







Languages
Pages
Legal


1
UNIVERSITE JOSEPH FOURIER FACULTE DE MEDECINE DE GRENOBLE
Déficits hypophysaires Néonataux.
A propos de 10 cas cliniques.
Le 3 Avril 2009
Mémoire présenté par Dominique JAYET En vue de l’obtention du DES de Pédiatrie

2
SOMMAIRE
1 INTRODUCTION : ............................................................................................................................. 4
1.1 Définition : ............................................................................................................................... 4
1.2 Epidémiologie : ........................................................................................................................ 4
1.3 Embryologie - Anatomie – Physiologie : ................................................................................. 5
1.4 Diagnostic clinique et biologique, et traitement :................................................................... 6
1.4.1 Déficit somatotrope : ...................................................................................................... 6
1.4.2 Déficit thyréotrope : ........................................................................................................ 8
1.4.3 Déficit corticotrope : ....................................................................................................... 9
1.4.4 Déficit gonadotrope : .................................................................................................... 11
1.4.5 Déficit lactotrope : ......................................................................................................... 12
1.4.6 Diabète insipide central : .............................................................................................. 13
1.5 Diagnostics différentiels : ...................................................................................................... 14
1.6 Diagnostic étiologique : ......................................................................................................... 15
1.7 Surveillance : ......................................................................................................................... 19
2 MATERIEL ET METHODE : .............................................................................................................. 20
2.1 Le type d’enquête :. .............................................................................................................. 20
2.2 Les dossiers étudiés : ............................................................................................................. 20
2.3 Les éléments recueillis : ........................................................................................................ 20
3 RESULTATS : ................................................................................................................................... 21
3.1 Cas cliniques : ........................................................................................................................ 21
3.1.1 Cas clinique n°1 (CN née en novembre 2004) : ............................................................. 21
3.1.2 Cas clinique n°2 (DS né en janvier 2005) : ..................................................................... 23
3.1.3 Cas clinique n°3 (PV né en novembre 2005) : ............................................................... 25
3.1.4 Cas clinique n°4 (NS né en février 2006) ....................................................................... 27
3.1.5 Cas cliniques n°5 (DOCC née en octobre 2006) :........................................................... 29

3
3.1.6 Cas clinique n°6 (MAE né en novembre 2006) : ............................................................ 31
3.1.7 Cas clinique n°7 (RJ née en juin 2007) ........................................................................... 35
3.1.8 Cas clinique n°8 (MM née en juin 2007) : ..................................................................... 38
3.1.9 Cas clinique n°9 (CK née en juin 2007) : ........................................................................ 41
3.1.10 Cas Clinique n°10 (CA né en janvier 2009) - frère du cas clinique n°9 : ........................ 43
3.2 Analyse : ................................................................................................................................ 44
3.2.1 Les données cliniques initiales : .................................................................................... 44
3.2.2 Les données biologiques : ............................................................................................. 47
3.2.3 Les résultats de l’IRM : .................................................................................................. 50
4 Discussion : .................................................................................................................................... 51
4.1 Un diagnostic difficile : .......................................................................................................... 51
4.1.1 Incidence en Isère : ....................................................................................................... 51
4.1.2 Le délai diagnostic : ....................................................................................................... 51
4.1.3 L’hypoglycémie néonatale : .......................................................................................... 52
4.1.4 L’intérêt des tests dynamiques de contrôle dans l’enfance : ....................................... 53
4.2 Des éléments cliniques morphologiques évocateurs à ne pas négliger : ............................. 53
4.2.1 Le micropénis : .............................................................................................................. 53
4.2.2 Les anomalies de la ligne médiane : .............................................................................. 53
4.3 La cholestase néonatale et les troubles hépatiques : ........................................................... 54
4.4 L’IRM cérébrale : ................................................................................................................... 54
4.5 Dans l’enfance jusqu’à l’âge adulte : ..................................................................................... 54
5 Conclusion : ................................................................................................................................... 55
6 Annexes : ....................................................................................................................................... 56
7 Bibliographie ................................................................................................................................. 58

4
1 INTRODUCTION :
1.1 Définition :
Le déficit hypophysaire combiné multiple est l’association de déficits portant sur la production de
deux ou plusieurs hormones produites par l’hypophyse : soit l’antéhypophyse (déficit
somatotrope, thyréotrope, corticotrope, lactotrope ou gonadotrope) soit la posthypophyse
(diabète insipide) (1).
Le déficit hypophysaire néonatal est défini par l’apparition de ces déficits hypophysaires en
période néonatale. Il n’y a cependant pas de consensus qui ait déterminé un âge limite pour
pouvoir parler de déficit néonatal. Dans notre étude, nous avons considéré que l’enfant devait
avoir présenté des signes cliniques dans les trois premiers mois de vie, pour pouvoir parler de
déficit hypophysaire multiple néonatal.
1.2 Epidémiologie :
L’incidence des déficits hypophysaires congénitaux est faible. Elle est estimée à 1/3000-4000
naissances, mais elle pourrait être surestimée par le caractère transitoire de certains déficits (1).
Cependant, ces déficits congénitaux ne sont pas tous diagnostiqués en période néonatale : Ils
peuvent apparaitre plus tard durant l’enfance ou l’adolescence, et peuvent toucher initialement
un seul axe hypophysaire puis ensuite se compléter avec l’apparition de déficits dans les autres
axes (1). Les déficits hypophysaires multiples néonataux toucheraient environ 1/53000 nouveau-
nés (2).
Le déficit hypophysaire le plus fréquent est le déficit somatotrope. Lorsqu’il se révèle en période
néonatale, il est le plus souvent associé (50 à 70% des cas) à un autre déficit (gonadotrope,
corticotrope ou thyréotrope). (1; 3; 4).

5
1.3 Embryologie - Anatomie – Physiologie :
L’hypophyse est une glande endocrine logée dans la selle turcique de l’os sphénoïde, sous la face
inférieure du cerveau, et comprenant deux lobes, antérieur et postérieur, qui sont reliés
directement à l'hypothalamus par la tige pituitaire (5) :
l’antéhypophyse ou adénohypophyse :
o Elle est issue embryologiquement de la poche de Rathke.
o Elle est composée de tissu glandulaire.
o Elle est commandée par l’hypothalamus par l’intermédiaire du système porte
hypophysaire (vasculaire) qui véhicule les hormones hypothalamiques vers
l’hypophyse (Annexe 1).
o Elle produit des hormones :
L’hormone de croissance ou GH (Growth Hormone). La corticotrophine ou ACTH (Adrenocorticotropin hormone). La thyrotropine ou TSH (Thyroid Stimulating Hormone). L’hormone folliculo-stimulante ou FSH (Follicle Stimulating hormone). L’hormone lutéinisante ou LH (luteinizing hormone). La prolactine ou PRL.
la posthypophyse ou neurohypophyse :
o Son origine embryologique provient de l’infundibulum issu du lui-même du
diencéphale.
o Elle est composée de tissu nerveux, où se terminent des faisceaux nerveux
provenant du noyau supra-optique et du noyau para-ventriculaire.
o Elle joue un rôle endocrinien dans le stockage et la libération de l’hormone
antidiurétique (ADH) et de l’ocytocine qui proviennent de l’hypothalamus via la
tige pituitaire.

6
1.4 Diagnostic clinique et biologique, et traitement :
1.4.1 Déficit somatotrope (1) :
Il s’agit d’un défaut de production hypophysaire en hormone de croissance (GH).
1.4.1.1 Diagnostic clinique :
Chez l’enfant :
Il se manifeste par un retard de croissance staturo-pondéral : il n’y a pas la vitesse de croissance
importante des deux premières années de vie, mais dès la naissance, on retrouve une croissance
linéaire de type prépubertaire (+4 à 6cm/an) (6).
L’aspect clinique est parfois évocateur avec:
Un visage poupin,
Une ensellure nasale large,
Une saillie des bosses frontales,
L’enfant peut présenter aussi un faible développement musculaire et une adiposité tronculaire.
En période néonatale :
Le diagnostic est parfois difficile. Les symptômes qui peuvent être présents sont:
L’hypoglycémie (7) : liée à un déficit de la lipolyse secondaire
o Elle est souvent banalisée ou peut passer inaperçue : ses manifestations sont
parfois frustres ou atypiques, mêmes si l’hypoglycémie est profonde.
o Elle peut entrainer des convulsions.
o Elle survient après un jeûne court (avant la tétée, ou en fin de nuit).
Les anomalies de la ligne médiane et crânio-faciales (7) :
o Colobome rétinien, irien
o Fente palatine, labio-palatine
o Incisive médiane unique, agénésie des incisives latérales
o Sténose des sinus piriformes
o Nystagmus, malvoyance
o Dysplasie septo-optique
Un micropénis
Un ictère néonatal persistant

7
1.4.1.2 Diagnostic biologique :
Le diagnostic peut être fortement suspecté si le taux de GH est insuffisant lors d’une
hypoglycémie de jeûne spontanée ou lors d’un test dynamique de sécrétion (l’objectif du test est
une chute de la glycémie de 50% par rapport à la valeur de base) (3)
Chez l’enfant, après test de stimulation (3) :
Le déficit est dit complet si le pic est inférieur à 10mUI/L
Le déficit est partiel si le pic est compris entre 10 et 20 mUI/L
En période néonatale (1) :
Le déficit est dit complet si le taux est inférieur à 15 mUI/L
Le déficit est partiel si le taux est compris entre 15 et 30 mUI/L.
Associé à ce taux insuffisant, le dosage de l’IGF1 (Insuline-like Growth Factor 1) inférieur à
50ng/ml (sachant qu’il est tributaire de l’état nutritionnel de l’enfant (7) est un élément
diagnostique supplémentaire.
1.4.1.3 Traitement :
Le traitement substitutif consiste en l’administration quotidienne d’hormone de croissance
biosynthétique par voie sous-cutanée. La posologie initiale est de 0.025 à 0.035 mg/kg/j (1). Il est
recommandé, avant l’âge de 12 mois, de l’administrer en 2 injections quotidiennes afin de couvrir
le nycthémère et éviter les hypoglycémies.
Après 12 mois, l’administration se fait en une seule injection quotidienne. La posologie est ensuite
adaptée à la croissance et aux dosages de l’IGF1 qui doivent rester entre la moyenne et +2DS des
valeurs normales pour l’âge et le stade pubertaire.
Sa prescription (et son remboursement en France) nécessite des critères cliniques et biologiques :
1. Critères biologiques (chez l’enfant et le nouveau-né) :
Deux tests d’exploration dynamique des réserves mobilisables en GH avec au moins
une épreuve combinée (insuline/arginine, glucagon/propanolol, glucagon/betaxolol,
clonidine/betaxolol) (enfant)
ou 2 dosages de GH en hypoglycémie
ou 1 dosage en hypoglycémie et une anomalie de la ligne médiane
ou 1 dosage en hypoglycémie et un autre déficit hypophysaire associé.
2. Critères cliniques (chez l’enfant) :
Une taille ≤ -2DS par rapport aux normes pour l’âge à la mise sous traitement
et/ou une vitesse de croissance ≤-1DS par rapport aux normes pour l’âge, l’année
précédente.

8
1.4.2 Déficit thyréotrope (1) :
Il s’agit d’un défaut de production hypophysaire de TSH qui stimule la production des hormones
thyroïdiennes dont la forme active est la forme libre de la tétraïodothyronine (T4L).
Il n’est pas diagnostiqué par le dépistage néonatal qui ne détecte que les déficits thyroïdiens
périphériques, qui entrainent une élévation de la TSH.
Il est exceptionnellement isolé, et représente environ 1/100000 naissances (7).
1.4.2.1 Diagnostic clinique :
Chez l’enfant :
Il se traduit par une asthénie, une constipation, une frilosité, une prise de poids importante, une
cassure de la courbe staturale.
Ces symptômes peuvent être frustres et le diagnostic parfois retardé. Si le déficit est ancien, il
existe un retentissement squelettique avec une maturation osseuse retardée, se manifestant par
un âge osseux en retard par rapport à l’âge chronologique.
En période néonatale :
Le déficit thyréotrope se manifeste habituellement par un myxœdème, une hypotonie, des
troubles de la succion, une constipation, une hypothermie, un ictère néonatal prolongé, une
fontanelle large, un retard de la chute du cordon ombilical, une prise pondérale insuffisante, puis
un retard statural.
En l’absence de traitement substitutif précoce, le développement psychomoteur peut être
fortement altéré. En effet, les hormones thyroïdiennes jouent un rôle fondamental dans le
développement neuronal avant l’âge de 2 ans.
1.4.2.2 Diagnostic biologique :
Le diagnostic est confirmé par un taux de TSH bas ou une absence d’élévation du taux de TSH en
regard d’un taux de T4L bas, selon les normes pour l’âge.
1.4.2.3 Traitement :
Le traitement substitutif par L-Thyroxine doit être débuté en urgence, afin d’éviter un retard du
développement psychomoteur pouvant amener à un déficit intellectuel.
La posologie initiale est de 5 à 15µg/kg/jour (8), en une prise unique le matin, directement à la
cuillère. Le flacon se conserve au réfrigérateur.
La posologie est ensuite adaptée aux dosages de T4L et TSH en fonction des normes pour l’âge.

9
1.4.3 Déficit corticotrope (1) :
Il s’agit d’un défaut de production hypophysaire en ACTH qui stimule la fonction glucocorticoïde,
c'est-à-dire la production de cortisol surrénalien.
Il peut être masqué par un déficit thyréotrope, et peut ne se révéler qu’après sa correction (7).
Quand il existe un déficit hypophysaire, il est fondamental d’éliminer un déficit corticotrope
associé, en raison du risque vital qu’il engendre.
1.4.3.1 Diagnostic clinique :
Chez l’enfant (1; 7) :
Il peut associer une asthénie, une hypotension artérielle, des malaises avec hypoglycémie, une
pâleur et un retard de croissance statural. En cas de décompensation aiguë, on peut retrouver des
douleurs abdominales et un état de choc hémodynamique (avec déshydratation, tachycardie et
hypotension artérielle).
En période néonatale (1; 7) :
Il peut se manifester par :
Des hypoglycémies répétées,
Un ictère néonatal cholestatique,
Une hypotonie,
Une hypotension artérielle sévère, un état de choc hémodynamique inexpliqué,
Un syndrome de perte de sel.
1.4.3.2 Diagnostic biologique :
Chez l’enfant, on ne peut se contenter du dosage de la cortisolémie à 8h, en effet le nouveau-né
n’a pas encore de cycle du cortisol : il faut effectuer un cycle du cortisol, c'est-à-dire des dosages
répétés toutes les 4 heures.
L’élément diagnostic principal est le dosage de la cortisolémie en situation de stress : chez le
nouveau-né, lors d’une hypoglycémie, et chez l’enfant lors d’un test dynamique de stimulation.
La cortisolémie sera alors inférieure à 500 nmol/L (1).

10
1.4.3.3 Traitement :
Le traitement substitutif par hydrocortisone doit être débuté en urgence. La voie d’administration
et la posologie initiale dépendront de l’état clinique de l’enfant : en cas de mauvaise tolérance du
déficit, la posologie pourra être supra-physiologique comme en situation de stress et par voie
parentérale.
La posologie moyenne est ensuite de 5 à 10 mg/m²/jour, en 3 prises réparties en doses
décroissantes dans la journée, afin de mimer le cycle physiologique du cortisol.
Comme le déficit central est un déficit glucocorticoïde, il n’est pas nécessaire de réaliser une
substitution minérallocorticoïde, ni une supplémentation en sel.
L’éducation du patient et des parents est importante, et les consignes en cas de stress doivent
être expliquées : Il faut doubler, voire tripler la dose en cas de stress (fièvre, accident, chirurgie,
voire sport>2h) et administrer par voie intramusculaire en cas de vomissements.
Le patient doit porter une carte d’insuffisant surrénal afin qu’en situation d’urgence, le personnel
médical puisse en être informé du déficit, même si le patient est inconscient.

11
1.4.4 Déficit gonadotrope (1) :
Il s’agit d’un défaut de production en FSH et LH.
1.4.4.1 Diagnostic clinique :
En période néonatale, on peut retrouver chez le garçon, une cryptorchidie ou un micropénis.
Mais son retentissement se manifeste principalement en période pubertaire : absence de
développement pubertaire spontané après 12 ans chez la fille et après 15 ans chez le garçon.
1.4.4.2 Diagnostic biologique :
Il peut être suspecté devant des taux sériques en FSH, LH et en testostérone bas au moment du
pic post-natal, si d’autres déficits hypophysaires ont pu être mis en évidence au préalable.
En période pubertaire, les taux d’œstradiol et de testostérone seront prépubères, et les taux de
FSH et de LH seront bas.
Il n’y aura pas de réponse hypophysaire gonadotrope après test de stimulation au LH-RH.
1.4.4.3 Traitement :
Chez le garçon en cas de micropénis, il consiste dans la petite enfance en l’administration de
testostérone par voie injectable, dans le but d’obtenir un développement normal du pénis (9; 10).
A l’âge de la puberté normale (en fonction de l’âge osseux, 11 ans chez la fille et 13 ans chez le
garçon), le traitement consiste en l’administration des hormones sexuelles périphériques
(testostérone ou oestroprogestatif) pour induire la puberté avec une posologie progressivement
croissante pour mimer le développement pubertaire normal.

12
1.4.5 Déficit lactotrope (1) :
Il s’agit d’un défaut de production hypophysaire en prolactine (PRL).
1.4.5.1 Diagnostic clinique :
Il se manifeste uniquement chez la femme par une absence de montée de lait après
l’accouchement.
1.4.5.2 Diagnostic biologique :
Le diagnostic est affirmé devant une prolactine plasmatique basse, et une absence de réponse au
test dynamique de stimulation (métoclopramide).
Cependant, dans la situation du déficit hypophysaire multiple, le taux de prolactine peut être
explosif, il signifie alors qu’il existe une déconnexion entre l’hypothalamus et l’hypophyse qui
empêche le rétrocontrôle négatif.
1.4.5.3 Traitement :
Il n’y a aucun traitement

13
1.4.6 Diabète insipide central :
Il s’agit d’un défaut de libération de l’hormone antidiurétique (ADH)
1.4.6.1 Diagnostic clinique :
L’élément clinique majeur est le syndrome polyuro-polydipsique, avec des signes de
déshydratation (3).
L’enfant présente aussi une stagnation pondérale.
Il est rarement associé aux déficits hypophysaires congénitaux excepté en cas d’anomalie de la
ligne médiane (7).
1.4.6.2 Diagnostic biologique :
La natrémie et l’osmolarité sanguine sont élevées, tandis que la natriurie et l’osmolarité urinaire
sont diminuées (3).
Un test de restriction hydrique va permettre de confirmer le diagnostic (3) :
Un enfant normal restreint sa diurèse quelques heures après avoir été privé d’apport
hydrique. Sa natrémie et son osmolarité sanguine restent stables, tandis que l’osmolarité
urinaire va augmenter pour devenir supérieure à l’osmolarité sanguine (jusqu’à
500mosm/kg chez l’enfant de moins de deux ans et 800mosm/kg chez l’enfant de plus de
deux ans).
Dans le diabète insipide d’origine centrale, la production d’ADH est diminuée, si bien
qu’en restriction hydrique, l’enfant a une diurèse conservée avec apparition de signes de
déshydratation intracellulaire, d’une hypernatrémie et d’une hyperosmolarité sanguine
tandis que les urines restent diluées avec une natriurie et une osmolarité urinaire basses.
A la fin du test de restriction hydrique, un test thérapeutique au MINIRIN® est réalisé.
Dans le cadre du diabète insipide d’origine centrale, celui-ci corrige les symptômes.
1.4.6.3 Traitement :
Le traitement substitutif par MINIRIN® est débuté la dose de 0.5 µg x 2 / jour, par voie intra nasale
ou par voie sublinguale (qui est plus efficace du fait des rhinites fréquentes à cet âge). La
posologie est ensuite adaptée au bilan des entrées et des sorties, et aux ionogrammes sanguins et
urinaires.

14
1.5 Diagnostics différentiels :
Le diagnostic de déficit hypophysaire congénital est un diagnostic d’élimination.
Il convient de réaliser une IRM cérébrale et hypothalamo-hypophysaire pour écarter un déficit
secondaire à une tumeur, et pour rechercher des anomalies morphologiques hypothalamo-
hypophysaires, et rechercher des anomalies associées (dysplasie septo-optique) (1; 3).
1.5.1 Causes tumorales :
La plus fréquente est le craniopharyngiome, à développement intrasellaire. Il peut s’agir aussi
d’un adénome hypophysaire, d’un méningiome, d’un gliome ou d’une métastase de tumeur
périphérique.
Leur mode d’action peut être la compression de la tige pituitaire, ou l’envahissement par la
tumeur.
1.5.2 Causes infectieuses :
Encéphalites, méningites
1.5.3 Causes Iatrogènes :
L’hypopituitarisme peut survenir aussi après une radiothérapie, après une chirurgie concernant la
région hypothalamo-hypophysaire
1.5.4 Autres causes :
Il peut s’agir aussi d’une anoxo-ischémie, d’une hydrocéphalie, d’une anomalie vasculaire, d’un
traumatisme crânien grave, d’une hémochromatose, etc.

15
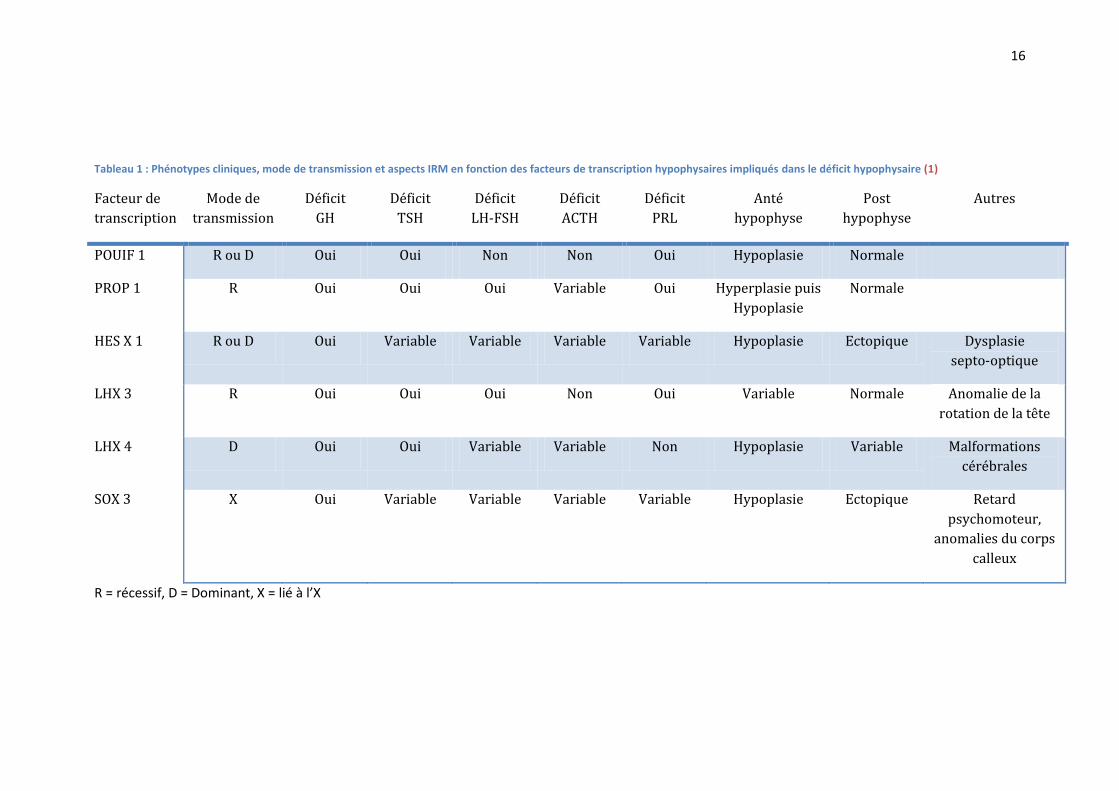
1.6 Diagnostic étiologique (1; 3; 8) : Le déficit hypophysaire combiné multiple est souvent d’origine génétique, lié à une mutation d’un
facteur de transcription impliqué dans la différenciation et la fonction de lignées cellulaires
antéhypophysaires. Cependant la mise en évidence d’une mutation précise reste à ce jour
difficile, même si de nouvelles mutations sont trouvées régulièrement.
Les modes de transmission, les phénotypes cliniques, et les aspects IRM en fonction des facteurs
de transcription hypophysaires impliqués dans le déficit hypophysaire, sont résumés dans le
TABLEAU 1.
L’algorithme décisionnel simplifié pour déterminer les facteurs de transcription hypophysaires à
séquencer est reproduit dans la FIGURE 1.
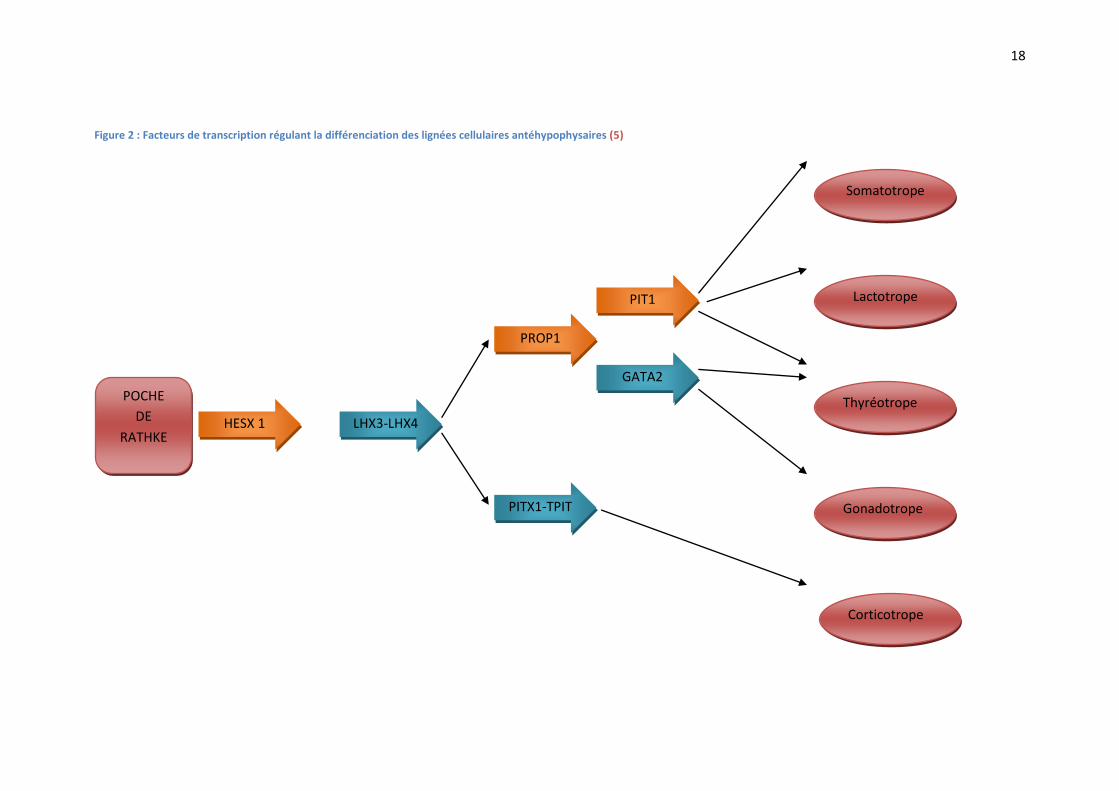
La régulation de la différenciation des lignées cellulaires hypophysaires par les différents facteurs
de transcription est résumée dans la FIGURE 2.
1.6.1.1 POU1F1 (ou PIT1) :
Une anomalie du facteur de transcription peut entrainer un déficit somatotrope, lactotrope, et
thyréotrope.
L’hypophyse est habituellement normale, ou hypoplasique. Il n’y a pas d’anomalie
posthypophysaire, ni d’interruption de tige.
1.6.1.2 PROP1 :
Une anomalie du facteur de transcription peut entrainer un déficit somatotrope, lactotrope,
thyréotrope, gonadotrope, et parfois corticotrope. Le phénotype peut être très variable, sans
corrélation entre le génotype et le phénotype.
L’hypophyse peut être hyperplasique, normale ou hypoplasique.
1.6.1.3 HESX1 :
Une anomalie du facteur de transcription peut entrainer un déficit pouvant aller du simple déficit
somatotrope au panhypopituitarisme, associé à une dysplasie septo-optique.
La dysplasie septo-optique associe une hypoplasie des nerfs optiques, une hypoplasie
hypophysaire et des anomalies de la ligne médiane (corps calleux, septum pellucidum,
posthypophyse ectopique). Elle touche 1/10000 à 1/50000 nouveau-nés (ORPHANET) (11).
Même si les mutations de HSEX1 sont reconnues pour être responsables de dysplasies septo-
optiques, elles ne sont retrouvées que pour un faible nombre de patients (1; 11). De plus, le
phénotype est très variable, et des mutations de HESX1 peuvent ne pas donner de dysplasie
septo-optique (12).
Enfin, des facteurs environnementaux semblent aussi intervenir dans la pathogénèse de la
dysplasie septo-optique (13; 14).
16
Tableau 1 : Phénotypes cliniques, mode de transmission et aspects IRM en fonction des facteurs de transcription hypophysaires impliqués dans le déficit hypophysaire (1)
Facteur de
transcription
Mode de
transmission
Déficit
GH
Déficit
TSH
Déficit
LH-FSH
Déficit
ACTH
Déficit
PRL
Anté
hypophyse
Post
hypophyse
Autres
POUIF 1 R ou D Oui Oui Non Non Oui Hypoplasie Normale
PROP 1 R Oui Oui Oui Variable Oui Hyperplasie puis
Hypoplasie
Normale
HES X 1 R ou D Oui Variable Variable Variable Variable Hypoplasie Ectopique Dysplasie
septo-optique
LHX 3 R Oui Oui Oui Non Oui Variable Normale Anomalie de la
rotation de la tête
LHX 4 D Oui Oui Variable Variable Non Hypoplasie Variable Malformations
cérébrales
SOX 3 X Oui Variable Variable Variable Variable Hypoplasie Ectopique Retard
psychomoteur,
anomalies du corps
calleux
R = récessif, D = Dominant, X = lié à l’X

17
Figure 1 : Algorithme décisionnel simplifié pour déterminer les facteurs de transcription hypophysaires à séquencer. (1)
Gène probable
Déficit corticotrope?
Déficit gonadotrope?
Malformation cérébrale?
D'abord éliminer une cause secondaire
IRM
OUI
Anomalie de la rotation de la tête et du cou
LHX 3
Dysplasiesepto-optique
HES X 1
Autres malformations
cérébrales, interruption de
tige
LHX 4
NON
NONou
prépubertaire
OUI
PROP 1
NON
Pit 1 PROP 1
OUI
PROP 1
18
Figure 2 : Facteurs de transcription régulant la différenciation des lignées cellulaires antéhypophysaires (5)
POCHE
DE
RATHKE
Somatotrope
Lactotrope
Thyréotrope
Gonadotrope
Corticotrope
HESX 1 LHX3-LHX4
PITX1-TPIT
GATA2
PROP1
PIT1

19
1.7 Surveillance (1) :
La surveillance est ciblée :
Sur la recherche de l’apparition de nouveaux déficits avec l’âge
Sur l’équilibre des traitements de substitution, et l’adaptation de leur posologie en
fonction de la clinique, de la croissance staturo-pondérale et de la biologie.
L’objectif du traitement est d’obtenir un développement staturo-pondéral conforme au canal
génétique, et un développement pubertaire harmonieux à un âge normal.
Il est important de prévenir les épisodes de déséquilibre du traitement, ou les décompensations
aiguës surtout quand il existe un déficit corticotrope ou un diabète insipide, en éduquant les
parents et l’enfant lorsqu’il est en âge de comprendre.

20
2 MATERIEL ET METHODE :
2.1 Le type d’enquête : Il s’agit d’une enquête épidémiologique descriptive, rétrospective réalisée au CHU de Grenoble.
2.2 Les dossiers étudiés : Les dossiers étudiés étaient ceux des patients suivis au CHU de Grenoble pour un déficit
hypophysaire multiple de découverte néonatale.
Les patients inclus étaient ceux dont le diagnostic a été posé sur des symptômes présents dans les
trois premiers mois de vie.
2.3 Les éléments recueillis :
2.3.1 Les éléments cliniques qui étaient relevés étaient les suivants :
Le déroulement de la grossesse
Le terme, le poids (P), la taille (T) et le périmètre crânien (PC) de naissance, leur valeur en
percentile pour déterminer l’existence d’un retard de croissance intra-utérin (RCIU) a été
établie selon les courbes de Leroy et Lefort (15)
La présentation clinique : existence de difficultés alimentaires, d’une stagnation
pondérale, d’une hypotonie, d’une détresse respiratoire, d’une constipation, d’un ictère
cutanéo-conjonctival, et d’une dysmorphie faciale. Etait relevée aussi la constatation
d’hypoglycémies récidivantes.
2.3.2 Les paramètres biologiques relevés étaient les suivants :
Pour l’axe thyréotrope : les taux sériques de TSH et de T4L
Pour l’axe corticotrope : le taux de cortisol stimulé lors d’un stress hypoglycémique.
Pour l’axe somatotrope : le taux sérique d’IGF1 de base, et le taux sérique de GH lors d’un
stress hypoglycémique.
Pour l’axe lactotrope : le taux sérique de PRL.
Pour l’axe gonadotrope, les taux sériques de FSH, LH et Testostérone lors du pic post-
natal (entre 4 et 6 semaines)
Pour la post hypophyse : les ionogrammes et osmolarités sanguines et urinaires, ainsi que
les résultats du test de restriction hydrique, si réalisé.
Les valeurs de la glycémie lors des dosages hormonaux et le test éventuellement utilisé
pour obtenir l’hypoglycémie.
Les normes utilisées pour les différents paramètres biologiques sont résumées en ANNEXE 2.
2.3.3 Les résultats de l’IRM cérébrale et de l’analyse génétique :
Les résultats des IRM réalisées ont été recueillis. Nous avons joint les images des IRM pour
lesquelles les anomalies étaient probantes et pour lesquelles les examens ont été conservés
informatiquement, permettant une qualité d’image suffisante.
Les résultats génétiques actuellement en notre possession ont été recueillis.

21
3 RESULTATS :
3.1 Cas cliniques :
3.1.1 Cas clinique n°1 (CN née en novembre 2004) :
3.1.1.1 Grossesse et mensurations à la naissance :
La grossesse a été marquée par un RCIU avec anamnios, le caryotype anténatal était normal 46XX.
Naissance d’une fille à 37 SA + 4 jours, avec un poids à 2230g (< 10èmep) pour une taille à 44cm
(<3èmep) et une PC à 31cm (10èmep), donc un RCIU harmonieux.
3.1.1.2 Circonstances de prise en charge :
La période néonatale a été marquée à 3 jours de vie par un ictère à bilirubine conjuguée et par
des hypoglycémies à répétition (1.4 mmol/L). L’examen clinique était sans particularité.
3.1.1.3 Bilan endocrinien :
Le bilan endocrinien a été réalisé à 4 jours de vie, en hypoglycémie spontanée (1.4 mmol/L).
La TSH était à 2.03 mUI/L pour une T4L à 20.8 pmol/L, normales. La cortisolémie était basse à 280 nmol/L en hypoglycémie. Le taux de GH était insuffisant à 13.56 µUI/mL en hypoglycémie et le dosage d’IGF1 de base était bas à 24 ng/mL. Les ionogrammes sanguins et urinaires étaient normaux.
3.1.1.4 Diagnostic retenu et traitement :
L’enfant présentait donc un déficit somatotrope complet associé à un déficit corticotrope partiel.
Un traitement substitutif par hormone de croissance et hydrocortisone a été débuté en urgence,
permettant l’amendement rapide des hypoglycémies.
3.1.1.5 Résultats de l’IRM :
L’IRM à 4 mois retrouvait une tige pituitaire grêle sans interruption, l’antéhypophyse et la
posthypophyse étaient d’aspect normal.
3.1.1.6 Evolution
A 3 ans de vie, un bilan hormonal de surveillance montre, par un test à l’ornithine, la persistance
du déficit somatotrope partiel (pic à 18.3 µUI/mL), mais une bonne stimulation de l’axe
corticotrope (cortisolémie qui passe de 253 nmol/L à 601 nmol/L) : la substitution en
hydrocortisone a été suspendue.
Le développement psychomoteur est normal.
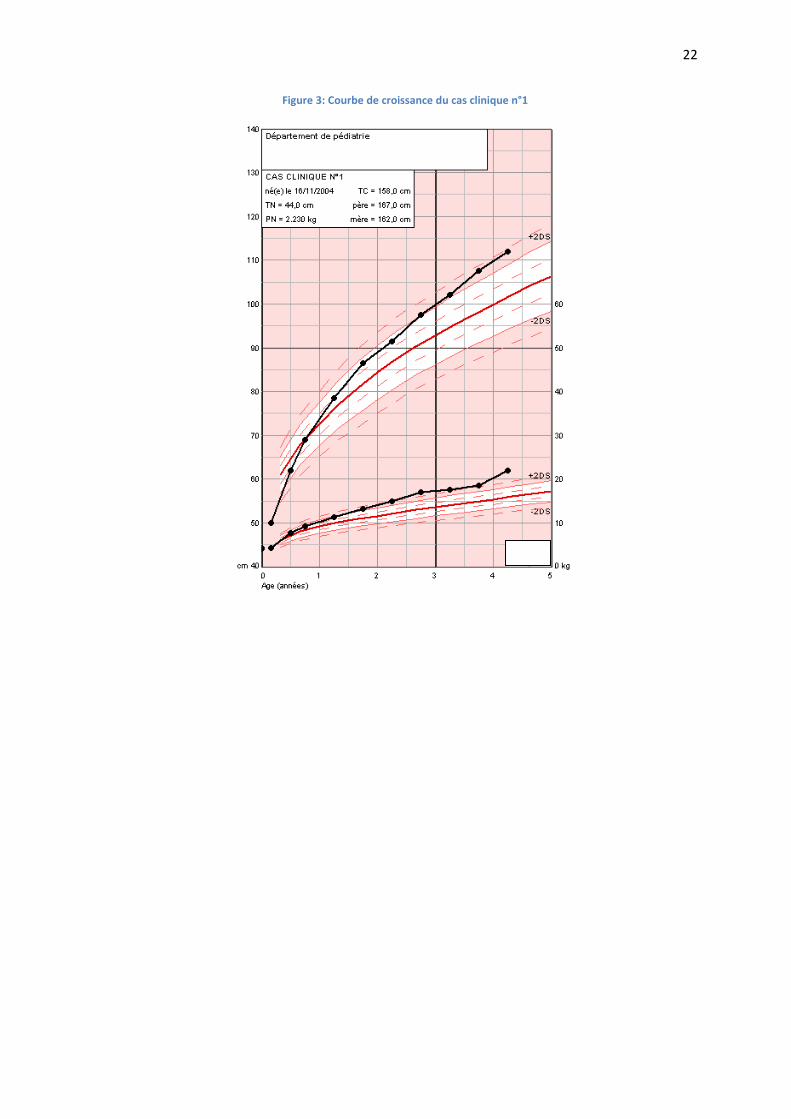
La croissance staturo-pondérale est très satisfaisante à +3DS (FIGURE 3).
22
Figure 3: Courbe de croissance du cas clinique n°1

23
3.1.2 Cas clinique n°2 (DS né en janvier 2005) :
3.1.2.1 Grossesse et mensurations à la naissance :
La grossesse avait été marquée par un hématome rétro-placentaire à 3 mois, puis une rupture
prématurée des membranes. Naissance d’un garçon eutrophique à 32 SA, avec un poids à 2100g
(50èmep), une taille à 46 cm (>50èmep) et un PC à 32.5cm (>90èmep).
3.1.2.2 Circonstance de prise en charge :
L’évolution initiale a été marquée par la fréquence anormale d’épisodes de bradycardie et de
cyanose, mis sur le compte d’un reflux gastro-oesophagien, d’une plicature gastrique traités
symptomatiquement. L’enfant sort du service de néonatalogie à 6 semaines de vie (38 SA).
L’enfant a été hospitalisé à 2 mois et 2 mois ½ de vie pour des malaises répétés (avec hypotonie,
pâleur, pauses respiratoires, bradycardies), lors d’épisodes infectieux viraux.
L’enfant a été à nouveau hospitalisé à 3 mois pour un malaise sévère sur fausse route lors de
l’administration de vitamine D. L’examen clinique retrouvait alors une macroglossie, un
myxœdème, une constipation, une hernie ombilicale et inguinale, une fontanelle antérieure large,
une hypotonie axiale et un retard du développement psychomoteur. Une maladie métabolique
avait alors été évoquée, et le bilan biologique était négatif, notamment la recherche d’une
mucopolysaccharidose. La deuxième hypothèse était une pathologie endocrinienne.
3.1.2.3 Bilan endocrinien
Le bilan endocrinien a été réalisé à 3 mois de vie.
La TSH était inadaptée car non augmentée à 4.23 mUI/L pour une T4L basse à 9.9 pmol/L.
La cortisolémie passait de 414 nmol/L à 900nmol/L après test au Glucagon (hypoglycémie à
1.9mmol/L).
Le maximum du pic de GH était insuffisant après test au glucagon à 22 µUI/mL, avec un taux
d’IGF1 de base bas à 24 ng/mL.
Les taux de FSH à 0.3 mUI/mL, de LH < 0.2 mUI/L et de testostérone à 0.6 nmol/L (norme entre
0.36 et 5.2) sont à interpréter avec prudence car réalisés à la fin du pic post-natal.
La prolactine était explosive à 2327 µUI/mL.
Les ionogrammes sanguins et urinaires étaient normaux.
3.1.2.4 Diagnostic retenu et traitement :
L’enfant présentait donc un déficit somatotrope partiel associé à un déficit thyréotrope. Un
traitement substitutif par hormone de croissance et L-thyroxine a donc été débuté en urgence. La
symptomatologie s’est rapidement amendée.
3.1.2.5 Résultats de l’IRM :
L’IRM à 7 mois retrouvait une post hypophyse en place, une antéhypophyse de petite taille, la tige
pituitaire était grêle ainsi que la région hypothalamique. Il n’y avait pas d’anomalie des lobes
olfactifs.
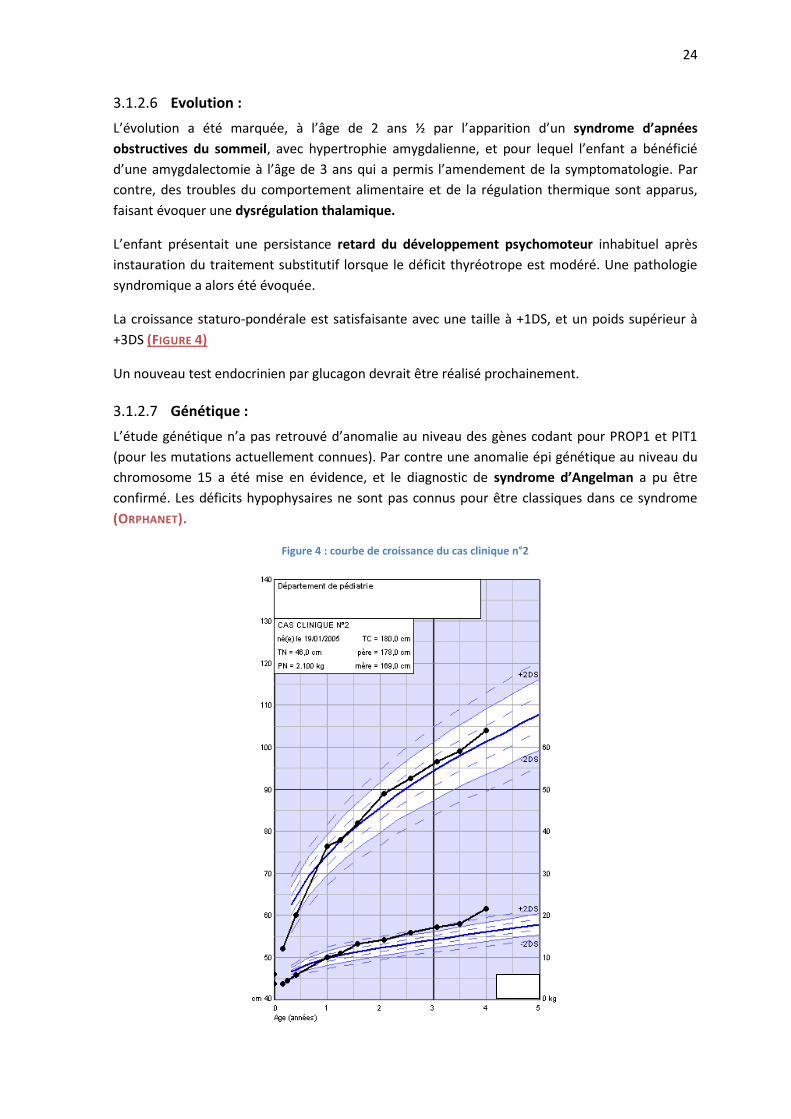
24
3.1.2.6 Evolution :
L’évolution a été marquée, à l’âge de 2 ans ½ par l’apparition d’un syndrome d’apnées
obstructives du sommeil, avec hypertrophie amygdalienne, et pour lequel l’enfant a bénéficié
d’une amygdalectomie à l’âge de 3 ans qui a permis l’amendement de la symptomatologie. Par
contre, des troubles du comportement alimentaire et de la régulation thermique sont apparus,
faisant évoquer une dysrégulation thalamique.
L’enfant présentait une persistance retard du développement psychomoteur inhabituel après
instauration du traitement substitutif lorsque le déficit thyréotrope est modéré. Une pathologie
syndromique a alors été évoquée.
La croissance staturo-pondérale est satisfaisante avec une taille à +1DS, et un poids supérieur à
+3DS (FIGURE 4)
Un nouveau test endocrinien par glucagon devrait être réalisé prochainement.
3.1.2.7 Génétique :
L’étude génétique n’a pas retrouvé d’anomalie au niveau des gènes codant pour PROP1 et PIT1
(pour les mutations actuellement connues). Par contre une anomalie épi génétique au niveau du
chromosome 15 a été mise en évidence, et le diagnostic de syndrome d’Angelman a pu être
confirmé. Les déficits hypophysaires ne sont pas connus pour être classiques dans ce syndrome
(ORPHANET).
Figure 4 : courbe de croissance du cas clinique n°2

25
3.1.3 Cas clinique n°3 (PV né en novembre 2005) :
3.1.3.1 Grossesse :
La grossesse avait été marquée par un RCIU (fémur < 3ème percentile), le doppler des artères
utérines était normal, le caryotype à l’amniocentèse était normal (46 XY), une TDM réalisée à
28SA ne met pas en évidence de chondrodysplasie. Naissance d’un garçon à 36 SA + 5 jours avec
un poids à 1700g (<3èmep) pour une taille à 42.5cm (<3èmep) et un PC à 31cm (<25èmep) : donc un
RCIU disharmonieux.
3.1.3.2 Circonstance de prise en charge :
L’enfant a présenté une détresse respiratoire néonatale. Dans le service de néonatalogie, des
épisodes répétés d’hypoglycémie ont alors été constatés (jusqu’à 2.4 mmol/L) malgré une
perfusion. L’enfant a présenté un ictère à bilirubine libre. L’examen clinique retrouvait une
malformation des organes génitaux externes à type d’hypospadias antérieur sans micropénis ni
cryptorchidie.
3.1.3.3 Bilan endocrinien :
A 22 jours de vie, une épreuve de jeûne a été réalisée : hypoglycémie à 2.9 mmol/L à H9.
La TSH était normale à 9.14 mUI/L, et la T4L était normale à 12.2 pmol/L.
La cortisolémie était basse à 342 mmol/L en stress hypoglycémique.
Le taux de GH était insuffisant à 19.4µUI/mL en hypoglycémie, et le taux d’IGF1 de base était à
52ng/mL (à la limite inférieure).
L’insulinémie était basse à 0.27 µUI/mL en hypoglycémie, permettant d’éliminer un
hyperinsulinisme.
Le taux sérique de FSH était à 1.3 mUI/mL, LH à 6.5 mUI/mL et testostérone à 7.1nmol/L éliminant
un déficit gonadotrope néonatal.
Les ionogrammes sanguins et urinaires étaient normaux.
3.1.3.4 Diagnostic retenu et traitement :
Cet enfant présentait donc un déficit somatotrope partiel associé à un déficit corticotrope
partiel. Le traitement substitutif par hormone de croissance et hydrocortisone a été débuté en
urgence, et les hypoglycémies se sont rapidement corrigées. Un traitement par androgène retard
a été débuté à 10 mois pour faciliter la prise en charge chirurgicale de son hypospadias à 17 mois.
3.1.3.5 Résultats de l’IRM :
L’IRM à 3 mois retrouvait une antéhypophyse et posthypophyse en place et d’aspect normal. La
tige pituitaire était bien visualisée.
3.1.3.6 Evolution :
A l’âge de 12 mois, un nouveau test de stimulation au glucagon a été réalisé, il confirmait le déficit
somatotrope complet (maximum du pic de GH à 7.26µUI/mL) et montrait la correction du déficit
corticotrope puisque la cortisolémie s’élevait à 674 nmol/L. Le traitement par hydrocortisone a
alors été arrêté.

26
Le développement psychomoteur est normal
La croissance staturo-pondérale est satisfaisante à +1DS (FIGURE 5).
Figure 5: Courbe de croissance du cas clinique n°3

27
3.1.4 Cas clinique n°4 (NS né en février 2006)
3.1.4.1 Grossesse et mensurations à la naissance :
La grossesse avait été marquée par un ralentissement de la vitesse de croissance au dernier
trimestre, les trois échographies anténatales étaient conformes à l’âge gestationnel. Naissance
d’un garçon, à 40 SA+1 jour avec un poids à 2110g (<<3èmep), pour une taille à 47cm (<3èmep) et un
PC à 32cm (10èmep), donc un RCIU harmonieux.
3.1.4.2 Circonstances de prise en charge :
A 8 heures de vie, des hypoglycémies sont apparues (0.8 mmol/L) persistantes malgré
enrichissement de l’alimentation. Les hypoglycémies étaient asymptomatiques. L’examen clinique
retrouvait des éléments dysmorphiques (avec un front bombé, une ensellure nasale marquée, un
visage arrondi), une absence de micropénis, et des testicules en place. La persistance des
hypoglycémies malgré l’augmentation des apports glucidiques par voie entérale et parentérale a
fait suspecter un déficit antéhypophysaire. Le caryotype était normal 46 XY
3.1.4.3 Bilan endocrinien :
Le bilan à 5 jours de vie, a été réalisé en hypoglycémie lors d’une épreuve de jeûne (1.8 mmol/L)
La TSH était normale à 3.89 mUI/L pour une T4L normale à 17 pmol/L.
La cortisolémie était basse à 276 mmol/L en stress hypoglycémique.
Le maximum du pic de GH était insuffisant à 13.8µUI/mL, et le taux d’IGF1 de base était bas à
23ng/mL.
L’insulinémie était basse à 0.77µUI/mL.
Le taux sérique de FSH était à 1.2mUI/mL, LH à 7.3mUI/mL, et testostérone à 5.5 nmol/L :
normaux.
Les ionogrammes sanguins et urinaires étaient normaux.
3.1.4.4 Diagnostic retenu et traitement :
Cet enfant présentait donc un déficit somatotrope complet associé à déficit corticotrope partiel.
Le traitement substitutif par hormone de croissance et hydrocortisone a été débuté en urgence,
permettant la correction rapide des hypoglycémies.
3.1.4.5 Résultats de l’IRM :
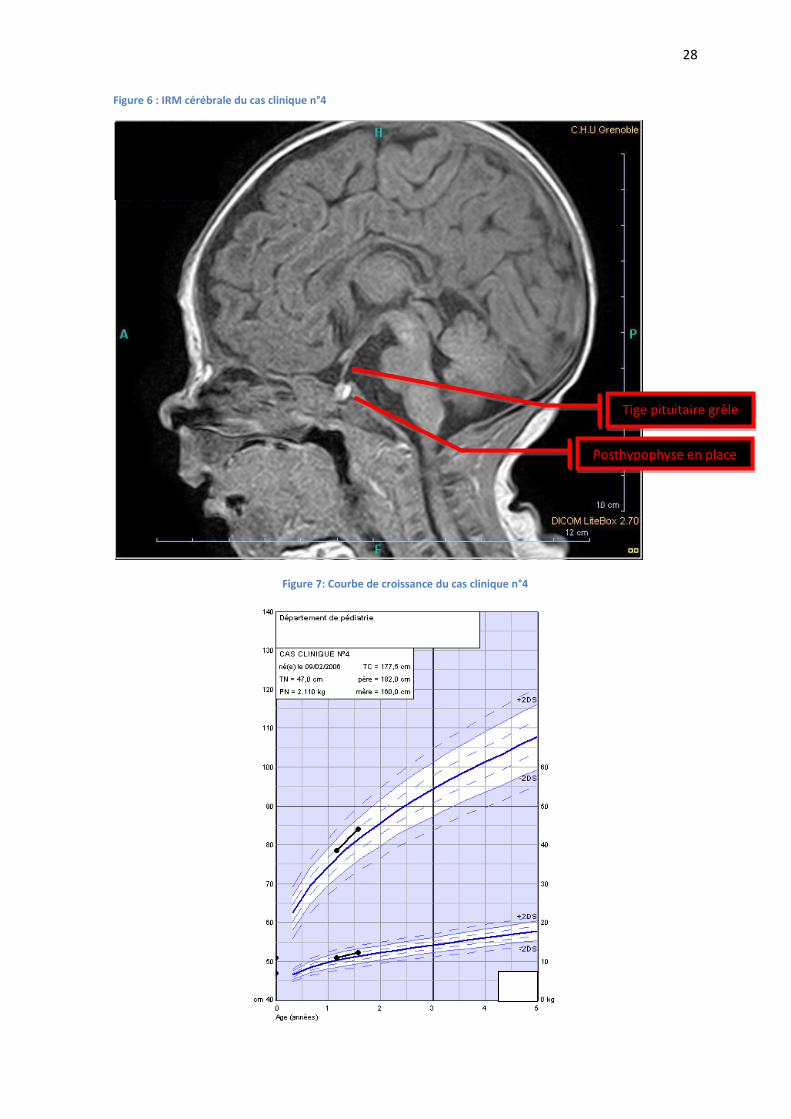
L’IRM à 1 mois retrouvait une antéhypophyse d’apparence normale, une tige pituitaire grêle, et
une posthypophyse normale en place (FIGURE 6).
3.1.4.6 Evolution :
A l’âge de 15 mois, un nouveau test dynamique au glucagon a été réalisé, il ne retrouvait pas de
déficit somatotrope ni corticotrope : la cortisolémie passait de 186 à 710 nmol/L, le maximum du
pic de GH était à 21.84 µUI/mL, pour une hypoglycémie à 2.4 mmol/L. Le traitement substitutif a
été arrêté, avec poursuite d’une bonne croissance.
Le développement psychomoteur est normal
L’évolution de la courbe de croissance staturo-pondérale est satisfaisante à +1DS (FIGURE 7).
28
Figure 6 : IRM cérébrale du cas clinique n°4
Figure 7: Courbe de croissance du cas clinique n°4
Tige pituitaire grêle
Posthypophyse en place

29
3.1.5 Cas cliniques n°5 (DOCC née en octobre 2006) :
3.1.5.1 Grossesse et mensurations à la naissance :
Les parents étaient consanguins (cousins germains)
La grossesse a été marquée par un RCIU. Naissance d’une fille à 41 SA + 4 jours, avec un poids de
2980g (<10èmep), une taille de 49cm (10èmep) et un PC de 33.5cm (25èmep), donc un RCIU
disharmonieux.
3.1.5.2 Circonstances de prise en charge :
Depuis la naissance, l’enfant présentait des difficultés alimentaires avec épuisement au sein,
absence d’amélioration sous préparation pour nourrisson.
A 5 semaines de vie, l’enfant a été hospitalisée pour prise en charge de la stagnation pondérale
avec des difficultés alimentaires majeures. L’interrogatoire retrouvait une constipation, une
stagnation pondérale depuis 2 semaines, et l’examen clinique retrouvait une somnolence, un
ictère cutanéo-conjonctival, un œdème des paupières, une macroglossie, une hypotonie axiale et
segmentaire franche, une hernie ombilicale réductible, un stridor, un souffle cardiaque systolique
peu intense et une constipation. Les bilans infectieux et métaboliques (ammoniémie et lactates)
étaient négatifs.
3.1.5.3 Bilan endocrinien :
La TSH était effondrée à 0.5pmol/L et la T4L était effondrée à 0.039mUI/mL. Le traitement
substitutif par L-Thyroxine a été débuté en urgence, et les autres axes hypophysaires ont alors été
explorés :
La cortisolémie était normale à 500 nmol/L après test au glucagon.
Le maximum du pic de GH sous Glucagon était insuffisant à 12 µUI/mL et le taux d’IGF1 de base
était bas à 29 ng/mL.
La Prolactine était explosive à 1363 UI/mL.
Le taux de FSH était à 11.7 mUI/mL, LH à 0.9 mUI/mL : normaux.
Les ionogrammes sanguins et urinaires étaient normaux.
3.1.5.4 Diagnostic retenu et traitement :
Cet enfant présentait donc un déficit thyréotrope profond associé à un déficit somatotrope
partiel. Un traitement par hormone de croissance à doses substitutives a été associé à la
L-Thyroxine, permettant l’amélioration rapide de l’état général et l’amendement des symptômes.
3.1.5.5 Les résultats de l’IRM :
Une IRM hypothalamo-hypophysaire à 2 mois ½ ne retrouvait pas d’interruption de la tige
pituitaire, mais ne pouvait éliminer une tige pituitaire grêle.

30
3.1.5.6 Evolution
A l’âge de 2 ans, un nouveau test de stimulation au Glucagon a été réalisé, montrant la
persistance du déficit somatotrope partiel, avec un maximum du pic de GH à 17.8 µUI/mL pour
une glycémie à 2.3 mmol/L).
Le développement psychomoteur est satisfaisant.
La croissance staturo-pondérale est satisfaisante avec une taille sur la moyenne et un poids à -1DS
(FIGURE 8).
3.1.5.7 Génétique :
L’étude génétique n’a pas retrouvé d’anomalie au niveau des gènes PIT1 et PROP1, concernant les
mutations actuellement connues.
Figure 8: Courbe de croissance du cas clinique n°5

31
3.1.6 Cas clinique n°6 (MAE né en novembre 2006) :
3.1.6.1 Grossesse et mensurations à la naissance :
La grossesse avait été sans particularité. Naissance d’un garçon eutrophique à 37 SA avec un poids
à 2590g (<25èmep), pour un taille à 50cm (>50èmep) et un PC à 34cm (>50èmep).
3.1.6.2 Circonstance de prise en charge :
L’enfant a présenté une détresse respiratoire et un état de choc hémodynamique d’origine
septique, probablement aggravé par le déficit qui était alors méconnu.
A 3 semaines de vie, est apparue une hypernatrémie (156 mmol/L) avec une polyurie
(7.4mL/kg/h) hyposmotique (54mosm/kg). Un test au Minirin® a confirmé le diagnostic de diabète
insipide en permettant de normaliser la natrémie et la diurèse.
3.1.6.3 Bilan endocrinien
Les axes antéhypophysaires ont donc été explorés, après test au glucagon.
La TSH était à 4.63 mUI/mL, et la T4L à 13.5 pmol/L, soit à la limite inférieure de la normale.
La cortisolémie passait de 218 à 652 nmol/L après test au glucagon.
Le maximum du pic de GH sous glucagon était insuffisant à 10.88µUI/mL et le taux d’IGF1 de base
était à la limite inférieure de la normale à 57 ng/mL.
Le taux sérique de FSH était à 1.0mUI/mL, la LH à 437mUI/mL, et la testostérone à 4nmol/L
(normal).
3.1.6.4 Diagnostic retenu et traitement :
L’enfant présentait donc un diabète insipide, associé à un déficit somatotrope complet, et
thyréotrope. Un traitement substitutif par L-thyroxine, et hormone de croissance a été ajouté en
urgence au Minirin.
3.1.6.5 Résultats de l’IRM :
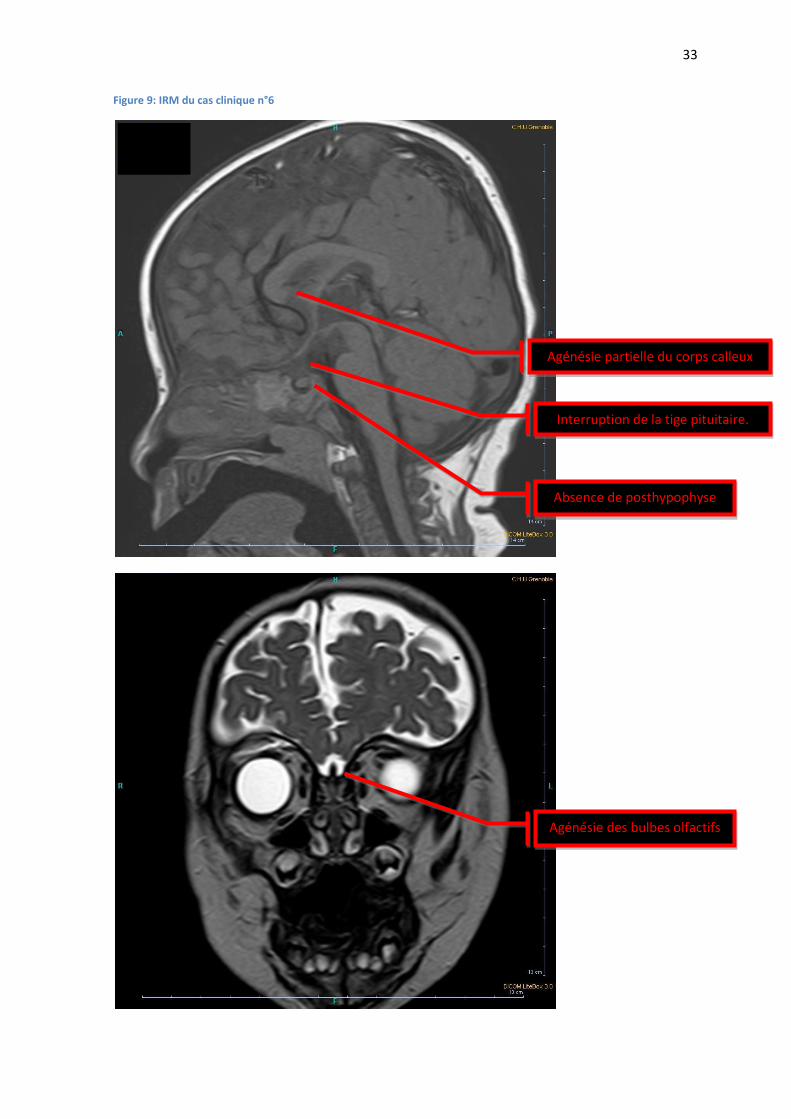
Une IRM à 1 mois ½ retrouvait une antéhypophyse de petite taille, une interruption de la tige
pituitaire, associées à une absence de posthypophyse (FIGURE 9).

32
3.1.6.6 Evolution :
Le suivi clinique a mis en évidence une absence de poursuite oculaire, et un examen
ophtalmologique a identifié une atrophie papillaire bilatérale au fond d’œil.
Une IRM à 10 mois permettait de mettre en évidence, outre les anomalies connues, une agénésie
partielle du corps calleux, associée à une agénésie des bulbes olfactifs et une hypoplasie du
chiasma optique et des nerfs optiques sur tout leur trajet avec absence de visualisation des
bandelettes optiques. Le diagnostic de dysplasie septo-optique a été posé.
A l’âge de 16 mois, l’enfant a présenté un syndrome d’apnées obstructives du sommeil, avec
hypertrophie amygdalienne. Une amygdalectomie a été pratiquée à l’âge de 17 mois, avec une
amélioration partielle de la symptomatologie. Une ventilation non invasive nocturne a donc été
débutée à l’âge de 19 mois
L’enfant présente un retard du développement psychomoteur avec des difficultés motrices et
alimentaires.
La croissance staturo-pondérale est satisfaisante avec un poids à +3DS une taille à +2DS
(FIGURE 10)
3.1.6.7 Génétique :
L’étude génétique n’a pas retrouvé d’anomalie au niveau du gène HESX1, pour les mutations
actuellement connues.
33
Figure 9: IRM du cas clinique n°6
Absence de posthypophyse
Interruption de la tige pituitaire.
Agénésie partielle du corps calleux
Agénésie des bulbes olfactifs

34
Figure 10 : courbe de croissance du cas clinique n°6
Hypoplasie du chiasma optique

35
3.1.7 Cas clinique n°7 (RJ née en juin 2007)
3.1.7.1 Grossesse et mensurations à la naissance :
La grossesse a été marquée par la prise de Cytotec® à 12 SA pour suspicion de fausse couche
spontanée, et un utérus myxœdémateux. Naissance d’une fille eutrophique à 40 SA + 2 jours, avec
un poids à 3090g (<25èmep) pour une taille à 50cm (<50èmep) et un PC à 34cm (<50èmep).
3.1.7.2 Circonstances de prise en charge :
A 17 heures de vie, sont apparues des difficultés alimentaires, chez une enfant hypotonique,
ictérique, avec un syndrome occlusif (absence d’émission du méconium, ballonnement). On
retrouvait alors une hypoglycémie (<2.2 mmol/L). L’alimentation a été arrêtée et une perfusion a
été débutée. Les examens complémentaires à visée digestive retrouvaient un iléus fonctionnel.
A 4 jours de vie, la perfusion a été arrêtée accidentellement, l’hypoglycémie récidivant
immédiatement (0.55 mmol/l), accompagnée de trémulations. Aucun bilan endocrinologique n’a
alors été effectué, la perfusion a été reprise et un avis endocrinologique a été demandé.
3.1.7.3 Bilan endocrinien :
Le bilan a été réalisé à 4 jours de vie, pendant une hypoglycémie spontanée à 0.7 mmol/L.
La TSH était inadaptée à 6 µUI/mL pour une T4L basse à 6 pmol/L.
La cortisolémie était effondrée à 11 nmol/L en stress hypoglycémique.
Le maximum du pic de GH en hypoglycémie était effondré à 0.94 µUI/mL et l’IGF1 était indosable.
L’insulinémie était à 0.83 µUI/mL.
Les ionogrammes sanguin et urinaire étaient normaux.
3.1.7.4 Diagnostic retenu et traitement :
L’enfant présentait donc un déficit somatotrope complet, associé à un déficit thyréotrope. Un
traitement substitutif par hydrocortisone, L-thyroxine et hormone de croissance a été débuté en
urgence, permettant la disparition des hypoglycémies, et la reprise d’un transit normal.
3.1.7.5 Résultats de l’IRM :
L’IRM à 11 mois retrouvait une interruption de la tige pituitaire avec une antéhypophyse
hypoplasique, et une posthypophyse ectopique (FIGURE 11).
3.1.7.6 Evolution
A 12 mois de vie, le traitement par hormone de croissance avait été interrompu afin de réaliser
une nouvelle exploration hormonale : 48h après l’arrêt, l’enfant a présenté des convulsions sur
hypoglycémies. L’enfant a été hospitalisée, les hypoglycémies ont été confirmées (2.5mmol/L) et
les dosages hormonaux en hypoglycémie retrouvaient un taux de GH effondré à 1.45µUI/mL, et
une cortisolémie basse à 180nmol/L.
Par ailleurs, un syndrome polyuro-polydipsique s’est révélé à 14 mois de vie, la courbe de
croissance staturo-pondérale s’infléchissait d’ailleurs depuis les 6 derniers mois. Un test de
restriction hydrique montrait alors une capacité à concentrer les urines (diurèse qui est passée de
7 ml/kg/heure à 1,5ml/kg/heure en fin de test) sans signe de déshydratation clinique, mais une

36
mauvaise tolérance avec agitation et soif intense : Un test au Minirin® a été effectué, améliorant
la symptomatologie, permettant de conclure à un diabète insipide partiel.
Le développement psychomoteur est normal.
La croissance staturo-pondérale est satisfaisante avec une taille sur la moyenne et un poids à -2DS
(FIGURE 12).
Figure 11 : IRM du cas clinique n°7.
Posthypophyse ectopique
Posthypophyse ectopique
Interruption de la tige pituitaire
Interruption de la tige pituitaire

37
Figure 12 : courbe de croissance du cas clinique n°7

38
3.1.8 Cas clinique n°8 (MM née en juin 2007) :
3.1.8.1 Grossesse et mensurations à la naissance :
La grossesse avait été sans particularité. Naissance d’une fille à 39 SA, avec un poids de 2790g
(<10èmep) pour une taille à 46cm (<3èmep) et un PC à 34cm (50èmep), soit un RCIU disharmonieux.
3.1.8.2 Circonstances de prise en charge :
La période néonatale était marquée dès 2 jours de vie, par une hypotonie avec des mouvements
anormaux, des difficultés alimentaires, une hypoglycémie à 1 mmol/L et un ictère cutanéo-
conjonctival. L’enfant est alors transférée dans le service de néonatalogie. L’examen clinique
d’entrée ne retrouve pas de dysmorphie, une volumineuse bosse séro-sanguine, le reste de
l’examen clinique est sans particularité. Le bilan paraclinique à l’entrée ne retrouve pas
d’argument pour une pathologie infectieuse, ou métabolique (hyperammoniémie transitoire,
Amino-acidogramme plasmatique normal) mais une souffrance fœtale aiguë (atteintes rénale,
hépatique, et cardiaque transitoires et anomalies de la substance blanche sous corticale pariétale
et occipitale en IRM). Aucun bilan endocrinien n’a été réalisé dans le bilan initial.
3.1.8.3 Bilan endocrinien :
Le bilan a été réalisé à 17 jours de vie, avec hypoglycémie de jeûne :
La TSH était inadaptée à 6.2 mUI/L pour une T4L basse à 9.2 pmol/L.
La cortisolémie était effondrée à 18nmol/L, en hypoglycémie modérée.
Le taux de GH en hypoglycémie modérée était insuffisant à 6.3 µUI/mL, et le taux d’GF1 de base
était bas à 44 ng/mL.
L’Insulinémie était à 2.05 µUI/mL.
Prolactine explosive à 1438 µU/mL
Les ionogrammes sanguins et urinaires étaient normaux.
3.1.8.4 Diagnostic retenu et traitement :
L’enfant présentait donc un déficit somatotrope complet associé à un déficit thyréotrope et
corticotrope profond. Un traitement substitutif par hydrocortisone, L-Thyroxine et hormone de
croissance a été débuté en urgence, permettant l’amendement de la symptomatologie.
3.1.8.5 Les résultats de l’IRM :
L’IRM à J15 retrouvait une tige pituitaire interrompue, une posthypophyse ectopique, une
antéhypophyse hypoplasique et une hypoplasie du nerf optique gauche (FIGURE 13).
3.1.8.6 Evolution :
L’examen ophtalmologique retrouvait une tête du nerf optique gauche de taille réduite avec
probable amblyopie de cet œil.
A 3 mois, les dosages de FSH LH sont respectivement à 4.3 et 0.4mUI/mL, normaux.

39
A l’âge de 12 mois, un test dynamique au Glucagon a été réalisé, il confirmait le déficit
somatotrope complet et corticotrope profond (en hypoglycémie à 2.5 mmol/L, le maximum du pic
de GH était à 6.7µUI/mL et la cortisolémie passait de 35 à 85nmol/L).
L’IRM à 12 mois confirmait l’hypoplasie de l’antéhypophyse, l’interruption de la tige pituitaire, et
l’ectopie de la posthypophyse, et retrouvait des lobes olfactifs présents normaux, une hypoplasie
du chiasma optique et du nerf optique gauche, et une agénésie de la paroi gauche de kyste du
septum pellucidum sans communication entre les ventricules latéraux. Elle retrouvait également
des hétérotopies nodulaires de la corne ventriculaire occipitale gauche, probablement sans
rapport avec la maladie.
Le diagnostic de Dysplasie septo-optique est donc possible.
Le développement psychomoteur est normal
La croissance staturo-pondérale est satisfaisante avec une taille à +1DS et un poids à -1DS
(FIGURE 14),
3.1.8.7 Génétique :
La recherche d’une anomalie génétique, notamment sur HESX1 est en cours.
Figure 13 : IRM du cas clinique n°8
Posthypophyse ectopique
Interruption de la tige pituitaire

40
Figure 14 : Courbe de croissance du cas clinique n°8
Nerf optique droit
normal
Nerf optique gauche hypoplasique

41
3.1.9 Cas clinique n°9 (CK née en juin 2007) :
3.1.9.1 Grossesse et mensurations à la naissance :
La grossesse avait été marquée par un RCIU non confirmé à la naissance. Naissance d’une fille
eutrophique, à 41 SA + 6 jours avec un poids à 3150g (25èmep) pour une taille à 45cm (<3èmep) et
un PC à 34cm (>25èmep).
3.1.9.2 Circonstances de prise en charge :
L’examen clinique à la naissance retrouvait une dysmorphie faciale avec un nez aplati retroussé,
un front large, une micro-rétro-mandibulie, un hypertélorisme, un épicanthus bilatéral et des
fentes palpébrales étroites. Il existait aussi une hypotonie axiale et des troubles de la succion.
Devant des hypoglycémies (0.5 mmol/L), l’enfant a été transférée dans le service de néonatalogie
pour le bilan étiologique.
3.1.9.3 Bilan endocrinien :
Le bilan a été réalisé à 6 jours de vie, en hypoglycémie (1.8 mmol/L).
La TSH était effondrée à 0.027 mUI/L, avec une T4L effondrée à 1.5 pmol/L.
La cortisolémie en stress hypoglycémique, était normale à 1041 nmol/L.
Le taux de GH en hypoglycémie était effondré <0.03µUI/mL et le taux d’IGF1 de base < 5 ng/mL.
La prolactine était à 15 µUI/mL.
Les ionogrammes sanguins et urinaires étaient normaux.
3.1.9.4 Diagnostic retenu et traitement :
L’enfant présentait donc un déficit somatotrope complet, associé à un déficit thyréotrope
profond. Le traitement substitutif par L-thyroxine et hormone de croissance a été débuté en
urgence, permettant la correction progressive des hypoglycémies et de l’hypotonie.
3.1.9.5 Résultats de l’IRM :
L’IRM à 1 semaine de vie ne retrouvait pas d’anomalie au niveau de la région hypothalamo-
hypophysaire, excepté une posthypophyse non vue, lié aux difficultés d’interprétation de
l’examen à cet âge.
3.1.9.6 Evolution :
L’IRM à 12 mois retrouve une hypoplasie de antéhypophysaire et posthypophysaire, une
interruption de la tige pituitaire, absence d’anomalie des lobes olfactifs, absence d’anomalie de la
ligne médiane. Elle retrouve également un aspect pathologique mais localisé de la substance
blanche pariétale de manière bilatérale, pouvant évoquer un retard de myélinisation ou une
gliose, sans rapport avec la pathologie hypophysaire.
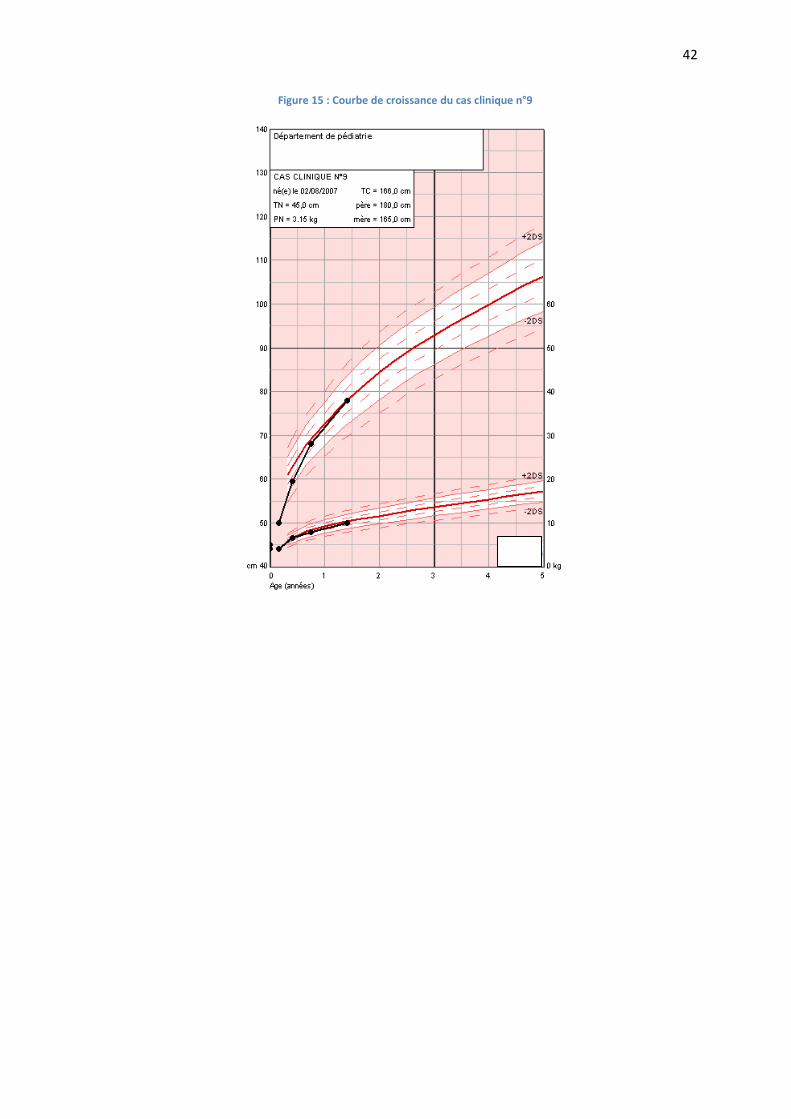
Le développement psychomoteur est normal.
Le développement staturo-pondéral est satisfaisant évoluant sur la moyenne (FIGURE N°15).
42
Figure 15 : Courbe de croissance du cas clinique n°9

43
3.1.10 Cas Clinique n°10 (CA né en janvier 2009) - frère du cas n°9 :
3.1.10.1 Grossesse et mensurations à la naissance :
La découverte de la grossesse avait été tardive à 5 mois sous contraception orale. La grossesse
avait été marquée par un diabète gestationnel sous régime simple. Naissance à terme d’un
garçon, avec un poids à 3420g (50èmep), pour une taille à 46cm (<3èmep) et un périmètre crânien à
38cm (50èmep).
3.1.10.2 Circonstances de prise en charge :
La naissance est marquée par des hypoglycémies (asymptomatiques) récidivantes, au minimum à
2.2 mmol/L malgré enrichissement de l’alimentation, l’enfant a donc été mis sous perfusion.
L’examen clinique retrouvait une dysmorphie faciale (avec un front large fuyant, une ensellure
nasale marquée, un hypertélorisme et une excroissance pré-tragienne) associée à une hypotonie
franche, et enfin un ictère à bilirubine libre.
3.1.10.3 Bilan endocrinien
Devant les antécédents dans la fratrie, les dosages hormonaux ont rapidement effectués (3 jours
de vie) en hypoglycémie spontanée (2 mmol/L).
La TSH était effondrée à 0.02 mUI/L, et la T4L était basse à 2.2 pmol/L.
Le cortisolémie en stress hypoglycémique était normale à 701nmol/L.
Le taux de GH en hypoglycémie était effondré à 0.03µUI/mL, et le taux d’IGF1 était bas à
10ng/mL.
L’insulinémie était < 0.2 µUI/mL.
Les ionogrammes sanguins et urinaires étaient normaux.
3.1.10.4 Diagnostic retenu et traitement :
L’enfant présentait donc comme sa sœur, un déficit somatotrope complet associé à un déficit
thyréotrope profond. Un traitement substitutif par L-Thyroxine et hormone de croissance a été
débuté en urgence. L’évolution a été favorable, avec disparition des hypoglycémies.
L’IRM sera réalisée ultérieurement, ainsi qu’une analyse génétique, étant donné la forme familiale
qui est exceptionnelle.

44
3.2 Analyse :
3.2.1 Les données cliniques initiales :
Les données cliniques sont résumées dans les TABLEAUX 2 ET 3.
3.2.1.1 Délai d’apparition des symptômes, et délai diagnostic :
Pour huit enfants (80%), les premiers symptômes sont apparus dans les deux premiers jours de
vie. Les deux autres patients ont eu des symptômes dans la première semaine et le premier mois
de vie, qui n’ont pas été rattachés initialement à une pathologie endocrinienne.
Le délai diagnostic était très variable puisqu’il s’étendait entre 3 jours et 3 mois, avec une
médiane de 10.5 jours avec un intervalle interquartile à [4 – 22 jours].
3.2.1.2 Les manifestations cliniques :
Elles pouvaient être très variables mais les signes cliniques retrouvés le plus fréquemment
étaient :
Les hypoglycémies récidivantes et profondes (70%). Cependant, sur les six enfants ayant
présenté des hypoglycémies récidivantes, deux (33.3%) seulement ont entrainé des
convulsions.
L’ictère prolongé (70%)
L’hypotonie (70%)
Les difficultés alimentaires (70%)
Et le retard de croissance intra-utérin (50%)
3.2.1.3 Les anomalies morphologiques :
4 enfants présentaient une dysmorphie faciale, dont 2 étaient frère et sœur.
3 parmi les 6 enfants restants présentaient des anomalies morphologiques directement liées au
déficit hypophysaire (macroglossie, fontanelle large, œdème palpébral, etc.)
45
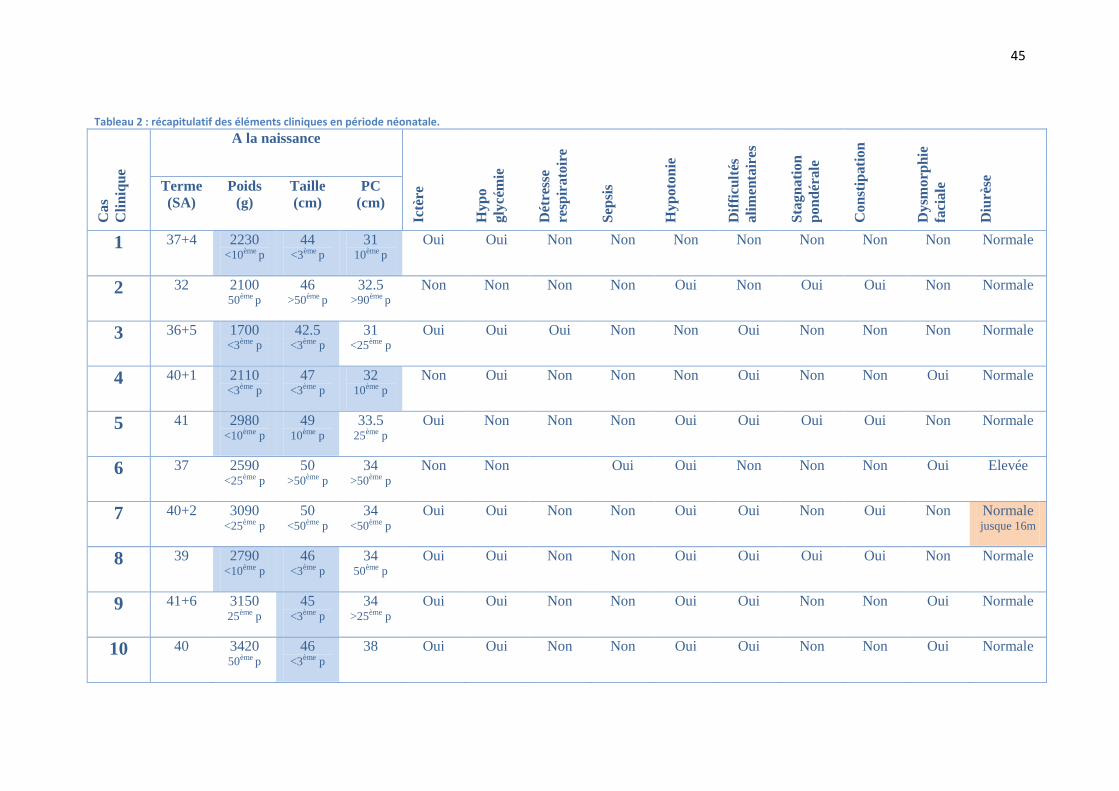
Tableau 2 : récapitulatif des éléments cliniques en période néonatale.
Cas
Cli
niq
ue
A la naissance
Ictè
re
Hyp
o
gly
cém
ie
Dét
ress
e
resp
irato
ire
Sep
sis
Hyp
oto
nie
Dif
ficu
ltés
ali
men
tair
es
Sta
gn
ati
on
pon
dér
ale
Con
stip
ati
on
Dysm
orp
hie
faci
ale
Diu
rèse
Terme
(SA)
Poids
(g)
Taille
(cm)
PC
(cm)
1 37+4 2230 <10ème p
44 <3ème p
31 10ème p
Oui Oui Non Non Non Non Non Non Non Normale
2 32 2100 50ème p
46 >50ème p
32.5 >90ème p
Non Non Non Non Oui Non Oui Oui Non Normale
3 36+5 1700 <3ème p
42.5 <3ème p
31 <25ème p
Oui Oui Oui Non Non Oui Non Non Non Normale
4 40+1 2110 <3ème p
47 <3ème p
32 10ème p
Non Oui Non Non Non Oui Non Non Oui Normale
5 41 2980 <10ème p
49 10ème p
33.5 25ème p
Oui Non Non Non Oui Oui Oui Oui Non Normale
6 37 2590 <25ème p
50 >50ème p
34 >50ème p
Non Non Oui Oui Non Non Non Oui Elevée
7 40+2 3090 <25ème p
50 <50ème p
34 <50ème p
Oui Oui Non Non Oui Oui Non Oui Non Normale jusque 16m
8 39 2790 <10ème p
46 <3ème p
34 50ème p
Oui Oui Non Non Oui Oui Oui Oui Non Normale
9 41+6 3150 25ème p
45 <3ème p
34 >25ème p
Oui Oui Non Non Oui Oui Non Non Oui Normale
10 40 3420 50ème p
46 <3ème p
38 Oui Oui Non Non Oui Oui Non Non Oui Normale

46
Tableau 3: fréquence des manifestations cliniques.
RCIU Ictère Hypoglycémie Détresse
respiratoire
Sepsis Hypotonie Difficultés
alimentaires
Stagnation
pondérale
constipation
nombre 5 7 7 1 1 7 7 3 4
Pourcentage 50% 70% 70% 10% 10% 70% 70% 30% 40%

47
3.2.2 Les données biologiques :
Les données biologiques sont résumées dans les TABLEAUX 4 ET 5.
3.2.2.1 Au moment du diagnostic :
A. Les axes antéhypophysaires :
L’axe somatotrope : Il s’agissait du déficit le plus représenté, puisqu’il touchait la
totalité des enfants. 8 enfants (80%) présentaient un déficit complet
L’axe thyréotrope : 70% des enfants présentaient un déficit thyréotrope, dont 3 (43%)
étaient profonds.
L’axe corticotrope : 50% des enfants présentaient un déficit corticotrope, dont 2
(40%) étaient profonds. Pour les 3 enfants présentant un déficit partiel, donc suspect,
un traitement substitutif a été prescrit devant le risque vital encouru.
L’axe lactotrope : 3 enfants sur 4 présentaient un taux de prolactine explosif,
indiquant une déconnection hypothalamo-hypophysaire.
L’axe gonadotrope : il n’a pas été mis en évidence de déficit gonadotrope pour les
enfants explorés, mais la période pubertaire sera importante à surveiller.
B. La posthypophyse :
Un seul enfant a présenté un diabète insipide au moment du diagnostic initial.
3.2.2.2 Au moment du test endocrinien de contrôle :
A. Les axes antéhypophysaires :
Pour deux patients ayant un diagnostic de déficits somatotrope et corticotrope associés, le déficit
corticotrope s’est avéré être transitoire.
Pour un troisième patient ayant un diagnostic de déficits somatotrope et corticotrope associés,
les deux déficits étaient transitoires, permettant l’arrêt de tout traitement substitutif jusqu’à ce
jour.
B. La posthypophyse :
Pour une enfant, un diabète insipide partiel s’est révélé tardivement à 16 mois de vie.
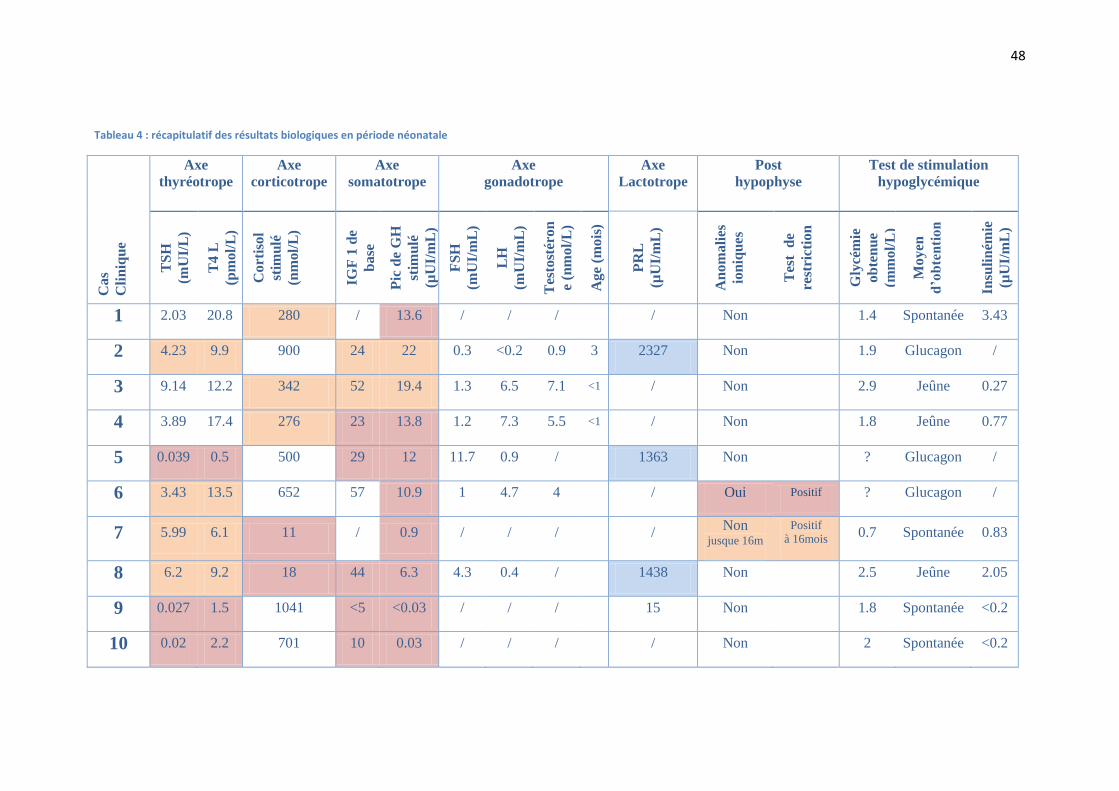
48
Tableau 4 : récapitulatif des résultats biologiques en période néonatale
Cas
Cli
niq
ue
Axe
thyréotrope
Axe
corticotrope
Axe
somatotrope
Axe
gonadotrope
Axe
Lactotrope
Post
hypophyse
Test de stimulation
hypoglycémique
TS
H
(mU
I/L
)
T4 L
(pm
ol/
L)
Cort
isol
stim
ulé
(nm
ol/
L)
IGF
1 d
e
base
(ng/m
L)
Pic
de
GH
stim
ulé
(µU
I/m
L)
FS
H
(mU
I/m
L)
LH
(mU
I/m
L)
Tes
tost
éron
e (n
mol/
L)
Age
(mois
)
PR
L
(µU
I/m
L)
An
om
ali
es
ion
iqu
es
Tes
t d
e
rest
rict
ion
Gly
cém
ie
ob
ten
ue
(mm
ol/
L)
Moyen
d’obtention
Insu
lin
émie
(µU
I/m
L)
1 2.03 20.8 280 / 13.6 / / / / Non 1.4 Spontanée 3.43
2 4.23 9.9 900 24 22 0.3 <0.2 0.9 3 2327 Non 1.9 Glucagon /
3 9.14 12.2 342 52 19.4 1.3 6.5 7.1 <1 / Non 2.9 Jeûne 0.27
4 3.89 17.4 276 23 13.8 1.2 7.3 5.5 <1 / Non 1.8 Jeûne 0.77
5 0.039 0.5 500 29 12 11.7 0.9 / 1363 Non ? Glucagon /
6 3.43 13.5 652 57 10.9 1 4.7 4 / Oui Positif ? Glucagon /
7 5.99 6.1 11 / 0.9 / / / / Non
jusque 16m Positif
à 16mois 0.7 Spontanée 0.83
8 6.2 9.2 18 44 6.3 4.3 0.4 / 1438 Non 2.5 Jeûne 2.05
9 0.027 1.5 1041 <5 <0.03 / / / 15 Non 1.8 Spontanée <0.2
10 0.02 2.2 701 10 0.03 / / / / Non 2 Spontanée <0.2

49
Tableau 5: fréquence des anomalies biologiques inaugurales.
Déficit thyréotrope Déficit corticotrope Déficit somatotrope Taux de prolactine
explosif
Diabète insipide
Nombre 7 5 10 3 / 4 1
Pourcentage 70% 50% 100% 75% 10%

50
3.2.3 Les résultats de l’IRM :
Le délai de réalisation de la première IRM s’étendait de 1 semaine à 11 mois avec une médiane à
2.5mois.
Sur les neuf enfants ayant bénéficié d’une IRM, huit (88.8%) avaient au moins une anomalie
morphologique de la région hypothalamo hypophysaire :
3.2.3.1 Antéhypophyse :
Cinq enfants (55%) avaient une antéhypophyse hypoplasique
3.2.3.2 Tige pituitaire :
Huit enfants (88.8%) avaient une anomalie de la tige pituitaire :
Quatre (44.4%) avaient une tige pituitaire grêle.
Quatre (44.4%) avaient une interruption de la tige pituitaire.
3.2.3.3 Posthypophyse :
Quatre enfants (44.4%) avaient une anomalie posthypophysaire :
Un enfant (11.1%) avait une posthypophyse hypoplasique.
Deux (22.2%) avaient une posthypophyse ectopique.
Et un (11.1%) avait une agénésie de la posthypophyse.
3.2.3.4 Anomalies associées :
Quatre enfants (44.4%) avaient des anomalies cérébrales associées :
Deux dysplasies septo-optique.
Et pour deux enfants, les anomalies cérébrales mises en évidence ne sont pas connues
pour être liées particulièrement aux déficits hypophysaires.

51
4 DISCUSSION :
4.1 Un diagnostic difficile :
4.1.1 Incidence en Isère :
Dans la littérature, on retrouve une incidence des hypopituitarismes congénitaux, de 1/3000
naissances (déficits congénitaux quelque soit l’âge de découverte) (ORPHANET). Cette incidence
diminue à 1/53000 naissances si on se limite aux déficits congénitaux de découverte néonatale
(2).
Sur la période de cinq ans pendant laquelle les 10 cas de déficits hypophysaires multiples
néonataux, il y a eu en Isère 49285 naissances vivantes. L’incidence en Isère est donc
approximativement de 1/5000 naissances.
L’incidence des déficits hypophysaires néonataux est donc probablement sous évaluée du fait des
formes pauci-symptomatiques qui retardent l’âge du diagnostic. Les enfants adressés en
consultation dans la première année de vie pour un retard de croissance staturale, pour lesquels
un hypopituitarisme est alors diagnostiqué, sont probablement en parti, des formes néonatales
passées inaperçues.
4.1.2 Le délai diagnostic :
Le délai diagnostic variable de notre série montre bien la difficulté du diagnostic de déficit
hypophysaire multiple en période néonatale.
Il est important d’être vigilant devant des hypoglycémies répétées persistantes et profondes :
dans ces circonstances, Il est important de poser les indications des bilans hormonaux (dosage du
cortisol et de l’hormone de croissance au moment des hypoglycémies pour tester les axes
corticotrope et somatotrope, et dosage de la TSH et de la T4L et contrôle des ionogrammes
sanguins et urinaires pour explorer l’axe thyréotrope et la posthypophyse).
En effet le retard diagnostic peut engager le pronostic et vital (16; 17) et neurologique.

52
4.1.3 L’hypoglycémie néonatale :
La première difficulté de l’hypoglycémie est de l’identifier :
En effet, même s’il s’agit du symptôme le plus fréquent, elle peut être asymptomatique.
La deuxième difficulté est de ne pas la négliger et de poursuivre les investigations :
Elle est en effet souvent mise sous le compte d’autres problèmes courants en période néonatale
(prématurité, hypotrophie, ou insuffisance des apports liés aux difficultés d’alimentation) (18)
L’horaire de survenue d’une hypoglycémie par rapport au repas est un élément d’orientation
diagnostic important chez l’enfant (19; 20). Chez le nouveau-né avec un déficit hypophysaire, les
hypoglycémies peuvent survenir après un jeûne court, le matin au réveil ou avant la tétée (7).
La troisième difficulté est la réalisation du bilan hormonal au moment de l’hypoglycémie :
Chez le nouveau-né, les dosages sont réalisés au moment d’une hypoglycémie (soit spontanée,
soit provoquée par un jeûne). Il est cependant difficile de connaitre le moment exact de
l’hypoglycémie par les mesures répétées des glycémies capillaires. L’utilisation du Holter
glycémique peut s’avérer intéressante, car celui-ci permet de détecter des hypoglycémies
asymptomatiques, d’en connaitre leur fréquence, leur horaire de survenue par rapport au repas
et enfin d’effectuer les dosages hormonaux au moment exact de l’hypoglycémie grâce au réglage
des alarmes.
Les limites de la glycémie en période néonatale et chez le prématuré sont controversées, et
l’interprétation des dosages hormonaux en GH et en cortisol est dépendante de la profondeur de
l’hypoglycémie : en cas de doute important, il est donc préférable de débuter le traitement
substitutif, devant le risque vital encouru, et de réaliser plus tard dans l’enfance, de nouveaux
tests dynamiques.
Les tests dynamiques de stimulation chez le nouveau-né ne sont en règle générale pas effectués,
car la réalisation de prélèvements répétés en période néonatale est toujours délicate. Si un
prélèvement ne peut être réalisé, notamment au moment du nadir de la glycémie, on risque de
diagnostiquer à tort un déficit somatotrope ou corticotrope, car on n’aura pas détecté le
maximum du pic de GH ou de cortisol. De plus, la réalisation du test nécessite un laboratoire
capable de réaliser ces analyses rapidement, ce qui n’est pas toujours facile dans les petites
structures.

53
4.1.4 L’intérêt des tests dynamiques de contrôle dans l’enfance :
4.1.4.1 Déficits partiels :
Le diagnostic d’un déficit partiel, portant notamment sur les axes somatotrope et corticotrope,
doit amener au plus tard vers l’âge de 2-3 ans, à réaliser un test dynamique de contrôle. Pour se
faire, il faudra interrompre le traitement substitutif par hormone de croissance et par
hydrocortisone (Après dosage de la cortisolémie le matin avant administration du traitement,
pour vérifier la production endogène glucocorticoïde).
Après ce test dynamique, si l’on ne retrouve plus de déficit, on pourra parler de déficit transitoire
et interrompre le traitement substitutif, en poursuivant la surveillance clinique et biologique.
4.1.4.2 Les déficits complets :
En cas de déficits complets avec des dosages effondrés en période néonatale en hypoglycémie
profonde, il ne semble pas nécessaire de les contrôler dans la petite enfance, notamment devant
le risque de mettre en jeu le pronostic vital lors de l’arrêt des traitements.
Le cas clinique n°7 illustre bien cette situation.
Dans ce cas, les traitements sont maintenus jusqu’à la fin de la croissance pubertaire. Les
épreuves de stimulation hypophysaire sont alors réalisés pour discuter de la nécessité ou non de
poursuivre une substitution hormonale à l’âge adulte.
4.2 Des éléments cliniques morphologiques évocateurs à ne pas
négliger :
4.2.1 Le micropénis :
Le micropénis, chez le garçon est aussi un signe classique de déficit somatotrope. Le déficit
anténatal en gonadotrophines peut également aboutir à un micropénis sans malformation (17).
4.2.2 Les anomalies de la ligne médiane :
Les anomalies de la ligne médiane doivent aussi faire rechercher un hypopituitarisme, et un
hypopituitarisme doit faire rechercher des anomalies de la ligne médiane, par la réalisation d’un
examen ophtalmologique (hypoplasie du nerf optique), et d’une IRM cérébrale (21). Les enfants
présentant des anomalies de la ligne médiane présentent plus fréquemment des convulsions, des
anomalies du développement psychomoteur, pour lesquels il faudra être vigilant (21).

54
4.3 La cholestase néonatale et les troubles hépatiques : L’ictère néonatal prolongé est aussi un signe d’appel très fréquent. Il s’agit classiquement d’une
cholestase avec ictère à bilirubine conjuguée dont le mécanisme est mal connu. Il semblerait que
l’hormone de croissance et le cortisol participeraient à la stimulation de la maturation normale de
la synthèse des acides biliaires et du transport des acides biliaires (2; 22)L’ictère peut parfois aussi
être à bilirubine libre (23). La cholestase s’améliore généralement rapidement après le début du
traitement substitutif.
4.4 L’IRM cérébrale : L’IRM cérébrale est aussi importante afin de rechercher des anomalies morphologiques de
l’hypophyse : dans notre série, comme dans la littérature (24). Les anomalies hypophysaires sont
fréquentes. Cependant, le premier examen a souvent été réalisé précocement, avec une
interprétation de l’IRM parfois difficile à cet âge. Ceci nous fait penser qu’il est intéressant de
contrôler l’IRM cérébrale à distance afin de pouvoir détecter les anomalies morphologiques
éventuelles. Le cas clinique n°8 illustre bien ce propos.
Il semble exister des relations entre les résultats de l’IRM et le phénotype : dans le contexte
d’interruption de la tige pituitaire par rapport à l’hypoplasie simple hypophysaire, le diagnostic est
souvent plus précoce, on retrouve plus fréquemment des hypoglycémies, le déficit somatotrope
est souvent plus profond, et la présence d’un déficit hypophysaire multiple est plus fréquent (25).
4.5 Dans l’enfance jusqu’à l’âge adulte : L’objectif du traitement substitutif est d’obtenir une croissance, et un développement pubertaire
normaux. Pour cela il faut adapter les posologies régulièrement aux contrôles biologiques et à
l’évolution clinique.
Il convient du contrôler régulièrement l’ensemble des axes hypophysaires pour ne pas
méconnaitre l’apparition de nouveaux déficits hypophysaires et pour les traiter sans délai.
Les déficits corticotropes confirmés et les déficits thyréotropes sont définitifs et doivent être
substitués à vie du fait du risque vital. Concernant l’axe somatotrope, à la fin de la croissance,
il est indispensable de réaliser de nouveaux tests dynamiques pour contrôler la persistance du
déficit et évaluer sa profondeur dans l’objectif de discuter la poursuite du traitement par
hormone de croissance avec l’AMM adulte.

55
5 CONCLUSION :
Le déficit hypophysaire multiple néonatal est une pathologie rare mais non exceptionnelle.
Son diagnostic est difficile et souvent retardé par rapport aux premiers signes cliniques.
Notre série est intéressante par la diversité des présentations cliniques néonatales, par les
différentes combinaisons des atteintes hormonales, et par la diversité des anomalies
morphologiques découvertes à l’IRM. Le recul sur plusieurs années pour certains patients est
également intéressant.
Dans la littérature, sont décrites des séries plus conséquentes, mais avec souvent des recueils sur
des périodes plus longues, ou des recueils internationaux, avec des prises en charge
probablement différentes. Les 10 cas cliniques présentés portent sur des diagnostics qui
s’échelonnent uniquement sur une période de 5 ans, et limités au département de l’Isère :
La prise en charge par la même équipe de néonatologues, d’endocrinologues pédiatres, et de
généticiens, nous permet d’avoir des données homogènes, notamment pour les critères
diagnostiques, pour les éléments du suivi et pour la prise en charge thérapeutique.
Il serait intéressant de réaliser une étude plus large, avec un recul sur l’évolution clinique, afin de
pouvoir établir des recommandations de prise en charge diagnostique.
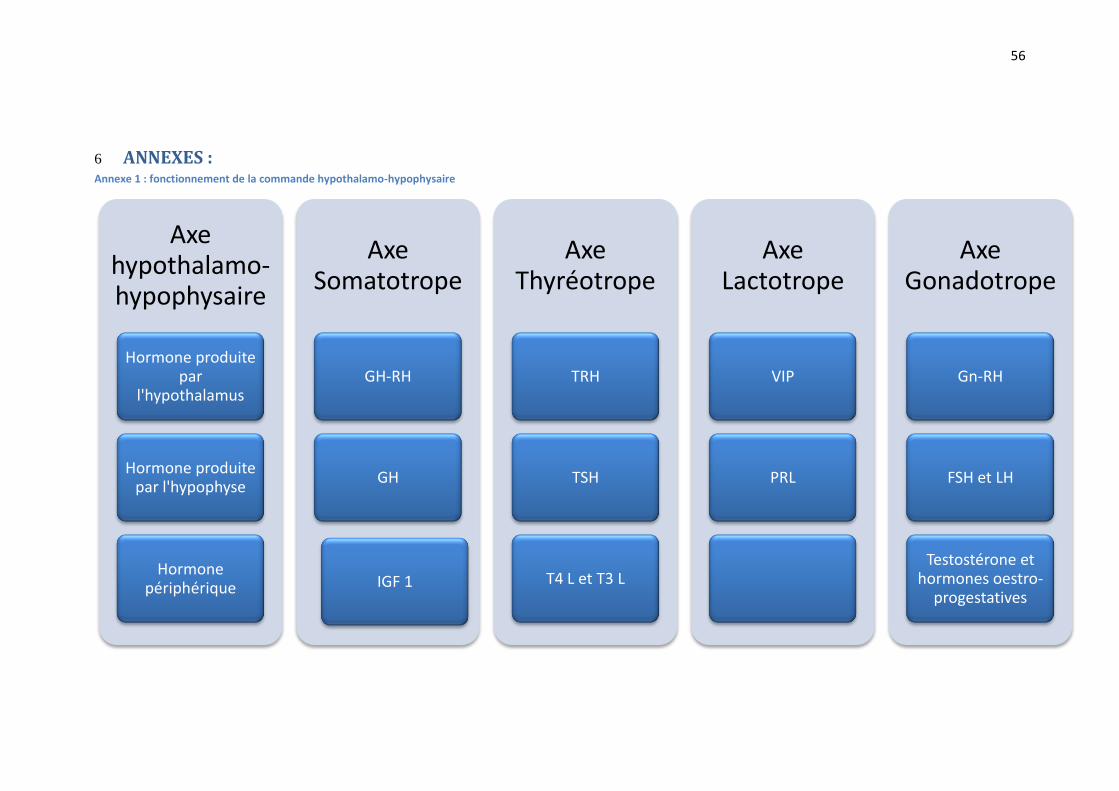
56
6 ANNEXES : Annexe 1 : fonctionnement de la commande hypothalamo-hypophysaire
Axe hypothalamo-hypophysaire
Hormone produite par
l'hypothalamus
Hormone produite par l'hypophyse
Hormone périphérique
Axe Somatotrope
GH-RH
GH
IGF 1
Axe Thyréotrope
TRH
TSH
T4 L et T3 L
Axe Lactotrope
VIP
PRL
Axe Gonadotrope
Gn-RH
FSH et LH
Testostérone et hormones oestro-
progestatives
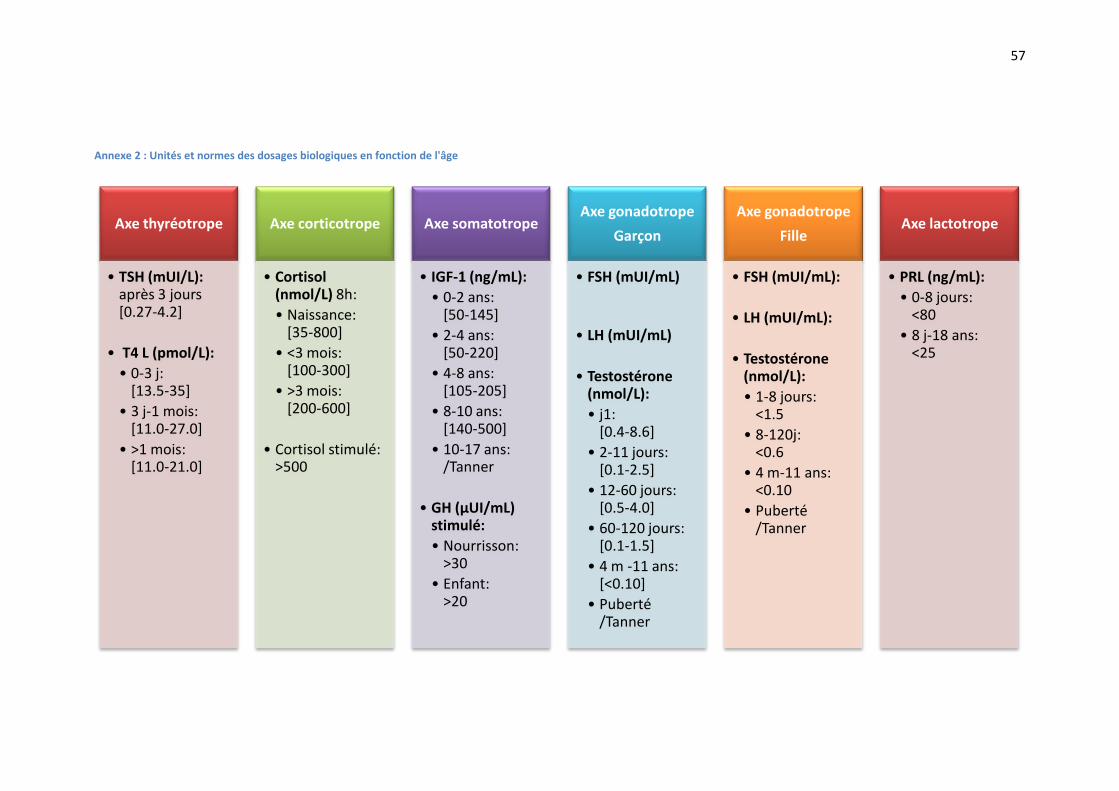
57
Annexe 2 : Unités et normes des dosages biologiques en fonction de l'âge
Axe thyréotrope
• TSH (mUI/L):après 3 jours[0.27-4.2]
• T4 L (pmol/L):
• 0-3 j:[13.5-35]
• 3 j-1 mois:[11.0-27.0]
• >1 mois:[11.0-21.0]
Axe corticotrope
• Cortisol (nmol/L) 8h:
• Naissance:[35-800]
• <3 mois:[100-300]
• >3 mois:[200-600]
• Cortisol stimulé:>500
Axe somatotrope
• IGF-1 (ng/mL):
• 0-2 ans:[50-145]
• 2-4 ans:[50-220]
• 4-8 ans:[105-205]
• 8-10 ans:[140-500]
• 10-17 ans:/Tanner
• GH (µUI/mL) stimulé:
• Nourrisson:>30
• Enfant:>20
Axe gonadotrope
Garçon
• FSH (mUI/mL)
• LH (mUI/mL)
• Testostérone (nmol/L):
• j1:[0.4-8.6]
• 2-11 jours:[0.1-2.5]
• 12-60 jours:[0.5-4.0]
• 60-120 jours:[0.1-1.5]
• 4 m -11 ans:[<0.10]
• Puberté/Tanner
Axe gonadotrope
Fille
• FSH (mUI/mL):
• LH (mUI/mL):
• Testostérone (nmol/L):
• 1-8 jours:<1.5
• 8-120j:<0.6
• 4 m-11 ans:<0.10
• Puberté/Tanner
Axe lactotrope
• PRL (ng/mL):
• 0-8 jours:<80
• 8 j-18 ans:<25

58
7 BIBLIOGRAPHIE 1. Castinetti F, Reynaud R, Saveanu A, Quentien MH, Albarel F, Barlier A. Déficit hypophysaire
combiné multiple: aspects cliniques et génétiques. Ann Endocrinol. 2008, Vol. 69, pp. 7-17.
2. Karnsakul W, Sawathiparnich P, Nimkarn S, Likitmaskul S, Santiprabhob J, Aanpreung P.
Anterior pituitary hormone effects on hepatic functions in infants with congenital
hypopituitarism. Ann Hepatol. 2007, Vol. 6, 2, pp. 97-103.
3. Geffner, ME. Hypopituitarism in childhood. Cancer Control. 2002, Vol. 9, pp. 212-22.
4. Pombo M., Barreiro J., Penalva A., Peino R., Dieguez C., Casanueva FF. Absence of growth
hormone (GH) secretion after the administration of either GH-Releasing hormone (GHRH), GH-
Releasing peptide (GHRP-6), or GHRH plus GHRP-6 in children with neonatal pituitary stalk
transection. J Clin Endocrinol Metab. 1995, Vol. 80, pp. 3180-4.
5. Netchine, I. Embryologie de l'hypophyse et pathologie moléculaire des déficits en hormone de
croissance. Endocrinologie périnatale. s.l. : Doin, 2005, pp. 61-75.
6. Pena-Almazan S, Buchlis J, Miller S, Shine B, MacGillivray M. Linear growth characteristics of
congenitally GH-deficient infants from birth to one year of age. J Clin Endocrinol Metab. 2001, Vol.
86, pp. 5691-4.
7. Coutant R., Bouvattier C., Bouhours-Nouet N., Limal JM. Déficits hypophysaires congénitaux:
physiologie, aspects cliniques et thérapeutiques. Endocrinologie périnatale. s.l. : Doin, 2005, pp.
77-104.
8. Palma Sisto, PA. Endocrine disorders in the neonate. Pediatr Clin North Am. 2004, Vol. 51, pp.
1141-68.
9. Grumbach, MM. A window of opportunity: the diagnosis of gonadotropin deficiency in the
male infant. J Clin Endocrinol Metab. 2005, Vol. 90, pp. 3122-7.
10. Main KM, Schmidt IM, Skakkebaek NE. A possible role for reproductive hormones in newborn
boys: progressive hypogonadism without the postnatal testosterone peak. J Clin Endocrinol
Metab. 2000, Vol. 85, pp. 4905-7.
11. Rainbow LA, Rees SA, Shaikh MG, Shaw NJ, Cole T, Barrett TG, Kirk JM. Mutation analysis of
POUF-1, PROP-1 and HESX-1 show low frequency of mutations in children with sporadic forms of
combined pituitary hormone deficiency and septo-optic dysplasia. Clin Endocrinol (Oxf). 2005, Vol.
62, pp. 163-8.
12. Sobrier ML, Maghnie M, Vié-Luton MP, Secco A, di Iorgi N, Lorini R. Novel HESX1 mutations
associated with a life-threatening neonatal phenotype, pituitary aplasia, but normally located
posterior pituitary and no optic nerve abnormalities. J Clin Endocrinol Metab. 2006, Vol. 91, pp.
4528-36.

59
13. McNay DE, Turton JP, Kelberman D, Woods KS, Brauner R, Papadimitriou A. HESX1
mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J Clin Endocrinol
Metab. 2007, Vol. 92, pp. 691-7.
14. Acers, TE. Optic nerve hypoplasia: septo-optic-pituitary dysplasia syndrome. Trans Am
Ophthalmol Soc. 1981, Vol. 79, pp. 425-57.
15. Leroy B, Lefort F. The weight and size of newborn infants at birth. Rev Fr Gynecol Obstet.
1971, Vol. 66, pp. 391-6.
16. Choo-Kang LR, Sun CC, Counts DR. Cholestasis and hypoglycemia: manifestations of
congenital anterior hypopituitarism. J Clin Endocrinol Metab. 1996, Vol. 81, pp. 2786-9.
17. Salisbury DM, Leonard JV, Dezateux CA, Savage MO. Micropenis: an important early sign of
congenital hypopituitarism. Br Med J (Clin Res Ed). 1984, Vol. 288, pp. 621-2.
18. Ehrlich, RM. Hypoglycaemia in infancy and childhood. Arch Dis child. 1971, Vol. 46, pp. 716-9.
19. Saudubray JM., De Lonlay P., Touati G., Martin D., Nassogne MC., Castelnau P. Genetic
hypoglycemia in infancy and childhood: physiopathology and diagnosis. J. Inherit. Metab. Dis.
2000, Vol. 23, pp. 197-214.
20. De Lonlay P., Giurgea I., Touati G., Saudubray JM. Neonatal hypoglycemia: aetiologies. Semin
neonatal. 2004, Vol. 9, pp. 49-58.
21. Cameron FJ, Khadilkar VV, Stanhope R. Pituitary dysfunction, morbidity and mortality with
congenital midline malformation of the cerebrum. Eur J Pediatr. 1999, Vol. 158, pp. 97-102.
22. Spray CH, Mckiernan P, Waldron KE, Shaw N, Kirk J, Kelly DA. Investigation and outcome of
neonatal hepatitis in infants with hypopituitarism. Acta Paediatr. 2000, Vol. 89, pp. 951-4.
23. Scott R., Aladangady N., Maalouf E. Neonatal hypopituitarism presenting with poor feeding,
hypoglycemia and prolonged unconjugated hyperbilirubinemia. J Matern Fetal Neonatal Med.
2004, Vol. 16, pp. 131-3.
24. Hamilton J, Blaser S, Daneman D. MR imaging in idiopathic growth hormone deficiency. Am J
Neuroradiol. 1998, Vol. 19, pp. 1609-15.
25. Arrigo T, De Luca F, Maghnie M, Blandino A, Lombardo F, Messina MF. Relationships
between neuroradiological and clinical features in apparently idiopathic hypopituitarism. Eur J
Endocrinol. 1998, Vol. 139, pp. 84-8.
Top Related