· Web viewMedical Devices – Substantial Change Review Form. Applicant Information and...
69
Medical Devices – Substantial Change Review Form Applicant Information and Instructions Significant/ Substantial Change Notification Covering Product Family- MDD 252. NSAI Use Only – Amendment Number-/ AIMD 253. NSAI Use Only – Amendment Number-/ TSE Human Blood Medicinal Substances P/O number: This is a: Regular/ Standard Review Fast Track (expedited) Modular submission (prior agreement only) If OBLs apply to this product, please state the relevant product families below: / ; / ; / DECLARATION(s) BY APPLICANT In signing this form, the manufacturer is verifying that the requirements of the Directive will be applied in full when the change has been implemented. Signed on behalf of the Manufacturer: Date: Name (please print): Position / Title: Contact person (if GRF-25-08 Rev. 16.1 Page 1 of 51
Transcript of · Web viewMedical Devices – Substantial Change Review Form. Applicant Information and...

Medical Devices – Substantial Change Review Form
Applicant Information and InstructionsSignificant/ Substantial Change Notification Covering Product Family-
MDD 252. NSAI Use Only – Amendment Number-/ AIMD 253. NSAI Use Only – Amendment Number-/ TSE Human Blood Medicinal Substances
P/O number:
This is a: Regular/ Standard Review Fast Track (expedited) Modular submission (prior agreement only)
If OBLs apply to this product, please state the relevant product families below: / ; / ; /
DECLARATION(s) BY APPLICANTIn signing this form, the manufacturer is verifying that the requirements of the Directive will be applied in full when the change has been implemented.
Signed on behalf of the Manufacturer:
Date:
Name (please print):Position / Title:Contact person (if different to Manufacturer):e-mail: Phone:
GRF-25-08 Rev. 16.1 Page 1 of 51

INSTRUCTIONS
1. Please complete all relevant sections of the form (excluding the NSAI Review sections).
2. Please enter as much information onto the form as possible - avoid entering “see Technical File/Design Dossier”. If the data is in supporting documentation, please ensure that there is a clear reference to the exact location of this information.
3. Please submit an unsigned version of this Application in Word as well as a signed copy - either scanned/secured (pdf) copy.
4. All application forms and supporting data to be forwarded in soft copy via one of the following (Hard copies not required)
a. NSAI upload facility : see http://www.nsaiinc.com/b. CD or Memory stick to the appropriate address
Europe N. AmericaNSAI1 Swift Square,Northwood,Santry, Dublin 9IrelandPhone : (01) 807 3929Fax : (01) 807 [email protected]
NSAI Inc.402 Amherst StreetNashuaNH 03063USAPhone : (603) 882 4412Fax : (603) 882 [email protected]
5. Supporting documents should be in SEARCHABLE format
6. Applications and supporting documentation must be in English
7. Please send a representative sample of the device(s). This is particularly important for new/novel devices. Any video of procedures/simulated use would also be helpful, if available.
GRF-25-08 Rev. 16 Page 2 of 54

APPLICANTS’ SUBMISSION CHECKLIST Completed application form (Word format, .doc or .docx) Application (min. Signed Declaration page(s)) scanned Sample of device(s) QMS certificates for any sites in Table 1 NOT registered with NSAI Type Examination Certificate if required Declaration of Conformity Labelling & IFU Essential Requirements Checklist Performance/Complaint Analysis Risk Management documentation Sterilisation Validation(s) – if sterile/intended to be sterilised Stability data – if necessary Biocompatibility data – if necessary Electrical Safety Testing data – if necessary Software/firmware lifecycle documents – if necessary Bench Testing data – if necessary Clinical investigation(s) report(s) and supporting documents per MEDDEV 2.7.1.
NSAI Equivalence table RF-25-28 if using equivalent route NSAI Post Market Surveillance (PMS) and Post Market Clinical Follow Up (PMCF) RF-25-27
Note – NSAI do not accept “For Information Only Data” (FIO); all attributes tested shall have clinically relevant specifications
For Transfers Copy of existing Notified Body Certificate(s)
(If not already supplied) Transition Plan Contact details for existing Notified Body
(NSAI will not contact the existing Notified Body prior to agreement with the Manufacturer)
For Own Brand Labeller (OBL) Copy of the OEM (maker’s) CE Certificate and last assessment reports
from their Notified Body Copy of the OEM’s Declaration of Conformity Traceability between OEM cert & devices covered in this application Copy of the contract between OBL & OEM outlining responsibilities Index for Technical documentation/Design dossier (version/date), listing
supporting documents Copy of the OEM’s Instructions for Use/Labelling
For Tissue of Animal Origin falling under Commission Regulation 722/2012Please complete Section 16.1For Human Blood DerivativesPlease complete Section 16.2For Medicinal Substances.Please complete Section 16.3
GRF-25-08 Rev. 16 Page 3 of 54

Table of Contents: Applicant Information and Instructions 1
DECLARATION(s) BY APPLICANT 1
INSTRUCTIONS 2
APPLICANTS’ SUBMISSION CHECKLIST 3
Section 1 – Manufacturer and Product Details 7
Section 2 – Nature of Change 10
Section 3 – Intended Use of the Device 12
Section 4 – Correct Classification and Appropriate Annex 13
Section 5 – Existing Legislation 14
Section 6 – Labelling and IFU 15
Section 7 – Solutions to Essential Requirements and Harmonised Standards 17
Section 8 – Performance/ Complaint Analysis 19
Section 9 – Risk Management 21
Section 10 – Sterilisation 23
10.1 – Sterilisation Validation 23
10.2 – Maintenance of Sterility over shelf life 24
Section 11 –Biocompatibility 26
Section 12 –Medical Electrical Equipment and Systems 29
Section 13 – Device Testing 34
13.1 – Device Design Testing 34
13.2 - Device Stability 36
Section 14 - Clinical Testing (Animal Model) 37
14.1 In Vivo Clinical Performance 37
14.2 In Vivo Device Performance 38
Section 15 – Clinical Performance (Human) 39
15.1 Clinical Evaluation 39
Section 16.0-Appendices 46
16.1 – Regulation 722/2012 : TSE 46
16.2 – Human Blood Derivative(s) 46
16.3 – Devices incorporating Medicinal Substances 47
Section 17 – Design Dossier 49
Section 18 – Critical Process Changes 50
GRF-25-08 Rev. 16 Page 4 of 54

Section 19 - NSAI Queries 52
19.1 Queries, Responses and Dispositions. 52
19.2 Query Status Table 14: 56
Section 20 - Conclusion of Assessment 57
Approval 58
GRF-25-08 Rev. 16 Page 5 of 54

Section 1 – Manufacturer and Product Details
Table 1 – Manufacturers Information & Summary Product data(Legal) Manufacturer’s Name (Legal) Manufacturer’s Address Design Site(s): Manufacturing Site(s):(i.e. sites of actual manufacture)
Assembly Site(s) if applic.: Sterilisation Site(s) if applic.: Scope of Site(s):(i.e. as shown on the QMS cert)
Name and address of EU Authorised Representative(if applicable)
Product/Product Family Name:(In compliance with NB/MED/2.5.1/REC4 & NBOG’S Best Practice Guide 2006-2)
GMDN Reference Number: See www.gmdnagency.com
Declaration of Conformity included - Location within submission : MDD ONLY :Class III IIb IIa Is Im Rule(s)RationaleConformity Assessment
Annex II V (+VII) V + III VIFull QA Prodn QA + Type Testing Product QA
AIMD ONLY :Conformity Assessment
Annex 2 Annex 3 + 5Full QA Production QA + Type Testing
ALL DEVICES :Clinical Strategy-- Clinical data from:
Clinical Investigation Literature (Equivalence) Combination
Date of this application (i.e. date of Declaration of Applicant)
GRF-25-08 Rev. 16 Page 6 of 54

NSAI REVIEW NSAI registration(s) if new site(s) -
If Annex II is “Design” included in scope Yes No Scope of registration(s) adequate to cover the product family Client QMS Certificates are valid and scope of registration(s) remains adequate for product family under review Product family name in line with GMDN & remains valid DoC reviewed - lists model numbers
Type Testing Certificate required – No Yes if YES – Supplied , Valid
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer (if applicable) Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 7 of 54
Section 2 – Nature of Change
1. Device Description:
2. Please provide a clear, detailed description of the change(s):
3. Did the change(s) arise from a vigilance or performance issue Yes No
If “Yes” – please advise -
If”No” what was the rationale/motivation for each change(s) made:
4. Does the change involve an addition to the members of the product family Yes NoIf “Yes” – please complete Table 2 below:
Table 2 – New Model/Catalogue NumbersSub-Family NEW
Model/Catalogue Number(s)
Description Class
5. Please advise what models are affected by the change
Table 3 – Affected Model/Catalogue NumberSub-Family AFFECTED
Model/Catalogue Number(s)
Description Class
6. Impact of changeTable 4-Impact of Change
Impact Yes No1. Is there a change to the solutions (documented evidence )
to the ERs?2. Is there a change to the Harmonised Standards?3.Does the change introduce new hazards which have not
been previously addressed?
4.Does the change adversely affect the risk profile associated with the existing hazards?
5.Does the change alter the details on intended use and /or compliance with the essential requirements given in the design/type approval dossier?
GRF-25-08 Rev. 16 Page 8 of 54

6.Does the change mean that the device will have different end users or be used in a different manner?
7.Does the change mean that the clinical data /performance evaluation data for the original device is not sufficient to confirm conformity of the changed device with the required characteristics and performance?
NSAI REVIEW Change adequately described Modifications/changes considered vs. approved product family Validity of reason(s) for change
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer (if applicable) Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 9 of 54
Section 3 – Intended Use of the Device
1. Please state the current Intended Use –
2.For class III and Class IIb implantables ONLY,Please outline the “Indications for use” (i.e. specific patient population intended for this device/family).
3. Is there a change in Intended Use Yes No
4. Is there a change in Indications for Use Yes No
NSAI REVIEWAgreed that this section is not required Yes No Reviewer Date: Comments:
5. Please enter a full description of the revised intended use and/ or indications for use of the device-
6. Does this change impact the classification/rule Yes No
If “No” please justify -
NSAI REVIEW Revised Intended Use clearly stated Revised Intended Use in line with labelling Impact of Revised Intended Use assessed – including classification/rule
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer (if applicable) Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 10 of 54

Section 4 – Correct Classification and Appropriate Annex
1.Please state the current MDD Classification- Rule: 2. Is there a change in Classification Yes No
NSAI REVIEWAgreed that this section is not required Yes No Reviewer: Date: Comments:
3.MDD Only – Please enter the revised Classification of the device based on the change, along with the Rule & Rationale for the choice
Class Rule Rationale
NSAI REVIEW All Rules reviewed Intended Use described supports Classification
Agree with Classification - Yes No
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 11 of 54

Section 5 – Existing Legislation1. Is the product OBL (Own Brand Labelled) Yes No*You are not the actual Maker of the device – and the identical product carries CE marking by the Maker.
NSAI REVIEWAgreed that this section is not required Yes No Reviewer: Date: Comments: Please complete all questions in this section:
2.Has there been a change to the Maker’s CE cert Yes NoIf “Yes” – please provide the Notified Body’s review of the changeReport/ Document Ref.
3. If the Maker uses NSAI as their Notified Body, please provide the file reference for the review of the change(s) –
MDD : 252. / AIMD: 253. /
NSAI REVIEW Revised OEM cert Report provide Review adequate
NSAI REVIEWTechnical Reviewer: Date: COMMENTS:
Additional Reviewer (if applicable): Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 12 of 54

Section 6 – Labelling and IFU1. Is there a change to the Labelling/IFU Yes NoIf “No” please justify -
NSAI REVIEWAgreed that this section is not required Yes No Reviewer: Date: Comments:
Please supply a sample of the revised draft labelling & IFU in English.
2. Location of the sample Label(s) & IFU in the supporting documentation
3. Please clarify the exact nature of change(s) to the labelling/IFU based on the proposed change(s) under review –
4. Are the requirements of EN 15223-1 & EN 1041 being met Yes No
Version of Standard –If “No” Please justify -
5. Is the device Medical Electrical Equipment Yes NoIf “Yes”a) Are the requirements of EN 60601-1 being met:
Yes No N/AVersion of Standard: If “No” please justify -
b) Are the requirements of EN 60601-1-6 being met: Yes No N/A
Version of Standard: If “No” please justify -
6. Do any vertical labelling standards apply Yes No(i.e. device-specific standards that require labelling)If “Yes”, please list (including current version) - If compliance with these vertical labelling standards is not claimed, please justify -
NSAI REVIEW Labels reviewed – details All relevant parts of ER 13, particularly 13.3 (MDD) or ER 14 (AIMD) met IFU reviewed – details All relevant parts of ER 13, particularly 13.6 (MDD) or ER 15 (AIMD) met Ensure traceability between the Risk Management File and the CER and labelling
GRF-25-08 Rev. 16 Page 13 of 54

Symbols & Markings on ME Equipment/System are per Table D.1, D.2, D.3 of EN 60601-1
Markings & IFU in line with EN 60601-1-2 (EMC) For OBL – OEM IFU claims in line with OBL IFU Vertical labelling standards -
NOTE – If an issue is identified with the sample labelling, ensure that the corrective action(s) are applied to all labelling operations.
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer (if applicable) Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 14 of 54

Section 7 – Solutions to Essential Requirements and Harmonised Standards
Please indicate how relevant Essential Requirements (Annex I) of the Directive are met for the proposed changes.1. Location of the revised solutions to Essential Requirements in the
supporting documentation FOR CLASS I STERILE devices: In particular ER 8FOR CLASS I MEASURING devices: In particular ER 10
2. For devices other than Class I Sterile &/or Measuring please ensure there is specific reference to the design elements associated with this application and demonstrate how the design outputs meet the design inputs through design verification and validation
3. Please list the relevant revised/updated Harmonised Standards in Table 5 below
Table 5 – Applicable Harmonised Standards ListStandard Year Full
ComplianceYes/No
FOR MDD CLASS I STERILE devices: In particular Harmonised Standards relating to sterilityFOR MDD CLASS I MEASURING device: In particular Harmonised Standards relating to the measuring function.
NSAI REVIEWClass I Sterile
ER 8 adequately addressed - Harmonised Standards for Sterilisation -
Class I Measuring ER 10 adequately addressed - Harmonised Standard(s) for Measuring -
Other classes of MDD devices & AIMDsERC Document Rev. ERs identified as N/A
Agree with non-applicability Has each subpart in the ERs been adequately addressed, if applicable
GRF-25-08 Rev. 16 Page 15 of 54
Solutions to ERs complete Ref to the supporting documentation adequate Harmonised standards used Adequately demonstrates that design outputs meet design inputs
through design verification & validation activities
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer (if applicable) Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 16 of 54

Section 8 – Performance/ Complaint Analysis
1. Is the change due to Vigilance or Performance Issue Yes No
NSAI REVIEWAgreed that this section is not required Yes No Reviewer Date: Comments:
2. If “Yes” please give details -
3. Has NSAI received the Vigilance Report(s) Yes NoIf “Yes” please provide the relevant Unique Identifier number(s) –
` If “No” please:(a) Justify: (b) If applicable, please submit a copy of the Competent Authority
report(s) along with the completed NSAI Vigilance Form located at [http://www.nsaiinc.com/services/MedicalDevice -“Vigilance Reporting”] to [email protected]
4. Has this product been the subject of product recalls or Incident Reports in other Regulatory geographies outside EU? If yes, please summarize and provide details with supporting documentation.
5. For those failure modes associated with the identified Root Causes, please clarify if the Occurrence Rates outlined in the Risk Management File required an update based on the observed real world rates.
NSAI REVIEW Vigilance reports reviewed Trending data supplied reviewed Negative review/refusal assessed
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer: Date: COMMENTS:
Additional Reviewer Date:
GRF-25-08 Rev. 16 Page 17 of 54

COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 18 of 54

Section 9 – Risk Management1. Did the proposed change affect or change any existing risks
Yes NoIf “No” please justify -
2. Did the proposed change introduce any new risks Yes NoIf “No” please justify -
3. Was the Risk review documented Yes No(e.g. during change control process, update to FMEA, Memo to file etc.)If “No” please justify -
4. If no update to Risk Management File, please provide rationale:
NSAI REVIEWAgreed that this section is not required Yes No Reviewer Date: Comments:
MANUFACTURERS – Where reviewed and updated, please provideRisk Mgmt File - Risk Analysis, Risk Management Report, including a signed and dated conclusion regarding residual/remaining risks, which clearly identifies or highlights risk(s) in relation to the proposed change(s).
NOTE: Additional information may be required for devices containing tissue of animal origin, human blood derivatives or medicinal substances: see APPENDICES.
FOR CLASS I STERILE devices: In particular risks related to sterilityFOR CLASS I MEASURING devices: In particular risks related to measuring function.
5. Risk management per EN ISO 14971 latest version Yes NoVersion of Standard: If “No” please justify -
6. Which of the multifunctional team provided the clinical input – i.e. risks associated with the clinical use of the devicePlease justify the appropriateness of their expertise -
7. Please provide a traceability matrix linking the contraindications, warnings and precautions from Risk Management File to the Instructions For Use and Clinical Evaluation Report.
8. If applicable, have relevant risk documentation for Medical Electrical Equipment, systems &/or Software been included Yes NoIf “No” please justify -
GRF-25-08 Rev. 16 Page 19 of 54

NSAI REVIEW Signed & dated conclusion regarding residual/remaining risks Residual/remaining risks acceptable, based on intended application of
the device Compliance with ISO 14971 – version: Suitable cross-functional team Contra-indications/Warning/Precautions in IFU originate in Risk file Risk analysis adequate Solutions adopted by Manufacturer conform to safety principles, taking
account of the generally accepted state of the art Adequately covers the proposed changes
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer (if applicable) Date: Clinical risks (actual risks involved in the use, application of the
treatment or deployment of the device) identified by suitably qualified personCOMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 20 of 54

Section 10 – Sterilisation10.1 – Sterilisation Validation
1. Is the product provided sterile or intended to be sterilised prior to use Yes No
2.Does the proposed change affect sterilisation Yes NoIf “No” please justify -
NSAI REVIEWAgreed that this section is not required Yes No Reviewer: Date: Comments:
MANUFACTURERS - Please provide the necessary sterilisation validation protocol(s) & report(s) and populate Table 6 below for the proposed changes
3. Is a full validation/revalidation required Yes No
4. If a full validation/revalidation not completed, please provide an Adoption justification/rationale report -
Table 6 – Sterilisation Information Summary for ChangesDevice sub-family
Cat. Number
Sterilisation Method
Sterilisation Location
Protocol/ Report No.
Site resp for Release
5. Is EtO used for Sterilisation of the device(s) Yes NoIf “No” please go to Section 11.
6. For EtO Sterilisation OnlyIs compliance with EN ISO 10993-7 latest version claimed Yes NoIf “No” please explain -
7. Please categorise the device according to the duration of contact A – Limited Exposure B – Prolonged Exposure C – Permanent Contact
GRF-25-08 Rev. 16 Page 21 of 54

10.2 – Maintenance of Sterility over shelf life1. Does the product have a shelf life Yes No
If “No” please justify: NSAI REVIEWAgreed that this section is not required Yes NoReviewer Date: Comments:
2. Please define the shelf life/expiry date – Current yearsProposed years
3. Is the aging based on Accelerated Real Time data
4. Is the packaging stability affected Yes NoIf “No”, please justify - If “Yes”, is compliance with EN ISO 11607 latest version claimed
Yes NoIf “No” please explain -
5. Confirm start date of real time studies
6. Please supply the relevant report(s) to justify the proposed change – Protocol # Report #
NSAI REVIEW Compliance with relevant harmonised standard for sterilisation - EtO Residuals compliant with EN ISO 10993-7 – version Adoption rationale adequate (if appropriate) Packaging stability data meets EN ISO 11607-1/-2 – version Packaging real time data planned Packaging real time testing available & reviewed Data supports the proposed change(s)
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 22 of 54

Section 11 –Biocompatibility
(NOT REVIEWED BY NSAI for Class I Devices)
1.Does the product contact the patient’s body directly or indirectly Yes No
2.Does the proposed change affect biocompatibility Yes NoIf “No” please justify -
NSAI REVIEWAgreed that this section is not required Yes No Reviewer Date: Comments:
Please confirm the categorisation of the devices with respect to Body Contact and Duration of Contact in Table 7 below & the testing conducted in Table 8
3. Is compliance with EN ISO 10993 series latest versions claimed Yes No
If “No” please explain -
Table 7a – Biocompatibility Categorisation: Body ContactSurface-contacting devices External communicating
devicesImplant devices
Skin Blood path, indirect Tissue/boneMucosal membranes Tissue/bone/dentin BloodBreached/compromised surfaces
Circulating blood
Table 7b – Duration of ContactLimited exposure (< 24hrs)
Prolonged exposure(>24hrs <30 days)
Permanent contact (>30 days)
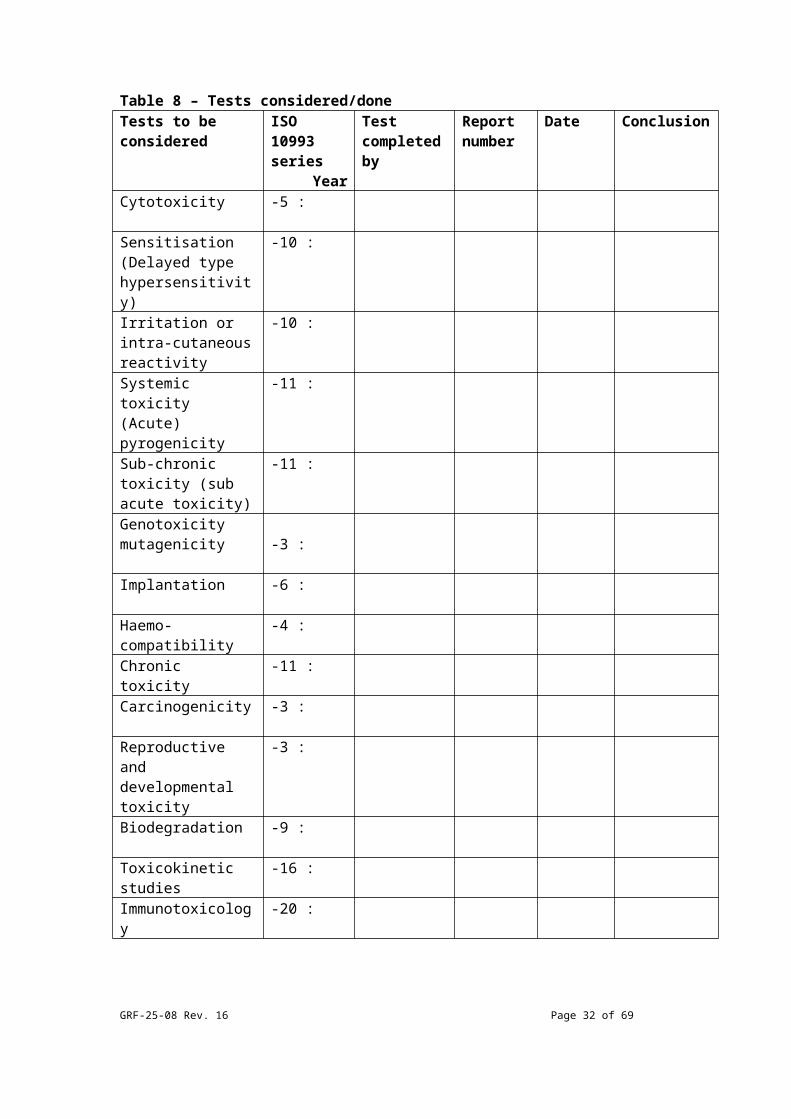
Table 8 – Tests considered/doneTests to be considered
ISO 10993 series
Year
Test completed by
Report number
Date Conclusion
Cytotoxicity -5 : Sensitisation(Delayed type hypersensitivity)
-10 :
Irritation or intra-cutaneous reactivity
-10 :
GRF-25-08 Rev. 16 Page 23 of 54

Systemic toxicity (Acute)pyrogenicity
-11 :
Sub-chronic toxicity (sub acute toxicity)
-11 :
Genotoxicitymutagenicity -3 : Implantation -6 : Haemo-compatibility
-4 :
Chronic toxicity -11 : Carcinogenicity -3 : Reproductive and developmental toxicity
-3 :
Biodegradation -9 : Toxicokinetic studies
-16 :
Immunotoxicology -20 : PLEASE NOTE : Testing should be done on finished/sterilized device(s) or on materials that have been processed in the same manner, including sterilisation
4. Please provide rationale for selecting &/or waiving tests
5. Who provided this rationale -
6. Please provide evidence of their expertise/competence -
NSAI REVIEW EN ISO 10993-1 – version applied Test results acceptable Rationale appropriate for tests selected Competence of expert
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer (if applicable) Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
GRF-25-08 Rev. 16 Page 24 of 54

QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 25 of 54

Section 12 –Medical Electrical Equipment and Systems
1. Is the product ME Equipment or System* Yes No
*Definitions: ME Equipment – electrical equipment having an applied part or transferring energy to or from the PATIENT or detecting such energy transfer to or from the patient and which is:Provided with not more than one connection to a particular supply mains; and Intended by its manufacturer to be used:In the diagnosis, treatment, or monitoring of a patient; or For compensation or alleviation of disease, injury or disability
ME System – combination, as specified by its manufacturer, of items of equipment, at least one of which is ME Equipment to be inter-connected by functional connection or by us of a multiple socket-outlet
NSAI REVIEWAgreed that this section is not required Yes No Reviewer Date: Comments:
2. Please answer all questions below and complete Tables 9, 10 & 11.Please provide all relevant Test Reports, and EN 62304 Software Development Process & Validation Report, as well as Software Risk Assessment.
3. Have the applicable requirements of EN 60601-1 latest version, including the mandatory risk assessment to EN 14971 been applied
Yes No
(a) If “Yes” – please list all applicable “Part 2’s” in Table 11 below(b) (i) If “No”, is a particular standard (60601-2-xx) applicable that refers
to a prior 60601-1 (ex. 2nd edition)? Yes No
(ii) If “No” – please provide rationale for not applying the latest version of EN 60601-1 –
4. Please list the document(s) submitted substantiating conformance to the edition of EN 60601-1 claimed – Note – the electrical review will include a review of the document(s) in which conformance with all applicable EN 60601-1 requirements as well as EN 60601-2-x if applicable are tested. Please ensure the tester understands and is familiar with a comprehensive test report/checklist format addressing each applicable requirement. Abbreviated reports and summaries are NOT acceptable.
GRF-25-08 Rev. 16 Page 26 of 54

5. What is the expected Service Life of the device years
6. What is the Essential Performance of the device –
7. Does the product incorporate Software/Firmware or meets the Definition of Standalone Software per MEDDEV 2.1/6? Yes NoIf “Yes” Have the requirements of EN 62304, including the mandatory risk assessment to EN 14971 been applied in submitted software documents Yes NoVersion of Standard : If not the latest version, please explain -
8. Please provide the safety classification (A, B, C) and rationale for each software or firmware unit.Please also provide all documentation to demonstrate compliance with EN 62304: as shown below
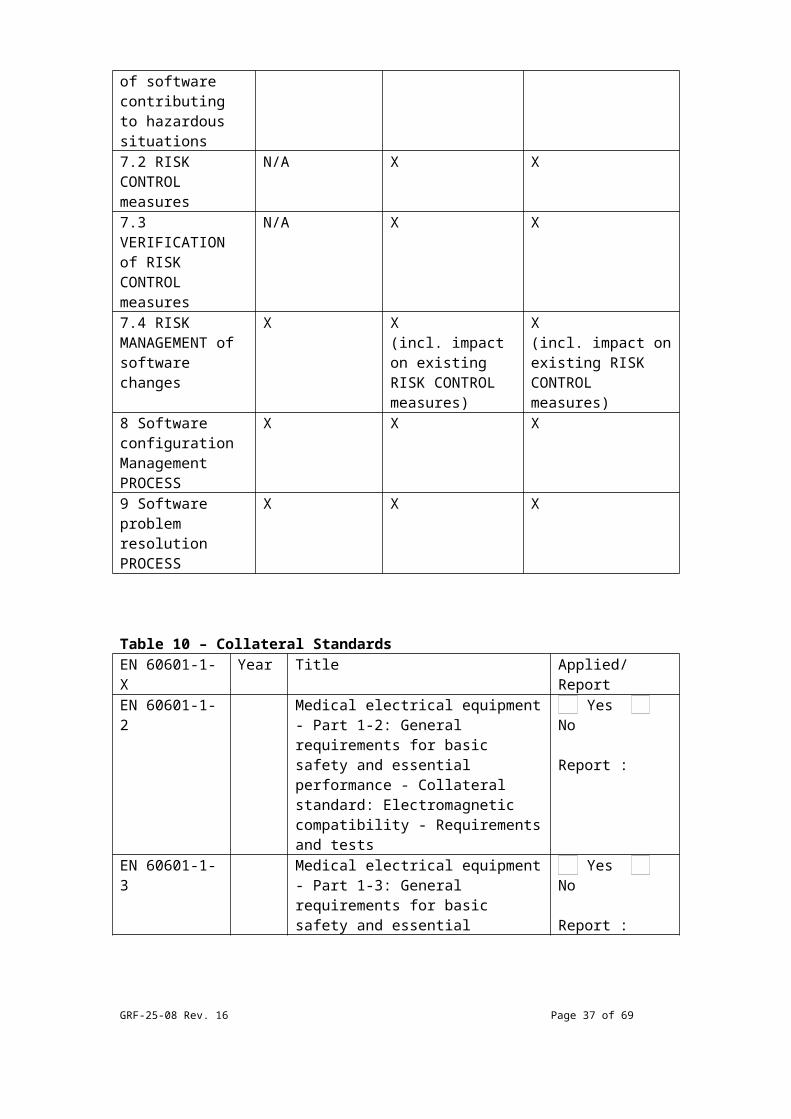
Table 9 – EN 62304 ComplianceEN 62304 requirement
Class A Class B Class C
4.3 Software safety classification
X X X
5.1 Software development plan
X X X
5.2 Softwarerequirements
X X(incl. RISK CONTROL measures)
X(incl. RISK CONTROL measures)
5.3 SoftwareARCHITECTURAL design
N/A X X(incl. segregation for RISK CONTROL)
5.4 Software detaileddesign
N/A X X(incl. detailed design of SOFTWARE UNIT & interfaces)
5.5 SOFTWARE UNITimplementation
X X(incl. verification & acceptance criteria)
X(incl. verification & acceptance criteria)
5.6 Software integration& integration testing
N/A X X
5.7 SOFTWARE SYSTEMtesting
N/A X X
5.8 Software X X X
GRF-25-08 Rev. 16 Page 27 of 54

release (VERSION)
(incl. ANOMALIES, how created, archive, repeatability)
(incl. ANOMALIES, how created, archive, repeatability)
6.1 Softwaremaintenance plan
X X X
6.2 Problem &modification analysis
X X(incl. analysis of CHANGE REQUESTS)
X(incl. analysis of CHANGE REQUESTS)
6.3 Modificationimplementation
X X X
7.1 Analysis of softwarecontributing to hazardoussituations
N/A X X
7.2 RISK CONTROLmeasures
N/A X X
7.3 VERIFICATION of RISK CONTROL measures
N/A X X
7.4 RISK MANAGEMENT of software changes
X X(incl. impact on existing RISK CONTROL measures)
X(incl. impact on existing RISK CONTROL measures)
8 Software configurationManagement PROCESS
X X X
9 Software problemresolution PROCESS
X X X
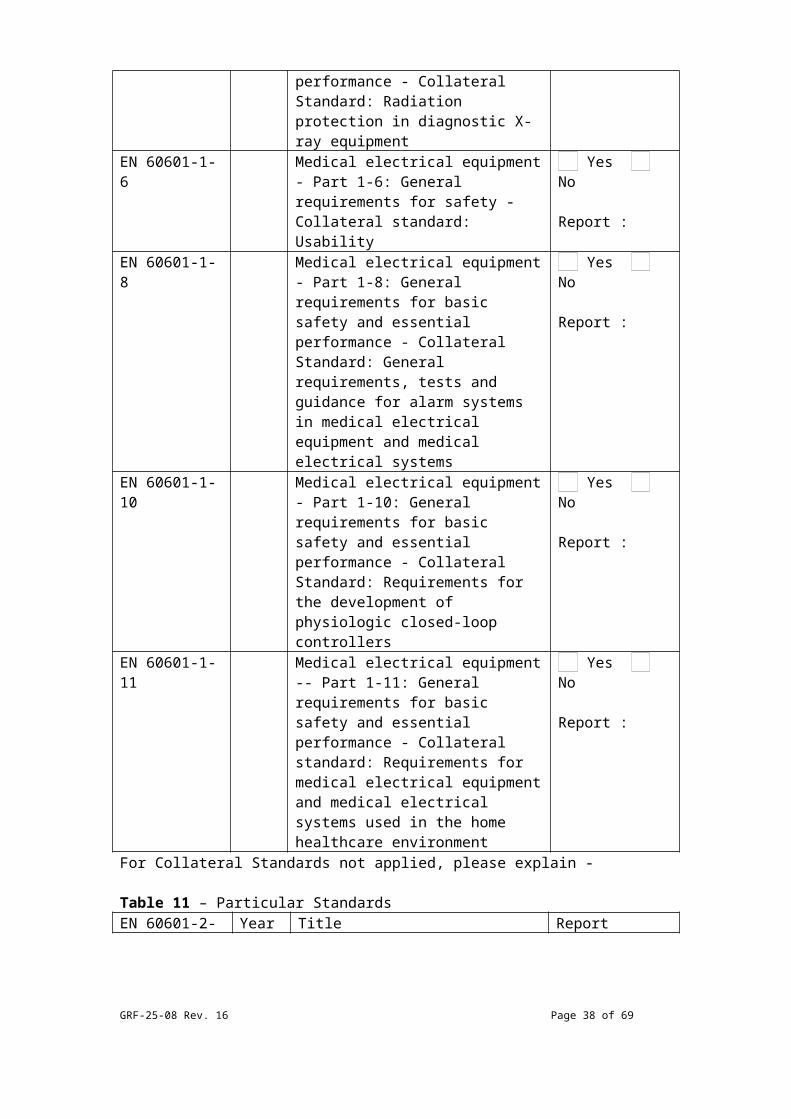
Table 10 – Collateral StandardsEN 60601-1-X Year Title Applied/ ReportEN 60601-1-2 Medical electrical equipment -
Part 1-2: General requirements for basic safety and essential performance - Collateral standard: Electromagnetic compatibility - Requirements and tests
Yes No
Report :
EN 60601-1-3 Medical electrical equipment - Part 1-3: General requirements
Yes No
GRF-25-08 Rev. 16 Page 28 of 54

for basic safety and essential performance - Collateral Standard: Radiation protection in diagnostic X-ray equipment
Report :
EN 60601-1-6 Medical electrical equipment - Part 1-6: General requirements for safety - Collateral standard: Usability
Yes No
Report :
EN 60601-1-8 Medical electrical equipment - Part 1-8: General requirements for basic safety and essential performance - Collateral Standard: General requirements, tests and guidance for alarm systems in medical electrical equipment and medical electrical systems
Yes No
Report :
EN 60601-1-10
Medical electrical equipment - Part 1-10: General requirements for basic safety and essential performance - Collateral Standard: Requirements for the development of physiologic closed-loop controllers
Yes No
Report :
EN 60601-1-11
Medical electrical equipment -- Part 1-11: General requirements for basic safety and essential performance - Collateral standard: Requirements for medical electrical equipment and medical electrical systems used in the home healthcare environment
Yes No
Report :
For Collateral Standards not applied, please explain -
Table 11 – Particular StandardsEN 60601-2-X*
Year Title Report
*Also includes EN 80601-2-x.For Particular Standards not applied, please explain -
NSAI REVIEW EN 60601-1 – 3rd edition applied. If not : 60601-2- permits 2nd
edition until - Test results demonstrate all applicable requirements acceptable
GRF-25-08 Rev. 16 Page 29 of 54

Compliance with EN 62304 submitted. Highest EN 62304 safety class of software module = overall system
safety class All appropriate Collateral Standards applied All appropriate Particular Standards applied Additional directives (ex. Telephony) apply and test certifications (ex.
RTTL) submitted
NSAI REVIEWTechnical Reviewer Date: COMMENTS:
Clinical Reviewer (if applicable) Date: COMMENTS:
Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 30 of 54

Section 13 – Device Testing 13.1 – Device Design Testing
1. Is the product Class IIb implantable or Class III Yes No
2. Do the changes require additional bench testing Yes NoIf “No” please justify -
NSAI REVIEWAgreed that this section is not required Yes No Reviewer: Date: Comments:
3. For the proposed change(s), please supply a Design Traceability Matrix or Design Input/ Output document and verify that the following have been included:
- Design input/ user need
- Specification for each input
- Source of each specification
- Justification of the source (via use of a standard: Harmonised, Non-Harmonised, ASTM, AAMI), internally validated specification with clinical feedback, etc.)
- Design Output/ Documented Evidence - Comment on whether D/I was met or not
4. Depending of the type of the device, “bench testing” can include, but is not limited to:
- Static and dynamic accelerated durability- Bond strength- Coating integrity- Coating durability- Corrosion testing- Simulated use testing
Note: NSAI shall not accept “For Information Only” data (FIO); all attributes tested shall have clinically relevant specifications.
Note: If safety and performance of the proposed device is being demonstrated via equivalence to a predicate device, please supply side-by-side bench data of the devices in question.
5. Please verify that the following has been supplied: Representative sample of the device Video of procedures/simulated use, (if available Computer modeling reports- Finite Element Analysis (FEA), Computational Fluid Dynamics (CFD), etc. (if applicable.)
GRF-25-08 Rev. 16 Page 31 of 54
All relevant bench test protocols and reports.
6. Please verify that the protocols and reports include: Justified test parameters per relevant standards Acceptance Criteria Sample size methods, justification and documented source Justified deviations (if applicable)
If not, please justify:
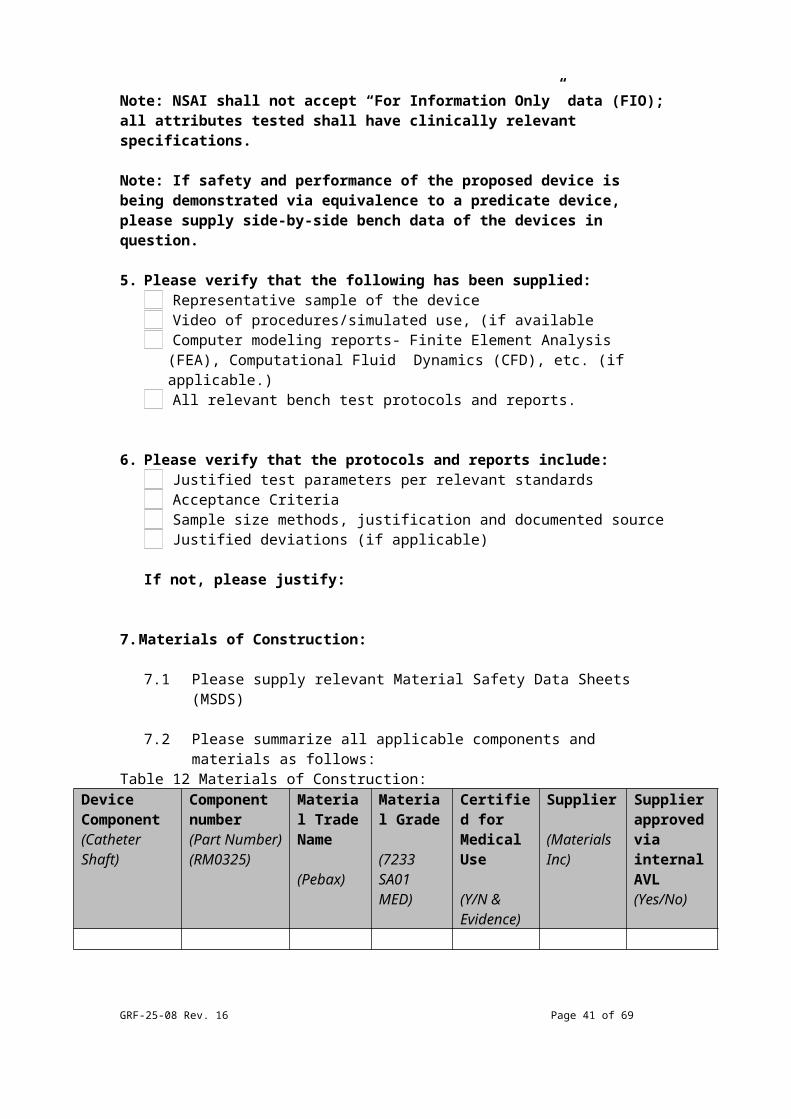
7. Materials of Construction:
7.1 Please supply relevant Material Safety Data Sheets (MSDS)
7.2 Please summarize all applicable components and materials as follows:
Table 12 Materials of Construction:Device Component(Catheter Shaft)
Component number(Part Number)(RM0325)
Material Trade Name
(Pebax)
Material Grade
(7233 SA01 MED)
Certified for Medical Use
(Y/N & Evidence)
Supplier
(Materials Inc)
Supplier approved via internal AVL(Yes/No)
Note: Please supply this table as an attachment to the submission
7.3. Please describe the design characteristics that address the interaction of the device surface and its environment of use.
8. Please describe the device lot release criteria:
NSAI REVIEW Device Design testing dataset meets design requirements/
specifications Dataset adequate (justified sample size/ testing per Risk Mgmt) Relevant Standards applied Adequate materials information supplied
Technical Reviewer Date: COMMENTS:
Additional Reviewer (if applicable) Date:
GRF-25-08 Rev. 16 Page 32 of 54

COMMENTS:
QUERIES No Yes Number(s)
13.2 - Device Stability 1. Is there a change in the product shelf life Yes No
NSAI REVIEWAgreed that this section is not required Yes No Reviewer Date: Comments:
2. Current shelf life: Proposed shelf life: 3. Is the aging based on Accelerated Real Time data4. Confirm start date of real time studies 5. Please supply the relevant report(s) which substantiate device shelf life
Protocol # Report # 6. Please confirm the number of sterilisation cycles that the devices have
undergone prior to testing- 7. Please note the conditioning applied (eg. Ageing, transport etc) 8. Were all device attributes assessed at the proposed shelf life.
Yes NoIf “No” please justify reason for omitting other attributes-
NSAI REVIEW Device stability data provided Adequate justification for attributes tested/omitted Standard(s) used - Device real time data planned Device real time testing available & reviewed Data supports the proposed change(s)
Technical Reviewer Date: COMMENTS: Additional Reviewer (if applicable) Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 33 of 54

Section 14 - Clinical Testing (Animal Model)1. Have acute/chronic animal studies been completed to substantiate the
in vivo safety and/or effectiveness of the change Yes NoIf “No” please explain -
NSAI REVIEWAgreed that animal test is not required Yes No Reviewer Date: Comments:
2. Does the data provide detailed information on all studies in animal models which substantiate the stated intended use, i.e.:
- Study objectives- Methodology- Results, Analysis and Conclusions- Rationale for selection of the model(s)
If “No” please explain-
3. Please provide summary data of acute in vivo device performance and indicate the document number(s) of all studies in animal models-
4. Please provide summary data of acute in vivo device performance against pre-defined requirements, and/or clearly define the location of the relevant data within each animal report-
5. Was the product design assessed in the animal studies equivalent to the device design of this submission design Yes NoIf “No” please explain-
14.1 In Vivo Clinical PerformanceNSAI REVIEW
Pre-clinical studies adequate. Documentation valid – supporting pre-clinical evaluation.
Clinical Reviewer Date: COMMENTS:
GRF-25-08 Rev. 16 Page 34 of 54

QUERIES No Yes Number(s)
14.2 In Vivo Device Performance NSAI REVIEW
Pre-clinical studies adequate to substantiate design validation. Appropriate pre-clinical investigators.
NSAI REVIEWTechnical Expert Reviewer Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 35 of 54

Section 15 – Clinical Performance (Human)
(NOT REVIEWED by NSAI for Class I Devices) 15.1 Clinical Evaluation1. Do the changes require additional clinical data Yes No
If “No” please justify -
NSAI REVIEWAgreed that this section is not required Yes No Reviewer Date: Comments
If “Yes”, this section is not required.
Revisions to the MDD 93/42/EC by 2007/47/EC have implications for the clinical data & the evaluation of the data to be provided by Manufacturers to the Notified Body, to demonstrate the clinical safety & performance of the medical device.
Clinical data must always be documented for all medical device classifications.
MedDev 2.7.1 latest version provides guidance on the procedure to be adopted by the Manufacturer to evaluate clinical data.See NSAI Guidance document for further information.
Please supply a Clinical Evaluation Report to support the safe use of the device as per MedDev 2.7.1.
2. Please provide the document number and location of the updated Clinical Evaluation Report (CER) -
3. Does the supporting documentation submitted to NSAI include: Literature search protocol Full text of articles referenced in the CER.
If not please explain -
4. Does the CER comply with MedDev 2.7.1 Yes NoVersion of MedDev used: If “No” please explain -
5. Please identify the individual(s) who performed the clinical evaluation - Is their CV included? Yes NoIf “No” please explain -
6. Please provide justification of the choice of evaluator(s) -
GRF-25-08 Rev. 16 Page 36 of 54
7. For Class III or Class IIb Implantables, was a clinical investigation completed to demonstrate the safety and performance of the device
Yes NoIf “No” please explain -
8. For this device :a) Are any clinical investigations planned Yes Nob) Are any clinical investigations on-going Yes Noc) Are any clinical investigations completed Yes No
If “Yes” please provide additional information and status –
9. For clinical investigations, does the supporting documentation submitted to NSAI include
Letter of no objection from Competent Authority/ies (CAs) or other regulatory agency/ies as appropriate
Clinical investigation plan and amendments for which no grounds for objection were raised
Ethics committee opinion(s) and comments arising from their review
Signed and dated final report (signed by the sponsor, the co-coordinating clinical investigator – if appointed – and principal investigator at each site).If not please explain -
NSAI REVIEWTO BE COMPLETED BY NSAI CLINICAL REVIEWER ONLY
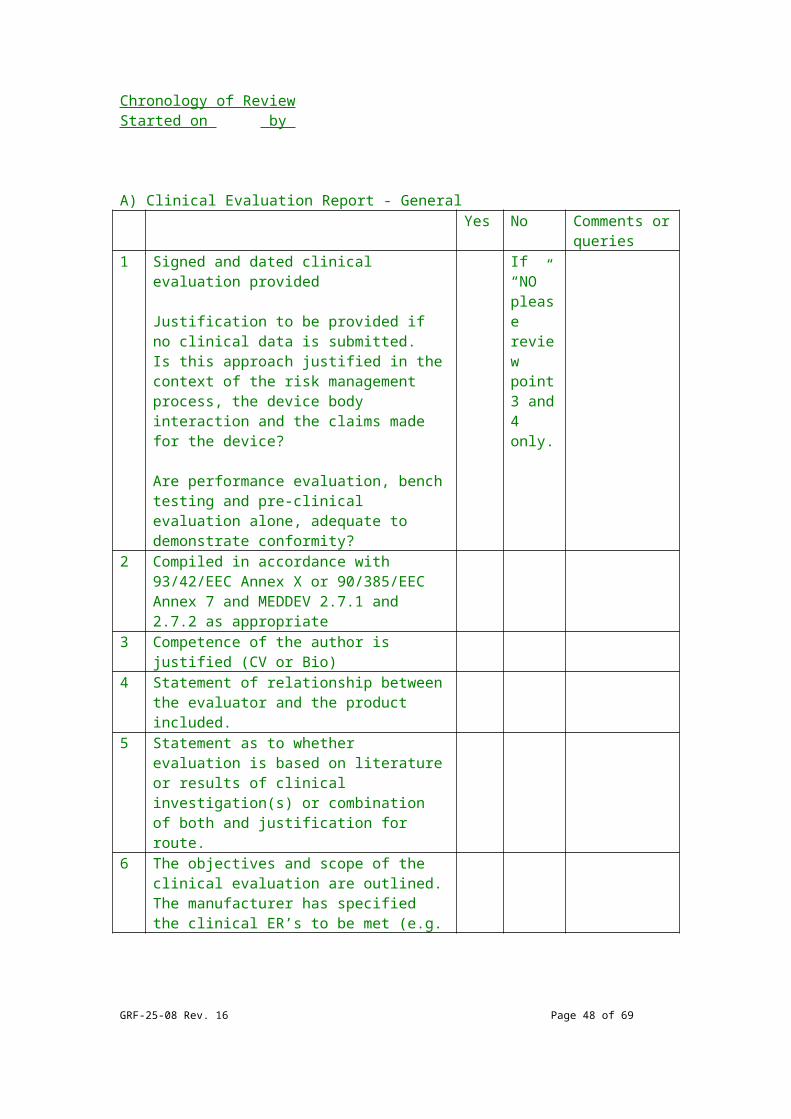
Chronology of ReviewStarted on by
A) Clinical Evaluation Report - GeneralYes No Comments or
queries 1 Signed and dated clinical evaluation
provided
Justification to be provided if no clinical data is submitted. Is this approach justified in the context of the risk management process, the device body interaction and the claims made for the device?
Are performance evaluation, bench testing and pre-clinical evaluation alone, adequate to demonstrate conformity?
If “NO” please review point 3 and 4 only.
2 Compiled in accordance with 93/42/EEC
GRF-25-08 Rev. 16 Page 37 of 54
Annex X or 90/385/EEC Annex 7 and MEDDEV 2.7.1 and 2.7.2 as appropriate
3 Competence of the author is justified (CV or Bio)
4 Statement of relationship between the evaluator and the product included.
5 Statement as to whether evaluation is based on literature or results of clinical investigation(s) or combination of both and justification for route.
6 The objectives and scope of the clinical evaluation are outlined. The manufacturer has specified the clinical ER’s to be met (e.g. clinical performance(s), safety, risks and favourable benefit/risk ratio related to intended use, target group(s) and indication(s) to be met).
7 The procedures and steps (sections 5-9 of MEDDEV 2.7.1) are outlined in report.
8 Is data presented from clinical experience of the device in question or equivalent devices (e.g. PMS reports, studies, registries, adverse event databases, FCAs etc)?
9 Has the opinion of all experts involved been documented? Has the opinion of the expert(s) been observed? If not please justify.
10 Is there a clinical data analysis & conclusion provided, to address safety & performance of device, compliance with ERs & the risk benefit analysis of device? Are the conclusions of the manufacturer valid?
B) Clinical literature reviewTO BE COMPLETED BY NSAI CLINICAL REVIEWER ONLY
Yes No Comments or queries
1 Literature search protocol document submitted.
Source of data and justificationDatabase search strategySelection criteria and justification
2 Full copies of published articles identified as being relevant to the device in question submitted.
3 Clinical data appraisal criteria pre-defined4 Literature reflects current state of the art
GRF-25-08 Rev. 16 Page 38 of 54
for technology and medical practice5 Favourable and unfavourable data
considered6 Rationale for selection/relevance of
published literature provided7 Validity of data with respect to author,
study design & state of the art technologies, addressed by the manufacture
8 Analysis of market experience of same or similar devices included
9 Are gaps identified in demonstration of compliance with ERs
10 Are gaps identified in demonstration of equivalencei.e. technical, biological and clinical? If differences are identified has the manufacturer analysed thesignificance these might have on safety and performance?
11 Are labelling claims substantiated by the clinical data together with the pre-clinical data provided?
12 Is there a clinical data analysis & conclusion provided, to address safety & performance of device, compliance with ERs & the risk benefit analysis of device?
13 Is the pre-clinical with clinical data sufficient to demonstrate compliance with ERs (safety & performance under normal conditions of use)?
14 If a manufacturer deems that a clinical investigation is not required for a Class IIb implantable or Class III device is this justified? Is the justification to rely on existing clinical data valid? Is the data presented in the literature review sufficient to address the hazards identified in the risk analysis?
15 Does the manufacturer include a conclusion with a risk benefit analysis for use of the device as intended taking account of the “state of the art”? Does he identify any gaps in the evidence to cover all relevant aspects of safety and performance in the conclusion? Are the manufacturer’s conclusions valid?
16 Has the manufacturer submitted a post market clinical follow up plan for the device e.g. study or registry? If not is this adequately justified?
GRF-25-08 Rev. 16 Page 39 of 54

C) Clinical investigation reviewTO BE COMPLETED BY NSAI CLINICAL REVIEWER ONLY
Yes No Comments or queries.
1 Was Annex X of 93/42/EEC or Annex VII of 90/385/EEC taken into account, as appropriate?
2 Does the manufacturer claim compliance with ISO 14155 ?
3 Has a copy of the protocol submitted to the CA or other regulatory agency for which no grounds for objection were raised been submitted and reviewed? Is this the same as that submitted to the CA(s)
4 Has a copy (ies) of the letter of “no objection”/ approval from the CA(s), (if available) or other approval from the relevant regulatory agency (ies) together with any comments made arising from regulatory review been submitted and reviewed?
5 Has a copy (ies) of the Ethics Committee(s) opinion(s) and comments arising from their review or a summary of all EC opinions and any comments/conditions arising from their reviews been submitted and reviewed?
6 Have there been any changes to the protocol? What is the rationale given? Have these been approved by the CA(s)/regulatory authority (ies) and EC(s)?
7 Has the sponsor fulfilled any conditions before starting the investigation?
8 Where the investigation(s) was performed outside the EU has the manufacturer demonstrated that the use of the device (clinical practice and techniques) and patient population are equivalent to those for which the device will be used within the EU (if relevant)?
9 For drug device combinations have any issues or concerns raised as part of the clinical assessment of the medicinal substance by the medicinal CA or the EMA been considered and / or resolved?
10 Has a copy of the signed and dated final report been submitted and reviewed?
11 Does the final report contain the
GRF-25-08 Rev. 16 Page 40 of 54
following information?Summary – presenting essentials
of studyIntroduction- including context of
development of device and guidelines followed in development of protocol (e.g. ISO 14155, Declaration of Helsinki etc)
Materials and methodsSummary of Clinical investigation
planResults- analysis and resultsDiscussion and conclusionsSignature- should be signed off by
sponsor, the co-ordinating clinical investigator (if appointed) and principal investigator at each centre.
12 Is the investigation as notified consistent in terms of design and end points with the investigational results presented?
13. Have pass/fail criteria of the investigation been met?
14 Have all serious adverse events been reported to the appropriate regulatory/ ethics committees?
15 If a clinical investigation is terminated is there follow up of patients? Has manufactured agreed to submit this data and PMS data to NSAI for review?
16 If a clinical investigation is terminated prematurely, is a valid justification provided?
17 Have the results and conclusions of the clinical investigation(s) demonstrated compliance with the identified relevant essential requirements?
18 Are the labelling claims substantiated by the clinical data when taken together with the relevant pre-clinical data?
19 Has the manufacturer submitted a post-market clinical follow up plan (e.g. study or registry)? If not is this adequately justified?
OVERALL CLINICAL EVALUATION ASSESSMENT :TO BE COMPLETED BY NSAI CLINICAL REVIEWER ONLY
Yes NoAre the conclusions of the manufacturer’s clinical evaluation valid (taking account of documents NBOG CL 2010-1 and MEDDEV 2.7.1 rev 3 of 2009)?
GRF-25-08 Rev. 16 Page 41 of 54

Clinical reviewer(s): Date:
Comments:
QUERIES No Yes Number(s)
Conclusion:
Internal & External Assessors used: No YesIf Yes – Details: Opinion/Comments:
Signature: Date:
GRF-25-08 Rev. 16 Page 42 of 54

Section 16.0-Appendices
16.1 – Regulation 722/2012: TSE1. Does the product contain tissue of animal origin covered by Regulation
722/2012 Yes No
2. Do the changes impact the tissue of animal origin Yes NoIf “No” please justify -
If “No” please go to Section 16.2NSAI REVIEWAgreed Yes No NSAI Reviewer Date: Comments If “Yes”, this section is not required.
Please complete Appendix A, highlighting the change(s) from the originally approved data (see separate document for Appendices)
NSAI REVIEWReviewer Date: COMMENTS:
QUERIES No Yes Number(s)
16.2 – Human Blood Derivative(s)1. Does the product contain human blood derivative(s) covered by Directive 2000/70/EC Yes No
Do the changes impact the human blood derivative(s) Yes NoIf “No” please justify -
If “No” please go to Section 16.3NSAI REVIEWAgreed Yes No NSAI Reviewer Date: Comments If “Yes”, this section is not required.
Please complete Appendix B, highlighting the change(s) from the originally approved data (see separate document for Appendices)
NSAI REVIEWReviewer Date:
COMMENTS:
GRF-25-08 Rev. 16 Page 43 of 54

QUERIES No Yes Number(s)
16.3 – Devices incorporating Medicinal Substances1. Does the product incorporate a medicinal substance liable to act on the body with ancillary action Yes No
If “No” – no additional sections required.
Please confirm if the proposed change impacts the sections in Table 13 below:
SECTION YES
NO
Agreed by NSAI
Reviewer Date
Med
icin
al s
ubst
ance
Requ
irem
ents
Qualitative and quantitative particulars of the constituentsDrug Master FileE.Ph. monographs or methodsE.D.Q.M. certificateDescription of method of manufactureControl of starting materialsControl of tests carried out at intermediate stages of the manufacturing process of the medical deviceControl tests on finished productToxicityReproductive FunctionEmbryo/foetal and perinatal ToxicityMutagenic PotentialCarcinogenicity Potential
GRF-25-08 Rev. 16 Page 44 of 54

PharmacodynamicsPharmacokineticsLocal toleranceRisk Analysis – additionalStability – additional
1. Do the changes require a supplementary Competent Authority review per MDD ER 7.4 paragraph 3 Yes NoIf “No” please justify -
If “No” – no additional sections required.
NSAI REVIEWAgreed Yes No NSAI Reviewer Date: Comments If “Yes”, this section is not required.
Changes to the ancillary substance incorporated in a device, in particular related to its manufacturing process, need to be reviewed by the relevant Competent Authority to confirm that the Quality and Safety of the ancillary substance are maintained.Please complete Appendix C, highlighting the change(s) from the originally approved data (see separate document for Appendices).
NSAI REVIEWReviewer Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 45 of 54

Section 17 – Design Dossier
1. Is the product Class III using Annex II Yes No
If “Yes” - Does the product use particles with at least one dimension below 100nm Yes No
NSAI REVIEWAgreed that review not required Yes No Reviewer Date: Comments
2. Please provide information regarding Agglomeration state/aggregation Composition (e.g. chemical composition and structure) Particle size/size distribution Purity/impurity Shape Solubility (hydrophobicity, liposolubility, water solubility) Stability Surface area Surface chemistry Surface charge Coating characteristics
-
NSAI REVIEWTechnical Expert Reviewer Date: COMMENTS:
QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 46 of 54

Section 18 – Critical Process Changes
1. Is the product MDD Class III Annex II or AIMD Annex II Yes No2. Has there been a change to manufacturing processes Yes No
If No, please justify -
NSAI REVIEWAgreed that this section is not required Yes No Reviewer Date: Comments:
3. Has there been a change in critical processes and/or materials Yes No
If Yes, please supply a detailed overview - If No, please justify -
NOTE – for devices containing medicinal substances, human blood derivatives or using tissue of animal origin, additional information may be requested in the appropriate Appendix/Appendices.
4. Have there been any changes in key laboratory studies and testing – Yes No
If Yes, please supply a summary of changes - If No, please justify -
NSAI REVIEW (add Query # if raised) Design specs including solutions to ERs Safety and efficacy of the device as demonstrated by the Risk Analysis Clinical safety of the device Design outputs meet input requirements for design and development Laboratory results obtained and conclusions made are applicable to the design specifications and performances claimed
Design validation demonstrated that the product is capable of meeting the requirements for its intended use
Validation program and the results obtained are consistent with all the claimed properties and the intended uses and conform to the applicable essential requirements.
Manufacturing processes overview reviewed
Are further tests/proof required to allow assessment of conformity with the requirements of the Directive Yes No
NSAI REVIEWReviewer Date: COMMENTS: QUERIES No Yes Number(s)
GRF-25-08 Rev. 16 Page 47 of 54
Section 19 - NSAI Queries
19.1 Queries, Responses and Dispositions.Company Query #1 – Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
Company Query #2 – Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
Company Query #3 – Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
GRF-25-08 Rev. 16 Page 48 of 54

Company Query #4 –
Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
Company Query #5 – Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
Company Query #6 – Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
Company Query #7 – Company Response: Date:
GRF-25-08 Rev. 16 Page 49 of 54

Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
Company Query #8 – Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
Company Query #9 – Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
If not accepted:FOLLOW-ON QUERY:
Company Query #10 – Company Response: Date:
Response Disposition: By: Date: Accepted Yes NoRationale:
GRF-25-08 Rev. 16 Page 50 of 54

If not accepted:FOLLOW-ON QUERY:
GRF-25-08 Rev. 16 Page 51 of 54

19.2 Query Status Table 14:
Query # Disposition By Date
GRF-25-08 Rev. 16 Page 52 of 54

Section 20 - Conclusion of Assessment
For MDD Class III Annex II or AIMD Annex II:
NSAI confirms the following: Design specs and solutions to the ERs (all design verification &
validation testing) are appropriate based on the proposed intended use
Design outputs meet the input requirements for design and development
Manufacturing processes overview has been reviewed. The Risk Analysis has demonstrated compliance to EN ISO 14971 The clinical safety of the device. Inspection process overview has been reviewed.
No further tests/proof required to allow assessment of conformity with the requirements of the Directive
NOTIFIED BODY STATEMENT:Conclusion –
(The following details shall appear on the Approval Letter)
1. Details of the change:
2. Approval conditions:
ROLE Name DateFile ManagerExpert OpinionBiomechanical ReviewElectrical ReviewSoftware ReviewClinical ReviewTSE ExpertOutcome of Competent Authority Review(where applicable)
GRF-25-08 Rev. 16 Page 53 of 54

(Remarks: By signing this report, the reviewer confirms that he / she has no conflict of interest with the above named company (e.g. training, consultancy, financial, personal or political) that would affect the integrity of the technical review process and hence the review results.)
Approval
PRESENTED TO THE TECHNICAL REVIEW COMMITTEE MEETING ON:
Outcome of Meeting:
Follow Up required No YesIf “Yes” please provide details –
APPROVAL DEMONSTRATED by SIGNING APPROVAL AMENDMENT RECORD
CA Notified of Notified Body DecisionDevices incorporating Medicinal Substances/Human Blood Derivatives ONLY
The decision of the NSAI to grant approval / not to grant approval of the file has been communicated to the Competent Authority:
Personnel in C.A. notified of decision Date of Notification Notification performed by
If the NSAI decided to grant approval of the file despite the receipt of a negative opinion from the Competent Authority please enter the justification for this decision.
GRF-25-08 Rev. 16 Page 54 of 54