




Presentation Download · PDF fileUSP-NF unless superseded by a chapter or monograph ... USP...
127
Transcript of Presentation Download · PDF fileUSP-NF unless superseded by a chapter or monograph ... USP...


USP-NF Up to Date Activities
Compendial Initiatives
Robert Femia, Ph.D.
Senior Vice President
Science, Chemical Medicines and General Chapters

Introduction

5
Works with more than
900 scientists,
practitioners, and
regulators to revise
standards that help
protect public health
Values-driven
organization focused
on empowering its
staff and volunteers
About USP
Internationally
recognized and
globally focused
More than 900
employees worldwide
Laboratory facilities in
U.S., India, China,
Brazil, and Ghana
Offices in Switzerland,
Ethiopia, Indonesia, the
Philippines, and
Nigeria
Founded in 1820,
nonprofit, private,
independent, and
self-funded
USP is cited in US
Law
US FDA (product
approval)
USP Documentary
Standard and
Reference Standards
(product compliance
in US)
Mission: USP’s mission is to improve global health through
public standards and related programs that help ensure the
quality, safety, and benefit of medicines and foods.

6
USP: The Organization
What We Do:
Establish and disseminate public written standards for the
quality, purity, identity, strength, and labeling of medicines
Provide physical Reference Standards to support tests and
assays in the USP–NF and FCC
Educate producers, practitioners, and others seeking
information on quality and USP standards
Verify products for manufacturers and award
the USP Verified Mark to those that meet
our standards
Work with international health agencies
to improve the quality of medicines
Worldwide
Work with FDA and Industry Stakeholders

FDA reviews proposed standards in Pharmacopeial
Forum and provides comments
Participates as delegates, provides resolutions,
provides members to the council of the Convention
and other Convention committees
Provides FDA liaisons to USP’s expert committees
Participates in workshops and stakeholder forums
Works with USP through cooperative research and
development agreements
Collaborates with USP through quarterly meetings,
pharmacopeial discussion group and special topics
such as OTC monograph modernization,
compounding, structured product labeling,
pharmacologic classes
7
FDA Engagement Goes Beyond Enforcing USP Standards
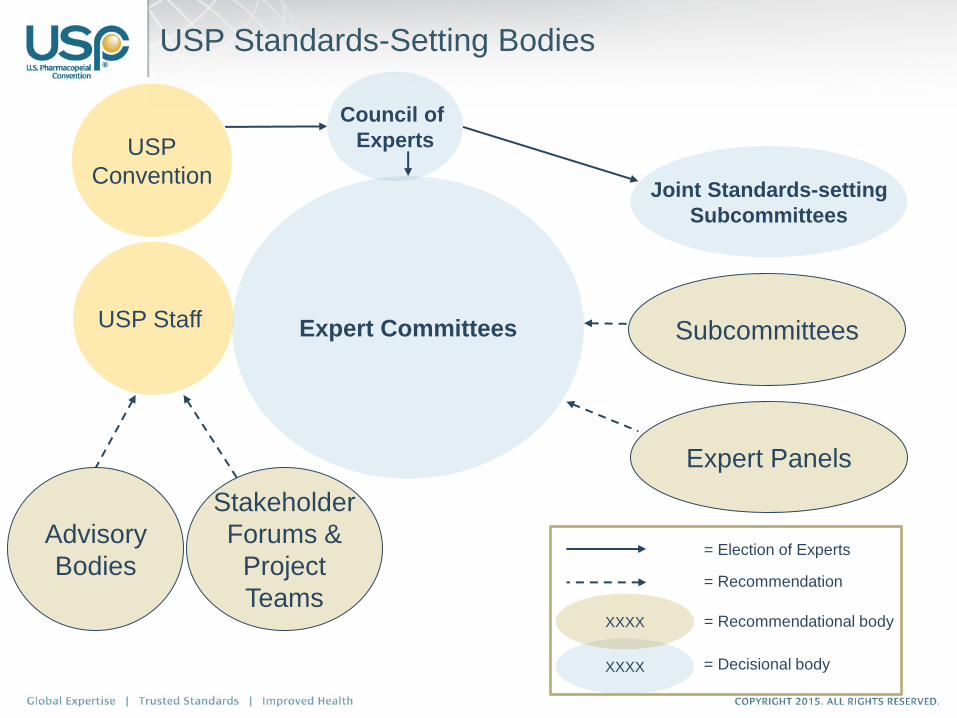
USP Standards-Setting Bodies
Expert Committees
Advisory
Bodies
Stakeholder
Forums &
Project
Teams
USP Staff
Expert Panels
Subcommittees
Joint Standards-setting
Subcommittees
USP
Convention
XXXX
XXXX
Council of
Experts
= Recommendation
= Recommendational body
= Decisional body
= Election of Experts

724 volunteers serving on 24 Expert Committees, 58 Expert Panels
‒ 415 Expert Committee members
‒ 309 Expert Panel members*
144 Government Liaisons from FDA
‒ CDER: 99 ‒ ORA: 4
‒ CFSAN: 8 ‒ OC/ONDQA: 3
‒ CBER: 9 ‒ CDRH: 1
‒ CVM: 20
8 other Government Liaisons (US and international)
Industry and consultants comprise almost 70% of volunteers
35% EC members are new, 25% are international
876 Participants in Standards-setting Activities
* Does not include Expert Committee members also serving on Expert Panels

USP CONVENTION MEMBERS Kazakhstan (1) Uzbekistan (1)
Russia (2) Belarus (1)
Ukraine (2) Canada (11)
Sweden (1)
Belgium (2)
U.K. (2) Switzerland (2)
France (1)
U.S. (388) Turkey (1) Japan (1)
South Korea (1)
Taiwan (1)
China (3)
Vietnam (1)
Philippines (1)
Morocco (1)
India (4)
Jordan (2)
Mexico (3) Colombia (1)
Thailand (1)
Sierra Leone (1)
Ghana (1) Peru (2)
Egypt (1) Indonesia
(1) Saudi Arabia (1)
Nigeria (1)
Ethiopia (1) Brazil (7) Chile (1)
Tanzania (1) Uganda (1) Australia (1) Argentina (3)
Total Categories* Countries Delegates at the 2015
INTERNATIONAL REPRESENTATION *COMPOSITION BY CATEGORY
15%
7%
4%
2%
72%
7 17 33 68
Nongovernmental Standards-Setting and Conformity Assessment Bodies
Consumer and Other Organizations
Representing the Public Interest
Manufacturer, Trade, and Affiliated
Associations
Governmental Bodies or Divisions or Associations Thereof
Health Practitioner Professional and Scientific Associations
Academic Institutions and
Associations Thereof
86% 1% EUROPE
1%
NORTH AMERICA
4% LATIN AMERIC SUB-SAHARAN
AFRICA AND CARIBBEAN
2% 2% 1% 137 196
NEWLY INDEPENDENT STATES
EAST ASIA MIDDLE EAST AND NORTH AFRICA
w w w.usp.org LEG101G_2015 -10
PR
AC
TITI
ON
ER
S
A
S
O
2% WORLD
SU
1% UTH ASIA
30%
42%
FAST FACTS 458 Members 6 (based on Bylaws) 39 Represented 262 Convention Meeting

11
Council of Experts
6 Collaborative Groups and 24 Expert Committees

USP Standard Setting
Process

13
Science is the Base of USP Standard Setting

General Notices, General Chapters,
and
Monographs
• General Notices – contain requirements applicable throughout
USP-NF unless superseded by a chapter or monograph
• General Chapters
– Contain requirements applicable to multiple monographs referencing
the specific general chapter(s)
– General Chapter requirements supersede General Notice
requirements
• Below <1000> - Enforceable Chapters (when referenced in
General Notices, General Chapters or individual monographs)
• Above <1000> - Information Chapters
• Monograph
– Requirements are specific to the article
– Monograph requirements supersede General Notices and General
Chapter requirements.

New monographs:
Proposed tests, limits, and validation (according to <1225>)
Packaging, storage, and labeling requirements
Reference Standard commitments
Revisions:
Reasonable justification
Adequate supporting data for methods and specifications showing
improvement
USP-initiated revisions:
Modernizations and other revisions can be based on other
pharmacopoeias’ standards, ICH specifications, etc.
Types of Standards Development

Standards-setting Process: How to Develop a Monograph
Monograph development is initiated
Scientific Liaison performs technical review and drafts monograph
USP evaluates procedures requiring RS prior to publication and
RS collaborative testing (optional)
Proposal is published for 90-day public comment period
Scientific Liaison and Expert Committee reviews comments
Expert Committee ballots to adopt proposal
Monograph is published in USP-NF and becomes official six
months after publication unless otherwise indicated
Manufacturer submits proposal USP initiates development
Approved Not Approved Next
steps?
3-6
m*
15-1
8m
*
*Shorter if Accelerated Revision

General Chapters describe the basics of commonly used
analytical techniques or common requirements and how they
apply in compendial articles.
Originated by need (to avoid repetition of a test in
monographs); input from stakeholders; ECs or EPs; Staff, etc.
Early input is requested from stakeholders through USP
website announcement (“Prospectus”)
Stimuli articles and workshops maybe used to collect input and
asses impact of new or revised general chapters
EC’s workplans are accessible through www.usp.org
General Chapters Development Process

– Monograph Development
– Bulk Procurement – Donation from pharmaceutical industry
– Purchase
– In-house custom synthesis
– Protocol & Test Kit Preparation
– Bulk Sampling/Sample Shipping
– Collaborative Testing – USP Laboratories, Contract Laboratories, Agencies,
Industrial Laboratories
– Data Analysis/Value Assignment
– Internal Review/Approval
How Does USP Develop a Reference Standard?

19
Need for Ref
Std
Hand-off Meeting (RMA,
Science, RSE)
RSS Drafts PPD,
TP, TK
Testing Labs
Results received
from labs
Results analyzed
by RSS
RSS generates
RSCEP
RSCEP Peer
review (Stage 2)
OK Peer review
comment
Internal ballot QA
review (Stage 3)
Packaging
QA batch record review
(complete, accurate,
compliant)
QA/marketing Release
Testing issue
Additional testing if required
Comments to be addressed
Approved
Not approved
RSE Reference Standard Development Process
Collecting info (Documents)
Stage 1 Stage 2
Stage 3
RMA – Reference Material Acquisition
RSE – Reference Standard Evaluation
PPD – Product Planning Document
TP – Test Protocol
TK – Test Kit
RSS – Reference Standard Scientist
RSCEP – Reference Standard Candidate Evaluation Package

20
Uses of USP Reference Standards
There are two main types of USP Reference Standards:
Standards with Quantitative Applications
Assays (for drug substances and for formulations)
Limit tests (e.g., Impurity Reference Standards)
Standards with only Qualitative Applications
Identification tests
Elution markers
System Suitability tests

Up-to-Date Activities

22
Core Compendial Programs
The United States Pharmacopeia and
the National Formulary (USP–NF)
Food Chemicals Codex (FCC)
USP Dietary Supplements
Compendium (DSC)
Reference Standards
Other Resources
Pharmacopeial Forum (PF)
FCC Forum
USP Dictionary
Chromatographic Columns

23
United States Pharmacopeia – National Formulary
A compendia of internationally recognized standards for drug substances, dosage
forms, compound preparations, excipients, and dietary supplements.
It contains:
More than 4,900 monographs with specifications for identity, purity, quality, and
strength.
More than 320 General Chapters, with step-by-step tests and methods.
Standards that when deemed official by USP, are FDA-enforceable
for drugs manufactured or marketed in the U.S.

24
Expert Committees Therapeutic Category
Number of
Official
Monographs
Total
Number of
Volunteers
% new
volunteers
Chemical
Medicines 1
Antiviral, Antimicrobials
& Antibiotics 977 15 53
Chemical
Medicines 2
Cardiovascular, Cough,
Cold, & Analgesic 678 16 38
Chemical
Medicines 3
Gastrointestinal, Renal,
Endocrine and
Ophthalmology, Oncology,
Dermatology & Veterinary
959 17 41
Chemical
Medicines 4
Psychiatric, Psychoactive,
Neuromuscular, Aerosol, &
Imaging Drugs 799 18 39
Chemical
Medicines 5 Pulmonary & Steroids 570 16 38
Chemical
Medicines 6 Over the Counter (OTC) 475 18 27
Chemical Medicines – Demographics

2005: Initial evaluation of its documentary standards (in
USP-NF) identified ~700 outdated monographs.
2010: FDA established a Monograph Modernization Task
Group that interfaced with the USP Monograph
Modernization to address this need.
- The goal of the Task Group was to identify monographs
in need of modernization, especially regarding outdated
analytical methodologies.
2010-2015: Multiple approaches—apart from relying
solely on method submissions from industry
sponsors—have been used in developing modernized
monographs.
USP Monograph Modernization History
25

USP-NF Up to Date
USP Convention Resolution 2:
USP will meet the needs of U.S. Food and Drug Administration (FDA), industry, and other stakeholders for modern monographs within USP–NF. USP will work to: • eliminate the existing backlog of monographs in need of
modernization, and • proactively evaluate and update monographs to maintain their
relevance given scientific advances and evolving manufacturing and regulatory approaches.
USP will work with industry and FDA to explore new strategies for sharing analytical methods and specifications needed to modernize monographs.
26

27
New Strategy Aligned With Existing Mission
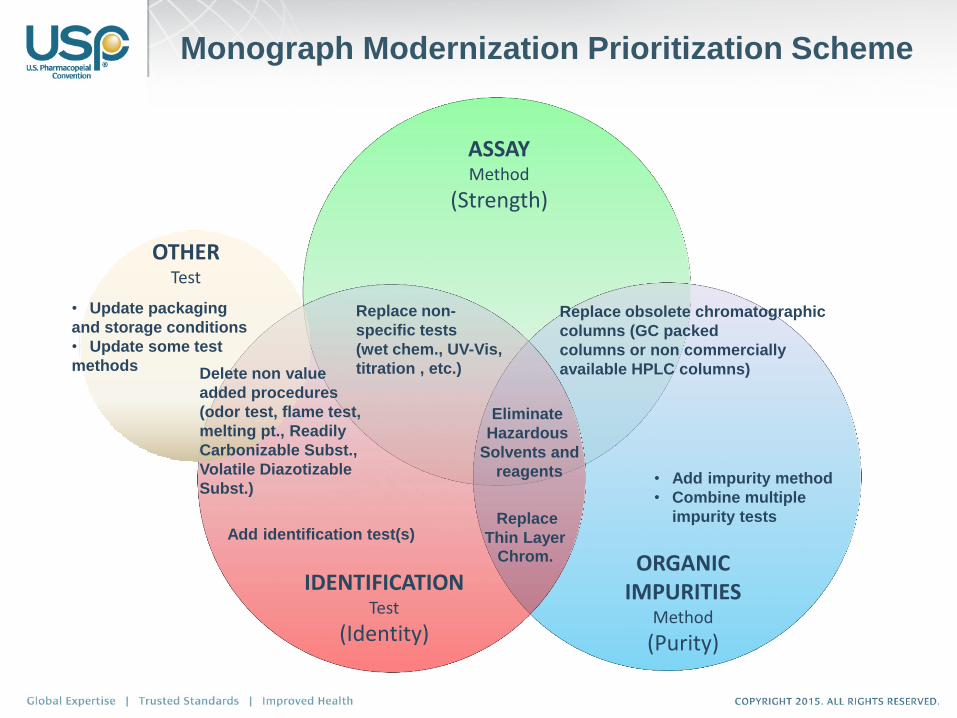
ASSAY Method
(Strength)
ORGANIC IMPURITIES
Method
(Purity)
Replace obsolete chromatographic
columns (GC packed
columns or non commercially
available HPLC columns)
• Add impurity method
• Combine multiple
impurity tests
Eliminate
Hazardous
Solvents and
reagents
Replace non-
specific tests
(wet chem., UV-Vis,
titration , etc.)
Add identification test(s) Replace
Thin Layer Chrom.
IDENTIFICATION Test
(Identity)
Monograph Modernization Prioritization Scheme
OTHER Test
• Update packaging
and storage conditions
• Update some test
methods Delete non value
added procedures
(odor test, flame test,
melting pt., Readily
Carbonizable Subst.,
Volatile Diazotizable
Subst.)

Add Identification Test
Add Organic Impurities
Replace TLC with HPLC or UHPLC
Replace outdated equipment (i.e., not commercially available)
Eliminate hazardous procedures and materials such as solvents, reagents,
and other chemicals
Replace non-specific tests with stability indicating procedures
Delete non value added procedures
Replace organoleptic tests
Provide alternative procedures (e.g. <197K. or <197A>)
Update on Reference Standards
Up-to-date
Omission
Reagents tagging
Additional monograph information (e.g., chemical structure, chemical
name. etc.)
Chapter dependencies and changes in General Notices
Update Description and Solubility
Monograph Modernization - Identification,
Assay, Organic Impurities, Other Sections
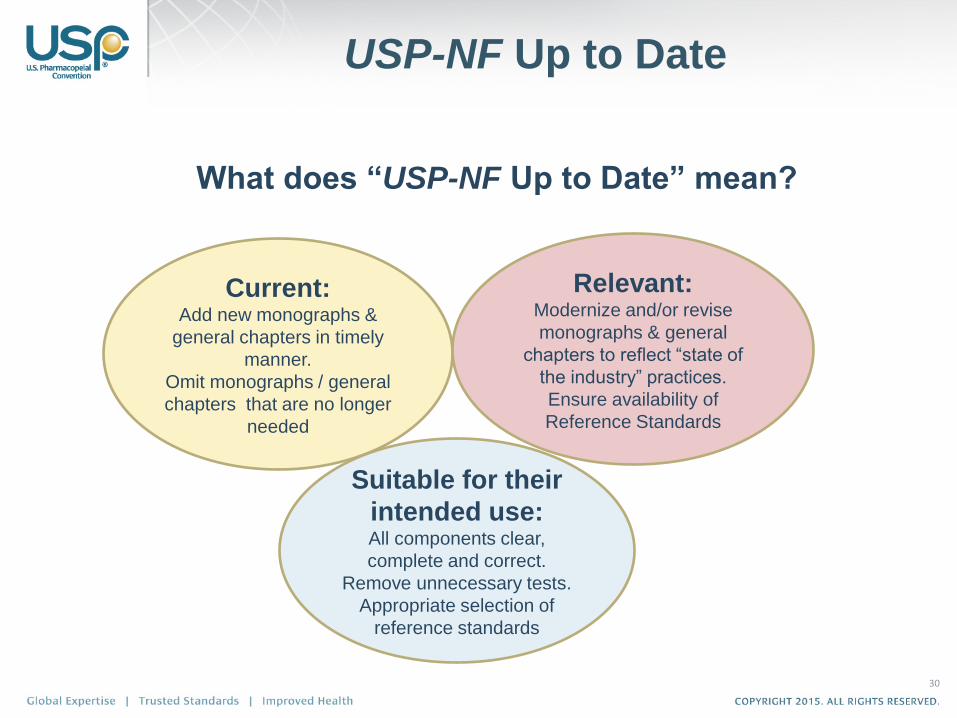
USP-NF Up to Date
30
What does “USP-NF Up to Date” mean?
Current: Add new monographs &
general chapters in timely
manner.
Omit monographs / general
chapters that are no longer
needed
Suitable for their
intended use: All components clear,
complete and correct.
Remove unnecessary tests.
Appropriate selection of
reference standards
Relevant: Modernize and/or revise
monographs & general
chapters to reflect “state of
the industry” practices.
Ensure availability of
Reference Standards
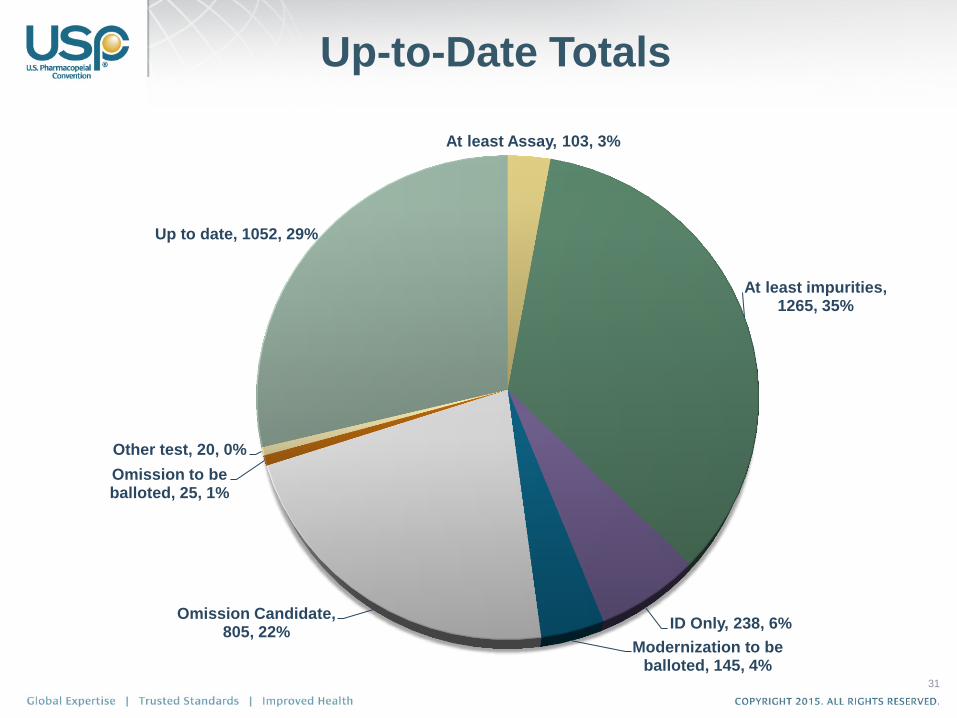
31
At least Assay, 103, 3%
At least impurities, 1265, 35%
ID Only, 238, 6%
Modernization to be balloted, 145, 4%
Omission Candidate, 805, 22%
Omission to be balloted, 25, 1%
Other test, 20, 0%
Up to date, 1052, 29%
Up-to-Date Totals
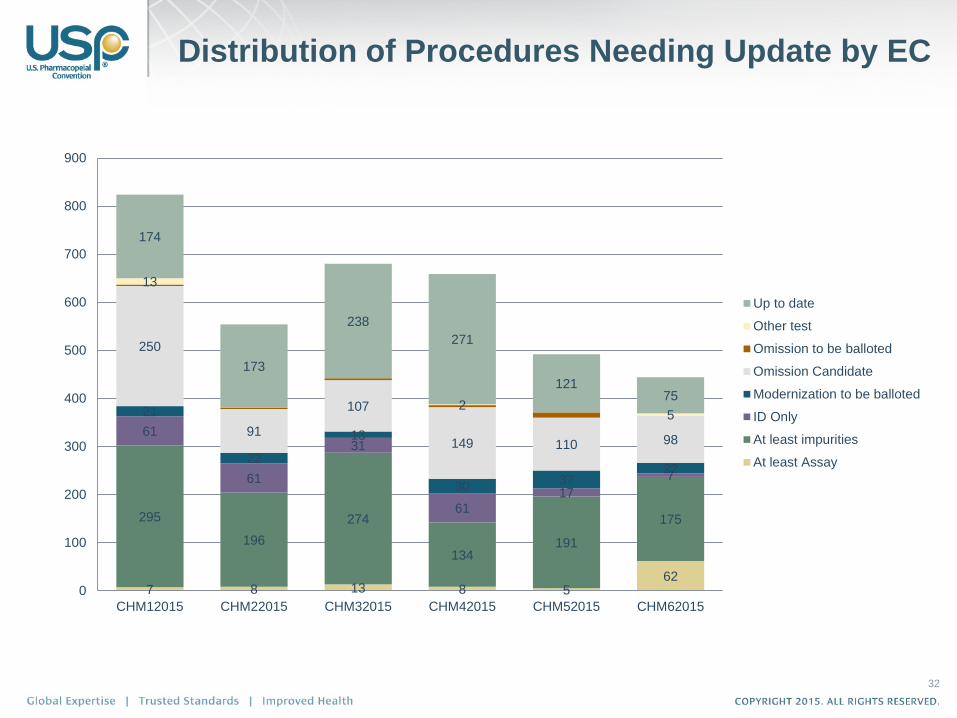
Distribution of Procedures Needing Update by EC
32
7 8 13 8 5 62
295
196
274
134 191
175
61
61
31
61 17
7
21
22
13
30 37
22
250
91
107
149 110 98
13
2 5
174
173
238
271
121 75
0
100
200
300
400
500
600
700
800
900
CHM12015 CHM22015 CHM32015 CHM42015 CHM52015 CHM62015
Up to date
Other test
Omission to be balloted
Omission Candidate
Modernization to be balloted
ID Only
At least impurities
At least Assay

• Traditional donor model (‘externally sourced’)
• Need to engage sponsors
• USP laboratories (‘internally sourced’)
• Extensive testing facilities for procedure development
• Laboratory sites in US, India, China and Brazil
• FDA (CRADA ORA Labs; OTC Working Group)
• Trade organizations (CHPA; Excipient Trade Association)
• Expert panels to leverage industry expertise, gain early stakeholder input and buy-in
• Other Pharmacopeias
• Identify alternate source procedures (i.e., procedures that can be used from other pharmacopeias)
USP-NF Up to Date Strategies
USP-NF Up to Date Opportunities and Challenges
• Opportunities:
• Stakeholder collaborations and global expert panels can stimulate additional avenues for both update and harmonization.
• Sourcing procedures from other compendia, literature, etc.
• Use of global USP laboratory facilities to develop and validate procedures.
• With FDA involvement, prioritizing and requesting submissions - the hope is that industry is much more likely to submit a proposal.
• Challenges:
• Prioritization of monographs and chapters in need of updating
• Obtaining procedures and acceptance criteria from sponsors
• Balancing the need to introduce modern methodology with the feasibility to implement by users globally

Global Health
Monographs

The Global Health Standards Program: Objectives
36
USP Mission: To improve global health through public standards and related programs
that help ensure the quality, safety and benefit of medicines and foods.
USP Vision: USP envisions a world in which all have access to high quality, safe, and
beneficial medicines and foods.
Global Health Standards Program Objectives: The global health standards
program’s objectives are to:
• Ensure availability of relevant, modern standards for the world’s most essential
medicines;
• Enable accessibility of these standards in non-US settings; and
• Engage stakeholders for development, adoption, and implementation of these
standards for improved public health outcomes

Prepared by Gaurvika Nayyar 37
GHS Program will focus on Medicines of global health
importance Marketed Outside the United States

The Global Health Monographs will have a New Section in
the USP-NF
38
New section of USP-NF
Starting USP 40-NF35
Section clearly distinguished
and designated to indicate non-
US status
Section Preface will explain the
regulatory status of GH monographs
Preface
“This section contains monographs for
articles which are not currently legally
marketed in the United States, but
which have been approved by a
stringent regulatory authority as defined
by the World Health Organization and
are used for essential purposes in other
parts of the world. Selection and
prioritization of new entries to this
section will be accomplished in close
collaboration with stakeholders
throughout the global health
community. These monographs are not
applicable to articles marketed for use
in the United States.”

Over the Counter
Monographs

40
Shared Goals … but difficult to balance
FDA
• Public Safety (maximize)
• Unambiguous enforceable specifications
• Referee method(s)
• U.S. scope, participate globally

Chemical Medicines 6 Expert Committee
Prescription Non-Prescription Drug Stakeholder Forum (OTC Team) – FDA/CHPA/USP
USP OTC Team – FDA OTC WG
Acetaminophen Expert Panel
CHPA
USP Industry Collaboration with HPLC Column Manufacturers
41
OTC Focused Teams

Three meetings conducted: April 6, July 1, and Sept. 7
Discussion topics include:
1. Update on Monograph Modernization list
2. GC <321> and GC <327>
Currently finalizing CDA to work collaboratively on OTC
projects
Case Study - To work on two diphenhydramine
monographs
42
USP OTC Team – FDA OTC WG
Collaboration

43
Prioritization Need Help from FDA,
Industry Stakeholders,
USP Expert Panels

Continuous Manufacturing
45
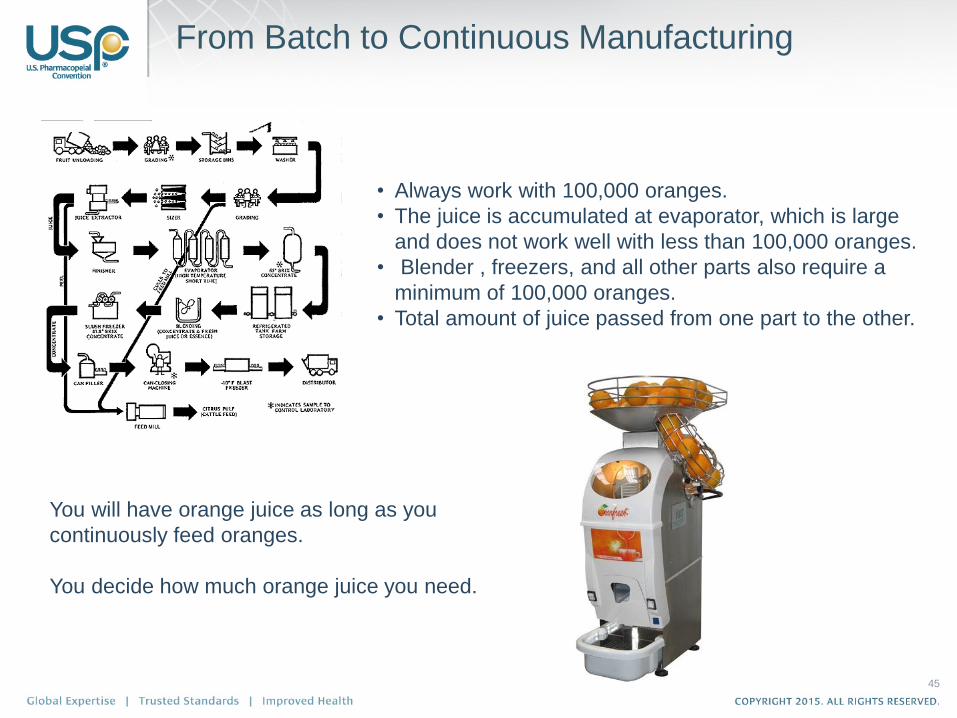
From Batch to Continuous Manufacturing
• Always work with 100,000 oranges.
• The juice is accumulated at evaporator, which is large
and does not work well with less than 100,000 oranges.
• Blender , freezers, and all other parts also require a
minimum of 100,000 oranges.
• Total amount of juice passed from one part to the other.
You will have orange juice as long as you
continuously feed oranges.
You decide how much orange juice you need.

46
Continuous Manufacturing---Why?

47
Control Challenges of Continuous Manufacturing
1. Material Control • Characterization of in-coming materials
• In-process measurements
• Selection of sampling frequency
• In-process parameters and material attributes
• Setting of appropriate acceptance criteria
2. System Control • System design
• Equipment integration
• Verification
3. Process Control • Analytical tools to the control system to support implementation of feed-back or feed-forward
control
• Sophisticated data management tools
• Defining representative sampling to consistently assure product quality over time
• Location of sampling probes
• Sample size and sampling frequency
• Need for enhanced process understanding
• Availability of mechanistic models for all processing steps
• Implementation of multivariate analysis for determination of product quality
4. Product release control

Potential USP roles in Pharmaceutical Continuous
Manufacturing development
• Control strategy standardization
• Ingredient characterization standardization (monograph and
reference standard)
• Product release standardization
• Equipment/system requirement standardization
• Sensing technology standardization
• System modeling standardization
• Other?
48

49
Interest in Continuous Manufacturing from FDA is Strong
50
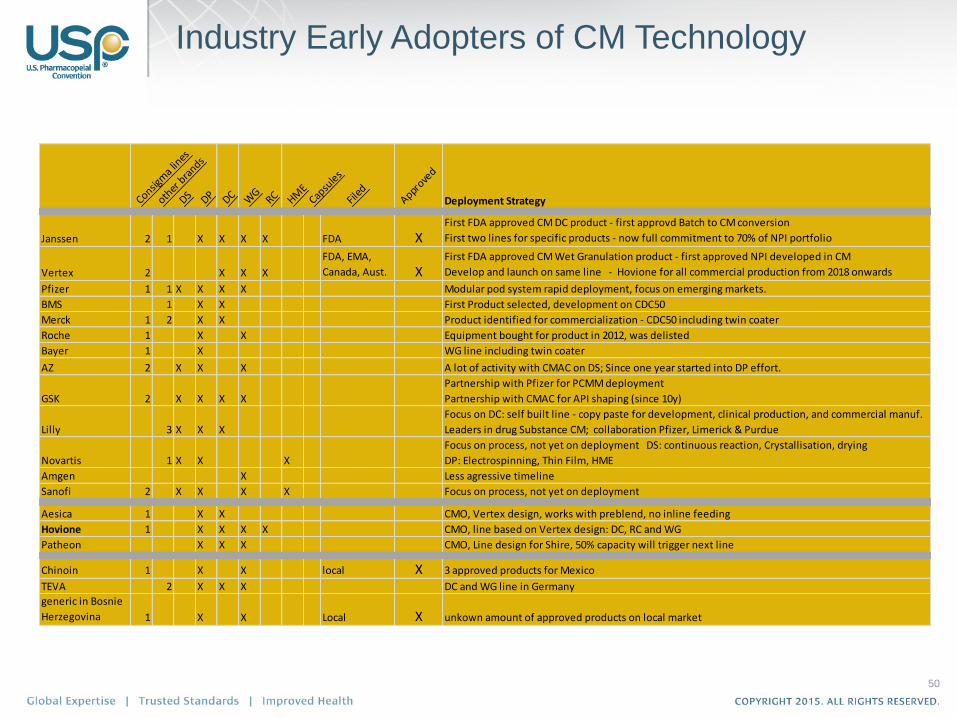
Industry Early Adopters of CM Technology
Consigm
a lin
es
other
bra
nds
DS DP DC WG
RC HME
Capsu
les
Filed
Approve
d
Deployment Strategy
Janssen 2 1 X X X X FDA XFirst FDA approved CM DC product - first approvd Batch to CM conversion
First two lines for specific products - now full commitment to 70% of NPI portfolio
Vertex 2 X X X
FDA, EMA,
Canada, Aust. XFirst FDA approved CM Wet Granulation product - first approved NPI developed in CM
Develop and launch on same line - Hovione for all commercial production from 2018 onwards
Pfizer 1 1 X X X X Modular pod system rapid deployment, focus on emerging markets.
BMS 1 X X First Product selected, development on CDC50
Merck 1 2 X X Product identified for commercialization - CDC50 including twin coater
Roche 1 X X Equipment bought for product in 2012, was delisted
Bayer 1 X WG line including twin coater
AZ 2 X X X A lot of activity with CMAC on DS; Since one year started into DP effort.
GSK 2 X X X X
Partnership with Pfizer for PCMM deployment
Partnership with CMAC for API shaping (since 10y)
Lilly 3 X X X
Focus on DC: self built line - copy paste for development, clinical production, and commercial manuf.
Leaders in drug Substance CM; collaboration Pfizer, Limerick & Purdue
Novartis 1 X X X
Focus on process, not yet on deployment DS: continuous reaction, Crystallisation, drying
DP: Electrospinning, Thin Film, HME
Amgen X Less agressive timeline
Sanofi 2 X X X X Focus on process, not yet on deployment
Aesica 1 X X CMO, Vertex design, works with preblend, no inline feeding
Hovione 1 X X X X CMO, line based on Vertex design: DC, RC and WG
Patheon X X X CMO, Line design for Shire, 50% capacity will trigger next line
Chinoin 1 X X local X 3 approved products for Mexico
TEVA 2 X X X DC and WG line in Germany
generic in Bosnie
Herzegovina 1 X X Local X unkown amount of approved products on local market

Monograph Prioritization

1. Objective, readily available, and relevant measurement factors
2. Usable output – getting all factors down to one priority index
score
3. Customized to the program unit
4. Priority is set at the start of the process and carried through the
lifecycle
Requirements
52

Priority Index Score
53
WS * S
WS Sustainability Weighting
WI Public Health Impact DALY Weighting
WI-Exp Public Health Impact Exposure Risk Weighting
S Sustainability Factor- Normalized
I Public Health Impact DALY Factor- Normalized
I-Exp Public Health Impact Exposure Risk Factor- Normalized
PI Priority Index Score
*All inputs will be normalized to
one universal scale
WI * I PI WI-Exp * I-Exp

1. The inputs are value measures as determined by our current portfolio
i.e. – Use data on what drives the need for RS to as one decision point.
2. Mission- provide insight on monographs that are needed for mission that might not generate $ but will enable improvements in public health
3. Urgency- increase responsiveness to market by considering time to LOE (and time elapsed from LOE).
Chemical Medicines Objective: Increase Availability to Market
54

How does Monograph Volume Relate to API in the US?

How does Monograph Volume Relate to API Worldwide?
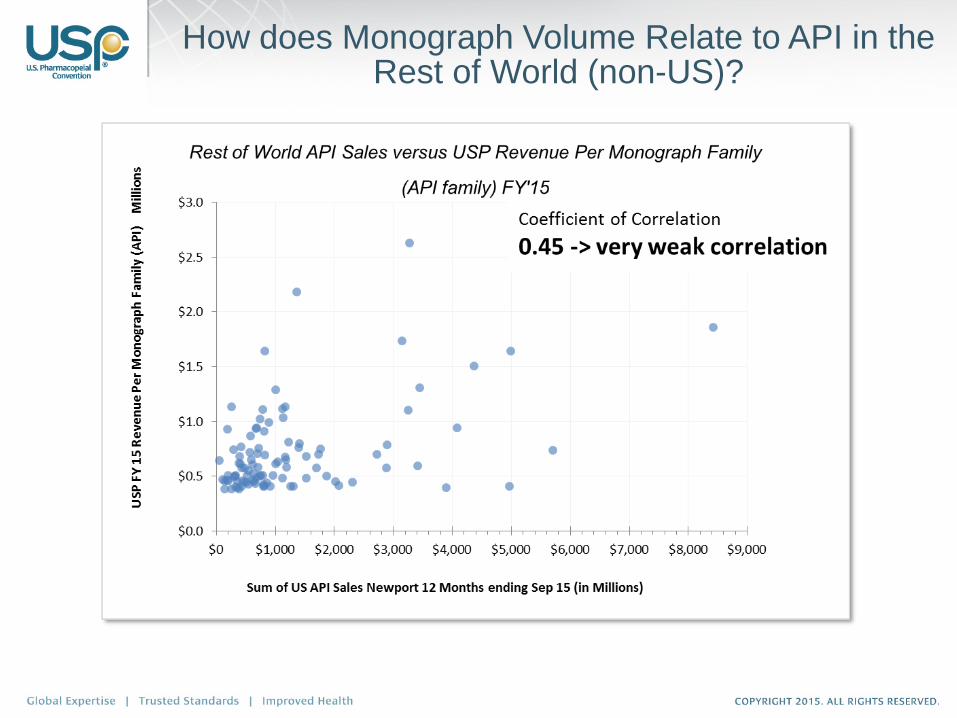
How does Monograph Volume Relate to API in the Rest of World (non-US)?

USP Donations

Many USP monographs need to be updated to modern analytical
methodologies
Replace organoleptic tests, TLC impurity tests, etc.
Addition of a dissolution test, specified impurities with different limits, etc.
FDA approved product not covered by current monograph
(USP won’t know unless you tell us)
Why submit a monograph revision?
59

Need to protect what is seen as trade secrets/intellectual property
Lack of resources within a company to devote to providing
monographs/materials
Product which may have long since been genericized
being seen as very low value to devote any resources
Can’t connect how USP fits into the overall
global public health interest
Too far in advance of patent expiry to donate
60
What We’ve Heard – Barriers to Donating

If a standard is based on another method, you will still need to comply
with the standard. Why shouldn’t that standard be based on your
company’s science?
As the foundation for a USP standard, your science will be viewed by
industry peers as the “science of choice” for establishing quality.
If your company does not have the resources to devote to providing
monographs and materials, there are other ways to be involved:
– Providing comments to a developing standard through Pharmacopeial Forum
– Applying to serve on an Expert Committee or Panel
61
What We’ve Learned – The Benefits

Without quality standards, consequences include:
– Adverse public health events
– Inadequate treatment
– Therapeutic failure
– Breakdown in trust in the health system
By working with USP in advance of your loss of exclusivity, you can get
a jump-start in trying to establish a standard that can be harmonized
with other pharmacopeias.
– Harmonized standards can help eliminate/minimize redundant testing
Your company can mitigate risks associated with noncompliance.
If a USP standard is based on your company’s method, your company
can save time and resources.
62
What We’ve Learned – The Benefits

What would personally motivate you to partner with USP on this effort?
Your company?
What are the biggest barriers for you to participate? For your company?
How can we overcome these barriers?
How important is it to your company to be publicly recognized for its
commitment to quality?
What kind of information and outreach does your company need from
USP in order to make your ability to donate easier?
If you donate, what kind of input would your company want from USP
regarding the status of the standard in development?
63
Question & Answer



USP Chapters <232> and <233>
Implementation Strategy
Kahkashan Zaidi, Ph.D.
Principal Scientific Liaison
Science, General Chapters

Delete <231> Heavy Metals
Over 1200 references in the USP-NF
Introduce Three New Chapters: 1. <232>Elemental Impurities—Limits (Official But Not Implemented)
2. <2232>Elemental Contaminants in Dietary Supplements (Official
But Not Implemented )
3. <233> Elemental Impurities—Procedures (Official)
USP’s Approach
68

<231> Heavy Metals <231> Deletion Date o Jan 1, 2018
Publish Omission of General
Chapter <231>
o Published in USP 38–NF 33 with an
official date of December 1, 2015
USP to publish/Post list of
monographs and Chapters with
cross reference to <231>
o Accomplished---July 2014 and Jan
14, 2015
Delete cross-references to General
Chapter <231> Heavy metals from all
individual monographs
o Accomplished---USP 38 and 39 and
following publications with delayed
implementation on Jan 1, 2018
69

<231> Heavy Metals
70

<232> Harmonization with Q3D---Today
USP 39
Q3D USP <232>
Scope Harmonized Harmonized
(Exception: TPNs)
List of Elements 24 15
Not Included: Tl, Au, Se,
Co, Ba, Sn, Li, Sb and Ag
PDEs Harmonized For 15
Elements
Harmonized For 15
Elements
Other Routes Harmonized
Harmonized
Options 4 options
3 options
Implementation Harmonized Harmonized
71

Table 1: Elemental Impurities for Drug Products USP 39--Official
Element Oral Daily
Dose PDE
(µg/day)
Parenteral
Daily Dose PDE
(µg/day)
Inhalational
Daily Dose
PDE (µg/day)
Inorganic Arsenic 15 15 2
Cadmium 5 2 2
Lead 5 5 5
Inorganic
Mercury
30 3 1
Chromium 11000 1100 3
Copper 3000 300 30
Molybdenum 3000 1500 10
Nickel 200 20 5
Palladium 100 10 1
Platinum 100 10 1
Vanadium 100 10 1
Osmium 100 10 1
Rhodium 100 10 1
Ruthenium 100 10 1
Iridium 100 10 1 72

Table 2. Example Concentration Limits for Components of
Drug Products with a 10-g Maximum Daily Dose
Elements
Concentration Limits (µg/g) for Components
Used in Oral Drug Products
Concentration Limits
(µg/g) for Components
Used in
Parenteral Drug Products
Concentration Limits
(µg/g) for Components
Used In
Inhalation Drug Products
Cadmium 0.5 0.2 0.2
Lead 0.5 0.5 0.5
Inorganic arsenica 1.5 1.5 0.2
Inorganic mercurya 3 0.3 0.1
Iridium 10 1 0.1
Osmium 10 1 0.1
Palladium 10 1 0.1
Platinum 10 1 0.1
Rhodium 10 1 0.1
Ruthenium 10 1 0.1
Chromium 1100 110 0.3
Molybdenum 300 150 1
Nickel 20 2 0.5
Vanadium 10 1 0.1
Copper 300 30 3
a See Speciation section.
73

Key changes Proposed in PF 42(2)
Requirements/language for Drug Substance
and excipients
Tables 1 & 3 (previously Table 2) revised to
add additional elements
Added a new section and new table (Table 2)
to clarify risk assessment
Analytical testing
Format changes
74

<232> Revision in PF 42(2)
Drug substances and Excipients
The limits presented in this chapter do not apply to excipients and drug substances, except where specified in an individual monograph. However, elemental impurity levels present in drug substances and excipients must be known, documented, and made
available upon request.▪However, manufacturers of pharmaceutical products need
certain information about the content of elemental impurities in drug substances or excipients in order to meet the criteria of this chapter. Drug product manufacturers can use elemental impurity test data on components from tests performed by drug substance or excipient manufacturers, who may provide test data, or if applicable, risk assessments. Elemental impurity data generated by a qualified supplier of drug product components are acceptable for use by a drug product manufacturer to demonstrate compliance with this chapter in the final drug product. Drug substance or excipient manufacturers who choose to perform a risk assessment must conduct that risk assessment using Table 2 in this chapter. Elements that are inherent in the nature of the material, as in the case of some naturally-sourced materials, must be considered in the risk assessment.▪1S (USP40)
75
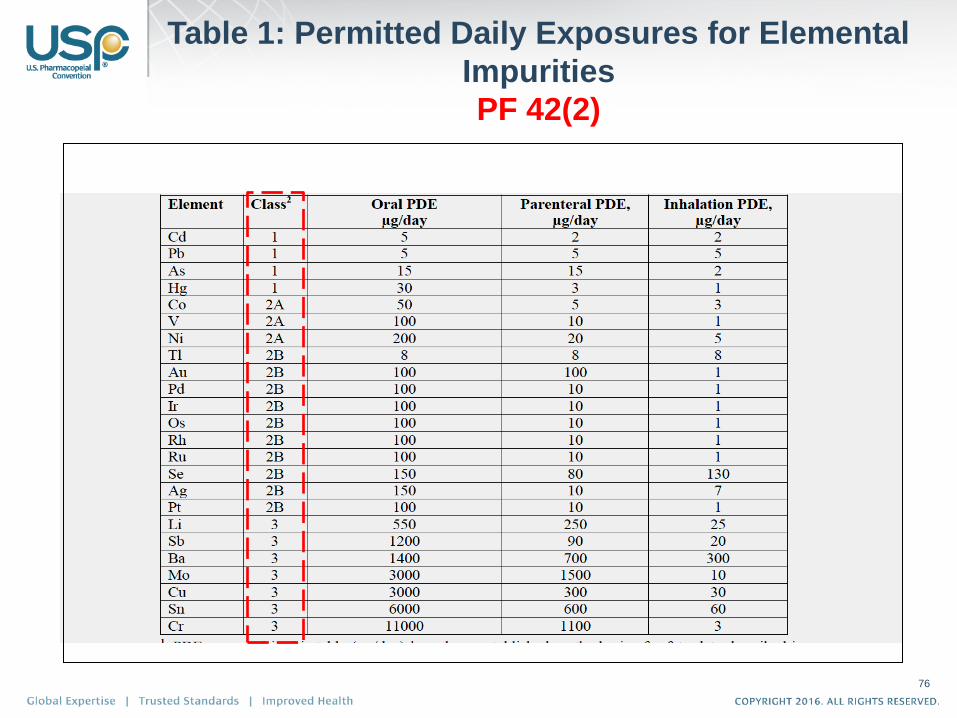
Table 1: Permitted Daily Exposures for Elemental
Impurities
PF 42(2)
76
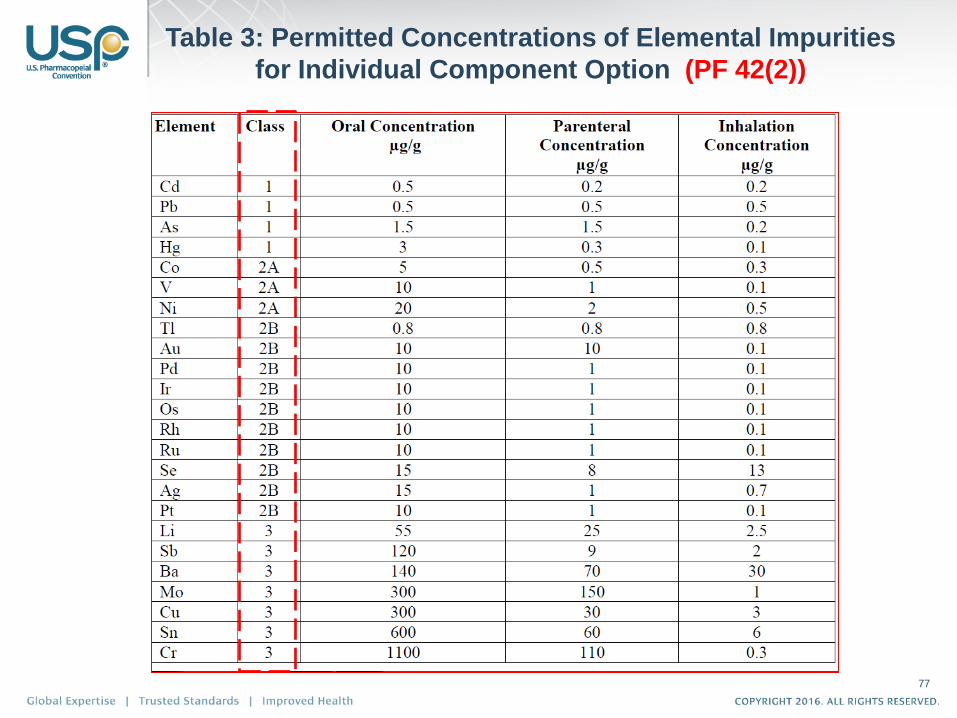
Table 3: Permitted Concentrations of Elemental Impurities
for Individual Component Option (PF 42(2))
77

Table 2: Elements to be Considered in
the Risk Assessment
Element Class
If Intentionally Added
(All Routes)
If Not Intentionally Added
Oral Parenteral Inhalation
Cd 1 yes yes yes yes
Pb 1 yes yes yes yes
As 1 yes yes yes yes
Hg 1 yes yes yes yes
Co 2A yes yes yes yes
V 2A yes yes yes yes
Ni 2A yes yes yes yes
Tl 2B yes no no no
Au 2B yes no no no
Pd 2B yes no no no
Ir 2B yes no no no
Os 2B yes no no no
Rh 2B yes no no no
Ru 2B yes no no no
Se 2B yes no no no
Ag 2B yes no no no
Pt 2B yes no no no
Li 3 yes no yes yes
Sb 3 yes no yes yes
Ba 3 yes no no yes
Mo 3 yes no no yes
Cu 3 yes no yes yes
Sn 3 yes no no yes
Cr 3 yes no no yes
78

<232> Elemental Impurities
• ANALYTICAL TESTING
– If, by process monitoring and supply-chain
control, manufacturers can demonstrate
compliance, then further testing may not be
needed. When testing is done to
demonstrate compliance, proceed as
directed in Elemental Impurities—Procedures
⟨233⟩. and minimally include arsenic,
cadmium, lead, and mercury in the Target
Elements evaluation.▪▪1S (USP40)
79

Harmonization with Q3D---Future
Q3D USP <232>
Scope Harmonized Harmonized
(Exception: TPNs)
List of Elements Harmonized (24) Harmonized (24)
PDEs Harmonized Harmonized
Other Routes Harmonized
Harmonized
Options 4 options
3 options
Implementation Harmonized Harmonized
80

<231> and Veterinary Product Monographs
• Veterinary Products are out of scope
• Should we remove heavy metals testing from these
monographs?
– 197 official monographs
• 76 are drug substance monographs.
• Not all of these have labeling to indicate for vet use
only.
– Many vet drug products contain drug substances
that are also used in human formulations
– Human drug product may also have an
approved vet product.
81

New Chapters
• <730> Plasma Spectrochemistry
• <1730> Plasma Spectrochemistry—
Theory and Practice
• <735> X-Ray Fluorescence
• <1735> X-Ray Fluorescence Spectrometry
82

Chapters Impacted by <231> Deletion
1. <381> ELASTOMERIC CLOSURES FOR INJECTIONS
2. <661> PLASTIC PACKAGING SYSTEMS AND THEIR MATERIALS OF CONSTRUCTION
3. <661.1> PLASTIC MATERIALS OF CONSTRUCTION
4. <661.2> PLASTIC PACKAGING SYSTEMS FOR PHARMACEUTICAL USE
5. <661.3> PLASTIC COMPONENTS AND SYSTEMS USED IN PHARMACEUTICAL MANUFACTURING [NEW--- In PF 42(3)]
83

Arsenic ⟨211⟩
Lead ⟨251⟩
Selenium ⟨291⟩
Mercury ⟨261⟩
84
Other Element Specific Chapters

Limit tests and references to element specific chapters are included in about 1000 monographs?
What is the future of element specific chapters?
Are these specific element chapters and limit tests in monographs unnecessary unless there is a known quality- or safety-related reason to maintain the specific elemental impurity limit(s) in place for selected components (drug substances or excipients)?
With ⟨233⟩ in place, analytical procedures specific to individual elements are no longer
necessary? Removing references and (special) limits from drug product monographs would
align those monographs with ⟨232⟩, providing industry with only one set of elements and limits, as well as one analytical procedure.
Other Chapters
85

Other Element Specific chapters
86
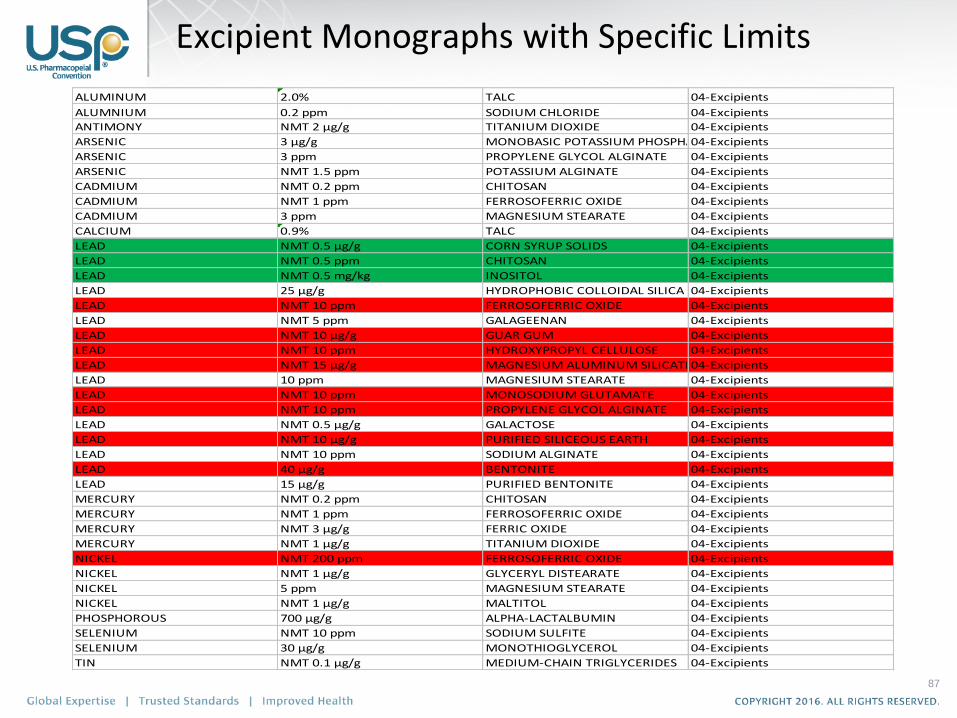
87
ALUMINUM 2.0% TALC 04-Excipients
ALUMNIUM 0.2 ppm SODIUM CHLORIDE 04-Excipients
ANTIMONY NMT 2 µg/g TITANIUM DIOXIDE 04-Excipients
ARSENIC 3 µg/g MONOBASIC POTASSIUM PHOSPHATE04-Excipients
ARSENIC 3 ppm PROPYLENE GLYCOL ALGINATE 04-Excipients
ARSENIC NMT 1.5 ppm POTASSIUM ALGINATE 04-Excipients
CADMIUM NMT 0.2 ppm CHITOSAN 04-Excipients
CADMIUM NMT 1 ppm FERROSOFERRIC OXIDE 04-Excipients
CADMIUM 3 ppm MAGNESIUM STEARATE 04-Excipients
CALCIUM 0.9% TALC 04-Excipients
LEAD NMT 0.5 µg/g CORN SYRUP SOLIDS 04-Excipients
LEAD NMT 0.5 ppm CHITOSAN 04-Excipients
LEAD NMT 0.5 mg/kg INOSITOL 04-Excipients
LEAD 25 µg/g HYDROPHOBIC COLLOIDAL SILICA 04-Excipients
LEAD NMT 10 ppm FERROSOFERRIC OXIDE 04-Excipients
LEAD NMT 5 ppm GALAGEENAN 04-Excipients
LEAD NMT 10 µg/g GUAR GUM 04-Excipients
LEAD NMT 10 ppm HYDROXYPROPYL CELLULOSE 04-Excipients
LEAD NMT 15 µg/g MAGNESIUM ALUMINUM SILICATE04-Excipients
LEAD 10 ppm MAGNESIUM STEARATE 04-Excipients
LEAD NMT 10 ppm MONOSODIUM GLUTAMATE 04-Excipients
LEAD NMT 10 ppm PROPYLENE GLYCOL ALGINATE 04-Excipients
LEAD NMT 0.5 µg/g GALACTOSE 04-Excipients
LEAD NMT 10 µg/g PURIFIED SILICEOUS EARTH 04-Excipients
LEAD NMT 10 ppm SODIUM ALGINATE 04-Excipients
LEAD 40 µg/g BENTONITE 04-Excipients
LEAD 15 µg/g PURIFIED BENTONITE 04-Excipients
MERCURY NMT 0.2 ppm CHITOSAN 04-Excipients
MERCURY NMT 1 ppm FERROSOFERRIC OXIDE 04-Excipients
MERCURY NMT 3 µg/g FERRIC OXIDE 04-Excipients
MERCURY NMT 1 µg/g TITANIUM DIOXIDE 04-Excipients
NICKEL NMT 200 ppm FERROSOFERRIC OXIDE 04-Excipients
NICKEL NMT 1 µg/g GLYCERYL DISTEARATE 04-Excipients
NICKEL 5 ppm MAGNESIUM STEARATE 04-Excipients
NICKEL NMT 1 µg/g MALTITOL 04-Excipients
PHOSPHOROUS 700 µg/g ALPHA-LACTALBUMIN 04-Excipients
SELENIUM NMT 10 ppm SODIUM SULFITE 04-Excipients
SELENIUM 30 µg/g MONOTHIOGLYCEROL 04-Excipients
TIN NMT 0.1 µg/g MEDIUM-CHAIN TRIGLYCERIDES 04-Excipients
Excipient Monographs with Specific Limits

Stim Article in PF 42(4)
88
Other Element Specific chapters in the USP-NF
STIMULI TO THE REVISION PROCESS Stimuli articles do not necessarily reflect the policies
of the USPC or the USP Council of Experts
Future of Element-Specific Chapters in the USP–NF
USP’s Chemical Analysis Expert Committee and Kahkashan Zaidia ABSTRACT
The Chemical Analysis Expert Committee (CAEC) is evaluating the idea of removing element-specific chapters and limit tests in monographs from the USP–NF. The CAEC is considering the effect of this proposal, as well as the effect of retaining these chapters and limit tests. The CAEC strongly encourages comments and discussions regarding this proposal.

89
Element Specific Chapters
Questions:
a. Are these element specific limit tests in monographs
necessary?
b. Are there known quality- or safety-related reasons to keep
specific elemental impurity limit(s) in the monographs (drug
substances, excipients and Drug Products)?
USP needs stakeholder’s
Feedback

90
Element Specific Chapters
It will not be an Easy Task
BUT IF IT CAN BE DONE THEN,
a. Removing references and (special) limits from drug product monographs would:
a. Align those monographs with ⟨232⟩, providing industry with only one set of elements and limits, as well as one analytical procedure.
a. Will simplify compliance and establishment of consistent, safety-based elemental impurity limits with the focus on the drug product rather than component-specific limits.
b. With ⟨233⟩ in place, analytical procedures specific to individual elements
are no longer necessary

General Notices (GN)
– Overarching – Apply to all chapters and monographs
General Test Chapters
– Tests and assays applying to multiple monographs
– Supersede GN if conflicting
Monographs: API, Excipients, Drug Products
– Supersede both GN and Chapters if conflicting
General Information Chapters
– Guidance
– Do not contain specifications
Structural Hierarchy
91

Implementation
• Implementation through General
Notices
• No reference to <232> will be in
monographs
92

<232> Implementation USP General Notices:
5.60.30. Elemental Impurities in USP Drug Products and Dietary Supplements Effective January 1, 2018 • Elemental impurities will be controlled in official drug products
according to the principles defined and requirements specified in Elemental Impurities—Limits ⟨232⟩. Effective January 1, 2018, elemental contaminants are controlled in official dietary supplements according to the principles defined and requirements specified in Elemental Contaminants in Dietary Supplements ⟨2232⟩. Also effective January 1, 2018, Heavy Metals ⟨231⟩ will be omitted and all references to it in general chapters and monographs will be deleted. Early adoption of the requirements in ⟨232⟩ and ⟨2232⟩ are permitted by USP, and if ⟨232⟩ or ⟨2232⟩, as applicable, is fully implemented with respect to a particular drug product or dietary supplement in advance of the January 1, 2018 date, that product and its ingredients will no longer need to comply with applicable ⟨231⟩ requirements to be considered by USP to be in conformance with USP–NF requirements.(RB 1-Apr-2015)
93

USP General Notices
3. CONFORMANCE TO STANDARDS
– 3.10. Applicability of Standards
• Early adoption of revised standards in advance
of the official date is allowed by USP unless
specified otherwise at the time of publication.
94

Analytical Procedures Harmonized analytical procedures should be
established by the pharmacopoeias for determining
levels of metal impurities, with allowance for use of any appropriate
validated procedure for a particular application.
USP Chapter <233> Elemental Impurities—Procedures
Proposed in PF 36(1) (2010)
Sample Preparation
Procedures
Validation requirements
Harmonization through PDG 95

USP Website
• March 27, 2015 • General Chapters and Related Information
• To be Published in Second Supplement to USP 38-NF 33: (official
on December 1, 2015)
– <232> Elemental Impurities—Limits
– <233> Elemental Impurities—Procedures
• Revision Plan (updated March 27, 2015)
• Frequently Asked Questions
• FAQs on the Implementation of USP General Chapters <232>
Elemental Impurities—Limits <233> Elemental Impurities—
Procedures, and <2232> Elemental Contaminants in Dietary
Supplements (updated 27–Mar–2015)
96

Implementation – USP
ICH Q3D step 4 published Dec 16, 2014
Implementation of <232> and <2232> o Jan 1, 2018
Via USP General Notices
Omission of Chapter <231> o Jan 1, 2018
Delete cross-references to General
Chapter <231> Heavy metals from all
individual monographs
o Jan 1, 2018
o Deletion Marked up---USP 38 and
following publications with delayed
implementation on Jan 1, 2018
97



United State Pharmacopeia:
Plastic Packaging Standards: <661> Chapters
Desmond G. Hunt, M.S., Ph.D. Principle Scientific Liaison Science, General Chapters

Identification
• Infrared Spectroscopy (IR)
• Differential Scanning Calorimetry (DSC)
Biological Tests
• USP <87>; Biological Reactivity Test, In Vitro
• USP <88>; Biological Reactivity Test, In Vivo (if a class designation is required).
Physicochemical Tests (water extract)
• Nonvolatile Residue (NVR); not required for PET/PETG
• Residue on Ignition (RoI, if NVR > 5 mg); not required for PET/PETG
• Heavy Metals (HM)
• Buffering Capacity (BC); not required for PET/PETG
Other Tests (for PET and PETG)
Test Methods Required in the Current USP <661>
USP <661> Containers—Plastics: “The Old”

A Starting Point for the USP Standards for Plastics
Building safety into a system by using well-characterized
and safe materials of construction (Quality by Design).
The testing required to select and qualify a system or a
system component is correlated to the risk that the system’s
finished output is adversely effected by its interaction with
the component or system (Risk-based Approach).
The USP standard serves as a baseline (Aim for the
Middle).

Scope and Objectives of USP Standards for Plastics
Scope Focus on:
• Safety
• Material and component-derived foreign impurities
Objectives To provide tests and specifications for the characterization of
plastic materials • So materials can be rationally selected for use and so that the selection
can be justified
To provide tests and specifications for the characterization of
plastic components • So plastic components/systems can be rationally selected for use and
that the selection can be justified
To provide tests and specifications for the safety qualification of
manufacturing, packaging and delivery systems (or their
components)

The Essence of the Current Strategy for Plastics
Standardize at the Materials
of Construction level
Customize at the Component
or System level
Characterization of
Materials of Construction
<661.1>
Component or System Testing for Packaging
<661.2>
Component or System Testing
for Manufacturing
Component or System Testing
for Devices
Extractable Guidance
<1663>
Leachable Guidance
<1664>

The Foundation

United States Pharmacopeia:
USP GENERAL CHAPTER <1663> Assessment of
Extractables Associated with Pharmaceutical
Packaging/Delivery Systems

1. PURPOSE
2. KEY TERMS
3. SCOPE
4. BACKGROUND INFORMATION
5. GENERATING THE EXTRACT 1. General Concepts and Critical Experimental Design Parameters
2. Chemical Nature of the Extracting Medium
3. Extraction Time and Temperature
4. Extraction Stoichiometry
5. Mechanism of Extraction—Extraction Technique
6. Extractions That are Not Solvent Mediated
6. CHARACTERIZING THE EXTRACT 1. Objectives and Challenges
2. Processes Involved in Extract Characterization
3. Preparation of Extracts for Analysis
7. SUMMARY 1. Assessing the Completeness of an Extractables Assessment
2. Example Extractables Profiles—Materials Characterization
10
8
<1663> Extractables Associated with
Packaging/Delivery Systems

United States Pharmacopeia:
USP GENERAL CHAPTER <1664> Assessment of
Leachables Associated with Pharmaceutical
Packaging and Delivery Systems

<1664.1>
Orally Inhaled
Nasal Drug
Products
<1664.2>
Parenteral and
Ophthalmic Drug
Products
(To Come)
<1664> Assessment of Leachables Associated with
Pharmaceutical Packaging and Delivery Systems
<1664> Leachables Associated with
Packaging/Delivery Systems

1. PURPOSE
2. KEY TERMS
3. SCOPE
4. BACKGROUND INFORMATION
5. CONCEPTS 1. General Concepts for Leachables Assessment
2. Safety Thresholds
3. Information Sharing
6. LEACHABLES STUDY DESIGN
7. LEACHABLES CHARACTERIZATION 1. Analytical Thresholds
2. Analytical Method Requirements
3. Preparing the Drug Product for Analysis—Sample Preparation
4. Analytical Techniques
5. Quantitative Methods—Validation Considerations
8. ESTABLISHING A LEACHABLES–EXTRACTABLES CORRELATION
9. CONSIDERATIONS IN DEVELOPING LEACHABLES SPECIFICATIONS AND
ACCEPTANCE CRITERIA
10.ADDITIONAL CONSIDERATIONS 1. Simulation Studies
2. Inorganic (Elemental) Leachables
<1664> Leachables Associated with
Packaging/Delivery Systems

Overview: <661> Plastic Packaging Systems and their Materials of Construction
<661.1> Plastic Material of
Construction
<661.2> Plastic Packaging Systems for
Pharmaceutical Use
- Identification
- Biological Activity
- Physico-chemical Tests
- Extractable Metals
- Plastic Additives
- Biological Activity
- Physico-chemical Tests
- Safety Assessment
(Extractables/Leachables)
<661> Plastic Packaging Systems and their Materials of Construction

<661.1> Plastic Materials of Construction
Comparison of Testing Required for Various Dosage Forms
Test Parameter Oral and Topical Dosage Forms All Other Dosage Forms
Chemical Tests
Identification DSC/IR DSC/IR
Physicochemical Tests Water extraction: • UV absorbance, • Acidity/alkalinity • TOC
Water extraction: • UV absorbance, • Acidity/alkalinity • TOC
Extractable Metals Acid extraction: • ICP analysis for targeted and
relevant metals
Acid extraction: • ICP analysis for targeted and
relevant metals
Polymer Additives Proper Reference to Indirect Food Additive Regulations, CFR 174-186
Direct chemical testing
Biological Reactivity
In Vitro per USP <87> Required Required
In Vivo per USP <88> Not required Required as needed to obtain plastic classification

Objective: To gain as much information about a
material of construction to determine potential
suitably
• Development
• Packaging System Change
Requirement of <661.1> can be met by :
• The materials of construction meeting requirement of
<661.1>
• The component or system meeting the requirement of
<661.2>
<661.1> Material Characterization Chapters
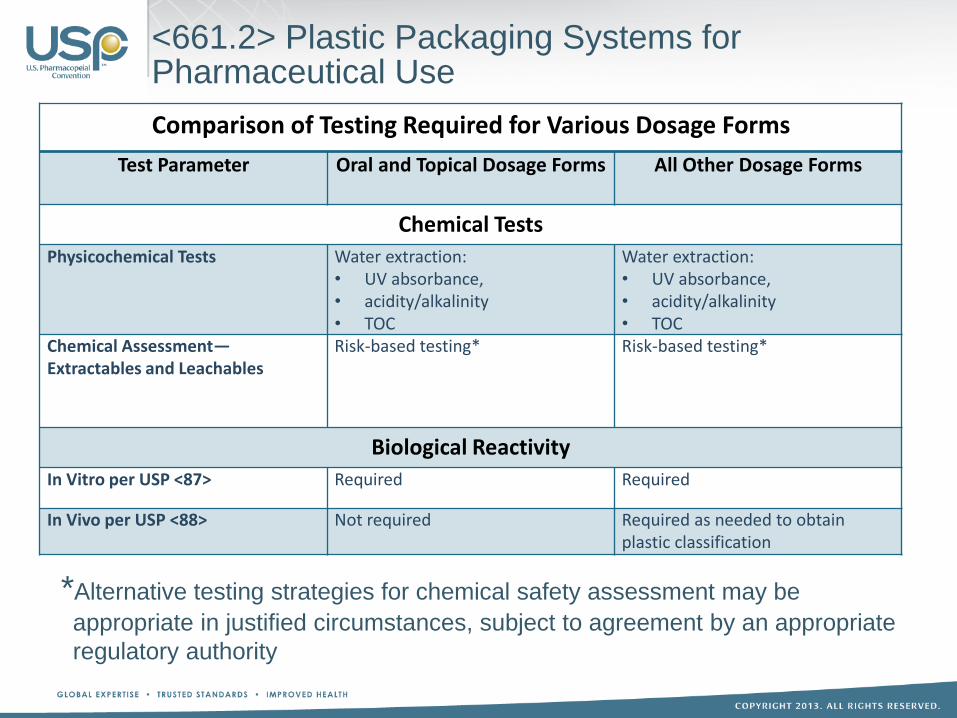
Comparison of Testing Required for Various Dosage Forms
Test Parameter Oral and Topical Dosage Forms All Other Dosage Forms
Chemical Tests
Physicochemical Tests Water extraction: • UV absorbance, • acidity/alkalinity • TOC
Water extraction: • UV absorbance, • acidity/alkalinity • TOC
Chemical Assessment— Extractables and Leachables
Risk-based testing*
Risk-based testing*
Biological Reactivity
In Vitro per USP <87> Required Required
In Vivo per USP <88> Not required Required as needed to obtain plastic classification
<661.2> Plastic Packaging Systems for Pharmaceutical Use
*Alternative testing strategies for chemical safety assessment may be
appropriate in justified circumstances, subject to agreement by an appropriate
regulatory authority

Revision Impact
50 year standard being revised
Added new polymers
Added new testing procedures and specifications
New testing paradigm
• Materials of Construction
• Packaging System
Currently approved drug products that uses a
plastic component in it’s packaging system could
be impacted.

Qualification: Packaging Material/System (High-
Risk Dosage Form)
1 month: selection of alternative material/component
8 months: develop extractable profile with toxicological
assessment
24+months: leachable migration study
• If sponsor wants to submit actual shelf-life data
− 2 year self-life
− Control room temperature product
~3Years: qualify a new packaging systems for one drug
product
A company can have many products that use the same
packaging component, which increase the issue
exponentially.

Currently Approved Products
Rationale
Goal of <661> revision
• “To aid in the selection and qualification of packaging
materials and components that are deemed suitable
and safe”
Suitability and safety of a packaging system, with it’s
drug product, is determined by regulatory authorities
• A packaging system, with a specific drug product,
would meet the requirements of <661.1> and <661.2>,
if it had gained regulatory approval.

New Products
Existing Products
– Material/Component Change
• New raw material/component supplier
• Process changes
–Reaction conditions, cycle times, equipment, etc.
• Stock outs or product withdrawal
Incoming material and component quality control

Situation Required Testing
General Situation Specific Circumstance <661.1> <661.2>
Changes to a packaging system used with a currently marketed pharmaceutical product
A new material is introduced into the packaging system
Yes (for the new material)
Yes
A material of construction in the packaging system is changed in either composition or process
Yes (for the changed material)
Yes
The packaging system is changed, in either composition or process, in a manner that does not involve a change in its materials or to its materials (for example, changing the thicknesses of individual layers in a multi-layered film)
No Yes
Packaging System Change

Situation Required Testing
General Situation Specific Circumstance <661.1> <661.2>
Packaging system used with a currently marketed pharmaceutical product is to be applied to a different pharmaceutical product
Dosage form and conditions of use are similar for the current and different pharmaceutical products
No No
Dosage form and/or conditions of use are different from the current pharmaceutical products (moving from “high risk” to “low risk”)
No No
Dosage form and/or conditions of use are different from the current pharmaceutical products (moving from “low risk” to “high risk”)
Yes Yes
Packaging System Applied to New Product
Application and Applicability

Current Materials
• Polyvinyl Chloride, Plasticized
• Polyethylene
• Cyclic Olefin
• Polyethylene Terephthalate
• Polyethylene Terephthalate G
• Polyamide 6 (Nylon)
• Polycarbonate
• Polyethylene vinyl acetate
• Polyvinyl Chloride, Non
Plasticized
Future Revision: <661.1> PF 42 (4) July 2016
Future Materials
• Acrylonitrile butadiene styrene
• Polybutylene terephthalate
• Poly(methylmethacrylate)
• Polystyrene
• Polysulfone
• Polytetrafluoroethylen
• Polyurethane

Proposed revision of chapter’s Scope to better clarify intent • <661.1>
• <661.2>
Proposed revision to remove of the Biological Reactivity
requirement for materials and packaging used for oral and topical
drug products • <661.1>
• <661.2>
Proposed addition of the Spectral Transmission Test in <661.2> • Test currently resides in <671> Container−Performance Testing
Topic of Discussion
• Should specific date be proposed when the grandfathering clause should be
removed from the chapter
Future Revision: <661.1> and <661.2> PF 42 (4) July
2016

Upcoming Packaging Revisions/Development
<381> Elastomeric Closure for Injection
• The Expert Panel began working on the revision of <381> in May 2014
• Workshop June 19-20, 2017
<87> Biological Reactivity, In vitro, <88> Biological Reactivity, In
Vivo3200-
• The Expert Panel began working on the revision of <87> and <88> in March
2015
<660> Container−Glass
• The USP Glass Expert Panel began work on revising <660> and <1660> in
September 2016
• Workshop June 19-20, 2017
Metal Packaging Systems and Their Materials of Construction
• Stakeholder Webinar April 2015
• To form Expert Pane early 2017




















