
D. Régulation des protéines tyrosines kinases
151
VETAGRO SUP CAMPUS VETERINAIRE DE LYON Année 2011 - Thèse n° LES INHIBITEURS DE TYROSINES KINASES EN CANCEROLOGIE VETERINAIRE : ACTUALITE CHEZ LE CHIEN ET COMPARAISON AVEC L’HOMME THESE Présentée à VETAGRO SUP, CAMPUS VETERINAIRE DE LYON et soutenue publiquement le 01 juillet 2011 pour obtenir le grade de Docteur Vétérinaire par Cindy MORGANT Née le 01 juin 1985 à Valence (26)
Transcript of D. Régulation des protéines tyrosines kinases

VETAGRO SUP CAMPUS VETERINAIRE DE LYON
Année 2011 - Thèse n°
LES INHIBITEURS DE TYROSINES KINASES EN
CANCEROLOGIE VETERINAIRE : ACTUALITE CHEZ LE
CHIEN ET COMPARAISON AVEC L’HOMME
THESE
Présentée à VETAGRO SUP, CAMPUS VETERINAIRE DE LYON
et soutenue publiquement le 01 juillet 2011
pour obtenir le grade de Docteur Vétérinaire
par
Cindy MORGANT Née le 01 juin 1985
à Valence (26)

1
VETAGRO SUP CAMPUS VETERINAIRE DE LYON
Année 2011 - Thèse n°
LES INHIBITEURS DE TYROSINES KINASES EN
CANCEROLOGIE VETERINAIRE : ACTUALITE CHEZ LE
CHIEN ET COMPARAISON AVEC L’HOMME
THESE
Présentée à VETAGRO SUP, CAMPUS VETERINAIRE DE LYON
et soutenue publiquement le 01 juillet 2011
pour obtenir le grade de Docteur Vétérinaire
par
Cindy MORGANT Née le 01 juin 1985
à Valence (26)

2
3
Département et corps enseignant du Campus Vétérinaire de Lyon
de VetAgro Sup
4

5
Remerciements
A Monsieur le Professeur Claude GHARIB,
De la Faculté de Médecine de Lyon,
Qui nous a fait l’immense honneur d’accepter la présidence de ce jury de thèse.
Hommages respectueux.
A Madame le Docteur Frédérique PONCE,
De VetAgro Sup, Campus vétérinaire de Lyon,
Qui m'a permis de mener à bien ce travail et m'a encadrée tout au long de son élaboration.
Veuillez trouver ici l'expression de ma profonde reconnaissance.
A Monsieur le Docteur Jean-Jacques THIEBAULT,
De VetAgro Sup, Campus vétérinaire de Lyon,
Pour accepter de juger ce travail et de faire partie de ce jury de thèse.
Veuillez trouver ici l'expression de ma sincère gratitude.
A Messieurs les Docteurs Tom CHAPUIS et Damien LEROUX,
Pour leurs avis éclairés sur le sujet et leurs conseils qui m’ont été d’une aide précieuse.
Sincères remerciements.

6

7
A mes parents, pour m’avoir « coachée » pendant toute ma scolarité de la maternelle à
l’école véto en passant par la prépa, pour votre soutien inconditionnel, votre amour et vos
conseils avisés qui me seront toujours utiles. Pour toutes les concessions que vous avez
toujours faites pour nous…Ce diplôme est un peu le vôtre aussi ! Je vous aime très fort
A Aurélie, ma « soeurette », la niçoise, parisienne et bientôt Chinoise ! Ta spontanéité et ta
folie font de toi quelqu’un d’exceptionnel ! Malgré nos différences et la distance qui nous
sépare, notre complicité reste inébranlable. Quand est-ce qu’on retourne vivre chez les
parents ??
A David, mon petit chou, devenu en peu de temps éleveur de Triops et dresseur de chien ! Ta
joie de vivre me manque tous les jours. Surtout ne change pas et arrête de grandir trop vite car
c’est bientôt toi qui va devoir m’apprendre les équations ! Je t’aime trrroooppp
A mes grand-mères, pour toutes leurs petites attentions et leurs bons petits plats ! Ca y est, je
suis enfin « vitrinaire » !
A Jocelyne et Robert, pour m’avoir appris que des vacances sans le soleil de Ste Maxime et
l’air de la montagne de Crest-Voland ne sont pas de vraies vacances ! Et parce qu’un week-
end sans vous voir n’est pas un vrai week-end ! Votre présence est toujours un plaisir.
A Cathy, sans qui je n’aurais peut-être pas réussi le concours ! Mes révisions à Nice, sur la
Croisette et à l’île de Ste Marguerite avaient un petit air de vacances ! Continue à profiter de
la vie comme tu sais si bien le faire !
A toute ma famille, parce que tous les instants passés ensemble sont un vrai bonheur !
A Maxime, pour ton humour dans les bons comme dans les mauvais moments, pour m’avoir
donné le courage de me lever le matin pendant plusieurs mois. Pour la vie ensemble qui nous
attend et parce qu’il n’y a rien de plus agréable que la simplicité du quotidien à deux.
Je t’aime
A Maryse, Gilles, Séverine, Lionel et Léa, pour votre accueil chaleureux au sein de votre
famille.
A Marie, pour tous ces bons moments passés ensemble à Châteauneuf : les jeux de pistes
dans le jardin, le grand tour à vélo, les commandes Yves Rocher et nos goûters devant les
Razmokets pour n’en citer que quelque uns. Ces souvenirs resteront gravés dans ma mémoire.
Tu comptes beaucoup pour moi.

8
A Héléna, parce que es et tu resteras ma meilleure amie. Pour nos souvenirs inoubliables à
Lyon, Marcy, en Ardèche, au Grau du Roi et à Parga ! Pour ton entrain en toute circonstance
et parce qu’avec toi je n’ai même pas honte d’écouter Skyrock…et A Toc, pour m’avoir fait
apprécier le footing de 7h du matin, qu’il pleuve ou qu’il vente…
A Océane, pour ton enthousiasme de tous les instants et ta motivation à toute épreuve ! Entre
les séances photos en maillot de bain, les promenades à la « rez », la descente de l’Ardèche en
canoë, et pleins d’autres moments, nos fous rires restent inoubliables ! Profite à fond de ton
tour du monde avec un ami… !
A Dorothée, pour notre complicité depuis la prépa ! Ca y est, la vraie vie va enfin
commencer et je ferais tout pour que l’on ne se perde pas de vue ! Je te souhaite beaucoup de
bonheur avec Kévin.
A Dinette, Marie Kir, Prachette et Liloute, pour tous nos bons moments passés ensemble
en clinique et en dehors ! Nos repas de groupe de clinique étaient tellement bien ! A quand le
prochain ??
A Fanny qui m’a sauvé un bounty que je croyais définitivement perdu…je n’oublierai jamais
ton geste ! Et Amandine, ça te dit une petite séance photo ? On est un groupe de clinique au
top !
A l’équipe « Villefontaine » (Céline, Maxime, Cha, Laëticia, Schnaps et Marion), pour
tous nos week-ends passés ensemble dans cette clinique à l’ambiance…si particulière !
A tous ceux qui ont su rendre ces 5 années d’école inoubliables : Laure, Roxanne,
Gaspard, Pioupiou, Perrine, …
A Idole, Nala, Simba et maintenant Snoopy, pour tous ses moments de bonheur qui me
manquent tant, et parce que c’est avec vous que j’ai commencé à apprendre mon métier !

9
Table des matières
Remerciements ......................................................................................................................... 5
Table des illustrations ............................................................................................................ 15
Liste des abréviations ............................................................................................................. 17
Introduction ............................................................................................................................ 19
I. Les protéines tyrosines kinases ................................................................................ 21
A. Structure et classification ............................................................................................. 21
1. Définition d’une protéine tyrosine kinase ................................................................ 21
2. Structure générale ...................................................................................................... 21
a. Structure ................................................................................................................... 21
b. Structure tertiaire ...................................................................................................... 24
c. Forme active et forme inactive ................................................................................. 25
3. Classification ............................................................................................................... 26
4. Exemple du récepteur tyrosine kinase c-Kit et de son ligand ................................ 30
a. Structure de la protéine c-Kit ................................................................................... 30 b. Les différentes formes de la protéine c-Kit .............................................................. 31
c. Etude du gène Kit ..................................................................................................... 32 d. Le ligand de c-Kit, le Stem Cell Factor (SCF) ......................................................... 33
B. Localisation des PTK : Exemple de la protéine c-Kit ................................................ 36
1. Localisation normale .................................................................................................. 36
2. Localisation tumorale ................................................................................................ 36
C. Mode d’action des protéines tyrosines kinases ........................................................... 37
1. Cas des récepteurs tyrosines kinases ........................................................................ 37
2. Cas des non récepteurs tyrosines kinases ................................................................. 40

10
D. Régulation des protéines tyrosines kinases ................................................................. 41
1. Régulation de la transcription du gène Kit .............................................................. 41
2. Régulation par des facteurs de croissance ............................................................... 41
3. Autorégulation via le domaine juxtamembranaire, la boucle d’activation et la
partie C-terminale .......................................................................................................... 41
4. Rôle des phosphatases ................................................................................................ 43
5. Autres mécanismes régulateurs ................................................................................ 43
E. Fonctions des protéines tyrosines kinases ................................................................... 44
1. Exemple de la protéine c-Kit ..................................................................................... 44
2. Exemple du VEGFR ................................................................................................... 44
3. Exemple du PDGFR ................................................................................................... 44
4. Exemple de la protéine Met (=HGFR) ..................................................................... 45
5. Exemple du EGFR ..................................................................................................... 45
II. Implication des protéines tyrosines kinases en cancérologie ....................... 47
A. Dysfonctionnement des protéines tyrosines kinases ................................................... 47
1. Surexpression d’un récepteur tyrosine kinase ......................................................... 48
2. Mutations du gène codant pour un récepteur tyrosine kinase ............................... 48
3. Boucle autocrine/paracrine ....................................................................................... 49
4. Formation de protéines fusions ................................................................................. 49
5. Expression inappropriée d’une kinase ..................................................................... 50
6. Conséquences : activation constitutive des tyrosines kinases………….…………50
B. Intérêt pronostique des tyrosines kinases .................................................................... 51
1. Chez le chien ............................................................................................................... 51
2. Chez l’homme ............................................................................................................. 53

11
C. Les facteurs impliqués dans l’angiogenèse : VEGFR, PDGFR, FGFR et Tie 1/2 ... 54
1. Le VEGFR .................................................................................................................. 54 a. VEGF et cancer du sein/tumeur mammaire ............................................................. 55
b. VEGF et mastocytose/mastocytome ........................................................................ 56
2. Le PDGFR ................................................................................................................... 56
3. Le FGFR ...................................................................................................................... 56
4. Le Tie 1/2 ..................................................................................................................... 56
D. Exemple de la protéine c-Kit ........................................................................................ 58
1. Protéine c-Kit et mastocytome/mastocytose ............................................................ 58
a. Mise en évidence de la présence de la protéine c-Kit dans les mastocytomes ......... 58 b. Méthode de détection des mutations de Kit ............................................................. 58 c. Les différentes mutations du gène Kit associées aux mastocytoses/mastocytomes . 60 d. Mise en évidence d’un hot spot et d’un polymorphisme du gène Kit ...................... 65
e. Conséquences de ces mutations ................................................................................ 65 f. Localisation anormale de la protéine c-Kit ............................................................... 65 g. Intérêt de la détection des mutations du gène Kit .................................................... 66
h. Intérêt diagnostique de la protéine c-Kit .................................................................. 66
2. Protéine c-Kit et GIST ............................................................................................... 67
3. Protéine c-Kit et tumeur mammaire/cancer du sein ............................................... 69
4. Protéine c-Kit et hémangiosarcome .......................................................................... 69
E. Exemple de la protéine Met (=HGFR) ......................................................................... 70
F. Exemple de la protéine PTEN....................................................................................... 72
G. Exemple de la protéine EGFR ..................................................................................... 73

12
III. Les inhibiteurs des tyrosines kinases .................................................................. 75
A. Les inhibiteurs de tyrosines kinases ayant une AMM chez le chien ......................... 76
1. Le Masivet® (masitinib) ............................................................................................ 76 a. Structure ................................................................................................................... 76
b. Cibles ........................................................................................................................ 76 c. Mode d’action ........................................................................................................... 78 d. Protocole ................................................................................................................... 80 e. Efficacité du masitinib sur les mastocytomes .......................................................... 80 f. Autres indications du masitinib en cancérologie ...................................................... 86
i. Chez le chien .......................................................................................................... 86
ii. Chez l’homme ....................................................................................................... 87
g. Indication du masitinib sur les maladies inflammatoires ......................................... 89
i. Chez le chien .......................................................................................................... 89
ii. Chez l’homme ....................................................................................................... 90
h. Indications du masitinib sur les maladies du système nerveux central chez l’homme
...................................................................................................................................... 92 i. La sclérose en plaques ............................................................................................ 92
ii. La maladie d’Alzheimer ........................................................................................ 93
i. Effets secondaires ...................................................................................................... 94 j. Récapitulatif des voies de développement du masitinib chez le chien et chez
l’homme ....................................................................................................................... 95
2. Le Palladia® (toceranib) ............................................................................................ 96
a. Structure ................................................................................................................... 96 b. Cibles ........................................................................................................................ 96 c. Mode d’action ........................................................................................................... 97
d. Protocole ................................................................................................................... 98 e. Efficacité du toceranib sur les mastocytomes .......................................................... 98
f. Influence du statut de c-Kit ..................................................................................... 101 g. Action du toceranib sur les autres cancers chez le chien ....................................... 102 h. Effets secondaires ................................................................................................... 103
B. Les inhibiteurs de tyrosines kinases ayant une AMM chez l’homme ..................... 104
1. Le Gleevec® (imatinib) ............................................................................................ 104 a. Structure ................................................................................................................. 104 b. Cibles ...................................................................................................................... 104 c. Mode d’action ......................................................................................................... 105
d. Protocole ................................................................................................................. 105 e. Efficacité de l’imatinib dans la leucémie myéloïde chronique .............................. 106 f. Efficacité de l’imatinib dans les GIST .................................................................... 107 g. Efficacité de l’imatinib dans les mastocytoses ....................................................... 107
h. Efficacité de l’imatinib dans les mastocytomes chez le chien ............................... 108 i. Autres cibles de l’imatinib chez l’homme .............................................................. 110 j. Effets secondaires .................................................................................................... 111
k. Résistance à l’imatinib ........................................................................................... 111

13
2. L’Iressa® (gefitinib) ................................................................................................. 113
3. Le Tarceva® (erlotinib) ........................................................................................... 113
4. Le Nexavar® (sorafenib) ......................................................................................... 113 a. Chez l’homme ........................................................................................................ 113 b. Chez le chien .......................................................................................................... 114
5. Le Sprycel® (dasatinib) ........................................................................................... 115
6. Le Sutent® (sunitinib) .............................................................................................. 116 a. Structure ................................................................................................................. 116
b. Cibles ...................................................................................................................... 116 c. Mode d’action ......................................................................................................... 117 d. Protocole ................................................................................................................. 118 e. Etude in vitro et in vivo sur des souris ................................................................... 118
f. Efficacité du sunitinib dans les GIST ..................................................................... 118 g. Efficacité du sunitinib dans les carcinomes rénaux naïfs ....................................... 119 h. Efficacité du sunitinib dans les carcinomes rénaux réfractaires aux cytokines ..... 120 i. Efficacité du sunitinib dans les cancers neuroendocriniens du pancréas ................ 120
j. Effets secondaires .................................................................................................... 121
7. Le Tyverb® (lapatinib) ............................................................................................ 122
8. Le Tassigna® (nilotinib) .......................................................................................... 123
9. Récapitulatif .............................................................................................................. 124
C. Les autres types d’inhibiteurs de tyrosines kinases ................................................. 125
1. Les anticorps monoclonaux ..................................................................................... 126
2. Les nucléotides non-sens .......................................................................................... 126
3. Les immunotoxines ................................................................................................... 126
D. Comparaison des différents inhibiteurs de tyrosines kinases ................................. 127
Conclusion ............................................................................................................................. 133
Bibliographie ......................................................................................................................... 135
Annexes ................................................................................................................................. 149 Annexe 1 : Abréviations françaises et internationales des acides aminés (71) .................. 149

14

15
Table des illustrations
Liste des figures
Figure 1 : Organisation structurale de plusieurs RTK .............................................................. 23 Figure 2 : Organisation structurale de plusieurs NRTK ........................................................... 24
Figure 3 : Représentation schématique de la forme activée et auto-inhibée de c-Kit .............. 25 Figure 4 : Les différents types de RTK .................................................................................... 29 Figure 5 : Structure de la protéine c-Kit chez le chien ............................................................. 30 Figure 6 : Les différentes formes de la protéine c-Kit ............................................................. 31 Figure 7 : Structure du gène Kit ............................................................................................... 32
Figure 8 : Structure de l’ADNc du gène codant pour le SCF .................................................. 33
Figure 9 : Formation des différents isoformes du SCF ............................................................ 34 Figure 10 : Fonctionnement normal de la protéine c-Kit ........................................................ 38
Figure 11 : Activation du système PI 3 kinase par la protéine c-Kit ....................................... 39 Figure 12 : Modèle d’auto-inhibition/activation d’un RTK .................................................... 42 Figure 13 : Différentes voies d’activation constitutive des RTK ............................................. 47 Figure 14 : Comparaison de l’électrophorèse sur gel d’agarose et de la méthode PAGE ....... 59 Figure 15 : Résumé de l’ensemble des mutations de Kit trouvées dans l’étude de Letard et al.
.................................................................................................................................................. 62
Figure 16 : Localisation et fréquence des mutations de Kit identifiées dans l’étude de Letard
et al. .......................................................................................................................................... 62 Figure 17 : Mutations de c-Kit identifiées chez 2 chiens atteints de GIST .............................. 68
Figure 18 : Formule chimique du masitinib ............................................................................. 76
Figure 19 : Représentation graphique de l’action du masitinib sur différentes kinases ........... 78 Figure 20 : TTP des chiens atteints de mastocytome cutané et traités soit avec du masitinib
soit avec un placebo ................................................................................................................. 82
Figure 21 : Médiane du TTP chez les chiens atteints de mastocytome cutané et recevant en
première intention du masitinib ou un placebo ........................................................................ 83
Figure 22 : Pourcentage de survie de chiens atteints de mastocytome cutané en fonction de
leur réponse au traitement à 6 mois .......................................................................................... 84 Figure 23 : Structure chimique du toceranib ............................................................................ 96
Figure 24 : Représentation graphique de l’action du toceranib sur différentes kinases ........... 97 Figure 25 : Comparaison du TTP chez les chiens traités avec du Palladia® et ceux traités avec
un placebo ................................................................................................................................ 99
Figure 26 : Comparaison de la durée de survie et du TTP de chiens atteints de mastocytome
cutané avec ou sans mutation du gène Kit après traitement à base de toceranib ................... 101 Figure 27 : Structure chimique de l’imatinib mesylate .......................................................... 104
Figure 28 : Réponse des xénogreffes de mastocytomes à différentes concentrations d’imatinib
................................................................................................................................................ 108 Figure 31 : Représentation graphique de l’action du dasatinib sur différentes kinases ......... 115 Figure 29 : Structure chimique du sunitinib ........................................................................... 116 Figure 30 : Représentation graphique de l’action du sunitinib sur différentes kinases .......... 117
Figure 32 : Représentation graphique de l’action du lapatinib sur différentes kinases .......... 122 Figure 33 : Différentes stratégies de lutte sélective contre les RTK ...................................... 125

16
Liste des tableaux
Tableau 1: Classification des RTK ........................................................................................... 27 Tableau 2 : Classification des NRTK ....................................................................................... 28
Tableau 3 : Grade des mastocytomes selon Patnaik ................................................................ 52 Tableau 4 : Taux de survie à 50 mois de 83 chiens atteints de mastocytome de différents
grades et traités par chirurgie large, selon Patnaik ................................................................... 53 Tableau 5 : Tableau récapitulatif des mutations de Kit trouvées dans 7 études différentes .... 60 Tableau 6 : Tableau récapitulatif des mutations retrouvées chez l’homme lors de mastocytose
.................................................................................................................................................. 64 Tableau 7 : Tableau récapitulatif des différentes mutations de c-Kit retrouvées dans les GIST
chez l’homme ........................................................................................................................... 67 Tableau 8 : Résumé des tumeurs de l’homme pouvant exprimer EGFR et fréquence associée
.................................................................................................................................................. 73 Tableau 9 : IC50 du masitinib sur différentes tyrosines kinases .............................................. 77 Tableau 10 : Rôles du masitinib dans différents maladies ....................................................... 79 Tableau 11 : Comparaison des taux de réponse au Masivet® versus placebo à 6 mois chez des
chiens atteints de mastocytome cutané .................................................................................... 81 Tableau 12 : Comparaison des taux de survie à 12 et 24 mois de chiens atteints de
mastocytomes non opérables et traités avec du masitinib ou un placebo ................................ 82 Tableau 13 : Pourcentage de survie à 1 et 2 ans de chiens atteints de mastocytome cutané en
fonction de leur réponse après 6 mois de traitement avec du masitinib ................................... 84 Tableau 14 : Tableau récapitulatif des effets du masitinib sur le taux de réponse et sur la durée
de survie de chiens atteints de mastocytome cutané ............................................................... 85
Tableau 15 : Essais cliniques sur le masitinib en association avec certains agents de
chimiothérapie .......................................................................................................................... 86 Tableau 16 : Récapitulatif des voies de développement du masitinib chez le chien ............... 95 Tableau 17 : Récapitulatif des phases d’essais cliniques du masitinib chez l’homme ............ 95
Tableau 18 : IC50 du toceranib sur différentes TK .................................................................. 96 Tableau 19 : Action du toceranib sur différents cancers chez le chien .................................. 102
Tableau 20 : IC50 de l’imatinib sur différentes TK ............................................................... 105 Tableau 21 : IC50 du sunitinib sur différentes TK ................................................................. 117 Tableau 22 : Quelques exemples d’inhibiteurs et d’activateurs de l’enzyme CYP3A4 du
cytochrome P450 .................................................................................................................... 118
Tableau 23 : Efficacité du Sutent® sur 312 patients atteints de GIST ................................... 119 Tableau 24 : Comparaison de l’effet du Sutent par rapport à l’IFN-alpha lors de carcinome
rénal naïf ................................................................................................................................. 119 Tableau 25 : Comparaison de l’effet du Sutent lors de carcinome rénal réfractaires aux
cytokines ................................................................................................................................. 120 Tableau 26 : Récapitulatif des ITK ayant une AMM vétérinaire ou humaine ....................... 124 Tableau 27 : Comparaison des différents ITK ayant une AMM chez le chien ...................... 129
Tableau 28 : Comparaison des différents ITK ayant une AMM chez l’homme .................... 130
Liste des photos
Photo 1 : Réponse au toceranib de 2 chiens présentant un mastocytome cutané avec une ITD
sur le gène codant pour c-Kit ................................................................................................. 100

17
Liste des abréviations
ADCC : Antibody Dependent Cellular Cytotoxicity
AcM : Anti-corps Monoclonal
ADN : Acide DésoxyriboNucléique
ADNc : Acide DésoxyriboNucléique complémentaire
ADP : Adénosine DiPhosphate
AINS : Anti-Inflammatoire Non Stéroïdien
AMM : Autorisation de Mise sur le Marché
AMP : Adénosine MonoPhosphate
ARNm : Acide Ribo-Nucléique messager
Asp816val : Substitution de l’aspartate en valine au codon 816 de la protéine c-Kit
ATP: Adénosine TriPhosphate
CSF-1R : Colony Stimulating Factor 1 Receptor
EDSS : Expanded Disability Status Scale
EGF : Epidermal Growth Factor, facteur
EGFR: Epidermal Growth Factor Receptor
EMA : European Medicines Agency
FAK : Focal Adhesion Kinase
FDA : Food and Drug Administration
FGF : Fibroblast Growth Factor
FGFR : Fibroblast Growth Factor Receptor
FLT3 : Fms-Like Tyrosine kinase 3
HB-EGF : Heparin Binding-Epidermal Growth Factor
HGF : Hepatocyte Growth Factor
HGFA : Hepatocyte Growth Factor Activateur
HGFR (=Met) : Hepatocyte Growth Factor Receptor
HMC-l : Human Mast Cell 1
GIST : Tumeur Stromale Gastro-Intestinale
GM-CSF : Granulocyte Macrophage-Colony-Stimulating Factor
IC 50 : Concentration Inhibitrice à 50%
IFN : Interféron
IGF-1R: Insulin Like Growth Factor 1 Receptor
ITD : Duplication Interne en Tandem
ITK : Inhibiteur de Tyrosine Kinase
JM : Juxtamembranaire
Kb : Kilo base
kDa: Kilodalton
MAP : Mitogen-Activated Protein
Mg : Magnésium
MICI : Maladie Inflammatoire Chronique des Intestins
Mm : Millimètre
NRTK : Non Récepteur Tyrosine Kinase
ODN : OligoDésoxyNucléotide
OMS : Organisation Mondiale de la Santé
Pb: Paire de base
PCR : Polymerase Chain Reaction, réaction de polymérisation en chaîne
PDGF : Platelet-Derived Growth Factor
PDGFR : Platelet-Derived Growth Factor Receptor
PI3 kinase : PhosphatidylInositol-3 kinase

18
PIP2 : PhosphatidylInositol-3,4 biPhosphate
PIP3 : phosphatidylInositol-3,4,5 triPhosphate
PTEN : Phosphatase and TENsin homolog
PTK : Protéine Tyrosine Kinase
PTP : Non Récepteur Tyrosine Phosphatase
PTB : PhosphoTyrosine Binding
RPTP : Récepteur Tyrosine Phosphatase
RTK : Récepteur Tyrosine Kinase
RT-PCR : Reverse Transcription PCR
SAM : Sterile Alpha Motif
SCF : Stem Cell Factor
Src : Sarcoma
STAT : Signal Transducers and Activators of Transcription
TGF : Transforming Growth Factor
TK : Tyrosine Kinase
TM : Trans-Membranaire
TNF: Tumor Necrosis Factor
TTP : Time To Progression
VEGF : Vascular Endothelial Growth Factor
VEGFR : Vascular Endothelial Growth Factor Receptor
VHL : Von Hippel-Lindau
WT : Wild Type

19
Introduction
Avec le développement de la gériatrie en médecine animale, le vétérinaire est de plus en plus
souvent confronté aux cancers chez les animaux de compagnie. Dans le cadre d’une prise en
charge globale, il se doit de proposer toutes les options thérapeutiques disponibles pour
améliorer la qualité de vie de l’animal.
Des progrès dans le domaine de la biologie moléculaire ont permis d’améliorer la
connaissance de la cellule et particulièrement de la cellule cancéreuse. Grâce à de nouvelles
technologies comme le microarray (puce à ADN), il est aujourd’hui possible d’analyser le
génome d’une cellule et de visualiser l’expression de différents gènes afin d’avoir le véritable
profil génétique d’une cellule. Il s’agit d’un outil extrêmement intéressant, mis à disposition
des cancérologues.
Il est ainsi possible d’identifier des molécules impliquées dans le processus de cancérisation
et de mieux les cibler. On parle alors de « thérapie ciblée ».
Alors que les agents de chimiothérapie sont cytotoxiques pour toutes les cellules en division,
qu’elles soient cancéreuses ou non, entraînant de nombreux effets secondaires, cette nouvelle
approche de thérapie ciblée permet de mieux viser la cellule anormale à travers la
connaissance des gènes impliqués dans la transformation tumorale. D’une thérapeutique
antiproliférative cytotoxique, on évolue vers une thérapeutique ciblée dont l’objectif n’est
plus de faire disparaître la tumeur mais de la contrôler tout en améliorant la durée de vie du
patient.
La thérapie ciblée apporte de nombreux avantages : en étant plus sélective, elle permet d’être
plus efficace sur la cellule cancéreuse, la dose utilisée peut être augmentée sans risque de
toxicité sur les cellules saines. Les effets secondaires étant moindres, le confort du patient est
donc amélioré et une utilisation à long terme est possible.
Parmi les molécules ciblées, les protéines tyrosines kinases sont souvent impliquées dans la
cancérogenèse chez l’homme et chez le chien. Ces protéines participent à de nombreuses
fonctions cellulaires comme la prolifération et la différentiation cellulaire, la survie cellulaire
et la transduction d’un signal.
Les inhibiteurs de tyrosines kinases se sont alors développés chez l’homme. L’imatinib est le
premier à avoir obtenu une AMM en 2001 pour le traitement de la leucémie myéloïde
chronique (102). De nombreux autres inhibiteurs de tyrosines kinases sont actuellement
disponibles en cancérologie humaine. Leur utilisation chez le chien est très récente et deux
molécules possèdent à ce jour une AMM, le masitinib et le toceranib (81, 82). Ces molécules
sont indiquées dans le traitement du mastocytome, tumeur cutanée fréquente chez le chien.
Le but de cette thèse est de synthétiser l’ensemble des connaissances actuelles sur les
inhibiteurs de tyrosines kinases utilisés chez le chien et de les comparer à la médecine
humaine.

20
Nous présenterons dans une première partie les protéines tyrosines kinases en décrivant
précisément leur mode d’action, leur régulation et leur fonction. La deuxième partie de notre
travail est consacrée à l’étude des dysfonctionnements de ces protéines et leur implication
dans un certains nombre de cancers chez le chien et chez l’homme. Ces pré-requis sont
nécessaires pour comprendre l’utilisation des inhibiteurs de tyrosines kinases.
Nous étudierons et comparerons ensuite les différents inhibiteurs de tyrosines kinases
disponibles en médecine humaine et vétérinaire.

21
I. Les protéines tyrosines kinases
A. Structure et classification
1. Définition d’une protéine tyrosine kinase
Les protéines kinases sont des protéines dont l’activité enzymatique consiste à transférer un
groupement phosphate d’une molécule d’ATP à une autre protéine entraînant un changement
de conformation de celle-ci et de ce fait une modification de son activité. Elles sont
impliquées dans la transduction des signaux.
Les protéines kinases sont divisées en 2 groupes en fonction de l’acide aminé qu’elles
phosphorylent :
-les protéines tyrosines kinases (PTK), si elles transfèrent un groupement phosphate
uniquement à un résidu tyrosine
-les protéines sérine-thréonines kinases, si elles transfèrent un groupement phosphate
uniquement à des résidus sérines et/ou thréonines.
De plus, on distingue 2 types de protéines kinases selon qu’elles s’expriment ou non à la
surface cellulaire :
-les récepteurs tyrosines kinases (RTK), localisés à la surface cellulaire
-les non récepteurs tyrosines kinases (NRTK), localisés dans le cytoplasme ou dans le noyau
de la cellule (55, 136).
Les réactions de déphosphorylation sont catalysées par des protéines spécifiques appelées
phosphatases.
Nous nous intéresserons dans cette thèse uniquement aux PTK qui sont les protéines le plus
souvent impliquées dans les processus cancéreux et les mieux connues.
2. Structure générale
a. Structure
Les RTK ont tous une structure commune, ce sont des glycoprotéines composées d’une partie
extracellulaire où se situe le site de liaison au ligand, une partie transmembranaire et une
partie cytoplasmique qui comprend un domaine juxtamembranaire (JM) et un domaine kinase.
La partie extracellulaire permet la fixation et la spécificité du ligand.
La partie transmembranaire est constituée d’une hélice et permet l’ancrage de la protéine à la
membrane cellulaire.

22
La partie cytoplasmique comprend :
-un domaine JM impliqué dans la régulation négative de la protéine (51)
-un domaine kinase lui-même divisé en
un site de liaison de l’ATP détaillé ci-dessous
un site de liaison du substrat
un site responsable de l’activité catalytique contenant un résidu
aspartate.
Le site de liaison de l’ATP est une zone très importante car les inhibiteurs de tyrosines
kinases (ITK) sont des molécules mimant l’ATP et qui se lient à la PTK de façon compétitive
avec l’ATP.
Ce site est divisé en 5 parties :
-la région adénine : il s’agit d’une région hydrophobe qui est lié à l’ATP
via des ponts hydrogènes entre les groupes amines N-1 et N-6 de l’adénine
et NH et le groupe carbonyl de la protéine kinase. Certains inhibiteurs de
tyrosines kinases se lient à la protéine kinase via ces ponts hydrogènes.
-la région du sucre : il s’agit d’une région hydrophile dans la plupart des
PTK.
-la poche hydrophobique : elle n’est pas utilisée pour la liaison avec l’ATP
mais est exploitée par la plupart des ITK.
-la chaîne hydrophobique : elle est commandée par l’hélice C-alpha. Elle
n’est pas utilisée par l’ATP.
-la région de liaison des phosphates : le groupe triphosphate de l’ATP est
contenu dans cette région par la zone riche en glycine et est lié à plusieurs
acides aminés impliqués dans l’activité catalytique (32, 136).
Bien que toutes les PTK aient une structure similaire au site de liaison de l’ATP, quelques
différences sont toutefois présentes et ont leur importance pour la spécificité des ITK.

23
Figure 1 : Organisation structurale de plusieurs RTK (52)
Le domaine L participe à la liaison du ligand.
Le domaine kringle est caractérisé par une large boucle stabilisée par 3 ponts disulfures. Il
intervient dans l’interaction avec d’autres protéines.
Le domaine SAM est un domaine d’environ 70 acides aminés qui participe à la transduction
du signal et à la répression de la transcription.
On remarque que la grande majorité des RTK est constituée d’une simple chaîne
polypeptidique à l’exception des PTK Met et IGF-1R et leurs familles respectives. Certains
ont un domaine tyrosine kinase en 2 parties séparées par un insert, il s’agit par exemple du
PDGFR et de la protéine c-Kit.
La plupart des RTK sont à l’état de monomère en l’absence de ligand.
Les NRTK ont un domaine catalytique commun similaire à celui des RTK. Ils sont dépourvus
de domaine extracellulaire et de domaine transmembranaire. Ils sont localisés dans le
cytoplasme. Cependant, quelques NRTK sont ancrés à la membrane cellulaire via des
modifications dans leur séquence en acides aminés.
En plus d’un domaine kinase, ils ont un domaine qui permet des interactions avec une autre
protéine (via un domaine SH2 et/ou SH3), avec un lipide ou avec l’ADN (52, 92).

24
Figure 2 : Organisation structurale de plusieurs NRTK (55)
b. Structure tertiaire
La cristallographie a permis l’étude de la structure tertiaire des PTK. La zone cytoplasmique
d’une protéine kinase est bilobée, elle est composée d’un lobe N-terminal et d’un lobe C-
terminal séparés par une région charnière qui permet la rotation des 2 domaines. Entre les 2
lobes se trouve le site (ou poche) catalytique.
Le lobe N-terminal est composé majoritairement de feuillets béta et d’une hélice C-alpha et il
est impliqué dans la fixation et l’orientation de l’ATP et du magnésium. Le lobe C-terminal
est majoritairement composé d’hélices alpha et il est impliqué dans la fixation du substrat. Il
contient une zone appelée boucle d’activation (81, 133).
Le site de liaison du substrat est beaucoup moins connu et est très peu souvent utilisé comme
cible par les ITK malgré les avantages qu’il pourrait apporter. En effet, la grande majorité des
ITK miment l’ATP et agissent de façon compétitive avec celui-ci. Or la haute concentration
d’ATP intracellulaire peut réduire l’efficacité de ces ITK. De plus, un ITK se liant au site de
liaison du substrat aurait une meilleure spécificité et sélectivité.

25
c. Forme active et forme inactive
Les 2 lobes peuvent bouger l’un par rapport à l’autre et déterminent ainsi 2 formes à la
protéine:
-une forme active où la poche catalytique est « ouverte »
-une forme inactive où la poche est « fermée »
Dans chaque lobe, un segment est responsable de la forme active ou inactive. Dans le lobe N-
terminal, il s’agit de l’hélice C-alpha et dans le lobe C-terminal, il s’agit de la boucle
d’activation. Les 2 segments sont couplés et l’activation de l’un entraîne l’activation de
l’autre.
La boucle d’activation est constituée de 20 à 30 acides aminés. Sa séquence nucléotidique
contient 2 régions importantes hautement conservées : DFG…..APE. Le motif DFG
correspondant à la séquence Aspartate-Phényalanine-Glycine est impliqué dans la liaison à
l’ATP-Mg. Il se situe au début de la boucle et contient un aspartate impliqué dans l’activité
enzymatique. La fin de la boucle représentée par le motif APE (Alanine-Proline-Glutamate)
permet la liaison au substrat. Le nombre de résidu tyrosine localisé sur la boucle d’activation
est variable en fonction des protéines.
La forme active est nécessaire pour permettre à l’ATP de se lier à la PTK et à l’ADP d’être
relâché. Dans la forme inactive, la protéine est le plus souvent monomérique et elle n’est pas
phosphorylée. Cette forme inactive est stabilisée par la région JM qui empêche la boucle
d’activation de se positionner en forme active (32, 57,109).
Figure 3 : Représentation schématique de la forme activée et auto-inhibée de c-Kit (132)
Le domaine JM est représenté en rouge. La boucle d’activation (BA) est représentée en vert.
Dans le schéma A, l’hélice C-alpha est dans sa conformation active et la boucle d’activation
est sous forme étendue ; la protéine est active.
Dans le schéma B, l’hélice C-alpha est dans sa conformation inactive, le domaine JM s’insère
entre les 2 lobes et la boucle d’activation n’est pas étendue ; la protéine est inactive.

26
Une protéine kinase peut être divisée en 12 régions très conservées. Le contrôle de l’activité
de la protéine kinase peut être réalisé par des sous-unités ou des domaines qui peuvent se
rajouter :
-sous-unité qui répondent à un 2nd
messager (ex : AMP)
-sous-unité dont le niveau d’activité dépend de l’état fonctionnel de la cellule (protéine kinase
cycline dépendante)
-sous-unité qui cible la kinase à différentes molécules (domaines SH2 ou SH3)
-sous-unité qui inhibe la kinase par un processus d’autorégulation (60).
Les différentes kinases sont similaires lorsqu’elles sont sous forme active mais sont
différentes dans leur conformation inactive.
En résumé, les PTK sont des enzymes essentielles au bon fonctionnement cellulaire. Les
RTK sont des glycoprotéines composées d’une partie extracellulaire sur laquelle se lie le
ligand, d’une partie transmembranaire permettant l’ancrage de la protéine à la
membrane cellulaire, d’une partie JM ayant un rôle prépondérant dans la régulation
négative de la protéine et d’une partie kinase permettant la réalisation de l’activité
enzymatique. Ces protéines existent sous 2 formes, active et inactive, en fonction de la
présence de substrat, d’ATP et du ligand.
3. Classification
Les PTK ont été découvertes en 1979 à travers l’étude de virus. Actuellement, les
scientifiques estiment qu’il y a 518 protéines kinases codées par le génome humain dont 90
sont des PTK. Parmi ces PTK, il y a 58 RTK et 32 NRTK (55,92).
Les PTK sont classées en famille selon leur homologie structurale basée sur la séquence de
leur domaine kinase (20 familles pour les RTK et 10 familles pour les NRTK).

27
Tableau 1: Classification des RTK (130)

28
Tableau 2 : Classification des NRTK (130)
La famille des split kinases est composée des protéines FGFR, VEGFR, PDGFR, c-Kit et
FLT3.
On peut également classer les RTK en différents types en fonction de la structure de leur
domaine intracellulaire :
-Les RTK de type I sont constitués de deux séquences riches en cystéine répétées dans le
domaine extracellulaire et un domaine intracellulaire possédant l’activité tyrosine kinase.
Exemple : EGFR.
-Les RTK de type II ont également deux séquences riches en cystéine dans le domaine
extracellulaire mais ont une structure hétéro-tétramérique avec 2 chaînes alpha et 2
chaînes béta. Chaque chaîne alpha est unie par une liaison disulfure à une chaîne béta et à
une autre chaîne alpha. Ce groupe inclut les récepteurs IGF-1R et HGFR également
nommé Met.
-Les RTK de type III ont un domaine kinase interrompu par un insert et 5 domaines de
type immunoglobuline en région extracellulaire.
Exemples : PDGFR, FLT3, c-Kit. Ces RTK sont souvent impliqués dans les cancers
malins chez l’homme.

29
-Les RTK de type IV ont aussi un domaine kinase interrompu par un insert et 3 domaines
de type immunoglobulines en région extracellulaire (66). Un exemple est le FGFR.
Les types I, III et IV sont monomériques, tandis que le type II est un tétramère alpha2-béta2.
Figure 4 : Les différents types de RTK (66)

30
4. Exemple du récepteur tyrosine kinase c-Kit et de son ligand
a. Structure de la protéine c-Kit
Le récepteur c-Kit est bien connu car il est impliqué dans de nombreux cancers chez l’homme
(dont les GIST, la mastocytose et le cancer du sein), et chez le chien (en particulier les GIST,
le mastocytome et les tumeurs mammaires).
Le récepteur c-Kit appartient au type III de RTK, il est caractérisé par un domaine
extracellulaire constitué de 5 domaines de type immunoglobuline et la présence d’un insert de
70 à 100 acides aminés situé entre les 2 domaines kinases. Chez l’homme, l’insert a une
longueur de 80 acides aminés (94, 153).
Figure 5 : Structure de la protéine c-Kit chez le chien (91)
Les trois premiers domaines de type immunoglobuline permettent la fixation du ligand, le
stem cell factor (SCF), tandis que les deux derniers facilitent la dimérisation de la protéine. La
fonction de la partie du domaine de type immunoglobuline située proche de la membrane
n’est pas encore connue.

31
b. Les différentes formes de la protéine c-Kit
Des isoformes de la protéine c-Kit existent ainsi qu’une forme soluble et une forme tronquée
(cf. fig.6).
Deux isoformes de c-Kit chez la souris et 4 isoformes de c-Kit chez l’homme ont été
identifiées. Elles résultent d’un épissage alternatif de l’ARNm codant pour la protéine c-Kit.
Chez l’homme, les isoformes sont caractérisées par la présence ou l’absence d’une séquence
de 4 acides aminés appelés GNNK (glycine-aspargine-aspargine-lysine) située dans la région
extracellulaire proche du domaine transmembranaire et la présence ou l’absence d’un résidu
sérine dans l’insert situé entre les 2 domaines kinases. Chez la souris, les deux isoformes sont
caractérisées par la présence ou l’absence d’une séquence de quatre acides aminés GNNK
situé dans la région extracellulaire proche du domaine transmembranaire (10).
Figure 6 : Les différentes formes de la protéine c-Kit (10)
A : les 4 isoformes de c-Kit résultent d’un épissage alternatif de l’ARNm.
B : une forme soluble de c-Kit est générée par protéolyse et est retrouvée dans le sérum
normal.
C : une forme tronquée de c-Kit dans laquelle il manque les domaines extracellulaire et
transmembranaire est décrite. Elle serait due à un promoteur inconnu localisé sur les introns
15 et 16.

32
La fonction des 2 isoformes de la protéine c-Kit chez la souris a été étudiée par plusieurs
auteurs. Reith et al. ont montré que l’isoforme de c-Kit GNNK- chez la souris présente une
autophosphorylation en l’absence de ligand plus importante que pour l’isoforme GNNK+ et
de ce fait une légère activation de la voie de signalement en aval (126). De plus, les 2
isoformes GNNK chez l’homme et chez la souris ont la même affinité pour le ligand SCF.
Lors de la fixation du ligand SCF sur les cellules c-Kit GNNK-, la phosphorylation des
résidus tyrosines est plus importante, la protéine c-Kit est internalisée plus rapidement et les
kinases MAP sont activées plus fortement que lors de l’activation des cellules c-Kit GNNK+.
Mais les isoformes GNNK- et GNNK+ activent de façon similaire la PI 3 kinase et la protéine
Akt.
Il semblerait donc que les isoformes diffèrent dans le signal qu’ils induisent et donc dans leur
fonction. Aucune étude n’a été faite concernant les 2 isoformes du résidu sérine (10).
c. Etude du gène Kit
Le gène Kit a été identifié pour la première fois en tant qu’oncogène dans le virus du sarcome
félin de Hardy-Zuckerman 4 qui induit chez les chats infectés des fibrosarcomes
multicentriques. La séquence correspondante au proto-oncogène chez l’homme et la souris a
été clonée et on a montré qu’elle code pour un récepteur protéine kinase (12).
Le gène a été localisé en premier lieu chez la souris sur le locus W du chromosome 5. Chez
l’homme, il est localisé sur le chromosome 4 dans la région péricentromérique du bras long. Il
fait 89 kb et contient 21 exons. Il code pour la protéine c-Kit (976 acides aminés) dont le
poids moléculaire est de 145 kDa (112).
Chaque exon code pour une partie précise de la protéine : le domaine extracellulaire est codé
par les exons 1 à 9, l’hélice transmembranaire est codée par l’exon 10, le domaine JM est
codé par les exons 11 et 12, le premier domaine kinase contenant le site de liaison de l’ATP
est codé par les exons 12, 13 et 14 et le deuxième domaine kinase contenant le site
phosphotransférase est codé par les exons 16, 17, 18 et 19. L’insert est codé par l’exon 15 (74,
132).
Figure 7 : Structure du gène Kit (83)
TM : partie transmembranaire TGA : codon stop
Tk1 : domaine tyrosine kinase 1 ATP représente le site de liaison de l’ATP
Tk2 : domaine tyrosine kinase 2 ATG : codon d’initiation
KI : insert SP : peptide signal

33
Chez le chien, l’analyse par RT-PCR de la séquence ADN du gène Kit montre une bande de
756 pb de même longueur que celle de la souris mais plus courte que celle de l’homme, du
chat et du rat qui fait 768 pb. Chez le chien, la séquence du gène montre 90.4% d’homologie
avec le chat, 87.2% avec l’homme, 80.3% avec la souris et 79.6% avec le rat (67).
La structure et la séquence en acides aminés de la protéine sont hautement conservées dans les
différentes espèces. Le domaine JM montre 100 % d’homologie entre l’homme et le chien
(36).
d. Le ligand de c-Kit, le Stem Cell Factor (SCF)
Son nom est à mettre en relation avec son rôle dans la survie, l’auto-renouvellement et la
différentiation des cellules souches hématopoïétiques. Le SCF a été localisé chez la souris sur
le locus Sl du chromosome 10 et sur le chromosome 12 de l’homme. Des mutations sur ce
locus entraînent des déficiences de plusieurs types de cellules. L’absence complète du SCF ou
de c-Kit entraîne la mort causée par une anémie sévère in utero ou en péri natalité. Une perte
de fonction partielle de c-Kit ou du SCF entraîne un déficit en mastocytes, une anémie, une
hypo pigmentation et une stérilité (8).
On a ainsi pu en déduire le rôle du SCF : ce facteur de croissance est essentiel pour le
déroulement normal de l’hématopoïèse, le développement des mastocytes et des mélanocytes
ainsi que pour la fertilité (72).
Figure 8 : Structure de l’ADNc du gène codant pour le SCF (66)
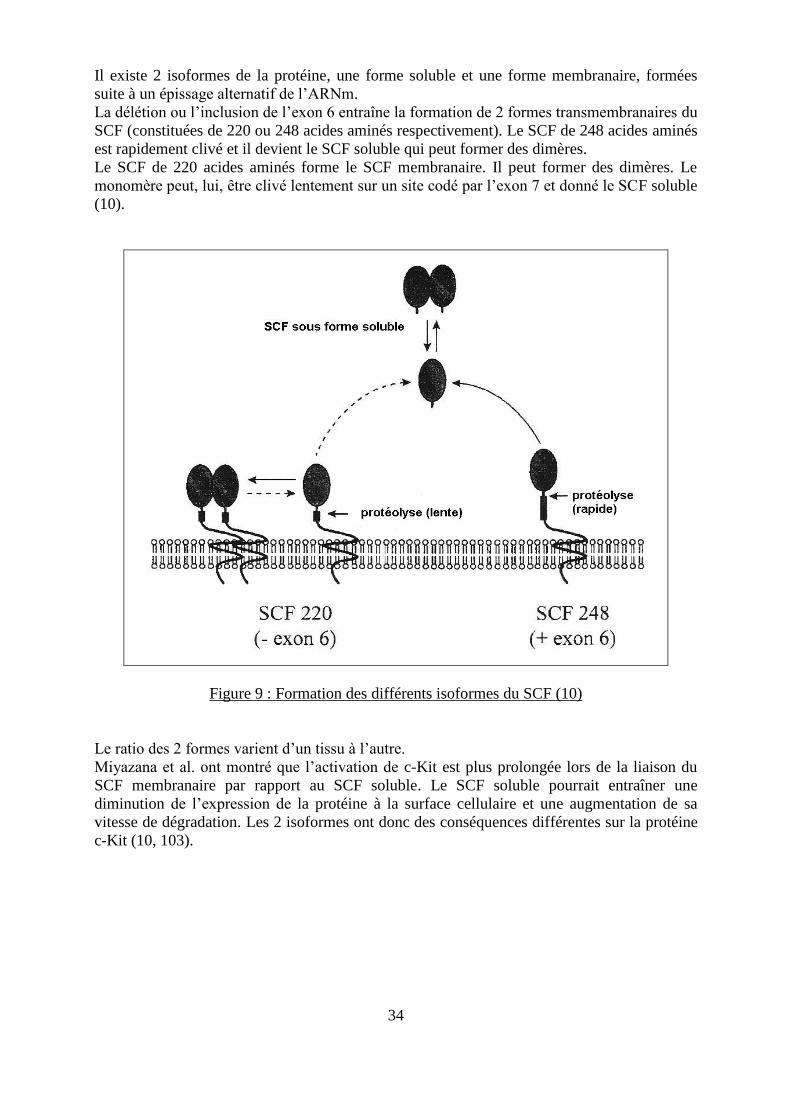
34
Il existe 2 isoformes de la protéine, une forme soluble et une forme membranaire, formées
suite à un épissage alternatif de l’ARNm.
La délétion ou l’inclusion de l’exon 6 entraîne la formation de 2 formes transmembranaires du
SCF (constituées de 220 ou 248 acides aminés respectivement). Le SCF de 248 acides aminés
est rapidement clivé et il devient le SCF soluble qui peut former des dimères.
Le SCF de 220 acides aminés forme le SCF membranaire. Il peut former des dimères. Le
monomère peut, lui, être clivé lentement sur un site codé par l’exon 7 et donné le SCF soluble
(10).
Figure 9 : Formation des différents isoformes du SCF (10)
Le ratio des 2 formes varient d’un tissu à l’autre.
Miyazana et al. ont montré que l’activation de c-Kit est plus prolongée lors de la liaison du
SCF membranaire par rapport au SCF soluble. Le SCF soluble pourrait entraîner une
diminution de l’expression de la protéine à la surface cellulaire et une augmentation de sa
vitesse de dégradation. Les 2 isoformes ont donc des conséquences différentes sur la protéine
c-Kit (10, 103).

35
Zhang et al. ont analysé la structure du SCF. La forme soluble du SCF est constituée de 165
acides aminés mais la région se liant à la protéine c-Kit est constituée des acides aminés 1 à
141. Le SCF peut exister sous forme de monomère ou de dimère. Chaque protomère est
constitué d’une courte chaîne de 4 hélices anti-parallèles. Les 2 protomères peuvent interagir
entre eux via des interactions polaires et non polaires et former un homodimère non covalent.
La région du SCF qui se lie à c-Kit est composée d’une poche hydrophobe entourée d’une
région chargée positivement et négativement localisée sur la partie terminale de chaque
protomère. La bivalence du SCF est la seule force responsable de la dimérisation du domaine
extracellulaire de c-Kit (156).
En résumé, la protéine c-Kit est un RTK de type III codée par un gène constitué 21
exons. Elle présente plusieurs isoformes qui diffèrent dans le signal émis. Cette protéine
est impliquée dans de nombreux cancers chez l’homme et chez le chien et présente une
forte homologie dans la séquence en acides aminés entre ces 2 espèces. Le SCF est le
ligand de la protéine c-Kit et entraîne son activation. Il présente 2 isoformes ayant des
conséquences différentes sur la protéine c-Kit.

36
B. Localisation des PTK : Exemple de la protéine c-Kit
1. Localisation normale
Dans les tissus normaux, des scientifiques ont montré l’expression de la protéine c-Kit ou de
son ARNm dans les cellules souches hématopoïétiques, les mastocytes, les cellules
germinales, les cellules épithéliales de la glande mammaire, les cellules interstitielles de Cajal
situées dans l’intestin, les mélanocytes, les cellules de moelle osseuse, les cellules vasculaires
endothéliales, les astrocytes, les tubules rénaux, les glandes sudoripares, le mégacaryocyte, le
système nerveux central et plus particulièrement le cervelet (10, 95, 136).
2. Localisation tumorale
La présence de c-Kit dans des tissus tumoraux et plus précisément sa quantité et sa forme
(normale ou mutée) permettent de suspecter son rôle étiologique en cancérologie.
Son étude dans certains cancers peut permettre de corréler le taux de c-Kit à un grade tumoral
ou une mutation donnée de c-Kit et le type tumoral.
Chez l’homme, c-Kit a été retrouvée dans un certains nombre de tumeurs : GIST, séminome,
cancer du poumon à petites cellules, cancer du côlon, neuroblastome, cancer du sein, leucémie
myéloïde sévère, leucémie lymphoblastique sévère à cellule T, myélome multiple, syndrome
myélodysplasique, désordre myéloprolifératif, lymphome non hodgkin à cellule B, lymphome
T sinonasal, mélanome (92, 112).
Actuellement, chez le chien, c-Kit est impliquée dans les mastocytomes (85,120), les GIST
(36) et les tumeurs mammaires (67).
En effet, CA. London a montré que les mastocytes tumoraux provenant de mastocytomes de
grade 2 chez le chien expriment la protéine c-Kit, les ARNm codant pour c-Kit et SCF, ce qui
amène à s’interroger sur le rôle de c-Kit dans l’apparition des mastocytomes canins (85).
Chez l’homme, des scientifiques ont montré que 100% des patients ayant une mastocytose
associée à des désordres hématologiques ont une même mutation du gène Kit qui se traduit
par une substitution de l’aspartate en valine en position 816 (asp816val) (107).
Une autre étude a montré que sur 21 chiens présentant une GIST, 11 soit 52% exprimaient la
protéine c-Kit (36). Chez l’homme, Hirota et al. ont étudié 5 GIST et ont montré que les 5
tumeurs exprimaient la protéine c-Kit mutante, celle-ci est activée de façon indépendante à la
présence de ligand (50).
Une étude sur des tumeurs mammaires de chiennes a montré une augmentation de la quantité
d’ARNm codant pour c-Kit dans les tissus tumoraux mammaires (67).
Concernant le cancer du sein chez la femme, il a été montré que contrairement au chien
l’expression de c-Kit est plus faible dans le tissu tumoral que dans le tissu normal (108).
Il semble donc que la protéine c-Kit soit impliquée dans des cancers issus de cellules où elle
s’exprime de façon normale. D’autres PTK comme VEGFR, EGFR, Met etc… sont
impliquées dans certains types de tumeurs.

37
Nous reviendrons plus en détail sur le rôle de c-Kit et d’autres PTK dans la transformation
d’une cellule normale en cellule tumorale.
C. Mode d’action des protéines tyrosines kinases
1. Cas des récepteurs tyrosines kinases
On rappelle que les RTK existent sous forme active et inactive. Le RTK peut exercer son
activité enzymatique lorsqu’il est sous forme active.
Le ligand se lie sur un site spécifique de la partie extracellulaire du récepteur qui est sous
forme inactive, monomérique le plus souvent mais il peut également être dimérique. La
fixation du ligand entraîne une dimérisation du récepteur ou une stabilisation du dimère.
Puis, il y a autophosphorylation de résidus tyrosines (transphosphorylation) localisés sur la
boucle d’activation, cela entraîne la stimulation de l’activité kinase de la protéine,
l’enlèvement des gênes stériques sur les 2 sites de liaison (du substrat et de l’ATP) et le
positionnement correct des résidus impliqués dans la liaison du substrat et dans l’activité
catalytique. De plus, d’autres tyrosines localisées dans la région JM, l’insert, et la région C-
terminale sont phosphorylées (cisphosphorylation).
Au final, le RTK est sous forme active, les sites de fixation de l’ATP et du substrat sont
accessibles et le substrat peut se lier à la protéine kinase via des séquences qui reconnaissent
les phosphotyrosines (SH2 et PTB) et l’ATP peut se lier à son site spécifique. La protéine
kinase peut alors exercer son activité catalytique (51, 52, 134,136).

38
Figure 10 : Fonctionnement normal de la protéine c-Kit (106)
A : le récepteur c-Kit est monomérique et inactif (JM = domaine juxtamembranaire, TK =
domaine tyrosine kinase)
B : le ligand se lie à la protéine ce qui entraîne la dimérisation du RTK
C : la liaison du ligand entraîne l’autophosphorylation de c-Kit sur les domaines TK1 et TK2
et son activation
D : le substrat se lie au RTK activé au niveau des phosphotyrosines.
Il existe un niveau basal d’autophosphorylation sans liaison du ligand dû à la collision des
protéines dans la membrane et la formation spontanée de dimère ; ce niveau basal est contrôlé
par l’auto-inhibition des protéines et par l’action des phosphatases.
Chaque PTK active différentes protéines. Par exemple, la protéine c-Kit active 3 systèmes en
aval : le système Ras/ERK/MAP, le système PI3 kinase/Akt et le système JAK/STAT.

39
Nous allons étudier l’activation du système PI3 kinase/Akt par la protéine c-Kit :
La PI3 kinase/Akt est une des voies de signalisation les plus souvent activées par c-Kit suivie
par la voie impliquant les protéines Ras/MAP.
La PI3 kinase est composée de 2 sous unités, une sous-unité de 85 kDa (appelée p85) et une
sous-unité catalytique de 110 kDa (appelée p110). La sous unité p85 est associée à la protéine
activée c-Kit via un domaine SH2 et est ensuite phosphorylée par c-Kit sur un résidu tyrosine.
La phosphorylation entraîne l’activation de la PI3 kinase et ainsi la sous-unité p110
phosphoryle un PIP2 (phosphatidylinositol-3,4 biphosphate) qui devient un PIP3
(phosphatidylinositol-3,4,5 triphosphate). Celui-ci phosphoryle la protéine sérine-thréonine
kinase Akt qui s’active.
La protéine Akt active ou inhibe d’autres voies de signalisation impliquant entre autres les
protéines BAD, p27, mTOR et joue un rôle dans l’inhibition de l’apoptose, la croissance et la
prolifération cellulaire. La protéine phosphatase PTEN inactive la protéine Akt en
déphosphorylant le PIP3 qui devient PIP2.
La surexpression des protéines Akt et PI3 kinase ou des mutations de la protéine PTEN
entraînant sa perte de fonctions ont été mises en évidence dans certains cancers (par exemple
présence de mutations de PTEN dans certains ostéosarcomes chez le chien) (56, 80,75).
Figure 11 : Activation du système PI 3 kinase par la protéine c-Kit (80)

40
2. Cas des non récepteurs tyrosines kinases
Les NRTK sont maintenus sous forme inactive par des protéines et des lipides inhibiteurs et
par une auto-inhibition. Leur activation se fait lors de la dissociation des inhibiteurs, lors de
transphosphorylation, ou lors de phosphorylation par un NRTK différent (52).
L’activation des NRTK s’arrête lors de l’action des tyrosines phosphatases (136).
En résumé, les RTK passent de la forme inactive à la forme active par liaison avec un
ligand spécifique. Cela entraîne une dimérisation du récepteur, une
autophosphorylation et lui permet d’exercer son activité catalytique. L’action des
phosphatases permet le passage du RTK de sa forme active à sa forme inactive.

41
D. Régulation des protéines tyrosines kinases
1. Régulation de la transcription du gène Kit
De multiples facteurs de transcription influencent la transcription du gène codant pour la
protéine c-Kit : Sp1, AP-2, Ets, Myb… Ils agissent sur le promoteur du gène (10).
Dans l’état actuel des connaissances, la fonction des différents facteurs n’est pas encore
clairement définie.
2. Régulation par des facteurs de croissance
De nombreuses cytokines interviennent dans la régulation de la protéine c-Kit et de son
ARNm. La protéine GM-CSF régule négativement l’ARNm de c-Kit. L’interleukine 4 régule
négativement l’expression de c-Kit dans les mastocytes humains en ayant un effet complexe
sur leur maturation. Le TGF-béta régule négativement c-Kit en agissant sur la stabilité de
l’ARNm des cellules hématopoïétiques peu différenciées. Le TNF-alpha inhibe l’effet du SCF
sur la survie des cellules hématopoïétiques primitives. L’IFN-gamma entraîne une diminution
de la concentration en ARNm codant pour c-Kit (10).
3. Autorégulation via le domaine juxtamembranaire, la boucle
d’activation et la partie C-terminale
Le domaine JM se lie à l’interface entre les lobes N et C-terminaux et perturbe la
conformation de la boucle d’activation et du motif DFG (Apartate-Phénylalanine-Glycine). Il
stabilise le segment d’activation dans sa forme inactive. La protéine ne peut donc pas passer
en mode actif (104). De plus, des mutations dans la région codant pour le domaine JM et
modifiant sa structure entraînent une activation permanente de c-Kit indépendamment de la
liaison du ligand. Ce phénomène est à l’origine de nombreux processus cancéreux.
Une autre partie de la PTK intervient dans l’auto inhibition, il s’agit de la partie C-terminale.
Sa longueur et sa séquence en acides aminés sont variables et elle contient plusieurs résidus
tyrosines. Par exemple, la partie C-terminale du RTK Tie2, lorsqu’elle est non phosphorylée,
se lie à une zone proche du site de liaison du substrat, ce qui le rend inaccessible.
Des mutations de la région codant pour la partie C-terminale entraîne une augmentation de
l’activité kinase de la PTK ce qui prouve son rôle dans l’auto inhibition (51).
Lors de la phosphorylation du domaine JM et de la partie C-terminale, les changements de
forme induits permettent à la boucle d’activation d’adopter une conformation active.
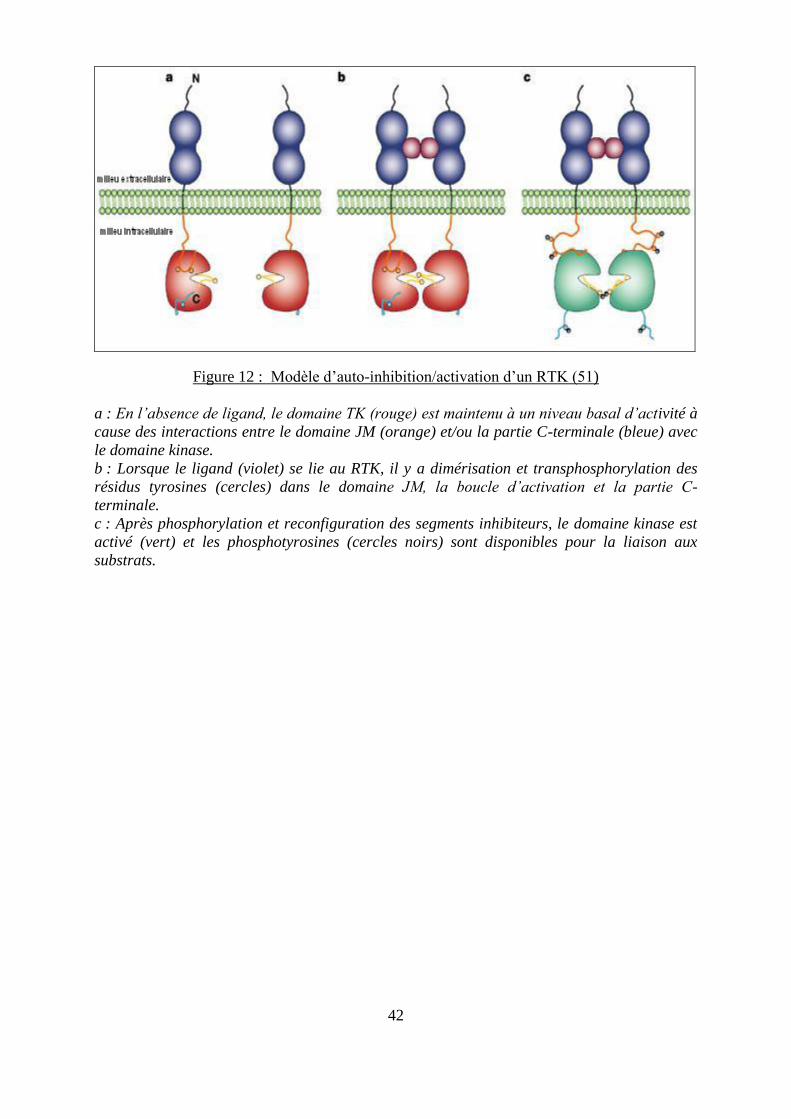
42
Figure 12 : Modèle d’auto-inhibition/activation d’un RTK (51)
a : En l’absence de ligand, le domaine TK (rouge) est maintenu à un niveau basal d’activité à
cause des interactions entre le domaine JM (orange) et/ou la partie C-terminale (bleue) avec
le domaine kinase.
b : Lorsque le ligand (violet) se lie au RTK, il y a dimérisation et transphosphorylation des
résidus tyrosines (cercles) dans le domaine JM, la boucle d’activation et la partie C-
terminale.
c : Après phosphorylation et reconfiguration des segments inhibiteurs, le domaine kinase est
activé (vert) et les phosphotyrosines (cercles noirs) sont disponibles pour la liaison aux
substrats.

43
4. Rôle des phosphatases
Les phosphatases catalysent les réactions de déphosphorylation des RTK et les rendent alors
inactifs.
On distingue 2 types de phosphatases : les récepteurs tyrosines phosphatases (RPTP) et les
non récepteurs tyrosines phosphatases (PTP).
Les PTP sont localisées dans le cytoplasme ou dans le noyau. La grande majorité a un
seul domaine catalytique sauf : Shp1 et Shp 2 qui en ont 2.
Shp1 est une enzyme phosphatase qui contient 2 domaines SH2 en tandem, un
domaine phosphatase et une partie C-terminale. Elle se lie à c-Kit via son domaine
SH2 sur des résidus tyrosines phosphorylés et les déphosphoryle. Elle agit également
en déphosphorylant des protéines kinases activées par c-Kit. Elle agit uniquement sur
les cellules hématopoïétiques et épithéliales.
Shp2 a la même structure que Shp1 mais agit sur tous les types cellulaires. Son mode
d’action est similaire à celui de Shp1 (131).
Les RPTP sont des protéines membranaires regroupées en famille selon la
conformation de leur domaine extracellulaire. Ils ont tous 2 domaines catalytiques
intracellulaires sauf une famille appelée RPTP béta. Cependant, le domaine
catalytique distal n’a pas ou très peu d’activité (55).
5. Autres mécanismes régulateurs
La liaison du SCF entraîne rapidement l’endocytose de c-Kit, son ubiquitinylation et sa
dégradation. L’ubiquitinylation est une modification biochimique nécessitant plusieurs étapes pour aboutir
in fine à la fixation covalente d'une ou de plusieurs protéines d’ubiquitine (8 kDa) sur un ou
plusieurs résidus lysines de la protéine substrat. Ces modifications entraînent la dégradation
de la protéine ubiquitinée par le protéasome, un complexe enzymatique.
En résumé, les PTK sont finement régulées tout au long de leur vie, depuis la
transcription de leur gène jusqu’à leur dégradation. La transcription du gène codant
pour les RTK est régulée ainsi que l’ARNm lui-même. La PTK est autorégulée via le
domaine JM, la boucle d’activation et la partie C-terminale. Elle est également sous
l’influence des phosphatases.

44
E. Fonctions des protéines tyrosines kinases
La phosphorylation des protéines joue un rôle important dans le contrôle de la division
cellulaire, le métabolisme cellulaire, la survie et l’apoptose des cellules. Il s’agit d’un
mécanisme finement régulé par lequel un signal extracellulaire peut être transmis à travers la
cellule jusqu’au noyau et exercer son action sur l’ADN.
1. Exemple de la protéine c-Kit
L’importance de la protéine c-Kit a été démontrée par des expériences sur des souris mutantes
qui n’expriment pas la protéine c-Kit ou le SCF. Ces souris présentent toutes un phénotype
particulier caractérisé par une altération de l’hématopoïèse, de la mélanogenèse et de la
gamétogenèse.
On dénombre donc plusieurs fonctions à cette protéine :
prolifération et différentiation des cellules souches
migration des cellules germinales
déroulement normal de l’hématopoïèse
survie, expansion et maturation des cellules NK (natural killers)
maintien d’une hématopoïèse, d’une mélanogenèse et d’une gamétogenèse normale
croissance et différentiation des mastocytes
croissance et différentiation des cellules interstitielles de Cajal (136).
2. Exemple du VEGFR
régulation de l’angiogenèse (136).
3. Exemple du PDGFR
migration et prolifération de cellule mésenchymateuse
développement des reins et du système cardiovasculaire
inflammation
angiogenèse
cicatrisation
maintenance des vaisseaux sanguins
régulation indirecte des fonctions des cellules endothéliales (136).

45
4. Exemple de la protéine Met (=HGFR)
développement embryonnaire
cicatrisation
régénération tissulaire
croissance cellulaire
différentiation cellulaire (79).
5. Exemple du EGFR
survie cellulaire
angiogenèse
prolifération cellulaire
différentiation cellulaire
inhibition de l’apoptose cellulaire
motilité cellulaire (136).
Les protéines kinases sont des protéines dont la structure est hautement conservée entre les
différentes espèces. Elles occupent une place importante dans la cellule de part leurs
différents rôles. Ce sont des protéines finement régulées et leur dérégulation peut avoir des
conséquences graves. Grâce à la biologie moléculaire, les scientifiques ont pu mettre en
évidence leur rôle dans l’apparition de certaines maladies telles que les cancers.
Nous allons étudier maintenant comment une PTK contribue à la formation d’un processus
cancéreux.

46

47
II. Implication des protéines tyrosines kinases
en cancérologie
A. Dysfonctionnement des protéines tyrosines kinases
La dérégulation des tyrosines kinases peut se faire selon différentes modalités. Il peut s’agir
d’une surexpression des RTK, d’une mutation du gène codant pour les RTK ou d’une
stimulation par une boucle autocrine/paracrine (cf. fig.13). On peut également avoir formation
de protéines fusion et expression inappropriée d’une kinase.
Ces différentes anomalies entraînent une activation du RTK ou un renforcement du signal
émis par le RTK et a donc des conséquences sur la croissance cellulaire.
Figure 13 : Différentes voies d’activation constitutive des RTK (157)
P = phosphotyrosine

48
Nous allons étudier plus en détail les causes de dysfonctionnement des RTK.
1. Surexpression d’un récepteur tyrosine kinase
Dans ce cas, le nombre de RTK est supérieur à la normale. Cela peut être lié soit à une
amplification du gène codant pour le RTK soit à un dysfonctionnement de la dégradation des
RTK. L’amplification des gènes peut être due à une augmentation du nombre de copies d’un
gène ou à une perte de contrôle d’un gène liée à sa translocation proche d’un autre gène
hautement exprimé. Cela entraîne la perte de la régulation transcriptionnelle. La réponse des
cellules cancéreuses au facteur de croissance est donc exacerbée.
De plus, il y a un niveau basal d’autophosphorylation plus élevé car la formation spontanée de
dimère due à la collision des molécules est plus importante du fait du plus grand nombre de
RTK (80). La prolifération cellulaire est donc plus importante et il y a formation de tumeur.
Un exemple chez la femme est la surexpression de HER2 dans le cancer du sein (157).
2. Mutations du gène codant pour un récepteur tyrosine kinase
Ce sont des modifications de la séquence nucléotidique de l’ADN sous forme de délétion,
substitution ou insertion. Ces anomalies génétiques peuvent entraîner des changements de la
séquence en acides aminés de la protéine au niveau de la partie extracellulaire ou dans la
partie catalytique et le plus souvent au niveau du site de liaison de l’ATP. Ces changements
dans la séquence d’acides aminés peuvent entraîner des changements de forme de la molécule
et des modifications de son activité (157).
Par exemple, chez le chien, l’ITD (duplication interne en tandem) est une duplication en
tandem localisée au niveau de l’exon 11 du gène codant pour la protéine c-Kit et retrouvée
lors de mastocytome. Elle entraîne une activation de la protéine c-Kit indépendamment de la
liaison du ligand (83).
Chez l’homme, le mutant EGFRvIII dans lequel il manque les acides aminés 6 à 273 dans le
domaine extracellulaire entraîne l’activation du RTK et induit une prolifération cellulaire sans
liaison au ligand. Il est retrouvé dans les glioblastomes, les tumeurs ovariennes, le cancer du
poumon non à petites cellules et les carcinomes mammaires (157).

49
On peut classer les mutations en fonction de leurs conséquences sur l’activité de la protéine
ou en fonction de leur localisation sur la protéine :
On parle de mutation “gain of function” si l’activité de la protéine est augmentée par
rapport à une protéine normale ou de mutation « loss of function » si l’activité de la
protéine est diminuée par rapport à la protéine normale.
Longley et al. ont classé les mutations de la protéine c-Kit en 2 types : type « poche
catalytique » lorsque la mutation concerne le site enzymatique de la protéine ou de
type « régulation » lorsqu’elle concerne une partie de la protéine impliquée dans la
régulation de celle-ci.
Un exemple de mutation de type « poche catalytique » est la substitution asp816val
localisée dans la boucle d’activation retrouvée dans la majorité des cas de
mastocytoses adultes. Un exemple de mutation de type « régulation » est l’ensemble
des mutations touchant le domaine JM dans les mastocytomes de chiens (88).
Un des avantages de classer ainsi les mutations est de pouvoir prévoir la réponse à un ITK en
fonction du type de mutation car chaque ITK agit préférentiellement sur un type de mutation.
Par exemple, l’imatinib inhibe seulement les cellules avec une mutation de type « régulation »
qui affecte le domaine JM, il n’est donc pas efficace sur les mastocytoses adultes qui
présentent une substitution au codon 816 (88).
3. Boucle autocrine/paracrine
Une boucle autocrine est caractérisée par l’expression du RTK et du ligand par une même
cellule et une surexpression de l’un des deux. Une boucle paracrine est caractérisée par une
surexpression du RTK dans une cellule et la présence de ligand synthétisé par une cellule
voisine. Les deux boucles entraînent une augmentation de l’activité des RTK et de ce fait une
transformation tumorale des cellules impliquées.
Citons pour exemple, chez l’homme, la boucle autocrine de l’EGFR et de son ligand TGF-
alpha qui est responsable de glioblastomes. Chez le chien, la boucle autocrine de HGFR et de
son ligand HGF est responsable d’ostéosarcome. De même, la boucle autocrine de c-Kit est
retrouvée dans les hémangiosarcomes (80, 157).
4. Formation de protéines fusions
Dans ce cas, une portion d’un gène (souvent la portion codant pour la partie kinase) se
retrouve liée à un autre gène ce qui entraîne une perte de la régulation de la protéine.
Par exemple chez l’homme, la translocation des bras longs des chromosomes 9 et 22
entraînent la formation de la protéine fusion Bcr-Abl responsable de la leucémie myéloïde
chronique. La protéine Abl a une activité catalytique excessive et entraîne la transformation
maligne des cellules (80).

50
5. Expression inappropriée d’une kinase
Dans ce cas, le tissu normal n’exprime pas la protéine kinase mais le tissu néoplasique
correspondant l’exprime. On peut noter, par exemple, la protéine c-Kit et le cancer du
poumon non à petites cellules chez l’homme.
6. Conséquences : activation constitutive des tyrosines kinases
Les conséquences de ces anomalies sont une perte de contrôle des TK ou une
autophosphorylation des TK responsables d’une augmentation de leur activité sans fixation du
ligand. Il en résulte une stimulation de la croissance cellulaire et augmentation de la survie
des cellules.
En résumé, les PTK peuvent être dérégulées de différentes manières. Il peut s’agir d’une
anomalie au niveau du gène (surexpression, mutations ou translocation entraînant la
formation de protéine fusion). La PTK peut également s’exprimer dans un tissu où elle
ne s’exprime pas dans les conditions normales. Des boucles autocrines-paracrines sont
également possibles. Toutes ces anomalies entraînent une activation constitutive de la
PTK. Il en résulte une stimulation de la croissance et de la survie cellulaire et de ce fait
une transformation néoplasique de la cellule.

51
B. Intérêt pronostique des tyrosines kinases
1. Chez le chien
Nous allons étudier l’intérêt pronostique de la protéine c-Kit dans le cas des mastocytomes
chez le chien.
Actuellement le pronostic du mastocytome est basé sur le grade histologique déterminé par
Patnaik (cf. tableau 3), sur le stade clinique de la maladie et sur différents marqueurs tels que
le Ki-67. Or la classification de Patnaik, largement utilisée, présente des lacunes car de
nombreux mastocytomes sont classés dans le grade 2 et les tumeurs de ce grade ont un
comportement différent ; on peut donc difficilement en déduire un pronostic fiable (115). De
plus, la détermination du grade histologique est subjective et des résultats différents pour une
même tumeur peuvent être obtenus par des anatomopathologistes différents.
Il semble donc intéressant de s’intéresser à la valeur pronostique de ces TK qui pourrait
permettre de prédire de manière plus fiable l’évolution de la tumeur.
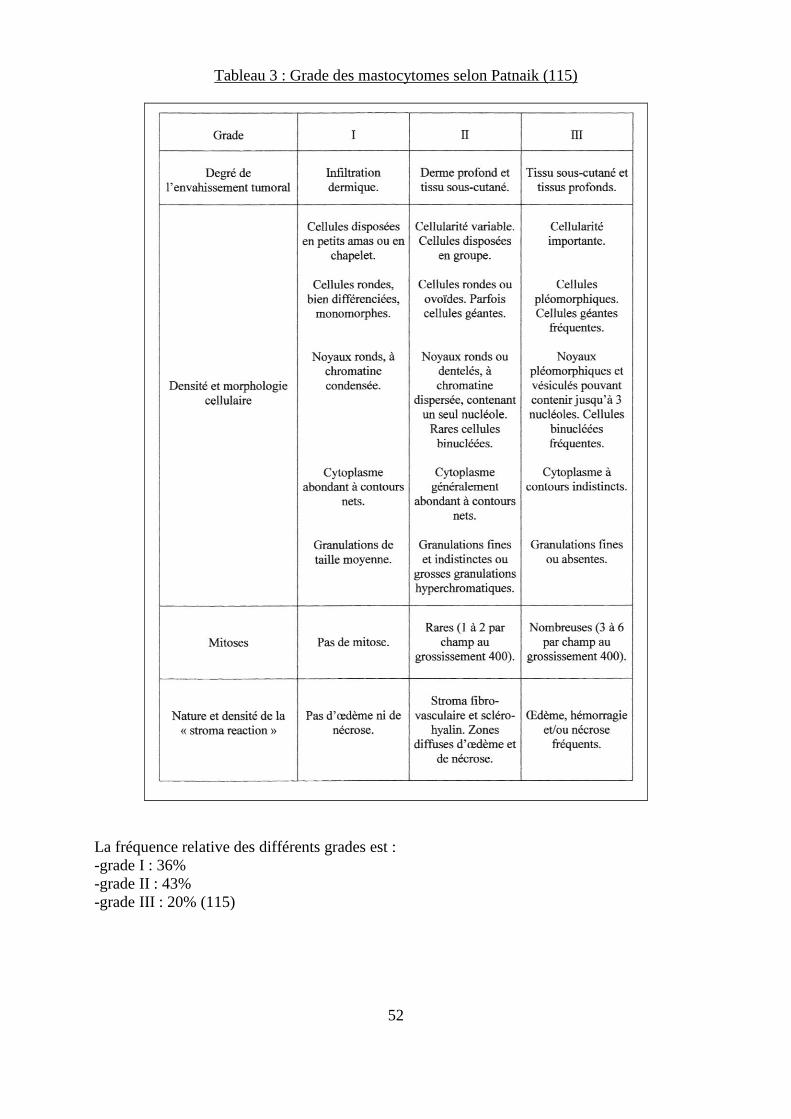
52
Tableau 3 : Grade des mastocytomes selon Patnaik (115)
La fréquence relative des différents grades est :
-grade I : 36%
-grade II : 43%
-grade III : 20% (115)

53
Tableau 4 : Taux de survie à 50 mois de 83 chiens atteints de mastocytome de différents
grades et traités par chirurgie large, selon Patnaik (115)
Dans les mastocytomes, la grande majorité des mutations identifiées concerne le domaine JM
de c-Kit comme nous le verrons au II.D.1.c. (83, 155). De plus, dans cette espèce, le
mastocytome est une tumeur souvent agressive par rapport à d’autres espèces telles que
l’homme où la mastocytose a le plus souvent un comportement bénin. Cela amène à
s’interroger sur le rôle des mutations du domaine JM dans l’agressivité de la maladie.
De nombreux auteurs ont pu établir une corrélation entre la présence d’une mutation de c-Kit
dans le JM et en particulier une ITD et un haut grade tumoral, un temps de survie diminué,
une augmentation du taux de mortalité et une augmentation du taux de récidive (148, 155).
Cependant, aucune corrélation entre la race et la présence d’un type de mutation ni entre la
race et le grade tumoral n’a pu être établie.
De même, une répartition anormale (c'est-à-dire cytoplasmique) de la protéine c-Kit dans la
cellule est associée à un taux de récidive plus élevé et une durée de vie plus faible (64,149).
Ce résultat pourrait permettre de créer une nouvelle classification permettant d’évaluer le
pronostic du mastocytome chez le chien en fonction de la répartition de c-Kit visualisée par
immunohistochimie.
D’autres marqueurs permettent d’affiner le pronostic, il s’agit du Ki-67 et de l’AgNOR (13,
149).
En conclusion, connaître le pronostic d’une tumeur est essentiel pour pouvoir
déterminer un traitement adapté. Dans le cas du mastocytome, des études ont permis de
montrer qu’une mutation de c-Kit et plus particulièrement une ITD, la localisation
anormale de la protéine c-Kit, un Ki-67 et un AgNOR élevés et un grade histologique
élevé sont associés à un mauvais pronostic.
L’intérêt de ces marqueurs pronostiques réside dans leur utilisation combinée afin
d’avoir une évaluation la plus précise et la plus fiable possible.
2. Chez l’homme
La famille EGFR a été très étudiée et de nombreuses études ont montré que la surexpression
du RTK HER1 est associée à un faible pronostic dans les cancers de l’ovaire, de l’œsophage,
cervical, de la vessie, du côlon, de l’estomac et de l’endomètre. La surexpression du RTK
HER2 est associée à un mauvais pronostic dans les cancers du sein, de l’ovaire, de la prostate,
du poumon et des os. Dans le cancer du poumon à petites cellules et les GIST, l’expression de
c-Kit est à relier à un mauvais pronostic. De même, une mutation du VEGFR a été associée à
un mauvais pronostic dans les cancers du poumon et du côlon et dans les cancers du sein (92).
Taux de survie
des grades I
Taux de survie
des grades II
Taux de survie
des grades III
93% 44% 6%

54
C. Les facteurs impliqués dans l’angiogenèse : VEGFR,
PDGFR, FGFR et Tie 1/2
L’angiogenèse est un processus qui décrit le développement de nouveaux vaisseaux sanguins
(néovascularisation) à partir de vaisseaux préexistants. C’est un mécanisme finement régulé
par des interactions entre inhibiteurs et activateurs. Il a lieu très tôt au cours de
l’embryogenèse, le système vasculaire étant le premier système à se mettre en place. A l’état
adulte, on observe très peu d’angiogenèse dans des conditions normales hormis pour le cycle
de croissance des poils, la cicatrisation, lors de la gestation et de la lactation.
La croissance tumorale et la dissémination des métastases dépendent de la néoformation de
vaisseaux. En effet, une tumeur ne peut pas croître de plus de quelques millimètres sans un
apport sanguin lui fournissant oxygène et nutriments. Elle est donc obligée de former son
propre réseau de micro vaisseaux sanguins.
Le déroulement de l’angiogenèse se fait en plusieurs étapes décrites ci-dessous :
activation des cellules endothéliales
dégradation protéolytique de la matrice sous endothéliale vasculaire
migration des cellules endothéliales
prolifération des cellules endothéliales
constitution d'une lumière vasculaire
stabilisation du vaisseau par des péricytes
De nombreux facteurs sont indispensables à la néovascularisation dont des RTK. Quatre RTK
ont été identifiés comme étant responsables de la régulation de l’angiogenèse : VEGFR,
PDGFR, FGFR et Tie 1/2 (81). Nous allons les détailler et mettre en évidence leur rôle
respectif dans le processus d’angiogenèse.
1. Le VEGFR
Il existe 2 récepteurs au VEGF : VEGFR 1 (Flk-1) et VEGFR 2 (KDR). VEGFR est exprimé
normalement uniquement par l’endothélium vasculaire. Son ligand est VEGF, il s’agit d’une
protéine de 46 kDa. Elle est produite normalement par les macrophages, les cellules
plasmatiques, les lymphocytes et exerce une action paracrine sur les cellules endothéliales.
L’interaction VEGF/VEGFR est impliquée dans l’induction de la prolifération des cellules
endothéliales, dans la migration endothéliale, la perméabilité vasculaire et dans la maturation
des vaisseaux (80,101). VEGF est le plus puissant inducteur de l’angiogenèse.

55
a. VEGF et cancer du sein/tumeur mammaire
Chez l’homme, le VEGF a été particulièrement étudié dans les cancers du sein et il a été
démontré que son expression est associée à un mauvais pronostic (38, 62, 145).
Plusieurs études ont été réalisées sur des tumeurs mammaires de chiennes afin d’étudier
l’association VEGF/VEGFR, son rôle dans l’angiogenèse et en tant que marqueur
pronostique. Chez le chien, l’expression de VEGF dans les tumeurs bénignes est faible
(granules petites et peu nombreuses, localisées au pôle luminal) par rapport à celle dans les
tumeurs malignes (granules larges et nombreuses, réparties de manière diffuse dans le
cytoplasme). De plus, le nombre de cellules positives au VEGF est corrélé positivement à la
densité de micro vaisseaux et ces 2 paramètres sont plus élevés dans les cellules malignes les
moins différenciées par rapport aux cellules bénignes. L’angiogenèse et la malignité
augmentent donc conjointement. Le nombre de cellules endothéliales positives au Flk-1 est
supérieur dans le cas des tumeurs malignes et il augmente en fonction du grade tumoral (40,
42, 127,128).
Les métastases sont favorisées par l’induction de nouveaux vaisseaux et par l’augmentation
de la perméabilité des vaisseaux. L’angiogenèse augmente donc le risque de diffusion
métastatique (127, 128).
Néanmoins, Millanta et al. ont étudié 28 tumeurs mammaires de chiennes et 48 tumeurs
mammaires de chattes et ont déduit que :
-il n’y a pas de corrélation entre l’expression de VEGF et les paramètres clinicopathologiques
d’une part ni avec le taux de survie d’autre part. Il n’y a donc pas de corrélation entre une
augmentation de l’expression de VEGF et un mauvais pronostic.
-la densité en micro vaisseaux est corrélée au type histologique de la tumeur, au grade de la
tumeur et au taux de survie mais pas à l’invasion lymphatique ni au nombre de cellules
positives au VEGF (contrairement à l’étude précédente ce qui peut s’expliquer par des
techniques différentes pour quantifier les 2 paramètres). Une forte densité en micro vaisseaux
est associée à un mauvais pronostic. De plus, l’étude de la répartition de VEGF et de VEGFR
permet de déduire qu’une boucle autocrine et/ou paracrine est possible (101).
Une étude sur des souris a consisté à injecter des lignées cellulaires de différentes tumeurs
humaines. Un traitement par un anticorps anti-VEGF montre une inhibition de la croissance
tumorale et une diminution de la densité en micro vaisseaux. Cela montre que VEGF est
impliqué dans la croissance tumorale via l’angiogenèse et constitue une cible thérapeutique
intéressante (63).
En résumé, l’interaction VEGF/VEGFR semble donc impliquée dans l’angiogenèse
tumorale. Celle-ci est associée à un mauvais pronostic dans les tumeurs mammaires chez
le chien et le cancer du sein chez la femme. VEGF agit via une boucle autocrine et
paracrine. Son rôle dans la croissance tumorale en fait donc une cible thérapeutique très
prometteuse.

56
b. VEGF et mastocytose/mastocytome
Chez l’homme, la mastocytose est une maladie peu fréquente. L’étiologie est souvent une
mutation de la protéine c-Kit. L’implication de VEGF/VEGFR n’a pas été démontrée.
Une étude portant sur 18 chiens a montré que les cellules mastocytaires néoplasiques
expriment VEGF et VEGFR mais VEGF n’est pas utilisé comme un facteur autocrine ;
malgré le fait que dans de nombreux cancers chez l’homme, les cellules utilisent VEGF
comme un facteur de croissance autocrine (124).
Comme dans le cas du cancer du sein et des tumeurs mammaires, la densité en
microvaisseaux, marqueur de l’angiogenèse, est corrélée positivement au grade tumoral et à la
présence de métastases dans les mastocytomes (99, 116,121).
En résumé, le rôle de l’interaction VEGF/VEGFR n’a pas été démontré dans la
mastocytose chez l’homme. Il semblerait que cette association soit impliquée dans le
mastocytome chez le chien mais en l’absence d’une boucle autocrine.
2. Le PDGFR
Le PDGFR est exprimé par le stroma et les péricytes. Son ligand est PDGF. Chez l’homme,
l’interaction PDGF-PDGFR est impliquée dans la prolifération et la migration endothéliale.
Une étude chez l’homme a montré que le PDGF-C (un ligand du PDGFR) joue un rôle
essentiel dans l’angiogenèse, indépendamment de la voie impliquant VEGF. Le PDGF-C agit
directement sur les cellules vasculaires (péricytes, cellules musculaires lisses, cellules
endothéliales), il stimule les fibroblastes du stroma et les macrophages (77).
3. Le FGFR
FGFR est exprimé dans l’endothélium vasculaire. Son ligand est FGF. Il a été montré que
chez l’homme, il agit de façon synergique avec le VEGF pour induire l’expression de VEGF.
4. Le Tie 1/2
Tie1 et Tie2 sont des récepteurs des angiopoïétines Ang1 et Ang2. Ils sont exprimés dans les
vaisseaux sanguins retrouvés dans différents types de tumeurs chez l’homme. Ils sont
impliqués dans le recrutement de péricytes et de cellules musculaires lisses ainsi que dans le
maintien de l’intégrité vasculaire.
VEGFR et Tie1/2 coordonnent le processus d’angiogenèse.

57
En résumé, plusieurs facteurs sont impliqués dans le contrôle de l’angiogenèse chez
l’homme et chez le chien. Il s’agit de VEGFR, PDFR, FGFR et Tie 1 et 2. Chez le chien,
l’homme et la souris, l’interaction VEGF/VEGFR est souvent impliquée dans
l’angiogenèse tumorale par la surexpression de VEGFR par les cellules néoplasiques.
L’intérêt de VEGFR est à la fois pronostique et thérapeutique.
La contribution des autres facteurs au processus cancérigène est suspectée chez
l’homme et chez le chien mais n’a pas été démontrée à ce jour.
Le processus d’angiogenèse tumorale est essentiel pour la croissance tumorale et pour la
dissémination métastatique.

58
D. Exemple de la protéine c-Kit
1. Protéine c-Kit et mastocytome/mastocytose
Chez l’homme et le chien, le rôle de l’oncogène codant pour c-Kit semble être déterminant
dans l’étiopathogénie des mastocytomes.
Il y a 88% d’homologie de la séquence en acides aminés de la protéine c-Kit entre l’homme et
le chien (91, 147).
a. Mise en évidence de la présence de la protéine c-Kit dans les
mastocytomes
L’étude de London et al. concerne uniquement des mastocytomes de chien de grade 2, les
méthodes utilisées sont l’immunohistochimie, la cytométrie de flux et la PCR. Les auteurs
concluent que les cellules mastocytaires néoplasiques de grade 2 expriment la protéine c-Kit
et son ARNm et qu’elle garde la possibilité de se lier à son ligand, le SCF. De plus, ils ont
montré que des sites antigéniques de c-Kit sont conservés entre l’homme et le chien, et des
homologies de l’ARNm codant pour c-Kit sont présentes entre le chien, l’homme, et la souris
(85).
L’étude de Reguera et al. a été réalisée sur des mastocytomes de différents grades. Ils ont mis
en évidence par immunohistochimie que les mastocytomes canins expriment la protéine c-Kit.
Plus le grade tumoral augmente, plus l’expression de la protéine est importante. Dans la
plupart des mastocytomes, la protéine c-Kit est localisée dans le cytoplasme contrairement
aux mastocytes normaux où elle se localise dans la membrane (125).
En conclusion, les mastocytes tumoraux expriment la protéine c-Kit et son ARNm mais
sa localisation est anormale dans la plupart des mastocytes néoplasiques.
b. Méthode de détection des mutations de Kit
Classiquement, la méthode utilisée pour détecter les mutations de c-Kit est l’amplification
d’une partie du gène par PCR suivis d’une électrophorèse sur gel d’agarose.
Les limites de cette technique sont :
- l’incapacité à détecter des insertions ou délétions dont la taille est inférieure à 9-10 pb
- la difficulté à détecter des bandes de faible intensité
- la nécessité d’utiliser un haut pourcentage de gel d’agarose avec un coût financier
important
- la nécessité de séquencer le produit de la PCR pour déterminer la taille exacte de chaque
ITD

59
Une nouvelle technique est proposée par Jones et al. : Elle consiste en l’utilisation de
l’électrophorèse sur gel polyacrylamide fluorescent appelée méthode PAGE (61).
Figure 14 : Comparaison de l’électrophorèse sur gel d’agarose et de la méthode PAGE (61)
a) : Electrophorèse sur gel d’agarose de produits PCR issus de 8 mastocytomes de grade II
avec et sans ITD. L’ITD est figurée par une flèche verticale.
b) : Des images typiques de la méthode PAGE sont représentées.
-A gauche, un gel avec uniquement des protéines c-Kit normales est représenté. Le
produit issu de la PCR est bleu, les marqueurs de poids moléculaire sont rouges.
-Au centre, un gel avec uniquement des protéines c-Kit présentant une ITD est
représenté. Les flèches verticales représentent le produit issu de la PCR généré par
l’ITD.
-A droite est figurée l’analyse du Genescan qui permet de calculer la taille de chaque
ITD. Les marqueurs de poids moléculaire sont représentés par des pics larges rouges
et les produits issus de la PCR sont représentés par des pics fins.
Ce nouveau procédé permet la détection d’insertion ou délétion de 1 à 2 pb, les bandes de
faible intensité peuvent être détectées et leur taille peut être déterminée.
Une comparaison des 2 méthodes sur plusieurs échantillons de mastocytomes de chien a
permis de montrer que la méthode PAGE est plus efficace, plus précise et plus sensible que la
méthode classique.
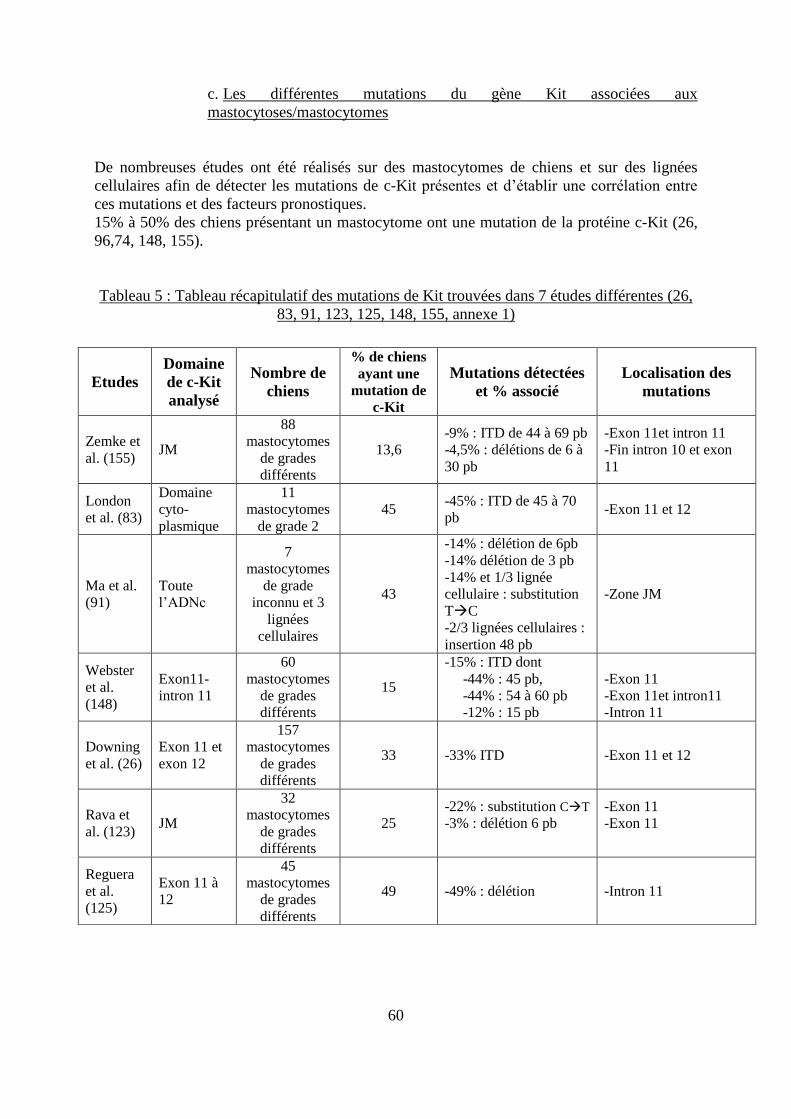
60
c. Les différentes mutations du gène Kit associées aux
mastocytoses/mastocytomes
De nombreuses études ont été réalisés sur des mastocytomes de chiens et sur des lignées
cellulaires afin de détecter les mutations de c-Kit présentes et d’établir une corrélation entre
ces mutations et des facteurs pronostiques.
15% à 50% des chiens présentant un mastocytome ont une mutation de la protéine c-Kit (26,
96,74, 148, 155).
Tableau 5 : Tableau récapitulatif des mutations de Kit trouvées dans 7 études différentes (26,
83, 91, 123, 125, 148, 155, annexe 1)
Etudes
Domaine
de c-Kit
analysé
Nombre de
chiens
% de chiens
ayant une
mutation de
c-Kit
Mutations détectées
et % associé
Localisation des
mutations
Zemke et
al. (155) JM
88
mastocytomes
de grades
différents
13,6
-9% : ITD de 44 à 69 pb
-4,5% : délétions de 6 à
30 pb
-Exon 11et intron 11
-Fin intron 10 et exon
11
London
et al. (83)
Domaine
cyto-
plasmique
11
mastocytomes
de grade 2
45 -45% : ITD de 45 à 70
pb -Exon 11 et 12
Ma et al.
(91)
Toute
l’ADNc
7
mastocytomes
de grade
inconnu et 3
lignées
cellulaires
43
-14% : délétion de 6pb
-14% délétion de 3 pb
-14% et 1/3 lignée
cellulaire : substitution
TC
-2/3 lignées cellulaires :
insertion 48 pb
-Zone JM
Webster
et al.
(148)
Exon11-
intron 11
60
mastocytomes
de grades
différents
15
-15% : ITD dont
-44% : 45 pb,
-44% : 54 à 60 pb
-12% : 15 pb
-Exon 11
-Exon 11et intron11
-Intron 11
Downing
et al. (26)
Exon 11 et
exon 12
157
mastocytomes
de grades
différents
33 -33% ITD -Exon 11 et 12
Rava et
al. (123) JM
32
mastocytomes
de grades
différents
25
-22% : substitution CT
-3% : délétion 6 pb
-Exon 11
-Exon 11
Reguera
et al.
(125)
Exon 11 à
12
45
mastocytomes
de grades
différents
49 -49% : délétion -Intron 11

61
Chez le chien, les mutations les plus fréquemment retrouvées sont localisées dans la partie du
gène codant pour le domaine JM c'est-à-dire les exons 11 et 12 et l’intron 11. Ces mutations
consistent en une duplication d’une partie du gène dont la taille varie de 3 à 79 pb. Cette
duplication est appelée ITD. On trouve également des insertions et des délétions dans l’exon
11 (26, 83, 91, 123, 125, 148, 155). Cependant, les études présentées dans ce tableau ne sont
pas toutes comparables car la zone analysée de l’ADN n’est pas la même.
D’autres mutations concernant le domaine extracellulaire ou le domaine phosphotranférase
ont été trouvées lors d’une vaste étude sur 191 chiens atteints de mastocytomes (74).
Cette étude a mise en évidence que :
-26,2% (50 chiens) présentent une mutation du gène Kit
-32 chiens ont une mutation dans l’exon 11 dont 25 sont des ITD entre les résidus 571 et 590.
Il y a également des insertions et délétions entre les résidus 555 et 559
-9 chiens ont une mutation dans l’exon 8 dont 8 sont des ITD (duplication de 12 pb entraînant
une insertion de 4 acides aminés) et une substitution
-8 chiens ont une mutation dans l’exon 9 (substitution)
-1 chien a une mutation dans l’exon 17, il s’agit d’une délétion couplée à une insertion (chez
l’homme, il s’agit de la majorité des cas).

62
Figure 15 : Résumé de l’ensemble des mutations de Kit trouvées dans l’étude de Letard et al.
(74)
Figure 16 : Localisation et fréquence des mutations de Kit identifiées dans l’étude de Letard
et al. (74)
TM : domaine transmembranaire
JM : domaine juxtamembranaire
TK : domaine tyrosine kinase
Ig : domaine de type immunoglobuline

63
Dans la mastocytose, la mutation retrouvée sur l’exon 17 est fréquente chez l’homme et des
mutations sur les exons 8 et 9 sont souvent retrouvées chez l’enfant.
En conclusion, les mutations du domaine JM sont fréquentes chez le chien et la plus fréquente
est une ITD impliquant les exons 11 voire 12. Elle est retrouvée plus fréquemment dans les
mastocytomes de grades élevés. On trouve également des mutations dans l’intron 11 et dans
les exons 8 et 9 (mutations commune dans les cas de mastocytoses chez l’enfant).
Les mutations dans le domaine kinase sont retrouvées très peu souvent et semblent de ce fait
peu impliquées dans la progression des mastocytomes canins (74, 147).
Chez certains chiens atteints de mastocytomes, aucune mutation du gène codant pour la
protéine c-Kit n’a été mise en évidence ce qui laisse penser que d’autres mécanismes
indépendants des mutations de c-Kit sont impliqués dans la transformation néoplasique des
mastocytes. En effet, Ohmori et al. ont étudié une lignée cellulaire canine issue d’un
mastocytome de grade 2. Ils n’ont retrouvé aucune mutation de c-Kit et ont montré que ces
cellules sont capables de proliférer en présence d’un ITK le STI575 à la dose de 10 µm (111).
Chez l’homme, la mutation la plus fréquente en cas de mastocytose de l’adulte est asp816val.
Elle n’est pas retrouvée chez le chien (112). Elle est localisée dans l’exon 17 codant pour la
boucle d’activation et est retrouvée chez plus de 90% des adultes atteints de mastocytose.
D’autres mutations nettement moins fréquentes sont retrouvées dans la boucle d’activation, la
zone JM, la partie transmembranaire et le domaine extracellulaire (<5%). On note que,
contrairement au chien, très peu de mutations sont retrouvées dans l’exon 11 (112). Chez
l’enfant, la mutation asp816val est très peu fréquente et lorsqu’on la retrouve elle est
responsable d’une maladie atypique agressive et persistante contrairement à la maladie
classique de l’enfant. Cette mutation au codon 816 a également été mise en évidence chez les
patients atteints de mastocytose avec désordre hématologique associé et dans la lignée
cellulaire HMC-l issue de leucémie mastocytaire (37, 107).

64
Tableau 6 : Tableau récapitulatif des mutations retrouvées chez l’homme lors de mastocytose
(112, annexe 1)
Domaine de c-Kit
correspondant
Exon Mutation Conséquence Fréquence Maladie fréquemment
associée
Extracellulaire 8
Délétion
D419
Inconnu
<5% Mastocytose
systémique familiale
9 Substitution
K509I
Inconnu <5% Mastocytose
systémique familiale
Transmembranaire 10 Substitution
F522C
Activatrice <5% Mastocytose
systémique
10 Substitution
A533D
Activatrice <5% Mastocytose cutané
familiale
JM 11 Substitution
V559I
Activatrice <5% Mastocytose
systémique agressive
11 Substitution
V560G
Activatrice <5% Mastocytose
systémique indolente et
leucémie mastocytaire
Boucle
d’activation
17 Substitution
R815K
Inconnu <5% Urticaire pigmentaire
pédiatrique
17 Substitution
D816V
Activatrice >90% Mastocytose
systémique adulte
17 Substitution
D816Y
Activatrice <5% Mastocytose
systémique
17 Substitution
D816H
Inconnu <5% Mastocytose
systémique et leucémie
myéloblastique aiguë
17 Substitution
D816F
Activatrice <5% Mastocytose
systémique
17 Substitution
I817V
Inconnu <5% Mastocytose
systémique bien
différenciée
17 Insertion
V815-I816
Inconnu <5% Mastocytose
systémique
17 Substitution
D820G
Inconnu <5% Mastocytose
systémique agressive
17 Substitution
E839K
Activatrice <5% Urticaire pigmentaire

65
d. Mise en évidence d’un hot spot et d’un polymorphisme du gène Kit
Des mutations du gène codant pour la protéine c-Kit sans conséquence sur la séquence en
acides aminés de la protéine sont retrouvées dans le domaine JM mais aussi dans d’autres
régions du gène, on parle de polymorphisme génétique (74, 111, 155).
Les mutations du gène codant pour c-Kit tel que les duplications, délétions et substitutions
ainsi que le polymorphisme de ce gène en font un hotspot pour les mutations chez le chien.
e. Conséquences de ces mutations
Chez l’homme
Une analyse de la mutation tyr814asp (équivalente à la mutation asp816val chez l’homme) de
la lignée cellulaire murine P-815 a montré que celle-ci est responsable d’une phosphorylation
constitutive de c-Kit en l’absence de ligand et d’une activation de l’activité kinase de la
protéine c-Kit. Cette activation n’est pas due à une stimulation autocrine car l’ARNm du SCF
n’est pas présent dans les cellules (144).
D’autres études et notamment une étude sur la lignée cellulaire HMC-l (leucémie
mastocytaire humaine) ont montré que la mutation asp816val entraîne une activation de la
protéine c-Kit en l’absence de ligand (37, 107).
Les conséquences de cette activation sont :
-une augmentation de la prolifération et de la survie des mastocytes
-une altération de la migration et de l’adhésion des mastocytes
-une augmentation de la dégranulation (112).
Chez le chien
La protéine c-Kit mutée est phosphorylée en l’absence de ligand et les cellules prolifèrent
indépendamment de la présence de ligand (74, 83, 91, 141).
Les mutations au niveau du domaine JM, qui sont les plus fréquentes, entraînent son
changement de conformation. Or le domaine JM est impliqué dans l’auto-inhibition de la
protéine. On comprend donc que sa perte de fonction entraîne une activation de la protéine.
L’activation de la protéine c-Kit est responsable d’une prolifération cellulaire incontrôlée. Les
mutations à l’origine de cette activation semble donc avoir un rôle essentiel dans la
pathogénie des mastocytomes chez l’homme et chez le chien.
f. Localisation anormale de la protéine c-Kit
La protéine c-Kit est localisée normalement au niveau de la membrane cellulaire. Or, lors de
mutations, on distingue différentes localisations de la protéine : distribution cytoplasmique
focale à ponctuelle ou diffuse. Des études ont montré que la distribution anormale de c-Kit est
associée à une augmentation de la prolifération cellulaire (147, 148, 149).

66
g. Intérêt de la détection des mutations du gène Kit
La mise en évidence d’une mutation d’un RTK à l’origine de la transformation tumorale
permet de sélectionner un ITK susceptible d’avoir une action sur la tumeur.
En fonction du type de mutation, les cellules sont plus ou moins sensibles à certains ITK.
Connaitre la localisation de la mutation permet de prédire l’effet d’un ITK et donc de pouvoir
choisir l’ITK le plus adapté.
De plus, certains ITK tels que le masitinib ont une activité plus importante sur les
mastocytomes dont la protéine c-Kit est mutée (30).
h. Intérêt diagnostique de la protéine c-Kit
Le diagnostic d’un mastocytome se fait par examen histopathologique et l’un des critères
diagnostiques est la présence de granules intra-cytoplasmiques.
Or dans le cas de mastocytomes très peu différenciés, les granules peuvent ne pas être
présentes et un mastocytome peut être confondu par exemple avec un lymphome.
La biologie moléculaire a donc tout son intérêt. En effet, la mise en évidence d’une ITD dans
l’exon 11 de c-Kit permet de différencier un mastocytome d’un lymphome car cette mutation
est absente dans les lymphomes (154).
A l’avenir, il serait donc intéressant de chercher des mutations c-Kit dans un but diagnostique,
pronostique et thérapeutique.
En résumé, la protéine c-Kit est impliquée dans les mastocytomes chez les chiens via des
mutations, la plus commune étant l’ITD localisée dans l’exon 11. Des mécanismes autres
que les mutations de la protéine c-Kit entrent en jeu dans l’étiologie des mastocytomes.
Chez l’homme, la mutation asp816val est la plus fréquente. Ces mutations sont détectées
habituellement par amplification par PCR d’une partie du gène suivi d’une
électrophorèse sur gel d’agarose. Une nouvelle technique est proposée, elle consiste en
l’utilisation de l’électrophorèse sur gel polyacrylamide fluorescent (méthode PAGE).
Ces mutations entraînent une activation constitutive de la protéine c-Kit responsable
d’une croissance cellulaire incontrôlée. L’intérêt de la détection de ces mutations est à la
fois diagnostique, pronostique et thérapeutique.
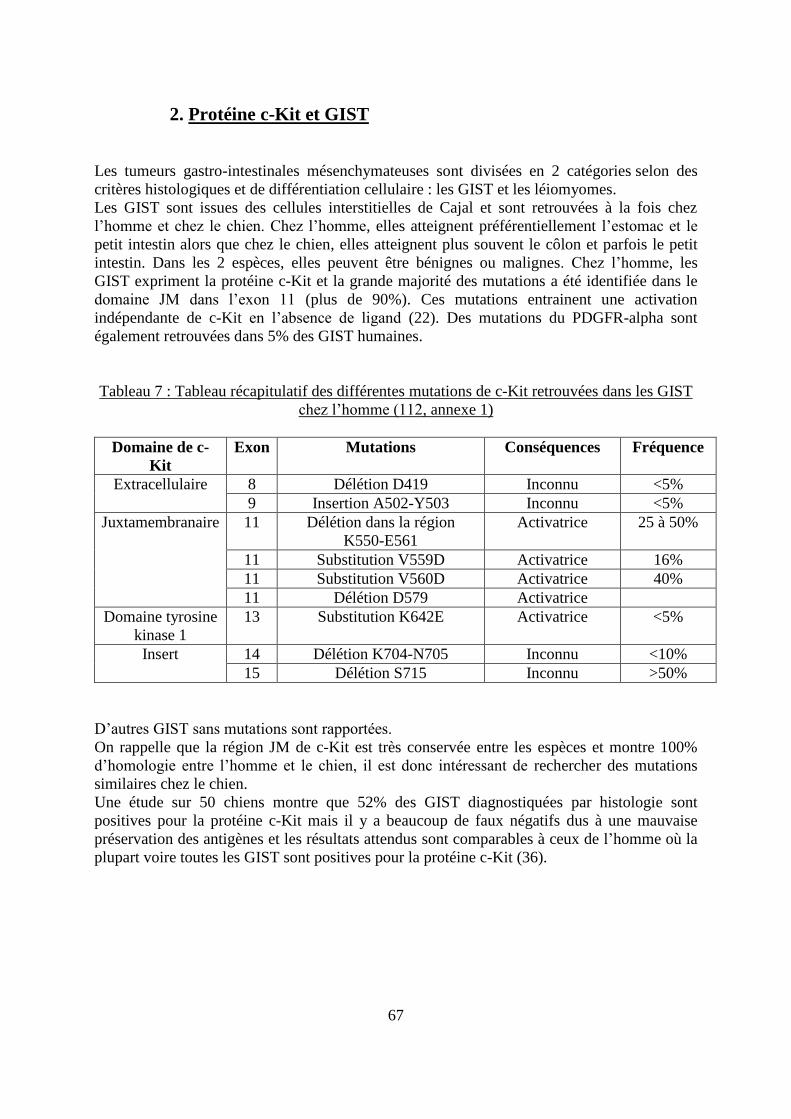
67
2. Protéine c-Kit et GIST
Les tumeurs gastro-intestinales mésenchymateuses sont divisées en 2 catégories selon des
critères histologiques et de différentiation cellulaire : les GIST et les léiomyomes.
Les GIST sont issues des cellules interstitielles de Cajal et sont retrouvées à la fois chez
l’homme et chez le chien. Chez l’homme, elles atteignent préférentiellement l’estomac et le
petit intestin alors que chez le chien, elles atteignent plus souvent le côlon et parfois le petit
intestin. Dans les 2 espèces, elles peuvent être bénignes ou malignes. Chez l’homme, les
GIST expriment la protéine c-Kit et la grande majorité des mutations a été identifiée dans le
domaine JM dans l’exon 11 (plus de 90%). Ces mutations entrainent une activation
indépendante de c-Kit en l’absence de ligand (22). Des mutations du PDGFR-alpha sont
également retrouvées dans 5% des GIST humaines.
Tableau 7 : Tableau récapitulatif des différentes mutations de c-Kit retrouvées dans les GIST
chez l’homme (112, annexe 1)
Domaine de c-
Kit
Exon Mutations Conséquences Fréquence
Extracellulaire 8 Délétion D419 Inconnu <5%
9 Insertion A502-Y503 Inconnu <5%
Juxtamembranaire 11 Délétion dans la région
K550-E561
Activatrice 25 à 50%
11 Substitution V559D Activatrice 16%
11 Substitution V560D Activatrice 40%
11 Délétion D579 Activatrice
Domaine tyrosine
kinase 1
13 Substitution K642E Activatrice <5%
Insert 14 Délétion K704-N705 Inconnu <10%
15 Délétion S715 Inconnu >50%
D’autres GIST sans mutations sont rapportées.
On rappelle que la région JM de c-Kit est très conservée entre les espèces et montre 100%
d’homologie entre l’homme et le chien, il est donc intéressant de rechercher des mutations
similaires chez le chien.
Une étude sur 50 chiens montre que 52% des GIST diagnostiquées par histologie sont
positives pour la protéine c-Kit mais il y a beaucoup de faux négatifs dus à une mauvaise
préservation des antigènes et les résultats attendus sont comparables à ceux de l’homme où la
plupart voire toutes les GIST sont positives pour la protéine c-Kit (36).

68
La recherche de mutations dans le domaine JM de la protéine c-Kit chez le chien met en
évidence (cf. fig.17) :
- une délétion du tryptophane au codon 556 et de la lysine au codon 557 (équivalente chez
l’homme à la délétion de tryptophane au codon 557 et de la lysine au codon 558)
associée à une duplication de la glutamine au codon 555 (équivalente chez l’homme à
la duplication de la glutamine au codon 556 trouvée sur 1/300 cas)
- une substitution de la proline par la leucine au codon 575 (équivalente chez l’homme à
la substitution de la proline par la leucine au codon 576 rapportée dans quelques cas
humains)
Figure 17 : Mutations de c-Kit identifiées chez 2 chiens atteints de GIST (36, annexe 1)
Deux chiens sur quatre atteints de GIST ne présentent pas de mutations du domaine JM.
Les mutations de c-Kit identifiées chez le chien sont similaires à celles trouvées chez
l’homme et suggèrent la même voie de cancérogenèse.
Dans les léiomyomes, aucune mutation de c-Kit ni chez l’homme ni chez le chien n’a été
identifiée d’où l’intérêt diagnostique de rechercher une mutation de c-Kit dans le cas d’une
tumeur gastro-intestinale mésenchymateuse dans le but de différencier une GIST d’un
léiomyome.
De plus, les GIST malignes ont plus fréquemment des mutations de c-Kit par rapport aux
GIST bénignes suggérant que la protéine c-Kit peut être un bon marqueur pronostique (88).
En résumé, dans les GIST humaines, 90% des patients présentent une mutation de la
protéine c-Kit et la mutation la plus fréquente concerne l’exon 11. Les mutations de c-
Kit chez les chiens atteints de GIST sont similaires. La recherche de ces mutations a un
intérêt diagnostique, pronostique et thérapeutique.

69
3. Protéine c-Kit et tumeur mammaire/cancer du sein
Les tumeurs mammaires chez la chienne sont très fréquentes, elles représentent 25 à 50% de
l’ensemble des tumeurs selon les auteurs. 65% de ces tumeurs sont bénignes (67).
On a vu précédemment que l’angiogenèse via VEGFR/VEGF intervient dans la croissance
des tumeurs mammaires.
Une étude de Kubo et al. sur 11 tumeurs mammaires de chiennes (classées en tumeurs
bénignes, adénocarcinomes et tumeurs mixtes malignes) montre que le niveau d’expression de
c-Kit est élevé dans les tumeurs bénignes et les adénocarcinomes (niveau comparable à celui
de la rate ou des mastocytes) et qu’il est faible dans les tumeurs mixtes malignes (67). Le
niveau d’expression de c-Kit semble donc être relié à la nature histologique de la tumeur. Les
tumeurs mixtes bénignes sont parfois difficiles à distinguer des tumeurs mixtes malignes d’où
l’intérêt d’étudier l’expression de c-Kit pour différencier les 2 types.
Contrairement au chien, chez la femme, le niveau d’expression de c-Kit est diminué dans les
tumeurs mammaires malignes par rapport au tissu mammaire normal et aux lésions bénignes
(67,108). De plus, le cancer du sein chez la femme est associé à la surexpression de HER2
ainsi qu’à l’interaction VEGFR/VEGF intervenant dans l’angiogenèse.
En résumé, la protéine c-Kit et l’interaction VEGFR/VEGF sont impliquées dans les
tumeurs mammaires de la chienne. Le niveau d’expression de c-Kit peut être relié à la
nature histologique de la tumeur. Contrairement à la chienne, chez la femme le niveau
d’expression de c-Kit diminue lorsque le grade de la tumeur augmente. Le cancer du
sein chez la femme est également associé à la surexpression de HER2 et à l’interaction
VEGFR/VEGF.
4. Protéine c-Kit et hémangiosarcome
L’hémangiosarcome chez le chien est un cancer difficile à traiter. Il atteint le plus souvent la
rate, l’oreillette droite et le tissu sous-cutané. Il ressemble à l’angiosarcome chez l’homme.
Chez le chien, la médiane de survie est de 6 mois. Il s’agit souvent d’un cancer qui métastase
et qui infiltre localement. Une étude de Fosmire et al. sur 8 lignées cellulaires issues
d’hémangiosarcomes de chiens montre que l’expression de c-Kit permet de distinguer une
lésion maligne d’une lésion bénigne. En effet, la protéine c-Kit est détectable sur les
hémangiosarcomes mais pas sur les hémangiomes bénins (35).
De plus, la protéine c-Kit est présente uniquement sur les vaisseaux capillaires fœtaux
humains et non sur les cellules endothéliales adultes. La présence de c-Kit sur les
hémangiosarcomes de chiens et les angiosarcomes humains permet de déduire qu’ils
proviennent de cellules endothéliales primitives peu différenciées alors que les hémangiomes
humains et les hématomes canins caractérisés par l’absence de la protéine c-Kit proviennent
de cellules endothéliales matures différenciées.
En résumé, la protéine c-Kit permet de distinguer une lésion maligne d’une lésion
bénigne.

70
E. Exemple de la protéine Met (=HGFR)
La protéine Met est une PTK qui appartient à la famille des RTK Met (cf. Tableau 1). Elle est
composée de 2 chaînes liées par des ponts disulfures. Chaque chaîne est issue d’un précurseur
de 170 kDa qui est glycosylé et clivé ce qui donne un hétérodimère mature.
Le ligand de Met est HGF. Il est synthétisé et sécrété comme une simple chaîne puis converti
en hétérodimère mature par une protéase sérine, appelée HGFA. Met est exprimée
normalement dans une grande variété de cellules épithéliales et HGF est produit par les
cellules stromales.
La protéine Met canine est composée de 1382 acides aminés, elle présente 84% homologie
avec la souris et 88% d’homologie avec l’homme. Les domaines JM et kinase montrent 100%
d’homologie avec la souris et l’homme. Le ligand HGF montre 87% d’homologie avec la
souris et 90% avec l’homme et HGFA montre 74% d’homologie avec la souris et 81% avec
l’homme (79). Met, HGF et HGFA présentent donc un fort pourcentage d’homologie entre
l’homme, la souris et le chien.
Les interactions Met/HGF induisent une prolifération cellulaire, une mobilité et une
morphogenèse cellulaire. Cette interaction est importante pendant le développement
embryonnaire, la cicatrisation, la régénération tissulaire, l’angiogenèse, l’invasion cellulaire et
la différentiation cellulaire.
La protéine Met est dérégulée dans un certains nombre de cancers chez l’homme comme le
carcinome des papilles rénales, le cancer du poumon, le cancer de l’estomac et les
ostéosarcomes. Cette dérégulation est liée à des mutations de Met, une surexpression ou la
présence d’une boucle autocrine. Ces dysfonctionnements entrainent une croissance cellulaire
anormale et la présence de métastases. De plus, l’expression et/ou un fonctionnement aberrant
de la protéine est associée à un mauvais pronostic et à un grade tumoral plus élevé (79). Met
semble donc associée au développement et à la progression tumorale.
De nombreuses lignées cellulaires issues de différentes tumeurs telles que les ostéosarcomes,
le mélanome et les hémangiosarcomes expriment la protéine Met et 76% de ces lignées
expriment également les protéines HGF et HGFA d’où la possibilité qu’une boucle autocrine
soit impliquée.
Les ostéosarcomes sont des cancers agressifs souvent métastatiques et dont le pronostic est
très défavorable. Chez le chien, ils atteignent des grandes races, d’âges moyens et sont
localisés préférentiellement aux métaphyses des os longs. Le surpoids et certaines lignées
familiales sont des facteurs de risques. Le comportement clinique et biologique des
ostéosarcomes est très similaire chez l’homme et le chien. Chez l’homme, la protéine Met est
impliquée dans les ostéosarcomes pour leur fort pouvoir invasif (98). Son implication dans les
ostéosarcomes chez le chien est donc recherchée.
Des analyses de lignées cellulaires issues d’ostéosarcomes de chiens montrent que 5 lignées
sur 7 sont positives pour la protéine Met. De plus, le niveau d’expression de Met est élevé et
comparable à celui des ostéosarcomes de l’homme. L’analyse de l’expression de Met dans des
métastases d’ostéosarcomes pulmonaires met en évidence que son niveau d’expression est
supérieur par rapport à la tumeur d’origine ce qui confirme son rôle dans le caractère invasif
et métastatique. Aucune corrélation n’a pu être établie entre l’expression de Met et le devenir
des chiens (34).

71
Les fortes similarités concernant la protéine Met et les ostéosarcomes entre l’homme et le
chien permettent de tester et de développer de nouvelles stratégies thérapeutiques telles que
les ITK sur les tumeurs du chien avant d’envisager des essais cliniques sur l’homme.
En résumé, la protéine Met est associée au développement, à la progression tumorale et
à un mauvais pronostic dans un certain nombre de cancers chez l’homme et en
particulier dans les ostéosarcomes. Elle est impliquée dans la dissémination
métastatique. Le comportement des ostéosarcomes est similaire chez le chien et
l’homme. Comme chez l’homme, la protéine Met est surexprimée dans les
ostéosarcomes chez le chien et associée au caractère invasif de la tumeur.

72
F. Exemple de la protéine PTEN
Le gène codant pour la protéine PTEN est appelée gène suppresseur de tumeur. Comme nous
l’avons vu précédemment, la protéine PTEN est une phosphatase qui agit en déphosphorylant
le PIP3. Elle régule les niveaux de PIP2 et PIP3 (cf. fig.11).
Or le PIP3 agit en activant des protéines kinases (ex : Akt) impliquées dans la régulation de la
prolifération et de la durée de vie cellulaire. Un dérèglement de PTEN peut donc avoir de
graves conséquences sur la cellule.
Il a été montré que la protéine PTEN inhibe la prolifération cellulaire, la migration et
l’invasion de cellules tumorales.
Dans de nombreux cancers chez l’homme tels que les carcinomes de l’endomètre, de la
prostate, du rein et du sein et les glioblastomes, PTEN est mutée. PTEN n’a pas été identifiée
dans les ostéosarcomes chez l’homme (81, 114).
Chez le chien, une étude sur 5 lignées cellulaires issues d’ostéosarcomes, indique que la
protéine PTEN est absente dans 3 sur 5 lignées (lignées CO2, CO5 et CO7) ; et dans 4 sur 5
lignées (lignées CO2, CO3, CO5 et CO7) la protéine Akt est hautement phosphorylée. Dans
les lignées où PTEN est absente, on observe une délétion d’une grande partie du gène. Dans la
lignée CO3, on observe une substitution de l’adénine en thymine au nucléotide 1126 du codon
340 entraînant une substitution d’une tyrosine en asparagine. L’étude de 15 ostéosarcomes de
chien montre que dans 10 cas la protéine est absente. L’évaluation par Southern blotting
d’une tumeur avec absence de PTEN indique une délétion d’une grande partie du gène codant
pour PTEN (75).
En conclusion, PTEN est régulée négativement ou mutée dans un grand nombre
d’ostéosarcomes de chien contrairement à l’homme.
Chez l’homme, on retrouve une délétion du gène PTEN souvent associée à la perte de
fonction de la protéine dans les cancers cités ci-dessus. La substitution d’une tyrosine n’a pas
été mise en évidence chez l’homme.
Une possible restauration de l’activité de PTEN peut donc être une piste thérapeutique.
En résumé, la phosphatase PTEN est mutée dans les ostéosarcomes chez le chien ce qui
entraîne sa perte de fonction, cela n’a pas été mis en évidence chez l’homme.
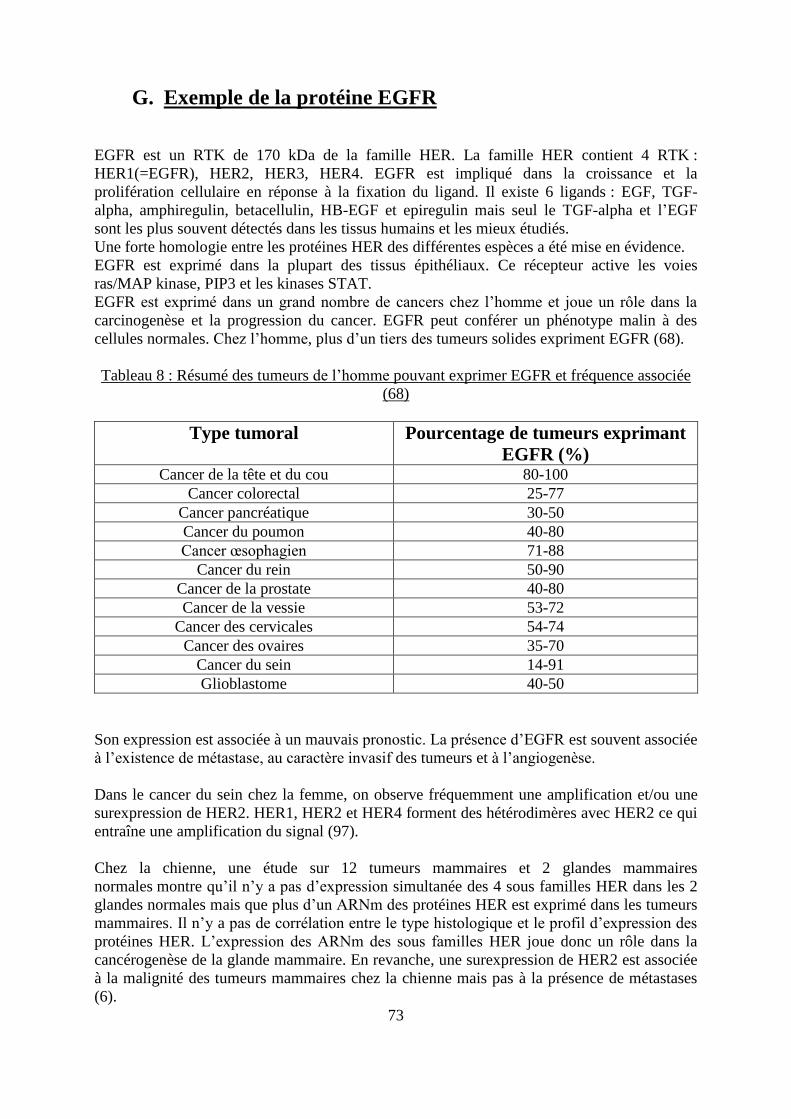
73
G. Exemple de la protéine EGFR
EGFR est un RTK de 170 kDa de la famille HER. La famille HER contient 4 RTK :
HER1(=EGFR), HER2, HER3, HER4. EGFR est impliqué dans la croissance et la
prolifération cellulaire en réponse à la fixation du ligand. Il existe 6 ligands : EGF, TGF-
alpha, amphiregulin, betacellulin, HB-EGF et epiregulin mais seul le TGF-alpha et l’EGF
sont les plus souvent détectés dans les tissus humains et les mieux étudiés.
Une forte homologie entre les protéines HER des différentes espèces a été mise en évidence.
EGFR est exprimé dans la plupart des tissus épithéliaux. Ce récepteur active les voies
ras/MAP kinase, PIP3 et les kinases STAT.
EGFR est exprimé dans un grand nombre de cancers chez l’homme et joue un rôle dans la
carcinogenèse et la progression du cancer. EGFR peut conférer un phénotype malin à des
cellules normales. Chez l’homme, plus d’un tiers des tumeurs solides expriment EGFR (68).
Tableau 8 : Résumé des tumeurs de l’homme pouvant exprimer EGFR et fréquence associée
(68)
Type tumoral Pourcentage de tumeurs exprimant
EGFR (%) Cancer de la tête et du cou 80-100
Cancer colorectal 25-77
Cancer pancréatique 30-50
Cancer du poumon 40-80
Cancer œsophagien 71-88
Cancer du rein 50-90
Cancer de la prostate 40-80
Cancer de la vessie 53-72
Cancer des cervicales 54-74
Cancer des ovaires 35-70
Cancer du sein 14-91
Glioblastome 40-50
Son expression est associée à un mauvais pronostic. La présence d’EGFR est souvent associée
à l’existence de métastase, au caractère invasif des tumeurs et à l’angiogenèse.
Dans le cancer du sein chez la femme, on observe fréquemment une amplification et/ou une
surexpression de HER2. HER1, HER2 et HER4 forment des hétérodimères avec HER2 ce qui
entraîne une amplification du signal (97).
Chez la chienne, une étude sur 12 tumeurs mammaires et 2 glandes mammaires
normales montre qu’il n’y a pas d’expression simultanée des 4 sous familles HER dans les 2
glandes normales mais que plus d’un ARNm des protéines HER est exprimé dans les tumeurs
mammaires. Il n’y a pas de corrélation entre le type histologique et le profil d’expression des
protéines HER. L’expression des ARNm des sous familles HER joue donc un rôle dans la
cancérogenèse de la glande mammaire. En revanche, une surexpression de HER2 est associée
à la malignité des tumeurs mammaires chez la chienne mais pas à la présence de métastases
(6).

74
En résumé, chez l’homme plus d’un tiers des tumeurs solides expriment EGFR. Son
expression est associée à un mauvais pronostic, à l’existence de métastases, et à
l’angiogenèse. Dans le cas des tumeurs mammaires chez la chienne, on retrouve la
présence des protéines EGFR mais sans corrélation avec le type histologique. De plus, la
surexpression de HER2 est corrélée à la malignité des tumeurs mammaires. Chez la
femme, on retrouve fréquemment une surexpression de HER2 dans le cancer du sein.
En conclusion, une tumeur peut être liée à la dérégulation d’une ou plusieurs tyrosines
kinases. On retrouve fréquemment le même comportement biologique entre les tumeurs chez
l’homme et le chien ce qui permet des études cliniques associées entre les 2 espèces.
En effet, le chien est un bon modèle animal qui peut permettre de développer de nouvelles
molécules anticancéreuses comme les ITK.

75
III. Les inhibiteurs des tyrosines kinases
Nous avons vu précédemment que de nombreux cancers sont associés à une dérégulation
d’une ou plusieurs tyrosines kinases. La stratégie thérapeutique consiste donc à cibler ces
protéines et à les inhiber. De nombreux ITK ont déjà prouvé leur efficacité chez l’homme et
sont aujourd’hui étudiés chez l’animal.
Un ITK idéal aurait comme caractéristiques :
-d’être efficace
-d’être spécifique afin de limiter les effets secondaires liés à l’action d’un ITK sur des
tyrosines kinases non ciblées
-d’atteindre facilement sa cible
-d’être facile à synthétiser
-d’avoir une bonne disponibilité orale
-d’avoir un large volume de distribution
-d’avoir une demi-vie d’élimination qui permet une prise quotidienne
-de ne pas déclencher de réponse immunitaire
-de ne pas engendrer de résistance
-d’être à un prix abordable, notamment en médecine vétérinaire
Chez l’homme, de nombreux ITK ont eu une AMM comme par exemple le Gleevec®
(imatinib), le Sutent® (sunitinib) ou l’Iressa® (gefitinib). Chez le chien, deux ITK ont obtenu
une AMM récemment ; il s’agit du Masivet® (masitinib) et du Palladia® (toceranib). Des
études sur d’autres ITK sont en cours.

76
A. Les inhibiteurs de tyrosines kinases ayant une AMM chez
le chien
1. Le Masivet® (masitinib)
Le Masivet® a obtenu l’AMM en novembre 2008 pour le traitement des mastocytomes
cutanés de grades 2 ou 3, non opérables ou récidivants après chirurgie sans atteinte
métastatique et avec mutation de la protéine c-Kit. Il a été commercialisé par le laboratoire
AB Science en 2009. Il s’agit du premier agent anticancéreux vétérinaire (41,59).
a. Structure
Le masitinib est le 2-amino-4-aryl-thiazole.
Figure 18 : Formule chimique du masitinib (30)
b. Cibles
Le masitinib agit à la fois sur des RTK et sur des NRTK. Il exerce principalement une forte
inhibition sur la protéine c-Kit, le PDGFR et les NRTK Lyn, Fyn et Lck.
L’IC50 (concentration inhibitrice à 50%) d’un ITK correspond à la concentration nécessaire
pour inhiber 50% de l’activité d’une tyrosine kinase. Elle permet de mesurer l’efficacité d’un
ITK. En effet, plus l’IC50 est faible, plus l’ITK est efficace.
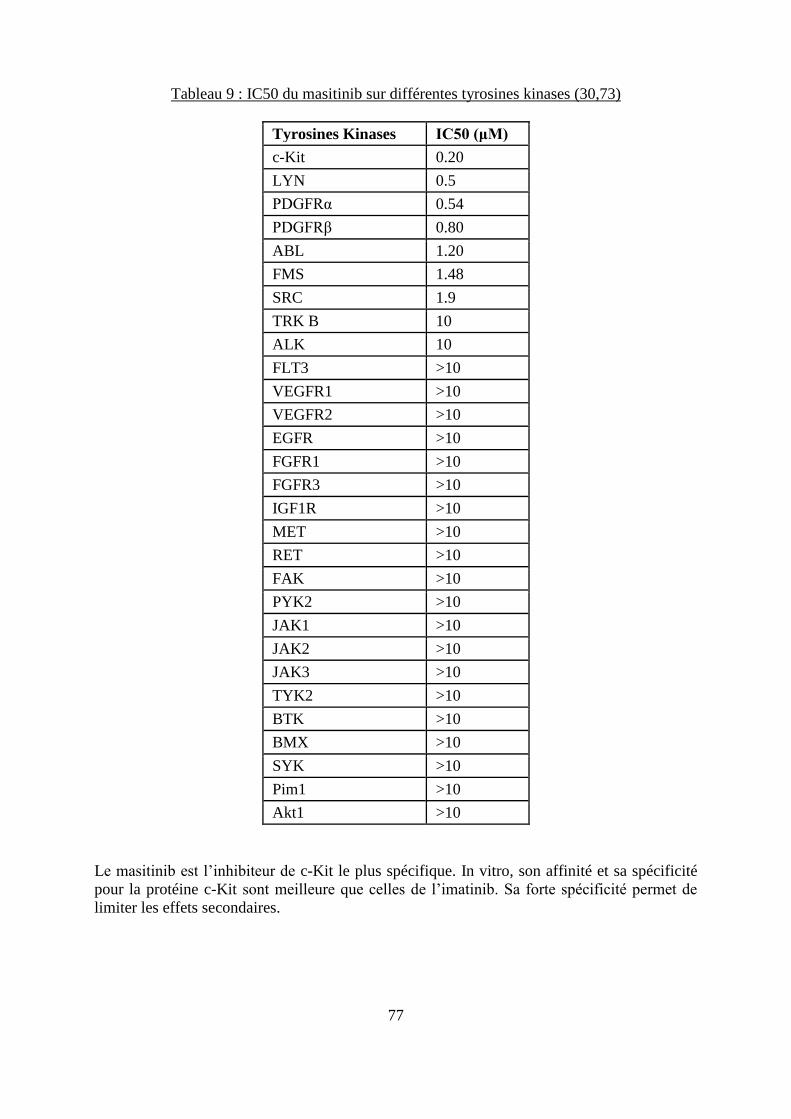
77
Tableau 9 : IC50 du masitinib sur différentes tyrosines kinases (30,73)
Tyrosines Kinases IC50 (μM)
c-Kit 0.20
LYN 0.5
PDGFRα 0.54
PDGFRβ 0.80
ABL 1.20
FMS 1.48
SRC 1.9
TRK B 10
ALK 10
FLT3 >10
VEGFR1 >10
VEGFR2 >10
EGFR >10
FGFR1 >10
FGFR3 >10
IGF1R >10
MET >10
RET >10
FAK >10
PYK2 >10
JAK1 >10
JAK2 >10
JAK3 >10
TYK2 >10
BTK >10
BMX >10
SYK >10
Pim1 >10
Akt1 >10
Le masitinib est l’inhibiteur de c-Kit le plus spécifique. In vitro, son affinité et sa spécificité
pour la protéine c-Kit sont meilleure que celles de l’imatinib. Sa forte spécificité permet de
limiter les effets secondaires.
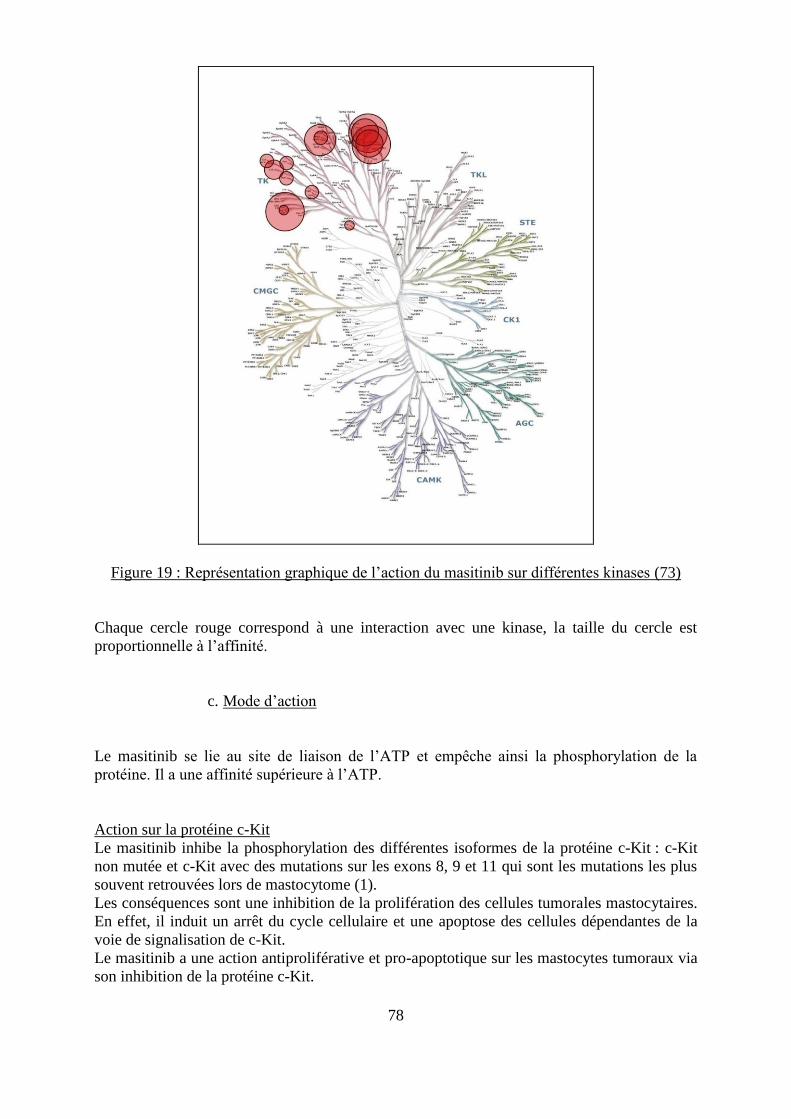
78
Figure 19 : Représentation graphique de l’action du masitinib sur différentes kinases (73)
Chaque cercle rouge correspond à une interaction avec une kinase, la taille du cercle est
proportionnelle à l’affinité.
c. Mode d’action
Le masitinib se lie au site de liaison de l’ATP et empêche ainsi la phosphorylation de la
protéine. Il a une affinité supérieure à l’ATP.
Action sur la protéine c-Kit
Le masitinib inhibe la phosphorylation des différentes isoformes de la protéine c-Kit : c-Kit
non mutée et c-Kit avec des mutations sur les exons 8, 9 et 11 qui sont les mutations les plus
souvent retrouvées lors de mastocytome (1).
Les conséquences sont une inhibition de la prolifération des cellules tumorales mastocytaires.
En effet, il induit un arrêt du cycle cellulaire et une apoptose des cellules dépendantes de la
voie de signalisation de c-Kit.
Le masitinib a une action antiproliférative et pro-apoptotique sur les mastocytes tumoraux via
son inhibition de la protéine c-Kit.

79
Action sur le système Lyn/FAK
Lyn appartient à la famille des Src, elle est exprimée essentiellement dans les cellules
hématopoïétiques et dans le tissu nerveux. Lyn entraîne la dégranulation des mastocytes
dépendante des immunoglobulines E. Elle active la protéine NRTK FAK. La protéine FAK
est impliquée dans l’adhésion, l’invasion et la formation de métastases ainsi que dans la
résistance aux chimiothérapies. Elle influence de plus la prolifération et la survie cellulaire
(110).
Lyn est sur-exprimée dans un certains nombres de cancers comme la leucémie myéloïde
chronique et la leucémie lymphocytaire chronique chez l’homme ; et cette protéine a été
associée à certaines formes de résistance à l’imatinib et à la formation de métastases.
En bloquant la protéine Lyn, le masitinib permet de limiter la formation de métastases et
l’apparition de résistance ainsi que la dégranulation des mastocytes.
Action sur le PDGFR
En agissant sur les PDGFR, le masitinib a une action anti-tumorale, anti-angiogénique et
permet de resensibiliser la tumeur aux agents de chimiothérapie. En effet, il a été montré que
l’inhibition du PDGFR-béta réduit la pression interstitielle intra-tumorale et augmente ainsi
l’assimilation des agents de chimiothérapie.
Voici en résumé les cibles du masitinib, son mode d’action et les maladies
potentiellement traitées :
Tableau 10 : Rôles du masitinib dans différents maladies (110)
Cibles Actions Maladies potentiellement
traitées
c-Kit, PDGFR, Lyn/FAK Inhibition des tyrosines
kinases
Mastocytome, lymphome à
cellule T, mélanome,
hémangiosarcome splénique
PDGFR, Lyn/FAK Potentialise les effets des
agents de chimiothérapies
Tumeurs traitées par
chimiothérapie
Mastocytes via c-Kit/Lyn Inhibition de l’activation des
mastocytes
Dermatite atopique, arthrite,
asthme, MICI

80
d. Protocole
La posologie recommandée est de 12.5 mg/kg une fois par jour per os. Le Masivet® est
conditionné en boite de 30 comprimés de 50 mg et 150 mg (1).
En fonction des effets adverses rencontrés, le traitement peut être interrompu, réduit à 9
mg/kg/jour ou 6 mg/kg/jour ou arrêté transitoirement.
Un arrêt définitif doit être entrepris en cas de :
• hausse anormale des facteurs rénaux
• anémie hémolytique non régénérative
• et si une neutropénie sévère, et/ou une diarrhée sévère, et/ou des
vomissements sévères persistent après diminution de la dose
journalière.
Lorsque la maladie est stable, le traitement doit être poursuivi.
Un suivi très régulier de la fonction rénale et hépatique est préconisé.
e. Efficacité du masitinib sur les mastocytomes
On rappelle que la réponse objective est une réponse complète ou partielle au traitement.
Une réponse complète est définie par une disparition des signes de la maladie évalués à au
moins 3 semaines d’intervalle. Une réponse partielle est définie par une réduction supérieure à
30% de la somme des diamètres les plus longs des lésions cibles mesurée à au moins 3
semaines d’intervalle. Une maladie est qualifiée de stable lorsque la maladie ne progresse pas.
Une progression de la maladie est définie par une augmentation d’au moins 20% de la somme
des diamètres des lésions cibles, de la progression de lésions non cibles ou de l’apparition de
nouvelle lésion.
Une maladie contrôlée correspond à une réponse complète, partielle ou une maladie stable.
Une maladie progressive est définie lorsque la maladie n’est pas contrôlée.
L’activité biologique est définie par le pourcentage de chiens ayant eu une réponse (complète
ou partielle) au traitement et ceux chez lesquels la maladie est stable.
Le TTP correspond à la durée entre le 1er
jour de traitement et le jour où la maladie progresse.
Une étude de grande envergure de Hahn et al. a été réalisé sur 202 chiens de races différentes
afin d’évaluer l’efficacité du masitinib pendant 6 mois (45). Puis le taux de survie à 12 et 24
mois après le début du traitement a été évalué sur 132 chiens de cette étude (44). Les chiens
présentaient un mastocytome cutané de grade 2 ou 3, non opérable ou récidivant après
chirurgie, sans atteinte métastatique.

81
Ces études ont permis de conclure que :
-le masitinib induit une bonne réponse tumorale (réponse complète ou partielle).
Tableau 11 : Comparaison des taux de réponse au Masivet® versus placebo à 6 mois chez des
chiens atteints de mastocytome cutané (45)
Masivet® (n=161) Placebo (n=41) Valeur de p
50% 29% 0.020
n=nombre de chiens
-le masitinib retarde la progression de la tumeur quelque soit le statut de c-Kit (mutée ou
non). La chirurgie reste l’option thérapeutique de choix lorsqu’elle est possible. Cependant,
lorsque la tumeur n’est pas opérable, le masitinib retarde significativement la progression du
mastocytome (TTP de 173 jours contre 75 jours ; p=0.001). Par ailleurs, le TTP est amélioré
sur les mastocytomes dont la protéine c-Kit est mutée (241 jours contre 83 jours ; p=0.002) ou
non-mutée (140 jours contre 75 jours ; p=0.027) (44, 45).
Son action sur les mastocytomes, dont la protéine c-Kit n’est pas mutée, peut s’expliquer par
l’affinité du masitinib pour la forme sauvage de c-Kit. L’inhibition de c-Kit entraîne alors une
baisse de la prolifération des mastocytes même si l’événement oncogénique se situe sur une
autre molécule que c-Kit.
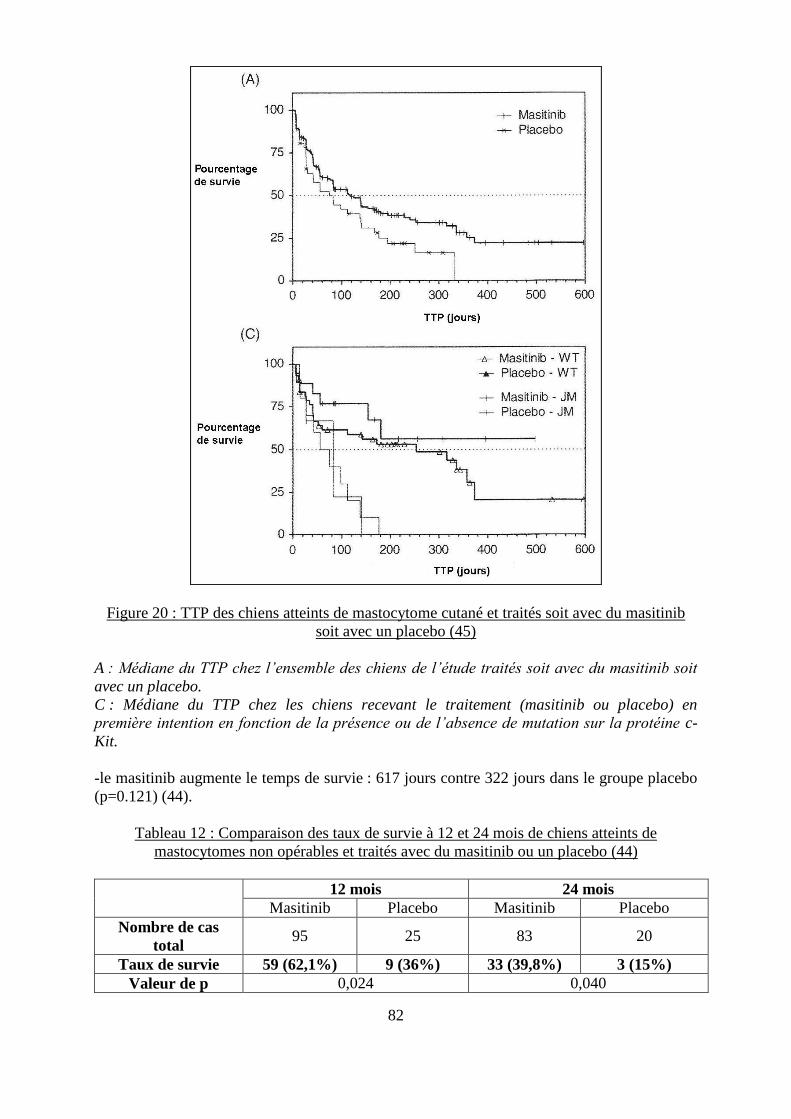
82
Figure 20 : TTP des chiens atteints de mastocytome cutané et traités soit avec du masitinib
soit avec un placebo (45)
A : Médiane du TTP chez l’ensemble des chiens de l’étude traités soit avec du masitinib soit
avec un placebo.
C : Médiane du TTP chez les chiens recevant le traitement (masitinib ou placebo) en
première intention en fonction de la présence ou de l’absence de mutation sur la protéine c-
Kit.
-le masitinib augmente le temps de survie : 617 jours contre 322 jours dans le groupe placebo
(p=0.121) (44).
Tableau 12 : Comparaison des taux de survie à 12 et 24 mois de chiens atteints de
mastocytomes non opérables et traités avec du masitinib ou un placebo (44)
12 mois 24 mois
Masitinib Placebo Masitinib Placebo
Nombre de cas
total 95 25 83 20
Taux de survie 59 (62,1%) 9 (36%) 33 (39,8%) 3 (15%)
Valeur de p 0,024 0,040
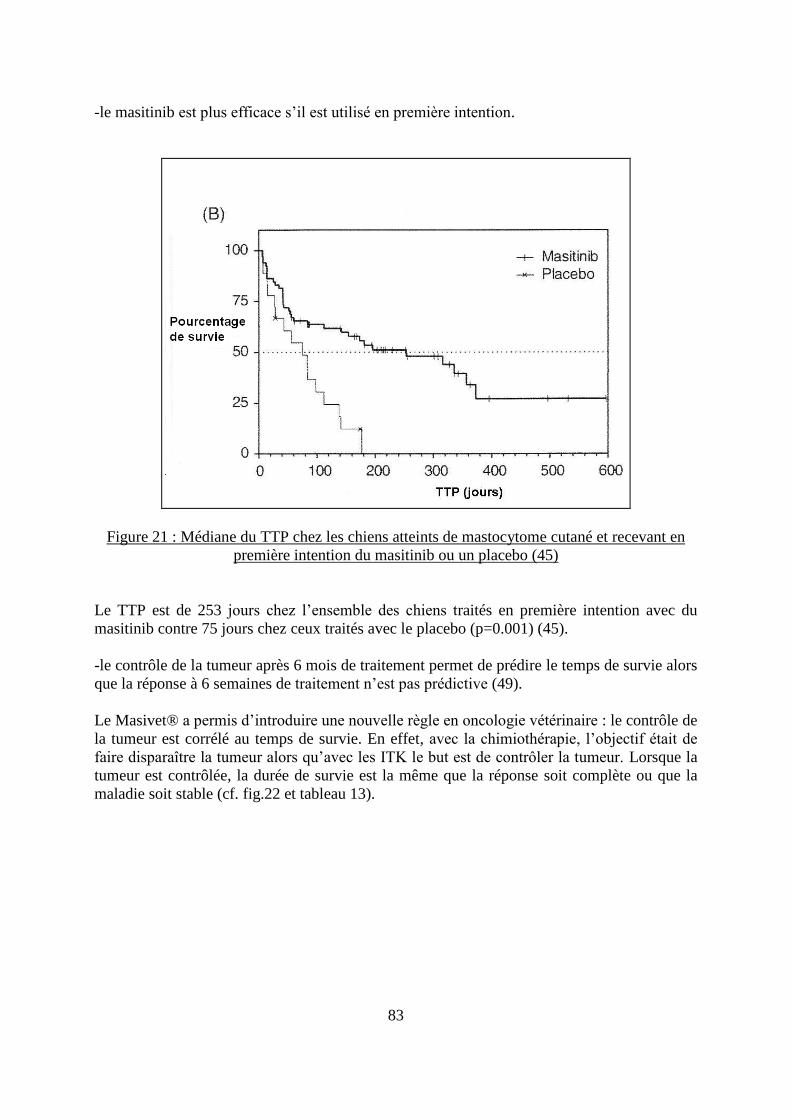
83
-le masitinib est plus efficace s’il est utilisé en première intention.
Figure 21 : Médiane du TTP chez les chiens atteints de mastocytome cutané et recevant en
première intention du masitinib ou un placebo (45)
Le TTP est de 253 jours chez l’ensemble des chiens traités en première intention avec du
masitinib contre 75 jours chez ceux traités avec le placebo (p=0.001) (45).
-le contrôle de la tumeur après 6 mois de traitement permet de prédire le temps de survie alors
que la réponse à 6 semaines de traitement n’est pas prédictive (49).
Le Masivet® a permis d’introduire une nouvelle règle en oncologie vétérinaire : le contrôle de
la tumeur est corrélé au temps de survie. En effet, avec la chimiothérapie, l’objectif était de
faire disparaître la tumeur alors qu’avec les ITK le but est de contrôler la tumeur. Lorsque la
tumeur est contrôlée, la durée de survie est la même que la réponse soit complète ou que la
maladie soit stable (cf. fig.22 et tableau 13).

84
Figure 22 : Pourcentage de survie de chiens atteints de mastocytome cutané en fonction de
leur réponse au traitement à 6 mois (44)
Tableau 13 : Pourcentage de survie à 1 et 2 ans de chiens atteints de mastocytome cutané en
fonction de leur réponse après 6 mois de traitement avec du masitinib (44)
Evaluation de la réponse à 6 mois Pourcentage de
survie à 1 an
Pourcentage de
survie à 2 ans
Population totale
Maladie contrôlée
Maladie progressive
95,1%
41,4%
77,1%
11,8%
Chiens avec une protéine c-Kit mutée
Maladie contrôlée
Maladie progressive
92,3%
45,5%
58,3%
10,5%
Chiens avec une protéine c-Kit
normale
Maladie contrôlée
Maladie progressive
96,0%
42,7%
90,5%
13,3%
L’évaluation de la tumeur à 6 mois permet de prédire si le temps de survie sera court ou long.
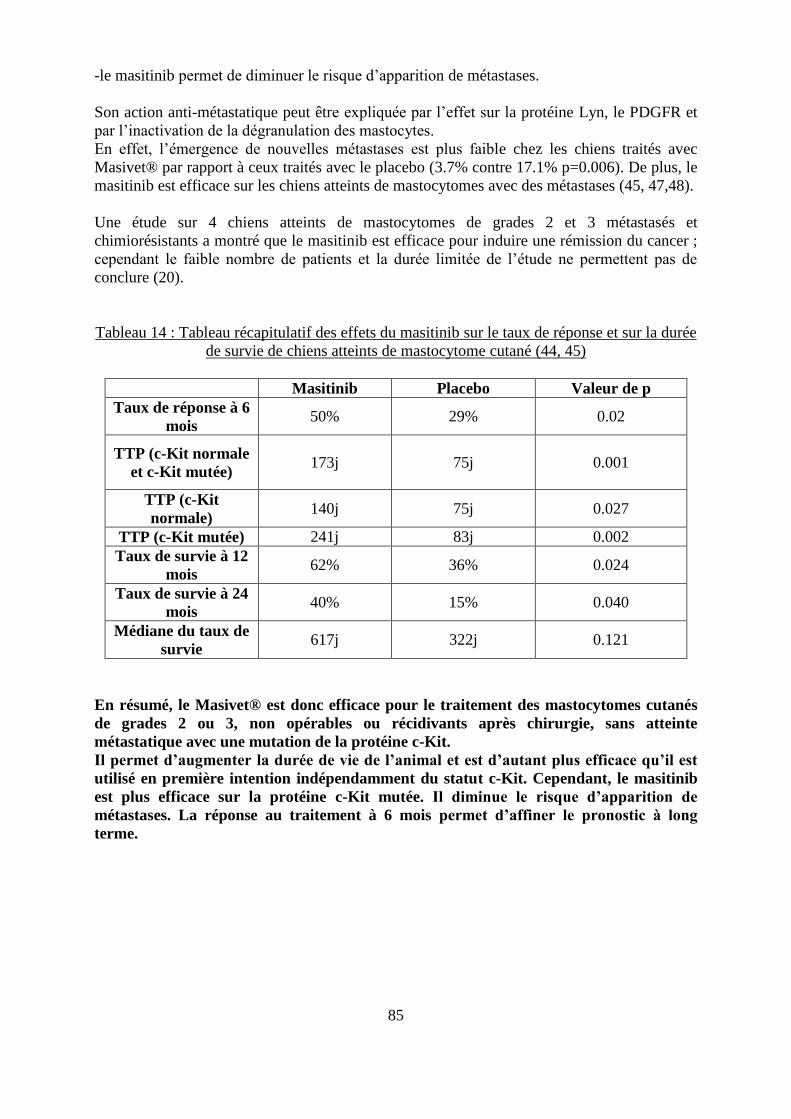
85
-le masitinib permet de diminuer le risque d’apparition de métastases.
Son action anti-métastatique peut être expliquée par l’effet sur la protéine Lyn, le PDGFR et
par l’inactivation de la dégranulation des mastocytes.
En effet, l’émergence de nouvelles métastases est plus faible chez les chiens traités avec
Masivet® par rapport à ceux traités avec le placebo (3.7% contre 17.1% p=0.006). De plus, le
masitinib est efficace sur les chiens atteints de mastocytomes avec des métastases (45, 47,48).
Une étude sur 4 chiens atteints de mastocytomes de grades 2 et 3 métastasés et
chimiorésistants a montré que le masitinib est efficace pour induire une rémission du cancer ;
cependant le faible nombre de patients et la durée limitée de l’étude ne permettent pas de
conclure (20).
Tableau 14 : Tableau récapitulatif des effets du masitinib sur le taux de réponse et sur la durée
de survie de chiens atteints de mastocytome cutané (44, 45)
Masitinib Placebo Valeur de p
Taux de réponse à 6
mois 50% 29% 0.02
TTP (c-Kit normale
et c-Kit mutée) 173j 75j 0.001
TTP (c-Kit
normale) 140j 75j 0.027
TTP (c-Kit mutée) 241j 83j 0.002
Taux de survie à 12
mois 62% 36% 0.024
Taux de survie à 24
mois 40% 15% 0.040
Médiane du taux de
survie 617j 322j 0.121
En résumé, le Masivet® est donc efficace pour le traitement des mastocytomes cutanés
de grades 2 ou 3, non opérables ou récidivants après chirurgie, sans atteinte
métastatique avec une mutation de la protéine c-Kit.
Il permet d’augmenter la durée de vie de l’animal et est d’autant plus efficace qu’il est
utilisé en première intention indépendamment du statut c-Kit. Cependant, le masitinib
est plus efficace sur la protéine c-Kit mutée. Il diminue le risque d’apparition de
métastases. La réponse au traitement à 6 mois permet d’affiner le pronostic à long
terme.

86
f. Autres indications du masitinib en cancérologie
i. Chez le chien
Lymphome T chez le chien
Le masitinib est capable de bloquer la prolifération de lignée cellulaire issue de lymphome T
via leur action sur le PDGFR (135).
Rôle de resensibilisation à la chimiothérapie
Le masitinib est capable de potentialiser les effets cytotoxiques de la chimiothérapie dans
différents cancers. En effet, Thamm et al. ont montré qu’il sensibilise les sarcomes
histiocytaires à la vinblastine, les ostéosarcomes et les carcinomes mammaires à la
gemcitabine et dans un moindre degré d’autres lignées cellulaires (issues d’hémangiosarcome
et de mélanome) à la doxorubicine (143).
Cette sensibilisation permet de réduire les doses d’agents de chimiothérapie et ainsi de
minimiser leurs effets toxiques. Les mécanismes, par lesquels le masitinib sensibilise les
cellules cancéreuses aux agents cytotoxiques, ne sont pas encore clairement définis. Le
PDGFR et la voie de signalement Lyn/FAK pourraient avoir un rôle.
Des études sont actuellement en cours afin de tester l’efficacité du masitinib en association
avec certains agents de chimiothérapie.
Tableau 15 : Essais cliniques sur le masitinib en association avec certains agents de
chimiothérapie (73)
Agent unique
Lymphome T
Masitinib (agent simple)
Doxorubicine (agent simple)
Masitinib associé à la doxorubicine
Mélanome avec mutation de
c-Kit dans le domaine JM
Masitinib (agent simple)
Carboplatine (agent simple)
Protocoles combinés
Hémangiosarcome splénique
Masitinib (agent simple)
Doxorubicine (agent simple)
Masitinib associé à la doxorubicine
Mélanome sans mutation de
c-Kit dans le domaine JM
Carboplatine (agent simple)
Masitinib associé au carboplatine
Masitinib associé à la doxorubicine
Ostéosarcome (post
amputation)
Carboplatine (agent simple)
Masitinib associé au carboplatine
Masitinib associé à la gemcitabine
Sarcome histiocytaire Masitinib associé à la vinblastine

87
Autres tumeurs
Dans d’autres cancers comme les tumeurs mammaires, les ostéosarcomes et les
hémangiosarcomes, nous avons vu précédemment que la protéine c-Kit était impliquée dans
le processus cancérigène. Des études sur l’efficacité du masitinib sur ces tumeurs sont en
cours (cf. tableau 16).
En résumé, le masitinib a montré son efficacité contre les mastocytomes cutanés du
chien. Il permet de plus une resensibilisation à la chimiothérapie de certaines lignées
cellulaires cancéreuses. Des études sont en cours sur l’efficacité du masitinib contre
d’autres tumeurs.
ii. Chez l’homme
Le cancer du pancréas
Le cancer du pancréas est un cancer de mauvais pronostic. Le traitement de référence fait
intervenir une chirurgie lorsqu’elle est réalisable, une radiothérapie et une chimiothérapie à
base de gemcitabine. Cependant, l’efficacité de cette molécule en agent unique est modérée.
Les causes de ce cancer ne sont pas totalement connues mais il semblerait que certaines
tyrosines kinases soient surexprimées comme EGFR, VEGFR, FAK, Src et Lyn. De plus le
recrutement de cellules inflammatoires telles que les mastocytes facilite la croissance de la
tumeur et la formation de métastases. Le masitinib pourrait donc avoir un potentiel
thérapeutique dans ce cas.
Différentes études ont déjà montré que le masitinib permet de sensibiliser les cellules
tumorales réfractaires à la gemcitabine (53, 102). De plus, l’association masitinib/gemcitabine
a une action anti tumorale prometteuse.
L’objectif de l’étude en phase 3 est d’étudier l’efficacité et l’innocuité du masitinib en
association avec la gemcitabine par rapport à un placebo en association avec la gemcitabine.
Les GIST
Les GIST sont liées, dans la grande majorité des cas, à une mutation de la protéine c-Kit
comme nous l’avons vu au II.D.2.
Récemment, l’imatinib (Gleevec®) a prouvé son efficacité dans le traitement des GIST mais
des résistances sont apparues comme nous le verrons au III.B.1.k. (16, 69,70, 137).
Le masitinib a prouvé son efficacité en premier traitement sur des GIST. Il est actuellement
testé sur des GIST résistantes à l’imatinib.

88
Le myélome multiple
L’incidence du myélome multiple est de 50 000 nouveaux cas par an dans les pays
industrialisés (Amérique du nord, Europe du Nord, de l’Ouest et du Sud, Japon, République
de Corée, Australie et Nouvelle Zélande). Dans 15% des myélomes multiples, une
translocation t(4 ;14) est présente et entraine une surexpression du FGFR3 qui représente
donc une cible thérapeutique. De plus, les cellules cancéreuses plasmatiques expriment c-Kit
qui est impliquée dans la production d’interleukine 6, un stimulant de la croissance de ces
cellules.
La première ligne de traitement du myélome multiple est représentée par une
polychimiothérapie suivie d’une autogreffe de moelle. Les patients qui rechutent reçoivent
une nouvelle chimiothérapie à base de bortezomid (Velcade®) considéré comme le traitement
standard ou à base de lénalidomide (Revlimid®).
Actuellement, une étude sur le masitinib en phase 3 est en cours (2). Il s’agit d’une étude de
phase 3, multicentrique randomisée, en 2 groupes parallèles comparant l’efficacité et la
tolérance du masitinib à 9 mg/kg/jour en association avec le bortezomib (Velcade®) et de la
dexaméthasone versus placebo en association avec le bortezomib (Velcade®) et de la
dexaméthasone dans le traitement de patients atteints de myélome multiple en rechute après
une première ligne de traitement.
Les mélanomes
Une étude de phase 3 est en cours sur le mélanome métastasé présentant une mutation du
domaine JM de c-Kit. Des études en phase 2 sont également prévues pour les autres types de
mélanomes (2).
La mastocytose
Dans une étude de phase 2, le masitinib a prouvé son efficacité sur les mastocytoses
systémiques indolentes quelque soit le statut de la protéine c-Kit (117). Une étude en phase 3
est en cours.
En résumé, dans le domaine de la cancérologie chez l’homme le masitinib montre un
intérêt thérapeutique dans le cancer du pancréas, les GIST, le myélome multiple, les
mélanomes et la mastocytose.

89
g. Indication du masitinib sur les maladies inflammatoires
Les pathologies inflammatoires sont liées à une dérégulation du système immunitaire qui
conduit à une infiltration de nombreuses cellules de l’inflammation (mastocyte, macrophage,
lymphocyte, polynucléaire, cellule dendritique, etc…) dans les tissus. Cette infiltration peut
être la conséquence d’une réponse immunitaire anormale liée à la présence d’anticorps dirigés
contre soi-même ou contre des agents extérieurs (allergènes, agents infectieux).
Les cellules inflammatoires, en libérant de nombreux facteurs, peuvent induire la destruction
et le remodelage de l’organe infiltré pouvant aller jusqu’à la fibrose, comme les articulations
dans la polyarthrite rhumatoïde, les bronches et bronchioles pulmonaires dans l’asthme.
L’activation des cellules inflammatoires et le remodelage tissulaire fibrosant est également
sous la dépendance de kinases (PDGFR, c-Kit, Lyn, etc…).
Par ailleurs, des résultats expérimentaux récents concluent que le mastocyte joue un rôle clé
dans l’organisation et l’activation de l’inflammation, en participant directement à la
destruction tissulaire, mais aussi en recrutant et en activant les autres cellules de l’immunité
(macrophages, lymphocytes, etc…). L’inhibition de kinases comme c-Kit et Lyn diminue la
migration et l’activation des mastocytes, pouvant ainsi conduire à une diminution de
l’inflammation tissulaire dans les pathologies inflammatoires.
Chez les animaux, les mastocytes sont impliqués dans de nombreuses maladies
inflammatoires telles que la dermatite atopique chez le chien, l’asthme chez le chat, l’arthrite
et les MICI. Il apparait donc intéressant de tester le masitinib sur ces maladies.
Une étude préclinique sur des souris a montré un impact bénéfique de l’utilisation du
masitinib dans le cas de désordre inflammatoire chronique impliquant des mastocytes (46).
i. Chez le chien
La dermatite atopique
La dermatite atopique est dermatose inflammatoire chronique prurigineuse due à une
anomalie de la barrière cutanée et à une prédisposition génétique à développer une allergie de
type IV (médiée par les lymphocytes) aux allergènes de l’environnement.
La pathogénie de la dermatite atopique chez le chien implique diverses cellules
inflammatoires dont les mastocytes qui jouent un rôle principal. L’interaction SCF/c-Kit est
impliquée dans la survie, la différentiation, la prolifération, l’adhésion et la dégranulation des
mastocytes comme nous l’avons vu précédemment. De plus, c-Kit agit en synergie avec les
récepteurs aux immunoglobulines E pour induire la libération de médiateurs et de cytokines
impliqués dans la dermatite atopique. Le masitinib est donc supposé avoir un rôle dans le
contrôle de cette maladie.

90
Une étude de phase 2 sur la dermatite atopique chez 11 chiens a été réalisée. L’index de
sévérité et d’étendue de la dermatite atopique chez le chien (score CADESI), la surface des
lésions, un score de prurit et la toxicité ont été évaluées pendant 28 jours de traitement à base
de masitinib à 11 mg/kg/j. Le score CADESI s’est amélioré de 38.2% à 14 jours (+-29.2%,
p=0.0025) et, à 28 jours sa réduction moyenne était de 50.7% (+-29.8%, p=0.0004). Par
ailleurs, 50% des chiens sous traitement ont vu leur score de prurit diminuer de plus de 50%.
Cette étude, bien que réalisée sur un faible effectif, a été concluante et a démontré que le
masitinib est un traitement prometteur de la dermatite atopique (19).
Une étude de phase 3 réalisée sur plus de 300 chiens a mise en évidence l’efficacité du
masitinib, mesurée par une amélioration significative du score CADESI, dans le traitement de
la dermatite atopique. Ainsi, le taux de chiens répondant après trois mois de traitement, défini
par une amélioration du score CADESI de plus de 50% était de 43% (86/202) pour les chiens
traités par masitinib contre 26% (27/104) pour les chiens sous placebo (p<0.001).
Dans cette étude, 190 chiens étaient en échec de traitement par la ciclosporine et/ou les
corticostéroïdes. Dans cette population, le taux de chiens répondant après trois mois de
traitement, défini par une amélioration du score CADESI de plus de 50% était de 41 %
(49/121) pour les chiens traités par masitinib versus 23% (16/69) pour les chiens sous placebo
(p=0.029) (11).
ii. Chez l’homme
L’arthrite rhumatoïde
L’arthrite rhumatoïde est une maladie inflammatoire chronique très répandue, on évalue sa
prévalence à 0.8% dans les pays industrialisés.
Un dérèglement du système immunitaire entraîne l’attaque de la synovie et crée des troubles
articulaires et rhumatismaux. La polyarthrite rhumatoïde est une pathologie inflammatoire des
articulations qui conduit à la destruction des articulations touchées. Elle est due à un
recrutement et une activation anormale des cellules immunitaires dans l’articulation. Le
processus inflammatoire est sans doute lié à une réaction auto-immune au niveau de la
synoviale. Dans la polyarthrite, le mastocyte joue probablement un rôle important en
produisant des chimiokines qui recrutent les autres cellules inflammatoires, des cytokines
(telles que le TNF) responsables de l’inflammation, et des métalloprotéases en partie
responsables de la destruction articulaire.
Le traitement de base associe des anti-inflammatoires. La ligne de traitement standard après
les anti-inflammatoires est un modificateur de l’inflammation comme la chloroquine ou la
salazopyrine. En cas d’échec le méthotrexate, un immunosuppresseur non dénué de toxicité à
long terme, est utilisé. Lorsque les patients résistent au méthotrexate, la deuxième ligne de
traitement est constituée par le méthotrexate en association avec des anticorps dirigés contre
le TNF-alpha. En cas de résistance, la troisième ligne de traitement est composée du
méthotrexate en combinaison avec un anticorps monoclonal dirigé contre le CD20 (antigène
spécifique des cellules B).
Certains médicaments utilisés ont engendré des résistances et cela représente un obstacle
majeur à la prise en charge thérapeutique.
Nous rappelons que l’association SCF/c-Kit est impliquée dans la survie, la prolifération, la
différentiation, l’adhésion et la dégranulation des mastocytes, le masitinib en inhibant cette
voie de signalement pourrait permettre de contrôler la maladie.

91
Dans un modèle expérimental de polyarthrite rhumatoïde à l’aide de souris transgénique, le
masitinib a démontré une capacité à prévenir le développement de cette pathologie.
L’indication du masitinib porte sur le traitement des formes modérées à sévères en échec au
méthotrexate soit environ 1/3 des polyarthrites rhumatoïdes.
Une étude de phase 2 a été réalisée pendant 12 semaines sur 43 patients n’ayant pas de
réponse satisfaisante aux traitements classiques. Une amélioration de la maladie a été mise en
évidence chez la plupart des patients. Quelques effets secondaires légers à modérés sont
cependant apparus mais ont été bien tolérés (142).
Une étude en phase 3 est en cours. Il s’agit d’une étude multicentrique, randomisée, en double
aveugle, contrôlée avec 3 groupes parallèles sur 24 semaines comparant l’efficacité et la
tolérance du masitinib à 3 ou 6 mg/kg/jour à celles du méthotrexate chez des patients atteints
de polyarthrite rhumatoïde.
L’asthme
L’asthme touche 4 à 7% de la population des pays industrialisés. Il s’agit d’une maladie
inflammatoire chronique dont les acteurs principaux sont les mastocytes et les cellules
dendritiques. La protéine c-Kit est exprimée à la fois dans les mastocytes et dans les cellules
bronchiales structurales. De plus, une autre PTK, le PDGFR, contribue au remodelage des
bronches ayant lieu lors d’asthme sévère.
Cette maladie est prise en charge de façon satisfaisante via des anti-inflammatoires et des
bronchodilatateurs chez la plupart des patients mais dans les cas d’asthme sévère la maladie
reste symptomatique malgré un traitement avec des fortes doses de corticostéroïdes inhalés et
par voie systémique. Les patients font fréquemment des crises qui peuvent donner lieu à une
hospitalisation. De plus, on peut observer des effets secondaires majeurs liés aux
corticostéroïdes à long terme.
L’indication du masitinib porte sur le traitement de l’asthme permanent sévère soit environ
10% des patients asthmatiques. Le masitinib cible à la fois c-Kit et le PDGFR et une étude de
phase 2 a montré qu’il est efficace sur les patients cortico-dépendant atteints d’asthme sévère.
Il permet en effet une diminution de la dose de corticoïde administrée et parallèlement une
amélioration de l’asthme (54).
Une étude en phase 3 est en cours. Il s’agit d’une étude prospective multicentrique,
randomisée, en double aveugle en 2 groupes parallèles pour évaluer l’efficacité et la tolérance
du masitinib à 6 mg/kg/j chez des patients présentant un asthme sévère permanent traité par
des corticoïdes par voie orale.
En résumé, concernant les maladies inflammatoires, le masitinib a prouvé son efficacité
dans le traitement de la dermatite atopique chez le chien. Chez l’homme, il a présenté un
intérêt dans la prise en charge de l’arthrite rhumatoïde et de l’asthme. On notera que
chez le chat, des études ont montré un effet bénéfique du masitinib sur l’asthme félin, les
MICI et les mastocytomes.

92
h. Indications du masitinib sur les maladies du système nerveux central chez
l’homme
Le cerveau et la moelle épinière constituent le système nerveux central. Ils peuvent être lésés
suite à des pathologies inflammatoire (sclérose en plaques, par exemple) et/ou dégénératives
(telles que la maladie d’Alzheimer). La distinction entre ces deux types de pathologie n’est
plus aussi claire aujourd’hui. Il existe des formes de sclérose en plaques qui prennent un
aspect dégénératif (sclérose en plaques primitivement ou secondairement progressive) et des
données expérimentales suggèrent qu’il existe une composante inflammatoire dans la maladie
d’Alzheimer.
Le mastocyte est présent en quantité relativement abondante dans le cerveau et la moelle
épinière, notamment autour des vaisseaux. Certaines études estiment qu’il jouerait un rôle
majeur pour permettre le passage des cellules de l’inflammation du sang vers le cerveau,
susceptible de provoquer ainsi une destruction tissulaire. Il pourrait être également recruté
dans des maladies dégénératives et contribuer à la destruction tissulaire. Par ailleurs,
l’activation de kinases comme le PDGFR pourrait contribuer au processus dégénératif de la
maladie d’Alzheimer en augmentant l’expression de la gamma-sécrétase, une enzyme
participant à la production des fibrilles amyloïdes qui, en s’accumulant dans le cerveau, sont
responsables de la désorganisation du réseau neuronal.
Si ces résultats étaient avérés, il est possible que l’inhibition des kinases c-Kit, Lyn, et
PDGFR puisse contribuer à l’amélioration de la condition des patients atteints de ces maladies
i. La sclérose en plaques
La sclérose en plaques est la première cause de handicap chez le jeune adulte. Il existe 2
formes de sclérose en plaques : la forme rechute/rémission et la forme progressive (60% des
patients). Dans la forme progressive, on distingue la forme d’emblée progressive et la forme
secondairement progressive après avoir débuté en forme rechute/rémission.
Les mastocytes sont impliqués dans la pathogénie de la sclérose en plaques car :
-ils régulent le passage de la barrière hémato encéphalique
-le cerveau est l’organe qui contient le plus de mastocytes et cette cellule participe au
recrutement des lymphocytes T
-le mastocyte peut aussi par dégranulation contribuer à la dégradation de la myéline.
Cinq produits (sous forme injectable) sont enregistrés dans la forme rechute/rémission : 3
IFN, le Copaxone® (glatiramère) et le Tysabri® (natalizumab). Aucun médicament n’est
enregistré dans la forme progressive d’emblée. Dans la forme secondairement progressive,
aucun médicament n’est enregistré aux USA et seule une chimiothérapie avec le mitoxantrone
est enregistrée en Europe avec cependant des risques non négligeables de cancer secondaire.

93
Une étude de phase 2 a montré que le masitinib est efficace et bien toléré contre la sclérose en
plaques progressive d’emblée et secondairement progressive (146).
Une étude de phase 3 est en cours. Il s’agit d’une étude prospective, multicentrique,
randomisée, en double aveugle, en 2 groupes parallèles sur 96 semaines évaluant l’efficacité
et la tolérance du masitinib à 6 mg/kg/j dans le traitement de patients souffrant de sclérose en
plaques progressive d’emblée ou secondairement progressive et présentant à l’inclusion un
score EDSS (Expanded Disability Status Scale) entre 2 et 6 inclus.
L'échelle EDSS, cotée de 0 (pas de handicap) à 10 (décès) est très utilisée pour coter le niveau
de handicap des patients atteints de sclérose en plaques. Elle fait référence à la notion de
fonctions neurologiques. Il s'agit des fonctions suivantes : pyramidale (marche), cérébelleuse
(coordination), parole et déglutition, sensitive (toucher et douleur), intestinale et urinaire,
visuelle, mentale, autres.
ii. La maladie d’Alzheimer
Une étude de phase 2 a montré que la masitinib permet une diminution voire un arrêt de la
perte cognitive liée à la maladie d’Alzheimer chez des patients traités pendant au moins 24
semaines avec une bonne tolérance (119).
Une phase 3 doit être initiée.
En résumé, le masitinib représente un réel espoir thérapeutique dans certaines maladies
du système nerveux chez l’homme telles que la sclérose en plaques et la maladie
d’Alzheimer.

94
i. Effets secondaires
Les principaux effets secondaires mis en évidence lors de ces différentes études sont :
vomissements et diarrhée (très fréquents mais d’intensité modérée et souvent transitoire)
fatigue, léthargie, anorexie (fréquents)
neutropénie, hausse des enzymes hépatiques, anémie hémolytique, syndrome de fuite
protéique
Ces effets secondaires sont à relier à l’inhibition de c-Kit dans les cellules normales. En effet,
l’inhibition de c-Kit dans les cellules de Cajal dans l’intestin est responsable de diarrhée.
L’apoptose des mastocytes entraîne un relargage de médiateurs pouvant entraîner des effets
systémiques tels que diarrhée et vomissements.
Ces effets sont en général d’intensité faible à modérée, transitoires et peuvent être contrôlés
par un traitement symptomatique.
Un syndrome de fuite protéique peut être présent chez un faible pourcentage de chiens. Le
mécanisme d’apparition de ce syndrome est le suivant : l’inhibition du PDGFR est
responsable d’une dysjonction des podocytes entrainant une fuite d’albumine hors des
vaisseaux. La fraction de masitinib non liée aux protéines augmente alors et l’activité du
masitinib est donc plus importante ce qui peut entraîner des diarrhées et des vomissements à
l’origine d’une déshydratation (73).
Il s’agit d’un syndrome prévisible, d’apparition progressive et réversible. Le suivi de
l’albuminémie doit donc être fait régulièrement toutes les 2 semaines. En cas albuminémie
inférieure à 20g/L, le traitement doit être interrompu.
En résumé, le masitinib présente des effets secondaires liés à l’action de la molécule sur
des PTK (c-Kit, PDGFR principalement) de cellules non cancéreuses. On peut observer
des signes gastro-intestinaux, des signes généraux tels que la fatigue, des modifications
hématologiques et biochimiques (anémie, neutropénie, élévation des enzymes
hépatiques, syndrome de fuite protéique). Ces signes sont en général d’intensité faible à
modérée et peuvent être contrôlés par un traitement symptomatique.
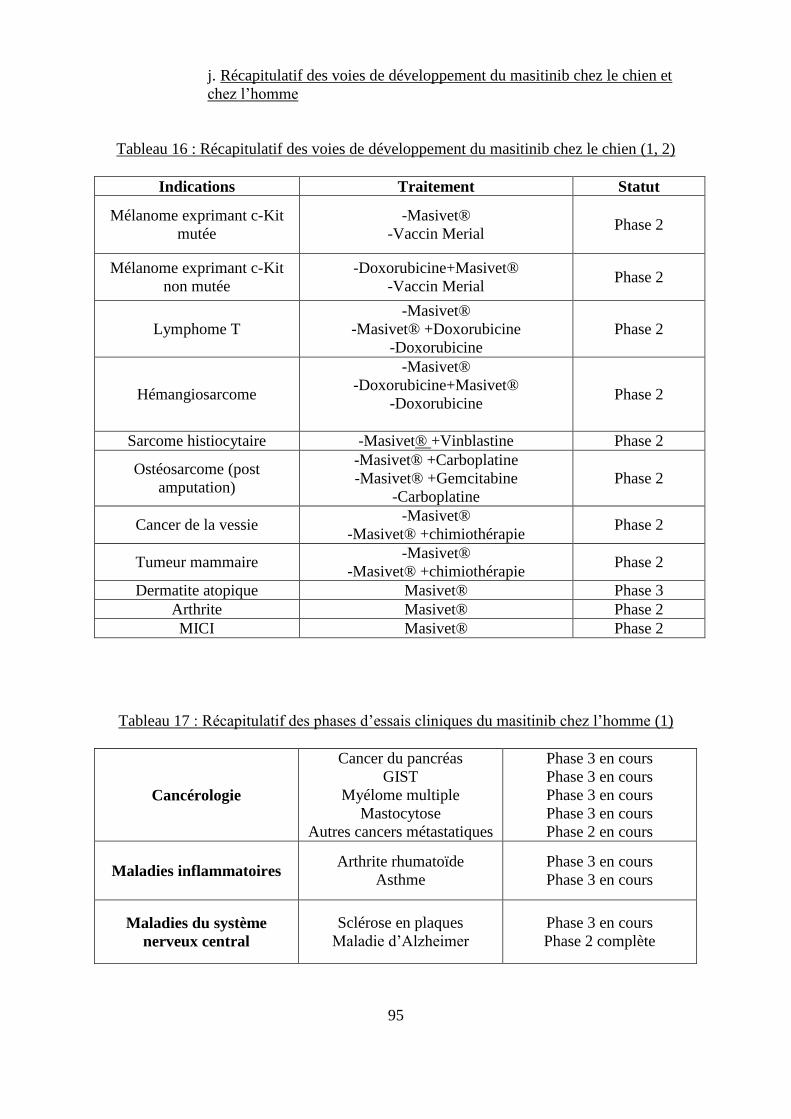
95
j. Récapitulatif des voies de développement du masitinib chez le chien et
chez l’homme
Tableau 16 : Récapitulatif des voies de développement du masitinib chez le chien (1, 2)
Indications Traitement Statut
Mélanome exprimant c-Kit
mutée
-Masivet®
-Vaccin Merial Phase 2
Mélanome exprimant c-Kit
non mutée
-Doxorubicine+Masivet®
-Vaccin Merial Phase 2
Lymphome T
-Masivet®
-Masivet® +Doxorubicine
-Doxorubicine
Phase 2
Hémangiosarcome
-Masivet®
-Doxorubicine+Masivet®
-Doxorubicine
Phase 2
Sarcome histiocytaire -Masivet® +Vinblastine Phase 2
Ostéosarcome (post
amputation)
-Masivet® +Carboplatine
-Masivet® +Gemcitabine
-Carboplatine
Phase 2
Cancer de la vessie -Masivet®
-Masivet® +chimiothérapie Phase 2
Tumeur mammaire -Masivet®
-Masivet® +chimiothérapie Phase 2
Dermatite atopique Masivet® Phase 3
Arthrite Masivet® Phase 2
MICI Masivet® Phase 2
Tableau 17 : Récapitulatif des phases d’essais cliniques du masitinib chez l’homme (1)
Cancérologie
Cancer du pancréas
GIST
Myélome multiple
Mastocytose
Autres cancers métastatiques
Phase 3 en cours
Phase 3 en cours
Phase 3 en cours
Phase 3 en cours
Phase 2 en cours
Maladies inflammatoires Arthrite rhumatoïde
Asthme
Phase 3 en cours
Phase 3 en cours
Maladies du système
nerveux central
Sclérose en plaques
Maladie d’Alzheimer
Phase 3 en cours
Phase 2 complète

96
2. Le Palladia® (toceranib)
Le Palladia® a obtenu l’AMM en 2009 et a été commercialisé par le laboratoire Pfizer Santé
Animale en début d’année 2010. Il est indiqué chez le chien pour le traitement des
mastocytomes cutanés de grades 2 ou 3 selon Patnaik, récidivants avec ou sans atteinte des
nœuds lymphatiques régionaux. L’équivalent humain est le Sutent® (sunitinib).
a. Structure
Le toceranib a une structure chimique de type indolinone. Les indolinones sont des composés
polycycliques (139). Les 3-substitut indolin-2-ones sont des composés inventés et synthétisés
en tant qu’ITK qui peuvent exercer leur sélectivité sur différents types de RTK. En modifiant
la composition chimique de ces composés, des molécules distinctes ciblant des RTK
particuliers ont été synthétisées (9,89).
L’analyse cristallographique de ces molécules a permis de montrer qu’elles peuvent se lier au
site de liaison de l’ATP des RTK.
Le toceranib est le 5-[(Z)-(5-fluoro-2-oxo-1,2-dihydro-3H-indol-3-ylidene)methyl]-2,4-
dimethyl-N-(2-pyrrolidin-1-ylethyl)-1H-pyrrole-3-carboxamide.
Figure 23 : Structure chimique du toceranib (84)
b. Cibles
Le toceranib cible le VEGFR2, le PDGFR et c-Kit et d’autres molécules de la famille des split
kinases. Il est moins spécifique que le masitinib.
Tableau 18 : IC50 du toceranib sur différentes TK (78)
Tyrosines kinases IC 50 (µM)
PDGFR 0,01
VEGFR2 0,03
c-Kit 0,05
EGFR >10

97
Figure 24 : Représentation graphique de l’action du toceranib sur différentes kinases (73)
On remarque que le toceranib inhibe de nombreuses kinases. Son action est donc moins
prévisible que celle du masitinib.
c. Mode d’action
Le toceranib a une action anti-angiogénique et anti-tumorale (3).
Action du toceranib sur la protéine c-Kit des mastocytes in vitro
L’effet de 3 ITK de type indolinone (SU11652, SU11654 et SU11655) dont le toceranib
(SU11654) a été testé sur 4 lignées cellulaires mastocytaires présentant une protéine c-Kit
normale ou avec des mutations affectant soit le domaine JM soit le domaine catalytique.
Les 3 ITK inhibent les formes normales ou mutées de la protéine c-Kit de façon dose-
dépendante en induisant l’arrêt de leur cycle cellulaire et leur apoptose mais ils sont plus
efficaces sur les protéines c-Kit présentant une mutation du domaine JM (78).
Ceci peut être expliqué par le fait qu’une mutation de c-Kit dans le domaine catalytique induit
une plus forte activation de la PI3 kinase et de ce fait un plus fort pouvoir cancérigène qu’une
mutation du domaine JM.

98
Action du toceranib sur la protéine c-Kit de mastocytomes in vivo
Une étude sur 11 chiens a permis de montrer que le toceranib à la dose de 3.25 mg/kg inhibe
la phosphorylation de la protéine c-Kit dans les mastocytomes et affecte ainsi la voie de
signalisation en aval de c-Kit. En effet, 73% des chiens ont une inhibition de la
phosphorylation de la protéine c-Kit après administration d’une simple dose orale de
toceranib. Les 27% restant ont un faible taux de phosphorylation de la protéine c-Kit avant
l’administration de toceranib. Et de ce fait, la sensibilité des anticorps dirigés contre la
protéine c-Kit phosphorylée n’est pas suffisante pour détecter une modulation de la
phosphorylation induite par le toceranib (122).
d. Protocole
Le Palladia® se présente sous forme de comprimés de 10, 15 et 50 mg. La posologie
recommandée est de 3.25 mg/kg tous les 2 jours. Elle peut être diminuée de 0.5 mg/kg avec
un minimum de 2.2 mg/kg en fonction des effets secondaires. Un arrêt transitoire du
traitement est possible.
Il est conseillé d’interrompre le traitement en cas d’anémie, hypo-albuminémie et
hyperphosphatémie simultanée, en cas d’hématocrite inférieur à 26%, si la créatinémie
dépasse 2 mg/dl ou en cas de neutropénie inférieure à 1000/µl.
L’utilisation conjointe de cimétidine peut être indiquée pour éviter les effets de l’histamine en
cas de dégranulation mastocytaire massive et pour lutter contre les érosions et ulcérations
digestives observées chez quelques animaux traités avec Palladia®.
Un suivi très régulier des animaux traités doit être entrepris.
e. Efficacité du toceranib sur les mastocytomes
Une étude multicentrique randomisée, en double aveugle de grande envergure sur 153 chiens
présentant un mastocytome récidivant de grade 2 ou 3 avec ou sans atteinte des nœuds
lymphatiques régionaux a été réalisée pendant 6 semaines (86).
Le taux de réponse objective (réponse complète ou partielle) est de 7.9 % pour les chiens
traités avec le placebo et de 37.2% pour les chiens traités avec le toceranib à la dose de 3.25
mg/kg/j (p<0.001).
La médiane du TTP est de 3 semaines pour les chiens ayant reçu le placebo et est supérieur à
6 semaines pour ceux ayant reçu le toceranib (p<0.0001) (cf. fig.25).
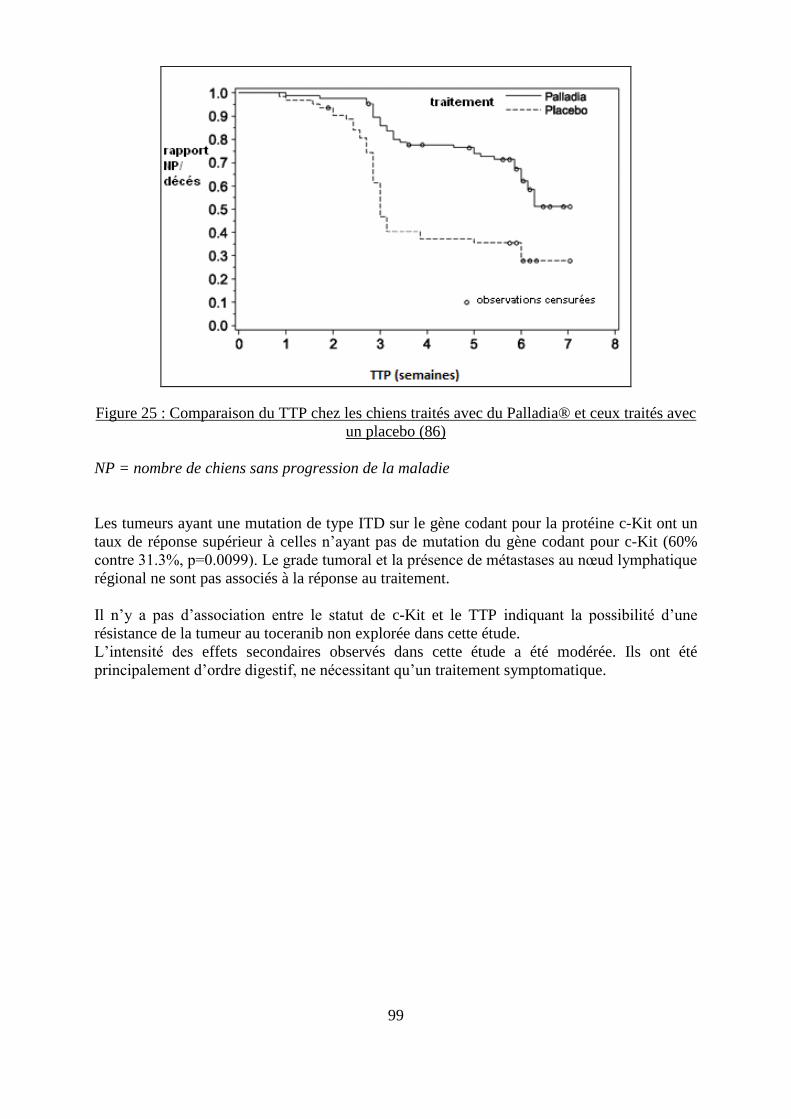
99
Figure 25 : Comparaison du TTP chez les chiens traités avec du Palladia® et ceux traités avec
un placebo (86)
NP = nombre de chiens sans progression de la maladie
Les tumeurs ayant une mutation de type ITD sur le gène codant pour la protéine c-Kit ont un
taux de réponse supérieur à celles n’ayant pas de mutation du gène codant pour c-Kit (60%
contre 31.3%, p=0.0099). Le grade tumoral et la présence de métastases au nœud lymphatique
régional ne sont pas associés à la réponse au traitement.
Il n’y a pas d’association entre le statut de c-Kit et le TTP indiquant la possibilité d’une
résistance de la tumeur au toceranib non explorée dans cette étude.
L’intensité des effets secondaires observés dans cette étude a été modérée. Ils ont été
principalement d’ordre digestif, ne nécessitant qu’un traitement symptomatique.
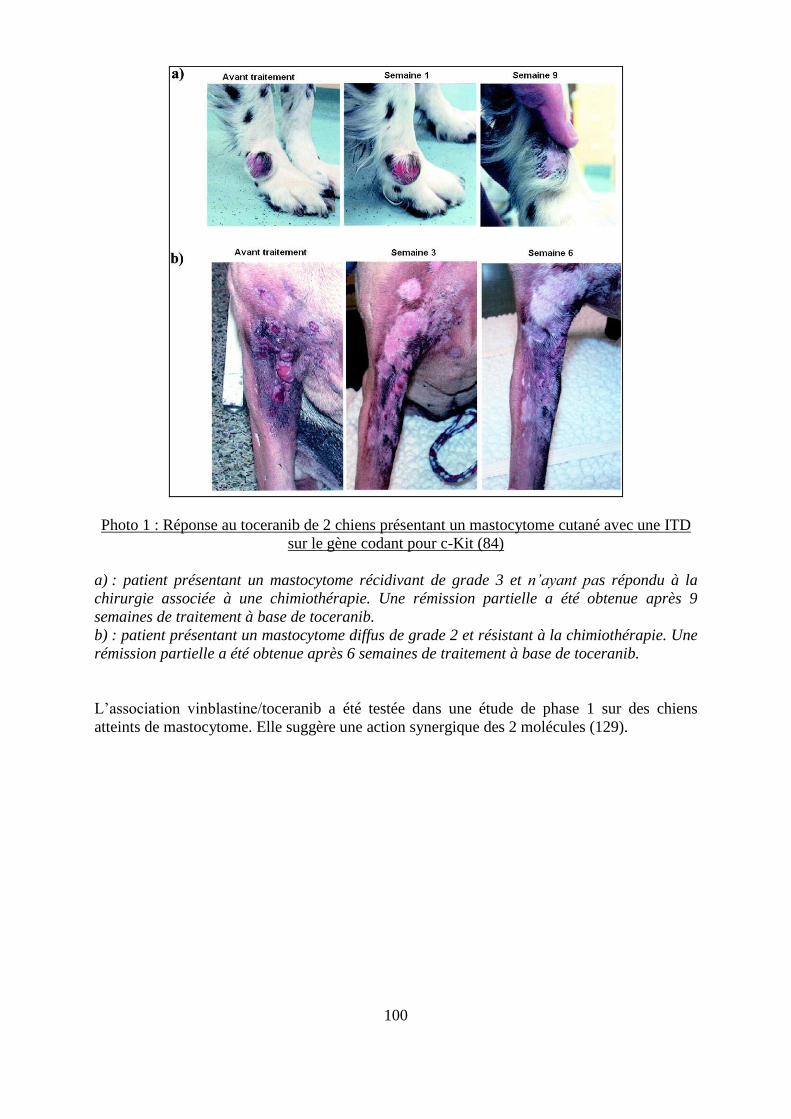
100
Photo 1 : Réponse au toceranib de 2 chiens présentant un mastocytome cutané avec une ITD
sur le gène codant pour c-Kit (84)
a) : patient présentant un mastocytome récidivant de grade 3 et n’ayant pas répondu à la
chirurgie associée à une chimiothérapie. Une rémission partielle a été obtenue après 9
semaines de traitement à base de toceranib.
b) : patient présentant un mastocytome diffus de grade 2 et résistant à la chimiothérapie. Une
rémission partielle a été obtenue après 6 semaines de traitement à base de toceranib.
L’association vinblastine/toceranib a été testée dans une étude de phase 1 sur des chiens
atteints de mastocytome. Elle suggère une action synergique des 2 molécules (129).
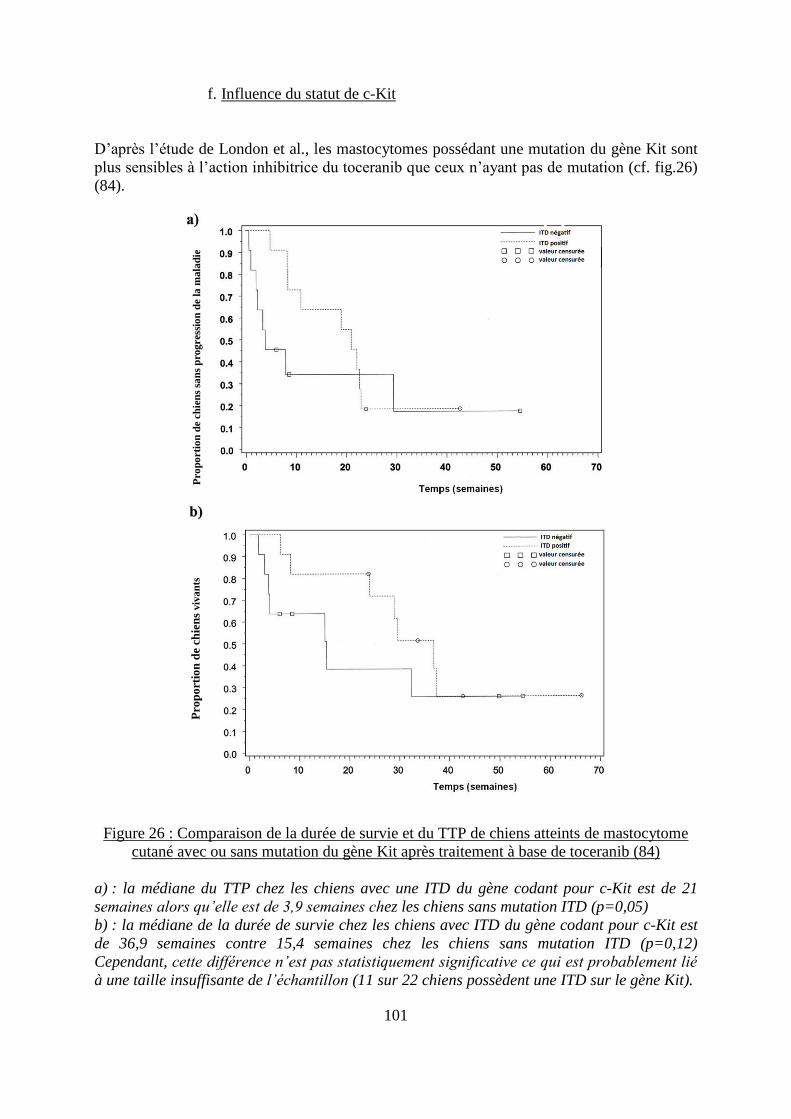
101
f. Influence du statut de c-Kit
D’après l’étude de London et al., les mastocytomes possédant une mutation du gène Kit sont
plus sensibles à l’action inhibitrice du toceranib que ceux n’ayant pas de mutation (cf. fig.26)
(84).
Figure 26 : Comparaison de la durée de survie et du TTP de chiens atteints de mastocytome
cutané avec ou sans mutation du gène Kit après traitement à base de toceranib (84)
a) : la médiane du TTP chez les chiens avec une ITD du gène codant pour c-Kit est de 21
semaines alors qu’elle est de 3,9 semaines chez les chiens sans mutation ITD (p=0,05)
b) : la médiane de la durée de survie chez les chiens avec ITD du gène codant pour c-Kit est
de 36,9 semaines contre 15,4 semaines chez les chiens sans mutation ITD (p=0,12)
Cependant, cette différence n’est pas statistiquement significative ce qui est probablement lié
à une taille insuffisante de l’échantillon (11 sur 22 chiens possèdent une ITD sur le gène Kit).
Pro
po
rtio
n d
e ch
ien
s viv
an
ts
Pro
po
rti
on
de c
hie
ns
san
s p
rogress
ion
de
la m
ala
die
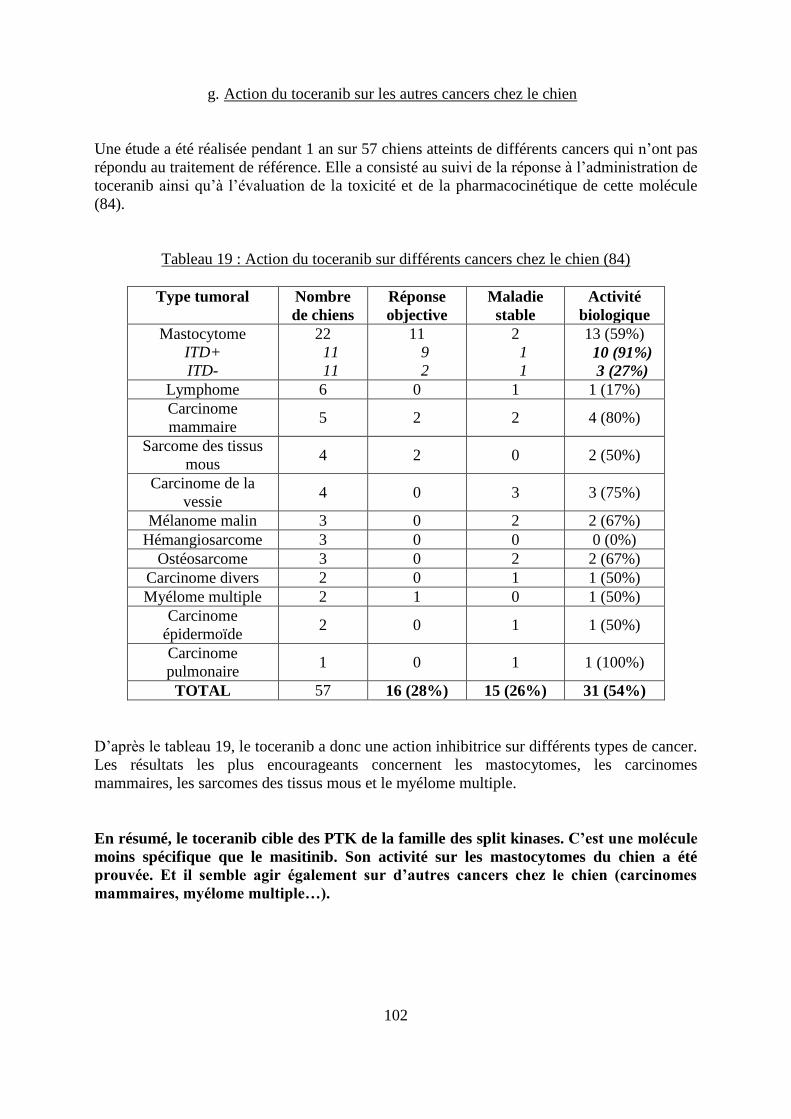
102
g. Action du toceranib sur les autres cancers chez le chien
Une étude a été réalisée pendant 1 an sur 57 chiens atteints de différents cancers qui n’ont pas
répondu au traitement de référence. Elle a consisté au suivi de la réponse à l’administration de
toceranib ainsi qu’à l’évaluation de la toxicité et de la pharmacocinétique de cette molécule
(84).
Tableau 19 : Action du toceranib sur différents cancers chez le chien (84)
Type tumoral Nombre
de chiens
Réponse
objective
Maladie
stable
Activité
biologique
Mastocytome
ITD+
ITD-
22
11
11
11
9
2
2
1
1
13 (59%)
10 (91%)
3 (27%)
Lymphome 6 0 1 1 (17%)
Carcinome
mammaire 5 2 2 4 (80%)
Sarcome des tissus
mous 4 2 0 2 (50%)
Carcinome de la
vessie 4 0 3 3 (75%)
Mélanome malin 3 0 2 2 (67%)
Hémangiosarcome 3 0 0 0 (0%)
Ostéosarcome 3 0 2 2 (67%)
Carcinome divers 2 0 1 1 (50%)
Myélome multiple 2 1 0 1 (50%)
Carcinome
épidermoïde 2 0 1 1 (50%)
Carcinome
pulmonaire 1 0 1 1 (100%)
TOTAL 57 16 (28%) 15 (26%) 31 (54%)
D’après le tableau 19, le toceranib a donc une action inhibitrice sur différents types de cancer.
Les résultats les plus encourageants concernent les mastocytomes, les carcinomes
mammaires, les sarcomes des tissus mous et le myélome multiple.
En résumé, le toceranib cible des PTK de la famille des split kinases. C’est une molécule
moins spécifique que le masitinib. Son activité sur les mastocytomes du chien a été
prouvée. Et il semble agir également sur d’autres cancers chez le chien (carcinomes
mammaires, myélome multiple…).

103
h. Effets secondaires
Les effets secondaires les plus souvent rencontrés sont :
-signes gastro-intestinaux (diarrhée, anorexie, vomissements), léthargie, neutropénie,
thrombocytopénie, hypo-albuminémie et augmentation de l’alanine aminotransférase très
fréquemment
-boiterie, perte de poids, sang dans les selles, désordre musculosquelettique moins
fréquemment
Des effets secondaires sévères sont apparus chez plus d’un tiers des chiens et 5 chiens sont
morts potentiellement en relation avec la molécule (33). Le toceranib, par son action sur le
VEGFR, entraîne une ischémie digestive ce qui explique les signes digestifs observés chez
certains chiens.

104
B. Les inhibiteurs de tyrosines kinases ayant une AMM chez
l’homme
1. Le Gleevec® (imatinib)
Le Gleevec® a obtenu l’AMM en mai 2001 pour le traitement de la leucémie myéloïde
chronique chez l’homme et est distribué par le laboratoire Novartis. Il a été approuvé en 2008
pour le traitement des GIST malignes métastatiques et/ou non opérables.
a. Structure
La formule chimique de l’imatinib mesylate est le 4-[(4-methyl-1-piperazinyl)methyl]-N-[4-
methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]aminophenyl] benzamide methanesulfonate.
Figure 27 : Structure chimique de l’imatinib mesylate (18)
b. Cibles
L’imatinib montre une activité contre la protéine Abl avec une IC50 autour de 0,25µM. Son
activité contre PDGFR et c-Kit est similaire avec des valeurs d’IC50 proches. Les valeurs de
l’IC50 pour de nombreuses autres PTK sont 100 fois plus fortes ce qui démontre que
l’imatinib présente une forte spécificité pour les protéines Abl, c-Kit et PDGFR.

105
Tableau 20 : IC50 de l’imatinib sur différentes TK (21,14)
Tyrosines kinases IC 50 (µm)
v-ABL 0,1 à 0,3
BCR-ABL 0,25
TEL-ABL 0,35
TEL-ARG 0,5
PDGFR 0,1
TEL-PDGFR 0,15
c-Kit 0,15
Flt-3 >10
c-Fms et v-Fms >10
KDR >10
HER1 et HER2 >100
IGF-1R >100
v-Src >10
Jak-2 >100
c. Mode d’action
L’imatinib agit de façon compétitive avec l’ATP.
d. Protocole
La posologie recommandée est de :
400 mg / jour en une prise unique en phase chronique de leucémie myéloïde
chronique, 600 mg (en une ou deux prises) dans les phases d’accélération ou en crise
blastique aiguë, avec augmentation respective à 600 mg et 800 mg si la réponse est
jugée insuffisante, la tolérance restant correcte et sous surveillance rapprochée. La
dose minimale efficace permettant une réponse thérapeutique optimale dans la
leucémie myéloïde chronique est supérieure ou égale à 300 mg.
400 mg / jour dans les GIST
100 mg suffisent souvent dans les syndromes hyper-éosinophiliques mais 400 mg est
une dose optimale dans les leucémies myélomonocytaires chroniques avec
translocation t(5 ;12).

106
e. Efficacité de l’imatinib dans la leucémie myéloïde chronique
La leucémie myéloïde chronique résulte de la transformation néoplasique d’une cellule
souche hématopoïétique primitive. La maladie est due à une translocation appelée
translocation de Philadelphie impliquant les bras longs des chromosomes 9 (gène Abl) et 22
(gène Bcr).
Il y a formation d’une protéine kinase fusion Bcr-Abl qui est activée de façon constitutive et
entraîne une augmentation de la prolifération cellulaire, une diminution de l’apoptose et une
perturbation des interactions avec le domaine extracellulaire.
La maladie se déroule en 3 phases : la phase initiale chronique, la phase d’accélération et la
phase de leucémie aiguë.
Sans traitement, la médiane de survie des patients atteints de leucémie myéloïde chronique est
comprise entre 2 et 3 ans.
Il existe 3 niveaux de contrôle de l’évolution de la maladie :
-la réponse hématologique complète : normalisation des comptages cellulaires sanguins et
présence de globules blancs différenciés ainsi que disparition des symptômes de la maladie
-réponse cytogénétique complète : aucune métaphase avec chromosome de Philadelphie
positif (au moins 20 métaphases pour l’analyse). La réponse cytogénétique majeure
correspond à la présence de moins de 35% de métaphases avec chromosome de Philadelphie
positif.
-la rémission moléculaire : pas de transcrit Bcr-Abl détectable par PCR
La réponse cytogénétique est fréquemment utilisée comme marqueur de l’efficacité d’un
traitement. Les traitements conventionnels sont les agents cytotoxiques tels que le busulfan,
l’hydroxycarbamide et la cytarabine mais aucun de ces agents n’entraîne une rémission
cytogénétique chez un nombre significatif de cas. Les polychimiothérapies ont des effets
secondaires considérables et ont été abandonnées. L’IFN-alpha a été introduit dans les années
1980. Il permet une rémission cytogénétique durable chez plus de 1/3 des patients et est plus
efficace que le busulfan et l’hydroxycarbamide. L’association IFN-alpha et cytarabine donne
de meilleures résultats que l’utilisation d’IFN-alpha seule mais cela n’a pas été confirmé dans
d’autres études. La greffe de cellule souche permet l’éradication de la maladie, la survie à
long terme varie entre 50 et 80%. Elle est réservée aux patients ayant un donneur compatible
et implique de fortes doses de chimiothérapies et une irradiation totale du corps (21).
L’imatinib est un ITK qui cible la protéine Abl et a été développé pour traiter la leucémie
myéloïde chronique (18, 28, 29).
La phase 2 d’essai clinique a montré que l’imatinib est efficace pour traiter les patients en
phase aiguë, en phase d’accélération et en phase chronique. Les meilleurs résultats sont
obtenus pour la phase chronique avec 95% des patients qui ont obtenu une réponse
hématologique complète avec un taux de survie sans progression de la maladie à 18 mois de
89%. Le taux de réponse cytogénétique complète est de 41% avec 60% de réponse
cytogénétique majeure (21).

107
La phase 3 d’essai clinique a permis de montrer que l’imatinib est nettement plus efficace que
l’association IFN-alpha/cytarabine. Après dix-huit mois de traitement, 74 % des patients
traités par l'imatinib étaient en rémission cytogénétique complète contre 8 % des patients
traités par l’association IFN-alpha/cytarabine. La FDA a donc approuvé l’imatinib comme
traitement de première ligne de la leucémie myéloïde chronique en 2001.
Cependant, en association, l’imatinib agit de façon synergique avec IFN-alpha et la
cytarabine.
La réponse au traitement est suivie à l’aide de numérations formules sanguines et d’analyses
cytogénétiques de la moelle osseuse. La détection précoce de mutation dans le domaine
kinase de la protéine Bcr-Abl est primordiale afin d’adapter le traitement en vue d’une
éventuelle résistance des cellules à l’imatinib.
Le traitement à base d’imatinib peut être interrompu chez les patients étant en rémission
moléculaire complète depuis au moins 2 ans (93).
f. Efficacité de l’imatinib dans les GIST
Les GIST sont liées à des mutations de la protéine c-Kit le plus souvent mais peuvent
également être liées à des mutations du PDGFR alpha.
75 à 90% des patients bénéficient d’un traitement à base d’imatinib. Dans les études
précliniques, l’imatinib inhibe la prolifération et induit l’apoptose des cellules issues de GIST.
Dans l’étude de phase II, 147 patients ayant une GIST avancée ont participé afin d’évaluer
l’activité de l’imatinib. Ils ont reçu une dose quotidienne de 400 mg ou 600 mg. 53,7% ont
présenté une réponse partielle, 27,9% ont présenté une maladie stable. Aucun patient n’a eu
de réponse complète. 13,6% des patients ont présenté une résistance à l’imatinib. La thérapie
a été bien tolérée malgré quelques effets secondaires d’intensité faible à modéré (œdème,
fatigue, diarrhée). Dans 5% des cas, des hémorragies intra-abdominales ou gastro-intestinales
ont été présentes. Il n’y a pas eu de différences significatives entre les effets secondaires et la
réponse aux 2 doses d’imatinib (23).
L’étude permet de conclure que l’imatinib est un traitement efficace des GIST à un stade
avancé qui sont des maladies résistantes aux agents conventionnels de chimiothérapie.
Suite à cette étude, la FDA a approuvé l’imatinib en 2008 pour le traitement des GIST
malignes métastatiques et/ou non opérables.
g. Efficacité de l’imatinib dans les mastocytoses
Les mastocytoses systémiques sont liées le plus souvent à la mutation asp816val de c-Kit. In
vitro, cette mutation engendre une absence de réponse à l’imatinib. La plupart des études in
vivo ne montrent pas d’efficacité de l’imatinib sur les malades ayant la mutation asp816val
mais les patients n’ayant pas cette mutation (présence d’une protéine c-Kit sauvage ou d’une
mutation de la zone JM) répondent au traitement.
L’intérêt du génotypage des patients avant le début du traitement trouve donc tout son intérêt.
Cependant, une étude sur 14 patients atteints de mastocytoses systémiques a révélé que
l’imatinib à la dose de 400 mg/j était tout aussi efficace chez des patients ayant la mutation
asp816val sur la protéine c-Kit que ceux n’ayant pas cette mutation (27). Une étude de phase
3 est attendue pour comparer l’efficacité de l’imatinib seul ou associé à la prednisone.
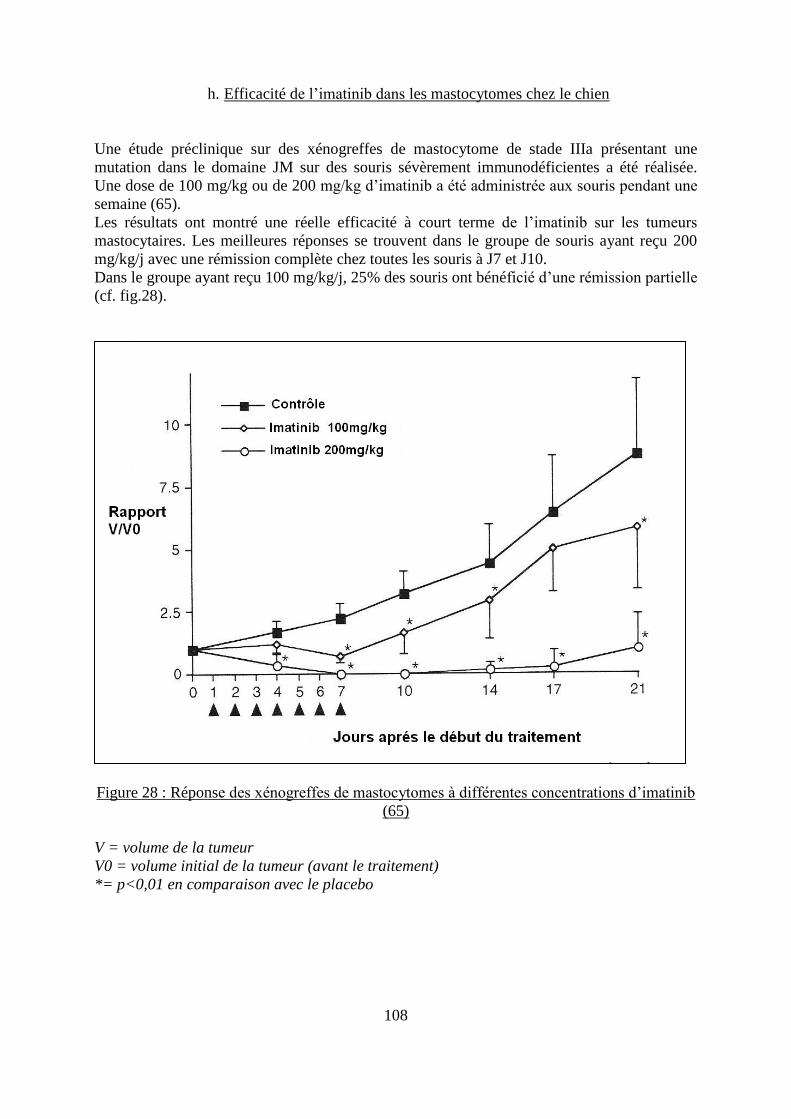
108
h. Efficacité de l’imatinib dans les mastocytomes chez le chien
Une étude préclinique sur des xénogreffes de mastocytome de stade IIIa présentant une
mutation dans le domaine JM sur des souris sévèrement immunodéficientes a été réalisée.
Une dose de 100 mg/kg ou de 200 mg/kg d’imatinib a été administrée aux souris pendant une
semaine (65).
Les résultats ont montré une réelle efficacité à court terme de l’imatinib sur les tumeurs
mastocytaires. Les meilleures réponses se trouvent dans le groupe de souris ayant reçu 200
mg/kg/j avec une rémission complète chez toutes les souris à J7 et J10.
Dans le groupe ayant reçu 100 mg/kg/j, 25% des souris ont bénéficié d’une rémission partielle
(cf. fig.28).
Figure 28 : Réponse des xénogreffes de mastocytomes à différentes concentrations d’imatinib
(65)
V = volume de la tumeur
V0 = volume initial de la tumeur (avant le traitement)
*= p<0,01 en comparaison avec le placebo

109
Isotani et al. ont réalisé une étude clinique sur 21 chiens présentant un mastocytome cutané,
48% (soit 10 chiens) ont répondu à l’imatinib au 14ème
jour à la dose de 10 mg/kg/j. Parmi
eux, 5 chiens présentaient une ITD dans l’exon 11 du gène codant pour c-Kit. Malgré le faible
nombre de chiens, l’efficacité de l’imatinib a été prouvée sur une courte période avec des
réponses partielles ou complètes (58). Mais la réponse au traitement ne peut pas être prédite
par la présence ou l’absence de mutation ITD sur l’exon 11. En effet, d’autres mutations de c-
Kit peuvent être responsables de la transformation tumorale des mastocytes (par exemple des
mutations sur les exons 9, 11 et 13) et l’imatinib est également efficace sur ce type de
mutations.
Cette étude est intéressante mais présente plusieurs limites : en effet, le nombre de chiens est
faible ; de plus, certains chiens ayant répondu aux traitements à base d’imatinib étaient
également sous prednisone. Nous ne pouvons donc pas exclure que la prednisone ait contribué
à la réponse.
Une autre étude a montré l’efficacité de l’imatinib sur des chiens atteints de mastocytome
avec atteinte de la moelle osseuse. Cette affection est rare, progresse rapidement et est
souvent fatale. Les chiens atteints présentent souvent une neutropénie ce qui rend l’utilisation
des agents cytotoxiques difficiles.
14 chiens ont été inclus dans l’étude :
-3 ont reçu de la prednisone : la maladie a progressé jusqu’à l’euthanasie.
-8 ont reçu de la prednisone et de la lomustine : la maladie a progressé chez 7 chiens et un
chien a répondu partiellement, la médiane de survie est de 43 jours.
-3 ont reçu de la prednisone et de l’imatinib : une rémission complète a été observé chez tous
les chiens sans effets secondaires, la lomustine a été ajoutée chez 2 chiens.
Les résultats les plus concluants ont été obtenus avec l’association : prednisone, lomustine et
imatinib. Cependant, seulement 2 chiens ont bénéficié de cette association et des études plus
importantes doivent être menées afin de pouvoir conclure de manière fiable (95).
L’intérêt de combiner un ITK et un agent cytotoxique est de contourner une éventuelle
apparition de résistance et d’augmenter le pouvoir anticancéreux par des effets synergiques ou
additifs.
Chez le chien, le problème principal réside dans l’hépatotoxicité de l’imatinib entraînant une
élévation des enzymes hépatiques et une interruption du traitement. Dans les études
précédentes, la toxicité hépatique n’a pas été mise en évidence. Cela peut s’expliquer par la
courte durée de la thérapie liée au refus de plusieurs propriétaires de continuer le traitement
étant donné le coût important. A l’inverse, chez le chat, l’imatinib est bien toléré.

110
i. Autres cibles de l’imatinib chez l’homme
La leucémie myélomonocytaire chronique est liée à une translocation t(5 ;12) qui résulte en
l’activation du PDGFR. L’imatinib a montré une réponse forte et rapide chez les patients
atteints de cette maladie.
Le syndrome hyper-éosinophilique est un diagnostic d’exclusion à évoquer devant une hyper-
éosinophilie importante et persistante pour laquelle aucune étiologie secondaire n’est
retrouvée. Les progrès récents en biologie moléculaire ont permis d’identifier un transcrit de
fusion FIP1L1–PDGFR-alpha généré par une délétion interstitielle sur le chromosome 4q12
retrouvé dans certains syndromes hyper-éosinophiliques. La détection de ce réarrangement est
primordiale puisque les malades positifs pour ce transcrit peuvent bénéficier d’un traitement
par l’imatinib avec une évolution spectaculaire.
Le syndrome hyper-éosinophilique peut être associé à une mastocytose systémique avec
désordres hématologiques.
Le dermatofibrosarcome protubérant est du à une translocation t(17;22) qui conduit à une
surexpression du gène PDGFR . L’imatinib a un effet thérapeutique sur cette maladie.
Les cancers du poumon à petites cellules sont également sensibles à l’imatinib.
Le PDGF est impliqué dans l’hypertension pulmonaire idiopatique et l’imatinib a montré son
efficacité contre cette maladie à travers des études effectuées sur des souris (21).
En résumé, chez l’homme l’imatinib a été initialement développé pour traiter la
leucémie myéloïde chronique. Cette molécule a ensuite permis le traitement des GIST
malignes métastatiques et/ou non opérables mais des résistances sont apparues. Elle est
également prometteuse dans les traitements de la leucémie myélomonocytaire
chronique, du syndrome hyper-éosinophilique, du dermatofibrosarcome protubérant,
du cancer du poumon à petites cellules et dans l’hypertension pulmonaire idiopathique.
Cependant, elle n’est pas efficace contre les mastocytoses dues à la mutation asp816val.
Chez le chien, l’étude de l’imatinib a montré une bonne efficacité dans le traitement des
mastocytomes, seul ou en association avec un agent cytotoxique. Toutefois, le problème
principal réside dans l’hépatotoxicité de la molécule qui ne permet pas à l’heure actuelle
son utilisation dans cette espèce.

111
j. Effets secondaires
L’imatinib est en général bien toléré et les effets secondaires présents sont souvent d’intensité
faible à modérée et nécessitent rarement un arrêt transitoire du traitement. Ils sont cependant
plus fréquents dans les stades avancés de leucémie myéloïde chronique. On les divise en
effets secondaires hématologiques et non hématologiques.
Effets secondaires non-hématologiques :
-œdème et rétention de fluide (souvent superficiels et atteinte de la zone périorbitaire dans
50% des cas)
-signes gastro-intestinaux (nausée fréquente, diarrhée, douleur abdominale)
-réactions cutanées (éruptions cutanées chez 1/3 des patients, urticaire)
-arthralgie, myalgie et douleur osseuse (fréquent)
-toxicité hépatique (rare)
Effets secondaires hématologiques :
-myélosuppression
k. Résistance à l’imatinib
Les résistances secondaires à l’imatinib sont relativement fréquentes. Elles sont observées
dans la leucémie myéloïde chronique et les GIST. On observe également des résistances
primaires comme par exemple la mutation asp816val du gène codant pour la protéine c-Kit
présente dans la majorité des mastocytoses systémiques.
Les résistances secondaires sont souvent des mutations dans le domaine kinase qui empêche
la liaison de l’imatinib avec la tyrosine kinase.
In vitro, le mécanisme de résistance le plus commun est l’amplification du gène Bcr-Abl ou
une augmentation de la transcription de ce gène (21, 150).
In vivo, on retrouve une mutation dans le domaine kinase de la protéine Abl qui empêche la
liaison de l’imatinib, cette mutation serait présente avant la thérapie et la pression de sélection
exercée par l’imatinib favoriserait son émergence. On retrouve également une surexpression
de la protéine Bcr-Abl.
L’observation de résistances a conduit à rechercher d’autres molécules efficaces contre la
leucémie myéloïde chronique :
-des agents qui ciblent la voie de signalisation en aval de la protéine Bcr-Abl : les inhibiteurs
de farnesyl transférase, les inhibiteurs de la protéine MAP et les inhibiteurs du PIP3
-des agents qui affectent la protéine Bcr-Abl : la geldanamycin et ses dérivés inhibent la
protéine chaperone HSP90 qui a pour rôle de stabiliser la protéine Bcr-Abl. Le trioxyde
d’arsenic entraîne la dégradation de la protéine Bcr-Abl
-d’autres inhibiteurs de la protéine Abl : PD173955 appartient à la famille des pyrido (2,3-d)
pyrimidine et se lie à la protéine Bcr-Abl sous sa forme active et inactive. Il inhibe la PTK à
des concentrations 100 fois inférieures à celle de l’imatinib et il agit sur des protéines Abl
ayant une mutation leur permettant d’acquérir une résistance à l’imatinib. Ce composé n’est
actuellement pas utilisé car ces propriétés pharmacologiques ne sont pas adaptées mais il reste
prometteur pour cibler les cellules résistantes à l’imatinib.

112
De plus, la combinaison de l’imatinib avec une seconde génération d’ITK telle que le
nilotinib ou le dasatinib efficace sur la plupart des mutations résistantes à l’imatinib permet de
contrer l’apparition de résistances (138).
Concernant la résistance à l’imatinib observée dans les GIST, des mutations dans le domaine
kinase de la protéine c-Kit ou des mutations du PDGFR seraient en cause.
Les mutations correspondant à une substitution de la thréonine en isoleucine au codon 670 de
c-Kit, au codon 674 du PDGFR-alpha ou au codon 681 du PDGFR-béta causent une
résistance à l’imatinib. Ces mutations correspondent à la même substitution au codon 315
dans la protéine Bcr-Abl qui cause fréquemment des résistances dans la leucémie myéloïde
chronique. Ces mutations sont appelées gatekeeper. La thréonine permet la liaison de
l’imatinib via un pont hydrogène et sa substitution par de l’isoleucine empêche la liaison de
l’ITK (43).
Le sorafenib, un ITK ciblant les tyrosines kinases VEGFR, PDGFR et Raf, a montré son
efficacité contre ce type de mutations.
En résumé, malgré la bonne efficacité de l’imatinib, des résistances sont apparues chez
l’homme liées fréquemment à des mutations dans le domaine kinase empêchant la
liaison de l’imatinib. Ces résistances ont conduit à la recherche de nouvelles molécules
permettant de limiter ce phénomène.

113
2. L’Iressa® (gefitinib)
Il s’agit d’un ITK qui inhibe la famille des tyrosines kinases EGFR. C’est un inhibiteur
compétitif de l’ATP. Il a obtenu l’AMM en 2003. Des essais cliniques ont montré que cet ITK
est efficace dans le traitement du cancer du poumon non à petites cellules. Des taux de
réponses jusqu'à 19% et une stabilisation de la maladie chez 34% des patients ont été obtenus.
Les effets secondaires les plus courants sont une toxicité gastro-intestinale et des effets sur la
peau. En mai 2003, la FDA a approuvé le gefitinib pour le traitement des cancers avancés du
poumon non à petites cellules traités précédemment avec une chimiothérapie.
Le gefitinib induit un arrêt du cycle cellulaire et l’apoptose dans une variété de lignée
cellulaire in vitro et a une activité sur les cancers du poumon, du côlon, ovarien, du sein et
prostatique dans des modèles de xénogreffes sur des souris (80,92).
3. Le Tarceva® (erlotinib)
Le Tarceva ® a obtenu l’AMM en 2004. L’erlotinib est un ITK qui inhibe les protéines
kinases de la famille de l’EGFR. Il est indiqué dans le traitement des patients atteints du
cancer du poumon non à petites cellules à un stade avancé ou métastatique après échec à une
chimiothérapie. Il est également indiqué en première ligne dans le traitement des patients
atteints de cancer pancréatique non opérable, métastatique ou avancé en association avec la
gemcitabine.
Dans le cas du cancer du pancréas, la médiane de survie est de 6,4 mois avec l’erlotinib et le
gemcitabine alors qu’elle est de 6 mois avec le placebo et le gemcitabine.
Dans le cas du cancer du poumon, la médiane de survie est de 6,7 mois avec l’erlotinib alors
qu’elle est de 4,7 mois avec le placebo. Un test pour la mutation EGFR a été développé par
une société américaine Genzyme et permet de détecter les patients susceptibles de répondre au
traitement (80). Des résistances au traitement ont été mises en évidence.
4. Le Nexavar® (sorafenib)
a. Chez l’homme
Il s’agit d’un ITK ciblant VEGFR, PDGFR et Raf. Il a une action à la fois anti-tumorale et
anti-angiogénique. Il a été approuvé par la FDA en décembre 2005 pour le traitement du
cancer rénal avancé et en novembre 2007 pour le carcinome hépatocellulaire.
Des études sont en cours sur le cancer de la thyroïde et du cerveau.
Il a également montré son efficacité sur les mutations gatekeeper des protéines c-Kit et
PDGFR responsables d’une résistance à l’imatinib (43).

114
b. Chez le chien
Le sorafenib a été étudié en comparaison avec le carboplatine sur une lignée cellulaire
d’ostéosarcome (D-17) issue d’une chienne caniche de 11 ans chez laquelle l’ostéosarcome a
métastasé au poumon. Le carboplatine est souvent utilisé lors d’ostéosarcome chez le chien.
Le sorafenib entraîne l’apoptose des cellules de façon dose dépendante. L’IC50 du sorafenib
est de 1,3 µm alors que celle du carboplatine est de 179,1µm à 72h. L’association du
sorafenib et du carboplatine montre un effet antagoniste.
In vitro, la lignée cellulaire canine D-17 est plus sensible à l’action inhibitrice du sorafenib
que d’autres tumeurs humaines telles que le carcinome hépatocellulaire ou l’adénocarcinome
du conduit biliaire (152).
En conclusion, le sorafenib montre une puissante activité anti-tumorale in vitro sur cette
lignée cellulaire, il est plus efficace que le carboplatine. D’autres études sont nécessaires pour
confirmer son efficacité in vivo lors d’ostéosarcome chez le chien.
Les ostéosarcomes chez l’homme et le chien sont très similaires et des études dans le
traitement des ostéosarcomes chez le chien pourraient permettre des avancées dans la prise en
charge de ce cancer chez l’homme.
En résumé, le sorafenib est approuvé chez l’homme pour le traitement du cancer rénal
avancé et du carcinome hépatocellulaire. Une étude in vitro sur une lignée cellulaire
d’ostéosarcome de chien a montré son fort pouvoir anti-tumoral, des études in vivo sont
attendues.

115
5. Le Sprycel® (dasatinib)
Le Sprycel® a été enregistré en 2006. Il s’agit d’un inhibiteur de la protéine Bbr-Abl, de la
famille des Src, de c-Kit et d’autres PTK. Il a été approuvé pour le traitement des patients
atteints de leucémie myéloïde chronique et pour les patients atteints de leucémie
lymphoblastique aiguë positive au chromosome de Philadelphie après une résistance ou une
intolérance à l’imatinib. Il est actuellement évalué pour d’autres cancers tels que le cancer
avancé de la prostate.
Figure 29 : Représentation graphique de l’action du dasatinib sur différentes kinases (73)

116
6. Le Sutent® (sunitinib)
Le Sutent® a été développé par le laboratoire Pfizer ; il est indiqué dans le carcinome rénal
métastatique et dans le traitement des GIST malignes non opérables et/ou métastatiques après
échec à une thérapie à base d’imatinib due à une résistance ou une intolérance. Il a été
approuvé par la FDA en 2006. Il vient également d’avoir un avis positif pour le traitement des
tumeurs neuroendocrines pancréatiques par le comité scientifique de l’Agence Européenne du
médicament en mai 2011.
a. Structure
Sa formule chimique est : l’acide butanedioique hydroxy-, (2S)-, composé avec N-[2-
(diethylamino)ethyl]-5-[(Z)-(5-fluoro-1,2-dihydro-2-oxo-3H-indol-3-ylidine)methyl]-2,4-
dimethyl-1H-pyrrole-3-carboxamide.
Figure 30 : Structure chimique du sunitinib (5)
b. Cibles
Le sunitinib cible particulièrement le PDGFR-alpha et béta, le VEGFR1, 2 et 3, c-Kit, Flt-3,
CSF-1R et RET.
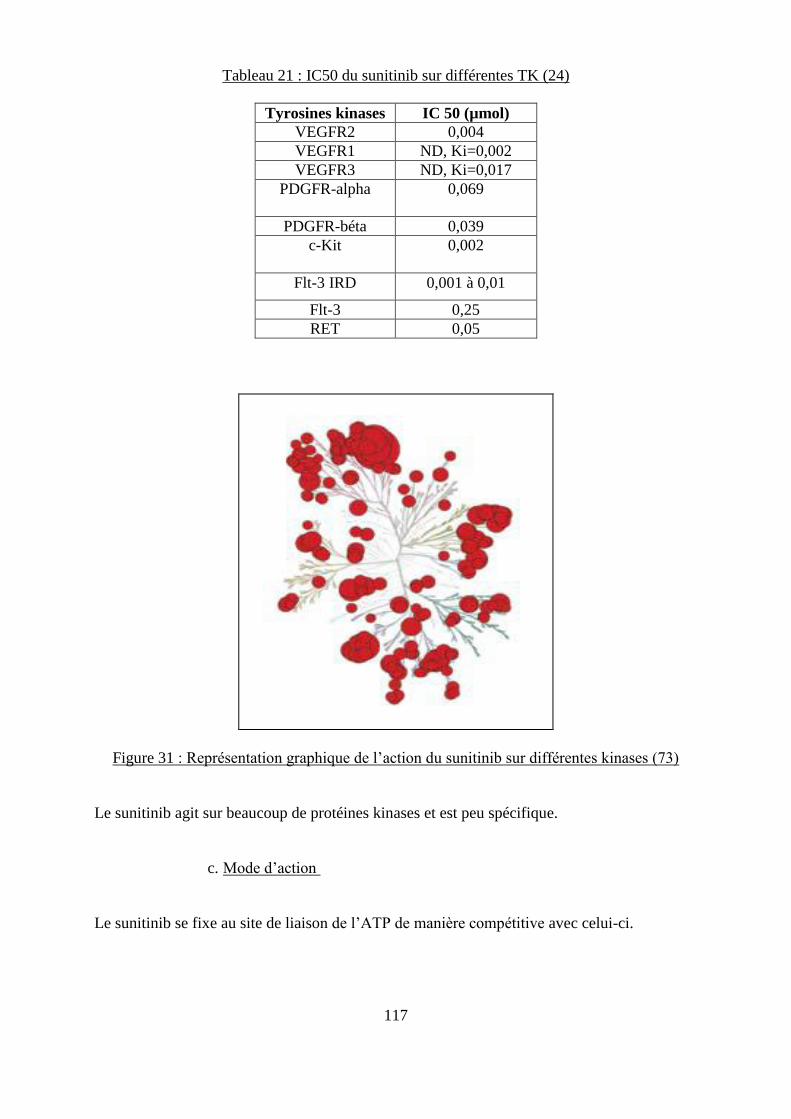
117
Tableau 21 : IC50 du sunitinib sur différentes TK (24)
Tyrosines kinases IC 50 (µmol)
VEGFR2 0,004
VEGFR1 ND, Ki=0,002
VEGFR3 ND, Ki=0,017
PDGFR-alpha
0,069
PDGFR-béta 0,039
c-Kit
0,002
Flt-3 IRD 0,001 à 0,01
Flt-3 0,25
RET 0,05
Figure 31 : Représentation graphique de l’action du sunitinib sur différentes kinases (73)
Le sunitinib agit sur beaucoup de protéines kinases et est peu spécifique.
c. Mode d’action
Le sunitinib se fixe au site de liaison de l’ATP de manière compétitive avec celui-ci.

118
d. Protocole
La dose recommandée est de 50 mg par jour selon un schéma d’administration de 4 semaines
de traitement suivies de 2 semaines sans traitement. Une dose de 37,5 mg/kg par jour peut être
administrée de façon continue chez les patients ayant un changement cyclique de symptômes
lors du schéma d’administration 4/2.
Le sunitinib est métabolisé par l’enzyme CYP3A4 du cytochrome P450. La dose doit être
modifiée selon l’administration d’inhibiteur ou d’activateur de cette enzyme.
Tableau 22 : Quelques exemples d’inhibiteurs et d’activateurs de l’enzyme CYP3A4 du
cytochrome P450 (24)
Inhibiteurs du CYP3A4 Kétoconazole, itraconazole, clarithromycine,
atazanavir, indinavir
Activateurs du CYP3A4 Dexaméthasone, phenytoïne, carbamazépine,
rifampine, phénobarbital
e. Etude in vitro et in vivo sur des souris
Des souris avec des greffes de différents types de tumeurs humains ont été utilisées. Le
Sutent® montre une puissante activité anti-tumorale et cause la régression, l’arrêt du cycle
cellulaire ou réduit la croissance des différentes tumeurs. Le sunitinib inhibe sélectivement la
phosphorylation du VEGFR2 et du PDGFR-béta de façon dose et temps dépendante lorsque
la concentration plasmatique est supérieure ou égale à 50-100 ng/ml. Une constante inhibition
des 2 récepteurs n’est pas requise pour que l’inhibiteur soit efficace (100).
f. Efficacité du sunitinib dans les GIST
Le premier traitement des GIST est l’imatinib mais une résistance primaire est rapportée chez
12 à 20% des patients et une résistance secondaire est fréquente après une durée médiane de 2
ans. Le mécanisme à l’origine d’une résistance est le plus souvent une mutation secondaire de
c-Kit dans le domaine kinase qui empêche la liaison de l’imatinib.
Une première étude randomisée en double aveugle, contrôlée par un placebo sur des patients
atteints d’une GIST qui progresse pendant le traitement à base d’imatinib ou qui sont
intolérants à l’imatinib a été réalisée. Les 312 patients ont reçu 50 mg de Sutent® (207
patients) ou un placebo (105 patients) pendant 4 semaines puis aucun traitement pendant 2
semaines (118).
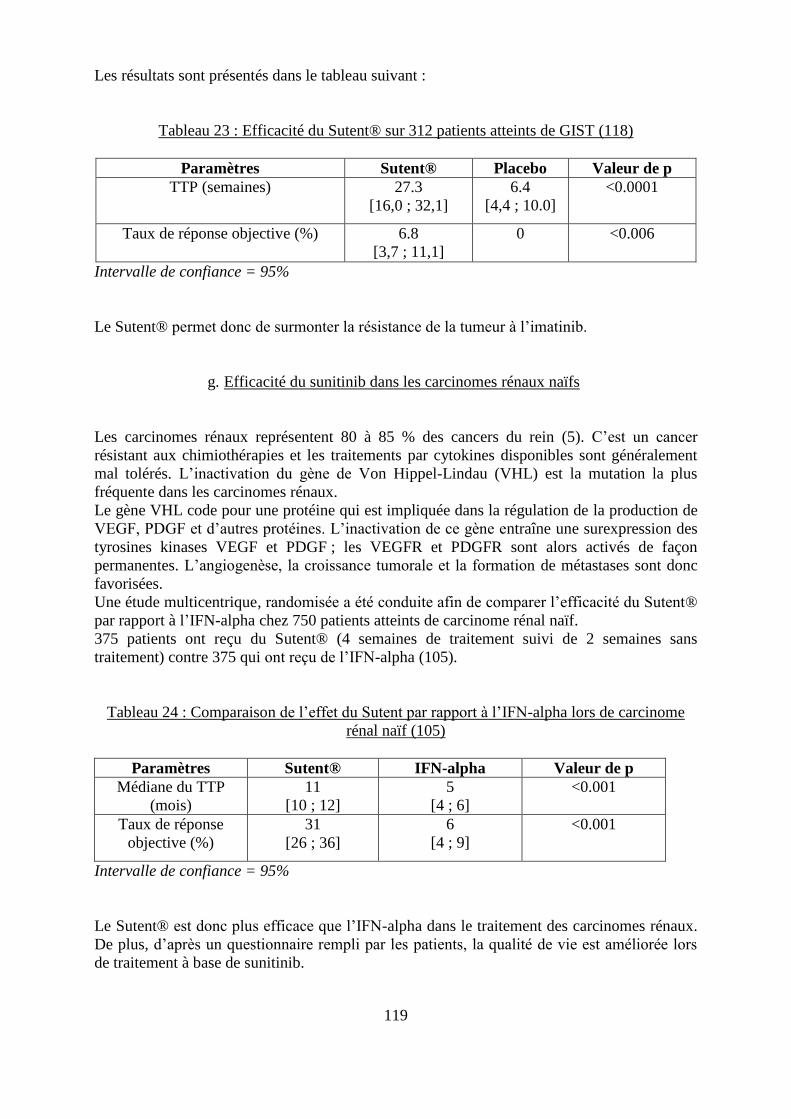
119
Les résultats sont présentés dans le tableau suivant :
Tableau 23 : Efficacité du Sutent® sur 312 patients atteints de GIST (118)
Paramètres Sutent® Placebo Valeur de p
TTP (semaines) 27.3
[16,0 ; 32,1]
6.4
[4,4 ; 10.0]
<0.0001
Taux de réponse objective (%) 6.8
[3,7 ; 11,1]
0 <0.006
Intervalle de confiance = 95%
Le Sutent® permet donc de surmonter la résistance de la tumeur à l’imatinib.
g. Efficacité du sunitinib dans les carcinomes rénaux naïfs
Les carcinomes rénaux représentent 80 à 85 % des cancers du rein (5). C’est un cancer
résistant aux chimiothérapies et les traitements par cytokines disponibles sont généralement
mal tolérés. L’inactivation du gène de Von Hippel-Lindau (VHL) est la mutation la plus
fréquente dans les carcinomes rénaux.
Le gène VHL code pour une protéine qui est impliquée dans la régulation de la production de
VEGF, PDGF et d’autres protéines. L’inactivation de ce gène entraîne une surexpression des
tyrosines kinases VEGF et PDGF ; les VEGFR et PDGFR sont alors activés de façon
permanentes. L’angiogenèse, la croissance tumorale et la formation de métastases sont donc
favorisées.
Une étude multicentrique, randomisée a été conduite afin de comparer l’efficacité du Sutent®
par rapport à l’IFN-alpha chez 750 patients atteints de carcinome rénal naïf.
375 patients ont reçu du Sutent® (4 semaines de traitement suivi de 2 semaines sans
traitement) contre 375 qui ont reçu de l’IFN-alpha (105).
Tableau 24 : Comparaison de l’effet du Sutent par rapport à l’IFN-alpha lors de carcinome
rénal naïf (105)
Paramètres Sutent® IFN-alpha Valeur de p
Médiane du TTP
(mois)
11
[10 ; 12]
5
[4 ; 6]
<0.001
Taux de réponse
objective (%)
31
[26 ; 36]
6
[4 ; 9]
<0.001
Intervalle de confiance = 95%
Le Sutent® est donc plus efficace que l’IFN-alpha dans le traitement des carcinomes rénaux.
De plus, d’après un questionnaire rempli par les patients, la qualité de vie est améliorée lors
de traitement à base de sunitinib.

120
h. Efficacité du sunitinib dans les carcinomes rénaux réfractaires aux cytokines
Deux études ont été réalisées avec des patients atteints de carcinomes rénaux réfractaires aux
traitements à base de cytokines (5).
Dans l’étude 1, l’échec au traitement est basé sur une évidence radiographique de signes de
progression de la maladie définis par les critères du RECIST (Response Evaluation Criteria In
Solid Tumors) ou de l’OMS (Organisation Mondiale de la Santé) pendant une période de
traitement de 9 mois avec des cytokines (IFN-alpha, interleukine 2 ou les 2).
Dans l’étude 2, l’échec au traitement est défini comme une progression de la maladie ou une
toxicité inacceptable du traitement. Le taux de réponse objectif est calculé.
106 patients sont impliqués dans l’étude 1 et 63 patients dans l’étude 2.
Les patients reçoivent 50 mg de Sutent® pendant plusieurs cycles de 4 semaines de thérapie
suivis de 2 semaines sans traitement.
Tableau 25 : Comparaison de l’effet du Sutent lors de carcinome rénal réfractaires aux
cytokines (5)
Paramètres Etude 1 Etude 2
Taux de réponse objectif (%) 34.0
[25,0 ; 43,8]
36.5
[24,7 ; 49,6]
Médiane de la durée de
réponse (semaines)
Non atteinte 54
[34,3 ; 70,1]
Intervalle de confiance = 95%.
Les résultats de ces deux études suggèrent que le sunitinib exerce une activité anti-tumorale
importante dans le carcinome rénal métastatique en tant que traitement de seconde ligne, et
forment la base du lancement d'un programme phase III à grande échelle visant à déterminer
les bienfaits potentiels du sunitinib dans le stade plus précoce de la maladie.
i. Efficacité du sunitinib dans les cancers neuroendocriniens du pancréas
L’effet anti-angiogénique du sunitinib via son action anti-VEGF a été testé sur les tumeurs
neuroendocrines du pancréas.
Une étude de phase 3, multinationale, en double aveugle sur des patients atteints d’un cancer
neuroendocrinien du pancréas a été réalisée de juin 2007 à avril 2009. 86 patients ont reçu du
sunitinib à la dose de 37,5 mg par jour et 85 autres patients ont reçu un placebo.

121
Le TTP est de 11,4 mois pour le groupe ayant pris le sunitinib et de 5,5 mois pour le groupe
placebo (p=0,001). Le taux de réponse objective est de 9,3% dans le groupe sunitinib (2
réponses complètes et 6 réponses partielles). La durée médiane de la réponse au traitement est
de 8,1 mois. Il n’y a eu aucune réponse complète ni partielle dans le groupe placebo. Ces
résultats permettent de montrer l’efficacité clinique et la sécurité du sunitinib chez des
patients atteints de cancer pancréatique neuroendocrinien (151).
Le sunitinib a également une activité anticancéreuse sur le cancer du sein métastatique, le
cancer du côlon, le cancer urothéliale, le carcinome hépatocellulaire non opérable et le cancer
du poumon non à petites cellules récurrent et à un stade avancé (24).
L’effet anti-angiogénique du sunitinib via son action sur le VEGFR a été étudié sur des souris.
L’étude a montré qu’un traitement à base de sunitinib lors de tumeur pancréatique
neuroendocrine entraîne une augmentation du nombre de micrométastases hépatiques. Le
traitement anti-angiogénique est responsable d’une adaptation de la tumeur qui augmente
alors son potentiel métastatique. Ce résultat est observé plus particulièrement quand le
traitement anti-angiogénique est de courte durée (31).
Ce phénomène a également été mis en évidence lors de l’utilisation de différents traitements
anti-VEGFR (113).
En résumé, le sunitinib est efficace dans le traitement des GIST malignes non résécables
et/ou métastatiques après échec à une thérapie à base d’imatinib due à une résistance ou
une intolérance et dans le cancer du rein métastatique naïf ou réfractaire aux cytokines.
Une étude de phase 3 met en évidence l’intérêt thérapeutique de cette molécule sur le
cancer du pancréas neuroendocrinien. Des études ont montré l’activité anticancéreuse
du sunitinib dans différents cancers. Cependant, le bénéfice de l’inhibition du VEGFR
peut être transitoire. Il y a en effet un risque métastatique plus important lors de
traitement de courte durée.
j. Effets secondaires
Le sunitinib est en général bien toléré. Les effets secondaires sont d’intensité faible à modérée
et sont contrôlés avec des traitements symptomatiques. Un arrêt du traitement ou une
diminution de la dose de sunitinib sont possibles (24).
Les effets secondaires les plus communs sont :
-signes gastro-intestinaux (diarrhée, vomissements, nausée et inflammation des muqueuses)
-effet sur la thyroïde (hypothyroïdie le plus souvent)
-hémorragie (le sunitinib a un effet anti-angiogénique et peut compromettre la cicatrisation)
-hypertension par l’action anti-VEGF
-effets cardiaques : dysfonctionnement du ventricule gauche et insuffisance cardiaque
congestive associée, arythmie ventriculaire
-effets dermatologiques : changement de couleur de la peau, érythrodysesthésie palmo-
plantaire
-myélosuppression
-autres (toxicité sur les surrénales, syndrome du mal de tête)

122
7. Le Tyverb® (lapatinib)
Il s’agit d’un ITK très spécifique qui inhibe EGFR et HER2. Il a été approuvé en 2007 par la
FDA pour le traitement du cancer du sein chez des femmes traitées avec la capecitabine
(chimiothérapie).
En 2010, il a été approuvé pour le traitement en première ligne des cancers du sein triple
positif (récepteur hormonal, EGFR, HER2).
Figure 32 : Représentation graphique de l’action du lapatinib sur différentes kinases (73)

123
8. Le Tassigna® (nilotinib)
Il s’agit d’un ITK qui cible la protéine Bcr-Abl. Il a été développé et commercialisé en 2007
pour les patients atteints de leucémie myéloïde chronique résistante à l’imatinib ou n’y
répondant pas. Dans une étude de phase 1, 92% des patients résistant ou ne répondant pas à
l’imatinib ont montré un comptage cellulaire sanguin normal après 5 mois de traitement avec
le nilotinib. Il présente cependant des risques de complications cardiaques (92).
En résumé, de nombreux ITK sont à l’étude chez l’homme. Ces ITK sont développés
dans le but de traiter de nouveaux cancers ou de contrer l’apparition de résistances.
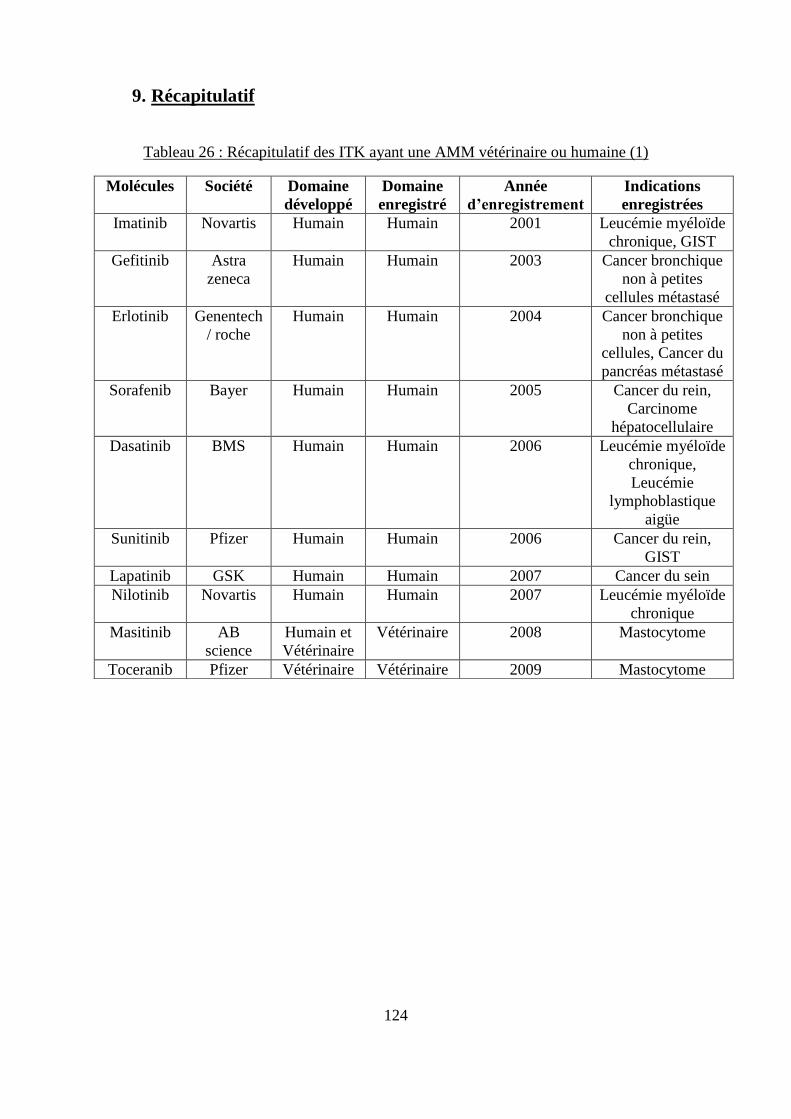
124
9. Récapitulatif
Tableau 26 : Récapitulatif des ITK ayant une AMM vétérinaire ou humaine (1)
Molécules Société Domaine
développé
Domaine
enregistré
Année
d’enregistrement
Indications
enregistrées
Imatinib Novartis Humain Humain 2001 Leucémie myéloïde
chronique, GIST
Gefitinib Astra
zeneca
Humain Humain 2003 Cancer bronchique
non à petites
cellules métastasé
Erlotinib Genentech
/ roche
Humain Humain 2004 Cancer bronchique
non à petites
cellules, Cancer du
pancréas métastasé
Sorafenib Bayer Humain Humain 2005 Cancer du rein,
Carcinome
hépatocellulaire
Dasatinib BMS Humain Humain 2006 Leucémie myéloïde
chronique,
Leucémie
lymphoblastique
aigüe
Sunitinib Pfizer Humain Humain 2006 Cancer du rein,
GIST
Lapatinib GSK Humain Humain 2007 Cancer du sein
Nilotinib Novartis Humain Humain 2007 Leucémie myéloïde
chronique
Masitinib AB
science
Humain et
Vétérinaire
Vétérinaire 2008 Mastocytome
Toceranib Pfizer Vétérinaire Vétérinaire 2009 Mastocytome
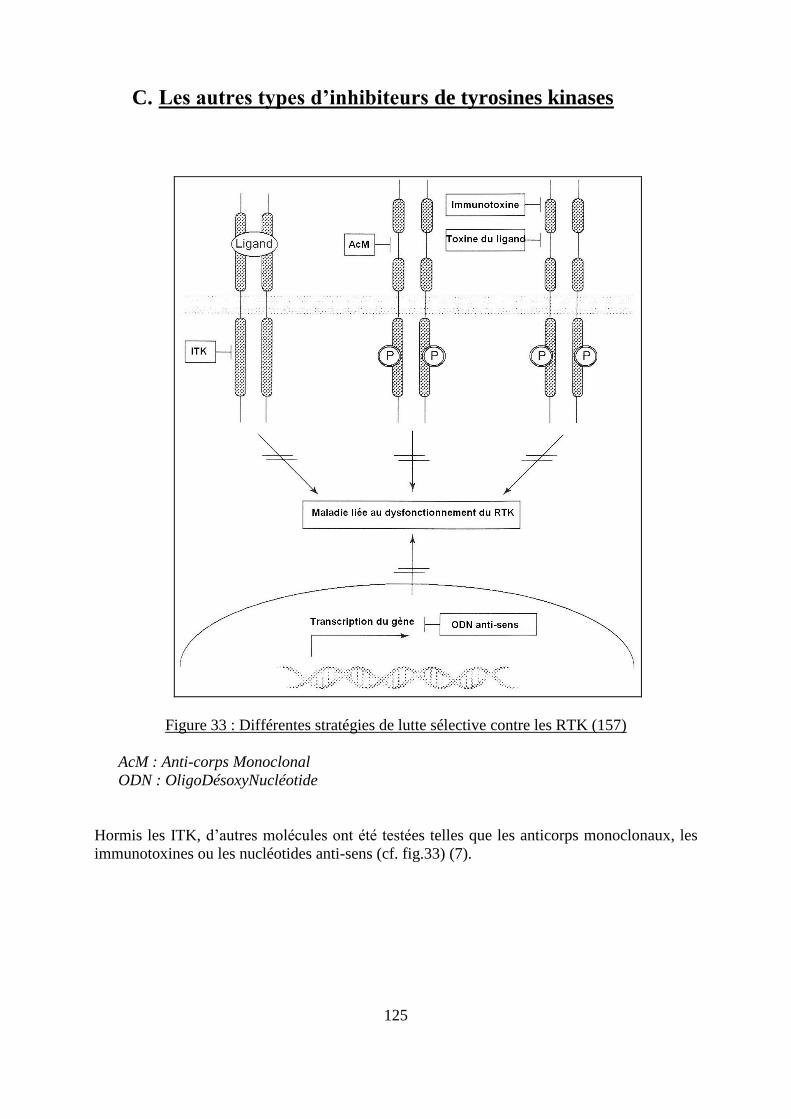
125
C. Les autres types d’inhibiteurs de tyrosines kinases
Figure 33 : Différentes stratégies de lutte sélective contre les RTK (157)
AcM : Anti-corps Monoclonal
ODN : OligoDésoxyNucléotide
Hormis les ITK, d’autres molécules ont été testées telles que les anticorps monoclonaux, les
immunotoxines ou les nucléotides anti-sens (cf. fig.33) (7).

126
1. Les anticorps monoclonaux
Les anticorps monoclonaux sont spécifiquement dirigés contre le domaine extracellulaire des
RTK. Ils empêchent l’interaction ligand-RTK et de ce fait aucun signal n’est transmis par le
RTK. Ils entraînent également une augmentation de l’internalisation du RTK. De plus, ils
induisent une réaction immunitaire via le complément ou par ADCC entrainant la lyse des
cellules tumorales.
Par exemple, l’Herceptin® (transtuzumab) a été développé pour cibler la protéine HER2 dans
les cancers du sein. Il a été approuvé en 1998 par la FDA. En tant que premier traitement dans
les cancers du sein métastatique, le taux de réponse est de 25% et lorsqu’il est combiné à une
chimiothérapie le taux de réponse est d’environ 50%.
Un autre exemple est celui de l’Erbitux® (cetuximab) ciblant la protéine EGFR dans un
certain nombre de cancers tels que le cancer du pancréas, le cancer du rein et le carcinome du
sein.
L’inconvénient de cette stratégie est qu’elle requiert la génération d’anticorps humanisé et le
développement potentiel par l’hôte d’anticorps dirigés contre l’anticorps monoclonal limite la
durée d’efficacité du traitement (80, 157).
2. Les nucléotides non-sens
Ce sont des oligodésoxynucléotides qui interagissent avec l’ARNm et bloquent la
transcription et donc l’expression de protéines spécifiques. On peut noter par exemple
l’oligodésoxynucléotides qui cible IGF-1R et qui induit l’apoptose des cellules de mélanomes
malins.
Une nouvelle technique d’interférence par l’ARN permet de mettre sous silence l’expression
de gène ciblé de façon puissante et spécifique.
Cette technique est investiguée pour les isoformes de VEGF et pourrait avoir de futures
applications (92).
3. Les immunotoxines
Ce sont des molécules qui contiennent à la fois une toxine et un conjugué leur permettant de
se lier au RTK. Le conjugué peut être une chaîne de l’anticorps dirigé contre le RTK ou une
partie du facteur de croissance se liant au RTK.
Les immunotoxines ciblent spécifiquement un antigène des cellules cancéreuses. Lors de la
liaison de l’immunotoxine au RTK, la toxine est internalisée dans un endosome puis libérée
dans le cytoplasme, elle inhibe la synthèse protéique ce qui entraine l’apoptose de la cellule
cancéreuse.
Un exemple est la protéine DAB389EGF qui contient une partie de la toxine diphtérique et
une séquence du ligand EGF. Cette protéine cible les EGFR de cancers tels que le cancer du
sein ou le cancer du poumon non à petites cellules et entraîne la mort des cellules cancéreuses.

127
En résumé, différents types d’inhibiteurs existent tels que les anticorps monoclonaux, les
immunotoxines, et les nucléotides non-sens. Les anticorps monoclonaux tels que
l’Herceptin® et l’Erbitux® ont été développé et représentent la 1ère
génération de
thérapie ciblée. Leur principal inconvénient est leur durée d’efficacité limitée.
D. Comparaison des différents inhibiteurs de tyrosines kinases
L’ensemble des ITK présentés dans cette thèse est récapitulé dans les tableaux 27 et 28.

128
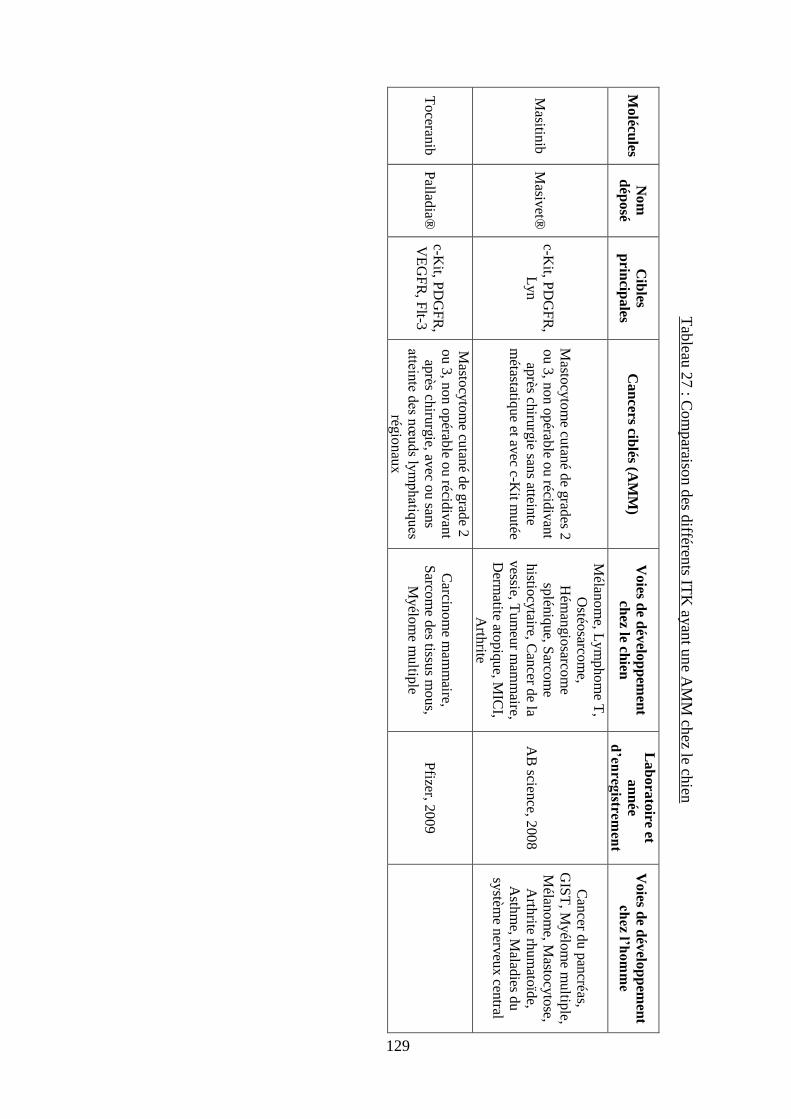
129
Tab
leau 2
7 : C
om
paraiso
n d
es différen
ts ITK
ayan
t un
e AM
M ch
ez le chien
Molécu
les N
om
dép
osé
Cib
les
prin
cipa
les C
an
cers ciblés (A
MM
) V
oies d
e dév
elop
pem
ent
chez le ch
ien
La
bo
rato
ire et
an
née
d’e
nreg
istremen
t
Vo
ies de d
évelo
pp
emen
t
chez l’h
om
me
Masitin
ib
Masiv
et®
c-Kit, P
DG
FR
,
Lyn
Masto
cyto
me cu
tané d
e grad
es 2
ou 3
, non o
pérab
le ou récid
ivan
t
après ch
irurg
ie sans attein
te
métastatiq
ue et av
ec c-Kit m
utée
Mélan
om
e, Lym
pho
me T
,
Ostéo
sarcom
e,
Hém
angio
sarcom
e
splén
ique, S
arcom
e
histio
cytaire, C
ancer d
e la
vessie, T
um
eur m
amm
aire,
Derm
atite atopiq
ue, M
ICI,
Arth
rite
AB
science, 2
008
AB
science, 2
00
8
Can
cer du
pan
créas,
GIS
T, M
yélo
me m
ultip
le,
Mélan
om
e, Masto
cyto
se,
Arth
rite rhu
mato
ïde,
Asth
me, M
aladies d
u
systèm
e nerv
eux
central
Toceran
ib
Pallad
ia®
c-Kit, P
DG
FR
,
VE
GF
R, F
lt-3
Masto
cyto
me cu
tané d
e grad
e 2
ou 3
, non o
pérab
le ou récid
ivan
t
après ch
irurg
ie, avec o
u san
s
atteinte d
es nœ
uds ly
mphatiq
ues
régio
nau
x
Carcin
om
e mam
maire,
Sarco
me d
es tissus m
ou
s,
Myélo
me m
ultip
le
Pfizer, 2
00
9
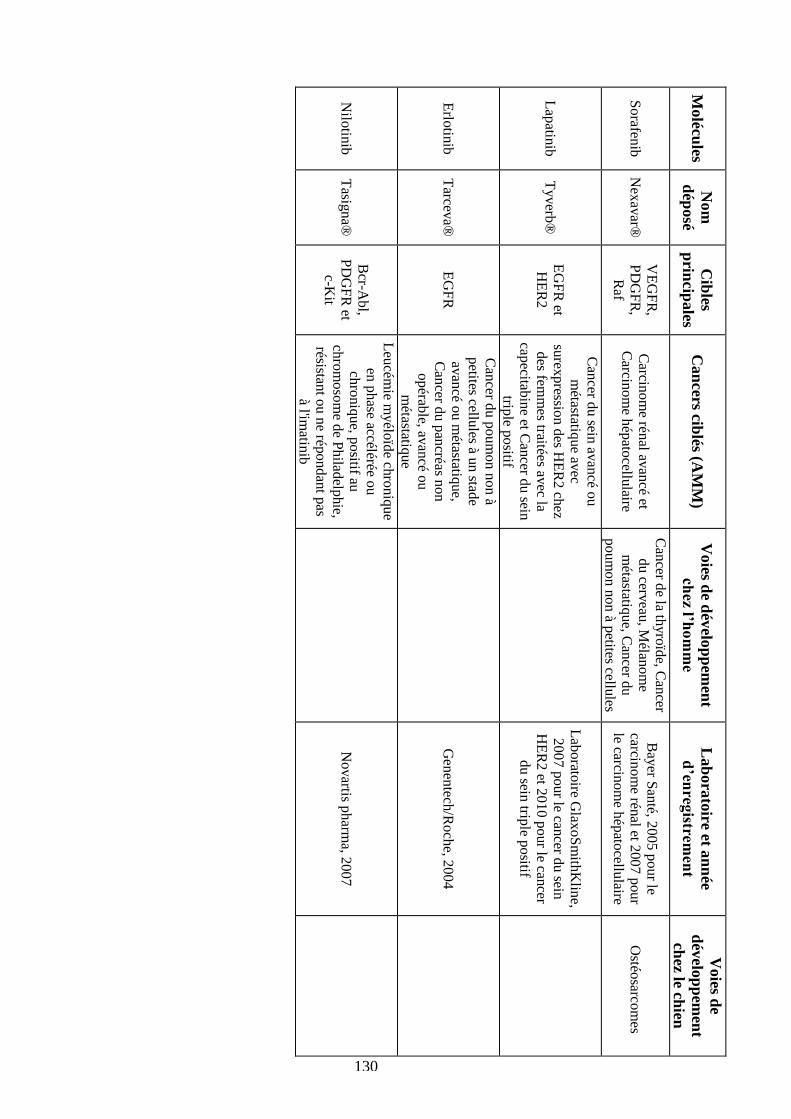
130
Molécu
les N
om
dép
osé
Cib
les
prin
cipales
Can
cers ciblés (A
MM
) V
oies d
e dév
elop
pem
ent
chez l’h
om
me
Lab
ora
toire et a
nn
ée
d’en
registre
men
t
Voies d
e
dév
elop
pem
ent
chez le ch
ien
Sorafen
ib
Nex
avar®
VE
GF
R,
PD
GF
R,
Raf
Carcin
om
e rénal av
ancé et
Carcin
om
e hép
atocellu
laire
Can
cer de la th
yro
ïde, C
ancer
du cerv
eau, M
élanom
e
métastatiq
ue, C
ancer d
u
poum
on n
on à p
etites cellules
Bay
er San
té, 20
05
po
ur le
carcino
me rén
al et 200
7 p
ou
r
le carcino
me h
épato
cellulaire
Ostéo
sarcom
es
Lap
atinib
T
yverb
®
EG
FR
et
HE
R2
Can
cer du sein
avan
cé ou
métastatiq
ue av
ec
surex
pressio
n d
es HE
R2 ch
ez
des fem
mes traitées av
ec la
capecitab
ine et C
ancer d
u sein
triple p
ositif
Lab
orato
ire Glax
oS
mith
Klin
e,
20
07
po
ur le can
cer du
sein
HE
R2
et 20
10 p
our le can
cer
du
sein trip
le po
sitif
Erlo
tinib
T
arceva®
E
GF
R
Can
cer du p
oum
on n
on à
petites cellu
les à un stad
e
avan
cé ou m
étastatique,
Can
cer du p
ancréas n
on
opérab
le, avan
cé ou
métastatiq
ue
G
enen
tech/R
och
e, 20
04
Nilo
tinib
T
asign
a®
Bcr-A
bl,
PD
GF
R et
c-Kit
Leu
cémie m
yélo
ïde ch
roniq
ue
en p
hase accélérée o
u
chro
niq
ue, p
ositif au
chro
moso
me d
e Philad
elphie,
résistant o
u n
e répondan
t pas
à l'imatin
ib
N
ovartis p
harm
a, 20
07
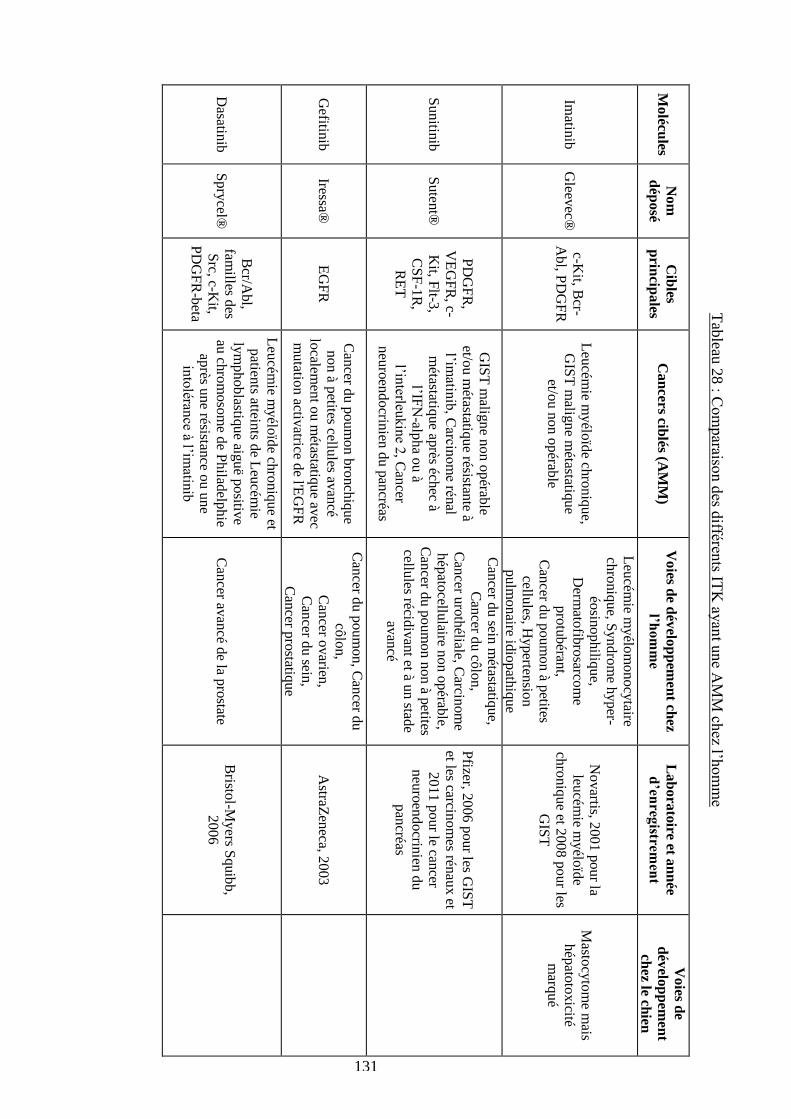
131
Tab
leau 2
8 : C
om
paraiso
n d
es différen
ts ITK
ayan
t une A
MM
chez l’h
om
me
Molécu
les N
om
dép
osé
Cib
les
prin
cipa
les C
an
cers ciblés (A
MM
) V
oies d
e dév
elop
pem
ent ch
ez
l’hom
me
La
bo
rato
ire et an
née
d’e
nreg
istremen
t
Vo
ies de
dév
elop
pem
ent
chez le ch
ien
Imatin
ib
Gleev
ec®
c-Kit, B
cr-
Ab
l, PD
GF
R
Leu
cémie m
yélo
ïde ch
roniq
ue,
GIS
T m
aligne m
étastatique
et/ou n
on o
pérab
le
Leu
cémie m
yélo
mono
cytaire
chro
niq
ue, S
yndro
me h
yp
er-
éosin
ophiliq
ue,
Derm
atofib
rosarco
me
pro
tubéran
t,
Can
cer du p
oum
on à p
etites
cellules, H
yperten
sion
pulm
onaire id
iopath
iqu
e
No
vartis, 2
00
1 p
ou
r la
leucém
ie myélo
ïde
chro
niq
ue et 2
00
8 p
ou
r les
GIS
T
Masto
cyto
me m
ais
hép
atoto
xicité
marq
ué
Sunitin
ib
Su
tent®
PD
GF
R,
VE
GF
R, c-
Kit, F
lt-3,
CS
F-1
R,
RE
T
GIS
T m
aligne n
on o
pérab
le
et/ou m
étastatique résistan
te à
l’imatin
ib, C
arcinom
e rénal
métastatiq
ue ap
rès échec à
l’IFN
-alpha o
u à
l’interleu
kin
e 2, C
ancer
neu
roen
docrin
ien d
u p
ancréas
Can
cer du sein
métastatiq
ue,
Can
cer du cô
lon
,
Can
cer uro
théliale, C
arcinom
e
hép
atocellu
laire non o
pérab
le,
Can
cer du p
oum
on n
on
à petites
cellules récid
ivan
t et à un
stade
avan
cé
Pfizer, 2
00
6 p
our les G
IST
et les carcino
mes rén
aux et
20
11
po
ur le can
cer
neu
roen
do
crinien
du
pan
créas
Gefitin
ib
Iressa®
EG
FR
Can
cer du p
oum
on b
ronch
ique
non à p
etites cellules av
ancé
localem
ent o
u m
étastatique av
ec
mutatio
n activ
atrice de l'E
GF
R
Can
cer du p
oum
on, C
ancer d
u
côlo
n,
Can
cer ovarien
,
Can
cer du sein
,
Can
cer pro
statiqu
e
AstraZ
eneca, 2
003
Dasatin
ib
Sp
rycel®
Bcr/A
bl,
familles d
es
Src, c-K
it,
PD
GF
R-b
eta
Leu
cémie m
yélo
ïde ch
roniq
ue et
patien
ts atteints d
e Leu
cémie
lym
phoblastiq
ue aig
uë p
ositiv
e
au ch
rom
oso
me d
e Philad
elphie
après u
ne résistan
ce ou u
ne
into
lérance à l’im
atinib
Can
cer avan
cé de la p
rostate
Bristo
l-Myers S
quib
b,
20
06

132

133
Conclusion
Le concept de thérapie ciblée et la découverte des ITK ont révolutionné le domaine de la
cancérologie. La meilleure compréhension de l’étiologie moléculaire du cancer a permis le
développement de nouvelles thérapies qui sont rassemblées sous le terme de « thérapie ciblée ».
Contrairement aux agents de chimiothérapie, ces thérapies interfèrent spécifiquement avec des
molécules-clés qui interviennent dans la transformation tumorale. Ces molécules sont très souvent
des RTK.
Cette approche ciblée laisse envisager des options thérapeutiques plus efficaces que la thérapie
cytotoxique. De plus, elle offre une unique opportunité de thérapie combinée avec d’autres agents
anticancéreux afin de diminuer les toxicités individuelles de chaque molécule (39). Chaque ITK
possède en effet un profil de toxicité propre lié à son action sur des protéines non cibles. Les
études s’orientent donc actuellement vers la recherche de molécules très spécifiques afin de limiter
au maximum les effets secondaires.
L’objectif est d’identifier les anomalies moléculaires qui permettent la sélection de patients aptes à
bénéficier du traitement. Il faut « cibler » le patient qui sera susceptible de répondre à une thérapie
à base d’ITK afin d’éviter de traiter des malades inutilement.
L’apparition de résistance lors de thérapie ciblée est un problème fréquent mais la caractérisation
moléculaire des mécanismes de résistances ouvre de nouvelles opportunités pour la création d’une
2nde
génération d’ITK ou la combinaison de plusieurs ITK afin de les contourner.
Le problème du coût financier important de ces thérapies se pose en médecine vétérinaire et
humaine.
Chez l’homme, les ITK peuvent être utilisés dans différents cancers. Chez le chien, seul le
mastocytome peut actuellement être traité avec un ITK : le masitinib ou le toceranib.
L’utilisation des ITK n’est pas seulement limitée au domaine de l’oncologie. En effet, certaines
maladies inflammatoires (telles que l’asthme ou l’arthrite rhumatoïde chez l’homme et la
dermatite atopique chez le chien) et les maladies du système nerveux central telles que la maladie
d’Alzheimer bénéficient d’un traitement à base d’ITK.

134
A moyen terme, les ITK offrent de nouvelles options thérapeutiques extrêmement intéressantes en
médecine humaine et vétérinaire. De nombreux ITK sont actuellement en cours d’études pour
l’homme et pour le chien.

135
Bibliographie
1. AB SCIENCE. (page consultée le 15 décembre 2010). http://masivet.com/.
2. AB SCIENCE Pharmaceutical compagny. (page consultée le 10 décembre 2010). http://www.ab-science.com/.
3. ABADIE J., ARMAND J.P., BORDEAU W., DECROUY D., DEVAUCHELLE P. juin
2010. Symposium sur "Regards croisés sur le mastocytome". Paris : Pfizer, juin 2010.
4. ABADIE J., IBISH C. 2009. Comment traiter un mastocytome chez le chien. Nouv. Prat.
Vet. 2009, Vol. 9, 42, pp. 23-28.
5. ADAMS V.R., LEGGAS M. 2007. Sunitinib malate for the treatment of metastatic renal
cell carcinoma and gastrointestinal stromal tumors. Clin. Ther. 2007. Vol. 29, 7. pp. 1338-
1353.
6. AHERN T.E., BIRD R.C., BIRD A.E., WOLFE L.G. 1996. Expression of the oncogene
c-erbB-2 in canine mammary cancers and tumor-derived cell lines. Am. J. Vet. Res. 1996.
Vol. 57, 5. pp. 693-696.
7. AL-OBEIDI F.A., LAM K.S. 2000. Development of inhibitors for protein tyrosine kinases.
Oncogene. 2000, Vol. 19, pp. 5690-5701.
8. ANDERSON D.M., LYMAN S.D., BAIRD A., WIGNALL J.M., EISENMAN J.,
RAUCH C., et al. 1990. Molecular cloning of mast cell growth factor, a hematopoietin
that is active in both membrane bound and soluble forms. Cell. 1990, Vol. 63, 1, pp. 235-
243.
9. ANNIE T., FONG T., SHAWVER L.K., SUN L., TANG C., APP H., et al. 1999. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor
receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization and
growth of multiple tumor types. Cancer Res. 1999, Vol. 59, pp. 99-106.
10. ASHMAN L.K. 1999. The biology of stem cell factor and its receptor c-kit. Int. J.
Biochem. Cell Biol. 1999, Vol. 31, pp. 1037-1051.
11. BEALE, et al. 2008. Efficacy and safety of masitinib in the treatment of atopic dermatitis
in dogs. North. Am. Vet. Dermatol. Forum. 2008. Sous presse.
12. BESMER P., MURPHY J.E., GEORGE P.C., QIU F., BERGOLD P.J., LEDERMAN
L., et al. 1986. A new acute transforming feline retrovirus and relationship of its oncogene
v-kit with the protein kinase gene family. Nature. 1986, Vol. 320, pp. 415-421.
13. BOKOBZA A. 2006. Le mastocytome cutané canin : actualités pronostiques et
thérapeutiques (2000-2005). Thèse de doctorat vétérinaire. Université Paul-Sabatier de
Toulouse, 2006. 68 pp.

136
14. BUCHDUNGER E., O'REILLY T., WOOD J. 2002. Pharmacology of imatinib
(STI571). Eur. J. Cancer. 2002, Vol. 38, 5, pp. 28-36.
15. BÜTTNER C., HENZ B.M., WELKER P., SEPP N.T., GRABBE J. 1998. Identification of activating c-kit mutations in adult-, but not in childhood-onset indolent
mastocytosis: a possible explanation for divergent clinical behavior. J. invest. Dermatol.
1998. Vol. 111, 6. pp. 1227-1231.
16. BUI B.N., BLAY J., DUFFAUD F., HERMINE O., A. LE CESNE. 2007. Preliminary
efficacy and safety results of masitinib administered, front line in patients with advanced
GIST. A phase II study. J. Clin. Oncol. 2007. Vol. 25, 18. Abstract.
17. CHAN P.M., ILANGUMARAN S., LA ROSE J., CHAKRABARTTY A.,
ROTTAPEL R. 2003. Autoinhibition of the kit receptor tyrosine kinase by the cytosolic
juxtamembrane region. Mol. Cell. Biol. 2003, Vol. 23, 9, pp. 3067-3078.
18. COHEN M.H., WILLIAMS G., JOHNSON J.R., DUAN J., GOBBURU J.,RAHMAN
A., et al. 2002. Approval summary for imatinib mesylate capsule in the treatment of
chronic myelogenous leukemia. Clin. Cancer Res. 2002, Vol. 8, pp. 935-942.
19. DAIGLE J., MOUSSY A., MANSFIELD C., HERMINE O. 2010. Masitinib for the
treatment of canine atopic dermatits : a pilot study. Vet. Res. Commun. 2010. Vol. 34, 1.
pp. 51-63.
20. DE VOS J.P., MOUSSY A., BURM A., VAN KUIJK L., HERMINE O. 26 au 29 mars
2009. Assessment of response to the treatment with masitinib of chemotherapy resistant,
grade 2 and 3, metastasized canine cutaneous mast cell tumors. Report of four cases.
Proceedings of the Annual Congress of the European Society of Veterinary Oncology. 26
au 29 mars 2009.
21. DEININGER M.W.N., DRUKER B.J. 2003. Specific targeted therapy of chronic
myelogenous leukemia with imatinib. Pharmacol. Rev. 2003. Vol. 55, pp. 401-423.
22. DEMETRI G.D. 2001. Targeting c-kit mutations in solid tumors: scientific rationale and
novel therapeutic options. Semin. Oncol. 2001. Vol. 28, pp. 19-26.
23. DEMETRI G.D., VON MEHREN M., BLANKE C.D., VAN DER ABBEELE A.D.,
EISENBERG B., ROBERTS P.J., et al. 2002. Efficacy and safety of imatinib mesylate
in advanced gastrointestinal stromal tumors. New Engl. J. Med. 2002. Vol. 347, 7. pp. 472-
480.
24. DESAI J., GURNEY H., PAVLAKIS N., McARTHUR G.A., DAVIS I.D. 2007. Sunitinib malate in the treatment of renal cell carcinoma and gastrointestinal stromal
tumor: Recommendations for patient management. J. Clin. Oncol. 2007. Vol. 3, pp. 167-
176.
25. DIN O.S., WOLL P.J. 2008. Treatment of gastrointestinal stromal tumor : focus on
imatinib mesylate. Ther. Clin. Risk Manag. 2008. Vol. 4, pp. 149-162.

137
26. DOWNING S., CHIEN M.B., MOORE P.E., LONDON C.A. 2002. Prevalence and
importance of internal tandem duplications in exons 11 and 12 of c-kit in mast cell tumors
of dogs. Am. J. Vet. Res. 2002, Vol. 63, 12, pp. 1718-1723. Abstract.
27. DROOGENDIJK H.J., KLUIN-NELEMANS H.J.C.,VAN DOORMAAL J.J.,
ORANJE A.P., VAN DE LOOSDRECHT A.A., VAN DAELE P.L.A. 2006. Imatinib
mesylate in the treatment of systemic mastocytosis : a phase II trial. Cancer. 2006. Vol.
107, 2. pp. 345-351.
28. DRUKER B.J., LYDON N.B. 2000. Lessons learned from the development of an Abl
tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin. Invest. 2000. Vol.
105, 1. pp. 3-7.
29. DRUKER B.J., TALPAZ M., RESTA D.J., PENG B., BUCHDUNGER E., FORD
J.M., et al. 2001. Efficacy and safety of a specific inhibitor of the BCR/ABL tyrosine
kinase in chronic myeloid leukemia. New Engl. J. Med. 2001. Vol. 344, 14. pp. 1031-1037.
30. DUBREUIL P., LETARD S., CIUFOLINI M., GROS L., HUMBERT M.,
CASTERAN N., et al. 2009. Masitinib (AB1010), a potent and selective tyrosine kinase
inhibitor targeting KIT. Plos One. 2009, Vol. 4, 9.
31. EBOS J.M.L., LEE C.R., CRUZ-MUNOZ W., BJARNASON G.A., CHRISTENSEN
J.G., KERBEL R.S. 2009. Accelerated metastasis after short-term treatment with a potent
inhibitor of tumor angiogenesis. Cancer Cell. 2009. Vol. 15, pp. 232-239.
32. ENGH R.A., BOSSEMEYER D. 2002. Structural aspects of protein kinase control-role
of conformational flexibility. Pharmacol. Ther. 2002, Vol. 93, pp. 99-111.
33. EUROPEAN MEDICINES AGENCY. Page consultée le 03 avril 2011. http://www.ema.europa.eu.
34. FERRACINI R., ANGELINI P., CAGLIERO E., LINARI A., MARTANO M.,
WUNDER, et al. 2000. Met oncogene aberrant expression in canine osteosarcoma. J.
Orthop. Res. 2000, Vol. 18, 2, pp. 253-257.
35. FOSMIRE S.P., DICKERSON E.B., SCOTT A.M., BIANCO S.R.,PETTENGILL
M.J., MEYLEMANS H., et al. 2004. Canine malignant hemangiosarcoma as a model of
primitive angiogenic endothelium. Lab. Invest. 2004, Vol. 84, pp. 562-572.
36. FROST D., LASOTA J., MIETTINEN M. 2003. Gastrointestinal stromal tumors and
leiomyomas in the dog : a histopathologic, immunohistochemical and molecular genetic
study of 50 cases. Vet. Pathol. 2003, Vol. 40, 1, pp. 42-54.
37. FURITSU T., TSUJIMURA T., TONO T., IKEDA H., KITAYAMA H.,
KOSHIMIZU U., et al. 1993. Identification of mutations in the coding sequence of the
proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent
activation of c-kit product. J. Clin. Invest. 1993, Vol. 92, pp. 1736-1744.
38. GASPARINIG. 2000. Pronostic value of vascular endothelial growth factor in breast
cancer. The Oncologist. 2000. Vol. 5, pp. 37-44.

138
39. GLEIXNER K.V., REBUZZI L., MAYERHOFER M., GRUZE A.,
HADZIJUSUFOVIC E., SONNECK K., et al. 2007. Synergistic antiproliferative effects
of KIT tyrosine kinase inhibitors on neoplastic canine mast cells. Exp. Hematol. 2007, Vol.
35, 10, pp. 1510-1521.
40. GRAHAM J.C., MYERS R.K. 1999. The pronostic significance of angiogenesis in
canine mammary tumors. J. Vet. Intern. Med. 1999. Vol. 13, pp. 416-418.
41. GRAND J.G. 2010. Le masitinib : nouvelle approche dans le traitement du mastocytome.
Point Vet. 2010. 303. pp. 14-15.
42. GRIFFEY S.M., VERSTRAETE F.J., KRAEGEL S.A., LUCROY M.D.,
MADEWELL B.R. 1998. Computer-assisted image analysis of intratumoral vessel
density in mammary tumors from dogs. Am. J. Vet. Res. 1998. Vol. 59, 10. pp. 1238-1242.
43. GUIDA T., ANAGANTI S., PROVITERA L., GEDRICH R., SULLIVAN
E.,WILHELM S.M., et al. 2007. Sorafenib inhibits imatinib-resistant KIT and platelet-
derived growth factor receptor β gatekeeper mutants. Clin. Cancer Res. 2007. Vol. 13, 11.
pp. 3363-3369.
44. HAHN K.A., LEGENDRE A.M., SHAW N.G., B.PHILLIPS, OGILVIE G.K.,
PRESCOTT D.M., et al. 2010. Evaluation of 12- and 24-month survival rates after
treatment with masitinib in dogs with nonresectable mast cell tumors. Am. J. Vet. Res.
2010. Vol. 71, 11. pp. 1354-1361.
45. HAHN K.A., OGLIVIE G., RUSK T., DEVAUCHELLE P., LEBLANC A.,
LEGENDRE A., et al. 2008. Masitinib is safe and effective for the treatment of canine
mast cell tumors. J. Vet. Intern. Med. 2008, Vol. 22, pp. 1301-1309.
46. HERMINE O. juin 2009. A tyrosine kinase inhibitor targeting c-kit for chronic
inflammatory diseases involving mast cells. North Am. Vet. Dermatol. Forum. juin 2009,
Vol. 20, 3, pp. 214-230. Abstract.
47. HERMINE O. 16 au 19 octobre 2009. Impact of masitinib on metastatic mast cell tumors
and on the emergence of metastasis from non-metastatic tumors. Proceedings of the 29th
Annual Conference of the Veterinary Cancer Society. 16 au 19 octobre 2009.
48. HERMINE O., KINET J.P., DUBREUIL P., MOUSSY A., HAHN K.A. 2009. Masitinib reduces the onset of metastasis and improves long-term survival in dogs with
mesurable grade 2 and grade 3 mast cell tumours. Proceedings of the British Small Animal
Veterinary Association Congress. 2009.
49. HERMINE O., KINET J.P., DUBREUIL P., MOUSSY A., HAHN K.A.. 26 au 29
mars 2009. Evaluation of masitinib in the treatment of canine mast cell tumors : long-term
follow-up efficacy data from phase 3 clinical study. Proceedings of the Annual Congress
of the European Society of Veterinary Oncology. 26 au 29 mars 2009.
50. HIROTA S., ISOZAKI K., MORIYAMA Y., HASHIMOTO K., NISHIDA T.,
ISHIGURO S., et al. 1998. Gain-of-function mutations of c-kit in human gastrointestinal
stromal tumors. Science. 1998, Vol. 279, pp. 577-580.

139
51. HUBBARD S.R. 2004. Juxtamembrane autoinhibition in receptor tyrosine kinases.
Nature. 2004, Vol. 5, pp. 464-470.
52. HUBBARD S.R., TILL J.H. 2000. Protein tyrosine kinase structure and function. Annu.
Rev. Biochem. 2000, Vol. 69, pp. 373-398.
53. HUMBERT M., CASTERAN N., LETARD S., HANSSENS K., IOVANNA J.,
FINETTI P., et al. 2010. Masitinib combined with standard gemcitabine chemotherapy:
In vitro and in vivo studies in human pancreatic tumour cell lines and ectopic mouse
model. Plos one. 2010. Vol. 5, 3.
54. HUMBERT M., DE BLAY F., GARCIA G., PRUD'HOMME A., LEROYER C.,
MAGNAN A., et al. 2009. Masitinib, a c-kit/PDGF receptor tyrosine kinase inhibitor,
improves disease control in severe corticosteroid-dependent asthmatics. Allergy. 2009.
Vol. 64, 8. pp. 1194-1201.
55. HUNTER T. 1998. The phosphorylation of proteins on tyrosine : its role in cell growth
and disease. The Croonian Lecture. 1998, Vol. 353, pp. 583-605.
56. HUNTER T., BLUME-JENSEN P., JANKNECHT R. 1998. The Kit receptor promotes
cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of
Bad on Ser136. Curr. Biol. 1998. Vol. 8, pp. 779-785.
57. HUSE M., KURIYAN J. 2002. The conformational plasticity of protein kinases. Cell.
2002, Vol. 109, pp. 275-282.
58. ISOTANI M., ISHIDA N., TOMINAGA M., YAGIHARA H., OCHI S., KATO R., et
al. 2008. Effect of tyrosine kinase inhibition by imatinib mesylate on mast cell tumors in
dogs. J. Vet. Intern. Med. 2008, Vol. 22, pp. 985-988.
59. JEANNEY M. 2008. Le premier anticancéreux vétérinaire traitera les mastocytomes et
sera disponible en février. Science et Pratique. 2008, 1012.
60. JOHNSON L.N., NOBLE M.E, OWEN D.J. 1996. Active and inactive protein kinases :
structural basis for regulation. Cell. 1996, Vol. 85, pp. 149-158.
61. JONES C.L.R, GRAHN R.A, CHIEN M.B., LYONS L.A., LONDON C.A. 2004. Detection of c-kit mutations in canine mast cell tumors using fluorescent polyacrylamide
gel electrophoresis. J. Vet. Diagn. Invest. 2004, Vol. 16, pp. 95-100.
62. JONG J.S., VAN DIEST P.J., VAN DER VALK P., BAAK J.P.. 1998. Expression of
growth factors, growth inhibiting factors, and their receptors in invasive breast cancer. I:
An inventory in search of autocrine and paracrine loops. J. Pathol. 1998. Vol. 184, pp. 44-
52.
63. KIM K.J., LI B., WINER J., ARMANINI M., GILLETT N., PHILLIPS H.S.,
FERRARA N. 1993. Inhibition of vascular endothelial growth factor-induced
angiogenesis suppresses tumour growth in vivo. Nature. 1993. Vol. 362, pp. 841-844.

140
64. KIUPEL M., WEBSTER J.D., KANEENE J.B., MILLER R., YUZBASIYAN-
GURKAN V. 2004. The use of kit and tryptase expression patterns as pronostic tools for
canine cutaneous mast cell tumors. Vet. Pathol. 2004, Vol. 41, pp. 371-377.
65. KOBIE K., KAWABATA M., HIOKI K., TANAKA A., MATSUDA H., MORI T.,
MARUO K. 2007. The tyrosine kinase inhibitor imatinib (STI571) induces regression of
xenografted canine mast cell tumors in SCID mice. Res. Vet. Sci. 2007. Vol. 82, pp. 239-
241.
66. KOLLER J. 2004. Protéine c-Kit : données structurales, fonctions, et implication en
pathologie tumorale chez l'homme et le chien. Thèse de doctorat vétérinaire. Université
Paul Sabatier de Toulouse, 2004. 94 pp.
67. KUBO K., MATSUYAMA S., KATAYAMA K., TSUTSUMI C., YONEZAWA K.,
SHIMADA T., et al. 1998. Frequent expression of the c-kit proto-oncogene in canine
malignant mammary tumor. J. Vet. Med. Sci. 1998, Vol. 12, pp. 1335-1340.
68. LASKIN J.L., SANDLER A.B. 2004. Epidermal growth factor receptor : a promising
target in solid tumours. Cancer Treat. Rev. 2004, Vol. 30, 1, pp. 1-17.
69. LE CESNE A., BLAY J., BUI B., BOUCHE O., ADENIS A et DOMONT J., et al.
2009. Masitinib mesylate in imatinib-naive locally advanced or metastatic gastrointestinal
stromal tumor (GIST): Results of the french sarcoma group phase II trial. J. Clin. Oncol.
2009. Abstract.
70. LE CESNE A., BLAY J.Y., BINH NGUYEN B., BOUCHE O., ADENIS A.,
DOMONT J., et al. 2010. Phase II study of oral masitinib mesilate in imatinib-naïve
patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur.
J. Cancer. 2010. Vol. 46, 8. pp.1344-1351.
71. LEDER P., CLAYTON D.A., RUBENSTEIN E. 1994. Introduction to molecular
medicine. U.S : Scientific American Medicine, 1994. 333 pp.
72. LEMURA A., TSAY M., ANDO A., WERSHIL B.K., GALLI S.J. 1994. The c-kit
ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am. J.
Pathol. 1994, Vol. 144, 2, pp. 321-328.
73. LEROUX D. 2010. Les Inhibiteurs de tyrosine kinase en médecine vétérinaire : état des
lieux et perspectives. [Présentation powerpoint]. VetAgro Sup, Campus vétérinaire de
Lyon, CEAV Médecine interne. 2010
74. LETARD S., YANGY., HANSSENS K., PALMERINI F., LEVENTHAL P.S.,
GUERY S., et al. 2008. Gain-of-function mutations in the extracellular domain of kit are
common in canine mast cell tumors. Mol. Cancer Res. 2008, Vol. 6, 7, pp. 1137-1145.
75. LEVINE R.A., FOREST T., SMITH C. 2002. Tumor suppressor PTEN is mutated in
canine osteosarcoma cell lines and tumors. Vet. Pathol. 2002, Vol. 39, pp. 372-378.
76. LEVITZKI A. 1999. Protein tyrosine kinase inhibitors as novel therapeutic agents.
Pharmacol. Ther. 1999, Vol. 82, pp. 231-239.

141
77. LI X., KUMAR A., ZHANG F., LEE C., LI Y., TANG Z., ARJUNAN P. 2010. VEGF-
independent angiogenic pathways induced by PDGF-C. Oncotarget. 2010. Vol. 1, 4, pp.
309-314.
78. LIAO A.T., CHIEN M.B., SHENOY N., MENDEL D.B., McMAHON G.,
CHERRINGTON J.M., LONDON C.A. 2002. Inhibition of constitutively active forms
of mutant kit by multitargeted indolinone tyrosine kinase inhibitors. Blood. 2002, Vol. 100,
2, pp. 585-593.
79. LIAO A.T., McMAHON M., LONDON C.A. 2005. Characterization, expression and
function of c-met in canine spontaneous cancers. Vet. Comp. Oncol. 2005, Vol. 3, 2, pp.
61-72.
80. LONDON C.A. 2004. Kinase inhibitors in cancer therapy. Vet. Comp. Oncol. 2004, Vol.
2, pp. 177-193.
81. LONDON C.A. 2007. The role of small molecule inhibitors for veterinary patients. Vet.
Clin. North Am. Small. Anim. Pract. 2007, Vol. 37, pp. 1121-1136.
82. LONDON C.A. 2009. Tyrosine kinase inhibitors in veterinary medicine. Topics
Compagnion Anim. Med. 2009, Vol. 24, pp. 106-112.
83. LONDON C.A., GALLI S.J., YUUKI T., HU Z., HELFAND S.C., GEISSLER E.N.
1999. Spontaneous canine mast cell tumors express tandem duplications in the proto-
oncogene c-kit. Exp. Hematol. 1999, Vol. 27, 4, pp. 689-697.
84. LONDON C.A., HANNAH A.L., ZADOVOSKAYA R., CHIEN M.B., KOLLIAS-
BAKER C., ROSENBERG M., et al. 2003. Phase I dose escalating study of SU11654, a
small molecule receptor tyrosine kinase inhibitor, in dogs with spontaneous malignancies.
Clin. Cancer. Res. 2003, Vol. 9, pp. 2755-2768.
85. LONDON C.A., KISSEBERTH W.C., GALLI S.J., GEISSLER E.N., HELFAND
S.C. 1996. Expression of stem cell factor receptor (c-kit) by the malignant mast cells from
spontaneous canine mast cell tumours. J. Comp. Pathol. 1996, Vol. 115, pp. 399-414.
86. LONDON C.A., MALPAS P.B., WOOD-FOLLIS S.L., BOUCHER J.F., RUSK A.W.,
ROSENBERG M., et al. 2009. Multi-center, placebo-controlled, double-blind,
randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase
inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor
following surgical excision. Clin. Cancer. Res. 2009, Vol. 15, 11, pp. 3856-3865.
87. LONDON C.A., SEGUIN B. 2003. Mast cell tumors in the dog. Vet. Clin. North Am.
Small Anim. Pract. 2003, Vol. 33, pp. 473-489.
88. LONGLEY B.J., REGUERA M.J., MA Y. 2001. Classes of c-kit activating mutations :
proposed mechanisms of action and implications for disease classification and therapy.
Leuk. Res. 2001, Vol. 25, pp. 571-576.

142
89. MA Y., CARTER E., WANG X., SHU C., McMAHON G., LONGLEY B.J. 2000. Indolinone derivatives inhibit constituvely activated kit mutants and kill neoplastic mast
cells. J. Invest. Dermatol. 2000, Vol. 114, pp. 392-394.
90. MA Y., CUNNINGHAM M.E., WANG X., GHOSH I., REGAN L., LONGLEY B.J.
1999. Inhibition of spontaneous receptor phosphorylation by residues in a putative alpha-
helix in the kit intracellular juxtamembrane region. J. Biol. Chem. 1999, Vol. 274, 19, pp.
13399-13402.
91. MA Y., LONGLEY B.J., WANG X., BLOUNT J.L., LANGLEY K., CAUGHEY G.H.
1999. Clustering of activating mutations in c-kit's juxtamembrane coding region in canine
mast cell neoplasms. J. Invest. Dermatol. 1999, Vol. 112, pp. 165-170.
92. MADHUSUDAN S., GANESAN T.S. 2004. Tyrosine kinase inhibitors in cancer therapy.
Clin. Biochem. 2004, Vol. 37, 7, pp. 618-635.
93. MAHON F.X., REA D., GUILHOT D., GUILHOT F., HUGUET F., NICOLINI F., et
al. 2010. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have
maintained complete molecular remission for at least 2 years: the prospective, Multicentre
Stop Imatinib (STIM) Trial. Lancet Oncol. 2010. Vol. 11, 11. pp. 1029-1035.
94. MAJUMDER S., BROWN K., QIU F.H., BESMER P. 1988. C-kit protein, a
transmembrane kinase : identification in tissues and characterization. Mol. Cell. Biol. 1988,
Vol. 8, 11, pp. 4896-4903.
95. MARCONATO L., BETTINI G., GIACOBONI C., ROMANELLI G., CESARI A.,
ZATELLI A., et al. 2008. Clinicopathological features and outcome for dogs with mast
cell tumors and bone marrow involvement. J. Vet. Intern. Med. 2008, Vol. 22, pp. 1001-
1007.
96. MASSON K., RÖNNSTRAND L. 2009. Oncogenic signaling from the hematopoietic
growth factor receptors c-kit and Flt3. Cell. Signal. 2009. Vol. 21, pp. 1717-1726.
97. MATSUYAMA S., NAKAMURA M., YONEZAWA K., SHIMADA T., OHASHI F.,
TAKAMORI Y., KUBO K. 2001. Expression patterns of the erbB subfamily mRNA in
canine benign and malignant mammary tumors. J. Vet. Med. Sci. 2001, Vol. 63, 9, pp. 949-
954.
98. McEWEN E.G., KUTZKE J., CAREW J., PASTOR J., SCHMIDT J.A., TSAN R., et
al. 2003. C-Met tyrosine kinase receptor expression and function in human and canine
osteosarcoma cells. Clin. Exp. Metastasis. 2003, Vol. 20, pp. 421-430.
99. MEDERLE O., MEDERLE N., BOCAN E.V., CEAUSU R., RAICA M. 2010. VEGF
expression in dog mastocytoma. Rev. Med. Chir. Soc. Med. Nat. Iasi. 2010. Vol. 114, 1.
pp. 185-188.
100. MENDEL D.B., LAIRD A.D., XIN X., LOUIE S.G., CHRISTENSEN J.G., LI G., et
al. 2003. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting
vascular endothelial growth factor and platelet-derived growth factor receptors. Clin.
Cancer Res. 2003. Vol. 9. pp. 327-337.

143
101. MILLANTA F., SILVESTRI G., VASELLI C., CITI S., PISANI G., LORENZI D.,
et al. 2006. The role of vascular endothelial growth factor and its receptor Flk-1/KDR in
promoting tumour angiogenesis in feline and canine mammary carcinomas : a preliminary
study of autocrine and paracrine loops. Res. Vet. Sci. 2006, Vol. 81, pp. 350-357.
102. MITRY E., HAMMEL P., DEPLANQUE G., MORNEX F.,LEVY P., SEITZ J., et
al. 2009. Oral tyrosine kinase inhibitor masitinib in combination with gemcitabine in
patients with advanced pancreatic cancer: A multicenter phase II study. J. Clin. Oncol.
2009. Vol. 27, 15. Abstract.
103. MIYAZAWA K., WILLIAMS D.A., GOTOH A., NISHIMAKI J., BROXMEYER
H.E.,TOYAMA K. 1995. Membrane-bound steel factor induces more persistent tyrosine
kinase activation and longer life span of c-kit gene-encoded protein than its soluble form.
Blood. 1995. Vol. 85, 3. pp. 641-649.
104. MOL C.D., DOUGAN D.R., SCHNEIDER T.R., SKENE R.J., KRAUS M.L.,
SCHEIBE D.N, .et al. 2004. Structural basis for the autoinhibition and STI-571 inhibition
of c-kit tyrosine kinase. J. Biol. Chem. 2004, Vol. 279, 30, pp. 31655-31663.
105. MOTZER R.J., HUTSON T.E., TOMCZAK P., MICHAELSON M.D.,
BUKOWSKI R.M, RIXE O., et al. 2007. Sunitinib versus interferon alfa in metastatic
renal-cell carcinoma. New Engl. J. Med. 2007. Vol. 356, 2. pp. 115-124.
106. MULLER C. 2008. Les tumeurs gastro-intestinales (GIST) : classification, diagnostic,
intêret thérapeutique des inhibiteurs de tyrosine kinase (ITK). Synthèse bibliographique
chez l'homme et l'animal. Thèse de doctorat vétérinaire. Faculté de Médecine de Créteil,
2008. 122 pp.
107. NAGATA H., WOROBEC A.S., OH C.K., CHOWDHURY B.A., TANNENBAUM
S., SUZUKI Y., et al. 1995. Identification of a point mutation in the catalytic domain of
the proto-oncogene c-kit in peripheral blood mononuclear cells of patients who have
mastocytosis with an associated hematologic disorder. Natl. Acad. Sci. USA. 1995. Vol. 92,
pp. 10560-10564.
108. NATALI P.G., NICOTRA M.R., MOTTOLESE M., BOTTI C., ULLRICH A. 1992. Breast cancer is associated with loss of the c-kit oncogene product. Int. J. Cancer. 1992,
Vol. 52, 5, pp. 713-717.
109. NOLEN B., TAYLOR S., GHOSH G. 2004. Regulation of protein kinases : controlling
activity through activation segment conformation. Mol. Cell. 2004, Vol. 15, pp. 661-675.
110. OGILVIE G., AHN A. 2010. Masitinib-The efficacy of the targeted therapy in
veterinary medicine. Vet. Cancer Society Newsletter. 2010. Vol. 34, 2.
111. OHMORI K., KAWARAI S., YASUDA N., TANAKA A., MATSUDA H.,
NISHIMURA R., et al. 2008. Identification of c-kit mutations-independent neoplastic cell
proliferation of canine mast cells. Vet. Immunol. and Immunopathol. 2008, Vol. 126, pp.
43-53.

144
112. ORFAO A., GARCIA-MONERO A.C., SANCHEZ L., ESCRIBANO L. 2007. Recent advances in the understanding of mastocytosis : the role of kit mutations. Br. J.
Haematol. 2007, Vol. 138, pp. 12-30.
113. PAEZ-RIBES M., ALLEN E., HUDOCK J., TAKEDA T., OKUYAMA H., VINALS
F., et al. 2009. Antiangiogenic therapy elicits malignant progression of tumors to increased
local invasion and distant metastasis. Cancer cell. 2009. Vol. 15, pp. 220-231.
114. PARSONS R., SIMPSON L. 2001. PTEN : life as a tumor suppressor. Exp. Cell Res.
2001. Vol. 264, pp. 29-41.
115. PATNAIK A.K., et al. 1984. Canine cutaneous mast cell tumor: morphologic grading
and survival time in 83 dogs. Vet. Pathol. 1984. Vol. 21, 5. pp. 469-474.
116. PATRUNO R., ARPAIA N., GADALETA C.D., PASSANTIN L., ZIZZO N.,
MISINO A., et al. 2009. VEGF concentration from plasma-activated platelets rich
correlates with microvascular density and grading in canine mast cell tumour spontaneous
model. J. Cell. Mol. Med. 2009. Vol. 13, 3. pp. 555-561.
117. PAUL C., SANS B., SUAREZ F., CASSASUS P., BARETE S., LANTERNIER F., et
al. 2010. Masitinib for the treatment of systemic and cutaneous mastocytosis with
handicap : a phase 2a study. Am. J. Hematol. 2010. Vol. 85. pp. 921-925.
118. PFIZER. (pages consultée le 8 janvier 2011). http://pfizer.com/home/.
119. PIETTE F., BELMIN J., VINCENT H., SCHMIDT N., PARIEL S., VERNY M., et
al. Avril 2011. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer's disease:
a randomised, placebo-controlled phase 2 trial. Alzheimer's Research & Therapy. Avril
2011, Vol. 3, 2, pp. 3-16.
120. PREZIOSI R., MORINI M., SARLI G. 2004. Expression of the Kit protein (CD117) in
primary cutaneous mast cell tumors of the dog. J. Vet. Diagn. Invest. 2004. Vol. 16, pp.
554-561.
121. PREZIOSI R., SARLI G., PALTRINIERI M. 2004. Prognostic value of intratumoral
vessel density in cutaneous mast cell tumors of the dog. J. Comp. Pathol. 2004. Vol. 130,
2-3. pp. 143-151.
122. PRYER N.K., LEE L.B., ZADOVASKAYA R., YU X., SUKBUNTHERNG J.,
CHERRINGTON J.M., LONDON C.A. 2003. Proof of target for SU11654 : inhibition
of kit phosphorylation in canine mast cell tumors. Clin. Cancer. Res. 2003, Vol. 9, pp.
5729-5734.
123. RAVA F., BRIZZOLA S., STEFANELLO D., CREMA S., TURIN L. 2005. A study
of mutations in the c-kit gene of 32 dogs with mastocytoma. J. Vet. Diagn. Invest. 2005,
Vol. 17, pp. 385-388.

145
124. REBUZZI L., WILLMANN M., SONNECK K., GLEIXNER K.V., FLORIAN S.,
KONDO R., et al. 2007. Detection of vascular endothelial growth factor (VEGF) and
VEGF receptors Flt-1 and KDR in canine mastocytoma cells. Vet. Immunol. and
Immunopathol. 2007, Vol. 115, pp. 320-333.
125. REGUERA M.J., FERRER L., RABANAL R.M. 2002. Evaluation of an intron
deletion in the c-kit gene of canine mast cell tumors. Am. J. Vet. Res. 2002, Vol. 63, 9, pp.
1257-1261.
126. REITH A.D., ELLIS C., LYMAN S.D., ANDERSON D.M., WILLIAMS D.E.,
BERNSTEIN A., et al. 1991. Signal transduction by normal isoforms and W mutant
variants of the Kit receptor tyrosine kinase. EMBO J. 1991. Vol. 10, pp. 2451-2459.
127. RESTUCCI B., BORZACCHIELLO G., MAIOLINO P., MARTANO M.,
PACIELLO O., PAPPARELLA S. 2004. Expression of vascular endothelial growth
factor receptor Flk-1 in canine mammary tumours. J. Comp. Pathol. 2004, Vol. 130, pp.
99-104.
128. RESTUCCI B., PAPPARELLA S., MAIOLINO P., DE VICO G. 2002. Expression of
vascular endothelial growth factor in canine mammary tumors. Vet. Pathol. 2002, Vol. 39,
pp. 488-493.
129. ROBAT C., LONDON C.A., BUNTING L., McCARTAN L., STINGLE N.,
SELTING K., et al. 2011. Safety evaluation of combination vinblastine and toceranib
phosphate (Palladia) in dogs : a phase I dose-finding study. Vet. Comp. Oncol. 2011. pp. 1-
10.
130. ROBINSON D.R., WU Y.M., LIN S.F. 2000. The protein tyrosine kinase family of the
human genome. Oncogene. 2000, Vol. 19, pp. 5548-5557.
131. ROSKOSKI R. Jr. 2005. Signaling by kit protein-tyrosine kinase-the stem cell factor
receptor. Biochem. Biophys. Res. Commun. 2005, Vol. 337, pp. 1-13.
132. ROSKOSKI R. Jr. 2005. Structure and regulation of kit protein-tyrosine kinase-The
stem cell factor receptor. Biochem. Biophys. Res. Commun. 2005, Vol. 338, pp. 1307-1315.
133. SCAPIN G. 2002. Structural biology in drug design: selective protein kinase inhibitors.
Drug Discov. Today. 2002, Vol. 7, 11, pp. 601-611.
134. SCHLESSINGER J. 2000. Cell signaling by receptor tyrosine kinases. Cell. 2000. Vol.
13, 103. pp. 193-200.
135. SERRES F., MEYER C., TIERNY D, HIDALGO A., HAELEWYN C.,
MARESCAUX L. 18 au 20 mars 2010. Masitinib for maintenance chemotherapy of two
dogs with T-cell multicentric lymphoma. Proceedings of the Annual Congress of the
European Society of Veterinary Oncology . 18 au 20 mars 2010.
136. SHCHEMELININ I., SEFC L., NECAS E. 2006. Protein kinases, their function and
implication in cancer and other diseases. Folia Biol. 2006, Vol. 52, pp. 81-101.

146
137. SORIA J.C., MASSARD C., MAGNE N., BADER T., MANSFIELD C. D., BLAY
J.Y., et al. 2009. Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib
in advanced and/or metastatic solid cancers. Eur. J. Cancer. 2009. Vol. 45, 13. pp. 2333-
2341.
138. STEGMEIER F., WARMUTH M., SELLERS W.R., DORSCHM. 2010. Targeted
cancer therapies in the twenty-first century : lessons from imatinib. Clin. Pharmacol. Ther.
2010. Vol. 87, 5. pp. 543-552.
139. SUN L., TRAN N., TANG F., APP H., HIRTH P., McMAHON G., TANG C. 1998. Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of
tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine
kinases. J. Med. Chem. 1998. Vol. 41, 14. pp. 2588-2603.
140. TAKEUCHI Y., FUJINO Y., FUKUSHIMA K., WATANABE M., NAKAGAWA T.,
et al. 2011. Biological effect of tyrosine kinase inhibitors on three canine mast cell. J. Vet.
Pharmacol. Therap. 2011. Vol. 34, 2. pp.1-8.
141. TAKEUCHI Y., FUJINO Y., WATANABE M., NAKAGAWA T., OHNO K.,
SASAKI N., et al. 2010. Aberrant autophosphorylation of c-kit receptor in canine mast
cell tumor cell lines. Vet. Immunol. and Immunopathol. 2010, Vol. 137, pp. 208-216.
142. TEBIB J., MARIETTE X., BOURGEOIS P., FLIPO R.M, GAUDIN P., LE LOËT
X., et al. 2009. Masitinib in the treatment of active rheumatoid arthritis: results of a
multicentre, open-label, dose-ranging, phase 2a study. Arthritis Res. Ther. 2009. Vol. 11,
3. pp. 1-12.
143. THAMM D.H., ROSE B., KOW K., HUMBERT M., MANSFIELD C.D., MOUSSY
A., et al. 2011. Masitinib as a chemosensitizer of canine tumor cell lines: A proof of
concept study. Vet. J. 2011. Sous presse.
144. TSUJIMURA T., FURITSU T., MORIMOTO M., ISOZAKI K., NOMURA S.,
MATSUZAWA Y., et al. 1994. Ligand-Independent activation of c-kit receptor tyrosine
kinase in a murine mastocytoma cell line P-815 generated by a point mutation. Blood.
1994, Vol. 83, 9, pp. 2619-2626.
145. VERHEUL H.M., PINEDO H.M. 2000. The role of vascular endothelial growth factor
(VEGF) in tumor angiogenesis and early clinical development of VEGF-receptor kinase
inhibitors. Clin. Breast Cancer. 2000. Vol. 1, Supp.1. pp. 80-84.
146. VERMERSCH P., BENRABAH R., SCHMIDT N., ZEPHIR H.,CLAVELOU
P.,VONGSOUTHI C.,et al. Avril 2009. Oral masitinib in patients with primary
progressive or secondary progressive multiple sclerosis. American Academy of Neurology.
Avril 2009. Abstract.
147. WEBSTER J.D., KIUPEL M.,YUSBASIYAN-GURKAN V. 2006. Evaluation of the
kinase domain of c-kit in canine cutaneous mast cell tumors. BMC Cancer. 2006, Vol. 6,
85, pp. 1-8.

147
148. WEBSTER J.D., YUZBASIYAN-GURKAN V., KANEENE J.B., MILLER R.,
RESAU J.H., KIUPEL M. 2006. The role of c-kit in tumorigenesis : evaluation in canine
cutaneous mast cell tumors. Neoplasia. 2006, Vol. 8, 2, pp. 104-111.
149. WEBSTER J.D., YUZBASIYAN-GURKAN V., MILLER R.A., KANEENE J.B.,
KIUPEL M. 2007. Cellular proliferation in canine cutaneous mast cell tumors :
associations with c-kit and its role in prognostication. Vet. Pathol. 2007, Vol. 44, pp. 298-
308.
150. WEISBER G. E., GRIFFIN J.D. 2003. Resistance of imatinib (Glivec) : update on
clinical mechanisms. Drug Resist. Updates. 2003, Vol. 6, pp. 231-238.
151. WIEDENMANN B., RAYMOND E., DAHAN L., RAOUL J.L., BANG Y.J.,
BORBATH I., et al. 2011. Sunitinib malate for the treatment of pancreatic
neuroendocrine tumors. New Engl. J. Med. 2011, Vol. 364, pp. 501-513.
152. WOLFESBERGER B., TONAR Z., GERNER W., SKALICKY M., HEIDUSCHKA
G., EGERBACHER M., et al. 2010. The tyrosine kinase inhibitor sorafenib decreases
cell number and induces apoptosis in a canine osteosarcoma cell line. Res. Vet. Sci. 2010,
Vol. 88, pp. 94-100.
153. YARDEN Y., KUANG W.J., YANG-FENG T., COUSSENS L., MUNEMITSU S.,
DULL T.J., et al. 1987. Human proto-oncogene c-kit : a new cell surface receptor tyrosine
kinase for an unidentified ligand. EMBO J. 1987, Vol. 6, 11, pp. 3341-3351.
154. ZEMKE D., YAMINI B., YUZBASIYAN-GURKAN V. 2001. Characterization of an
undifferentiated malignacy as a mast cell tumor using mutation analysis in the proto-
oncogene c-kit. J. Vet. Diagn. Invest. 2001, Vol. 13, pp. 341-345.
155. ZEMKE D., YAMINI B., YUZBASIYAN-GURKAN V. 2002. Mutations in the
juxtamembrane domain of c-kit are associated with higher grade mast cell tumors in dogs.
Vet. Pathol. 2002, Vol. 39, pp. 529-535.
156. ZHANG Z., ZHANG R., JOACHIMIAK A., SCHLESSINGER J., KONG X.P.
2000. Crystal structure of human stem cell factor : implication for stem cell factor receptor
dimerization and activation. Proc. Natl. Acad. Sci. USA . 2000, Vol. 97, 14, pp. 7732-7737.
157. ZWICK E., BANGE J., ULLRICH A. 2002. Receptor tyrosine kinases as targets for
anticancer drugs. Trends Mol. Med. 2002, Vol. 8, 1.

148
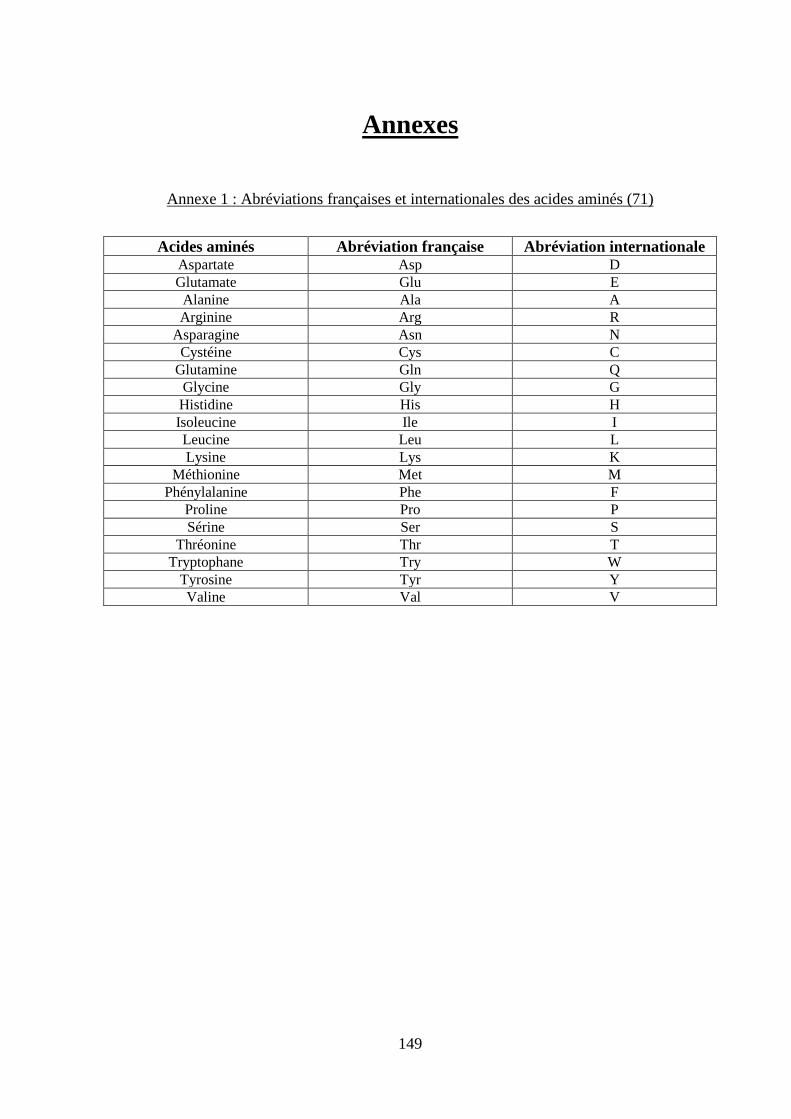
149
Annexes
Annexe 1 : Abréviations françaises et internationales des acides aminés (71)
Acides aminés Abréviation française Abréviation internationale
Aspartate Asp D
Glutamate Glu E
Alanine Ala A
Arginine Arg R
Asparagine Asn N
Cystéine Cys C
Glutamine Gln Q
Glycine Gly G
Histidine His H
Isoleucine Ile I
Leucine Leu L
Lysine Lys K
Méthionine Met M
Phénylalanine Phe F
Proline Pro P
Sérine Ser S
Thréonine Thr T
Tryptophane Try W
Tyrosine Tyr Y
Valine Val V

150
MORGANT Cindy
Les inhibiteurs de tyrosines kinases en cancérologie vétérinaire : actualité chez
le chien et comparaison avec l’homme
Thèse d’Etat de Doctorat Vétérinaire : Lyon, le 1
er juillet 2011
RESUME :
Le concept de thérapie ciblée avec la découverte des inhibiteurs de tyrosines kinases a
révolutionné la médecine humaine et vétérinaire. Les tyrosines kinases sont des protéines
souvent impliquées dans la transformation tumorale chez l’homme et le chien. Leur
identification présente un rôle à la fois thérapeutique et pronostique.
Chez le chien, deux molécules possèdent actuellement une AMM pour le traitement des
mastocytomes cutanés non opérables : le masitinib (Masivet®) et le toceranib (Palladia®).
Chez l’homme, de nombreux ITK se sont développés depuis le succès de l’imatinib
(Gleevec®) dans le traitement de la leucémie myéloïde chronique mis sur le marché en 2001.
Cependant, des résistances à ces molécules sont apparues.
L’intérêt des ITK réside dans leur utilisation combinée et associée aux traitements anti-
cancéreux classiques et dans la nécessité d’identifier le patient susceptible de bénéficier de
cette thérapie.
MOTS CLES : Inhibiteur de tyrosine kinase, Chien, Mastocytome, Homme, Protéine c-Kit
JURY :
Président : Monsieur le Professeur Claude GHARIB
1er Assesseur : Madame le Docteur Frédérique PONCE
2ème Assesseur : Monsieur le Docteur Jean-Jacques THIEBAULT
Membre invité : Monsieur le Docteur Tom CHAPUIS
DATE DE SOUTENANCE :
Vendredi 1
er juillet 2011
ADRESSE DE L’AUTEUR :
Quartier la Grenette
26300 Châteauneuf sur Isère


















