TESI DI LAUREA SPECIALISTICA - to.camcom.it · Meccanismi di difesa dell’ospite Pag. 51...
135
UNIVERSITÀ DEGLI STUDI DI TORINO Facoltà di Medicina e Chirurgia Corso di Laurea Specialistica in Biotecnologie Mediche TESI DI LAUREA SPECIALISTICA “Sviluppo di un saggio cellulare ad alta resa per l’identificazione di inibitori dell’espressione delle oncoproteine del Papillomavirus umano di tipo 16” Candidata: Relatore: Valentina Dell’Oste Dott. David Lembo Anno Accademico 2004-2005
Transcript of TESI DI LAUREA SPECIALISTICA - to.camcom.it · Meccanismi di difesa dell’ospite Pag. 51...

UNIVERSITÀ DEGLI STUDI DI TORINO
Facoltà di Medicina e Chirurgia
Corso di Laurea Specialistica in Biotecnologie Mediche
TESI DI LAUREA SPECIALISTICA
“Sviluppo di un saggio cellulare ad alta resa
per l’identificazione di inibitori dell’espressione delle
oncoproteine del Papillomavirus umano di tipo 16”
Candidata: Relatore:
Valentina Dell’Oste Dott. David Lembo
Anno Accademico 2004-2005

IINNDDIICCEE
INTRODUZIONE Pag. 1 I PAPILLOMAVIRUS Pag. 1
Caratteristiche generali Pag. 1
Struttura del virione Pag. 2
Ciclo replicativo del virus Pag. 5
Meccanismi patogenetici Pag. 13
Coltivazione del virus Pag. 25
CARATTERISTICHE CLINICHE DELLE INFEZIONI
DEI PAPILLOMAVIRUS Pag. 27
Epidemiologia Pag. 27
Manifestazioni cliniche Pag. 31
Profilassi Pag. 36
Diagnosi delle infezioni Pag. 38
Terapie Pag. 44
HPV E MODULAZIONE DEL SISTEMA IMMUNITARIO Pag. 51
Meccanismi di difesa dell’ospite Pag. 51
Meccanismi di evasione dal sistema immunitario Pag. 55
CONTESTO E RAZIONALE DELLA RICERCA Pag. 61 Il processo di ricerca e sviluppo di un farmaco antivirale Pag. 61
Sviluppo di un farmaco anti HPV: presupposti Pag. 63
Individuazione del bersaglio Pag. 65
La prova del principio Pag. 66
Sviluppo di saggi per lo screening di antivirali Pag. 70
SCOPO DEL LAVORO Pag. 83
MATERIALI E METODI Pag. 84
RISULTATI Pag. 91
Realizzazione e caratterizzazione di una linea cellulare
“reporter” contenente la regione LCR del genoma di HPV-16 Pag. 91

Analisi di un pannello di citochine utilizzando il clone
indicatore P13 Pag. 97
Effetto di IL-4 e IL-13 sui livelli di mRNA di E6 e E7 di
HPV-16 in cellule CaSki Pag. 101
DISCUSSIONE Pag. 105
BIBLIOGRAFIA Pag. 119
RINGRAZIAMENTI Pag. 133

INTRODUZIONE
I PAPILLOMAVIRUS
Caratteristiche generali
I papillomavirus (dal Latino “papilla”, pustola e dal Greco “oma”, tumore) sono
classificati nella famiglia Papillomaviridae, di cui sono noti 118 tipi (de Villiers et al.,
2004).
I papillomavirus umani (HPV) sono piccoli virus nudi a DNA, ubiquitari,
suddivisi in più di 200 genotipi, che inducono lesioni iperproliferative della pelle
(verruche) o delle membrane mucose (condilomi). I virus cutanei sono epidermotropici
e infettano principalmente la cute a livello di mani e piedi; le cellule bersaglio degli
HPV mucosali sono invece principalmente localizzate a livello della mucosa labiale, del
tratto respiratorio e dei genitali.
Gli HPV sono ulteriormente suddivisi in virus ad alto, medio e basso rischio
sulla base delle lesioni a cui sono associati (Tabella 1).
Gruppo di HPV Genotipo di HPV
Alto rischio
Rischio intermedio
Basso rischio
16, 18, 45, 56
31, 33, 35, 51, 52, 58
6, 11, 42, 43, 44
I gruppi ad alto rischio sono implicati nello sviluppo di tumori altamente
maligni, in particolare cancro alla cervice uterina e altri tumori del tratto anogenitale,
mentre quelli a basso rischio sono comunemente rilevabili nelle verruche a livello
Tabella 1: Sottogruppi di HPV, in base al rischio di indurre cancro anogenitale e lesioni precancerose.

genitale (Sanclemente et al., 2002). La comprensione del coinvolgimento degli HPV in
queste patologie ne ha sottolineato l’importanza medica e ha stimolato lo studio della
messa a punto di vaccini e farmaci per la loro prevenzione e cura.
Struttura del virione
Gli HPV sono piccoli virus icosaedrici nudi a DNA, che si replicano nel nucleo
di cellule epiteliali squamose.
Le particelle virali presentano un diametro di 52-55 nm ed un coefficiente di
sedimentazione di 300. Il virione è costituito da un genoma a singola molecola di DNA
circolare, a doppia elica, avente un peso molecolare di 7900 paia di basi, di cui il 42% è
formato da guanina e citosina. Il DNA costituisce il 12% del peso del virione ed è
associato a istoni cellulari, formando un complesso simile a cromatina (Figura 1).
Una caratteristica comune dei diversi sottotipi di HPV è la presenza di un
genoma formato da otto sequenze codificanti (ORF), localizzate su una singola elica di
Figura 1: Rappresentazione schematica del virione degli HPV.

DNA virale, che rappresenta lo stampo per la trascrizione di messaggeri policistronici
(Figura 2). Esiste inoltre una regione in cui non sono presenti ORF, chiamata LCR
(“long control region”) o URR (“upstream regulatory region”) o “noncoding region”, le
cui dimensioni sono notevolmente diverse tra i genomi dei diversi sottotipi di HPV (in
media rappresenta il 5% del genoma virale, con un peso molecolare che varia da 400 a
1000 paia di basi). All’interno di questa regione sono concentrate le sequenze
regolatorie richieste per la replicazione e trascrizione virale.
Le ORF degli HPV sono suddivise in regioni precoci (E, “early”, costituenti il
45% del genoma virale) e tardive (L, “late”, rappresentanti il 40% del genoma virale);
tali regioni codificano rispettivamente per proteine precoci (E1, E2, E3, E4, E5, E6, E7,
E8) e tardive (L1, L2). I geni E3 e E8 non presentano una funzione chiaramente
definita.
I geni precoci, espressi sia in cellule infettate in modo non produttivo sia in
cellule trasformate, sono trascritti in numerose sequenze codificanti parzialmente
sovrapposte, che subiscono uno “splicing” alternativo e presentano una comune
sequenza finale in 3’, definita da un segnale di poliadenilazione. Le regioni precoci
codificano per proteine virali regolatorie, tra le quali quelle necessarie per dare inizio
alla replicazione del DNA virale (Sanclemente et al., 2002). Negli HPV a elevato
rischio le proteine precoci sono codificate a partire da promotori precoci, quale, ad
esempio, P97 in HPV-31, prima dell’inizio della replicazione virale.

Le regioni tardive del genoma, L1 e L2, codificano invece per le proteine virali
del capside e sono espresse solo in cellule infettate in modo produttivo. La trascrizione
di proteine tardive è sotto il controllo di promotori tardivi, quale, ad esempio, P742 in
HPV-31 e si verifica durante l’assemblaggio di nuovi virioni maturi (Fehrmann et al.,
2003).
Il capside, che racchiude il genoma virale, presenta una simmetria icosaedrica
ed è composto da 72 capsomeri. Ogni capsomero è un pentamero di L1 (55 kD), una
proteina strutturale del capside, che costituisce l’80% delle proteine virali totali. L1 è
coinvolta principalmente nel legame al DNA virale, come dimostrano studi su virus
mutanti per L1, i quali sono ancora in grado di formare il capside, ma non possono
incorporare il DNA.
Il capside di ogni virione contiene inoltre circa 12 copie di L2 (70 kD), la
proteina minore del capside (Burd et al., 2003). L2 svolge principalmente ruoli di tipo
strutturale, ma anche diverse funzioni regolatorie durante il ciclo vitale degli HPV, tra le
quali il legame a recettori secondari, la determinazione della localizzazione nucleare del
Figura 2: Struttura del genoma degli HPV

virus e l’incapsidazione selettiva del DNA nel capside virale, aumentando così
l’infettività dei virioni.
Ciclo replicativo del virus
Il virus ha un tropismo molto ristretto per l’epitelio squamoso pluristratificato e
si replica pertanto solo nelle cellule epiteliali in differenziamento della cute e delle
mucose, come dimostra il fatto che le funzioni replicative del virus, come la sintesi del
DNA virale, la produzione di proteine capsidiche e il montaggio dei virioni, hanno
luogo unicamente a livello di cheratinociti in differenziamento terminale.
Le fasi iniziali della replicazione (adsorbimento a recettori cellulari,
penetrazione e scapsidazione) sono poco conosciute, date le difficoltà di propagazione
del virus, che avviene solo in colture organotipiche che mimano in vitro la struttura
dell’epidermide (Dianzani et al., 2001). L’infezione virale inizia con il legame del
virione alla superficie della cellula bersaglio, in questo caso lo strato basale dei
cheratinociti, in seguito a piccole ferite o traumi che ne permettano l’esposizione. I
recettori utilizzati dal virus per prendere contatto con le cellule e penetrare all’interno di
esse non sono stati ancora chiaramente identificati. Studi di legame con virioni marcati
radioattivamente hanno dimostrato che gli HPV possono legarsi non solo a cellule
epiteliali squamose, ma anche ad altri tipi di cellule. Questo indica che lo spiccato
tropismo degli HPV per i cheratinociti non è dovuto alla specificità del recettore.
L’integrina α6 è stato il primo candidato come possibile recettore degli HPV,
secondo studi che utilizzano le VLP (“Virus like particles”). Le VLP si legano
all’integrina e l’uso di anticorpi diretti contro α6 blocca il legame del virus alla cellula. Il
ruolo di questa integrina come recettore è stato confermato anche da studi in cui, su
linee cellulari dove l’espressione recettoriale era stata soppressa, veniva fatta esprimere

unicamente α6: questo era sufficiente per rendere possibile l’ingresso del virus nella
cellula. L’integrina α6 coopera con le subunità β1 e β4 dell’integrina β, situate sulla
superficie cellulare. L’integrina α6β1 viene espressa su un’ampia varietà di cellule, tra
cui piastrine, linfociti, cellule endoteliali, mentre α6β4 si trova su cellule epiteliali,
mesenchimali e neuronali. Gli HPV si possono legare ad entrambi i tipi di cellule, ma
preferenzialmente a quelle con profilo α6β4. L’espressione di queste integrine non
risulta però necessaria per l’ingresso del virus nelle cellule, come dimostra il fatto che
alcuni HPV entrano in cellule prive di questi recettori. E’ stato osservato che tali virus, a
livello di cheratinociti, si legano ad eparina e glicosaminoglicani, seguito da successivo
legame al recettore e internalizzazione (Howley et al., 2001).
Le particelle virali internalizzate sono trasportate in fagosomi, processo che può
essere inibito da citoclasina B e taxolo: questo implica un possibile coinvolgimento di
microtubuli e microfilamenti, inibiti da tali sostanze. Nonostante il legame alla
membrana plasmatica e l’ingresso del virione in grandi vescicole citoplasmatiche possa
essere monitorato con un microscopio elettronico, non si osservano virioni completi nel
nucleo delle cellule infettate, mentre si riscontra un segnale molto forte per proteine L1
e L2. Questa osservazione indica che la scapsidazione del virione si verifica nel
citoplasma e che le proteine L1 e L2 migrano nel nucleo grazie a segnali di
localizzazione nucleare.
Il ciclo replicativo del virus può essere suddiviso in uno stadio non produttivo o
precoce e uno produttivo, più tardivo, correlati allo stadio differenziativo della cellula
ospite. La fase non produttiva implica lo stabilirsi del genoma virale come plasmide
nucleare a livello dello strato basale dell’epitelio. Le cellule appartenenti a questo strato
sono costituite da cellule staminali in continua divisione, che rappresentano un serbatoio
per gli strati superiori (Hummel et al., 1992). Dato che la cellula basale è l’unica cellula

dell’epitelio in grado di dividersi, il virus deve realizzare un’infezione a questo livello
per indurre una lesione persistente, ma l’espressione dei geni tardivi, la sintesi del DNA
e delle proteine capsidiche, avvengono solo nelle cellule in differenziamento terminale
degli strati superiori (Dianzani et al., 2000). L’infezione delle cellule basali porta
all’attivazione della cascata di espressione dei geni virali, che permette la produzione di
20-100 copie per cellula di DNA virale in forma episomale, che viene mantenuto
stabilmente replicandosi in sincronia con il DNA cellulare. A livello basale
l’espressione dei geni virali è limitata a specifici geni precoci; alcuni di questi, in
particolare E5, E6, E7, stimolano la cellula infettata a proliferare e ad espandersi
lateralmente. Un gruppo di cellule figlie (in cui le copie di DNA virale sono state
equamente ripartite, assicurando l’infezione persistente delle cellule staminali
dell’epidermide) abbandona la membrana basale per stratificare e differenziare,
rendendo possibile l’ingresso del virus negli strati superiori dell’epidermide. A questo
livello inizia la fase tardiva del ciclo replicativo degli HPV, in assenza di replicazione
del DNA virale, con l’espressione di geni virali tardivi e la traduzione di proteine
strutturali. Successivamente, nelle cellule differenziate completamente viene perso il
controllo del numero di copie genomiche e il DNA è amplificato fino ad avere migliaia
di copie per cellula. Infine, negli strati più superiori dell’epitelio si verifica
l’assemblaggio e il rilascio nell’ambiente extracellulare di particelle virali mature
(Figura 3) ( Lambert, 1991; Laimins, 1996; Sanclemente et al., 2002; zur Hausen
2002).

La trascrizione dei geni virali è un processo complesso per la presenza di
promotori multipli, di meccanismi di “splicing” alternativi e per la produzione
diversificata di mRNA nelle differenti linee cellulari. Come gli herpesvirus e gli
adenovirus, gli HPV possono utilizzare per la trascrizione le RNA polimerasi DNA-
dipendenti della cellula e pertanto devono iniziare la trascrizione nel nucleo della cellula
ospite, dove si trovano gli enzimi necessari. Il genoma degli HPV viene trascritto in due
tempi, precoce e tardivo, come riportato in Figura 4.
Figura 3: Ciclo vitale degli HPV
(modificata da: zur Hausen, H. 2002. Nat. Rev. Cancer. 2:342–350).

Nella produzione dei diversi tipi di mRNA sono coinvolti promotori multipli ed
in particolare si distingue un promotore precoce ed uno tardivo. Nel caso di HPV-31,
P97 è il promotore maggiore, attivo in cellule differenziate in maniera non terminale;
questo promotore dirige l’espressione di E6 e E7 e corrisponde a P97 di HPV-16 e P105
di HPV-18.
La regione regolatrice LCR contiene diversi “enhancer” costitutivi, con
specificità di tessuto e di tipo cellulare che possono essere indotti da un’ampia varietà di
molecole, come sarà ampiamente discusso in questa tesi. Tali elementi sono essenziali
per l’inizio dell’espressione genica virale e per il mantenimento della latenza. Un
importante regolatore della trascrizione è il prodotto del gene E2, descritto
successivamente (Dianzani et al., 2001).
La regolazione dell’espressione dei geni virali è controllata da diversi fattori di
trascrizione cellulari, che si legano all’LCR tra i quali ricordiamo AP-1 (“activator
protein-1”), YY1 (“yin yang-1”), alcuni membri della famiglia degli “octamer binding
factor”, NF-1 (“nuclear factor-1”), elementi responsivi ai glucocorticoidi e Sp1
(Saunders et al., 1998).
DNA parentale mRNA Proteine precoci
DNA progenie mRNA Proteine tardive
Progenie virale
Figura 4: Meccanismo di replicazione e trascrizione degli HPV.

Le prime proteine virali ad essere espresse sono i fattori di replicazione E1 e E2;
esse formano un complesso che si lega a sequenze all’origine della replicazione virale,
che recluta la polimerasi cellulare e le proteine accessorie che mediano la replicazione
(Conger et al., 1999).
La proteina E1 svolge un ruolo importante nella fase plasmidica dell’infezione.
Le sue dimensioni variano dai 593 (HPV-48) ai 681 (HPV-10) aminoacidi, con un peso
molecolare compreso tra 67,5 kD (HPV-47) a 76,2 kD (HPV-10). Viene codificata a
partire dall’ORF E1, che è la più grande e meglio conservata ORF tra i diversi sottotipi
degli HPV, a dimostrazione del suo ruolo cruciale durante la replicazione virale, svolto
in collaborazione con E2. La proteina svolge un’attività ATPasica e 3’-5’elicasica ed è
necessaria per l’inizio della sintesi e per l’allungamento del DNA virale, in quanto
riconosce regioni ricche di AT situate all’origine della replicazione. Studi di mutagenesi
hanno dimostrato che E1 è indispensabile per il mantenimento del DNA in forma
plasmidica e, in assenza di E1, il genoma virale può essere espresso solo in forma
integrata.
E1 ha un’organizzazione strutturale tripartita: una regione N-terminale ad attività
non ben definita, uno spaziatore di lunghezza variabile e una zona C-terminale correlata
all’attività ATPasica ed elicasica. Il dominio di legame al DNA è stato caratterizzato
attraverso studi di cristallizzazione: è costituito da un’estesa ansa e da una regione ad α
elica, importanti per il riconoscimento del DNA.
E1 interagisce, oltre che con E2, con numerose altre proteine cellulari, quali la
subunità p180 della DNA polimerasi α cellulare e recluta il macchinario di replicazione
cellulare a livello del sito di origine della replicazione del genoma virale. E1 lega inoltre
il complesso ciclina E-Cdk2, specifico della fase S del ciclo cellulare, contribuendo a

un’efficiente replicazione virale in associazione alla replicazione cellulare.
L’interazione E1-Ubc9 è importante per l’accumulo intra nucleare di E1, dovuto
all’aggiunta di un gruppo SUMO su E1 da parte di Ubc9.
La proteina E2 è un’ importante regolatore della replicazione e della trascrizione
virale; la proteina è conservata tra le diverse sottofamiglie degli HPV, ha un peso
molecolare di 50 kD ed è attiva sotto forma di dimeri. Presenta diversi domini: nella
regione C-terminale si trova una sequenza che codifica per specifici domini di legame al
DNA; in seguito a cristallizzazione si è visto che tali domini sono costituiti da una
struttura dimerica a barile β, che lega il DNA; è inoltre presente una sequenza di legame
a E1 (Hedge et al., 1992). Nella porzione N-terminale della proteina è situato un
dominio di transattivazione, formato da una regione ad α elica affacciata ad una zona a
foglietti β; questi due domini sono separati da una regione cerniera interna che, al
contrario, non è ben conservata tra i diversi HPV né per quanto riguarda le dimensioni
né per la composizione aminoacidica.
E2, così come E1, è espresso a partire da un promotore precoce, svolgendo
pertanto un ruolo attivo nel controllare il numero di copie di genoma virale nelle cellule
indifferenziate. Lega il DNA come dimero in modo specifico a livello della sequenza
consenso palindromica ACCN6GGT: quattro di queste sequenze sono presenti nella
regione URR e tre fiancheggiano le regioni che riconoscono E2 presenti all’origine
della replicazione.
E2 è importante anche nel regolare la trascrizione virale a livello di promotori
precoci; esiste in due forme: a piena lunghezza, E2TA, con attività di attivatore o
repressore della trascrizione e troncato, E2TR e E8E2, repressori della trascrizione. Il
ruolo di E2TA nel mantenimento dei plasmidi virali è dovuto alla sua capacità di legare

tali plasmidi ai cromosomi implicati nella mitosi cellulare, in modo tale che il genoma
virale sia racchiuso nel nucleo cellulare quando questo si riforma durante la telofase. I
siti di legame per E2 sono localizzati vicino ai siti di legame per i fattori di trascrizione
cellulari che attivano i promotori precoci. A bassi livelli, E2 si lega a specifiche
sequenze di riconoscimento e attiva i promotori precoci, mentre a concentrazioni
elevate reprime la trascrizione bloccando il legame dei fattori di trascrizione (Steger et
al., 1997). Il ruolo di E2 come repressore è molto importante nel regolare i livelli di E6
e E7, le due principali oncoproteine virali e la sua perdita è il primo stadio di
trasformazione neoplastica. Ci sono due principali meccanismi di repressione messi in
atto da E2: E2TA spiazza i fattori Sp1 e TFIID dal promotore di E6, mentre E8E2 è in
grado, attraverso meccanismi non ancora del tutto chiari, di reprimere la trascrizione a
grandi distanze (Villa et al., 2002).
Le ultime fasi del ciclo replicativo degli HPV sono caratterizzate dalla
trascrizione dei geni virali tardivi (L1 e L2), tra i quali è compreso anche E4, nonostante
questo gene sia localizzato all’interno della regione precoce. Questi geni sono sotto il
controllo di un promotore specifico, situato all’interno dell’ORF di E7 degli HPV che
danno infezioni a livello genitale, attivo solo in cheratinociti differenziati. Questo
promotore, a differenza del precoce, non viene modulato negativamente da E2 e quindi
presenta elevati livelli di espressione in cellule differenziate. Lo stretto legame tra
l’espressione di geni tardivi e lo stadio di differenziazione degli epiteli indica che questo
processo è controllato da diversi fattori cellulari specifici, la cui modalità d’azione non è
ancora stata chiarita completamente (Longworth et al., 2004).
Il ruolo della proteina E4 non è del tutto chiaro: infatti, nonostante sia espressa
ad alti livelli in tessuti infettati e non sia rilevabile all’interno delle particelle virali

mature, non si sa quale sia la sua funzione nel ciclo vitale del virus. Studi di mutagenesi
effettuati sul gene E4 in BPV-1 evidenziano che E4 non è essenziale alla replicazione o
alla trasformazione virale. E’ stato osservato che E4 di HPV-16 svolge un ruolo
nell’infezione produttiva: la proteina si trova associata al citoscheletro di citocheratina,
di cui induce il collasso, che presumibilmente contribuisce alla liberazione della
progenie virale. Non si è tuttavia osservato il collasso di filamenti intermedi di cheratina
in cellule esprimenti E4 di HPV-1, mettendo così in discussione la generalità di questi
effetti. I dati attualmente disponibili sono compatibili con la possibilità che E4 potrebbe
essere coinvolta nella replicazione del DNA virale, attraverso l’alterazione
dell’ambiente extracellulare in modo tale da favorire i processi di sintesi o il rilascio del
virus (Howley et al., 2001).
Poche informazioni sono note riguardo l’assemblaggio e rilascio degli HPV. I
virioni si osservano solo nello strato granuloso dell’epitelio, mai in strati inferiori e
senza alcun effetto citolitico.
Meccanismi patogenetici
I meccanismi patogenetici degli HPV differiscono da quelli di altre famiglie
virali in quanto l’infezione richiede cellule epiteliali proliferanti, situate nello strato
basale dell’epidermide e delle mucose. In queste cellule l’espressione dei geni virali è
limitata a specifici geni precoci, che inducono la cellula a proliferare; l’espressione di
geni virali tardivi, con conseguente assemblaggio e rilascio del virione maturo, si
verifica invece negli strati sovrastanti.
Il ciclo replicativo completo del virus, con produzione di una progenie virale
matura è tipica delle infezioni da HPV a basso o medio rischio o delle lesioni a basso

grado degli HPV ad alto rischio, in cui il genoma degli HPV permane in forma
plasmidica.
Invece, nelle lesioni ad alto grado indotte dall’infezione da HPV ad alto rischio,
quali HPV-16 e HPV-18, avviene l’integrazione del DNA virale nel DNA cellulare, con
conseguente mancata produzione di una progenie virale completa (Figura 5).
Tale integrazione è determinante nei meccanismi di trasformazione e
immortalizzazione cellulare, poiché si verifica a livello della ORF E2, con conseguente
perdita dell’azione repressiva di E2 sulle oncoproteine virali E6 e E7, che svolgono un
ruolo fondamentale nei meccanismi di tumorigenesi (Longworth et al., 2004). Pertanto
nelle cellule dei tumori indotti da HPV ad alto rischio, i geni E1, E6 e E7 sono integrati
e funzionali, con conseguente stimolo alla proliferazione cellulare, mentre i geni E2, E4
e E5 vengono persi o non sono trascritti.
Figura 5: Meccanismo di integrazione del DNA virale in quello cellulare
(modificata da zur Hausen. 2002. Nat. Rev. Cancer. 2:342–350).

Oltre alle proteine E6 e E7, anche E5 è coinvolta, in misura minore, nei processi
di stimolazione della proliferazione. Verranno ora descritte in modo maggiormente
dettagliato queste proteine virali.
Il gene E5 degli HPV codifica per una proteina altamente idrofobica di circa 80
aminoacidi, localizzata a livello delle membrane endosomali, dell’apparato di Golgi e
della membrana plasmatica. Essa può avere un ruolo trasformante nelle fasi iniziali
dell’infezione, durante le quali il virus è presente in forma episomale, predisponendo la
cellula a successivi stimoli mitogeni. E5 contrasta i meccanismi di inibizione di crescita,
formando un complesso con il recettore per EGF, per il fattore di crescita derivato dalle
piastrine e per il CSF-1 (“colony stimulating factor”); inoltre la proteina E5 si lega a una
subunità di 16 kD dell’ATPasi vacuolare, interferendo con l’acidificazione degli
endosomi. Vengono così inibite le variazioni di pH, con conseguente aumento del
ricambio del recettore di EGF a livello della membrana plasmatica che porta ad un
aumento del segnale dato dal complesso EGFR/EGF. E5 svolge molteplici funzioni:
attiva la trasduzione del segnale per la mitosi tramite fattori di trascrizione come c-jun e
c-fos, inattiva la p21 e previene inoltre l’apoptosi in seguito a danno al DNA. Tuttavia il
suo ruolo nei processi di trasformazione è secondario, in quanto la sua espressione viene
persa in seguito all’integrazione del genoma virale (zur Hausen, 2002).
Un ruolo di primaria importanza nei processi di trasformazione è svolto dalle
oncoproteine virali E6 e E7, grazie alla capacità di agire sui meccanismi di regolazione
del ciclo cellulare. La sola espressione di E6 e E7, soprattutto se concomitante, degli
HPV a elevato rischio è sufficiente a indurre l’immortalizzazione di cheratinociti
primari umani in coltura (Hawley-Nelson et al., 1989). Queste linee cellulari non sono

tuttavia sufficienti per innescare il processo di tumorigenesi in topi nudi e richiedono
altri eventi, come ad esempio la presenza dell’oncogene Ras attivato (Hurlin et al.,
1991). Queste osservazioni testimoniano che, in vivo, è necessario un processo a più fasi
per favorire la progressione tumorale indotta dagli HPV. A differenza degli HPV ad alto
rischio, E6 e E7 dei papillomavirus umani a basso rischio sono incapaci di
immortalizzare cheratinociti in vitro, nonostante possano prolungarne il tempo di vita
(Thomas et al., 2001).
Il gene E6 è uno dei primi ad essere espresso durante l’infezione dei
papillomavirus. Codifica per una proteina costituita da 150 aminoacidi che presenta due
domini Cys-X-X-Cys; la proteina E6 è distribuita nel nucleo e nel citoplasma; è priva di
attività enzimatica intrinseca e, per esercitare la sua funzione, deve legarsi a diverse
proteine cellulari (zur Hausen 2002). La sua espressione porta alla trasformazione di
cellule NHI 3T3 (fibroblasti murini) e all’immortalizzazione di cellule epiteliali umane
della mammella (Liu et al., 1999). L’efficiente immortalizzazione di cheratinociti umani
richiede invece la coespressione di E6 e E7 (Hawley-Nelson et al., 1989). I meccanismi
d’azione di questa proteina virale sono stati chiariti soprattutto grazie a studi
sull’interazione con p53.
p53 è uno dei primi geni oncosoppressori ad essere stato caratterizzato. Svolge
un ruolo di primaria importanza nel regolare l’espressione di proteine coinvolte nel
controllo del ciclo cellulare: viene attivata in seguito a danno al DNA e induce
l’espressione di p21, un inibitore delle chinasi dipendenti dalle cicline, con conseguente
blocco del ciclo cellulare e induzione di apoptosi.
Generalmente uno dei meccanismi di risposta dell’organismo ad un’infezione
virale è l’innesco di apoptosi, in modo tale da limitare la diffusione dell’infezione
virale. Molti virus, tra i quali gli HPV, hanno evoluto un sistema di evasione

dall’apoptosi, contribuendo così alla progressione tumorale. Per bloccare l’attività pro
apoptotica di p53 e rendere possibile la progressione del ciclo cellulare, E6 lega p53
attraverso l’ubiquitina ligasi E6AP, formando un complesso ternario. Questo porta
all’ubiquitinazione di p53 da parte di E6AP e alla sua successiva degradazione
attraverso il sistema del proteosoma 26S, con conseguente diminuzione della vita media
di p53 nei cheratinociti, da diverse ore a meno di 20 minuti (Huibregtse et al., 1997). E6
regola p53 anche indirettamente, associandosi a p300/CBP, un coattivatore di p53. In
seguito all’inattivazione funzionale p53, vengono deregolati i principali meccanismi di
controllo del ciclo cellulare in G1/S e G2/M, con conseguenti anomalie a livello della
duplicazione e della struttura dei cromosomi.
E’ interessante notare che il legame di E6 a E6AP ha come conseguenza
l’ubiquitinazione di E6AP stessa (Kao et al., 2000). Quindi E6 potrebbe regolare i
livelli dei substrati naturali di E6AP attraverso la sua degradazione. Alcune delle
proteine bersaglio di E6AP appartengono alla famiglia Src delle tirosine chinasi, che
interagiscono con diverse cascate di trasduzione del segnale (Frame, 2002).
E6 presenta anche un’attività indipendente da p53, importante per
l’immortalizzazione di cellule umane. Sono state identificate proteine E6 di HPV-16
incapaci di degradare p53, ma che immortalizzano cellule epiteliali mammarie umane.
Viceversa, altri mutanti mantengono la capacità di degradare p53, ma non sono in grado
di immortalizzare le cellule (Liu et al., 1999).
Questi dati dimostrano che per l’immortalizzazione cellulare è importante
l’interazione di E6 con altre proteine, oltre a p53. Ad esempio E6 interagisce con
proteine appartenenti alla famiglia dei PDZ; queste presentano un dominio conservato
che si trova spesso in proteine situate nelle aree di contatto tra le cellule, come le
giunzioni strette tra le cellule epiteliali o le giunzioni sinaptiche delle cellule neurali. Le

proteine PDZ sono importanti per il mantenimento dell’architettura molecolare al fine di
rendere possibile la trasmissione del segnale (Craven et al., 1998). Il legame dei
membri della famiglia dei PDZ, MUPP-1, hDLG e hSCRIB, all’estremità C-terminale
delle proteine E6 ad alto rischio porta alla loro degradazione (Kiyono et al., 1998).
L’importanza di questa interazione è stata confermata in esperimenti su topi transgenici
esprimenti la proteina E6 priva del dominio di legame a PDZ. In questi topi viene
mantenuta la capacità di inattivare p53, ma essi non sviluppano la iperdisplasia
epidermica, che invece si osserva frequentemente in topi transgenici per E6 normale
(Nguyen et al., 2003). Non è chiaro quali siano i meccanismi attivati in seguito al
legame di E6 alle proteine PDZ e quali membri di tale famiglia siano più importanti per
questi fenotipi.
Un’altra funzione fondamentale della proteina E6 ad alto rischio
nell’immortalizzazione cellulare è la capacità di attivare l’espressione della subunità
catalitica della telomerasi, hTERT. Si tratta di un enzima formato da quattro subunità,
che addiziona ripetizioni esameriche all’estremità telomerica dei cromosomi. L’attività
telomerasica è solitamente limitata a cellule embrionali ed è assente in cellule
somatiche. La perdita di questa attività porta ad accorciamento dei telomeri, con
successive divisioni cellulari e induzione di senescenza (Liu et al., 1999). E6 attiva la
trascrizione di hTERT attraverso l’azione combinata di Myc e Sp1. E6 lega Myc e il suo
cofattore Max, portando all’attivazione del promotore di hTERT (Veldman et al., 2003).
Per determinare l’immortalizzazione delle cellule è più importante l’azione su hTERT
rispetto a quella su p53. Tuttavia l’immortalizzazione dei cheratinociti umani del derma
(HFK) richiede la presenza di E7, con conseguente inattivazione della proteina
retinoblastoma, che porta a deregolazione del ciclo cellulare (Flores et al., 2000).

Quindi la degradazione di p53 è fondamentale per una completa trasformazione, mentre
il legame a proteine PDZ e l’attivazione di hTERT è necessaria per l’immortalizzazione.
Il ruolo primario di E6 nel ciclo degli HPV non è di per se indurre
trasformazione o immortalizzazione, ma rendere più semplice alcune fasi del ciclo
replicativo del virus. Usando un sistema genetico che permetta la trasfezione in
cheratinociti del genoma dell’HPV clonato, è stato dimostrato che l’espressione della
proteina E6 funzionale è necessaria per mantenere i genomi di HPV-31 e HPV-11 come
episomi, in grado di replicarsi stabilmente (Thomas et al., 1999)
La seconda oncoproteina degli HPV, importante per l’immortalizzazione delle
cellule infettate e per la patogenesi virale, è E7. Le proteine E7 degli HPV ad alto e
basso rischio si trovano prevalentemente nel nucleo e hanno una dimensione di 100
aminoacidi. L’espressione di E7 porta alla trasformazione di cellule NIH 3T3 murine
immortalizzate e, meno frequentemente, di cheratinociti umani (Munger et al., 1989).
Un’efficiente immortalizzazione di cheratinociti umani richiede però l’azione
concomitante di E6 e E7. Topi transgenici che esprimono solo E7 sviluppano lesioni a
basso grado e displasie cervicali ad elevato grado che possono andare incontro a
progressione maligna, mentre topi transgenici per E6 sviluppano solo lesioni
iperproliferative a basso grado (Riley et al., 2003). La caratteristica peculiare di E7
riguarda la sua capacità di associarsi alle proteine appartenenti alla famiglia del
retinoblastoma (Rb) (Dyson et al., 1989). Il legame a Rb si verifica a livello di una delle
tre regioni conservate presenti in tutte le proteine E7 degli HPV a elevato rischio: CR1
nella porzione N-terminale; CR2, che contiene una sequenza LXCXE che lega Rb;
CR3, in C-terminale, che contiene due domini a dita di zinco, importanti per la
dimerizzazione. I domini CR1 e CR2 di E7 presentano un’ omologia di sequenza alle

regioni CR1 e CR2 della proteina E1A di adenovirus, anch’essa capace di legare la
proteina Rb (Phelps et al., 1988).
La famiglia di proteine del retinoblastoma comprende Rb, p107 e p130, espresse
in modo diverso durante il ciclo cellulare (Berezutskaya et al., 1997). Mentre Rb è
espresso costitutivamente durante tutte le fasi del ciclo, p107 viene sintetizzato
soprattutto durante la fase S, mentre p130 predomina durante la fase G0 (Classon et al.,
2001).
Rb, nella forma ipofosforilata, presente durante la fase G1 precoce, forma
complessi con i fattori di trascrizione appartenenti alla famiglia E2F/DP1, che si legano
ai promotori di geni coinvolti nella progressione del ciclo cellulare verso la fase S
(come la DNA polimerasi α, la timidina chinasi e la timidilato sintetasi) o nei
meccanismi apoptotici e questo ha come conseguenza la repressione della trascrizione
(Edmonds et al., 1989). Per rendere possibile la progressione dalla fase G1 a S,
complessi ciclina chinasi fosforilano Rb, con conseguente rilascio di Rb dal complesso
con E2F, che rende possibile la trascrizione di geni coinvolti nella sintesi del DNA.
E7 recluta Rb lontano dal complesso E2F/DP1, con conseguente attivazione
costitutiva dei geni bersaglio di E2F (Berezutskaya et al., 1997). Oltre a legare Rb, E7
ne media anche la degradazione attraverso il sistema del proteosoma. I membri della
famiglia dei Rb sono i principali regolatori dell’uscita dal ciclo cellulare, che avviene
durante la differenziazione degli epiteli. La perdita della funzione di Rb rende possibile
la replicazione produttiva in cellule soprabasali differenziate (Chellappan et al., 1992).
Il legame di E7 a Rb è importante per il mantenimento di un adeguato numero di copie
di HPV-31 in forma episomica in cellule indifferenziate (Longworth et al., 2004).
Questo è legato alla perdita dei punti di controllo del ciclo cellulare, che bloccano il
mantenimento di DNA in forma extracromosomale.

Il legame dei membri della famiglia di Rb a E7 non è limitato agli HPV ad
elevato rischio, in quanto anche proteine E7 a basso rischio si associano a Rb, anche se
con una ridotta affinità di legame. Le proteine E7 degli HPV ad alto e basso rischio
presentano sequenze aminoacidiche simili, ma non identiche, a livello del dominio CR2,
che media il legame a Rb. La mutazione di un singolo aminoacido a livello del dominio
CR2 di E7 appartenenti agli HPV a basso rischio ha come conseguenza una maggiore
affinità di legame di E7 e l’acquisizione della capacità di trasformare cellule di roditore.
Inoltre, la proteina E7 di HPV-1 a basso rischio non è in grado di degradare Rb e questo
spiega la sua incapacità di attivare geni regolati da E2F. Questo implica che, nonostante
il legame tra E7 e Rb sia molto importante, altri fattori partecipano alla trasformazione e
immortalizzazione cellulare (Ciccolini et al., 1994). E7 degli HPV ad elevato rischio
induce inoltre la degradazione di Rb mediata da meccanismi di ubiquitinazione,
importante per il superamento del blocco del ciclo cellulare, mentre gli HPV a basso
rischio non presentano questa capacità (Fehrmann et al., 2003).
Oltre a legare i membri della famiglia di Rb, le proteine E7 si associano alle
cicline A e E, così come gli inibitori dipendenti dalle cicline chinasi, p21 e p27. Dato
che le cicline e le chinasi ad esse associate guidano la progressione nel ciclo cellulare
fosforilando la proteina Rb, non sorprende che E7 agisca aumentando l’attività di queste
proteine. Le proteine E7 ad elevato rischio legano direttamente i complessi ciclina A-
cdk2 e E7 di HPV-18 lega anche la ciclina E indirettamente, attraverso p107 (McIntyre
et al., 1996). La proteina E7 ad elevato rischio aumenta i livelli delle cicline A e E,
mente le E7 a basso rischio non hanno questo effetto. p21 e p27, due inibitori delle
cicline chinasi, sono legate da E7, che ne blocca l’azione e aumenta ulteriormente
l’attività delle cicline chinasi (Funk et al., 1997) Questo punto è molto importante, in
quanto chiarisce perché l’azione associata di E6 e E7 porta a un’immortalizzazione dei

cheratinociti più efficiente rispetto a quando le due proteine agiscono separatamente.
Infatti, come si è affermato precedentemente, E6 viene neutralizzato da INK4, mentre
E7 supera questa inibizione attivando direttamente le cicline A e E. E6, a sua volta,
impedisce l’apoptosi indotta da E7 degradando proteine proapoptotiche.
Il terzo gruppo di proteine legate da E7 sono le istone deacetilasi (HDAC).
Generalmente la repressione di promotori indotti da E2F è mediata non solo dal legame
a Rb, ma anche dall’azione di HDAC (Figura 6) (Brehm et al., 1998).
Rb
Ciclina
D/ C
DK4/6
G0
G2
G1
S
HDACHDAC
HDACHDAC
Rb DP
HDAC
E2F
Rb
P
P
P
Rb
P
P
P
E2F
Rb
E7
E2F
DP
In cellule che non presentano il papillomavirus umano, Rb lega le HDAC e le
recluta a livello di promotori inducibili da E2F. Recentemente si è visto che le proteine
E7 legano HDAC indipendentemente dal loro legame a Rb e questa associazione è
importante per permettere a E7 di svolgere un ruolo nell’immortalizzazione e nel
mantenimento del virus in forma episomale. Le proteine HDAC sono espresse in tutti i
Figura 6: Meccanismo regolazione del ciclo cellulare mediato dalle proteine Rb, HDAC e E2F/DP1
(modificata da: Longworth, M. S., and L. A. Laimins. 2004. J. Virol. 78: 3533–3541).

tessuti e agiscono per rimuovere i gruppi acetile da code N-terminali degli istoni che
formano i nucleosomi, bloccando così la replicazione. In più le HDAC deacetilano
direttamente i fattori E2F, con conseguente perdita della loro funzione (Munger et al.,
1989). Sono state identificate tre classi di HDAC, ma sono stati studiati principalmente i
membri delle prime due classi. Le HDAC di classe I sono attive solo quando legate a
cofattori che ne modulano l’attività o le dirigono al sito dove verranno deacetilate.
Fanno parte di questa classe le HDAC umane 1, 2, 3, 8, localizzate esclusivamente nel
nucleo. Le HDAC di classe II entrano ed escono dal nucleo. Le proteine E7 degli HPV
ad elevato rischio si legano alle HDAC 1 e 2 attraverso MIP2β, che si lega direttamente
a E7 (Brehm et al., 1999). In particolare, la proteina E7 di HPV-16 e HPV-31 spiazza
HDAC da Rb e lega questa proteina indipendentemente da Rb (Brehm et al., 1998).
Nella proteina E7 di HPV-31, la mutazione del sito di legame per HDAC, porta
all’incapacità di mantenere stabilmente il genoma virale in forma episomale e
all’impossibilità di aumentare la vita media delle cellule trasfettate. Non è chiaro il
motivo per cui il legame di HDAC a E7 sia necessario per il mantenimento del genoma
virale, ma sono state formulate diverse ipotesi.
La prima di queste afferma che, poiché il legame di E7 alle HDAC impedisce a
queste ultime di legare Rb, E7 potrebbe agire bloccando alcune importanti attività di
Rb. Una seconda possibilità è che il legame di HDAC a E7 ne blocchi la capacità di
deacetilare i fattori di trascrizione E2F, portando alla loro rilocalizzazione all’esterno
del nucleo. La rimozione dell’attività di HDAC dai promotori ne rende possibile
l’acetilazione e la successiva attivazione (Hurford et al., 1997). In esperimenti di
trasfezione transiente, si è visto che E7 transattiva il promotore della fosfatasi cdc25A,
attraverso i siti di legame a E2F presenti su tale promotore e questa attività dipende dal
legame sia di Rb sia di HDAC (Nguyen et al., 2003). cdc25A è importante per la

defosforilazione e attivazione delle cdk ed è necessario per la progressione nel ciclo
cellulare (Jinno et al., 1994). Questo è un altro importante bersaglio di E7, necessario
per lo svolgimento del suo ruolo nel contesto della patogenesi virale. Infine E7 silenzia i
geni reclutando HDAC, come nel caso di IRF1 (“interferon regulatory factor 1”), la cui
espressione è importante per la risposta del sistema immnunitario e degli interferoni
verso infezioni di papillomavirus persistenti (Park et al., 2000).
La regione C-terminale di proteine E7 appartenenti a papillomavirus ad alto e
basso rischio presenta domini Cys-X-X-Cys, simili a domini a dita di zinco. La
mutazione di una o di entrambe le cisteine in uno di questi domini porta alla perdita
della capacità di immortalizzare cellule HFK e di trasformare cellule di roditore (Dyson
et al., 1989). Inoltre la stabilità di E7 diminuisce notevolmente in seguito alla mutazione
di queste cisteine, dimostrando la loro importanza nel mantenere l’integrità strutturale di
E7 (Longworth et al., 2004).
Una delle proprietà più particolari della proteina E7 degli HPV ad elevato
rischio è la loro capacità di indurre instabilità genomica. Molti tipi di cancro positivi per
il papillomavirus umano contengono diverse aneuploidie, ad indicare che variazioni nel
numero di cromosomi sono eventi importanti nella progressione tumorale (Hashida et
al., 1991). L’espressione unicamente di E7 è sufficiente ad indurre un aumento anomalo
nel numero di cromosomi in cheratinociti umani primari (Duensing et al., 2000). I
centrosomi sono i principali centri di organizzazione dei microtubuli e guidano la
segregazione dei cromosomi in cellule figlie durante la divisione cellulare. Proteine E7
mutate che non legano o degradano Rb, ma si associano a p107, mantengono la capacità
di indurre anomalie a livello dei centrosomi (Duensing et al., 2003). Tali anomalie si
osservano anche in cellule prive di Rb e p53 e in fibroblasti embrionali di topi “knock
out” per Rb, p130 e p107. E’ possibile che il legame di una combinazione di membri

della famiglia di Rb o di altri fattori sia richiesta per mediare le anomalie del
centrosoma (Duensing et al., 2003).
Coltivazione del virus
Gli HPV sono difficilmente coltivabili in vitro, in quanto si replicano in vivo in
epiteli squamosi stratificati, le cui condizioni non sono completamente riproducibili in
coltura su monostrati cellulari.
L’infezione di monostrati cellulari (fibroblasti o cellule epiteliali) con HPV,
induce la replicazione del DNA virale in forma episomale e l’espressione di proteine
non strutturali, ad eccezione di E4. Il principale limite di questa tecnica è la mancata
espressione di proteine virali strutturali. In monostrati cellulari è possibile tuttavia
produrre il virus in forma infettiva se i geni codificanti per le due proteine virali
strutturali vengono forniti attraverso altri vettori virali attenuati.
Date le difficoltà dell’isolamento dell’HPV da monostrati cellulari, per la
diagnosi clinica di questo tipo di infezione, si utilizzano tecniche per la ricerca del DNA
virale, quali PCR o ibridazione molecolare.
Il tropismo specie-specifico di HPV ne ha limitato lo studio attraverso l’uso di
modelli animali. Tuttavia un possibile approccio per la propagazione degli HPV è stato
esporre colture di cellule epiteliali primarie all’azione del virus e in seguito porle sotto
la capsula renale di topi nudi. Quest’ultimo è un sito anatomico immunologicamente
protetto, che può sostenere la crescita di cellule eterologhe e la formazione di un epitelio
pluristratificato, in grado di riprodurre le caratteristiche di un epitelio squamoso
stratificato. Questo tipo di approccio ha reso possibile la propagazione in coltura di
diversi tipi di HPV, ma non viene applicato abitualmente nella pratica clinica; infatti si

tratta di un sistema non ottimizzabile per la produzione del virus su larga scala o per
l’analisi della replicazione virale.
Il ciclo replicativo completo del virus può essere riprodotto su colture cellulari
utilizzando sistemi di colture “raft”, formati da epiteli squamosi stratificati posti su
un’interfaccia aria-acqua. Questa tecnica è più complicata rispetto alla coltivazione su
monostrato cellulare e la replicazione virale completa avviene solo per una piccola
percentuale di cellule. Tuttavia è possibile ricorrere a questo sistema per studiare aspetti
biologici, genetici e biochimici del virus (Lowy et al., 2001).

CARATTERISTICHE CLINICHE DELLE INFEZIONI DA
PAPILLOMAVIRUS
Epidemiologia
Gli HPV sono patogeni ampiamente distribuiti nella specie umana, che si
trasmettono prevalentemente per via sessuale. Sono virus molto resistenti al calore e a
condizioni di aridità, pertanto si può verificare anche un tipo di trasmissione non
sessuale, ad esempio per mezzo di fomiti, in seguito a prolungato contatto con abiti
contaminati (Roden et al., 1997).
Gli HPV ad alto rischio sono strettamente associati a carcinomi alla cervice
uterina, che costituiscono la seconda causa di cancro mortale nella donna, con 288 000
vittime ogni anno nel mondo. In particolare sono coinvolti nell’80-90% dei casi di
neoplasia intraepiteliale della cervice uterina (CIN). Uno studio recente, condotto su
donne latino-americane, ha rilevato che l’infezione con HPV-16 e HPV-18 aumenta
significativamente il rischio di sviluppare cancro alla cervice uterina, con una
prevalenza di HPV-16 circa 10 volte superiore a quella di HPV-18; anche la
progressione delle lesioni precancerose è maggiore in seguito ad infezione di HPV-16
rispetto a quanto avviene per altri tipi di HPV (Swan et al., 1999).
Ogni anno sono riportati circa 510 000 nuovi casi, di cui l’80% in paesi in via di
sviluppo: 68 000 in Africa, 77 000 in America Latina e 245 000 in Asia.
In assenza di programmi di “screening” (quali il Pap test) che identifichino
precocemente la malattia, il cancro alla cervice uterina viene diagnosticato tardivamente
e porta a morte nella maggior parte dei casi. La più elevata incidenza annua si rileva
nell’America del Sud (93.8 su 100 000 donne ad Haiti, IARC 1996) e in Asia (30 su 100
000 donne in India) e in Sud Africa (61.4 su 100 000 donne in Tanzania). La prevalenza

di infezioni da HPV nel mondo è di 630 milioni di casi e quella delle infezioni cliniche
di 190 milioni. La prevalenza di cancro alla cervice uterina è invece di 2 274 000: 1 300
000 in Asia, 409 000 in Europa, 218 000 in Africa, 172 000 in America Latina, 167 000
in America del Nord e 8 000 in Australia. Studi epidemiologici hanno rilevato che in
USA, il 75% della popolazione compresa tra i 15 e 50 anni viene infettata da HPV, di
cui il 60% manifesta infezione di tipo transiente (rilevazione con anticorpi), il 10%
infezione persistente (rilevazione del DNA virale), il 4% anomalie citologiche e l’1%
lesioni cliniche. La prevalenza dell’infezione di HPV in donne sessualmente attive è del
18-25%, in particolare tra adolescenti. Queste donne possono trasmettere l’infezione al
proprio compagno o ai loro bambini. Nei neonati l’infezione da HPV può causare
papillomi nella cavità orale e nel tratto respiratorio superiore. In individui infettati da
HIV, l’infezione da HPV causa verruche estese e patologie rapidamente progressive
(www.who.int).
Ci sono diversi fattori che costituiscono un rischio per lo sviluppo del tumore
oltre all’infezione degli HPV, quali l’attività sessuale, l’età, la co-infezione con i virus
HIV, CMV, HHV-6, HHV-7 e HSV-2, condizioni di immunodepressione,
probabilmente anche il consumo di ormoni steroidei (principio attivo dei contraccettivi
orali) e il fumo di sigaretta. Per quanto concerne l’età si è osservato che la maggior
parte dei tumori cervicali origina a livello delle giunzioni situate tra l’epitelio colonnare
dell’endocervice e l’epitelio squamoso dell’esocervice. In questi siti avvengono continui
cambiamenti e il rischio maggiore di contrarre l’infezione coincide con il periodo di
maggiore plasticità, che si verifica durante la pubertà e in gravidanza, mentre
diminuisce in menopausa. L’infezione degli HPV è più diffusa in giovani donne attive
sessualmente, in un’età compresa tra i 18 e i 30 anni, anche se il tumore colpisce

soprattutto donne aventi più di 35 anni, ad indicare che l’infezione è probabilmente
contratta in giovane età e progredisce lentamente verso il cancro.
La risposta immunitaria principale verso il virus è di tipo cellulare; pertanto
situazioni di immunodepressione, come trapianti o sindrome da immunodeficienza
acquisita, aumentano il rischio di contrarre l’infezione (Calore et al., 2001).
La regione LCR del genoma virale presenta sequenze simili agli elementi
responsivi agli ormoni glucocorticoidi e pertanto la loro attività è aumentata dagli
ormoni steroidei, come il progesterone e il desametasone. L’uso prolungato di
contraccettivi orali è quindi un altro importante fattore di rischio. Un ulteriore elemento
da considerare è il fumo, che è però indipendente dall’infezione virale per lo sviluppo di
cancro cervicale (Adam et al., 2000). Anche gravidanze multiple sono un fattore di
rischio tra donne che presentano infezione da HPV, così come il consumo di alcol e le
abitudini alimentari (Burd et al., 2003).
Altri virus trasmessi sessualmente potrebbero agire come cofattori nello sviluppo
di cancro cervicale; tra questi, ad esempio HSV-2, CMV, HHV-6 e HHV-7 (zur
Hausen, 1982). Una forte riduzione del rischio di sviluppare neoplasie cervicali è invece
la coinfezione con virus adeno-associati (Coker et al., 2001). Il prodotto del gene Rep78
di adenovirus blocca la trascrizione degli oncogeni E6 e E7, interferendo con il legame
delle proteine che legano la sequenza TATA a livello del promotore P97 nella regione
LCR degli HPV.
Il titolo virale è direttamente correlato alla gravità della malattia. In particolare è
stato dimostrato che HPV-16 raggiunge livelli maggiori rispetto agli altri HPV e, solo
per HPV16, tale dose è direttamente proporzionale alla gravità della malattia. Gli altri
tipi di virus ad alto rischio, anche a basso titolo, sono in grado di indurre tumori maligni
(Swan et al., 1999).

Un altro fattore emergente nello sviluppo di neoplasie cervicali è il ruolo svolto
dalle diverse varianti virali (Giannoudis et al., 2001). Le varianti virali differiscono per
quanto riguarda proprietà chimiche e patogenicità. Sulla base di variazioni di sequenza
delle regioni L1, L2, LCR del genoma virale, è stato possibile individuare cinque
varianti filogenetiche di HPV-16: europeo (E), asiatico (As), asiatico-americano (AA),
africano-1 (Af1) e africano-2 (Af2). La loro oncogenicità varia in base alla distribuzione
geografica e all’origine etnica della popolazione virale. In uno studio è stato dimostrato
che, a causa dell’aumento dell’attività trascrizionale e delle variazioni subite a livello
delle sequenze responsive ai progesteroni, la variante asiatica-americana presenta
un’attività maggiormente oncogena rispetto alla variante europea.
Anche la predisposizione genetica svolge un ruolo importante nel favorire
l’insorgenza di cancro alla cervice uterina (Magnusson et al., 2001). Essa determina la
suscettibilità all’infezione, l’abilità di eliminarla e il tempo necessario per lo sviluppo
della malattia. Gli effetti di un ambiente famigliare condiviso incidono solo per un 2%,
soprattutto tra sorelle piuttosto che tra madre e figlia. Numerosi studi hanno inoltre
riscontrato un’associazione tra carcinomi contenenti HPV-16 o HPV-18 e la presenza di
particolari antigeni leucocitari umani (HLA). Individui con alcuni tipi di antigeni
tessutali, come, ad esempio, HLA-DQB1*03 (DQ3), corrono maggiori rischi di
sviluppare cancro alla cervice uterina. Allo stesso modo pazienti con HLA-B7 tendono
a sviluppare carcinomi più aggressivi con una diagnosi peggiore. L’antigene HLA-II
Dqw3 è espresso dal 67-88% delle donne con cancro alla cervice, mentre solo dal 50%
della popolazione di controllo (Burd et al., 2003). Infine, all’interno della popolazione,
è possibile riscontrare un polimorfismo a livello del codone 72 della proteina p53, che
può codificare alternativamente per un’arginina o una prolina. La variante con l’arginina

è più suscettibile alla degradazione da parte della proteina E6: infatti, è maggiormente
presente nei tumori associati agli HPV (Sanclemente et al., 2002).
La gravità della malattia tende inoltre ad aumentare per il verificasi della
contemporanea co-infezione di diversi tipi di HPV. Queste sono state riscontrate
nell’11,8% di pazienti con analisi citologiche normali e nel 34,5% di pazienti con lieve
o moderata discariosi. La maggior parte delle infezioni multiple è caratterizzata dalla
presenza di due genotipi virali, anche se sono già stati riscontrati casi con tre, quattro,
cinque genotipi (Kleter et al., 1999).
Manifestazioni cliniche
Gli HPV possono essere suddivisi in tre sottotipi: a basso, intermedio ed alto
rischio, in base alla gravità della patologia ad essi associata.
Gli HPV a basso rischio sono associati a lesioni proliferative della pelle e delle
mucose, in genere benigne. Le manifestazioni cliniche comprendono verruche comuni,
piane e plantari, condilomi acuminati genitali e anali, condilomi piani cervicali (Figura
7), lesioni maculari pitiriasiformi in pazienti con epidermodisplasia verruciforme (EV),
papillomi laringei. Questi ultimi si presentano nell’infanzia e, nonostante siano benigni,
possono causare ostruzione respiratoria acuta e danno spesso recidive (Tabella 2) (Dolei
et al., 2002).

Figura 7: Condiloma cervicale.

Tipi di lesioni Genotipi di papillomavirus
Lesioni cutanee
Verruche volgari, piane e plantari
Verruche in soggetti con EV
Carcinomi cutanei in soggetti con EV
1, 2, 3, 4, 7, 10, 27, 28, 29, 41
5, 8, 9, 12, 14, 15, 17, 19, 20, 47, 49
5, 8, 14, 17, 20, 47
Lesioni mucose
Condilomi acuminati e piani
Papulosi Bowenoide
Condiloma gigante
Papillomi delle vie respiratorie
Papillomi congiuntivali
Lesioni della mucosa orale:
Iperplasia focale epiteliale
Infezioni con papillomavirus del tratto genitale
Lesioni sulle labbra
Carcinomi della cervice uterina:
- alta associazione
- moderata associazione
- scarsa associazione
Cancro vulvare
6, 11, 42, 43, 44, 54, 55
16
6, 11
6, 11
6, 11
13, 32
6, 11, 16,
2
16, 18, 45, 56
31, 33, 35, 51, 52
6, 11, 42, 43, 44
16
Tra le lesioni cutanee, le verruche comuni o volgari sono le forme più diffuse e
si manifestano in forma di papule bianche-grigiastre o brune, piatte o rilevate, che si
Tabella 2: Associazioni prevalenti tra genotipi degli HPV e manifestazioni cliniche.

localizzano più frequentemente a livello delle mani, in particolare sulle superfici dorsali
e nelle regioni periungueali. Vi sono inoltre verruche piane, che hanno l’aspetto di
papule rosse, lievemente rilevate, che insorgono a livello del viso o delle mani e le
verruche plantari e palmari, che si localizzano rispettivamente nella pianta dei piedi e
nel palmo delle mani. I genotipi degli HPV più frequentemente riscontrati in verruche
sono i tipi 1, 2, 3, 4 e 7.
La maggior parte dei restanti tipi cutanei degli HPV (genotipi 5, 8, 9, 12, 14, 15,
17, 19, 20, 47, 49) è stata ritrovata nelle lesioni della epidermodisplasia verruciforme
(EV), un’affezione caratterizzata dalla diffusione delle lesioni da HPV disseminate in
gran parte della superficie corporea, simili a verruche piane e macule rossastre, che si
manifesta in soggetti con profonde alterazioni dell’immunità cellulare. Non è
infrequente la degenerazione in carcinoma a cellule squamose. Tra gli HPV di tipo
cutaneo, i genotipi 5 e 8 e meno frequentemente il 14, 17, 20 e 47, sono stati identificati
in carcinomi a cellule squamose che possono insorgere in tali individui.
Le lesioni mucose benigne da HPV comprendono prevalentemente condilomi
acuminati e piani, che sono conseguenti a trasmissione sessuale del virus e insorgono a
livello del pene, dei genitali femminili, dell’uretra, dell’area perianale e del retto. Si
manifestano come masse esofitiche verrucose di consistenza molle (condilomi piani) o
modestamente rilevate (condilomi acuminati). Sono generalmente associati ad infezioni
dei genotipi 6 e 11 di HPV a basso rischio e non portano a cancro. La maggior parte
delle lesioni è asintomatica e si può risolvere spontaneamente in 3-4 mesi, rimanere
invariata o aumentare di dimensione e numero. Quando le verruche sono di colore
rosso-marrone devono essere sottoposte a biopsia, in quanto potrebbe trattarsi di
papulosi Bowenoide, causata da HPV-16 e HPV-18 e, dal punto di vista istologico,

presentare la stessa configurazione delle neoplasie intraepiteliali. Queste lesioni
potrebbero evolvere in carcinoma.
Altre sedi mucose infettate dagli HPV, caratterizzate da lesioni benigne di tipo
papillomatoso, si trovano a livello respiratorio, congiuntivale e orale.
L’infezione può verificarsi in forma latente o inattiva, con andamento
asintomatico, che presenta la zona epiteliale infettata citologicamente normale. Il DNA
dei papillomavirus umani (solitamente HPV-6 e 11) è riscontrabile generalmente nel
10% dei casi.
L’infezione attiva, caratterizzata da lesioni squamose intraepiteliali (SIL), si
manifesta con grandi cellule arrotondate dette coilociti. Le SIL sono classificate in
rapporto alla gravità delle lesioni istologiche. In Europa è possibile distinguere tre gradi
di neoplasie intraepiteliali cervicali (CIN): CIN1 (lieve), CIN2 (moderata), CIN3
(severa). Negli Stati Uniti le SIL sono suddivise in SIL a basso grado (LGSIL, che
corrisponde a CIN1) e SIL ad alto grado (HGSIL, paragonabile a CIN2 e CIN3). A
livello di vagina, vulva, ano e pene si riscontrano lesioni simili ma non identiche:
neoplasie intraepiteliali vaginali (VAIN), vulvari (VIN), anali (AIN) e penili (PIN). La
maggior parte di queste lesioni è associata ad infezione da HPV. In particolare, nelle
LGSIL si riscontrano soprattutto HPV ad alto rischio. La maggior parte delle lesioni
mantiene il virus in forma episomica e sostiene un ciclo di replicazione completo: sono
espressi anche geni tardivi e origina una particella virale completa. Le HGSIL sono
prevalentemente associate a HPV ad alto rischio, che non possono però compiere un
ciclo di replicazione completo, a causa di difetti differenziativi tipici di queste lesioni. I
geni tardivi non sono espressi, il DNA virale è integrato in quello cellulare e
l’espressione degli oncogeni E6 e E7 è deregolata. CIN3 è caratterizzata da aneuploidia
cromosomica ed evolve verso forme maligne (Stanley et al., 2003).

Profilassi
Igiene
La comprensione del fatto che le lesioni a livello cervicale, vulvare, vaginale e
perianale sono di origine infettiva ha portato a notevoli miglioramenti in materia di
prevenzione del cancro alla cervice uterina. In primo luogo sono state consigliate misure
legate all’igiene, per evitare la trasmissione iatrogena tra i pazienti affetti da disturbi
ginecologici.
La prevenzione meccanica di infezioni anogenitali trasmesse sessualmente è
invece impraticabile. Infatti, le infezioni da HPV sono molto diffuse all’interno della
popolazione sessualmente attiva e l’uso del preservativo o di sostanze spermicide offre
solo una protezione limitata, in quanto le infezioni possono essere trasmesse in seguito
al contatto con altre parti del corpo, come labbra, ano, scroto, che non sono protetti dal
preservativo (Burd et al., 2003).
Vaccini
Una possibile strategia per la prevenzione e cura delle infezioni degli HPV è la
vaccinazione; in questo contesto, LGSIL e HGSIL devono essere considerate
separatamente. Nella LGSIL, infatti, è disponibile un numero maggiore di potenziali
bersagli antigenici, in quanto il ciclo replicativo del virus si svolge in modo completo.
In un individuo immunocompetente l’utilizzo del vaccino dovrebbe portare alla
guarigione della lesione, senza rischio di ricorrenze. Sono stati eseguiti studi riguardanti
l’infezione di HPV mucosali in cani (un valido modello di LGSIL), i quali indicano che
E7 non è un antigene ottimale per la realizzazione di un vaccino. Al contrario,
l’immunizzazione con E1 e E2 è più efficace dal punto di vista profilattico e
terapeutico. Per massimizzare la risposta, il vaccino può essere somministrato in

associazione ad immunomodulatori. I primi trial clinici per la sperimentazione di questo
tipo di vaccinazione hanno dato buoni risultati nel caso di lesioni a basso grado. Il punto
da chiarire è se una vaccinazione terapeutica sarà in grado di eliminare anche
l’infezione latente.
Nel caso di infezioni con HPV ad alto rischio la realizzazione di un vaccino è
più complessa. HGSIL è una patologia eterogenea, le lesioni sono instabili, la risposta
del sistema immunitario non è regolata e il virus mette in atto sistemi di evasione; infine
l’espressione di HPV è molto variabile; pertanto la risposta al vaccino varierà
dall’eliminazione completa del virus all’assenza di eliminazione. Ci sono solo due
possibili bersagli antigenici: E6 e E7, ovvero le uniche proteine ad essere espresse in
HGSIL. Tuttavia, le lesioni classificate come CIN3 regrediscono attraverso un
meccanismo mediato dal sistema immunitario e in questo caso un vaccino potrebbe
essere efficace, ma in associazione ad altri farmaci (Stanley et al., 2003).
I vaccini diretti verso i virus ad alto rischio sono in fase I e II di
sperimentazione, ma attualmente non sono ancora disponibili in commercio. Sono
costituiti da VLP, formate dalle proteine strutturali del capside L1 e L2 ma prive di
DNA virale all’interno. Sono realizzate esprimendo le ORF L1 e L2 in cellule eucariote.
Queste si assemblano spontaneamente in VLP, altamente immunogene. Data l’elevata
specificità antigenica delle proteine del capside degli HPV, non è possibile ottenere una
protezione crociata verso diversi genotipi; pertanto sarebbe necessario effettuare una
vaccinazione specifica per ogni tipo di HPV. I vaccini migliori sono costituiti da un
insieme di VLP dei principali genotipi ad alto rischio.
L’immunogenicità e la sicurezza del vaccino di HPV-16 realizzato con VLP
contenenti la proteina maggiore del capside L1, sono state valutate in un trial di fase I, a
doppio cieco, randomizzato e supportato da un controllo con placebo (Harro et al.,

2001). Il vaccino è stato realizzato inserendo il gene codificante per L1 in un
baculovirus, usato come vettore. Questo è stato poi fatto esprimere in cellule di insetto.
Una dose ottimale di 50 µg di vaccino è stata somministrata mediante iniezione nel
muscolo deltoide a distanza di 0, 1, 4 mesi. Questo vaccino è ben tollerato, anche senza
adiuvanti; il titolo anticorpale è 10 volte maggiore rispetto a quello sviluppato durante
un’infezione naturale e persiste per un lungo periodo di tempo.
Anche se i dati disponibili attualmente non permettono di arrivare a conclusioni
sicure riguardo gli effetti protettivi del vaccino, studi su modelli animali permetteranno
di appurare meglio questi punti. Se si riuscirà a sviluppare un vaccino efficace per
l’uomo sarà teoricamente possibile prevenire più di 300 000 casi di cancro all’anno
(Burd et al., 2003).
Diagnosi delle infezioni
Per la diagnosi di laboratorio delle infezioni causate dagli HPV, dato che
mancano efficaci metodi colturali, si ricorre in primo luogo a saggi di tipo citologico e
istologico. Sono state introdotte anche tecniche molecolari, come l’ibridazione in situ e
soprattutto la PCR. Tra le alterazioni che possono essere confuse con le verruche
causate dagli HPV, vi sono quelle indotte dal poxvirus del mollusco contagioso.
Citologia convenzionale
Il principale metodo per la rilevazione degli HPV ad alto rischio è la colorazione
di Papanicolau degli strisci di campioni prelevati dalla cervice uterina (chiamata
comunemente “Pap test”); è stata introdotta nel 1949 dal patologo George Papanicolau,
prima che si sapesse quale fosse la causa del cancro alla cervice uterina. Questa tecnica
si basa sull’osservazione dei cambiamenti morfologici subiti dalle cellule nelle zone

trasformate della cervice. In conformità a questo tipo di colorazione è stata realizzata la
classificazione di Bethesda, introdotta nel 1988 e approvata nel 1991, in sostituzione al
sistema CIN (introdotto nel 1973 e basato sull’analisi dell’architettura dei tessuti). Un
aggiornamento della classificazione di Bethesda è stato realizzato nel 2001, per
integrarvi i miglioramenti nella comprensione di questi tipi di patologie. Le anomalie
delle cellule squamose sono suddivise in quattro categorie: 1) ASC, atipia delle cellule
squamose 2) LGSIL, lesioni a cellule squamose intraepiteliali di basso grado 3) HGSIL,
lesioni a cellule squamose intraepiteliali ad alto grado 4) carcinoma alle cellule
squamose.
Il “Pap test” presenta alcuni limiti. I campioni prelevati sono, nell’8% dei casi,
inadeguati. Si possono verificare risultati falsamente negativi nel 20-30% dei casi, in
quanto, talvolta, il campione è contaminato da sangue, funghi, batteri presenti a livello
della cervice, impedendo la rilevazione di cellule anomale. Inoltre, se le cellule
rimangono esposte all’aria troppo a lungo prima della fissazione, si alterano (Burd et
al., 2003).
Citologia su monostrato
Sono stati sviluppati nuovi metodi per la raccolta e l’analisi di campioni destinati
al “Pap test”, al fine di ridurre il numero di risultati falsamente negativi. Con tali sistemi
i campioni sono raccolti in una soluzione conservante, anziché essere posti direttamente
sul vetrino manualmente. La struttura cellulare è mantenuta più correttamente, in quanto
le cellule sono immediatamente fissate. Inoltre si utilizza una particolare spatola
cervicale per la raccolta del campione, che permette di raccogliere un numero di cellule
doppio rispetto agli altri strumenti. Il monostrato cellulare uniforme che si ottiene viene
così esaminato più facilmente dai tecnici, si evita la seccatura del campione e viene

rimossa la maggior parte dei contaminanti (muco, proteine, globuli rossi, batteri e
lieviti) Attualmente sono disponibili due sistemi di citologia su monostrato approvati
dalla FDA: il sistema “PrepStain” e il “ThinPrep Pap” (Figura 8).
I costi sono maggiori rispetto al “Pap test” tradizionale, ma numerose
pubblicazioni in questo campo consigliano l’utilizzo della citologia su monostrato per la
rilevazione di lesioni precancerose. La sensibilità della diagnosi è maggiore per tutti i
tipi di patologie, in una percentuale che varia dal 4 al 117% in base al tipo di paziente.
Quando si paragonano i risultati della citologia su monostrato e della colorazione di Pap
tradizionale al “gold standard”, costituito da biopsia effettuata con colposcopia diretta,
la citologia su monostrato è più efficace nell’identificazione di un quadro displastico
(Sheets et al., 1995).
La FDA ha approvato due nuovi dispositivi: “AutoPap” 300QC (NeoPath,
Redmond, Wash) e “PapNet” (Neuromedical Systems, Suffern, N.Y.), realizzati al fine
di rendere le procedure diagnostiche maggiormente automatizzabili. Sistemi
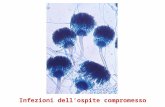
Figura 8: Striscio realizzato con la tecnica “ThinPrep Pap”, che mette in evidenza cellule squamose
anomale con effetto citopatico causato dagli HPV (freccia), in accordo con il quadro istologico della
LSIL.

computerizzati mostrano, su uno schermo, cellule potenzialmente anomale, destinate a
successiva analisi. Con questo sistema si possono studiare sia campioni ottenuti con il
“Pap test” tradizionale sia quelli ottenuti con la tecnica del monostrato cellulare.
Un altro sistema per migliorare la diagnosi effettuata con il Pap test è realizzare
una colorazione specifica per gli HPV. “BenchMark” (Ventana Medical Systems,
Tucson, Ariz) è un sistema automatizzato modulare che realizza una colorazione
immunoistochimica su campioni ottenuti con il sistema “ThinPrep Pap”. Sono
disponibili sonde per alcuni sottotipi di HPV ad alto rischio (HPV-16, 18, 31, 33, 35,
39, 45, 51, 52, 56, 59, 70) e a basso rischio (HPV-6, 11, 42, 43, 44) (Burd et al., 2003).
Istopatologia
I pazienti che, in seguito ad analisi con “Pap test”, presentano anomalie, ma
senza evidenti lesioni cervicali, sono solitamente sottoposti a colposcopia e a biopsia
con colposcopia. La colposcopia può rilevare displasie ad alto e basso grado, ma non
patologie microinvasive. Se non vengono rilevate anomalie, viene effettuata una
biopsia. Essa è utilizzata per confermare la maggior parte delle diagnosi attraverso
l’osservazione delle alterazioni morfologiche tipiche delle infezioni da HPV, come
iperplasia epiteliale (acantosi), vacuolizzazione citoplasmatica degenerativa (coilocitosi)
in cheratinociti differenziati con nuclei atipici. Inoltre può essere utilizzata una
colorazione che evidenzi gli antigeni o gli acidi nucleici degli HPV. Sono disponibili
anticorpi monoclonali o policlonali diretti verso un antigene comune tra gli HPV,
ovvero un epitopo lineare situato al centro della proteina maggiore del capside, molto
espressa tra i diversi genotipi virali. Il legame dell’anticorpo viene rilevato attraverso la
marcatura con perossidasi-antiperossidasi. La colorazione è solitamente localizzata nel

nucleo delle cellule infettate, anche se talvolta può essere osservata nel citoplasma dei
coilociti.
Il DNA o l’RNA degli HPV può essere evidenziato in tessuti derivanti da
biopsia attraverso metodi di ibridazione in situ, che prevedono l’uso di sonde marcate
con radioisotopi o con ligandi chimicamente reattivi e rilevati con autoradiografia,
fluorescenza o con una reazione colorimetrica. Le tecniche di ibridazione in situ
localizzano le sequenze degli acidi nucleici dell’HPV all’interno di singole cellule,
mantenendo la morfologia delle cellule e del tessuto al fine di valutare le alterazioni
morfologiche associate alle lesioni. Per la rilevazione degli HPV, sono preferibili sonde
che non utilizzano isotopi e metodi enzimatici sono più adatti rispetto a quelli basati
sulla fluorescenza per agevolare l’interpretazione. Le caratteristiche del segnale
(confluente o appuntito) riflettono la forma episomale o integrata del DNA virale.
L’intensità del segnale è direttamente proporzionale al numero di copie. Le tecniche in
situ di amplificazione del segnale o del bersaglio sono state sviluppate per rilevare
enzimaticamente un basso numero di copie delle sequenze di acido nucleico degli HPV
(Burd et al., 2003).
Rilevazione del DNA degli HPV
PCR con primer specifici: si basa sulle variazioni di sequenza all’interno dei
geni E6 e E7 nell’ambito dei diversi genotipi di HPV. Sono stati realizzati quattordici
primer specifici per cento paia di basi della sequenza codificante per E7 di diversi
genotipi di HPV ad alto rischio. La sensibilità analitica del saggio è tra le dieci e le
duecento copie di HPV per campione. Questo tipo di PCR viene utilizzata
prevalentemente a scopo di ricerca, in quanto la resa è limitata dalla necessità di usare,
per ogni campione, amplificazioni multiple con PCR (Burd et al., 2003).

PCR con primer aspecifici: la maggior parte degli studi basati sulla PCR
utilizzano primer consenso per amplificare un ampio spettro di tipi di HPV in una
singola reazione di PCR. I primer sono rivolti verso regioni conservate del genoma
degli HPV, come il gene che codifica per la proteina capsidica L1. Ad esempio i primer
MY09-MY11, sono diretti verso un frammento di 450 paia di basi all’interno della
sequenza codificante L1 (Bosch et al., 1995). Questi primer tuttavia falliscono nel
rilevare il DNA degli HPV nel 7% dei casi di cancro cervicale. Questo potrebbe essere
dovuto all’assenza di DNA virale nelle cellule tumorali o a risultati della PCR
falsamente negativi dovuti all’integrazione del DNA dei papillomavirus nel genoma
cellulare, che potrebbe aver distrutto le sequenze verso le quali sono diretti i primer.
Sono stati sviluppati diversi metodi per identificare i diversi genotipi di HPV
dopo l’amplificazione con primer generali e consenso. Tra queste ricordiamo analisi di
sequenziamento, dei polimorfismi di lunghezza dei frammenti di restrizione e
ibridazione con sonde specifiche usando “Dot blot” (Burd et al., 2003).
Ibridazione liquida: viene realizzata utilizzando il kit “Hybrid Capture” (Digene,
Beltsville, Md.), approvato dalla FDA per la rilevazione del DNA degli HPV su
campioni prelevati dalla cervice. Si tratta di un saggio che rende possibile
l’amplificazione del segnale di ibridazione e che determina quantitativamente la
presenza di virus nel campione attraverso tecniche di chemioluminescenza basate
sull’uso di anticorpi. Il DNA proveniente da un paziente viene denaturato e unito a un
gruppo di sonde a RNA all’interno di una soluzione. Si usano due gruppi di sonde a
RNA: il gruppo A riconosce alcuni sottotipi di HPV a basso rischio, mentre il gruppo B
riconosce quelli ad alto rischio. Si forma un ibrido RNA-DNA, che viene immobilizzato
all’interno di pozzetti situati in una piastra e decorato con anticorpi primari e
successivamente secondari, diversamente marcati. Il segnale luminoso, misurato con un

luminometro ed espresso come unità di luminosità relativa, viene paragonato a un
valore limite. La sensibilità analitica varia da 6,6 a 17,6 pg/ml in base al tipo di HPV.
Questa tecnica non presenta una forte rilevanza clinica, ma è ampiamente usata
in studi di ricerca ed epidemiologia. Non viene utilizzata per analisi di screening sulla
popolazione generale, ma è indicata come supporto alla diagnosi di infezione da HPV e
per discriminare tra papillomavirus umani ad alto e basso rischio; è utilizzato inoltre per
un’analisi più approfondita di pazienti risultati positivi al “Pap test”, al fine di valutare il
rischio di sviluppare cancro alla cervice uterina. Tuttavia questo saggio non può essere
realizzato indipendentemente dal “Pap test”. I limiti sono legati alla reattività crociata
degli anticorpi utilizzati per la rilevazione del segnale, che può portare ad ottenere
risultati falsamente positivi (Burd et al., 2003).
Rilevazione dell’mRNA degli HPV: viene realizzata utilizzando il test “In-Cell”
(Invirion, Frankfurt, Mich), che permette di identificare l’RNA dei geni E6 e E7. E’ così
possibile determinare se i geni oncogeni degli HPV sono presenti e attivi. Il saggio può
essere automatizzato su ogni sistema in grado di rilevare la fluorescenza. La citometria
di flusso può essere adattata a questo saggio utilizzando campioni derivanti da analisi
citologiche. Quest’analisi può essere eseguita anche direttamente su strisci destinati al
“Pap test” e visualizzata utilizzando un microscopio a fluorescenza. La sensibilità del
saggio è del 100%, la specificità del 70% (Burd et al., 2003).
Terapie
Gli obiettivi delle terapie verso le patologie associate a HPV sono:
- l’eradicazione delle infezioni;
- la guarigione dei sintomi;
- la prevenzione delle malattie a lungo termine;

- la prevenzione delle infezioni.
Le terapie attualmente disponibili hanno raggiunto questi obiettivi solo in parte.
Infatti, non si tratta di terapie antivirali specifiche, ma di trattamenti ablativi che non
risolvono l’infezione virale e il problema della trasmissione. E’ possibile suddividere le
terapie disponibili in quattro categorie: 1) ablative (trattamenti chirurgici) 2) agenti
citotossici 3) terapia fotodinamica 4) terapie immunomodulatorie.
Trattamenti chirurgici
Le lesioni non invasive intraepiteliali, identificabili solo utilizzando il
microscopio, sono solitamente trattate con procedure ablative superficiali, come la
crioterapia o la terapia laser. Sono procedure ambulatoriali che garantiscono il
mantenimento della fertilità; sono efficaci, a breve termine, ma il tasso di ricorrenza è
elevato, in quanto si rimuove unicamente la lesione superficiale, mentre persistono in
profondità i cheratinociti infettati.
Con la crioterapia, il tessuto anomalo e cinque millimetri di tessuto circostante
sono raffreddati con una sonda per diverse volte consecutive, al fine di indurre necrosi.
Un’alternativa è la rimozione del tessuto attraverso un laser di biossido di carbonio: è
efficace come la crioterapia e la zona cicatrizza più velocemente, subendo meno
deformazioni, ma i costi sono più elevati. Le procedure di rimozione che sfruttano
un’ansa elettronica sono attualmente considerate il trattamento più adatto per la cura di
lesioni non invasive. Si utilizza un filo carico elettricamente per l’ablazione della zona
alterata e del canale endocervicale distale. E’ meno cara rispetto alla terapia con il laser
e preserva il tessuto rimosso per un successivo esame istologico.
I tumori di dimensione inferiore ai tre millimetri sono trattati utilizzando la
biopsia conica.

I tumori precocemente invasivi sono affrontati ricorrendo ad isterectomia o
radioterapia. L’obiettivo è distruggere le cellule maligne della cervice, dei tessuti
paracervicali e dei linfonodi regionali. I tumori in stadio già avanzato sono trattati con
radioterapia diretta verso il tumore primario e verso i potenziali siti di metastatizzazione
(Burd et al., 2003).
Agenti citotossici
Gli agenti citotossici sono utilizzati per il trattamento delle verruche genitali. Si
tratta di preparati ad uso topico, che distruggono le cellule al momento del contatto,
indipendentemente dalla presenza di HPV. Sono caratterizzati da forti effetti collaterali
e da un elevato tasso di ricorrenza della malattia.
Un esempio di tali agenti è costituito dalla Podofilina, un derivato del
Podophyllum, ad uso topico. Agisce legando le proteine dei microtubuli, con
conseguente arresto della cellula in metafase. E’ controindicato in gravidanza.
L’acido tricloroacetico è un altro agente utilizzato a livello topico, ma che può
provocare effetti collaterali quali ulcerazioni, dermatiti, infezioni secondarie. Il
vantaggio rispetto alla Podofilina è che non dà effetti collaterali sistemici e pertanto può
essere utilizzato in gravidanza. Questi farmaci sono stati analizzati in ristrette
sperimentazioni cliniche, al fine di valutare la loro efficacia nel trattamento della CIN e
della VIN, ma non sono abitualmente utilizzati per la cura di infezioni di HPV a livello
mucosale.
Il 5-fluorouracile, un antitumorale antimetabolita, è disponibile sotto forma di
crema, ma la sua applicazione sulle lesioni genitali esterne è limitata, in quanto provoca
notevoli reazioni infiammatorie. Il meccanismo d’azione si basa sul blocco del trasporto
della timidina extracellulare, inibendo la via di salvataggio della sintesi dei nucleotidi.

Presenta rischio di teratogenesi e il suo uso è controindicato in gravidanza. E’ utilizzato
per il trattamento della VIN, ma i risultati sono variabili (Stanley et al., 2003).
Terapia fotodinamica
La terapia fotodinamica presenta un’efficacia variabile nel trattamento delle
infezioni di HPV. Il meccanismo d’azione si basa sull’attivazione di un fotosensore (ad
esempio l’acido aminolevulinico) da parte della luce, portando al rilascio di specie
dell’ossigeno molto reattive. Questo provoca la distruzione dei tessuti, con conseguente
attivazione del sistema immunitario dell’ospite. Il fotosensore è somministrato in modo
sistemico o topico (nel caso di CIN, VIN e verruche genitali). Il fallimento della
risposta alla terapia si associa ad una diminuzione dell’espressione di MHC di classe I
su cellule neoplastiche, indicando che la terapia fotodinamica agisce come
immunomodulatore (Stanley et al., 2003).
Terapia immunomodulatoria
Una strategia per il controllo delle infezioni da HPV consiste nell’indurre o
potenziare la risposta immunitaria, in particolare cellulo-mediata. Questo rende
possibile il trattamento anche di infezioni silenti. Gli anticorpi sono invece scarsamente
coinvolti nel controllo delle infezioni di HPV, ma potrebbero prevenire infezioni
successive.
La terapia con Interferone, caratterizzato da attività antiproliferativa, antivirale e
immunomodulatoria, è molto importante, soprattutto nella papillomatosi respiratoria
ricorrente, ma anche in altre lesioni papillomatose, quali i condilomi genitali. Non sono
bloccate le recidive, tuttavia il trattamento ha il vantaggio di non danneggiare i tessuti e
può inoltre aiutare a ridurre la massa delle lesioni prima dell’intervento di distruzione

con mezzi fisici o chirurgici. L’efficacia terapeutica migliore si è osservata con IFN-α
somministrato per via parenterale e con IFN-β intralesionale, che possono indurre
remissione completa dei condilomi genitali recidivanti in gran parte dei pazienti trattati.
Tuttavia si verificano effetti collaterali, quali febbre e mialgia. Il trattamento è costoso,
di efficacia limitata e non è applicato nei trattamenti di routine di queste lesioni.
Sono particolarmente importanti gli Imidazochinoloni, agenti farmacologici che
modulano le cellule dendritiche e i macrofagi. Un esempio è l’Aldara (Imiquimod),
applicato in modo topico. Agisce come ligando di recettori situati su macrofagi e cellule
dendritiche, provocando il rilascio di citochine e IFN-α. E’ efficace nel trattamento di
verruche genitali. Non è ancora riconosciuto per il trattamento della CIN, mentre è stato
già realizzato un “trial” per la sua applicazione nella cura della VIN. Il suo utilizzo deve
essere strettamente controllato, in quanto il rilascio di citochine può causare potenti
effetti collaterali di tipo infiammatorio.
La Cimetidina è un altro farmaco ad azione immunomodulatoria. Si tratta di un
antagonista del recettore H2 istaminico. E’ stata somministrata per via orale, ad alte
dosi, per il trattamento di verruche genitali e cutanee, papillomatosi respiratoria e
congiuntivale, soprattutto nei bambini. La cimetidina è stata testata in trial clinici in
doppio cieco e con il placebo, ma non è risultata efficace nel trattamento di patologie
indotte da HPV.
Terapia antivirale
Attualmente non sono disponibili in commercio chemioterapici attivi contro
HPV, ma il loro sviluppo è auspicabile per diverse ragioni. Infatti, una terapia antivirale
ha le potenzialità di essere efficace verso infezioni sia inapparenti sia visibili
clinicamente. Ci sono numerosi pazienti immunodepressi infettati da HPV, che non

possono essere trattati con immunoterapia e per i quali i farmaci antivirali sarebbero
l’unica alternativa. Inoltre, lesioni multifocali, come la VIN, che non possono essere
sottoposte a terapie ablative e potrebbero essere resistenti a terapie immunomodulatorie,
sarebbero trattate efficacemente con chemioterapici. Per queste motivazioni lo sviluppo
di un antivirale attivo verso HPV è un obiettivo prioritario per aziende e gruppi di
ricerca. Esistono tuttavia una serie di ostacoli, che saranno maggiormente approfonditi
nel corso di questa tesi (Stanley et al., 2003).
Retinoidi
I retinoidi sono composti naturali o sintetici simili alla vitamina A, che svolgono
una funzione preventiva verso la CIN e il cancro cervicale. Presentano un effetto
antiproliferativo, inducono differenziazione in cellule epiteliali legandosi ad un
recettore nucleare, ma non presentano una specifica attività anti HPV. Tuttavia si è
osservato che, in vitro, portano ad una diminuzione dei livelli di trascrizione di E6/E7 e
delle molecole coinvolte nell’apoptosi. La potenzialità dei retinoidi come agenti
preventivi verso le infezioni da HPV è discutibile: in studi sperimentali sono risultati
efficaci solo in combinazione con agenti citotossici, come il Cisplatino (Stanley et al.,
2003).
Indolo 3-carbinolo (I-3-C)
E’ un derivato vegetale che ha effetti chemiopreventivi, come dimostrano studi
in fase pre-clinica. Presenta un’attività anti estrogenica e induce apoptosi. Topi
transgenici per E6/E7, esposti cronicamente a 17β-estradiolo, sviluppano cancro
cervicale; tuttavia, quando questi animali sono sottoposti ad una dieta supplementata
con I-3-C, l’incidenza di cancro si riduce. Questi risultati sono senza dubbio importanti,

ma necessitano di conferme su trial estesi a un maggior numero di pazienti (Stanley et
al., 2003).
Cidofovir
Tra i farmaci utilizzati per la cura delle infezioni degli HPV è necessario citare
anche il Cidofovir, un analogo nucleosidico, che presenta un’attività ad ampio spettro
verso i virus a DNA, in particolare il citomegalovirus, ed è relativamente efficace anche
verso gli HPV, inducendo apoptosi quando è applicato localmente a livello delle cellule
infettate (Andrei et al., 2001). Durante una sperimentazione, è stato applicato un gel,
costituito per l’1% da Cidofovir, in modo topico, per un mese su 15 donne affette da
una grave forma di CIN. Nell’80% delle pazienti è stata osservata una risposta completa
o parziale, che è stata in seguito verificata anche attraverso analisi istologiche e PCR
(Snoeck et al., 2000).

HPV E MODULAZIONE DEL SISTEMA IMMUNITARIO
Meccanismi di difesa dell’ospite
Il sistema immunitario è importante nel controllo delle infezioni degli HPV.
Questo concorda con il fatto che, in donne immunodepresse, la SIL (“lesione squamosa
intraepiteliale”) presenta una maggiore incidenza e persistenza. In particolare si osserva
l’intervento del sistema immunitario cellulare ed umorale. Le infezioni di HPV sono
simili a quelle mediate da virus non litici, in quanto non causano distruzione cellulare,
ma sono rilasciati dalle cellule infettate mediante desquamazione. La difesa ideale verso
questo tipo di infezione è una combinazione di anticorpi neutralizzanti e lisi cellulare
mediata dai linfociti T citotossici (CTL). I CTL agiscono su cheratinociti presenti negli
strati intermedi degli epiteli squamosi, dove si verifica la trascrizione e replicazione del
virus e dove le proteine precoci sono espresse abbondantemente. Nonostante le proteine
tardive L1 e L2 non siano bersaglio dei CTL, in quanto sono espresse negli strati
superficiali, gli anticorpi neutralizzanti sono diretti proprio verso queste proteine, per
prevenire nuove infezioni (Sanclemente et al., 2002).
La neutralizzazione mediata da anticorpi coinvolge in particolare IgG sieriche e
IgA secretorie, rivolte verso antigeni di superficie del virus, al fine di bloccarne
l’ingresso nelle mucose epiteliali. Gli anticorpi circolanti e il complemento possono
opsonizzare e agglutinare le particelle virali, facilitando la fagocitosi mediata dai
recettori C3b e Fc. Non è chiaro se la risposta anticorpale sia sufficiente a proteggere le
donne dall’infezione e se tale protezione sia duratura. La persistenza delle verruche,
anche in individui immunocompetenti, indica che il sistema immunitario trova difficoltà
nell’innescare una risposta effettiva, a causa di immunodepressione locale o mancato
riconoscimento delle proteine virali.

Gli anticorpi non sono sempre in grado di eliminare il virus, soprattutto quando
entra in stato di latenza, in cui il DNA virale è integrato in quello cellulare. E’
particolarmente importante, a questo punto, l’intervento del sistema dell’immunità
cellulare, mediato da linfociti T citotossici (CTL) CD8+ e da linfociti T “helper” di tipo
1 (Th1) CD4+. Questo sistema mette in atto strategie di sorveglianza intercellulari e
intracellulari, che impediscono l’accumulo di cellule tumorali, sia abolendo
l’espressione degli oncogeni virali, sia eliminando le cellule infettate attraverso
l’apoptosi (Figura 9) (zur Hausen et al., 2000).
LSIL
HSIL
Carcinoma in situ
Carcinoma invasivo
Diminuzione delcontrollo immunitario
Controllo intracellulare
Contro intercellulare
Instabilità cromosomica indotta da E6/E7, aneuploidia, alterazioni del DNA cellulare
Persistenza del DNA virale
Infezione
La sintesi endogenadi IFN-β è bloccata
HPV
HPV
HPV
I meccanismi di regolazione intracellulari prevengono l’immortalizzazione di
cellule infettate con HPV ad alto rischio. I meccanismi molecolari alla base di questo
processo implicano la modificazione di oncoproteine virali, attraverso fosforilazione o
Figura 9: Controllo intercellulare e intracellulare dell’ospite durante il processo di progressione
tumorale causato dagli HPV (modificata da zur Hausen. 2002. . Nat. Rev. Cancer. 2:342–350).

defosforilazione. L’oncoproteina E7, ad esempio, è fosforilata dalla caseina chinasi II.
Un altro meccanismo di inibizione è mediato dall’interazione tra oncoproteine virali e
proteine dell’ospite, quali gli inibitori delle cicline chinasi (p21 e p27), che solitamente
sono inattivate da E7, ma la cui overespressione porta a risultati opposti, bloccando E7.
La regolazione intercellulare, invece, previene la conversione verso un fenotipo
maligno di cellule immortalizzate (che dal punto di vista clinico equivalgono a lesioni
epiteliali di basso grado) da HPV. Questo meccanismo è mediato principalmente
dall’azione di citochine secrete dai Th1 e Th2. Specifiche citochine mediano la
repressione della trascrizione di oncogeni virali, a differenza di quanto accade nel caso
di IL-6 e IL-17, che potenziano la tumorigenicità di cellule di cancro alla cervice uterina
in topi nudi.
TGF-β rientra tra le citochine che bloccano la trascrizione di geni precoci di
HPV in cellule immortalizzate, mentre in cellule maligne l’attività trascrizionale rimane
invariata. La resistenza all’inibizione di TGF-β è un evento tardivo nel corso dello
sviluppo di carcinoma alla cervice. Altre citochine che bloccano la trascrizione di HPV
in cellule immortalizzate sono IL-1 e TNF-α (Woodworth et al., 1990).
Alcuni studi hanno dimostrato che anche TNF-α, secreto dai macrofagi, agisce
reprimendo la trascrizione dei geni di HPV in cellule immortalizzate, ma non in cellule
maligne. Questo è dovuto principalmente a modificazioni del complesso trascrizionale
AP-1. L’espressione di AP-1 nel corso della differenziazione cellulare e determina
l’espressione di E6 e E7 in epiteli stratificati. Mentre in cellule immortalizzate il
complesso trascrizionale AP-1 a livello del promotore di HPV è costituito da omodimeri
c-jun/c-jun, il trattamento con TNF-α porta invece alla formazione di eterodimeri c-
jun/fraI (quest’ultimo è anche attivato dall’acido retinoico, altro potente inibitore di
HPV), in grado di sopprimere la trascrizione di HPV. In cellule maligne il complesso

AP-1 è invece formato da eterodimeri c-jun/c-fos, che svolgono un ruolo importante
nella conversione di cellule immortalizzate verso un fenotipo maligno. (zur Hausen,
2000).
Altre importanti citochine inibitorie sono gli interferoni e IL-2, prodotti da
cellule Th1 attivate. IL-2 svolge un’azione indiretta, mediando l’attivazione dei
precursori dei CTL. IL-2 e IFN-α attivano le cellule NK, importanti nei primi giorni
dell’infezione finché si sviluppa, in 3-4 giorni, una risposta specifica mediata da CTL:
essi distruggono le cellule infettate, eliminando potenziali fonti di infezione.
Gli interferoni, soprattutto β e γ (α è più specifico per alcune linee cellulari),
agiscono sia su cellule immortalizzate sia su cellule maligne, eliminando il virus
attraverso l’induzione di uno stato antivirale nelle cellule. Il tumore può presentare
meccanismi di resistenza agli interferoni, attraverso l’azione di E6 e E7, che
interagiscono con fattori coinvolti nella sintesi di IFN-α, quali p48, bloccandone così la
trascrizione.
La dimostrazione di un’ inibizione selettiva, in vivo, mediata da IFN, della
trascrizione di HPV in cellule immortalizzate, deriva da esperimenti di ibridazione in
situ, in seguito al trapianto di cellule immortalizzate in topi nudi e trattamento con IFN.
Dopo tre giorni la trascrizione di E6 e E7 si riduce maggiormente rispetto a quanto
accade in vitro. Se lo stesso trattamento è realizzato su cellule maligne, non si ottengono
gli stessi risultati. Questi dati concordano anche con le osservazioni cliniche, in cui si
vede che, in lesioni a basso grado l’espressione di E6 e E7 è notevolmente inferiore
rispetto alle lesioni ad alto grado (Durst et al., 1991).
Da questi dati emerge un importante ruolo del controllo intercellulare di HPV
mediato dalle citochine, che viene perso con la progressione maligna del tumore (zur
Hausen et al., 2000).

Meccanismi di evasione dal sistema immunitario
Gli HPV, in modo particolare i sottotipi ad elevato rischio, hanno evoluto una
serie di meccanismi per evadere dai sistemi di difesa della cellula bersaglio, i quali sono
alla base della progressione dei tumori in cui è coinvolto HPV:
- portano a diminuzione dell’espressione cellulare del complesso maggiore di
istocompatibilità di classe I (MHCI) e della proteina di trasporto TAP-1, con
conseguente alterazioni nella presentazione dell’antigene (Cromme et al., 1994);
- modificano l’espressione di chemochine e citochine, bloccando i processi di
comunicazione tra i cheratinociti infettati da HPV e gli effettori del sistema
immunitario (Rosl et al.,1999);
- guidano la cellula all’evasione dagli stimoli apoptotici mediati dai complessi
CD95/TNF, TRAIL/TNF-α e i loro relativi meccanismi di trasmissione del
segnale (Walczak et al., 2000).
L’interazione tra HPV e il sistema immunitario si differenzia da quella di altri
virus. Infatti, mentre patogeni quali il citomegalovirus producono specifici antigeni che
interferiscono con i meccanismi di processazione dell’antigene, nessuna proteina di
HPV svolge questa funzione come scopo primario. E’ la concomitanza di più fattori a
minimizzare la presentazione del virus al sistema immunitario.
Diverse alterazioni del sistema di presentazione dell’antigene, che coinvolgono i
meccanismi di trasporto al proteosoma, i recettori HLA e i sistemi cellulari di
riconoscimento dell’antigene, si verificano, ad esempio, nel corso di lesioni squamose
intraepiteliali a elevato grado e carcinoma in situ. Questo insieme di meccanismi di

evasione dal sistema immunitario è determinante per la progressione delle infezioni
verso la malignità.
Un altro meccanismo coinvolge le cellule presentanti l’antigene. Infatti, per
essere riconosciute dai CTL, gli antigeni virali devono essere rilasciati dalla cellula
infettata e presentati da cellule presentanti l’antigene (APC), tra le quali importanti sono
le cellule di Langerhans. In seguito all’infezione, si osserva una diminuzione e
un’alterazione morfologica di queste cellule, fenomeno che potrebbe spiegare la lunga
persistenza del virus nelle cellule. La causa potrebbe essere una diminuzione di TNF-α,
che è un potente attivatore delle cellule di Langerhans, o l’espressione dell’oncoproteina
E7, che interferisce con la loro differenziazione. Inoltre, poiché l’infezione degli HPV
non comprende una fase viremica, diminuisce la possibilità di un’efficace presentazione
antigenica da parte delle APC.
Anche l’evasione dai meccanismi apoptotici è particolarmente importante nel
corso della patogenesi virale. Per definizione, l’apoptosi è un programma geneticamente
determinato che porta all’attivazione di deossiribonucleasi attivate da caspasi. Di
conseguenza, non solo viene clivato il DNA a elevato peso molecolare, ma anche il
genoma virale che non è ancora stato incapsidato. Quindi è un processo importante nel
limitare la diffusione della progenie virale nell’organismo e nel mantenere un adeguato
rapporto tra il numero di cellule perse e acquisite; l’alterazione di questo equilibrio può
favorire la progressione tumorale.
Gli oncogeni di alcuni tipi di HPV interferiscono con la capacità della cellula di
andare incontro a morte cellulare programmata, come è stato dimostrato utilizzando la
tecnica “TUNEL” (che misura il livello di frammentazione del DNA direttamente su
sezioni di tessuto primario) (Gavrieli et al., 1992).

L’apoptosi è generalmente innescata nelle cellule da un’ampia gamma di diversi
segnali intrinseci, come mancanza di nutrienti, ipossia o agenti chemioterapici. Tuttavia
il principale evento fisiologico che rende possibile l’accensione del macchinario che
porta a morte cellulare programmata è il legame tra i ligandi naturali in grado di attivare
l’apoptosi (CD95L, TRAIL, TNF-α) ai recettori suicidi corrispondenti presenti sulla
superficie cellulare, che appartengono alla famiglia dei recettori transmembrana TNF. In
seguito al legame con il ligando, si verifica l’oligomerizzazione del recettore, seguita
dal reclutamento del dominio FADD (“Fas-associated-death domain”) e della
procaspasi 8, in modo tale da realizzare il complesso DISC (“Death-inducing signaling
complex”). La formazione di DISC stimola il taglio proteolitico caspasi a valle e porta
ad alterazioni morfologiche come condensazione dei nuclei, frammentazione del DNA
intranucleosomale (Figura 10).
Caspasi 8
APOPTOSI
Degradazione enzimatica delle proteine del nucleo
Attivazione delle caspasi
DISC
FADD
Dominio di morte
Legame di CD95LOligomerizzazione di CD95
CD95CD95L
Caspasi 8
APOPTOSI
Degradazione enzimatica delle proteine del nucleo
Attivazione delle caspasi
DISC
FADD
Dominio di morte
Legame di CD95LOligomerizzazione di CD95
CD95CD95L
Figura 10: Formazione di DISC mediata da CD95
(modificata da: Finzer, P., A. et al., 2002. Cancer Letter. 188: 15-24).

Per quanto riguarda l’apoptosi mediata da CD95, si possono verificare due
diversi meccanismi di trasmissione del segnale (Figura 11): in cellule di tipo I l’apoptosi
viene innescata dalla formazione di DISC, con conseguente attivazione delle caspasi 8 e
3. In cellule di tipo II, viene temporaneamente inibita la formazione di DISC, mentre le
procaspasi 3 e 9 sono attivate in seguito alla distruzione delle membrane mitocondriali e
il rilascio di fattori stimolanti l’apoptosi, come il citocromo c. La morte di cellule di tipo
II viene inibita in seguito all’overespressione di Bcl-2 o Bcl-xl. L’apoptosi mediata da
CD95/TNF-α è regolata negativamente da FLIP (“FLICE-caspase 8 inhibitory
proteins”), che comprende fattori virali e cellulari coinvolti nella trasformazione e nella
permanenza del virus nelle cellule. Questi fattori competono con la procaspasi 8, in
quanto presentano una struttura simile, ma sono prive del dominio catalitico essenziale
per la proteolisi (Scaffidi et al., 1999).
APOPTOSI
Tipo I Tipo II
Attivazione diCasp-8
c-Flip
DISC
CD95L
CD95
Bcl-2Bcl-XL
Casp-3Casp-9
Cyt c, Apaf-1
APOPTOSI
Tipo I Tipo II
Attivazione diCasp-8
c-Flip
DISC
CD95L
CD95
Bcl-2Bcl-XL
Casp-3Casp-9
Cyt c, Apaf-1
Figura 11: Meccanismi apoptotici di tipo I e II
(modificata da: Finzer, P., A. et al., 2002. Cancer Letter. 188: 15-24).

Gli HPV hanno sviluppato strategie efficienti per modulare la risposta apoptotica
in seguito a infezione. La proteina E5 dei papillomavirus umani riduce la quantità del
recettore CD95 sulla superficie cellulare. Tuttavia, poiché E5 non è espressa nelle
cellule cancerose, la rilevanza fisiologica della diminuzione dell’espressione di CD95 è
probabilmente limitata al ciclo di vita naturale, in cui il temporaneo blocco dell’apoptosi
protegge gli intermedi di replicazione e i virioni immaturi dal clivaggio mediato dalle
caspasi. Questo dato è supportato dalla scoperta che l’espressione di superficie del
recettore I di CD95/TNF non viene alterata quantitativamente in cellule contenenti
l’HPV immortalizzate rispetto ai cheratinociti (Aguilar-Lemarroy et al., 2002).
Invece le oncoproteine E6/E7 di HPV-16 e HPV-18 modulano efficientemente
altri aspetti dei meccanismi apoptotici. Ad esempio E6 di HPV-16 sensibilizza
all’apoptosi cellule epiteliali mammarie immortalizzate in presenza di tamoxifene e di
altri agenti che recano danno al DNA, ma protegge le cellule fibrose durante la
differenziazione naturale in maniera dipendente o meno da p53. Al contrario fibroblasti
umani immortalizzati da E7 sono resistenti alla morte cellulare mediata da TNF-
α/cicloesimide (Thompson et al., 2001), ma non a quella mediata da TRAIL in
cheratinociti (Basile et al., 2001). Entrambi gli oncogeni portano ad un aumento di
apoptosi in cellule epiteliali umane in seguito a esposizione a condizioni di ipossia (Kim
et al., 1997), mentre cellule di carcinoma cervicale HPV-16 positive sono meno
suscettibili all’apoptosi mediata da CD95 (Aguilar-Lemarroy et al., 2001).
Il principale effetto di E6 sui meccanismi apoptotici è l’inattivazione proteolitica
di alcuni fattori pro apoptotici come p53, Bak, Bax o c-Myc, attraverso il sistema del
proteosoma, secondo le modalità precedentemente descritte. In particolare,
l’inattivazione di p53 è un punto chiave nella regolazione dell’apoptosi indotta da
CD95. La sola applicazione del CD95-L induce morte cellulare in cellule

immortalizzate da E7, mentre i cheratinociti esprimenti E6 oppure E6/E7 sono resistenti
(Aguilar-Lemarroy et al., 2002). Questi ultimi possono essere sensibilizzati all’apoptosi
indotta da CD95-L in seguito al blocco della degradazione mediata dal proteosoma,
preceduta dalla riespressione di p53 e c-Myc. Questa osservazione concorda con l’idea
che l’attivazione del recettore CD95 necessita di modificazioni aggiuntive da parte di
p53 nella sua forma fosforilata.
E6 compie la sua funzione antiapoptotica anche ad altri livelli, in particolare
agendo su TNF-α, secondo un meccanismo mediato da p53. TNF-α, a differenza di
CD95, induce altri meccanismi protettivi per la cellula, come quelli mediati da NF-κB o
dalle MAP chinasi. E6 impedisce al dominio di morte di TNF R1 di interagire con
FADD. Viene infatti bloccata la porzione C-terminale del recettore, impedendo così
l’attivazione delle procaspasi 8 e 3, entrambe necessarie per un’efficiente trasmissione
del segnale apoptotico (Finzer et al., 2002).
E7, com’è stato precedentemente descritto, agisce inattivando Rb, che svolge
un’azione anti-apoptotica; pertanto la diminuzione dei livelli intracellulari di Rb,
causata da E7, favorisce l’innesco di meccanismi apoptotici (Jones et al., 1997).
Quando E7 viene introdotto in fibroblasti umani porta a diminuzione della procaspasi 8
che protegge da morte cellulare indotta da TNF-α/CHX. E7 svolge inoltre un ruolo
importante nel modulare l’equilibrio tra l’istone deacetilasi (HDAC) e l’istone acetilasi
(HAT), in quanto lega HDAC attraverso la proteina Mi2β; anche E6 influenza questo
equilibrio, abrogando la funzione costimolatoria di CBP e p300. E’ stato dimostrato che
l’inibizione di HDAC induce blocco della crescita e apoptosi in cellule esprimenti E7,
ma non quando viene espressa solo la proteina E6 (Finzer et al., 2001).

CONTESTO E RAZIONALE DELLA RICERCA
Il processo di ricerca e sviluppo di un farmaco antivirale
La realizzazione di un nuovo farmaco richiede un percorso molto complesso e di
lunga durata, diviso fondamentalmente in due fasi: ricerca e sviluppo (Figura 1).
Necessità medica
Studi di biologiamolecolare sul virus
Disegno razionale basatosulla struttura
Identificazione dei meccanismi rilevanti per patogenesi e
replicazione virale
Sviluppo si saggi “high-throuput”per lo “screening” di librerie di
prodotti naturali e derivanti dalla chimica combinatoriale
“Lead compound”
Farmaco candidato
Farmaco commercializzabile
Sviluppo pre-clinico
“Hit compound”
Sviluppo clinico
Necessità medica
Studi di biologiamolecolare sul virus
Disegno razionale basatosulla struttura
Identificazione dei meccanismi rilevanti per patogenesi e
replicazione virale
Sviluppo si saggi “high-throuput”per lo “screening” di librerie di
prodotti naturali e derivanti dalla chimica combinatoriale
“Lead compound”
Farmaco candidato
Farmaco commercializzabile
Sviluppo pre-clinico
“Hit compound”
Sviluppo clinico
Anche per la messa a punto di farmaci antivirali è necessario intraprendere
questo lungo percorso di ricerca, ma con una serie di difficoltà aggiuntive:
- i virus sono parassiti endocellulari obbligati, che utilizzano enzimi della cellula ospite
per compiere il loro ciclo riproduttivo. Tali enzimi non possono essere colpiti dal
farmaco, perchè anche la cellula stessa sarebbe danneggiata;
- i farmaci antivirali devono presentare un’efficacia del 100% per non rischiare di
trasformare l’infezione acuta in persistente;
- alcuni patogeni sono difficilmente coltivabili in vitro (es. HBV, HCV, HPV) e
pertanto non esiste un valido modello cellulare utilizzabile nelle fasi di ricerca. Per altri
virus non ci sono invece validi modelli animali che riproducono la patologia umana (es.
HCV): diventa così difficile valutare la sicurezza e l’efficacia di un farmaco;
Figura 1: Percorso per la realizzazione di un farmaco.

- per alcune infezioni virali sono disponibili farmaci, ma che spesso sono attivi nelle
prime 24-48 ore dell’infezione, quando è ancora difficile formulare una diagnosi
precisa.
La fase di ricerca prevede l’invenzione o l’esplorazione di nuove molecole con
attività antivirale. In primo luogo è necessario identificare, sulla base delle conoscenze
disponibili riguardo la struttura e il ciclo replicativo del virus, il bersaglio virale da
inibire. Tale bersaglio, ad esempio un enzima o un anti-recettore virale, deve svolgere
un ruolo rilevante nei meccanismi patogenetici e replicativi del patogeno e deve
verosimilmente consentire lo sviluppo di un farmaco dotato di tossicità selettiva nei suoi
confronti.
In seguito bisogna confermare la validità del bersaglio, in altre parole fornire la
cosiddetta prova del principio: le recenti innovazioni tecnologiche permettono di
inattivare singoli geni nel contesto di un genoma virale o di bloccarne l’espressione
consentendo di verificare direttamente le disponibilità di una proteina virale e il suo
ruolo nel processo patogenetico. Questi approcci sperimentali possono confermare con
buona approssimazione la validità del bersaglio proposto.
A questo punto si allestisce il saggio biologico più appropriato per analizzare
una serie di molecole (derivanti dall’archivio di composti chimici, prodotti della
chimica combinatoriale, fonti naturali o molecole endogene) che potrebbero inibire il
bersaglio. Al termine di questo processo si arriva ad identificare i “lead compounds”,
vale a dire molecole che potrebbero essere efficaci candidati per le successive fasi di
ricerca.
In seguito si realizzano una serie di esperimenti in vitro su modelli cellulari e
quindi in vivo su modelli animali, al fine di valutare e ottimizzare le caratteristiche
farmacocinetiche e la tossicità dei composti: al termine di questa fase verranno

identificate le molecole candidate per le successive fasi di sviluppo (da migliaia di
molecole di partenza si otterranno al massimo due, tre molecole ottimali).
Il processo di sviluppo clinico di un farmaco comprende quattro fasi, realizzate
sull’uomo:
- Fase I: viene in genere realizzata su una decina di soggetti sani e volontari (ad
eccezione dei farmaci antineoplastici e dei prodotti biotecnologici), ricoverati in idonee
unità attrezzate e sotto la sorveglianza di personale medico; comprende lo studio della
farmacocinetica, della tollerabilità e del metabolismo del farmaco.
- Fase II: gli studi sono eseguiti su 200-400 pazienti, il più possibile omogenei, al fine
di confermare l’efficacia terapeutica e la tollerabilità del farmaco. Come trattamenti di
controllo si utilizzano il placebo e un farmaco sicuramente attivo già presente in
commercio.
- Fase III: consiste nell’estensione degli studi controllati a casistiche più ampie e meno
selezionate per una più accurata determinazione dell’efficacia terapeutica e della
tollerabilità. Spesso tali studi sono multicentrici, al fine di assicurare una maggiore
variabilità delle casistiche.
- Fase IV: comprende tutti gli studi eseguiti dopo la commercializzazione del farmaco.
Si propone di individuare eventi avversi anche gravi, ma rari, non legati all’attività
farmacologica specifica della sostanza, ma soprattutto a condizioni particolari di
reattività del singolo paziente (Ongini, 2001).
Sviluppo di un farmaco anti HPV: presupposti
Per capire se è possibile sviluppare un farmaco anti HPV, così come per altri
antivirali, è necessario soddisfare alcune esigenze sia commerciali sia virologiche.

Per quanto riguarda le esigenze commerciali, deve esserci una richiesta sul
mercato e il farmaco deve produrre un profitto. Per adempiere alle esigenze virologiche
di un farmaco antivirale bisogna invece fornire la prova del principio.
La necessità medica di avere un farmaco anti HPV è sicuramente molto elevata.
Infatti gli HPV sono stati inclusi nella lista degli agenti carcinogenici di gruppo 1
dall’Agenzia Internazionale per la Ricerca sul Cancro (IARC), essendo la principale
causa di tumori anogenitali. In particolare HPV-16 si riscontra nel 60% delle neoplasie
maligne, HPV-18 nel 10-12%, HPV-31 e HPV-45 nel 4-5% dei casi. Il test di
“screening” di Papanicolaou ha significativamente ridotto l’incidenza dell’infezione nei
paesi industrializzati, ma in molti paesi questo tipo di cancro è una delle principali cause
di morte.
L’incidenza del cancro alla cervice uterina, nel mondo, è di circa 470 600 nuovi
casi l’anno, mentre in Italia è di 3500; la mortalità nel mondo è di 233 400, in Italia di
1500.
Le terapie disponibili sono poche e di tipo ablativo: rimozione dell’utero,
conizzazione, isterectomia. Non esiste un farmaco e tutti gli approcci sono ad elevato
grado di ricorrenza, nel senso che il virus non viene in realtà eliminato.
In base ad una recente analisi di mercato, effettuata dall’agenzia
DATAMONITOR, si riscontra la forte esigenza di sviluppare, nel corso dei cinque,
dieci anni futuri, prodotti non invasivi per il trattamento di infezioni da HPV, che
vengano poi commercializzati gradualmente.
In primo luogo, le aziende farmaceutiche Merck & GSK attualmente stanno
sviluppando i vaccini profilattici anti HPV, soprattutto attivi contro i tipi 16 e 18. In
base alle popolazione destinatarie di questo vaccino, DATAMONITOR crede che
l'immunizzazione potrebbe fornire un mercato potenziale di 1-3 miliardi di dollari.

Tuttavia si ritiene che, accanto a strategie profilattiche, sarebbe necessario
possedere un farmaco (antivirale o biologico) che abbia come bersaglio terapeutico
direttamente HPV, che potrebbe fruttare un guadagno annuale di 1.4 bilioni di dollari
all’interno dei sette maggiori mercati mondiali (www.piribo.com).
Nel loro insieme queste considerazioni giustificano la necessità di un processo di
ricerca e sviluppo di un farmaco antivirale per i trattamenti dei carcinomi anogenitali
associati all’infezione da HPV ad alto rischio.
Individuazione del bersaglio
Nel corso degli ultimi anni, grazie al miglioramento delle tecniche
biotecnologiche, sono stati compresi maggiormente i meccanismi patogenetici di HPV e
sono iniziati gli studi per la messa a punto di farmaci antivirali e vaccini. Tuttavia i
problemi rimangono molti, per diverse ragioni: HPV non cresce in colture cellulari
standard e conseguentemente non è possibile ottenere virus a elevato titolo; questo
impedisce l’allestimento dei classici saggi antivirali, quali ad esempio i sistemi per
inibire polimerasi o proteasi, che tradizionalmente costituiscono dei bersagli selettivi
per i farmaci antivirali. Infine l’infezione latente non si risolve.
Sia le proteine E6 e E7 che E1 e E2, rappresentano dei potenziali bersagli di
farmaci antivirali. Tuttavia ci sono una serie di ostacoli per il loro sviluppo.
Dal punto di vista strutturale, è noto che E6 e E7 legano rispettivamente due e
uno ione zinco, dando origine a strutture a dita di zinco con una dimensione anomala di
29 aminoacidi, ma non è ancora stata determinata la struttura di nessuna di queste due
proteine mediante risonanza magnetica nucleare e spettroscopia o cristallografia a raggi
x e questo rappresenta un ostacolo importante per lo sviluppo di un farmaco. E’ tuttavia

possibile sfruttare le proprietà di queste proteine per legare ioni zinco e interagire con
ligandi cellulari per la messa a punto di saggi ad alta resa, quantificando l’effetto di
piccole molecole sul rilascio di ioni zinco da E6 e E7 (Beerheide et al., 1999).
Un altro problema riguarda il fattore di trascrizione virale E2. Esso è un
bersaglio per alcuni aspetti inadeguato, in quanto presenta funzioni sia di attivatore sia
di repressore e potrebbe non essere possibile determinare quale di queste due funzioni
sia alterata da un farmaco. Tuttavia si è visto che in cellule di cancro alla cervice uterina
infettate con un virus ricombinante esprimente E2, quest’ultimo lega il promotore
precoce di HPV, reprimendo la trascrizione di E6 e E7, con conseguente stabilizzazione
di p53 e delle altre proteine che controllano il ciclo cellulare. E1 invece è un bersaglio
più adatto in quanto è il principale regolatore della replicazione virale, presenta attività
ATPasica ed elicasica. Potrebbe pertanto essere sfruttato in saggi di “screening” per
l’individuazione di molecole antivirali. Tuttavia nessun tentativo di inibire E1 o E2 ha
data fino ad ora buoni risultati e inoltre, dato che queste due proteine non sono espresse
quando il virus si presenta in forma integrata nel genoma cellulare, la loro inibizione
rappresenterebbe un approccio funzionante solo per infezioni virali produttive. E6 e E7
risultano quindi il bersaglio più adatto per lo sviluppo di un farmaco anti HPV, ma
occorre fornire la prova del principio, al fine di dimostrare che l’inibizione
dell’espressione di E6 e E7 modifica effettivamente il fenotipo trasformato (Bernard et
al., 2004).
La prova del principio
Esiste un’ampia letteratura in cui si descrive come, in molti casi, i trascritti di E6
e E7 siano stati efficacemente inibiti in vitro mediante macromolecole come ribozimi,
attameri e “RNA interference” (siRNA), con conseguente diminuzione della crescita di

linee cellulari HPV positive. Il problema comune di questi approcci, basati sullo
sviluppo di macromolecole e subunità terapeutiche biodegradabili, consiste nel riuscire
ad indirizzare questi composti in specifici siti anatomici a concentrazioni attive dal
punto di vista terapeutico, superando i blocchi imposti dal sistema immunitario.
Per quanto concerne le strategie d’inibizione basate sui ribozimi, è interessante
analizzare lo studio effettuato dal gruppo di ricerca di Chen (Chen et al., 1996). Tre
ribozimi diretti contro l’RNA di HPV-18 sono stati clonati in un vettore di espressione
plasmidico. Ogni plasmide è stato in seguito trasfettato in cellule HeLa HPV-18 positive
e in cellule non esprimenti HPV, come cellule Tu167 di cancro orale. L’effetto dei
ribozimi sulle cellule HeLa è una notevole riduzione della crescita (particolarmente
efficace è il ribozima diretto contro il nucleotide 309 del trascritto di HPV-18) e un
aumento della dipendenza verso i fattori di crescita. Una molecola con la stessa
sequenza antisenso del ribozima, ma priva della subunità catalitica altera in misura
minore il fenotipo delle HeLa. L’effetto dei ribozimi deve essere attribuito all’aumento
della concentrazione intracellulare di p53.
Un altro sistema per inibire l’espressione delle oncoproteine di HPV è basato
sull’uso di attameri, molecole peptidiche selezionate per legarsi, in vivo, a una proteina
bersaglio e bloccarne selettivamente l’attività intracellulare, in modo tale da valutare le
conseguenze dell’inattivazione di una proteina nel suo contesto cellulare naturale. Per
isolare molecole in grado di legare la proteina E6 di HPV-16 è stata analizzata una
libreria di attameri peptidici, attraverso una variante della tecnica del doppio ibrido, in
un ceppo di lievito. Sono stati ottenuti 17 attameri, la cui specificità per E6 e per altre
proteine (come E7) è stata testata mediante saggi d’ibridazione. Alcuni sono specifici
inibitori di E6 di HPV16, altri sono attivi anche su E6 di altri HPV. Per analizzare il
legame degli attameri a E6 in vitro è stato eseguito un “GST pull down”: alcuni attameri

non legano E6 in vitro, mentre in vivo presentano importanti effetti biologici,
probabilmente a causa di un’ alterata conformazione della proteina in vitro. Le
conseguenze dell’espressione di attameri su colture cellulari sono state valutate
attraverso un saggio di formazione di colonie e un “TUNEL”. Si osserva che solo un
numero limitato di attameri inibisce la crescita di colonie e induce apoptosi, nonostante
siano numerosi gli attameri in grado di legarsi a E6. Questo potrebbe essere dovuto a
una minore affinità di alcuni attameri rispetto ai ligandi cellulari di E6, oppure al fatto
che tali attameri si legano a domini di E6 non coinvolti nell’inibizione della crescita
cellulare. Infine si è visto che l’inibizione di E6 si associa a un aumento di p53 in
cellule HPV16 positive. Lo svantaggio degli attameri, che si è cercato di risolvere con
siRNA, è di possedere un’attività in vitro non molto prolungata (Butz et al., 2000).
Per quanto riguarda la tecnologia dell’”RNA interference”, è stato realizzato uno
studio dal gruppo di ricerca guidato da F. Hoppe-Seyler (Butz et al., 2003) per inibire
selettivamente l’ oncoproteina E6 in cellule HeLa HPV18 positive. Il vettore contenente
la sequenza da inibire (pSUPER18E6, specifico per una sequenza di 19 nucleotidi
all’interno dell’ORF codificante per E6) è stato trasfettato nelle cellule con il sistema
della precipitazione con calcio fosfato. Per studiare il grado d’inibizione di E6 è stata
effettuata una RT-PCR: si osserva una notevole riduzione dei livelli di E6, a
dimostrazione della specificità del costrutto. E’ stata poi valutata l’espressione di p53,
un noto inibitore di E6 e di p21, attivata da p53. Si osserva un notevole aumento sia di
p53 (in quanto non è più degradata da E6), sia di p21, a dimostrazione che p53 è anche
funzionale. I livelli di E7 sono invece normali, a dimostrazione della selettività del
vettore pSUPER18E6. Per studiare le conseguenze dell’inibizione di E6 sulla crescita
cellulare è stato effettuato un saggio che analizza la formazione delle colonie: la
capacità di formare colonie di cellule HeLa HPV-18 positive trasfettate con

pSUPER18E6 è completamente alterata, mentre su cellule prive di HPV il vettore non
ha effetto. Attraverso il saggio TUNEL si è visto inoltre che l’inibizione di E6 con
siRNA supera la resistenza delle cellule cancerose all’apoptosi. Questa strategia
d’inibizione è più vantaggiosa rispetto alla tecnica dell’antisenso, attraverso la quale si
osserva solo una certa diminuzione della crescita, ma non un’induzione così marcata di
apoptosi. Il principale ostacolo da superare per l’utilizzo di siRNA in terapia consiste
nel trovare efficienti sistemi di trasferimento dei vettori e nel superare i blocchi del
sistema immunitario.
Un altro tipo di approccio per l’inibizione dell’espressione delle oncoproteine E6
e E7 di HPV è stato far esprimere in modo ectopico la proteina E2 di HPV in cellule
HeLa HPV-18 positive. Quando il genoma virale è presente in forma episomale nella
cellula, E2 si lega al promotore virale precoce del virus, regolando negativamente
l’espressione delle oncoproteine E6 e E7. Tuttavia durante il processo di integrazione
del DNA virale in quello cellulare, l’ORF codificante per la proteina E2 viene persa,
con alterazione della regolazione di E6 e E7 e conseguente overespressione delle
oncoproteine. E’ stato dimostrato che l’espressione ectopica della proteina E2 bovina in
linee di carcinoma cervicale reprime l’espressione degli oncogeni E6 e E7 con
conseguente induzione di senescenza cellulare e ripristino dei meccanismi di
segnalazione intracellulare mediati dalle proteine p53 e pRb (Goodwin et al., 2000;
Wells et al., 2000; DeFilippis et al., 2003).
Sulla base dei risultati ottenuti da questi e da altri lavori, è possibile affermare
che l’inibizione dell’espressione o dell’attività delle oncoproteine E6 e E7 rappresenta
un valido bersaglio farmacologico.

Sviluppo di saggi per lo “screening” di antivirali
Nel corso degli anni ’60 e ’70, sulla scia del successo ottenuto nella cura delle
infezioni batteriche attraverso l’uso di antibiotici, le aziende farmaceutiche intrapresero
un programma di “screening” alla “cieca” (il cosiddetto “blind screening”), allo scopo
di individuare composti chimici dotati di attività antivirale. Tale approccio era poco
efficace, poiché non veniva identificato a priori un meccanismo o un enzima da inibire.
Di conseguenza i successi ottenuti furono pochi, ad eccezione della scoperta della
Amantadina, approvata alla fine degli anni ’60 per il trattamento del virus dell’influenza
A. In realtà, il meccanismo di inibizione di questi prodotti spesso non era noto (nel caso
specifico dell’Amantadina, solo intorno al 1990 ne fu compreso il meccanismo
d’azione).
Con l’introduzione della tecnologia del DNA ricombinante, i sistemi di
“screening” alla “cieca” sono stati abbandonati. I geni essenziali alla crescita del virus
possono essere identificati, clonati ed espressi in organismi e i prodotti dei geni virali
possono essere purificati e analizzati a livello molecolare. In questo modo è possibile
effettuare una ricerca del composto farmacologicamente attivo mirata a uno specifico
bersaglio virologico. Sono stati quindi sviluppati saggi di “screening” basati sul
meccanismo e saggi cellulari (“mechanism-based screens” e “cell-based screens”),
caratterizzati dal fatto che i loro bersagli sono enzimi codificati dal virus o processi
replicativi virali.
Nel corso degli anni ’90, grazie ai numerosi progressi scientifici e alle pressioni
economiche per la riduzione dei costi di sviluppo (al fine di ottenere un aumento della
competitività tra le case farmaceutiche), è emersa la necessità di migliorare le
metodiche di “screening” per la messa a punto di nuovi farmaci. Sono stati così
sviluppati nuovi sistemi di screening, i cosiddetti saggi “high-throughput”. Si tratta di

saggi basati sul meccanismo o saggi cellulari, attraverso i quali è possibile saggiare un
altissimo numero di composti, in modo automatizzato, per valutarne l’attività di
attivatori o inibitori di un particolare bersaglio biologico, come nel caso di recettori di
superficie o enzimi metabolici.
E’ cresciuto il numero di potenziali bersagli terapeutici, derivati da studi di
genomica funzionale e sono state realizzate librerie di composti da analizzare, derivati
dalle tecniche di sintesi, parallele e combinatoriali, di molecole chimiche. La stima del
numero di geni che compongono il genoma umano (circa 30 000) e il numero di singole
strutture chimiche ottenibili in linea teorica usando le metodiche attualmente disponibili
(circa 100 milioni) indicano che sarebbero necessari più di 1012 saggi per la mappatura
completa dei possibili bersagli terapeutici, a livello strutturale e funzionale.
Inizialmente si osservava una maggiore tendenza all’adozione di piastre a 96
pozzetti e di sistemi robotizzati, liquidi e su piastra. Molti gruppi di ricerca tendono ora
a preferire formati con piastre a 384 pozzetti, a densità più elevata e volumi più ridotti.
Un tipico programma di screening ad alta resa, all’interno di industrie farmaceutiche,
opera nell’ordine di 10 000 composti esaminati al giorno; nel caso di laboratori che
lavorano con sistemi “ultra-high-throughput”, si ragiona invece con dimensioni
dell’ordine di 100 000 molecole al giorno.
Per andare incontro alla necessità di ridurre i costi di sviluppo e di manodopera,
si è verificata una tendenza alla miniaturizzazione dei saggi, simultaneamente alla loro
automatizzazione. La miniaturizzazione riduce la quantità di reagenti chimici e biologici
necessari e facilita l’analisi parallela di numerosi campioni. Per rendere possibile tutto
questo sono stati seguiti due approcci generali: in primo luogo, sistemi basati su piastra
sono stati sostituiti da sistemi basati su pozzetti a maggiore densità e minore volume;

successivamente sono stati progettati dispositivi che rendono possibile la continua
realizzazione delle analisi di screening.
I tentativi di raggiungere un’adeguata automazione, miniaturizzazione ed alta
resa ha avuto come conseguenza l’aumento dell’enfasi nello sviluppo di formati più
omogenei e di tecniche di rilevazione a maggiore sensibilità. Ad esempio, i saggi
omogenei “mix and measure” sono adatti per soddisfare l’elevata resa, in quanto
permettono di evitare i passaggi di filtrazione, separazione, lavaggio, molto laboriose e
difficili da automatizzare.
Un’altra possibilità è la costruzione di un farmaco anche in base a studi
strutturistici. Questo dipende dal fatto di possedere la struttura atomica del bersaglio
d’inibizione, spesso ottenuta con raggi x o cristallografia. E’ così possibile costruire
ligandi che legano il sito attivo dell’enzima o un altro punto critico e ne inibiscono
l’attività. Con questo sistema sono stati progettati, ad esempio, inibitori della proteasi di
HIV.
Stanno assumendo una sempre maggiore importanza le tecniche di rilevazione
basate sulla fluorescenza, grazie alla loro elevata sensibilità e viene sempre più
abbandonato l’uso di radioisotopi. Un notevole contributo è stato dato dalla messa a
punto di saggi basati sulla tecnologia dei geni “reporter”.
Tecnologia dei geni “reporter”
La tecnologia dei geni “reporter” è ampiamente sfruttata per monitorare gli
eventi cellulari associati ai meccanismi di trasduzione del segnale e di espressione
genica.
Il termine “gene reporter” è utilizzato per definire un gene con un fenotipo
facilmente distinguibile rispetto alle proteine endogene. L’attività di questi geni può

essere controllata da precise sequenze regolatorie, responsive all’alterazione delle
condizioni all’interno di cellule ospiti. Ad esempio, segnali extracellulari sono rilevati
dai diversi tipi di recettori presenti sulla superficie cellulare, i quali permettono
l’attivazione di un meccanismo di trasduzione del segnale, attraverso il reclutamento di
fattori di trascrizione che, dopo essersi legati a specifiche sequenze responsive sul
DNA, attivano la trascrizione del gene. Per capire la relazione tra l’attivazione e
l’inibizione delle diverse vie e i loro effetti sull’espressione genica, sono stati fusi
specifici elementi responsivi del gene di interesse con geni codificanti per proteine
“reporter”.
L’unità “reporter” è formata dal promotore e dal gene “reporter”. Il promotore
lega fattori di trascrizione attivati in risposta a una cascata di attivazione del segnale. Il
legame con il fattore di trascrizione rende possibile l’espressione del gene “reporter.
Spesso vengono utilizzati promotori endogeni, quali c-fos, ma è necessario prestare
attenzione ai diversi tipi di segnali che possono attivare il promotore e quindi falsificare
i risultati del saggio. I promotori che contengono un unico sito di legame per fattori di
trascrizione, come CRE (“cAMP responsive element”), riducono i rischi di interferenza,
anche se sono sottoposti all’azione di segnali endogeni. Una valida alternativa è
utilizzare promotori che non leghino fattori di trascrizione nativi del sistema cellulare in
studio, quali Gal4 di lievito: viene trasfettato il dominio di legame per i fattori di
trascrizione di Gal4, accoppiato a un dominio regolatore e al gene “reporter”, la cui
espressione è regolata da elementi responsivi a Gal4.
I geni reporter maggiormente utilizzati sono descritti in tabella 1. In generale
hanno il vantaggio di presentare una bassa attività di fondo nelle cellule e di amplificare
i segnali provenienti dalla superficie cellulare per produrre una risposta molto sensibile
e facilmente rilevabile. La scelta del “reporter” dipende dalla linea cellulare utilizzata,

dal tipo di esperimento e dal sistema di rilevazione. I geni “reporter” attualmente
disponibili sono classificati in geni intracellulari ed extracellulari. I geni intracellulari
(ad esempio CAT, β-galattosidasi, GFP, luciferasi) sono prodotti e trattenuti nelle
cellule; i geni extracellulari (quali fosfatasi alcalina, β-lattamasi) codificano per proteine
che sono secrete nel mezzo di coltura: pertanto è possibile rilevare la proteina “reporter”
nel mezzo di coltura senza alterare la cellula (New et al., 2003).

Gene reporter Vantaggi Svantaggi
CAT
(“Chloramphenicol
acetyltransferase”, batterico)
Non presenta attività endogena;
disponibile test ELISA
automatizzato.
Uso di radioisotopi.
β-Galattosidasi (batterica)
Ben caratterizzata; stabile; sono
disponibili saggi bio o
chemioluminescenti
Attività endogena in cellule di
mammifero.
Luciferasi (lucciola)
Attività specifica; non presenta
attività endogena; conveniente.
Richiede un substrato, O2 e
ATP.
Luciferasi (batterica)
Adatta per l’analisi della
trascrizione genica in procarioti.
Meno sensibile rispetto alla
luciferasi di lucciola; non adatta
all’uso in cellule di mammifero.
Fosfatasi alcalina
(placenta umana)
Proteina secreta; sono
disponibili saggi colorimetrici
poco costosi e saggi
luminescenti molto sensibili.
Attività endogena in alcune
cellule; interferisce con i
composti che devono essere
testati.
GFP
(“Green fluorescent protein”)
Autofluorescente (non necessità
di un substrato); non presenta
attività endogena.
Richiede modifiche post
traduzionali; bassa sensibilità
(non c’è amplificazione del
segnale).
Per la realizzazione del saggio cellulare descritto in questa tesi è stato utilizzato
come gene “reporter” la luciferasi di lucciola (Photinus pyralis). Si tratta di una
famiglia di enzimi che catalizzano l’ossidazione di diversi substrati (come la proteina
luciferina), emettendo luce. La quantità di luce emessa è proporzionale all’attività
dell’enzima e fornisce un’indicazione immediata relativa all’attività trascrizionale del
Tabella 1: Geni reporter maggiormente utilizzati.

promotore del gene e che ne guida l’espressione. Esistono diversi tipi di luciferasi: la
luciferasi batterica, ad esempio, è una proteina dimerica, labile al calore, che la rende
meno adatta all’uso in cellule di mammifero. La luciferasi della lucciola è uno dei
principali geni “reporter” utilizzati in cellule di mammifero, per la sua elevata
sensibilità (10-20 moli di enzima). I problemi di quest’ultimo tipo di luciferasi derivano
dal fatto che è necessaria la lisi della cellula prima dell’aggiunta del substrato luciferina
e il fascio di luce emessa dura pochi secondi. Tuttavia, l’uso di membrane permeabili e
di esteri di luciferina lisabili dalla luce ha risolto il problema della lisi cellulare. Sono
inoltre disponibili reagenti che prolungano la vita media del fascio di luce per diverse
ore, rendendo possibile l’applicazione di questo saggio su formati multi pozzetto. La
luciferasi “Renilla” è particolarmente adatta per saggi su cellule viventi, in quanto
catalizza l’ossidazione della celenterazina, che è permeabile alla membrana. Tutte le
luciferasi non presentano attività endogena in cellule di mammifero.
La tecnologia dei geni “reporter” è stata applicata in numerosi settori di studio,
quali:
Analisi dei promotori: la tecnologia dei geni reporter è stata utilizzata in un
primo momento soprattutto per lo studio della responsività di promotori e “enhancer”
situati a monte dei geni. Questo sistema è stato utilizzato, ad esempio, per lo studio di
fattori di trascrizione responsabili dell’espressione basale e tessuto specifica di alcuni
recettori (es. β2 adrenergici, recettore di LH, IL-2 ecc...), delle sequenze responsive a
diversi ormoni, al calcio, all’ossido nitrico e ad altre sostanze.
Trasferimento genico: i geni reporter sono stati utilizzati come marcatori del
trasferimento genico effettuato attraverso diverse tecniche di trasformazione. A questo
scopo si utilizzano vettori contenenti il “reporter” e il gene di interesse. Esempi di tale
applicazione sono la tecnologia del FACS (“Fluorescence activated cell sorting”), che

permette di rilevare il trasferimento di geni utilizzando come marcatore la GFP. I geni
“reporter” sono anche utilizzati nel monitoraggio dell’espressione genica in animali e
piante transgeniche e nel campo della terapia genica, per il trattamento di malattie quali
Parkinson, fibrosi cistica e cancro.
“Imaging” dell’espressione genica: storicamente l’organizzazione spaziale
dell’espressione genica in piante e animali veniva determinata usando saggi
colorimetrici o basati sulla fluorescenza, che utilizzavano come marcatori la β-
glucuronidasi e la β-galattosidasi. Tuttavia lo sviluppo della GFP e della luciferasi come
marcatori non invasivi dell’espressione genica, in associazione a tecnologie quali la
microscopia a fluorescenza, ha reso possibile l’acquisizione di informazioni temporali e
spaziali dell’espressione genica anche a livello della singola cellula. E’ possibile
identificare la localizzazione subcellulare, l’interazione tra specifiche proteine,
l’interazione tra diverse proteine nello stesso istante, compresi i cambiamenti nel loro
ambiente in risposta a eventi intra o extra cellulari.
“Screening” a elevata resa: l’applicazione della tecnologia dei geni “reporter” a
sistemi di “screening” ad alta resa ha permesso l’utilizzo dei saggi cellulari come valida
alternativa ai saggi biochimici in vitro. Il principale vantaggio di questi saggi è che
riproducono in parte le condizioni fisiologiche e forniscono informazioni riguardanti la
biodisponibilità e la tossicità del composto; la possibilità di mantenere queste cellule in
coltura per molte settimane permette un’osservazione a lungo termine dei cambiamenti
adattativi associati alla resistenza ai farmaci e agli effetti collaterali.
Caratterizzazione di recettori: la tecnologia dei geni “reporter” è stata utilizzata
per la clonazione, l’espressione funzionale e la caratterizzazione di recettori di
membrana e intracellulari. Questo ha reso possibile l’identificazione di ligandi agonisti
e antagonisti che alterano l’attività recettoriale in cellule viventi. Il principale vantaggio

dei saggi cellulari con i geni reporter è la loro adattabilità a strategie a elevata resa, non
radioattive e semplici da realizzare. Tali saggi sono effettuati su piastre a 96 o 384
pozzetti e sono spesso associati a sistemi computerizzati. Questo permette l’analisi di
milioni di composti con potenziale interesse terapeutico per le aziende farmaceutiche.
Sarà inoltre possibile identificare nuovi bersagli terapeutici, coinvolti nei meccanismi
patogenetici di numerose malattie (Naylor, 1999).
Chimica combinatoriale
Una possibile risposta alle sempre crescenti esigenze imposte dal mercato
farmacologico, è rappresenta dalla chimica combinatoriale. Si tratta di un procedimento
che permette la sintesi di un gran numero di sostanze (anche milioni di molecole)
strutturalmente correlate, che vengono poi esaminate al fine di individuare quelle di
potenziale utilità terapeutica, da avviare alla sperimentazione clinica. La sintesi non è
casuale: infatti, essendo note le caratteristiche delle sostanze di partenza, ci si può
attendere che i prodotti finali possiedano le proprietà desiderate. I tempi e i costi del
processo sono notevolmente inferiori rispetto a tutti gli approcci precedentemente usati.
Gli insiemi (o biblioteche) di composti combinatori da sperimentare vengono
realizzati in modo relativamente semplice. Ci si basa su reazioni chimiche standard per
assemblare specifici gruppi di unità strutturali fino a ottenere un’enorme varietà di
molecole più grandi. Ad esempio, si possono considerare quattro molecole: A1, A2, B1,
B2. Le molecole A1 e A2 sono strutturalmente correlate e si definiscono come
appartenenti alla stessa classe di composti; B1 e B2 derivano invece da una seconda
classe. Supponiamo che queste due classi di composti possano reagire per formare una
nuova molecola, una variante della quale potrebbe essere un farmaco efficace. Le
tecniche combinatorie ci permettono di costruire tutte le possibili combinazioni: A1-B1,

A1-B2, A2-B1 e A2-B2. In realtà il ricercatore lavora con un numero di molecole molto
maggiore.
Le tecniche combinatorie attualmente disponibili sono fondamentalmente di due
tipi. La prima, nota come sintesi parallela (Figura 2), fu introdotta a metà degli anni
’80 da H. M. Geysen.
Tutti i prodotti vengono assemblati separatamente, in recipienti di reazione
distinti. A questo scopo si utilizza una micropiastra da
titolazione, munita di pozzetti (tipicamente 96 pozzetti),
che sono parzialmente riempiti con una soluzione
contenente sferette inerti di polistirene (pallini grigi in
figura 2). Si aggiunge il primo insieme di molecole
(gruppo A-quadrati-, ad esempio diversi tipi di ammine)
alle sferette e si filtra il contenuto di ogni pozzetto per
eliminare le sostanze che non hanno reagito (cioè che
non si sono fissate alle sferette). Si unisce il secondo
insieme di molecole (gruppo B –triangoli-, ad esempio
diversi acidi carbossilici), quindi si filtra per eliminare il
materiale che non ha reagito. Una volta prodotta una
biblioteca di 96 composti, si staccano le strutture finali
dalle sferette per poterne analizzare l’attività biologica.
Sebbene le reazioni chimiche necessarie per legare i
composti alle sferette e per poi staccarli introducano una
complicazione nel processo di sintesi, la disponibilità di
Figura 2: Sintesi parallela (Le Scienze quaderni n. 102, 1997).

semplici metodi di purificazione consente di superare questi problemi. Oggi in molti
laboratori apparecchiature robotizzate sono d’aiuto nel lavoro di routine della sintesi
parallela, per esempio introducendo piccole quantità di molecole reattive nel pozzetto
opportuno, al fine di rendere più veloce e accurato il processo.
La seconda tecnica per generare una biblioteca di composti è conosciuta come
sintesi “taglia e mescola” (Fig. 3).
Fu introdotta nella seconda metà degli anni ’80
da Arpad Furka. A differenza della sintesi parallela,
nella quale ogni composto rimane nel proprio recipiente,
la sintesi “taglia e mescola” genera una miscela di
composti correlati all’interno dello stesso contenitore.
Questo metodo riduce il numero di recipienti necessari e
aumenta il numero di composti che si possono ottenere
(fino a diversi milioni). Tuttavia, prendere in
considerazione un numero così elevato di prodotti e
provarne l’attività biologica può diventare assai
complicato. Si inizia con provette riempite con una
soluzione di sferette inerti di polistirene. Si aggiunge il
primo insieme di molecole (gruppo A) alle provette,
mettendo le molecole A1 nella prima provetta, le
molecole A2 nella seconda e così via.
Figura 3: Sintesi “taglia e mescola”(Le Scienze quaderni n. 102, 1997).

Successivamente si uniscono i contenuti di tutte le provette. Si divide la miscela
in parti uguali, quindi si aggiunge la seconda serie di molecole (gruppo B), mettendo la
molecola B1 nella prima provetta, B2 nella seconda e così di seguito. Si separano le
sferette dal materiale non reagito e si staccano le strutture finali.
Spesso si analizza il contenuto di ogni provetta per determinare l’attività
biologica media della miscela. Poiché ogni miscela è costituita dagli stessi componenti
finali, è possibile individuare la variante che funziona meglio. A questo punto si ripete
la sintesi, aggiungendo solo il composto più potente (ad esempio B2) in nuove provette
contenenti i composti dell’altro gruppo (A), per capire quale delle combinazioni A-B2
sia la più attiva biologicamente. Molte industrie farmaceutiche hanno automatizzato
anche questa tecnica, attraverso l’uso di sistemi robotizzati che introducono reagenti ed
effettuano le operazioni di miscelatura. Sono stati inoltre ideati sistemi che rendono
possibile l’identificazione del composto reattivo in una miscela di molti prodotti. E’
stato visto che, alla fine di una sintesi, tutte le molecole fissate a una singola sferetta
hanno la stessa struttura. E’ pertanto possibile separare dalla miscela le sferette che
recano molecole biologicamente attive e poi, attraverso tecniche di rilevazione molto
sensibili, determinare la struttura molecolare dei composti fissati. Purtroppo questo
sistema funziona solo per certi composti, come peptidi e piccoli frammenti di DNA.
Altri ricercatori hanno messo a punto sistemi per addizionare a ciascuna sferetta un
tracciante chimico, che indica essenzialmente l’ordine con cui specifiche unità sono
state aggiunte alla struttura. Nonostante questi metodi innovativi, a causa delle difficoltà
nell’identificazione dei composti realizzati attraverso la sintesi “taglia e mescola”,
attualmente la maggior parte delle industrie farmaceutiche continua ad affidarsi alla
sintesi parallela.

La chimica combinatoriale ha dato validi risultati nell’applicazione alle
benzodiazepine, importanti farmaci che modulano positivamente il recettore GABAA.
Dato l’ampio spettro delle loro attività (ansiolitiche, anti convulsivanti, ipnotico
sedative…) le benzodiazepine furono i primi composti sottoposti a sintesi combinatoria
per la ricerca di nuovi farmaci. Successivamente le industrie farmaceutiche hanno
applicato le tecniche di chimica combinatoria a un’ampia gamma di sostanze di
partenza. In generale, i chimici utilizzano biblioteche combinatorie di piccole molecole
organiche come fonte di composti guida promettenti o per ottimizzare l’attività di un
composto già identificato. Questa tecnica potrebbe quindi essere efficacemente sfruttata
per la realizzazione di molecole in grado di inibire selettivamente l’attività di HPV
(Plunkett et al., 1997).

SCOPO DEL LAVORO
Gli HPV ad alto rischio sono carcinogeni umani di gruppo I e rappresentano la
principale causa di tumori alla cervice uterina. I meccanismi patogenetici attraverso i
quali gli HPV inducono la progressione maligna delle cellule infettate sono stati chiariti
in modo esaustivo e implicano il coinvolgimento delle oncoproteine virali E6 e E7, le
quali sono responsabili del mantenimento del fenotipo cellulare trasformato.
Tuttavia, non sono attualmente disponibili farmaci per il trattamento di questi
tumori e la terapia è prevalentemente di tipo chirurgico. La necessità clinica, le
considerazioni relative al mercato potenziale, l’esistenza di un bersaglio farmacologico
validato, indicano che la ricerca e lo sviluppo di un farmaco antivirale per il trattamento
dei tumori associati all’infezione da HPV ad alto rischio è necessario, vantaggioso e
fattibile.
Pertanto l’obiettivo di questa tesi è stato sviluppare e validare un saggio
cellulare, che permetta l’analisi ad alta resa di banche di molecole sintetiche o
biologiche al fine di identificare inibitori dell’espressione delle oncoproteine E6 e E7 di
HPV ad alto rischio.
Inoltre, per verificare se il saggio da noi sviluppato potesse essere realmente
utilizzato in un processo di “screening”, abbiano intrapreso un progetto dimostratore
volto ad identificare fattori inibitori in una collezione di 32 citochine appartenenti a
diverse classi funzionali.

MATERIALI E METODI
Linee cellulari e plasmidi
Per la realizzazione degli esperimenti sono state utilizzate le cellule HaCat, una
linea cellulare di cheratinociti umani spontaneamente immortalizzati e la linea cellulare
CaSki, che deriva da un carcinoma cervicale umano positivo per HPV-16.
Entrambe le linee cellulari sono state coltivate nel terreno EAGLE modificato da
Dulbecco (DMEM) (Gibco-BRL), contenente 0,06 mg/ml di gentamicina, 2 mM di
glutammina ed il 10% di siero di vitello (Gibco-BRL); le cellule sono state mantenute a
37°C e al 5% di CO2.
Sono inoltre state utilizzate le cellule P13 e P21, cloni stabili derivati da cellule
HaCat, coltivati in un terreno a cui sono stati aggiunti 0.2 mg/ml di antibiotico G418
(Gibco-BRL) per la selezione.
Il plasmide “reporter” pALuc HPV-16 LCR contiene la sequenza LCR completa
(compresa tra i nucleotidi 7009 e 7024) del ceppo di riferimento europeo di HPV-16
(HPV-16R, contenuto nella banca dati di HPV), clonata a monte del gene della
luciferasi della lucciola. pSTneoB è un vettore di espressione per cellule di mammifero,
che conferisce resistenza all’antibiotico G418.
Citochine
Per la realizzazione degli esperimenti sono state utilizzate le seguenti citochine:
Activina A, GDF-15, GM-CSF, IL-1β, IL-3, IL-4, IL-6, IL-7, IL-8 (CXCL8), IL-9, IL-
10, IL-11, IL-13, IL-15, IL-17, IL-18, IL-19, IL-20, IL-21, IL-22, I-309 (CCL1), IP-10
(CXCL10), IFN α, β, γ, LEC (CCL16), MIP1α (CCL3), MIP1β (CCL4), NAP-2
(CXCL7), SDF-1 (CXCL12) SPARC, TGFβ1/ β2/ β3 umano e TNF-α .

Esse sono state acquistate dalle aziende: Bender MedSystems (Burlingame CA),
Peprotech (RochyHill NJ), R&DSystems (Minneapolis MN).
Trasfezione e selezione di cloni cellulari trasfettati stabilmente
Per ottenere una linea cellulare stabile, le cellule HaCat, piastrate ad una densità
di 2 x 105 cellule per piastra (sono state utilizzate capsule Petri dal diametro di 60 mm),
sono state trasfettate con i plasmidi pALuc HPV-16 LCR e pSTneo, mediante il metodo
della coprecipitazione con fosfato di calcio.
Quattro ore prima della trasfezione, il terreno è stato sostituito con terreno
fresco. In seguito, per ogni trasfezione, successivamente per ogni trasfezione sono state
preparate la soluzione HBS 2X (Na2HPO4 1.5 mM, NaCl 280 mM, HEPES -acido 4-2-
idrossietil-1-piperazina-etansulfonico- pH 7.1 50 mM) e la soluzione contenente il DNA
(10 µg del plasmide pALuc HPV16 LCR, 50 ng di plasmide pSTneo, 37 µl CaCl2 2M,
H2O per arrivare a un volume finale di 300 µl).
La soluzione contenente il DNA plasmidico è stata addizionata, goccia a goccia
e in agitazione, alla soluzione HBS 2X. Il tutto è stato incubato per 1 minuto a
temperatura ambiente, per consentire la formazione dei precipitati di DNA-fosfato di
calcio. Trascorso questo periodo di tempo, la soluzione è stata aggiunta alla coltura
cellulare, dove è stata lasciata per una notte. Il giorno successivo il terreno è stato
rimosso e sostituito da altro terreno, contenente il 10% di siero.
24 ore dopo la trasfezione, le cellule sono state staccate dal substrato mediante
una soluzione di tripsina/EDTA in tampone fosfato PBS (“phosphate-buffered saline”),
seminate in tre piastre, dal diametro di 100 mm e selezionate in terreno contenente
l’antibiotico G418 (0.4 mg/ml) per 2-3 settimane, fino alla comparsa delle colonie
cellulari persistenti.

Le colonie sono state tripsinizzate e raccolte come “pool”. Singoli cloni sono
stati ottenuti attraverso il metodo della diluizione al limite seminando le cellule del
“pool” in piastre a 96 pozzetti ad una densità di 0.5 cellule per pozzetto.
25 cloni sono stati espansi e saggiati per l’attività luciferasica basale. Per gli
studi successivi sono stati scelti due di questi cloni, nominati P13 e P21, in quanto
presentavano l’attività luciferasica più elevata.
Saggio della Luciferasi
I cloni trasfettati stabilmente sono stati seminati in piastre di coltura a 24 o 96
pozzetti, a una densità rispettivamente di 6 x 104 e di 2 x 104 cellule per pozzetto.
Il giorno successivo, alcune cellule sono state trattate con diverse citochine,
mentre altre, usate come controllo, non sono state trattate. 24 ore dopo il trattamento, i
monostrati di cellule sono stati lavati due volte con il tampone fosfato PBS, lisate in
ghiaccio con 40 µl (nel caso di piastre a 24 pozzetti) o 25 µl (per piastre a 96 pozzetti)
di tampone di lisi “reporter” (Promega); infine le proteine solubili sono state recuperate
in seguito a centrifugazione.
E’ poi stata valutata la concentrazione delle proteine presenti nel surnatante
(attraverso il saggio colorimetrico “Biorad DC Protein Assay”) e la loro attività
luciferasica mediante il saggio luciferasico (Promega), utilizzando come strumentazione
il luminometro Lumino (Startec Medical System). Per la realizzazione del saggio
luciferasico sono stati addizionati 100 µl di “Luciferase Assay Reagent” (Promega) a 20
µl di lisato cellulare.

Saggio per la valutazione della vitalità cellulare
Le cellule sono state seminate a una densità di 6 x 104 cellule in piastre a 24
pozzetti e il giorno successivo alcune di queste sono state trattate con citochine, mentre
altre, utilizzate come controllo, non sono state trattate.
Dopo 24 e 48 ore dal trattamento è stata determinata la vitalità cellulare
mediante il saggio colorimetrico CellTiter 96R AQueous One Solution Cell Proliferation.
Esso si basa sull’utilizzo di un reagente (CellTiter 96R AQueous One Solution Cell
Proliferation Reagent) contenente i composti tetrazolio MTS e PES (fenazina
etansolfato, un reagente di accoppiamento elettronico): PES presenta un’elevata stabilità
chimica, che rende possibile la reazione con MTS, per formare una soluzione stabile.
MTS viene ridotto nelle cellule in un prodotto colorato, il formazano, solubile nei
terreni di coltura tessutali. Questa conversione si accompagna alla produzione di
NADPH o NADH da parte degli enzimi deidrogenasi, presenti in cellule attive dal
punto di vista metabolico
E’ necessario in primo luogo scongelare il reagente 96R AQueous One Solution
Cell Proliferation lasciandolo per 90 minuti a temperatura ambiente o 10 minuti nel
bagno riscaldato a 37°C. In ogni pozzetto della piastra a 96 pozzetti, contenente il
campione diluito in 100 µl di terreno di coltura, si aggiungono 20 µl di reagente. La
piastra viene incubata per 1-4 ore a 37°C in un ambiente umidificato, al 5% di CO2.
L’assorbanza del prodotto colorato viene letta a 409 nm. La quantità di
formazano prodotta è direttamente proporzionale al numero di cellule vive presenti in
coltura.

Real-time PCR quantitativa su DNA
Il DNA genomico delle cellule HaCat, CaSki e dei cloni P13 e P21 è stato
isolato lisando le cellule con una soluzione costituita da 10 mM Tris/HCl pH 8.0, 25
mM EDTA, 100 mM NaCl, 0.5% SDS, 100 µg/ml di proteinasi K e incubando la
soluzione a 50°C per 4-5 ore. La digestione è seguita da un’estrazione con fenolo
cloroformio, precipitazione del DNA in etanolo 100%, ammonio acetato 7.5 M e alcol
isoamilico. Infine è stato effettuato un trattamento con RNasi (1 µg/ml di RNasi e 0.1%
di SDS in tampone TE per 1 ora a 37° C). L’estrazione con fenolo, cloroformio, alcol
isoamilico viene ripetuta una seconda volta. Successivamente si centrifuga e si lava il
lisato cellulare con etanolo 70%. Si risospende tale lisato in tampone TE e si incuba la
soluzione a 65°C per 1 ora.
La real-time PCR quantitativa su DNA è stata allestita in un volume di reazione
di 25 µl, utilizzando il reagente “Brillant SYBR Green QPCR Master Mix”
(Stratagene). In particolare, la miscela di reazione conteneva 12.5 µl di Syber Mix, 0.75
µl di Rox (diluita 1 a 1000), 1.5 µl di primer senso 10 µM, 1.5 µl di primer antisenso 10
µM e acqua distillata sterile fino a un volume finale di 20 µl. In un secondo momento
sono stati aggiunti 5 µl di campione o di acqua (come controllo).
Per amplificare la regione LCR di HPV-16 sono stati disegnati i seguenti primer:
primer senso: 5’-CCAAATCCCTGTTTTCCT-3’.
primer antisenso: 5’-ATGTGCCTAACAGCGGTA-3’.
Per avere un controllo positivo interno, il saggio è stato effettuato anche sul gene
della β-actina, utilizzando i seguenti primer:
primer senso: 5’-GTTGCTATCCAGGCTGTG-3’;
primer antisenso: 5’-TGTCCACGTCACACTTCA-3’.

L’amplificazione del DNA è stata effettuata su una piastra a 96 pozzetti,
utilizzando come strumentazione il modello Mx3000P “Real-time Machine PCR
System” (Stratagene).
La real-time PCR realizzata è di tipo comparativo-quantitativo. Per ogni analisi è
stata delineata in parallelo una curva standard di riferimento, sulla base di diluizioni
seriali di DNA estratto da cellule CaSki, da 0.005 ng a 50 ng di DNA. Le cellule CaSki
contengono 600 copie di DNA genomico integrato di HPV-16 per cellula. Per esprimere
i risultati quantitativi ottenuti in equivalente genomico per i campioni sconosciuti, è
stato utilizzato un fattore di conversione pari a 6.6 pg DNA per cellula diploide.
Quantificazione dei trascritti E6-E7
L’RNA totale è stato estratto a partire da 1 x 106 cellule HaCat (usate come
controllo negativo) e cellule CaSki, utilizzando il kit NucleoSpin II (Machery-Nagel).
Successivamente 2 µg di RNA sono stati retrotrascritti con il kit RevertAid cDNA
Synthesis (Fermentas), utilizzando “Random exhamer primer”. Il cDNA risultante è
stato quantificato mediante una “Real-Time PCR” utilizzando i seguenti primer:
1) specifici per il gene E6 di HPV-16:
primer senso: 5’-GCAAGCAACAGTTACTGAGACGT-3’;
primer antisenso: 5’-GCAACAAGACATACATCGACCGG-3’;
2) primer specifici per il gene E7 di HPV-16:
primer senso: 5’-GATGGTCCAGCTGGACAAGC-3’;
primer antisenso: 5’-GTGCCCATTAACAGGTCTTC-3’;
3) primer specifici per il gene della β-actina:
primer senso: 5’-GTTGCTATCCAGGCTGTG-3’;
primer antisenso: 5’-TGTCCACGTCACACTTCA-3’.

L’amplificazione del DNA è stata realizzata utilizzando la miscela di reazione
“Master Mix Brillant SYBR Green QPCR” (Stratagene), in una piastra di reazione a 96
pozzetti, utilizzando come strumentazione il modello Mx3000P “Real-Time Machine
PCR System” (Stratagene). I risultati sono stati normalizzati sulla base dei livelli di β-
actina ed espressi come percentuale di inibizione rispetto ai controlli non trattati (usati
come calibratori)
Citofluorimetria
1 x 106 cellule HaCat e Caski sono state raccolte, lavate due volte con PBS
contenente l’1% di albumina di siero bovino (BSA) e lo 0.01% di NaN3.
Successivamente sono state incubate con anticorpi diretti verso IL-13Rα1 (IgG1
murina), IL-4R (IgG2a murina) o anticorpi anti isotipo (R&D Systems, Minneapolis,
MN) per 30 minuti a 4°C. Le cellule sono state quindi lavate per due volte e incubate
con anticorpi di capra diretti verso IgG-FITC murina (Immunotech, Marsiglia, Francia)
per 30 minuti a 4°C e analizzate con lo strumento FACSort (Becton-Dickinson,
Rutherford, NJ).

RISULTATI
Realizzazione e caratterizzazione di una linea cellulare “reporter”
contenente la regione LCR del genoma di HPV-16
Per la realizzazione di una linea cellulare “reporter”, utilizzabile per l’analisi di
inibitori della trascrizione dei geni E6 e E7 di HPV-16, è stata scelta la linea di
cheratinociti umani HaCat, in quanto supporta una forte attività del promotore LCR
(Sailaja e Bernard, 1999) del genoma di HPV-16, ma è priva di qualsiasi sequenza del
DNA degli HPV in grado di competere con il sistema “reporter”.
Le cellule sono state cotrasfettate con il plasmide pALuc HPV-16 LCR, che
contiene la sequenza LCR completa, clonata a monte del gene della luciferasi di
lucciola e con il plasmide pSTneoB, che conferisce resistenza all’antibiotico G418. In
seguito le cellule sono state sottoposte a selezione con l’antibiotico G418 e clonati con
il sistema della diluizione al limite.
Venticinque cloni sono stati espansi e analizzati per l’attività luciferasica basale.
Nove di questi sono risultati positivi e, per la realizzazione degli esperimenti successivi,
sono stati scelti due cloni, chiamati P13 e P21, poiché manifestano un’elevata attività
luciferasica (Figura 1).

050
100150200250300350400450
Atti
vità
luci
fera
sica
(RLU
/µg
di p
rote
ina)
P1 P6 P9 P11 P12 P13 P14 P18 P21 P23
* *0
50100150200250300350400450
Atti
vità
luci
fera
sica
(RLU
/µg
di p
rote
ina)
P1 P6 P9 P11 P12 P13 P14 P18 P21 P23
* *
Per verificare e dimostrare la presenza della sequenza LCR all’interno dei cloni
saggiati, è stata eseguita una PCR, a partire dal DNA estratto dalle cellule HaCat
parentali (come controllo negativo), dai cloni P13 e P21 e da cellule CaSki (cellule
umane di cancro alla cervice uterina, HPV-16 positive, usate come controllo positivo).
L’amplificato è stato quindi visualizzato attraverso una corsa elettroforetica, eseguita su
gel di agarosio (allo 0.8%): è stato possibile evidenziare un frammento di dimensioni
attese (152 nucleotidi) nelle cellule P13, P21 e Caski, mentre non si osserva alcun
segnale nei campioni di cellule HaCat (Figura 2).
Figura 1: Analisi dell’attività luciferasica dei cloni “reporter” ottenuti
attraverso il sistema della diluizione al limite

400
300
200
100
bp Mw 1 2 3 4
Successivamente, per quantificare il numero di copie del plasmide “reporter”
integrato è stata realizzata una “Real-Time PCR” quantitativa sul DNA genomico
estratto dalle stesse cellule.
E’ noto che le cellule CaSki contengono 600 copie di genoma di HPV-16
integrato per cellula (Capone et al., 2000); pertanto esse sono state scelte per la
costruzione di una curva standard di riferimento. Il numero ottenuto di copie di LCR per
cellula è una copia per genoma, in entrambi i cloni.
Questi risultati dimostrano che sono stati prodotti cloni contenenti un sistema
“reporter” integrato.
I cloni P13 e P21 sono stati poi validati per l’uso in un saggio cellulare,
analizzando i seguenti parametri:
1) Stabilità dell’espressione della luciferasi nel tempo;
Figura 2: Corsa elettroforetica dell’amplificato della regione LCR del
genoma di HPV-16, ottenuto mediante PCR, in cellule HaCat, P13, P21,
Caski. In cellule HaCat (pozzetto 1), non si osserva alcun segnale. Nei cloni
P13, P21 (pozzetti rispettivamente 2 e 3) e in cellule Caski (pozzetto 4), è
possibile rilevare il segnale di un amplificato di 152 nucleotidi, indicativo
della presenza della regione LCR integrata nel genoma cellulare.

2) Risposta a stimoli inibitori noti;
3) Compatibilità con un formato ad alta resa;
1) Per valutare la stabilità dei cloni selezionati, ne sono stati analizzati i livelli di
attività luciferasica a tempi diversi, successivi alla selezione. Nella figura 3 si osserva
che è possibile rilevare livelli stabili di attività luciferasica nei cloni P13 e P21, raccolti
a 30, 40, 50, 60, 60 e 80 giorni di coltura continua dopo la selezione, con un valore
medio di circa 400 unità luminose relative (RLU)/µg di proteina per il clone P13 e 370
RLU/µg di proteina per il clone P21.
0
100
200
300
400
500
30 dps 40 dps 50 dps 60 dps 70 dps 80 dps
Luci
fera
se a
ctiv
ity (R
LU/ µ
g)
P13P21
Att
ività
luci
fera
sica
(RL
U/m
g)
0
100
200
300
400
500
30 dps 40 dps 50 dps 60 dps 70 dps 80 dps
Luci
fera
se a
ctiv
ity (R
LU/ µ
g)
P13P21
Att
ività
luci
fera
sica
(RL
U/m
g)
2) Per valutare se le linee cellulari “reporter” possano essere utilizzate per
Figura 3: Validazione del saggio. Stabilità dell’espressione della luciferasi nel tempo.
Livelli di attività luciferasica nei cloni P13 e P21, a 30, 40, 50, 60, 70, 80 giorni
successivi alla selezione con l’antibiotico G418, espressi in unità luminose relative per
microgrammo di proteina. Si calcola con un valore medio di circa 400 RLU/µg di
proteina per il clone P13 e 370 RLU/µg di proteina per il clone P21. I risultati mostrati
nel grafico rappresentano la media ± SD di tre esperimenti eseguiti in duplicato.

selezionare gli inibitori di LCR, è stata analizzata la risposta di P13 e P21 ad inibitori
noti della regione LCR di HPV-16: le citochine TGF-β1 e TNF-α (Baldwin et al., 2004;
Kyo et al., 1994). Le cellule, seminate in una piastra a 24 pozzetti, sono state trattate per
24 ore con 50 ng/ml di ciascuna citochina, mentre alcuni campioni, usati come
controllo, non sono stati trattati. Come si osserva in figura 4, è possibile evidenziare il
67% e il 62% di inibizione da parte di TGF-β e il 55% e 47% di inibizione da parte di
TNF-α, rispettivamente, sui cloni P13 e P21. Questo dato dimostra che il sistema
“reporter” integrato risponde in maniera attesa a stimoli noti e che quindi le linee
cellulari “reporter” presentano le potenzialità di identificare nuovi inibitori dell’attività
della regione LCR.
0
20
40
60
80
untreated TGF-b1 TNF-a
Luci
fera
se a
ctiv
ity (%
of i
nhib
ition
)
P13P21
Att
ività
luci
fera
sica
(% d
i ini
bizi
one)
Non trattate TGF-β1 TNF-α
0
20
40
60
80
untreated TGF-b1 TNF-a
Luci
fera
se a
ctiv
ity (%
of i
nhib
ition
)
P13P21
Att
ività
luci
fera
sica
(% d
i ini
bizi
one)
Non trattate TGF-β1 TNF-α
Figura 4: Validazione del saggio. Risposta a stimoli inibitori noti della regione LCR
di HPV-16. I cloni P13 e P21 sono stati trattati con 50 ng/ml di TGF-β1 e TNF-α o
non sono stati trattati. L’attività luciferasica è stata misurata 24 ore dopo il
trattamento. I risultati mostrati nel grafico rappresentano la media ± SD di tre
esperimenti eseguiti in duplicato.

3) Per verificare se l’attività luciferasica espressa dai cloni P13 e P21 fosse
compatibile con un formato miniaturizzato ad alta resa, si sono seminate 6000, 10 000 e
20 000 cellule per pozzetto, in una piastra a 96 pozzetti.
In figura 5 è possibile osservare che l’aumento del numero di cellule da 5000 a
20 000 per pozzetto è accompagnato da un progressivo aumento dell’attività luciferasica
(rispettivamente 1700, 2700, 6000 RLU). Tenendo conto del valore basale registrato dal
luminometro (circa 40 RLU), il segnale emesso da 20 000 cellule per pozzetto è più che
sufficiente per la rilevazione, ad indicare che l’attività luciferasica generata dai cloni
cellulari “reporter” rende possibile l’impiego del saggio in un sistema ad alta resa.
0
1000
2000
3000
4000
5000
6000
7000
5000 10000 20000
cells seeded/well
Lucu
fera
se a
ctiv
ity (R
LU)
P13P21
Atti
vità
luci
fera
sica
(RL
U)
Cellule seminate/pozzetto
0
1000
2000
3000
4000
5000
6000
7000
5000 10000 20000
cells seeded/well
Lucu
fera
se a
ctiv
ity (R
LU)
P13P21
Atti
vità
luci
fera
sica
(RL
U)
Cellule seminate/pozzetto
Figura 5: Validazione del saggio. Compatibilità con un formato ad alta resa. Si sono
seminate 5000, 10 000, 20 000 cellule per pozzetto, in un formato a 96 pozzetti, dei
cloni P13 e P21; il giorno successivo ne è stata analizzata l’attività luciferasica. I
risultati mostrati nel grafico rappresentano la media ± SD di tre esperimenti eseguiti in
duplicato.

Analisi di un pannello di citochine utilizzando il clone indicatore P13
Per verificare se il saggio cellulare messo a punto potesse essere
vantaggiosamente utilizzato in un processo di “screening” ad alta resa, abbiamo
analizzato, in un formato di piastra a 24 pozzetti, un pannello di trentadue citochine,
appartenenti a diverse classi funzionali, utilizzando il clone P13 come linea cellulare
“reporter”.
I cloni sono stati trattati con le diverse le citochine, ad una concentrazione di 50
ng/ml, per 24 ore. L’analisi è stata ripetuta tre volte, in duplicato. In base ai risultati
ottenuti è stato possibile suddividere le citochine in tre gruppi, sulla base della
percentuale di inibizione dell’attività luciferasica relativa alla regione LCR (Tabella1).

Citochine % di inibizione di LCR (valori medi ± SD)
Gruppo*
Tabella 1: Effetto delle citochine sull’attività della regione LCR del genoma di HPV-16.
* I gruppi sono formati in base alla percentuale di inibizione di LCR.
Gruppo I: 0-29%; Gruppo II: 30%-49%; Gruppo III: 50%-70%

Anti-infiammatorie
IL-4 56.6 ± 7.2 III
IL-10 0 I
IL-13 64.3 ± 5.8 III
TGF-β1 61.4 ± 8.4 III
TGF-β2 60.1± 6.5 III
TGF-β3 58.9 ± 5.9 III
Activina 32.4 ± 4.3 II
GDF-15 16.3 ± 2.1 I
Osteonectina 0.2 ± 0.003 I
Pro-infiammatorie
IL-1β 33.3 ± 4.6 II
IL-15 12.6 ± 1.8 I
IL-17 29.6 ± 3.6 I
IL-18 3 ± 0.5 I
IL-19 9 ± 1.2 I
IL-20 10.2 ± 0.8 I
IL-22 10.3 ± 1.7 I
TNF-α 53.7 ± 4.3 III
GM-CSF 12 ± 0.7 I
Fattori di crescita
IL-3 2.1 ± 0.09 I
IL-6 5.2 ± 0.3 I
IL-7 2.5 ± 0.5 I
IL-21 2.4 ± 0.4 I
Fattori chemotattici
IL-8 0 I
IP-10 5.2 ± 0.9 I
LEC 16.1 ± 2.3 I
MIP-1α 0 I
MIP-1β 27.3 ± 3.8 I
NAP-2 0 I
I-309 0.9 ± 0.2 I
Interferoni
IFN-α 58.2 ± 7.6 III
IFN-β 63.1 ± 4.8 III
IFN-γ 35.5 ± 4.3 II
Tabella 1: Effetto delle citochine sull’attività della regione LCR del genoma di HPV-16

Sono state classificate nel gruppo I ventuno citochine, che hanno mostrato
un’attività inibitoria nulla o molto bassa (da 0% a 29%). Tre citochine (activina, IL-1β,
e IFN-γ), caratterizzate da una modesta attività inibitoria (dal 30% al 49%), sono state
classificate nel gruppo II, mentre otto citochine (IL-4, IL-13, TGF-β1, TGF-β2, TGF-
β3, IFN-α e IFN-β), a maggiore attività inibitoria (dal 50% al 70%), sono state
classificate nel gruppo III. Attraverso questo saggio non è stata evidenziata alcuna
citochina con attività stimolatoria sull’LCR di HPV-16.
Dato che, in generale, l’ esito di un saggio di questo tipo, dipende in parte
dall’attività citostatica o citotossica del composto somministrato, abbiamo analizzato
l’effetto di tutte le citochine classificate nel gruppo III sulla proliferazione del clone P13
attraverso il saggio di proliferazione cellulare descritto in Materiali e Metodi. Nessuna
delle citochine studiate presentava un’attività antiproliferativa alla concentrazione e nei
tempi utilizzati per il saggio della luciferasi. Questi risultati dimostrano che la riduzione
dell’attività luciferasica non è una conseguenza della ridotta vitalità cellulare.
All’interno del gruppo III, le citochine IL-4 e IL-13 sono particolarmente
interessanti, in quanto non erano ancora state descritte, in studi precedenti, come
inibitori dell’attività trascrizionale del promotore LCR degli HPV e per questo motivo
sono state oggetto di analisi più dettagliate.
Per stabilire le dosi di IL-4 e IL-13 in grado di produrre il 50% di inibizione
dell’attività di LCR (ID50), il clone P13 è stato trattato con diverse concentrazioni di
citochine, comprese tra 0.1 ng/ml e 250 ng/ml. La figura 6 evidenzia che entrambe le
citochine riducono l’attività di LCR in modo dipendente dalla concentrazione. Infine,
un’analisi di regressione non lineare dei dati ha permesso di calcolare la ID50 come pari
a 4 ng/ml per IL-4 e 11.3 ng/ml per IL-13.

A
B
10 -1 10 0 10 1 10 2 10 30
10
20
30
40
50
60
70IL 4
ng/ml
% d
i ini
bizi
one
10 -1 10 0 10 1 10 2 10 30
10
20
30
40
50
60
70IL 13
ng/ml
% d
i ini
bizi
one
A
B
10 -1 10 0 10 1 10 2 10 30
10
20
30
40
50
60
70IL 4
ng/ml
% d
i ini
bizi
one
10 -1 10 0 10 1 10 2 10 30
10
20
30
40
50
60
70IL 4
ng/ml
% d
i ini
bizi
one
10 -1 10 0 10 1 10 2 10 30
10
20
30
40
50
60
70IL 13
ng/ml
% d
i ini
bizi
one
10 -1 10 0 10 1 10 2 10 30
10
20
30
40
50
60
70IL 13
ng/ml
% d
i ini
bizi
one
Figura 6: Percentuale di inibizione dell’attività luciferasica dei cloni P13
(A) e P21 (B) in seguito a trattamento con dosi crescenti di citochine IL-4 e
IL-13. Si osserva che entrambe le citochine riducono l’attività di LCR in
modo dipendente dalla concentrazione.

Effetto di IL-4 e IL-13 sui livelli di mRNA di E6 e E7 di HPV-16 in
cellule CaSki
Per verificare se la riduzione dell’attività luciferasica provocata da IL-4 e IL-13
nel clone “reporter” P13 rifletta una diminuzione dei livelli dell’mRNA di E6 e E7 in
cellule di cancro alla cervice uterina positive per HPV-16, è stata scelta la linea cellulare
CaSki come modello di studio.
Per prima cosa abbiamo esaminato l’espressione delle componenti recettoriali di
IL-4 e IL-13, in particolare la catena α del recettore di IL-4 e la catena α1 del recettore
di IL-13, in cellule HaCat, Caski e in monociti THP-1 (usati come controllo positivo)
(Hart et al., 1999).
Come si può osservare in figura 7, l’analisi al citofluorimetro rivela che le
cellule HaCat e CaSki esprimono entrambe le catene del recettore di IL-4 e IL-13 e sono
quindi potenzialmente responsive a queste citochine.

Figura 7: Analisi al citofluorimetro dell’espressione superficiale delle componenti
recettoriali IL-4R e IL-13Rα1 in monociti THP-1 (usati come controllo positivo),
in cellule Caski e HaCat. Le cellule THP-1, HaCat e CaSki esprimono entrambe le
catene del recettore di IL-4 e IL-13 e sono quindi potenzialmente responsive a
queste citochine. Ogni pannello costituisce un istogramma dell’espressione di IL-
4R e IL-13Rα1 (curva ombreggiata). Le curve non ombreggiate rappresentano la
colorazione con un anticorpo secondario, usato come controllo.
Intensità della fluorescenza
Numero di cellule

Successivamente abbiamo studiato l’effetto di IL-4 e IL-13 sull’espressione dei
livelli di mRNA dei geni E6 e E7 in cellule CaSki. Il TGF-β1 è stato utilizzato in questi
esperimenti in quanto è un noto inibitore dell’attività del promotore LCR e il suo
recettore è espresso in cellule CaSki (Kang et al., 1998). A questo scopo le cellule sono
state trattate rispettivamente con 50 ng/ml di TGF-β, IL-4 e IL-13 per 24 ore e i livelli
di mRNA sono stati determinati mediante la “Real-Time PCR”.
I risultati illustrati in figura 8 dimostrano che tutte le citochine esaminate
riducono i livelli di mRNA di E6 e E7 sebbene in misura diversa. Il trattamento con
TGF-β1 esercita un forte effetto inibitorio riducendo rispettivamente dell’86,2% e dell’
85,5% i livelli dei trascritti di E6 ed E7; il trattamento con IL-4 provoca un riduzione
del 56.3% dei livelli del trascritto E6 e del 62% dei livelli del trascritto E7, mentre dopo
trattamento con IL-13 le cellule Caski presentano una riduzione dei livelli del trascritto
E6 pari al 50% ed una riduzione dei livelli del trascritto E7 pari al 53%.
Questi dati confermano l’effetto inibitorio esercitato sull’attività trascrizionale
del LCR, che è stato osservato nella linea cellulare reporter dopo trattamento con TGF-
β, IL-4 ed IL-13 ed esclude la possibilità che la riduzione dell’attività luciferasica possa
essere una conseguenza di un effetto aspecifico esercitato dalle citochine sull’attività
enzimatica o sulla stabilità.

0
0,2
0,4
0,6
0,8
1
1,2
Non trattato TGF β1 IL-4 IL-13
Live
lli d
i mR
NA
rela
tivi
E6E7
0
0,2
0,4
0,6
0,8
1
1,2
Non trattato TGF β1 IL-4 IL-13
Live
lli d
i mR
NA
rela
tivi
E6E7
Figura 8: Effetto delle citochine TGF-β1, IL-4 e IL-13 (50 ng/ml per 24 ore)
sull’espressione dei geni E6 e E7 in cellule CaSki, analizzato attraverso “Real-time PCR”. I
risultati sono espressi come livelli relativi di mRNA. Se si pone pari a 1 la quantità di
mRNA estratto da cellule non trattate, in seguito a trattamento con il TGF-β1 si registrano
livelli relativi di mRNA del trascritto E6 e E7 pari a circa 0.2, mentre livelli pari a circa 0,5
dopo trattamento con IL-4 e IL-13.

DISCUSSIONE
Il carcinoma della cervice uterina, ampiamente diffuso a livello mondiale,
rappresenta la seconda causa di morte per tumore nella donna. Gli HPV ad alto rischio,
quali HPV-16, HPV-18, HPV-31 e HPV-45, sono stati identificati come gli agenti
causali dei carcinomi cervicali ed in particolare il DNA dell’HPV-16 è stato evidenziato
nella maggior parte di essi.
Attualmente il trattamento di lesioni associate a infezione da HPV viene
realizzato chirurgicamente. Tuttavia, nonostante la rimozione chirurgica della neoplasia,
spesso la crescita del tumore riprende, a causa della persistenza del virus nei tessuti
sani. La prevenzione delle infezioni da HPV attraverso vaccinazione o immunoterapia è
in corso di studi, ma non è ancora stata introdotta nella pratica clinica. Pertanto risulta
necessario lo sviluppo di strategie antivirali finalizzate al trattamento di neoplasie
indotte da HPV e tali da ridurre la loro ricorrenza. Nonostante siano stati elucidati i
principali meccanismi coinvolti nella patogenesi dei tumori causati da HPV, la
realizzazione di farmaci per il loro trattamento presenta notevoli ostacoli: in primo
luogo i sistemi per lo studio del virus in vitro sono tecnicamente complessi e non
convenienti per l’analisi di molecole antivirali; inoltre il genoma di HPV non codifica
per proteine enzimatiche, quali polimerasi o proteasi, che rappresentano i bersagli
tradizionali per le terapie antivirali. Pertanto nuovi bersagli rilevanti per la patogenesi
virale devono essere identificati e validati per poter essere inclusi nel programma di
sviluppo di un farmaco.
Potenziale bersaglio di farmaci anti-HPV sono le oncoproteine E6 e E7. I geni
E6 ed E7 degli HPV ad alto rischio sono i principali oncogeni virali, essendo in grado,
cooperativamente, di immortalizzare e trasformare un’ampia varietà di cellule primarie
umane (Munger et al., 1989). Inoltre è necessaria la loro continua espressione in cellule

di carcinomi della cervice uterina per il mantenimento del fenotipo trasformato
(DeFilippis et al., 2003; Goodwin et al., 2000; Wells et al., 2000).
Le oncoproteine E6 ed E7 innescano il processo di trasformazione della cellula
infettata interagendo funzionalmente e fisicamente con numerose proteine regolatorie
cellulari (p53, la subunità catalitica della telomerasi hTERT, la proteina del
retinoblastoma p105Rb, le cicline A e E, p21, p27 e le deacetilasi istoniche) sovvertendo
funzioni cellulari, quali il controllo del ciclo cellulare, l’apoptosi, la senescenza, il
riparo del DNA e la stabilità genomica (Finzer et al., 2002; zur Hausen, 2002;
Longworth et al., 2004).
A differenza di molti altri tipi di tumore, la maggior parte dei carcinomi della
cervice uterina associati ad HPV mantengono i geni oncosoppressori p53 e p105Rb in
forma non mutata (Scheffner et al., 1991) e sono quindi potenzialmente suscettibili alle
strategie terapeutiche finalizzate al recupero del controllo della crescita cellulare in
seguito alla repressione delle oncoproteine E6 ed E7 di HPV. Sistemi basati sull’utilizzo
di macromolecole, quali ribozimi, RNA antisenso, RNA “interference”, per reprimere l’
espressione delle oncoproteine di HPV in cellule di carcinoma della cervice uterina,
hanno dato come risultato una forte inibizione della proliferazione. (DeFilippis et al.,
2003; Butz et al., 2003; Chen et al., 1996). Recentemente è stato osservato che
l’espressione ectopica della proteina E2 di HPV, capace di legare il promotore maggiore
precoce (LCR) del virus e inibire la trascrizione dei geni di E6 ed E7, causa una forte
inibizione della sintesi del DNA cellulare, un’acquisizione rapida del fenotipo
senescente con il recupero delle vie di segnale mediate da p53 e p105Rb in cellule del
carcinoma cervicale (HeLa) (Goodwin et al., 2000; Wells et al., 2000). Questi studi
hanno fornito la prova del principio che l’inibizione delle due oncoproteine virali possa
bloccare la crescita di tumori associati a HPV. Tuttavia, questi approcci basati sull’uso

di macromolecole sono limitati da problemi associati al rilascio di concentrazioni attive
a livello di specifici siti anatomici, in seguito a somministrazione sistemica. Per poter
allestire strategie terapeutiche sulla base di queste conoscenze, sono necessari metodi
capaci di identificare piccole molecole chimiche o di origine biologica in grado di
inibire selettivamente l’attività trascrizionale della sequenza LCR di HPV.
La porzione LCR, che regola la trascrizione di E6 ed E7, presenta numerosi siti
di legame per fattori di trascrizione virali e cellulari e dipende dalla loro azione
coordinata. Pertanto è improbabile realizzare un inibitore diretto verso tale regione
disegnando il farmaco sulla base della struttura del promotore. Questo approccio
richiede infatti bersagli precisi e ben caratterizzati, come i siti catalitici di enzimi virali
o le interazioni specifiche proteina-proteina. Un approccio alternativo è utilizzare come
potenziale risorsa di farmaci candidati, librerie di composti generate con la chimica
combinatoriale, estratti di prodotti naturali o collezioni di citochine.
Per analizzare queste collezioni di prodotti bioattivi abbiamo generato e validato
un saggio cellulare ad alta resa, che rappresenta l’oggetto di questa tesi.
Come bersaglio è stata scelta la sequenza LCR dell’HPV ad alto rischio di tipo
16, essendo il principale virus isolato dai carcinomi della cervice uterina con una
prevalenza del 50-60% nella maggioranza dei paesi (Bosch et al., 2003).
Il sistema indicatore che abbiamo sviluppato è costituito da una linea cellulare
“reporter” di cheratinociti umani (HaCat), trasfettati stabilmente con un plasmide
ricombinante contenente il gene Luciferasi posto sotto il controllo trascrizionale della
sequenza LCR di HPV-16, la quale possiede la sequenza promotore e gli elementi
”enhancer” degli oncogeni E6 ed E7. In questo sistema, l’attività luciferasica è una
misura proporzionale dell’attività trascrizionale della sequenza LCR.

La linea cellulare “reporter” è stata validata per l’utilizzo in un saggio cellulare
misurando:
1) La stabilità dell’espressione luciferasica nel tempo;
2) La risposta a inibitori noti della regione LCR, quali TGF-β1 e TNF-α;
3) La compatibilità con un formato ad alta resa.
I nostri risultati hanno dimostrato che il saggio ad alta resa generato permette
una facile, rapida e sicura identificazione di inibitori dell’ espressione delle
oncoproteine E6 ed E7 di HPV-16 in banche di composti sintetici o biologici. Inoltre, il
vantaggio di un saggio cellulare rispetto a uno non cellulare, è il maggiore grado di
successo che si può riscontrare utilizzando gli inibitori identificati con il primo saggio,
in modelli animali, poiché tale saggio verifica l’entrata e potenzialmente la stabilità del
composto all’interno della cellula.
Anche in studi precedenti (Craigo et al., 2000) si descrive un procedimento
volto a identificare molecole inibitorie dell’espressione dei geni codificanti per le
oncoproteine di HPV, basato però sulla tecnica della trasfezione transiente. In questo
caso, come sistema indicatore, è stata utilizzata la linea cellulare di carcinoma cellulare
C33A (HPV negativa), trasfettata transientemente con un plasmide ricombinante posto
sotto il controllo trascrizionale del promotore della sequenza LCR di HPV-16.
La realizzazione del saggio transiente richiede una serie di passaggi tecnici, così
articolati:
• Le cellule vengono seminate (giorno 1) in un formato non compatibile con un
saggio ad alta resa (ad esempio piastre da 60 o 35 mm di diametro).
• In seguito le cellule sono trasfettate transientemente con il plasmide
ricombinante (giorno 2). Questo passaggio richiede l’uso di reagenti specifici tra
i quali, oltre al plasmide ricombinante purificato, anche un plasmide di

normalizzazione purificato e i reagenti necessari per la trasfezione. Tale
passaggio inoltre deve essere eseguito da un operatore dotato di competenze e
manualità specializzate.
• Successivamente vengono aggiunte le molecole da analizzare (giorno 3).
• Infine viene misurata l’attività luciferasica specifica (giorno 4). A questo scopo
il terreno di coltura viene rimosso, le cellule vengono lavate, staccate dal
substrato, centrifugate e il surnatante viene eliminato. Il sedimento cellulare
viene lisato con un tampone di lisi appropriato. I lisati cellulari sono nuovamente
centrifugati per rimuovere i componenti subcellulari e il campione ottenuto è
sottoposto al dosaggio del contenuto proteico, ad esempio attraverso il metodo
di Bradford. Successivamente vengono misurate l’attività luciferasica e l’attività
del gene indicatore del plasmide di normalizzazione (per esempio Renilla
luciferasi o β-galattosidasi). I campioni vengono letti uno alla volta utilizzando
un luminometro. I risultati ottenuti con il dosaggio della luciferasi devono essere
normalizzati con quelli ottenuti misurando l’attività dell’indicatore del plasmide
di normalizzazione, per annullare le differenze tra diversi campioni dovute a
eventuali variazioni nell’efficienza di trasfezione. Per ottenere l’attività
luciferasica specifica, l’attività luciferasica normalizzata viene a sua volta divisa
per la concentrazione delle proteine ottenuta con il metodo di Bradford.
E’ evidente che il saggio descritto non è adatto per l’analisi di numerose molecole
poiché richiede tempi lunghi, deve essere eseguito da operatori specializzati (la
manualità necessaria rappresenta un limite e aumenta il rischio di imprecisioni) e
soprattutto implica la ripetizione delle operazioni di trasfezione ad ogni esperimento, il
che determina la continua necessità di plasmidi ricombinanti purificati e di numerosi
reagenti e kit commerciali. Inoltre questo tipo di procedura non è compatibile con un

formato ad alta resa, ovvero un formato a 96 pozzetti. Di conseguenza, questo
procedimento consente di analizzare solo poche decine di sostanze la settimana.
Il saggio oggetto di questa tesi, basato invece sulla trasfezione stabile, supera molti
dei problemi riscontrati durante la realizzazione degli esperimenti sopra descritti. In
primo luogo l’applicazione del nostro saggio riduce notevolmente il tempo necessario
per il completamento dell’analisi di inibitori dell’espressione delle oncoproteine di
HPV. Il saggio può essere effettuato secondo lo schema temporale descritto in figura 1:
• La linea cellulare “reporter” viene seminata in piastre a 96 pozzetti (giorno 1);
• Successivamente vengono aggiunte le molecola da saggiare (giorno 2);
• Infine viene addizionato l’agente di rivelazione dell’attività luciferasica
direttamente nei pozzetti, che vengono letti tutti simultaneamente mediante un
luminometro (giorno 3).
1) La linea cellulare “reporter” viene seminatasu una piastra con formato a 96 pozzetti
2) Screening di piccole librerie di molecole
3) Misura dell’attività luciferasicamediante un lettore di micropiastre
4) Identificazione dell’ “Hit compound”
Giorno 1
Giorno 2
Giorno 3
1) La linea cellulare “reporter” viene seminatasu una piastra con formato a 96 pozzetti
2) Screening di piccole librerie di molecole
3) Misura dell’attività luciferasicamediante un lettore di micropiastre
4) Identificazione dell’ “Hit compound”
Giorno 1
Giorno 2
Giorno 3
Figura 1: Sviluppo di un saggio cellulare ad alta resa.

I principali vantaggi del saggio che abbiamo sviluppato sono determinati
dall’utilizzo, come indicatore, di una linea cellulare trasfettata stabilmente. Infatti
questo implica che:
- Non è necessario un evento di trasfezione, con conseguente risparmio di almeno un
giorno di lavoro e di molte ore di manodopera. Inoltre si consegue un notevole
risparmio economico, a livello dei diversi plasmidi e reagenti che sarebbero richiesti per
ripetere ogni volta la trasfezione.
- Non ci sono problemi connessi con eventuali differenze nell’efficienza di trasfezione
tra i diversi campioni. Pertanto non è necessario utilizzare un sistema di
normalizzazione.
- Utilizzando come agente “reporter” il gene codificante per la luciferasi, non sono
richieste procedure particolari per la preparazione del campione (come ad esempio la
rimozione del terreno, il lavaggio, la raccolta e la centrifugazione delle cellule, il
dosaggio proteico) perchè è disponibile un kit commerciale (Steady-Glo assay system,
Promega) che permette l’analisi direttamente in un formato a 96 pozzetti. L’unica
procedura richiesta è l’aggiunta del reagente direttamente nel pozzetto contenente le
cellule senza rimuovere il terreno di coltura. Tale manovra può essere eseguita con una
pipettatrice multicanale o un sistema automatizzato. Il reagente lisa le cellule e
contemporaneamente fornisce il substrato per la luciferasi.
- L’attività luciferasica può essere rilevata contemporaneamente nei 96 pozzetti della
piastra con un luminometro adatto, riducendo quindi i tempi di analisi.
Pertanto l’utilizzo del saggio da noi realizzato consente di esaminare centinaia di
molecole al giorno e migliaia di molecole alla settimana in maniera rapida e con costi
contenuti rispetto alla tecnologia precedentemente descritta.

La risposta immunitaria dell’ospite all’infezione da HPV è fondamentale
nell’eliminazione del virus e nel controllo di infezioni virali persistenti. Meccanismi di
regolazione intercellulari svolgono un ruolo importante nel prevenire la conversione di
cellule infette da HPV verso un fenotipo maligno; si ritiene che tali meccanismi siano
mediati da citochine, che vengono secrete da cellule epiteliali mucosali infette,
bloccando la trascrizione genica del virus (Altmann et al., 1994; zur Hausen, 2000). In
particolare, alcune citochine pro-infiammatorie ed anti-infiammatorie, come la famiglia
degli interferoni, IL-1, TNF-α e TGF-β, reprimono la trascrizione degli oncogeni virali
(Kim et al., 2000; Fontaine et al., 2001; Woodworth et al., 1990; Baldwin et al., 2004;
Kyo et al., 1994).
Il saggio cellulare descritto è stato impiegato in un formato a 24 pozzetti per
analizzare un pannello di 32 citochine appartenenti a diverse classi funzionali,
comprendenti citochine anti- e pro-infiammatorie, chemochine, fattori di crescita o
interferoni. Le citochine sono state classificate in tre gruppi in base alla percentuale di
inibizione dell’attività luciferasica: il gruppo I comprende 21 citochine che esercitano
attività inibitoria nulla o bassa (0-29%), il gruppo II è costituito da 3 citochine (activina,
IL-1β, e IFN-γ) che mostrano una modesta attività inibitoria (30-49%), mentre il gruppo
III comprende 8 citochine, precisamente IL-4, IL-13, TGF-β1, TGF-β2, TGF- β3, TNF-
α, IFN-α e IFN-β, che esercitano un’alta attività inibitoria (50-70%) sull’attività
luciferasica. Nessuna citochina ha mostrato attività antiproliferativa alla concentrazione
e al tempo post-trattamento scelto per il saggio luciferasico, indicando che la riduzione
dell’attività luciferasica non è una conseguenza della ridotta proliferazione cellulare o
della ridotta vitalità.

I risultati dell’analisi hanno confermato l’attività inibitoria degli interferoni, di
TNF-α e di TGF-β1 sulla regione LCR dell’HPV-16; questi dati dimostrano
l’affidabilità del saggio.
La superfamiglia TGF-β comprende tre isoforme TGF-β (TGF-β1, TGF-
β2, TGF-β3), le activine, le inibine, fattori di crescita e di differenziazione (GDFs) e le
proteine BMP (“Bone morphogenetic protein”) (Johnson et al., 2002). Queste molecole
agiscono attraverso complessi recettoriali costituiti da due polipeptidi (recettori di tipo I
e II) ad alta affinità, che possono formare eterodimeri; il dominio intracellulare serina-
treonina chinasico, presente nel recettore di tipo II, attiva le proteine Smad, che
trasmettono il segnale nel nucleo, regolando l’espressione dei geni bersaglio (Roberts,
1999; Mizono et al., 2000).
L’analisi che abbiamo eseguito, rivela che oltre al TGF-β1, anche il TGF-β2 ed
il TGF-β3 inibiscono l’attività trascrizionale della regione LCR di HPV-16, mentre un
effetto moderato o nullo è stato osservato con l’ Activina o con GDF-15,
rispettivamente. La differente attività dei membri della superfamiglia TGF-β, sulla
sequenza LCR, è giustificabile considerando che Activina e TGF-β regolano in maniera
differente alcuni marcatori di differenziamento delle cellule HaCat e pertanto si ritiene
che possano trasmettere, almeno in parte, differenti segnali all’interno della cellula
(Shimizu et al., 1998).
Diversi studi sono stati compiuti per comprendere il ruolo del TGF-β nel
controllo delle infezioni da HPV. In particolare, è stato osservato che il TGF-β inibisce
la proliferazione cellulare e l’espressione a livello trascrizionale degli oncogeni E6 ed
E7 di HPV-16 in cellule immortalizzate pre-cancerose (Woodworth et al., 1990).
Recentemente è stato proposto un modello per chiarire tale effetto inibitorio sull’attività

della regione LCR di HPV-16: il trattamento con TGF-β riduce i livelli
dell’oncoproteina Ski, impedendone il legame e l’attivazione del fattore di trascrizione
nucleare NFI presente nella regione LCR dell’HPV-16, con conseguente blocco
dell’attività trascrizionale di LCR (Figura2) (Baldwin et al., 2004).
citoplasma citoplasma
TGFTGF--ββ
citoplasma citoplasma
TGFTGF--ββ
A dispetto dell’effetto inibitorio esercitato dal TGF-β su cheratinociti
immortalizzati HPV-16 positivi, alcune linee cellulari del carcinoma cervicale,come le
cellule HeLa HPV-18 positive, non risultano responsive alla citochina (Hasskarl et al.,
2000). La resistenza di tali cellule maligne all’attività antiproliferativa del TGF-β è
interpretabile come meccanismo virale di evasione dai sistemi di difesa della cellula
bersaglio, manifestabile nel corso dello sviluppo del carcinoma cervicale; le stesse
oncoproteine virali, E6 ed E7, svolgono un ruolo di mediatori di tale resistenza,
modificando l’espressione delle citochine con il conseguente blocco dei processi di
comunicazione tra i cheratinociti infettati da HPV e gli effettori del sistema
Figura 2: Modello che illustra l’inibizione della regione LTR di HPV-16 mediata da TFG-β in
cheratinociti immortalizzati (Modificata da: Baldwin, A. et al., 2004. Journal of Virology. 78
(8): 3953-64).

immunitario. In fibroblasti embrionali murini, E7 di HPV-16 è in grado di diminuire
l’attività del promotore del TGF-β2 e di inibire la via di trasduzione del segnale della
citochina, bloccando direttamente l’attività trascrizionale delle proteine Smad (Smad2,
Smad3, Smad4) (Lee et al., 2002; Murvai et al., 2004). La proteina E6 di HPV-18
invece è in grado di mediare la degradazione proteolitica di TIP-2/GIPC, proteina
coinvolta nell’espressione del recettore di TGF-β di tipo III a livello delle membrane
cellulari (Bonvin et al., 2005); queste azioni svolte dalle oncoproteine favoriscono il
processo di trasformazione cellulare associato a HPV.
L’impiego del saggio cellulare ad alta resa ha inoltre identificato due citochine,
IL-4 ed IL-13, dotate di un’alta attività inibitoria nei confronti della regione LCR di
HPV-16. Non essendo mai state descritte in letteratura come inibitori dell’attività
trascrizionale della sequenza LCR, IL-4 ed IL-13 sono state oggetto di analisi più
dettagliate. Entrambe le citochine riducono l’attività di LCR in modo dipendente dalla
concentrazione e la ID50 è risultata pari a 4 ng/ml per IL-4 e 11.3 ng/ml per IL-13.
Poiché la regione LCR guida la trascrizione dei geni E6 ed E7 nel genoma di
HPV, abbiamo utilizzato una RT Real Time PCR per verificare se la riduzione
dell’attività luciferasica indotta da IL-4 ed IL-13 nella linea cellulare reporter (HaCat)
riflettesse un’inibizione dei livelli dei trascritti di E6 ed E7 in una linea cellulare umana
derivata dal carcinoma della cervice uterina e positiva per HPV-16 (CaSki). Il saggio
RT-Real Time PCR ha evidenziato, in seguito al trattamento delle cellule CaSki con IL-
4, una inibizione del trascritto di E6 e E7 rispettivamente del 56,3% e del 62%, mentre
in seguito al trattamento delle cellule CaSki con IL-13, un’ inibizione del trascritto di
E6 e E7 rispettivamente del 50% e del 53%. Questi dati dimostrano che IL-4 ed IL-13
inibiscono la trascrizione mediata da LCR nel “contesto naturale”, validando il saggio
cellulare come strumento per identificare inibitori della trascrizione di E6 ed E7.

IL-4 ed IL-13 sono citochine strutturalmente e funzionalmente correlate, secrete
principalmente dai linfociti Th2 attivati; le due citochine condividono molte funzioni
biologiche ed esercitano una varietà di effetti immunoregolatori su differenti tipi
cellulari, quali linfociti B, monociti, cheratinociti, cellule dendritiche.
I recettori di IL-4 e IL-13 sono multimerici e condividono alcune componenti
recettoriali : la catena alfa del recettore dell’IL-4 (IL-4Rα) e la catena alfa 1 del
recettore dell’IL-13 (IL-13Rα1) possono eterodimerizzare e formare un recettore che
lega sia IL-4 sia IL-13 (Murata et al., 1999). Questo recettore, chiamato IL-13R o anche
IL-4R di tipo II, è espresso su numerosi tipi di cellule, sia emopoietiche sia di altro tipo,
ad eccezione di cellule T. L’associazione di IL-4Rα con IL-2R γc forma IL-4R di tipo I,
che viene espresso su cellule ematopoietiche e può legare solo IL-4 (Mueller et al.,
2002).
Le due citochine condividono anche la via di trasduzione del segnale, poiché è la
comune componente recettoriale IL-4Rα a presentare residui tirosin-chinasici
intracitoplasmatici a cui si associa la chinasi JAK in seguito alla dimerizzazione dei
complessi recettoriali. Nella forma fosforilata JAK recluta Stat6, che viene
successivamente fosforilato e dimerizza per migrare al nucleo e attivare promotori
bersaglio.
IL-4 ed IL-13 condividono diverse funzioni biologiche: intervengono come
mediatori delle reazioni allergiche promuovendo lo scambio isotipico verso la classe
IgE, aumentano l’espressione del complesso maggiore di istocompatibilità di classe II
sulle cellule e sotto-regolano l’espressione delle citochine proinfiammatorie secrete dai
monociti (Chomarat et al., 1998). Le due citochine hanno inoltre effetti
proinfiammatori su altri tipi di cellule ed in particolare aumentano l’espressione di IL-6
da parte di cellule endoteliali e cheratinociti (Derocq et al., 1994).
Finora IL-4 ed IL-13 non erano state considerate citochine antivirali, ma
recentemente è stata dimostrata la loro capacità di reprimere l’espressione di HIV in
monociti e macrofagi umani (Naif et al, 1997; Montaner et al., 1997). In uno studio
condotto su una linea cellulare di pro-monociti, IL-4 è in grado di inibire l’espressione

di HIV, indotta dagli esteri del forbolo, a livello trascrizionale (inibizione della regione
LCR) attraverso l’azione di NFkB. Si ritiene pertanto che IL-4 possa indurre uno stato
di senescenza cellulare in monociti infettati, impedendo l’espressione del virus in
seguito allo stimolo dato da citochine infiammatorie (Goletti et al., 2002).
Inoltre, è stata evidenziata la capacità di IL-4 di sopprimere l’espressione e la
replicazione del virus dell’Epatite B a livello trascrizionale in linee cellulare del
carcinoma epatocellulare (Lin et al., 2003).
Questi osservazioni, insieme ai risultati presentati in questa tesi, evidenziano un
ruolo emergente di IL-4 e IL-13 nella difesa dell’ospite contro le infezioni virali,
sebbene il loro significato clinico sia ancora da chiarire.
Concludendo, il saggio, oggetto di questa tesi, costituisce un valido strumento
per l’identificazione di composti inibenti la trascrizione delle oncoproteine E6 ed E7 di
HPV-16. Data la sua adattabilità ad un formato ad alta resa, il suo impiego può
concretamente facilitare la ricerca e lo sviluppo di farmaci per il trattamento di
carcinomi umani associati a HPV.
Il saggio cellulare ci ha permesso di eseguire la prima analisi sistematica degli
effetti esercitati dalle citochine sull’ attività trascrizionale della regione LCR di HPV-
16. I dati ottenuti rappresentano quindi una fonte di informazioni utili per guidare gli
studi futuri sul ruolo svolto dalle citochine nella “clearence” del virus dai tessuti infetti
e nella sorveglianza delle infezioni persistenti causate da HPV.
La scoperta di nuove citochine, IL-4 e IL-13, inibitorie dell’espressione di E6 ed
E7, giustifica inoltre indagini future finalizzate a verificare se il trattamento delle cellule
del carcinoma cervicale con queste citochine possa permettere il recupero dell’attività
degli oncosoppressori p53 e p105Rb ed il controllo della crescita cellulare.

BIBLIOGRAFIA Adam, E., Z. Berkova, Z. Daxnerova, J. Icenogle, W. C. Reeves, and R. H.
Kaufman. 2000. Papillomavirus detection: demographic and behavioral characteristics
influencing the identification of cervical disease. Am. J. Obstet. Gynecol. 182: 257-
264.
Aguilar-Lemarroy, A., S. Kirchhoff, N.J. Whitaker, P. Gariglio, H. zur Hausen,
P.H. Krammer, F. Rosl. 2001. Differential sensitivity of HPV 16 and HPV 18
positive cervical carcinoma cells to CD95-mediated apoptosis: complementation of a
CD95-sensitive phenotype by somatic cell hybridization. Int. J. Cancer. 93: 823–831.
Aguilar-Lemarroy, A., P. Gariglio, N.J. Whitaker, S.T. Eich- horst, H. zur
Hausen, P.H. Krammer, Thompson, D. A., V. Zacny, G.S. Belinsky, M. Classon,
D.L. Jones, R. Schlegel, K. Munger. 2001. The HPV E7 oncoprotein inhibits tumor
necrosis factor alpha-mediated apoptosis in normal human fibroblasts. Oncogene. 20:
3629–3640.
Altmann, A., I. Jochmus, F. Rosl. (1994). Intra- and extracellular control mechanisms
of human papillomavirus infection. Intervirology. 37: 180-188
Andrei, G., R. Snoek, D. Schols, and E. De Clerq. Induction of apoptosis by
cidofovir in human papillomavirus (HPV)-positive cells. 2001. Oncol. Res. 12: 397-
408.
Arany, I., KJ. Grattendick, WE. Whitehead, IA. Ember, SK. Tyring. 2003. A
functional interferon regulatory factor-1 (IRF-1)-binding site in the upstream
regulatory region (URR) of human papillomavirus type 16. Virology. 310 (2): 280-6.
Baldwin, A., L. Pirisi, KE. Creek. 2004. NFI-Ski interactions mediate Transforming
Growth Factor modulation of Human Papillomavirus type 16 early gene expression.
Journal of Virology. 78 (8): 3953-64.
Balmelli, C. Nasal immunization of mice with human papillomavirus type 16 virus-
like particles elicits neutralizing antibodies in mucosal secretions. 1998. J. Virol. 72:
8220-8229.
Basile, J. R., V. Zacny, K. Munger. 2001. The cytokines tumor necrosis factor-a
(TNF-a) and TNF-related apoptosis-inducing ligand differentially modulate
proliferation and apoptotic pathways in human keratinocytes expressing the human

papillomavirus-16 E7 oncoprotein. J. Biol. Chem. 276: 22522–22528.
Bedell, M. A., K. H., Jones, S. R., Grossman, and L. A. Laimins. 1989.
Identification of human papillomavirus type 18 transforming genes in immortalized
and primary cells. J. Virol. 63:1247–1255.
Beerheide, V., H. U. Bernard, Y. J. Tan. 1999. Novel screens for potential drugs
against cervical cancer identify zinc ejecting inhibitors of the HPV-16 E6 oncoprotein.
Journal of the national cancer institute. 91: 1211-20.
Berezutskaya, E., B. Yu, A. Morozov, P. Raychaudhuri, and S. Bagchi. 1997.
Differential regulation of the pocket domains of the retinoblastoma family proteins by
the HPV16 E7 oncoprotein. Cell Growth Differ. 8:1277–1286.
Bernard, H. U. 2004. Established and potential strategies against papillomavirus
infections. Journal of Antimicrobial Chemotherapy. 53: 137-139.
Bosch, F., M. M. Manos, N. Munoz, M. Sherman, A. M. Jansen, J. Peto, M. H.
Schiffman, V. Moreno, R. Kurman, K. V. Shah, and International Biological
Study on Cervical Cancer (IBSCC) Study Group. 1995. Prevalence of human
papillomavirus in cervical cancer: a worldwide perspective. J. Natl. Cancer Inst.
87:796–802.
Bosch, F. X., and S. de Sanjose. (2003). Chapter 1: Human papillomavirus and
cervical cancer-burden and assessment of casuality. J. Natl.Cancer. Inst. Monogr. 31:
3-13
Bonvin, AF., C. Reynaud, C. Kretz-Remy, P. Jalinot. 2005. Human Papillomavirus
type 18 E6 binds the cellular PDZ protein TIP-2/GIPC, which is involved in
transforming growth factor β signaling and triggers its degradation the proteasome.
Journal of Virology. 79 (7): 4229-37.
Braun, L., M. Dürst, R. Mikumo, and P. Gruppuso. Differential response of non
tumorigenic and tumorigenic human papillomavirus type 16-positive epithelial cells to
transforming growth factor- I. 1990. Cancer Res. 50: 7324-7332.
Brehm, A., E. A. Miska, D. J. McCance, J. L. Reid, A. J. Bannister, and T.
Kouzarides. 1998. Retinoblastoma protein recruits histone deacetylase to repress
transcription. Nature. 391:597–601.
Brehm, A., S. J. Nielsen, E. A. Miska, D. J. McCance, J. L. Reid, A. J., Bannister,
and T. Kouzarides. 1999. The E7 oncoprotein associates with Mi2 and histone
deacetylase activity to promote cell growth. EMBO J. 18:2449–2458.

Burd, E. M. Human Papillomavirus and cervical cancer. 2003. Clinical Microbiology.
16: 1-17.
Butz, K., C. Denk, A. Ullmann, M. Scheffner and F. Hoppe-Seyler. Induction of
apoptosis in human papillomavirus-positive cancer cells by peptide aptamers targeting
the viral E6 oncoprotein. 2000. Proc. Natl Acad. Sci. USA. 97: 6693-6697.
Butz, K., T. Ristriani, A. Hengstermann, C. Denk, M. Scheffner, F. Hoppe-Seyler.
2003. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-
positive cancer cells. Oncogene. 22: 5938-5945
Calore, E. E., S. M. M. Pereira, and M. J. Cavaliere. 2001. Progression of cervical
lesions in HIV-seropositive women: a cytological study. Diagn. Cytopathol. 24:117–
119.
Capone, RB., SI Pai, WM Koch, ML Gillison, HN Danish, WH Westra, R. Daniel,
KV Shah, D. Sidransky. 2000. Detection and quantitation of Human Papillomavirus
(HPV) DNA in the sera of patients with HPV-associated head and neck squamous cell
carcinoma. Clinical Cancer Research. 6: 4171-75.
Chellappan, S., V. B. Kraus, B. Kroger, K. Munger, P. M. Howley, W. C. Phelps,
and J. R. Nevins. 1992. Adenovirus E1A, simian virus 40 tumor antigen, and human
papillomavirus E7 protein share the capacity to disrupt the interaction between
transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci.
USA 89:4549–4553.
Chen, Z., P. Kamath, S. Zhang, L. St John, K. Adler-Storthz, E. J. Shillitoe.
(1996). Effects on tumor cells of ribozymes that cleave the RNA transcripts of human
papillomavirus type 18. Cancer Gene Ther. 3: 18-23
Chen, G., and A. Stenlund. 2000. Two patches of amino acids on the E2 DNA
binding domain define the surface for interaction with E1. J. Virol. 74:1506–1512.
Chomarat, P., J. Banchereau. 1998. Interleukin-4 and interleukin-13: their
similarities and discrepancies. Int. Rev. Immunol. 17: 1-52
Ciccolini, F., G. Di Pasquale, F. Carlotti, L. Crawford, and M. Tommasino. 1994.
Functional studies of E7 proteins from different HPV types. Oncogene 9:2633–2638.
Coker, A. L., R. B. Russell, S. M. Bond, L. Pirisi, Y. Liu, M. Mane, N. Kokorina,
T. Gerasimova, and P. L. Hermonat. 2001. Adeno-associated virus is associated with
a lower risk of high-grade cervical neoplasia. Exp. Mol. Pathol. 70:83–89.
Conger, K. L., J. S. Liu, S. R. Kuo, L. T. Chow, and T. S. Wang. 1999. Human

papillomavirus DNA replication. Interactions between the viral E1 protein and two
subunits of human dna polymerase alpha/primase. J. Biol. Chem. 274:2696–2705.
Craigo, J., M. Callahan, R. C. Huang, A. L. DeLucia. 2000. Inhibition of human
papillomavirus type 16 gene expression by nordihydroguaiaretic acid plant lignan
derivatives. Antiviral Res. 47 (1): 19-28.
Craven, S. E., and D. S. Bredt. 1998. PDZ proteins organize synaptic signaling
pathways. Cell. 93:495–498.
Cromme, F. V., J. Airey, M. T. Heemels, H. L. Ploegh, P.J. Keating, P. L. Stern,
C. J. Meijer, J. M. Walboomers. 1994. Loss of transporter protein, encoded by the
TAP-1 gene, is highly correlated with loss of HLA expression in cervical carcinomas.
J. Exp. Med. 179: 335–340.
DeFilippis, R. A., E. C. Goodwin, L. Wu, DiMaio D. 2003. Endogenous human
papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and
apoptosis in HeLa cervical carcinoma cells. J. Virol. 77: 1551-1563
Derocq, J. M., M. Segui, C. Poinot-Chazel, A. Minty, D. Caput, P. Ferrara, P.
Casellas. 1994. Interleukin-13 stimulates interleukin-6 production by human
keratinocytes. Similarity with interleukin-4. FEBS Lett. 343:32-6
Dolei, A., Papovavirus. Manuale di Virologia Medica. Seconda edizione. Ed. The
McGraw-Hill Companies. 2001. 205-215. [a cura di: Ferdinando Dianzani, Guido
Antonelli, Maria Rosaria Capobianchi, Antonina Dolei].
Duensing, S., and K. Munger. 2003. Human papillomavirus type 16 E7 oncoprotein
can induce abnormal centrosome duplication through a mechanism independent of
inactivation of retinoblastoma protein family members. J. Virol. 77:12331–12335.
Durst, M., FX Bosch, D. Glitz, A. Schneider, H. Zur Hausen. 1991. Inverse
relationship between human papillomavirus type 16 early gene expression and cell
differentiation in nude mouse epithelial cysts and tumors induced by HPV-positive
human cell lines. J. Virology. 65: 796-804.
Dyson, N., P. M. Howley, K. Munger, and E. Harlow. 1989. The human papilloma
virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science
243:934–937.
Edmonds, C., and K. H. Vousden. 1989. A point mutational analysis of human
papillomavirus type 16 E7 protein. J. Virol. 63:2650–2656.
Evander, M., I. H. Frazer, E. Payne. 1997. Identification of the alpha-6-integrin as a

candidate receptor for papillomaviruses. J Virol. 71(3): 2449–2456.
Fehrmann, F. and L. A. Laimins, 2003. Human papillomaviruses: targeting
differentiating epithelial cells for malignant transformation. Oncogene. 22: 5201-5207.
Filippova, M., H. Song, JL. Connolly, TS. Dermody, PJ. Duerksen-Hunghes. 2002.
The Human Papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and
protects cells from TNF-induced apoptosis. The journal of biological chemistry. 277
(24): 21730-39.
Finzer, P., A. Aguilar-Lemarroy, F. Rosl. 2002. The role of human papillomavirus
oncoproteins E6 and E7 in apoptosis. Cancer Letter. 188: 15-24.
Finzer, P., C. Kuntzen, H. zur Hausen, and F. Rösl. Inhibitors of histone deacetylase
arrest cell cycle and induce apoptosis in cervical carcinoma cells circumventing human
papillomavirus oncogene expression. 2001. Oncogene. 20: 4768-4776.
Finzer, P., C. Kuntzen, U. Soto, H. zur Hausen, F. Rosl. 2001. Inhibitors of histone
deacetylase arrest cell cycle and induce apoptosis in cervical carcinoma cells
circumventing human papillomavirus oncogene expression. Oncogene. 20: 4768–4776.
Flores, E., B. L. Allen-Hoffman, D. Lee, and P. F. Lambert. 2000. The human
papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life
cycle. J. Virol. 74:6622–6631.
Fontaine V., E. van der Meijden, J. ter Schegget. 2001. Inhibition of human
papillomavirus-16 long control region activity by interferon-gamma overcome by p300
overexpression. Mol. Carcinog. 31: 27-36
Frame, M. C. 2002. Src in cancer: deregulation and consequences for cell behaviour.
Biochim. Biophys. Acta. 1602:114–130.
Funk, J. O., S. Waga, J. B. Harry, E. Espling, B. Stillman, and D. A. Galloway.
1997. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is
blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 11:2090–2100.
Gavrieli, Y., Y. Sherman, S.A. Ben-Sasson. 1999. Identification of programmed cell
death in situ via specific labeling of nuclear DNA fragmentation. J. Cell. Biol. 119:
493–501.
Giannoudis, A., and C. S. Herrington. 2001. Human papillomavirus variants and
squamous neoplasia of the cervix. J. Pathol. 193:295–302.
Giroglou, T., L. Florin, F. Schafer. 2001. Human papillomavirus infection requires
cell surface heparan sulfate. J Virol. 75(3): 1565–1570.

Goletti, D., A. L. Kinter, E. M. Coccia, A. Battistini, N. Petrosillo, G. Ippolito, G.
Poli. 2002. Interleukin (IL)-4 inhibits phorbol-ester induced HIV-1 expression in
chronically infected U1 cells independently from the autocrine effect of endogenous
tumor necrosis factor-α, IL-1β and IL-1 receptor antagonist. Citokine. 17: 28-35.
Goodwin, E. C., E. Yang, C. J. Lee, H. W. Lee, DiMaio D., E. S. Hwang. (2000).
Rapid induction of senescence in human cervical carcinoma cells. PNAS USA. 97:
10978-10983
Goodwin, E. C., and D. DiMaio. (2000). Repression of human papillomavirus
oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant
tumor suppressor pathways. PNAS USA. 97: 12513-12518
Grimm, D. and J. A. Kleinschmidt. Progress in adeno-associated virus type 2 vector
production: promises and prospects for clinical use. 1999. Hum. Gene Ther. 10: 2445-
2450.
Harro, C. D. Safety and immunogenicity trial in adult volunteers of a human
papillomavirus 16 L1 virus-like particle vaccine. 2001. J. Natl Cancer Inst. 93: 284-
292.
Hart, P. H., C. S. Bonder, J. Balogh, H. L. Dickensheets, N. Vazquez, K. V. Davies,
J. J. Finlay-Jones, R. P. Donnelly. 1999. Diminished responses to IL-13 by human
monocytes differentiated in vitro: role of the IL-13Ralpha1 chain and STAT6. Eur. J.
Immunol. 29: 2087-2097
Hashida, T., and S. Yasumoto. 1991. Induction of chromosome abnormalities in
mouse and human epidermal keratinocytes by the human papillomavirus type 16 E7
oncogene. J. Gen. Virol. 72:1569–1577.
Hasskarl, J., K. Butz, N. Whitaker, A. Ullmann, M. Durst, F. Hoppe-Seyler. 2000.
Differential cell cycle response of nontumorigenic and tumorigenic human
papillomavirus-positive keratinocytes towards transforming growth factor-beta 1. J.
Mol. Med. 78(2): 94-101
Hawley-Nelson, P., K. H. Vousden, N. L. Hubbert, D. R. Lowy, and J. T. Shiller.
1989. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin
keratinocytes. EMBO J. 8: 3905-3910
Hegde, R. S., S. R. Grossman, L. A. Laimins, and P. B. Sigler. 1992. Crystal
structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its
DNA target. Nature. 359:505 512.

Hershey, GK. 2003. IL-13 receptors and signaling pathways: an evolving web.
Journal of allergy clinical immunology. 11 (4): 677-85.
Ho, G. Y. Persistent genital human papillomavirus infection as a risk factor for
persistent cervical dysplasia. 1995. J. Natl Cancer Inst. 87: 1345-1347.
Howley, P. M. and D. R. Lowy. Papillomaviruses and their replication. In: Fields, B.
N., Knipe D. M., Howley, P. M., editors. Fields Virology, Philadelphia (PA):
Lippincott-Raven; 2001. 2197-2229.
Huibregtse, J. M., M. Scheffner, and P. M. Howley. 1993. Cloning and expression
of the cDNA for E6-AP, a protein that mediates the interaction of the human
papillomavirus E6 oncoprotein with p53. Mol. Cell. Biol. 13:775–784.
Hummel, M., J. B. Hudson, and L. A. Laimins. 1992. Differentiation-induced and
constitutive transcription of human papillomavirus type 31b in cell lines containing
viral episomes. J. Virol. 66:6070–6080.
Hurford, R. K., Jr., D. Cobrinik, M. H. Lee, and N. Dyson. 1997. pRB and
p107/p130 are required for the regulated expression of different sets of E2F responsive
genes. Genes Dev. 11:1447–1463.
Johnson, A. N., S. J. Newfeld. 2002. The TGF-beta family: signaling pathways,
developmental roles, and tumor suppressor activities. Scientific World Journal. 2: 892-
925
Jones, D. L., D. Thompson, K. Munger. 1997. Destabilization of the RB tumor
suppressor and stabilization of p53 contribute to HPV 16 type 16 E7-induced
apoptosis. Virology. 239: 97–107.
Joyce, J. G., J. S. Tung, C. T. Przysiecki, J. C. Cook, E. D. Lehman, J. A. Sands,
K. U. Jansen, and P. M. Keller. 1999. The L1 major capsid protein of human
papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-
surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 274:5810–5822.
Lowy, D. R. and P. M. Howley. Papillomaviruses. In: Fields, B. N., Knipe D. M.,
Howley, P. M., editors. Fields Virology, Philadelphia (PA): Lippincott-Raven; 2001.
2231-2264.
Kang S. H., K. Won, H. W. Chung, H. S. Jong, Y. S. Song, S. J. Kim, Y. J. Bang,
N. K. Kim. (1998). Genetic integrity of trasforming growth factor beta (TGF-beta)
receptors in cervical carcinoma cell lines: loss of growth sensitivity but conserved
transcriptional response to TGF-beta. Int. J. Cancer. 77: 620-625

Kao, W. H., S. L. Beaudenon, A. L. Talis, J. M. Huibregtse, and P. M. Howley.
2000. Human papillomavirus type 16 E6 induces self-ubiquitination of the E6AP
ubiquitin-protein ligase. J. Virol. 74:6408–6417.
Kim, C. Y., M.H. Tsai, C. Osmanian, T.G. Graeber, J.E. Lee, R.G. Giffard, J.A.
DiPaolo, D.M. Peehl, A.J. Giaccia. 1997. Selection of human cervical epithelial cells
that possess reduced apoptotic potential to low-oxygen conditions. Cancer Res. 57:
4200–4204.
Kim, KY., L. Blatt, MW. Taylor. 2000. The effects of interferon on the expression of
human papillomavirus oncogenes. Journal of General Virology. 81: 695-700.
Kiyono, T., A. Hiraiwa, M. Fujita, Y. Hayashi, T. Akiyama, and M. Ishibashi.
1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human
homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad.
Sci. USA 94:11612–11616.
Kiyono, T., S. A. Foster, J. I. Koop, J. K. McDougall, D. A. Galloway, and A. J.
Klingelhutz. 1998. Both Rb/p16INK4a inactivation and telomerase activity are
required to immortalize human epithelial cells. Nature 396:84–88.
Kleter, B., L. J. van Doorn, L. Schrauwen, A. Molijn, S. Sastrowijoto, J.
TerSchegget, J. Lindeman, B. Ter Harmsel, M. Burger, and W. Quint. 1999.
Development and clinical evaluation of a highly sensitive PCR-reverse hybridization
line probe assay for detection and identification of anogenital human papillomavirus. J.
Clin. Microbiol. 37:2508–2517.
Kyo, S., M. Inoue, N. Hayasaka, T. Inoue, M. Yutsudo, O. Tanizawa, A. Hakura.
(1994). Regulation of early gene expression of human papillomavirus type 16 by in
inflammatory cytokines. Virology. 200: 130-139
Lee, DK., BC. Kim, IY. Kim, E. Cho, DJ. Satterwhite, SJ. Kim. 2002. The Human
Papilloma Virus E7 oncoprotein inhibits Transforming Growth Factor signaling by
blocking binding of the Smad complex to Its target sequence. The journal of biological
chemistry. 277 (41): 38557-64.
Lin, SJ., PJ. Shu, C. Chang, AK. Ng, C. Hu2. 2003. IL-4 suppresses the expression
and the replication of Hepatitis B Virus in the hepatocellular carcinoma cell line
Hep3B1. The Journal of Immunology. 171: 4708–16.
Liu, J. P. 1999. Studies of the molecular mechanisms in the regulation of telomerase
activity. FASEB J. 13:2091–2104.

Liu, J. S., S. R. Kuo, A. M. Makhov, D. M. Cyr, J. D. Griffith, T. R. Broker, and
L. T. Chow. 1998. Human Hsp70 and Hsp40 chaperone proteins facilitate human
papillomavirus-11 E1 protein binding to the origin and stimulate cell-free DNA
replication. J. Biol. Chem. 273:30704–30712.
Liu, Y., J. J. Chen, Q. Gao, S. Dalal, Y. Hong, C. P. Mansur, V. Band, and E. J.
Androphy. 1999. Multiple functions of human papillomavirus type 16 E6 contribute to
the immortalization of mammary epithelial cells. J. Virol. 73:7297–7307.
Longworth, M. S. and L. A. Laimins, 2004. Pathogenesis of Human
Papillomaviruses in Differentiating Epithelia. Microbiology and Molecular Biology.
68: 362-372.
Longworth, M. S., and L. A. Laimins. 2004. The binding of histone deacetylases and
the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle
of human papillomavirus type 31. J. Virol. 78: 3533–3541.
Maehama, T. Selective down-regulation of human papillomavirus transcription by 2-
deoxyglucose. 1998. Int. J. Cancer. 76: 639-646.
Magnusson, P. K. E., P. Lichtenstein, and U. B. Gyllenstein. 2000. Heritability of
cervical tumors. Int. J. Cancer 88:698–701.
Malejczyk, M., J. Jozwiak, A. Osiecka, PI. Roszkowski, W. Mazurkiewicz-
Smoktunowicz, TT. Rogozinski, L. Walczak, S. Jablonska, S. Majewski, J.
Malejczyk. 1997. Serum levels of soluble tumor-necrosis-factor receptors in patients
with benign and malignant HPV-associated anogenital lesions. Int J Cancer. 73 (1):
16-9.
McIntyre, M. C., M. N. Ruesch, and L. A. Laimins. 1996. Human papillomavirus E7
oncoprotein bind a single form of cyclin E in a complex with cdk2 and p107. Virology.
215: 73-82
Mizono, K., P. ten Dijke, C. H. Heldin. 2000. TGF-beta signalling by Smad proteins.
Adv. Immunol. 75: 115-57
Montaner, LJ., RT. Bailer, S. Gordon. 1997. IL-13 acts on macrophages to block the
completion of reverse transcription, inhibit virus production, and reduce virus
infectivity. Journal Leukocite Biology. 62 (1): 126-32.
Muderspach, L. A phase I trial of human papillomavirus (HPV) peptide vaccine for

women with high-grade cervical and vulvar intraepithelial neoplasia who are HPV 16
positive. 2000. Clin. Cancer Res. 6: 3406-3416.
Mueller, T.D., J. L. Zhang, W. Sebald, A. Duschl. 2002. Structure, binding, and
antagonists in the IL-4/IL-13 receptor system. Biochim. Biophys Acta. 1592: 237-50
Munger, K., B. Werness, N. Dyson, W. Phelps, E. Harlow, and P. Howley. 1989.
Complex formation of human papillomavirus E7 proteins with the retinoblastoma
tumor suppressor gene product. EMBO J. 8:4099–4105.
Munger, K., W. C. Phelps, V. Bubb, P. M. Howley, and R. Schlegel. 1989. The E6
and E7 genes of the human papillomavirus type 16 together are necessary and
sufficient for transformation of primary human keratinocytes. J. Virol. 63:4417–4421.
Murata, T., J. Tagushi, R. K. Puri, H. Mohri. 1999. Sharing of receptor subunits and
signal transduction pathway between the IL-4 and IL-13 receptor system. Int. J.
Hematol. 69: 13-20
Murvai, M., AA. Borbély, J. Kònya, L. Gergely, G. Veress. 2004. Effect of human
papillomavirus type 16 E6 and E7 oncogenes on the activity of the trasforming growth
factor-β2 (TGF-β2) promoter. Archives Virology. 149: 2379-92.
Naylor, L. H., 1999. Reporter gene technology: the future looks bright. Biochemical
Pharmacology. 58: 749-757.
New, D. C., Y. H. Miller-MartiniWong, 2003. Reporter gene assay and their
applications to bioassays of natural products. Phytotherapy research. 17: 439-448.
Nguyen, D. X., T. F. Westbrook, and D. J. McCance. 2002. Human papillomavirus
type 16 E7 maintains elevated levels of the cdc25A tyrosine phosphatase during
deregulation of cell cycle arrest. J. Virol. 76:619–632.
Nguyen, M. L., M. M. Nguyen, D. Lee, A. E. Griep, and P. F. Lambert. 2003. The
PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for
E6’s induction of epithelial hyperplasia in vivo. J. Virol. 77: 6957–6964.
Okamoto, A., C. D. Woodworth, K. Yen, J. Chung, S. Isonishi, T. Nikaido, T.
Kiyokawa, H. Seo, Y. Kitahara, K. Ochiai, and T. Tanaka. 1999. Combination
therapy with podophyllin and vidarabine for human papillomavirus positive cervical
intraepithelial neoplasia. Oncol. Rep. 6:269–276.
Ongini, E. Ricerca e sviluppo di nuovi farmaci. Farmacologia generale e molecolare

Seconda edizione. Ed. UTET, 2001. 18- 39. [a cura di Francesco Clementi e Guido
Fumagalli].
Ozbun, M., C. Meyers. 1996. Transforming Growth Factor b1 induces differentiation
in Human Papillomavirus-positive keratinocytes. Journal of Virology. 70 (8): 5437-46.
Park, J. S., E. J. Kim, H. J. Kwon, E. S. Hwang, S. E. Namkoong, and S. J. Um.
2000. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV
E7 oncoprotein. implication for the E7-mediated immune evasion mechanism in
cervical carcinogenesis. J. Biol. Chem. 275:6764– 6769.
Phelps, W. C., C. L. Yee, K. Munger, and P. M. Howley. 1988. The human
papillomavirus type 16 E7 gene encodes transactivation and transformation functions
similar to those of adenovirus E1A. Cell. 53:539–547.
Pim, D., A. Storey, M. Thomas, P. Massimi, and L. Banks. 1994. Mutational
analysis of HPV-18 E6 identifies domains required for p53 degradation in vitro,
abolition of p53 transactivation in vivo and immortalisation of primary BMK cells.
Oncogene 9:1869–1876.
Plunkett, M. J. and Ellman, J. N., 1997. La chimica combinatoriale e i nuovi farmaci.
Le Scienze quaderni. 102: 16-21.
Riley, R. R., S. Duensing, T. Brake, K. Munger, P. F. Lambert, and J. M. Arbeit.
2003. Dissection of human papillomavirus E6 and E7 function in transgenic mouse
models of cervical carcinogenesis. Cancer Res. 63:4862–4871.
Roberts, A. B. 1999. TGF-beta signalling from receptors to the nucleus. Microbes
Infect. 1: 1265-73
Roden, R. B., D. R. Lowy, and J. T. Schiller. 1997. Papillomavirus is resistant to
dessication. J. Infect. Dis. 176:1076–1079.
Rosl, F. 2001. Restoration of p53 expression sensibilize human papillomavirus type 16
immortalized human keratinocytes to CD95-mediated apoptosis. Oncogene. 21: 165–
175.
Rosl, F., B. C. Das, M. Lengert, K.Geletneky, and H. zur Hausen. 1997.
Antioxidant-induced changes of the AP-1 transcription complex are paralleled by a
selective suppression of human papillomavirus transcription. J. Virol. 71: 362-370.
Rosl, F., K. Kleine-Lowinski, H. zur Hausen. 1999. The possible role of chemokines
in HPV-linked carcinogenesis, in: B.J. Rollins (Ed.), Chemokines and Cancer, Humana
Press Inc, Totoxa, NJ, pp. 207–225.

Saez, E., SE. Rutberg, E. Muller, H. Oppenheim, J. Smoluk, SH. Yuspa. 1995. c-
fos is required for malignant progression of skin tumors. Cell. 82: 721-32.
Sailaja, G., RM. Watts, HU Bernard. 1999. Many different papillomaviruses have
low transcriptional activity in spite of strong epithelial specific enhancers. Journal of
General Virology. 80: 1715-24.
Sanclemente, G. and D. K. Gill, 2002. Human papillomavirus molecular biology and
pathogenesis. JEADV. 16: 231-240.
Saunders, N., H. I. Frazer. 1998. Simplifying the molecular mechanisms of human
papillomavirus. Dermatol Clin. 16(4): 823–827.
Scheffner, M., K. Munger, J. C. Byrne, P. M. Howley. 1991. The state of the p53
and retinoblastoma genes in human cervical carcinoma cell lines. PNAS USA. 88:
5523-5527
Scheffner, M., J. M. Huibregtse, and P. M. Howley. 1994. Identification of a human
ubiquitin-conjugating enzyme that mediates the E6-AP-dependent ubiquitination of
p53. Proc. Natl. Acad. Sci. USA 91:8797–8801.
Sheets, E. E., N. M. Constantine, S. Sinisco, B. Dean, and E. S. Cibas. 1995.
Colposcopically-directed biopsies provide a basis for comparing the accuracy of
ThinPrep and Papanicoloau smears. J. Gynecol. Technol. 1:27–34.
Shimizu, A., M. Kato, A. Nakao, T. Imamura, P. ten Dijke, C. H. Heldin, M.
Kawabata, S. Shimada, K. Miyazono. 1998. Identification of receptors and Smad
proteins involved in activin signalling in a human epidermal keratinocyte cell line.
Genes Cells. 3: 125-34
Snoeck, R., J. C. Noel, C. Muller, E. De Clercq, and M. Bossens. 2000. Cidofovir, a
new approach for the treatment of cervix intraepithelial neoplasia grade III (CIN III). J.
Med. Virol. 60: 205–209.
Steger, G., and S. Corbach. 1997. Dose-dependent regulation ofte early promoter of
HPV 18 by the viral E2 protein. J. Virol. 71:50–58.
Suzich, J. A. Systemic immunization with papillomavirus L1 protein completely
prevents the development of viral mucosal papillomas. 1995. Proc. Natl Acad. Sci.
USA. 92: 11553-11557. infection. J. Virol. 69, 3959-3963 (1995).
Swan, D. C., R. A. Tucker, G. Tortolero-Luna, M. F. Mitchell, L. Wideroff, E. R.
Unger, R. A. Nisenbaum, W. C. Reeves, and J. P. Icenogle. 1999. Human papilloma
(HPV) DNA copy number is dependent on grade of cervical disease and HPV type. J.

Clin. Microbiol. 37:1030–1034.
Thomas, J. T., W. G. Hubert, M. N. Ruesch, and L. A. Laimins. 1999. Human
papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of
episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad.
Sci. USA 96:8449–8454.
Um, SJ., EJ. Kim, ES. Hwang, SJ. Kim, SE. Namkoong, JS. Park. 2000.
Antiproliferative effects of retinoic acid/interferon in cervical carcinoma cell lines:
cooperative growth suppression of IRF-1 and p53. Int J Cancer. 85(3):416-23.
Underwood, M. R., L. M. Shewchuk, A. M. Hassell. 2000. Searching for antiviral
drugs for human papillomaviruses. Antiviral Therapy. 5: 229-42.
van Wilson, G., M. West, K. Woytek and D. Rangasamy. 2002. Papillomavirus E1
proteins: form, function and features. Virus Genes. 24: 275-290.
Veldman, T., X. Liu, H. Yuan, and R. Schlegel. 2003. Human papillomavirus E6 and
Myc proteins associate in vivo and bind to and cooperatively activate the telomerase
reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA. 100:8211–8216.
Vieira, KB., DJ. Goldstein, LL. Villa. 1996. Tumor necrosis factor alpha interferes
with the cell cycle of normal and papillomavirus-immortalized human keratinocytes.
Cancer Res. 56(10): 2452-7..SYMPOSIUM IN
Villa, L. L., H. U. Bernard, M. Kast, A. Hildesheim, G. Amestoy, E. L. Franco.
2002. Past, present and future of HPV research: highlights from the 19th international
papillomavirus conference-HPV 2001. Virus Res. 89: 163-173.
von Knebel Doeberitz, M., C. Rittmüller, H. zur Hausen, and M. Dürst, 1992.
Inhibition of tumorigenicity of cervical cancer cells in nude mice by HPV E6-E7
antisense RNA. Int. J. Cancer. 51: 831-834.
Walczak, H., P.H. Krammer. 2000. The CD95 (APO-1/Fas) and the TRAIL (APO-
2L) apoptosis system. Exp. Cell Res. 256: 58–66.
Wells, S. I., D. A. Francis, A. Y. Karpova, J. J. Dowhanick, J. D. Benson, P. M.
Howley. 2000. Papillomavirus E2 induces senescence in HPV-positive cells via pRB-
and p21(CIP)-dependent pathways. EMBO J. 19: 5762-5771
Woodworth, CD., V. Notario, JA DiPaolo. 1990. Trasforming growth factors beta 1
and 2 trascriptionally regulate human papillomavirus type 16 early gene expression in

HPV-immortalized human genital epithelial cells. J. Virology. 64: 4767-75.
Zhang, L. F. HPV 6b virus like particles are potent immunogens without adjuvant in
man. 2000. Vaccine. 18: 1051-1058.
zur Hausen, H. 1982. Human genital cancer: synergism between two virus infections
and or synergism between a virus infection and initiating events? Lancet. ii:1370–
1372.
Zur Hausen, H. 2000. Papillomavirus causing cancer: evasion from host-cell control
in early events in carcinogenesis. Journal of the National Cancer Institute. 92: 690-98
zur Hausen, H. 2002. Papillomaviruses and cancer: from basic studies to clinical
application. Nat. Rev. Cancer. 2:342–350.