
第 10 章 高效液相色谱法
82
description
第 10 章 高效液相色谱法. high performance liquid chromatogrphy HPLC. 第 10 章 高效液相色谱法. 10.1 概述 10.2 HPLC 的分类与基本原理 10.3 各类高效液相色谱法 10.4 固定相 10.5 流动相 10.6 高效液相色谱仪 10.7 定性、定量分析方法 10.8 应用与示例. 10.1 概述. 1. 基本概念与特点 2. HPLC 与经典 LC、GC 的比较 3. HPLC 仪器结构. 10.1 概述 >> 1. 基本概念与特点. - PowerPoint PPT Presentation
Transcript of 第 10 章 高效液相色谱法


10.1 概述10.2 HPLC 的分类与基本原理10.3 各类高效液相色谱法10.4 固定相10.5 流动相10.6 高效液相色谱仪10.7 定性、定量分析方法10.8 应用与示例
第 10 章 高效液相色谱法

10.1 概述
1. 基本概念与特点2. HPLC 与经典 LC、 GC 的比较3. HPLC 仪器结构

概念:高效液相色谱法 (HPLC) 是在 20 世纪 60 年代末,以经典液相色谱为基础,引入了气相色谱的理论,在技术上采用了高效固定相、高压输液系统和高灵敏度的在线检测器,从而发展起来的一种新型分离分析技术。随着科学和技术的不断改进与发展,目前已成为应用极为重要、广泛的分离分析手段 .
特点:分离效率高、分析速度快、应用范围广、操作自动化。
10.1 概述 >>1. 基本概念与特点

10.1 概述 >>2.HLPC 与经典液相色谱和气相的比较
高效液相色谱与经典液相色谱比较
经典液相色谱 高效液相色谱 常压或减压 高压, 40~ 50MPa
填料颗粒大 填料颗粒小, 2~ 50μm
柱效低 柱效高 ,40000~ 60000块 /m
分析速度慢 分析速度快色谱柱只用一次 色谱柱可重复多次使用不能在线检测 能在线检测

10.1 概述 >>2.HLPC 与经典液相色谱和气相的比较
高效液相色谱与气相色谱比较气相色谱 高效液相色谱
只能分析挥发性物质,只能分析20% 的化合物 几乎可以分析各种物质
不能分析热不稳定物质 可以分析热不稳定物质用毛细管色谱可得到很高的柱效 色谱柱不能很长,柱效不会很高有很灵敏的检测器如 ECD 和较灵敏的通用检测器 (FID和 TCD) 没有较高灵敏的通用检测器
流动相为气体,无毒,易于处理 流动相有毒,费用较高 运行和操作容易 运行和操作比 GC 难一些仪器制造难度较小 仪器制造难度大
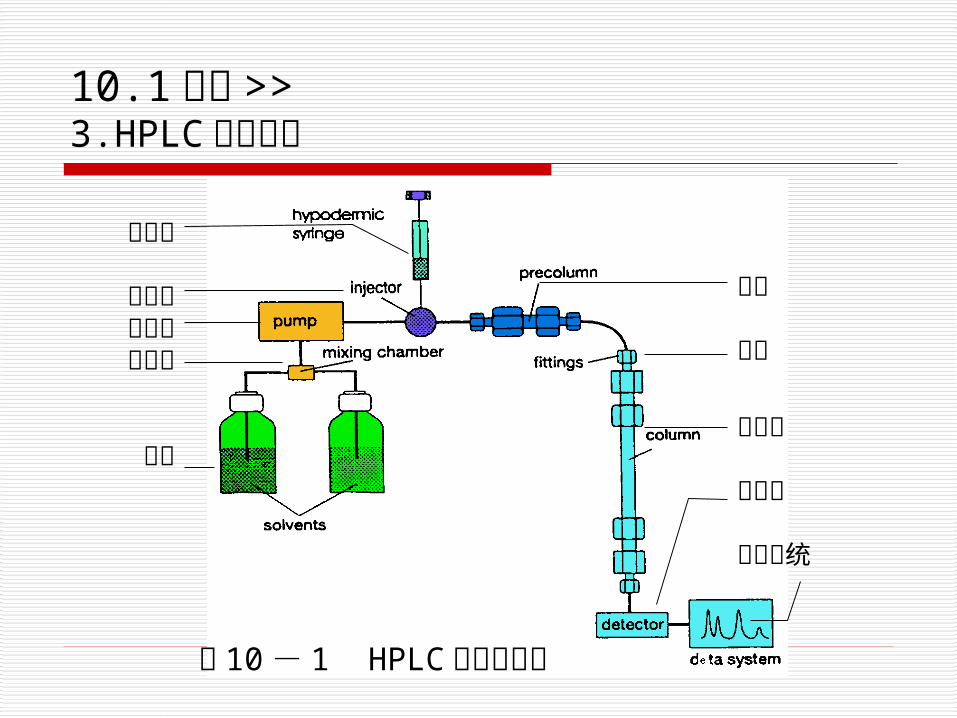
图 10 - 1 HPLC 仪器结构图
注射器
进样器高压泵混合室
溶剂
预柱
接头
色谱柱
检测器
数据系统
10.1 概述 >>3.HPLC 仪器结构

10.2 HPLC 的分类与基本原理10.2.1 高效液相色谱法的分类10.2.2 基本原理
化合物的极性、电荷、分子大小、旋光性四种主要的性质可以用来产生高效液相色谱分离。
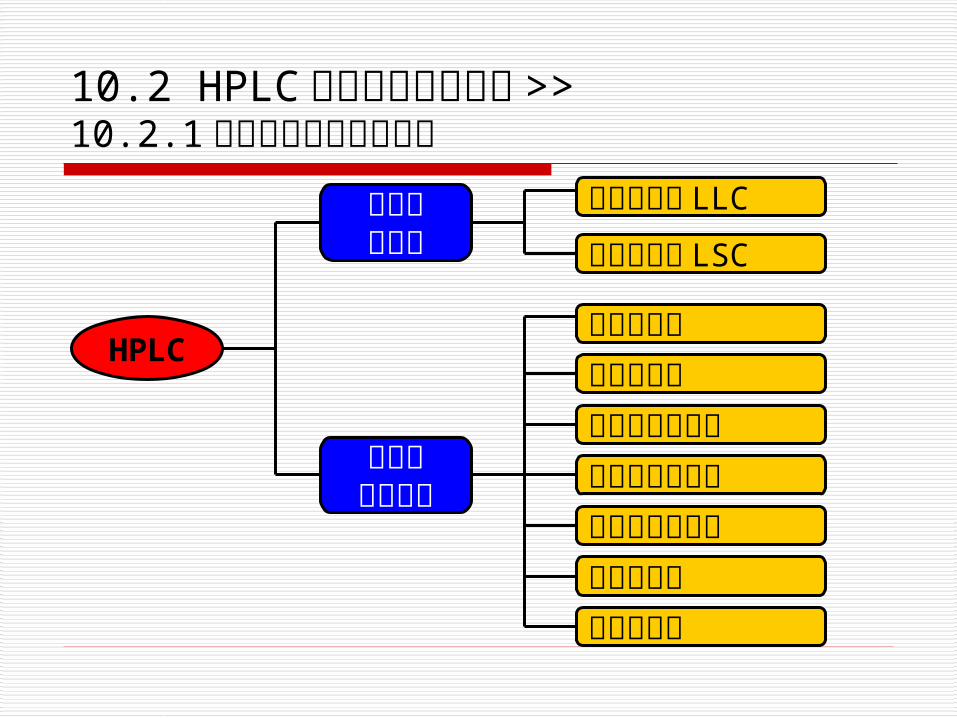
按固定相分类
按分离机理分类
液液色谱法 LLC
液固色谱法 LSC
分配色谱法吸附色谱法离子交换色谱法分子排阻色谱法化学键合色谱法亲合色谱法胶束色谱法
HPLC
10.2 HPLC 的分类与基本原理 >>10.2.1 高效液相色谱法的分类

详细分类图1 液-固吸附色谱法 LSC
2 液-液分配色谱法 LLC
3 化学键合色谱法 BPC
4 离子交换色谱法 IEC
5 空间排阻色谱法 SEC
6 亲合色谱法 AC7 胶束色谱法 MC8 手性色谱法 CC
9 毛细管电泳色谱法 CE
正相~ NLLC
反相~ RLLC
正相~ NBPC
反相~ RBPC
一般~ IEC
离子色谱法 IC
氨基酸色谱法 AA
凝胶渗透色谱法 GPC
凝胶过滤色谱法 GFC
开管~ OTCEC
填充~ PCEC
一般~ RBPC
离子对色谱法 PIC
离子抑制色谱法ISC
胶束~ MEKC
壁处理~ CEC
键合相~ CEC
毛细管凝胶~ CGE
HP
LC
HPLC 与经典 LC和 GC 在基本原理、概念和方法基本上相同,主要差别是流动相的性质不同。因此,某些公式的表现形式或参数的含意有些差别。
1. Van Deemter 方程式 2. Giddings 偶合式简介
10.2 HPLC 的分类与基本原理 >> 10.2.2 基本原理

10.2 HPLC 的分类与基本原理 >> 10.2.2 基本原理 >>1. Van Deemter 方程式
⑴Van Deemter 方程在气相色谱中的表现形式
⑵Van Deemter 方程在 HPLC 的表现形式: H=A+Cu
流动相为液体,组分的纵向扩散系数 B 很小,流速u 较高,故纵向扩散相 B/u 可以忽略。可以近似地认为 H与 u成线性关系, A为截距, C为斜率。
0
ACuu
BH
Cuu
BAH
,无多径项,无涡流扩散毛细管柱
填充柱
,:
:
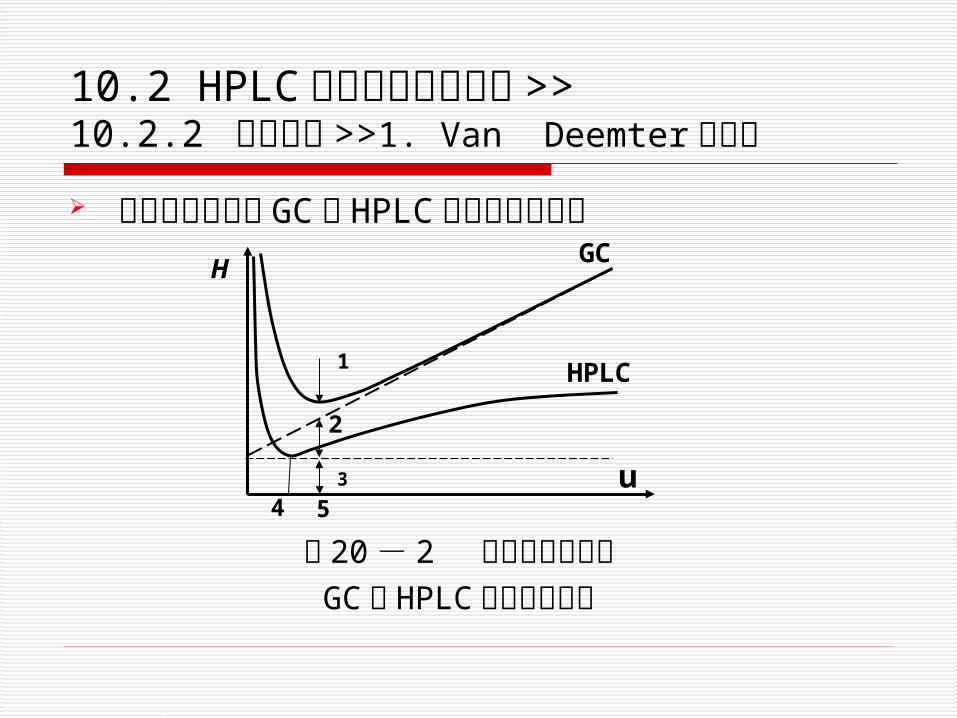
10.2 HPLC 的分类与基本原理 >> 10.2.2 基本原理 >>1. Van Deemter 方程式
流动相的流速对 GC与 HPLC 板高影响的差别
3
1
4 5u
HPLC
GCH
图 20 - 2 流动相的流速对GC与 HPLC 板高影响对比
2
10.2 HPLC 的分类与基本原理 >> 10.2.2 基本原理 >>1. Van Deemter 方程式
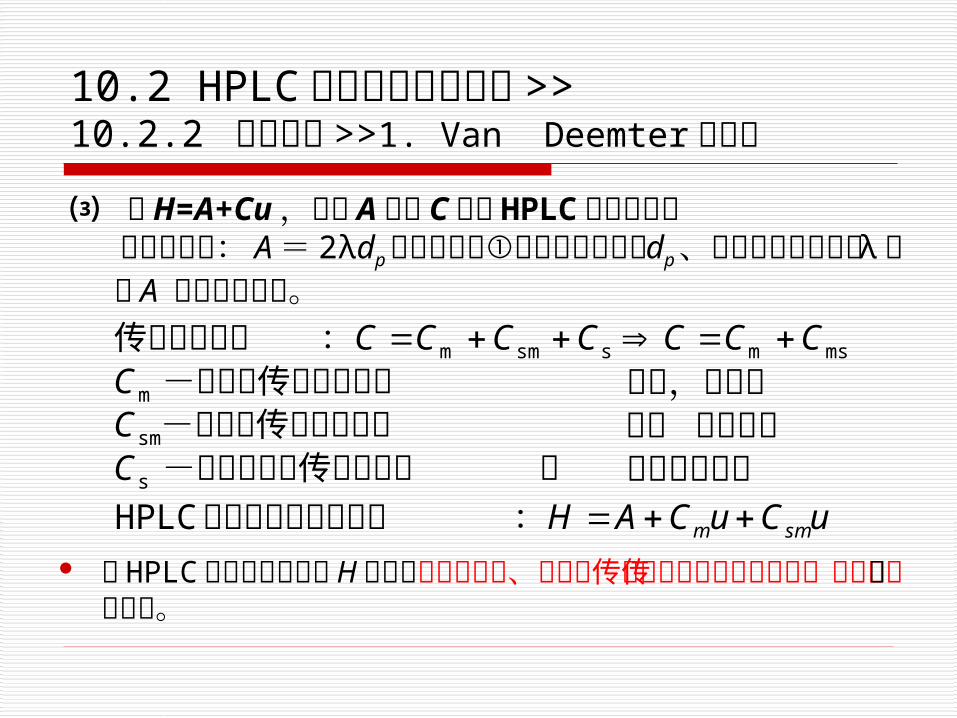
⑶ 由H=A+Cu,讨论 A项与 C项对 HPLC 柱效的影响 涡流扩散项: A= 2λdp ,可以通过①降低固定相粒径
dp 、②降低不规则因子 λ ,使 A 变小柱效提高。
故 HPLC 的塔板理论高度 H 主要由涡流扩散项、流动相传质阻抗项和静态流动相传质阻抗项三项构成。
数静态流动相传质阻抗系-流动相传质阻抗系数-固定液传质阻抗系数-
:传质阻抗系数
s
sm
m
msmssmm
CCC
CCCCCCC
uCuCAH smm :的范氏方程表达形式HPLC
静态阻抗系数阻抗系数动态
很小,可忽略
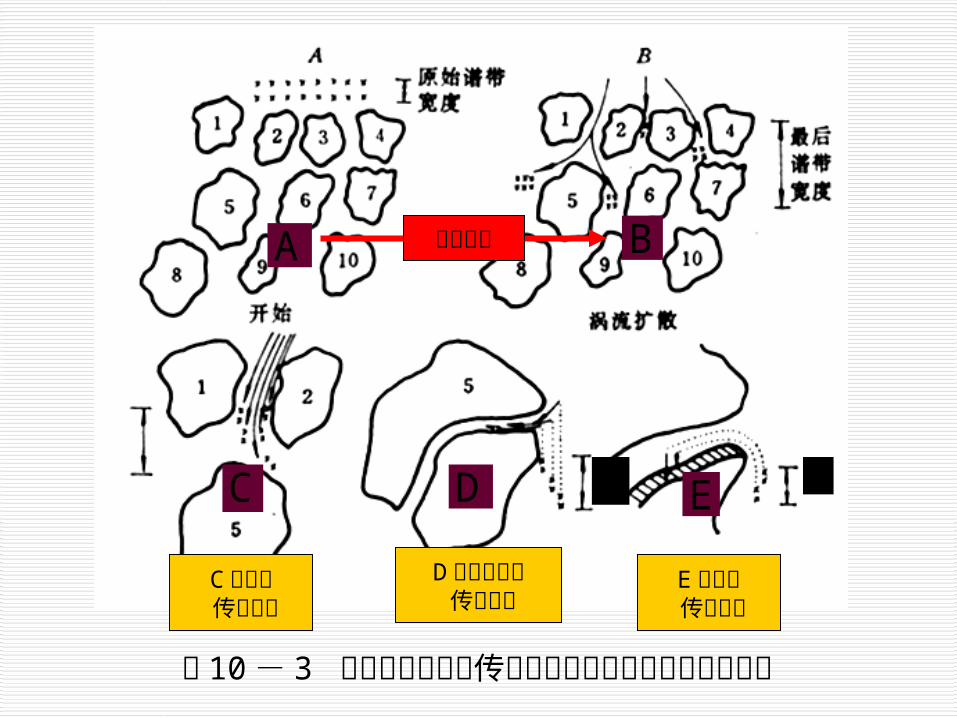
A
C
B
d e
C 流动相传质阻抗
D静态流动相传质阻抗
E固定相传质阻抗
D E
图 10- 3 涡流扩散与各种传质阻抗对液相色谱峰展宽的影响
涡流扩散
10.2 HPLC 的分类与基本原理 >> 10.2.2 基本原理 >>1. Van Deemter 方程式
⑷ 应用范氏方程选择 HPLC 的分离条件① 采用粒径小而均匀的球形固定相,首选化学键
合相,用匀浆法装柱。② 采用低黏度流动相,低流量 (1mL/min) ,首选甲醇。
③ 采用柱温箱,避免室温波动,增加实验重复性,柱温以 25~ 30℃为宜。

Giddings认为:影响塔板高度的各项因素,并不都是孤立的。而且证明,涡流扩散项 A 与流动相传质阻抗项是偶合的( HA 为偶合相)。
)()(
中在填充中在一般
GC HPLC
111
sdA
smA
ssmdA
ssm
m
HHHHHHH
HHHHH
uCuCu
B
uCAH
10.2 HPLC 的分类与基本原理 >> 10.2.2 基本原理 >>2.Giddings 偶合式简介
10.3 各类高效液相色谱法
10.3.1 吸附色谱法 (LSC)
10.3.2 分配色谱法 (LLC)
10.3.3 化学键合色谱法 (BPC)
10.3.4 其他色谱法
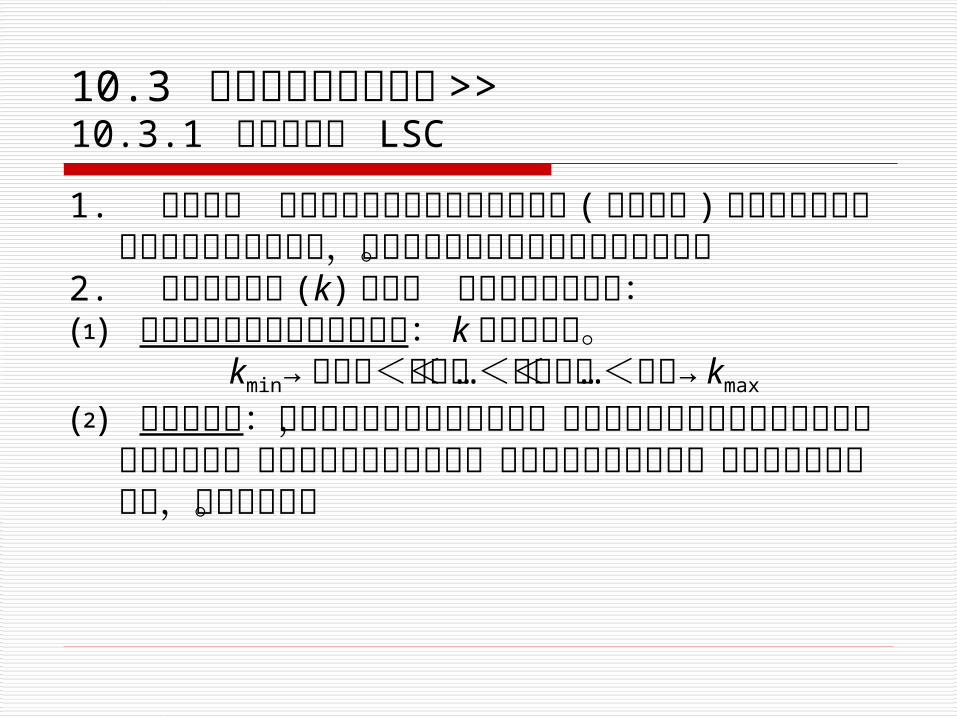
10.3 各类高效液相色谱法 >>10.3.1 吸附色谱法 LSC
1. 分离机理 吸附色谱法是被分离的组分分子 ( 溶质分子 ) 与流动相分子争夺吸附剂表面活性中心,因溶质分子的吸附系数的差别而分离。
2. 影响容量因子 (k) 的因素 以硅胶吸附剂为例:⑴ 硅胶与溶质分子的亲和力顺序: k 大者后出峰。 kmin→饱和烃<芳烃<…<酯醛酮<…<羧酸→ kmax
⑵ 溶质的极性:常用流动相是以烷烃为底剂,加入适当的极性溶剂组成二元或多元溶剂系统,从而能调整溶解的极性,控制组分的保留时间。溶剂系统的极性越大,洗脱力越强。

10.3 各类高效液相色谱法 >>10.3.2 分配色谱法 (LLC)
1. 分离机理:分配色谱法是由于样品组分溶入固定相(s) 与流动相 (m)达到“平衡”后的分配系数的差别而分离。组分在固定相与流动相中的溶解度差别越大(K、 k 差别也大 ) 分离效果越好。
2. 正相液-液色谱法 (NLLC) :流动相极性小于固定相极性的 LLC称为 NLLC。正相洗脱时样品中极性小的组分先流出色谱柱,极性大的组分后流出色谱柱 .
3. 反相液-液色谱法 (RLLC) :流动相极性大于固定相极性的 LLC称为 RLLC。反相洗脱时样品中极性大的组分先流出色谱柱,极性小的组分后流出色谱柱 .
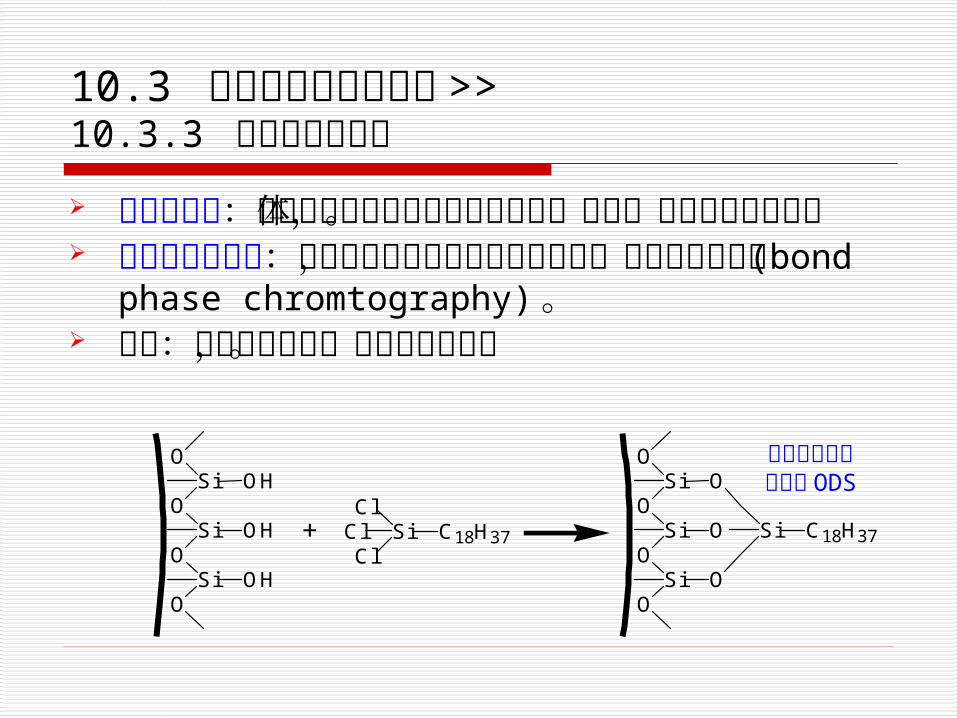
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 化学键合相:将具有官能团的固定相键合到载体表面,构成化学键合相。
化学键合色谱法:以化学键合相为固定相的色谱法,简称键合色谱法 (bond phase chromtography)。
特点:既有分配作用,又有吸附性能。
SiO
OOH
Si
SiO
OOH
OH
+ SiClCl
ClC18H37 Si
O
OO
Si
SiO
OO
O
Si C18H37
硅胶表面
十八烷基硅烷键合相 ODS
1. 反相键合相色谱法, RBPC2. 正相键合色谱法, NBPC3. 离子对色谱法, IPC or. PIC 4. 离子抑制色谱法, ISC5. 其他色谱法
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法

1. 反相键合相色谱法 RBPC 典型的反相键合色谱法 (RBPC) ,是用非极性的
键合固定相和极性流动相组成的色谱体系。固定相常用十八烷基硅烷键合相 (ODS或 C18);流动相常用甲醇-水、乙腈-水。
非典型反相键合色谱系统,用弱极性或中等极性键合相与极性大于固定相的流动相组成。
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>1.反相键合相色谱法
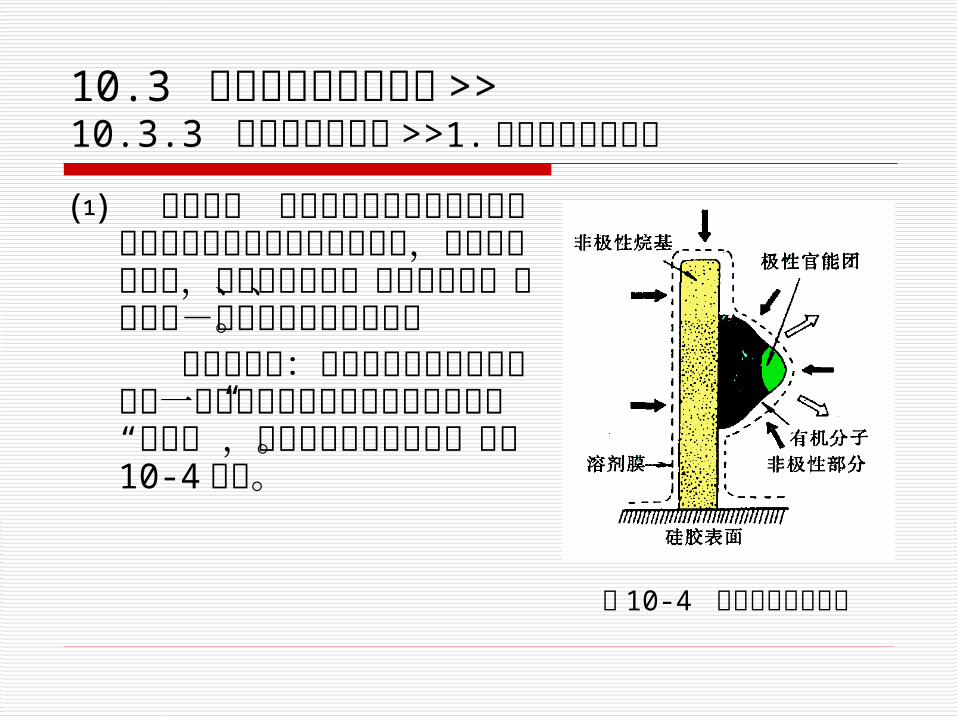
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>1.反相键合相色谱法
⑴ 分离机理 反相键合相表面具有非极性烷基官能团和未被取代的硅醇级,分离机理较复杂,有疏溶剂理论、双保留机理、顶替吸附-液相相互作用模型等。
疏溶剂理论:把非极性的烷基键合相看作一层键合在硅胶表面上的十八烷基的“分子毛”,它有较强的疏水特性。如图 10-4所示。 图 10-4 疏水剂缔合示意图
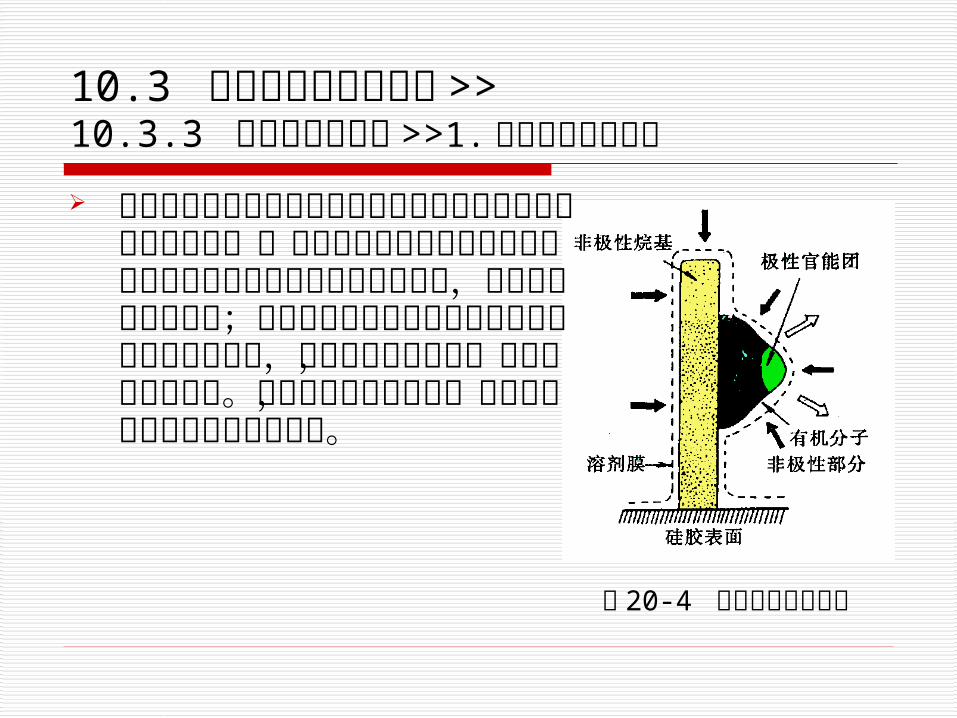
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>1.反相键合相色谱法
当用极性溶剂为流动相来分离含有极性官能团的有机化合物时,一方面分子中的非极性部分与固定相表面上的疏水烷基产生缔合作用,使它保留在固定相中;另方面被分离物的极性部分受到极性流动相的作用,促使它离开固定相,并减小其保留作用。结果两种作用力之差,决定了分子在色谱中的保留行为。 图 20-4 疏水剂缔合示意图

10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>1.反相键合相色谱法
⑵ 流动相的极性与容量因子的关系 流动相极性增大,洗脱能力降低,溶质的 k增大,
tR增大;反之, k 减小, tR也减小。作反相洗脱时,对于结构相近的组分,极性大的组分先出峰。

正相键合色谱法 (NBPC):固定相一般以氰基或氨基等极性基团作为键合相;流动相以烷烃 ( 正己烷 )中加入适量的极性调整剂 (如氯仿、甲醇、乙腈等 )为流动相。用途主要用于分离极性不同的化合物、异构体,特别适用于分离不同类型的化合物。
⑴ 分离机理 主要靠组分分子与固定相之间的范德华力、氢键作用力的差别而进行分离。
⑵ 流动相的极性与容量因子的关系 作正相洗脱时,流动相的极性增大,洗脱能力增加,
k 减小, tR 减小;反之, k 与 tR增大。分离结构相近的组分时,极性大的组分后出峰。
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>2.正相键合色谱法
离子对色谱 (ion pair chormatography, IPC) 在流动相中加入与被测离子相反电荷的离子对试剂,
使之形成中性的离子对化合物,从而增加样品离子在非极性固定相中的溶解度,使分配系数增加,分离改善。分配系数的大小主要取决于离子对化合物的解离平衡常数和离子对试剂的浓度。
⑴ 常用离子对试剂 分离碱类:用烷基磺酸盐为离子对试剂。如十二烷
基磺酸钠,正戊 /己 /庚 /辛磺酸钠 (PIC-B5/6/7/8)等。 分离酸类:用四丁基季铵盐 (PIC-A) ,如四丁基胺磷酸盐 (TBA)等。
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>3. 离子对色谱法

10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>3. 离子对色谱法
⑵ 分离机理 以 RPIC 分离碱类物质为例,用离子对模式说明分离机理
⑶ 分配系数
⑷ 主要用途 有机酸碱盐的分离,避免流动相 ( 酸碱 )对泵和流路的腐蚀;药物分析,如生物碱、有机酸、磺胺类药物、某些抗生素和维生素,体内药物分析等。缺点是价格昂贵。
固定相流动相
3333
RSOBHRSOBHRSONaNaRSOBHH)B(碱
中性离子对
ms
m3s32
PICARCOO/PICARCOO7.5pH:(RCOOH)分离PICBRNH/PICBRNH,3.5~3pH:)(RNH分离
K
K
,酸碱

在反相色谱法中,通过调节流动相的 pH 值,抑制样品组分的解离,增加它在固定相中的溶解度,以达到分离有机弱酸、弱碱的目的,这种技术称为离子抑制色谱法。
⑴ 适用范围 适用于 3.0≤pKa≤7.0弱酸; 7.0≤pKa≤8.0弱碱。⑵ 抑制剂 流动相中加入少量的弱酸、弱碱或缓冲溶液为抑制
剂 ( 常用乙酸、氨水、磷酸盐、乙酸盐 ) 。
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>4. 离子抑制色谱法

⑶ 影响容量因子的因素 对于弱酸,当流动相 pH< pKa时,组分以分子形
式为主, k值增大, tR增大;反之,组分以离子形式为主, k值变小, tR 减小。
对于弱碱,以上两种情况正好相反。⑷ 用途 可用于有机弱酸、弱碱与两性化合物的分离,以及它们与分子型化合物共存时的分离。
不适用于 pKa< 3 的酸、 pKa> 8 的碱,这些可采用离子对色谱法、离子交换色谱法进行分离。
10.3 各类高效液相色谱法 >> 10.3.3 化学键合色谱法 >>4. 离子抑制色谱法

10.3 各类高效液相色谱法 >> 10.3.5 其他色谱法 (阅读 )
离子色谱法 手性色谱法 环糊精色谱法 胶束色谱法 亲和色谱

1. 根据相对分子质量选择2. 根据溶解度选择3. 根据分子结构选择
相对分子质量十分低的样品,其挥发性好,适用于气相色谱。标准液相色谱类型 (液 - 固、液 - 液、及离子交换色谱 )最适合的相对分子质量范围是200~ 2000 。对于相对分子质量大于 2000 的样品,则用尺寸排阻法为最佳。
10.3 各类高效液相色谱法 >> 10.3.6 分离类型的选择 >>1. 根据相对分子质量选择

弄清样品在水、异辛烷、异丙醇、苯、四氯化碳中的溶解度是很有用的。
⑴ 如果样品可溶于水并属于能离解物质,以采用离子交换色谱为佳;
⑵ 如样品可溶于烃类 ( 如苯或异辛烷 ) ,则可采用液 - 固吸附色谱;
⑶ 如果样品溶解于四氯化碳,则多采用常规的分配和吸附色谱分离;
⑷ 如果样品既溶于水又溶于异丙醇时,常用水和异丙醇的混合液作液 - 液分配色谱的流动相,以憎水性化合物作固定相。
10.3 各类高效液相色谱法 >> 10.3.6 分离类型的选择 >>2.根据溶解度选择

用红外光谱法,可预先简单地判断样品中存在什么官能团。然后,确定采用什么方法合适。
例如:酸、碱化合物用离子交换色谱;脂肪族或芳香族用液 - 液分配色谱、液 - 固吸附色谱;异构体用液 - 固吸附色谱;同系物不同官能团及强氢键的用液 - 液分配色谱。
10.3 各类高效液相色谱法 >> 10.3.6 分离类型的选择 >>3.根据分子结构选择

液相色谱分离类型选择参考表
相对分子质量> 2000
相对分子质量< 2000
排阻色谱,水为流动相
排阻色谱,非水流动相
同系物——分配色谱异构体——吸附色谱分子大小差异——排阻色谱反相液一液色谱排阻色谱,水为流动相
碱——阳离子交换色谱
酸——阴离子交换色谱
反相离子对色谱
溶于水
不溶于水
溶于水
不离解
可离解
不溶于水
离子与非离子
样品

10.4 固定相10.4.1 液-固色谱固定相10.4.2 化学键合相10.4.3 其他固定相

1.硅胶 ⑴无定形多孔硅胶,国产代号 YWG, dp5~10μm
⑵球形全多孔硅胶,国产代号 YQG , dp3~10μm
⑶堆积硅珠,国产代号 YQG , dp3~5μm
2.氧化铝 ⑴球形 (5~ 10μm);⑵无定形 (5~ 10μm)。3. 高分子微孔多球 ( 有机胶 ) ⑴国产 YSG 系列;⑵日立 3010 胶等。4.其它 ⑴分子筛;⑵聚酰胺等。
10.4 固定相 >>10.4.1 液-固色谱固定相

用化学反应的方法将固定液的官能团键合在载体 ( 硅胶 ) 表面上,所形成的固定相,称为化学键合相。具有分配和吸附作用。优点:
①使用过程不易流失; ②化学性质稳定,在 pH2~8 的溶液不变质; ③热稳定性好,一般在 70℃ 以下稳定; ④载样量大,比硅胶高约一个数量级; ⑤适于作梯度洗脱。
10.4 固定相 >>10.4.2 化学键合相

化学键合相一般用硅胶作载体,具有分配作用和一定的吸附作用。吸附作用的大小视键合覆盖率而定。用化学反应方法将载体表面上残存的硅醇基除去,称为封尾 ( 封顶或遮盖 ) ,所形成的键合相称为封尾键合相,完全封尾的键合相无吸附作用,缺点是疏水强。
按极性分类:非极性、中等极性、极性键合相 3 类
10.4 固定相 >>10.4.2 化学键合相

10.4 固定相 >>10.4.2 化学键合相
1. 非极性键合相:十八烷基硅烷键合相 (ODS) 是最常用的非极性键合相。
2. 中等极性键合相:常见的有醚基键合相。3. 极性键合相:强极性-氨基键合相 (分析糖类 ) ;
中强极性-氰基键合相。
SiO
OOH
Si
SiO
OOH
OH
+ SiClCl
ClC18H37 Si
O
OO
Si
SiO
OO
O
Si C18H37

10.4 固定相 >>10.4.3其他固定相
1. 离子交换剂2. 凝胶3. 手性固定相4. 亲合色谱固定相

10.5 流动相 ( 溶剂系统 )
10.5.1 分离方程式10.5.2 Snyder 溶剂分类10.5.3 正相洗脱与反相洗脱10.5.4 洗脱方式10.5.5 流动相溶剂的选择

10.5 流动相 ( 溶剂系统 )>>10.5.1 分离方程式 依据分离方程式讨论溶
剂系统对分离度的影响
分离度受三项因素左右: a 项由色谱柱的质量决定;b、 c两项由流动相决定。分配系数比 α 与容量因子 k虽然相互关联,但溶剂的种类主要改变 α 。这是因为溶剂的种类不同,分子间的作用力的性质、强度不同,选择性不同。在溶剂种类确定以后,配比的调整只是改变极性,改变洗脱能力,改变保留时间,而分子间作用力的性质不变 ,因此主要是改变容量因子 k。概括为:溶剂种类主要影响分配系数比 α( 主要改变峰间距 ) ,溶剂配比主要影响容量因子 k( 主要改变保留时间 ) 。
c b a 1
1
4 2
2
k
knR

组分的分配系数,由样品组分、溶剂、固定相三者间的作用力 ( 分子间作用力 )综合作用所决定 (表 10-2)
Snyder 选用了乙醇、二氧六环、硝基甲苯 3个参考物,用于检验 3 种分子间作用力,分别用 Xe、 Xd、 Xn 表述。测定、计算出溶剂的极性参数 P′值列于表 10- 3。 P′
值大者,极性大,洗脱能力大。表 10-2 参考物与被检溶剂的作用力关系
参考物乙醇
( 质子给予体 )
二氧六环( 质子受体 )
硝基甲烷( 强偶极 )
被检溶剂的作用力类型
质子受体作用力 (Xe)
质子给予作用力 (Xd)
强偶极作用力 (Xn)
10.5 流动相 ( 溶剂系统 )>> 10.5.2 Snyder 溶剂分类

表 10-3 常用溶剂的极性参数 与分子间作用力溶剂 P′ Xe Xd Xn
苯 2.7 0.23 0.32 0.45
乙醚 2.8 0.53 0.13 0.34
二氯甲烷 3.1 0.29 0.18 0.53
正丙烷 4.0 0.53 0.21 0.26
四氢呋喃 4.0 0.38 0.20 0.42
氯仿 4.1 0.25 0.41 0.33
乙醇 4.3 0.52 0.19 0.29
乙酸乙酯 4.4 0.34 0.23 0.43
丙酮 5.1 0.35 0.23 0.42
甲醇 5.1 0.48 0.22 0.31
乙腈 5.8 0.31 0.27 0.42
乙酸 6.0 0.39 0.31 0.30
水 10.2 0.37 0.37 0.25
。,正己烷注:正戊烷 1.00.0 PP
P

1. 正相洗脱 用极性小于固定相的流动相的洗脱方式,称为正相洗脱、正相展开。适用于正相色谱法。参阅 P507表 18-3 常用溶剂的极性参数 P’ 与分子间作用力。 P ’ 大,极性大,洗脱能力强。例如,水、正己烷、正戊烷的值分别为 10.2、 0.1、 0.0 ,作正相洗脱时,水洗脱能力最强,正己烷和正戊烷的洗脱能力最弱。
10.5 流动相 ( 溶剂系统 )>> 10.5.3正相洗脱与反相洗脱

2. 反相洗脱 在反相洗脱中,溶剂的洗脱能力用强度因子 S值表示, S值大者洗脱能力大。反相色谱的首选纯溶剂为甲醇、乙腈与四氢呋喃,常以水为底剂。
10.5 流动相 ( 溶剂系统 )>> 10.5.3正相洗脱与反相洗脱
表 10-5 反相洗脱溶剂的强度因子 S值
溶剂 水 甲醇 乙腈 丙酮 二噁烷 乙醇 异丙醇 四氢呋喃
S值 0 3.0 3.2 3.4 3.5 3.6 4.2 4.5
组别 Ⅷ Ⅱ Ⅵ Ⅵ Ⅵ Ⅱ Ⅱ Ⅲ

3. 无论正相或反相洗脱,一般均采用多元溶剂系统。
为溶剂的百分浓度。
为溶剂的强度因子;
为溶剂的极性参数;式中:。反相洗脱强度因子:;正相洗脱极性参数:
极性参数混合溶剂(流动相)的
S
P
SSS
PPP
bbaaab
abaaab
10.5 流动相 ( 溶剂系统 )>> 10.5.3正相洗脱与反相洗脱

1. 等度洗脱 (isocratic elution) 用恒定配比的溶剂系统的洗脱方式,称为等度洗脱 优点:简便、重复性好、色谱柱易再生。 缺点:对于成分复杂的样品,效果欠佳。
2. 梯度洗脱 (gradient elution) 梯度洗脱就是在分离过程中使两种或两种以上不
同极性的溶剂按一定程序连续改变它们之间的比例,从而使流动相的强度、极性、 pH值、离子强度相应地变化,达到提高分离效果、缩短分析时间的目的。
10.5 流动相 ( 溶剂系统 )>> 10.5.4洗脱方式

10.5 流动相 ( 溶剂系统 )>> 10.5.4洗脱方式
梯度洗脱的实质:是通过不断地变化流动相的浓度配比,来调整混合样品中各组分的 K值,使所有谱带都以最佳平均 K值通过色谱柱。
比较梯度洗脱与程序升温:在梯度洗脱中溶质 K值的变化是通过溶剂的极性、 pH值和离子强度来实现的,用于分离组分性质差别较大的复杂样品;而程序升温是借改变温度来影响溶质的 K值,用于分离宽沸程的混合样品。
优点:①缩短分析周期;②提高分离效果;③改善峰形,很少拖尾;④增加检测灵敏度。

高效液相色谱中流动相是液体,它对组分有亲和力,并参与固定相对组分的竞争。流动相溶剂的选择将直接影响组分的分离度 .
1. 基本要求⑴ 溶剂必须具有合适的极性和良好的
选择性 ( 相对于待测样品 ) ,参阅Snyder 溶剂分类;
⑵ 溶剂要与检测器匹配,对于紫外吸收检测器,应注意选用检测波长比溶剂的紫外截止波长要长;
溶剂名称 紫外截止波长 /nm
正己烷 190二硫化碳 380四氯化碳 265苯 210氯仿 245
二氯甲烷 233四氢呋喃 212丙酮 330乙腈 190甲醇 205水 187
10.5 流动相 ( 溶剂系统 )>> 10.5.5 流动相溶剂的选择

10.5 流动相 ( 溶剂系统 )>> 10.5.5 流动相溶剂的选择
⑶ 高纯度。由于高效液相灵敏度高,对流动相溶剂的纯度也要求高。不纯的溶剂会引起基线不稳,或产生“伪峰”。痕量杂质的存在,将使截止波长值增加 50~ 100nm。
⑷ 化学稳定性好。不与样品发生反应或聚合的溶剂。⑸ 低粘度。若使用高粘度溶剂,势必增高压力,不利
于分离。常用的低粘度溶剂有丙酮、乙醇、乙晴等。但粘度过于低的溶剂也不宜采用,例戊烷、乙醚等,它们易在色谱柱或检测器内形成气泡,影响分离。

2. 一般原则 ⑴首选溶剂;⑵多元系统的组成;⑶洗脱能力;⑷
溶剂系统对样品的洗脱能力;⑸替代溶剂系统;⑹离子抑制色谱法。
3. 简要步骤 ( 以正相色谱法为例 ) ⑴配制初试二元溶剂系统;⑵调整溶剂系统的洗脱
能力;⑶按初始系统的 S值,配制相同 S值的替代溶剂系统;⑷若二元溶剂系统效果不好,可增加元数或加极性调整剂;⑸对组分性质差别较大的样品,等度洗脱无法兼顾两头,可用梯度洗脱。
10.5 流动相 ( 溶剂系统 )>> 10.5.5 流动相溶剂的选择

10.6 高效液相色谱仪10.6.0 仪器组成概述10.6.1 高压输液系统10.6.2 进样系统10.6.3 分离系统-色谱柱10.6.4 检测系统

工作站
自动进样器
预柱 分析柱 检测器
分步收集器
进样器
高压泵
贮液瓶
恒温箱
高效液相色谱仪由五大部分组成:高压输液系统、进样系统、分离系统、检测系统和色谱工作站
10.6 高效液相色谱仪 >>10.6.0 仪器组成概述

10.6 高效液相色谱仪 >>10.6.0 仪器组成概述
高效液相色谱分析过程 分析前,选择适当的色谱柱和流动相。开泵,冲洗
柱子,待柱子达到平衡而且基线平直后,用微量注射器把样品注入进样口,流动相把试样带入色谱柱进行分离,分离后的组分依次流入检测器的流通池,最后和洗脱液一起排入流出物收集器。当有样品组分流过流通池时,检测器把组分浓度转变成电信号,经过放大输入计算机,经过工作站数据处理,就得到色谱图。色谱图是定性、定量和评价柱效高低的依据。

10.6 高效液相色谱仪 >> 10.6.1 高压输液系统
由于高效液相色谱所用固定相颗粒极细,因此对流动相阻力很大,为使流动相较快流动,必须配备有高压输液系统。高压输液系统由储液罐、高压输液泵、过滤器、梯度洗脱装置等组成。
1. 储液罐 一般由玻璃、不锈钢或氟塑料制成 , 容量为 1~ 2L ,
用来储存足够数量、符合要求的流动相。 注意:流动相在进入高压泵之前,应先进行脱气和
过滤处理。

10.6 高效液相色谱仪 >> 10.6.1 高压输液系统
2. 输液泵 有恒流泵和恒压泵两种。 高压输液泵是核心部件,要求:密封性好,输出流
量恒定,压力平稳,可调范围宽,便于迅速更换溶剂及耐腐蚀等要求。
恒流泵特点 输出流量保持恒定而与色谱柱引起阻力变化无关。分为机械注射泵和机械往复泵 (常用 ) 两种。
恒压泵特点 能保持输出压力恒定,但其流量则随色谱系统阻力而变化,故保留时间的重视性差。

10.6 高效液相色谱仪 >> 10.6.1 高压输液系统
图 10-7 柱塞往复泵示意图
电动机 偏心轮 柱塞
柱 球形单向阀
溶剂

10.6 高效液相色谱仪 >> 10.6.1 高压输液系统
图 10-8 柱塞往复泵的两种连接方式
A 泵 B 泵吸排
贮液瓶
色谱柱
并联
A 泵 B 泵吸排
贮液瓶
色谱柱
串联

10.6 高效液相色谱仪 >> 10.6.1 高压输液系统
3. 梯度洗脱装置 梯度洗脱就是在分离过程中使两种或两种以上不同
极性的溶剂按一定程序连续改变它们之间的比例,从而使流动相的强度、极性、 pH值或离子强度相应地变化,达到提高分离效果,缩短分析时间的目的。梯度洗脱装置分为两类:
外梯度装置 又称低压梯度,流动相在常温常压下混合,用高压泵压至柱系统,仅需一台泵即可。
内梯度装置 又称高压梯度,将两种溶剂分别用泵增压后,按电器部件设置的程序,注入梯度混合室混合。

10.6 高效液相色谱仪 >> 10.6.2 进样系统
进样系统是将被分离的样品导入色谱柱的装置,一般包括进样口、注射器和进样阀等。要求密封性、重复性好,死体积小,便于实现自动化。
1. 注射器 微量注射器, 5μL、 10 μL等。2. 进样阀 六通阀进样器,如下图所示。
进样系统是引起柱前展宽的主要因素

10.6 高效液相色谱仪 >> 10.6.3 分离系统-色谱柱 色谱柱是系统的心脏部件,包括柱管与固定相两部分。⑴ 柱管材料 有玻璃、不锈钢、铝、铜及内衬聚合材料的其他金属。玻璃管耐压有限,故金属管用得较多。
⑵ 柱长 /内径 分析型柱长 10~ 25cm ,内径为 2~5mm ,实验制备型柱长 10~ 30cm ,内径 20~ 40mm 。
⑶ 前置柱 ( 预柱 ) 一般在分析柱前端,其内填充物和分析柱完全一样,淋洗溶剂由于先经过前置柱而被其中的固定相饱和,它流过分析柱时就不再洗脱其中固定相,保证分离性能不受影响。
⑷ 装柱方法 一般采用匀浆填充法装柱,先将固定相填料调成匀浆然后在高压泵作用下,快速将其压入装有洗脱液的色谱柱内,经冲洗后,即可备用。
⑸ 柱的再生 有反相洗脱再生和正相洗脱再生两种。

10.6 高效液相色谱仪 >> 10.6.3 分离系统-色谱柱
色谱柱的柱效评价 按规定,用 HPLC建立分析方法时,需进行“系统适用性试验”,给出分析状态下最小理论板数、分离度、重复性和拖尾因子 ( 对称因子 ) 。分析样品时,需检验柱性能是否合乎要求,可见评价柱效的重要意义。
评价的推荐条件:流动相流量 1mL/min ,进样量10μL ,检定波长 254nm ,其它参见下表。

10.6 高效液相色谱仪 >> 10.6.3 分离系统-色谱柱 >> 色谱柱的柱效评价
色谱柱名称 样品 流动相 (V/V) 测死时间 柱效指标
烷基键合相柱
苯、萘、联苯、菲。
甲醇 / 水(83/17) 反相色谱
柱用苯磺酸水溶液
正相色谱柱用四氯
乙烯
dp
/μm
3
4
5
7
10
n
>个 /m 80000
60000
50000
40000
25000
苯基键合相柱 甲醇 / 水(57/43)
硅胶柱 正己烷 ( 无水 )
氨基键合相柱正庚烷 / 异丙
醇(93/7)氰基键合相柱
三苯甲醇苯甲醇苯乙醇

检测器是反映色谱过程中组分浓度随时间变化的重要部件。要求灵敏度高、重复性好、线性范围宽、死体积小以及对温度和流量的变化不敏感等。有两种类型:
⑴ 溶质性检测器 它仅对被分离组分的物理或物理化学特性有响应,此类检测器有紫外、荧光、电化学检测器等
⑵ 总体检测器 它对试样和洗脱液总的物理和化学性质响应。此类检测器有示差折光检测器 ( 已少用 )等。
1. 紫外 - 可见光检测器 (UV-Vis)2. 荧光检测器3. 激发光散射检测器4. 化学发光检测器5. 安培检测器
10.6 高效液相色谱仪 >> 10.6.3 检测系统

10.6 高效液相色谱仪 >> 10.6.3 检测系统 >>1.紫外 - 可见光检测器
⑴ 检测原理:基于 Lambert-Beer 定律,即被测组分对紫外光或可见光具有吸收,且吸收强度与组分浓度成正比。液相色谱仪一般都配置有 UV-Vis 检测器 .
⑵ 组成:光源、流通池、检测元件、工作站等。
⑶ 特点:①灵敏度高,噪声低;②不破坏样品,可用于制备;③对温度、流动相流速波动不敏感,可用于梯度淋洗;④属于浓度型检测器。

⑷缺点 ①只能检测有紫外吸收的样品; ②检测波长>流动相的截止波长。⑸波长选择 ①选择 λmax 作检测波长,以得到较高的灵敏度; ②检测波长>流动相的截止波长。⑹紫外检测器的类型 ①固定波长型 ( 已淘汰 ); ②可变波长型 (最常用 ); ③二极管阵列检测器 (最新型 ) 。
10.6 高效液相色谱仪 >> 10.6.3 检测系统 >>1.紫外 - 可见光检测器

二极管阵列检测器 (diode-array detector,DAD) 以光电二极管阵列作为检测元件的紫外检测器 ( 图 10-13)它可构成多通道并行工作,同时检测由光栅分光,再入射到阵列式接受器上的全部波长的信号,然后,对二极管阵列快速扫描采集数据,得到的是时间、光强度和波长的三维谱图。
DAD与 UV 检测器的区别 普通 UV 检测器是先用单色器分光,然后让特定波
长的光进入流通池。 DAD 检测器是先让所有波长的光都通过流通池,然后通过一系列分光技术,使所有波长的光在接受器上被检测。
10.6 高效液相色谱仪 >> 10.6.3 检测系统 >>2.二极管阵列检测器简介

• 图 10- 13 光电二极管阵列检测器示意图
• 时间、光强度和波长的三维谱图
10.6 高效液相色谱仪 >> 10.6.3 检测系统 >>2.二极管阵列检测器简介

直接紫外检测:所使用的流动相为在检测波长下无紫外吸收的溶剂,检测器直接测定被测组分的紫外吸收强度。多数情况下采用直接紫外检测。
间接紫外检测:使用具有紫外吸收的溶液作流动相,间接检测无紫外吸收的组分。在离子色谱中使用较多,如以具有紫外吸收的邻苯二甲酸氢钾溶液作阴离子分离的流动相,当无紫外吸收的无机阴离子被洗脱到流动相中时,会使流动相的紫外吸收减小。
柱后衍生化光度检测:对于那些可以与显色剂反应生成有色配合物的组分 (过渡金属离子、氨基酸等 ),可以在组分从色谱柱中洗脱出来之后与合适的显色剂反应,在可见光区检测生成的有色配合物。
10.6 高效液相色谱仪 >> 10.6.3 检测系统 >>3. 检测方法

10.7.1 定性分析方法10.7.2 定量分析方法
10.7 定性、定量分析方法

1 色谱鉴定 保留值对比鉴定2 化学鉴定 分离制备后鉴定3 色谱-光谱联用鉴定
10.7 定性、定量分析方法 >>10.7.1 定性分析方法

工作曲线法
外标一点法
外标二点法采用对照品外标法
内标法 采用内标物
内标一点法
内标二点法
内标对比法
校正因子法
工作曲线法
10.7 定性、定量分析方法 >>10.7.2 定量分析方法

(1)内标对比法不需校正因子。药物分析常用示量%表示
(ms) 样品 —样品溶液中内标物的含量; (ms) 对照 —对照品溶液中内标物的含量; w—平均片重 ( 或丸重 ); m—取样量;(Ai/As) 样品—样品溶液中,待测物 A/内标物 A;(Ai/As) 对照—对照品溶液中,待测物 A/内标物 A 。
%100
/
/
m
w
m
m
AA
AA
s
s
si
si
对照
样品
对照
样品标示量%=
10.7 定性、定量分析方法 >>10.7.2 定量分析方法

例 10-3 用内标法测定复方乙酰水杨酸片 (APC) 中阿司匹林 A 、非那西汀 P 、咖啡因 C 三组分的质量分数,扑热息痛 S 作内标物。实验条件:色谱0.4×50cm日立 3010 胶;流动相:甲醇 / 三乙醇胺=1/500 ,流量 1mL/min; UV273 nm 检测。实验结果如下表。样品溶液:称取 1片量的药品粉末0.5005g 经多次萃取后配制成 100mL 溶液。 wA、 wP、wC 为样品中三组分的未知质量 (g/片 ), 0.5005g 为加入内标物的质量。试用内标对比法求:
⑴每片中 A、 P和 C 的含量; ⑵每片中 A、 P和 C 的质量分数。
10.7 定性、定量分析方法 >>10.7.2 定量分析方法 >>例 10-3

10.7 定性、定量分析方法 >>10.7.2 定量分析方法 >>例 10-3
P
A
SC
图 10-17 APC 的分析
被测溶液所含组分
阿司匹林A
内标物S
非那西汀P
咖啡因C
相对保留时间 /min
0.53 1.00 1.45 2.50
对照溶液
c 0.220 0.035 0.150 0.035
A 154856 171222 692272 372221
样品溶液
c wA 0.035 wP wC
A 178024 202694 820968 407792
单位: c:g/100mL , A:μV·s

10.7 定性、定量分析方法 >>10.7.2 定量分析方法 >>例 10-3
P
A
SC
图 18-17 APC 的分析
标准
标准
样品样品 i
si
sii m
AA
AAw
/
/ )1(
)/g(214.0220.0
171222/154856
202694/178024片
标准
样品 Aw
)/g(150.0150.0
171222/692272
202694/820968片
标准
样品 Aw
)/g(0324.0035.0
171222/372221
202694/407792片
标准
样品 Aw

10.7 定性、定量分析方法 >>10.7.2 定量分析方法 >>例 10-3
P
A
SC
图 10-17 APC 的分析
⑵每片中 A、 P和 C 的质量分数 ωi :
%8.42%1005005.0
214.0A
%0.30%1005005.0
150.0P
%5.6%1005005.0
0324.0C

10.8 应用与示例
1. 多环芳烃的分析2.磺胺类药物的分析3.氨基酸的分析4. 中药分析5. 离子抑制色谱法6.正相色谱法

课后作业
思考题: 2、 4、 5
习 题: 1、 2、 3、 7
(第 7题,参阅教材第 2章 2.4.5 相关与回归简介, P31-32)


















