VASKÜLİTLER - baskent-adn.edu.trbaskent-adn.edu.tr/nefroloji/VASKULITLER.pdf · Sitokinler:...
77
VASKÜLİTLER Dr. Nurcan Cengiz
Transcript of VASKÜLİTLER - baskent-adn.edu.trbaskent-adn.edu.tr/nefroloji/VASKULITLER.pdf · Sitokinler:...

VASKÜLİTLER
Dr. Nurcan Cengiz

Vaskülit terimi damar duvarlarında
inflamasyonun varlığını ifade eder.
İnflamatuar infiltrasyon nötrofil, eozinofil
ya da mononükleer hücrelerden oluşabilir.
Pediatrik romatolog, pediatrik nefrolog,
kardiyolog, dermatolog.


Büyük damarlar: aorta ve onun, direkt
olarak büyük vücut bölümlerine giden
major dalları.
Orta büyüklükteki damarlar: Renal,
hepatik, kononer ve mezenterik arterler.
Küçük damarlar: İntraparankimal distal
arterler, kapillerler, venüller.
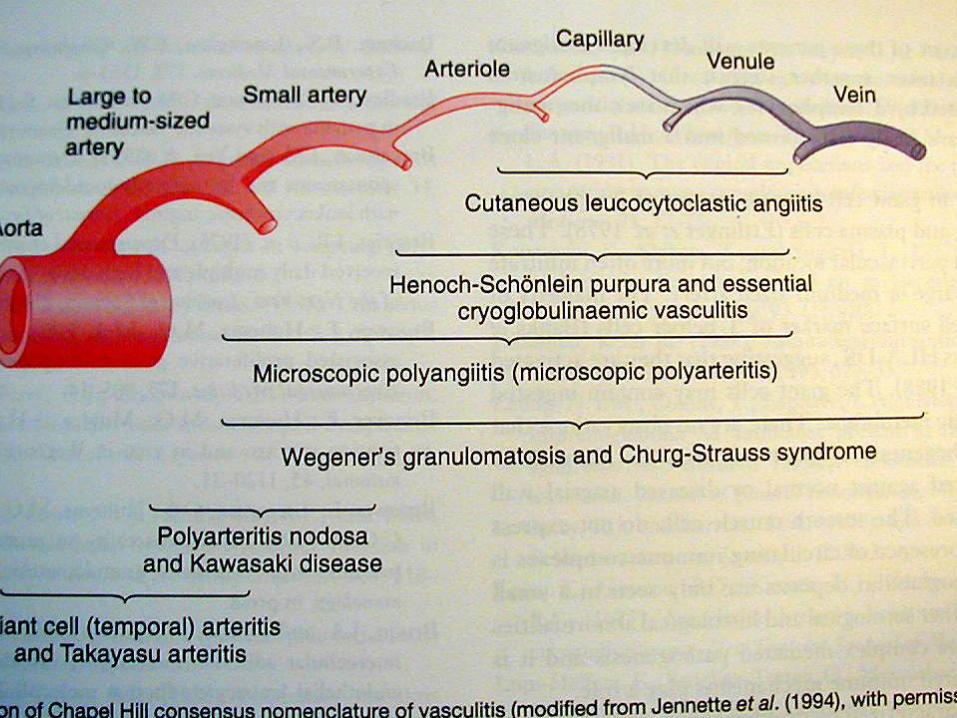

Damar boyutlarına göre vaskülitler ( 1997)
Küçük damar vasküliti
- Wegener granülomatozis
- Churg-Strauss sendromu
- Mikroskopik Polianjiitis
- Henoch-Schönlein purpura
- Esansiyel kriyoglobulinemik vaskülit
- Kutanöz lökositoklastik anjiitis
Orta çaplı damar vasküliti
- Poliarteritis nodosa
- Kawasaki hastalığı
Büyük damar vasküliti
- Dev hücreli ( temporal) arterit
- Takayasu arteriti

Chapel Hill Consensus Conference on
Nomenclature of Systemic vasculitis
Large Vessel Vasculitis
Giant cell (temporal) arteritis: Aortu ve onunmajör dallarını tutan granulomatöz arterit.Karotid arterin ekstrakranial dalları sık tutulur.(Özellikle temporal arter) Genellikel 50 yaşüzerinde görülür ve polimyalgia rheumaticaile ilişkilidir.
Takayasu’s arteritis: Genellikle 50 yaşaltında, genç kadınlarda aorta ve onun majordallarını etkiler


Medium Vessel Vasculitis
Polyarteritis Nodosa: orta ve küçük
boyutlu damarların nekrotizan
inflamasyonu
Kawasaki Hastalığı. Large medium ve
small sized arterlerin tutulumu,
mukokutanöz lenf nodu sendromu.
Koroner arterler sıklıkla tutulur ve
genellikle çocuklarda görülür.

Small – Vessel Vasculitis:
Wegener’s granulomatosis
Churg-Strauss sendrom: Solunum sisteminitutan, eosinofilden zengin granulomatözinflamasyon, küçük, orta boylu damarlarıetkileyen necrotizan vaskülitis. Astım veeosinofili ile birliktelik
Microscopic polyangiitis: Küçük damarlarıetkileyen, immun depositlerin çok az veya hiçolmadığı nekrotizan vaskülit. Necrotizanglomerülonefrit ve pulmoner kapillaritis sıktır.

Essential cryoglobulinemic vasculitis:
Küçük damarları etkileyen cryoglobulin
içeren immun depositlerle karakterize
vaskülit. Sonunda cryoglobulinler tespit
edilir. Deri ve glomerüller sıklıkla tutulur.
Kutanöz lökositoklastik angiitis.
Sistemik vaskülit ve GN olmaksızın
izole kutanöz lökositoklastik angiitis.
Henoch-Schönlein Purpura:


Vaskülit Sendromlarını düşündüren
klinik ve laboratuar bulguları
Klinik :
Ateş, kilo kaybı, yorgunluk, halsizlik
Deri lezyonları (palpabl purpura, vaskülitik
urtiker, livedo retikülaris, nodüller, ülserler)
Nörolojik bulgular (başağrısı, fokal
lezyonlar...)
Artralji, artrit, myalji, serozit
Hipertansiyon
Pulmoner infiltrasyon veya hemoraji

Laboratuar:
CRP , Sedimantasyon
Lökozitoz, anemi
Eosinofili
ANCA (+)
Factor VIII – related antigen
(Von Willebrand faktör)
Cryoglobulinemia
Dolaşan immun komplexler
Hematüri


LÖKOSİTOKLASTİK VASKULİTLER
Lökositoklastik vaskülit (LV), küçük
damarları, özellikle postkapiller venüller,
kapillerleri, arteriolleri tutan inflamasyon
ve nekroz ile karakterizedir.
Nekrotik damar duvarını nötrofiller
infiltre eder, ayrıca nükleer debrisler,
hemoraji, fibrin depositler bulunur.

IF mikroskopide immunglobulin, kompleman,
fibrin depositleri görülür. Palpabl purpura en
sık klinik bulgudur. Ödem, ürtiker, nodüller ve
büller de görülebilir.
Lökositoklastik vaskülitin en sık görülen 2
klinik formu HSP ve hipersensitivite
vaskulitidir. Bu tür vaskülit çocuklarda bazı
konnektif doku hst ve granulamatöz
hastalıklarda da nadiren görülebilir.


LV ile ilişkili hastalıklar:
- Henoch-Schönlein purpura
- Hipersensitivite angiitisi
- Kutanöz poliartriti
- ANCA ilişkili vaskülitler: WG, Mik.PA
Churg- strauss sendromu
- Konnektif doku hst: SLE,
Dermatomyozis,JRA
- Diğer: Ürtikerial vaskülit
Esansiyel mikst kriyoglobulinemi
Malignancy

Patogenez: Postkapiller venüller hasarın başlangıç yeridir. İmmun komplex depolanması patogenezin temelini oluşturur.
Antijen ve antikor komplexleri vasküler kompartmanda dolaştıktan sonra damar duvarlarında depolanmaya başlar.
Bunun sonucunda lokal kompleman aktivasyonu başlar ve adezyon molekülleri ortaya çıkar.
Bunlar nötrofil ve makrofajların endotel hücrelerine doğru migrasyon ve adezyonuna neden olur.
Bunu takiben proteolitik enzimler lökotrienler, NO ve reaktif 02 molekülleri salınarak damar duvarını ve alttaki dokuları hasara uğratır.

İmmun kompleksleri : Uzun yıllardır, damar
duvarlarında immun komplexlerin depolanmasının
vaskülitle ilişkisi bilinmektedir. Bu teoriye göre solubl
immun komplexler damar duvarlarında depolanarak
vasküler permeabilitenin artmasına neden olur.
Ayrıca kompleman yolu aktive olur. C5a gibi
kompleman yolunun ürünleri nötrofil ve makrofajlar
için kemoatraktandır ve bu hücrelerin infiltrasyonuna
neden olur.
İnflamasyon bölgesinde nötrofillerin birikimi
enzimlerin ve serbest radikallerin salınımına ve bu
yolla damar duvarı hasarına neden olur.

Otoantikorlar: Birçok otoantikor vaskülitle ilişkili bulunmuştur.
ANCA ( Antinötrofilic sitoplazmik ab)
AECA (Antiendotelial cell ab.)
ANCA’ların etanolle fixe edilmiş normal nötrofillere bağlama
paternlerine göre tipleri vardır:
- cANCA (granüler sitoplazmik): Özellikle Wegener
granülomatozisde bulunur. Proteinaz 3’e karşı oluşur.
- pANCA (perinükleer): Mikroskopik poliarteritisde tespit
edilir ve myeloperoksidaza karşı oluşur.
- ANCAnötrofilleri aktive eder, endotele adezyonlarını
artırır. Nötrofille ilişkili endotelial hasarı artırır.

ANCA(+) Hastalıklar AntiPR3(cANCA) AntiMPO(pANCA)
Wegener Granülomatozis 85 10
Mikroskopik polianjiitis 45 45
PAN 5 15
Churg-Strauss 10 60

Adezyon Molekülleri ve Sitokinler: Adezyon
moleküllerinin inflamasyonda kritik rolleri
vardır. Vasküler endotel ile lökositler arasında
etkileşimi sağlarlar. Endotel hücreleri ve
lökositlerin yüzeyinde bulunurlar. Bu
moleküller 4 yapısal gruba aittirler:
- İntegrinler - Selectinler
- Ig superailesi - kadherinler

Sitokinler: Sitokinler, normal immun sistem fonksiyonlarınıidame ettiren ve inflamatuar cevap oluşumunda kritik roloynayan değişik hücre tipleri tarafından üretilen solublproteinlerdir. Bunlar arasında TNF ve IL-1 vaskülitpatogenezinde bilhassa önemli etkileri olan sitokinlerdir.
1. Endotel hücrelerinde adezyon moleküllerini ekspresyonunartırarak lökosit adezyon, aktivasyon ve migrasyonunuilerletmek.
2. Endotel hücreleri tarafından salınan IL-6 ve IL-8’i artırarak,komşu endotel hücreleri ve lökositlerin lokal stimulasyonunkolaylaştırmak.
3. Antitrombotik protein C/S yolunun down regulasyonu vedoku faktörüprokoagulan aktivitede artış.
Sistemik vaskülitli hastalarda, serumda IL-1, IL-2, IL-6 ve TNFdüzeyleri artmıştır.


HENOCH-SCHÖNLEİN PURPURA
Çocuklarda en sık görülen sistemik vaskülittir.
Nontrombositopenik purpura, artrit veyaartralji, karın ağrısı, gastrointestinal kanamave glomerülonefrit ile karakterizedir.
Küçük damarlarda, başlıca postkapillervenüllerde IgA dominant immun depositlerbulunur.
Schönlein (1837) ve Henoch (1874)tarafından tanımlanmıştır. Ancak sendromlailgili ilk rapor 1801’de Haberden tarafından 5yaşında bir erkek çocukta bildirilmiştir.

Epidemiyoloji:
Daha çok çocukluk çağında görülen birhastalıktır.
İnsidansı 100.000’de 13.5’dir.
Sıklıkla 3-15 yaşları arasında görülür.
Erkeklerde biraz daha sıktır. E: K= 1.5:1.0
Daha çok kış aylarında ve ÜSYE’larınıtakiben oraya çıkar.
HSP’yi tetikleyen enfeksiyonlar arasında ensık hemolitik streptokoklar suçlanmış olupdiğer ilişkili ajanlar varicella, rubella, rubeola,hepatit B, parvovirüs B19, Mycoplasmapnömonia. Helicobacter pylori, staf. Aureusve campılobacterdir.
HSP nadiren, insekt bite, ilaçlar ve bazı gıdaallerjenleri ile de ortaya çıkabilir.

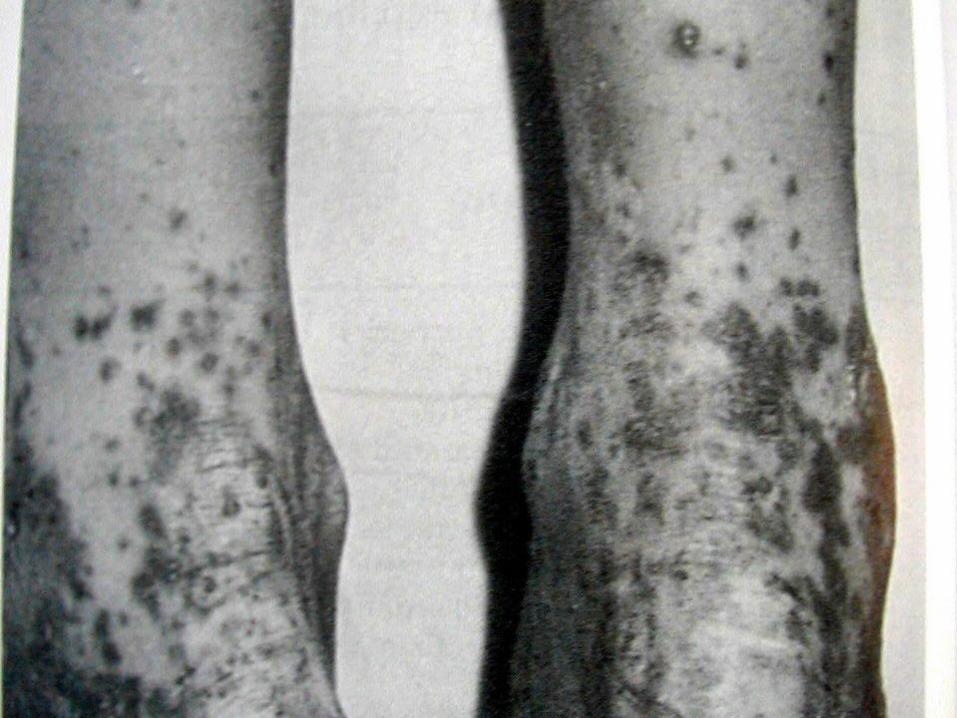
Deri Tutulumu:
Palpabl purpurik lezyonlar özellikle basınca maruz
kalan bölgelere yerleşir. Ayak bileği malleol çevresi,
bacak dorsal yüzü, gluteal bölge gibi. Yüz ve skalp
tutulumu azdır.
Akut, simetrik, eritematöz makuler veya urtikerial raş
şeklinde başlar. 12-24 saat sonra lezyonlar birleşir,
palpabl purpura oluşturur. Rengi eflatun,
kahverengiye dönüşme eğilimdedir. Geniş ekimotik
alanlarda nadiren ulserasyonlar oluşabilir. Hemorajik
buller de tanımlanmıştır.
3 yaş altındaki çocuklarda, skalp, periorbital bölge, el
ve ayakların dorsal yüzleri ve scrotumda subkutan
ödem görülebilir.
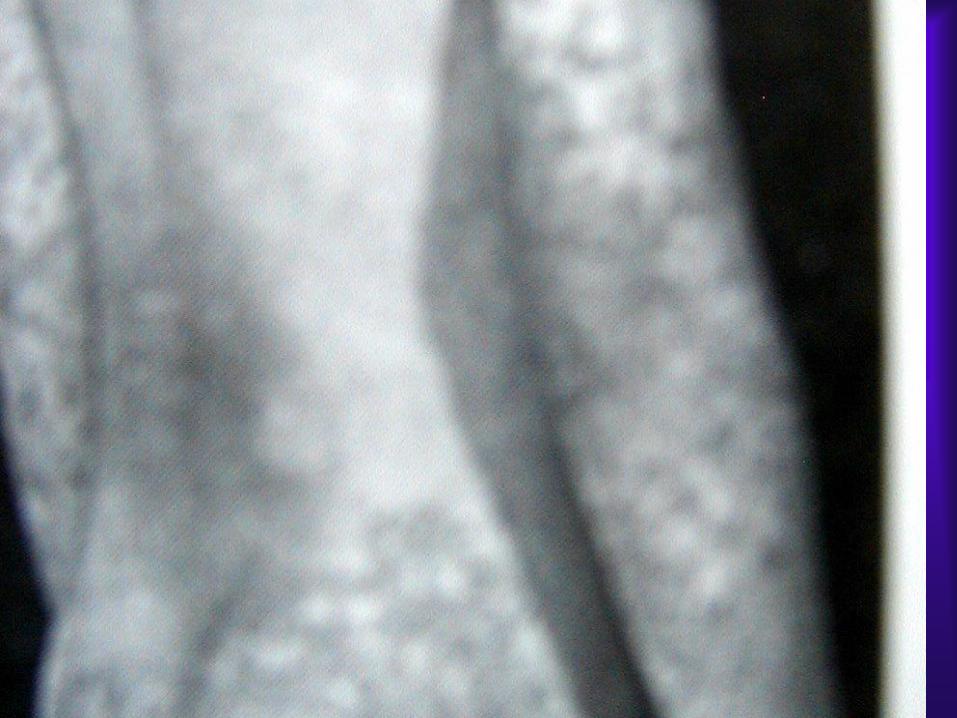

Gastrointestinal Tutulumu: Döküntülerin başlamasından
itibaren 1-4 hafta içinde yaklaşık vakaların 2/3’ünde GI
semptomlar görülür.
Submukozal ve subserozal hemoraji ve ödem nedeniyle karın
ağrısı görülür. Ağrı kolik tarzında periumblikal yerleşimlidir. Hafif
abdominal distansiyon ve kusma eşlik edebilir. Gizli veya
semptomatik GI kanama ( melena, hematemez, hematokezya)
görülebilir.
Sıklıkla kendi kendini sınırlar. Nadiren masif GI kanama
görülebilir.
%14-36 vakada bulgular akut cerrahi karını taklit edebilir.
GI tutulumunun en önemli komplikasyonu intussussepsiyondur.
(%4.6). Vakaların çoğunda ince barsakta olur.
Barsak iskemi ve infarktı, intestinal perforasyon, fistül oluşumu,
gaz ileal striktür, hemorajik pankreatit, pseudomembanöz kolit
daha nadiren görülür.

Renal Tutulum: % 20-40 vakada görülür. Renal tutulum sık görülen mikroskopik hematüri ve hafif proteinuriden daha az sıklıkla görülen nefrotik sendrom, nefritik sendrom, hipertansiyon ve böbrek yetmezliğine kadar değişir. Başlangıç yaşı >7 olanlar, persistan purpurik lezyonların varlığı, ciddi abdominal semptomları olanlar ve faktör XIII aktivitesinin düşük olması renal hastalık riskini artırmaktadır. Vakaların çoğunda renal tutulum purpuranın başlangıcından sonra 1 ay içinde gelişir.


HSP Renal tutulumunun sınıflanması
I. Minimal değişiklik
II. Pure mesengial proliferasyon (kresent )
a. Fokal
b. Diffüz
III. Mezengial proliferatif GN (Kresent oranı < %50)
a.Fokal
b.Diffüz
IV. Mezengial proliferatif GN, kresent oranı %50-75
a.Fokal
b.Diffüz
V. Mezengial proliferatif GN, kresent oranı > %75
a.Fokal
b.Diffüz
VI. Membranoproliferatif (mezengiokapiller) GN

HSP Nefritinin Klinik Sınıflaması
A. Normal: FM normal, idrar anomalisi yok, renal
fonksiyon normal
B. Minor idrar anormalliği: Hematüri (mikroskopik +
İntermitten makroskopik)ve/veya proteinüri (1g/24h),
renal fonksiyon normal.
C. Aktif renal hastalık: Proteinüri (1g/24h) ve/veya
HT renal fonksiyon normal.
D. Renal yetmezlik. GFR< 60ml/dk/1.73m2

Eklem Tutulumu: %50-80 vakada görülür. Dizler,ayak bilekleri daha az olarak wrists, dirsekler, parmakeklemleri tutulur. Periartiküler şişme ve hassasiyet,ağrı ve hareket kısıtlığı vardır. Eritem genellikleyoktur. Eklem semptomları geçicidir, rezidüel anomalibırakmaksızın birkaç gün içinde iyileşir.
Diğer Klinik Bulgular: CNS tutulumu nöbetler vekomaya neden olabilir. MR’da bilateral multifokalserebral lezyonlar görülür. Nadir komplikasyonlarsubaraknoid kanama, Guillian Barre sendromu,oküler tutulum, rekürrent epistaksis, parotitis vekardittir. Pulmoner hemoraji nadir fakat fatal seyirlidir.

Patoloji:
Cilt biyopsisinde dermal kapillerler ve postkapillervenüllerde IgA depositleri içeren lökositoklastikvaskülit görülür.
Böbrekte mezengial proliferatif GN’den kresentoluşumuna kadar değişen bulgular vardır. Epitelialhücrelerin proliferasyonu kresent oluşumu ilesonuçlanır.
IF mikroskopide, mezengium ve periferal kapillerinduvarında IgA depolanması görülür. IgA depositleribaşlıca IgA1 subklasında olur. Ayrıca depositler C3,IgG, properdin, fibrin içerir.



Laboratuvar Bulguları: Laboratuvar
bulguları diagnostik değildir. Trombosit
sayısı normal veya artmış, orta
derecede trombositoz hafif normokrom
anemi olabilir.


Tedavi:
HSP tedavisi supportive tedavidir. Hidrasyon, beslenme ve
elektrolit dengesine dikkat edilir. Ağrı basit analjeziklerle
(asetaminofen) kontrol edilir.
Glukokotrikoidler eklem ve cilt bulgularını dramatik bir şekilde
düzeltmekle beraber bu bulguların tedavisinde kullanılması
endike değildir. Sistemik steroidler GI tutulumu ve kanaması
olan hastalarda endikedir. Prednison 1-2 mg/kg 7 gün kullanılıp
2-3 haftada azaltılarak kesilir. Bazı araştırmacılar kısa süreli
steroid kullanımının renal tutulumu önleyip önlemediği üzerinde
durmuşlardır ancak sonuçlar değişkendir.
Ciddi renal tutulumu olan hastalar da steroidle tedavi edilmelidir.
Puls metil prednisolon ciddi vakalarda kullanılabilir. Erken
dönemde kullanılırsa etkilidir.
Prednison, siklofosfamid ve heparin, varfarin ya da dipridamol
ile kombine tedaviler de ağır vakalarda etkilidir.
IVIG ve plazma exchange ile iyisonuçlar bildirilmiş olmakla
beraber geniş çalışmalar yoktur.
Persistan proteinürisi olan hastalara ACE inhibitörleri
kullanılabilir.

Prognoz:
Vakaların 2/3’ünde genellikle 1 ay içinde iyileşme görülür. Küçük
çocuklarda büyüklere göre daha kısa ve iyi seyirli, rekürrensler
daha azdır.
%16-40 vakada en az 1 rekürrens bildirilmiştir. Epizodlar
genellikle rash ve karın ağrısı ile karakterize ve ilk ataktan daha
hafif ve kısa sürelidir. Rekürrenslerin büyük kısmı ilk 6 haftada
görülürken başlangıçtan 2 yıl sonrasına kadar da olabilir.
Epizodlar spontan olabildiği gibi ÜSYE ile birlikte de ortaya
çıkabilir.
Prognoz genellikle iyidir ve supportive tedavi yeterlidir. Önemli
morbiditeler akut dönemde GIS tutulumuna, uzun dönemde ise
renal tutuluma bağlı ortaya çıkar.
Renal tutulumu olan çocukların prognozu değişken olmakla
beraber klinik ve histopatolojik bulgularla koreledir. %1-5 vakada
son dönem böbrek yetmezliğine progresyon bildirilmiştir. Renal
tx başarılıdır, rekürrens ve graft kaybı sık değildir.


HİPERSENSİTİVİTE VASKÜLİTİ
İlaçlara ve diğer antijenlere (örn;infeksiyonlar) karşı
hipersensitiviteye bağlı polianjiitis çocukluk çağı
küçük damar vaskülitlerinin en önemli
nedenlerindendir.
Penisilinler, sulfonamidler, kinolonlar, allopurinol,
tiazid diüretikleri, NSAIDs, fenitoin gibi ilaçların
hipersensitiviteye yol açma potansiyeli vardır.
Streptokinaz, rekombinan growth hormon, sitokinler
ve monoklonal antikorlar da immun kompleks
formasyonu ve vaskülite neden olabilir. Hepatit B ve
C, HIV ve streptokoklar da benzer tabloya neden
olabilir.
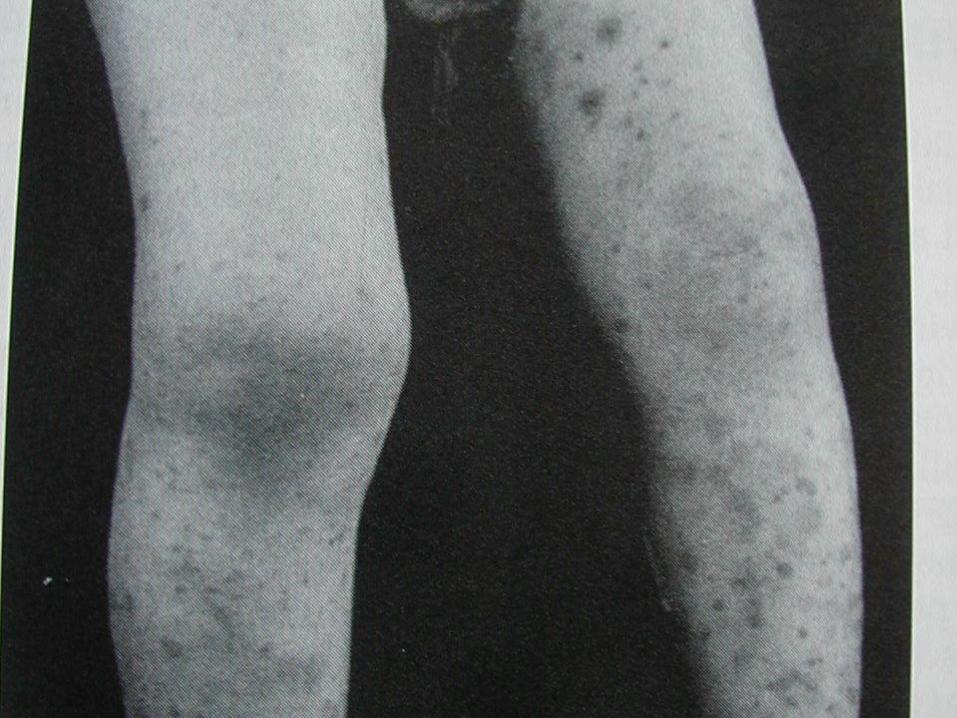

Hipersensitivite vasküliti antijenle karşılaşmadan 7-14
gün sonra ateş, artralji, myalji, LAP ve rash gibi
bulgularla başlar. Cild bulguları palpabl nodül, ürtiker
ve purpurayı içerir. Lezyonlar daha çok bacaklarda
yerleşmiştir. Diğer organ tutulumları nadirdir.
HSP ile ayırıcı tanısı bazen güç olabilir. Lökositoz,
eozinofili, dolaşan immun kompleksler ve ESR artar,
C3 azalır. Cilt biyopsisinde lökositoklastik vaskülit
bulguları vardır.

Tedavi: Hastalık sıklıkla akut ve self-limitingözelliktedir. Ancak bazen relapslar ve kronikseyir gösterebilir.
Sorumlu ajanın uzaklaştırılması tedavide ilkbasamaktır.
Antihistaminikler ve NSAIDs kutanözsemptomları ve artraljiyi düzeltmedeyeterlidir.
Ciddi sistemik vaskülit bulguları varsa steroidendikasyonu vardır.


POLİARTERİTİS NODOSA
PAN deri abdominal organlar, böbrekler, CNS
ve kasların vasküler inflamasyonu sonucu
oluşan bir multisistem hastalığı iken CPA
sadece deri, kaslar, eklemler ve sinirleri tutar.
Klasik PAN 1866’da Kusmaull ve Meier
tarafından tanımlanmıştır. Küçük ve orta
boyutlu arterlerin nekrotizan inflamasyonu
olup tutulan arterlerde irregüler olarak nodül
ve anevrizmalar vardır.

ACR PAN Tanı Kriterleri (1990)
Lighfoot kriterleri
Kilo kaybı
BUN veya cre yüksekliği
Livedo retikülaris
HbsAg(+)
Testis ağrısı
Arteriografik değişiklikler
Myalji
Biyopsi
Mono ve polinöropati
Diastolik KB>90

PAN Tanı kriterleri (Özen ve ark)
(ANA, AntiDNA (-) olmak koşulu ile)
Major: Renal hastalık
Muskuloskeletal bulgular
Minör: Cilt bulguları Kardiak hast
GİS tutulumu Akc hast
Periferal nöropati Akut faz reaktanları
CNS tutulumu HbsAg(+)
Hipertansiyon
Konstitusyonel semptomlar


Epidemioloji: Nadir görülen, her yaş grubunu
tutabilen bir hastalıktır. En çok 5. ve 6. dekadlarda
ortaya çıkan bir hastalıktır.
Kız ve erkek çocukları eşit olarak etkiler.
Küçük yaşlarda da görülebilmekle beraber pik yaptığı
yaşlar 9-11 yaşlardır.
Hastalığın nedeni bilinmemekle beraber ÜSYE,
streptokokal enf ve hepatit B sonrası ortaya çıkması
nedeni ile duyarlı bireylerde postenfesiyöz otoimmun
bir cevap olarak geliştiği düşünülmektedir.
Enfeksiyöz mononükleoz, tbc, CMV ve parvovirus
enfeksiyonları da PAN ile ilişkili bulunmuştur.

Patoloji: Küçük ve orta boyutlu arterlerin tüm
tabakalarında lenfosit infiltrasyonu ve nekrotizan
vaskülit görünümü vardır.
Hafif inflamasyondan yoğun fibrinoid nekroz, tromboz
ve infarktlara kadar değişen görünümleri içerir.
Anevrizma oluşumu sıktır.
Renal arter tutulumu sıktır.
Glomerüler tutulum değişkendir.


Klinik: Klinik prezentasyon değişkendir. Tutulan damarların
lokalizasyonuna göre bulgular değişebilir.
Semptomlar başlamadan önce nedeni bilinmeyen ateş
başlayabilir.
Ciddi abdominal ağrı, mezenterik arterial tutulumu hatta
trombozu düşündürür.
Renovasküler tutulum hipertansiyon, hematüri ve proteinüriye
neden olur.
Ciltte purpura, ödem, tutulan arterlerin seyri boyunca palpabl
nodüller görülür. CNS’de transient iskemik atak, psikoz, periferal
nöropati (parestezi ve güçsüzlük) görülür.
Kalpte myokardial iskemi, myokardit, buna bağlı kalp yetmezliği,
perikardit ve aritmiler olabilir.
Daha az sıklıkta testiküler ağrı, kemik ağrıları, körlüğe yol açan
retinal arteritis, artralji, artrit, myalji görülebilir.


Laboratuvar:
ESR yüksek, anemi, lökositoz,
hipergamaglobulinemi vardır.
Renal tutulum varlığında hematüri ve
proteinüri vardır.
KCFT yüksekliği Hepatit B
enfeksiyonunu düşündürür ve erişkinde
çocuklardan daha sıktır.

Tanı: Biyopsi ve anjiografide vaskülitbulgularının gösterilmesi ile konur.
Renal biyopside necrotizan arteritisgörülür.
Anjiografide tutulan arterlerdesegmental stenoz ve anevrizmaldilatasyonlar görülür.
Renal ve mezenterik arterler sıklıklatutulur.

Ayırıcı tanı: HSP, WG, Good Pasture send, SLE,
Malignensi
Tedavi: Oral ve IV steroidler kullanılır. Oral ve IV
siklofosfamid birlikte kullanılabilir. Ekstremitelerin
vasküler yetmezliği ile sonuçlanan endarterit
varlığında İloprost (prostasiklin analoğu) kullanılabilir.
Prognoz: Değişkendir. Hafif hastalıkta daha az
komplikasyon gelişir ve prognoz iyidir. Ancak ölüme
kadar giden multiorgan tutulumları olabilir. Agresif
immunsupresif tedavinin prognoz üzerine olumlu
etkisi vardır.



WEGENER GRANULOMATOZİS
Küçük, orta çaplı damarları etkileyen nekrotizanvaskülite respiratuar trakt tutulumunun eşlik ettiğigranülomatöz inflamasyonla karakterizedir.
Üst solunum yolu bulguları: Hastaların %90’ında ÜSYhastalığı görülür. Burunda inflamasyon veülserasyona neden olduğunda epistaksis, rinore venazolakrimal kanal tıkanıklığı görülür. Yumuşak dokunekrozunu kıkırdak destrüksiyonu izler. Karakteristikeğer burnu deformitesi ortaya çıkar. Yüz ağrısı vepürülan sinüzit semptomları, otitis media ve ileti tipiişitme kaybı sık görülür. İnflamasyon larinks, trakeave büyük hava yollarını tutabilir.

Pulmoner hastalık: Vakaların %90’ında tanısırasında alt solunum yolu bulguları vardır.Semptomlar nonspesifik olup öksürük,dispne, hemoptizi, plörezi görülebilir.
Akciğer biyopsi bulguları:
Major:1. Parankimal nekroz ve nötrofilikmikroabseler 2. vaskülit 3. mikst inflamatuarinfiltratın eşlik ettiği granulomlar
Minör:1. interstisyel fibrozis 2. alveolerkanama 3. eozinofili 4. bronşial lezyonlar
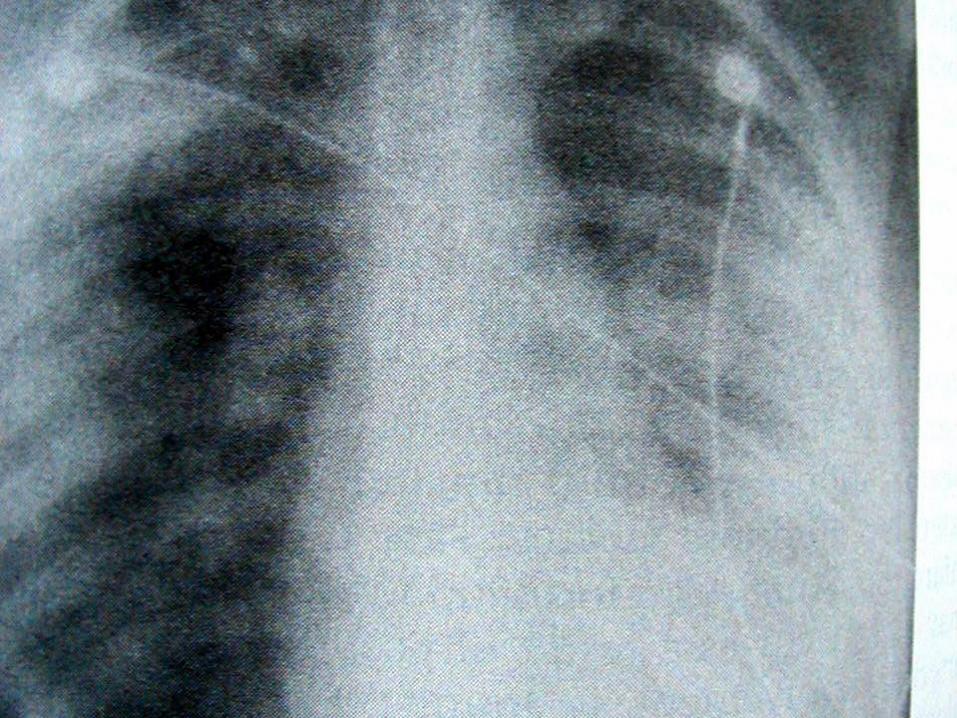

Solunum sistemi dışı granülomatöz
inflamasyon: Böbrek dahil olmak üzere
birçok sistem tutulabilir. Hipofiz
tutulumu, vertebra, cilt, orbita,
mediasten ve meme dokusu tutulumları
bildirilmiştir.

CHURG-STRAUSS SENDROMU
1951 yılında Churg ve Strauss tarafından PAN’ınbir varyantı olarak tanımlanmıştır.
Ekstrarenal hastalık: Geç başlangıçlı astımlakarakterizedir. Göğüs grafisinde geçici infiltratlarsıktır. Bol eozinofil içeren plevral efüzyonlar görülür.Solunum sistemine ait semptomlar vaskülitten yıllarcaönce görülebilir. Eozinofili sıktır. Vaskülit bulgularıözellikle kalp, cilt, barsaklar, kasiskelet ve sinirsistemlerini etkiler. Mononöritis multiplex 2/3 vakadagörülür. Optik nöropati ve retinal arter oklüzyonu,ventrikül disfonksiyonu ve aritmiler, ciddi alveolerkanamalar görülebilir.
Renal tutulum: Mikroskopik hematüri, granülersilendirler, proteinüri, nefrotik sendrom, böbrekyetmezliği bildirilmiştir. Histolojik patern fokalsegmental glomerülonefrittir.

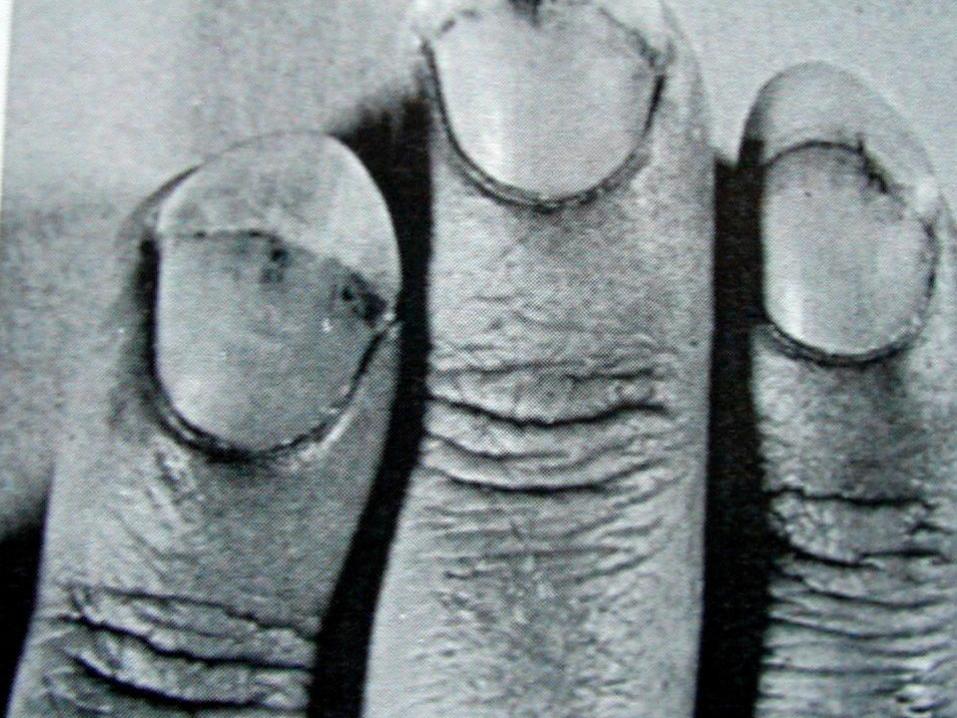
MİKROSKOPİK POLİANJİİTİS
Mikroskopik polianjiitis granülomatöz inflamasyon
olmaksızın, küçük damarları tutan ve immun
birikimlerin görülmediği nekrotizan vaskülittir.
Klinik: Kırgınlık, kilo kaybı, ateş ve grip benzeri
semptomlarla başlar.
Cilt tutulumu: Palpabl purpura, izole nekrotik
lezyonlar, tırnak yatağı infarktları ve splinter
hemorajiler sıktır.
Kas ve eklem tutulumu: Artrit, artralji, myalji
Gastrointestinal tutulum: Karın ağrısı, ishal,
kanama ve perforasyon görülebilir.Ağız mukozasında
ağır ülserasyonlar.

SSS tutulumu: Beyin, kranial ve periferik sinirleri
etkileyen nekrotik vaskülit gelişebilir. Konvülziyonlar,
fokal nörolojik defisid gelişebilir. Asimetrik
mononöritis multipleks ve distal duyusal nöropati ile
karakterize periferik sinir tutulumu santral bulgularda
daha fazla görülür.
Göz bulguları: Konjonktivit, episklerit, korneal
ülserasyonlar,üveit,retinal vaskülit, orbital tutulum
Kardiak tutulum: Disritmi, kalp yetmezliği, dilate
kardiomyopati, perikardit, valvüler lezyonlar.
Böbrek hastalığı: Sık görülür. Mikroskopik
hematüri, proteinüri, eritrosit vr granüler silendirler
böbrek hastalığının habercileridir. Hastalık ilerledikçe
böbrek fonksiyonları hızla bozulur. Hipertansiyon
olabilir. Hızlı ilerleyen GN ve ekstrakapiller
proliferasyon ve kresent oluşumu olabilir.

KAWASAKİ HASTALIĞI
(MUKOKUTANÖZ LENF NODU SENDROMU)
Genellikle 5 yaşın altında görülen,
etiyolojisi bilinmeyen, LAP,rash, el ve
ayaklarda ödem ve diffüz mukozal
inflamasyonla karakterize bir vaskülit
sendromudur. Hastalığın en önemli
tutulum yeri koroner arterler olup %15-
20 vakada görülür. Hastalığın tanısı 5
kriterin varlığı ile konur.

Kawasaki hast. tanı kriterleri
1. Beş gün ve daha uzun süren nedeniaçıklanamayan ateş
2. Bilateral eksudatif olmayan konjonktival konjesyon
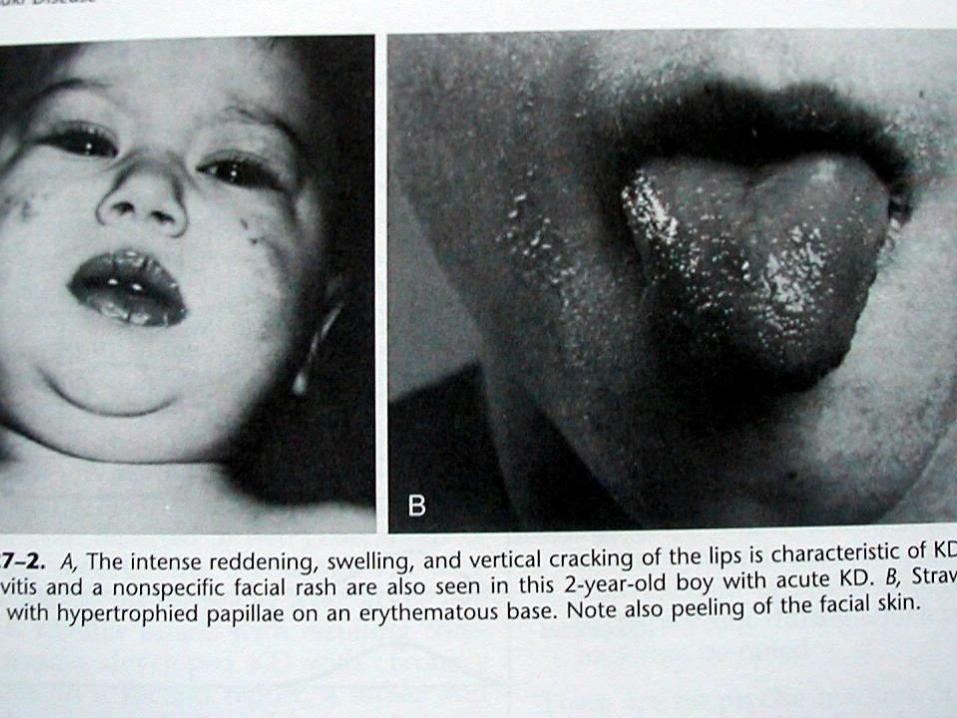
3. Orofarengeal mukozal eritem, kırmızı veya kuruçatlak dudaklar, çilek dili
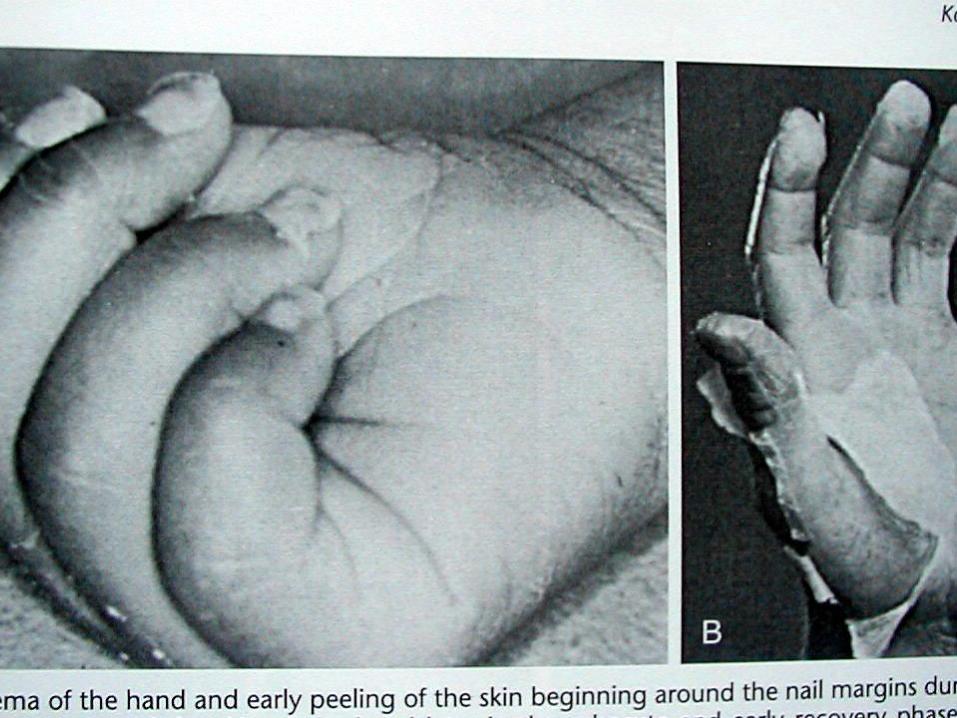
4. Avuç içleri, ayak tabanlarında eritem, eller veayaklarda endürasyon, ödem ve periungualdeskuamasyon
5. Polimorf döküntü (daha çok gövdede, vezikülerdeğil)
6. Akut nonsüpüratif sevikal LAP (genellikle tektaraflı, en az 1.5 cm çaplı bir veya daha fazla lenfnodu)