
Phenylalanine and Phenylglycine Analogues as Arginine Mimetics...
15
Phenylalanine and Phenylglycine Analogues as Arginine Mimetics in Dengue Protease Inhibitors Lena F. Weigel, † Christoph Nitsche, † Dominik Graf, † Ralf Bartenschlager, §,‡ and Christian D. Klein* ,† † Medicinal Chemistry, Institute of Pharmacy and Molecular Biotechnology IPMB, Heidelberg University, Im Neuenheimer Feld 364, D-69120 Heidelberg, Germany ‡ Department of Infectious Diseases, Molecular Virology, Heidelberg University, Im Neuenheimer Feld 345, D-69120 Heidelberg, Germany § German Centre for Infection Research, Heidelberg University, D-69120 Heidelberg, Germany * S Supporting Information ABSTRACT: Dengue virus is an increasingly global pathogen. One of the promising targets for antiviral drug discovery against dengue and related flaviviruses such as West Nile virus is the viral serine protease NS2B-NS3. We here report the synthesis and in vitro characterization of potent peptidic inhibitors of dengue virus protease that incorporate phenyl- alanine and phenylglycine derivatives as arginine-mimicking groups with modulated basicity. The most promising compounds were (4-amidino)-L-phenylalanine-containing in- hibitors, which reached nanomolar affinities against dengue virus protease. The type and position of the substituents on the phenylglycine and phenylalanine side chains has a significant effect on the inhibitory activity against dengue virus protease and selectivity against other proteases. In addition, the non-natural, basic amino acids described here may have relevance for the development of other peptidic and peptidomimetic drugs such as inhibitors of the blood clotting cascade. ■ INTRODUCTION The dengue virus (DENV) belongs to the family of flaviviridae which also includes the West Nile virus (WNV), hepatitis C virus (HCV), and yellow fever virus (YFV). With 3.97 billion people living in areas with a high risk of DENV transmission 1 and an estimated 390 million infections in 2010, 2 DENV constitutes a global health problem. Since 2010, there were local transmissions reported in Croatia 3 and France, 4 indicating that DENV is spreading to new areas. Therefore, DENV is one of the most important mosquito-borne viruses. In severe cases, infection with DENV can cause hemorrhagic fever and dengue shock syndrome. Up to now, there is no specific drug treatment available. Four serotypes of the virus (DENV 1−4) have been extensively characterized, 5 and a fifth serotype was described recently. 6 The viral genome is an 11 kb positive-sense, single- stranded RNA which encodes a polyprotein with three structural (capsid, envelope protein, membrane protein precursor) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). 7 The nonstructural protein NS3 contains a protease and a helicase domain. The protease domain of NS3, also denoted as NS3pro, has a trypsin- like fold with a catalytic triad formed by Ser135, His51, and Asp75. Catalytic activity of NS3pro depends on the formation of a complex with the NS2B protein, which forms a part of the substrate recognition site. 8 For biochemical studies, the NS2B- NS3pro complex is usually produced as a fusion protein in which the two constituents are connected by a glycine−serine linker. Viral proteases are successful targets for the discovery and development of antiviral drugs. With protease inhibitors as key components in HIV therapy 9,10 and novel drugs such as boceprevir, telaprevir, and simeprevir as HCV NS3 protease inhibitors, 11,12 the clinical efficacy of viral protease inhibitors is established. Recently, nonpeptidic DENV protease inhibitors were described with good activity against DENV serotype 2 (DENV-2) in the low micromolar range. 13,14 Behnam et al. described peptide hybrid inhibitors with activity in the nanomolar range. 15 These peptidic inhibitors are derived from the protease recognition sequence. Peptidic inhibitors of DENV protease usually include the basic amino acids arginine and lysine as preferred residues in the P 1 ,P 2 , and P 3 positions. 16,17 The guanidino group of arginine is a major determinant of specificity and activity. Replacement of this basic side chain with the intention to improve cellular permeability and metabolic stability of the inhibitors is usually detrimental to activity, 17 and previous SAR studies demon- strated a loss of activity if arginine is replaced by nonbasic amino acids. 18 Another approach to improve the inhibitory activity in vitro and in cellulo is the combination of a peptide sequence (RKn-NH 2 ) with N-terminal “capping” groups, Received: April 21, 2015 Published: September 14, 2015 Article pubs.acs.org/jmc © 2015 American Chemical Society 7719 DOI: 10.1021/acs.jmedchem.5b00612 J. Med. Chem. 2015, 58, 7719−7733
Transcript of Phenylalanine and Phenylglycine Analogues as Arginine Mimetics...

Phenylalanine and Phenylglycine Analogues as Arginine Mimetics inDengue Protease InhibitorsLena F. Weigel,† Christoph Nitsche,† Dominik Graf,† Ralf Bartenschlager,§,‡ and Christian D. Klein*,†
†Medicinal Chemistry, Institute of Pharmacy and Molecular Biotechnology IPMB, Heidelberg University, Im Neuenheimer Feld 364,D-69120 Heidelberg, Germany‡Department of Infectious Diseases, Molecular Virology, Heidelberg University, Im Neuenheimer Feld 345, D-69120 Heidelberg,Germany§German Centre for Infection Research, Heidelberg University, D-69120 Heidelberg, Germany
*S Supporting Information
ABSTRACT: Dengue virus is an increasingly global pathogen.One of the promising targets for antiviral drug discoveryagainst dengue and related flaviviruses such as West Nile virusis the viral serine protease NS2B-NS3. We here report thesynthesis and in vitro characterization of potent peptidicinhibitors of dengue virus protease that incorporate phenyl-alanine and phenylglycine derivatives as arginine-mimickinggroups with modulated basicity. The most promisingcompounds were (4-amidino)-L-phenylalanine-containing in-hibitors, which reached nanomolar affinities against denguevirus protease. The type and position of the substituents onthe phenylglycine and phenylalanine side chains has a significant effect on the inhibitory activity against dengue virus proteaseand selectivity against other proteases. In addition, the non-natural, basic amino acids described here may have relevance for thedevelopment of other peptidic and peptidomimetic drugs such as inhibitors of the blood clotting cascade.
■ INTRODUCTION
The dengue virus (DENV) belongs to the family of flaviviridaewhich also includes the West Nile virus (WNV), hepatitis Cvirus (HCV), and yellow fever virus (YFV). With 3.97 billionpeople living in areas with a high risk of DENV transmission1
and an estimated 390 million infections in 2010,2 DENVconstitutes a global health problem. Since 2010, there werelocal transmissions reported in Croatia3 and France,4 indicatingthat DENV is spreading to new areas. Therefore, DENV is oneof the most important mosquito-borne viruses. In severe cases,infection with DENV can cause hemorrhagic fever and dengueshock syndrome. Up to now, there is no specific drug treatmentavailable. Four serotypes of the virus (DENV 1−4) have beenextensively characterized,5 and a fifth serotype was describedrecently.6 The viral genome is an 11 kb positive-sense, single-stranded RNA which encodes a polyprotein with threestructural (capsid, envelope protein, membrane proteinprecursor) and seven nonstructural proteins (NS1, NS2A,NS2B, NS3, NS4A, NS4B, and NS5).7 The nonstructuralprotein NS3 contains a protease and a helicase domain. Theprotease domain of NS3, also denoted as NS3pro, has a trypsin-like fold with a catalytic triad formed by Ser135, His51, andAsp75. Catalytic activity of NS3pro depends on the formationof a complex with the NS2B protein, which forms a part of thesubstrate recognition site.8 For biochemical studies, the NS2B-NS3pro complex is usually produced as a fusion protein in
which the two constituents are connected by a glycine−serinelinker.Viral proteases are successful targets for the discovery and
development of antiviral drugs. With protease inhibitors as keycomponents in HIV therapy9,10 and novel drugs such asboceprevir, telaprevir, and simeprevir as HCV NS3 proteaseinhibitors,11,12 the clinical efficacy of viral protease inhibitors isestablished. Recently, nonpeptidic DENV protease inhibitorswere described with good activity against DENV serotype 2(DENV-2) in the low micromolar range.13,14 Behnam et al.described peptide hybrid inhibitors with activity in thenanomolar range.15 These peptidic inhibitors are derivedfrom the protease recognition sequence. Peptidic inhibitors ofDENV protease usually include the basic amino acids arginineand lysine as preferred residues in the P1, P2, and P3positions.16,17 The guanidino group of arginine is a majordeterminant of specificity and activity. Replacement of thisbasic side chain with the intention to improve cellularpermeability and metabolic stability of the inhibitors is usuallydetrimental to activity,17 and previous SAR studies demon-strated a loss of activity if arginine is replaced by nonbasicamino acids.18 Another approach to improve the inhibitoryactivity in vitro and in cellulo is the combination of a peptidesequence (RKn-NH2) with N-terminal “capping” groups,
Received: April 21, 2015Published: September 14, 2015
Article
pubs.acs.org/jmc
© 2015 American Chemical Society 7719 DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733

leading to peptide hybrids.18,19 The tripeptide sequence Arg-Lys-Nle-NH2 capped with 2,4-thiazolidinedione derivativesshowed increased activity and membrane permeability.19
Further exploration of the C-terminal residue identifiedphenylglycine as a non-natural amino acid that resulted inhigh inhibitory activity. By merging this optimized sequence toa 2,4-thiazolidinedione “cap”, the in vitro activity of the peptidehybrid inhibitors could be enhanced into the nanomolarrange,15 and dual inhibitors of DENV and WNV protease canbe obtained.20 However, these peptide hybrids were still notoptimized for the S2 pocket with respect to target affinity. Inaddition, previously published docking simulations showed theimportance of two basic residues, such as lysine in S1 andarginine in S2.
19 Considering these studies, we screened forarginine-mimicking moieties to identify alternative fragmentsthat favorably fit into the S2 pocket. We herein present thesynthesis and structure−activity relationship (SAR) of DENVprotease inhibitors using phenylalanines and phenylglycineanalogues as substructure for arginine mimetics. Side chainmodifications were performed on the peptides coupled to solidsupport, which increased yields and diminished formation ofbyproducts. These SAR explorations led to the discovery ofnovel and potent peptidic inhibitors of dengue protease thatincorporate non-natural amino acids.
■ RESULTS AND DISCUSSION
Chemistry. The chemical structure of the reported peptidicinhibitors here is based on the recently published inhibitorsequence R-Arg-Lys-Phg-NH2.
15 All peptides were obtained bysolid-phase peptide synthesis according to the Fmoc protocol.
The unnatural amino acids 30b, 32a−b, and 34a−b weresynthesized by protecting the α-amine position with a benzoylsubstituent. With the benzoyl moiety as N-terminal cap, allsynthesized non-natural amino acids could be coupled to thepresynthesized Lys-Phg-sequence with good synthetic effi-ciency. Scheme 1 shows the synthetic approach for inhibitors30b and 34b. Benzoyl protection of (3-nitro)-L-phenylalaninefollowed by esterification21 gave the methyl ester 2, which wasreduced to provide 3 as key intermediate for further syntheses.Compounds 4 and 6 were obtained by reaction of 3 with di-tert-butyl dicarbonate and bis-boc-pyrazole-1-carboxamidine,17
respectively. After methyl ester cleavage, the benzoyl-cappedamino acids 5 and 7 were coupled to the presynthesized Lys-Phg-sequence according to the Fmoc protocol. An analogoussynthetic strategy was applied for the synthesis of inhibitors32a−b as shown in Scheme 2. Compounds 9a−b wereobtained after benzoyl protection and esterification. Thefollowing synthetic steps: palladium-catalyzed reaction at 40°C, boc protection of the side chain, and hydrolysis of methylester afforded the amino acids 12a−b, which were successfullycoupled to the indicated sequence. The syntheses ofbenzamidine-containing peptidic inhibitors as C-terminalarginine mimetics have already been described for furin22 andWest Nile virus protease.23 For dengue protease inhibitors,there are currently no peptidic or peptidomimetic inhibitorsthat include amidinophenylalanine and amidinophenylglycineas arginine mimicking residues at the N-terminal position.Previous work by Cesar et al.24 described a method tosynthesize amidines on Wang resin by solid phase support.According to this procedure, benzamidine-containing inhibitors
Scheme 1. Synthetic Approach for Compounds 30b and 34ba
a(a) SOCl2, MeOH; (b) BzCl, DIPEA, DCM; (c) Pd/C, H2, MeOH, HCl (aq); (d) Boc2O, DIPEA, DCM; (e) LiOH, H2O/THF, 0 °C; (f) bis-boc-pyrazole-1-carboxamidine, DMAP, DIPEA, MeOH.
Scheme 2. Synthesis of (3-/4-Aminomethyl)-L-phenylalanine-Containing Peptides Listed in Table 1a
aNumbering of ring carbons indicates the substitutions (3 = meta-substituted, 4 = para-substituted). (a) SOCl2, MeOH; (b) BzCl, DIPEA, DCM;(c) Pd/C, H2, acetic acid, 40 °C; (d) Boc2O, NaHCO3, MeOH; (e) LiOH, H2O/THF, 0 °C.
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7720
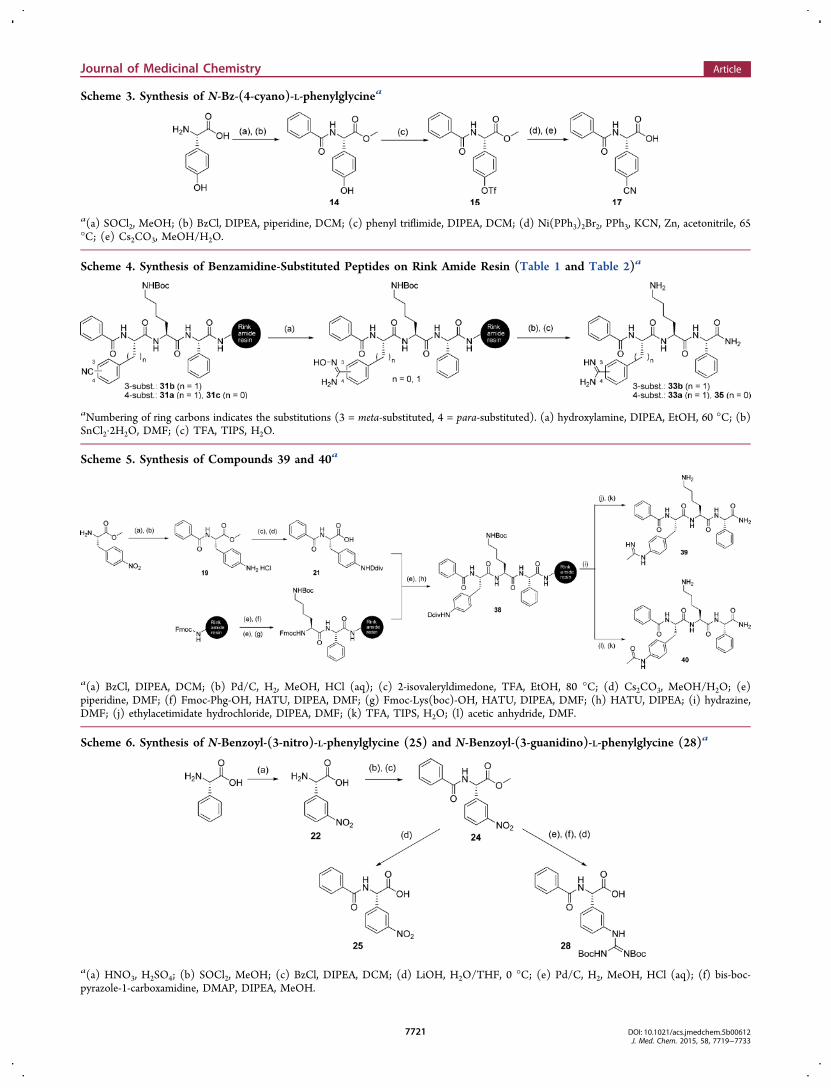
Scheme 3. Synthesis of N-Bz-(4-cyano)-L-phenylglycinea
a(a) SOCl2, MeOH; (b) BzCl, DIPEA, piperidine, DCM; (c) phenyl triflimide, DIPEA, DCM; (d) Ni(PPh3)2Br2, PPh3, KCN, Zn, acetonitrile, 65°C; (e) Cs2CO3, MeOH/H2O.
Scheme 4. Synthesis of Benzamidine-Substituted Peptides on Rink Amide Resin (Table 1 and Table 2)a
aNumbering of ring carbons indicates the substitutions (3 = meta-substituted, 4 = para-substituted). (a) hydroxylamine, DIPEA, EtOH, 60 °C; (b)SnCl2·2H2O, DMF; (c) TFA, TIPS, H2O.
Scheme 5. Synthesis of Compounds 39 and 40a
a(a) BzCl, DIPEA, DCM; (b) Pd/C, H2, MeOH, HCl (aq); (c) 2-isovaleryldimedone, TFA, EtOH, 80 °C; (d) Cs2CO3, MeOH/H2O; (e)piperidine, DMF; (f) Fmoc-Phg-OH, HATU, DIPEA, DMF; (g) Fmoc-Lys(boc)-OH, HATU, DIPEA, DMF; (h) HATU, DIPEA; (i) hydrazine,DMF; (j) ethylacetimidate hydrochloride, DIPEA, DMF; (k) TFA, TIPS, H2O; (l) acetic anhydride, DMF.
Scheme 6. Synthesis of N-Benzoyl-(3-nitro)-L-phenylglycine (25) and N-Benzoyl-(3-guanidino)-L-phenylglycine (28)a
a(a) HNO3, H2SO4; (b) SOCl2, MeOH; (c) BzCl, DIPEA, DCM; (d) LiOH, H2O/THF, 0 °C; (e) Pd/C, H2, MeOH, HCl (aq); (f) bis-boc-pyrazole-1-carboxamidine, DMAP, DIPEA, MeOH.
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7721

were synthesized by loading Fmoc-protected cyano-L-phenyl-alanines and 17 on Lys-Phg substituted resin. The preparationof 17 was performed according to procedures for phenylalaninesynthesis described previously (Scheme 3).25,26 Starting from(4-hydroxy)-L-phenylglycine, benzoyl and methyl ester protec-tion furnished 14, followed by conversion to 15 using phenyltriflimide. Nickel catalyzed cyanation25−27 of 15 with potassiumcyanide and final methyl ester hydrolysis afforded 17. To obtainbenzamidine-containing inhibitors, the resin-bound peptides31a−c were treated with hydroxylamine and diisopropylethyl-amine to achieve the amidoxime as intermediate (Scheme 4).The reduction of amidoximes with tin(II) chloride dihydrateafforded the benzamidine-containing compounds 33a−b and35. Intermediate 19 was prepared by benzoyl protection of (4-nitro)-L-phenylalanine methyl ester followed by hydrogenation.19 was further treated with isovaleryldimedone28 and cesiumcarbonate29 to obtain 21. Inhibitors 39 and 40 were synthesizedby coupling 21 to Lys-Phg on Rink amide resin. After removalof the protecting group 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl (ddiv) by 2% hydrazine in DMF, thepeptide-substituted resin was treated with ethylacetimidate toafford compound 39 (Scheme 5). 40 was prepared as shown inScheme 5 by using acetic anhydride after ddiv removal.Compound 28 was obtained as follows: 24 was prepared bynitration of L-phenylgylcine followed by benzoyl and methylester protections (Scheme 6). This intermediate was convertedto 27 by reduction and subsequent reaction with bis-boc-pyrazole-1-carboxamidine. The cleavage of methyl esterafforded 28. Although different conditions and reagents wereevaluated, coupling of 28 to the Lys-Phg sequence on solidsupport remained unsuccessful. However, coupling of 25 to thissequence using standard coupling conditions, followed byreduction on the solid support using tin(II) chloride andsubsequent reaction with bis-boc-pyrazole-1-carboxamidinefurnished the desired peptide 37 with a (3-guanidino)-L-phenylglycine moiety as arginine mimetic (Scheme 7).Derivatives 42a−b and 45a−b were synthesized as shown inScheme 2 by using 3-trifluoromethyl benzoate and 3-phenyl-propionate as N-terminal caps.Structure−Activity Relationships. A series of 16
tripeptides with the general sequence Bz-X-Lys-Phg-NH2
(Figure 1), in which X refers to phenylalanine and phenyl-glycine analogues as arginine mimetics, was evaluated against
DENV protease serotype 2. The SAR explorations were focusedon the side chain of these residues and in particular on varyingtheir basic functionalities. To confirm selectivity for DENVprotease, all compounds were also evaluated against WNVprotease, thrombin, and trypsin (see Table 1).Nearly all compounds have a negligible activity against
trypsin (see Tables 1 and 2). Compounds 34b and 37 possess amoderate inhibitory activity against trypsin, with IC50 values of18.2 and 7.1 μM, respectively.For most compounds, the activity against WNV protease was
below 90% relative inhibition. The compounds 32a, 34b, and40 have a notable activity against WNV protease with IC50values ranging from 1.6 to 3.9 μM. We observed for 32a a 3-fold selectivity toward WNV protease compared to DENVprotease. This underlines that WNV protease prefers substratescontaining lysine at the P2 residue, which could be mimickedmore convincingly by the aminomethylphenylalanine moiety.30
Moderate activity against WNV protease was also observed forcompound 40.In contrast, inhibition of dengue protease was generally much
more pronounced by the compounds presented here (Table 1).However, the pyridyl substituent (29a) caused a decrease ofinhibitory activity against DENV protease in comparison toarginine. A further extension of the phenylalanine substitutionto para-substituted aniline (30a) and nitrile (31a) derivativesalso led to practically inactive analogues. For the correspondingaminomethyl-substituted analogue (32a), an IC50 value (4.3μM) in the range of the reference compound I (Table 1) was
Scheme 7. Synthesis of Compound 37a
a(a) Piperidine, DMF; (b) HATU, HOAt, DIPEA, DMF; (c) TFFH, DIPEA, DMF; (d) SnCl2·2H2O, DIPEA, DMF; (e) bis-boc-pyrazole-1-carboxamidine, DMAP, DIPEA, DMF; (f) TFA, TIPS, H2O.
Figure 1. Common structural motif Bz-X-Lys-Phg-NH2.
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7722

observed. Although (4-aminomethyl)-L-phenylalanine is con-sidered as one of the suitable replacements for basic sidechains,31 there is no improvement of inhibitory potencycompared to the previously published peptide hybrid byBehnam et al.15 Nevertheless, the aminomethyl moiety is lessbasic than the guanidino function of arginine. Previousapproaches to replace arginine by lysine or less basic residuesresulted in a loss of activity.19,32 The use of (4-guanidino)-L-phenylalanine as arginine replacement has already beendescribed for a tetrapeptidic aldehyde dengue proteaseinhibitor, which, however, cannot be considered as a drug-likelead compound.17 With an IC50 value of 580 nM, the (4-guanidino)-L-phenylalanine analogue 34a had somewhat lowerinhibitory activity than the benzamidine analogue (33a, IC50 =
440 nM, Ki = 232 nM, Table 1). This 10-fold increase ininhibitory activity compared to reference I suggests apreference for basic aromatic residues in S2. Besides this, theinsertion of amidinophenylalanine, as a more drug-like residuethan 4-guanidinophenylalanine, offers the opportunity todevelop amidoxime prodrugs. Amidoximes are less basic thanthe corresponding benzamidines and possess improvedpharmacokinetic properties, in particular with respect togastrointestinal bioavailability.33,34 Although benzamidines areoften used as arginine mimicking groups, particularly inthrombin inhibitors35 and other protease inhibitors of thetrypsin family,36,37 compound 33a showed only minor effectson thrombin. The known serine protease inhibitor p-aminobenzamidine was previously tested against the protease from
Table 1. Inhibitory Activity of N-Benzoyl-Capped Tripeptides with Several Phenylalanine Analogues as Arginine Mimetics;Effect of Modifications by Extending the Side Chain and Increasing Basicity (General Formula: Bz-X-Lys-Phg-NH2)
h
aPercent inhibition of the DENV NS2B-NS3pro protease serotype 2 (compd, 50 μM; substrate, 50 μM; Km, 105 μM; enzyme, 100 nM). bFurtherinhibitor concentrations were assayed for compounds percent inhibition ≥70%; IC50 values against DENV NS2B-NS3pro protease serotype 2(substrate concentration: 50 μM). cPercent inhibition of the West Nile virus (WNV) NS2B-NS3pro protease (compd, 50 μM; substrate, 50 μM; Km,212 μM; enzyme, 150 nM). dFurther inhibitor concentrations were assayed for compounds percent inhibition ≥90%; IC50 values against WNVNS2B-NS3pro protease (substrate concentration, 50 μM) ePercent inhibition of thrombin (compd, 25 μM; substrate, 50 μM; Km, 16 μM; enzyme,10 nM). fPercent inhibition of trypsin (compd, 50 μM; substrate, 50 μM, enzyme, 1 nM). gLigand efficiency values are given in kcal/mol per non-hydrogen atom.40 hAll measurements were carried out in triplicate. Standard deviations were ≤10%, n.d. = not determined.
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7723

DENV serotype 2, without notable effect even at highconcentration (20% inhibition at 100 μM).38 Ekonomiuk etal. identified two compounds with bisbenzamidine andguanidino-benzyl group by a fragment-based docking withIC50 values of 2.8 and 34.2 μM against WNV NS2B-NS3pro,respectively.39 These small nonpeptidic compounds arepotential lead candidates with ligand efficiencies of 0.34 and0.28 for WNV protease. Compounds 33a and 34a showed onlynegligible activity against WNV, but we obtained ligandefficiencies of 0.21 and 0.2040 against DENV NS2B-NS3pro.Therefore, 33a and 34a have lower ligand efficiencies than thecompounds reported by Ekonomiuk et al., however, regardingtargets like DENV protease, larger compounds appear to be theonly option to obtain significant binding energies due to thelarge and predominantly solvent-exposed active site.41
To confirm that para-amidinophenylalanine (33a) is themost promising N-terminal residue, we also investigatedvariations at the meta-position of the phenylalanine substituent.Shifting the side chain moieties from para- to meta-positionresulted in loss of activity for the most active analogues (32b,33b, and 34b). The effect of meta-substitution for pyridyl-(29b), amino- (30b), and nitrile residues (31b) in relation tothe para-substituted derivatives is low. For compound 32b, a 3-fold lower target affinity compared to the para-substitutedanalogue 32a was observed. Surprisingly, the activity profile ofthe meta-substituted phenylalanine analogues 33b and 34b is
significantly different. Whereas all para-substituted compoundshave only negligible affinity against thrombin, 33b and 34bshow an increase in thrombin activity, accompanied by a 7-foldand 3-fold loss of inhibitory potency against DENV proteasecompared to the para analogues. Previous work bySturzebecher et al. already identified m-amidinophenylalaninederivatives as promising scaffolds for thrombin inhibitors,42
underlining these findings. In addition, we shortened the sidechain of phenylalanine to phenylglycine, a (so far) underex-plored non-natural amino acid. We synthesized 37, whereby (3-guanidino)-L-phenylglycine closely resembles the side chainlength of arginine and includes the aromatic residue ofphenylalanine analogues, which showed promising activitiesfor DENV protease in this study. This exchange of the basicresidue resulted in an inhibitory potency (IC50 = 3.09 μM,Table 1) comparable to the reference compound I. Therefore,the (3-guanidino)-L-phenylglycine moiety can be considered assuitable replacement for the arginine side chain and may berelevant for the design of peptide-based inhibitors of othertargets with arginine recognition motifs. Unexpectedly, theexchange of (4-amidino)-L-phenylalanine to (4-amidino)-L-phenylglycine resulted in the practically inactive compound35 (relative inhibition: 24.8%, Table 1). A comparison of 35and 37 underlines the preferred length of the side chain. Toconfirm that basic phenylalanine analogues are the mostsuitable alternative for arginine residues, we synthesized 39 and
Table 2. Inhibitory Activity of Peptide Hybrids with Various N-Terminal Caps (General Formula R1-R2-Lys-Phg-NH2) againstDENV and WNV Proteases and Thrombing
aPercent inhibition of the DENV NS2B-NS3pro protease serotype 2 (compd, 50 μM; substrate, 50 μM; Km, 105 μM; enzyme, 100 nM). bIC50values against DENV NS2B-NS3pro protease serotype 2 (substrate concentration, 50 μM). cPercent inhibition of the West Nile virus (WNV)NS2B-NS3pro protease (compd, 50 μM; substrate, 50 μM; Km, 212 μM; enzyme, 150 nM). dPercent inhibition of thrombin (compd, 25 μM;substrate, 50 μM; Km, 16 μM; enzyme, 10 nM). eIC50 values against thrombin (substrate concentration, 50 μM). fPercent inhibition of trypsin(compd, 50 μM; substrate, 50 μM; enzyme, 1 nM). gAll measurements were carried out in triplicate. Standard deviations were ≤10%, n.d. = notdetermined.
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7724

40. Compound 39 with a less basic functionality showed a 20-fold loss of activity, and the nonbasic (4-acetylamino)-L-phenylalanine analogue 40 was completely inactive againstDENV protease.The most promising tripeptide sequences H-(4-amidino)-
Phe-Lys-Phg-NH2 and H-(4-guanidino)Phe-Lys-Phg-NH2 weremerged with 3-trifluoromethyl benzoic acid and 3-phenylpropionic acid as alternative N-terminal caps. The combinationof 3-trifluoromethyl benzoic acid and the sequence R-(4-amidino)Phe-Lys-Phg-NH2 resulted in the most potentcompound of this series (42a, IC50 = 210 nM, Ki = 139 nMagainst dengue protease, Table 2). A phenylpropionic acid capcaused a slight loss of activity (45a, IC50 = 260 nM, Ki = 160nM, Table 2) but still gave a 1.5-fold more active inhibitor than33a. Compared to the reference sequences 41 and 44, a 5- and10-fold improvement by exchanging arginine to (4-amidino)-L-phenylalanine was achieved. Combination of the sequence R-(4-guanidino)Phe-Lys-Phg-NH2 with the selected caps led to amoderate decrease in activity (43 and 46 vs 34a). These resultslend further support to the hypothesis that 4-guanidinophenyl-alanine is less tolerated by dengue protease.Almost all investigated compounds show negligible inhibition
of thrombin, with the highly active dengue protease inhibitor42a displaying an inhibition of 50.3% at 25 μM againstthrombin. Replacement of the benzyl group by the above-mentioned, alternative caps in combination with the sequenceR-(3-amidino)Phe-Lys-Phg-NH2 led to a minor drop in activityagainst dengue protease (42b and 45 b, Table 2). Moreconspicuous is the change of selectivity between dengueprotease and thrombin. Whereas 42b is a very weak thrombininhibitor, a significantly higher thrombin affinity was observedfor the sequence R-(3-amidino)Phe-Lys-Phg-NH2 with 3-phenyl propionate cap (45b, IC50 value of 4.59 μM againstthrombin, Table 2).Docking Studies. For the docking studies, we used the
structure of DENV protease serotype 3 (pdb: 3U1I) reportedby Noble et al.43 to obtain an idea of possible interactions ofthe inhibitors with the target protein. 3U1I is the only availablestructure of the NS2B-NS3 complex of dengue protease with asmall-molecular cocrystallized inhibitor. The serotype 2 and 3proteases have an identical substrate specificity and a highdegree of conservation of the active sites. We thereforeconsidered it appropriate to use the 3U1I structure for
modeling studies. Docking simulations were performed with aselection of compounds covering a wide range of activities.However, no significant differences in the binding mode ordocking scores could be observed. We therefore conclude thatdocking/scoring simulations are unable to capture subtledifferences in ligand recognition that are responsible for thevariation of affinity, at least in the relatively high affinity rangewhich we observe in the present data set. For visualizationpurposes, and as representative examples, we chose compounds42a (highest affinity) and 43 (see Figure 2). As expected, thebasic residues of both arginine mimetics are located in the S2pocket to interact with Asp75 by electrostatic interactions. Thisinteraction is comparable to the placement of basic residuesobserved in previous docking studies15,19 and the inhibitor−enzyme complex in the 3U1I structure.43 This finding alsomatches the SAR results that a basic residue is required in theS2 pocket. Moreover, the docking study shows the necessity ofthe phenylalanine residue. Whereas a p-amidinophenylglycineresidue (Table 1, compound 35) shows almost no inhibitoryactivity, residues like p-amidinophenylalanines completelyoccupy the S2 pocket, suggesting that the (4-amidino)-L-phenylglycine residue could be sterically hindered. This canexplain the remarkable preference for the substituted phenyl-alanine residues. The side chain of lysine is placed in S1 and theN-terminal cap is in the region of S3. The phenylglycine residueis placed near the S1′ pocket and can interact with ahydrophobic surface.
Aprotinin Assay. To obtain information about the bindingmode and to exclude nonspecific binding, the competitivetryptophan fluorescence assay reported by Bodenreider et al.was used.44 This assay discriminates between specific andnonspecific binders and relies on the intrinsic fluorescence ofTrp50 near the active site of dengue protease. An inhibitor thatbinds specifically to the active site of the enzyme quenches thetryptophan fluorescence if the inhibitor absorbs radiation in therange of the tryptophan emission (305−360 nm). Thefluorescence will be restored if aprotinin displaces the inhibitorfrom the active site. Aprotinin is a known competitive ligand ofdengue protease with relatively high affinity and without anytryptophan residues,43,45 therefore a restoration of Trp50fluorescence by aprotinin indicates a competition between theinhibitors.18,19 However, none of the compounds listed inTable 1 and Table 2 showed sufficient absorption between 305
Figure 2. Docking results for compounds 42a (green) and 43 (magenta) with crystal structure of DENV NS2B-NS3 protease (pdb: 3U1I). (A)Solvent-accessible surface, colored according to the Kyte−Doolittle hydrophobicity of the underlying amino acid (hydrophobic, red; hydrophilic,blue). S1, S2, S3, and S1′ stand for protease subsites occupied by ligand moieties. (B) Ribbon model of the protein in the same orientation as in (A)with highlighted side chains of the catalytic triad. The protease domain is colored in red and the cofactor in blue. The figures were generated usingthe Chimera software.58
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7725

and 360 nm, and these were therefore not suitable for theaprotinin competition assay. We therefore synthesized inhibitor47 (Table S1), with the most active sequence of (4-amidino)Phe-Lys-Phg-NH2 and 4-aminobenzoic acid as N-terminal cap. Abz is a commonly used building block ofsubstrates for fluorescence-based assays. With an IC50 value of1.8 μM, the activity is weaker than the most active compounds42a and 45a, but nevertheless 47 can be considered as arepresentative example. 47 causes a concentration-dependentnonlinear decrease of the Trp fluorescence in dengue protease(Figure S1). Addition of aprotinin causes a restoration offluorescence. This indicates a specific binding of the inhibitorsat the active site. Furthermore, the Hill slopes between 0.8 and1.7 (Table 1 and 2) indicate a specific inhibition.Antiviral Activity in Cell Culture and Membrane
Permeability. The antiviral activity of five selected com-pounds, shown in Table 3, was determined in dengue virusserotype 2 infected human hepatocarcinoma cells (Huh-7) at acompound concentration of 50 μM (plaque assay). Nocytotoxic effects were observed at this concentration of testcompounds. The highest antiviral activity was observed for 45awith a reduction of virus replication of 58.8%, whereas thereference I and 33a, 42a, and 46 showed no relevant antiviralactivity (percent inhibition <50%). This may be due to thebasic residues of (4-amidino)-L-phenylalanine and (4-guanidi-no)-L-phenylalanine which increase the polarity of thecompounds and may hinder cellular uptake.Fifteen compounds, including the five compounds tested in
cells, were screened for their permeability with the parallelartificial membrane permeability assay (PAMPA) procedure.PAMPA is an easily applicable, in vitro permeability model thatpredicts the passive biomembrane permeability of substances.46
None of the tested compounds had a detectable, passivemembrane permeability (Table S2). The weak permeability canbe explained by the polarity of peptidic compounds with twopositively charged residues in P1 and P2. Lack of passivepermeability may partially explain the low antiviral activity incells and suggests to orient further exploration toward residueswith lower polarity. For example, p-amidinophenylalanines maybe masked as amidoxime prodrugs.Metabolic Stability of Selected Compounds in Liver
Microsomes. Peptidic compounds are sensitive toward
metabolic clearance by enzymes in blood, kidney, liver, andother organs.47 Hepatic metabolism is a major eliminationroute and can be assayed with liver microsomes. These containphase 1 metabolic enzymes which generate polar metabolites byhydrolysis, reduction, and oxidation.48 We studied themetabolic stability of compounds 45a and 46 in vitro usingliver microsomes from rats, along with testosterone as referencecompound. The samples were incubated for 30 min at 37 °C,and the loss of the parent compound was monitored. The half-times were: 45a, 22 min; 46, 18 min; testosterone, 63 min.Given the short halftime of the compounds in liver micro-somes, the lack of activity in the hepatocyte-based viralreplication assay may be due to fast metabolic clearance inthese cells. This suggests that further consideration should begiven to structural changes which increase the metabolicstability of the compounds such as modifications within thepeptide backbone structure.
■ CONCLUSIONIn this study, we evaluated the suitability of para- and meta-substituted phenylalanine and phenylglycine analogues as non-natural fragments for the discovery of dengue proteaseinhibitors. Straightforward syntheses on Rink amide resinfurnished novel, peptidic dengue protease inhibitors witharginine-mimicking side chains. Using reactions on solidsupport, we obtained all synthesized peptides in high puritythrough a fast and elegant method. We found that the exchangeof arginine to p-amidinophenylalanine results in a 10-fold morepotent peptidic inhibitor (33a) for dengue protease comparedto reference I. Incorporation of the (4-amidino)-L-phenyl-alanine residue leads to a more potent inhibitor with higherligand efficiency that does not contain the potentiallypromiscuous 2,4-thiazolidinedione “cap”.15,19 The most potentdengue protease inhibitors 42a and 45a showed lower activityagainst WNV protease and no significant activity againstthrombin and trypsin. An unexpected but significant observa-tion is the reduced activity of meta-substituted phenylalanineanalogues. Despite this, we identified (4-acetylamino)-L-phenylalanine as potential, nonbasic arginine replacement forWest Nile virus protease inhibitors. In summary, we presentoptimized peptidic inhibitors of dengue protease with lowpromiscuity. Future work will aim on the further increase of
Table 3. Results of Dengue Virus Plaque Assay for Selected Compounds (General Formula R1-R2-Lys-Phg-NH2)
aIC50 values against DENV NS2B-NS3pro protease serotype 2 (substrate concentration, 50 μM). bPercent inhibition of viral replication in Huh-7cells (compd, 50 μM).
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7726

target affinity and selectivity, along with the development of aprodrug strategy for arginine mimetics to improve pharmaco-kinetic properties and antiviral activity in cells.
■ EXPERIMENTAL SECTIONDENV and WNV Protease Expression and Purification. The
dengue virus (serotype 2) and West Nile virus NS2B-NS3proproteases were expressed and purified according to the protocoldescribed before.49,50
Protease Assays. All protease assays were performed in 96 well V-bottom plates (Greiner Bio-One, Germany) and monitored using aBMG Labtech Fluostar OPTIMA microtiter fluorescence plate reader.The activity of the enzymes were determined as the slope per second(RFU/s) and monitored for 15 min. All determinations of percentageinhibition were calculated in relation to a positive control (withoutinhibitor) and performed in triplicate.DENV and WNV protease assays were performed as reported
before.18,19,50−53 In short, the continuous enzymatic assay wasperfomed at an excitation wavelength of 320 nm and monitored atan emission wavelength of 405 nm. The assay buffer was used asdescribed before as 50 mM Tris-HCl (pH 9.0), 10% ethylene glycol,and 0.0016% Brij 58.50 The inhibitors (final concentration: 50 μM)were preincubated for 15 min with DEN protease (100 nM) or WNVprotease (150 nM), followed by the addition of substrate to initiate thereaction (final concentration: 50 μM). FRET substrates Abz-Nle-Lys-Arg-Arg-Ser-3-(NO2)Tyr and Abz-Gly-Leu-Lys-Arg-Gly-Gly-3-(NO2)-Tyr were used for DEN and WNV protease, respectively.The thrombin assay was performed as described previously.19,54 In
brief, continuous fluorimetric assay were performed at an excitationwavelength of 355 nm and emission wavelength of 460 nm. The assaybuffer was used according to the literature as 50 mM Tris-HCl (pH7.5), 150 mM NaCl, and 0.05% Tween 20.54 The inhibitors (finalconcentration: 25 μM) were preincubated with thrombin (10 nM) for15 min. The addition of boc-Val-Pro-Arg-AMC substrate (finalconcentration: 50 μM; Bachem, Germany) initiated the reaction.The inhibition of catalytic activity of trypsin was determined by a
continuous fluorimetric assay at an excitation wavelength of 355 nmand emission wavelength of 460 nm. The inhibitors (finalconcentration: 50 μM) were preincubated with trypsin (1 nM) for15 min by using the buffer described for the thrombin assay. Theaddition of boc-Val-Pro-Arg-AMC substrate (final concentration: 50μM; Bachem, Germany) initiated the reaction.IC50 and Ki Determination. The assays were generally performed
as described before. Further inhibitor concentrations (IC50) wereperformed for compounds with a percent inhibition of at least 70%.The determination of IC50 was performed by using different inhibitorconcentrations (method A: 0, 0.125, 0.25, 0.5, 1, 1.5, 2, 2.5 μM;method B: 0, 0.5, 1, 2, 3, 4, 5, 6 μM; method C: 0, 2.5, 5, 10, 15, 20,25, 30 μM) and 50 μM substrate in triplicate. IC50 values werecalculated by Prism 6.0 (GraphPad Software, Inc.). Ki determinationwas performed by using inhibitor concentrations of method A andsubstrate concentrations of 50, 100, 150, and 200 μM in triplicate. Kivalues were determined by plotting IC50 values against substrateconcentrations (Cheng−Prusoff equation).51,55,56
Docking. The calculations were performed on an Intel(R)Core(TM)2 Quad CPU Q9450 @ 2.66 GHz running open SuSE11.0, using GOLD 5.2 and its graphical interface Hermes 1.6.57 Theligands were prepared using Chem3D Pro (PerkinElmer). The crystalstructure of the NS2B-NS3 protease of DEN serotype 3 in complexwith a tetrapeptidyl aldehyde was extracted from the dimer structure3U1I.43 All waters and sulfate ions were removed, and the structurewas further prepared using Dock Prep (Chimera).58 All hydrogenswere added, and the tetrapeptidyl aldehyde ligand was extracted fromthe structure. The binding site was defined in a radius of 10 Å aroundthe extracted ligand. Ten different solutions were calculated for everyligand, and the docking results with the highest (42a) and second-highest (43) GOLD scores were aligned and visualized usingChimera.58
Aprotinin Competition Assay. The tryptophan quenching assaywas performed as described by Bodenreider et al.44 DENV protease (4μM) was incubated with inhibitor 47 at different concentrations (5, 10,20, 30, and 50 μM) by using the assay buffer as described previously.Moreover, the protease was incubated with the inhibitor (50 μM) andaprotinin (10 μM) together. Furthermore, as a negative control, theauto fluorescence of the enzyme and aprotinin without inhibitor (0μM) at the same concentrations was determined. The fluorescence ofthe assay samples was monitored on a Tecan Safire II instrument usingan excitation wavelength of 280 nm and an emission wavelength of320 nm. All determinations were performed in triplicate.
Cell Viability Assay (Cytotoxicity Assay). Huh-7 cells wereseeded into 96-well plates (Greiner BioOne, Germany) in 50 μL ofDMEM (Dulbecco’s Modified Minimal Essential Medium (LifeTechnologies, Germany)) (104 cells/well) supplemented with 10%fetal bovine serum. After overnight incubation at 37 °C, the mediumwas replaced with DMEM containing the respective concentration ofthe tested compound. Each concentration was assayed in triplicate.Cells were infected immediately with WT DENV serotype 2 with anMOI (multiplicity of infection) of 1 in the presence of the compound.After incubation for 48 h at 37 °C, the medium was harvested and thetriplicates were pooled and stored at −80 °C. Then 50 μL of freshDMEM was added to the cells and cell viability was determined usingCell-Titer Glo Luminescent Viability Assay. The highest nontoxicconcentration was used for determination of the virus yield reductionby plaque assay using Vero E6 cells.
Virus Titer Reduction Assay (Plaque Assay). Vero E6 cells wereseeded into 24-well plates (Greiner BioOne, Germany) with a densityof 2.5 × 105 cells per well in DMEM supplemented with 10% fetalbovine serum. After overnight incubation at 37 °C, the cells wereinoculated with the virus-containing supernatants that were dilutedwith DMEM ranging from 10−1 to 10−6 prior to infection. Afterincubation of the cells with 100 μL of the virus-containing dilution at37 °C with agitation for 1 h, the medium was removed and 1 mL ofplaque medium was added. After further incubation for 7 days at 37°C, the cells were fixed with 5% (v/v) formaldehyde for 2 h, stainedwith 1% (w/v) crystal violet in 10% (v/v) ethanol, and plaques werecounted. The virus titer reduction was calculated as percentage ofplaques relative to an untreated control.
Parallel Artificial Membrane Permeability Assay (PAMPA).PAMPA was performed as described before.19 In short, precoatedPAMPA plate systems (BD Bioscience, Germany) were used toanalyze permeability coefficient of compunds 32a−b, 33a−b, 34a−b,37, 39, 40, 42a−b, 43, 44, 45a−b, and 46. The measurements wereperformed on Jasco HPLC-system with UV detector and RP-18column (ReproSil-Pur-ODS, Dr. Maisch GmbH, Germany, 3 μm, 50mm × 2 mm) using the method: eluent A, water (+0.1% TFA); eluentB, acetonitrile (+0.1% TFA); flow rate, 1 mL/min; gradient, 1% B (0.2min), 100% B (3.5 min), 100% B (4.5 min), 1% B (4.6 min), 1% B (5min). Phosphate buffered saline (PBS) was used for all experiments.Calibration curves (correlation coefficients were found to be at least0.9) were generated for all compounds and references (amiloride,caffeine, famotidine, furosemide, and phenytoin46) at 10, 25, 50, 100,150, and 200 μM. PAMPA donor plate were filled with 300 μL of the200 μM compound solutions in PBS in triplicate and 200 μL of PBSwere added to wells of PAMPA acceptor plate. The plate system wasincubated at room temperature for 5 h. The analysis was performedwith HPLC by transferring the solutions of donor and acceptor platesinto in 96-well polystyrene U-bottom plates (Greiner Bio-One,Germany) before. The concentrations were calculated using thegenerated calibration curves. Permeability (Pe) and the mass retention(R) were determined as described in literature.46
Metabolic Stability in Liver Microsomes. Pooled liver micro-somes from male Sprague−Dawley were purchased from Sigma-Aldrich (Germany). To determine the metabolic stability, livermicrosomal proteins (0.2 mg/mL) were supplemented withNADPH (5 mM) in Dulbecco’s Phosphate Buffered Saline (DPBS(Life Technologies, Germany)) and preincubated at 37 °C for 15 min.Additionally, test compounds (100 μM) were added and incubated at37 °C for 30 min. Aliquots were removed at various time points (0, 2,
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7727

5, 10, 20, 30 min). The reaction was terminated by the addition ofacetonitrile, and the samples were cooled with ice for 15 min beforecentrifugation (4500g at 4 °C for 15 min). The supernatants were usedfor further analysis. The loss of parent compound was monitored byHPLC on a Jasco HPLC system with UV detector and RP-18 column(ReproSil-Pur-ODS, Dr. Maisch GmbH, Germany, 3 μm, 50 mm × 2mm) using the method: eluent A, water (+0.1% TFA); eluent B.acetonitrile (+ 0.1% TFA); flow rate, 1 mL/min; gradient, 1% B (0.2min), 100% B (3.5 min), 100% B (4.5 min), 1% B (4.6 min), 1% B (5min). Calculation of the metabolic stability was determined by dividingthe peak areas of the unaltered parent compound in the metabolismsample by the peak areas of the parent compound in the referencesample. The activity of the microsomal preparations was verified byusing a positive control (testosterone).Chemistry. All chemicals were purchased from Sigma-Aldrich
(Germany) and Alfa Aesar (Germany) with analytical grade. Aminoacids were purchased from Alfa Aesar (Germany), CarbolutionChemicals (Germany), Orpegen (Germany), Iris Biotech (Germany),and Sigma-Aldrich (Germany). Reaction controls were performed bythin layer chromatography on Merck silica gel plates 60 F254 with UVdetection and ninhydrin reagent. Flash chromatography wasperformed on a Biotage IsoleraOne purification system. NMR spectrawere recorded on Varian NMR instruments at 300 or 500 MHz inCDCl3, CD3OD, acetone-d6 or D2O. Chemical shifts are given in partsper million (ppm), and nondeuterated solvents were used as internalstandard. Coupling constants (J) are given in hertz (Hz). HR-ESI massspectrometry was performed on a Bruker micrOTOF-Q II. Peptidepurity was characterized by analytical HPLC on a Jasco HPLC systemwith UV detector (method A) and an Agilent 1200 HPLC system withMWD detector combined with a Bruker micrOTOF-Q II instrument(method B). A ReproSil-Pur-ODS-3 column, Dr. Maisch GmbH,Germany, 5 μm, 50 mm × 2 mm was used for HPLC. The conditionsfor method A were: eluent A, water (+0.1% TFA); eluent B,acetonitrile (+0.1% TFA); flow rate, 1 mL/min; gradient, 1% B (0.2min), 100% B (3.5 min), 100% B (4.5 min), 1% B (4.6 min), 1% B (5min). The conditions for method B were: eluent A, water (+0.1%formic acid); eluent B, acetonitrile (+0.1% formic acid); flow rate 0.3mL/min, gradient, 5% B (1 min), 95% B (6 min), 95% B (10 min), 5%B (10.1 min), 5% B (12 min). UV detection was performed at 254 nmfor both methods.General Procedures. Procedure A: Synthesis of Peptide Hybrids.
N-Terminal peptide hybrids were synthesized as described previouslywith some modifications.18,53 In short, the peptide sequence wassynthesized analogously to the Fmoc protocol using Rink amide resinand 3.0 equiv of Fmoc protected amino acids, 5.0 equivdiisopropylethylamine, and 3.0 equiv HATU as coupling reagent.Then 25% piperidine in DMF was used for Fmoc removal. After Fmocremoval and coupling steps, the resin was washed with DMF, DCM,and again DMF, respectively. The benzoyl protected amino acids (2.0equiv) were coupled with HATU (2.0 equiv) and diisopropylethyl-amine (2.2 equiv) to the dipeptide sequence. The peptides werecleaved from the resin by standard procedure (92.5% TFA, 5.0% TIPS,and 2.5% water), precipitated in cold diethyl ether, and purified bypreparative RP-HPLC on an AKTApurifier, GE Healthcare(Germany) with an RP-18 column (Rephrospher, Dr. MaischGmbH, Germany, C18-DE, 5 μm, 30 mm × 16 mm and 120 mm ×16 mm). All peptides were freeze-dried and characterized by HR-ESIand 1H NMR for selected compounds (analytical data are provided inthe Supporting Information). All evaluated peptide hybrids wereobtained with a purity of at least 95% unless indicated otherwise.Procedure B: Synthesis of Benzamidine-Containing Peptides
(33a−b, 35, 42a−b, 45a−b). Benzamidine-containing peptides weresynthesized in analogy to the synthesis of amidines describedpreviously.24 In short, peptide synthesis was performed according toprocedure A. The peptide-substituted resin was swelled for 1 h inTHF. Then 10 equiv of a 1 M hydroxylamine hydrochloride solutionwas added and stirred at 60 °C for 16 h. The resin was washed withMeOH, THF, and DMF. Then 15 equiv mL of a 1 M SnCl2/H2O wasadded under nitrogen atmosphere and stirred gently for 3 days at 80°C. The substituted resin was washed with DMF, 5% DIPEA in
MeOH, MeOH, DCM, and DEE. After cleavage and precipitation incold diethyl ether, the peptides were purified by preparative RP-HPLCas described in procedure A. All peptides were freeze-dried andcharacterized by HR-ESI and 1H NMR for selected compounds(analytical data are provided in the Supporting Information). Thepurity was determined by analytical HPLC with method A. Allevaluated peptides were obtained with a purity of at least 95% unlessindicated otherwise.
Procedure C: Synthesis of Bz-(3-guanidino)-Phg-Lys-Phg-NH2(37). The peptide synthesis of 36 (Bz-(3-nitro)-Phg-Lys(boc)-Phg-NH2) was performed according to procedure A with somemodifications. 25 was coupled with HATU (3.5 equiv), HOAt (3.5equiv), and diisopropylethylamine (5.0 equiv) to the dipeptidesequence on solid support. After 1.5 h coupling time, the resin waswashed with DMF, DCM, and again DMF to obtain 36. Then tin(II)chloride dihydrate (60.0 equiv, 815 mg) and diisopropylethylamine(20.0 equiv, 200 μL) in 2 mL of DMF was added to the resin andshaken overnight. The resin was washed with DMF, DCM, and DMF.A mixture of bis-boc-pyrazole-1-carboxamidine (8.0 equiv, 150 mg)and DMAP (3.0 equiv, 20 mg) in 2 mL of DMF was added and shakenovernight. The resin was washed with DMF, DCM, and DEE. Theresin was dried overnight, and the peptide was cleaved using thestandard procedure (92.5% TFA, 5.0% TIPS, and 2.5% water). Theproduct was purified by preparative RP-HPLC as described inprocedure A. The peptide was freeze-dried and characterized byHR-ESI and 1H NMR (analytical data are provided in the SupportingInformation). The purity was determined by analytical HPLC withmethod A. Compound 37 was obtained with a purity of at least 95%.
Synthesis of Bz-(4-acetamidino)-Phe-Lys-Phg-NH2 (39). Bz-(4-DdivNH)Phe-(NHboc)Lys-Phg-NH2 (38) was synthesized accordingto the Fmoc protocol described in procedure A. 37-substituted resin(120 mg; scale, 0.063 mmol) was swelled for 1 h in DCM. Then 2%hydrazine-DMF solution was used to cleave the ddiv group (4 × 15min). Ethylacetimidate hydrochloride59 (16 mg, 0.13 mmol) anddiisopropylethylamine (43 μL, 0.25 mmol) were dissolved in 1 mL ofDMF and filled into the syringe. The reaction mixture was stirred atroom temperature overnight. The substituted resin was washed withDMF, DCM, and DEE. The resin was dried overnight, and the peptidewas cleaved by the standard procedure (92.5% TFA, 5.0% TIPS, and2.5% water). The product was purified by preparative RP-HPLC asdescribed in procedure A. The peptide was freeze-dried andcharacterized by HR-ESI. The purity was determined by analyticalHPLC with method A (analytical data are provided in the SupportingInformation).
Synthesis of Bz-(4-acetylamino)-Phe-Lys-Phg-NH2 (40). 38 wassynthesized according to the Fmoc protocol described in procedure A.The 38-substituted resin (117 mg, scale: 0.063 mmol) was swelled for1 h in DCM. Then 2% hydrazine−DMF solution was used to cleavethe Ddiv group (4 × 15 min). Acetic anhydride (24 μL, 0.25 mmol)and diisopropylethylamine (43 μL, 0.25 mmol) were dissolved in 0.5mL of DMF and filled into the syringe. The reaction mixture wasstirred for 1 h. The substituted resin was washed with DMF, DCM,and DEE. The resin was dried overnight, and the peptide was cleavedby the standard procedure (92.5% TFA, 5.0% TIPS, and 2.5% water).The product was purified by preparative RP-HPLC as described inprocedure A. The peptide was freeze-dried and characterized by HR-ESI and 1H NMR. The purity was determined by analytical HPLCwith method A (analytical data are provided in the SupportingInformation).
Amino Acid Synthesis. Synthesis of (3-Nitro)-L-phenylglycine(22).60,61 To a solution of L-phenylglycine (25 g, 165 mmol) inconcentrated sulfuric acid (80 mL) was added a mixture of nitric acid(65%, 30 mL) and concentrated sulfuric acid (25 mL) dropwise at 0°C. The resulting mixture was kept at 0 °C overnight and then pouredon ice. Aqueous ammonia solution (25%) was added until the pHreached a value of 5. The resulting precipitate was collected andwashed with water and diethyl ether. After recrystallization fromethanol (500 mL), crude 22 was obtained as a colorless solid (yield:15.1 g, 47%). 1H NMR (300 MHz, DMSO-d6): δ = 4.25 (d, J = 6.2
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7728

Hz, 1H), 7.40−7.88 (m, 2H), 8.08−8.40 (m, 2H) ppm. HRMS (ESI):m/z [M + H]+ calcd for C8H9N2O4, 197.0557; found, 197.0559.Procedure D: Synthesis of Methyl Ester Amino Acids.21 (4-
Cyano)-L-phenylalanine Methyl Ester Hydrochloride (8a). Theamino acid (0.50 g, 2.63 mmol) was dissolved in methanol (10 mL)and cooled on ice. Thionyl chloride (0.95 mL, 13.14 mmol) was addeddropwise. The reaction mixture was warmed to room temperature andstirred overnight. The mixture was then concentrated in vacuo. Nofurther purification was necessary (yield: 626 mg, quant). 1H NMR(300 MHz, CD3OD): δ = 7.75 (d, J = 8.37 Hz, 2 H), 7.48 (d, J = 8.37Hz, 2 H), 4.40−4.46 (m, 1 H), 3.81 (s, 3 H), 5.07−5.17 (m, 1 H), 3.38(d, J = 6.17 Hz, 1 H), 3.25 (d, J = 7.19 Hz, 1 H) ppm. HRMS (ESI)m/z [M + H]+ calcd, 205.0972; found, 205.0975.(3-Nitro)-L-phenylglycine Methyl Ester Hydrochloride (23).60
Thionyl chloride (6 mL, 83 mmol) was added dropwise to methanol(60 mL) at 0 °C. The mixture was allowed to warm to roomtemperature before 22 (11.7 mg, 59 mmol) was added. The solutionwas refluxed for 1 h, and the solvent was totally evaporated. Theresulting residue was recrystallized from methanol/diethyl ether toobtain the hydrochloride of 22 as a colorless solid (yield: 8.2 g, 56%).1H NMR (300 MHz, CD3OD): δ = 3.84 (s, 3H), 5.48 (s, 1H), 7.78 (t,J = 8.2 Hz, 1H), 7.92 (m, 1H), 8.37 (ddd, J = 8.2, 2.3, 1.1 Hz, 1H),8.44 (t, J = 2.2 Hz, 1H) ppm. HRMS (ESI): m/z [M + H]+ calcd forC9H11N2O4, 211.0713; found, 211.0718.(4-OH)-L-Phenylglycine Methyl Ester Hydrochloride (13). Synthe-
sized in analogy to 8a. Yellow−orange solid (yield: 736 mg, quant). 1HNMR (300 MHz, CD3OD): δ = 6.87−7.36 (m, 4 H), 5.18 (s, 1 H),3.78 (s, 3 H) ppm. HRMS (ESI) m/z [M + H]+ calcd, 182.0812;found, 182.0812.Procedure E: Synthesis of N-benzoyl Methyl Ester Amino Acids.
N-Benzoyl-(4-cyano)-L-phenylalanine Methyl Ester (9a). A solutionof 8a (720 mg, 2.99 mmol) in 50 mL of dry DCM was chilled on ice-bath (0 °C, N2), and diisopropylethylamine (2.61 mL, 14.96 mmol)and benzoyl chloride (0.43 mL, 3.74 mmol) were added. The reactionmixture was warmed to rt and stirred overnight. The solvent wasremoved in vacuo. The residue was redissolved in water and acidifiedto a pH value of 3. The aqueous phase was extracted with ethyl acetate,and the combined organic layers were washed with brine. Then theorganic solvent was dried under reduced pressure and purified by flashchromatography on silica gel. Yellowish oil (yield: 655 mg, 81%). 1HNMR (300 MHz, CDCl3): δ = 7.70−7.77 (m, 1 H), 7.42−7.62 (m, 3H), 7.27 (d, J = 8.37 Hz, 2 H), 7.26 (d, J = 8.37 Hz, 2 H), 6.65 (d, J =7.05 Hz, 1 H), 5.07−5.17 (m, 1 H), 3.79 (s, 2 H), 3.21−3.47 (m, 1 H).HRMS (ESI) m/z [M + Na]+ calcd, 331.1053; found, 331.1064.N-Benzoyl-(3-nitro)-L-phenylalanine Methyl Ester (2). Synthesized
according to procedure E. Colorless solid (yield: 1.1 g, 40%). 1H NMR(300 MHz, CDCl3): δ = 3.32 (dd, J = 13.8, 5.4 Hz, 1H), 3.47 (dd, J =13.9, 5.7 Hz, 1H), 3.82 (s, 3H), 5.14 (dd, J = 12.7, 5.6 Hz, 1H), 6.71(d, J = 7.1 Hz, 1H), 7.42−7.56 (m, 5H), 7.75 (m, 2H), 8.04 (m, 1H),8.13 (dt, J = 7.3, 2.1 Hz, 1H) ppm. HRMS (ESI): m/z [M + Na]+
calcd for C17H16N2NaO5, 351.0951; found, 351.0957.N-Benzoyl-(3-cyano)-L-phenylalanine Methyl Ester (9b). Synthe-
sized according to procedure E. Yellowish oil, crystallized on standing(yield: 1031 mg, 85%). 1H NMR (300 MHz, CDCl3): δ = 7.71−7.78(m, 2 H), 7.51−7.60 (m, 2 H), 7.48 (s, 1 H), 7.39−7.46 (m, 4 H), 6.69(d, J = 7.05 Hz, 1 H), 5.05−5.14 (m, 1 H), 3.80 (s, 3 H), 3.20−3.42(m, 2 H) ppm. HRMS (ESI) m/z [M + Na]+ calcd, 331.1053; found,331.1054.N-Benzoyl-(3-nitro)-L-phenylglycine Methyl Ester (24). Synthe-
sized according to procedure E. Colorless solid (yield: 1.96 g, 83%).1H NMR (300 MHz, CDCl3): δ = 3.82 (s, 3H), 5.38 (d, J = 6.3 Hz,1H), 7.44 (br d, J = 6.2 Hz, 1H), 7.45−7.60 (m, 4H), 7.85 (m, 3H),8.21 (ddd, J = 8.2, 2.3, 1.1 Hz, 1H), 8.32 (t, J = 2.2 Hz, 1H) ppm.HRMS (ESI): m/z [M + Na]+ calcd for C16H14N2NaO5, 337.0795;found, 337.0802.N-Benzoyl-(4-OH)-L-phenylglycine Methyl Ester (14). Synthesized
according to the general procedure E with some modifications. Asolution of 13 (645 mg, 2.96 mmol) in 50 mL of dry DCM was chilledon ice (0 °C, N2), and DIPEA (2.58 mL, 14.82 mmol) and benzoylchloride (0.43 mL, 3.70 mmol) were added. The reaction mixture was
warmed to room temperature and stirred overnight. The resultingproduct was dried under reduced pressure and dissolved in 25%piperidine/DCM to cleave the benzoyl group. The reaction mixturewas stirred for 3 h and dried under reduced pressure. The residue wasdissolved in water and acidified to a pH value of 3. The aqueous phasewas extracted with ethyl acetate, and the combined organic layers werewashed with brine. Then the organic solvent was dried under reducedpressure and purified by column chromatography on silica gel toobtain a yellowish oil. The oil was crystallized from methylenechloride−hexane to afford the desired product as white solid (yield:504 mg, 60%). 1H NMR (300 MHz, CDCl3): δ = 7.79−7.87 (m, 2 H),7.41−7.58 (m, 3), 7.29 (s, 1 H), 7.19 (d, J = 6.61 Hz, 1 H) 7.25−7.27(m, 1 H), 6.73−6.81 (m, 2 H), 5.69 (d, J = 6.75 Hz, 1 H), 3.78 (s, 3H) ppm. HRMS (ESI) m/z [M + Na]+ calcd, 308.0893; found,308.0894.
N-Benzoyl-(4-aminomethyl)-L-phenylalanine Methyl Ester Ac-etate (10a). 9a (0.35 g, 0.85 mmol) was dissolved in 30 mL of aceticacid and hydrogenated with palladium on carbon (10%, 0.18 g, 0.17mmol) at 40 °C for 2 h. The mixture was filtered through Celite andwashed with acetic acid. The solvent was evaporated under reducedpressure. The resulting product was redissolved in 3 mL of methanolto which cold diethyl ether was added to precipitate the product.Brownish solid (yield: 130 mg, 41%). 1H NMR (300 MHz, acetone-d6): δ = 7.79−7.89 (m, 2 H), 7.37−7.56 (m, 4 H), 7.22−7.30 (m, 3H), 4.88 (dd, J = 8.88, 5.36 Hz, 1 H), 4.40 (s, 2 H), 3.09- 3.31 (m, 2H), 1.95 (s, 3 H) ppm. HRMS (ESI) m/z [M + H]+ calcd, 313.1547;found, 313.1550.
N-Benzoyl-(3-aminomethyl)-L-phenylalanine Methyl Ester Tri-fluoroacetate (10b). 9b (605 mg, 1.96 mmol) was dissolved in 30mL of acetic acid and hydrogenated with palladium on carbon (10%,0.42 g, 0.39 mmol) at 40 °C for 3 h. The mixture was filtered throughCelite and washed with methanol. The solvent was evaporated underreduced pressure. The resulting product was redissolved in water/methanol and purified with RP18-flash chromatography. Colorless oil(yield: 421 mg, 50%). 1H NMR (300 MHz, D2O): δ = 7.71−7.78 (m,2 H), 7.51−7.60 (m, 2 H), 7.48 (s, 1 H), 7.39−7.46 (m, 4 H), 6.69 (d,J = 7.05 Hz, 1 H), 5.05−5.14 (m, 1 H), 3.80 (s, 3 H), 3.20−3.42 (m, 2H) ppm. 19F NMR (282 MHz, D2O): δ = −75.65. HRMS (ESI) m/z[M + Na]+ calcd, 335.1366; found, 335.1370.
Procedure F: Synthesis of N-Benzoyl-(4-amino)-L-phenylalanineMethyl Ester Hydrochloride (19). A mixture of 18 (0.83 g, 2.53 mmol)and palladium on carbon (10%, 0.15 mmol) in 50 mL of methanol andaqueous hydrochloric acid (1N, 5 mL) was stirred at roomtemperature overnight. The mixture was filtered through Celite andwashed with methanol. The solvent was evaporated under reducedpressure. No further purification was necessary. White-yellowish solid(yield: 750 mg, 89%). 1H NMR (300 MHz, CD3OD): δ = 7.69−7.77(m, 2 H), 7.48−7.57 (m, 1 H), 7.39−7.48 (m, 2 H), 7.34 (d, J = 8.37Hz, 2 H), 7.14 (d, J = 8.37 Hz, 2 H), 4.90 (s, 1 H) overlapped bysolvent peak, 3.74 (s, 3 H), 3.35 (d, J = 5.43 Hz, 1 H), 3.11 (dd, J =13.95, 9.84 Hz, 1 H) ppm. HRMS (ESI) m/z [M + H]+ calcd,299.1390; found, 299.1393.
N-Benzoyl-(3-amino)-L-phenylalanine Methyl Ester Hydrochlor-ide (3). Synthesized according to procedure F. Pale-red solid (yield:550 mg, quant). 1H NMR (300 MHz, CD3OD): δ = 3.20 (ddd, J =14.0, 9.9, 1.5 Hz, 1H), 3.42 (dd, J = 14.0, 5.4 Hz, 1H), 3.77 (s, 3H),4.96 (ddd, J = 9.8, 5.4, 1.5 Hz, 1H), 7.27 (m, 1H), 7.36 (m, 1H),7.41−7.58 (m, 5H), 7.74 (m, 2H) ppm. HRMS (ESI): m/z [M + H]+
calcd for C17H19N2O3, 299.1390; found, 299.1395.N-Benzoyl-(3-amino)-L-phenylglycine Methyl Ester Hydrochloride
(26). Synthesized according to procedure F with some modifications.A mixture of 24 (1.7 g, 5.4 mmol) and palladium on carbon (10%, 400mg, 0.38 mmol) in methanol (50 mL) and aqueous hydrochloric acid(1N, 5 mL) was stirred at room temperature under hydrogenatmosphere (1 bar) overnight. The mixture was filtered using Celite,the solvent was evaporated, and the crude hydrochloride 26 wasobtained as a pale-red solid (yield: 1.71 g, 85%). 1H NMR (300 MHz,CD3OD): δ = 3.78 (s, 3H), 5.84 (s, 1H), 7.39 (dt, J = 7.3, 2.0 Hz, 1H),7.44−7.52 (m, 2H), 7.54−7.62 (m, 4H), 7.87 (m, 2H) ppm. HRMS
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7729

(ESI): m/z [M + H]+ calcd for C16H17N2O3, 285.1234; found,285.1237.N-Benzoyl-(4-OTf)-L-phenylglycine Methyl Ester (15). Phenyl
triflimide (524 mg, 1.47 mmol) was added to a solution of 14 (490mg, 1.33 mmol) and diisopropylethylamine (580 μL, 3.33 mmol) in 50mL of dry DCM on ice at 0 °C under N2 atmosphere. The reactionmixture was warmed to room temperature and stirred overnight. Theresulting product was dried under reduced pressure and purified bycolumn chromatography on silica gel to give a colorless oil whichcrystallized on standing at room temperature. Colorless oil (yield: 544mg, quant). 1H NMR (300 MHz, CDCl3): δ = 7.80−7.88 (m, 2 H),7.51−7.60 (m, 3 H), 7.43−7.50 (m, 2 H), 7.28−7.33 (m, 2 H), 5.82(d, J = 6.61 Hz, 1 H), 3.81 (s, 3 H) ppm. 19F NMR (282MHz,CDCl3): δ = −72.87 (CF3) ppm. HRMS (ESI) m/z [M + Na]+
calcd, 440.0386; found, 440.0385.N-Benzoyl-(4-cyano)-L-phenylglycine Methyl Ester (16)..25,26 Bis-
(triphenylphosphine)nickel bromide (94 mg, 0.13 mmol), triphenyl-phosphine (99 mg, 0.38 mmol), zinc powder (91 mg, 1.39 mmol), andpotassium cyanide (98 mg, 1.51 mmol) were added to a Schlenk flaskand heated to 65 °C under a nitrogen atmosphere. 15 (526 mg, 1.26mmol) was dissolved in dry acetonitrile (4 mL) and added dropwise.The reaction mixture was stirred at 65 °C for 3 h. The reactionmixture was then filtered through Celite and diluted with dichloro-methane. The organic layer was washed with brine, dried withmagnesium sulfate, and evaporated under reduced pressure. Theresulting product was purified by flash chromatography. Colorless oil(yield: 90 mg, 24%). 1H NMR (300 MHz, CDCl3): δ = 7.82 (d, J =7.05 Hz, 2 H), 7.54−7.66 (m, 4 H), 7.50 (s, 1 H), 7.39−7.46 (m, 2 H),5.81 (d, J = 6.46 Hz, 1 H), 3.77 (s, 3 H) ppm. HRMS (ESI) m/z [M +Na]+ calcd, 317.0897; found, 317.0887.N-Benzoyl-(3-bis-boc-guanidino)-L-phenylglycine Methyl Ester
(27). A mixture of crude 26 (225 mg, 0.7 mmol), bis-boc-pyrazole-1-carboxamidine (235 mg, 0.75 mmol), DMAP (50 mg, 0.4 mmol),and DIPEA (0.15 mL, 0.9 mmol) in methanol (25 mL) was stirred 72h at room temperature. The solvent was evaporated, and the residuewas purified by flash chromatography (cyclohexane/ethyl acetate) toobtain 27 as colorless solid (yield: 300 mg, 81%). 1H NMR (300 MHz,CDCl3): δ = 1.50 (s, 9H), 1.55 (s, 9H), 3.79 (s, 3H), 5.76 (d, J = 7.1Hz, 1H), 7.17 (br d, J = 7.2 Hz, 1H), 7.22 (br d, J = 7.8 Hz, 1H), 7.36(t, J = 7.8 Hz, 1H), 7.42−7.59 (m, 4H), 7.69 (br d, J = 8.1 Hz, 1H),7.84 (m, 2H) ppm. HRMS (ESI): m/z [M + Na]+ calcd forC27H34N4NaO7, 549.2320; found, 549.2317.N-Benzoyl-(4-DdivNH)-L-phenylalanine Methyl Ester (20).28 To a
stirred suspension of 19 (0.74 g, 2.48 mmol) and 2-isovaleryldimedone(1.6 mL, 7.44 mmol) in 15 mL of ethanol, trifluoracetic acid (19 μL,0.25 mmol) was added. The reaction mixture was refluxed at 80 °C for3 days. The mixture was then concentrated in vacuo. The residue wasredissolved in ethyl acetate and washed with brine, dried withmagnesium sulfate, and evaporated under reduced pressure. Theresulting product was purified by flash chromatography (yield: 1.23 g,quant). 1H NMR (300 MHz, CDCl3): δ = 7.73 (d, J = 8.07 Hz, 2 H),7.41−7.55 (m, 3 H), 7.05−7.24 (m, 3 H), 6.63 (d, J = 6.90 Hz, 1 H),4.99−5.17 (m, 1 H), 3.78 (s, 3 H), 3.20−3.42 (m, 2 H), 2.90−3.02(m, 1 H), 2.35−2.53 (m, 3 H), 1.82 (dt, J = 13.43, 6.64 Hz, 1 H),1.23−1.35 (m, 2 H), 1.08 (s, 4 H), 0.92−1.03 (m, 2 H), 0.74 (dd, J =6.68, 2.86 Hz, 3 H) ppm; HRMS (ESI) m/z [M + Na]+ calcd,527.2516; found, 527.2522.Procedure G: Boc Protection of N-Benzoyl-(aminomethyl)-L-
phenylalanine Methyl Ester. N-Benzoyl-(4-NHboc-methyl)-L-phenyl-alanine Methyl Ester (11a). 10a (0.156 g, 0.42 mmol) was dissolvedin 10 mL of methanol, and di-tert-butyl dicarbonate (180 μL, 0.84mmol) and sodium hydrogen carbonate (0.15 g, 1.76 mmol) wereadded. The reaction mixture was stirred for 3 days at roomtemperature. The reaction mixture was then evaporated under reducedpressure. The resulting product was dissolved in water and extractedwith ethyl acetate. The combined organic layers were dried withmagnesium sulfate and then evaporated under reduced pressure toafford 11a. Brownish oil (yield: 170 mg, quant). 1H NMR (300 MHz,CDCl3): δ = 7.74 (dd, J = 8.22, 1.03 Hz, 2 H), 7.38−7.57 (m, 3 H),7.16−7.25 (m, 2 H), 7.06−7.14 (m, 2 H), 6.61 (d, J = 7.49 Hz, 1 H),
5.09 (dt, J = 7.45, 5.52 Hz, 1 H), 4.86 (br s, 1 H), 4.28 (s, 2 H), 3.77(s, 3 H), 3.15−3.35 (m, 2 H), 1.46 (s, 8 H) ppm. HRMS (ESI) m/z[M + Na]+ calcd, 435.1890; found, 435.1887.
Procedure H: Methyl Ester Deprotection. N-Benzoyl-(3-NHboc)-L-phenylalanine (5). To a solution of 4 (80 mg, 0.2 mmol) in THF (5mL) at 0 °C was added a cold solution of lithium hydroxide (25 mg, 1mmol) in water (5 mL), and the mixture was stirred at 0 °C for 2 h.After addition of aqueous monosodium phosphate solution (1N, 10mL), aqueous hydrochloric acid (1N) was added dropwise until thepH reached a value of 3. After extraction with ethyl acetate, dryingover magnesium sulfate, and solvent evaporation, crude 5 wasobtained. Yellow oil (yield: 80 mg, quant). 1H NMR (300 MHz,CD3OD): δ = 1.50 (s, 9H), 3.10 (dd, J = 13.8, 9.4 Hz, 1H), 3.30 (m,1H), 4.83 (m, 1H), 6.93 (m, 1H), 7.19 (m, 2H), 7.39−7.54 (m, 4H),7.73 (m, 2H) ppm. HRMS (ESI): m/z [M − H]− calcd forC21H23N2O5, 383.1612; found, 383.1617.
N-Benzoyl-(3-bis-boc-guanidino)-L-phenylalanine (7). Synthe-sized according to 5. Yellow oil (yield: 180 mg, quant). 1H NMR(300 MHz, CD3OD): δ = 1.52 (s, 9H), 1.58 (s, 9H), 3.14 (m, 1H),3.35 (m, 1H), 4.76 (m, 1H), 7.06−7.54 (m, 7H), 7.73 (m, 2H) ppm.HRMS (ESI): m/z [M − H]− calcd for C27H33N4O7, 525.2355; found,525.2341.
N-Benzoyl-(4-NHboc-methyl)-L-phenylalanine (12a). 11a (0.158g, 0.38 mmol) was dissolved in 5 mL of THF and cooled on ice (0°C). Lithium hydroxide (0.05 g, 1.92 mmol) was dissolved in 5 mL ofwater (0 °C). The lithium hydroxide solution was added to the aminoacid solution and stirred on ice for 30 min and then overnight at 4 °C.The reaction mixture was then evaporated under reduced pressure.The resulting product was dissolved in sodium hydrogen phosphate(1.2 g/100 mL). Aqueous hydrochloric acid (1N) was added dropwiseuntil the pH reached a value of 3. The aqueous phase was extractedwith ethyl acetate. The combined organic layers were washed withbrine, dried with magnesium sulfate, and the solvent evaporated underreduced pressure to afford a white-yellowish oil. The oil wascrystallized from methylene chloride−hexane to afford the desiredproduct as a white-yellowish solid (yield: 129 mg, 85%). 1H NMR(300 MHz, CD3OD): δ = 7.67−7.76 (m, 2 H), 7.36−7.55 (m, 3 H),7.15−7.29 (m, 4 H), 4.80−4.86 (m, 1 H), 4.17 (s, 2 H), 3.35 (d, J =4.99 Hz, 1 H), 3.10 (dd, J = 13.87, 9.62 Hz, 1 H), 1.43 (s, 8 H) ppm.HRMS (ESI) m/z [M + Na]+ calcd, 421.1734; found, 421.1733.
N-Benzoyl-(3-NHboc-methyl)-L-phenylalanine (12b). Synthesizedaccording to 12a. White solid (yield: 316 mg, 91%). 1H NMR (300MHz, acetone-d6): δ = 7.83 (d, J = 6.90 Hz, 2 H), 7.75 (d, J = 7.93 Hz,1 H), 7.39−7.56 (m, 3 H), 7.30 (s, 1 H), 7.24 (d, J = 4.55 Hz, 2 H),7.17 (d, J = 4.40 Hz, 1 H), 6.38 (br s, 1 H), 4.86−4.96 (m, 1 H), 4.24(s, 1 H), 3.13−3.37 (m, 2 H), 1.41 (s, 9 H) ppm. HRMS (ESI) m/z[M + Na]+ calcd, 421.1734; found, 421.1737.
N-Benzoyl-(4-cyano)-L-phenylglycine (17). 16 (0.085 g, 0.29mmol) was dissolved in 5 mL of methanol and cooled on ice (0°C). Then 5 mL of a 1 M cesium carbonate solution (0 °C) was addedto the amino acid solution and stirred on ice for 90 min and storedovernight at 4 °C. Methanol was then evaporated under reducedpressure. The remaining aqueous phase was acidified with 1 N HCland extracted with dichloromethane. The combined organic layerswere washed with brine, dried with magnesium sulfate, and the solventevaporated under reduced pressure to afford a colorless oil. The oilwas crystallized from methylene chloride−hexane to afford the desiredproduct as a white solid (yield: 79.3 mg, 98%). 1H NMR (300 MHz,CDCl3): δ = 9.58 (br s, 1 H), 7.77 (d, J = 7.19 Hz, 2 H), 7.57 (q, J =8.61 Hz, 5 H), 7.37−7.46 (m, 2 H), 5.78 (d, J = 6.61 Hz, 1 H) ppm.HRMS (ESI) m/z [M + Na]+ calcd, 303.0740; found, 303.0731.
N-Benzoyl-(4-DdivNH)-L-phenylalanine (21).29 20 (1.2 g, 0.38mmol) was dissolved in 15 mL of methanol and cooled on ice (0 °C).Then 15 mL of a 1 M cesium carbonate solution (0 °C) was added tothe amino acid solution and stirred on ice for 90 min and then at 4 °Covernight. The reaction mixture was evaporated until methanol wasevaporated. The resulting aqueous phase was acidified with 1 N HCland extracted with dichloromethane. The combined organic layerswere washed with brine, dried with magnesium sulfate, and the solventevaporated under reduced pressure to obtain 21 as white-yellowish oil
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7730

(yield: 426 mg, 36%). 1H NMR (300 MHz, CDCl3): δ = 7.72 (d, J =7.93 Hz, 2 H), 7.39−7.56 (m, 3 H), 7.30 (s, 1 H), 7.07 (s, 2 H), 6.70(d, J = 7.05 Hz, 1 H), 5.06−5.16 (m, 1 H), 3.26−3.50 (m, 2 H), 2.91−3.03 (m, 2H), 2.47 (s, 3 H), 1.74−1.90 (m, 1 H), 1.08 (s, 4 H), 0.83−1.00 (m, 5 H), 0.74 (d, J = 6.61 Hz, 4 H) ppm. HRMS (ESI) m/z [M− H]+ calcd, 489.2395; found, 489.2422.N-Benzoyl-(3-NO2)-L-phenylglycine (25). To a solution of 24 (200
mg, 0.64 mmol) in THF (20 mL) at 0 °C was added a cold solution oflithium hydroxide (100 mg, 4 mmol) in water (20 mL), and themixture was stirred at 0 °C for 1 h. Aqueous hydrochloric acid (1N, 10mL) and water (10 mL) were added, and the remaining THF wasevaporated. The resulting precipitate was collected, washed with water,and dried to obtain 25 as a pale-yellow solid (yield: 140 mg, 73%). 1HNMR (300 MHz, CD3OD): δ = 5.88 (s, 1H), 7.48 (m, 2H), 7.57 (m,1H), 7.65 (t, J = 8.0 Hz, 1H), 7.90 (m, 3H), 8.23 (ddd, J = 8.2, 2.3, 1.1Hz, 1H), 8.42 (t, J = 2.2 Hz, 1H) ppm. HRMS (ESI): m/z [M − H]−
calcd for C15H11N2O5, 299.0673; found, 299.0675.N-Benzoyl-(3-bis-boc-guanidino)-L-phenylglycine (28). Synthe-
sized according to 5. Colorless oil (yield: 350 mg, quant). 1H NMR(300 MHz, CD3OD): δ = 1.52 (s, 18H), 5.59 (s, 1H), 7.35−761 (m,7H), 7.88 (m, 2H) ppm. HRMS (ESI): m/z [M − H]− calcd forC26H31N4O7, 511.2198; found, 511.2201.
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.jmed-chem.5b00612.
Inhibitory activity of compound 47 against DENV andWNV proteases, thrombin and trypsin, and the resultsfor the aprotinin competition assay; results of thepermeability testing (PAMPA); detailed procedure forthe synthesis of peptide hybrids, HR-MS and HPLC datafor all synthesized peptides used in biochemical andbiological evaluations, NMR data for selected peptides,and analytical data for synthetic intermediates (PDF)
■ AUTHOR INFORMATIONCorresponding Author*Phone: (+)49-6221-544875. Fax: (+)49-6221-546430. E-mail:[email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe thank Heiko Rudy for performing high resolution MS-ESI,Natascha Stefan for technical support and Mira Behnam forhelpful hints in enzymatic assays and useful discussions. Theauthors appreciate support by the Deutsche Forschungsge-meinschaft, KL-1356/3-1.
■ ABBREVIATIONS USEDAbz, aminobenzoic acid; Ddiv, 1-(4,4-dimethyl-2,6-dioxocyclo-hex-1-ylidene)-3-methylbutyl; DENV, dengue virus; DIPEA,diisopropylethylamine; HATU, O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate; HOAt,1-hydroxy-7-azabenzotriazole; MOI, multiplicity of infection;PAMPA, parallel artificial permeability assay; TFFH, fluoro-N,N,N′,N′-tetramethylformamidinium hexafluorophosphate;THR, thrombin; TRY, trypsin; WNV, West Nile virus
■ REFERENCES(1) Brady, O. J.; Gething, P. W.; Bhatt, S.; Messina, J. P.; Brownstein,J. S.; Hoen, A. G.; Moyes, C. L.; Farlow, A. W.; Scott, T. W.; Hay, S. I.
Refining the global spatial limits of dengue virus transmission byevidence-based consensus. PLoS Neglected Trop. Dis. 2012, 6 (8),e1760.(2) Bhatt, S.; Gething, P. W.; Brady, O. J.; Messina, J. P.; Farlow, A.W.; Moyes, C. L.; Drake, J. M.; Brownstein, J. S.; Hoen, A. G.; Sankoh,O.; Myers, M. F.; George, D. B.; Jaenisch, T.; Wint, G. R. W.;Simmons, C. P.; Scott, T. W.; Farrar, J. J.; Hay, S. I. The globaldistribution and burden of dengue. Nature 2013, 496 (7446), 504−507.(3) Schmidt-Chanasit, J.; Haditsch, M.; Schoneberg, I.; Gunther, S.;Stark, K.; Frank, C. Dengue virus infection in a traveller returning fromCroatia to Germany. Eurosurveillance 2010, 15 (40), 19677.(4) La Ruche, G.; Souares, Y.; Armengaud, A.; Peloux-Petiot, F.;Delaunay, P.; Despres, P.; Lenglet, A.; Jourdain, F.; Leparc-Goffart, I.;Charlet, F. First two autochthonous dengue virus infections inmetropolitan France, September 2010. Eurosurveillance 2010, 15 (39),19676.(5) Nitsche, C.; Holloway, S.; Schirmeister, T.; Klein, C. D.Biochemistry and medicinal chemistry of the dengue virus protease.Chem. Rev. 2014, 114 (22), 11348−11381.(6) Vasilakis, N.; Ooi, M.; Rabaa, M.; Mayer, S.; Widen; Perera, D.;Hickey, A.; Travassos Da Rosa, A.; Bossart, K.; Tesh, R.; Holmes, E.;Cardos, J. The daemon in the forest-emergence of a new dengue serotype inSouth East Asia. In 3rd International Conference on Dengue andDengue Haemorrhagic Fever, Bangkok, 2013.(7) Lim, S. P.; Wang, Q.-Y.; Noble, C. G.; Chen, Y.-L.; Dong, H.;Zou, B.; Yokokawa, F.; Nilar, S.; Smith, P.; Beer, D.; Lescar, J.; Shi, P.-Y. Ten years of dengue drug discovery: Progress and prospects.Antiviral Res. 2013, 100 (2), 500−519.(8) Noble, C. G.; Shi, P.-Y. Structural biology of dengue virusenzymes: Towards rational design of therapeutics. Antiviral Res. 2012,96 (2), 115−126.(9) De Clercq, E. Anti-HIV drugs: 25 compounds approved within25 years after the discovery of HIV. Int. J. Antimicrob. Agents 2009, 33(4), 307−320.(10) Wensing, A. M. J.; van Maarseveen, N. M.; Nijhuis, M. Fifteenyears of HIV protease inhibitors: Raising the barrier to resistance.Antiviral Res. 2010, 85 (1), 59−74.(11) Poordad, F.; McCone, J.; Bacon, B. R.; Bruno, S.; Manns, M. P.;Sulkowski, M. S.; Jacobson, I. M.; Reddy, K. R.; Goodman, Z. D.;Boparai, N.; DiNubile, M. J.; Sniukiene, V.; Brass, C. A.; Albrecht, J.K.; Bronowicki, J.-P. Boceprevir for untreated chronic HCV Genotype1 infection. N. Engl. J. Med. 2011, 364 (13), 1195−1206.(12) Zeuzem, S.; Andreone, P.; Pol, S.; Lawitz, E.; Diago, M.;Roberts, S.; Focaccia, R.; Younossi, Z.; Foster, G. R.; Horban, A.;Ferenci, P.; Nevens, F.; Mullhaupt, B.; Pockros, P.; Terg, R.; Shouval,D.; van Hoek, B.; Weiland, O.; Van Heeswijk, R.; De Meyer, S.; Luo,D.; Boogaerts, G.; Polo, R.; Picchio, G.; Beumont, M. Telaprevir forretreatment of HCV infection. N. Engl. J. Med. 2011, 364 (25), 2417−2428.(13) Liu, H.; Wu, R.; Sun, Y.; Ye, Y.; Chen, J.; Luo, X.; Shen, X.; Liu,H. Identification of novel thiadiazoloacrylamide analogues as inhibitorsof dengue-2 virus NS2B/NS3 protease. Bioorg. Med. Chem. 2014, 22(22), 6344−6352.(14) Tomlinson, S. M.; Watowich, S. J. Anthracene-based inhibitorsof dengue virus NS2B−NS3 protease. Antiviral Res. 2011, 89 (2),127−135.(15) Behnam, M. A. M.; Nitsche, C.; Vechi, S. M.; Klein, C. D. C-Terminal residue optimization and fragment merging: Discovery of apotent peptide-hybrid inhibitor of dengue protease. ACS Med. Chem.Lett. 2014, 5 (9), 1037−1042.(16) Erbel, P.; Schiering, N.; D'Arcy, A.; Renatus, M.; Kroemer, M.;Lim, S. P.; Yin, Z.; Keller, T. H.; Vasudevan, S. G.; Hommel, U.Structural basis for the activation of flaviviral NS3 proteases fromdengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13 (4), 372−373.(17) Yin, Z.; Patel, S. J.; Wang, W.-L.; Chan, W.-L.; Ranga Rao, K. R.;Wang, G.; Ngew, X.; Patel, V.; Beer, D.; Knox, J. E.; Ma, N. L.;Ehrhardt, C.; Lim, S. P.; Vasudevan, S. G.; Keller, T. H. Peptide
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7731

inhibitors of dengue virus NS3 protease. Part 2: SAR study oftetrapeptide aldehyde inhibitors. Bioorg. Med. Chem. Lett. 2006, 16 (1),40−43.(18) Nitsche, C.; Behnam, M. A. M.; Steuer, C.; Klein, C. D. Retropeptide-hybrids as selective inhibitors of the dengue virus NS2B-NS3protease. Antiviral Res. 2012, 94 (1), 72−79.(19) Nitsche, C.; Schreier, V. N.; Behnam, M. A. M.; Kumar, A.;Bartenschlager, R.; Klein, C. D. Thiazolidinone−peptide hybrids asdengue virus protease inhibitors with antiviral activity in cell culture. J.Med. Chem. 2013, 56 (21), 8389−8403.(20) Bastos Lima, A.; Behnam, M. A. M.; El Sherif, Y.; Nitsche, C.;Vechi, S. M.; Klein, C. D. Dual inhibitors of the dengue and West Nilevirus NS2B-NS3 proteases: Synthesis, biological evaluation anddocking studies of novel peptide-hybrids. Bioorg. Med. Chem. 2015,23, 5748−5755.(21) Brenner, M.; Huber, W. Herstellung von α-Aminosaureesterndurch Alkoholyse der Methylester. Helv. Chim. Acta 1953, 36 (5),1109−1115.(22) Steinmetzer, T.; Becker, G.; Garten, W. N-Terminally modifiedtetrapeptide derivatives having a c-terminal arginine mimetic. U.S.Patent 0,312,873, December 22, 2011.(23) Hammamy, M. Z.; Haase, C.; Hammami, M.; Hilgenfeld, R.;Steinmetzer, T. Development and characterization of new peptidomi-metic inhibitors of the West Nile virus NS2B−NS3 Protease.ChemMedChem 2013, 8 (2), 231−241.(24) Cesar, J.; Nadrah, K.; Sollner Dolenc, M. Solid-phase synthesisof amidines by the reduction of amidoximes. Tetrahedron Lett. 2004,45 (40), 7445−7449.(25) Herzner, H.; Kunz, H. (p-Sulfomethyl)phenylalanine as a mimicof O-sulfatyl-tyrosine in synthetic partial sequences of P-Selectinglycoprotein ligand 1 (PSGL-1). Tetrahedron 2007, 63 (28), 6423−6436.(26) Bazewicz, C. G.; Lipkin, J. S.; Lozinak, K. A.; Watson, M. D.;Brewer, S. H. Synthesis of isotopomers of N-(tert-butoxycarbonyl)-4-cyano-l-phenylalanine methyl ester: choice of cyanation solvent.Tetrahedron Lett. 2011, 52 (51), 6865−6868.(27) Takagi, K.; Sakakibara, Y. Nickel(0) or palladium(0)-catalyzedcyanation of aryl triflates. Chem. Lett. 1989, 18 (11), 1957−1958.(28) Chhabra, S. R.; Hothi, B.; Evans, D. J.; White, P. D.; Bycroft, B.W.; Chan, W. C. An appraisal of new variants of Dde amine protectinggroup for solid phase peptide synthesis. Tetrahedron Lett. 1998, 39(12), 1603−1606.(29) Díaz-Mochon, J. J.; Bialy, L.; Bradley, M. Full orthogonalitybetween Dde and Fmoc: The direct Synthesis of PNA−Peptideconjugates. Org. Lett. 2004, 6 (7), 1127−1129.(30) Chappell, K. J.; Stoermer, M. J.; Fairlie, D. P.; Young, P. R.Insights to substrate binding and processing by West Nile virus NS3Protease through combined modeling, protease mutagenesis, andkinetic studies. J. Biol. Chem. 2006, 281 (50), 38448−38458.(31) Gouvea, I. E.; Izidoro, M. A.; Judice, W. A. S.; Cezari, M. H. S.;Caliendo, G.; Santagada, V.; dos Santos, C. N. D.; Queiroz, M. H.;Juliano, M. A.; Young, P. R.; Fairlie, D. P.; Juliano, L. Substratespecificity of recombinant dengue 2 virus NS2B-NS3 protease:Influence of natural and unnatural basic amino acids on hydrolysisof synthetic fluorescent substrates. Arch. Biochem. Biophys. 2007, 457(2), 187−196.(32) Schuller, A.; Yin, Z.; Brian Chia, C. S.; Doan, D. N. P.; Kim, H.-K.; Shang, L.; Loh, T. P.; Hill, J.; Vasudevan, S. G. Tripeptideinhibitors of dengue and West Nile virus NS2B−NS3 protease.Antiviral Res. 2011, 92 (1), 96−101.(33) Clement, B. Reduction of N-hydroxylated compounds:Amidoximes (N-Hydroxyamidines) as Pro-Drugs of amidines. DrugMetab. Rev. 2002, 34 (3), 565−579.(34) Clement, B.; Lopian, K. Characterization of in vitrobiotransformation of new, orally active, direct thrombin inhibitorXimelagatran, an amidoxime and ester Prodrug. Drug Metab. Dispos.2003, 31 (5), 645−651.(35) Brandstetter, H.; Turk, D.; Hoeffken, H. W.; Grosse, D.;Sturzebecher, J.; Martin, P. D.; Edwards, B. F. P.; Bode, W. Refined 2·
3ÅX-ray crystal structure of bovine thrombin complexes formed withthe benzamidine and arginine-based thrombin inhibitors NAPAP, 4-TAPAP and MQPA: A starting point for improving antithrombotics. J.Mol. Biol. 1992, 226 (4), 1085−1099.(36) Sturzebecher, J.; Vieweg, H.; Steinmetzer, T.; Schweinitz, A.;Stubbs, M. T.; Renatus, M.; Wikstrom, P. 3-Amidinophenylalanine-based inhibitors of urokinase. Bioorg. Med. Chem. Lett. 1999, 9 (21),3147−3152.(37) Hammami, M.; Ruhmann, E.; Maurer, E.; Heine, A.; Gutschow,M.; Klebe, G.; Steinmetzer, T. New 3-amidinophenylalanine-derivedinhibitors of matriptase. MedChemComm 2012, 3 (7), 807−813.(38) Mueller, N. H.; Yon, C.; Ganesh, V. K.; Padmanabhan, R.Characterization of the West Nile virus protease substrate specificityand inhibitors. Int. J. Biochem. Cell Biol. 2007, 39 (3), 606−614.(39) Ekonomiuk, D.; Su, X.-C.; Ozawa, K.; Bodenreider, C.; Lim, S.P.; Otting, G.; Huang, D.; Caflisch, A. Flaviviral protease inhibitorsidentified by fragment-based library docking into a structure generatedby molecular dynamics. J. Med. Chem. 2009, 52 (15), 4860−4868.(40) Ligand efficiency was calculated as LE = 1.4(−log IC50)/(non-hydrogen bonds) in kcal/mol according to Hopkins, A. L.; Groom, C.R.; Alex, A. Ligand efficiency: a useful metric for lead selection. DrugDiscovery Today 2004, 9 (10), 430−431.(41) Perola, E. An analysis of the binding efficiencies of drugs andtheir leads in successful drug discovery programs. J. Med. Chem. 2010,53 (7), 2986−2997.(42) Sturzebecher, J.; Prasa, D.; Hauptmann, J.; Vieweg, H.;Wikstrom, P. Synthesis and structure−activity relationships of potentthrombin inhibitors: piperazides of 3-amidinophenylalanine. J. Med.Chem. 1997, 40 (19), 3091−3099.(43) Noble, C. G.; Seh, C. C.; Chao, A. T.; Shi, P. Y. Ligand-boundstructures of the dengue virus protease reveal the active conformation.J. Virol. 2012, 86 (1), 438−446.(44) Bodenreider, C.; Beer, D.; Keller, T. H.; Sonntag, S.; Wen, D.;Yap, L.; Yau, Y. H.; Shochat, S. G.; Huang, D.; Zhou, T.; Caflisch, A.;Su, X.-C.; Ozawa, K.; Otting, G.; Vasudevan, S. G.; Lescar, J.; Lim, S.P. A fluorescence quenching assay to discriminate between specific andnonspecific inhibitors of dengue virus protease. Anal. Biochem. 2009,395 (2), 195−204.(45) Aleshin, A. E.; Shiryaev, S. A.; Strongin, A. Y.; Liddington, R. C.Structural evidence for regulation and specificity of flaviviral proteasesand evolution of the Flaviviridae fold. Protein Sci. 2007, 16 (5), 795−806.(46) Chen, X.; Murawski, A.; Patel, K.; Crespi, C.; Balimane, P. Anovel design of artificial membrane for improving the PAMPA model.Pharm. Res. 2008, 25 (7), 1511−1520.(47) Craik, D. J.; Fairlie, D. P.; Liras, S.; Price, D. The future ofpeptide-based drugs. Chem. Biol. Drug Des. 2013, 81 (1), 136−147.(48) Yan, Z.; Caldwell, G. W. Metabolic assessment in livermicrosomes by co-activating cytochrome P450s and UDP-glycosyl-transferases. Eur. J. Drug Metab. Pharmacokinet. 2003, 28 (3), 223−232.(49) Steuer, C.; Gege, C.; Fischl, W.; Heinonen, K. H.;Bartenschlager, R.; Klein, C. D. Synthesis and biological evaluationof α-ketoamides as inhibitors of the dengue virus protease withantiviral activity in cell-culture. Bioorg. Med. Chem. 2011, 19 (13),4067−4074.(50) Steuer, C.; Heinonen, K. H.; Kattner, L.; Klein, C. D.Optimization of assay conditions for dengue virus protease: Effect ofvarious polyols and nonionic detergents. J. Biomol. Screening 2009, 14(9), 1102−1108.(51) Copeland, R. A. Enzymes: A Practical Introduction to Structure,Mechanism, And Data Analysis. 2nd ed.; John Wiley & Sons: NewYork, 2000; pp 305−317.(52) Nitsche, C.; Steuer, C.; Klein, C. D. Arylcyanoacrylamides asinhibitors of the dengue and West Nile virus proteases. Bioorg. Med.Chem. 2011, 19 (24), 7318−7337.(53) Nitsche, C.; Klein, C., Fluorimetric and HPLC-based denguevirus protease assays using a FRET substrate. In Antiviral Methods and
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7732

Protocols; Gong, E. Y., Ed.; Humana Press: Totowa, NJ, 2013; Vol.1030, pp 221−236.(54) Diamond, S. L. Thrombin 1536 HTS, Assay ID 1046. PubChemOpen Chemistry Database; National Center for BiotechnologyInformation, U.S. National Library of Medicine: Bethesda, MD,2008; http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1046 (ac-cessed May 26, 2015).(55) Cheng, Y.-C.; Prusoff, W. H. Relationship between theinhibition constant (KI) and the concentration of inhibitor whichcauses 50% inhibition (I50) of an enzymatic reaction. Biochem.Pharmacol. 1973, 22 (23), 3099−3108.(56) Copeland, R. A.; Lombardo, D.; Giannaras, J.; Decicco, C. P.Estimating KI values for tight binding inhibitors from dose-responseplots. Bioorg. Med. Chem. Lett. 1995, 5 (17), 1947−1952.(57) Jones, G.; Willett, P.; Glen, R. C.; Leach, A. R.; Taylor, R.Development and validation of a genetic algorithm for flexibledocking1. J. Mol. Biol. 1997, 267 (3), 727−748.(58) Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.;Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. UCSF ChimeraAvisualization system for exploratory research and analysis. J. Comput.Chem. 2004, 25 (13), 1605−1612.(59) Jernigan, F. E.; Sieracki, N. A.; Taylor, M. T.; Jenkins, A. S.;Engel, S. E.; Rowe, B. W.; Jove, F. A.; Yap, G. P. A.; Papish, E. T.;Ferrence, G. M. Sterically bulky tris(triazolyl)borate ligands as water-soluble analogues of tris(pyrazolyl)borate. Inorg. Chem. 2007, 46 (2),360−362.(60) Qiu, J.; Xu, B.; Huang, Z.; Pan, W.; Cao, P.; Liu, C.; Hao, X.;Song, B.; Liang, G. Synthesis and biological evaluation of Matijing-Suderivatives as potent anti-HBV agents. Bioorg. Med. Chem. 2011, 19(18), 5352−5360.(61) Sassatelli, M.; Debiton, E.; Aboab, B.; Prudhomme, M.; Moreau,P. Synthesis and antiproliferative activities of indolin-2-one derivativesbearing amino acid moieties. Eur. J. Med. Chem. 2006, 41 (6), 709−716.
Journal of Medicinal Chemistry Article
DOI: 10.1021/acs.jmedchem.5b00612J. Med. Chem. 2015, 58, 7719−7733
7733





![RESEARCH ARTICLE Open Access Arginine deiminase …L-arginine following a modified method using diacetyl monoxime thiosemicarbazide [32]. One unit of ADI ac-tivity is defined as the](https://static.fdocument.pub/doc/165x107/607a00dc980f9628c81f6843/research-article-open-access-arginine-deiminase-l-arginine-following-a-modified.jpg)












