







Languages
Pages
Legal


Inleidende begrippen van de chemischethermodynamicaMortier, T.
Copyright 2017 - Tom Mortier
Gecompileerd met LATEX op 18 juli 2017


Voorwoord
De voorliggende tekst is grotendeels gebaseerd op het boek ‘Basic Chemical Thermodynamics’ vanE. Brian Smith (Referentie [1]) dat als uitgangspunt werd genomen. Bovendien werd er gebruikgemaakt van verschillende uitstekende handboeken zoals ‘Physical Chemistry’ van Gilbert W. Ca-stellan (Referentie [2]), ‘Physical Chemistry’ van Thomas Engel en Philip Reid (Referentie [3])alsook ‘Physical Chemistry’ van P.W. Atkins (Referentie [4]).
Het studeren van fysische scheikunde is een reis doorheen de tijd. Verschillende namen van eminentewetenschappers uit een ver verleden komen aan bod. Verschillende verwijzingen naar de origineleliteratuur zullen in de tekst zelf voorkomen. Een prachtig en aan te raden boek dat kan helpen bijhet studeren is ‘The World of Physical Chemistry’ van Keith J. Laidler (Referentie [5]) om diepereinzichten te verkrijgen in de achtergronden van verschillende fysicochemische ontdekkingen.
De tekst werd gecompileerd met LATEX. Alle figuren die in de tekst voorkomen, zijn origineel. Desimulaties werden gemaakt met behulp van Gnuplot (http://www.gnuplot.info). Voor sommigetekeningen werd het package TikZ gebruikt. Veel noodzakelijke informatie om figuren te kunnenmaken, werd gevonden via http://www.texample.net/tikz.
Na reeds meer dan tien jaar in het onderwijs te hebben gestaan, ben ik vele van mijn studentendankbaar. Ik leer nog elke dag van hen bij en hoop dat ik sommigen onder hen ook een beetjeverder op weg hebben kunnen helpen in hun eigen denken. Opmerkingen en aanvullingen zijn danook altijd welkom. Eventuele fouten en/of onnauwkeurigheden mogen ook altijd gemeld wordenvia [email protected].


Inhoudsopgave
Voorwoord . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . iiiNotatie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . viii
1 Fundamentele concepten 11.1 Algemene inleiding . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11.2 Evenwicht in mechanische systemen . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2.1 Systeem verricht geen arbeid . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.2.2 Systeem verricht arbeid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
1.3 Reversibiliteit en evenwicht . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41.4 Het belang van de thermodynamica voor chemici . . . . . . . . . . . . . . . . . . . . 51.5 De mol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.6 Het ideale gas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
1.6.1 Voorwaarden . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91.6.2 Het concept van de partieeldrukken . . . . . . . . . . . . . . . . . . . . . . . 10
1.7 De barometrische verdelingswet . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2 Energie & de eerste hoofdwet 172.1 Arbeid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.2 Warmte en temperatuur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192.3 Temperatuursmetingen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192.4 Warmte en moleculaire bewegingen . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202.5 Behoud van energie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202.6 Toestandsfuncties en padfuncties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212.7 De wiskundige eigenschappen van toestandsfuncties . . . . . . . . . . . . . . . . . . . 222.8 Enthalpie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 272.9 De warmtecapaciteit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
3 Entropie, evenwicht en de Tweede Hoofdwet 313.1 Reversibiliteit en evenwicht . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 313.2 Evenwichtscondities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 323.3 Entropie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 333.4 Entropie als een toestandsfunctie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 353.5 Entropie voor een expansie van een gas . . . . . . . . . . . . . . . . . . . . . . . . . . 363.6 Entropieveranderingen die de warmtestroom begeleiden . . . . . . . . . . . . . . . . 373.7 Entropie en evenwicht . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 373.8 Een cosmologische uitwijding . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
v

vi Inhoudsopgave
3.9 Entropie als een functie van druk en temperatuur . . . . . . . . . . . . . . . . . . . . 383.9.1 Druk . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 383.9.2 Temperatuur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
3.10 Moleculaire basis van entropie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 393.11 Statistische basis van de Tweede Hoofdwet . . . . . . . . . . . . . . . . . . . . . . . . 413.12 De grootte van entropieveranderingen . . . . . . . . . . . . . . . . . . . . . . . . . . 42
4 Evenwicht in chemische systemen 454.1 De vrije energie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454.2 Gibbs vrije energie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
4.2.1 De drukafhankelijkheid van de Gibbs vrije energie . . . . . . . . . . . . . . . 484.2.2 De temperatuursafhankelijkheid van de Gibbs vrije energie . . . . . . . . . . 49
4.3 Fase-evenwichten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 504.3.1 De evenwichtsvoorwaarden tussen de materietoestanden . . . . . . . . . . . . 504.3.2 De Clapeyron vergelijking . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 524.3.3 De Clausius-Clapeyron vergelijking . . . . . . . . . . . . . . . . . . . . . . . . 53
4.4 De dampdruk van vloeistoffen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 544.5 Fasediagrammen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 594.6 De chemische potentiaal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
4.6.1 De fundamentele vergelijking . . . . . . . . . . . . . . . . . . . . . . . . . . . 604.6.2 De eigenschappen van de chemische potentiaal . . . . . . . . . . . . . . . . . 614.6.3 De chemische potentiaal en de vrije energie . . . . . . . . . . . . . . . . . . . 624.6.4 De chemische potentiaal van een ideaal gas in een mengsel van ideale gassen . 644.6.5 De vrije Gibbs energie en de entropie van het mengen . . . . . . . . . . . . . 65
4.7 Evenwicht tussen gasvormige reagentia . . . . . . . . . . . . . . . . . . . . . . . . . . 684.8 Eigenschappen van de evenwichtsconstante . . . . . . . . . . . . . . . . . . . . . . . 71
4.8.1 Temperatuursafhankelijkheid van de evenwichtsconstante . . . . . . . . . . . 714.8.2 Het berekenen van de evenwichtspartieeldrukken in een mengsel van ideale
gassen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 744.9 Basisresultaten van de chemische thermodynamica . . . . . . . . . . . . . . . . . . . 79
4.9.1 De vrije Gibbs energieverandering . . . . . . . . . . . . . . . . . . . . . . . . 794.9.2 De standaard enthalpieverandering . . . . . . . . . . . . . . . . . . . . . . . . 794.9.3 De invloed van de druk . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 794.9.4 De chemische potentiaal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
4.10 Het principe van Le Chatelier . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
5 Bepaling van thermodynamische grootheden 815.1 De wet van Hess . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 815.2 Standaard vormingsenthalpieën . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
6 Ideale en Reële Oplossingen 856.1 De ideale oplossing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
6.1.1 Algemene kenmerken van de ideale oplossing . . . . . . . . . . . . . . . . . . 856.1.2 De chemische potentiaal in ideale oplossingen . . . . . . . . . . . . . . . . . . 866.1.3 Binaire oplossingen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87

Inhoudsopgave vii
6.1.4 Het benzeen/tolueen systeem . . . . . . . . . . . . . . . . . . . . . . . . . . . 926.1.5 Temperatuur-samenstellingsdiagrammen en gefractioneerde destillatie . . . . 956.1.6 Ideale oplossingen van vaste stoffen in vloeistoffen . . . . . . . . . . . . . . . 976.1.7 Ideaal verdunde oplossingen . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
6.2 Colligatieve eigenschappen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 986.2.1 Vriespuntsverlaging . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99
7 Niet-ideale oplossingen 1017.1 Het concept van de activiteit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1017.2 Activiteit van vaste stoffen in vloeistoffen . . . . . . . . . . . . . . . . . . . . . . . . 1037.3 Elektrochemische cellen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
7.3.1 Een eenvoudige elektrochemische cel . . . . . . . . . . . . . . . . . . . . . . . 1037.3.2 Standaard elektrode potentialen . . . . . . . . . . . . . . . . . . . . . . . . . 104
8 Thermodynamica van gassen 1098.1 Expansie van een ideaal gas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1098.2 Irreversibele expansie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1108.3 Toestandsvergelijkingen voor gassen . . . . . . . . . . . . . . . . . . . . . . . . . . . 1138.4 Het Joule-Thomson experiment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1148.5 Niet-ideale gassen: fugaciteit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1158.6 Berekeningen van fugaciteiten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
9 De moleculaire basis van de thermodynamica 1199.1 Energieniveaus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1199.2 Microtoestanden . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1229.3 De Boltzmann factor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1249.4 Het gedrag van de warmtecapaciteit . . . . . . . . . . . . . . . . . . . . . . . . . . . 126
Bibliografie 128

Notatie
Voor de notatie van de thermodynamische grootheden worden de aanbevelingen uitgeschreven doorde International Union of Pure and Applied Chemistry (IUPAC) gevolgd zoals deze werden gepu-bliceerd in Quantities, Units and Symbols in Physical Chemistry, IUPAC Green Book [6].
Symbolena activiteit q warmteA Helmholtz vrije energie Q ladingc concentratie S entropieC warmtecapaciteit T temperatuurE elektromotorische kracht U interne energief fugaciteit v snelheidG Gibbs vrije energie V VolumeH enthalpie w arbeidK evenwichtsconstante W aantal microtoestandenm molaliteit x molfractieM molmassa z partitiefunctien hoeveelheid stof ε moleculaire energiep druk µ chemische potentiaalξ reactievordering

Hoofdstuk 1
Fundamentele concepten
De thermodynamica is de tak van de wetenschap die het gedrag van de materie en de transformatiestussen de verschillende vormen van energie beschrijft op macroscopische schaal. In dit hoofdstukwordt het belang van de thermodynamica voor chemici beschreven en worden enkele inleidendeconcepten besproken.
1.1 Algemene inleiding
Thermodynamica is één van de meest krachtige technieken die we kunnen gebruiken om natuur-lijke fenomenen te bestuderen. Ondanks de naam houdt de thermodynamica zich niet bezig metde dynamica van systemen, maar wel met evenwichten. Meer in het bijzonder bestudeert de ther-modynamica juist die evenwichten waarbij er geen neiging meer wordt vertoond dat er nog eenverandering zou gaan optreden. Het is zelfs niet nodig om veronderstellingen te doen over defundamentele natuur van de moleculen die een systeem uitmaken zodat de besluiten bijgevolg vrijalgemeen zijn. De thermodynamica biedt aan wetenschappers een reeks van relaties aan tussenmacroscopische eigenschappen die kunnen worden gemeten in het laboratorium zoals bijvoorbeeldde temperatuur, de evenwichtsconstante, het volume en de oplosbaarheid. Deze relaties kunnenallen worden afgeleid uit enkele initiële postulaten die de wetten van de thermodynamica wordengenoemd.
Beschouwen we bijvoorbeeld de belangrijke chemische evenwichtsreactie
N2 + 3H2 −−−− 2NH3
dat de basis vormt van het Haber-Bosch procédé voor de vorming van ammoniak. Als chemiciwillen we in staat zijn om een aantal belangrijke vragen te kunnen beantwoorden over dergelijkeevenwichten. Wat zijn bijvoorbeeld de eigenschappen van N2, H2 en NH3 om de positie van hetevenwicht bij een welbepaalde temperatuur en druk te bepalen? In andere woorden, uit hetgeenwe weten of wat we te weten kunnen komen over de eigenschappen van deze gassen, willen wevoorspellen hoeveel NH3 er kan gevormd worden onder een gegeven reeks van voorwaarden. Een
1
2 Hoofdstuk 1. Fundamentele concepten
tweede belangrijke vraag die we ons als chemici stellen is hoe het evenwicht zal gaan verschuivenwanneer we de temperatuur en de druk veranderen? De evenwichtsposities zijn in de praktijk nietaltijd zoals ze zijn voorspeld door de thermodynamica. In het geval van het Haber-Bosch procédéis voor de synthese van ammoniak een katalysator essentieel gebleken om het evenwicht te kunnenbereiken. Bovendien is de opbrengst van ammoniak bij evenwicht groter wanneer de temperatuurwordt verlaagd en de druk wordt verhoogd.
1.2 Evenwicht in mechanische systemen
Onze ervaringen met de wereld rondom ons, biedt ons reeds een goed inzicht in de positie vanevenwichten in mechanische systemen. Het woord systeem wordt gebruikt waarmee dat deel vanhet universum wordt bedoeld dat we aan het onderzoeken zijn op een welbepaald tijdstip. Hetsysteem is gescheiden van de rest van het universum of de omgeving door grenzen die fysiek kunnenzijn zoals de wanden in een container.
1.2.1 Systeem verricht geen arbeid
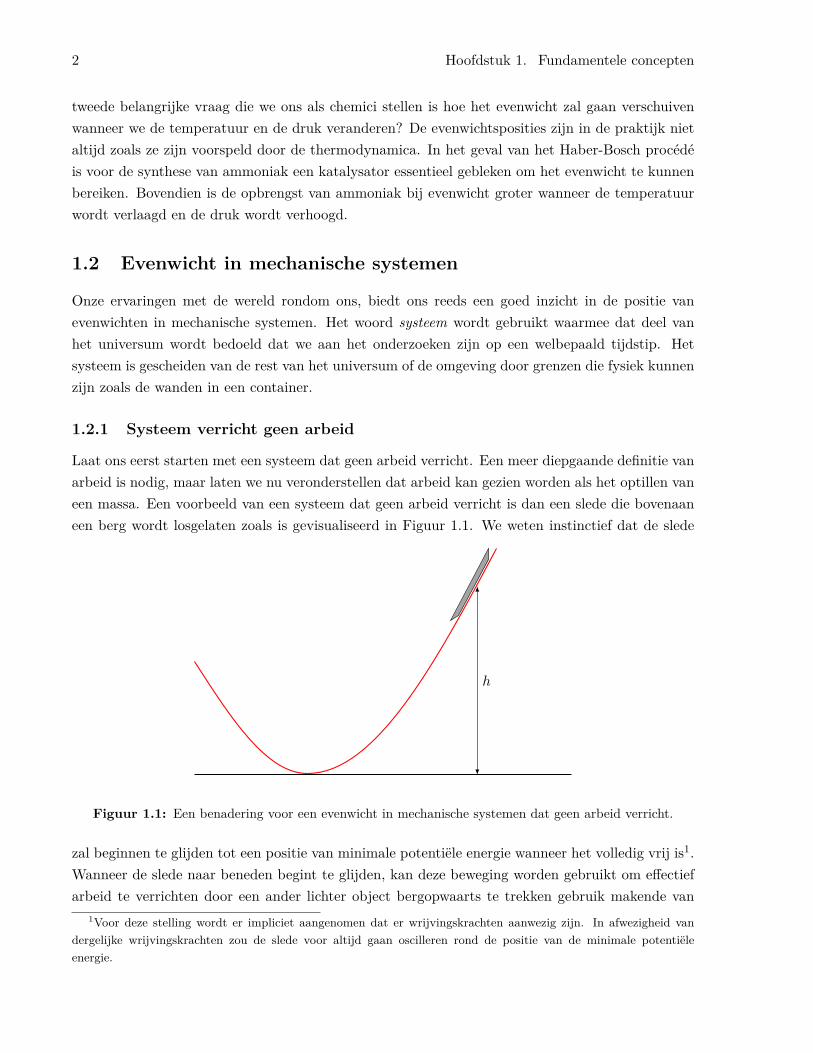
Laat ons eerst starten met een systeem dat geen arbeid verricht. Een meer diepgaande definitie vanarbeid is nodig, maar laten we nu veronderstellen dat arbeid kan gezien worden als het optillen vaneen massa. Een voorbeeld van een systeem dat geen arbeid verricht is dan een slede die bovenaaneen berg wordt losgelaten zoals is gevisualiseerd in Figuur 1.1. We weten instinctief dat de slede
h
Figuur 1.1: Een benadering voor een evenwicht in mechanische systemen dat geen arbeid verricht.
zal beginnen te glijden tot een positie van minimale potentiële energie wanneer het volledig vrij is1.Wanneer de slede naar beneden begint te glijden, kan deze beweging worden gebruikt om effectiefarbeid te verrichten door een ander lichter object bergopwaarts te trekken gebruik makende van
1Voor deze stelling wordt er impliciet aangenomen dat er wrijvingskrachten aanwezig zijn. In afwezigheid vandergelijke wrijvingskrachten zou de slede voor altijd gaan oscilleren rond de positie van de minimale potentiëleenergie.

1.2. Evenwicht in mechanische systemen 3
een touw en een katrol. Wanneer de slede echter naar beneden glijdt totaan de onderkant vande heuvel, verliest het weliswaar de mogelijkheid om arbeid te verrichten dat in ons eenvoudigmechanisch systeem de potentiële energie is
U = mgh
waarbij m de massa, g de valversnelling en h de hoogte boven de evenwichtspositie. Op de even-wichtspositie is de potentiële energie minimaal en zal dU = 0. Bovendien zal elke spontane bewegingvan de slede op de heuvel zodanig zijn dat het de potentiële energie reduceert en bijgevolg de mo-gelijkheid om arbeid te verrichten. De slede zal nooit spontaan bergopwaarts glijden.
1.2.2 Systeem verricht arbeid
Beschouwen we nu een systeem dat uitwendige arbeid verricht. Opnieuw kijken we hiervoor naarde slede op de helling als ons systeem, maar de slede is nu gebonden aan een touw zodat wanneerhet bergafwaarts glijdt, het via een katrol een massa kan doen verhogen zoals is gevisualiseerd inFiguur 1.2. Het is misschien nog eenvoudiger om goed te verstaan wat er wordt bedoeld met defactoren die het evenwicht controleren wanneer we het systeem verder vereenvoudigen door bij-
katrol
Ons ‘systeem’
Figuur 1.2: Evenwicht voor een mechanisch systeem aangewend om arbeid te verrichten.
voorbeeld twee massa’s (m1 en m2) te hangen over een wrijvingsloze katrol zoals wordt getoond inFiguur 1.3. Het systeem zal zich in rust bevinden wanneer m1 = m2 daar de krachten gebalanceerdzijn. Deze evenwichtspositie kan zodanig worden gedefinieerd dat het bruikbaar is in thermodyna-mische studies. Wanneer we toestaan dat de massa m1 begint te vallen over een afstand dh, dan

4 Hoofdstuk 1. Fundamentele concepten
m2m1
Katrol
Systeem
Figuur 1.3: Een mechanisch systeem aangewend om arbeid te verrichten.
zal m1 arbeid verrichten volgens m2gdh door m2 omhoog te tillen wanneer m2 < m1. Wanneer m2
dichter in grootte komt te liggen van m1, dan verkrijgen we overeenkomstig meer arbeid totdat bijm1 = m2 een infinitesimale verplaatsing zal leiden tot een arbeid die gelijk is aan m1gdh. Dit is demaximale arbeid die m1 kan verrichten daar het nu een gelijke massa optilt. Wanneer m1 = m2, ishet systeem in evenwicht. De evenwichtsvoorwaarde van het systeem kan nu worden gedefinieerdals datgene waarvoor een kleine verplaatsing zal leiden tot het systeem die de maximaal mogelijkearbeid verricht.
De arbeid verricht door een dergelijk systeem is gelijk aan het verlies van de potentiële energie,−dU , van het systeem. Wanneer het systeem zodanig is ingesteld dat het niet in staat is om externearbeid te verrichten, dan zal bij evenwicht de potentiële energie minimaal zijn of dU = 0 daar dearbeid op het systeem gelijk is aan −dU .
1.3 Reversibiliteit en evenwicht
De processen die we observeren in de natuur zijn irreversibel. De slede die bergafwaarts glijdt,verdrijft zijn potentiële energie als wrijvingswarmte. Om de slede terug te brengen naar de top vande berg, moeten we arbeid verrichten daar het niet spontaan zal terugkeren. Bovendien kunnen wede warmte die wordt gegenereerd door de slede niet verzamelen en gebruiken in bijvoorbeeld eenmotor om genoeg arbeid te genereren om de situatie te herstellen.We kunnen ons echter wel inbeelden wat reversibele processen zijn. In Figuur 1.4 zal wanneerm1 = m2 + ∆m de zwaarste massa vallen met toenemende snelheid. Wanneer de massa valt overeen afstand h zal de minimale arbeid die nodig is om het systeem te herstellen naar de originele

1.4. Het belang van de thermodynamica voor chemici 5
m1
m2
(a) m1 = m2 + ∆mIrreversibele verandering
m1m2
(b) m1 = m2 + dmReversibele verandering
Figuur 1.4: Massa’s zijn verbonden aan een draad die gespannen is over een frictieloze katrol om irre-versibele en reversibele veranderingen in een mechanisch systeem aan te tonen. Wanneer een systeem inmechanisch evenwicht is, zijn de massa’s stationair.
situatie gelijk zijn aan ∆mgh. Wanneer echter m1 = m2 + dm, zodat de massa’s slechts vanelkaar verschillen door een infinitesimaal kleine hoeveelheid, zal m1 infinitesimaal traag vallen enbijgevolg de maximale hoeveelheid arbeid verrichten. Enkel een infinitesimaal kleine hoeveelheidarbeid; dmgh is nodig om het systeem te herstellen of om de massa m1 terug te brengen naar destartpositie. Op elk punt gedurende het reversibel proces geldt dat
m1 = m2 + dm (1.1)
een voorwaarde die virtueel niet te onderscheiden is vanm1 = m2, de voorwaarde voor het evenwichtin het systeem. Een reversibele verandering is dan ook een verandering die wordt uitgevoerdzodat het systeem altijd bij evenwicht is en is bijgevolg, verschillend van natuurlijke observeerbareprocessen, oneindig traag.
1.4 Het belang van de thermodynamica voor chemici
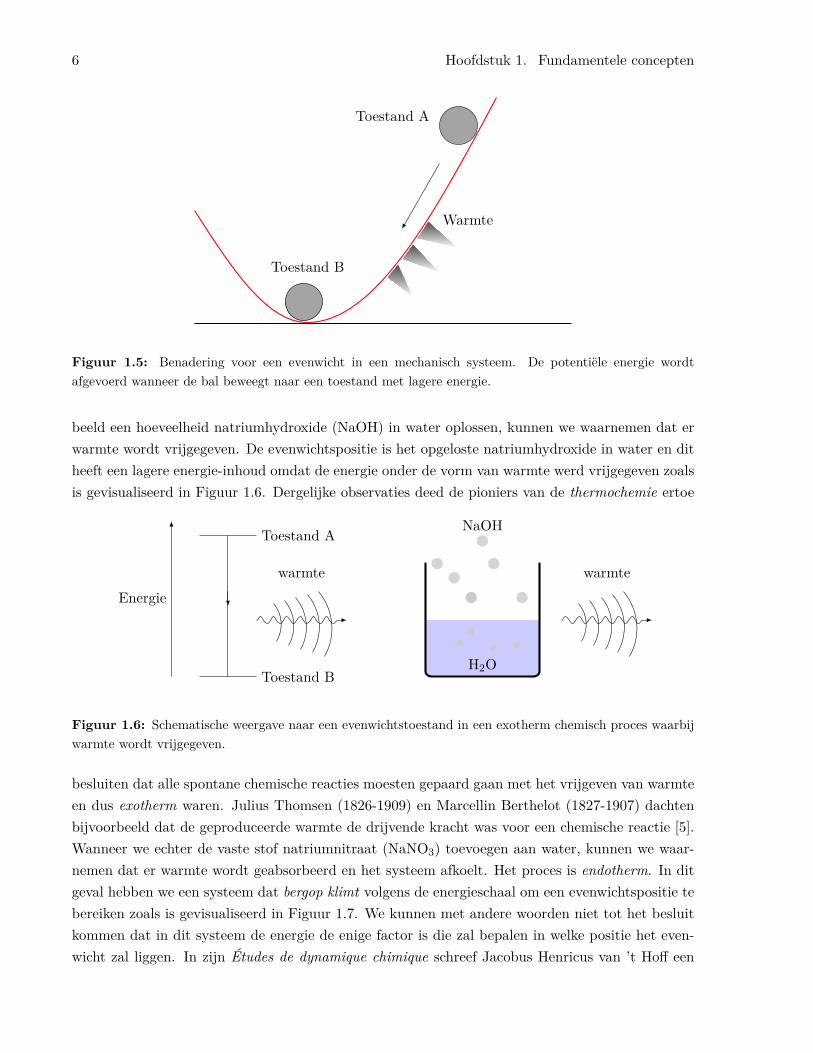
In een mechanisch systeem wordt er warmte vrijgegeven wanneer lichamen over een oppervlak glij-den of rollen zoals is weergegeven in Figuur 1.5. Moesten chemische processen gelijkaardige regelsvolgen, dan zouden we verwachten dat er altijd warmte zou vrijkomen wanneer ze tot een evenwichtzouden komen2. De vrijgekomen warmte zou het systeem doen opwarmen. Wanneer we bijvoor-
2Dit is enkel waar als chemische processen geen arbeid verrichten.
6 Hoofdstuk 1. Fundamentele concepten
Toestand A
Toestand B
Warmte
Figuur 1.5: Benadering voor een evenwicht in een mechanisch systeem. De potentiële energie wordtafgevoerd wanneer de bal beweegt naar een toestand met lagere energie.
beeld een hoeveelheid natriumhydroxide (NaOH) in water oplossen, kunnen we waarnemen dat erwarmte wordt vrijgegeven. De evenwichtspositie is het opgeloste natriumhydroxide in water en ditheeft een lagere energie-inhoud omdat de energie onder de vorm van warmte werd vrijgegeven zoalsis gevisualiseerd in Figuur 1.6. Dergelijke observaties deed de pioniers van de thermochemie ertoe
Energie
Toestand B
Toestand A
warmte
H2O
NaOH
warmte
Figuur 1.6: Schematische weergave naar een evenwichtstoestand in een exotherm chemisch proces waarbijwarmte wordt vrijgegeven.
besluiten dat alle spontane chemische reacties moesten gepaard gaan met het vrijgeven van warmteen dus exotherm waren. Julius Thomsen (1826-1909) en Marcellin Berthelot (1827-1907) dachtenbijvoorbeeld dat de geproduceerde warmte de drijvende kracht was voor een chemische reactie [5].Wanneer we echter de vaste stof natriumnitraat (NaNO3) toevoegen aan water, kunnen we waar-nemen dat er warmte wordt geabsorbeerd en het systeem afkoelt. Het proces is endotherm. In ditgeval hebben we een systeem dat bergop klimt volgens de energieschaal om een evenwichtspositie tebereiken zoals is gevisualiseerd in Figuur 1.7. We kunnen met andere woorden niet tot het besluitkommen dat in dit systeem de energie de enige factor is die zal bepalen in welke positie het even-wicht zal liggen. In zijn Études de dynamique chimique schreef Jacobus Henricus van ’t Hoff een

1.4. Het belang van de thermodynamica voor chemici 7
Energie
Toestand A
Toestand B
warmte
H2O
NaNO3
warmte
Figuur 1.7: Schematische weergave naar een evenwichtstoestand in een endotherm chemisch proces waarbijwarmte wordt opgenomen.
kritiek op het principe van Thomsen-Berthelot [7]. Hij noteerde dat het principe enkel waar wasbij het absolute nulpunt, maar niet bij andere temperaturen. Hij beschreef een belangrijke kwali-tatieve discussie waarbij de evenwichtsconstante K werd beïnvloed door de temperatuur. Als dereactie opging van links naar rechts en er hierbij warmte vrijkwam, dan zou de evenwichtsconstanteverlagen wanneer de temperatuur werd verhoogd. Wanneer er echter warmte werd opgenomentijdens een reactie die opging van links naar rechts dan zou een verhoging in de temperatuur deevenwichtsconstante juist doen verhogen. Het was enkele maanden later in 1884 dat de Fransechemicus Henri Louis Le Chatelier (1850-1936) deze bevindingen van van’t Hoff citeerde en werddit het principe van Le Chatelier genoemd.
Het bestaan van die andere kracht die een fysicochemische systemen drijft naar een evenwichtkan verder worden geïllustreerd door systemen te beschouwen waarbij de energie constant is. Webeschouwen bijvoorbeeld een willekeurig gas dat zich volledig in één van twee glazen bollen bevindten waarvan de andere glazen bol vacuüm werd gezogen zoals is weergegeven in Figuur 1.8a. De tweeglazen bollen zijn gescheiden van elkaar, maar zijn door middel van een afsluitkraan aan elkaarverbonden. Wanneer de afsluitkraan wordt geopend, zal het gas zodanig stromen dat het zichzelf
vacuüm
(a) (b)
Figuur 1.8: De expansie van een gas in een vacuüm.
gelijkmatig verdeelt tussen de twee containers. Voor een ideaal gas3 is er geen verandering inenergie die deze expansie zal begeleiden niettegenstaande er een duidelijke drijvende kracht bestaat
3De meeste reële gassen zijn bijna altijd ideaal onder normale omstandigheden.

8 Hoofdstuk 1. Fundamentele concepten
die ervoor zorgt dat het gas zichzelf doet verdelen tussen de twee containers.
Een tweede voorbeeld is de warmtestroom die optreedt wanneer een stuk warm metaal in thermischcontact wordt geplaatst met een kouder stuk metaal zoals schematisch wordt getoond in Figuur 1.9.De energie zal onder de vorm van warmte stromen tot de beide lichamen dezelfde temperatuur
warm lichaam
koud lichaam
warmtestroom
Figuur 1.9: De warmtestroom die optreedt wanneer een warm lichaam in contact wordt gebracht met eenkouder lichaam.
bezitten. Wanneer een dergelijk systeem wordt geïsoleerd van de omgeving, zal er geen veranderingzijn in de totale energie. Dit is opnieuw een eigenschap verschillend van de energie die bepaalt hoehet evenwicht zal worden bereikt.
Het bestaan van deze extra factor verklaart waarom het noodzakelijk is om een meer gesofisticeerdeanalyse op te bouwen om het evenwicht te beschrijven in fysicochemische systemen dan datgenewat volstonden voor de beschrijving van mechanische systemen.
1.5 De mol
Het concept van de hoeveelheid stof is centraal in fysicochemische berekeningen en metingen als-ook in andere takken van de scheikunde. De hoeveelheid stof van een systeem is evenredig methet aantal elementaire entiteiten van de aanwezige stof in het systeem. De elementaire entiteitenmoeten worden beschreven. Ze kunnen atomen, moleculen, ionen of specifieke groepen van derge-lijke deeltjes zijn. De entiteit is zelf een natuurlijke eenheid voor het meten van de hoeveelheidstof. We kunnen bijvoorbeeld de hoeveelheid stof beschrijven in een staal ijzer door te zeggen dat

1.6. Het ideale gas 9
er 2, !0 × 1024 Fe atomen zijn in het staal. De hoeveelheid stof in een kristal NaCl kan wordenbeschreven door te zeggen dat er 8,0× 1020 ionenparen Na+ en Cl− aanwezig zijn in een kristal.
Daar elk tastbaaar staal van de materie een dergelijk gigantisch aantal atomen of moleculen bevat,is een eenheid groter dan de entiteit zelf nodig om de hoeveelheid stof te meten. De SI eenheid voorde hoeveelheid stof is de mol. De mol is gedefinieerd als de hoeveelheid stof die er exact aanwezigis in 0,012 kg van het koolstof-12 (12C) isotoop. Eén mol van eender welke stof bevat hetzelfdeaantal elementaire entiteiten als er koolstofatomen zijn in exact 0,012 kg van het koolstof-12. Ditgetal is de constante van Avogadro4
NA = 6,022045× 1023 mol−1
De massa van een mol moleculen van een stof wordt de molmassa genoemd en wordt uitgedrukt inde eenheden g mol−1.
1.6 Het ideale gas
We dienen eerst de eigenschappen van de toestand van een ideaal gas te beschrijven. Deze toe-stand, waar reële gassen naar neigen bij lage temperaturen, speelt een belangrijke rol in chemischethermodynamica.
1.6.1 Voorwaarden
Een ideaal gas kan zodanig worden gedefinieerd als het voldoet aan de twee voorwaarden.
1. De druk, volume en de absolute temperatuur zijn gerelateerd aan elkaar door de vergelijking
pV = nRT (1.2)
waarbij n het aantal mol van de stof is en R de gasconstante.
2. De energie van het gas is enkel afhankelijk van de temperatuur en niet van de druk of hetvolume.
Hoewel we geen kennis nodig hebben van de moleculaire natuur van de materie om de besluitenvan de chemische thermodynamica te begrijpen, is het wel handig om dit toch te doen in hetgeval van een ideaal gas. De ideale gastoestand komt voor wanneer de moleculen zich gedragen alseenvoudige massapunten die niet met elkaar interageren. Ze worden niet door elkaar afgestoten enzullen elkaar ook niet aantrekken. De totale energie van een dergelijk gas is bijgevolg de kinetische
4Amadeo Avogadro (1776–1856) behaalde een doctoraat in de rechten in 1796, maar volgde nadien lessen overwiskunde, fysica alsook scheikunde. Uiteindelijk werd hij professor in de fysische scheikunde aan de Universiteitvan Turijn. Zijn hypothese dat dezelfde volumes van alle gassen bij dezelfde temperatuur en druk een gelijk aantalmoleculen zou bezitten, werd lang genegeerd door de wetenschappelijke wereld totdat Stanislao Cannizzaro (1826–1910) tijdens een chemisch congres in Karlsrühe in 1858 een brochure uitdeelde hoe de hypothese van Avogadro deverwarring over atomaire massa’s kon ophelderen [5, 8].

10 Hoofdstuk 1. Fundamentele concepten
energie en het kan worden aangetoond dat dit evenredig is met de absolute temperatuur. Daarer geen potentiële energie voortkomt uit de krachten tussen de moleculen, zal de energie van eenideaal gas niet veranderen wanneer het volume wordt veranderd en de afstand tussen de moleculenworden gewijzigd.
1.6.2 Het concept van de partieeldrukken
Daar de moleculen geen ruimte innemen en niet interageren, zal in een mengsel van ideale gassenelk gas zich zodanig gedragen alsof het zich alleen in de container zou bevinden. De totale druk isdan ook de totale som van de drukken die elk van de gassen zou uitoefenen alsof het alleen zou zijnin hetzelfde volume. Deze drukken worden de partieeldrukken genoemd.
Wanneer nA moleculen van een ideaal gas A en nB moleculen van een ideaal gas B worden gemengd,dan kunnen we schrijven dat de totale druk p gelijk is aan
p = pA + pB
De bijdragen van A en B tot de totale druk zal afhangen van het aantal mol van elk van dezeaanwezige stoffen. De partieeldrukken pA en pB worden bijgevolg gegeven door
pA =(
nAnA + nB
)p en pB =
(nB
nA + nB
)p
met nA/(nA + nB) = xA de molfractie van de stof A in het mengsel.
1.7 De barometrische verdelingswet
In de discussie over het gedrag van ideale gassen wordt er meestal stilzwijgend aangenomen dat dedruk van het gas overal in de container dezelfde waarde bezit. Strict genomen is deze benaderingenkel correct in de afwezigheid van een zwaartekrachtveld. Daar alle metingen gebeuren in labo-ratoriumomgevingen waar steeds een zwaartekrachtveld aanwezig is, is het handig om een zekerinzicht te krijgen welk effect er wordt voortgebracht door de invloed van dit veld. Het mag wordengezegd dat voor gasvormige systemen van gewone afmetingen, de invloed van het zwaartekracht-veld zo klein is dat het niet zal worden opgemerkt, zelfs niet met extreem verfijnde experimentelemethoden. Voor een fluïdum5 met een hogere dichtheid zoals een vloeistof, zal het effect vrij uit-gesproken zijn en zal de druk verschillend zijn bij verschillende verticale posities in een container.In Figuur 1.10 wordt een cilindrische kolom gevisualiseerd waarin zich een fluïdum bevindt. Dedwarsdoorsnede van de cilindrische kolom heeft een oppervlakte A. Het fluïdum is bij een constantetemperatuur T onderhevig aan een zwaartekrachtveld die neerwaarts is gericht om een deeltje eenvalversnelling g te geven. De verticale coördinaat z wordt opwaarts gemeten vanaf grondniveauwaarbij z = 0. De druk bij eendere welke hoogte z in de kolom wordt bepaald door de totale massa
5De term “fluïdum” wordt hier gebruikt om een medium te beschrijven dat bij een constante temperatuur en drukeen welbepaalde massa en volume heeft, maar geen vaste vorm.

1.7. De barometrische verdelingswet 11
A z = 0
z
dz
g
Figuur 1.10: Cilindrische kolom waarin zich een fluïdum bevindt onderhevig aan een zwaartekrachtveld.
m van het fluïdum boven die hoogte. De neerwaartse kracht op deze massa is mg. Deze krachtgedeeld door de oppervlakte is de druk bij een hoogte z en kan worden geschreven als
p = F
A= mg
A(1.3)
Wanneer we de druk bij een hoogte z + dz gelijkstellen aan p+ dp, dan is
p+ dp = m′g
A
waarbij m′ de massa van het fluïdum is boven de hoogte z + dz. We hebben echter de druk bijeendere welke hoogte z in de kolom gedefinieerd door de totale massa m van het fluïdum boven diehoogte zodat bijgevolg geldt dat
m′ + dm = m of m′ = m− dm
Als dm de massa van het fluïdum is in het stukje tussen z en z + dz, dan kunnen we schrijven dat
p+ dp = (m− dm) gA
= mg
A− gdm
A
Met het oog op Vergelijking 1.3, wordt dit dan
dp = −gdmA
Als ρ de dichtheid is van het fluïdum, dan is dm = ρAdz. Gebruik makende van deze uitdrukkingin dp levert op dat
dp = −ρgdz (1.4)
Vergelijking 1.4 is een differentiaalvergelijking die de verandering in de druk dp relateert aan dedichtheid van het fluïdum, de gravitationele versnelling (of hier de valversnelling) en de vermeer-dering in hoogte dz. Het negatieve teken betekent dat als de hoogte toeneemt (dz is +), de druk

12 Hoofdstuk 1. Fundamentele concepten
van het fluïdum zal afnemen (dp is −). Het effect van de hoogteverandering op de druk is evenre-dig aan de dichtheid van het fluïdum. Bijgevolg zal dit effect belangrijk zijn voor vloeistoffen enverwaarloosbaar zijn voor gassen.
Wanneer de dichtheid van een fluïdum onafhankelijk is van de druk zoals dit het geval is voorvloeistoffen, dan kan Vergelijking 1.4 onmiddellijk worden geïntegreerd. Daar ρ en g constantenzijn, worden ze uit de integraal gelaten en bekomen we dat∫ p
p0= −ρg
∫ z
0dz
en na integratie levert dit op datp− p0 = −ρgz (1.5)
met p0 de druk op de bodem en p de druk bij een hoogte z boven de bodem van de cilindrischekolom. Vergelijking 1.5 is de gebruikelijke vergelijking voor de hydrostatische druk in een vloeistof.
Om Vergelijking 1.4 toe te passen op gassen, moet het worden erkend dat de dichtheid van het gaseen functie is van de druk. Wanneer we vervolgens het gas als ideaal beschouwen, dan volgt uitVergelijking 1.2 dat6
ρ = pM
RT
Gebruik makende van deze uitdrukking in Vergelijking 1.4, hebben we vervolgens
dp = −pMg
RTdz
en na het scheiden van veranderlijken7 levert dit op dat
dpp
= −Mg
RTdz (1.6)
Vergelijking 1.6 is opnieuw een differentiaalvergelijking die we rechtstreeks kunnen integreren zodatwe bekomen dat
ln p = −Mg
RTz + C (1.7)
met C de integratieconstante die wordt geëvalueerd in termen van de druk op het grondniveau zodatwanneer z = 0 is p = p0. Invullen van deze waarden in Vergelijking 1.7 vinden we dat ln p0 = C.Het substitueren van deze waarde voor C en na het herschikken, reduceert Vergelijking 1.7 tot
ln(p
p0
)= −Mgz
RT(1.8)
ofp = p0e
−Mgz/RT (1.9)
6pV = nRT = m
MRT ⇒ ρ = m
V= pM
RT7Het scheiden van veranderlijken is een veel gebruikte wiskundige techniek voor het oplossen van differentiaalver-gelijkingen. Zie “Wiskunde voor Chemici” via https://www.slideshare.net/tomor/wiskunde-voor-chemici.

1.7. De barometrische verdelingswet 13
Vergelijking 1.9 wordt de barometrische verdelingswet genoemd en relateert de druk aan eenderwelke hoogte z, de temperatuur van de cilindrische kolom, de molmassa van het gas en de versnellingdie wordt veroorzaakt door het zwaartekrachtveld. Daar de dichtheid evenredig is aan de druk enhet aantal mol per kubieke meter evenredig is aan de druk, kan Vergelijking 1.9 worden geschrevenin twee equivalente vormen, namelijk
ρ = ρ0e−Mgz/RT of c = c0e
−Mgz/RT (1.10)
waarbij ρ en ρ0 de dichtheden zijn en waarbij c en c0 de concentraties in mol/m3 bij z en op hetgrondniveau. Vergelijking 1.10 wordt de gravitationele verdelingswet genoemd. Vergelijkingen 1.9en 1.10 worden verdelingswetten genoemd omdat ze de verdeling van het gas in de cilindrischekolom beschrijven. In Figuur 1.11 wordt, gebruik makend van Vergelijking 1.9, een plot getoond
0
0,2
0,4
0,6
0,8
1,0
0 5 10 15 20
100 K
300 K
1000 K
p/p
0
z (km)
Figuur 1.11: Een plot van p/p0 versus z voor stikstofgas.
waarbij p/p0 wordt uitgezet in functie van z voor stikstofgas bij drie temperaturen. Bij de hoogstetemperatuur (1000 K) is de verdeling meer gladgestreken dan bij de laagste temperatuur (100 K).De verandering van de druk in functie van de hoogte is bijgevolg minder uitgesproken als detemperatuur hoger wordt. Als de temperatuur oneindig zou zijn, dan zou de druk overal in decilindrische kolom dezelfde zijn.
Het is aan te raden om verdelingswetten van het exponentiële type zoals in Vergelijking 1.9 vannaderbij te bestuderen daar deze zo vaak voorkomen in fysische scheikunde in een meer algemenevorm als de Boltzmann verdeling. Vergelijking 1.6 is bijzonder leerzaam in het bespreken van de
14 Hoofdstuk 1. Fundamentele concepten
exponentiële verdeling en kan worden geschreven als
− dpp
= Mg
RTdz (1.11)
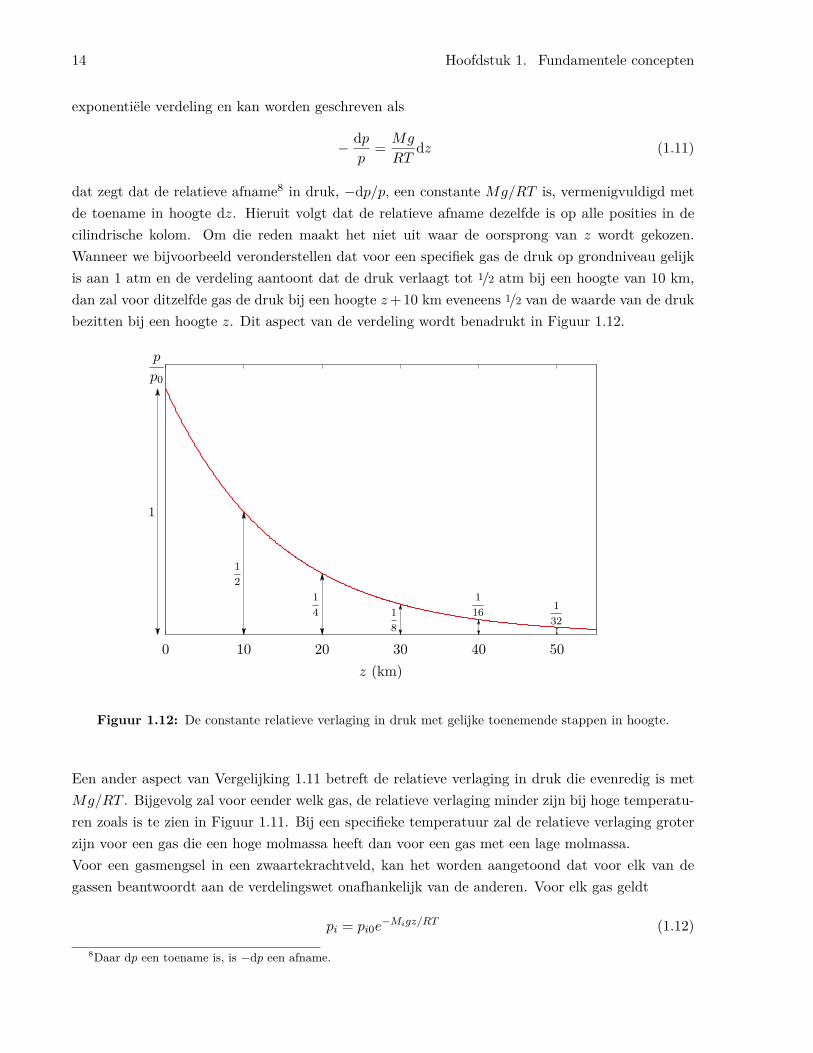
dat zegt dat de relatieve afname8 in druk, −dp/p, een constante Mg/RT is, vermenigvuldigd metde toename in hoogte dz. Hieruit volgt dat de relatieve afname dezelfde is op alle posities in decilindrische kolom. Om die reden maakt het niet uit waar de oorsprong van z wordt gekozen.Wanneer we bijvoorbeeld veronderstellen dat voor een specifiek gas de druk op grondniveau gelijkis aan 1 atm en de verdeling aantoont dat de druk verlaagt tot 1/2 atm bij een hoogte van 10 km,dan zal voor ditzelfde gas de druk bij een hoogte z+10 km eveneens 1/2 van de waarde van de drukbezitten bij een hoogte z. Dit aspect van de verdeling wordt benadrukt in Figuur 1.12.
0 10 20 30 40 50
1
12
14 1
8
116 1
32
p
p0
z (km)
Figuur 1.12: De constante relatieve verlaging in druk met gelijke toenemende stappen in hoogte.
Een ander aspect van Vergelijking 1.11 betreft de relatieve verlaging in druk die evenredig is metMg/RT . Bijgevolg zal voor eender welk gas, de relatieve verlaging minder zijn bij hoge temperatu-ren zoals is te zien in Figuur 1.11. Bij een specifieke temperatuur zal de relatieve verlaging groterzijn voor een gas die een hoge molmassa heeft dan voor een gas met een lage molmassa.Voor een gasmengsel in een zwaartekrachtveld, kan het worden aangetoond dat voor elk van degassen beantwoordt aan de verdelingswet onafhankelijk van de anderen. Voor elk gas geldt
pi = pi0e−Migz/RT (1.12)
8Daar dp een toename is, is −dp een afname.

1.7. De barometrische verdelingswet 15
waarbij pi de partieeldruk is van het ide gas in het mengsel bij een hoogte z, pi0 de partieeldrukvan het gas op grondniveau en Mi de molmassa van het gas. Het interessant gevolg van deze wetis dat de partieeldrukken van zeer lichte gassen snel dalen met de hoogte dan die van zwaarderegassen. In de aardatmosfeer zal het samenstellingspercentage gassen bij zeer grote hoogten dan ookverschillend zijn dan deze bij grondniveau. Bij een hoogte van 100 km zullen lichtere gassen zoalshelium en neon een hoger percentage van de atmosfeer inhouden dan dat ze doen bij grondniveau.Gebruik makend van Vergelijking 1.12 kunnen we de samenstellilng van de aardatmosfeer schattenbij verschillende hoogten. Zelfs als is de atmosfeer niet isothermisch en niet in evenwicht zijn dezebenaderingen vrij behoorlijk.
Voorbeeld 1.1De partieeldruk van argon in de atmosfeer is 0,0093 atm. Wat is de druk van argon op een hoogtevan 50 km als de temperatuur gelijk is aan 20 °C? Stel g = 9,807 m/s2.
Oplossing
In SI-eenheden is MAr = 39,9 g/mol = 0,0399 kg/mol, T = (20 + 273,15) K = 293,15 K enz = 50 km = 50× 103 m. Bijgevolg is
Mgz
RT= 0,0399 kg/mol · 9,807 m/s2 · 50× 103 m
8,314 J/mol K · 293,15 K = 8,03
zodatp = p0e
−Mgz/RT = 0,0093 atm · e−8,03 = 3,0× 10−6 atm


Hoofdstuk 2
Energie & de eerste hoofdwet
2.1 Arbeid
We hebben reeds een onderscheid gemaakt tussen twee types van energie die kunnen worden over-gedragen van één systeem in een ander systeem, namelijk arbeid en warmte. Wanneer energiewordt toegepast op iemands teen door hierop een massa te laten vallen of door de teen in eenkom met heet water te brengen, kan men het onderscheid zeer goed verstaan. In thermodynamicaverstaat men onder de formele definitie van arbeid dat “arbeid is de verplaatsing van energie vanéén mechanisch systeem naar een ander. Het is steeds volledig converteerbaar door een massa teverplaatsen. Arbeid kan worden uitgedrukt in termen van een kracht en de verplaatsing vanuit hetpunt van actie.
w =∫ l2
l1Fdl
Expansie is een voorbeeld van arbeid die regelmatig voorkomt in chemische problemen. Dit is de
p puit
l
Figuur 2.1: Expansie van een gas tegen een uitwendige druk.
arbeid die wordt uitgevoerd door de atmosfeer terug te dringen wanneer een systeem van volumeverandert. We definiëren de arbeid op het systeem als positief omdat dit zal resulteren naareen systeem dat energie wint en de arbeid die wordt verricht door het systeem wordt vervolgens
17

18 Hoofdstuk 2. Energie & de eerste hoofdwet
gedefinieerd als negatief. Wanneer een gas expandeert tegen een uitwendige druk puit (Figuur 2.1)is de arbeid die wordt uitgevoerd
w =∫ l2
l1− (puitA) dl
waarbij A de oppervlakte is van de zuiger en l de afstand over de welke de zuiger beweegt. WanneeerAdl = dV dan is
w = −∫ V2
V1puitdV
Wanneer het systeem van een initieel volume V1 naar een finaal volume V2 wordt teruggedrongenwaarbij er gebruik werd gemaakt van een constante externe druk puit, dan wordt de arbeid gegevendoor
w = −∫ V2
V1puitdV = −puit
∫ V2
V1dV = −puit (V2 − V1) = −puit∆V
Wanneer de uitwendige druk voortdurend wordt aangepast zodat het gelijk wordt gehouden aan p,de druk van het gas in de cilinder, dan is het systeem altijd in evenwicht en is de expansie reversibel.Onder deze omstandigheden is
wrev = −∫ V2
V1pdV (2.1)
Het negatieve teken staat voor het gegeven dat we de arbeid beschouwen die wordt uitgevoerd doorhet systeem dat we aanzien als een negatieve hoeveelheid omdat de energie is ‘verloren’ door hetsysteem. Voor een expansie, ∆V is een positieve hoeveelheid en het systeem doet arbeid dat zalleiden tot een negatieve waarde van w.
Voorbeeld 2.1Bereken de arbeid die wordt uitgevoerd door één mol gas dat expandeert van 5 dm3 tot 10 dm3
tegen een constante druk van 2 atm.
Voor een constante druk is de arbeid gelijk aan
w = p∆V
We converteren de data naar SI-eenheden
w = 2 · 1,013× 105 Pa ·(10−3 − 5× 10−3
)m3
= 1013 J
Er zijn verschillende typen van arbeid. Eéntje die regelmatig voorkomt in fysische scheikunde iselektrische arbeid waarbij een lading Q wordt verplaatst door een elektrisch potentiaalverschil E1.In dit geval is
w = −∫EdQ
Dit is de arbeid die kan worden bekommen uit elektrochemische cellen. Het vermogen is de snelheidwaarbij de arbeid wordt verricht en wordt gemeten in joules per seconde of Watt.
1We merken op dat de IUPAC voor het elektrisch potentiaalverschil de symbolen U , ∆V of ∆φ aanbeveelt. Wekiezen hier echter voor E om geen verwarring te creëren met volume of inwendige energie.

2.2. Warmte en temperatuur 19
2.2 Warmte en temperatuur
Begrijpen wat er wordt verstaan onder warmte was een uitdaging voor de pioniers van de thermo-dynamica. De equivalentie tussen warmte en arbeid als verschillende vormen van energie was nietondubbelzinnig tot relatief recente tijden. In 1842 poneerde Julius Robert Mayer (1814–1878), eenDuitse geneesheer, de wet van behoud van energie in zijn moderne vorm waarbij alle vormen vanenergie naast warmte werden opgenomen [9]. In onze tijd definiëren we warmte als ‘de overgangvan energie die resulteert uit temperatuursverschillen’.
De moeilijkheid in het begrijpen van warmte kwam grotendeels uit de verwarring met betrekkingtot het concept van de temperatuur. Vroegere wetenschappers hadden de neiging om te geloven datobjecten tot thermisch evenwicht kwamen wanneer ze allen een evenredige hoeveelheid van warmteper eenheid van volume bezaten. Het was de wetenschapper Joseph Black (1728–1799)2 die veeldeed om deze stelling te verklaren [10]. Hij toonde aan dat verschillende substanties verschillendewarmtecapaciteiten bezaten. Het thermische evenwicht werd bereikt tussen twee lichamen wanneerhun temperaturen gelijkwaardig werden. Warmte bleef stromen totdat de temperatuursgradiëntenverdwenen. Het temperatuursverschil was de drijvende kracht voor de warmtestroom. De relatietussen de hoeveelheid warmte getransfereerd door een lichaam en de daaropvolgende veranderingin zijn temperatuur was afhankelijk van de warmtecapaciteit
C = dqdT
waarbij C de warmtecapaciteit is, q de warmte en T de temperatuur.
2.3 Temperatuursmetingen
Temperatuur kan worden gemeten gebruik makende van eender welk lichaam die een geschikteeigenschap heeft die afhankelijk is van zijn ‘warmte’. De expansie van een geschikte vloeistof ineen glazen container is de meest gebruikte methode om de temperatuur te meten. Traditionelethermometers werden gekalibreerd door ze te plaatsen in kokend water bij een druk van één atmos-feer. De aflezing werd de waarde van 100 °C. De oorsprong van deze schaal, 0 °C werd gedefinieerddoor de temperatuur van smeltend ijs. Tussenliggende temperaturen werden gemeten door eenlineair gedrag aan te nemen. Deze aanname is echter niet exact en de temperatuur die we metenis afhankelijk van welk type thermometer dat er wordt gebruikt. We zullen de temperatuur echterdefiniëren op basis van een perfecte gasschaal.
2Joseph Black werd professor chemie aan de universiteit van Glasgow in 1756 en professor chemie in Edinburgh in1766. Vier jaar na zijn dood, in 1803, werd Lectures on the Elements of Chemistry gepubliceerd door John Robison(1739–1805) die één van zijn voormalige studenten was [5].
20 Hoofdstuk 2. Energie & de eerste hoofdwet
0 273,15 373,15
ijspunt
damppuntp
T
Figuur 2.2: Temperatuur (T ) gedefinieerd in termen van een ijspunt bij 273,15 K en een druk van eenideaal gas (p).
2.4 Warmte en moleculaire bewegingen
Hoewel het niet noodzakelijk is om de moleculaire natuur van de materie in te roepen om ther-modynamica te begrijpen, is het vaak handig om dit toch te doen. Het is zeker handig in hetonderscheid tussen warmte en arbeid. Arbeid kan worden gezien als de energie geassocieerd metgeordende bewegingen van lichamen of van de deeltjes waaruit de lichamen bestaan die bijvoorbeeldde grenzen terugduwen. Bewegingen van het massacentrum van een stroom elektronen in een draadzijn van dit type. Aan de andere kant zijn warmtestromen de bewegingen van thermische energie.Thermische energie komt van de wanordelijke willekeurige bewegingen van moleculen. Dergelijkewillekeurige bewegingen kunnen niet allemaal worden geconverteerd in arbeid tenzij we in staat zijnomm de moleculen in de rusttoestand te brengen– wat onmogelijk is onder normale omstandighe-den. Enkel bij het absoluut temperatuursnulpunt kunnen we ons een toestand voorstellen waarbijde bewegingen van de moleculen niet verder kunnen worden verlaagd en waarbij al hun energie isomgezet in arbeid.
2.5 Behoud van energie
Het principe van het behoud van energie staat gekend als de Eerste hoofdwet van de Thermody-namica. De algebraïsche som van alle energieveranderingen in een geïsoleerd systeem is gelijk aannul. Een geïsoleerd systeem is een systeem dat geen energie of materie kan uitwisselen met zijn

2.6. Toestandsfuncties en padfuncties 21
omgeving.De eerste wet van de thermodynamica vertelt ons dat energie kan worden geconverteerd van eenvorm naar een ander, maar dat er geen energie kan worden gecreëerd of vernietigd. Wanneer eenchemisch systeem verandert van een toestand in een ander, zal de netto energietransfer naar zijnommgeving gebalanceerd moeten zijn door een overeenkomstige verandering in interne energie vanhet systeem. Wanneer het systeem start in een toestand A en verandert naar een toestand B,kunnen we schrijven dat
∆U = UB − UA = q + w (2.2)
waarbij U de energie is die het systeem bevat en de interne energie wordt genoemd, q is de warmtedie wordt geabsorbeerd door het systeem en w is de arbeid die wordt gedaan op het systeem. ∆Uis enkel afhankelijk van de initiële en finale toestanden van het systeem en niet van het gevolgdpad.
2.6 Toestandsfuncties en padfuncties
Een alternatieve verklaring van de eerste wet van de thermodynamica is dat ∆U onafhankelijkis van het gevolgde pad tussen de initiële en finale toestanden, en bijgevolg enkel afhankelijk isvan de initiële en finale toestanden. We maken deze verklaring logisch door de kinetische energiete beschouwen. Beschouw één enkel molecule in het systeem. Beeld je in dat het molecule eenmassa m heeft met een initiële snelheid v1. We kunnen de snelheid stelselmatig opdrijven volgensv1 → v2 → v3 → v4. De verandering in kinetische energie volgens deze sequentie wordt gegevendoor
∆Ek =(1
2m (v2)2 − 12m (v1)2
)+(1
2m (v3)2 − 12m (v2)2
)+(1
2m (v4)2 − 12m (v3)2
)= 1
2m (v4)2 − 12m (v1)2
Zelfs wanneer v2 en v3 eender welke arbitraire waarden aannemen, zullen ze geen invloed hebbenop het resultaat. We besluiten dat de verandering in kinetische energie enkel afhankelijk is vande initiële en finale snelheid en dat het onafhankelijk is van het gevolgde pad tussen deze tweewaarden. Ons besluit blijft hetzelfde wanneer we het aantal snelheidsniveaus binnen een bepaaldinterval laten toenemen. Dit besluit geldt voor alle moleculen in het systeem en bijgevolg ook voor∆U .
Eender welke functie die beantwoordt aan deze eigenschap wordt een toestandsfunctie genoemdomdat het enkel afhankelijk is van de toestand van het systeem en niet van het gevolgde pad omdeze toestand te bereiken. Elke toestandsfunctie, zoals U , kan wiskundig worden uitgedrukt volgens
∆U =∫ f
idU = Uf − Ui (2.3)
Deze vergelijking stelt dat om ∆U te laten afhangen van de initiële en finale toestanden die wordengekarakteriseerd door i en f, de waarde van de integraal onafhankelijk moet zijn van het gevolgde

22 Hoofdstuk 2. Energie & de eerste hoofdwet
pad. Wanneer dit het geval is, dan kan U worden uitgedrukt door een infinitesimale hoeveelheiddU dat wanneer het wordt geïntegreerd enkel afhankelijk is van de initiële en finale toestanden. Dehoeveelheid dU wordt een exacte differentiaal genoemd.Het is handig om een cyclische integraal te definiëren, dat wordt aangeduid met het symbool
∮,
om een cyclisch pad aan te duiden zodat de initiële en finale toestanden identiek zijn. Voor U ofeender welke andere toestandsfunctie geldt dan dat∮
dU = Uf − Ui = 0 (2.4)
omdat de initiële en finale toestanden dezelfde zijn in een cyclisch proces.
De toestand van een perfect gas kan worden gedefinieerd door p, V en T te specifiëren. WanneerpV = nRT voor een vaste massa gas, moeten we enkel twee van de p, V en T specifiëren daar ditvoldoende zal zijn om de overige variabele te fixeren. Dit is het geval voor eender welke substantie(mengsel of een vaste samenstelling) zelfs wanneer dit niet de perfecte gas vergelijking zal volgen.We mogen schrijven dat
T = f(p, V )
Dergelijke vergelijkingen die p, V en T linken, worden toestandsvergelijkingen genoemd.
We hebben gezien dat U verschilt van q en w daar het enkel afhangt van de toestand van hetsysteem. We kunnen dus de toestand van een stof definiëren door bijvoorbeeld p en T constant tehouden waarbij U een bepaalde waarde zal hebben. U zoals p, T en V worden toestandsfunctiesgenoemd.
Toestandsfuncties kunnen afhankelijk zijn van de massa materiaal die we beschikbaar hebben.Bijgevolg kunnen V en U tweemaal zo groot zijn wanneer we de hoeveelheid materiaal in het systeemverdubbelen (waarbij andere zaken gelijk worden gehouden). Deze worden extensieve eigenschappengenoemd. Anderzijds zijn T en p onafhankelijk van de hoeveelheid materiaal waarmee we te makenhebben. Deze worden intensieve eigenschappen genoemd. Wanneer we een systeem verdelen inkleinere delen, dan zullen de intensieve eigenschappen van elk deel dezelfde waarde bezitten alsvoor het ganse systeem.
Om De toestand van een enkelfasige toestand in text đ and math đ
2.7 De wiskundige eigenschappen van toestandsfuncties
We zullen nu iets dieper ingaan op de wiskundige eigenschappen van toestandsfuncties. De ther-modynamische toestandsfuncties worden gedefinieerd door twee variabelen van de reeks p, V enT . In het formuleren van veranderingen in toestandsfuncties zullen we gebruik maken van partiëleafgeleiden. De volgende discussie is bijgevolg niet toepasbaar op padfuncties zoals w en q.
Beschouw één mol van een ideaal gas zodat
p = f (V, T ) = RT
V

2.7. De wiskundige eigenschappen van toestandsfuncties 23
Merk op dat p kan geschreven worden als een functie van twee variabelen V en T . De veranderingin p resulteert in een verandering in V of T dat evenredig is aan de volgende partiële afgeleiden:(
∂p
∂V
)T
= lim∆V→0
p (V + ∆V, T )− p (V, T )∆V = −RT
V 2(∂p
∂T
)V
= lim∆T→0
p (V,∆T + T )− p (V, T )∆T = R
V
(2.5)
Het subscript T in (∂p/∂T )T duidt erop dat T constant wordt gehouden in het afleiden met be-trekking tot V . De partiële afgeleiden in Vergelijking 2.5 staan toe om te bepalen hoe een functieverandert wanneer de variabelen veranderen. De verandering van p naar dp wanneer de waardenvan zowel T als V veranderen, kan geschreven worden als
dp =(∂p
∂T
)VdT +
(∂p
∂V
)TdV (2.6)
Tweede orde of hogere afgeleiden in relatie tot één van de beide variabelen kunnen eveneens wordengenomen. De gemengde tweede partiële afgeleiden zijn van bijzonder belang. We beschouwen degemengde partiële afgeleiden van p.
∂ ( ∂p∂V)T
∂T
V
= ∂2p
∂T∂V=
∂∂ (RTV
)∂V
T
∂T
V
=
∂ (−RTV 2
)∂T
V
= − R
V 2
∂ ( ∂p∂T)V
∂V
T
= ∂2p
∂T∂V=
∂∂ (RTV
)∂T
V
∂V
T
=
∂ (RV)
∂V
T
= − R
V 2
(2.7)
Voor alle toestandsfuncties f en dus ook voor ons bijzonder geval p, zal de volgorde naar waar defunctie zal worden afgeleid geen invloed hebben op de uitkomst. Omwille van deze reden mogenwe schrijven dat ∂ (∂f (V, T )
∂V
)T
∂T
V
=
∂ (∂f(V, T )∂T
)V
∂V
T
(2.8)
Vergelijking 2.8 is enkel geldig voor toestandsfuncties f . We hebben dan ook een krachtig wiskundiginstrument gevonden om na te gaan of f al dan niet een toestandsfunctie is. Wanneer f eentoestandsfunctie is, kunnen we schrijven dat
∆f =∫ f
i= ffinaal − finitieel (2.9)
Vergelijking 2.9 stelt dat f kan worden uitgedrukt als een infinitesimale hoeveelheid, df , dat wan-neer het wordt geïntegreerd enkel afhankelijk is van de initiële en finale toestanden. df wordt eenexacte differentiaal genoemd. Een voorbeeld van een toestandsfunctie en zijn exacte differentiaalzijn U en dU = đq − puitdV .
Voorbeeld 2.2

24 Hoofdstuk 2. Energie & de eerste hoofdwet
a) Bereken
(∂f
∂x
)y,
(∂f
∂y
)x
,
(∂2f
∂x2
)y
,
(∂2f
∂y2
)x
,
∂(∂f
∂x
)y
∂y
x
, en
∂ (∂f∂y)x
∂x
y
voor de functie f(x, y) = yex + xy + x ln y
b) Bepaal of f(x, y) een toestandsfunctie is van de variabelen x en y.
c) Als f(x, y) een toestandsfunctie is van de variabelen x en y, wat is de totale differentiaal df?
Oplossing
a) De afgeleiden zijn(∂f
∂x
)y
= yex + y + ln y,(∂f
∂y
)x
= ex + x+ x
y(∂2f
∂x2
)y
= yex,(∂2f
∂y2
)x
= − x
y2∂(∂f
∂x
)y
∂y
x
= ex + 1 + 1y,
∂ (∂f∂y)x
∂x
y
= ex + 1 + 1y
b) We hebben aangetoond dat ∂(∂f
∂x
)y
∂y
x
=
∂ (∂f∂y)x
∂x
y
Bijgevolg is f(x, y) een toestandsfunctie van de variabelen x en y.
c) De totale differentiaal wordt gegeven door
df =(∂f
∂x
)ydx+
(∂f
∂y
)x
dy
= (yex + y + ln y) dx+(ex + x+ x
y
)dy
Twee andere belangrijke resultaten uit het differentiaalrekenen worden veelvuldig gebruikt in dethermodynamica. Beschouw bijvoorbeeld een functie z = f(x, y) dat kan worden herschikt tot x =g(y, z) of y = h(x, z) zoals bijvoorbeeld wanneer p = nRT/V , dan is V = nRT/p en T = pV/nR.In dit geval is (
∂x
∂y
)z
= 1(∂y
∂x
)z
(2.10)
De cyclische kettingregel wordt eveneens gebruikt(∂x
∂y
)z
(∂y
∂z
)x
(∂z
∂x
)y
= −1 (2.11)

2.7. De wiskundige eigenschappen van toestandsfuncties 25
Bewijs
De totale differentiaal voor y = h(x, z) is
dy =(∂y
∂x
)zdx+
(∂y
∂z
)xdz (2.12)
De totale differentiaal voor x = g(y, z) is
dx =(∂x
∂y
)z
dy +(∂x
∂z
)ydz (2.13)
Substitueren van de uitdrukking voor dx uit Vergelijking 2.13 in het rechterlid van Vergelijking 2.12 levert op dat
dy =(∂y
∂x
)z
[(∂x
∂y
)z
dy +(∂x
∂z
)ydz]
+(∂y
∂z
)xdz
of na herschikken dat [(∂y
∂x
)z
(∂x
∂y
)z
− 1]dy +
[(∂y
∂x
)z
(∂x
∂z
)y
+(∂y
∂z
)x
]dz = 0 (2.14)
Daar dy en dz willekeurige infinitesimale veranderingen zijn, is de enige manier dat het linkerlid uit Vergelijking 2.14gelijk is aan nul voor alle keuzes van dy en dz als de coëfficiënten voor dy en dz elk afzonderlijk gelijk zijn aan nul.Door de coëfficiënten van dy gelijk te stellen aan nul, bekomen we(
∂y
∂x
)z
(∂x
∂y
)z
= 1 of(∂x
∂y
)z
= 1(∂y
∂x
)z
zodat we Vergelijking 2.10 hebben bewezen. Bovendien moet het gelden dat eender welke van de drie veranderlijkenx, y en z kunnen worden gekozen als afhankelijke veranderlijke. Bijgevolg moet het eveneens gelden dat(
∂y
∂x
)z
= 1(∂x
∂y
)z
en(∂x
∂z
)y
= 1(∂z
∂x
)y
Door nu de coëfficiënten van dz uit Vergelijking 2.14 gelijk te stellen aan nul, bekomen we(∂y
∂x
)z
(∂x
∂z
)y
+(∂y
∂z
)x
= 0(∂y
∂x
)z
(∂x
∂z
)y
= −(∂y
∂z
)x(
∂y
∂x
)z
(∂x
∂z
)y(
∂y
∂z
)x
= −1
1(∂x
∂y
)z
(∂y
∂z
)x
(∂z
∂x
)y
= −1
Door het omgekeerde te nemen van de laatste uitdrukking bekomen we Vergelijking 2.11.
Met behulp van de bewezen Vergelijkingen 2.10 en 2.11 kunnen we Vergelijking 2.6 herformuleren.
dp =(∂p
∂T
)VdT +
(∂p
∂V
)TdV
Stel dat deze uitdrukking geëvalueerd moet worden voor een specifieke stof zoals stikstofgas. Watzijn dan de grootheden die moeten worden gemeten in het laboratorium om numerische waardenvoor (∂p/∂T )V en (∂p/∂V )T te bekomen? Door gebruik te maken van Vergelijkingen 2.10 en 2.11bekomen we (
∂p
∂V
)T
(∂V
∂T
)p
(∂T
∂p
)V
= −1 (2.15)

26 Hoofdstuk 2. Energie & de eerste hoofdwet
zodat (∂p
∂T
)V
= −(∂p
∂V
)T
(∂V
∂T
)p
= −
(∂V
∂T
)p(
∂V
∂p
)T
= β
κen
(∂p
∂V
)T
= − 1κV
waarbij β en κ de respectievelijk eenvoudig te meten volumetrische thermische expansiecoëfficiënten de isothermische compressibiliteit zijn die worden gedefinieerd door
β = 1V
(∂V
∂T
)p
(2.16)
enκ = − 1
V
(∂V
∂p
)T
(2.17)
Zowel (∂V /∂T )p en (∂V /∂p)T kunnen worden gemeten door de verandering in volume van hetsysteem te bepalen wanneer de druk en de temperatuur worden gevarieerd terwijl de tweede veran-derlijke constant wordt gehouden. Het min-teken in de vergelijking voor κ is zodanig gekozen datde waarden van de isothermische compressibiliteit positief zijn. Waarden voor β en κ voor enkelevloeistoffen zijn weergegeven in Tabellen 2.1 en 2.2.
Tabel 2.1: Isothermische expansiecoëfficiënt van enkele vloeistoffen bij 298 K
Stof β × 104/K−1 Stof β × 104/K−1
H2O 2,06 CH3OH 14,9CH3COCH3 14,6 CH3CH2OH 14,0CS2 11,2 CH3COOH 10,8Hg 18,11 CHCl3 12,1CCl4 11,4 C2H4Cl2 11,4CH3CH2OCH2CH3 16,5 C6H5CH3 10,5
Bron: Lide, D.R.,ed., Handbook of Chemistry and Physics, CRC Press, 84th ed.,2003 (Referentie [11]).
Tabel 2.2: Isothermische compressibiliteit van enkele vloeistoffen bij 298 K
Stof κ× 104/MPa−1 Stof κ× 104/MPa−1
H2O 4,591 CH3OH 12,14CH3COCH3 12,62 CH3CH2OH 11,19CS2 9,38 CH3COOH 9,08Hg 0,401 CHCl3 9,96CCl4 10,50 C2H4Cl2 7,97CH3CH2OCH2CH3 18,65 C6H5CH3 8,96
Bron: Lide, D.R.,ed., Handbook of Chemistry and Physics, CRC Press, 84th ed.,2003 (Referentie [11]).

2.8. Enthalpie 27
Met behulp van de definities voor β en κ kan Vergelijking 2.6 worden geschreven in de vorm
dp = β
κdT − 1
κVdV (2.18)
dat kan worden geïntegreerd zodat∫ p2
p1dp = ∆p =
∫ Tf
Ti
β
κdT −
∫ Vf
Vi
1κV
dV ≈ β
κ(Tf − Ti)−
1κ
ln VfVi
(2.19)
2.8 Enthalpie
Chemische systemen worden over het algemeen niet gebruikt in het laboratorium om arbeid teverrichten. De enige vorm van arbeid dat ze kunnen doen is pV arbeid die komt uit de expansie ofde samentrekking3. Wanneer we een chemisch systeem bij een constant volume beschouwen, dankan het geen arbeid verrichten daar đw = −pdV = 0. Daar dU = đq + đw, is
dU = (đq)V en ∆U = (q)V (2.20)
De verhoging in interne energie van het systeem is om die reden gelijk aan de warmte die wordtgeabsorbeerd bij een constant volume of voor een systeem dat geen arbeid verricht.De meeste chemische experimenten worden eerder uitgevoerd bij een constante druk dan bij eenconstant volume. Door de expansie zal onder deze voorwaarden de arbeid verricht door het systeemniet gelijk zijn aan nul.
đw = −pdV en w = −p∆V
∆U = q + w = UB − UA = (q)p − p (VB − VA)
zodat(q)p = (UB + pVB)− (UA + pVA)
U + pV is net zoals U een toestandsfunctie daar U , p en V toestandsfuncties zijn. We definiërende functie U + pV als de enthalpie zodat
H = U + pV (2.21)
∆H = (q)p en dH = (đq)p (2.22)
De verhoging in enthalpie van een systeem is gelijk aan de warmte die bij een constante druk wordtgeabsorbeerd als wordt aangenomen dat het systeem enkel pV arbeid verricht.Zelfs voor veranderingen die voorkomen bij een andere voorwaarden dan de constante druk, zal∆H een begrensde waarde bezitten hoewel het onder deze voorwaarden niet gelijk zal zijn als dewarmte die wordt geabsorbeerd. Op een gelijkaardige wijze zal ∆U een begrensde waarde bezittenvoor eender welke verandering ongeacht of het bij een constant volume zal zijn, maar het is enkel
3Een heel belangrijke uitzondering waarbij chemische reacties worden toegestaan om arbeid verschillend van pVarbeid, te verrichten, is de elektrochemische cel.

28 Hoofdstuk 2. Energie & de eerste hoofdwet
voor een verandering bij constant volume dat ∆U = q.Het belang van deze nieuwe toestandsfunctie, de enthalpie, wordt duidelijk wanneer thermochemiewordt bestudeerd. De thermochemie is die tak van de thermodynamica die zich bezighoudt metwarmteveranderingen geassocieerd met chemische reacties. Wanneer we bijvoorbeeld zien bij eenchemische reactie
CS2 + 3O2 −−→ CO2 + 2SO2 ∆H = −1108 kJ mol−1
dan betekent dit dat voor één m ol van de reactie de enthalpie van het systeem verlaagt met1108 kJ en dat deze hoeveelheid warmte door de reactie wordt vrijgelaten bij een constante T en p.Eén mol reactie is wanneer het toegewezen aantal mol stof zoals is bepaald door de stoichiometri-sche coëfficiënten aan de linkerkant van de reactievergelijking worden omgezet naar de stoffen aande rechterkant van de vergelijking. Wanneer de verandering in thermodynamische eigenschappenwordt gegeven voor een specifieke chemische reactie of proces, dan zal het altijd refereren naar eenmol reactie tenzij een uitzondering tot deze regel specifiek staat aangegeven.∆H en ∆U zijn gewoonlijk vrij gelijk voor processen waarin vaste stoffen en vloeistoffen zijn betrok-ken, maar voor gassen kunnen ze significant verschillen. Wanneer een gasreactie een veranderingvan ∆n mol gassen in het systeem innhouden, dan zal ∆H = ∆U + ∆(pV ), en voor ideale gassen∆(pV ) = (∆n)RT , zodat
∆H = ∆U + ∆nRT
Bij 298 K is RT = 2,5 kJ mol−1 een niet geringe hoeveelheid.
2.9 De warmtecapaciteit
De warmtecapaciteit van een lichaam kan worden gedefinieerd door
C = đqdT
Wanneer de warmtecapaciteit wordt bepaald bij een constant volume door de warmte te meten dienodig is om één eenheid in temperatuur te stijgen, dan zal
dU = (đq)V
zodatCV =
(∂U
∂T
)V
(2.23)
Wanneer de warmtecapaciteit wordt gemeten bij een constante temperatuur is, daar dH = (đq)p
Cp =(∂H
∂T
)p
(2.24)
Voor vaste stoffen en vloeistoffen, zijn Cp en CV gewoonlijk vrij gelijkaardig in grootte, maar voorgassen kunnen deze waarden significant verschillen. Daar U = U+pV en voor n mol van een ideaalgas, pV = nRT , hebben we dat
H = U + nRT

2.9. De warmtecapaciteit 29
Door nu af te leiden, bekomen we dat dH = dU+nRdT of (đq)p = (đq)V +nRdT . Daar C = đq/dTkunnen we schrijven dat
Cp = CV + nR (2.25)
Voor argon is bij kamertemperatuur Cp = 20,8 J K mol−1, CV = 12,5 J K mol−1 zodat Cp−CV =12,5 J K mol−1 dat gelijk is aan R.
Uitgewerkte oefeningen
1. In 1845 suggereerde James Joule tijdens het beschrijven van zijn werk over warmte en tempe-ratuur dat het water beneden aan de Niagarawatervallen die 50 m hoog zijn, warmer was danaan de top [12]. Schat de verhoging in temperatuur. De warmtecapaciteit van één mol water,0,018 kg, is 80 J K−1. De valversnelling veroorzaakt door de zwaartekracht is 9,81 ms−2.Joule zijn eigen schatting was ongeveer 0,10 K.
OplossingWe maken gebruik van de eerste hoofdwet van de thermodynamica en berekenen de verande-ring in energie.
∆U = mgh = 0,018 kg · 9, 81 ms−2 · 50 m = 8,83 J
We kunnen nu de verhoging in temperatuur schatten door gebruik te maken van de relate
CV = ∆U∆T
⇒ ∆T = ∆UCV
= 8,83 J80 J K−1 = 0,11 K
2. Bereken de arbeid verricht tegen een standaard atmosferische druk wanneer een substantieexpandeert met 1 cm3.
OplossingDaar 1 atm = 105 N m−2 en 1 cm3 = 10−6 m3, kunnen we de verrichte arbeid berekenen uit
w = p∆V = 105 N m−2 · 10−6 m3 = 10−1 Nm = 0,10 J
3. Bereken het verschil tussen ∆H en ∆U wanneer één mol water wordt gekookt bij 373 K en1 atm. Het volume van één mol van een ideaal gas bij 373 K is 0,03 m3 en het volume vanvloeibaar water mag worden verwaarloosd.
OplossingDaar het volume van vloeibaar water mag worden verwaarloosd, kunnen we gebruik makenvan de relatie
∆H = ∆U + ∆nRT = ∆U + 1 mol · 8,31 J mol−1K−1 · 373 K = ∆U + 3100 J
We bekomen bijgevolg een verschil tussen ∆H en ∆U van 3,1 kJ.

30 Hoofdstuk 2. Energie & de eerste hoofdwet
4. Een blok metaal met een massa van 1,0 kg wordt verwarmd tot 400 K en vervolgens on-dergedompeld in 0,30 kg water. De temperatuur van het water stijgt van 294 K tot 300 K.Bereken de warmtecapaciteit van het metaal. Stel de warmtecapaciteit van water gelijk aan4200 J K−1 kg−1.
OplossingWe gaan uit van een constant volume. Daar qverloren door metaal = qgewonnen door water, kunnenwe schrijven dat
mmetaal∆TmetaalCV,metaal = mwater∆TwaterCV,water
zodat
CV,metaal = mwater∆TwaterCV,watermmetaal∆Tmetaal
= 0,30 kg · (300 K− 294 K) · 4200 J K−1 kg−1
1,0 kg · (400 K− 300 K)= 75,6 J K−1 ≈ 76 J K−1

Hoofdstuk 3
Entropie, evenwicht en de TweedeHoofdwet
3.1 Reversibiliteit en evenwicht
Wanneer een chemische reactie optreedt, kunnen we experimenteel vaststellen dat volgens de eerstehoofdwet van de thermodynamica de energie wordt behouden, maar we hebben nog geen maniergevonden in welke richting de reactie zal opgaan. We hebben met andere woorden nog geen ge-schikte definitie gevonden voor de positie van het chemisch evenwicht. Voor moleculaire systemenwaarvan het evenwicht kan worden bereikt door endotherme processen zal de energie, verschillendvan de potentiële energie in mechanische systemen, geen afdoende criterium zijn voor het chemischevenwicht. Een nieuwe en noodzakelijke factor moet worden geïntroduceerd die ons in staat steltom te begrijpen waarom warmte altijd van een ‘heet’ naar een ‘koude’ lichaam stroomt en waaromeen ideaal gas steeds zal trachten te expanderen om de container te willen vullen zelfs wanneer ditzonder energieverlies (door het systeem) tijdens deze processen zal gepaard gaan.
We hebben geobserveerd in onze beschouwing van mechanische systemen dat wanneer er een ver-andering optreedt, zodat het systeem steeds in evenwicht zal zijn, de verandering oneindig traagzal optreden en in staat zal zijn om de maximale hoeveelheid arbeid te verrichten. Een dergelijkeverandering noemen we reversibel of omkeerbaar. De voorwaarden waaraan reversibele veranderin-gen moeten voldoen zijn dezelfde voorwaarden waaraan moet worden voldaan wanneer het systeemin een toestand van evenwicht zal zijn.
Voor een reversibele verandering zal de arbeid uitgevoerd door het systeem maximaal zijn. Bijgevolgzal voor een reversibele verandering đw meer negatief zijn dan voor de equivalente irreversibelespontane verandering1. De verandering in inwendige energie, dU moet dezelfde zijn voor eenderwelke verandering die zal optreden. Bijgevolg zal voor een reversibele verandering, daar đwrev <
đwirr, dat đqrev < đqirr. Gedurende een reversibele verandering zal het systeem de maximale warmte
1Arbeid uitgevoerd door het systeem is negatief.
31

32 Hoofdstuk 3. Entropie, evenwicht en de Tweede Hoofdwet
absorberen uit de omgeving en de maximale hoeveelheid arbeid uitvoeren op de omgeving. Het isobserveerbaar dat spontane processen minder warmte absorberen en minder arbeid verrichten dande overeenkomstige reversibele processen (Figuren 3.1 en 3.2).
Figuur 3.1: Een spontane verandering gebeurt bij een eindige snelheid en vereist een eindige hoeveelheidarbeid verricht op het systeem als het systeem moet worden hersteld naar de originele toestand.
m+ dm m
Figuur 3.2: Een reversibele verandering waarbij het proces infinitesimaal traag verloopt en de origineletoestand van het systeem kan worden hersteld door een infinetisemale hoeveelheid arbeid.
3.2 Evenwichtscondities
We zijn nu in een positie gekomen om de algemene voorwaarde van evenwicht uiteen te zetten diezowel toepasbaar is op mechanische alsook moleculaire systemen. Spontane veranderingen zijn dezedie wanneer ze worden uitgevoerd onder de gepaste omstandigheden, gebruikt kunnen worden omarbeid te verrichten. Wanneer ze reversibel worden uitgevoerd, brengen ze een maximale hoeveelheid

3.3. Entropie 33
arbeid op. In natuurlijke processen wordt de maximale arbeid nooit bereikt. Dit is één van de veleequivalente stellingen van de Tweede Wet van de Theromodynamica2. Zoals de Eerste Wet, is deTweede Wet gebaseerd op ervaring of op experimentele waarnemingen. Desalniettemin is het in depraktijk soms moeilijk te zeggen hoeveel arbeid er gepaard gaat met vele spontane processen. Hetmengen van twee ideale gassen is een bijzonder moeilijke casus.
In een eenvoudig mechanisch systeem zal de capaciteit om arbeid te verrichten eenvoudigweg depotentiële energie zijn en het evenwicht wordt gedefinieerd als de positie van de minimale potentiëleenergie. De totale inwendige energie van een moleculair systeem kan echter niet volledig getransfor-meerd worden in arbeid en de positie van de minimale inwendige energie definieert niet het chemischevenwicht in moleculaire systemen. We hebben een hoeveelheid nodig van de capaciteit van eendergelijk systeem om arbeid te verrichten alsook een toestandsfunctie die het verlies reflecteert vande capaciteit om arbeid te verrichten.
3.3 Entropie
In beide voorbeelden die we reeds eerder hebben beschouwd (de expansie van een ideaal gas in eenvacuüm en de warmtestroom van een warm lichaam naar een koud lichaam) zal het systeem decapaciteit verliezen om arbeid te verrichten. De verloren capaciteit is duidelijk gerelateerd aan đq,daar dU = đq+ đw. De verloren capaciteit is eveneens gerelateerd aan de temperatuur want als wede warmtestroom q beschouwen van een heet naar een koud alsook naar een warm reservoir zoalsgeïllustreerd in Figuur 3.3, zal het verlies aan capaciteit om arbeid te verrichten duidelijk groterzijn dan wanneer men vertrekt van een heet naar een warm reservoir. In het laatste geval zal dearbeid bekomen worden door de warmtestroom van Tw → Tk omdat dit een spontaan proces blijktte zijn.
We hebben gezien dat đqrev geen geschikte maat is voor de ‘niet-beschikbare’ arbeid omdat hetafhankelijk is van het gevolgde pad zodat qrev geen toestandsfunctie is. We hebben eveneens geziendat onze maat de temperatuur moet mee betrokken worden omdat een vaste hoeveelheid warmtestroomt over een groot temperatuursverschil een groter verlies in de capaciteit van het systeeminhoudt om arbeid te verrichten dan dezelfde warmte die over een kleiner temperatuursverschilstroomt.
Het zal worden aangetoond dat hoewel qrev niet onafhankelijk van het gevolgde pad, de integraal vande warmte đqrev gedeeld door de temperatuur bij dewelke het wordt getransfereerd wel onafhankelijkzal zijn van het gevolgde pad en enkel afhangt van de initiële en finale toestanden van het systeem.We kunnen de entropie S zodanig definiëren dat
∆S = SB − SA =∫ B
A
đqrevT
(3.1)
2Een andere, misschien meer gekende, stelling van de Tweede Wet is dat warmte nooit spontaan kan stromen vaneen koud naar een warm lichaam.

34 Hoofdstuk 3. Entropie, evenwicht en de Tweede Hoofdwet
T
warm (Tw)
q
heet (Th)
koud (Tk)
q
q
Figuur 3.3: De stroom van de warmte van een heet reservoir naar een koud reservoir. De warmtestroomkan rechtstreeks gebeuren of via een warm reservoir.
De entropie is een toestandsfunctie en het is om die reden een geschikte maat voor het verlies vande capaciteit van het systeem om arbeid te verrichten. Bij constante temperatuur đqrev = TdS enomdat dU = đqrev + đwrev, bekomen we dat
đwrev = dU − TdSarbeid = verandering inwendige energie− ‘niet-beschikbare energie’
Wanneer van een toestand A naar een toestand B wordt overgegaan, zal ∆S steeds hetzelfde zijn.Het zal echter enkel gelijk zijn aan
∫ đqrevT
voor een reversibel pad. We hebben gezien dat de enigevoorwaarde onder dewelke we een verandering kunnen beschouwen van een reversibel pad is dathet systeem in evenwicht moet zijn. Om die reden is dS = đq/T een voorwaarde voor evenwicht.
Voor spontane veranderingen dq < dqrev, en
dS > đqT
een voorwaarde die houdbaar is voor observeerbare processen. De vergelijking
dS = đqT
(3.2)
is de meest algemene definitie van evenwicht die er beschikbaar is. Wanneer we een ‘geïsoleerd’systeem – een systeem dat geen energie kan uitwisselen met zijn omgeving, beschouwen zodat het

3.4. Entropie als een toestandsfunctie 35
noch arbeid kan verrichten noch warmte kan absorberen en waarvoor đq = 0, kunnen we bekomenvoor de evenwichtsvoorwaarden dat
dS = 0, S = constant
Voor een observeerbare verandering dS > 0 wanneer dS > đq/T .
Voor een geïsoleerd systeem eender welke spontane verandering zal neigen om hogere entropietoe-standen te produceren totdat de entropie een maximumwaarde bereikt. Bij dit punt zal het systeemin evenwicht zijn en zal de entropie constant blijven bij zijn maximale waarde.
3.4 Entropie als een toestandsfunctie
De entropie is een toestandsfunctie, maar we dienen deze stelling te rechtvaardigen. Traditioneelwordt dit in de meeste handboeken over thermodynamica gedaan wanneer men de efficiëntie vanwarmtemotoren beschouwt. Deze beschouwing was een hoofdbezigheid voor de pioniers van dethermodynamica. Als chemici zijnde, zullen we onszelf met behulp van een shortcut toelaten omde stelling te rechtvaardigen dat de entropie een toestandsfunctie is gebruik makende van enkel eenideaal gas.
Uit de Eerste Wet volgt dat dU = đqrev − pdV wanneer het systeem enkel pV verricht. Voor éénmol van een ideaal gas, mogen we schrijven dat dU = CV dT zodat de inwendige energie van eenideaal gas onafhankelijk is van zijn volume. Bovendien, omdat p = RT/V , is
dS = đqT
= dU + pdVT
= CVdTT
+RdVV
Door nu het rechterlid van deze vergelijking te integreren, bekomen we∫ SB
SAdS = CV
∫ TB
TA
dTT
+R
∫ VB
VA
dVV
zodatSB − SA = CV ln TB
TA+R ln VB
VA(3.3)
Dit houdt in dat đqrev/T een exacte differentiaal is en dat S een toestandsfunctie is voor een ideaalgas.
Voorbeeld 3.1Eén mol zuurstofgas wordt geëxpandeerd van 10 dm3 bij 298 K tot 20 dm3 bij 400 K. WanneerCp = 29,4 JK−1mol−1, bepaal dan de entropieverandering die geassocieerd wordt met een dergelijkeexpansie.
Voor een ideaal gas geldt dat
∆S = CV ln TBTA
+R ln VBVA

36 Hoofdstuk 3. Entropie, evenwicht en de Tweede Hoofdwet
en omdatCP = CV +R
geldt datCV = CP −R = 29,4− 8,31 = 21,1 JK−1 mol−1
Om die reden is
∆S =(
21,1 ln 400298 + 8,31 ln 20
10
)JK−1mol−1
= 12,0 JK−1mol−1
3.5 Entropie voor een expansie van een gas
Als een voorbeeld van de entropieverandering beschouwen we de isothermische expansie van eenideaal gas. We weten reeds dat ∆U = q + w. Daar voor een ideaal gas U onafhankelijk is van hetvolume, ∆U = 0 en q = −w, zal de warmte die uit de omgeving wordt gewonnen gelijk zijn aan dearbeid die door het systeem wordt uitgevoerd. Wanneer het gas expandeert en de druk verlaagt,wordt de externe druk continu aangepast zodat p = pex + dp waardoor de expansie kan reversibelkan worden uitgevoerd om de maximale arbeid te verrichten, en
−wrev =∫ B
ApdV
Voor n mol van een ideaal gas isp = nRT
V
zodat−wrev = nRT
∫ B
A
dVV
= nRT ln VBVA
en∆S = qrev
T= nR ln VB
VA(3.4)
Wanneer één mol van een ideaal gas expandeert in een volume van 0,01 m3 naar 0,10 m3, danis de entropieverandering ∆S = 8,3 ln 10 = 19,1 JK−1mol−1. Deze relatie zal correct zijn voorzowel reversibele als irreversibele veranderingen daar de entropie een toestandsfunctie is en ∆Sonafhankelijk van het pad tussen de toestanden A en B. Het warmteverlies door de omgeving isgelijk aan de warmte die wordt gewonnen door het ga en bijgevolg voor een reversibele expansie∆Sglobaal = 0 (daar ∆Sglobaal = qrev/T − qrev/T ).Wanneer we echter een irreversibele expansie van een ideaal gas in een vacuüm (dat geen arbeidverricht) beschouwen, dan is ∆Sglobaal niet langer gelijk aan nul. Voor het gas zelf geldt dat∆S = nR lnVB/VA, maar de veranderingen in de omgeving zullen verschillend zijn. Het gas verrichtgeen arbeid en omdat q = −w = 0, zal het systeem geen warmte absorberen uit de omgeving, zodatdit onveranderd blijft. Bijgevolg
∆Sglobaal = nR ln VBVA
+ 0T
= nR ln VBVA

3.6. Entropieveranderingen die de warmtestroom begeleiden 37
Daar VB > VA, zal de totale entropie van het systeem en zijn omgeving verhoogd zijn.
Wanneer de entropieverandering in een gas hetzelfde is in beide gevallen, moeten we de omgevingbestuderen vooraleer we kunnen beslissen of een verandering is opgetreden in een reversibele ofirreversibele manier. Dit beperkt de bruikbaarheid van entropie in het definiëren van evenwichts-voorwaarden.
3.6 Entropieveranderingen die de warmtestroom begeleiden
We beschouwen de warmtestroom van een reservoir dat wordt gehouden op een temperatuur Thnaar een reservoir bij Tk. Wanneer we ons inbeelden dat de warmtestroom reversibel is bij elketemperatuur, dan wordt de globale entropie gegeven door
dS = −đqTh
+ đqTk
ofdS = đq
(Th − TkThTk
)Dus wanneer een proces een observeerbaar proces is waarvoor geldt dat Th > Tl, dan is dS > 0 ende entropie van het systeem zal verhogen. Enkel wanneer Th = Tk zullen reversibele evenwichts-voorwaarden worden bekomen.
3.7 Entropie en evenwicht
Wanneer we een geïsoleerd systeem hebben dat geen energie kan uitwisselen met zijn omgeving,hebben we gezien dat entropie constant zal blijven bij evenwicht of zal verhogen wanneer eenobserveerbare verandering voorkomt. Observeerbare veranderingen zullen zich blijven voordoentotdat de entropie een maximale waarde bereikt op een tijdstip dat het systeem in evenwicht zalzijn. Het gas uit ons voorbeeld expandeert totdat het uniform is verdeeld en er zich geen verdereveranderingen meer voordoen. Dit kan worden uitgedrukt als volgt: in een systeem bij constanteenergie en volume (en dewelke geen arbeid kan verrichten) is de entropie maximaal bij evenwichtzodat (dS)U,V = 0.
Hoewel geen van deze reeks voorwaarden voorkomen in een serie van gewone chemische experimen-ten, stellen zez ons wel in staat om twee factoren te identificeren die chemische processen drijvennaar een evenwicht. Ten eerste is er de neiging om de energie te minimaliseren en ten tweede iser de neiging om de entropie te maximaliseren. In het algemeen moet er een compromis wordenbereikt tussen deze twee neigingen.
3.8 Een cosmologische uitwijding
Een geïsoleerd systeem zal neigen naar zijn maximale entropie, maar de meeste systemen zijn nietgeïsoleerd en zijn dan ook vrij om energie uit te wisselen met de omgeving. Wanneer we echter

38 Hoofdstuk 3. Entropie, evenwicht en de Tweede Hoofdwet
zowel het systeem alsook de omgeving tezamen beschouwen, hebben we in principe een geïsoleerdsysteem waarvoor geldt dat dS > 0 voor eender welk spontaan proces. Deze definitie van hetsysteem tezamen met de omgeving kan zelfs worden uitgebreid tot het ganse universum. Dit zalleiden tot een algemene stelling dat de energie van het universum is constant, maar de entropie isvoortdurend aan het stijgen. De implicaties dat wanneer de entropie van het universum finaal demaximale waarde zou bereiken zodat er geen observeerbare veranderingen meer zullen voorkommen,heeft veel voldoening gegeven aan pessimisten. Wanneer deze toestand voorkomt, zal alle energieen materiaal van het universum uniform verdeeld zijn en volledig onbeschikbaar om arbeid teverrichten.
3.9 Entropie als een functie van druk en temperatuur
3.9.1 Druk
We hebben gesteld dat voor n mol van een ideaal gas bij een constante temperatuur geldt dat
∆S = SB − SA = nR ln VBVA
Daar p ∝ 1/V voor een vaste hoeveelheid van een ideaal gas onder isothermische voorwaarden,geldt dat
∆S = SB − SA = −nR ln pBpA
Wanneer we S definiëren als de entropie van één mol van een ideaal gas bij een druk van éénatmosfeer bij een welbepaalde temperatuur, dan is
S = S −R ln p
p= S −R ln p
1 atm (3.5)
Deze uitdrukking wordt vaak geschreven als
S = S −R ln p
waarbij p dan de nnumerische waarde van de druk is die wordt uitgedrukt in atmosfeer. Dezepraktijk kan soms leiden tot verwarring over de dimensies van de termen in thermodynamischevergelijkingen en we zullen de notatie S = S −R ln (p/atm) aannemen.De uitdrukking vertelt ons dat wanneer de druk wordt verhoogd, de entropie van een gas zalverlagen.
3.9.2 Temperatuur
We weten reeds datdS = đqrev
T
en omdatCV =
(đqrevT
)V
en Cp =(đqrev
T
)p

3.10. Moleculaire basis van entropie 39
hebben we dS = (CV /T ) dT bij constant volume en dS = (Cp/T )dT bij een constante druk.Wanneer we nu de uitdrukking bij een constante druk integreren van een temperatuur TA naar eentemperatuur TB en waarbij we aannemen dat Cp een constante is, dan bekomen we dat
∫ B
AdS = SB − SA = Cp
∫ TB
TA
dTT
= Cp lnT |TBTA
zodat∆S = Cp ln TB
TA
Wanneer we nu stellen dat S0 de entropie is van een substantie bij een absolute nulpuntstempera-tuur, dan mogen we schrijven voor de entropie bij een temperatuur T , S (T ) dat
S (T ) = S0 +∫ T
0
CpT
dT (3.6)
Deze uitdrukking kan geïntegreerd worden als Cp gekend is als een functie van de temperatuur.Soms wordt de uitdrukking ook geschreven als
S (T ) = S0 +∫ T
0Cpd(lnT )
Voorbeeld 3.2De warmtecapaciteit van vloeibare kwik bij een constante temperatuur is bijna constant bij28 JK−1mol−1 tussen het vriespunt bij 234 K en 298 K. Schat de entropie van vloeibaar kwikbij het vriespunt. De entropie van vloeibare kwik bij 298 K is 77,4 JK−1mol−1.
Wanneer Cp kan beschouwd worden als een constante, zal de verandering van entropie met zijntemperatuur gegeven worden door
∆S = Cp ln TBTA
Invullen van de data geeft
∆S = S298K − S234K = 28 ln 298234 = 6,8 JK−1mol−1
zodatS234K = S298K − 6,8 = 77,4− 6,8 = 70,6 JK−1mol−1
3.10 Moleculaire basis van entropie
De entropie is gerelateerd aan de uniformiteit van systemen. Wanneer gradiënten in de temperatuurof concentratie worden geëlimineerd, zal de entropie verhogen. We kunnen dit idee kwantitatiefuitwerken wanneer we de expansie van een ideaal gas beschouwen. De disscussie houdt een over-weging in van de moleculaire natuur van de materie. Het is niet essentieel in de ontwikkeling vande klassieke thermodynamica, maar deze discussie biedt wel waardevolle inzichten.

40 Hoofdstuk 3. Entropie, evenwicht en de Tweede Hoofdwet
Toestand A Toestand B
Figuur 3.4: De expansie van een gas. In Toestand A bezet het gas slechts de helft van het reservoir. Intoestand B is het gas uniform verdeeld over het ganse reservoir.
Beschouw M gasmoleculen die zich in de helft van een container bevinden zoals geïllustreerd in Fi-guur 3.4 en vervolgens toegelaten worden om het ganse volume te bezetten. De waarschijnlijkheiddat Toestand A toevallig zou kunnen voorkomen is (1/2)M dat hetzelfde is als de kans dat M ob-jecten zich allemaal in één van de twee containers zou bevinden tussen dewelke ze lukraak zijnverdeeld. We kunnen de relatieve waarschijnlijkheid van Toestand A (PA) in relatie tot ToestandB (PB) schrijven als
PAPB
=(1
2
)MWanneer we in plaats van VA/VB = 1/2 te maken, we willekeurige waarden zouden selecteren, dankunnen we aantonen dat
PAPB
=(VAVB
)Mzodat
ln PBPA
= M ln(VBVA
)In het overgaan van Toestand A naar Toestand B zijn we van een toestand met lage waarschijn-lijkheid naar een toestand met hoge waarschijnlijkheid overgegaan. We weten bovendien dat
∆S = SB − SA = R ln VBVA
voor één mol gas. Wanneer M numerisch gelijk is aan de constante van Avogadro, NA, dan is deentropieverandering per mol gelijk aan
SB − SA = R
NA(lnPB − lnPA) = R
NAln PBPA
De entropie van een systeem in eender welke toestand is evenredig aan lnP waarbij P de ‘waar-schijnlijkheid’ is van het systeem. We kunnen, Boltzmann3 volgend, schrijven dat
S = kB lnW (3.7)3Ludwig Boltzmann (1844–1906) gaf in 1877 de nu beroemde relatie tussen de entropie en de waarschijnlijkheid [5].
Boltzmann had ook andere vele belangrijke bijdragen aan onderwerpen in de wiskunde, fysica, chemie en filosofie.Naar het einde van zijn leven werd Boltzmann echter meer en meer moedeloos. Het echte bestaan van atomen werdzwaar in vraag gesteld op het einde van de negentiende eeuw en dit raakte aan de wortels van Boltzmanns gehelelevenswerk. Na een aantal mislukte zelfmoordpogingen, hing hij zich in 1906 op tijdens een vakantie in Triëste, eenstad in Italië.

3.11. Statistische basis van de Tweede Hoofdwet 41
met kB de constante van Boltzmann (R/NA) en W het aantal ‘microtoestanden’ van het systeem.Dit laatste is een moeilijk concept. Het is het aantal manieren waarop de toestand kan wordenopgebouwd door de posities te specifiëren en de snelheden van de atomen waaruit het bestaat.Richard Feynman drukte het als volgt uit: “W is het ‘aantal manieren waarop het binnenstevan het systeem kan worden opgebouwd wanneer de buitenkant hetzelfde blijft". Voor systemendie ongeveer 1023 moleculen bezitten, kan W zeer groot zijn, in de grootte-orde van 1023. Eenbelangrijke tak van de fysicochemie bestaat uit statistische mechanica die zich bezighoudt metde berekening van W en bijgevolg de evaluatie van de thermodynamischhe eigenschappen vanmoleculaire systemen zonder toevlucht te zoeken tot het thermodynamisch experiment.
Voor een eenvoudig mechanisch systeem zal het entropieverschil tussen één toestand en een anderetoestand gewoonlijk verwaarloosbaar zijn. Stel bijvoorbeeld een object dat kan geworpen wordenvanaf een zekere afstand en willekeurig kan verdeeld worden tussen twee dozen waarvan de ene doostweemaal de grootte is van de ander. De waarschijnlijkheid dat het object landt in de grotere doos(B) zal tweemaal zo groot zijn dan dat het object zou landen in de kleinere doos (A). We kunnenschrijven dat
∆S = SB − SA = k ln 2 = 1,38× 10−23 J K−1 · ln 2 ≈ 1× 10−24 J K−1
Dit is een zeer klein entropieverschil dat kan worden verwaarloosd zonder dat er enig significanteonnauwkeurigheid wordt geïntroduceerd wanneer berekeningen worden uitgevoerd op een dergelijksysteem. Dit is bovendien de reden waarom enkel de energie moet worden beschouwd wanneer mende positie van het evenwicht gaat bepalen in mechanische systemen.Wanneer we echter twee toestanden beschouwen voor een mol gas, waarbij in de ene toestand hetgas wordt gehouden in een volume dat tweemaal zo groot is dan het andere volume, dan bekomenwe
∆S = SB − SA = k ln 2NA = R ln 2 = 8,314 J K−1 · ln 2 = 5,8 J K−1
dat een substantiële en ver van verwaarloosbare bijdrage is.
3.11 Statistische basis van de Tweede Hoofdwet
Wanneer we de entropie beschouwen vanuit moleculair oogpunt, realiseren we dat het in principeniet onmogelijk zou zijn dat alle moleculen in een container zich tegelijkertijd in één helft zoudenbevinden, maar dat dit weliswaar zeer onwaarschijnlijk is. Het is dan ook zeer onwaarschijnlijkdat alle luchtmoleculen in een kamer waarin iemand zich bevindt, zich zouden concentreren in eenhoek zodat er in de rest van de kamer geen lucht meer aanwezig is om te kunnen ademen, maarhet is strict genomen niet onmogelijk. Om in te zien hoe onwaarschijnlijk, beschouwen we een molgas dat zich bevindt in twee volumes waarvan één de helft van de grootte is van de andere. Voorhet proces B→ A (Figuur 3.4) geldt dat
∆S = −5,8 J K−1 = R
Lln pApB

42 Hoofdstuk 3. Entropie, evenwicht en de Tweede Hoofdwet
zodatpApB
= exp(−5,8 · 6× 1023
8,3
)≈ exp(−1023)
Het is dus zeer onwaarschijnlijk dat een mol gas zich uit zichzelf in één helft van de container zoubevinden. Het is zelfs nog meer onwaarschijnlijk dat een dergelijke storende gebeurtenis zich zouvoordoen in een kamer daar het vele hoeveelheden gassen zal bevatten.
3.12 De grootte van entropieveranderingen
De entropie van een chemisch systeem wordt grotendeels bepaald door de vrijheid die de moleculenin een systeem bezitten. In vaste stoffen waar de moleculen of de atomen strak aan elkaar gebondenzijn, zal de entropie laag zijn. In gassen waar de moleculen vrij zijn om te bewegen in een grootvolume, zal de entropie hoog zijn. Vloeistoffen bezitten eigenschappen die tussen deze van vastestoffen en gassen liggen.
Wanneer een vloestof verdampt, zullen de moleculen van een toestand van bescheiden vrijheid naareen toestand gaan met hoge vrijheid. De entropie die gepaard gaat met verdamping, zal bijgevolgpositief zijn en wordt uitgedrukt als
∆Svap = qrevT
= ∆HvapT
(3.8)
Voor benzeen is ∆Hvap = 30,7 kJmol−1 en het gewone kookpunt is 353,3 K zodat ∆Svap =87,0 J K−1mol−1. Voor de meeste niet-polaire vloeistoffen zal de verdampingsentropie bij hetgewone kookpunt ongeveer gelijk zijn aan 90 J K−1mol−1. Deze laatste veralgemening staat gekendals de regel van Trouton [13].
De entropieverandering geassocieerd met een chemische reactie hangt af van de natuur van dereagentia en de producten. De reactie
CaCO3(s) −−−− CaO(s) + CO2(g)
heeft een hoge en positieve entropieverandering van 160 J K−1mol−1 daar de vaste stoffen eenlage entropie bezitten, maar het koolstofdioxide als gas een hoge entropie. Zelfs in een reactiewaarin twee gassen betrokken zijn, kunnen er significante entropieveranderingen voorkomen. Voorde evenwichtsreactie
N2O4(g) −−−− 2NO2(g)
is ∆S = 177 J K−1mol−1 bij een gasdruk van 1 atm. De entropie van NO2 is groter wanneerhet aanwezig is als onafhankelijke moleculen dan wanneer het gebonden is om N2O4 moleculen tevormen. De positieve entropieverandering verloopt ten gunste van de dissociatie van het dimeer datongeveer voor 20 procent is gedissocieerd bij kamertemperatuur ondanks de relatief hoge bindings-energie dat zou veronderstellen dat er relatief weinig dissociatie zou verwacht worden bij dergelijketemperaturen in afwezigheid van entropie-overwegingen.

3.12. De grootte van entropieveranderingen 43
Uitgewerkte oefeningen
1. Bereken de entropieverandering wanneer 0,011 m3 ideaal gas bij 273 K en een druk van 1 atmwordt samengedrukt tot een druk van 10 atm.
OplossingWe stellen dat de temperatuur constant blijft en berekenen eerst het aantal mol ideaal gasmet behulp van de ideale gaswet waarbij 1 atm = 105 N m−2.
n = pAVART
= 105 N m−2 · 0,011 m3
8,314 J mol−1K−1 · 273 K= 0,485 mol
We berekenen vervolgens het volume tot hetwelke het gas wordt samengedrukt uit de idealegaswet.
VB = nRT
pB= 0,485 mol · 8,314 J mol−1K−1 · 273 K
10× 105 N m−2 = 0,0011 m3
De entropieverandering kan nu berekend worden via
∆S = nR ln VBVA
= 0,485 mol · 8,314 J molK−1 · ln 0,0011 m3
0,011 m3 = −9,3 J K−1
2. De warmtecapaciteit van gasvormig argon bij een constante druk is 20, 8 J mol−1K−1. Schatde entropieverandering wanneer één mol argon wordt opgewarmd van 300 K tot 1200 K bijeen druk van 1 atm.
OplossingInvullen van de gegevens in de vergelijking voor de entropieverandering bij een constante druklevert op dat
∆S = Cp ln TBTA
= 20,8 J mol−1K−1 · ln 1200 K300 K = 28,8 J mol−1K−1
Voor één mol argon is de entropieverandering dan 28,8 J K−1
3. Schat de entropieverandering wanneer één mol water wordt verdampt bij 373 K. De enthal-pieverandering bij verdamping is 40,7 kJmol−1 bij deze temperatuur.
OplossingWe maken hiervoor gebruik van de relatie
∆Svap = ∆HvapT
zodat, na invullen van de gegevens, we bekomen dat
∆Svap = 40700 Jmol−1
373 K = 109 Jmol−1K−1
Voor één mol water is ∆Svap = 109 JK−1


Hoofdstuk 4
Evenwicht in chemische systemen
4.1 De vrije energie
Voor systemen bij een constante energie kan de positie van het evenwicht worden gedefinieerd doorde voorwaarde van de maximale entropie. Daar een systeem en zijn omgeving tezamen moetenbeschouwd worden als een nieuw systeem waarbij de energie constant is, zal de positie die zal leidentot een maximale entropie voor het systeem en de omgeving de evenwichtspositie betekenen. Het isechter meer geschikt omm een definitie te hebben over de positie van het evenwicht die kan wordenuitgedrukt in termen van de eigenschappen van het systeem alleen en dewelke geen wetenschap vergtvan de veranderingen in de omgeving. Om een dergelijke voorwaarde te hebben, keren we terugnaar het concept van de maximale arbeid. We definieerden de positie van het evenwicht in termenvan de capaciteit van het systeem om arbeid te verrichten. Wanneer gedurende een veranderinghet systeem de maximale arbeid zal verrichten zodat de verandering reversibel is en het systeem inelk stadium in evenwicht zal zijn. We introduceren de thermodynamische functies die gerelateerdzijn aan de maximale hoeveelheid arbeid die kan worden bekomen uit een systeem bij constantetemperatuur. Deze bieden een geschikte definitie voor de positie van het evenwicht in chemischen fysische systemen. Voor reversibele voorwaarden, zoals dU = đqrev + đwrev en dS = đqrev/Thebben we
dU = TdS + đwrev
endwrev = dU − TdS
Wanneer we een toestandsfunctie, de Helmholtz vrije energie A definiëren zodat
A = U − TS (4.1)
hebben we bij een constante temperatuur dat
dA = dU − TdS
enđwrev = dU − TdS = dA
45

46 Hoofdstuk 4. Evenwicht in chemische systemen
Aan deze voorwaarde moet worden voldaan door een reversibel proces en bijgevolg is dw = dAeveneens een voorwaarde voor het systeem om in evenwicht te zijn. Wanneer gedurende een rever-sibele verandering het systeem arbeid verricht, zal đwrev negatief zijn. Daar dA ook negatief zalzijn, zal A verlagen. A is de equivalente functie in het moleculaire systeem bij constante T en V ,tot de energie U in een mechanisch systeem. Het is de maat van de maximale hoeveelheid arbeidhet systeem kan doen op zijn omgeving.
Voor een spontaan proces, kan het systeem arbeid verrichten, maar de arbeid zal lager zijn voor deequivalente reversibele verandering en bijgevolg zal đw minder negatief zijn dan đwrev.
4.2 Gibbs vrije energie
Omdat we chemici zijn, zijn we vaker geïnteresseerd in systemen bij constante druk en temperatuurdan in systemen bij constant volume. Onder voorwaarden van een constante druk mogen weschrijven dat
đwrev = −pdV + đwtoegevoegd
waarbij đwtoegevoegd de arbeid is die verschilt van de pV -arbeid die wordt uitgevoerd op het systeem.
Elektrische arbeid die wordt uitgevoerd op een oplossing die wordt geëlektrolyseerd kan een voor-beeld zijn van deze toegevoegde arbeid. Bij evenwicht mogen we schrijven dat
đwrev = dU − đqrev
enđqrev = TdS
Bijgevolg isđwtoegevoegd − pdV = dU − TdS
ofđwtoegevoegd = dU + pdV − TdS
Omdat de combinatie van de variabelen U+pV−TS veelvuldig voorkomen, werd de toestandsfunctievoor de Gibbs vrije energie met symbool G gedefinieerd zodat
G = U + pV − TS = H − TS (4.2)
en daar A = U − TS, kunnen we eveneens schrijven dat
G = U + pV − TS = A+ pV (4.3)
Bij constante p en T geldt datdG = dU + pdV − TdS

4.2. Gibbs vrije energie 47
Daar đwtoegevoegd = dU + pdV − TdS voor een reversibele verandering, hebben we dat
đwtoegevoegd = dG
We hebben bijgevolg een voorwaarde voor evenwicht gevonden in een systeem bij constante tem-peratuur en druk. Voor een spontaan proces zal đwtoegevoegd minder negatief zijn voor de overeen-komstige reversibele veranderinbg en
G is een toestandsfunctie en bijgevolg heeft ∆G een definitieve waarde voor eender welke verande-ring.
In de vergelijking∆G = ∆H − T∆S
hebben we gevonden dat de balans bij constante druk en temperatuur tussen de neigingen vanhet systeem om tot een maximale entropie te komen en om de energie te minimaliseren (of bijconstante temperatuur meer strict de enthalpie). Bij hogere temperaturen zal de bijdrage vande entropieverandering tot de vrij energie verandering (−T∆S) relatief meer belangrijk worden.Beschouwen we de reactie
N2O4 −−−− 2NO2
waarbij ∆H positief omdat er een energie nodig is om het dimeer (N2O4) samen te houden en ∆Sis positief omdat de gescheiden monomeren meer vrijheid hebben om te bewegen dan wanneer hetgebonden is in dimeren. Bij lage temperaturen is ∆G positief en er zal slechts weinig dissociatieoptreden. Bij hogere temperaturen zal de meer gunstige entropieterm (−T∆S < 0) domineren eneen verhoogde dissociatie zal worden geobserveerd.
Nu we gevonden hebben hoe de positie van het evenwicht kan worden gedefinieerd bij constantetemperatuur en druk, zullen we ons nu beperken tot deze voorwaarden. We zullen de eigenschappenvan de vrije Gibbs energie G onderzoeken die ons het criterium van evenwicht zal bieden. Wedifferentiëren
G = U + pV − TS
zodatdG = dU + pdV + V dp− SdT − TdS
Voor een systeem dat enkel pV -arbeid doet, geldt dat
dU = đqrev + đwrev
zodatdU = TdS − pdV
Invullen van deze laatste uitdrukking in de vergelijking dG, bekomen we
dG = V dP − SdT (4.4)

48 Hoofdstuk 4. Evenwicht in chemische systemen
Deze vergelijking is één van de meest belangrijke thermodynamische vergelijkingen voor chemicidaar het ons vertelt hoe de vrije energie, en bijgevolg de evenwichtspositie, varieert met de druk ende temperatuur.
4.2.1 De drukafhankelijkheid van de Gibbs vrije energie
Bij een constante temperatuur dT = 0 is
dG = V dP en(∂G
∂P
)T
= V (4.5)
Voor n mol van een ideaal gas pV = nRT is
dG = nRTdpp
Voor een drukverandering van pA naar pB is
∆G = GB −GA = nRT
∫ pB
pA
dpp
= nRT ln p|pBpA
∆G = nRT ln pBpA
(4.6)
Meestal wordt de vrije energie van een gas gerelateerd aan de standaard vrije energie G− diegedefinieerd is als de vrije energie van één mol van het gas bij een druk van één atmosfeer1.Bijgevolg
G = G− +RT ln p
p−
Daar p− de waarde heeft van één atmosfeer is
G = G− +RT ln p/1 atm
We schrijvenG = G− +RT ln (p/atm) (4.7)
Soms wordt deze vergelijking uitgedrukt als
G = G− +RT ln p
waarbij p nu een dimensieloze verhouding representeert. Dit kan tot verwarring leiden als vergelij-kingen van dit type worden gebruikt!
Voorbeeld 4.1De standaard vrije energie van stikstofgas is gedefinieerd als nul bij 298 K en een druk van 1 at-mosfeer. Bereken de waarde bij 10 atm en 0,20 atm bij dezelfde temperatuur.
Voor een ideaal gas1We merken hier op dat volgens de IUPAC zowel het symbool als − worden gebruikt om de standaard aan te
geven [6]. Beide symbolen zijn in gelijke mate aanvaardbaar.

4.2. Gibbs vrije energie 49
a) Bij 10 atm
G = G− +RT ln (p/atm)= 0 + 8,31× 298× ln 10= 5702 Jmol−1
b) Bij 0,20 atm
G = 0 + 8,31× 298× ln 0,2= −3986 Jmol−1
Het is gebruikelijk om een speciaal symbool µ in te voeren voor de Gibbs vrije energie per mol. Wedefiniëren
µ = G
n
Voor de molaire Gibbs energie van een ideaal gas hebben we bijgevolg
µ = µ− +RT ln p
Ook hier zal het symbool p een zuiver getal representeren en zal dit getal moeten worden verme-nigvuldigd met 1 atm om de waarde ervan te kennen in atmosferische druk. De logaritmische termin bovenstaande uitdrukking is behoorlijk groot in de meeste omstandigheden en kan niet wordenverwaarloosd. Uit deze vergelijking zou het duidelijk moeten zijn dat bij een specifieke tempera-tuur, de druk de Gibbs vrij energie van een ideaal gas beschrijft. Hoe hoger de druk des te hogerezal de Gibbs vrije energie zijn zoals wordt geïllustreerd in Figuur 4.1
4.2.2 De temperatuursafhankelijkheid van de Gibbs vrije energie
We hernemen de basisvergelijking dG = V dP − SdT . Bij een constante druk dp = 0 geldt dat
dG = −SdT en(∂G
∂T
)p
= −S (4.8)
Uit de definitie G = H − TS volgt echter dat −S = (G−H) /T zodat Vergelijking 4.8 kan wordengeschreven als (
∂G
∂T
)p
= G−HT
(4.9)
Het kan belangrijk kan zijn om te weten hoe de functie G/T afhankelijk is van de temperatuur.Door de regels van afleiden (kettingregel) te volgen, bekomen we dat∂ (GT
)∂T
p
= 1T
(∂G
∂T
)p− G
T 2
Gebruik makende van Vergelijking 4.8 bekomen we∂ (GT)
∂T
p
= −ST− G
T 2 = −TS +G
T 2
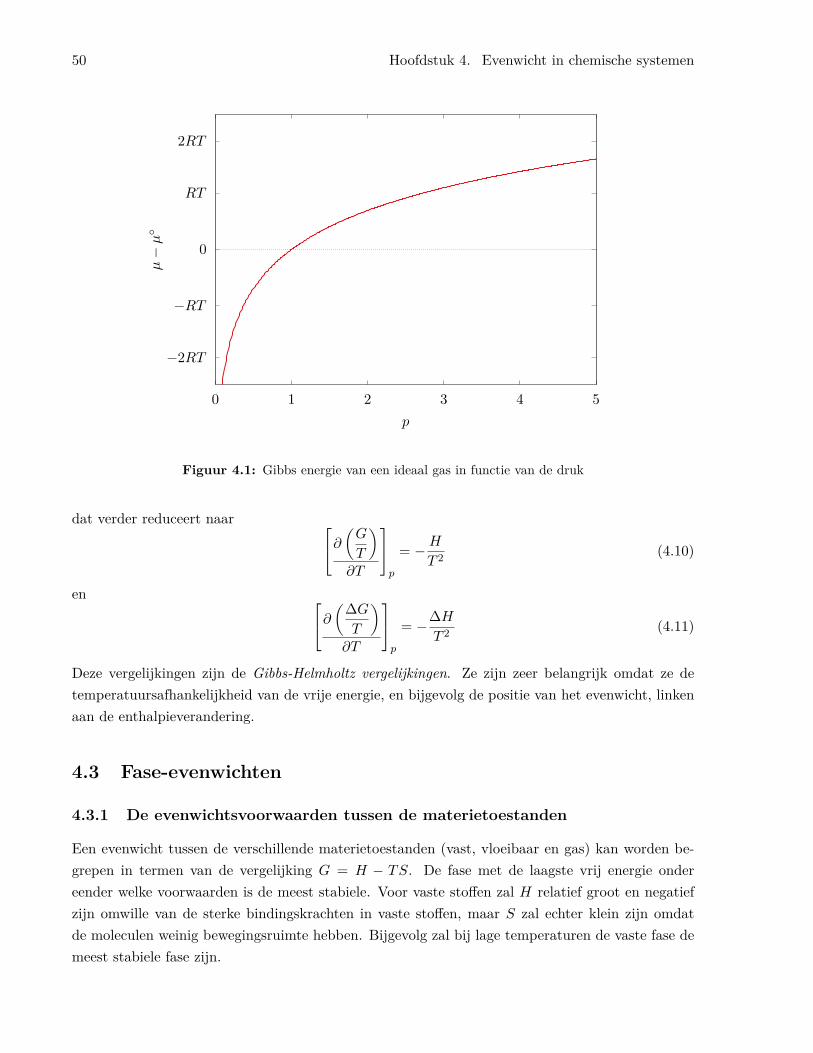
50 Hoofdstuk 4. Evenwicht in chemische systemen
−2RT
−RT
0
RT
2RT
0 1 2 3 4 5
µ−µ
p
Figuur 4.1: Gibbs energie van een ideaal gas in functie van de druk
dat verder reduceert naar ∂ (GT)
∂T
p
= −HT 2 (4.10)
en ∂ (∆GT
)∂T
p
= −∆HT 2 (4.11)
Deze vergelijkingen zijn de Gibbs-Helmholtz vergelijkingen. Ze zijn zeer belangrijk omdat ze detemperatuursafhankelijkheid van de vrije energie, en bijgevolg de positie van het evenwicht, linkenaan de enthalpieverandering.
4.3 Fase-evenwichten
4.3.1 De evenwichtsvoorwaarden tussen de materietoestanden
Een evenwicht tussen de verschillende materietoestanden (vast, vloeibaar en gas) kan worden be-grepen in termen van de vergelijking G = H − TS. De fase met de laagste vrij energie ondereender welke voorwaarden is de meest stabiele. Voor vaste stoffen zal H relatief groot en negatiefzijn omwille van de sterke bindingskrachten in vaste stoffen, maar S zal echter klein zijn omdatde moleculen weinig bewegingsruimte hebben. Bijgevolg zal bij lage temperaturen de vaste fase demeest stabiele fase zijn.

4.3. Fase-evenwichten 51
Bij gassen is H dicht bij nul omdat er geen sterke interacties zijn tussen de moleculen, maar S zaldaarentegen groot zijn omdat de moleculen een grotere hoeveelheid ruimte hebben om in te bewegen.Gassen zijn bijgevolg de meest stabiele fase bij hogere temperaturen. Er is een tussenliggende ruimtewaar vloeistoffen stabiel zijn. Vloeistoffen bezitten een minder negatieve H, maar een grotere Sdan vaste stoffen.
Een simulatie van deze drie materietoestanden waarbij de Gibbs vrij energie wordt uitgezet in
Tsmelt Tdamp
G
T
vast
vloeibaar
gas
Figuur 4.2: Simulatie voor de vrije Gibbs energie als een functie van de temperatuur voor een zuiveresubstantie.
functie van de temperatuur voor een zuivere stof, is gevisualiseerd in Figuur 4.2. De hellingen vande rechten worden bepaald door de entropie van de fasen omdat
(∂G
∂T
)p
= −S
De fase met de hoogste entropie, de gasfase, heeft de meest negatieve helling en de vrije Gibbsenergie is de laagste bij de hoogste temperaturen. Waar de rechten die de twee fasen representeren,snijden, zullen de vrije Gibbs energieën aan elkaar gelijk zijn. Wanneer ∆G = 0, en ∆G =∆H − T∆S, bekomen we voor de overgang tussen twee fasen in evenwicht dat ∆H = T∆S zodat
∆Ssmelt = ∆HsmeltTsmelt
en ∆Sdamp = ∆HdampTdamp
(4.12)

52 Hoofdstuk 4. Evenwicht in chemische systemen
4.3.2 De Clapeyron vergelijking
De thermodynamische beschouwingen uit de vorige sectie kunnen worden toegepast op een meerkwantitatieve wijze om een belangrijke relatie te bekomen. We beschouwen hiervoor twee fasen,bijvoorbeeld α voor de vloeibare fase en β voor de dampfase, die in evenwicht zijn bij een tem-peratuur T en een druk p. Wanneer we de voorwaarden veranderen naar T + dT en p + dp, danbekomen we (dG = V dp− SdT ) voor de vloeibare fase
dG(α) = V (α)dp− S(α)dT
en voor de gasfasedG(β) = V (β)dp− S(β)dT
Wanneer onder deze nieuwe voorwaarden de fasen nog steeds in evenwicht zijn, dan geldt dat
dG(α) = dG(β)
zodatV (α)dp− S(α)dT = V (β)dp− S(β)dT
Door te herschikken, vinden we
[S(β)− S(α)]dT = [V (β)− V (α)]dp
Wanneer de transformatie wordt geschreven als α→ β (van ‘vloeibaar’ naar ‘gas’), dan is ∆Sdamp =S(β)− S(α) en ∆Vdamp = V (β)− V (α) zodat
dTdp = ∆Vdamp
∆Sdampof dp
dT = ∆Sdamp∆Vdamp
(4.13)
Daar de twee fasen in evenwicht zijn, geldt dat
∆Gdamp = ∆Hdamp − T∆Sdamp = 0
zodat∆Sdamp = ∆Hdamp
T
en bijgevolgdpdT = ∆Hdamp
T∆Vdamp
waarbij T het kookpunt is bij de beschouwde druk. De vergelijking( dpdT
)evenwicht
= ∆HdampT∆Vdamp
(4.14)
staat gekend als de Clapeyron vergelijking2. Het is exact en wordt toegepast op een evenwichttussen twee fasen dat zowel het smeltproces of de verdamping zal voorstellen.
2Benoît Paul Émile Clapeyron (1799–1864) was een Frans ingenieur [14].

4.3. Fase-evenwichten 53
Voorbeeld 4.2Bij 273,16 K is de enthalpieverandering voor het smelten van water gelijk aan 6,0 kJ mol−1 en deovereenkomstige volumeverandering is −1,6× 10−6 m3 mol−1. Schat de temperatuur wanneer hetijs zal smelten bij een druk van 1000 atm (1 atm = 105 Pa).
We kunnen de Clapeyron vergelijking schrijven als
∆T∆p = ∆Vsmelt
∆Ssmelt= T∆Vsmelt
∆Hsmelt
⇒ ∆T = ∆p · T∆Vsmelt∆Hsmelt
= 103 × 105 · 273 · (−1,6× 10−6)6,0× 103 K
= −7,3 K
We kunnen bijgevolg schatten dat ijs zal smelten bij (273,16− 7,3) K = 265,9 K.
4.3.3 De Clausius-Clapeyron vergelijking
Wanneer het wordt toegepast op verdampen kan de Clapeyron vergelijking gewijzigd worden omeen andere belangrijke relatie te bekomen. We starten met
dpdT = ∆Hdamp
T∆Vdamp
waarbij∆Vdamp = V (g)− V (l)
Nu is bij kamertemperatuur en een druk van 1 atm V (g) ≈ 24000 cm3 en V (l) ≈ 100 cm3 voor éénmol substantie zodat V (g) V (l). Bijgevolg kan ∆Vdamp worden vervangen door V (g). Wanneerhet gas de ideale gaswet volgt, dan hebben we voor één mol dat
V (g) = RT
p
zodatdpdT = ∆Hdamp
T· 1V (g) = ∆Hdamp
T· p
RT= ∆Hdamp
RT 2 · p
Bijgevolg is3d ln pdT = ∆Hdamp
RT 2
Als ∆Hdamp onafhankelijk is van de temperatuur, dan kunnen we bovendien schrijven dat∫d ln p = ∆Hdamp
R
∫ 1T 2dT
ln p = −∆HdampRT
+ constante
3Herinner je dat d ln xdx = 1
x.

54 Hoofdstuk 4. Evenwicht in chemische systemen
Deze vergelijkingen, die de temperatuursafhankelijkheid van de dampdruk van een vloeistof linkenaan ∆Hdamp of de enthalpieverandering per mol op verdamping, worden de Clausius-Clapeyronvergelijkingen genoemd. In tegenstelling tot de Clapeyron vergelijking, zijn de Clausius-Clapeyronvergelijkingen niet exact daar er een aantal benaderingen werden geïntroduceerd in de afleiding.Desalniettemin zijn de Clausius-Clapeyron vergelijkingen bijzonder nuttig!
4.4 De dampdruk van vloeistoffen
We beschouwen het verdampen van een vloeistof vanuit het standpunt van de verandering van devrije energie op verdamping. De vrije energie van één mol van een ideale damp wordt gegeven door
G(g) = G− (g) +RT ln (p/atm)
waarbij p de dampdruk is van een vloeistof, G− (g) de vrije energie van één mol van een damp bijeen druk van één atmosfeer. De vrije energie van één mol van een vloeistof zal eenvoudig gelijkzijn aan G− (l) daar we kunnen aannemen dat de vrije energie van een gecondenseerde fase virtueelonafhankelijk is van de druk. De verandering in vrije energie die voorkomt wanneer één mol vaneen vloeistof wordt verdampt om één mol damp te vormen bij zijn evenwichtsdruk p is
∆G(g) = G(g)−G(l) = G− (g)−G− (l) +RT ln (p/atm)
Wanneer de vloeistof en zijn damp in evenwicht zijn, dan is er geen verandering in vrije energiewanneer een hoeveelheid vloeistof wordt verdampt. Bijgevolg is G(g)−G(l) en
∆G−damp = −RT ln (p/atm) (4.15)
Deze vergelijking vertelt ons dat de dampspanning van een vloeistof wordt bepaald door de veran-dering van de vrij energie wanneer één mol van een vloeistof wordt verdampt om één mol dampte produceren bij een druk van één atmosfeer. Bij een normaal kookpunt wanneer de vloeistof inevenwicht is met zijn damp bij één atmosfeer druk, p = 1 en G−damp = 0.
Om de temperatuursvariatie van de dampdruk te vinden, gebruiken we de Gibbs-Helmholtz verge-lijkingen ∂ (∆G
T
)∂T
p
= −∆HT 2
d ln pdT = − 1
R
∂(
∆G−vapT
)∂T
=∆H−vapRT 2
d ln pdT =
∆H−vapRT 2 (4.16)
We hebben bijgevolg opnieuw de Clausius-Clapeyron vergelijking afgeleid.
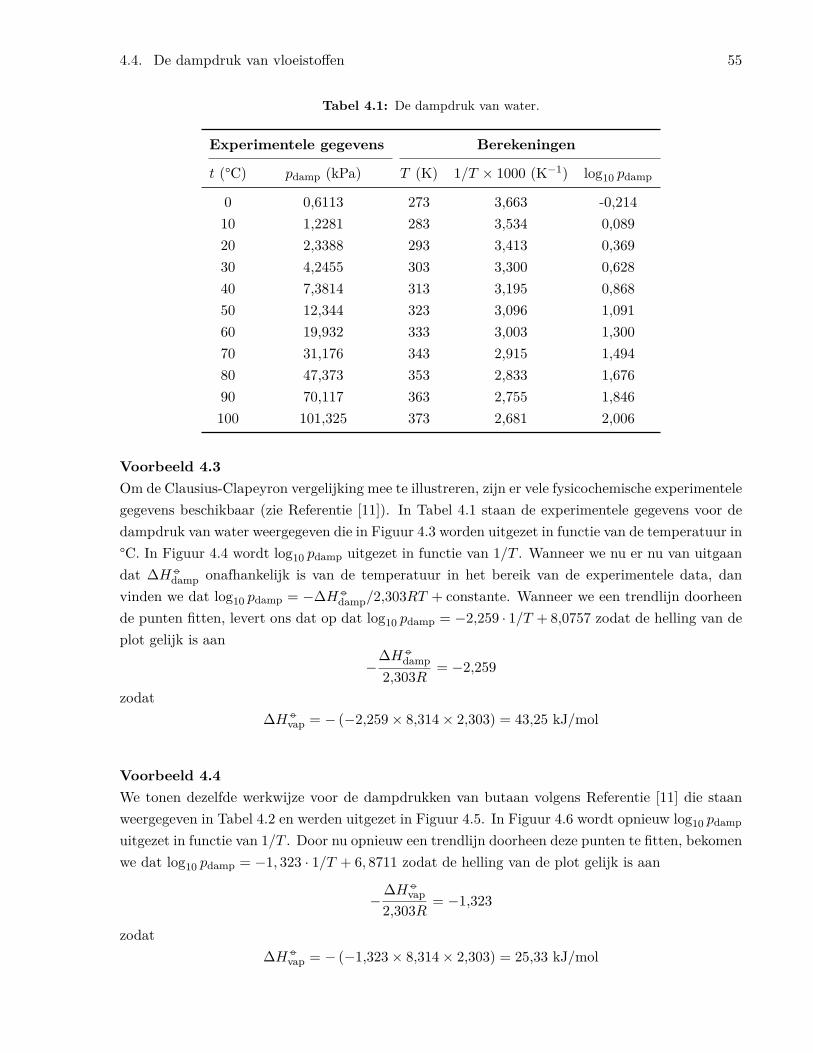
4.4. De dampdruk van vloeistoffen 55
Tabel 4.1: De dampdruk van water.
Experimentele gegevens Berekeningen
t (°C) pdamp (kPa) T (K) 1/T × 1000 (K−1) log10 pdamp
0 0,6113 273 3,663 -0,21410 1,2281 283 3,534 0,08920 2,3388 293 3,413 0,36930 4,2455 303 3,300 0,62840 7,3814 313 3,195 0,86850 12,344 323 3,096 1,09160 19,932 333 3,003 1,30070 31,176 343 2,915 1,49480 47,373 353 2,833 1,67690 70,117 363 2,755 1,846100 101,325 373 2,681 2,006
Voorbeeld 4.3Om de Clausius-Clapeyron vergelijking mee te illustreren, zijn er vele fysicochemische experimentelegegevens beschikbaar (zie Referentie [11]). In Tabel 4.1 staan de experimentele gegevens voor dedampdruk van water weergegeven die in Figuur 4.3 worden uitgezet in functie van de temperatuur in°C. In Figuur 4.4 wordt log10 pdamp uitgezet in functie van 1/T . Wanneer we nu er nu van uitgaandat ∆H−damp onafhankelijk is van de temperatuur in het bereik van de experimentele data, danvinden we dat log10 pdamp = −∆H−damp/2,303RT + constante. Wanneer we een trendlijn doorheende punten fitten, levert ons dat op dat log10 pdamp = −2,259 · 1/T + 8,0757 zodat de helling van deplot gelijk is aan
−∆H−damp2,303R = −2,259
zodat∆H−vap = − (−2,259× 8,314× 2,303) = 43,25 kJ/mol
Voorbeeld 4.4We tonen dezelfde werkwijze voor de dampdrukken van butaan volgens Referentie [11] die staanweergegeven in Tabel 4.2 en werden uitgezet in Figuur 4.5. In Figuur 4.6 wordt opnieuw log10 pdamp
uitgezet in functie van 1/T . Door nu opnieuw een trendlijn doorheen deze punten te fitten, bekomenwe dat log10 pdamp = −1, 323 · 1/T + 6, 8711 zodat de helling van de plot gelijk is aan
−∆H−vap2,303R = −1,323
zodat∆H−vap = − (−1,323× 8,314× 2,303) = 25,33 kJ/mol

56 Hoofdstuk 4. Evenwicht in chemische systemen
0
20
40
60
80
100
0 20 40 60 80 100
pda
mp(kPa
)
t (C)
Figuur 4.3: Het uitzetten van de experimentele gegevens voor de dampdruk pdamp van water in functievan de temperatuur (t).
−0,5
0
0,5
1
1,5
2
2,5
2,6 2,8 3 3,2 3,4 3,6 3,8 4
log 1
0p
dam
p
1000/T (K−1)
helling = −∆H−vap2,303R
Figuur 4.4: Het uitzetten van het tiendelig logaritme van de dampdruk van water in functie van hetreciproke van de absolute temperatuur (T ).
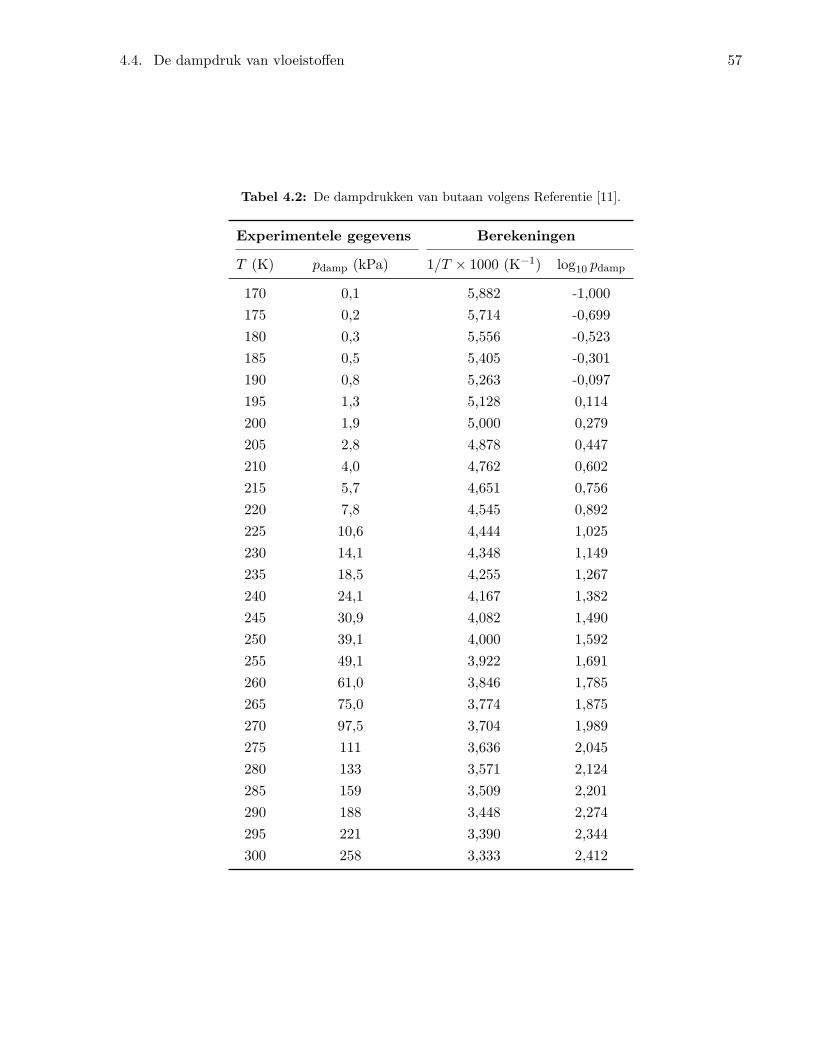
4.4. De dampdruk van vloeistoffen 57
Tabel 4.2: De dampdrukken van butaan volgens Referentie [11].
Experimentele gegevens Berekeningen
T (K) pdamp (kPa) 1/T × 1000 (K−1) log10 pdamp
170 0,1 5,882 -1,000175 0,2 5,714 -0,699180 0,3 5,556 -0,523185 0,5 5,405 -0,301190 0,8 5,263 -0,097195 1,3 5,128 0,114200 1,9 5,000 0,279205 2,8 4,878 0,447210 4,0 4,762 0,602215 5,7 4,651 0,756220 7,8 4,545 0,892225 10,6 4,444 1,025230 14,1 4,348 1,149235 18,5 4,255 1,267240 24,1 4,167 1,382245 30,9 4,082 1,490250 39,1 4,000 1,592255 49,1 3,922 1,691260 61,0 3,846 1,785265 75,0 3,774 1,875270 97,5 3,704 1,989275 111 3,636 2,045280 133 3,571 2,124285 159 3,509 2,201290 188 3,448 2,274295 221 3,390 2,344300 258 3,333 2,412
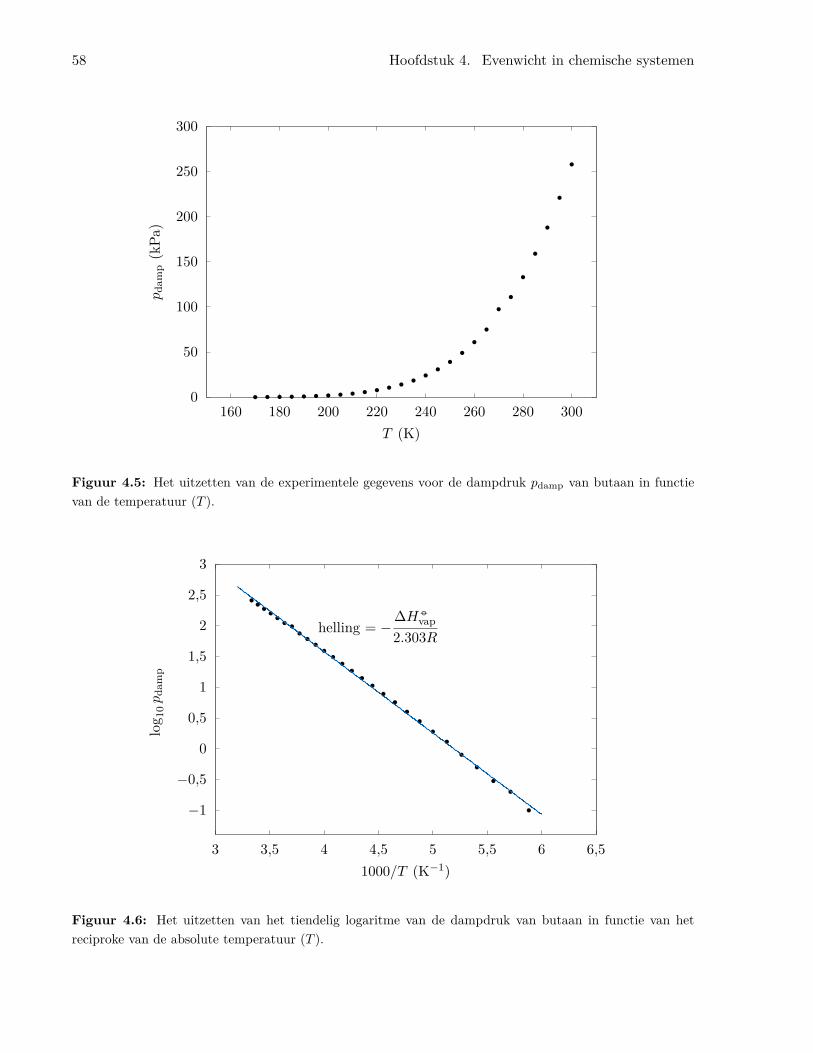
58 Hoofdstuk 4. Evenwicht in chemische systemen
0
50
100
150
200
250
300
160 180 200 220 240 260 280 300
pda
mp(kPa
)
T (K)
Figuur 4.5: Het uitzetten van de experimentele gegevens voor de dampdruk pdamp van butaan in functievan de temperatuur (T ).
−1
−0,5
0
0,5
1
1,5
2
2,5
3
3 3,5 4 4,5 5 5,5 6 6,5
log 1
0p
dam
p
1000/T (K−1)
helling = −∆H−vap2.303R
Figuur 4.6: Het uitzetten van het tiendelig logaritme van de dampdruk van butaan in functie van hetreciproke van de absolute temperatuur (T ).

4.5. Fasediagrammen 59
4.5 Fasediagrammen
We kunnen de Clapeyron vergelijking en de Clausius-Clapeyron vergelijkingen gebruiken om zeerhandige (p,T )-fasediagrammen mee te construeren. De Clapeyron vergelijking zal geïntegreerdworden om een curve te bekomen voor de vaste stof-vloeibare fase volgens∫ p2
p1dp = ∆Hsmelt
∆Vsmelt
∫ T2
T1
dTT
p2 − p1 = ∆Hsmelt∆Vsmelt
ln T2T1
De Clapeyron vergelijking kan niet gebruikt worden om een fasetransitie naar de gasfase mee tevisualiseren daar het molair volume van het gas een functie is van de druk. Door echter aan tenemen dat V (g) V (l), hebben we de Clausius-Clapeyron vergelijking kunnen afleiden die weopnieuw zullen integreren. ∫ p2
p1d ln p = ∆Hdamp
R
∫ T2
T1
1T 2dT
ln p2p1
= ∆HdampR
( 1T1− 1T2
)= ∆Hdamp
R
(T2 − T1T1T2
)
Tsmelt Tkook
Vast
Vloeibaar
Gas
p
T
Figuur 4.7: Een simulatie van een (p,T )-fasediagram voor een willekeurig eenvoudige substantie.
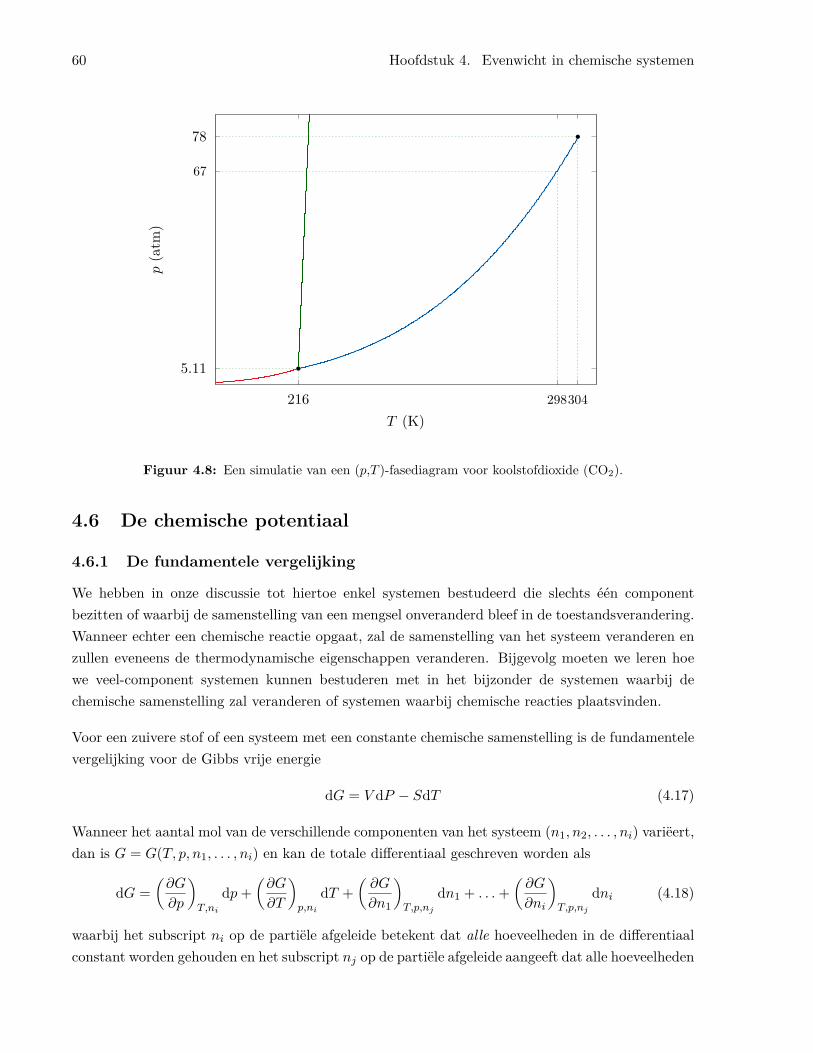
60 Hoofdstuk 4. Evenwicht in chemische systemen
5.11
67
78
216 298304
p(atm
)
T (K)
Figuur 4.8: Een simulatie van een (p,T )-fasediagram voor koolstofdioxide (CO2).
4.6 De chemische potentiaal
4.6.1 De fundamentele vergelijking
We hebben in onze discussie tot hiertoe enkel systemen bestudeerd die slechts één componentbezitten of waarbij de samenstelling van een mengsel onveranderd bleef in de toestandsverandering.Wanneer echter een chemische reactie opgaat, zal de samenstelling van het systeem veranderen enzullen eveneens de thermodynamische eigenschappen veranderen. Bijgevolg moeten we leren hoewe veel-component systemen kunnen bestuderen met in het bijzonder de systemen waarbij dechemische samenstelling zal veranderen of systemen waarbij chemische reacties plaatsvinden.
Voor een zuivere stof of een systeem met een constante chemische samenstelling is de fundamentelevergelijking voor de Gibbs vrije energie
dG = V dP − SdT (4.17)
Wanneer het aantal mol van de verschillende componenten van het systeem (n1, n2, . . . , ni) variëert,dan is G = G(T, p, n1, . . . , ni) en kan de totale differentiaal geschreven worden als
dG =(∂G
∂p
)T,ni
dp+(∂G
∂T
)p,ni
dT +(∂G
∂n1
)T,p,nj
dn1 + . . .+(∂G
∂ni
)T,p,nj
dni (4.18)
waarbij het subscript ni op de partiële afgeleide betekent dat alle hoeveelheden in de differentiaalconstant worden gehouden en het subscript nj op de partiële afgeleide aangeeft dat alle hoeveelheden

4.6. De chemische potentiaal 61
behalve één constant worden gehouden in de differentiaal. Zo zal bijvoorbeeld (∂G/∂n1)T,p,njdn1
betekenen dat T , p en alle hoeveelheden behalve n1 constant worden gehouden in de differentiaal.
Wanneer het systeem geen verandering in samenstelling ondergaat is
dn1 = 0, . . . ,dni = 0
zodat Vergelijking 4.18 reduceert naar
dG =(∂G
∂p
)T,ni
dp+(∂G
∂T
)p,ni
dT (4.19)
Door nu Vergelijking 4.19 met Vergelijking 4.17 te vergelijken, bekomen we dat(∂G
∂p
)T,ni
= V en(∂G
∂T
)p,ni
= −S (4.20)
Om het schrijven te vereenvoudigen, kunnen we nu de chemische potentiaal µi van de ide componentdefiniëren als
µi =(∂G
∂ni
)T,p,nj
(4.21)
De totale differentiaal van G in Vergelijking 4.18 kan nu worden herschreven als
dG = V dP − SdT + µ1dn1 + . . .+ µidni
dat gewoonlijk in een meer compacte vorm voorkomt
dG = V dP − SdT +∑i
µidni (4.22)
Vergelijking 4.22 wordt soms de meest fundamentele vergelijking van de chemische thermodynamicagenoemd.
4.6.2 De eigenschappen van de chemische potentiaal
De chemische potentiaal µi kan beschouwd worden als de stijging in de vrije energie van een systeemwanneer één mol van een component i wordt toegevoegd aan een oneindig grote hoeveelheid vaneen mengsel zodat het niet significant de totale samenstelling verandert.
Wanneer een kleine hoeveelheid van een stof i, dni mol wordt toegevoegd aan een systeem waarbijT , p en al de andere hoeveelheden constant worden gehouden, zal de stijging in Gibbs vrije energiegegeven worden door Vergelijking 4.22 en zal deze vergelijking reduceren naar dG = µidni. Destijging in Gibbs vrije energie per mol van een toegevoegde stof is(
∂G
∂ni
)T,p,nj
= µi
Deze vergelijking drukt de onmiddellijke betekenis van µi uit. Voor eender welke stof i in eenmengsel, is de waarde van µi gelijk aan de verhoging in de Gibbs vrije energie wanneer er een

62 Hoofdstuk 4. Evenwicht in chemische systemen
infinitesimale hoeveelheid van deze stof wordt toegevoegd aan het mengsel per mol van de toe-gevoegde stof. De toegevoegde hoeveelheid is beperkt tot een infinitesimale hoeveelheid zodat desamenstelling van het mengsel en bijgevolg de waarde van µi niet zal veranderen.
Een alternatieve benadering houdt een extreem groot systeem in. Stel bijvoorbeeld een grote kamervol waterige suikeroplossing. Wanneer dan één mol water wordt toegevoegd aan een dergelijk grootsysteem, zal de samenstelling van het systeem ongeveer gelijk blijven voor alle praktische doeleindenen zal bijgevolg µH2O constant zijn. De verhoging in Gibbs vrije energie die volgt bij het toevoegenvan één mol water aan een kamer vol oplossing is de waarde van µH2O in de oplossing.
Daar µi de afgeleide is van een extensieve variabele door een andere, is het een intensieve eigenschapvan het systeem en moet het overal dezelfde waarde hebben in een systeem bij evenwicht. Dechemische potentiaal kan bijgevolg gezien worden als de drijvende kracht die chemische systemennaar een evenwicht stuurt.
Veronderstel dat µi twee verschillende waarden, µAi en µBi , heeft in twee gebieden van het systeem, Aen B. Door T , p en alle andere hoeveelheden constant te houden, veronderstellen we een overdrachtdni mol van i van het gebied A naar een gebied B. Voor de verhoging in Gibbs vrije energie inde twee gebieden, kunnen we met behulp van Vergelijking 4.22 schrijven dat dGA = µAi (−dni) endGB = µBi dni daar +dni mol gaan naar B en −dni mol gaan naar A. De totale verandering inGibbs energie van het systeem is de som dG = dGA +GB of
dG =(µBi − µAi
)dni
Wanneer µBi lager is dan µAi , zal dG negatief zijn en zal de overdracht van materie de Gibbsvrije energie van het systeem verlagen zodat de overdracht spontaan gebeurt. Een stof i stroomtbijgevolg spontaan van een gebied met hoge µi naar een gebied met lage µi. Deze stroom blijft zichvoordoen totdat de waarde van µi gelijk is over het ganse systeem of met andere woorden totdathet systeem in evenwicht is. Het feit dat µi dezelfde waarde moet hebben over het ganse systeemis een belangrijke evenwichtsvoorwaarde die we telkens opnieuw zullen gebruiken!
De grootheid µi wordt de chemische potentiaal van een substantie i genoemd omdat de materiespontaan van een gebied met hoge chemische potentiaal naar een gebied met lage chemische po-tentiaal stroomt zoals de elektrische stroom spontaan stroomt van een gebied met hoge elektrischepotentiaal naar een gebied met lagere elektrische potentiaal.
4.6.3 De chemische potentiaal en de vrije energie
Voor een zuivere stof is de chemische potentiaal(∂G
∂ni
)T,p,nj
eenvoudig de molaire vrij energie G/ni. Voor één mol van een ideaal gas is
G = G− +RT ln(p/atm) (4.23)

4.6. De chemische potentiaal 63
en wordt de chemische potentiaal van een zuiver ideaal gas bijgevolg expliciet gegeven door
µ = µ− +RT ln(p/atm)
Mengsels van ideale gassen gedragen zich alsof elk gas zich alleen in de container zou bevinden.De thermodynamische eigenschappen van de gassen die een dergelijk systeem opmaken, kunnenuitgedrukt worden in termen van de partieeldrukken of de drukken die ze zouden genereren wanneerze zich alleen in de container zouden bevinden. Bijgevolg geldt dat
µi = µ−i +RT ln(pi/atm) (4.24)
met pi de numerische waarde van de partieeldruk van de i-de component wanneer het wordt uitge-druk in atmosfeer.
In een hypothetisch mengsel is de chemische potentiaal µi niet gelijk aan G/ni daar µi de verho-ging in vrij energie symboliseert die voorkomt wanneer een mol van de stof i is toegevoegd aaneen oneindig grote hoeveelheid van een mengsel zodat de samenstelling niet veranderd. Afgeleidehoeveelheden van dit type worden partiële molaire hoeveelheden genoemd. We schrijven dat
µi =(∂G
∂ni
)T,p,nj
= Gi (4.25)
met Gi de partiële molaire vrije energie van de component i in het systeem. Het subscript nj duidtaan dat de hoeveelheid van deeltjes die verchillend zijn van i, constant zijn. We definiëren de anderepartiële molaire hoeveelheden als(
∂V
∂ni
)T,p,nj
= Vi en(∂S
∂ni
)T,p,nj
= Si (4.26)
waarbij Vi en Si respectievelijk het partieel molair volume en de entropie zijn. Daar nu(∂G
∂p
)T
= V
kunnen we schrijven dat (∂µi∂p
)T
= ∂
∂ni
(∂G
∂p
)T
= Vi
Een analoge argumentatie leidt tot (∂µi∂T
)p
= −Si
Partiële molaire hoeveelheden spelen een belangrijke rol in de studie van niet-ideale mengsels.Ze worden gewoonlijk vervangen door de overeenkomstige molaire hoeveelheden. In eenvoudigeberekeningen voor ideale gassen of ideale oplossingen, kan Vi vervangen worden door het volumevan één mol van een zuivere stof i in de bestemde fysische toestand.

64 Hoofdstuk 4. Evenwicht in chemische systemen
Palladium membraan
Zuiver H2pH2
N2 + H2p = pH2 + pN2
Figuur 4.9: De chemische potentiaal van een ideaal gas in een mengsel.
4.6.4 De chemische potentiaal van een ideaal gas in een mengsel van idealegassen
We beschouwen een systeem gevisualiseerd in Figuur 4.9. Het rechterhandcompartiment bevat eenmengsel van waterstofgas onder een partieeldruk pH2 en stikstofgas onder een partieeldruk ptextN2
zodat de totaaldruk zijnde p = pH2 + ptextN2 . Het mengsel wordt gescheiden van een linkercompar-timent door palladium membraan.4 Daar waterstofgas gemakkelijk doorheen het membraan kangaan, bevat het linkermembraan zuiver waterstofgas. Wanneer het evenwicht wordt bereikt, is dedruk van het zuivere waterstofgas in het linkercompartiment per definitie gelijk aan de partieeldrukvan het waterstofgas in het mengsel. De evenwichtsvoorwaarde vereist dat de chemische potentiaalvan het waterstofgas dezelfde waarde moet hebben in beide compartimenten van het vat.
µH2(zuiver) = µH2(mix)
De chemische potentiaal van het zuivere waterstofgas bij een druk pH2 is
µH2(zuiver) = µ−H2(zuiver) +RT ln pH2
De chemische potentiaal in het mengsel moet dan ook zodanig zijn dat
µH2(mix) = µ−H2(zuiver) +RT ln pH2
Deze vergelijking toont aan dat de chemische potentiaal van waterstofgas in een mengsel de loga-ritmische functie is van de partieeldruk van het waterstofgas in het mengsel. Door dit argument teherahalen gebruik makend van een mengsel van eender welk aantal ideale gassen en een geschiktmembraan dat doorlaatbaar is voor een substantie i, kan aangetoond worden dat de chemischepotentiaal van de stof i in het mengsel wordt gegeven door
µi = µ−i +RT ln pi (4.27)
waarbij pi de partieeldruk is van een substantie i in het mengsel. De term µ−i heeft dezelfdebetekenis als voor een zuiver gas; het is de chemische potentiaal van een zuiver gas bij een drukvan één atmosfeer en een temperatuur T .Door gebruik te maken van de relatie pi = xip waarbij xi de molfractie is van de stof i in het
4Lees het stukje ‘Palladium laat selectief Waterstof door’ uit Chemische feitelijkheden via http://www.chemischefeitelijkheden.nl/Uploads/Magazines/Membraan-253_1.pdf.

4.6. De chemische potentiaal 65
mengsel en p de totale druk, kunnen we voor pi in Vergelijking 4.27 bekomen door het logaritmeuit te zetten dat
µi = µ−i +RT ln p+RT ln xi (4.28)
De eerste twee termen in Vergelijking 4.28 zijn gelijk aan de chemische potentiaal voor de zuiverestof i bij een druk p zodat Vergelijking 4.28 reduceert tot
µi = µ−i(zuiver) +RT ln xi (4.29)
Daar xi een een fractie is en het logartime bijgevolg negatief zal zijn, toont Vergelijking 4.29 datde chemische potentiaal voor eender welk gas in een mengsel altijd lager zal zijn dan de chemischepotentiaal van het zuivere gas bij dezelfde totale druk. Wanneer bijgevolg een zuiver gas bij eendruk p in contact wordt gebracht met een mengsel onder dezelfde totale druk, dan zal het zuiveregas spontaan vloeien in het mengsel. Dit is de thermodynamische interpretatie van het feit datgassen alsook vloeistoffen en vaste stoffen, in elkaar diffunderen.
4.6.5 De vrije Gibbs energie en de entropie van het mengen
Daar de vorming van een mengsel uit zuivere bestanddelen altijd spontaan zal gebeuren, moet ditproces gepaard gaan met een verlaging van de vrije Gibbs energie. Ons objectief bestaat er nu inom de vrije Gibbs energie van het mengen te berekenen. De initiële toestand wordt gevisualiseerdin Figuur 4.10(a). Elk van de compartimenten bevat een zuiver bestanddeel onder een druk p.De scheidingswanden die de bestanddelen scheiden, worden weggetrokken en de finale toestand,getoond in Figuur 4.10, wordt bekomen dat het mengsel is onder dezelfde druk p. De temperatuur
(a)
T , pn1
T , pn2
T , pn3
(b)
T , pN = n1 + n2 + n3
Figuur 4.10: De vrije Gibbs energie van het mengen. (a) Initiële toestand. (b) Finale toestand.
is dezelfde in de initiële toestand en de finale toestand. Voor de zuivere bestanddelen, zijn de vrijeGibbsd energieën respectievelijk
G1 = n1µ−1 , G2 = n2µ
−2 , G3 = n3µ
−3
De vrije Gibbs energie van de initiële toestand is eenvoudigweg de som
Ginitieel = G1 +G2 +G3 = n1µ−1 + n2µ
−2 + n3µ
−3 =
∑i
niµ−i
De vrije Gibbs energie in de finale toestand wordt gegeven door de optellingsregel
Gfinaal = n1µ1 + n2µ2 + n3µ3 =∑i
niµi

66 Hoofdstuk 4. Evenwicht in chemische systemen
De vrije Gibbs energie van het mengen, ∆Gmix = Gfinaal−Ginitieel wordt gevonden door de waardenvan Gfinaal en Ginitieel in te vullen zodat
∆Gmix = n1(µ1 − µ−1
)+ n2
(µ2 − µ−2
)+ n3
(µ3 − µ−3
)=∑i
ni(µi − µ−i
)Gebruik makende van de relatie tussen µi − µ−i uit Vergelijking 4.29, vinden we dat
∆Gmix = RT (n1 ln x1 + n2 ln x2 + n3 ln x3) = RT∑i
ni ln xi
dat we in een iets handigere vorm kunnen gieten door de substitutie ni = xini door te voren waarbijn het totaal aantal mol is in het mengsel en xi de molfractie i. Bijgevolg bekomen we
∆Gmix = nRT (x1 ln x1 + x2 ln x2 + x3 ln x3) (4.30)
dat de finale uitdrukking is voor de Gibbs vrije energie van het mengen in termen van de molfractiesvan de bestanddelen in het mengsel. Elke term in het rechterlid is negatief zodat de totale somaltijd negatief zal zijn. Uit de afleiding kunnen we zien dat in het vormen van een ideaal mengselvan eender welk aantal bestanddelen de Gibbs vrije energie van het mengen altijd kan geschrevenworden als
∆Gmix = nRT∑i
xi ln xi (4.31)
Wanneer er slechts twee bestanddelen zijn in het mengsel, dan is x1 = x en x2 = 1 − x zodatVergelijking 4.31 kan geschreven worden als
∆Gmix = nRT [x ln x+ (1− x) ln (1− x)] (4.32)
Vergelijking 4.32 wordt uitgezet in Figuur 4.11. De kromme is symmetrisch rond x = 1/2. Degrootste afname in Gibbs vrije energie in het mengen wordt geassocieerd met de vorming van hetmengsel dat een gelijk aantal mol heeft van de twee bestanddelen. In een ternair systeem zal degrootste afname vann de Gibbs vrije energie van het mengen voorkomen wanneer de molfractie vanelk van de bestanddelen gelijk is aan 1/3 enzovoort. Het afleiden van ∆Gmix = Gfinaal − Ginitieel
naar de temperatuur levert meteen ∆Smix op.(∂∆Gmix∂T
)p,ni
=(∂Gfinaal∂T
)p,ni
−(∂∆Ginitieel
∂T
)p,ni
= − (Sfinaal − Sinitieel)
zodat (∂∆Gmix∂T
)p,ni
= −∆Smix (4.33)
Wanneer we beide leden van Vergelijking 4.31 afleiden naar de temperatuur, hebben we(∂∆Gmix∂T
)p,ni
= nR∑i
xi ln xi
zodat Vergelijking 4.33 kan worden geschreven als
∆Smix = −nR∑i
xi ln xi (4.34)
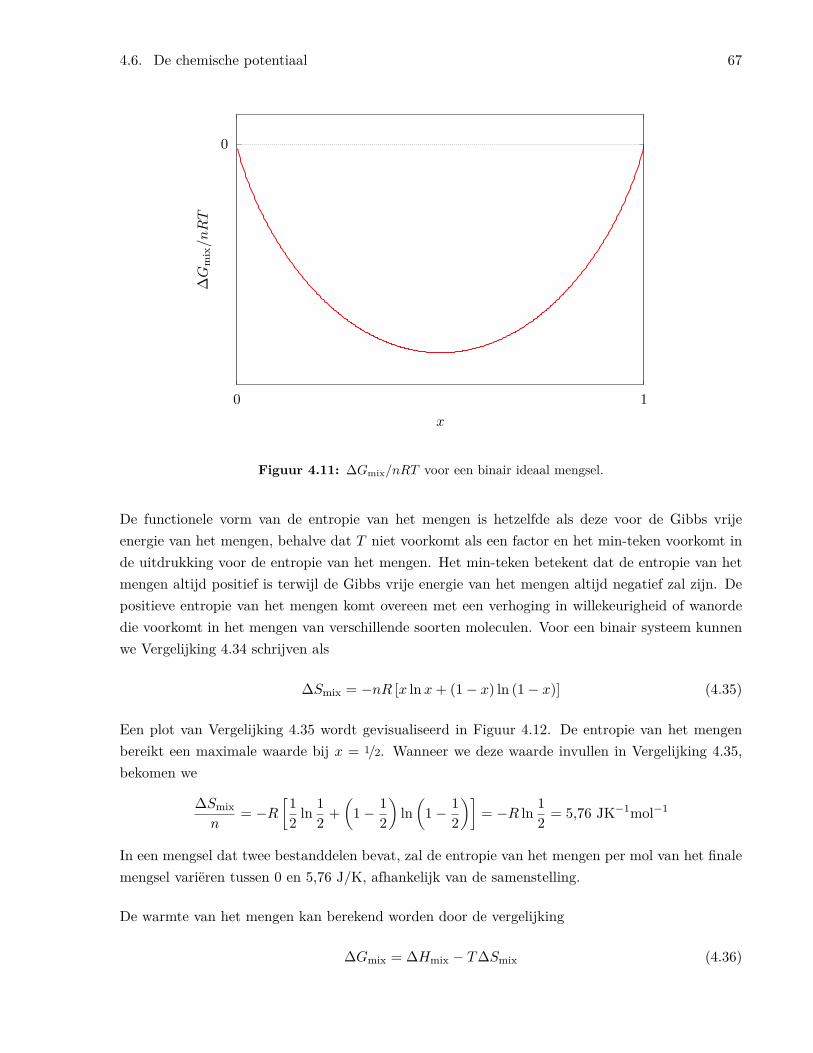
4.6. De chemische potentiaal 67
0
0 1
∆G
mix/nRT
x
Figuur 4.11: ∆Gmix/nRT voor een binair ideaal mengsel.
De functionele vorm van de entropie van het mengen is hetzelfde als deze voor de Gibbs vrijeenergie van het mengen, behalve dat T niet voorkomt als een factor en het min-teken voorkomt inde uitdrukking voor de entropie van het mengen. Het min-teken betekent dat de entropie van hetmengen altijd positief is terwijl de Gibbs vrije energie van het mengen altijd negatief zal zijn. Depositieve entropie van het mengen komt overeen met een verhoging in willekeurigheid of wanordedie voorkomt in het mengen van verschillende soorten moleculen. Voor een binair systeem kunnenwe Vergelijking 4.34 schrijven als
∆Smix = −nR [x ln x+ (1− x) ln (1− x)] (4.35)
Een plot van Vergelijking 4.35 wordt gevisualiseerd in Figuur 4.12. De entropie van het mengenbereikt een maximale waarde bij x = 1/2. Wanneer we deze waarde invullen in Vergelijking 4.35,bekomen we
∆Smixn
= −R[1
2 ln 12 +
(1− 1
2
)ln(
1− 12
)]= −R ln 1
2 = 5,76 JK−1mol−1
In een mengsel dat twee bestanddelen bevat, zal de entropie van het mengen per mol van het finalemengsel variëren tussen 0 en 5,76 J/K, afhankelijk van de samenstelling.
De warmte van het mengen kan berekend worden door de vergelijking
∆Gmix = ∆Hmix − T∆Smix (4.36)
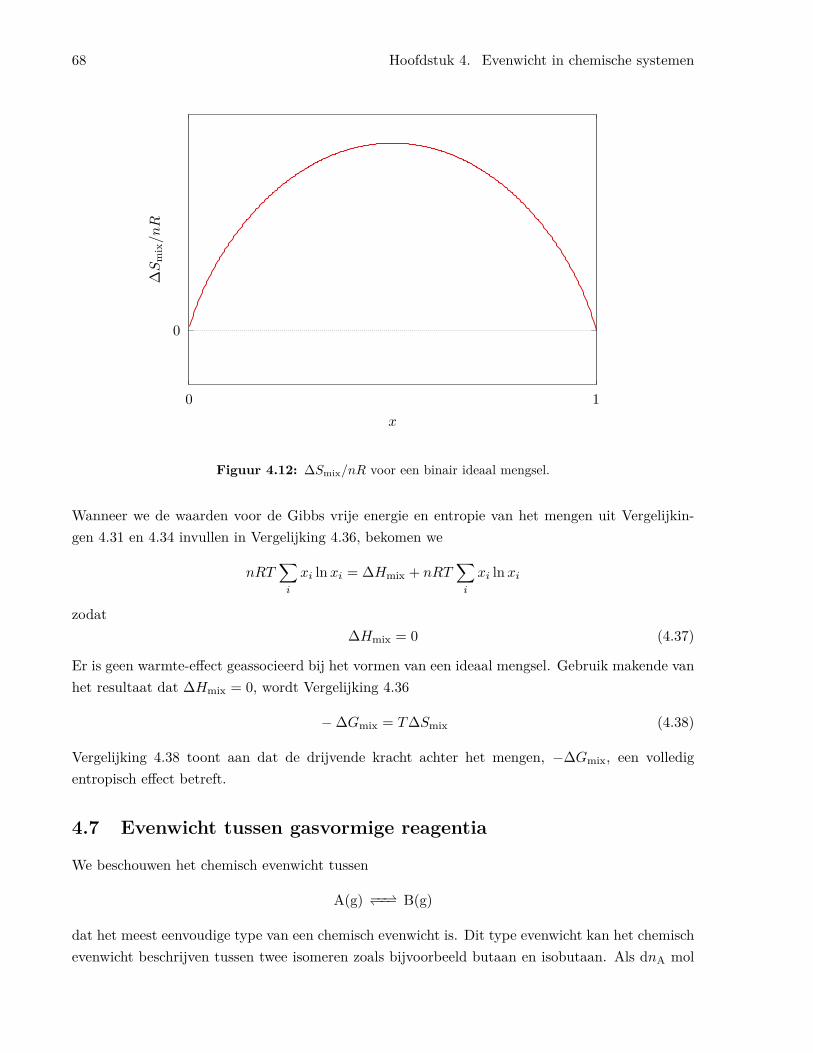
68 Hoofdstuk 4. Evenwicht in chemische systemen
0
0 1
∆S
mix/nR
x
Figuur 4.12: ∆Smix/nR voor een binair ideaal mengsel.
Wanneer we de waarden voor de Gibbs vrije energie en entropie van het mengen uit Vergelijkin-gen 4.31 en 4.34 invullen in Vergelijking 4.36, bekomen we
nRT∑i
xi ln xi = ∆Hmix + nRT∑i
xi ln xi
zodat∆Hmix = 0 (4.37)
Er is geen warmte-effect geassocieerd bij het vormen van een ideaal mengsel. Gebruik makende vanhet resultaat dat ∆Hmix = 0, wordt Vergelijking 4.36
−∆Gmix = T∆Smix (4.38)
Vergelijking 4.38 toont aan dat de drijvende kracht achter het mengen, −∆Gmix, een volledigentropisch effect betreft.
4.7 Evenwicht tussen gasvormige reagentia
We beschouwen het chemisch evenwicht tussen
A(g) −−−− B(g)
dat het meest eenvoudige type van een chemisch evenwicht is. Dit type evenwicht kan het chemischevenwicht beschrijven tussen twee isomeren zoals bijvoorbeeld butaan en isobutaan. Als dnA mol

4.7. Evenwicht tussen gasvormige reagentia 69
A wordt omgezet in dnB mol B bij een constante temperatuur en druk, hebben we voor dG =+µAdnA + µBdnB, waarbij dnA negatief is en dnB positief. We kunnen de reactievordering ξ
definiëren5 die gelijk zal zijn aan nul wanneer de reactie volledig naar de linkerkant zal verlopen engelijk aan één wanneer er één mol van een reagens volledig naar producten is omgezet. Voor heteenvoudig voorbeeld kunnen we schrijven
dξ = dnB = −dnA
zodat bij een constante temperatuur en druk geldt dat
dG = (µB − µA) dξ
De reactie zal doorgaan totdat G een evenwichtswaarde bereikt waar
0 ξev 1
∂G
∂ξ= −
∂G
∂ξ= 0
∂G
∂ξ= +
G
ξ
Figuur 4.13: Simulatie van de Gibbs vrije energie in functie van de reactievordering ξ van een chemischereactie.
(∂G
∂ξ
)T,p
= 0
zoals is geïllustreerd in Figuur 4.13. Daar(∂G
∂ξ
)T,p
= µB − µA
zal de evenwicchtspositie worden bereikt als µB = µA.5De IUPAC definieert de reactievordering (extent of reaction) als ξ en de reactiegraad (degree of reaction) als
α = ξ/ξmax [6].

70 Hoofdstuk 4. Evenwicht in chemische systemen
Wanneer de componenten de ideale gaswetten volgen, dan is
µi = µ−i +RT ln(pi/atm)
Om deze reden is (∂G
∂ξ
)T,p
= µB − µA = µ−B − µ−A +RT ln pBpA
met µ−B−µ−A = ∆G− de Gibbs vrije energieverandering voor één mol van een reactie die opgaat metzowel het reagens als het product blijvend in de standaard toestand bij een druk van één atmosfeerzodat (
∂G
∂ξ
)T,p
= ∆G− +RT ln pBpA
(∂G/∂ξ)T,p is de verandering in vrije Gibbs energie in functie van de reactievordering onder devoorwaarden gespecifieerd door pB en pA. Deze is gelijk aan de Gibbs vrije energieverandering vooréén mol van een reactie met A bij een partiële druk pA dat overgaat naar B bij een partiële druk pBwaarbij de partieeldrukken constant blijven. Het komt voor dat − (∂G/∂ξ)T,p de affiniteit van dereactie wordt genoemd terwijl (∂G/∂ξ)T,p soms de reactie vrije energie wordt genoemd en eenvoudigwordt geschreven als ∆G. We zullen echter ∆G′ schrijven waarbij de ′ ons eraan moet herinnerendat het in realiteit een afgeleide hoeveelheid is en overeenkomt met de vrije energieverandering vooréén mol van een reactie onder exact gedefinieerde voorwaarden.Bij evenwicht geldt dat
∆G′ =(∂G
∂ξ
)T,p
= 0
en omdat∆G′ = ∆G− +RT ln pB
pA(4.39)
is∆G− = −RT ln
(pBpA
)evenwicht
(4.40)
De waarde van pB/pA bij evenwicht wordt de evenwichtsconstante, Kp, van de reactie genoemd.
∆G− = −RT lnKp (4.41)
Vergelijking 4.41 is zeer belangrijk omdat het ons vertelt hoe de positie van het chemisch evenwichtkan worden gedefinieerd in termen van vrije energieën van de reagentia en de producten bij een drukvan één atmosfeer. Dergelijke standaard vrije energieën kunnen experimenteel worden bepaald. Devergelijking is eveneens waardevol voor een meer kwalitatieve beschouwing. Wanneer ∆G− negatiefis, weten we dat de richting van het evenwicht zal overeenkomen met de aanwezigheid van meerproducten dan reagentia (lnKp > 0) en wanneer ∆G− positief is, zal de reactie niet zover gaan enzullen de reagentia domineren in het evenwichtsmengsel. Met dit resultaat hebben we één van debelangrijke hoofddoelen van onze studie bereikt.
Stel dat de reactie ingewikkelder was geweest, zouden we in essentie dezelfde resultaten hebbenbekomen. Voor de reactie
aA + bB −−−− lL + mM

4.8. Eigenschappen van de evenwichtsconstante 71
kunnen we schrijven dat
dG = µLdnL + µMdnM + µAdnA + µBdnB =∑i
µidni
De reactievordering ξ zal op een meer ingewikkelde manier moeten gedefinieerd worden zodat
dξ = dnLl
= dnMm
= −dnAa
= −dnBb
= dniνi
waarbij νi de stoichiometrische coëfficiënten −a, −b, m en l representeert. De νi voor de reagentiazijn negatief gedefinieerd en deze voor de producten zijn positief gedefinieerd zodat
dG = (lµL +mµM − aµA − bµB) dξ
en∆G′ =
(∂G
∂ξ
)T,p
= lµL +mµM − aµA − bµB =∑i
νiµi
Daar µA = µ−A +RT ln (pA/atm), µB = µ−B +RT ln (pB/atm) etc, bekomen we dat
∆G′ = ∆G− +RT ln[
(pL/atm)l (pM/atm)m
(pA/atm)a (pB/atm)b
]
dat we op een meer beknopte manier kunnen schrijven als
∆G′ = ∆G− +RT ln∏i=1
(pi/atm)νi
Bij evenwicht geldt dat ∆G− = −RT lnKp waarbij
Kp =[
(pL/atm)l (pM/atm)m
(pA/atm)a (pB/atm)b
]evenwicht
=[∏i=1
(pi/atm)νi
]evenwicht
en∆G− =
∑i
νiµ−i
de standaard vrije energie verandering voor één mol van een reactie.Deze vergelijkingen zijn fundamenteel analoog aan de vergelijkingen die we hebben afgeleid vooreen illustratief voorbeeld van de isomerizatie van butaan waarbij Kp strict dimentieloos is zelfswanneer a+ b 6= l +m omdat alle drukken zelf ‘dimentieloos’ zijn of nog omdat p(atm)/1 atm.
4.8 Eigenschappen van de evenwichtsconstante
4.8.1 Temperatuursafhankelijkheid van de evenwichtsconstante
We kunnen de thermodynamische relaties die we zijn bekomen, gebruiken om na te gaan hoe depositie van het evenwicht zal veranderen wanneer we de temperatuur veranderen. We weten reeds,gebruik makende van (
∂G
∂T
)p
= −S

72 Hoofdstuk 4. Evenwicht in chemische systemen
enG = H − TS
dat we de Gibbs-Helmholtz vergelijking konden bekomen∂ (∆GT
)∂T
p
= −∆HT 2
We kunnen de vergelijking ∆G− = −RT lnKp nu afleiden naar de temperatuur om de belangrijkeuitdrukking te bekomen
(∂ lnKp
∂T
)p
= −
∂(
∆G−RT
)∂T
p
= − 1R
∂(
∆G−T
)∂T
p
= ∆H−RT 2 (4.42)
die de van ’t Hoff-vergelijking of de van ’t Hoff-isochoor wordt genoemd. Daar ∆G− en ∆H− geenfuncties zijn van de druk (daar ze per definitie de waarden bezitten bij 1 atm) kunnen we eveneensschrijven dat
d lnKp
dT = ∆H−RT 2 (4.43)
Wanneer we aannemen dat ∆H− niet afhangt van de temperatuur6, dan bekomen we na integratie∫ Kp(T2)
Kp(T1)d lnKp = ∆H−
R
∫ T2
T1
dTT 2
ln Kp(T2)Kp(T1) = −∆H−
R
( 1T2− 1T1
)Het uitzetten van log10Kp in functie van 1/T zal een rechte geven met een helling die gelijk isaan −∆H− /2,3R. Voor een exotherme reactie (∆H < 0) moet Kp dalen wanneer de temperatuurstijgt. Voor de reactie
N2 + 3H2 −−−− 2NH3 ∆H− = −92,4 kJmol−1
kunnen we voorspellen dat er minder ammoniak wordt gevormd in het evenwichtsmengsel bij hogeretemperaturen. Voor een endotherme reactie (∆H > 0) zal Kp stijgen bij stijgende temperatuur.Voor het evenwicht
N2O4 −−−− 2NO2 ∆H− = 58,0 kJmol−1
kunnen we voorspellen en observeren dat er meer dissociatie zal zijn bij hogere temperaturen.
Voorbeeld 4.5Voor de endotherme dissociatiereactie7
Cl2(g) −−−− 2Cl(g)6Dit zal vaak een goede benadering zijn. Hoewel H sterk afhangt van de temperatuur, zal ∆H− gecontroleerd
worden door een verschil in warmtecapaciteiten tussen de reagentia en de producten. Wanneer de warmtecapaciteitenvan de reagentia en de producten ongeveer hetzelfde zijn, dan zal ∆H− quasi onafhankelijk zijn van de temperatuur.
7De standaard Gibbs vrije vormingsenergieën (∆fG− ) en de standaard vormingsenthalpieën (∆fH
− ) bij 298,15 Kzijn gekend voor vele chemische stoffen en staan gedocumenteerd in Referentie [11]. Uit deze data werden zowel∆G−
reactie als ∆H−reactie berekend.

4.8. Eigenschappen van de evenwichtsconstante 73
is ∆G−reactie = 210,6 kJmol−1 en ∆H−reactie = 242,6 kJmol−1 bij 298,15 K.Bereken Kp bij 800, 1500 en 2000 K voor p = 0,010 bar.
Oplossing
We schrijven de van’t Hoff-vergelijking als
lnKp(Tf) = lnKp(298,15 K)− ∆H−reactieR
( 1Tf− 1
298,15 K
)en we weten dat
∆G−reactie = −RT lnKp(298,15 K)
zodatlnKp(298,15 K) = −∆G−reactie
RT= − 210,6× 103Jmol−1
8,314 JK−1mol−1 · 298,15 K= −84,96
Berekening Kp bij 800 K
lnKp(800 K) = −84,96− 242,6× 103 Jmol−1
8,314 JK−1mol−1
( 1800 K −
1298,15 K
)= −23,6
zodatKp(800 K) = exp(−23,6) = 5,83× 10−11
Berekening Kp bij 1500 K
lnKp(1500 K) = −84,96− 242,6× 103 Jmol−1
8,314 JK−1mol−1
( 11500 K −
1298,15 K
)= −6,54
zodatKp(1500 K) = exp(−6,54) = 1,44× 10−3
Berekening Kp bij 2000 K
lnKp(2000 K) = −84,96− 242,6× 103 Jmol−1
8,314 JK−1mol−1
( 12000 K −
1298,15 K
)= −1,68
zodatKp(2000 K) = exp(−1,68) = 0,186
We stellen vast dat Kp stijgt bij stijgende temperatuur voor een endotherme reactie. Bijgevolg zaler meer dissociatie optreden van Cl2 bij hogere temperaturen.

74 Hoofdstuk 4. Evenwicht in chemische systemen
4.8.2 Het berekenen van de evenwichtspartieeldrukken in een mengsel van ide-ale gassen
Daar Kp gedefinieerd kan worden in functie van de vrije energieën van de deelnemende stoffen in destandaardtoestanden bij een druk van één atmosfeer, moet Kp onafhankelijk zijn van de druk. Departieeldrukken van de reagentia en de producten kunnen bijgevolg geenszins arbitraire waardeninnemen omdat ze zijn gerelateerd door Kp. We kunnen de partieeldrukken in een mengsel vangassen bij evenwicht dan ook berekenen uit Kp. Het meest handig is om dit te doen in tabelvorm.We bespreken achtereenvolgens de dissociatie van dichloor, de dissociatie van distikstoftetraoxideen de synthese van ammoniak.
De dissociatie van dichloor
We beschouwen opnieuw de dissociatie van dichloor volgens de evenwichtsreactie
Cl2(g) −−−− 2Cl(g)
In tabelvorm zullen we de verschillende grootheden in kolommen oplijsten onder de chemischeformules van de stoffen in de uitgebalanceerde chemische reactie. We stellen n als het initieelaantal mol Cl2, ξev als de evenwichtsreactievordering en αev als de fractie die gedissocieerd is ophet evenwicht en gedefineerd als αev = ξev/n
Cl2(g) −−−− 2Cl(g)
Stoichiometrische coëfficiënt −1 +2Initieel aantal mol (ni ) n 0Aantal mol aanwezig bij evenwicht (ni) n − ξev 0 + 2ξevTotaal aantal mol (n) n = n − ξev + 0 + 2ξev = n + ξev
Molfracties (xi)n − ξevn + ξev
2ξevn + ξev
of, daar αev = ξev/n, zijn xi
1− αev1 + αev
2αev1 + αev
Partieeldrukken (pi = xip)(1− αev
1 + αev
)p
( 2αev1 + αev
)p
Gebruik makende van de waarden voor de partieeldrukken bekomen we
Kp = p2Cl
pCl2=
[( 2αev1 + αev
)p
]2
(1− αev1 + αev
)p
=
4α2ev
(1 + αev) · (1 + αev)p · p(1− αev1 + αev
)p
= 4α2evp
1− α2ev
(4.44)
Omdat Kp onafhankelijk is van de totale druk, zal de dissociatiegraad αev in principe moetenverlagen wanneer p stijgt. We kunnen Vergelijking 4.44 nu omvormen tot een vergelijking om αev
te berekenen.
Kp = 4α2evp
1− α2ev

4.8. Eigenschappen van de evenwichtsconstante 75
Kp ·(1− α2
ev
)= 4α2
evp
Kp −Kpα2ev = 4α2
evp
Kp = 4α2evp+Kpα
2ev
Kp = (4p+Kp)α2ev
Kp
4p+Kp= α2
ev
We bekomen uiteindelijk
αev =√
Kp
Kp + 4p (4.45)
Voorbeeld 4.6In Voorbeeld 4.5 werd Kp berekend bij p = 0,010 bar voor de dissociatiereactie Cl2(g) −−−− 2Cl(g)bij 800, 1500 en 2000 K. Bereken bij deze temperaturen telkens de dissociatiegraad αev.
Oplossing
Daar 1,01325 bar = 1 atm, is 0,010 bar ≈ 0,0099 atm. Besef dat deze druk gedeeld wordt door1 atm in de vergelijking!
Berekening αev bij 800 K
αev =√
Kp
Kp + 4p
=√
5,83× 10−11
5,83× 10−11 + 4 · 0,0099= 3,82× 10−5
Berekening αev bij 1500 K
αev =√
1,44× 10−3
1,44× 10−3 + 4 · 0,0099= 0,186
Berekening αev bij 2000 K
αev =√
0,1860,186 + 4 · 0,0099
= 0,907
De dissociatiegraad van Cl2 stijgt met de temperatuur zoals steeds het geval zal zijn voor eenendotherme reactie.

76 Hoofdstuk 4. Evenwicht in chemische systemen
De dissociatie van distikstoftetraoxide
De dissociatie van distikstoftetraoxide kan relatief eenvoudig in het laboratorium worden onderzochtdoor het meten van de dampdichtheden van het evenwichtsmengsel volgens
N2O4(g) −−−− 2NO2(g)
We merken op dat de theoretische beschrijving om de partieeldrukken te berekenen volledig analoogis als bij de dissociatie van dichloor. We kunnen opnieuw in tabelvorm de verschillende groothedenin kolommen oplijsten onder de chemische formules van de stoffen in de uitgebalanceerde chemischereactie.
N2O4(g) −−−− 2NO2(g)
Stoichiometrische coëfficiënt −1 +2Initieel aantal mol (ni ) n 0Aantal mol aanwezig bij evenwicht (ni) n − ξev 0 + 2ξevTotaal aantal mol (n) n = n − ξev + 0 + 2ξev = n + ξev
Molfracties (xi)n − ξevn + ξev
2ξevn + ξev
of, daar αev = ξev/n, zijn xi
1− αev1 + αev
2αev1 + αev
Partieeldrukken (pi = xip)(1− αev
1 + αev
)p
( 2αev1 + αev
)p
Voorbeeld 4.7Bij 298 K is Kp voor de dissociatie van distikstoftetraoxide 0,14. Bereken de fractie aan N2O4
moleculen die gedissocieerd zijn bij een druk van 1 atmosfeer en een druk van 10 atmosfeer.
Oplossing
De dissociatie van distikstoftetraoxide kan op analoge wijze beschreven worden als de dissociatievan dichloor. Bijgevolg kunnen we de formule voor de reeds afgeleide dissociatiegraad αev inVergelijking 4.45 invullen met de beschikbare gegevens.
Berekening αev bij 1 atm
αev =√
Kp
Kp + 4p
=√
0,140,14 + 4 · 1
= 0,184
Berekening αev bij 10 atm
αev =√
0,140,14 + 4 · 10

4.8. Eigenschappen van de evenwichtsconstante 77
= 0,0591
Bij een temperatuur van 298 K is de fractie aan N2O4 moleculen die gedissocieerd zijn bij een drukvan 1 atmosfeer duidelijk hoger dan de fractie aan N2O4 moleculen die gedissocieerd zijn bij eendruk van 10 atmosfeer.
De synthese van ammoniak
Een meer complex voorbeeld om te illustreren hoe we de partieeldrukken kunnen berekenen, is bijde synthese van ammoniak. Stel hiervoor dat we één mol N2 mengen met drie mol H2 volgens destoichiometrische verhouding. We beschouwen het volgend chemisch evenwicht
N2(g) + 3H2(g) −−−− 2NH3(g)
Stoichiometrische coëfficiënt −1 −3 +2Initieel aantal mol (ni ) 1 3 0Aantal mol aanwezig bij evenwicht (ni) 1− ξev 3− 3ξev 0 + 2ξevTotaal aantal mol (n) n = 1− ξev + 3− 3ξev + 2ξev = 4− 2ξev
Molfracties (xi)1− ξev
2 (2− ξev)3 (1− ξev)2 (2− ξev)
2ξev2 (2− ξev)
Partieeldrukken (pi = xip)1− ξev
2 (2− ξev)p3 (1− ξev)2 (2− ξev)p
2ξevp2 (2− ξev)
We zien meteen dat pH2 = 3pN2 . Wanneer we deze waarden invullen in Kp, bekomen we
Kp =p2NH3
pN2p3H2
=p2NH3
pN2(3pN2
)3 =p2NH3
33p4N2
We nemen nu de vierkantswortel en we bekomen
pNH3
p2N2
= 33/2√Kp
of gebruik makende van de partieeldrukken uit de tabel
33/2√Kp =
2ξevp2 (2− ξev)[ 1− ξev
2 (2− ξev)p]2 = 4ξ (2− ξev)
(1− ξev)2 p
De analyse van het mengsel levert de waarde van xNH3 op uit dewelke we de waarde van ξev kunnenbekomen. Uit de experimentele waarde van ξev kunnen we Kp berekenen en uit deze laatste ∆G− .We kunnen eveneens een uitdrukking vinden in termen van pNH3 en de totale druk. Daar p =pN2 + pH2 + pNH3 en pH2 = 3pN2 , vinden we dat p = 4pN2 + pNH3 of pN2 = 1/4(p− pNH3) zodat
pNH3
(p− pNH3)2 = 33/2√Kp
16

78 Hoofdstuk 4. Evenwicht in chemische systemen
We hebben een uitdrukking gevonden om de partieeldruk van NH3 te berekenen voor eender welketotale druk. Wanneer de omzetting naar NH3 laag is, is p−pNH3 ≈ p en pNH3 ≈ 0,325K1/2
p p2 zodatde partieeldruk van ammoniak ongeveer evenredig is aan de vierkantswortel van de druk. Wanneerde reagentia in het begin niet gemengd worden volgens de stoichiometrische verhouding, dan zal deuitdrukking nog meer complex worden.
Enkele besluiten
Als we de evenwichtsconstante uitdrukken in termen van de molfracties die worden gedefinieerddoor
xi = nin
waarbij n het totaal aantal mol in het systeem is, dan zal voor het evenwicht
aA + bB −−−− lL +mM
de evenwichtsconstante worden gedefinieerd als
Kx = xlLxmM
xaAxbB
Als de partieeldruk evenredig is aan de mofractie voor ideale gasmengsels (pi/atm = xi(p/atm),dan kunnen we schrijven dat
Kx = (pL/atm)l (pM/atm)m
(pA/atm)a (pB/atm)b· (p/atm)(a+b−l−m)
ofKx = Kp (p/atm)−∆n
waarbij ∆n = l + m − a − b de verandering in het aantal mol is van de gasvormige stoffen daarde reactie optreedt van links naar rechts. Bijgevolg zullen, tenzij ∆n = 0, de molfracties van decomponenten van het evenwichtsmengsel afhankelijk zijn van de totale druk zelfs wanneer Kp ditniet zal zijn. Wanneer de reactie zodanig is dat de verhoging van het aantal mol gas en bijgevolgeen verhoging in druk de molfractie van de producten zal reduceren in het finale evenwichtsmengsel.We kunnen de afhankelijkheid van Kx op de druk meer algemeen uitdrukken als(
∂ lnKx
∂p
)T
=( lnKp
∂p
)T
−∆n(∂ ln p∂p
)T
Voor ideale gassen is p∆V = ∆nRT . Bijgevolg daar (∂ lnKp/∂p)T = 0, bekomen we dat(∂ lnKx
∂p
)T
= −∆np
= −∆VRT
(4.46)
Deze vergelijking is niet alleen van toepassing op evenwichten waarin gassen zijn betrokken, maarook op evenwichten in oplossing en op elk evenwicht wanneer de evenwichtsconstante wordt uige-drukt in termen van de molfracties eerder dan van de partieeldrukken. In deze omstandigheden is∆V de volumeverandering die gepaard gaat met één mol van een reactie.

4.9. Basisresultaten van de chemische thermodynamica 79
4.9 Basisresultaten van de chemische thermodynamica
De besluiten van de thermodynamica zijn vrij algemeen. Wanneer we thermodynamica toepassenop een evenwicht, zullen we het juiste antwoord verkrijgen, zelfs als we een volledig verkeerd ideehebben over de natuur van de moleculen die het systeem opbouwen. Om die reden kunnen er eenrelatief weinig aantal belangrijke thermodynamische basisvergelijkingen worden gebruikt die eenruim toepasbaar zijn. We zullen deze vergelijkingen nu samenvatten.
4.9.1 De vrije Gibbs energieverandering
Voor eender welk systeem kunnen we bij evenwicht schrijven dat
∆G− = −RT lnK
waarbij K een hoeveelheid is die de evenwichtspositie karakteriseert in termen van de hoeveelheidmateriaal die aanwezig is in het evenwichtsmengsel. Bijgevolg kan K de evenwichtsconstante zijn,de dampdruk, of een oplosbaarheid. ∆G− is de standaard vrije Gibbs energieverandering voor éénmol reactie voor de vergelijking dat het beschrijft bij evenwicht.
4.9.2 De standaard enthalpieverandering
Een tweede basisvergelijking is (∂ lnK∂T
)p
= ∆H−RT 2
waarbij K opnieuw een hoeveelheid is die de postitie op het evenwicht karakteriseert en ∆H− destandaard enthalpieverandering is voor één mol van een reactie.
4.9.3 De invloed van de druk
De invloed van de druk op de positie van het evenwicht kan worden uitgedrukt als(∂ lnKx
∂p
)T
= −∆V −RT
waarbij ∆V − de volumeverandering is die gepaard gaat met één mol reactie onder standaardom-standigheden.
4.9.4 De chemische potentiaal
We kunnen voor de verandering van de vrije Gibbs energie naar de temperatuur en de druk schrijvendat
∆G′ =(∂G
∂ξ
)T,p
= ∆G− +RT ln∏i=1
(cic−
)νi

80 Hoofdstuk 4. Evenwicht in chemische systemen
waarbij (ci/c− ) een maat is voor de hoeveelheid van de ide bestanddeel van het reactiemengselrelatief aan dat in de standaardtoestand c− . Deze vergelijkingen zijn afhankelijk van het feit dat
µi = µ−i +RT ln(cic−
)waarbij (ci/c− ) een geschikte maat vormt van de concentratie i. Bijvoorbeeld (pi/atm) of xi, µ−ide chemische potentiaal is van de standaard toestand dat de toestand is waarvoor ci = c− .
4.10 Het principe van Le Chatelier
De richtingsverandering in de positie van het evenwicht veroorzaakt door de verandering in externeveranderlijken zoals T en p, kunnen algemeen worden gevonden door toepassing van het principevan Le Chatelier dat stelt dat een verstoring van een systeem bij evenwicht zal veroorzaken dat deevenwichtspositie op zo een manier verandert dat het neigt naar het verwijderen van de storing.Wanneer bijvoorbeeld warmte vrijkomt in een reactie ∆H < 0 zal het verlagen van de temperatuurervoor zorgen dat er meer reactie zal optreden zodat het evenwicht wordt verschoven naar dekant van de producten zodat het systeem neigt naar het verhogen van de temperatuur. Wanneereen reactie doorgaat met een positieve volumeverandering, dan zal het toepassen van de druk hetevenwicht verschuiven in de richting van de reagentia. Deze conclusies kunnen kwantitatief wordenuitgedrukt door de vergelijkingen (
∂ lnK∂T
)p
= ∆H−RT 2(
∂ lnKx
∂p
)T
= −∆V −RT
Het principe van Le Chatelier biedt een goede gids in de effecten van de druk en de temperatuurs-veranderingen. Om het universeel geldig te laten maken, moeten we dit principe meer rigoureusstellen. Bijgevolg is het aan te raden om het principe te gebruiken als een geheugensteun eerderdan als een fundamenteel thermodynamisch principe.

Hoofdstuk 5
Bepaling van thermodynamischegrootheden
5.1 De wet van Hess
In 1840 publiceerde Germain Henri Hess (1802–1850) in Sint-Petersburg een studie over de warmtenvan verschillende reacties die aantoonde dat steeds dezelfde hoeveelheid van de totale warmtewerd ontwikkeld waarbij het niet uitmaakte hoeveel tussenstappen er plaatsvonden vooraleer eeneindproduct werd bekomen [15]. Hess toonde bijgevolg voor de enthalpieverandering aan dat diteen toestandsfunctie is waarbij de veranderingen over een complete cyclus gelijk moet zijn aan nulen dat de verandering in elke toestandsfunctie tussen twee toestanden constant is en onafhankelijkvan het pad dat tussen de twee toestanden wordt gevolgd. De enthalpieveranderingen behorendebij sommige reacties zijn relatief gemakkelijk te meten waarbij het voor andere reacties moeilijkerzal zijn.
We nemen als voorbeeld de enthalpieverandering waarbij methaan wordt gevormd uit zijn elemen-ten. Zowel de reagentia als het eindproduct bevinden zich in de standaard toestanden zodat we destandaard vormingsenthalpie kunnen schrijven als ∆H−f
C(s) + 2H2(g) −−→ CH4(g)
De standaardtoestand van elk element wordt gedefinieerd als de meest stabiele vorm bij 1 atm ende vermelde temperatuur waarbij zeer vaak de vormingsenthalpieën worden gemeten en genoteerdbij 298 K. Het is niet gunstig om de rechtstreekse reactie uit te voeren, maar het is wel relatiefeenvoudig om de verbrandingsenthalpie van methaan in een appraat zoals een vlamcalorimeter temeten. Als methaan wordt verbrand met zuurstofgas, zal de geproduceerde warmte meteen deverbrandingsenthalpie geven daar ∆H = (q)p.
CH4(g) + 2O2(g) −−→ CO2(g) + 2H2O(l) ∆H = −890,4 kJ mol−1
C(s) + O2(g) −−→ CO2(g) ∆H = −393,5 kJ mol−1
81

82 Hoofdstuk 5. Bepaling van thermodynamische grootheden
2H2(g) + O2(g) −−→ 2H2O(l) ∆H = −571,6 kJ mol−1
Door de verbrandingsenthalpie van methaan te combineren met deze van koolstof alsook waterstof-gas, kunnen we de wet van Hess toepassen op het probleem. Uit Figuur 5.1 kunnen we inzien dat
CO2(g) + H2O(l)
C(s) + 2H2(g)
CH4(g)
∆Hf
∆H2
+2O2
∆H1
+2O2
Figuur 5.1: De vormingswarmte van methaan uit zijn elementen is equivalent met de twee verbrandings-processen die tezamen worden genomen.
∆H−f = ∆H1 −∆H2
∆H1 is de verbrandingsenthalpie van één mol koolstof en twee mol waterstofgas. Om die reden is
∆H−f (CH4) = [−393,5− 571,6 + 890,4] kJ mol−1
= 74,7 kJ mol−1
Het is niet altijd nodig om de alternatieve wegen uit te tekenen. Als we een reeks chemischereacties uitschrijven tezamen met de enthalpieveranderingen die hen begeleiden, zal de optellingde enthalpieverandering opleveren overeenkomstig met de algemene reactie. De algemene reactieen de samenstellende reacties tezamen representeren twee verschillende manieren van het uivoerenvan hetzelfde proces.
∆H/kJ mol−1
C(s) + O2(g) −−→ CO2(g) −393,52H2(g) + O2(g) −−→ 2H2O(l) −571,6
CO2(g) + 2H2O(l) −−→ CH4(g) + 2O2(g) +890,42H2(g) + C(s) −−→ CH4(g) −74,7
Als de omgekeerde reactie wordt beschouwd, zal de enthalpieverandering zijn teken omkeren.

5.2. Standaard vormingsenthalpieën 83
5.2 Standaard vormingsenthalpieën
De standaard vormingsenthalpieën van chemische stoffen zijn van grote praktische waarde voorchemici. Ze bieden een eenvoudige methode van het bepalen van entropieveranderingen die gepaardgaan met een chemische reactie daar
∆H− =∑
∆H−f (producten)−∑
∆H−f (reagentia) (5.1)


Hoofdstuk 6
Ideale en Reële Oplossingen
Een groot deel van chemische processen worden uitgevoerd in oplossing en toepassing van chemischethermodynamica aan oplossingen is een belangrijk deel van het onderwerp. Oplossingen kunnengasvormig, vloeibaar of vast zijn. Het bestanddeel dat overheerst in de oplossing wordt meestalaangeduid als het solvent. Het bestanddeel dat in mindere mate aanwezig is, wordt meestal deopgeloste stof genoemd. In sommige oplossingen zijn de componenten mengbaar in alle verhoudin-gen. Ethanol en water kunnen bijvoorbeeld mengen om een homogeen mengsel te vormen in eenderwelke relatieve hoeveelheden van ethanol en water. Andere componenten vertonen een gelimiteerdewederzijdse oplosbaarheid. Zo kan bijvoorbeeld slechts een gelimiteerde hoeveelheid natriumchlo-ride in water worden opgelost bij een bepaalde temperatuur. De concentratie van het zout zal dewaarde overeenkomend met een verzadigde oplossing niet overschrijden als er meer natriumchlo-ride wordt toegevoegd. Sommige niet-ionaire stoffen, zoals fenol en water, vertonen eveneens eengelimiteerde wederzijdse oplosbaarheid.
6.1 De ideale oplossing
6.1.1 Algemene kenmerken van de ideale oplossing
Het concept van de ideale oplossing is van groot belang in de thermodynamica. We beschouweneen oplossing bestaande uit verschillende vluchtige bestanddelen in een container dat initieel werdgeledigd. Daar alle componenten vluchtig zijn, zal een deel van de oplossing verdampen om deruimte boven de vloeistof met damp te vullen. Wanneer de oplossing en de damp tot een evenwichtkomen bij een temperatuur T , zal de totale druk in de container de som zijn van de partieeldrukkenvan de verschillende componenten van de oplossing zodat
p = p1 + p2 + · · ·+ pi + · · · (6.1)
De partieeldrukken zijn meetbaar net zoals de molfracties x1, . . . , xi, . . . , in de vloeistof. Wanneeréén van de bestanddelen i aanwezig is in relatief grote hoeveelheden vergeleken met de andere, dankan experimenteel worden gevonden dat
pi = xi p∗i (6.2)
85

86 Hoofdstuk 6. Ideale en Reële Oplossingen
waarbij pi de partieeldruk is van de i-de component in de dampfase in evenwicht met de oplossing,p∗i de dampdruk is van de zuivere vloeibare component i en xi de molfractie is van i in het vloeibaremengsel. Vergelijking 6.2 staat gekend als de wet van Raoult en experimenteel wordt deze wetgevolgd in eender welke oplossing als xi de eenheid zal benaderen. Wanneer de oplossing verdundis in alle componenten, behalve het solvent, zal het solvent steeds de wet van Raoult volgen. Daaralle componenten vluchtig zijn, zal er één worden aangewezen als het solvent. De ideale oplossingis gedefinieerd door de eis dat elke component de wet van Raoult zal volgen over het ganse bereikvan de samenstelling.
6.1.2 De chemische potentiaal in ideale oplossingen
Wanneer de vloeibare fase en dampfase in evenwicht zijn, kunnen we stellen dat voor elke componentin de oplossing geldt dat
µoplossingi = µdampi (6.3)
met µi de chemische potentiaal van de deeltjes i. De chemische potentiaal van een stof in de gasfaseis gerelateerd aan zijn partieeldruk volgens
µdampi = µ−i +RT ln(pi/atm) (6.4)
waarbij µ−i de chemische potentiaal is van de zuivere component i in de gasfase bij een standaardrukvan één atmosfeer. Vergelijking 6.3 kan dus worden geschreven als
µoplossingi = µ−i +RT ln(pi/atm) (6.5)
Voor een zuivere vloeistof i in evenwicht met zijn damp is µ∗i (vloeibaar) = µ∗i (damp) = µ∗i zodatde chemische potentiaal van een zuivere vloeistof wordt gegeven door
µ∗i = µ−i +RT ln(p∗i /atm) (6.6)
Wanneer Vergelijking 6.6 wordt afgetrokken van Vergelijking 6.5, bekomen we
µoplossingi = µ∗i +RT ln pip∗i
(6.7)
Voor een ideale oplossing geldt de wet van Raoult (pi = xi p∗i ) en wanneer we deze relatie invullen
in Vergelijking 6.7 wordt de belangrijke vergelijking voor ideale oplossingen bekomen.
µoplossingi = µ∗i +RT ln xi (6.8)
Vergelijking 6.8 brengt de chemische potentiaal van een component in een ideale oplossing in ver-band met de chemische potentiaal van een component i en de molfractie van deze component inde oplossing. Deze vergelijking is heel bruikbaar in het beschrijven van de thermodynamica vanoplossingen waarbij alle componenten vluchtig zijn en mengbaar in alle verhoudingen.

6.1. De ideale oplossing 87
6.1.3 Binaire oplossingen
Hoewel het ideale oplossingsmodel kan toegepast worden op eender welk aantal componenten, zullenwe de wiskunde vereenvoudigen door binaire oplossingen bestaande uit twee vluchtige componentente bestuderen. In een binaire oplossing is x1 + x2 = 1 en volgens de wet van Raoult kunnen weschrijven dat
p1 = x1p∗1 (6.9)
enp2 = x2p
∗2 = (1− x1) p∗2 (6.10)
De totale druk p over de oplossing wordt dan
p = p1 + p2 = x1p∗1 + (1− x1) p∗2 = p∗2 + (p∗1 − p∗2)x1 (6.11)
dat relateert aan de totale druk over het mengsel naar de molfractie van de eerste component inde vloeistof. In Figuur 6.1 worden simulaties getoond van Vergelijkingen 6.9 en 6.10 alsook van
0 1
p1
p∗1
p2
p∗2
Vloeistof
p
x1
Figuur 6.1: De totale dampdruk in evenwicht met een vloeibaar binair mengsel als een functie van desamenstelling volgens de wet van Raoult. De streepjeslijnen zijn de partiële dampdrukken van de tweecomponenten.
Vergelijking 6.11 voor de totale dampdruk van een willekeurig binair vloeibaar mengsel. De totaledampdruk p is een lineaire functie van x1. Het toevoegen van een opgeloste stof kan de damp-druk van het solvent doen verhogen of verlagen afhankelijk van dewelke het meest vluchtige zalzijn. In de meeste gevallen zullen er (kleine) afwijkingen van de wet van Raoult worden waar-genomen. In Figuur 6.2 worden de individuele partieeldrukken alsook de totale druk boven een
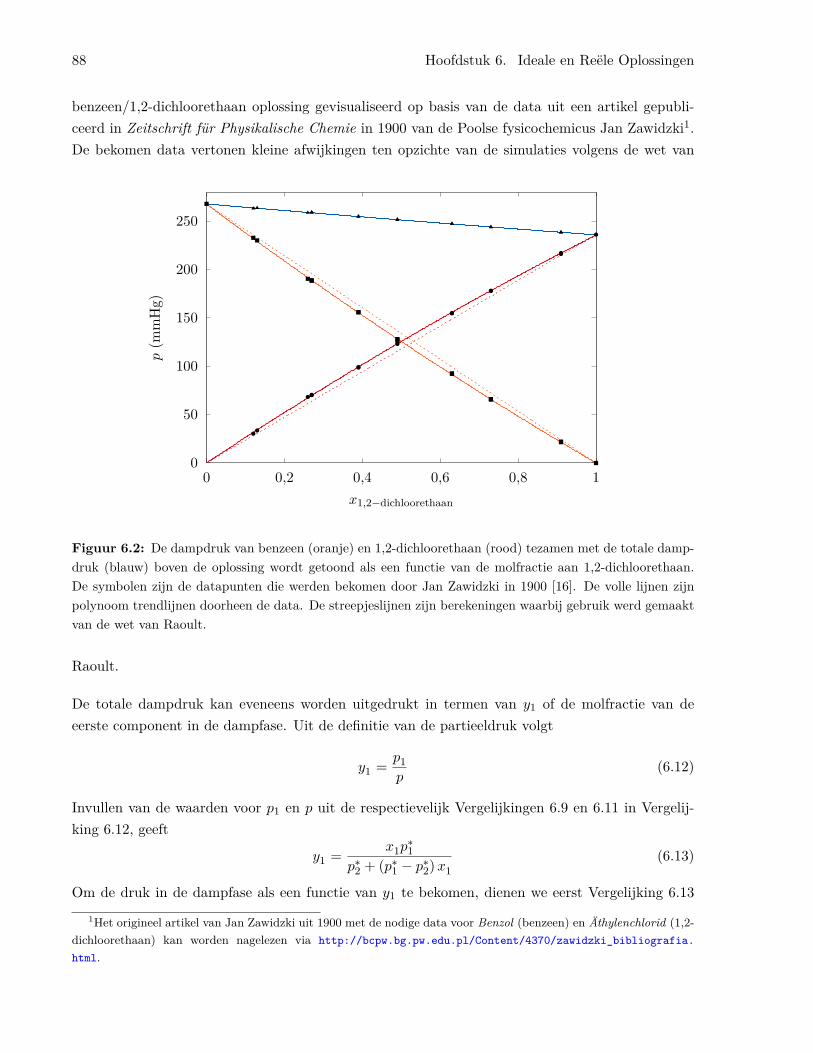
88 Hoofdstuk 6. Ideale en Reële Oplossingen
benzeen/1,2-dichloorethaan oplossing gevisualiseerd op basis van de data uit een artikel gepubli-ceerd in Zeitschrift für Physikalische Chemie in 1900 van de Poolse fysicochemicus Jan Zawidzki1.De bekomen data vertonen kleine afwijkingen ten opzichte van de simulaties volgens de wet van
0
50
100
150
200
250
0 0,2 0,4 0,6 0,8 1
p(m
mHg)
x1,2−dichloorethaan
Figuur 6.2: De dampdruk van benzeen (oranje) en 1,2-dichloorethaan (rood) tezamen met de totale damp-druk (blauw) boven de oplossing wordt getoond als een functie van de molfractie aan 1,2-dichloorethaan.De symbolen zijn de datapunten die werden bekomen door Jan Zawidzki in 1900 [16]. De volle lijnen zijnpolynoom trendlijnen doorheen de data. De streepjeslijnen zijn berekeningen waarbij gebruik werd gemaaktvan de wet van Raoult.
Raoult.
De totale dampdruk kan eveneens worden uitgedrukt in termen van y1 of de molfractie van deeerste component in de dampfase. Uit de definitie van de partieeldruk volgt
y1 = p1p
(6.12)
Invullen van de waarden voor p1 en p uit de respectievelijk Vergelijkingen 6.9 en 6.11 in Vergelij-king 6.12, geeft
y1 = x1p∗1
p∗2 + (p∗1 − p∗2)x1(6.13)
Om de druk in de dampfase als een functie van y1 te bekomen, dienen we eerst Vergelijking 6.131Het origineel artikel van Jan Zawidzki uit 1900 met de nodige data voor Benzol (benzeen) en Äthylenchlorid (1,2-
dichloorethaan) kan worden nagelezen via http://bcpw.bg.pw.edu.pl/Content/4370/zawidzki_bibliografia.html.

6.1. De ideale oplossing 89
uit te werken naar x1. Dit levert op dat
x1 = y1p∗2
p∗1 + (p∗2 − p∗1) y1(6.14)
Door Vergelijking 6.14 nu in te vullen in Vergelijking 6.11, bekomen we na algebraïsch omrekenendat
p = p∗1p∗2
p∗1 + (p∗2 − p∗1) y1(6.15)
Vergelijking 6.15 kan worden herschikt om een uitdrukking te bekomen voor y1 in termen van dedampdrukken van de zuivere bestanddelen en de totale druk volgens
y1 = p∗1p− p∗1p∗2p (p∗1 − p∗2) (6.16)
De verandering van de totale druk p in functie van respectievelijk x1 en y1 is niet hetzelfde. InFiguur 6.1 bestaat het systeem uit een enkelfasige vloeistof voor totaaldrukken boven de rechteen een tweefasig damp-vloeistof mengsel voor p − x1 punten die liggen op de rechte. Enkel depunten die boven de rechte liggen zijn betekenisvol, daar de punten die onder de curve liggenniet overeenkomen met evenwichtstoestanden waarvoor de vloeistof aanwezig is. Wanneer hetsysteem in een dergelijke onstabiele toestand wordt gebracht, zal de vloeistof verdampen zodat dedruk zal worden verhoogd om de waarde te bereiken zodat de vloeistof-damp fase wordt bereikt.Wanneer er niet voldoende vloeistof aanwezig is om de druk te verhogen totaan de rechte, zal allevloeistof verdampen en zal x1 niet langer een betekenisvolle variabele zijn. In Figuur 6.3 bestaat hetsysteem uit een enkelfasige damp voor drukken beneden de kromme en voor een tweefasige damp-vloeistof mengsel voor p− y1 punten die liggen op de kromme. Omwille van analoge redeneringenzoals hierboven beschreven, zullen punten die boven de kromme liggen niet overeenkomen metevenwichtstoestanden. De overmaat aan damp zou condenseren om een vloeistof te bekomen.
Voorbeeld 6.1Bij 353 K zijn de dampdrukken van benzeen en broombenzeen respectievelijk 757 en 66 mmHg.Bereken voor een equimolair mengsel (xbenzeen = xbroombenzeen = 0,5) de totale dampdruk alsookde molfractie aan benzeen in de dampfase. Neem aan dat het mengsel de wet van Raoult volgt.
Oplossing
De totale druk kan berekend worden volgens de wet van Raoult.
p = pbenzeen + pbroombenzeen
= xbenzeenp∗benzeen + xbroombenzeenp
∗broombenzeen
= 0,5 · 757 mmHg + 0,5 · 66 mmHg = 412 mmHg
De waarde van 412 mmHg ligt dicht tegen de experimentele waarde van 407 mmHg waardoor deoplossing de wet van Raoult relatief dicht benaderd.De mofractie benzeen in de dampfase kan worden geschat door
ybenzeen = xbenzeenp∗benzeen
p= 0,50 · 757 mmHg
412 mmHg = 0,92
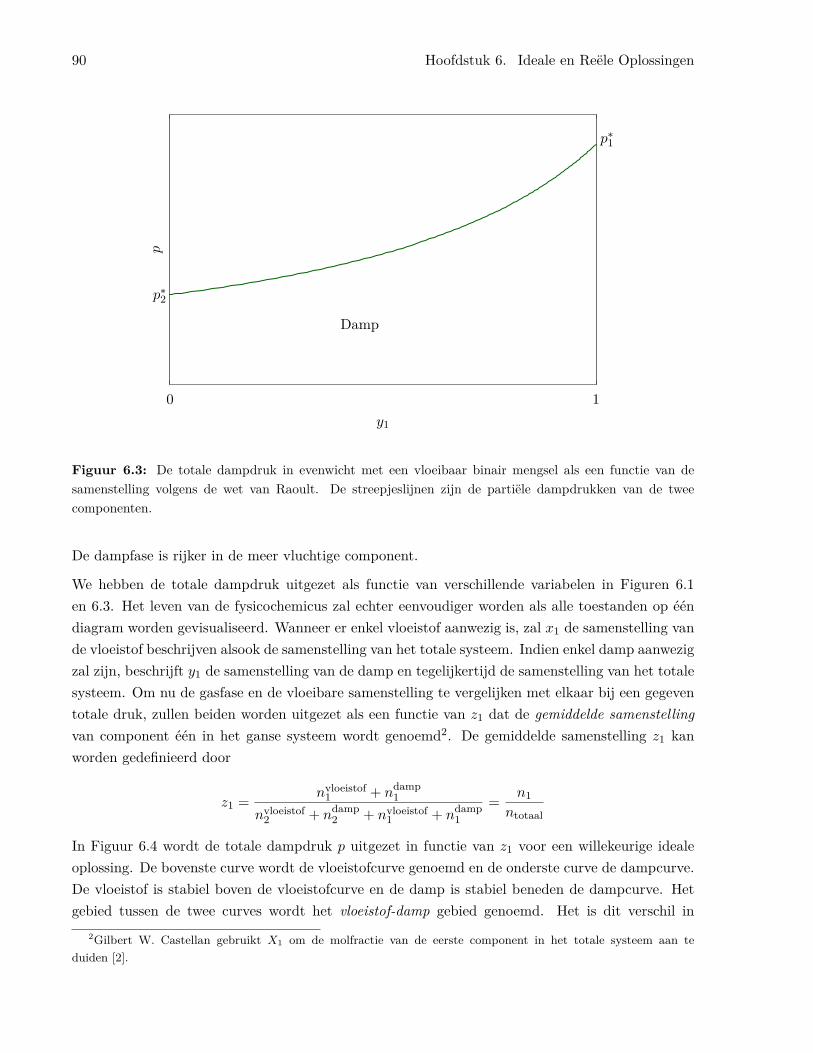
90 Hoofdstuk 6. Ideale en Reële Oplossingen
0 1
p∗1
p∗2
Damp
p
y1
Figuur 6.3: De totale dampdruk in evenwicht met een vloeibaar binair mengsel als een functie van desamenstelling volgens de wet van Raoult. De streepjeslijnen zijn de partiële dampdrukken van de tweecomponenten.
De dampfase is rijker in de meer vluchtige component.
We hebben de totale dampdruk uitgezet als functie van verschillende variabelen in Figuren 6.1en 6.3. Het leven van de fysicochemicus zal echter eenvoudiger worden als alle toestanden op ééndiagram worden gevisualiseerd. Wanneer er enkel vloeistof aanwezig is, zal x1 de samenstelling vande vloeistof beschrijven alsook de samenstelling van het totale systeem. Indien enkel damp aanwezigzal zijn, beschrijft y1 de samenstelling van de damp en tegelijkertijd de samenstelling van het totalesysteem. Om nu de gasfase en de vloeibare samenstelling te vergelijken met elkaar bij een gegeventotale druk, zullen beiden worden uitgezet als een functie van z1 dat de gemiddelde samenstellingvan component één in het ganse systeem wordt genoemd2. De gemiddelde samenstelling z1 kanworden gedefinieerd door
z1 = nvloeistof1 + ndamp1
nvloeistof2 + ndamp2 + nvloeistof1 + ndamp
1= n1ntotaal
In Figuur 6.4 wordt de totale dampdruk p uitgezet in functie van z1 voor een willekeurige idealeoplossing. De bovenste curve wordt de vloeistofcurve genoemd en de onderste curve de dampcurve.De vloeistof is stabiel boven de vloeistofcurve en de damp is stabiel beneden de dampcurve. Hetgebied tussen de twee curves wordt het vloeistof-damp gebied genoemd. Het is dit verschil in
2Gilbert W. Castellan gebruikt X1 om de molfractie van de eerste component in het totale systeem aan teduiden [2].
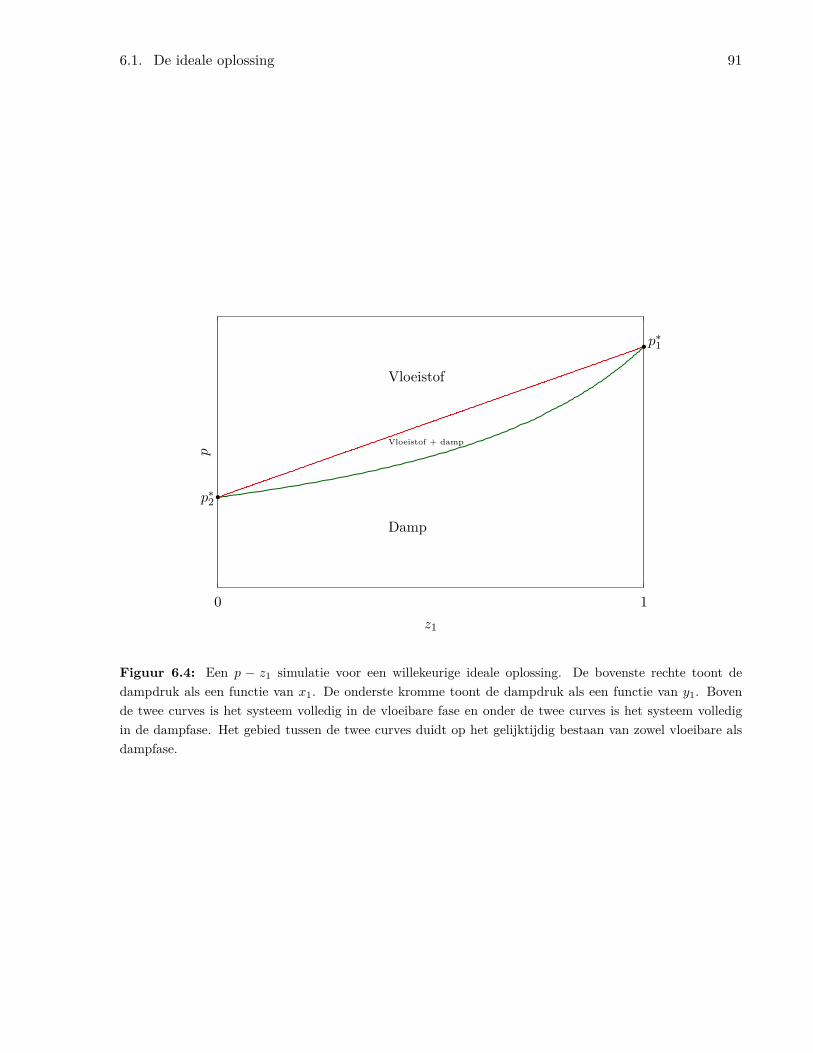
6.1. De ideale oplossing 91
0 1
p∗1
p∗2
Damp
Vloeistof
Vloeistof + damp
p
z1
Figuur 6.4: Een p − z1 simulatie voor een willekeurige ideale oplossing. De bovenste rechte toont dedampdruk als een functie van x1. De onderste kromme toont de dampdruk als een functie van y1. Bovende twee curves is het systeem volledig in de vloeibare fase en onder de twee curves is het systeem volledigin de dampfase. Het gebied tussen de twee curves duidt op het gelijktijdig bestaan van zowel vloeibare alsdampfase.

92 Hoofdstuk 6. Ideale en Reële Oplossingen
samenstelling van vloeistof en damp dat ons in staat stelt om bestanddelen in een mengsel tekunnen scheiden door middel van destillaties.
6.1.4 Het benzeen/tolueen systeem
Bij 298 K zijn de dampdrukken van de zuivere stoffen benzeen en tolueen respectievelijk p∗benzeen =96,4 mmHg en p∗tolueen = 28,9 mmHg. Mengsels van benzeen en tolueen benaderen heel sterkideale oplossingen. Een p − z-fasediagram wordt voor een benzeen-tolueen ideale oplossing wordtgevisualiseerd in Figuur 6.5. Om de bruikbaarheid van een druk-gemiddelde samenstelling (p− z)
20
30
40
50
60
70
80
90
100
0 0,2 0,4 0,6 0,8 1
a
b
c
l v
p(m
mHg)
zbenzeen
Figuur 6.5: Een p−z fasediagram voor een benzeen-tolueen ideale oplossing. Voor de simulatie werd gebruikgemaakt van p∗
benzeen = 96,4 mmHg en p∗tolueen = 28,9 mmHg. De bovenste rechte toont de dampdruk als
een functie van xbenzeen. De onderste kromme toont de dampdruk als een functie van ybenzeen. Boven detwee curves is het systeem volledig in de vloeibare fase en onder de twee curves is het systeem volledig inde dampfase. Het gebied tussen de twee curves duidt op het gelijktijdig bestaan van zowel vloeibare alsdampfase.
diagramma te demonstreren, beschouwen een proces bij een constante temperatuur waarbij de drukvan het benzeen/tolueen-systeem wordt verlaagd vanaf de waarde overeenkomend met het punt ain Figuur 6.5. Daar p > ptotaal, is het systeem initieel volledig in de vloeibare fase. Wanneerde druk wordt verlaagd bij een constante samenstelling, zal het systeem volleidg in de vloeibarefase blijven totdat de constante samenstellingslijn de p versus xbenzeen curve zal snijden. Vanaf ditpunt zal het systeem terechtkomen in het twee-fase gebied waar zowel damp als vloeistof samenvoorkomen. Wat zullen de waarden van de molfractie van de dampfase ( xbenzeen) en de molfractievan de vloeibare fase (ybenzeen) zijn wanneer het punt b wordt bereikt? Deze waarden kunnen

6.1. De ideale oplossing 93
worden bepaald door een verbindingslijn te construeren in het gebied waar de twee fasen samenvoorkomen. De verbindingslijn in Figuur 6.5 is een horizontale lijn bij de druk die ons interesseertdat de p versus xbenzeen en de p versus ybenzeen curven snijden. Voor alle waarden van de druk zalybenzeen steeds groter zijn dan xbenzeen dat ons aantoont dat de dampfase steeds verrijkt zal zijn inde meer vluchtigere of hogere dampdruk component in vergelijking met de vloeibare fase.
Voorbeeld 6.2Een ideale oplossing bestaat uit 5,00 mol benzeen en 3,25 mol tolueen. Bij 298 K zijn de damp-drukken van de zuivere stoffen respectievelijk 96,4 mmHg en 28,9 mmHg.
a) De druk boven de oplossing wordt gereduceerd vanaf 760 mmHg. Bij welke druk zal dedampfase voor het eerst verschijnen?
b) Wat is de samenstelling van de damp bij deze voorwaarden?
Oplossing
a) De molfracties van de componenten in de oplossing zijn
xbenzeen = 5,00 mol5,00 mol + 3,25 mol = 0,606 mol
xtolueen = 3,25 mol5,00 mol + 3,25 mol = 0,394 mol
De totale dampdruk boven de oplossing is bijgevolg
p = pbenzeen + ptolueen
= xbenzeenp∗benzeen + xtolueenp
∗tolueen
= 0,606 · 96,4 mmHg + 0,394 · 28,9 mmHg = 69,8 mmHg
Er zal geen damp worden gevormd totdat de druk zal verlaagd zijn totaan deze waarde.
b) De samenstelling van de damp bij een totale druk van 69,8 mmHg wordt met behulp vanVergelijking 6.16 gegeven door
ybenzeen = p∗benzeenp− p∗benzeenp∗tolueenp (p∗benzeen − p∗tolueen)
= 96,4 mmHg · 69,8 mmHg− 96,4 mmHg · 28,9 mmHg69,8 mmHg (96,4 mmHg− 28,9 mmHg) = 0,837
ytolueen = 1− ybenzeen = 0,163
Merk op dat de damp meer verrijkt is ten opzichte van de vloeistof in de meer vluchtigecomponent die de lagere kooktemperatuur bezit.
Om de relatieve hoeveelheid aan materiaal te berekenen van elk van de twee fasen in het gebiedwaar ze samen voorkomen, kunnen we een hefboomregel voor binaire oplossingen afleiden gebruik

94 Hoofdstuk 6. Ideale en Reële Oplossingen
makend van een geometrisch argument. De lengten van de lijnsegmenten lb en bv in Figuur 6.5worden gegeven door
lb = zbenzeen − xbenzeen = ntotaalbenzeenntotaal
− nvloeistofbenzeenntotaalvloeistof
(6.17)
bv = ybenzeen − zbenzeen = ndampbenzeenntotaaldamp
− ntotaalbenzeenntotaal
(6.18)
We vermenigvuldigen Vergelijking 6.17 met ntotaalvloeistof en Vergelijking 6.18 met ntotaaldamp . Vervolgenstrekken we de twee vergelijkingen van elkaar af en bekomen
lb · ntotaalvloeistof − bv · ntotaaldamp = ntotaalbenzeenntotaal
(ntotaalvloeistof + ntotaaldamp
)−(nvloeistofbenzeen + ndamp
benzeen
)= ntotaalbenzeen − ntotaalbenzeen = 0
We besluiten datlb · ntotaalvloeistof = bv · ntotaaldamp of ntotaalvloeistof
ntotaaldamp= bv
lb(6.19)
We kunnen dit resultaat ook herformuleren als
ntotaalvloeistof (zbenzeen − xbenzeen) = ntotaaldamp (ybenzeen − zbenzeen) (6.20)
Vergelijking 6.20 wordt de hefboomregel genoemd in analogie met rotationele krachten die optredenop een hefboom waarvan de lengte gelijk is aan lb+ bv met het steunpunt gepositioneerd op punt bin Figuur 6.5. De simulatie in dit specifieke voorbeeld werd uitgevoerd bij een druk van 65 mmHgvoor zbenzeen = 0,70 waarbij xbenzeen = 0,535 en ybenzeen = 0,793. Voor deze simulatie vinden wemet behulp van de hefboomregel dat
ntotaalvloeistofntotaaldamp
= bv
lb= ybenzeen − zbenzeenzbenzeen − xbenzeen
= 0,793− 0,700,70− 0,535 = 0,564
Dit betekent datntotaalvloeistof = 0,564 · ntotaaldamp = 0,564 ·
(ntotaal − ntotaalvloeistof
)zodat
1,564 · ntotaalvloeistof = 0,564 · ntotaal
ofntotaalvloeistof = 0,564
1,564 · ntotaal = 0,36 · ntotaal
Het totaal aantal mol dat zich in het benzeen/tolueen systeem zal bevinden in de vloeibare faseis bijgevolg 36% zodat het totaal aantal mol dat zich in de dampfase zal bevinden gelijk zal zijnaan 64%. We merken dus op dat wanneer b zeer dicht tegen v zou liggen, bv heel klein zal zijnen bijgevolg ntotaalvloeistof ntotaaldamp zodat het systeem hoofdzakelijk uit damp zal bestaan. Wanneer banderzijds dicht zal liggen bij l, is lb klein zodat ntotaaldamp ntotaalvloeistof en het systeem hoofdzakelijk zalbestaan uit vloeistof.

6.1. De ideale oplossing 95
Voorbeeld 6.3Voor de benzeen/tolueen oplossing in Voorbeeld 6.2, bereken
a) de totale druk;
b) de vloeistofsamenstelling;
c) de dampsamenstelling wanneer 1,50 mol van de oplossing omgezet is naar damp.
Oplossing
In totaal zijn er 8,25 mol bestanddelen zodat ntotaaldamp = 1,50 mol en ntotaalvloeistof = 6,75 mol. Invullen vandeze waarden in Vergelijking 6.20 voor de hefboomregel die de gemiddelde samenstelling zbenzeen =0,606 relateert aan de vloeibare en damp samenstellingen, levert
ntotaaldamp (ybenzeen − zbenzeen) = ntotaalvloeistof (zbenzeen − xbenzeen)1,50 · (ybenzeen − 0,606) = 6,75 · (0,606− xbenzeen)
zodat6,75 · xbenzeen + 1,50 · ybenzeen = 5,00
De totale druk wordt gegeven door
p = xbenzeenp∗benzeen + xtolueenp
∗tolueen
= xbenzeenp∗benzeen + (1− xbenzeen) p∗tolueen
= [96,4 · xbenzeen + 28,9 (1− xbenzeen)] mmHg
De dampsamenstelling wordt gegeven door
ybenzeen = p∗benzeenp− p∗benzeenp∗tolueenp (p∗benzeen − p∗tolueen)
= 96,4 mmHg · (p/mmHg)− 278667,5 mmHg · (p/mmHg)
We hebben drie vergelijkingen en drie onbekenden die opgelost kunnen worden door de variabelente elimineren door de vergelijkingen te combineren. De eerste vergelijking kan bijvoorbeeld wordengebruikt om ybenzeen uit te drukken in termen van xbenzeen en p. Dit resultaat kan vervolgensworden gesubstitueerd in de tweede en derde vergelijkingen zodat er twee vergelijkingen in termenvan xbenzeen en p worden bekomen. De oplossing voor xbenzeen bekomen uit deze twee vergelijkingenkan dan gesubstitueerd worden in de eerste vergelijking om ybenzeen te bekomen. De antwoordenzijn dan xbenzeen = 0,561, ybenzeen = 0,810 en p = 66,8 mmHg.
6.1.5 Temperatuur-samenstellingsdiagrammen en gefractioneerde destillatie
De verrijking van de dampfase boven een oplossing in de meer vluchtigere component is de basisvoor de gefractioneerde destillatie dat een belangrijke techniek is in scheikunde en in de chemi-sche industrie. Om gefractioneerde destillaties te bespreken is het echter meer geschikt om dit te
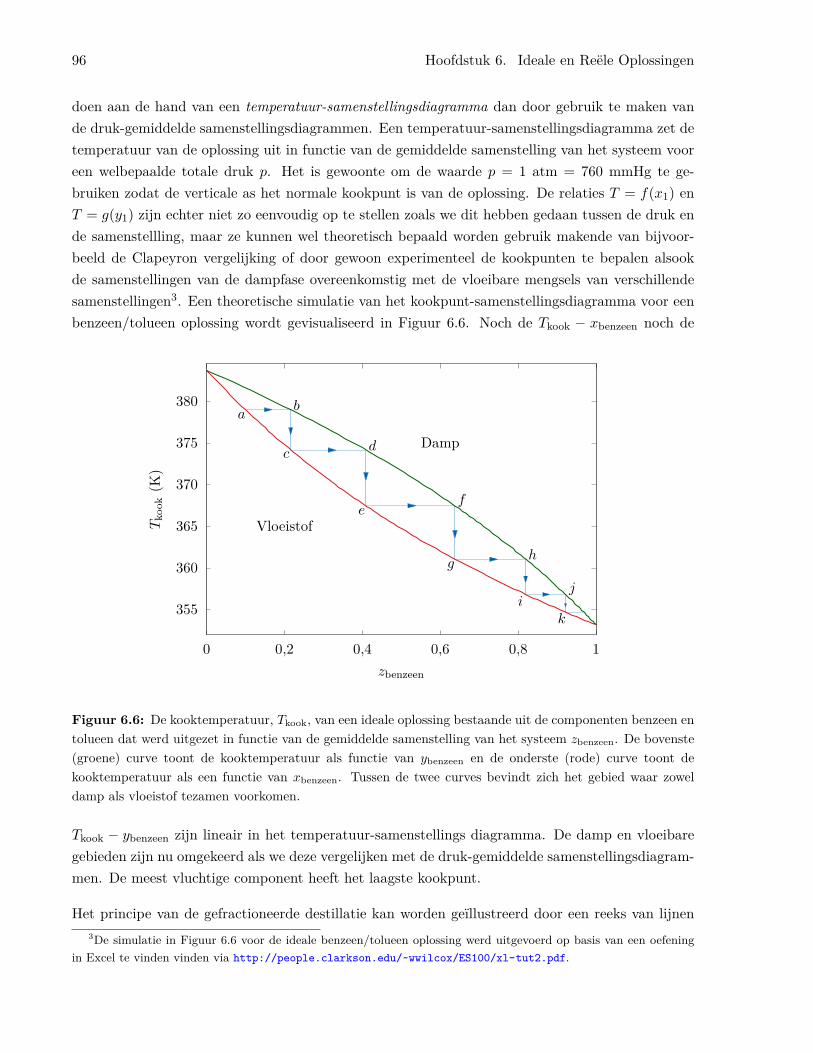
96 Hoofdstuk 6. Ideale en Reële Oplossingen
doen aan de hand van een temperatuur-samenstellingsdiagramma dan door gebruik te maken vande druk-gemiddelde samenstellingsdiagrammen. Een temperatuur-samenstellingsdiagramma zet detemperatuur van de oplossing uit in functie van de gemiddelde samenstelling van het systeem vooreen welbepaalde totale druk p. Het is gewoonte om de waarde p = 1 atm = 760 mmHg te ge-bruiken zodat de verticale as het normale kookpunt is van de oplossing. De relaties T = f(x1) enT = g(y1) zijn echter niet zo eenvoudig op te stellen zoals we dit hebben gedaan tussen de druk ende samenstellling, maar ze kunnen wel theoretisch bepaald worden gebruik makende van bijvoor-beeld de Clapeyron vergelijking of door gewoon experimenteel de kookpunten te bepalen alsookde samenstellingen van de dampfase overeenkomstig met de vloeibare mengsels van verschillendesamenstellingen3. Een theoretische simulatie van het kookpunt-samenstellingsdiagramma voor eenbenzeen/tolueen oplossing wordt gevisualiseerd in Figuur 6.6. Noch de Tkook − xbenzeen noch de
355
360
365
370
375
380
0 0,2 0,4 0,6 0,8 1
ab
cd
ef
gh
ij
k
Tko
ok(K
)
zbenzeen
Damp
Vloeistof
Figuur 6.6: De kooktemperatuur, Tkook, van een ideale oplossing bestaande uit de componenten benzeen entolueen dat werd uitgezet in functie van de gemiddelde samenstelling van het systeem zbenzeen. De bovenste(groene) curve toont de kooktemperatuur als functie van ybenzeen en de onderste (rode) curve toont dekooktemperatuur als een functie van xbenzeen. Tussen de twee curves bevindt zich het gebied waar zoweldamp als vloeistof tezamen voorkomen.
Tkook − ybenzeen zijn lineair in het temperatuur-samenstellings diagramma. De damp en vloeibaregebieden zijn nu omgekeerd als we deze vergelijken met de druk-gemiddelde samenstellingsdiagram-men. De meest vluchtige component heeft het laagste kookpunt.
Het principe van de gefractioneerde destillatie kan worden geïllustreerd door een reeks van lijnen3De simulatie in Figuur 6.6 voor de ideale benzeen/tolueen oplossing werd uitgevoerd op basis van een oefening
in Excel te vinden vinden via http://people.clarkson.edu/~wwilcox/ES100/xl-tut2.pdf.

6.1. De ideale oplossing 97
te volgen vertrekkende van a naar k in Figuur 6.6.
6.1.6 Ideale oplossingen van vaste stoffen in vloeistoffen
We beschouwen een vaste stof die we oplossen in een vloeistof om een ideale oplossing te vormenzoals is gevisualiseerd in Figuur 6.7. De opgeloste stof zal in de vaste fase de laagste energie
Vaste stof
Oplossing
Figuur 6.7: Evenwicht tussen een opgeloste stof in de vaste fase en in oplossing.
bezitten, maar het oplossen wordt begunstigd door een winst in entropie daar de opgeloste stofwordt gedispergeerd in de oplossing. Op het evenwicht zal de chemische potentiaal van de vastestof gelijk zijn aan de chemische potentiaal van dezelfde stof in de oplossing. Voor een idealeoplossing geldt dat
µ−2 (s) = µ−2 (oplossing) = µ−2 (l) +RT ln x2
(Het subscript 2 wordt gewoonlijk gebruikt om de vaste stof aan te geven en het subscript 1 hetsolvent.) Daarom kunnen we schrijven dat
RT ln x2 = µ−2 (s)− µ−2 (l) = −∆G−2fuswaarbij ∆G−2fus de vrije energieverandering is van het smelten (fusie). Uit de Gibbs-Helmholtzvergelijking ∂ (∆G2
T
)∂T
p
= −∆H2T 2
zodat (∂ ln x2∂T
)p
= ∆H−fusRT 2
met ∆H−fus de smeltenthalpie van de vaste stof. Op het smeltpunt van de vaste stof, Tfus, is deoplosbaarheid x2 gelijk aan een eenheid daar de twee vloeistoffen die een ideale oplossing vormenvolledig mengbaar zijn (ze zullen mengen in alle verhoudingen om een homogene oplossing tevormen). We kunnen de bovenstaande vergelijking vervolgens integreren en bekomen dat
ln x2 − ln(1) =∫ T
Tfus
∆H−fusRT 2 dT = −
[∆H−fusRT
]Tfus
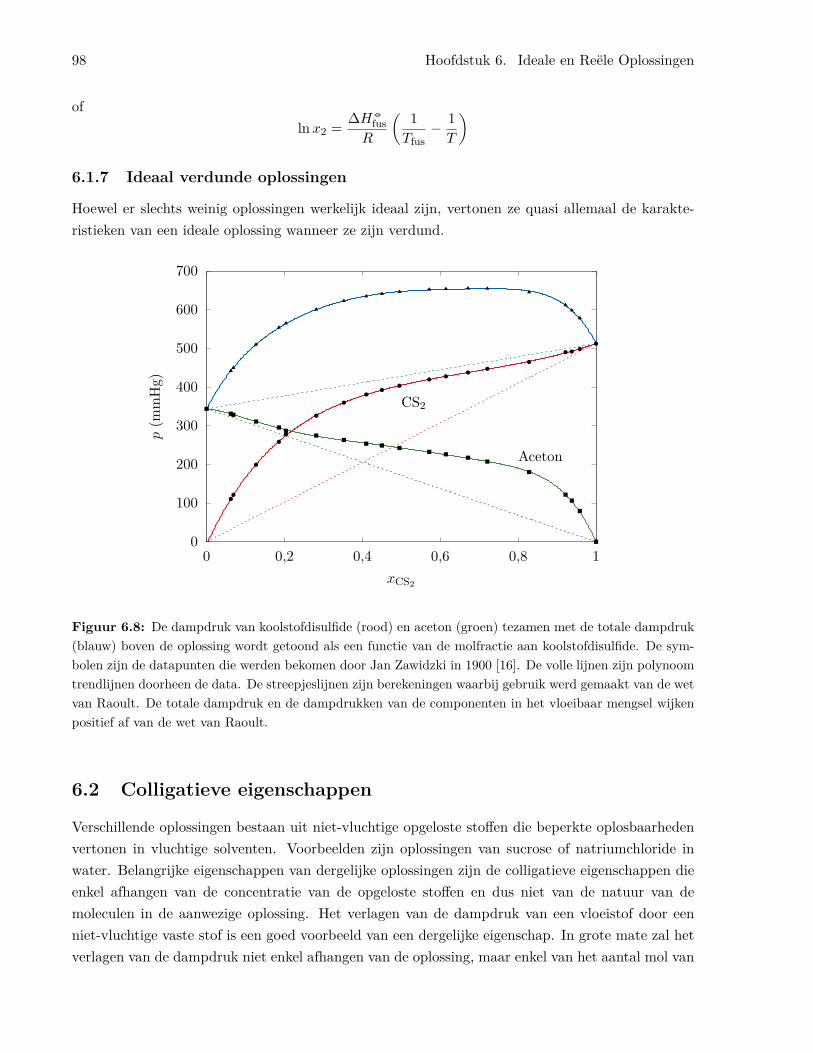
98 Hoofdstuk 6. Ideale en Reële Oplossingen
ofln x2 = ∆H−fus
R
( 1Tfus− 1T
)
6.1.7 Ideaal verdunde oplossingen
Hoewel er slechts weinig oplossingen werkelijk ideaal zijn, vertonen ze quasi allemaal de karakte-ristieken van een ideale oplossing wanneer ze zijn verdund.
0
100
200
300
400
500
600
700
0 0,2 0,4 0,6 0,8 1
CS2
Aceton
p(m
mHg)
xCS2
Figuur 6.8: De dampdruk van koolstofdisulfide (rood) en aceton (groen) tezamen met de totale dampdruk(blauw) boven de oplossing wordt getoond als een functie van de molfractie aan koolstofdisulfide. De sym-bolen zijn de datapunten die werden bekomen door Jan Zawidzki in 1900 [16]. De volle lijnen zijn polynoomtrendlijnen doorheen de data. De streepjeslijnen zijn berekeningen waarbij gebruik werd gemaakt van de wetvan Raoult. De totale dampdruk en de dampdrukken van de componenten in het vloeibaar mengsel wijkenpositief af van de wet van Raoult.
6.2 Colligatieve eigenschappen
Verschillende oplossingen bestaan uit niet-vluchtige opgeloste stoffen die beperkte oplosbaarhedenvertonen in vluchtige solventen. Voorbeelden zijn oplossingen van sucrose of natriumchloride inwater. Belangrijke eigenschappen van dergelijke oplossingen zijn de colligatieve eigenschappen dieenkel afhangen van de concentratie van de opgeloste stoffen en dus niet van de natuur van demoleculen in de aanwezige oplossing. Het verlagen van de dampdruk van een vloeistof door eenniet-vluchtige vaste stof is een goed voorbeeld van een dergelijke eigenschap. In grote mate zal hetverlagen van de dampdruk niet enkel afhangen van de oplossing, maar enkel van het aantal mol van

6.2. Colligatieve eigenschappen 99
de aanwezige opgeloste stoffen. Wanneer de dampdruk van het solvent de wet van Raoult volgt,dan zal p1 = p∗1x1. De opgeloste stof komt in deze vergelijking enkel inzoverre de molfractie x2 demolfractie x1, de molfractie van het solvent,
6.2.1 Vriespuntsverlaging
Wanneer een opgeloste stof wordt opgelost in een vloeistof, dan wordt het vriespunt van de vloeistofgewoonlijk verlaagd. De thermodynamica van dit proces is eenvoudig wanneer de opgeloste stofenkel oplost in de vloeibare fase van het solvent en geen vaste oplossingen ermee vormt.


Hoofdstuk 7
Niet-ideale oplossingen
7.1 Het concept van de activiteit
Voor veel reële oplossingen zal de vergelijking µi = µ−i (l) + RT ln xi niet standhouden voor hetgebruikte solvent en de uitdrukking die overeenkomt met de wet van Henry zal eveneens nietstandhouden voor de opgeloste stof. In dergelijke omstandigheden zal het geschikt zijn om hetconcept van de activiteit in te voeren. We mogen schrijven dat
µi = µ−i +RT ln ai (7.1)
Deze vergelijking definieert de activiteit ai van de ide component in termen van zijn chemischepotentiaal in de oplossing en in de standaard toestand. Deze activiteit kan worden gezien als deeffectieve concentratie van de component i relatief aan zijn standaardtoestand. De effectieve con-centratie kan in een reële oplossing enorm verschillen van de ware concentratie als gevolg van deinteracties tussen de moleculen. In hoeverre dit zo zal zijn, wordt gedefinieerd door de activiteits-coëfficiënt γi.
γu = aixi
= Effectieve concentratieReële concentratie (7.2)
Dit kan worden geïllustreerd door een reëel vloeibaar mengsel te beschouwen. In Figuur 7.1 wordende dampdrukken van een oplossing waarbij de dampdrukken van de componenten negatief afwijkenvan de wet van Raoult gevisualiseerd (de dampdrukken zijn lager dan deze zoals ze voorspeldworden door de wet). Het uitschrijven van de vergelijkingen voor de chemische potentiaal van ééncomponent in zowel de oplossing als in de dampfase,
µi(g) = µ−i (g) +RT ln (pi/atm)µi (oplossing) = µ−i (l) +RT ln ai
Daarµ−i (l) = µ−i (g) +RT ln (p∗i /atm)
als de zuivere vloeistof i in evenwicht moet zijn met zijn damp bij de verzadigde dampdruk. Bijge-volg bekomen we, daar µi(g) = µi(oplossing), dat
ai = pip∗i
101
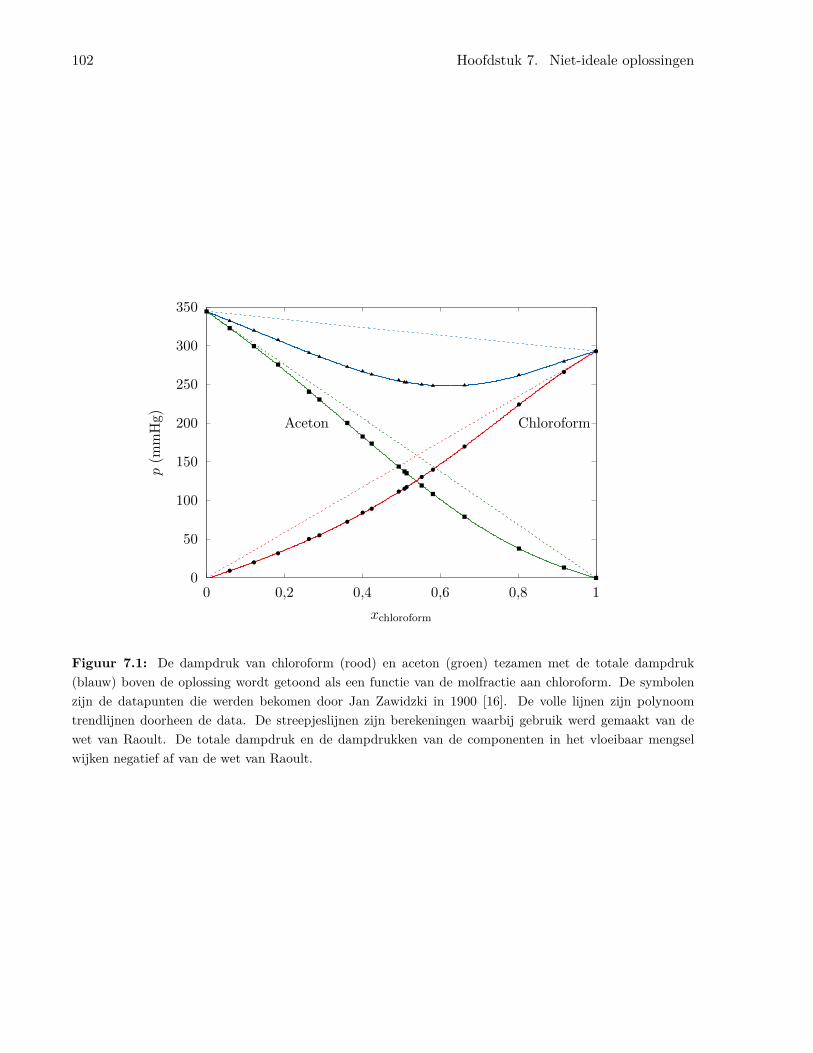
102 Hoofdstuk 7. Niet-ideale oplossingen
0
50
100
150
200
250
300
350
0 0,2 0,4 0,6 0,8 1
Aceton Chloroform
p(m
mHg)
xchloroform
Figuur 7.1: De dampdruk van chloroform (rood) en aceton (groen) tezamen met de totale dampdruk(blauw) boven de oplossing wordt getoond als een functie van de molfractie aan chloroform. De symbolenzijn de datapunten die werden bekomen door Jan Zawidzki in 1900 [16]. De volle lijnen zijn polynoomtrendlijnen doorheen de data. De streepjeslijnen zijn berekeningen waarbij gebruik werd gemaakt van dewet van Raoult. De totale dampdruk en de dampdrukken van de componenten in het vloeibaar mengselwijken negatief af van de wet van Raoult.

7.2. Activiteit van vaste stoffen in vloeistoffen 103
rekening houdend met de definitie van de activiteit als de effectieve concentratie. In het voorbeeldhebben we dat pi < pi(ideaal). De concentratie van i in de dampfase is lager dan dat we zoudenverwachten voor een oplossing van een dergelijke concentratie op basis van het ideale gedrag enai < xi. De effectieve concentratie is lager dan de reële concentratie en de activiteitscoëfficiëntγi = ai/xi is lager dan één. In een ideale oplossing is ai = x en γi = 1.
7.2 Activiteit van vaste stoffen in vloeistoffen
7.3 Elektrochemische cellen
7.3.1 Een eenvoudige elektrochemische cel
Een eenvoudige elektrochemische cel is geïllustreerd in Figuur 7.2. Eén van de elektroden is ver-vaardigd van zink in een oplossing van zinksulfaat en een andere elektrode bestaat uit koper on-dergedompeld in een oplossing van koper(II)sulfaat1. De oplossingen kunnen met elkaar verbondenworden zodat de ionen kunnen stromen van het ene compartiment naar het andere compartiment.
Zn
Zn2+ SO2−4
−
Zn→ Zn2+ + 2e−
inert elektrolyt
e−
+
Cu
Cu2+ SO2−4
Cu2+ + 2e−→ Cu
Figuur 7.2: Een eenvoudige elektrochemische cel.
De verbinding is een zoutbrug die zal verhinderen dat de oplossingen te snel zullen mengen. Deneiging in dergelijke cellen is voor zink om ionen te vormen waardoor het elektronen zal afgeven en
1Deze elektrochemische cel is gekend als het Daniell-element of de Daniell-cel. Het is een Galvanisch element dat in1836 werd beschreven in een artikel gepubliceerd in Philosophical Transactions met als titel On Voltaic Combinationsdoor John Frederic Daniell (1790–1845) [17]. Daniell was een goede vriend van Michael Faraday (1791–1867) [5].

104 Hoofdstuk 7. Niet-ideale oplossingen
waardoor de zinkelektrode negatief zal worden. Omgekeerd zullen de koper(II)-ionen in de oplos-sing de neiging vertonen om elektronen op te nemen en bijgevolg afzetten als metallisch koper. Dereactie wordt geschreven als
Zn + Cu2+ −−→ Zn2+ + Cu
Het zink wordt geoxideerd tot zinkionen en de koper(II)-ionen worden gereduceerd tot metallischkoper. Koper(II) treedt bijgevolg op als oxidator en het zink treedt op als een reductor. In de externestroomkring zullen de elektronen van de negatieve zinkelektrode naar de positieve koperelektrodestromen.
De maximale elektrische arbeid die wordt verricht door een dergelijke cel is gelijk aan het produktvan de lading die doorheen de cel zal stromen en het potentiaalverschil waardoorheen het zalstromen. De arbeid verricht op de cel is
w = −EQ (7.3)
waarbij E de elektromotorische kracht van de cel is en Q de lading die doorheen de cel stroomt.Voor één mol van de reactie die doorgaat in de cel kunnen we schrijven dat
Q = nNAe (7.4)
waarbij NA de constante van Avogadro is, −e de lading op het elektron en n het aantal molelektronen is dat wordt overgedragen per mol van de reactie. Het produkt NAe is de grootte vande totale lading van één mol elektronen en wordt de constante van Faraday genoemd, geschrevenals F . Bijgevolg
w = −nFE (7.5)
Voor de reactie die we hebben beschouwd, worden er twee elektronen overgedragen van het zinknaar het koper voor één mol van een reactie zodat n = 2.
Wanneer een cel werkt onder reversibele voorwaarden (dit wil zeggen dat de stroom oneindig traagzal verlopen) dan kunnen we het systeem beschouwen als een evenwicht volgens
dG = dwtoegevoegd(T, p) (7.6)
Voor één mol van de celreactie onder constante voorwaarden kunnen we schrijven
∆G′ = −nFE (7.7)
waarbij ∆G′ de reactie vrije energie is.
7.3.2 Standaard elektrode potentialen
Wanneer we de neiging om elektronen op te nemen of af te geven, zouden kunnen meten van elkemogelijke helft van een elektrochemische cel (elke elektrode en de oplossing waarin het is onderge-dompeld), dan zouden we de elektromotorische kracht van een cel kunnen berekenen die gemaakt

7.3. Elektrochemische cellen 105
is uit twee dergelijke elektroden. We kunnen deze neiging niet meten op een absolute basis, maarhet is voldoende wanneer we het kunnen meten relatief ten opzichte van referentie-elektroden. Destandaardelektrode is gekozen als de standaard waterstof elektrode (SHE)2 die een waarde E− = 0is toegekend. Deze elektrode wordt gemaakt door waterstofgas bij één atmosfeer in contactd tebrengen in een oplossing van zijn ionen bij een eenheid activiteit aan de oppervlakte van een platina-elektrode.Om de standaard elektrode potentiaal te bepalen van een metaal (of element), wordt de elektroche-mische cel opgesteld zoals schematisch is gevisualiseerd in Figuur7.3. Wanneer het metaal positief
Pt
H+ (a = 1)
H2
zoutbrug
Metaal
Mn+ (a = 1)
Figuur 7.3: Een elektrochemische cel met een waterstofgaselektrode om de standaard elektrode potentialenvan een metaal (of een element) te bepalen.
is ten opzichte van de waterstofgaselektrode, is de standaard elektrode potentiaal van het metaalpositief en vice versa. Als het metaal in de cel zink is, dan vinden we dat
E−Zn2+/Zn = −0,76 V
Voor koper vinden we datE−Cu2+/Cu = +0,34 V
Daar een zinkelektrode de neiging heeft om op te lossen zodat er zinkionen worden gevormd,zal de elektrode de neiging vertonen om negatief te worden omdat er in feite elektronen wordenteruggetrokken uit de oplossing volgens de halfreactie
Zn −−→ Zn2+ + 2e−
2SHE staata voor Standard Hydrogen Electrode.
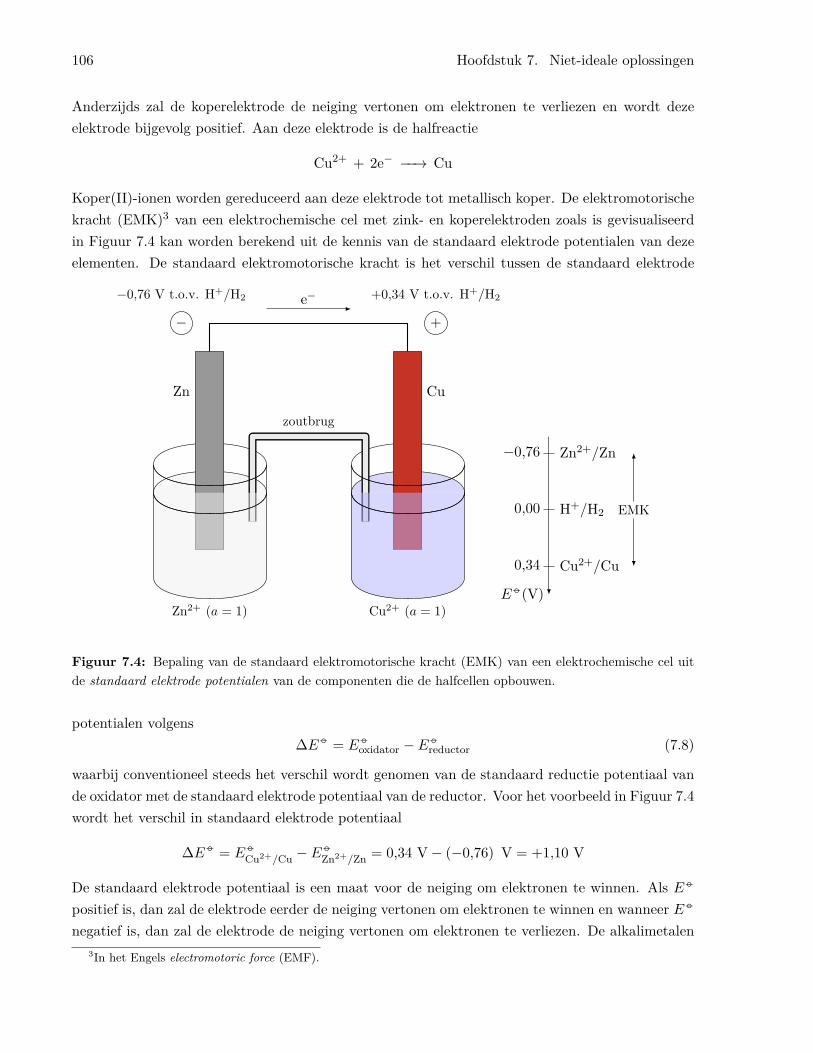
106 Hoofdstuk 7. Niet-ideale oplossingen
Anderzijds zal de koperelektrode de neiging vertonen om elektronen te verliezen en wordt dezeelektrode bijgevolg positief. Aan deze elektrode is de halfreactie
Cu2+ + 2e− −−→ Cu
Koper(II)-ionen worden gereduceerd aan deze elektrode tot metallisch koper. De elektromotorischekracht (EMK)3 van een elektrochemische cel met zink- en koperelektroden zoals is gevisualiseerdin Figuur 7.4 kan worden berekend uit de kennis van de standaard elektrode potentialen van dezeelementen. De standaard elektromotorische kracht is het verschil tussen de standaard elektrode
Zn
−
−0,76 V t.o.v. H+/H2
Zn2+ (a = 1)
zoutbrug
e−
+
+0,34 V t.o.v. H+/H2
Cu
Cu2+ (a = 1)E− (V)
−0,76 Zn2+/Zn
0,00 H+/H2
0,34 Cu2+/Cu
EMK
Figuur 7.4: Bepaling van de standaard elektromotorische kracht (EMK) van een elektrochemische cel uitde standaard elektrode potentialen van de componenten die de halfcellen opbouwen.
potentialen volgens∆E− = E−oxidator − E−reductor (7.8)
waarbij conventioneel steeds het verschil wordt genomen van de standaard reductie potentiaal vande oxidator met de standaard elektrode potentiaal van de reductor. Voor het voorbeeld in Figuur 7.4wordt het verschil in standaard elektrode potentiaal
∆E− = E−Cu2+/Cu − E−Zn2+/Zn = 0,34 V− (−0,76) V = +1,10 V
De standaard elektrode potentiaal is een maat voor de neiging om elektronen te winnen. Als E−
positief is, dan zal de elektrode eerder de neiging vertonen om elektronen te winnen en wanneer E−
negatief is, dan zal de elektrode de neiging vertonen om elektronen te verliezen. De alkalimetalen3In het Engels electromotoric force (EMF).
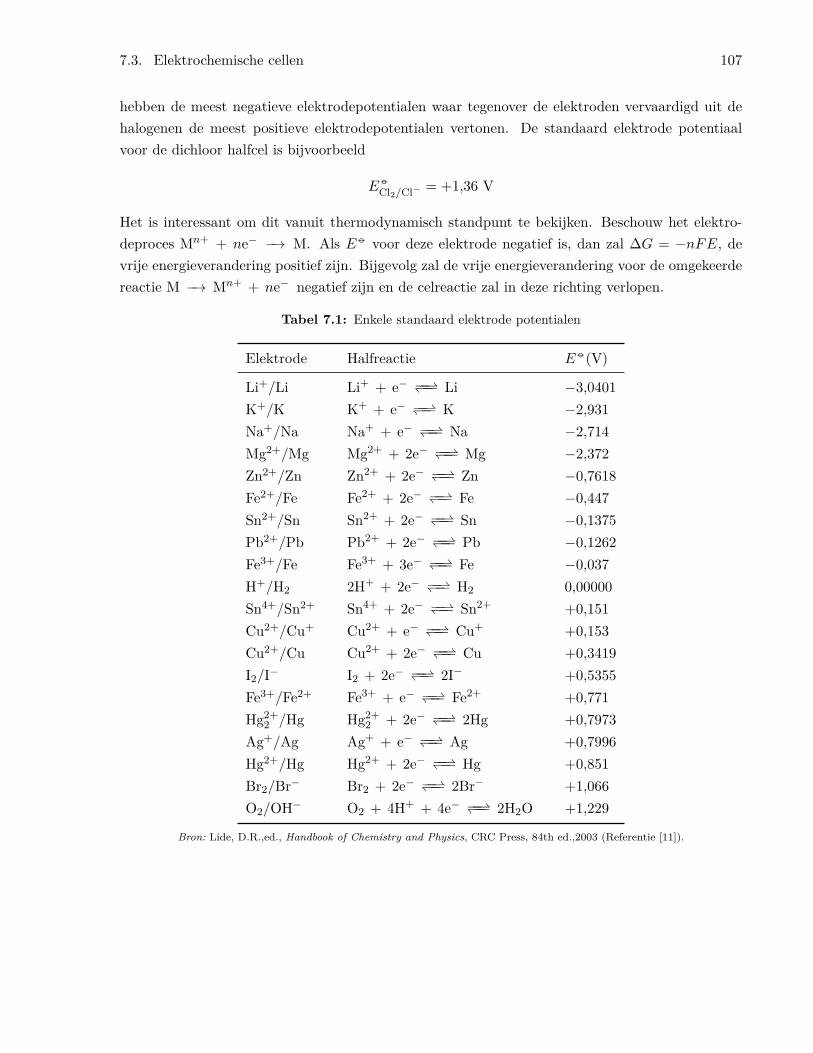
7.3. Elektrochemische cellen 107
hebben de meest negatieve elektrodepotentialen waar tegenover de elektroden vervaardigd uit dehalogenen de meest positieve elektrodepotentialen vertonen. De standaard elektrode potentiaalvoor de dichloor halfcel is bijvoorbeeld
E−Cl2/Cl− = +1,36 V
Het is interessant om dit vanuit thermodynamisch standpunt te bekijken. Beschouw het elektro-deproces Mn+ + ne− −→ M. Als E− voor deze elektrode negatief is, dan zal ∆G = −nFE, devrije energieverandering positief zijn. Bijgevolg zal de vrije energieverandering voor de omgekeerdereactie M −→ Mn+ + ne− negatief zijn en de celreactie zal in deze richting verlopen.
Tabel 7.1: Enkele standaard elektrode potentialen
Elektrode Halfreactie E− (V)
Li+/Li Li+ + e− −−−− Li −3,0401K+/K K+ + e− −−−− K −2,931Na+/Na Na+ + e− −−−− Na −2,714Mg2+/Mg Mg2+ + 2e− −−−− Mg −2,372Zn2+/Zn Zn2+ + 2e− −−−− Zn −0,7618Fe2+/Fe Fe2+ + 2e− −−−− Fe −0,447Sn2+/Sn Sn2+ + 2e− −−−− Sn −0,1375Pb2+/Pb Pb2+ + 2e− −−−− Pb −0,1262Fe3+/Fe Fe3+ + 3e− −−−− Fe −0,037H+/H2 2H+ + 2e− −−−− H2 0,00000Sn4+/Sn2+ Sn4+ + 2e− −−−− Sn2+ +0,151Cu2+/Cu+ Cu2+ + e− −−−− Cu+ +0,153Cu2+/Cu Cu2+ + 2e− −−−− Cu +0,3419I2/I− I2 + 2e− −−−− 2I− +0,5355Fe3+/Fe2+ Fe3+ + e− −−−− Fe2+ +0,771Hg2+
2 /Hg Hg2+2 + 2e− −−−− 2Hg +0,7973
Ag+/Ag Ag+ + e− −−−− Ag +0,7996Hg2+/Hg Hg2+ + 2e− −−−− Hg +0,851Br2/Br− Br2 + 2e− −−−− 2Br− +1,066O2/OH− O2 + 4H+ + 4e− −−−− 2H2O +1,229
Bron: Lide, D.R.,ed., Handbook of Chemistry and Physics, CRC Press, 84th ed.,2003 (Referentie [11]).


Hoofdstuk 8
Thermodynamica van gassen
In dit hoofdstuk vatten we de meeste thermodynamische relaties samen die de eigenschappen vanideale en niet-ideale gassen behandelen.
8.1 Expansie van een ideaal gas
We hebben reeds de veranderingen berekend in thermodynamische eigenschappen die samengaanmet de isotherme expansie van een ideaal gas. Daar(
∂U
∂V
)T
= 0
endU =
(∂U
∂T
)VdT +
(∂U
∂V
)TdV
hebben we voor een isothermische expansie dat dU = 0 en đq = −đw = +pdV . Bijgevolg is
q = −w =∫ V2
V1pdV = nRT ln
(V2V1
)= nRT ln
(p1p2
)We kunnen het gas eveneens adiabatisch laten expanderen zodat er geen warmte wordt uitgewisseldmet de omgeving tijdens de expansie. De temperatuur van het gas zal niet constant blijven tijdenseen adiabatische expansie. Daar
đw = −pdV
endU = CV dT
En omdatđq = 0 en dU − đw = 0
hebben we eveneensCV dT + pdV = 0
109

110 Hoofdstuk 8. Thermodynamica van gassen
Delen we beiden leden door T en vervangen we p = RT/V voor één mol van een ideaal gas, vindenwe dat
CVdTT
+RdVV
= 0
Wanneer het gas zich in de initiële toestand T1 alsook V1 bevindt en als het gas expandeert naareen finale toestand T2 alsook V2, kunnen we de vergelijking integreren als we aannemen dat CVonafhankelijk is van de temperatuur. We bekomen
CV ln T2T1
+R ln V2V1
= 0
Daar voor één mol gas geldt datCp − CV = R
kunnen we schrijven datCV ln T2
T1+ (Cp − CV ) ln V2
V1= 0
enT1T2
=(V2V1
)(Cp/CV −1)
Daar voor een ideaal gas de initiële en finale toestanden moeten voldoen aan de vergelijking van deideale gastoestand
T1T2
= p1V1p2V2
enp1V
Cp/CV
1 = p2VCp/CV
2 = constant
De verandering in interne energie voor een adiabatische expansie voor één mol gas van V1 naar V2
wordt berekend volgens de relatie∆U = CV (T2 − T1)
De initiële en finale temperaturen zijn gerelateerd aan de volumes door de hierboven afgeleidevergelijking.
8.2 Irreversibele expansie
Wanneer de druk op een gas plots wordt verlaagd van een waarde p1 naar p2 zoals schematischwordt gevisualiseerd in Figuur 8.1 en wanneer het gas wordt toegestaan om te expanderen tegendeze nieuwe druk naar een volume V2 dan zal het arbeid verrichten.
w = −∫ V2
V1pdV = −p2 (V2 − V1)
Als deze expansie adiabatisch wordt uitgevoerd zodat q = 0, dan is ∆U = q + w en bekomen we
w = ∆U = CV (T2 − T1)
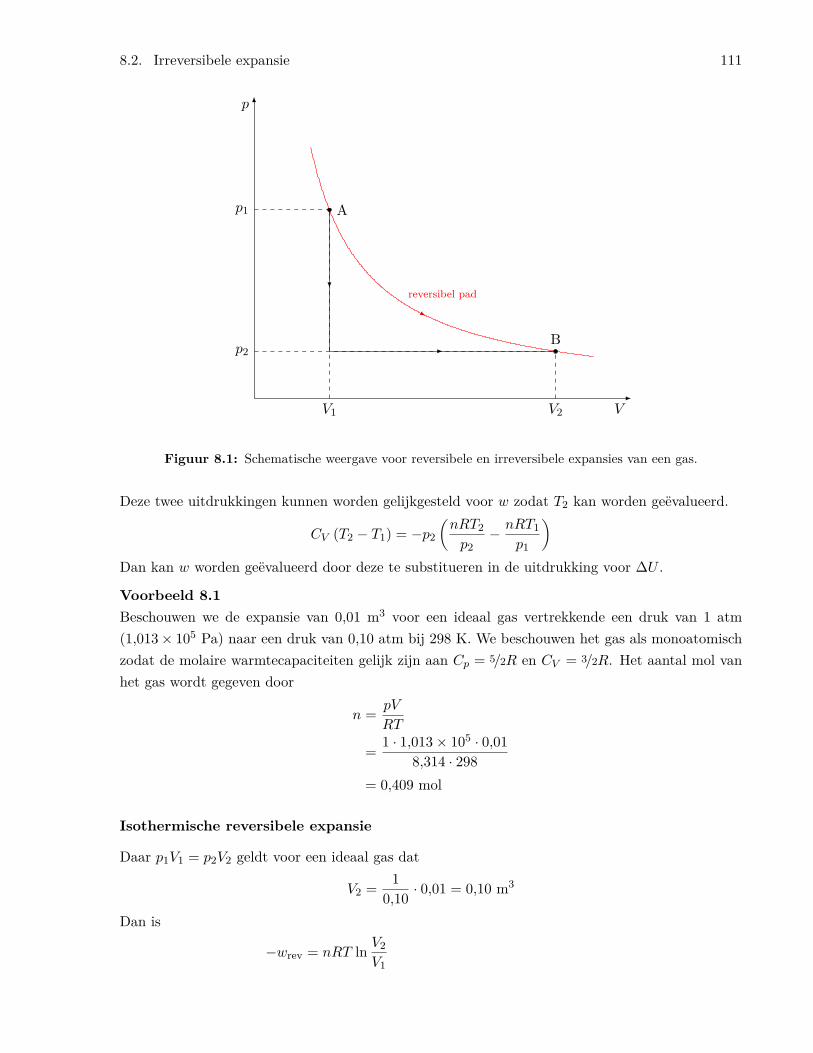
8.2. Irreversibele expansie 111
reversibel pad
p
V
p1
p2
V2V1
A
B
Figuur 8.1: Schematische weergave voor reversibele en irreversibele expansies van een gas.
Deze twee uitdrukkingen kunnen worden gelijkgesteld voor w zodat T2 kan worden geëvalueerd.
CV (T2 − T1) = −p2
(nRT2p2− nRT1
p1
)Dan kan w worden geëvalueerd door deze te substitueren in de uitdrukking voor ∆U .
Voorbeeld 8.1Beschouwen we de expansie van 0,01 m3 voor een ideaal gas vertrekkende een druk van 1 atm(1,013× 105 Pa) naar een druk van 0,10 atm bij 298 K. We beschouwen het gas als monoatomischzodat de molaire warmtecapaciteiten gelijk zijn aan Cp = 5/2R en CV = 3/2R. Het aantal mol vanhet gas wordt gegeven door
n = pV
RT
= 1 · 1,013× 105 · 0,018,314 · 298
= 0,409 mol
Isothermische reversibele expansie
Daar p1V1 = p2V2 geldt voor een ideaal gas dat
V2 = 10,10 · 0,01 = 0,10 m3
Dan is
−wrev = nRT ln V2V1

112 Hoofdstuk 8. Thermodynamica van gassen
wrev = −0,409 mol · 8,31 Jmol−1K−1 · 298 K · ln 10= −2331 J
Adiabatische reversibele expansie
Daar p1VCp/CV
1 = p2VCp/CV
2 , bekomen we
V2 =(p1p2
)CV /Cp
V1
= (10)3/5 · 0,01= 0,0398 m3
De finale temperatuur wordt nu gegeven door
T2 = p2V2nR
= 0,1 · 1,013× 105 Pa · 0,0398 m3
0,409 mol · 8,314 Jmol−1K−1
= 118,6 K
Daar voor dit adiabatisch proces q = 0, bekomen we ∆U = w = CV (T2 − T1). Met CV = n3/2R,bekomen we
w = 0,409 mol · 32 · 8,314 (118,6− 298) K = −915 J
Irreversibele adiabatische expansie
De arbeid verricht wordt gegeven door
w = −p2 (V2 − V1) = −p2
(nRT2p2− nRT1
p1
)Maar omdat q = 0, bekomen we ∆U = CV (T2 − T1) en bijgevolg
CV (T2 − T1) = −p2
(nRT2p2− nRT1
p1
)Dus
32R (T2 − 298) = RT2 +R · 2988 · 0,1
1,0zodat T2 = 190, 7 K en
w = ∆U = 32nR (190,7− 298)
= −547,2 J
We merken op dat het irreversibel proces minder arbeid verricht dan het equivalente reversibeleproces en dat het gas dus niet in dezelfde mate afkoelt.

8.3. Toestandsvergelijkingen voor gassen 113
8.3 Toestandsvergelijkingen voor gassen
De verandering van de interne energie van een gas dat arbeid verricht enkel tegen een atmosferischedruk, is dU = TdS − pdV . We kunnen deze vergelijking schrijven in de vorm
dUdV = T
( dSdV
)− p
Voor veranderingen bij een constante temperatuur kunnen we de vergelijking in een meer strictevorm schrijven. (
∂U
∂V
)T
= T
(∂S
∂V
)T− p
We weten dat dA = dU − TdS − SdT en dat dU = TdS − pdV , zodat we bekomen dat dA =−SdT − pdV . We zien bijgevolg(
∂A
∂T
)V
= −S en(∂A
∂V
)T
= −p
Omdat de orde van het afleiden van een toestandsfunctie er niet toe doet, geldt dat[∂
∂V
(∂A
∂T
)V
]T
= −(∂S
∂V
)T
en[∂
∂T
(∂A
∂V
)T
]V
= −(∂p
∂T
)V
zodat bijgevolg (∂S
∂V
)T
=(∂p
∂T
)V
(8.1)
Vergelijking 8.1 is één van een aantal Maxwell relaties die waardevol zijn in een aantal gebieden.Gebruik makende van deze relatie bekomen we dat(
∂U
∂V
)T
= T
(∂p
∂T
)V
= −p
een vergelijking die de verandering van de energie tot het volume relateert aan de toestandsverge-lijking, dat is p, V , T relaties van het gas (of inderdaad vloeistof of vaste stof).
Voor één mol van een ideaal gas geldt dat pV = RT en(∂p
∂T
)V
= R
V
Om die reden is (∂U
∂V
)T
= RT
V− p = p− p = 0
Voor een ideaal gas is (∂U/∂V )T = 0. De interne energie is onafhankelijk van het volume.We hebben eerder aangenomen dat dit besluit ingesloten ligt in de definitie van een ideaal gas.Nu hebben we aangetoond dat het een gevolg is voor de ideale gas toestandsvergelijking. Analogeargumenten tonen aan dat voor een ideaal gas ook geldt dat(
∂H
∂p
)T
= 0

114 Hoofdstuk 8. Thermodynamica van gassen
8.4 Het Joule-Thomson experiment
Binnen de grenzen van de fout van zijn apparaat toonde James Prescott Joule (1818–1889) in 1843aan dat er geen temperatuursverandering was tijdens de expansie van een gas in een vacuüm [18].Een schematische weergave van dit experiment is gevisualiseerd in Figuur 8.2. Tijdens het expe-
A B
Figuur 8.2: Het Joule experiment.
riment was đw = 0, en daar hij observeerde dat đq eveneens effectief nul bleek te zijn, kan menschrijven dat
dU = đq + đw = 0
en (∂U
∂V
)T
= 0
Meer zorgvuldigere metingen zouden hebben aangetoond dat voor reële gassen (∂U/∂V )T niet exactgelijk aan nul was.
Een meer superieure manier om deze effecten te onderzoeken, werd ontworpen door Joule en Thom-son. In hun apparaat, zoals is geïllustreerd in Figuur 8.3, stroomde gas bij een constante snelheiddoorheen een poreuze vulling. Het systeem was geïsoleerd zodat đq = 0. De kracht die werd uit-gevoerd op de zuiger om het gas doorheen de vulling te laten stromen is gelijk aan het productvan de druk p1 en de oppervlakte van de zuiger a. De afstand waarmee het punt van toepassingvan de kracht beweegt, is gelijk aan het volume van het gas dat doorheen de poreuze vulling wordtgedreven gedeeld door de oppervlakte. Wanneer één mol van het gas is bewogen, geldt dat
arbeid verricht op het gas = kracht× verplaatsing
= p1a
(V1a
)= p1V1
De netto arbeid is de arbeid van de compressie min de arbeid die wordt herwonnen wanneer hetgas expandeert aan de andere kant van de poreuze vulling. Bijgevolg
w = p1V1 − p2V2
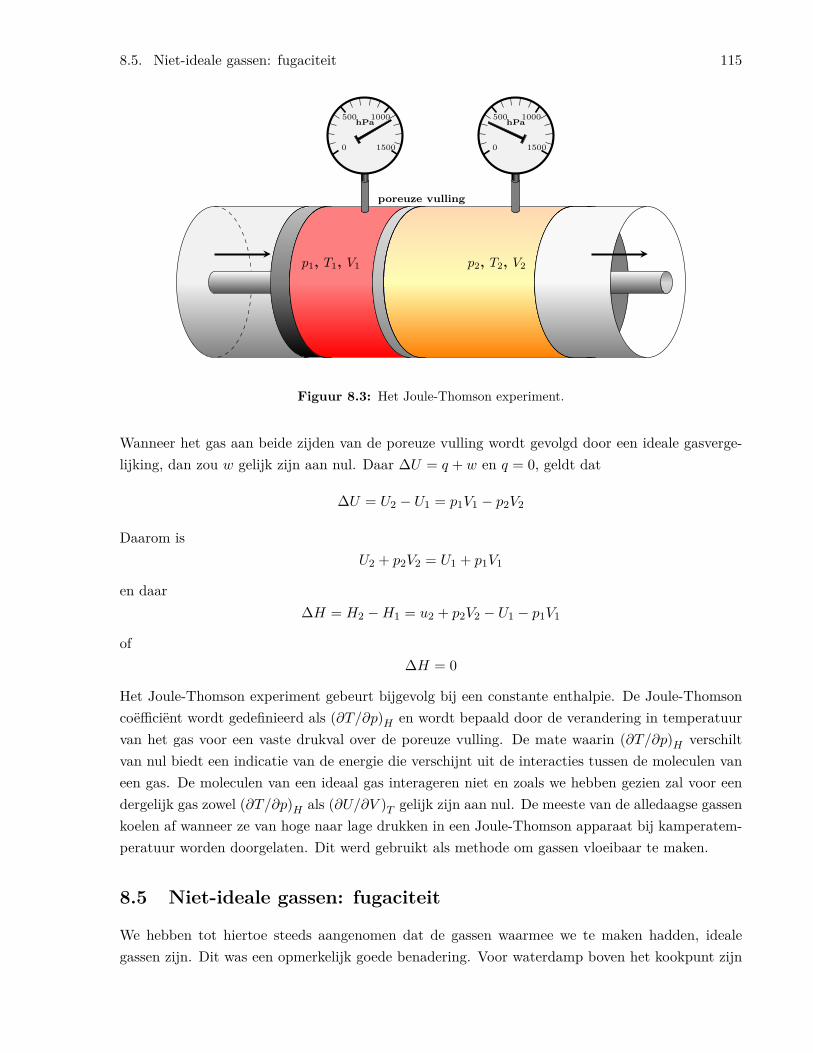
8.5. Niet-ideale gassen: fugaciteit 115
hPa
0
500 1000
1500
hPa
0
500 1000
1500
p1, T1, V1 p2, T2, V2
poreuze vulling
Figuur 8.3: Het Joule-Thomson experiment.
Wanneer het gas aan beide zijden van de poreuze vulling wordt gevolgd door een ideale gasverge-lijking, dan zou w gelijk zijn aan nul. Daar ∆U = q + w en q = 0, geldt dat
∆U = U2 − U1 = p1V1 − p2V2
Daarom isU2 + p2V2 = U1 + p1V1
en daar∆H = H2 −H1 = u2 + p2V2 − U1 − p1V1
of∆H = 0
Het Joule-Thomson experiment gebeurt bijgevolg bij een constante enthalpie. De Joule-Thomsoncoëfficiënt wordt gedefinieerd als (∂T/∂p)H en wordt bepaald door de verandering in temperatuurvan het gas voor een vaste drukval over de poreuze vulling. De mate waarin (∂T/∂p)H verschiltvan nul biedt een indicatie van de energie die verschijnt uit de interacties tussen de moleculen vaneen gas. De moleculen van een ideaal gas interageren niet en zoals we hebben gezien zal voor eendergelijk gas zowel (∂T/∂p)H als (∂U/∂V )T gelijk zijn aan nul. De meeste van de alledaagse gassenkoelen af wanneer ze van hoge naar lage drukken in een Joule-Thomson apparaat bij kamperatem-peratuur worden doorgelaten. Dit werd gebruikt als methode om gassen vloeibaar te maken.
8.5 Niet-ideale gassen: fugaciteit
We hebben tot hiertoe steeds aangenomen dat de gassen waarmee we te maken hadden, idealegassen zijn. Dit was een opmerkelijk goede benadering. Voor waterdamp boven het kookpunt zijn
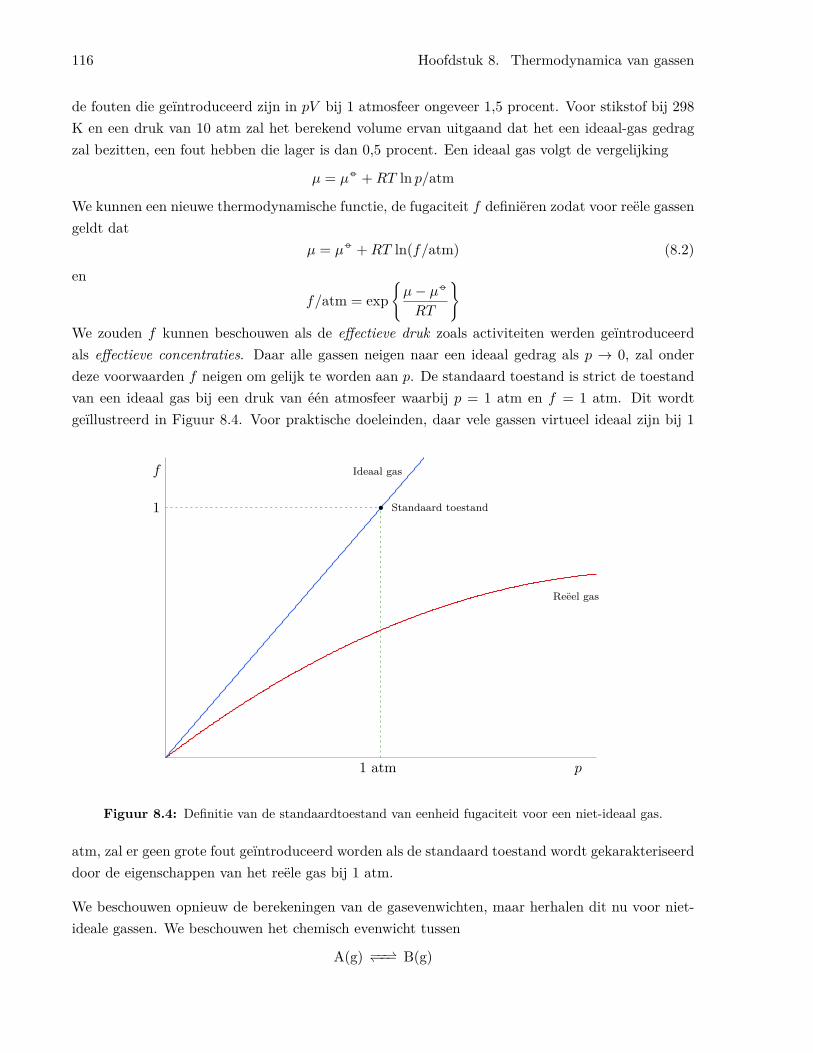
116 Hoofdstuk 8. Thermodynamica van gassen
de fouten die geïntroduceerd zijn in pV bij 1 atmosfeer ongeveer 1,5 procent. Voor stikstof bij 298K en een druk van 10 atm zal het berekend volume ervan uitgaand dat het een ideaal-gas gedragzal bezitten, een fout hebben die lager is dan 0,5 procent. Een ideaal gas volgt de vergelijking
µ = µ− +RT ln p/atm
We kunnen een nieuwe thermodynamische functie, de fugaciteit f definiëren zodat voor reële gassengeldt dat
µ = µ− +RT ln(f/atm) (8.2)
enf/atm = exp
µ− µ−
RT
We zouden f kunnen beschouwen als de effectieve druk zoals activiteiten werden geïntroduceerdals effectieve concentraties. Daar alle gassen neigen naar een ideaal gedrag als p → 0, zal onderdeze voorwaarden f neigen om gelijk te worden aan p. De standaard toestand is strict de toestandvan een ideaal gas bij een druk van één atmosfeer waarbij p = 1 atm en f = 1 atm. Dit wordtgeïllustreerd in Figuur 8.4. Voor praktische doeleinden, daar vele gassen virtueel ideaal zijn bij 1
p1 atm
f
1
Ideaal gas
Reëel gas
Standaard toestand
Figuur 8.4: Definitie van de standaardtoestand van eenheid fugaciteit voor een niet-ideaal gas.
atm, zal er geen grote fout geïntroduceerd worden als de standaard toestand wordt gekarakteriseerddoor de eigenschappen van het reële gas bij 1 atm.
We beschouwen opnieuw de berekeningen van de gasevenwichten, maar herhalen dit nu voor niet-ideale gassen. We beschouwen het chemisch evenwicht tussen
A(g) −−−− B(g)

8.6. Berekeningen van fugaciteiten 117
µA = µ−A +RT ln(fA/atm), µB = µ−B +RT ln(fB/atm)
∆G′ = ∆G− +RT ln(fB/fA)
Kf = ((fB/fA)evenwicht
in de stricte definitie van de evenwichtsconstante voor gasreacties, en
∆G− = −RT lnKf
8.6 Berekeningen van fugaciteiten
Daar voor één mol van een zuiver gas geldt dat µ = µ− +RT ln(f/atm) en µ− onafhankelijk is vande druk, hebben we voor een niet-ideaal gas dat(
∂µ
∂p
)T
= V = RT
(∂ ln f∂p
)T
waarbij V het volume is van een mol van het gas. Wanneer de toestandsvergelijking van het gas isgekend, dan kan de fugaciteit meteen worden bepaald gebruik makende van[
∂ ln (f/p)∂p
]T
=[∂ ln f∂p
]T
−[∂ ln p∂p
]T
= V
RT− 1p
Deze vergelijking kan worden geïntegreerd van een lage druk waarbij de fugaciteit gelijk is aan dedruk naar een hogere druk p zodat we bekomen
ln fp
=∫ p
0
(V
RT− 1p
)dp
Voor gassen die slechts matig afwijken van het ideaal gas gedrag, zal een meer geschikte benade-ringsrelatie kunnen worden gebruikt
f
p= p
pideaal
waarbij pideaal de druk is van een ideaal gas die hetzelfde volume zou bezitten. Fugaciteiten die be-rekend worden gebruik makende van deze vergelijking voor stikstofgas bij 273 K worden vergelekenmet de exacte waarden in Tabel 8.1.
Tabel 8.1: Berekeningen van fugaciteiten van stikstofgas bij 273 K
Druk/atm f/p p/pideaal
1 0,99955 0,9995510 0,9956 0,9957100 0,9703 0,98511000 1,839 2,070


Hoofdstuk 9
De moleculaire basis van dethermodynamica
De thermodynamica beschrijft de relaties tussen macroscopische eigenschappen en hoeft niet nood-zakelijk de moleculaire natuur van de materie in te roepen. Meer nog, de besluiten van de ther-modynamica lijken zelfs onafhankelijk van het bestaan van moleculen te kunnen standhouden.Desalniettemin is het mogelijk om de besluiten van de thermodynamica te relateren aan de ma-nier waarop moleculen zich gedragen. Deze manier van beschrijven staat gekend als de statistischethermodynamica.
9.1 Energieniveaus
Het is de kwantummechanica dat ons vertelt dat de energieën zijn ‘gekwantiseerd’. Dit betekentdat er enkel bepaalde waarden van de energie mogelijk zijn. Voor eenvoudige systemen, kunnen wedeze toegelaten waarden berekenen gebruik makende van de basisvergelijking uit de kwantumtheorieof de Schrödingervergelijking. De toegelaten waarden worden de energieniveaus van het systeemgenoemd. Elke toegelaten toestand en zijn overeenkomstige energie wordt gedefinieerd in termenvan de kwantumgetallen die een integraal bezitten of, in bijzondere gevallen, halve-integraalwaarden.
Het meest eenvoudige voorbeeld is een deeltje dat beperkt is tot een één-dimensionale doos. Deenergieniveaus voor een dergelijk systeem worden gevisualiseerd in Figuur 9.1. Als de doos kleinerwordt gemaakt of de deeltjes minder zwaar zijn, zullen de energieniveaus verder uit elkaar komente liggen. Voor een kubische doos kunnen de energieniveaus worden uitgedrukt als
E = h2
8ml2(n2x + n2
y + n2z
)(9.1)
waarbij l de lengte is van de doos, m de massa van het deeltje, h de constante van Planck en nx,ny en nz de kwantumgetallen zijn voor de beweging in de x-, y-, en z-richtingen. Wanneer eengasmolecule is beperkt tot een container, dan zal de energie geassocieerd met de beweging van hetzwaartepunt (dat de translatie-energie wordt genoemd) gekwantiseerd zijn.
119

120 Hoofdstuk 9. De moleculaire basis van de thermodynamica
123
4
5
6
7
8
9n
En
Figuur 9.1: De energieniveaus van een deeltje in een één-dimensionale doos.
Polyatomische moleculen kunnen eveneens rotationele en vibrationele energieën bezitten. Met eengoede benadering mogen we schrijven dat
Etotaal = Etranslatie + Erotatie + Evibratie
De rotationele en vibrationele energieën zijn eveneens gekwantiseerd. De energieniveaus geassoci-eerd met de rotatie van een linear molecule worden gegeven door de uitdrukking1
εrotatie = Erotatiehc
=(
h
8π2Ic
)J (J + 1) (9.2)
waarbij J het rotationeel kwantumgetal is, c de lichtsnelheid dat hier wordt uitgedrukt in cm s−1
en I het inertiemomentI =
∑mir
2i
voor massa’s mi bij een afstand ri ten opzichte van het massacentrum [19]. Voor vibrationelebewegingen, worden de energieniveaus gegeven door
Evibratie =(v + 1
2
)hν (9.3)
waarbij v het vibrationeel kwantumgetal is en ν de vibratiefrequentie. Door het omzetten naarspectroscopische eenheden (cm−1) verkrijgen we [19]
εvibratie = Evibratiehc
=(v + 1
2
)ν (9.4)
1Rotationele spectra worden vaak beschreven in termen van het golfgetal ν = ∆E/hc cm−1. Om die redenworden de energieën waarin men is geïnteresseerd eveneens in deze eenheden uitgedrukt en wordt geschreven datε = E/hc [19]. Het blijft belangrijk om goed de eenheden te blijven schrijven en de juiste notatie te gebruiken.

9.1. Energieniveaus 121
We zien dat zelfs in het laagste energieniveau wanneer v = 0 een vibrator een energie 1/2hν bezitwaarbij v het vibrationeel kwantumgetal is en ν de frequentie van de vibratie. Dit wordt denulpuntsenergie genoemd. Het bestaan ervan is te wijten aan het onzekerheidsbeginsel dat eenfundamenteel gevolg is van de kwantummechanica. Het vertelt ons dat we niet tegelijkertijd exactzowel het moment als de positie van een deeltje kunnen specifiëren. Wanneer deeltjes beperkt zijntot een beperkt ruimtegebied zoals dit zal voorkomen in een vibrationele beweging, kan hun lineairmoment niet exact gelijk zijn aan nul en bijgevolg zal de energie van de deeltjes ook niet gelijk zijnaan nul. Bijgevolg kan een oscillator nooit alle energie kunnen verliezen.Voor een typisch gasmolecule zullen onder normale voorwaarden de translatie energieniveaus zodicht op elkaar liggen dat we de translatie-energie kunnen beschouwen als continu veranderlijk. Ditis echter niet het geval voor rotationele en vibrationele niveaus. De afstand tussen de rotationeleniveaus is van de grootte-orde van 10 J mol−1 (Tabel 9.1) en de afstand tussen de vibrationele
Tabel 9.1: Rotationele en vibrationele energieniveaus van diatomaire moleculen
∆Erot = εJ=1 − εJ=0, ∆Evib = εv=1 − εv=0 = hν
θrot = h2/8π2l
k, θvib = hν
k
∆Erot(Jmol−1) 2θrot(K) ∆Evib(kJmol−1) θvib(K)
H2 1420 171 51,0 6140HCl 254 30 35,8 4300O2 34,6 4,2 18,8 2260N2 48,1 5,8 28,1 3380Cl2 5,8 0,70 6,8 810Br2 1,9 0,23 4,0 470I2 0,90 0,11 2,6 310CO 46,0 5,5 26,0 3120NO 41,0 4,9 22,8 2740
2θrot en θvib zijn de temperaturen bij welke kT gelijk wordt aan de energiedie overeenkomt aan respectievelijk de eerste rotationele en vibrationele transities.
niveaus is van de grootte-orde 10 kJ mol−1. Het patroon van energieniveaus dat vaak wordtgeobserveerd is geïllustreerd in Figuur 9.2.
Om nu in staat te zijn om onze kennis van de energieniveaus te kunnen gebruiken om de thermody-namische eigenschappen te kunnen berekenen, moeten we vinden hoe moleculen zichzelf verdelenover de beschikbare energieniveaus. Een moeilijkheid die ontstaat is dat verschillende toestanden,elk een afzonderlijke oplossing van de Schrödingervergelijking, dezelfde energie kan bezitten. Erwordt van dit energieniveau dan gezegd dat het is ontaard met een ontaarding die gelijk is aan hetaantal toestanden dat deze energie bezit. Voor elke rotationele energie εJ van een lineair molecule

122 Hoofdstuk 9. De moleculaire basis van de thermodynamica
01234
5
01234
5
01234
5
Jv
0
1
2
Figuur 9.2: Vibrationele en rotationele energieniveaus waarbij v en J de respectievelijke vibrationele enrotationele kwantumgetallen zijn.
zijn er 2J + 1 toestandsfuncties die overeenkomen met de 2J + 1 verschillende oriëntaties van hetmolecule dat de kwantumtheorie toelaat. Het is belangrijk om rekening te houden met ontaar-ding wanneer berekeningen worden gemaakt omtrent de manier waarop moleculen zichzelf kunnenverdelen volgens de energieniveaus. Het is vaak meer geschikt om het probleem te bespreken intermen van toestanden eerder dan energieniveaus. Elk atoom of molecule zal zich in één van deenergietoestanden bevinden. De totale energie van een aantal moleculen,
U =∑
toestandenεini
is afhankelijk van de energieën van de toestanden εi en het aantal moleculen die ze bezetten,ni. Als we een regel zouden kunnen vinden dat ons ni geeft, zouden we uit de wetenschap vande energieniveaus en hun ontaardingen de totale energie van het systeem kunnen berekenen. Meeralgemeen blijkt het dat een wetenschap van εi en ni dient om alle thermodynamische eigenschappente definiëren.
9.2 Microtoestanden
Microtoestanden2 worden geïdentificeerd als het aantal manieren waarop de algemene toestand vaneen systeem kan worden opgebouwd. Het aantal manieren waarop een toestand van een systeem
2In het Engels bestaat er voor microtoestanden (microstates) het mooie woord complexions.

9.2. Microtoestanden 123
zich kan voordoen, bepaalt de waarschijnlijkheid van zijn voorkomen. Wanneer we bijvoorbeeldtwee muntstukken omhoog gooien, is de waarschijnlijkheid om één kop (K) en één munt (M) tebekomen tweemaal de waarschijnlijkheid om twee kop te gooien. We kunnen alle mogelijkhedenuitschrijven als KM, MK, KK en MM zodat we kunnen inzien dat twee microtoestanden aanleidinggeven tot een gemengde configuratie. Beschouw nu drie moleculen verdeeld over energieniveausdie op gelijke afstand van elkaar gescheiden zijn zoals is gevisualiseerd in Figuur 9.3. We stellenbovendien dat de totale energie een vaste waarde van drie eenheden moet bezitten. Er zijn drieverschillende verdelingen mogelijk, namelijk I, II en III in Figuur 9.3, maar niet alle drie dezeverdelingen zijn even waarschijnlijk. Dit kunnen we inzien door aan te nemen dat de moleculen
3
a
bc
b
ac
c
ab
I
Ei
0123
6
a
b
c
a
c
b
b
a
c
c
a
b
c
b
a
b
c
a
II
1
III
abc
Figuur 9.3: Het rangschikken van drie moleculen over energieniveaus waarbij de totate energie wordtbeperkt tot drie eenheden.
individueel kunnen worden geïdentificeerd en we etiketteren elk molecule om de microtoestandente herkennen die elk van de drie verschillende verdelingen opbouwen. We vinden dat er zesmaalzoveel microtoestanden verdeling II opbouwen dan om verdeling III op te bouwen. Verdeling II isdus zes maal meer waarschijnlijk dan verdeling III.
De algemene formule voor het aantal microtoestanden,W , dat de verdeling opmaakt3 is hetzelfde alsdat voor het aantal manieren waarop N voorwerpen kunnen worden verdeeld over een aantal dozenzodat er n1 voorwerpen zich bevinden in de eerste doos, n2 voorwerpen in de tweede enzovoort,
W = N !n1!n2! . . . (9.5)
Het totaal aantal mogelijke rangschikkingen is N !, maar we moeten dit delen door n1!n2! . . . daar we3Voor het aantal microtoestanden wordt ook vaak het symbool Ω gebruikt [2].

124 Hoofdstuk 9. De moleculaire basis van de thermodynamica
niet in staat zijn om een onderscheid te maken tussen toestanden die enkel van elkaar verschillendoor een uitwisseling van deeltjes binnen een welbepaald energieniveau. Voor de verdeling I inFiguur 9.3 verkrijgen we op die manier dat (weet dat 0! = 1 en 1! = 1)
WI = 3!1!2! = 3
voor de verdeling IIWII = 3!
1!1!1! = 6
en voor de verdeling IIIWIII = 3!
3! = 1
zoals we hadden gevonden door een directe opsomming. De meest waarschijnlijke verdeling vaneen groot aantal objecten tussen een aantal dozen, als we geen andere beperkingen hebben, is eenuniforme verdeling daar dit zou overeenkomen met het maximum aantal microtoestanden. Echter,wanneer we de verdeling van de moleculen beschouwen over de energieniveaus willen we weten hoeze zullen worden verdeeld wanneer het systeem zich in een thermodynamisch evenwicht bevindt.Dit leidt tot een verschillend type van verdeling dat zal worden begunstigd door een compromistussen een brede verdeling over vele toestanden die de neiging hebben om W te maximaliseren ende beperking op de totale energie.
9.3 De Boltzmann factor
In 1877 beschreef Ludwig Boltzmann (1844–1906) dat er een nauwe link bestaat tussen de entropievan het systeem en het aantal microtoestanden waaruit het systeem is opgebouwd [20]. Hij druktedit uit als
S = k lnW (9.6)
waarbij k de constante van Boltzmann is. Voor een aantal moleculen verdeeld over energieniveausgeldt dan dat
S = k
N !
n1!n2! . . .
(9.7)
Beschouwen we nu N als een groot aantal moleculen die verdeeld zijn over een reeks energieniveauszodanig dat n0 moleculen zich bevinden in de laagste energietoestand en ni moleculen zich bevindenin de ide energietoestand. We nemen aan dat het aantal moleculen in elk energieniveau groot is.Laat ons nu onderzoeken wat de gevolgen zijn van het toevoegen van een kleine hoeveelheid energieεi aan het systeem om één molecule te promoveren van de laagste energietoestand naar de ide
energietoestand zoals is gevisualiseerd in Figuur 9.4. Het systeem dat we hebben gekozen is eenconstant volumesysteem en dat geen arbeid verricht. Wanneer het systeem in evenwicht is, geldtdat dA = dU −TdS = 0 en dS = dU/T . Het toevoegen van εi representeert een kleine veranderingin de energie van dit grote systeem waarvoor de bijbehorende entropieverandering wordt gegevendoor
dS = dUT
= εiT
(9.8)

9.3. De Boltzmann factor 125
0 n0
εi ni
Figuur 9.4: Het promoveren van één molecule van het laagste energieniveau naar de ide energietoestandwaarbij n0 en ni het aantal moleculen zijn in de twee toestanden voor de promotie.
De entropieverandering kan eveneens worden uitgedrukt in termen van het aantal microtoestandendie het systeeem opbouwen. De kleine verandering in entropie, dS, die resulteert uit het promoverenvan een molecule van de nulde naar de ide toestand in een zeer groot systeem wordt gegeven door
dS = k ln
N !(n0 − 1)!n1! . . . (ni + 1)! . . .
− k ln
N !
n0!n1! . . . ni! . . .
= −k ln
(ni + 1)!ni!
· (n0 − 1)!n0!
Bijgevolg is
dS = −k lnni + 1n0
≈ −k ln ni
n0(9.9)
daar ni 1. Door nu de twee Vergelijkingen 9.8 en 9.9 voor dS gelijk te stellen, bekomen we
dS = εiT
= −k ln nin0
ofnin0
= e−εi/kT (9.10)
waarbij de term e−εi/kT de Boltzmann factor van de ide energietoestand wordt genoemd en Vergelij-king 9.10 gekend staat als de Boltzmann verdeling. Deze vergelijking is één van de meest belangrijkerelaties in de fysische wetenschappen en biedt een enorm inzicht in systemen waarmee we in fysischechemie te maken hebben.
De Boltzmann factor vertelt ons dat bij lage temperaturen alle moleculen de neiging hebben om inde laagst mogelijke toestand te liggen terwijl bij hoge temperaturen de verdeling over de energie-toestanden ernaar neigt om uniform te worden zoals is gevisualiseerd in Figuur 9.5. De energie vaneen systeem neigt naar nul bij de absolute nulpuntstemperatuur en, als alle deeltjes liggen in delaagste toestand, zullen het aantal microtoestanden een eenheid vormen en is S = k lnW = 0. Wezijn gekomen tot de basis voor de Derde Wet van de Thermodynamica. De Boltzmann verdelingkan nu worden geschreven in termen van waarschijnlijkheid, P , van een energietoestand die bezetis. Wanneer het totaal aantal moleculen gelijk is aan N , zal de waarschijnlijkheid Pi van eenderwelk afzonderlijk molecule die de ide toestand bezet, kunnen geschreven worden als
Pi = niN
= ni/n0N/n0
= e−εi/kT∑e−εi/kT
(9.11)

126 Hoofdstuk 9. De moleculaire basis van de thermodynamica
Lage temperatuur
T → 0e−εi/kT → 0
Gemiddelde temperatuur Hoge temperatuur
T →∞e−εi/kT → 1
Figuur 9.5: Simulatie van de verdeling van de moleculen over de beschikbare energietoestanden bij ver-schillende temperaturen.
daar N = ∑ni.
Hoewel we de Boltzmann verdelingswet voor systemen zijn bekomen waarbij de waarden van ni
groot zijn, is de validiteit meer algemeen. Het kan worden aangetoond dat de Boltzmann verde-lingswet evengoed toegepast kan worden op systemen waarbij de waarschijnlijkheid zeer klein isvan eender welk afzonderlijke energietoestand die bezet is.
9.4 Het gedrag van de warmtecapaciteit
Het gedrag van de warmtecapaciteit van stoffen in de vaste fase en in de gasfase was een mysterievoor wetenschappers voor de ontdekking van de kwantumtheorie. In klassieke mechanica waar deenergie wordt beschouwd als continu veranderlijk, kan het worden aangetoond dat de energie van eensysteem gelijkmatig wordt verdeeld tussen de verschillende bewegingsmoden die de vrijheidsgradenworden genoemd. Bovendien draagt, volgens de klassieke fysica, elke bewegingsgraad RT/2 bijaan de molaire energie (het equipartitie principe) en R/2 (daar CV = (dU/dT )V aan de molairewarmtecapaciteit. Bijgevolg is4
εtranslatie = 12m〈v
2x〉+ 1
2m〈v2y〉+ 1
2m〈v2z〉 = 3
2RT (9.12)
4Deze bespreking is sterk vereenvoudigd. Voor een meer gedetailleerde en wiskundigere bespreking verwijzen wenaar Referentie [2].
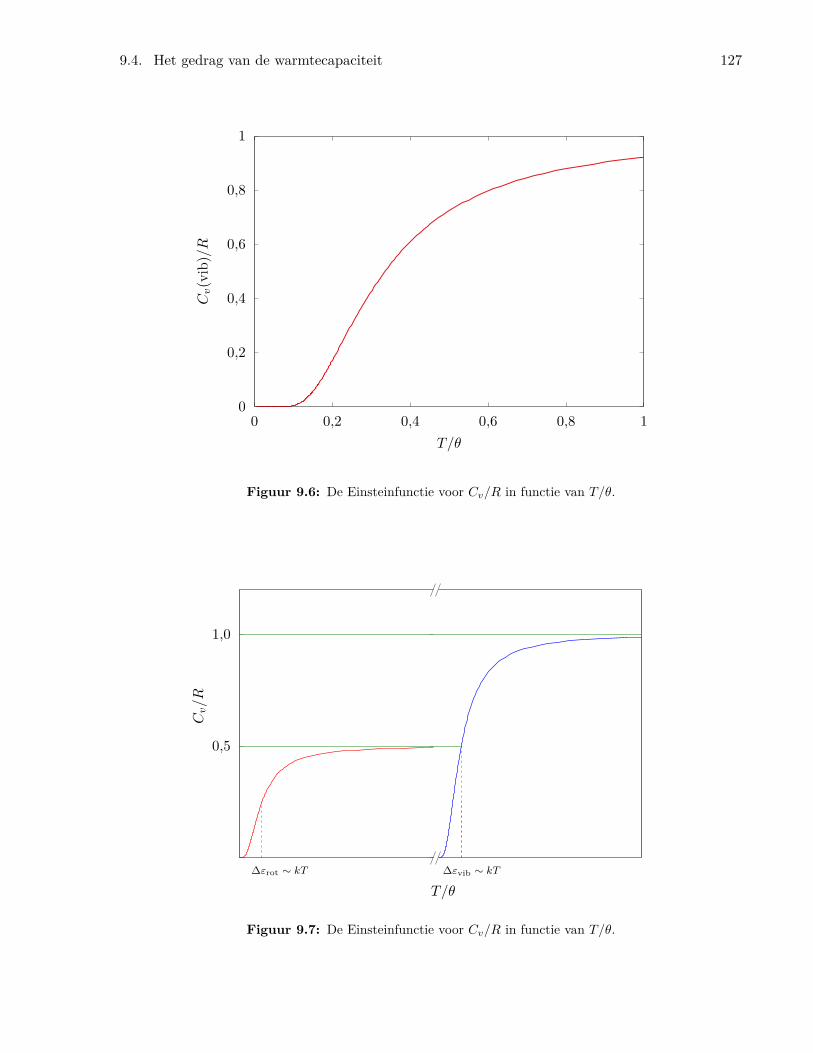
9.4. Het gedrag van de warmtecapaciteit 127
0
0,2
0,4
0,6
0,8
1
0 0,2 0,4 0,6 0,8 1
Cv(v
ib)/R
T/θ
Figuur 9.6: De Einsteinfunctie voor Cv/R in functie van T/θ.
0,5
1,0
∆εrot ∼ kT ∆εvib ∼ kT
Cv/R
T/θ
Figuur 9.7: De Einsteinfunctie voor Cv/R in functie van T/θ.


Bibliografie
[1] E. Brian Smith. Basic chemical thermodynamics. Oxford University Press, 4th edition, 1990.
[2] Gilbert W. Castellan. Physical Chemistry. Addison-Wesley Publishing Company, Inc., thirdedition, 1983.
[3] Thomas Engel and Philip Reid. Physical Chemistry. Pearson Education, Inc., 2006.
[4] Peter W. Atkins. Physical Chemistry. Oxford University Press, sixth edition, 1998.
[5] Keith J. Laidler. The World of Physical Chemistry. Oxford University Press, first edition,1993.
[6] E. R. Cohen, T. Cvitas, J. G. Frey, B. Holmström, K. Kuchitsu, R. Marquardt, I. Mills, F. Pa-vese, M. Quack, J. Stohner, H. L. Strauss, M. Takami, and A. J. Thor. Quantities, Unitsand Symbols in Physical Chemistry, IUPAC Green Book. IUPAC & RSC Publishing, 3rd edi-tion, 2008. http://www.iupac.org/fileadmin/user_upload/publications/e-resources/ONLINE-IUPAC-GB3-2ndPrinting-Online-Sep2012.pdf.
[7] J.H. van’t Hoff. Études de dynamique chimique. F. Muller & Company, 1884.
[8] https://www.chemheritage.org/historical-profile/amedeo-avogadro.
[9] J.R. Mayer. Bemerkungen über die Kräfte der unbelebten Natur. Annalen der Chemie undPharmazie, XLII:233–240, 1842.
[10] Joseph Black and John Robison. Lectures on the elements of chemistry: delivered in theUniversity of Edinburgh, by the late Joseph Black; now published from his manuscripts, byJohn Robison. Mundell for Longman and Rees, London, and Creech, Edinburgh, 1803.
[11] David R. Lide. CRC Handbook of Chemistry and Physics. CRC Press, 84th edition, 2003.
[12] J.P. Joule. On the existence of an equivalent relation between heat and the ordinary forms ofmechanical power. Philosophical Magazine, 27(179):205–207, 1845. http://dx.doi.org/10.1080/14786444508645256.
[13] Jaime Wisniak. Frederick Thomas Trouton: The Man, the Rule, and the Ratio. Chem.Educator, 6(1):55–61, 2001.
129

130 Bibliografie
[14] Jaime Wisniak. Benoit Paul Emile Clapeyron: A Short Bibliographical Sketch. Chem. Edu-cator, 5(2):83–87, 2000.
[15] Henry M. Leicester. Germain Henri Hess and the foundations of thermochemistry. Journal ofChemical Education, 28(11):581, 1951. http://dx.doi.org/10.1021/ed028p581.
[16] Jan Zawidzki. Über die Dampfdrucke binärer Flüssigkeitsgemische. Zeitschrift für Physikali-sche Chemie, 35:129–203, 1900.
[17] J. Frederic Daniell. On Voltaic Combinations. Philosophical Transactions of the Royal Societyof London, 126:107–124, 1836.
[18] J.P. Joule. On the calorific effects of magneto-electricity, and on the mechanical valueof heat. Philosophical Magazine, 23(152):263–276, 1843. http://dx.doi.org/10.1080/14786444308644730.
[19] Colin N. Banwell and Elaine M. McCash. Fundamentals of Molecular Spectroscopy. McGraw-Hill Publishing Company, 4th edition, 1994.
[20] Kim Sharp and Franz Matschinsky. Translation of Ludwig Boltzmann’s Paper “On the Re-lationship between the Second Fundamental Theorem of the Mechanical Theory of Heat andProbability Calculations Regarding the Conditions for Thermal Equilibrium” Sitzungberichteder Kaiserlichen Akademie der Wissenschaften. Mathematisch-Naturwissen Classe. Abt. II,LXXVI 1877, pp 373–435 (Wien. ber. 1877, 76:373–435). Reprinted in Wiss. Abhandlun-gen, Vol. II, reprint 42, p. 164–223, Barth, Leipzig, 1909. Entropy, 17(4):1971–2009, 2015.http://www.mdpi.com/1099-4300/17/4/1971.
Top Related