Paralelní výpočet SVD s aplikacemi pro vyhledávání informací

PARALELNÍ VÝPOČTYV MOLEKULÁRNÍ DYNAMICE
Vladimír Pelikán, Petr HoraÚstav termomechaniky AV ČR, v.v.i.

Paralelnívýpočtyvmolekulárnídynamice
Molekulární dynamika - co to je?
• technika počítačové simulace, kde je časový vývoj množiny interagujícíchatomů popsán integrací jejich pohybových rovnic.
• vychází ze zákonů klasické mechaniky,především Newtonových pohybových rovnic.
• jedná se o deterministickou techniku (rozdíl oproti metodě Monte Carlo).Počáteční množina pozic a rychlostí zcela určuje následný časový vývoj.
• jedna z metod statistické mechaniky.
1

Paralelnívýpočtyvmolekulárnídynamice
Historie molekulární dynamiky
1957 Alder a WainwrightPrvní článek referující o MD-simulaci.Fázový diagram systému tuhých koulí.Počítač UNIVAC a IBM 704.
1960 Gibson, Goland, Milgram a VineyardPrvní použití spojitého potenciálua časové integrační metody konečných diferencí.Počítač IBM 704, 500 atomů, jeden časový krok trval asi minutu.
1964 RahmanVlastnosti tekutého argonu s použitím Lennardova-Jonesova potenciálu.Počítač CDC 3600, 864 atomů.
1967 VerletFázový diagram argonu s pomocí Lennardovo-Jonesova potenciálu.Poprvé uveden Verletův seznam sousedů.Mimoto byl použit Verletův časový integrační algoritmus.
2

Paralelnívýpočtyvmolekulárnídynamice
Aplikace molekulární dynamiky
Tekutiny. Dostupnost nových realistických interakčních modelů umožňuje stu-dovat nové systémy, elementární i vícesložkové. Pomocí nerovnovážnýchtechnik jsou vyšetřovány transportní jevy (např. viskózita) a tepelné toky.
Defekty. Pozornost se přesouvá z bodových defektů (vakance, intersticiálníporucha) k lineárním (dislokace) a plošným defektům (hranice zrn, vrstevnéchyby). Reálnějších modelů je dosaženo díky lepším potenciálům.
Lomy. Lomový proces může v závislosti na příslušných parametrech nastatrůznými způsoby a probíhat různou rychlostí. Technická závažnost je zřejmáa simulace poskytuje proniknutí k podstatě.
Povrchy. Simulace stále hraje velkou roli při pochopení jevů jako např. rekon-strukce povrchu, povrchové tavení, povrchová difúze, zdrsňování, atd. Tytosimulace často vyžadují rozsáhlé vzorky a dlouhé simulační časy.
3

Paralelnívýpočtyvmolekulárnídynamice
Tření. Vývoj atomového silového mikroskopu (AFM) podnítil výzkumy adhezea tření mezi dvěma pevnými látkami. Souhrn makroskopických znalostí jekorigován a rozšiřován o mikroskopické základy.
Klastry. Klastry, tj. konglomeráty mnoha atomů, představují most mezi mole-kulárními systémy a pevnou fází a vykazují pozoruhodné vlastnosti. Klastrykovů jsou díky své úloze katalyzátoru v důležitých chemických reakcích (např.katalyzátory v automobilech) zvláště důležité z technologického hlediska.
Biomolekuly. Molekulární dynamika umožňuje studovat dynamiku rozsáhlýchmakromolekul, včetně biologických systémů jako např. proteiny, nukleovékyseliny (DNA, RNA) a membrány. Dynamické jevy mohou hrát klíčovouúlohu při regulačních procesech, které ovlivňují funkční vlastnosti biomolekul.Simulace léků je běžně používaná ve farmaceutickém průmyslu.
Elektronové vlastnosti a dynamika. Vývoj Carovy-Parrinellovy metody, přikteré jsou síly působící na atomy získány řešením problému elektronovéstruktury místo meziatomového potenciálu, umožňuje plně studovat vlast-nosti materiálů včetně jejich dynamiky (a tedy fázových přechodů a jinýchteplotně závislých jevů).
4

Paralelnívýpočtyvmolekulárnídynamice
Základní aparát molekulární dynamiky
• Hlavní součástí simulace je model fyzikálního systému.
• Pro simulace MD to znamená volbu potenciálu.
Potenciál: Funkce pozic atomů V (r1, . . . , rN), která představuje potenciálníenergii systému atomů uspořádaných v dané konfiguraci.Nejjednodušší tvar pro V je součet párových interakcí:
V (r1, . . . , rN) =∑
i
∑j>i
φ(|ri − rj|).
• Síly jsou odvozeny jako gradienty potenciálu vzhledem k výchylkám atomů:
F i = −∇riV (r1, . . . , rN).
5

Paralelnívýpočtyvmolekulárnídynamice
Lennardův-Jonesův potenciál 12-6
φLJ(r) = 4ε
[(σ
r
)12−
(σ
r
)6]
σ
−ε
0
r
φLJ
(r)
6√
2σ
r
6

Paralelnívýpočtyvmolekulárnídynamice
Postup simulace
1. Zahájení simulace
2. Řízení simulace
3. Získání výsledků ze simulace
7

Paralelnívýpočtyvmolekulárnídynamice
1. Zahájení simulace
1. Definovat pomyslný prostor, ve kterém bude simulace probíhat.
2. Přiřadit částicím počáteční pozice a rychlosti.
• Vytvořit zcela novou množinu počátečních pozic a rychlostí.• Převzít konečné pozice a rychlosti z předchozího běhu MD simulace.
8

Paralelnívýpočtyvmolekulárnídynamice
2. Řízení simulace
Jak lze přivést systém do požadovaného stavu?
• Hustota se řídí volbou objemu V simulačního boxu.
• Teplotních změn je obvykle dosaženo přeškálováním rychlostí.
• V pokročilejších simulacích se volí tlak a měří se objem boxu.
Indikátory stavu systému:
• stacionární, tj. kolísající okolo fixní hodnoty,
• relaxující, tj. kolísající okolo hodnoty, která se pomalu mění.
Stavová změna může být:
• vyvolána uměle, např. dojde-li k přenastavení parametrů simulace,
• samovolná, např. systém prochází fázovou transformací.
9

Paralelnívýpočtyvmolekulárnídynamice
3. Získání výsledků ze simulace
• prostá inspekce pozic atomů,
• jednoduché statistické veličiny,
• měření teploty tavení,
• prostorové korelace,
• dynamická analýza atd.
10

Paralelnívýpočtyvmolekulárnídynamice
Vícečásticový potenciál
G.J.Ackland, D.J.Bacon, A.F.Calder, T.Harry:Computer simulation of point defect properties in dilute Fe-Cu alloyusing a many-body interatomic potential.Philosophical Magazine A, 1997, Vol. 75, No. 3, 713–732
Energie souboru N atomů je dána výrazem
E =12
N∑i 6=j=1
V (rij) −N∑i=1
N∑j 6=i=1
φ (rij)
1/2
V (rij)− repulzivní potenciálφ (rij)− kohezivní potenciál
11

Paralelnívýpočtyvmolekulárnídynamice
Pohybové rovnice
Pro každý atom jsou definovány 3 pohybové rovnice:
mnd2sn
dt2+ C
dsn
dt+ Rsn = Fsn, pro s = x, y, z
kde t je čas, mn je hmotnost n-tého atomu, C je koeficient útlumu, Rsn jsousložky gradientu potenciálu a Fsn jsou složky vnější síly působící na n-tý atom.
Koeficient útlumu je různý od nuly pouze v případě relaxace.
12

Paralelnívýpočtyvmolekulárnídynamice
Metoda centrálních diferencí
dsn
dt
∣∣∣∣t=ti
∼=12∆t(sn(i + 1)− sn(i− 1))
d2sn
dt2
∣∣∣∣t=ti
∼=1∆t2(sn (i + 1)− 2sn (i) + sn (i− 1))
Dosazením této rovnice do pohybových rovnic dostaneme:
sn (i + 1) = sn (i− 1) +2
An(sn (i)− sn (i− 1)) +
∆t2
Anmn(Fsn −Rsn) ,
kdeAn = 1 +
C∆t
2mn.
13

Paralelnívýpočtyvmolekulárnídynamice
Popis paralelní úlohy
Úlohy molekulární dynamiky jsou postaveny na neustálém, ale předevšímmnohonásobném vyčíslování relativně jednoduché rekurentní formule,do které vstupují pouze údaje z nejbližšího okolí právě zpracovávaného atomu.
• U bcc železa se jedná např. pouze o 14 nejbližších sousedních atomů.
→ Takovéto úlohy jsou přímo předurčeny k paralelnímu zpracování.
PC PC PC PC PC PC
14

Paralelnívýpočtyvmolekulárnídynamice
Paralelní program
• Zdrojová verze programu je napsána v jazyce Fortran 90.
• Programový kód využívá systém MPI + OpenMP.
Používané funkce MPI:
• MPI INIT, MPI FINALIZE,
• MPI COMM RANK, MPI COMM SIZE,
• MPI SEND, MPI RECV, MPI BCAST,
• MPI ISSEND, MPI IRECV,
• MPI WAIT.
15

Paralelnívýpočtyvmolekulárnídynamice • Hash: metoda indexace buněk (cell index method)
M.P.Allen, D.J.Tildesley: Computer Simulation of Liquids.Oxford University Press, New York, 1987
D. Frenkel, B. Smit: Understanding Molecular Simulations.Academic Press, New York, 1996
• Požadavky na paměť:
6×8 + 2×8 = 64 bytů/atom → 6 GB (4 uzlů 2 GB RAM)
• Požadavky na diskový prostor:
6×8 = 48 bytů/atom → 4,5 GB
16

Paralelnívýpočtyvmolekulárnídynamice
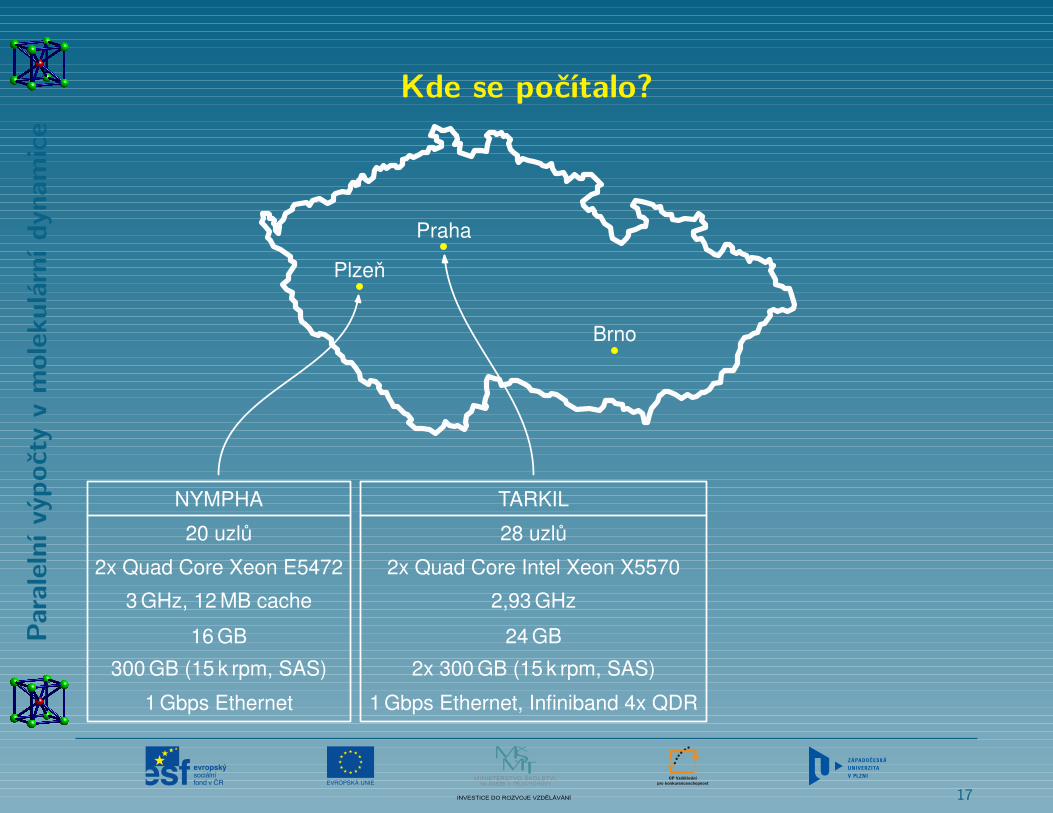
Kde se počítalo?
Plzen
Praha
Brno
17

Paralelnívýpočtyvmolekulárnídynamice
Kde se počítalo?
Plzen
Praha
Brno
NYMPHA
20 uzlu
2x Quad Core Xeon E5472
3 GHz, 12 MB cache
16 GB
300 GB (15 k rpm, SAS)
1 Gbps Ethernet
17
Paralelnívýpočtyvmolekulárnídynamice
Kde se počítalo?
Plzen
Praha
Brno
NYMPHA
20 uzlu
2x Quad Core Xeon E5472
3 GHz, 12 MB cache
16 GB
300 GB (15 k rpm, SAS)
1 Gbps Ethernet
TARKIL
28 uzlu
2x Quad Core Intel Xeon X5570
2,93 GHz
24 GB
2x 300 GB (15 k rpm, SAS)
1 Gbps Ethernet, Infiniband 4x QDR
17

Paralelnívýpočtyvmolekulárnídynamice
Kde se počítalo?
Plzen
Praha
Brno
NYMPHA
20 uzlu
2x Quad Core Xeon E5472
3 GHz, 12 MB cache
16 GB
300 GB (15 k rpm, SAS)
1 Gbps Ethernet
TARKIL
28 uzlu
2x Quad Core Intel Xeon X5570
2,93 GHz
24 GB
2x 300 GB (15 k rpm, SAS)
1 Gbps Ethernet, Infiniband 4x QDR
SKIRIT
35 uzlu
2x Dual Core Xeon 5160
3 GHz, 4 MB cache
4 GB
73 GB
1 Gbps Ethernet
17

Paralelnívýpočtyvmolekulárnídynamice
Kde se počítalo?
Plzen
Praha
Brno
NYMPHA
20 uzlu
2x Quad Core Xeon E5472
3 GHz, 12 MB cache
16 GB
300 GB (15 k rpm, SAS)
1 Gbps Ethernet
TARKIL
28 uzlu
2x Quad Core Intel Xeon X5570
2,93 GHz
24 GB
2x 300 GB (15 k rpm, SAS)
1 Gbps Ethernet, Infiniband 4x QDR
SKIRIT
35 uzlu
2x Dual Core Xeon 5160
3 GHz, 4 MB cache
4 GB
73 GB
1 Gbps Ethernet
17

Paralelnívýpočtyvmolekulárnídynamice
Závislost doby výpočtu na velikosti vzorkua na počtu procesorů
0 4 8 12 16 20 24 28 320
20
40
60
80
100
120
140
Np
Ts [s
]400350300240160 80
18

Paralelnívýpočtyvmolekulárnídynamice
Vizualizace výsledků
Vlastní nástroj pro vizualizaci v MATLABu. Proč?
• Existující programy jsou zbytečně složité.
• Výstupy našich programů mají specifický formát.
• Cizí program je cizí program.
Nejdůležitější možnosti vizualizačního programu:
• stanovení výřezu,
• stanovení pohledu,
• volba uvažované mřížky (základní,centrální nebo obě),
• volba konkrétního kroku simulace,
• filtrace atomů podle energie.
19

Paralelnívýpočtyvmolekulárnídynamice
Testovací úlohy
Klasický experiment → test přesnosti přístrojů
Počítačový experiment → test správnosti a přesnosti algoritmů a programů
Testování paralelního programu pro MD simulaci ve 3Dbylo prováděno na krystalu α-železa.
Šlo o následující typy ověřovacích experimentů:
• povrchová relaxace,
• teplotní roztažnost,
• Hookeův zákon.
20

Paralelnívýpočtyvmolekulárnídynamice
MD simulace trhlin ve 3D
1000× 50× 1000 + 999× 49× 999↓
99 miliónů atomů
Použitý potenciál: vícečásticový potenciál Finnisova–Sinclairova typu
Integrace pohybových rovnic: metoda centrálních diferencí
Integrační krok: 1× 10−14 s
21

Paralelnívýpočtyvmolekulárnídynamice
Popis trhliny
Velikost: 199× 49× 1
Metoda: vyjmutí centrálních atomů
Realizace: interakce přes počáteční rovinu trhliny jsou zakázány
22

Paralelnívýpočtyvmolekulárnídynamice
Zatěžování
a0
a 0
50 16514
2.331
0 K
Casovy krok
σA
[GP
a]
50 16514
2.331300 K
Casovy krok
σA
[GP
a]
23

Paralelnívýpočtyvmolekulárnídynamice
Příklad iniciace křehké trhliny při 0 K
• iniciace v časovém kroku 23 340
• ve střední rovině krystalu
24

Paralelnívýpočtyvmolekulárnídynamice
Časový krok:23300
25

Paralelnívýpočtyvmolekulárnídynamice
Časový krok:23350
25

Paralelnívýpočtyvmolekulárnídynamice
Časový krok:23400
25

Paralelnívýpočtyvmolekulárnídynamice
Časový krok:23450
25

Paralelnívýpočtyvmolekulárnídynamice
Časový krok:23500
25

Paralelnívýpočtyvmolekulárnídynamice
Časový krok:23550
25

Paralelnívýpočtyvmolekulárnídynamice
Časový krok:23600
25

Paralelnívýpočtyvmolekulárnídynamice
Šíření vln v krystalu bcc železa
qP
qSV
SH
kz /ω
kx /ω
26

Paralelnívýpočtyvmolekulárnídynamice
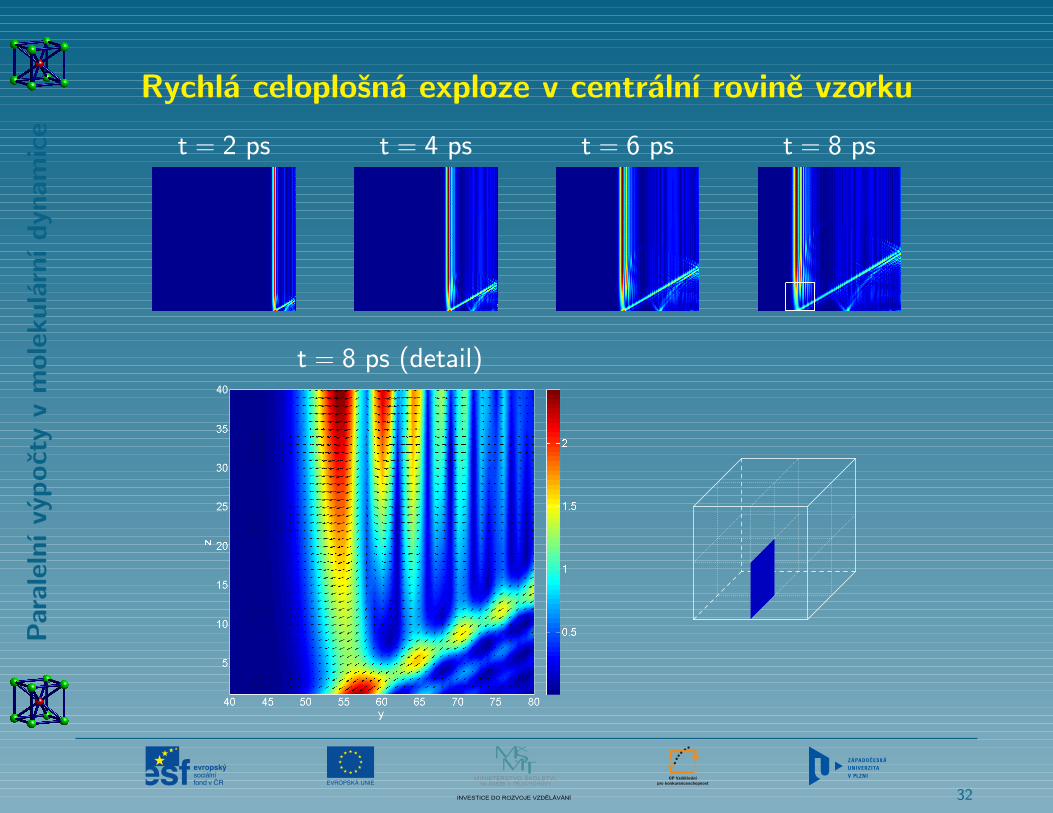
Rychlý celoplošný tah na vnější stěně krychlového vzorku
t = 2 ps t = 4 ps t = 6 ps t = 8 ps
t = 8 ps (detail)
27

Paralelnívýpočtyvmolekulárnídynamice
Moduly rychlostí jednotlivých atomů na ose y
v simulačních časech 2, 4, 6 a 8 ps
0 50 100 150 2000
5
10
15
20
25
30
35
40
y
|v|[
m/s
]
2 ps4 ps6 ps8 ps
4s 4s 4s
28

Paralelnívýpočtyvmolekulárnídynamice
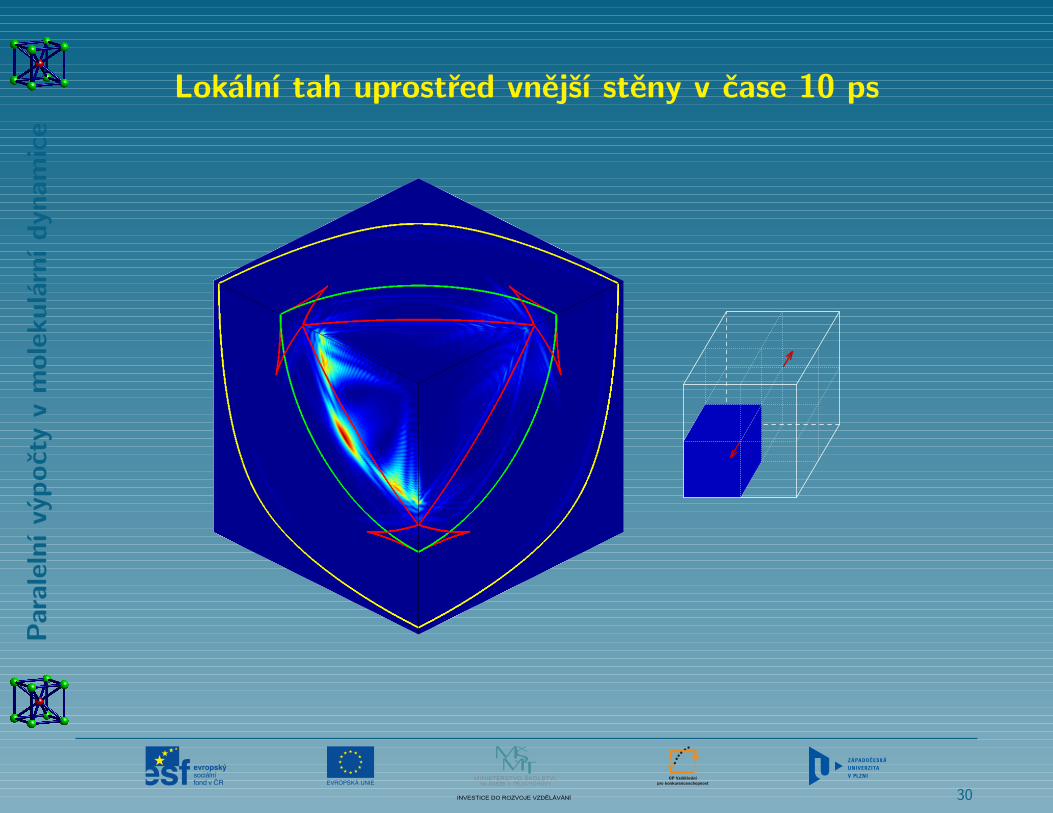
Rychlý lokální tah uprostřed vnější stěny vzorku
t = 2 ps t = 4 ps t = 6 ps t = 8 ps
t = 8 ps (detail)
29
Paralelnívýpočtyvmolekulárnídynamice
Lokální tah uprostřed vnější stěny v čase 10 ps
30

Paralelnívýpočtyvmolekulárnídynamice
Průběh vx vers. vy atomu na povrchu vzorku
-0.06 -0.04 -0.02 0 0.02 0.04 0.06-0.06
-0.04
-0.02
0
0.02
0.04
0.06
vx [m/s]
v y[m
/s]
31
Paralelnívýpočtyvmolekulárnídynamice
Rychlá celoplošná exploze v centrální rovině vzorku
t = 2 ps t = 4 ps t = 6 ps t = 8 ps
t = 8 ps (detail)
32

Paralelnívýpočtyvmolekulárnídynamice
Rychlá exploze ve středu krychlového vzorku
t = 2 ps t = 4 ps t = 6 ps t = 8 ps
t = 8 ps (detail)
33

Paralelnívýpočtyvmolekulárnídynamice
Děkuji za pozornost.
34

Paralelnívýpočtyvmolekulárnídynamice
OBSAH
Co to je?HistorieAplikaceMístoAparát
Lenardův–Jonesův potenciálSimulace – kroky
Vícečásticový potenciálPohybové rovnice
Metoda centrálních diferencíParalelní úlohaParalelní programKde se počítaloVizualizaceTestovací úlohyTrhlinyVlny
35