tesis aromaticos

of 125
-
Upload
fercho72169 -
Category
Documents
-
view
225 -
download
0
Transcript of tesis aromaticos
-
8/11/2019 tesis aromaticos
1/125
ESTANDARIZACIN DE LA TCNICA DE CROMATOGRAFA DE GASES
CON DETECTOR DE IONIZACIN POR LLAMA PARA LA DETERMINACIN
DE BTEX (BENCENO, TOLUENO, ETILBENCENO Y XILENO) EN MATRICES
ACUOSAS
JHON EDGAR ARROYAVE GARCA
LINA MARA VILLA FLREZ
UNIVERSIDAD TECNOLGICA DE PEREIRA
FACULTAD DE TECNOLOGA
ESCUELA DE QUMICA
QUMICA INDUSTRIAL
PEREIRA
2011
-
8/11/2019 tesis aromaticos
2/125
ESTANDARIZACIN DE LA TCNICA DE CROMATOGRAFA DE GASES
CON DETECTOR DE IONIZACIN POR LLAMA PARA LA DETERMINACIN
DE BTEX (BENCENO, TOLUENO, ETILBENCENO Y XILENO) EN MATRICES
ACUOSAS
JHON EDGAR ARROYAVE GARCA
LINA MARA VILLA FLREZ
TRABAJO DE GRADO
Requisito final para optar al ttulo de Qumico Industrial
DIRECTOR: JUAN PABLO ARRUBLA VLEZ Qco MSc.
ASESOR: CARLOS HUMBERTO MONTOYA N Qco Ind
GRUPO DE ESTUDIO DEL RECURSO HDRICO.
UNIVERSIDAD TECNOLGICA DE PEREIRA
FACULTAD DE TECNOLOGA
ESCUELA DE QUMICA
QUMICA INDUSTRIAL
PEREIRA
2011
-
8/11/2019 tesis aromaticos
3/125
NOTA DE ACEPTACIN DE TRABAJO DE GRADO
ESTANDARIZACIN DE LA TCNICA DE CROMATOGRAFA DE GASES
CON DETECTOR DE IONIZACIN POR LLAMA PARA LA DETERMINACIN
DE BTEX (BENCENO, TOLUENO, ETILBENCENO Y XILENO) EN MATRICES
ACUOSAS
Presentado por:
JHON EDGAR ARROYAVE GARCA
LINA MARA VILLA FLREZ
Los suscritos director y jurado del presente trabajo de grado, una vez realizada la
versin escrita y presenciado la sustentacin oral, decidimos otorgar:
La nota de ------------------------------------------------------------------
Con la connotacin: -----------------------------------------------------
Para constancia firmamos en la ciudad de Pereira hoy:
Director: JUAN PABLO ARRUBLA VLEZ
Firma: -------------------------------------------------------------
Asesor: CARLOS HUMBERTO MONTOYA N
Firma: -------------------------------------------------------------
Jurado:
Firma: -------------------------------------------------------------
-
8/11/2019 tesis aromaticos
4/125
DEDICATORIA
A Dios por darnos la vida y su amor infinito, por acompaarnos y darnos fortaleza en todo
momento de nuestras vidas y mostrarnos el camino que debemos seguir para alcanzar la
verdadera felicidad.
A la Virgen Mara por su amor, acompaamiento e intercesin ante su Hijo.
A nuestras familias por su amor, apoyo, comprensin y dedicacin en cada momento. Por
inculcar en nosotros valores y principios morales que nos hacen ser mejores personas.
Por estar siempre con nosotros especialmente en los momentos difciles ayudndonos a
superar las dificultades.
A nuestros amigos por el cario y apoyo que siempre nos brindaron.
-
8/11/2019 tesis aromaticos
5/125
AGRADECIMIENTOS
A Dios todopoderoso, por estar siempre con nosotros, cambiando nuestras vidas,
mostrndonos el camino que debemos seguir. Por darnos la fuerza necesaria para
cumplir nuestras metas y sueos a pesar de los obstculos que se pueden presentar. Por
ensearnos la felicidad que se alcanza al estar cerca de l.
A la Virgen Mara por su compaa en cada instante de nuestras vidas y por interceder entodo momento y lugar por nosotros ante su Hijo.
A nuestras familias por el cario, apoyo y amor incondicional que nos han brindado
durante el transcurso de nuestras vidas. Por el nimo y la motivacin en los momentos
difciles, haciendo posible la superacin de las dificultades.
A nuestro director Juan Pablo Arrubla y asesor Carlos Humberto Montoya por el apoyo,
tiempo, dedicacin y conocimientos que nos brindaron para la realizacin de este
proyecto.
A Hugo Fernando Arias, Jaime Alejandro Martnez y Paula Andrea Giraldo que con
paciencia nos ensearon a operar el cromatgrafo de gases y nos brindaron su
conocimiento y experiencia en el campo de la investigacin.
A los profesores de la escuela de qumica por los conocimientos aportados durante el
transcurso de nuestra carrera.
A Mara Victoria, Javier y Germn por su colaboracin cada que fue necesario.
A nuestros amigos por apoyarnos cuando lo necesitamos. Por hacer ms agradable
nuestra estada en la universidad.
A todas las entidades y personas que de alguna manera hicieron posible cumplir nuestra
meta.
-
8/11/2019 tesis aromaticos
6/125
-
8/11/2019 tesis aromaticos
7/125
1.1.5.1 Mtodo basado en la relacin seal/ruido 29
1.1.5.2 Mtodo basado en la desviacin estndar de la respuesta del blanco y 29
La pendiente de la recta de calibrado
1.1.5.2.1 Mtodos instrumentales que corrigen la seal frente a un blanco 30
1.1.5.2.2 Mtodos instrumentales que no corrigen la seal frente a un blanco 30
1.1.5.3 Mtodo basado en la extrapolacin de la recta de calibrado a 31
concentracin cero
1.1.6 Sensibilidad 31
1.2 CROMATOGRAFA DE GASES (GC) 31
1.2.1 Ventajas 32
1.2.2 Instrumentacin en cromatografa de gases 33
1.2.2.1 Gas portador 35
1.2.2.2 Sistema de inyeccin 36
1.2.2.3 Configuracin de la columna y del horno de la columna 37
1.2.2.4 Sistemas de deteccin 38
1.2.2.4.1 Detector de ionizacin por llama (FID) 39
1.2.3 Anlisis cualitativo y cuantitavivo 42
1.2.3.1 Anlisis cualitativo 42
1.2.3.2 Anlisis cuantitativo 42
1.2.3.2.1 Mtodo del patrn interno 43
1.2.3.2.2 Mtodo del estndar externo 43
1.2.3.2.3 Mtodo de la normalizacin de las reas 44
-
8/11/2019 tesis aromaticos
8/125
1.3 EXTRACCIN EN FASE SLIDA (SPE) 44
1.3.1 Consideraciones tericas 45
1.3.1.1 Interacciones polares 46
1.3.1.2 Interacciones apolares 47
1.3.1.3 Interacciones inicas 48
1.3.2 Etapas de la extraccin en fase slida 50
1.3.2.1 Acondicionamiento del adsorbente 50
1.3.2.2 Aplicacin de la muestra (adsorcin) 51
1.3.2.3 Lavado del adsorbente 51
1.3.2.4 Elucin 51
1.3.3 Ventajas 52
1.4 HIDROCARBUROS AROMTICOS (BTEX) 52
1.4.1 Estabilidad 52
1.4.2 Propiedades fsicas y qumicas 53
1.4.2.1 Comportamiento de los BTEX en agua 55
1.4.3 Fuente de hidrocarburos aromticos 56
1.4.3.1 Petrleo 56
1.4.3.1.1 Aromticos del reformado cataltico 56
1.4.3.1.2 Aromticos del craqueo con vapor 58
1.4.3.1.3 Separacin de hidrocarburos aromticos 58
1.4.4 Procesos de transformacin de aromticos 60
1.4.5 Refinacin del petrleo y composicin de la gasolina en Colombia 60
-
8/11/2019 tesis aromaticos
9/125
1.4.5.1 Refinacin nacional del petrleo 60
1.4.5.2 Composicin de la gasolina en Colombia 60
1.4.6 Usos de los BTEX 62
1.4.6.1 Benceno 62
1.4.6.2 Tolueno 63
1.4.6.3 Etilbenceno 63
1.4.6.4 p-Xileno 63
1.4.6.5 o-Xileno 64
1.4.6.6 m-Xileno 64
1.4.7 Toxicidad de los BTEX 64
1.4.7.1 Benceno 65
1.4.7.2 Tolueno 66
1.4.7.3 Etilbenceno 67
1.4.7.4 Xileno 67
1.4.8 Mtodos de anlisis de los BTEX 69
2 SECCIN EXPERIMENTAL 72
2.1 MUESTRA DE ANLISIS 72
2.2 MUESTREO 72
2.3 TRANSPORTE, PRESERVACIN Y ALMACENAMIENTO DE LA 72
MUESTRA
2.4 EXTRACCIN EN FASE SLIDA 73
2.5 COMPOSICIN DEL ESTNDAR DE BTEX 74
-
8/11/2019 tesis aromaticos
10/125
2.6 ANLISIS CROMATOGRFICO 75
2.6.1 Estndar 75
2.6.2 Patrones 76
2.6.3 Curvas de calibracin 77
2.6.4 Anlisis de las muestras 77
3 RESULTADOS Y DISCUSIN 78
3.1 ANLISIS CROMATOGRFICO DEL ESTNDAR 78
3.2 CALIBRACIN 80
3.3 TRATAMIENTO ESTADSTICO 84
3.3.1 Indicadores de relacin lineal 87
3.3.2 Exactitud 89
3.4 ESTANDARIZACIN DE LA TCNICA DE EXTRACCIN EN FASE 90
SLIDA (SPE)
3.4.1 Porcentaje de recuperacin 90
3.5 ANLISIS DE LAS MUESTRAS 92
3.5.1 Resultados de pH y temperatura en las trampas de grasas 92
3.5.2 Anlisis cromatogrfico 93
3.5.3 Anlisis de un humedal 97
3.5.4 Interferencias en el anlisis 98
4. CONCLUSIONES 99
5. BIBLIOGRAFA 101
6. ANEXOS 107
-
8/11/2019 tesis aromaticos
11/125
-
8/11/2019 tesis aromaticos
12/125
Tabla 18. reas de la cola del diclorometano medida por el equipo 80
Tabla 19. Datos de las reas obtenidos de cada patrn para la construccin de 81
las curvas de calibracin.
Tabla 20. Datos de las reas obtenidas para la elaboracin de la curva de 82
calibracin del benceno
Tabla 21. Datos para determinar la repetibilidad instrumental 84
Tabla 22. Datos para el clculo de la SD y el % RSD para el benceno 86
Tabla 23. Resultados estadsticos obtenidos de las curvas de calibracin 87
Tabla 24. Datos obtenidos para el test de linealidad 88
Tabla 25. Datos obtenidos para calcular la exactitud 89
Tabla 26. Datos obtenidos para el porcentaje de recuperacin de cada patrn 90
Tabla 27. Porcentajes de recuperacin de los BTEX 91
Tabla 28. Temperatura y pH en las trampas de grasas 93
Tabla 29. Concentraciones de BTEX en las estaciones de servicio 95
Tabla 30. Concentracin de BTEX en la salida del humedal 98
-
8/11/2019 tesis aromaticos
13/125
NDICE DE FIGURAS
Pg.
Figura 1. Diagrama de un sistema de cromatografa gas-lquido 35
Figura 2. Detector de ionizacin por llama caracterstico 41
Figura 3. Fases estacionarias polares y sus interacciones con los analitos 46
Figura 4. Fasesestacionarias apolares y sus interacciones con los analitos 47
Figura 5. Fases estacionarias de intercambio inico 49
Figura 6. Etapas de la extraccin en fase slida 51
Figura 7. Molcula del benceno 53
Figura 8. Principales reacciones de aromatizacin 57
Figura 9. Trampa de grasas 72
Figura 10. Cromatgrafo de gases utilizado en el anlisis 75
Figura 11. Cromatograma patrn 50ppm 78
Figura 12. Cromatograma del solvente de elucin (Diclorometano) 79
Figura 13. Grfica de barras de las concentraciones de BTEX 83
Figura 14. Curva de calibracin para el benceno 83
Figura 15. Grfica de barras de los porcentajes de recuperacin de los BTEX 91
Figura 16. Cromatograma del extracto obtenido en la muestra tomada de la 94
estacin de servicio centro
Figura 17. Cromatograma de la salida del humedal 97
-
8/11/2019 tesis aromaticos
14/125
NDICE DE ANEXOS
Pg.
Anexo 1 Reacciones de los BTEX para la produccin de diferentes productos 107
Anexo 2. Certificado del anlisis del estndar 110
Anexo 3. Datos de las reas obtenidas de cada patrn para la construccin de 111
las curvas de calibracin.
Anexo 4. Datos de las reas obtenidas para la elaboracin de la curva de 113
calibracin del benceno
Anexo 5. Curvas de calibracin de los BTEX 115
Anexo 6. Datos para la determinacin de la repetibilidad instrumental. 118
Anexo 7. Distribucin de t para diferentes niveles de confianza 119
Anexo 8. Cromatogramas de los extractos obtenidos en las muestras 120
tomadas de las estaciones de servicio
Anexo 9. Cromatogramas de los patrones de BTEX 123
-
8/11/2019 tesis aromaticos
15/125
GLOSARIO
BTEX: Acrnimo que define la mezcla de benceno, tolueno, etilbenceno y los tres
ismeros del xileno (orto, meta y para).
CROMATOGRAMA:Es un grfico de la respuesta del detector en funcin del tiempo.
DETECTOR: Dispositivo que responde a cierta caracterstica del sistema que est sujeto
a observacin y convierte esa respuesta en una seal susceptible de medirse.
ESTANDARIZACIN: Procedimiento estadstico que consiste en verificar y documentar,que exista un alto grado de seguridad en la obtencin de resultados que deberan ser
precisos y exactos dentro de las especificaciones y los atributos de calidad previamente
establecidos.
EXACTITUD: Expresa la cercana entre el valor que es aceptado, sea como un valor
convencional verdadero (Material de referencia interno de la firma), sea como un valor de
referencia aceptado (Material de referencia certificado o estndar de una farmacopea) y el
valor encontrado (Valor promedio) obtenido al aplicar el procedimiento de anlisis un
cierto nmero de veces.
FID (FLAME IONIZATION DETECTOR) DETECTOR DE IONIZACIN POR LLAMA:
Detector para cromatografa de gases que se basa en la captura de los iones producidos
durante la pirlisis de analitos orgnicos en una flama.
GC (GAS CHROMATOGRAPHY): CROMATOGRAFA DE GASES: Es una tcnica de
gran sensibilidad y exactitud que se utiliza para separacin, identificacin y cuantificacin
de compuestos voltiles. Se basa en la distribucin del analito entre un fase mvil gaseosa y
una fase liquida inmovilizada sobre la superficie. La fase mvil se denomina gas
transportador, ya que es un gas inerte cuya finalidad es transportar las molculas de la
muestra a travs de la columna. Los adsorbentes, tales como gel de slice, almina, sales
inorgnicas, polmeros porosos, tamices moleculares y carbn grafitizado, son las fases
estacionarias.
LMITE DE CUANTIFICACIN:Cantidad ms pequea del analito en una muestra que
puede ser cuantitativamente determinada con exactitud aceptable. Es un parmetro del
-
8/11/2019 tesis aromaticos
16/125
anlisis cuantitativo para niveles bajos de compuestos en matrices de muestra y se usa
particularmente para impurezas y productos de degradacin. Se expresa comoconcentracin del analito.
LMITE DE DETECCIN:Cantidad ms pequea de analito en una muestra que puede
ser detectada por una nica medicin, con un nivel de confianza determinado, pero no
necesariamente cuantificada con un valor exacto. Es comnmente expresado como
concentracin del analito.
LNEA BASE: Es la parte del cromatograma que registra la respuesta del detector en
ausencia de soluto o solvente.
PICO: Es la parte del cromatograma que registra la respuesta del detector mientras que
uno o ms componentes son eludos de la columna.
PRECISIN: Expresa la cercana de coincidencia (Grado de dispersin) entre una serie
de mediciones obtenidas de mltiples muestreos de una misma muestra homognea bajo
condiciones establecidas. Puede considerarse a tres niveles: repetibilidad, precisin
intermedia y reproducibilidad.
SPE (SOLID PHASE EXTRACTION) EXTRACCIN EN FASE SLIDA: La SPE es una
tcnica muy empleada en la preparacin de muestras para anlisis por cromatografa
lquida, de gases, electroforesis y an para espectrofotometra. Tambin, se ha convertido
en una de las tcnicas para clean-up y concentracin de muestras utilizadas por los
qumicos analticos.
SPLIT: Modo de inyeccin con divisin de flujo, en el cual rpidamente se vaporiza la
muestra antes de entrar en la columna. Una fraccin definida de la muestra de vapor entra
en la columna y el resto sale de la entrada a travs de un orificio de ventilacin.
SPLITLESS:Modo de inyeccin sin divisin de flujo, el cual utiliza una entrada divisora en
donde la abertura de divisin de flujo se bloquea durante el perodo de inyeccin de tal
manera que la mayor parte del vapor de la muestra entra en la columna
USEPA: (United States Environmental Protection Agency) Agencia de Proteccin
Ambiental de Estados Unidos cuya misin es la de proteger la salud de los humanos y la
del medio ambiente [1,17,25,41].
-
8/11/2019 tesis aromaticos
17/125
RESUMEN
La cromatografa de gases es una de las tcnicas ms ampliamente usadas en el anlisis
de contaminantes ambientales derivados del petrleo, ya que esta es una tcnica analtica
instrumental de alta sensibilidad capaz de identificar cualitativa y cuantitativamente
concentraciones muy bajas de estos componentes.
Se realiz la estandarizacin de la tcnica de Cromatografa de Gases con Detector de
Ionizacin por Llama (GC/FID) para la determinacin de BTEX en matrices acuosas. En el
proceso de estandarizacin se estimaron los valores para los parmetros que determinanel rendimiento del mtodo analtico como son: precisin, exactitud, linealidad, lmite de
deteccin, lmite de cuantificacin y sensibilidad; logrando valores aceptables de estas
medidas.
En la extraccin y preconcentracin de la matriz, se emple la extraccin en fase slida
(SPE) utilizando cartuchos C18y diclorometano como solvente de elucin. Este mtodo de
extraccin expuso buenos porcentajes de recuperacin, entre 69,22 y 80,01%, con
desviaciones estndar inferiores al 10%.
El mtodo fue aplicado para la determinacin de BTEX en matrices acuosas, usando
muestras reales procedentes de la trampa de grasas ms limpia de tres estaciones de
servicio de la ciudad de Pereira. Se encontraron concentraciones de 0,013ppm, 0,014ppm
y 0,018ppm de etilbenceno, m,p-xileno y o-xileno respectivamente en una de las tres
estaciones de servicio. Aunque en Colombia no existe una ley como tal que limite la
concentracin de BTEX en agua, los resultados obtenidos indican que se genera poca
contaminacin de agua con estos compuestos en las estaciones de servicio analizadas.
-
8/11/2019 tesis aromaticos
18/125
-
8/11/2019 tesis aromaticos
19/125
El benceno es una sustancia cancergena, razn por la cual el Ministerio de Trabajo, por
medio del Decreto 1214 del 6 de Julio de 1999, restringi el uso de ste en las industriasde Colombia [8].
A nivel internacional: el benceno, el tolueno y el etilbenceno son compuestos designados
como contaminantes prioritarios por la Agencia de Proteccin Ambiental de los Estados
Unidos (USEPA, por sus siglas en ingls). Y La accin y niveles de riesgo del benceno,
tolueno, etilbenceno y xilenos son descritos en las normas de calidad del gobierno
Holands para la evaluacin de la contaminacin del suelo y el agua [9].
La USEPA establece los siguientes lmites mximos permisibles en agua potable en mg/Lpara BTEX: benceno, 0.005; tolueno, 1.0; etilbenceno, 0.7 y mezcla de xilenos, 10 [10].
Los lmites mximos permisibles establecidos por la Secretara de Salud de Mxico
permisible en agua potable para BTEX en mg/L son: benceno, 0.01; tolueno, 0.3;
etilbenceno, 0.7 y mezcla de xilenos, 0.5 [11]. La unin europea (EU) establece el mximo
nivel contaminante de 0,001 mg/L para el benceno en agua potable [12].
Las consecuencias de la contaminacin con BTEX de las aguas sobre el abastecimiento
pblico, su uso agrcola e industrial, as como sus efectos ambientales pueden llegar a
tener influencias negativas de gran magnitud, es por esto que se necesita verificar laconcentracin de estos compuestos en las fuentes de captacin de plantas
potabilizadoras y efluentes de empresas como el de las estaciones de servicio.
Con este trabajo se pretende estandarizar por Cromatografa de Gases utilizando un
Detector de Ionizacin por Llama (GC/FID) el anlisis de BTEX en matrices acuosas, ya
que esta es una tcnica analtica instrumental de alta sensibilidad capaz de identificar
cualitativa y cuantitativamente concentraciones muy bajas de constituyentes voltiles y
semivoltiles del petrleo. El anlisis de estos compuestos con este mtodo analticorequiere de una extraccin y preconcentracin previa, para lo cual se han utilizado
tcnicas como el mtodo de purga y trampa con cromatografa de gases, extraccin en
fase slida (SPE), y la microextraccin en fase slida (SPME). Para el desarrollo de este
proyecto se busca la estandarizacin de la SPE [13].
Atendiendo a la demanda del sector industrial, organismos de control y a la comunidad en
general, el grupo de estudio del recurso hdrico implement un mtodo analtico
-
8/11/2019 tesis aromaticos
20/125
estandarizado para el anlisis de BTEX, como aporte de la universidad en el soporte
tcnico a la comunidad regional.
-
8/11/2019 tesis aromaticos
21/125
PLANTEAMIENTO DEL PROBLEMA
Las consecuencias de la contaminacin de aguas con BTEX sobre el abastecimiento
pblico, su uso agrcola e industrial, as como sus efectos ambientales, pueden llegar a
tener influencias negativas. Corporaciones como la Corporacin Autnoma Regional de
Risaralda (CARDER) y la Secretara de Salud de Risaralda son las encargadas de velar
por el cuidado del medio ambiente en la regin; es por esto que se requiere de una
tcnica analtica instrumental de alta sensibilidad para determinar de manera confiable el
grado de contaminacin y poder tomar as las medidas respectivas.
Teniendo en cuenta la gran demanda del sector industrial y comunidad en general para la
determinacin de BTEX en el agua, y ante la ausencia de un mtodo analtico
estandarizado para el anlisis de estos compuestos en el Laboratorio de Aguas y
Alimentos de la Universidad Tecnolgica de Pereira, es fundamental contar con una
tcnica analtica estandarizada como la cromatografa de gases con detector de
ionizacin por llama que permita implementar el mtodo analtico y sobre todo arrojar
datos con adecuado y comprobable grado de confianza. Igualmente esto ayudara aestudios posteriores del Grupo de Investigacin del Recurso Hdrico, brindndole ms
herramientas para profundizar en el estudio sobre BTEX.
Puede el Laboratorio de Aguas y Alimentos de la Universidad Tecnolgica de Pereira
suplir la necesidad regional para analizar BTEX en matrices acuosas a concentraciones
bajas por medio de la estandarizacin de una tcnica cromatogrfica de alta resolucin?
-
8/11/2019 tesis aromaticos
22/125
OBJETIVOS
1.1 OBJETIVO GENERAL
Estandarizar la tcnica de cromatografa de gases con detector de ionizacin por
llama (CG/FID) para la identificacin y cuantificacin de BTEX en matrices acuosas.
1.2 OBJETIVOS ESPECIFICOS Estandarizar la tcnica de extraccin en fase slida determinando los parmetros de:
porcentaje de recuperacin y precisin.
Obtener en forma experimental los parmetros de: precisin, exactitud, lmite de
deteccin y cuantificacin, linealidad y sensibilidad para la tcnica cromatogrfica.
Analizar el contenido de BTEX en los efluentes (trampas de grasas) de tres estaciones
de servicio de la ciudad de Pereira para determinar su grado de contaminacin.
-
8/11/2019 tesis aromaticos
23/125
1. MARCO TERICO
1.1 ESTANDARIZACIN
La necesidad de contar con mediciones exactas en la actualidad es una premisa
fundamental del desarrollo industrial y por ende, econmico y comercial de la sociedad.
Las mediciones en general, y particularmente las analticas, se utilizan para tomar
decisiones en diferentes campos: uno de ellos es la relacin compra/venta o aceptacin y
rechazo, como puede ser la aceptacin de un producto en otro pas o de un lote de
producto en una empresa; la eleccin de un proveedor o la evaluacin de la conformidadde un producto; en ciertos casos el establecimiento de multas si no se cumple con la
normatividad, etc. Claramente es importante determinar el resultado correcto y ser capaz
de demostrar que lo es, para que cualquier decisin basada en l pueda tomarse con
confianza; es all donde se hace importante la estandarizacin de mtodos de anlisis que
permite demostrar que un mtodo analtico cumple con los requisitos particulares para un
uso especfico en el laboratorio mediante el examen y provisin de evidencias objetivas
[14,15].
La estandarizacin de un mtodo analtico es un proceso riguroso que dependiendo de la
tcnica analtica a la que pertenezca el mtodo, la matriz, el analito, la cantidad de
parmetros de estandarizacin, y de la logstica empleada para su desarrollo, puede
requerir de un tiempo ms o menos considerable (en algunos casos puede superar los
seis meses) [15].
Muchos analistas consideran que un mtodo estndar o de referencia, que ha sido
estandarizado por algn organismo que posee una cierta reputacin, puede aplicarse
directamente al laboratorio. Siguiendo un mtodo previamente estandarizado se puedealcanzar buenos resultados, pero hace falta demostrar que funcionan en nuestro mbito
de trabajo.
Por tanto, un mtodo siempre debe estandarizarse cuando es necesario verificar que sus
parmetros de calidad se adecuan al problema analtico particular que se debe resolver
en el laboratorio [16].
-
8/11/2019 tesis aromaticos
24/125
Siguiendo un mtodo previamente estandarizado se puede alcanzar buenos resultados
pero hace falta demostrar que funcionan en nuestro mbito de trabajo. Por tanto, unmtodo siempre debe estandarizarse cuando es necesario verificar que sus parmetros
de calidad se adecuan al problema analtico particular que se debe resolver en el
laboratorio [16].
De acuerdo al mtodo de ensayo que se est estandarizando; volumtrico, gravimtrico,
instrumental, se debe establecer una metodologa especfica para encontrar los
parmetros que se quieran como son:
1.1.1 LINEALIDAD Y RANGO
La linealidad es la capacidad del mtodo para proporcionar resultados que son
directamente (o por medio de transformaciones matemticas) proporcionales a la
concentracin del analito en la muestra dentro de un rango establecido.
Siempre que sea posible se buscar una respuesta de tipo lineal que facilitar su trazado,
interpolacin e interpretacin. En el caso que la respuesta del mtodo no sea lineal pero si
proporcional a la concentracin son vlidos otros ajustes matemticos.
El rango se define como el intervalo comprendido entre la concentracin mnima y
mxima de analito para el cual se ha demostrado su correcta precisin, exactitud y
linealidad del mtodo descrito.
Para evaluar la linealidad se recomienda que dentro del rango establecido se estudien al
menos 5 niveles de concentracin las cuales se analicen por triplicado (K=5, nde
replicas=3); estadsticamente lo correcto sera analizar las muestras de forma aleatoria,
pero para minimizar posibles efectos de memoria en el equipo es conveniente analizarlasen sentido creciente de concentracin. Otro aspecto importante es realizar pesadas
independientes, ya que as se elimina el posible error sistemtico que se podra arrastrar
partiendo de una sola pesada y realizando diluciones; no obstante, para evaluar la
linealidad en las impurezas se suelen utilizar sucesivas diluciones ya que normalmente se
trabaja a niveles de concentracin muy bajos y esto dificultara las pesadas.
-
8/11/2019 tesis aromaticos
25/125
Con los resultados de estudio de la linealidad se hace una relacin entre las cantidades o
concentraciones X (variable independiente o predictiva) y la respuesta y (variabledependiente, por ejemplo reas, alturas, absorbancias, etc.). La relacin entre ambas
variables se expresa matemticamente como una recta de regresin del tipo y = bx + a,
donde b es el valor de la pendiente y a el trmino independiente. Esta regresin es
obtenida por un mtodo de ajuste (por lo general mnimos cuadrados); en algunos casos
podra ser necesaria alguna transformacin matemtica previa (uso de logaritmos,
recprocos de las variables, etc.) para obtener funciones lineales.
La pendiente bse encuentra relacionada con la sensibilidad del mtodo analtico de forma
que a mayor pendiente mayor sensibilidad (respuesta del mtodo frente a los cambios dela concentracin del analito).
El trmino independiente a, u ordenada en el origen, es la interseccin de la recta con el
eje de ordenadas y es indicativo del error sistemtico. La representacin grfica de la
recta de regresin en un sistema de coordenadas junto con los valores experimentales,
permite visualizar la bondad del ajuste. Si la recta no pasa cerca del origen de
coordenadas significa que el mtodo a evaluar est afectado por un error sistemtico por
defecto o exceso en el intervalo estudiado. Si existen diferencias apreciables entre losvalores experimentales y los puntos de la recta significa que la linealidad no es buena.
Independiente de la apariencia de la recta, resulta conveniente evaluar el coeficiente de
correlacin (r) y el coeficiente de determinacin (r2). El coeficiente de correlacin nos
indica el grado de relacin entre la variable x(concentracin), y la variable y(respuesta).
Su valor mximo es 1. Si res cercano a la unidad significa que existe correlacin con una
probabilidad elevada. Un valor nulo indica ausencia de relacin lineal entre las variables.
El valor recomendable para el coeficiente de correlacin es 0,999, aunque en el caso deimpurezas se admite 0,990.
La informacin obtenida mediante el clculo de res limitada y no justifica por s sola la
linealidad, siendo r2 coeficiente de determinacin el que aporta una mayor significacin
estadstica ya que representa la proporcin de la variacin total de y explicada por el
modelo [17].
-
8/11/2019 tesis aromaticos
26/125
1.1.2 PRECISIN
La precisin est relacionada con la dispersin de las medidas alrededor de su valormedio o central y corresponde al grado de concordancia entre ensayos individuales
cuando el mtodo se aplica repetidamente a mltiples alcuotas de una muestra
homognea.
La precisin se expresa matemticamente como la desviacin estndar (SD), o ms
comnmente como la desviacin estndar relativa (RSD) o coeficiente de variacin (CV)
[18].
El objetivo del estudio de la precisin es conocer la variabilidad o el ms-menos delmtodo de ensayo. Esta variabilidad es debida a errores aleatorios inherentes a todo
mtodo de ensayo. Como consecuencia de la existencia de estos errores, los anlisis
efectuados sobre muestras idnticas, en las mismas circunstancias, no conducen
generalmente a resultados idnticos. Los factores susceptibles que influirn sobre los
resultados de un ensayo no pueden ser siempre controlados (analista, equipo,
instrumental, reactivos, tiempo, etc.) de aqu la importancia del estudio de la precisin.
La precisin diferentes tipos de estudios:
1.1.2.1 Repetibilidad: se expresa matemticamente por el coeficiente de variacin
(desviacin estndar relativa) de una serie de medidas. Esta estudia la variabilidad del
mtodo efectuando una serie de anlisis sobre la misma muestra en las mismas
condiciones operativas (por un mismo analista, con los mismos aparatos y reactivos, etc.),
en un mismo laboratorio y en un periodo de tiempo corto.
Uno de los factores que ms puede influir en la Repetibilidad del mtodo de anlisis es la
concentracin del analito, ya que la desviacin estndar de las respuestas obtenidas
aumenta al disminuir la concentracin del analito.
1.1.2.1.1 Repetibilidad del sistema instrumental
Este parmetro estudia la variabilidad debida nicamente al instrumento, y se determina
analizando repetidamente una misma muestra de forma consecutiva de 6 a 10 veces. La
estimacin de esta se realiza con el clculo del coeficiente de variacin de las respuestas
obtenidas.
-
8/11/2019 tesis aromaticos
27/125
Los resultados obtenidos en la repetibilidad instrumental dependen del instrumento, por
ejemplo no se puede obtener el mismo coeficiente de variacin en un equipo coninyeccin automtica que con inyeccin manual.
1.1.2.1.2 Repetibilidad del mtodo
El ensayo de Repetibilidad del mtodo se efecta sobre una serie de alcuotas de una
muestra homognea que se analiza independientemente desde el principio (preparacin
de muestra) hasta el final (lectura de resultados) por el mismo instrumento y el mismo
analista.
La Repetibilidad del mtodo depende generalmente del proceso de preparacin de la
muestra. Es decir, cuanto mayor sea la manipulacin de la muestra ms probable es que
la variabilidad del mtodo aumente.
1.1.2.2 Precisin intermedia:Estudia la variabilidad del mtodo efectuando una serie de
anlisis sobre la misma muestra pero en condiciones operativas diferentes (diferentes
analistas, aparatos, das, etc.) y en un mismo laboratorio.
1.1.2.3 Reproducibilidad:Estudia la variabilidad del mtodo bajo condiciones operativas
diferentes y en distintos laboratorios [17].
1.1.3 EXACTITUD
La exactitud de un procedimiento analtico expresa la proximidad entre el valor que es
aceptado convencionalmente como valor verdadero o un valor de referencia y el valor
experimentalmente encontrado.
De esta definicin surge el problema de saber cul es el valor verdadero. No obstante,cuando se dispone de patrones de referencia certificados, el valor de dicho patrn es el
que se acepta como valor verdadero y la exactitud puede evaluarse aplicando el mtodo
sobre dicho patrn, o bien analizando muestras de placebo o de problema a las que se ha
aadido una cantidad conocida de dicho patrn. Tambin se acepta la comparacin de los
resultados con un mtodo de referencia validado del que se ha demostrado su exactitud;
entonces el valor verdadero es el que se obtiene con dicho mtodo de referencia.
-
8/11/2019 tesis aromaticos
28/125
La exactitud debe determinarse en todo el rango especificado para el mtodo analtico. Se
recomiendan un mnimo de 9 determinaciones sobre 3 niveles de concentracin delanalito que cubran el rango especificado (por ejemplo 3 determinaciones por 3 niveles de
concentracin, que podra ser la concentracin central y las concentraciones en los
extremos del rango).
La exactitud se expresar como porcentaje de recuperacin en la valoracin de una
cantidad conocida de analito aadida sobre la muestra o como la diferencia entre la media
obtenida y el valor aceptado como verdadero junto a los intervalos de confianza.
No siempre se obtienen valores de recuperacin cercanos al 100%, ya que sta dependede la matriz de la muestra, de la efectividad de mtodo de preparacin y extraccin y de la
concentracin del analito. Aunque es deseable alcanzar valores de recuperacin cercanos
al 100%, en algunas muestras de matrices complejas solo se obtienen valores del 50, 80
o 90%. En estos casos es importante que aunque la recuperacin sea baja, la precisin
del mtodo sea alta ya que entonces puede intentar aplicarse un factor de correccin.
La desviacin de la exactitud por exceso se produce cuando existen interferencias y la
selectividad del mtodo no es la adecuada, entonces se obtienen resultados superiores al
valor verdadero. En este caso, si es posible, se debera modificar las condiciones delmtodo para optimizar la selectividad o bien cambiar a otro alternativo que sea selectivo.
La desviacin de la exactitud por defecto suele producirse cuando la matriz de la muestra
es compleja y la extraccin del analito requiere varios pasos obtenindose recuperaciones
ms bajas. Cuando esto ocurre sera conveniente intentar optimizar la preparacin de la
muestra para mejorar el factor de recuperacin. Si esto es muy costoso o no es posible,
cuando la exactitud obtenida es repetible, es decir, tienen una precisin elevada y adems
es homognea en todos los niveles de concentracin estudiados, puede aplicarse unfactor de correccin en el clculo final para compensar las prdidas del analito debidas al
mtodo de extraccin [17].
1.1.4 LMITE DE DETECCIN (LD):
El lmite de deteccin (LD) corresponde a la mnima cantidad de analito en la muestra que
se puede detectar aunque no necesariamente cuantificar en las condiciones establecidas
-
8/11/2019 tesis aromaticos
29/125
y se expresa en unidades de concentracin (%, ppm, ppb, etc.). Su determinacin puede
efectuarse mediante la relacin entre el ruido y la seal debida al analito.
1.1.5 LMITE DE CUANTIFICACIN (LC):
Dado un mtodo analtico determinado, se entiende por lmite de cuantificacin (LC) de
dicho mtodo, la mnima cantidad de analito presente en la muestra que se puede
cuantificar, bajo las condiciones experimentales descritas, con una adecuada precisin y
exactitud; tambin se expresa en unidades de concentracin [17,18].
Existen diversos procedimientos de anlisis y sistemas instrumentales que dependiendo
de sus caractersticas definen en muchos casos cul es el mtodo ptimo para determinar
tanto el lmite de deteccin como el de cuantificacin. Entre los mtodos ms comunes se
tienen los siguientes:
1.1.5.1 Mtodo basado en la relacin seal/ruido
Este mtodo, uno de los ms conocidos y empleados, requiere que el procedimiento de
anlisis sea instrumental y que proporcione una seal blanco, un ruido de fondo o una
lnea de base, es decir una seal residual a concentracin cero de analito
(espectrofotometra UV-visible o la cromatografa de gases o lquida).
Este procedimiento presenta la desventaja de que en numerosas ocasiones al llevar a
cabo la comprobacin experimental del LC calculado, se observa que es posible obtener
resultados igualmente precisos y exactos an cuando se desciende ms en la
concentracin lmite
1.1.5.2 Mtodo basado en la desviacin estndar de la respuesta del blanco y la
pendiente de la recta de calibrado
De acuerdo a la IUPAC (International Union of Pure and Applied Chemistry), puede
calcularse el LD Y LC de un mtodo analtico a partir del conocimiento de la desviacin
atribuible a la respuesta de una muestra de placebo y la pendiente de la recta de
calibrado del analito.
La expresin a aplicar para este clculo vara en funcin de si el mtodo instrumental
empleado corrige la seal frente a un blanco o no.
-
8/11/2019 tesis aromaticos
30/125
1.1.5.2.1 Mtodos instrumentales que corrigen la seal frente a un blanco
Este primer caso correspondera a un mtodo espectrofotomtrico en el que se podra
calcular el LD y el LC tericos mediante la expresin:
Donde:
CL= Concentracin de analito en el lmite de cuantificacin o deteccin.
K= Constante que usualmente se considera igual a 10 para el LC e igual a 3 para el LD.
Sbl= desviacin estndar correspondiente a la seal del blanco o placebo.
pendiente de la curva de calibracin obtenida al representar la respuesta del mtodo
frente a la concentracin de analito. Evidentemente el rango de esta recta tiene que ser
cercano en concentraciones a los niveles lmite de cuantificacin [17].
Si el mtodo analtico realiza la lectura final por duplicado o triplicado, mejorando con ello
la precisin, se ha de introducir en la frmula el trmino correspondiente a las rplicas (n)
en la siguiente forma:
1.1.5.2.2 Mtodos instrumentales que no corrigen la seal frente a un blanco
El caso en que no se realiza correccin frente a un blanco es tpicamente el de mtodos
cromatogrficos GC o HPLC. En stos se ha de tener en cuenta tambin la seal media
obtenida del anlisis correspondiente al placebo, es decir, el ruido de fondo o backgrounddel sistema (Ybl) con lo que la expresin final ser entonces:
-
8/11/2019 tesis aromaticos
31/125
1.1.5.3Mtodo basado en la extrapolacin de la recta de calibrado a concentracin
cero
Se trata de un procedimiento aplicable tambin a mtodos analticos instrumentales que
proporcionan resultados numricos y dirigido a evitar el clculo, en ocasiones costoso en
tiempo, como se ha podido observar, de la seal media del blanco y su desviacin
estndar. El mtodo utiliza la pendiente de una recta de calibrado realizada a niveles de
concentracin cercanos a los lmites esperados, pero sustituye el valor real de la seal del
blanco por el resultante de la extrapolacin de dicha recta. La interseccin con el eje Y
corresponder tericamente al valor de la respuesta a concentracin cero de analito [17].
1.1.6 SENSIBILIDAD
La sensibilidad de un mtodo analtico corresponde a la mnima cantidad de analito que
puede producir un resultado significativo [18]. En contraste con el lmite de deteccin, la
sensibilidad de un mtodo est definida como la habilidad para distinguir entre diferentes
concentraciones, y se calcula as:
Para mtodos donde la respuesta con respecto a la concentracin es una funcin lineal, la
sensibilidad es constante con respecto a la concentracin y es igual a la pendiente de la
curva de calibracin. Contrariamente a las funciones lineales, la sensibilidad de mtodos
cuando su respuesta es no-lineal cambia con la concentracin del analito [19].
1.2 CROMATOGRAFA DE GASES
Desde sus inicios en los aos cincuenta, la cromatografa de gases (GC) se ha convertido
en la tcnica principal para la separacin y anlisis de compuestos voltiles. A partir de
entonces, las aplicaciones de sta tcnica han ido aumentado a medida que se han
mejorado los instrumentos de cromatografa; siendo factible la separacin, caracterizacin
y cuantificacin de una gran variedad de compuestos tanto en muestras ambientales,
biolgicas y mdicas, como en comidas, sabores y fragancias. En la actualidad es usada
-
8/11/2019 tesis aromaticos
32/125
rutinariamente en multitud de laboratorios universitarios, de investigacin e industriales,
debido a su alta resolucin, sensibilidad y selectividad. Adems de las aplicacionestpicamente analticas, la GC puede utilizarse a escala preparativa para la obtencin de
compuestos de elevada pureza [20,25].
En cromatografa de gases los analitos (siempre en estado gaseoso) se distribuyen entre
una fase mvil gaseosa y una fase estacionaria que puede ser un slido o una delgada
pelcula lquida que recubre al slido; dependiendo de la fase estacionaria que se utilice
(slida o lquida) la cromatografa en fase gaseosa se clasifica en: cromatografa gas-
slido(CGS) y cromatografa gas-lquido (CGL). De las dos modalidades, la CGL es la
forma ms selectiva de la cromatografa y la que se presta a mayores usos.
En cromatografa de gases, la fase mvil se denomina gas transportador, ya que es un
gas inerte cuya finalidad es transportar las molculas de la muestra a travs de la
columna. Los adsorbentes, tales como gel de slice, almina, sales inorgnicas, polmeros
porosos, tamices moleculares y carbn grafitizado, son las fases estacionarias en la CGS.
Esta se utiliza principalmente para la separacin de gases permanentes y compuestos
orgnicos muy voltiles. Los lquidos orgnicos de alto punto de ebullicin constituyen la
fase estacionaria de la CGL. La fase liquida se extiende como una pelcula delgada sobreun slido inerte llamado soporte slido. La base para la separacin es la particin de la
muestra dentro o fuera de esta pelcula lquida. Si se puede encontrar una fase lquida
que tenga solubilidad selectiva para dos compuestos, entonces estos dos pueden
separarse mediante cromatografa de gases.
1.2.1 VENTAJAS
Las siguientes son algunas ventajas generales de GC que cabe destacar:
Alta Resolucin: La eficiencia puede ser expresada en nmeros de platos, y lascolumnas capilares suelen tener nmeros de platos de cientos de miles. Los ismeros
con puntos de ebullicin muy prximos que no pueden separarse por destilacin se
separan fcilmente mediante la cromatografa de gases. Adems, el hecho de que las
concentraciones de soluto son muy diluidas, en las columnas de GC se elimina la
posibilidad de azetropos, que a menudo plaga las separaciones por destilacin.
-
8/11/2019 tesis aromaticos
33/125
La CG se presta a usos ms variados que la mejor columna de destilacin, ya que la
columna cromatogrfica puede sustituirse fcilmente. Esto permite la separacinselectiva debido a solubilidades diferentes, aun cuando los puntos de ebullicin estn
muy cercanos. Como hay numerosas columnas, se puede escoger entre ellas, lo que
confiere variedad a la gama de muestras que pueden manejarse [20].
Sensibilidad:Esta caracterstica del sistema de cromatografa de gases explica en
gran medida su uso extensivo. El ms simple detector de conductividad trmica
puede medir fcilmente microgramos. El detector de ionizacin por llama fcilmente
mide nanogramos (10-9g), y los detectores ms selectivos como el de captura de
electrones y el detector fotomtrico de llama alcanzan los picogramos (1012g). Este
nivel de sensibilidad es ms impresionante si se tiene en cuenta que el tamao de la
muestra utilizada es del orden de 1L o menos.
Tiempo de anlisis:La separacin de todos los componentes de una muestra puede
tardar desde varios segundos hasta 30 minutos. Anlisis que rutinariamente tardan
una hora o ms se pueden reducir a una cuestin de minutos, debido a la alta tasa de
difusin en fase gaseosa y el rpido equilibrio entre las fases mvil y estacionaria
[23].
Resultados cuantitativos: La GC permite obtener muy buenos resultados
cuantitativos. Sin embargo, la exactitud es funcin de muchos factores. Se puede
obtener buena exactitud en una amplia gama de concentraciones de la muestra,
desde miligramos hasta nanogramos [22].
Comodidad:El funcionamiento del GC es un procedimiento relativamente sencillo.
No es difcil para capacitar al personal no tcnico para llevar a cabo separaciones de
rutina [23].
Costos: En comparacin con muchos instrumentos de anlisis disponibles en la
actualidad, los cromatgrafos de gases representan un valor excelente [23].
1.2.2 INSTRUMENTACIN EN CROMATOGRAFA DE GASES
-
8/11/2019 tesis aromaticos
34/125
En 1954 se introdujo en el mercado el primer cromatgrafo de gases comercial.
Rpidamente fue aceptado como un importante instrumento para el anlisis qumico tantoen la investigacin como en la industria. Con igual rapidez se han ideado y desarrollado
perfeccionamientos, accesorios, complementos y variantes, que no han cesado de
aparecer hasta la fecha. En los aos sesenta, se hicieron habituales los integradores
electrnicos y los equipos para el procesamiento de datos basados en una computadora.
Los aos ochenta introdujeron la utilizacin de las computadoras para el control
automtico de la mayora de los parmetros instrumentales, tales como la temperatura de
la columna, caudales y la inyeccin de la muestra; el desarrollo de instrumentos de alto
rendimiento a un coste moderado; y tal vez lo ms importante, el desarrollo de las
columnas abiertas que son capaces de separar una multitud de analitos en un tiempo
relativamente corto [22,25].
Un cromatgrafo de gases funciona de la siguiente manera. Un gas portador inerte (como
el helio) fluye continuamente desde un cilindro de gas de gran tamao mediante el puerto
de inyeccin, la columna, y el detector. La tasa de flujo del gas portador es
cuidadosamente controlada para garantizar tiempos de retencin reproducibles y reducir
al mnimo el ruido y la deriva del detector. La muestra se inyecta (normalmente con una
microjeringa) en el puerto de inyeccin con calefaccin donde se vaporiza y es llevada ala columna; por lo general una columna capilar de 15 a 30 m de largo recubierta en su
interior con una delgada (0,2 m) pelcula de un lquido de alto punto de ebullicin (fase
estacionaria). Se da la particin de la muestra entre las fases mvil y estacionaria,
ocasionando la separacin en componentes individuales basados en la solubilidad relativa
en la fase lquida y presin de vapor relativa.
Despus de la columna, el gas portador y la muestra pasan por un detector. Este
dispositivo mide la cantidad de la muestra, y genera una seal elctrica. Esta seal va a
un sistema de datos integrador que genera un cromatograma (el acta de anlisis). En la
mayora de los casos el sistema informtico de gestin integra automticamente el rea
del pico, calcula el rendimiento e imprime un informe con los resultados cuantitativos y
tiempos de retencin. Cada uno de estos siete componentes se muestran en la figura 1.
-
8/11/2019 tesis aromaticos
35/125
Figura 1.Diagrama de un sistema de cromatografa gas-lquido.
1.2.2.1 GAS PORTADOR
El gas portador es la fase mvil en GC. Su objetivo principal es la de llevar la mezcla de
los solutos desde que se introduce en el sistema cromatogrfico hasta la salida del
detector, pasando a travs de la columna donde se produce la separacin. Debe ser
qumicamente inerte y no interaccionar ni con la columna ni con los componentes de la
mezcla, es decir, no debe afectar a los procesos de particin o de adsorcin. Un objetivo
secundario es proporcionar una matriz adecuada para el detector que permita medir los
componentes de la muestra [22].
Los gases ms utilizados son: nitrgeno, hidrgeno y helio. La eleccin de uno de ellos va
a depender de: la fase estacionaria, y el tipo de detector utilizado. Otros aspectos a
considerar seran costo, pureza y seguridad en su uso [22,24].
-
8/11/2019 tesis aromaticos
36/125
El Helio es el gas portador ms popular debido a que presenta mayor eficiencia a ratas de
flujo rpidas, se utiliza para los detectores de conductividad trmica y de ionizacin porllama. El Hidrgeno tambin es comnmente usado, aunque no se recomienda por su
potencial de explosin. El nitrgeno proporciona una sensibilidad un poco mayor, pero un
anlisis ms lento que el helio; se utiliza tanto para el detector de captura de electrones
como el detector de ionizacin por llama [20,22].
Es importante que el gas portador sea de alta pureza. Las impurezas (en especial oxigeno
y agua) pueden alterar qumicamente la fase lquida y, por ende, modificar los tiempos de
retencin. Las columnas de polisteres, poliglicoles y poliamidas son susceptibles de ser
degradadas por el oxigeno y el agua. Trazas de agua pueden des-adsorber otroscontaminantes en la columna y producir numerosas seales en el detector o hasta picos
fantasma [22].
1.2.2.2 SISTEMA DE INYECCIN
Las muestras para GC pueden ser gases, lquidos o slidos. La cantidad de muestra que
se debe introducir depende del tamao de la columna; pero, generalmente en
cromatografa de gases se utilizan muestras pequeas (entre 0,01 y 20 L) [22,25].
La eficacia de la columna requiere que la muestra sea de un tamao adecuado y que sea
introducida como un tapn de vapor; la inyeccin lenta de muestras demasiado grandes
provoca un ensanchamiento de las bandas y una pobre resolucin. El mtodo ms comn
de inyeccin de muestra implica el uso de una microjeringa para inyectar una muestra
liquida o gaseosa a travs de un septum de goma de silicona, en una cmara de
vaporizacin instantnea situada en la cabeza de la columna (esta cmara normalmente
est unos 50 C por encima del punto de ebullicin del componente menos voltil de la
muestra); el mbolo de la jeringa puede manejarse a mano (inyeccin manual) o mediante
un dispositivo electrnico o neumtico (inyeccin automtica). Para columnas capilares el
tamao de la muestra es de aproximadamente 103L; por lo cual, para mantener la
cantidad de muestra en el intervalo correcto, existe un inyector especial que reduce la
cantidad de muestra que llega a la cabeza de la columna capilar: el inyector split/splitless.
En el modo split la muestra se divide permitiendo pasar a la columna solamente una
pequea fraccin de la muestra, desechando el resto; mientras que en el modo splitless
toda la muestra se inyecta en la columna [25,27].
-
8/11/2019 tesis aromaticos
37/125
-
8/11/2019 tesis aromaticos
38/125
En general, la resolucin ptima se asocia con una temperatura mnima; en contrapartida
la reduccin de temperatura produce un aumento en el tiempo de elusin, y por lo tantodel tiempo que se necesita para completar el anlisis [25].
1.2.2.4 SISTEMAS DE DETECCIN
En el cromatgrafo de gases uno de los elementos ms importantes es el detector; este
es un dispositivo que indica y mide los solutos en la corriente del gas portador,
convirtiendo una seal no medible directamente en una seal elaborable de una
propiedad fsica. Esta seal es elaborada por una comparacin entre el gas portador puro
y el mismo gas llevando cada uno de los componentes previamente separados en la
columna, esto es traducido en una seal elctrica que es amplificada y registrada al
momento de salir de la columna [29].
Varias son las caractersticas generales que debe reunir un detector para ser utilizado en
cromatografa, y que se pondrn de manifiesto en la generacin y calidad de la seal del
mismo. Estas caractersticas son las siguientes:
Sensibilidad adecuada: La sensibilidad del detector indica la respuesta del mismo
ante un cambio de la propiedad fsica que mide; a su vez, este cambio de
propiedad fsica se deber a la presencia de una menor o mayor cantidad de
componente en el detector. Debe ser lo ms alta posible.
Buena estabilidad y reproducibilidad: La lnea base de un cromatograma est
sometida a fluctuaciones fortuitas, conocidas como ruido de fondo, el cual se
puede producir en los distintos componentes del cromatgrafo. Originar una seal
estable (lnea de base) y reproducible (pico) [21,25].
Respuesta lineal: la linealidad del detector considera que la respuesta del mismo,
seal, sea proporcional a la variacin en la cantidad de componente que en un
momento dado se encuentre en el detector. Esta debe extenderse a varios
rdenes de magnitud.
Intervalo de temperaturas de trabajo comprendido desde la temperatura ambiente
hasta al menos 400 C.
Tiempo de respuesta corto que sea independiente del caudal.
Respuesta semejante para todos los solutos o, por el contrario, una respuesta
selectiva y altamente predecible para uno o ms tipos de solutos [25].
-
8/11/2019 tesis aromaticos
39/125
Adems de las caractersticas indicadas, es deseable que el detector posea tambin:
tiempo de respuesta corto, resistencia mecnica y qumica, sencillez de manejo ymantenimiento.
Algunos detectores son universales, es decir que son sensibles a prcticamente todos los
compuestos que eluyen de la columna. Por otro lado, hay detectores discriminativos
(selectivos) que son sensibles solo a compuestos especficos, dando un cromatograma
muy sencillo. Tambin pueden ser clasificados como destructivos o no de los analitos.
Los detectores se clasifican en dos grupos dependiendo de si slo conducen a una
informacin nica, como el tiempo de retencin y los que producen, adems de tiempo deretencin, la informacin estructural del analito en cuestin. Por esta razn, algunos
cromatgrafos de gases estn equipados con dos o tres detectores vinculados en serie.
Sin embargo, la respuesta de todos los detectores depende de la concentracin molar o
de la masa de analito en la compaa de gas de arrastre [28].
En la tabla 1 puede observarse los detectores ms utilizados, con un breve resumen de
sus ventajas.
1.2.2.4.1 DETECTOR DE IONIZACIN POR LLAMA (FID)
El FID es el ms usado de los detectores, posee una alta sensibilidad y es de respuesta
universal, lo que significa que responde casi de la misma manera por unidad de masa de
analito sin que influya su estructura qumica mientras tenga carbonos orgnicos. Es
insensible a los gases no combustibles como H2O, CO2, SO2, y NOX.
El FID se basa en la conductividad elctrica de los gases. A temperatura y presin
normales, los gases se comportan como aislantes, pero si en su interior existen tomos omolculas cargadas elctricamente, o electrones libres, se produce un incremento en la
conductividad. Las molculas de la muestra, que estn presentes en el gas de arrastre,
llegan al detector y son quemadas por la llama producida por la combustin de aire e
hidrgeno, dando como resultado la formacin de iones los cuales son reunidos en un
electrodo colector que genera una corriente, que es convertida en voltaje, y
posteriormente amplificada para ser captada por el registrador [21]. Un detector de
ionizacin por llama caracterstico se observa en la figura 2.
-
8/11/2019 tesis aromaticos
40/125
-
8/11/2019 tesis aromaticos
41/125
que contienen carbono con alta sensibilidad (aproximadamente 10-13g/mL); (b) no
responde a las impurezas comunes del gas portador como agua y dixido de carbono; (c)cuenta con un amplio rango de respuesta lineal (aproximadamente 107) y una excelente
estabilidad de la lnea base; (d) es relativamente insensible a pequeos cambios de
flujo/rata en la columna durante la programacin de la temperatura; (e) es altamente
seguro, duradero, y fcil de usar; y (f) tiene detector bajo de volumen muerto y rpida
respuesta. Sus limitaciones son: (a) se da poca o ninguna respuesta a los gases no
combustibles y todos los gases nobles; y (b) es un detector destructivo que modifica las
propiedades fsicas y qumicas de la muestra de forma irreversible [13].
Figura 2.Detector de ionizacin por llama caracterstico [22].
El FID responde a compuestos que en la combustin ceden especies con carga elctrica
en una llama hidrgeno/aire, la reaccin de los radicales libres que se da es:
-
8/11/2019 tesis aromaticos
42/125
Estas especies cargadas, bajo la influencia de un campo elctrico, son capturadas en un
electrodo colector y medidas por un electrmetro, cuya salida es amplificada. El campo deaplicacin del FID es muy grande, ya que responde a casi todos los compuestos
orgnicos. Una desventaja es que a menudo es demasiado inespecfico y poco sensible
para el anlisis medioambiental y el anlisis de residuos [26].
1.2.3 ANLISIS CUALITATIVO Y CUANTITATIVO:
La cromatografa ha llegado a ser el principal mtodo para la separacin de especies
qumicas estrechamente relacionadas entre s. Adems, se puede emplear para laidentificacin cualitativa y cuantitativa de las especies separadas [25].
1.2.3.1 Anlisis cualitativo:
El tiempo de retencin o el volumen de retencin para un soluto dado se pueden utilizar
para su identificacin si se mantienen constantes las siguientes variables de la columna:
longitud, espesor, temperatura y presin (rata del flujo del gas portador). Sin embargo,
cuando se parte de una muestra desconocida, se hace difcil identificar sus componentes
por este procedimiento, ya que los miles de compuestos conocidos hacen que existandemasiadas posibilidades entre las que elegir [22].
El analista no siempre se enfrenta con muestras totalmente desconocidas, por lo que, en
muchos casos, el problema puede resolverse cromatogrficamente. El procedimiento ms
simple de anlisis cualitativo se realiza con ayuda de patrones: los tiempos de retencin
de los picos desconocidos se comparan con los tiempos de retencin de compuestos
conocidos, separados en la misma columna y en las mismas condiciones experimentales.
Como alternativa, cuando se sospecha que un componente ya identificado corresponde a
un pico dado se aade a la muestra problema algo de componente puro, y se realiza un
cromatograma. En el registro obtenido debe aparece el pico del componente aumentado
en su dimensin [21,30].
1.2.3.2 Anlisis cuantitativo:
En cromatografa de gases los parmetros cuantitativos son la altura, o el rea, del pico
del analito, las cuales son comparadas con la de uno o ms patrones.
-
8/11/2019 tesis aromaticos
43/125
Anlisis basados en la altura del pico:
El uso de la altura del pico presenta como ventaja la comodidad de medida, pero
solamente proporciona una exactitud aceptable en el caso de muestras sencillas de pocos
componentes que den lugar a picos agudos, estrechos y claramente separados. Adems
la altura del pico es muy sensible a variaciones en las condiciones de operacin, por lo
que el uso de este parmetro exige un control cuidadossimo de dichas condiciones. Las
medidas de altura de pico son interesantes para los anlisis de rutina, en los que se
puede sacrificar algo de la exactitud a favor de la sencillez y rapidez de las
determinaciones [20,25].
Anlisis basados en las reas de los picos:
El uso, como parmetro, del rea del pico es ms acertado cuando se requiere mayor
exactitud en las determinaciones cuantitativas. Como se sabe, el rea del pico es funcin
de la cantidad de componente o de la concentracin del mismo [21,25].
Los resultados de las reas o alturas de los picos de los analitos, se utilizan para
determinar las concentraciones exactas de cada una de las especies mediante los
siguientes mtodos:
1.2.3.2.1 Mtodo del patrn interno:
En cromatografa cuantitativa la mayor precisin se consigue por el uso de patrones
internos debido a que se evitan las incertidumbres asociadas a la inyeccin de la muestra.
En este procedimiento, se introduce en cada estndar y en la muestra una cantidad
exactamente medida del patrn interno, y la relacin de las reas (o alturas) del analito y
del patrn interno sirve como parmetro analtico [25].
1.2.3.2.2Mtodo del estndar externo:
Un estndar externo es el que se analiza separadamente de la rplica desconocida que
se est ensayando. Los estndares, que contienen distintas concentraciones conocidas
de analito junto a la matriz que es similar o idntica a la de la muestra, son inyectados. A
continuacin se obtienen los cromatogramas de los patrones y se representan las alturas
o reas de pico en funcin de la concentracin. La representacin grfica de los datos
-
8/11/2019 tesis aromaticos
44/125
-
8/11/2019 tesis aromaticos
45/125
Los principales objetivos de la SPE son: a) eliminacin de componentes que interfieren
con la matriz, b) concentracin selectiva y aislamiento de los analitos, y c) cambio de lamatriz del analito segn sea necesario para su posterior anlisis. El enriquecimiento
puede aumentar la sensibilidad de deteccin; a menudo este paso es necesario para
alcanzar el lmite de deteccin de la concentracin de analitos de inters para los anlisis
cualitativos y cuantitativos, sin enriquecimiento a menudo un anlisis fiable a nivel de
trazas no es posible [36].
Es esencialmente una tcnica de separacin basada en los mismos principios de la
cromatografa lquida, con un poder de resolucin menor, pero con buena selectividad. En
SPE se hace pasar una disolucin que contiene los analitos sobre una fase slida (o faseestacionaria) que los absorbe especficamente, la cual suele estar compactada en el
fondo de una pequea columna de plstico. Despus de la adsorcin, los analitos se
eluyen con una pequea cantidad de otro disolvente extractor, con el que interaccionan
ms fuertemente que con la fase estacionaria. Por tanto, la SPE no solo consigue un
cambio de matriz del analito, sino que reduce el volumen de la muestra [31,33].
1.3.1 CONSIDERACIONES TERICAS
En cualquier sistema de extraccin en fase slida se cuentan tres componentes a saber:1) la muestra problema que contiene los analitos en una matriz compleja, 2) la fase
estacionaria o soporte slido (cartucho) y 3) los solventes de acondicionamiento, lavado y
elucin.
Los componentes de la muestra se separan por migracin diferencial desde la fase
estacionaria hacia los solventes de lavado y elucin, atendiendo a las diferencias en
cuanto a propiedades fsicas y qumicas, las cuales favorecen las fuerzas de retencin
para algunos compuestos y las fuerzas de elucin para otros [33].
Las diferentes fases estacionarias se clasifican segn el tipo de interaccin que tengan
con el analito. Las interacciones son consecuencias de sus propiedades qumicas y
bsicamente son de tres tipos: interacciones polares, interacciones no polares e
interacciones de intercambio inico. Esta ltima clase se subdivide en intercambio
aninico e intercambio catinico.
-
8/11/2019 tesis aromaticos
46/125
Figura 3. Fases estacionarias polares y sus interacciones con los analitos [38].
1.3.1.1 Interacciones polares:
Las interacciones polares que se dan en la SPE incluyen puentes de hidrgeno,
interacciones dipolo-dipolo y otras interacciones entre tomos del analito y los grupos
polares de la fase estacionaria. La retencin de analitos por interacciones polares se
facilita mediante disolventes no polares. Por otra parte, la elucin de analitos desde
adsorbentes polares se facilita mediante disolventes polares con alta fuerza inica. Este
mecanismo de interaccin se denomina particin en fase normal [31].
Los adsorbentes polares ms comnmente utilizados para fase normal en SPE son slica
(SiO2)x, almina (Al2O3), silicato de magnesio (MgSiO3 o Florisil), y los adsorbentes de
slice enlazada en los que la slice reacciona con grupos funcionales altamente polares
para producir aminopropil [(SiO2)x-(CH2)3NH2]-, cianopropil [(SiO2)x-(CH2)3CN]-, y diol
-
8/11/2019 tesis aromaticos
47/125
-
8/11/2019 tesis aromaticos
48/125
ciclohexil, unidos covalentemente a la columna vertebral de la slica gel (Figura 4). Grupos
fenil aromticos tambin se pueden unir [38].
1.3.1.3 Interacciones inicas:
Las interacciones inicas ocurren entre molculas de analito con cargas opuestas a las
del adsorbente. Para que se d el intercambio inico se deben cumplir dos condiciones:
La matriz y el disolvente deben estar a un pH donde el analito y el adsorbente
estn cargados.
La matriz y el disolvente no deben contener altas concentraciones de iones de lamisma carga que el analito[31].
Los grupos inicos pueden estar cargados (positiva o negativamente) o no dependiendo
del pH. Cuando el adsorbente contiene grupos funcionales cargados positivamente y el
contrain intercambiable del analito en la matriz de la muestra lquida est cargado
negativamente, el proceso de acumulacin se llama intercambio aninico. Por el contrario,
si el grupo funcional en la superficie del adsorbente est cargado negativamente y el
contrain intercambiable del analito en la matriz de la muestra lquida est cargado
positivamente, el proceso de acumulacin se llama intercambio catinico [31,38].
Los adsorbentes de intercambio inico contienen grupos funcionales ionizados (aminas
cuaternarias o cidos sulfnicos), o grupos funcionales ionizables (aminas
primarias/secundarias o cidos carboxlicos). En el caso de los adsorbentes de
intercambio inico por lo general contienen grupos funcionales dbilmente bsicos como
las aminas primarias o secundarias (se cargan a condiciones de pH bajos) o grupos de
amonio cuaternario muy bsicos (se cargan a todos los pH). Los adsorbentes de
intercambio catinico contienen grupos funcionales dbilmente cidos como los cidoscarboxlicos (se cargan a condiciones de pH alto), aromticos muy cidos o cidos
sulfnicos alifticos (se cargan a todos los niveles de pH) [38].
La fuerza inica tambin juega un papel importante en las separaciones SPE de
intercambio inico. La fuerza inica es una medida de la concentracin total de iones
presentes en el medio. La retencin del analito en la fase estacionaria ser funcin del
nmero de otras especies inicas presentes en la matriz y disolvente capaces de competir
-
8/11/2019 tesis aromaticos
49/125
por los grupos cargados del adsorbente. As, bajas fuerzas inicas promueven la
retencin del analito, mientras que altas fuerzas inicas facilitan la elucin [31].
En la figura 5 se observan algunas fases estacionarios de intercambio inico.
Figura 5.Fases estacionarias de intercambio inico.
La mayora de las fases estacionarias utilizadas para SPE son de slica gel o
modificaciones de sta con tamaos y partculas en promedio de 40 m y tamaos de
poro por lo general de 60 , exceptuando aquellas usadas en las separaciones por
tamao molecular [33]. Las columnas para extraccin en fase slida tienen capacidad de
depsito de 1mL, 3mL, y 6mL, pero pueden ser adaptados depsitos de 15 75mL
cuando el volumen de la muestra es muy grande [39].
Algunas de las caractersticas estructurales de las fases estacionarias con su respectivo
mecanismo de separacin se resumen en la tabla 2.
-
8/11/2019 tesis aromaticos
50/125
Cod. Estructura Mecanismo retencin-
elucinSi Slicagel (Polar) Fase Normal
Florisil Silicato de Magnesio (Polar) Fase Normal
Diol -(CH2)3-O-CHOHCH2OH Fase Normal
NH2 -(CH2)3-NH2Aminopropil Fase Normal
Al-A Almina cida Fase Normal
Al-B Almina bsia Fase Normal
Al-N Almina neutra Fase Normal
CN -(CH2)3-CN Cianopropil Fase Normal/ReversaC-8 -(CH2)7-CH3 Octil Fase Reversa
C-18 -(CH2)17-CH3Octadecil Fase Reversa
Ph -(CH2)3-Phe Fenilpropil Fase Reversa
NH/NH2 -(CH2)3-NHCH2CH2NH2
Diamino (WAX)
Intercambio Aninico (Dbil)
SAX -(CH2)3-N+(CH3)3Cl
- Intercambio Aninico
(Fuerte)
SCX C6H4-SO2OH (Fuerte) Intercambio catinicoWCX -(CH2)3-COOH (Dbil) Intercambio catinico
G-25 Sephadex G-25 Exclusin
LC-PCN Cianopropilsilil Exclusin
Tabla 2.Fases estacionarias usadas en extraccin en fase slida.
1.3.2 ETAPAS DE LA EXTRACCIN EN FASE SLIDA
1.3.2.1. Acondicionamiento del adsorbente
Acondicionamiento del adsorbente es necesario a fin de garantizar una interaccin
reproducible con el analito. Acondicionado, tambin llamada la solvatacin, se traduce en
una humectacin de los grupos funcionales del adsorbente y por lo tanto produce un
ambiente que es adecuado para la adsorcin del analito. Adsorbentes polares suelen
estar acondicionados con 2-3 volmenes de columna de un disolvente, que es miscible
con agua (MeOH, THF, isopropanol, etc.), seguido por el disolvente en el que se disuelve
-
8/11/2019 tesis aromaticos
51/125
la sustancia analizada (matriz polar). Adsorbentes polares estn acondicionados con
solventes no polares.
Luego del acondicionamiento el adsorbente no debe funcionar en seco, porque de lo
contrario se destruye la solvatacin.
1.3.2.2 Aplicacin de la muestra (adsorcin)
Aplicacin de la muestra se puede realizar con presin positiva o negativa con un caudal
de ~3 mL/min.
1.3.2.3 Lavado del adsorbente
Lavado del absorbente se consigue normalmente con una solucin de lavado especial
que selectivamente eluye las impurezas, pero deja el analito en la columna. Sin embargo,
en algunos casos puede que no sea necesario. Si la diferencia de polaridad entre la
solucin de lavado y eluyente es muy grande, o si ambos no son miscibles, el secado del
adsorbente despus del lavado es recomendable.
1.3.2.4. Elucin
El analito purificado se eluye finalmente con un solvente fuerte, suficiente para desplazar
el analito del absorbente. La elucin no debe ser demasiada rpida, la velocidad de la
elucin depende de la columna o la dimensin del cartucho y de la cantidad de
adsorbente [31,36]. La figura 6 muestra las etapas de la extraccin en fase slida
Figura 6.Etapas de la extraccin en fase slida.
-
8/11/2019 tesis aromaticos
52/125
1.3.3 VENTAJAS
Bajo consumo de disolvente.
Tamao de muestra puede ser grande o pequeo
En algunos casos enorme ahorro de tiempo.
Posibilidades de automatizacin.
A menudo una preparacin de la muestra se puede resolver ms concretamente
mediante el uso de SPE, ya que es posible diferentes interacciones del analito con
la fase slida (absorbente), y los mtodos pueden ser optimizados mediante el
ajuste de las condiciones cromatogrficas. La SPE ofrece una multitud de
adsorbentes polares, hidrofbicos y o interacciones inicas, mientras que la
extraccin lquido-lquido se limita a la particin de equilibrios en la fase lquida
[36].
1.4 HIDROCARBUROS AROMTICOS (BTEX)
El acrnimo BTEX define la mezcla de benceno, tolueno, etilbenceno y los tres ismeros
del xileno (orto, meta y para) [41].
1.4.1 ESTABILIDAD
Los compuestos orgnicos voltiles (VOCs) son compuestos qumicos que tienen una
presin de vapor alta en condiciones normales para evaporarse significativamente y entrar
en la atmsfera. Los hidrocarburos aromticos como el benceno, tolueno, etilbenceno, y
los xilenos son algunos de estos compuestos los cuales se obtienen del carbn y delpetrleo.
Los hidrocarburos aromticos forman una gran familia de compuestos que tienen un
ncleo comn, el ncleo bencnico. El benceno contiene 92.3 por ciento de carbono y 7,7
por ciento de hidrgeno con la frmula qumica C6H6. La molcula de benceno se
representa mediante un hexgono formado por los seis conjuntos de tomos de carbono e
hidrgeno unidos con alternancia de enlaces simples y dobles (figura 7). La molcula de
-
8/11/2019 tesis aromaticos
53/125
benceno es la piedra angular de compuestos aromticos, la mayora de los cuales
contienen uno o ms anillos de benceno.
Figura 7.Molcula del benceno.
Los BTEX se caracterizan por poseer una gran energa de resonancia que desemboca enuna gran estabilidad termodinmica. Se trata de hidrocarburos orgnicos monoaromticos
de bajo peso molecular, poco solubles en agua [3].
El ncleo aromtico tiene una gran estabilidad (energa de resonancia); sin embargo, por
su acumulacin de electrones, reacciona fcilmente con varios tipos de agentes y tanto
mejor cuando no se pierde el carcter aromtico. Las reacciones de adicin ocurren con
dificultad y necesitan un gran aporte de energa, mientras que las de sustitucin son
fciles [42].
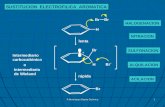
En las reacciones de sustitucin electroflica aromtica un electrfilo (E+) reacciona con
un anillo aromtico y sustituye uno de los hidrgenos. Se puede introducir en el anillo
aromtico muchos sustituyentes diferentes por reacciones de sustitucin electroflica; si se
seleccionan los reactivos adecuados es posible halogenar el anillo aromtico, nitrarlo,
sulfonarlo, alquilarlo o acilarlo. La estructura del compuesto aromtico determina su
reactividad y la regioselectividad de la reaccin [44].
1.4.2 PROPIEDADES FSICAS Y QUMICAS
La planaridad de las molculas de los hidrocarburos aromticos influye mucho en sus
propiedades fsicas: sus densidades son mayores que las de los alifticos y su punto de
ebullicin (Peb) y punto de fusin (Pf) son ms altos. Los Peb aumentan regularmente con
el peso molecular, pero los Pf dependen de la simetra de la molcula (Tabla 3).
-
8/11/2019 tesis aromaticos
54/125
-
8/11/2019 tesis aromaticos
55/125
-
8/11/2019 tesis aromaticos
56/125
para los BTEX en agua superficial y agua subterrnea, basados en la biodegradacin
aerobia y biodegradacin aerobia-anaerobia, respectivamente.
COMPUESTO AGUA SUPERFICIAL AGUA SUBTERR NEA
Benceno 5-16 10-730
Tolueno 4-22 7-28
Etilbenceno 3-10 6-228
o-Xileno 7-28 14-365
p-Xileno 7-28 14-56
m-Xileno 7-28 14-56
Tabla 5. Rango de valores de la vida media de los BTEX en das [3].
1.4.3 FUENTE DE HIDROCARBUROS AROMTICOS
Los hidrocarburos aromticos, principalmente benceno, tolueno, xilenos y etilbenceno,
tienen una importancia industrial extraordinaria; son materia prima para ms del 60% del
tonelaje de plsticos, elastmeros y fibras sintticas que se fabrican al igual que para
colorantes, insecticidas, medicamentos, etc. La mayor parte de hidrocarburos aromticos
se obtiene del petrleo (95%), y slo una pequea proporcin de la hulla(5%).
Aunque el contenido de aromticos originalmente presente tanto en el petrleo como en el
carbn es bajo, en determinados procesos de tratamiento trmico o cataltico de refineras
y coqueras se producen en proporciones significativas que hacen econmica su
separacin, obtenindose actualmente de la gasolina reformada, de la gasolina de
pirlisis y del alquitrn de la hulla [42].
1.4.3.1 PETRLEO
Actualmente, la mayor parte de las mezclas BTEX que se producen en las destileras de
petrleo se obtienen por los procesos de reformado cataltico y craqueo al vapor:
1.4.3.1.1 AROMTICOS DEL REFORMADO CATALTICO
El reformado cataltico de las fracciones C6-C8se realiza con catalizadores de Pt o Pt-Rh
sobre almina, a 500 C y 30 atm, con el fin de aum entar el ndice de octano de las
gasolinas y produce gran cantidad de hidrocarburos aromticos (BTEX), adems de
-
8/11/2019 tesis aromaticos
57/125
alcanos y alquenos. La gasolina vaporizada se diluye con H2para cortar la formacin de
carbn sobre catalizador; el proceso se llama tambin hydroforming. Las principalesreacciones de aromatizacin son [42]:
Figura 8. Principales reacciones de aromatizacin.
La gasolina reformada es la fuente ms importante de estos hidrocarburos, ya que
contiene hasta el 55% de ellos (Tabla 6).
COMPOSICI N DE GASOLINAS REFORMADAS
Benceno 3-6%
Tolueno 15-20%
Xilenos 19-21%
Etilbenceno 4-5%
Aromticos superiores 10-15%
No aromticos 35-45%
Tabla 6.Composicin de gasolinas reformadas.
-
8/11/2019 tesis aromaticos
58/125
-
8/11/2019 tesis aromaticos
59/125
COMPUESTO T (C)
Benceno 80
Tolueno 110
Etilbenceno 136
p- Xileno 138
m- Xileno 139
o- Xileo 145
Tabla 8.Puntos de ebullicin de los BTEX.
La volatilidad relativa del o-xileno es de 1,16 mientras que la del p- y m-xileno es de
alrededor de 1,02. Hacer una separacin para p- y m-xileno por destilacin est fuera de
toda lgica. El p-xileno es una molcula estrecha con los dos grupos metilo, uno en cada
extremo. El m-xileno es ms esfrico, debido a la posicin de los grupos metilo. El
proceso de separacin ms dependiente de la forma de la molcula, para un volumen
molecular fijo, es la cristalizacin. La diferencia en la forma molecular tiene dos efectos.
Primero, las molculas de p-xileno pueden agruparse juntas en una estructura cristalina
ms rpidamente, debido a su forma simtrica; como resultado de esto, el p-xileno tiene
un punto de congelacin mucho ms alto (13,3 C) qu e cualquiera de los otros ismeros.
Segundo, la diferencia de forma entre el p-xileno y el m-xileno significa que las molculas
de m-xileno no pueden ajustarse fcilmente dentro de la estructura cristalina del p-xileno
en la fase slida. Como resultado, la fase slida formada por congelacin parcial de una
mezcla de los dos ismeros contiene esencialmente p-xileno puro, y el factor de
separacin para un proceso de cristalizacin es realmente muy alto [37].
Tambin hay diferencias en las propiedades de adsorcin que se pueden utilizar para
aislar ismeros individuales del xileno. En la adsorcin, la estructura porosa del
adsorbente preferentemente retiene el ismero producto de inters. Un tratamiento
posterior con un lquido desorbente (por lo general otro orgnico como el tolueno) disocia
el producto del adsorbente. La separacin del ismero producto de xileno puede entonces
ser realizado utilizando una destilacin fraccionada simple [47].
-
8/11/2019 tesis aromaticos
60/125
1.4.4 PROCESOS DE TRANSFORMACIN DE AROMTICOS
Dado que la industria qumica tiene una demanda de hidrocarburos aromticos que nopuede satisfacerse con la distribucin de aromticos obtenida directamente de las
gasolinas reformadas, de pirlisis y del alquitrn de hulla, se han desarrollado procesos
de transformacin de hidrocarburos aromticos entre s. De un modo global, el objetivo de
estos procesos es contrarrestar el exceso de oferta de tolueno y la demanda de benceno
y xilenos. Los procesos ms significativos son:
Hidrodesalcohilacin de tolueno
Isomerizacin del m-xileno
Desproporcionamiento de tolueno y transalquilacin con trimetilbencenos.
1.4.5 REFINACIN DEL PETRLEO Y COMPOSICIN DE LA GASOLINA EN
COLOMBIA
1.4.5.1 REFINACIN NACIONAL DEL PETRLEO
En Colombia (2010), Ecopetrol fue capaz de refinar diariamente 315 mil barriles, es decir,
13.2 millones de galones de petrleo cada 24 horas. De esos, resultan unos 71500barriles por da de gasolina (3.6 millones de galones) y 97330 barriles de ACPM (4
millones de galones). El pas se autoabastece de crudo. Exporta e importa, dependiendo
de los requerimientos del mercado local e internacional y se surte fundamentalmente de
los yacimientos de La Cira-Infantas, en Barranca; Chuchupa en La Guajira; Cao Limn
en Arauca y Cusiana-Cupiagua, en Casanare [49].
1.4.5.2 COMPOSICIN DE LA GASOLINA EN COLOMBIA
La gasolina est compuesta por una mezcla de hidrocarburos que van desde los que
poseen 4 tomos de carbono hasta los que tienen 10-11 tomos de carbono; stos
hidrocarburos pueden ser parafnicos, isoparafnicos, olefnicos, naftnicos y aromticos,
obtenidos de diversos procesos de refinacin como destilacin, crackeo trmico y
cataltico, reformacin cataltica, alquilacin, e isomerizacin.
De las cuatro (4) clases en que se subdividen los hidrocarburos (parafnicos, naftnicos,
aromticos y olefnicos), la que predomina en el petrleo bruto es la clase de los
-
8/11/2019 tesis aromaticos
61/125
hidrocarburos parafnicos (parafinas), que pueden ser de cadena lineal (n-parafinas) o
ramificada (isoparafinas).
Los hidrocarburos aromticos se caracterizan por su elevado peso especfico y por un
poder antidetonante bastante elevado. Se encuentran en el petrleo bruto en cantidades
limitadas, salvo algn tipo que los contiene en mayor proporcin [50].
La empresa Colombiana de petrleos ECOPETROL sigue la normatividad de la American
Society for Testing and Materials (ASTM) y en las siguientes tablas se muestran las
principales caractersticas de las gasolinas distribuidas en Colombia.
Caractersticas Unidades Mtodos Mximo
Benceno mL/100 mLASTM D-5580 ASTM D-3606 ASTM D-6729
1,0
Aromticos mL/100 mL
ASTM D-5580 ASTM D-1319
Mtodo PIANO(ASTM D-6729)
28
Tabla 9.Gasolina corriente.
Caractersticas Unidades Mtodos Mximo
Benceno mL/100 mLASTM D-5580 ASTM D-3606 ASTM D-6729
2
Aromticos mL/100 mL
ASTM D-5580 ASTM D-1319 Mtodo PIANO(ASTM D-6729)
35
Tabla 10.Gasolina extra.
-
8/11/2019 tesis aromaticos
62/125
Caracterstica Unidades Mtodos Mximo
Aromticos mL/100 mL ASTM D-1319 ASTM D-5186
35
Tabla 11.Diesel corriente.
Caracterstica Unidades Mtodos Mximo
Aromticos mL/100 mLASTM D-1319 ASTM D-5186
35
Tabla 12.Diesel extra o diesel premium [51].
1.4.6 USOS DE LOS BTEX
El benceno y sus derivados se han utilizado en una variedad de productos, algunos de los
cuales incluyen: plaguicidas, detergentes, pinturas, colorantes, lubricantes, gomas, drogas
y explosivos.
1.4.6.1 Benceno
Desde que el benceno fue descubierto en 1825, se ha utilizado para una variedad de usos
industriales y comerciales. Uno de los primeros usos del benceno fue como locin de
afeitar, por el olor agradable distintivo del producto qumico. Tambin se empleo para
descafeinar el caf y como aditivo antidetonante en la gasolina.
El benceno se puede encontrar en una serie de productos elaborados por algunas de las
empresas ms grandes del mundo, como por ejemplo:
Coca-Cola
Pepsi
Cadbury Schweppes (Compaa Britnica de Confites)
Kraft Foods (Bebidas, queso, lcteos, snacks, confitera y cereales.)
Polar Beverages(Bebidas)
Algunos de los mayores productores de benceno en los Estados Unidos son:
-
8/11/2019 tesis aromaticos
63/125
Chevron
Shell Chemical Exxon
Dow Chemical
Amoco
El benceno es un componente integral en la produccin de polmeros, plsticos, resinas,
adhesivos, nylon, detergentes, colorantes, lubricantes, explosivos y plaguicidas. La
mayora de los materiales antes mencionados se producen a partir de tres usos derivados
del benceno: estireno, fenol y ciclohexano [52].
1.4.6.2 Tolueno
Ms del 50% del tolueno producido se convierte en benceno por hidrodesalquilacin. En
mucha menor escala se utiliza un proceso de desproporcin cataltica.
Alrededor del 10% de la produccin de tolueno se utiliza para obtener trilita o TNT
(2,4,6-trinitrotolueno), explosivo militar de gran potencia. Otro tanto se usa para
disolventes y el resto, resinas de poliuretano y diversas sntesis.
Una parte de las fracciones ricas en tolueno se incorporan a gasolinas para alcanzar altos
ndices de octano.
1.4.6.3 Etilbenceno
Si bien cerca del 99% del etilbenceno se consume en la produccin de estireno, una
pequea cantidad se utiliza en aplicaciones de solventes, remplazando algunas veces al
xileno.
1.4.6.4 p-Xileno
La mayor demanda en el mercado de los ismeros del xileno es de p-xileno, materia
prima para fibras sintticas, se utiliza para hacer el cido tereftlico (TPA) y dimetil
tereftalato (DMT), intermediarios en la fabricacin de fibras tereftalato de polietileno (PET),
plsticos moldeados y pelculas.
-
8/11/2019 tesis aromaticos
64/125
1.4.6.5 o-Xileno
El o-xileno se utiliza principalmente en la fabricacin de anhdrido ftlico que es la materia
prima para la fabricacin de plastificantes y resinas para pinturas. Menores usos
adicionales en la fabricacin de bactericidas, herbicidas para la soja, aditivos de aceite
lubricante.
1.4.6.6 m-Xileno
El m-xileno tiene menos aplicaciones, aunque el cido isoftlico se usa para obtener
algunos tipos de resinas de polister. Gran parte del excedente de m-xileno y sus mezclas
brutas se reincorporan a las gasolinas para aumentar el ndice de octano o se usan como
disolventes [42].
En el anexo 1 se muestras algunas reacciones de los BTEX para la produccin de
diferentes productos.
1.4.7 TOXICIDAD DE LOS BTEX
Todos los compuestos orgnicos voltiles son nocivos. Los efectos de la exposicin aestas sustancias incluyen los cambios en el hgado y los efectos dainos en los riones, el
corazn, los pulmones y el sistema nervioso [41].
Los BTEX estn en todas partes entre las muestras de inters ambiental (agua, aire,
tierra). Debido a que los BTEX son compuestos neutros, solubles en lpidos y de bajo
peso molecular, son absorbidos rpidamente por el organismo una vez inhalados o
ingeridos. La mayor fuente de riesgo de exposicin al benceno es por va area. La
exposicin humana a estos hidrocarburos aromticos, tambin ocurre por ingestin(consumo de agua o alimentos contaminados). Los BTEX al ser muy solubles en lpidos
tienden a acumularse en los tejidos grasos [3].
Se hace hincapi en el benceno, ya que este es el ms txico del grupo de los BTEX.
-
8/11/2019 tesis aromaticos
65/125
1.4.7.1 Benceno
El uso del benceno se ha regulado con el fin de proteger al pblico de niveles peligrosos
de e