condutividade com a temperatura de cada sistema aquoso. As amostras
de soluções de cloreto de sódio apresentaram um coeficiente de temperatura médio
82,45% menor que o coeficiente médio das amostras das salmouras características do Pré-
sal brasileiro. Essa diferença resulta do fato de que os coeficientes angulares das retas
ajustadas nos gráficos de condutividade versus temperatura são maiores no caso da
salmoura do Pré-sal em comparação às soluções de NaCl, como se observa nos gráficos
das Figuras 30 e 33, respectivamente. Esse resultado mostra que a salmoura é mais
sensível à variação de temperatura do que a da solução de NaCl, conforme descrito na
Seção 5.4. Tal fato também reforça a importância de se especificar adequadamente o
sistema aquoso em estudo, visto que a resposta da condutividade em relação à temperatura
também sofre uma influência significativa da composição iônica da solução em análise.
Além disso, este projeto logrou êxito em encontrar uma correlação experimental
para o TDS em função da condutividade elétrica medida a 25°C de amostras de uma
salmoura característica do Pré-sal brasileiro com composição igual à mostrada na Tabela
4. Essa correlação, expressa pela Equação (66), foi validada por meio de simulações
computacionais e também por testes experimentais de verificação. Entretanto, para as
demais temperaturas avaliadas neste trabalho, foram encontrados erros sistemáticos nos
valores de condutividade medidos. Esses erros podem ser explicados pela grande
dificuldade de se evitar a perda de água por evaporação durante as medições. Tal fato
também se refletiu na disparidade de valores entre o coeficiente de temperatura médio
obtido para as soluções experimentais e o resultante das simulações computacionais.
6.1. Propostas para Trabalhos Futuros
Como propostas para trabalhos futuros, baseado no que foi tratado anteriormente
e nos resultados deste trabalho, pode-se propor uma metodologia experimental padrão

87
para se desenvolverem correlações do TDS com a condutividade elétrica de uma salmoura
com uma composição específica. Tal metodologia consiste em primeiramente preparar
amostras com uma composição idêntica à do sistema aquoso que se deseja caracterizar.
Em seguida, deve-se medir a condutividade dessas amostras para uma faixa de
concentrações e de temperaturas de interesse. Para isso, sugere-se a utilização de mais de
três amostras para as mesmas configurações de medição, o que minimizaria ainda mais
os possíveis erros experimentais ao se utilizarem os resultados médios desses grupos de
amostras para as análises subsequentes. Ademais, seria de grande valia reduzir a distância
entre os pontos experimentais, pois, com isso, a maior quantidade de dados experimentais
contribuiria para o desenvolvimento de correlações mais precisas e tendências ainda mais
claras. Vale ressaltar ainda a necessidade primordial de se controlar rigorosamente a
evaporação de água ao longo dos processos de aquecimento e medição a fim de se evitar
alterações indesejadas na concentração das amostras, o que levaria à produção de
resultados que não sejam fidedignos da realidade.
Por fim, os dados experimentais obtidos devem ser ajustados preferencialmente
por retas a fim de serem geradas correlações da condutividade com a variação de
concentração, mantendo-se a temperatura fixa. Já as correlações da condutividade com a
variação de temperatura, mantendo-se a concentração fixa, devem ser utilizadas para se
determinar o coeficiente de temperatura para cada concentração e, em seguida, analisar
estatisticamente se é possível adotar um único valor médio que represente adequadamente
os demais. Dessa maneira, pode-se determinar o grau de dependência da condutividade
com a concentração e com a temperatura e, assim, conseguir desenvolver correlações para
o TDS que permitam estimar o valor dessa propriedade para outras condições dentro das
faixas consideradas para a solução em estudo. Para garantir a validação dos resultados
obtidos, sugere-se a utilização de um programa computacional apropriado para serem
simuladas as mesmas condições experimentais e, com isso, ser possível comparar os
resultados. Além disso, caso os erros experimentais se mostrem significativos,
recomenda-se utilizar o coeficiente de temperatura obtido nas simulações computacionais
a fim de se corrigir o valor da condutividade da amostra em função da temperatura na
qual foi medida, conforme foi feito neste trabalho.
Portanto, cumprindo-se essa metodologia padrão descrita acima e garantindo que
os desvios-padrão das condutividades de cada grupo de amostras com a mesma
configuração de medição sejam inferiores às diferenças das médias entre duas

88
temperaturas de medição consecutivas, estabelece-se um método confiável e passível de
ser repetido em laboratórios de caracterização de fluidos. Além disso, este método de
correlação do TDS com a condutividade elétrica exibe a vantagem de ser mais ágil do que
a tradicional gravimetria, já que, uma vez desenvolvidas as correlações para um
determinado sistema aquoso, pode-se calcular os valores de seu TDS a partir da medição
da condutividade em diferentes concentrações e temperaturas dentro das faixas
consideradas sem a necessidade de novas etapas laboratoriais.

89
REFERÊNCIAS BIBLIOGRÁFICAS
AFFONSO, B. F. & ANDRADE, C. P. C., 2017. Estudo sobre corrosão e incrustação
inorgânica na indústria de petróleo com ênfase no desenvolvimento de um combo
comercial para tratamento químico, Niteroi, RJ: s.n.
ALEXANDER, C. K. & SADIKU, M. N. O., 2013. Fundamentos de Circuitos Elétricos.
5ª ed. Porto Alegre: AMGH .
ANDRADE, F. P., 2016. "Atividade de um Soluto", Piracicaba: s.n.
ARAL, A. & DUARTE, L. R., 2010. Estudo da formação de incrustações carbonáticas,
Rio de Janeiro: s.n.
CARLSON, G., 2005. "Total Dissolved Solids from conductivity". Technical Support.
COLLINS, A. G., 1975. Geochemistry of Oilfield Waters. 1ª ed. Bartlesville(Oklahoma):
Elsevier.
FERNANDES, D. M. d. S., 2014. Análise Gravimétrica, Quixeramobim: Instituto
Federal de Eduação, Ciência e Tecnologia do Ceará.
FERREIRA, C. D. A. & CARDOSO, L. F., 2011. Estudo da Garantia de Escoamento em
um Campo de Óleo Pesado em Lâmina d'água Rasa, Rio de Janeiro: s.n.
FERREIRA, V. J. M. F., 2014. Gestão de Operações e Logística na Produção de
Petróleo: Fundamentos, Metodologia e Modelos Quantitativos. 1ª ed. s.l.:Campus.
FERREIRA, V. M., 2016. Avaliação de Métodos de Recuperação Melhorada de Petróleo
para Campos Marítimos no Brasil: O Caso da Bacia de Campos, Rio de Janeiro: s.n.
FRAUCHES-SANTOS, C., ALBUQUERQUE, M. A., OLIVEIRA, M. C. C. &
ECHEVARRI, A., 2014. "A Corrosão e os Agentes Anticorrosivos". Revista Virtual de
Química, 6(2), pp. 293-309.
FREIRE, A. S. & FLORIDO, W. L., 2016. Controle da Corrosão nos Dutos da Indústria
do Petróleo, Campos dos Goytacazes: s.n.
GENTIL, V., 2007. Corrosão. 3ª ed. Rio de Janeiro: LTC.

90
HUBERT, E. & WOLKERSDORFER, C., 2014. "Establishing a conversion factor
between electrical conductivity and total dissolved solids in South African mine waters".
Water SA, 41(4), pp. 490-500.
HUNTER, M. A., 1993. "Alkalinity and phosphonate studies for scale prediction and
prevention", Houston: s.n.
MARANDI, A., POLIKARPUS, M. & JÕELEHT, A., 2013. "A new approach for
describing the relationship between electrical conductivity and major anion concentration
in natural waters". Applied Geochemistry, Issue 38, pp. 103-109.
MARTÍNEZ, L., 2017. Condutividade de Soluções Iônicas, Campinas: s.n.
MURITO, M. M. C. & FINETE, V. d. L. M., 2009. "Capítulo 5 - Fundamentos em
química experimental". Em: Conceitos e Métodos para a Formação de Profissionais em
Laboratórios de Saúde. s.l.:IOC - Instituto Oswaldo Cruz, pp. 242-248.
NETZ, P. A. & ORTEGA, G. G., 2002. Fundamentos de Físico-Química: Uma
abordagem conceitual para as ciências farmacêuticas. 1ª ed. s.l.:ARTMED.
OGDEN, B. L., 2008. "Water Technology: Understanding, Interpreting and Utilizing
Water Anaysis Data". Water Technology SWPSC.
OHLWEILLER, O., 1981. "Capítulo13 - Condutometria". Em: Fundamentos de Análise
Instrumental. São Paulo: Livros Técnicos e Científicos, pp. 327-345.
OLI Systems, Inc., 2011. OLI Studio Stream Analyzer User Guide v9.5. [Online]
Available at:
http://wiki.olisystems.com/wiki/OLI_Studio_Stream_Analyzer_User_Guide_v9.5
[Acesso em 02 Agosto 2018].
PETROWIKI, 2016. Dissolved constituents in produced water. [Online]
Available at:
https://petrowiki.org/Dissolved_constituents_in_produced_water#cite_note-r6-6
[Acesso em 2018 Agosto 2018].
PILLING, S., s.d. "Determinação da Condutividade de Eletrólitos Fortes e Fracos da
Constante de Dissociação de Ácidos Fracos", São José dos Campos: s.n.

91
RADIOMETER ANALYTICAL, S., 2009. Conductivity Theory and Practice,
Villeurbanne Cedex, France: s.n.
RHOADES, J., KANDIAH, A. & MASHALI, A. M., 1992. "The use of salirle waters for
crop production". Irrigation and Drainage Paper.
RIOS, V. d. S., 2014. Recuperação de Óleo por Injeção de Polímeros - Abordagens
Experimental, Analítica e Numérica em Pequena Escala, Campinas: s.n.
RODRIGUES, M. O., s.d. Fundamentos de Ligação Química e Ligação Iônica, s.l.: s.n.
RUSYDI, A. F., 2018. "Correlation between conductivity and total dissolved solid in
various type of water: A review". IOP Conf. Series: Earth and Environmental Science,
118(012019).
SALAS, M. K. S., 2014. Estudo da Injeção de Água na Recuperação Melhorada de
Petróleo: Efeito da Salinidade e Surfactante, Rio de Janeiro: s.n.
SANABRIA, F. C. B., 2013. Avaliação da Injeção de Surfactantes como Método de
Recuperação Avançada em Reservatórios de Arenito, Campinas: s.n.
SANTANA, C. J. & MANZELA, A. A., 2016. "Incrustações Inorgânicas em Campos do
Pré-Sal". Revista de Engenharias da Faculdade Salesiana, Issue n.4, pp. 22-31.
SARTORELLI, R., 2015. Boas Práticas em Medição de Condutividade, s.l.: Mettler e
Toledo.
SHENG, J. P., 2010. Modern Chemical Enhanced Oil Recovery: Theory and Practice.
1ª ed. s.l.: Gulf Professional Publishing.
SIOSEMARDE, M. et al., 2010. "Determine of Constant Coefficients to Relate Total
Dissolved Solids to Electrical Conductivity". International Journal of Environmental,
Chemical, Ecological, Geological and Geophysical Engineering, 4(10).
SKOOG, D. A., WEST, D. M., HOLLER, F. J. & CROUCH, S. R., 2005. "Parte IV -
Métodos Eletroquímicos". Em: Fundamentos da Química Analítica. 8ª ed. São Paulo:
THOMSON, pp. 606-608.
SKOOG, WEST, HOLLER & CROUCH, 2005. "Parte III - Métodos Clássicos de
Analíse". Em: D. A. Skoog, ed. Fundamentos de Química Analítica. 8ª ed. São Paulo:
Thomson, pp. 294-320.

92
TÁVORA, M. P., 2007. Avaliação do Efeito de Particulados Sólidos na Eficiência de um
Inibidor de Corrosão Recomendado para Meios Salinos com CO2, Natal, RN: s.n.
TEIXEIRA, H. M. F., 2007. Desenvolvimento e aplicação de metodologias para
caracterização multielementar de água conata em amostras de petróleo, Rio de Janeiro:
s.n.
THIRUMALINI, S. & JOSEPH, K., 2009. "Correlation between Electrical Conductivity
and Total Dissolved Solids in Natural Waters". Malaysian Journal of Science, 28(1), pp.
55-61.
TODD, D. K. & MAYS, L. W., 2005. Groundwater Hydrology. 3ª ed. New Jersey: John
Wiley & Sons, Inc..
VASCONCELOS, J. S. & BROSEGUINI, M., 2013. Estudo de Caso da Corrosão em
Tubulação de Aço Carbono Provocada por Água Doce, Vitória, ES: s.n.
VIDAL, L. A., 2015. Estudo sobre as incrustações inorgânicas nos campos de petróleo,
Niterói, RJ: s.n.
WALTON, N., 1988. Electrical Conductivity and Total Dissolved Solids-What is Their
Precise Relationship?. Elsevier Science Publishers B.V., 25 Julho, Volume 12, pp. 4-5.
WARD, J. S. M. F. D., 1981. "Prediction of Viscosity for Partially Hydrolyzed
Polyacrylamide Solution in the Presence od Calcium and Magnesium Ion". SPE Journal,
21(5).
WEBB, P. J. & KUHN, O., 2004. "Enhanced Scale Management through the Application
of Inorganic Geochemistry and Statistics". SPE International Symposium on Oilfield
Scale, Volume SPE-87458-MS .


















