
TRabalho de Bioquimica
13
A Doença de Fabry Nosso organismo é feito de células, milhões delas. As células se multiplicam, vivem e morrem diariamente, sendo assim constantemente renovadas. Quando uma célula morre, é imediatamente destruída e seus componentes são reutilizados pelo organismo. Nessa reciclagem dos componentes das células é que se encontra o problema que causa os sintomas da Doença de Fabry. Quando uma célula é destruída, seus componentes são fagocitados ("comidos") por outras células, chamadas macrófagos, que são parentes dos glóbulos br ancos do sangue. Dentro dos macrófagos, o material da célula morta fica dentro de uma espécie de "bolha", chamada lisossomo. No lisossomo, a célula introduz enzimas, que são substâncias que "digerem" os restos celulares, quebrando substâncias grandes em pa rtes menores, que podem ser reutilizadas. As enzimas que agem nos lisossomos são fabricadas pela própria célula, seguindo uma "receita" que fica no núcleo: os genes. Se uma pessoa sofre alguma alteração em um gene, dizemos que ocorreu uma mutação. Quando ocorre uma mutação em um gene, a "receita" fica alterada e a enzima que é fabricada com base nesta receita alterada, geralmente não funciona. A doença de Fabry é uma doença hereditária causada por um gene deficiente do organismo. Por causa desse erro na estrutura genética do organismo, uma enzima essencial é produzida em quantidade insuficiente ou com uma estrutura que não funciona adequadamente. Sem esta enzima, certas substâncias (basicamente uma espécie de gordura chamada globotriaosilceramida ou GL-3), que deveriam ser removidas do organismo, permanecem nas células. O resultado é um acúmulo desse material. É esse depósito progressivo que causa a maioria dos problemas que aparecem na doença de Fabry. Com o tempo, à medida que o GL-3 vai se acumulando nas paredes dos vasos sangüíneos e em outros tecidos, o funcionamento do organismo sofre um grande prejuízo. Como o processo se dá nos vasos sanguíneos do corpo inteiro, os principais sistemas e órgãos, como o coração, o rim e o cérebro, deixam de funcionar devidamente, gerando problemas que podem acarretar risco de vida. Quando os sinais e sintomas da doença de Fabry são reconhecidos no início, os médicos são capazes de tratar melhor deles, e o resultado é uma melhora na qualidade de vida. Por isso é tão importante que seu médico o examine se você tiver algum sintoma sugestivo (especialmente se há algum caso da doença na sua família); e se você tiver a doença de Fabry, é importante passar por um exame feito por médico que entenda bem da doença. O que causa a Doença de Fabry? A doença de Fabry se estabelece porque o corpo é incapaz de produzir uma enzima denominada alfa-galactosid ase (alfa-galactosidase ou alfa-GAL) em quantidade ou estrutura adequada para realizar a sua função. O que a enzima alfa-GAL faz normalmente? A enzima alfa-GAL é uma das inúmeras enzimas normalmente presentes no lisossomo, que é uma estrutura que existe nas nossas células e parece funcionar como uma usina de reciclagem. A tarefa da enzima é quebrar certas substâncias para que possam ser reutilizadas pela célula ou eliminadas do corpo. Sem a enzima alfa-GAL em quantidade suficiente para realizar a tarefa adequadamente, aquelas substânci as (basicamente a GL-3) se acumulam nos lisossomos, principalmen te nas células encontradas nas paredes dos vasos sangüíneos. A doença de Fabry recebe o nome de doença de depósito lisossômico por causa desse acúmulo progressivo de substâncias nos lisossomos. Quais são os sintomas principais da Doença de Fabry? Como a GL-3 se acumula nas paredes dos vasos sangüíneos do corpo inteiro, os sintomas da doença são vários, diferentes e, caracteristicamente, pioram com o passar do tempo. Os sinais e sintomas mais comuns são citados abaixo. Os médicos conseguem tratar alguns dos sintomas, mas estes tratamentos não são dirigidos para a causa principal da doença, que é a falta da enzima e o depósito de GL-3 que resulta disso. Ao analisar a lista abaixo, lembre- se que nem todas as pessoas com doença de Fabry apresentam os mesmos sinais e sintomas, e que estes podem variar à medida que a doença progride e de pessoa para pessoa. A ausência de sintomas no momento não significa que eles não vão aparecer mais tarde. Dor e fadiga De modo geral, a dor é considerada o primeiro e o mais comum de todos os sintomas da doença de Fabry. Provavelmente a dor é causada pelo comprometimento do sistema nervoso, como resultado do depósito de GL-3; muitas vezes a dor é o primeiro sintoma que faz com que os pacientes sejam levados a um pediatra. Em muitos casos, a dor é causada por mudança do tempo, exposição a temperaturas altas, febre ou calor excessivo , estresse ou fadiga. A maioria das pe ssoas com a doe nça tem dois tipo s de dor: acro parestesia e “ crises de Fabr y”.
-
Upload
samuel-fernandes -
Category
Documents
-
view
43 -
download
0
Transcript of TRabalho de Bioquimica

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 1/13
A Doença de Fabry
Nosso organismo é feito de células, milhões delas. As células se multiplicam, vivem e morrem diariamente, sendoassim constantemente renovadas. Quando uma célula morre, é imediatamente destruída e seus componentes sãoreutilizados pelo organismo. Nessa reciclagem dos componentes das células é que se encontra o problema que causaos sintomas da Doença de Fabry.
Quando uma célula é destruída, seus componentes são fagocitados ("comidos") por outras células, chamadasmacrófagos, que são parentes dos glóbulos brancos do sangue. Dentro dos macrófagos, o material da célula morta ficadentro de uma espécie de "bolha", chamada lisossomo. No lisossomo, a célula introduz enzimas, que são substânciasque "digerem" os restos celulares, quebrando substâncias grandes em partes menores, que podem ser reutilizadas.
As enzimas que agem nos lisossomos são fabricadas pela própria célula, seguindo uma "receita" que fica no núcleo: osgenes. Se uma pessoa sofre alguma alteração em um gene, dizemos que ocorreu uma mutação. Quando ocorre umamutação em um gene, a "receita" fica alterada e a enzima que é fabricada com base nesta receita alterada, geralmentenão funciona.
A doença de Fabry é uma doença hereditária causada por um gene deficiente do organismo. Por causa desse erro naestrutura genética do organismo, uma enzima essencial é produzida em quantidade insuficiente ou com uma estruturaque não funciona adequadamente.
Sem esta enzima, certas substâncias (basicamente uma espécie de gordura chamada globotriaosilceramida ou GL-3),que deveriam ser removidas do organismo, permanecem nas células. O resultado é um acúmulo desse material. Éesse depósito progressivo que causa a maioria dos problemas que aparecem na doença de Fabry.
Com o tempo, à medida que o GL-3 vai se acumulando nas paredes dos vasos sangüíneos e em outros tecidos, ofuncionamento do organismo sofre um grande prejuízo. Como o processo se dá nos vasos sanguíneos do corpo inteiro,os principais sistemas e órgãos, como o coração, o rim e o cérebro, deixam de funcionar devidamente, gerandoproblemas que podem acarretar risco de vida.
Quando os sinais e sintomas da doença de Fabry são reconhecidos no início, os médicos são capazes de tratar melhordeles, e o resultado é uma melhora na qualidade de vida. Por isso é tão importante que seu médico o examine se vocêtiver algum sintoma sugestivo (especialmente se há algum caso da doença na sua família); e se você tiver a doença deFabry, é importante passar por um exame feito por médico que entenda bem da doença.
O que causa a Doença de Fabry? A doença de Fabry se estabelece porque o corpo é incapaz de produzir uma enzima denominada alfa-galactosidase(alfa-galactosidase ou alfa-GAL) em quantidade ou estrutura adequada para realizar a sua função.
O que a enzima alfa-GAL faz normalmente?
A enzima alfa-GAL é uma das inúmeras enzimas normalmente presentes no lisossomo, que é uma estrutura que existenas nossas células e parece funcionar como uma usina de reciclagem. A tarefa da enzima é quebrar certassubstâncias para que possam ser reutilizadas pela célula ou eliminadas do corpo. Sem a enzima alfa-GAL emquantidade suficiente para realizar a tarefa adequadamente, aquelas substâncias (basicamente a GL-3) se acumulamnos lisossomos, principalmente nas células encontradas nas paredes dos vasos sangüíneos. A doença de Fabryrecebe o nome de doença de depósito lisossômico por causa desse acúmulo progressivo de substâncias noslisossomos.
Quais são os sintomas principais da Doença de Fabry?
Como a GL-3 se acumula nas paredes dos vasos sangüíneos do corpo inteiro, os sintomas da doença são vários,diferentes e, caracteristicamente, pioram com o passar do tempo. Os sinais e sintomas mais comuns são citadosabaixo. Os médicos conseguem tratar alguns dos sintomas, mas estes tratamentos não são dirigidos para a causaprincipal da doença, que é a falta da enzima e o depósito de GL-3 que resulta disso. Ao analisar a lista abaixo, lembre-se que nem todas as pessoas com doença de Fabry apresentam os mesmos sinais e sintomas, e que estes podemvariar à medida que a doença progride e de pessoa para pessoa. A ausência de sintomas no momento não significaque eles não vão aparecer mais tarde.
Dor e fadiga
De modo geral, a dor é considerada o primeiro e o mais comum de todos os sintomas da doença de Fabry.Provavelmente a dor é causada pelo comprometimento do sistema nervoso, como resultado do depósito de GL-3;muitas vezes a dor é o primeiro sintoma que faz com que os pacientes sejam levados a um pediatra. Em muitos casos,a dor é causada por mudança do tempo, exposição a temperaturas altas, febre ou calor excessivo, estresse ou fadiga. A maioria das pessoas com a doença tem dois tipos de dor: acroparestesia e “crises de Fabry”.

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 2/13
A acroparestesia geralmente se apresenta como uma dor que afeta basicamente as mãos e os pés. Édescrita como uma dor ardente acompanhada de formigamento, que incomoda freqüentemente. Em geralessa dor aparece de forma intermitente ou diariamente. As “crises de Fabry” geralmente se apresentam como episódios de dor intensa, insuportável e ardente,inicialmente nas mãos e nos pés e que se irradia para outras partes do corpo. São crises debilitantes quepodem durar desde alguns minutos até alguns dias.
Muitas pessoas que têm a doença de Fabry acham que tratar da dor e da fadiga associadas com a doença significaque vão conseguir voltar a todas as atividades que realizavam normalmente. Em geral, isso implica evitar certasatividades, preparar-se para condições variáveis de clima, aumentar a ingestão de água ou líquidos, “economizar”energia ou tirar sonecas freqüentes. Existem medicações que trazem algum alívio, mas não resolvem a causa doproblema.
Transpiração prejudicada
Devido ao depósito de GL-3 nas glândulas sudoríparas (glândulas que produzem suor) e ao dano causado nos nervos,a maioria das pessoas que têm doença de Fabry transpira muito pouco (hipohidrose) ou simplesmente não transpira(anidrose). Isso pode causar febres freqüentes, superaquecimento com exercícios físicos e intolerância ao climaquente.
Erupção cutânea ou alteração da pele
A erupção cutânea roxo-avermelhada característica é o sinal mais visível da doença de Fabry e é conhecida comoangioqueratoma. Os angioqueratomas (“angio” se refere aos vasos sangüíneos e “queratoma” significa caloso ouendurecido) são encontrados comumente nas regiões que vão do umbigo até os joelhos e, em alguns casos, nos locaisde extensão (como os cotovelos ou joelhos). Geralmente, aparecem na adolescência e, muitas vezes, é por eles que apessoa é diagnosticada. Os angioqueratomas podem se apresentar com diversas dimensões, desde o tamanho deuma ponta de alfinete até alguns milímetros, e podem ser removidos por tratamento com laser de argônio. Como todosos sintomas da doença de Fabry, em última análise, os angioqueratomas resultam do acúmulo de GL-3.
Padrão corneano ou alteração ocular
Às vezes, a córnea das pessoas com doença de Fabry apresenta um desenho semelhante aos raios de uma roda debicicleta. Isso pode ser detectado por meio de um simples exame de olho chamado oftalmoscopia de lâmpada defenda, que quase todos os médicos de olhos (oftalmologistas) fazem. Esse desenho também é denominado córnea
verticilata e resulta da deposição de GL-3 nos vasos sangüíneos do olho. É por ele que algumas pessoas sãodiagnosticadas. A alteração corneana não afeta a visão.
Problemas gastrointestinais
Muitas pessoas com doença de Fabry têm problemas gastrointestinais tais como dor após ingerir uma refeição, diarréiae náusea; provavelmente em decorrência do comprometimento do sistema nervoso. Para ajudar a tratar dessessintomas, alguns médicos recomendam uma dieta pobre em gorduras ou receitam uma medicação para ser tomadaantes das refeições.
Prejuízo ou dano de órgãos e sistemas importantes
O depósito contínuo de GL-3 faz com que o caminho dos vasos sangüíneos fique estreitado com o passar do tempo.Isso significa que os rins, o coração e o cérebro não recebem alimento necessário para funcionar adequadamente. O
resultado disso é que as pessoas com doença de Fabry podem apresentar prejuízos muito sérios em órgãos esistemas importantes, chegando a apresentar risco de vida. Os problemas mais comuns são:
Problemas renais
diminuição da função renal (mostrada por excesso de proteína na urina) evolução para insuficiência renal
Uma diminuição leve da função renal pode ser tratada em parte com uma dieta pobre em sódio (sal) e proteínas. Ainsuficiência renal grave é tratada com diálise crônica ou então com transplante.
Problemas cardíacos
aumento do tamanho do coração

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 3/13
válvulas cardíacas defeituosas batimentos cardíacos irregulares ataque cardíaco insuficiência cardíaca
Alguns problemas cardíacos podem ser tratados com um marca-passo ou com cirurgia de revascularização miocárdica(ponte de safena).
Problemas do Sistema Cerebrovascular e do Sistema Nervoso Central
tontura ou vertigem dor de cabeça AVC (acidente vascular cerebral) ou derrame precoce
Se houver o risco de um derrame, seu médico pode receitar anticoagulantes (para afinar o sangue).
Fatores emocionais
Muitas pessoas com doença de Fabry têm depressão, desesperança, alienação e negação dos próprios sintomas.Talvez você ache conveniente conversar com mais alguém da comunidade Fabry, ou seja, alguém em situaçãosemelhante, com preocupações e temores similares aos seus.
O seu médico pode ajudá-lo a lidar com essas questões e também com assuntos relacionados com a transmissão dadoença para os seus filhos.
Quando aparecem os primeiros sintomas?
Muitas pessoas apresentam os sintomas da doença de Fabry no início da infância, em geral entre 6 e 9 anos. Noentanto, freqüentemente os sinais e sintomas da doença não são bem compreendidos pelo médico, chegando até aserem deixados de lado.
Como as pessoas com doença de Fabry são diagnosticadas? Talvez você não seja capaz de dizer se uma pessoa tem doença de Fabry simplesmente olhando para ela. Mas adoença pode ser diagnosticada em qualquer época, inclusive antes do nascimento. Os sinais e sintomas geralmente seapresentam na infância e só precisam ser reconhecidos. Os médicos e outros profissionais da saúde podem confirmaro diagnóstico por meio da dosagem da enzima (análise de sangue que mede a atividade da enzima alfa-GAL) e poranálise de mutação genética (análise de DNA).
Pergunte ao seu médico a respeito da necessidade de você ou alguém da sua família fazer um teste para detectar adoença de Fabry.
Como a doença de Fabry é transmitida?
A doença de Fabry é uma alteração hereditária. É uma herança recessiva ligada ao cromossomo X, o que significa que
o gene deficiente está localizado no cromossomo X. Para compreender melhor isto, saber um pouco de genética ajuda. Cada um de nós herda um cromossomo X da nossamãe e um cromossomo X ou Y do nosso pai. Quem herda um cromossomo X do pai é do sexo feminino (XX) e quemherda um cromossomo Y do pai é do sexo masculino (XY). Como o gene deficiente que causa a doença de Fabry estálocalizado no cromossomo X, tanto os homens como as mulheres podem ter esse gene (já que ambos têm pelo menosum cromossomo X).
Como o único exemplar do gene que os homens têm é deficiente, eles vão transmitir o gene a todas as filhas, mas anenhum dos filhos. Como as mulheres têm um gene deficiente e um gene normal, em cada gravidez elas têm 50% deprobabilidade de transmitir o gene deficiente tanto às filhas como aos filhos.
Um geneticista pode ajudar você a fazer um mapeamento do padrão de hereditariedade da sua família, utilizando umheredograma (árvore genealógica). Isso pode ajudar você a tomar decisões acertadas a respeito de planejamento
familiar e da possibilidade de transmitir a doença. O heredograma também pode ajudar você a entender como adoença de Fabry afetou seus parentes, tanto vivos como falecidos.

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 4/13
Quem pode herdar a doença de Fabry?
A doença de Fabry pode afetar qualquer pessoa que herdar o gene deficiente. Isso significa indivíduos do sexomasculino ou feminino de qualquer grupo étnico, filho de pai doente ou de mãe portadora (muitas vezes sem o saber)do gene deficiente. A manifestação dos sintomas da doença de Fabry depende da atividade da enzima a-GAL em cadapessoa.
As mulheres, que possuem um gene normal e um gene deficiente, podem apresentar qualquer nível de atividade daenzima a-GAL e, assim, não terem sintomas, ou terem sinais e sintomas leves ou graves.
Por outro lado, os homens têm sempre a atividade da enzima a-GAL diminuída e, por esta razão, apresentam maissinais e sintomas que as mulheres. Porém a gravidade destes sintomas é variável entre os pacientes.
Como a doença de Fabry é hereditária, se uma pessoa da família tiver a doença, é provável que outras também atenham. Por isso é tão importante montar o heredograma.
Se você tem a doença de Fabry, é importante consultar um médico que conheça bem a doença. Ligue para aGenzyme, que o ajudará a encontrar um especialista próximo de você.
As informações contidas neste material não substituem conversas periódicas com seu médico.
Que tipo de apoio está à disposição da comunidade de Fabry?
Além das informações contidas neste site, existe um número crescente de organizações dedicadas a fornecerinformações e apoio à comunidade de pacientes de Fabry e de outros que convivem com doenças genéticas. Convémnotar que as opiniões expressas pelas organizações mencionadas abaixo não necessariamente refletem os pontos devista da Genzyme.
A Genzyme não mantém nem é responsável pelo conteúdo das comunicações para as organizações que constam dalista, nem pelos respectivos sites na internet. A única exceção se refere ao site “Fabry Community”, que é elaborado emantido pela Genzyme Corporation.
Distúrbios do Metabolismo dos Carboidratos Distúrbios do Metabolismo do Piruvato Distúrbios do Metabolismo dos Aminoácidos
Distúrbios podem afetar o metabolismo, o qual constitui o meio através do qual oorganismo processa as substâncias necessárias para desempenhar suas funções.Freqüentemente, esses distúrbios são causados por anomalias genéticas que acarretam aausência de uma enzima específica necessária para estimular um processo metabólico.Dependendo do distúrbio, os efeitos podem ser graves ou inofensivos.
topo
Distúrbios do Metabolismo dos CarboidratosOs carboidratos são açúcares. Muitos açúcares, além dos bem conhecidos glicose,sacarose e frutose, estão presentes nos alimentos. Alguns açúcares (p.ex., sacarose)devem ser metabolizados (processados) por enzimas no organismo antes de seremutilizados como fonte de energia. Quando as enzimas necessárias para o seuprocessamento estão ausentes, pode ocorrer um acúmulo desses açúcares, acarretandoproblemas.
A galactosemia (concentração sérica elevada de galactose) é geralmente causada pelafalta de galactose 1-fosfatouridiltransferase, uma das enzimas necessárias para a
metabolização da galactose. Este distúrbio está presente desde o nascimento.

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 5/13
Um em cada 50.000 a 70.000 recém-nascidos nasce sem essa enzima. A princípio, orecémnascido parece normal, mas, após alguns dias ou semanas, ele apresentainapetência, vômito, icterícia e pára de crescer normalmente. Ele apresenta um fígadoaumentado de tamanho, presença excessiva de proteínas e aminoácidos na urina, edemados tecidos e retenção líquida. Quando o tratamento é iniciado tardiamente, as crianças
afetadas apresentam uma estatura baixa e retardo mental. Muitas apresentam catarata.Na maioria dos casos, a causa desses sintomas é desconhecida.
O médico suspeita de galactosemia quando exames laboratoriais revelam a presença degalactose e de galactose 1-fosfato na urina. O diagnóstico é confirmado pela ausência degalactose 1-fosfato-uridiltransferase no sangue e nas células hepáticas. Quando omédico e os pais da criança preocupam-se em relacão à possibilidade da galactosemiaporque alguém da família apresenta o distúrbio, ela pode ser diagnosticada aonascimento, através do exame de uma amostra de sangue.
O leite e os produtos laticínios, fontes de galactose, devem ser completamente
eliminados da dieta da criança afetada. A galactose também é encontrada em algumasfrutas, vegetais e frutos do mar (p.ex., algas) e estes alimentos também devem serevitados. Contudo, não se sabe se as pequenas quantidades de galactose presentesnesses alimentos causam problemas a longo prazo. Uma mulher sabidamente portadorade um gene para o distúrbio deve eliminar completamente a galactose de suaalimentação durante a gestação. Uma gestante que apresenta este distúrbio também deveevitá-la. Quando ela apresenta uma concentração elevada de galactose, este açúcar podeatravessar a placenta e atingir o feto, causando catarata. As mulheres com galactosemiadevem restringir a ingestão de galactose durante toda a vida.
Quando a galactosemia for adequadamente tratada, a maioria das crianças não apresentaretardo mental. Contudo, o seu quociente de inteligência (QI) será mais baixo que o deseus irmãos e essas crianças apresentam freqüentemente problemas de fala. As meninasfreqüentemente apresentam insuficiência ovariana na puberdade e na vida adulta esomente algumas poucas são capazes de conceber naturalmente.
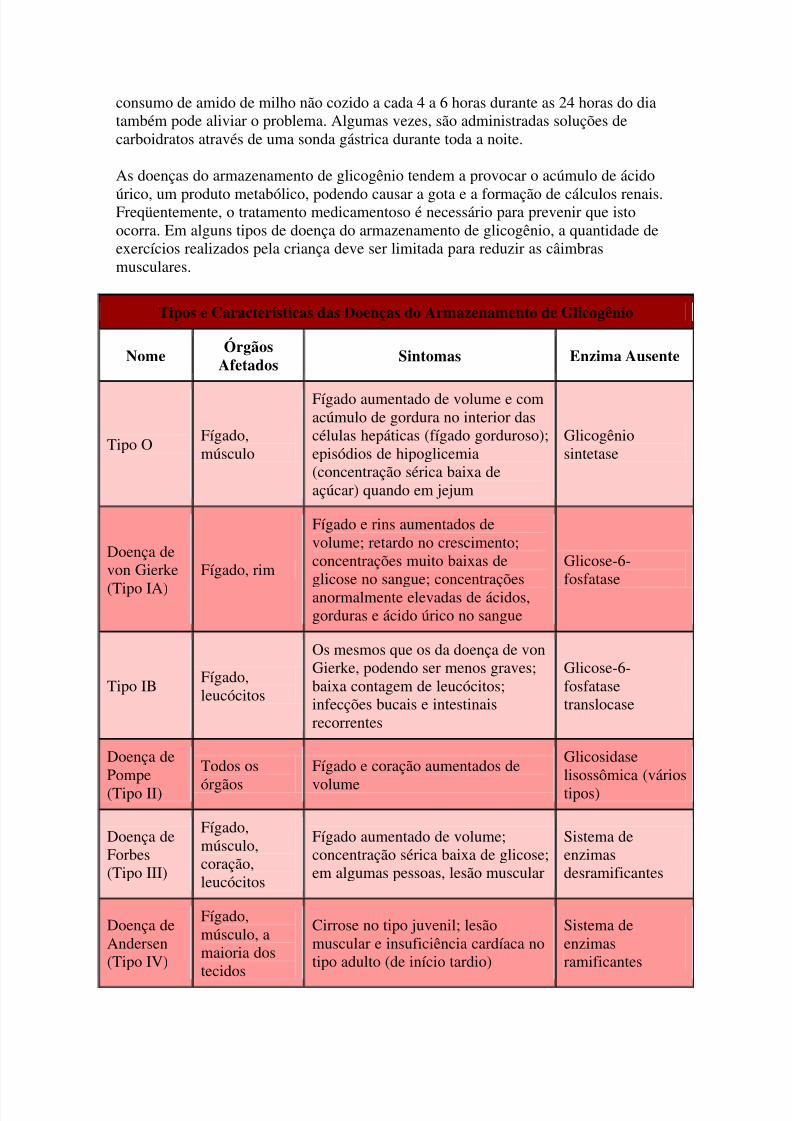
As doenças de armazenamento de glicogênio (glicogenoses) constituem um grupo dedistúrbios hereditários causados pela ausência de uma ou mais das muitas enzimasnecessárias para converter o açúcar (glicose) em sua forma de armazenamento, oglicogênio, ou para convertê-lo de volta em glicose e ser utilizado como energia. Nasdoenças do armazenamento de glicogênio, ocorrem depósitos de tipos ou quantidades
anormais de glicogênio nos tecidos do organismo, principalmente no fígado.Os sintomas são causados pelo acúmulo de glicogênio ou de seus subprodutos ou pelaincapacidade de produzir glicose quando esta é necessária. A idade em que os sintomasse manifestam e a gravidade dos mesmos varia consideravelmente de uma doença aoutra, pois, em cada doença, enzimas diferentes são afetadas.
O diagnóstico é estabelecido quando o exame de uma amostra de tecido, habitualmentedo músculo ou do fígado, revela a ausência de uma enzima específica.
O tratamento depende do tipo de doença do armazenamento de glicogênio. Para muitas
pessoas, a ingestão diária de várias refeições pequenas ricas em carboidratos ajuda aevitar que a concentração sérica de glicose caia. Para algumas crianças pequenas, o
5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 6/13
consumo de amido de milho não cozido a cada 4 a 6 horas durante as 24 horas do diatambém pode aliviar o problema. Algumas vezes, são administradas soluções decarboidratos através de uma sonda gástrica durante toda a noite.
As doenças do armazenamento de glicogênio tendem a provocar o acúmulo de ácido
úrico, um produto metabólico, podendo causar a gota e a formação de cálculos renais.Freqüentemente, o tratamento medicamentoso é necessário para prevenir que istoocorra. Em alguns tipos de doença do armazenamento de glicogênio, a quantidade deexercícios realizados pela criança deve ser limitada para reduzir as câimbrasmusculares.
Tipos e Características das Doenças do Armazenamento de Glicogênio
Nome Órgãos
Afetados Sintomas Enzima Ausente
Tipo OFígado,músculo
Fígado aumentado de volume e comacúmulo de gordura no interior dascélulas hepáticas (fígado gorduroso);episódios de hipoglicemia(concentração sérica baixa deaçúcar) quando em jejum
Glicogêniosintetase
Doença devon Gierke
(Tipo IA)
Fígado, rim
Fígado e rins aumentados devolume; retardo no crescimento;concentrações muito baixas de
glicose no sangue; concentraçõesanormalmente elevadas de ácidos,gorduras e ácido úrico no sangue
Glicose-6-
fosfatase
Tipo IBFígado,leucócitos
Os mesmos que os da doença de vonGierke, podendo ser menos graves;baixa contagem de leucócitos;infecções bucais e intestinaisrecorrentes
Glicose-6-fosfatasetranslocase
Doença de
Pompe(Tipo II)
Todos os
órgãos
Fígado e coração aumentados de
volume
Glicosidase
lisossômica (váriostipos)
Doença deForbes(Tipo III)
Fígado,músculo,coração,leucócitos
Fígado aumentado de volume;concentração sérica baixa de glicose;em algumas pessoas, lesão muscular
Sistema deenzimasdesramificantes
Doença deAndersen(Tipo IV)
Fígado,músculo, amaioria dos
tecidos
Cirrose no tipo juvenil; lesãomuscular e insuficiência cardíaca notipo adulto (de início tardio)
Sistema deenzimasramificantes

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 7/13
Doença deMcArdle(Tipo V)
MúsculoCâimbras musculares durante aatividade física
Fosforilasemuscular
Doença deHers (TipoVI)
Fígado
Fígado aumentado de volume;
episódios de hipoglicemia(concentração sérica baixa deglicose) quando em jejum;freqüentemente, sem sintomas
Fosforilasehepática
Doença deTarui (TipoVII)
Músculoesquelético,eritrócitos
Câimbras musculares durante aatividade física; destruição deeritrócitos (hemólise)
Fosfofrutocinase
A intolerância hereditária à frutose é um distúrbio hereditário no qual o organismo
não consegue utilizar a frutose porque a enzima fosfofrutoaldolase está ausente. Comoconseqüência, a frutose 1-fosfato, um subproduto da frutose, acumula- se no organismo,impedindo a formação de glicogênio e a sua conversão em glicose para ser utilizadacomo energia.
A ingestão de quantidades superiores à quantidade mínima de frutose ou sacarose(açúcar comum), a qual é convertida em frutose no organismo, acarreta hipoglicemia(concentração sérica baixa de glicose) acompanhada de sudorese, tremoresinvoluntários, confusão mental, náusea, vômito, dores abdominais e, algumas vezes,convulsões e coma. Podem ocorrer lesões renais e hepáticas e deterioração mentalquando a pessoa continua a consumir alimentos contendo frutose.
O diagnóstico é estabelecido quando o exame de uma amostra de tecido hepático revelaa ausência da enzima. O médico também avalia a resposta do organismo àadministração intravenosa de frutose e de glicose. Os portadores (pessoas que possuemum gene de um distúrbio mas que não o apresentam) podem ser identificados através daanálise do DNA (material genético) e de sua comparação com o DNA de portadores ede não portadores do distúrbio.
O tratamento inclui a eliminação da frutose (geralmente presente em frutas doces), dasacarose e do sorbitol (um substituto do açúcar) da dieta. Os episódios de hipoglicemiasão tratados com comprimidos de glicose. Todo indivíduo com uma intolerânciahereditária à frutose deve carregar consigo comprimidos de glicose.
A frutosúria é uma condição inofensiva na qual ocorre a excreção de frutose na urina.A frutosúria é causada por uma deficiência hereditária da enzima frutocinase.Aproximadamente 1 em cada 130.000 pessoas na população geral apresenta frutosúria.Ela não causa sintomas, mas a alta concentração de frutose no sangue e urina pode levara um diagnóstico errôneo de diabetes mellitus. A frutosúria não requer tratamento.
A pentosúria é uma condição inofensiva caracterizada pela excreção do açúcar xilulosena urina, devido à ausência da enzima necessária para metabolizá-lo.
Ela ocorre quase que exclusivamente em judeus; 1 em cada 2.500 judeus americanosapresenta pentosúria. Ela não causa problemas, mas a presença de xilulose na urina

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 8/13
pode levar a um diagnóstico errôneo de diabetes mellitus. Não há necessidade detratamento.
topo
Distúrbios do Metabolismo do Piruvato
O piruvato é formado no metabolismo dos carboidratos, gorduras e proteínas. Osproblemas hereditários relacionados ao metabolismo do piruvato podem causar umaampla variedade de distúrbios.
O piruvato é uma fonte de energia para as mitocôndrias, componentes celularesgeradores de energia. Um problema relacionado ao metabolismo do piruvato podecomprometer o funcionamento das mitocôndrias, causando qualquer um dos váriossintomas possíveis, como, por exemplo, lesão muscular, retardo mental, convulsões,acúmulo de ácido lático que acarreta acidose (excesso de ácido no organismo) ou falhasno funcionamento de um órgão (p.ex., coração, pulmões, rins ou fígado). Essesproblemas podem ocorrer em qualquer época, desde a primeira infância até a vidaadulta. O exercício, as infecções ou o consumo de álcool podem piorar os sintomas,causando uma acidose lática grave acompanhada de câimbras e fraqueza muscular.
A deficiência do complexo da piruvatodesidrogenase, um grupo de enzimasnecessárias para o metabolismo do piruvato, produz concentrações insuficientes deacetil coenzima A, a qual é essencial para a produção de energia. Os principais sintomasincluem a atividade muscular lenta, a má coordenação e um grave distúrbio doequilíbrio que praticamente impossibilita a marcha. Além disso, a pessoa pode
apresentar convulsões, retardo mental e malformação cerebral. Este distúrbio não temcura, mas algumas pessoas são beneficiadas com uma dieta rica em gordura.
A ausência de piruvatocarboxilase, uma enzima, interfere na produção de glicose ouimpede a sua produção no organismo. Ocorre um acúmulo de ácido lático e corposcetônicos no sangue, causando náusea e vômito. Ela é freqüentemente fatal. A síntese deaminoácidos, as unidades formadoras das proteínas, também depende dapiruvatocarboxilase. Quando essa enzima está ausente, a produção deneurotransmissores (substâncias que transmitem os impulsos nervosos) diminui,acarretando uma série de sintomas neurológicos (p.ex., retardo mental grave). Ahipoglicemia (concentração sérica baixa de glicose) e a acidose (acúmulo de ácidos no
sangue) podem ser aliviadas através da ingestão de refeições freqüentes ricas emcarboidratos. Contudo, não existe qualquer meio para se repor a ausência deneurotransmissores para tratar os sintomas neurológicos. Uma dieta com restriçãoprotéica pode ser benéfica para algumas pessoas que apresentam a forma leve dadoença.
topo
Distúrbios do Metabolismo dos Aminoácidos
Os aminoácidos, as unidades formadoras das proteínas, desempenham várias funções noorganismo. Os distúrbios hereditários do metabolismo dos aminoácidos podem serdefeitos na degradação dos aminoácidos ou no seu transporte para o interior das células.

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 9/13
Já foram identificados muitos desses distúrbios, incluindo a fenilcetonúria. Em todos osestados dos Estados Unidos, os recém-nascidos são submetidos à investigação dafenilcetonúria e também de outros distúrbios metabólicos.
FENILCETONÚRIA
A fenilcetonúria (FCU, fenilalaninemia, oligofrenia fenilpirúvica) é um distúrbio
hereditário no qual a enzima que processa o aminoácido fenilalanina está ausente,
acarretando em uma concentração perigosamente alta de fenilalanina no sangue.
Normalmente, a fenilalanina é convertida em tirosina, um outro aminoácido, eeliminada do organismo. Sem a enzima que a converte, a fenilalanina acumula-se noorganismo e é tóxica para o cérebro, causando retardo mental. A fenilcetonúria ocorrena maioria dos grupos geográficos, mas é rara em judeus com ascendentes oriundos daEuropa Oriental e em indivíduos da raça negra. Nos Estados Unidos, a sua incidência éde 1 em cada 16.000 recém-nascidos.
Sintomas
Os sintomas da fenilcetonúria geralmente estão ausentes nos recém-nascidos. Em raroscasos, o recém-nascido apresenta sonolência ou alimentase pouco. As crianças comfenilcetonúria tendem a ter a pele, o cabelo e os olhos mais claros que os membros dafamília que não a apresentam. Algumas apresentam uma erupção cutânea semelhante aoeczema. Quando não tratadas, as crianças afetadas desenvolvem rapidamente um certograu de retardo mental, geralmente grave.
Os sintomas em crianças com fenilcetonúria não diagnosticada ou não tratada incluemconvulsões, náusea e vômito, comportamento agressivo ou auto-agressivo,hiperatividade e, algumas vezes, sintomas psiquiátricos. As crianças afetadascomumente exalam um odor corpóreo tipo “cheiro de rato”, o qual é produzido por um
subproduto da fenilalanina (ácido fenilacético) na urina e no suor.
A fenilcetonúria em uma mulher grávida afeta profundamente o feto emdesenvolvimento, causando comumente retardo mental e físico. Muitas criançasapresentam microcefalia (uma cabeça anormalmente pequena que conduz ao retardomental) e doenças cardíacas. O controle rigoroso da concentração de fenilalanina namãe durante a gestação geralmente resulta em um feto normal.
Diagnóstico e Tratamento
O diagnóstico precoce é estabelecido quando o médico detecta uma concentraçãoelevada de fenilalanina e uma concentração baixa de tirosina durante a avaliação de umrecém-nascido. Quando a fenilcetonúria ocorre na família e quando o membro afetadopossui uma análise do DNA, pode ser realizada uma amniocentente ou uma coleta deamostra do vilo coriônico e a análise do DNA para se determinar se o feto apresenta odistúrbio.
O tratamento consiste em limitar a ingestão de fenilalanina, embora ela não deva ser
totalmente eliminada. Como todas as fontes naturais de proteínas contêmaproximadamente 4% de fenilalanina, é impossível o consumo de uma quantidade

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 10/13
suficiente de proteínas sem que seja excedida a quantidade aceitável de fenilalanina.Conseqüentemente, em vez de leite e carne, a pessoa deverá consumir diversosalimentos sintéticos que fornecem os demais aminoácidos. Podem ser ingeridosalimentos naturais com baixo teor protéico (p.ex., frutas e vegetais e quantidadeslimitadas de cereais em grãos). Existem disponíveis no comércio produtos sem
fenilalanina. Esses produtos ajudam a controlar a sua ingestão, dando à pessoa umpouco mais de liberdade para consumir alimentos naturais.
A ingestão de fenilalanina deve ser restringida desde as primeiras semanas de vida, paraque não ocorra retardo mental. Uma dieta restritiva iniciada precocemente e mantidaadequadamente possibilita um desenvolvimento normal da criança e previne aocorrência de lesão cerebral. Contudo, quando a dieta não for controlada rigorosamente,as crianças afetadas podem apresentar dificuldades na escola. As restrições dietéticasiniciadas após os 2 ou 3 anos de idade somente conseguem controlar a hiperatividadeextrema e as convulsões. Antigamente, pensava- se que a interrupção da dieta especialno momento em que o cérebro estava praticamente desenvolvido era uma medida
segura, mas vários relatos indicando a ocorrência de problemas de aprendizado ecomportamentais e uma diminuição da inteligência fizeram com que essa recomendaçãofosse reconsiderada. Atualmente, a maioria dos médicos acredita que a dieta comrestrição da fenilalanina deve ser mantida durante toda a vida.
topo
A Galactosemia pode ser descrita como uma concentração sanguínea elevada domonossacarídeo galactose (aldohexose, epímera da glicose em C-4), devido a umadesordem no metabolismo causada por atividade enzimática deficiente ou funçãohepática prejudicada.
Os seres humanos obtêm a galactose primariamente através do leite humano e bovino ede derivados lácteos, pela hidrólise da lactose, dissacarídeo que é composto por glicose e galactose unidas por ligação β-glicosídica. Galactose livre também está presente emalgumas frutas e vegetais, como tomates, bananas e maçãs. A digestão da lactose se dápor intermédio da enzima intestinal lactase, que a quebra nos dois monossacarídeos quea constituem. Após a quebra da lactose em glicose e galactose, se dá o processo demetabolização desses monossacarídeos, que envolve catálises enzimáticas que levarão,em seu final, à conversão da galactose em glicose para uso como fonte de energia. Afase de metabolização da galactose é a que apresenta problemas no paciente
galactosêmico, devido a deficiências enzimáticas em vários níveis possíveis.
Tipologia da doença
[editar] Galactosemia do Tipo 1
É a forma mais comum e grave de galactosemia. Nesse tipo, ocorre deficiência dagalactose-1-P uridil transferase (GALT), a enzima que converte galactose-1-fosfato(galactose-1-P) em uridina difosfato galactose (UDPgalactose), o que leva ao acúmulode galactose-1-fosfato e a sintomas variados.
A deficiência completa da atividade da GALT é conhecida como galactosemia clássica,e é frequentemente referida com o termo isolado "galactosemia". Pacientes não-tratados

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 11/13
tipicamente têm disfunção hepática e renal; tanto paciente tratados como não-tratadospodem ter catarata, neurodesenvolvimento anormal e insuficiência ovariana precoce.
[ editar ] Variante Duarte
Atividade parcial da GALT ocorre em numerosas variantes; a mais comum destas é avariante Duarte, na qual paciente têm um alelo duarte e um alelo clássico (D/G),resultando em uma atividade enzimática que corresponde a aproximadamente 25% donormal[4]. Pacientes com dois alelos Duarte (D/D) têm aproximadamente 50% daatividade enzimática normal.[4] Pacientes com atividade maior ou igual a 50% donormal se apresentam com pouca ou nenhuma evidência de morbidade neonatal ou alongo prazo se não tratados.[5][6]
[editar] Galactosemia do Tipo 2
Defeito da galactoquinase leva a acúmulo de galactose, tendo como principaiscaracterísticas os problemas oculares.
[editar] Galactosemia do Tipo 3
Causada por deficiência da uridil difosfo galactose-4-epimerase, a galactosemia do tipo3 é uma forma muito rara, com formas inicial e severa, que aparecem no curso dadoença.
[editar] Outros tipos
Existem outros tipos benignos de galactosemia que são assintomáticos como, porexemplo, a galactosemia Duarte e a variante da raça negra. Notamos, nessas variantes,deficiências enzimáticas que não levam a quadros sintomáticos graves, como os dagalactosemia clássica.
[editar] Diagnóstico
O diagnóstico de galactosemia é freqüentemente suspeitado quando um açúcar redutorque não reage no sistema glicose oxidase é encontrado na urina. Contudo, outrosaçúcares além da galactose podem dar o mesmo tipo de reação, e a própria enfermidade
hepática pode resultar em galactosúria (excreção de galactose através da urina).Portanto, a galactosemia clássica é diagnosticada pela demonstração de que a atividadeda galactose-1-P uridil transferase esteja ausente ou muito baixa nos eritrócitos.
No estado normal, é encontrada a transferase nas células hepáticas, nos leucócitos e noseritrócitos, e, na galactosemia, os ensaios permitem a verificação da carência da enzimanessas células. O diagnóstico pré-natal pode ser realizado por amniocentese, comcultura de fibroblastos derivados do líquido amniótico.
[editar] Tratamento
O tratamento para a galactose se dá pela restrição de galactose e lactose na dieta.Existem várias formas de galactosemia e as restrições alimentares variam de uma forma

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 12/13
para outra, havendo inclusive discordâncias em algumas formas. Nos casos mais graves,um monitoramento multidisciplinar, incluindo pediatria, neurologia, oftalmologia, endocrinologia, genética,nutrição e fonoaudiologia, minimiza os efeitos da doença.
A não-ingestão de galactose e lactose diminui bastante os sintomas, porém podem
ocorrer alguns problemas como deficiência de aprendizagem, distúrbios da fala e, nasmeninas, disfunções ovarianas. O grau em que os sintomas se manifestam em umpaciente submetido a dieta gera discordâncias e pesquisas, não havendo conclusõesprecisas e não pássiveis de contestação a esse respeito. Apesar da galactose ser utilizadana composição de várias estruturas celulares, a sua supressão da alimentação nãocausará maiores problemas pois, há um mecanismo metabólico para conversão daglicose em galactose.
O uso de suplementos à base de enzimas digestivas, como alternativa para osintolerantes à lactose, já é bem conhecido. O que muita gente ainda não sabe é que o usode enzimas pode amenizar também os sintomas causados pela alergia ao leite de vaca.Descubra, neste texto, as principais diferenças entre a intolerância à lactose e a alergia àproteína do leite e como utilizar os suplementos de enzimas a seu favor.
Intolerância à lactose
Dentre os distúrbios associados ao consumo do leite de vaca pode-se dizer que aintolerância à lactose é um dos mais comuns e amplamente divulgado e discutido. Estaintolerância pode ser definida como uma incapacidade do organismo em digerir alactose (açúcar presente no leite), resultando em alterações gastrintestinais, comodistensão e dores abdominais, gases e diarreia. Esses sintomas são mais evidentes nasprimeiras horas que sucedem o consumo de leite e derivados e são, geralmente, locais – sem comprometimentos sistêmicos graves.Essa dificuldade em digerir a lactose ocorre pela deficiência ou ausência da enzimalactase na mucosa intestinal – e pode surgir por um erro genético (o indivíduo nãoproduz a enzima) ou por consequência de distúrbios no intestino (diarreias infecciosas,
uso prolongado de antibióticos, Doença de Crohn, Doença Celíaca), fazendo com que oindivíduo diminua ou pare momentaneamente a produção da enzima. Alergia às proteínas do leite de vaca
Ao contrário da intolerância à lactose, distúrbio que envolve apenas a deficiência deuma enzima, a alergia às proteínas do leite envolve uma resposta exacerbada do sistemaimunológico. Tal resposta pode ser imediata (os sintomas aparecem poucas horas após oconsumo) ou tardia (os sintomas podem levar de 3 horas a 3 dias para aparecer). Aalergia tardia ao leite de vaca é a mais comum e, por seus sintomas aparecerem muitotempo depois do consumo do alimento, tem seu diagnóstico bastante dificultado. Poreste motivo é comum muitas pessoas apresentarem alergia tardia ao leite de vaca e nem
sequer imaginarem que alguns de seus problemas de saúde podem estar relacionados àela.

5/11/2018 TRabalho de Bioquimica - slidepdf.com
http://slidepdf.com/reader/full/trabalho-de-bioquimica-55a23386744c4 13/13
A alergia ocorre quando o organismo entra em contato com as proteínas intactas – maldigeridas – do leite, reconhece-as como corpos estranhos e reage contra elas,produzindo substâncias inflamatórias e causando processos inflamatórios crônicos. Ossintomas podem ser desencadeados em diversos órgãos-alvo e estudos já demonstrarama relação da alergia tardia ao leite de vaca com otite, dermatite, rinite, sinusite,
bronquite asmática, amigdalite, aumento na formação de muco, gastrite, esofagite,refluxo, obstipação intestinal, enxaqueca, artrite reumatoide, falta de concentração,hiperatividade (ADHD), ansiedade e até mesmo depressão.A causa da alergia tardia pode estar relacionada a fatores genéticos, dificuldade emdigerir as proteínas (possivelmente por deficiências enzimáticas) ou ainda por umahiperpermeabilidade intestinal, o que aumenta as chances de proteínas inteiras passaremdo intestino para a corrente sanguínea e assim, ativar a resposta inflamatória.Uso das enzimas digestivas
No caso da intolerância à lactose, o uso do suplemento da enzima lactase é muitocomum e eficaz, já que supre a deficiência desta enzima no organismo e possibilita,
assim, a digestão da lactose. A lactase pode ser encontrada sob a forma de pastilhasmastigáveis, em pó, comprimidos ou cápsulas em diversas farmácias convencionais, demanipulação ou ainda em lojas de produtos naturais. A dose administrada vai dependerda quantidade de lactose a ser ingerida e da capacidade do seu organismo em produzirmais ou menos lactase.Já no caso da alergia às proteínas do leite de vaca, o uso das enzimas digestivas aindanão é muito comum. Entretanto, alguns estudos já estão sendo realizados a fim deelucidar melhor o real papel de algumas enzimas na melhora dos sintomas causadospelos alimentos alergênicos.Os estudos sugerem que as enzimas, principalmente as proteases – que quebram asproteínas – melhoram a digestão das proteínas do leite, diminuem a permeabilidade daparede do intestino e ainda parecem regular o sistema imunológico. Assim, o risco deproteínas mal digeridas passarem pela barreira do intestino para a corrente sanguínea ede o sistema imune reagir de forma exacerbada contra elas diminui bastante,minimizando, dessa forma, os sintomas causados pela alergia.Atualmente, já existem no mercado suplementos com mix de enzimas digestivas(incluindo as proteases) que melhoram a digestão das proteínas e poderiam, assim,amenizar bastante os sintomas causados pela alergia às proteínas do leite de vaca.Independentemente do distúrbio – intolerância à lactose ou alergia às proteínas do leite
– só um profissional capacitado poderá te orientar nas substituições alimentares e no usodas enzimas digestivas como alternativa de tratamento.
Uma vez caracterizado o diagnóstico, pode se prevenir novos sintomas não usando leitee laticínios. Usando-os, a prevenção é mediante a tomada de fermento sintético prévia aqualquer ingestão de lactose. Cabe salientar que vários medicamentos, inclusiveantidiarréicos e anti-reumáticos contêm lactose no chamado excipiente, ou seja, no póou no líquido necessário para poder conter a substância básica num comprimido ousolução; isso é importante quando avaliamos os efeitos indesejáveis referidos pelosusuários.










![TEMAS DE BIOQUIMICA TEMA I: INTRODUCCION A LA BIOQUIMICA BIOQUIMICA [Tomo I].pdf · 1 TEMAS DE BIOQUIMICA TEMA I: INTRODUCCION A LA BIOQUIMICA Generalidades Concepto de materia, cuerpo,](https://static.fdocument.pub/doc/165x107/5a7707fe7f8b9a9c548dbcf5/temas-de-bioquimica-tema-i-introduccion-a-la-bioquimica-bioquimica-tomo-ipdfaa.jpg)







