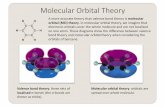
Overview to Molecular Modeling modeling intro.pdf‡Hückel molecular orbital theory ‡MOPAC theory...
40
Overview to Molecular Modeling
Transcript of Overview to Molecular Modeling modeling intro.pdf‡Hückel molecular orbital theory ‡MOPAC theory...

Overview toMolecular Modeling

�E of a molecular structure
�Geometry optimization
�Related properties� vibrational frequencies� nmr� e) density
�Energy method / Energy basis set // Geometry method / Geometry basis set
ComputationalChemistry

�Atoms obey laws of classical physics
�No e) structure
�MM2, MM3, MM+, others
�Useful� Large (bio) molecules� Small molecules
�NO energy value
ComputationalChemistry
MolecularMechanics

�E = 3 Ei
�Large number of parameters� C2H6
� C-C, 6 @ C-H� 6 @ C - C - H� 9 @ H - C - C - H
� C6H6
� 6 @ C - H, 6 @ C -/= C (not C - C or C = C)� 6 @ C - C - H, 24 torsion
�Parameters determined empirically
ComputationalChemistry
MolecularMechanics

�Electronic structure based on , ø = E ø
�, is known exactly
�ø is unknown except for simple systems (H-likeatoms, SHO, RR, particles in boxes, etc.)
ComputationalChemistry
MolecularMechanics
QuantumMechanics

�Overlap Integral
�Exchange Integral� Exchange Functional (HF theory)� Correlation Functional
Problems
ComputationalChemistry
MolecularMechanics
QuantumMechanics
�Ignore part of ,
�Hückel molecular orbital theory
�MOPAC theory
�ZINDO theory
ComputationalChemistry
MolecularMechanics
SemiempiricalMethods
QuantumMechanics
�HMO Hückel molecular orbital theory� Applied to conjugated hydrocarbons� Assumes ALL overlap integrals are zero
�EHT Extended Hückel theory� Applied to any molecule type
�Useful for “quick and dirty” calculations andstarting point for more advanced calculations
Hückel Theory
ComputationalChemistry
MolecularMechanics
SemiempiricalMethods
QuantumMechanics

� CNDO Complete Neglect of Differential Overlap
� INDO Intermediate Neglect of ...
� NDDO Neglect of Diatomic ...
� MINDO Modified INDO� MINDO/3
� MNDO Modified Neglect of ...� AM1 Austin Model 1� PM3 Parameterized Model Series 3� AM1/d and MNDO-d (MOPAC 2000, d e-’s)
� Useful for ground state energy and geometry
MOPACMolecular Orbital Package
ComputationalChemistry
MolecularMechanics
SemiempiricalMethods
QuantumMechanics

�ZINDO/1, ZINDO/3, ZINDO-d, etc
�Useful for� Transition states� Energies� Spectroscopy� Transition elements
�Not useful for optimizations
ZINDOZerner’s INDO
ComputationalChemistry
MolecularMechanics
SemiempiricalMethods
QuantumMechanics

�Use complete ,
�Estimate ø
�Variation Principle (Etrial $ Eexperimental)
ComputationalChemistry
MolecularMechanics
SemiempiricalMethods
ab initioMethods
QuantumMechanics

�HF-SCF� Hartree-Fock Self-Consistent Field
�B3LYP Density Function Theory (DFT)� Becke Exchange with Lee-Yang-Parr Correlation
�MP2/MP4� Second/Fourth Order Møller-Plesset perturbation
theory
�QCISD(T) Quadratic configurationinteraction
Level of Theory
ComputationalChemistry
MolecularMechanics
SemiempiricalMethods
ab initioMethods
QuantumMechanics

Trial Wave Functions(Basis Sets)
ComputationalChemistry
MolecularMechanics
SemiempiricalMethods
ab initioMethods
QuantumMechanics

�Open Shell (unrestricted)� Odd number of electrons� Excited states� 2 or more unpaired electrons� Bond dissociation processes
�Closed Shell (restricted)� Even number of electrons--all paired
Electron Spin
ComputationalChemistry
MolecularMechanics
SemiempiricalMethods
ab initioMethods
QuantumMechanics

Comparison of ab initio Methods (p 94)

Comparison of Models (F/F p 96)


Comparison of Commercial Software

�Capable of describing actual wave functionwell enough to give chemically useful results
�Can be used to evaluate I’s accurately and“cheaply”
Basis SetsBasis Set Criteria

Basis FunctionsHydrogenlike Orbitals

(n - l - 1) nodes
Hydrogenlike Orbitals

Basis FunctionsSlater-type Orbitals (STO’s)

Basis FunctionsGaussian-type Orbitals (GTO’s)

�Advantages� Complete� Favorable math properties
�Disadvantages� Not mutually orthogonal� Poor representation of electron probability near and
far away from nucleus (overcome using largenumber of GTO’s
GTO’s

�One or more STO on each nucleus
�Accuracy of calculation increases as� Orbital exponents chosen well� Number of STO’s used increases
Use of STO’s

�Use STO for occupied AO’s
�Examples� H 1s� C 1s 2s 2px 2py 2pz
Number of STO’s usedMinimal Basis Set

Number of STO’s usedSplit (Double Zeta æ) Basis Set
Linear combination of two similar orbitals withdifferent orbital exponents (different sizes)
ö2p = aö2p,inner + bö2p,outer
If a > b charge cloud contracted around nucleusIf b > a diffuse cloud

Examples:
H 1s, 1sN
C 1s, 2s, 2sN, 2px, 2py, 2pz, 2pxN, 2pyN, 2pzN
Triple Zeta basis sets are also used
Number of STO’s usedSplit (Double Zeta æ) Basis Set

�Extra s and p wave functions included thatare significantly larger than usual ones
�Useful for� Distant electrons� Molecules with lone pairs� Anions� Species with significant negative charge� Excited states� Species with low ionization potentials� Describing acidities
Number of STO’s usedDiffuse Basis Set

�Linear combination of different types oforbitals
�Examples� H 1s and 2p� C 1s, 2s, 2p and 3d
�Shifts charge in/out of bonding regions
Number of STO’s usedPolarized Basis Set

�Other attempts� Place STO’s in center of bonds instead of on only
nuclei
�Problems with increasing number of STO’sused� Number of I’s increases as N4 where N is the
number of basis functions� As minimization occurs, orbital exponents change
thus defining a new basis set to rebegin thecalculation
Number of STO’s used

�Wrong shape of GTO’s accounted for by� Choosing several á’s to get set of “primitive”
gaussians for compact and diffuse� Linear combination of primitives (usually 1-7) to get
STO� Optimize� “Freeze” as “contracted” gaussian function
�Use minimal, split/double zeta, polarization,diffuse sets
Use of STO’s/GTO’s

STO-NG
where N is the number of primitive gaussians
STO-3G
3 primitve gaussians per basis set
not the simplest minimal basis set
popular
Use of STO’s / GTO’sJargon: minimal basis set

K-LMG
where
K is the number of sp type inner shell primitive gaussians
L is the number of inner valence s and p primitive gaussians
M is the number of outer valence s and p primitive gaussians
Use of STO’s / GTO’sJargon: split basis set

3-21G
3 primitives for inner shell
2 sizes of basis functions for each valence orbital
6-311G
6 primitives for inner shell
3 sizes of basis functions for each valence orbital
Use of STO’s / GTO’sJargon: split basis set

* d-type orbital added to atoms with Z > 2
** d-type orbital added to atoms with Z > 2 and p-typeorbital added to H and He
d’s added:
STO-NG are 5 regular 3d’s
L-KMG are 6 3d’s dxx, dyy, dzz, dxy, dyz, dxz (formed bylinear combination of 5 regular 3d’s and 3s)
Use of STO’s / GTO’sJargon: polarization

6-31G* or 6-31G(d)
6-31G with d added for Z > 2 (FF choice)
6-31G** or 6-31G(d,p)
6-31G with d added for Z > 2 and p added to H
6-31G(2d)
6-31G with 2d functions added for Z > 2
Use of STO’s / GTO’sJargon: polarization

+ diffuse function included for Z > 2
++ diffuse function included for Z > 2 and for H
6-31+G(d)6-31G(d) with diffuse function added for Z > 2
6-31++G(d)6-31+G(d) with diffuse function added for H
Use of STO’s / GTO’sJargon: diffuse

SomeRecommendedStandardBasis Sets(F/F p 102)
~DZVP
~TZVP

Common Basis Sets