
Nephrotic Syndrome and Aberrant Expression of...
9
Nephrotic Syndrome and Aberrant Expression of Laminin Isoforms in Glomerular Basement Membranes for an Infant With Herlitz Junctional Epidermolysis Bullosa Daisuke Hata, MD*; Maki Miyazaki, MD‡; Shiro Seto, MD‡; Eiji Kadota, MD§; Eri Muso, MD; Kosho Takasu, MD¶; Aoi Nakano, MD#; Katsuto Tamai, MD#; Jouni Uitto, MD**; Michio Nagata, MD‡‡; Kayano Moriyama, BP§§; and Kaoru Miyazaki, PhD§§ ABSTRACT. Herlitz junctional epidermolysis bullosa (H-JEB) is a hereditary bullous disease caused by absent expression of laminin-5, a component of anchoring fila- ments within the dermal-epidermal basement membrane zone. Affected individuals usually die during the first 1 year of life. We studied an infant with H-JEB who pre- sented with nephrotic syndrome, a previously unre- ported complication that may contribute to early death in this disease. DNA analysis revealed a compound hetero- zygote for mutations 2379delG and Q995X in the LAMB3 gene. The patient had massive albuminuria, attributable to failure of the glomerular filtration barrier, and high urinary N-acetylglucosaminidase levels, indicating renal tubular involvement. Electron-microscopic examination of the renal tissue revealed diffuse fusion of the foot processes, irregular swelling of the lamina rara interna, and disappearance of endothelial cell fenestrations. Im- munohistopathologic analysis of the patient’s renal tis- sue revealed compositional changes in laminin isoforms of the glomerular basement membrane and no detectable laminin-5 in the renal tubular basement membrane, which suggests that laminin-5 may play an important role in renal function. Our findings strongly suggest that H-JEB should be considered in the spectrum of congen- ital nephrotic syndromes. Combination therapy with me- ticulous skin care and treatment strategies established for congenital nephrotic syndromes may rescue patients with this disease. Pediatrics 2005;116:e601–e607. URL: www.pediatrics.org/cgi/doi/10.1542/peds.2005-0160; Her- litz junctional epidermolysis bullosa, laminin-5, nephrotic syndrome, proteinuria, glomerular basement membrane. ABBREVIATIONS. H-JEB, Herlitz junctional epidermolysis bul- losa; PTC, premature termination codon; GBM, glomerular base- ment membrane; TBM, tubular basement membrane; NAG, N- acetylglucosaminidase; mAb, monoclonal antibody; PCR, polymerase chain reaction. J unctional epidermolysis bullosa is a group of autosomal recessive diseases characterized by profound skin fragility, with blister formation re- sulting from dermal-epidermal disadhesion at the level of the lamina lucida within the basement mem- brane zone. The most severe form, Herlitz junctional epidermolysis bullosa (H-JEB), manifests with gen- eralized blistering and erosions of the skin, with extracutaneous involvement, and is usually lethal during the first 1 year of life. 1 Immunohistochemical and molecular genetic stud- ies have demonstrated that the underlying cause of H-JEB is absent expression of laminin-5, a compo- nent of anchoring filaments traversing the lamina lucida of the dermal-epidermal basement membrane zone. A characteristic genetic lesion is a premature termination codon (PTC)-causing mutation in both alleles of 1 of the 3 genes (LAMA3, LAMB3, and LAMC2) encoding the subunit polypeptides of lami- nin-5 (ie, the 3, 3, and 2 chains, respectively). Because all 3 chains are necessary for the structural assembly of trimeric laminin-5 macromolecules, de- fects in any of the 3 genes can result in complete absence of laminin-5. 2,3 Laminin-5 is expressed in multiple tissues, including the skin and the respira- tory and gastrointestinal tracts, 2 and its absence in tissues other than skin accounts for the repertoire of extracutaneous manifestations. Laminins are heterotrimeric extracellular matrix proteins that are composed of , , and chains. At least 15 isoforms are assembled from 5 ,3 , and 3 chains. 4 In the kidney, laminin-11 (521) and laminin-1 (111)/laminin-10 (511) are the ma- jor laminin components of the glomerular basement membrane (GBM) and the renal tubular basement membrane (TBM), respectively. 5 Findings for lami- nin 2-null mice showing lethal congenital nephrotic syndrome suggested that laminin-11, which includes the laminin 2 chain, plays an important role in glomerular function. 6 Although a few studies have shown that laminin-5 is also expressed in the mu- From the Departments of *Pediatrics, Nephrology and Dialysis, and ¶Pa- thology, Kitano Hospital, Tazuke Kofukai Medical Institute, Osaka, Japan; Departments of ‡Pediatrics and §Pathology, Kishiwada City Hospital, Kishiwada, Osaka, Japan; #Department of Dermatology, Hirosaki Univer- sity School of Medicine, Hirosaki, Japan; **Department of Dermatology and Cutaneous Biology, Thomas Jefferson University, Philadelphia, Pennsylva- nia; ‡‡Department of Pathology, Institute of Clinical Medicine, University of Tsukuba, Tsukuba, Japan; and §§Division of Cell Biology, Kihara Insti- tute for Biological Research, Yokohama City University, Yokohama, Japan. Drs Hata and Miyazaki contributed equally to this work. Accepted for publication Apr 14, 2005. doi:10.1542/peds.2005-0160 No conflict of interest declared. Address correspondence to Daisuke Hata, MD, Department of Pediatrics, Kitano Hospital, Tazuke Kofukai Medical Institute, 2-4-20 Ohgimachi, Kita- ku, Osaka, 530 – 8480, Japan. E-mail: [email protected] PEDIATRICS (ISSN 0031 4005). Copyright © 2005 by the American Acad- emy of Pediatrics. www.pediatrics.org/cgi/doi/10.1542/peds.2005-0160 PEDIATRICS Vol. 116 No. 4 October 2005 e601 by guest on June 12, 2018 www.aappublications.org/news Downloaded from
-
Upload
phungquynh -
Category
Documents
-
view
224 -
download
1
Transcript of Nephrotic Syndrome and Aberrant Expression of...

Nephrotic Syndrome and Aberrant Expression of Laminin Isoforms inGlomerular Basement Membranes for an Infant With Herlitz Junctional
Epidermolysis Bullosa
Daisuke Hata, MD*; Maki Miyazaki, MD‡; Shiro Seto, MD‡; Eiji Kadota, MD§; Eri Muso, MD�;Kosho Takasu, MD¶; Aoi Nakano, MD#; Katsuto Tamai, MD#; Jouni Uitto, MD**; Michio Nagata, MD‡‡;
Kayano Moriyama, BP§§; and Kaoru Miyazaki, PhD§§
ABSTRACT. Herlitz junctional epidermolysis bullosa(H-JEB) is a hereditary bullous disease caused by absentexpression of laminin-5, a component of anchoring fila-ments within the dermal-epidermal basement membranezone. Affected individuals usually die during the first 1year of life. We studied an infant with H-JEB who pre-sented with nephrotic syndrome, a previously unre-ported complication that may contribute to early death inthis disease. DNA analysis revealed a compound hetero-zygote for mutations 2379delG and Q995X in the LAMB3gene. The patient had massive albuminuria, attributableto failure of the glomerular filtration barrier, and highurinary N-acetylglucosaminidase levels, indicating renaltubular involvement. Electron-microscopic examinationof the renal tissue revealed diffuse fusion of the footprocesses, irregular swelling of the lamina rara interna,and disappearance of endothelial cell fenestrations. Im-munohistopathologic analysis of the patient’s renal tis-sue revealed compositional changes in laminin isoformsof the glomerular basement membrane and no detectablelaminin-5 in the renal tubular basement membrane,which suggests that laminin-5 may play an importantrole in renal function. Our findings strongly suggest thatH-JEB should be considered in the spectrum of congen-ital nephrotic syndromes. Combination therapy with me-ticulous skin care and treatment strategies establishedfor congenital nephrotic syndromes may rescue patientswith this disease. Pediatrics 2005;116:e601–e607. URL:www.pediatrics.org/cgi/doi/10.1542/peds.2005-0160; Her-litz junctional epidermolysis bullosa, laminin-5, nephroticsyndrome, proteinuria, glomerular basement membrane.
ABBREVIATIONS. H-JEB, Herlitz junctional epidermolysis bul-losa; PTC, premature termination codon; GBM, glomerular base-ment membrane; TBM, tubular basement membrane; NAG, N-acetylglucosaminidase; mAb, monoclonal antibody; PCR,polymerase chain reaction.
Junctional epidermolysis bullosa is a group ofautosomal recessive diseases characterized byprofound skin fragility, with blister formation re-
sulting from dermal-epidermal disadhesion at thelevel of the lamina lucida within the basement mem-brane zone. The most severe form, Herlitz junctionalepidermolysis bullosa (H-JEB), manifests with gen-eralized blistering and erosions of the skin, withextracutaneous involvement, and is usually lethalduring the first 1 year of life.1
Immunohistochemical and molecular genetic stud-ies have demonstrated that the underlying cause ofH-JEB is absent expression of laminin-5, a compo-nent of anchoring filaments traversing the laminalucida of the dermal-epidermal basement membranezone. A characteristic genetic lesion is a prematuretermination codon (PTC)-causing mutation in bothalleles of 1 of the 3 genes (LAMA3, LAMB3, andLAMC2) encoding the subunit polypeptides of lami-nin-5 (ie, the �3, �3, and �2 chains, respectively).Because all 3 chains are necessary for the structuralassembly of trimeric laminin-5 macromolecules, de-fects in any of the 3 genes can result in completeabsence of laminin-5.2,3 Laminin-5 is expressed inmultiple tissues, including the skin and the respira-tory and gastrointestinal tracts,2 and its absence intissues other than skin accounts for the repertoire ofextracutaneous manifestations.
Laminins are heterotrimeric extracellular matrixproteins that are composed of �, �, and � chains. Atleast 15 isoforms are assembled from 5 �, 3 �, and 3� chains.4 In the kidney, laminin-11 (�5�2�1) andlaminin-1 (�1�1�1)/laminin-10 (�5�1�1) are the ma-jor laminin components of the glomerular basementmembrane (GBM) and the renal tubular basementmembrane (TBM), respectively.5 Findings for lami-nin �2-null mice showing lethal congenital nephroticsyndrome suggested that laminin-11, which includesthe laminin �2 chain, plays an important role inglomerular function.6 Although a few studies haveshown that laminin-5 is also expressed in the mu-
From the Departments of *Pediatrics, �Nephrology and Dialysis, and ¶Pa-thology, Kitano Hospital, Tazuke Kofukai Medical Institute, Osaka, Japan;Departments of ‡Pediatrics and §Pathology, Kishiwada City Hospital,Kishiwada, Osaka, Japan; #Department of Dermatology, Hirosaki Univer-sity School of Medicine, Hirosaki, Japan; **Department of Dermatology andCutaneous Biology, Thomas Jefferson University, Philadelphia, Pennsylva-nia; ‡‡Department of Pathology, Institute of Clinical Medicine, Universityof Tsukuba, Tsukuba, Japan; and §§Division of Cell Biology, Kihara Insti-tute for Biological Research, Yokohama City University, Yokohama, Japan.Drs Hata and Miyazaki contributed equally to this work.Accepted for publication Apr 14, 2005.doi:10.1542/peds.2005-0160No conflict of interest declared.Address correspondence to Daisuke Hata, MD, Department of Pediatrics,Kitano Hospital, Tazuke Kofukai Medical Institute, 2-4-20 Ohgimachi, Kita-ku, Osaka, 530–8480, Japan. E-mail: [email protected] (ISSN 0031 4005). Copyright © 2005 by the American Acad-emy of Pediatrics.
www.pediatrics.org/cgi/doi/10.1542/peds.2005-0160 PEDIATRICS Vol. 116 No. 4 October 2005 e601 by guest on June 12, 2018www.aappublications.org/newsDownloaded from

rine,7 rat,8 and human9–11 kidney, the functional im-portance of laminin-5 in the kidney is unknown.
In this article, we describe an infant with H-JEBcomplicated by nephrotic syndrome, compositionalchanges in laminin isoforms of the GBM, and nodetectable laminin-5 protein in the renal TBM. Thiscase suggests that laminin-5 may play an importantrole in renal function and that, in addition to theprotein loss from denuded skin and mucous sur-faces, massive proteinuria, a previously unreportedcomplication of H-JEB, may contribute to the highmortality rate for H-JEB in early postnatal life.
CASE REPORTA 3500-g male patient, born to unrelated Japanese parents after
an uncomplicated term pregnancy, developed widespread blister-ing of the skin and buccal mucous membranes in areas of frictionand trauma within 24 hours after birth. Despite nutritional sup-port and appropriate wound care, the skin lesions progressed, andthe patient was transferred to our hospital at 4 months of age.
At the time of admission, extensive denudation and erosionwere present on the patient’s occiput, back, buttocks, and extrem-ities, with erosions of the oral mucosa. The fingernails and toenailswere absent. Blood examination showed leukocytosis (11 500 cellsper mm3 [11.5 � 109 cells per L]), anemia (hemoglobin: 8.6 g/dL[86 g/L]), positive C-reactive protein results (12.1 mg/dL [0.121g/L]), and hypoalbuminemia (2.6 g/dL [26 g/L]). Blood cultureresults were negative. The patient was wrapped in silicon gauzeimpregnated with hydrated petrolatum, dimethylisopropyla-zulene, or topical antibiotics when necessary and was reared in atemperature- and humidity-controlled unit. He received intrave-nous hyperalimentation and repeated intravenous albumin trans-fusions. On the 11th hospital day, intravenous antibiotic therapywas started because of suspected sepsis. Despite repeated intra-venous albumin transfusions and gradually healing skin lesions,hypoalbuminemia (2.5 g/dL [25 g/L] on the 13th hospital day)persisted, which suggested significant protein loss from sites otherthan the denuded skin. Subsequently, urinalysis showed massivealbuminuria of 2.1 g/d, a high level of �2-microglobulin (104mg/L [0.104 g/L]; normal range: �0.5 mg/L [�0.5 � 10�3 g/L]),and a high level of �-N-acetylglucosaminidase (NAG) (38.4 IU/L;normal range: �7 IU/L), suggesting renal glomerular and tubularinvolvement. Despite the repeated albumin transfusions, hy-poalbuminemia progressed (1.1 g/dL [11 g/L]) and massive pro-teinuria continued (albuminuria: 18.9 g/d; urinary NAG level:99.3 IU/L) on the 21st hospital day. Throughout his hospitaliza-tion, the patient remained critically ill, with persistent suspected
sepsis, disseminated intravascular coagulation, severe anemia,electrolyte imbalance, and severe hypoproteinemia. The clinicalcondition continued to deteriorate, and the patient died on the25th hospital day.
Light-microscopic examination of a skin biopsy specimen takenfrom a newly developed blister on the dorsum of the right footdemonstrated a split at the dermal-epidermal junction, withoutassociated inflammatory cell infiltrates. Electron-microscopic ex-amination showed subepidermal cleft formation arising within thelamina lucida, as well as hemidesmosomal hypoplasia (Fig 1).
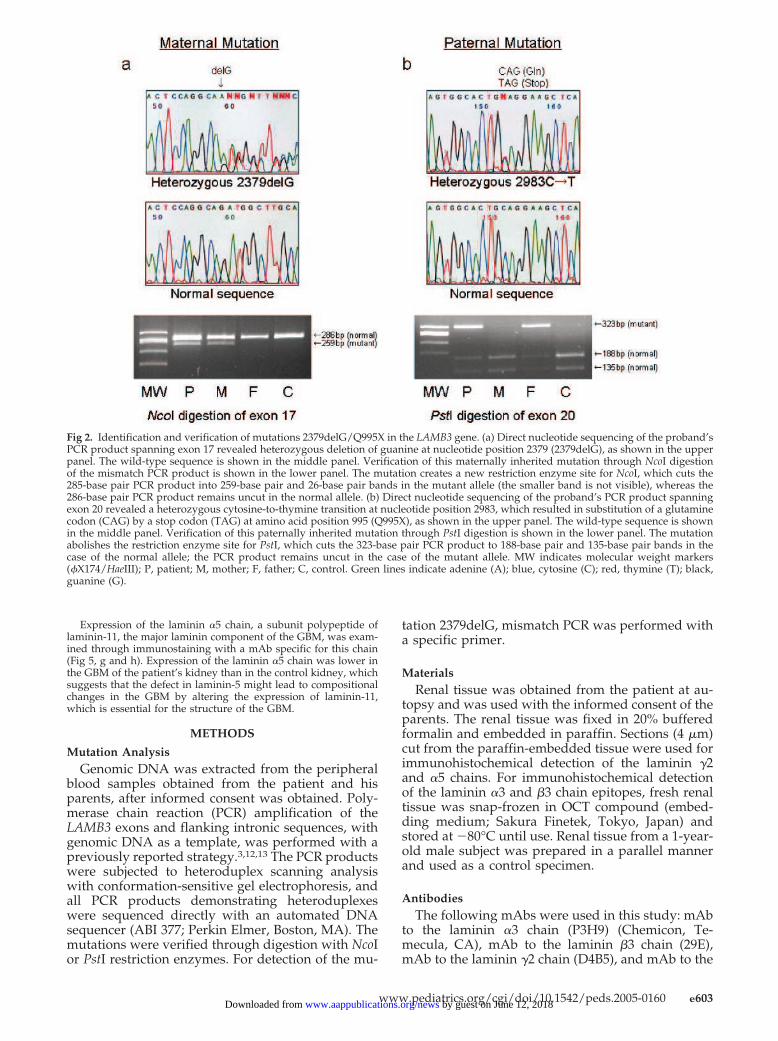
Two mutations were identified in the LAMB3 gene of the pa-tient, designated 2379delG and Q995X (Fig 2). The maternal mu-tation, 2379delG in exon 17 (Fig 2a), created a frame shift anddownstream PTC and predicted premature termination of proteintranslation. The paternal mutation, Q995X in exon 20 (Fig 2b),caused a stop codon at position 995 of the LAMB3 polypeptide.Both LAMB3 mutations were novel and not reported previouslyfor H-JEB. The results of the electron-microscopic examination ofthe skin and the PTC mutations in both alleles of the LAMB3 gene,together with the patient’s fatal clinical course, were diagnostic ofH-JEB.
Light-microscopic examination of the renal tissue obtained atautopsy showed glomeruli with obsolescence, with capillary base-ment membrane wrinkling and segmental shrinking. A wide uri-nary space was observed frequently. No apparent cell prolifera-tion was noted (Fig 3a). Denudation of the TBM was seenfrequently (Fig 3b). In the interstitium, focal mononuclear cellinfiltration was observed. Massive luminal dilation of tubuleswith flattened tubular epithelial cells was observed diffusely (Fig3c). Electron-microscopic examination of the renal tissue revealeddiffuse fusion of the foot processes and irregular swelling of thelamina rara interna (Fig 4a), as well as absent endothelial cellfenestrations (Fig 4b).
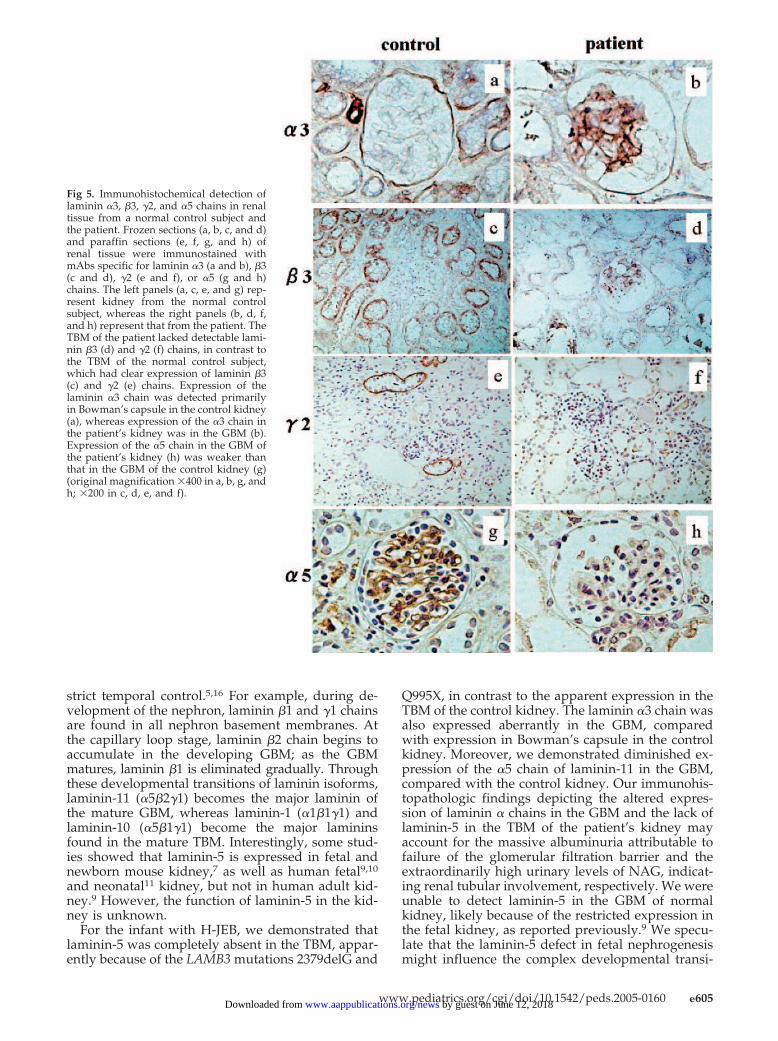
To examine expression of laminin-5 in the patient’s kidney,immunohistopathologic analysis was performed on the renal tis-sue obtained at autopsy. Expression of the laminin �3 and �2chains was examined through immunostaining with specificmonoclonal antibodies (mAbs) (Fig 5). Although these chains wereclearly localized to the TBM in the control specimen (Fig 5, c ande), they were completely absent in the patient’s kidney (Fig 5, dand f), which confirmed the lack of laminin-5 expression at thetissue level. Expression of the laminin �3 chain was also examinedthrough immunostaining with a specific mAb (Fig 5, a and b).Interestingly, the control kidney stained positively for the �3 chainin Bowman’s capsule (Fig 5a), whereas staining in the GBM wasapparent in the patient’s kidney (Fig 5b). Because the laminin �3chain is the subunit polypeptide not only of laminin-5 but also oflaminin-6 and laminin-7, expression of the �3 chain in the GBM ofthe patient’s kidney might indicate the expression of laminin-6 orlaminin-7 or the presence of an unreported novel laminin.
Fig 1. Electron micrograph of the pa-tient’s skin biopsy specimen, demon-strating cleavage within the lamina lu-cida and hemidesmosomal hypoplasia.e indicates epidermis; d, dermis; ld,lamina densa (original magnification�10 000).
e602 NEPHROTIC SYNDROME AND RENAL LAMININ ISOFORMS by guest on June 12, 2018www.aappublications.org/newsDownloaded from
Expression of the laminin �5 chain, a subunit polypeptide oflaminin-11, the major laminin component of the GBM, was exam-ined through immunostaining with a mAb specific for this chain(Fig 5, g and h). Expression of the laminin �5 chain was lower inthe GBM of the patient’s kidney than in the control kidney, whichsuggests that the defect in laminin-5 might lead to compositionalchanges in the GBM by altering the expression of laminin-11,which is essential for the structure of the GBM.
METHODS
Mutation AnalysisGenomic DNA was extracted from the peripheral
blood samples obtained from the patient and hisparents, after informed consent was obtained. Poly-merase chain reaction (PCR) amplification of theLAMB3 exons and flanking intronic sequences, withgenomic DNA as a template, was performed with apreviously reported strategy.3,12,13 The PCR productswere subjected to heteroduplex scanning analysiswith conformation-sensitive gel electrophoresis, andall PCR products demonstrating heteroduplexeswere sequenced directly with an automated DNAsequencer (ABI 377; Perkin Elmer, Boston, MA). Themutations were verified through digestion with NcoIor PstI restriction enzymes. For detection of the mu-
tation 2379delG, mismatch PCR was performed witha specific primer.
MaterialsRenal tissue was obtained from the patient at au-
topsy and was used with the informed consent of theparents. The renal tissue was fixed in 20% bufferedformalin and embedded in paraffin. Sections (4 �m)cut from the paraffin-embedded tissue were used forimmunohistochemical detection of the laminin �2and �5 chains. For immunohistochemical detectionof the laminin �3 and �3 chain epitopes, fresh renaltissue was snap-frozen in OCT compound (embed-ding medium; Sakura Finetek, Tokyo, Japan) andstored at �80°C until use. Renal tissue from a 1-year-old male subject was prepared in a parallel mannerand used as a control specimen.
AntibodiesThe following mAbs were used in this study: mAb
to the laminin �3 chain (P3H9) (Chemicon, Te-mecula, CA), mAb to the laminin �3 chain (29E),mAb to the laminin �2 chain (D4B5), and mAb to the
Fig 2. Identification and verification of mutations 2379delG/Q995X in the LAMB3 gene. (a) Direct nucleotide sequencing of the proband’sPCR product spanning exon 17 revealed heterozygous deletion of guanine at nucleotide position 2379 (2379delG), as shown in the upperpanel. The wild-type sequence is shown in the middle panel. Verification of this maternally inherited mutation through NcoI digestionof the mismatch PCR product is shown in the lower panel. The mutation creates a new restriction enzyme site for NcoI, which cuts the285-base pair PCR product into 259-base pair and 26-base pair bands in the mutant allele (the smaller band is not visible), whereas the286-base pair PCR product remains uncut in the normal allele. (b) Direct nucleotide sequencing of the proband’s PCR product spanningexon 20 revealed a heterozygous cytosine-to-thymine transition at nucleotide position 2983, which resulted in substitution of a glutaminecodon (CAG) by a stop codon (TAG) at amino acid position 995 (Q995X), as shown in the upper panel. The wild-type sequence is shownin the middle panel. Verification of this paternally inherited mutation through PstI digestion is shown in the lower panel. The mutationabolishes the restriction enzyme site for PstI, which cuts the 323-base pair PCR product to 188-base pair and 135-base pair bands in thecase of the normal allele; the PCR product remains uncut in the case of the mutant allele. MW indicates molecular weight markers(�X174/HaeIII); P, patient; M, mother; F, father; C, control. Green lines indicate adenine (A); blue, cytosine (C); red, thymine (T); black,guanine (G).
www.pediatrics.org/cgi/doi/10.1542/peds.2005-0160 e603 by guest on June 12, 2018www.aappublications.org/newsDownloaded from

laminin �5 chain (4C7) (Life Technologies, Gaithers-burg, MD). The mAbs 29E and D4B5 were raisedagainst purified human laminin-5 and human re-combinant laminin �2 chain (amino acid residues382–608), respectively, in our laboratory.10,14
Immunohistochemical AnalysesFor immunohistochemical analyses, the paraffin
sections were deparaffinized, rehydrated, immersedin 0.3% hydrogen peroxide-containing methanol forinactivation of intrinsic peroxidase, and treated withprotease XXIV (Sigma, St Louis, MO) for 20 minutesat room temperature. The frozen sections were im-mersed in 0.3% hydrogen peroxide-containing meth-
anol and treated with protease XXIV for 5 minutes.The paraffin sections were incubated with the anti-�2mAb (D4B5) or the anti-�5 mAb (4C7) at 4°C over-night, whereas frozen sections were incubated withanti-�3 mAb (P3H9) or anti-�3 mAb (29E) for 20 to 30minutes at room temperature. The labeled antigenwas detected with a HistoFine kit (Nichirei Pharma-ceutical, Tokyo, Japan) and observed through the3,3-diaminobenzidine reaction. Other experimentalconditions were as described previously.15
RESULTS AND DISCUSSIONMany of the laminin chains are expressed in the
GBM and TBM during renal development, under
Fig 3. (a) Obsolescence (arrowheads) with wrinkling of the capillary basement membrane (arrow), segmental shrinking of the glomerulartuft, and a wide urinary space (periodic acid-Schiff-methenamine-silver stain, counterstained with hematoxylin and eosin; originalmagnification �400). (b) Denudation of the TBM (arrows) (periodic acid-Schiff-methenamine-silver stain, counterstained with hematox-ylin and eosin; original magnification �200). (c) Massive luminal dilation of tubules with flattened tubular epithelial cells (arrow)(periodic acid-Schiff stain; original magnification �40).
Fig 4. (a) Electron micrograph of the patient’s kidney biopsy specimen, demonstrating diffuse fusion of the foot processes (arrowheads)and irregular swelling of the lamina rara interna (arrows) (original magnification �5000). (b) Area defined by the black box in a, showingabsent endothelial cell fenestrations (arrow) (original magnification �12 000).
e604 NEPHROTIC SYNDROME AND RENAL LAMININ ISOFORMS by guest on June 12, 2018www.aappublications.org/newsDownloaded from
strict temporal control.5,16 For example, during de-velopment of the nephron, laminin �1 and �1 chainsare found in all nephron basement membranes. Atthe capillary loop stage, laminin �2 chain begins toaccumulate in the developing GBM; as the GBMmatures, laminin �1 is eliminated gradually. Throughthese developmental transitions of laminin isoforms,laminin-11 (�5�2�1) becomes the major laminin ofthe mature GBM, whereas laminin-1 (�1�1�1) andlaminin-10 (�5�1�1) become the major lamininsfound in the mature TBM. Interestingly, some stud-ies showed that laminin-5 is expressed in fetal andnewborn mouse kidney,7 as well as human fetal9,10
and neonatal11 kidney, but not in human adult kid-ney.9 However, the function of laminin-5 in the kid-ney is unknown.
For the infant with H-JEB, we demonstrated thatlaminin-5 was completely absent in the TBM, appar-ently because of the LAMB3 mutations 2379delG and
Q995X, in contrast to the apparent expression in theTBM of the control kidney. The laminin �3 chain wasalso expressed aberrantly in the GBM, comparedwith expression in Bowman’s capsule in the controlkidney. Moreover, we demonstrated diminished ex-pression of the �5 chain of laminin-11 in the GBM,compared with the control kidney. Our immunohis-topathologic findings depicting the altered expres-sion of laminin � chains in the GBM and the lack oflaminin-5 in the TBM of the patient’s kidney mayaccount for the massive albuminuria attributable tofailure of the glomerular filtration barrier and theextraordinarily high urinary levels of NAG, indicat-ing renal tubular involvement, respectively. We wereunable to detect laminin-5 in the GBM of normalkidney, likely because of the restricted expression inthe fetal kidney, as reported previously.9 We specu-late that the laminin-5 defect in fetal nephrogenesismight influence the complex developmental transi-
Fig 5. Immunohistochemical detection oflaminin �3, �3, �2, and �5 chains in renaltissue from a normal control subject andthe patient. Frozen sections (a, b, c, and d)and paraffin sections (e, f, g, and h) ofrenal tissue were immunostained withmAbs specific for laminin �3 (a and b), �3(c and d), �2 (e and f), or �5 (g and h)chains. The left panels (a, c, e, and g) rep-resent kidney from the normal controlsubject, whereas the right panels (b, d, f,and h) represent that from the patient. TheTBM of the patient lacked detectable lami-nin �3 (d) and �2 (f) chains, in contrast tothe TBM of the normal control subject,which had clear expression of laminin �3(c) and �2 (e) chains. Expression of thelaminin �3 chain was detected primarilyin Bowman’s capsule in the control kidney(a), whereas expression of the �3 chain inthe patient’s kidney was in the GBM (b).Expression of the �5 chain in the GBM ofthe patient’s kidney (h) was weaker thanthat in the GBM of the control kidney (g)(original magnification �400 in a, b, g, andh; �200 in c, d, e, and f).
www.pediatrics.org/cgi/doi/10.1542/peds.2005-0160 e605 by guest on June 12, 2018www.aappublications.org/newsDownloaded from

tions of laminin isoforms in the GBM. It is wellestablished that a deficiency of the laminin �2 chainis accompanied by compensatory upregulation of thelaminin �4 chain in mouse skeletal muscle cells17 anda deficiency of the laminin �5 chain causes a strikingdefect in mouse glomerulogenesis.18 In addition, ithas been shown that laminin �2-null mice exhibit asevere nephrotic syndrome in the first postnatalweek; although the GBM appears ultrastructurallynormal and, at a molecular level, laminin �1 substi-tutes for �2 in the GBM, the �1 subunit seems to befunctionally inadequate.6 The findings for the �2-null mice are consistent with our present findings,which suggest that compositional changes in lamininisoforms in the GBM can result in failure of theglomerular filtration barrier. It was reported recentlythat human laminin �2 deficiency causes congenitalnephrosis with mesangial sclerosis and distinct eyeabnormalities.19 Our report is the second case studydemonstrating that a defect in a laminin subunitcauses dysfunction of human renal basement mem-branes. It is probable that massive proteinuria amongpatients with H-JEB has been overlooked, althoughthe possibility that the massive proteinuria for ourpatient is an unusual presentation of H-JEB has notbeen excluded completely. Collection of urine sam-ples with urine collection bags is almost impossible,because the bag attachment would hurt the fragileskin. We used soft gauze for collection of urine sam-ples. The histomorphologic findings for our patient’skidney seem more serious than those observed forthe kidneys of laminin �2-null mice or human pa-tients with laminin �2 deficiency, which might resultfrom the additional effects of many infectious, met-abolic, and/or pharmacologic insults on the fragileGBM of our patient.
The importance of GBM composition for glomer-ular function is also illustrated by Alport’s syn-drome, a human hereditary glomerular nephritis inwhich a mutation in the collagen �3, �4, or �5 chaingene leads to progressively altered glomerular func-tion. It has been shown that type IV collagen isolatedfrom human Alport’s syndrome kidney containing�1 and �2 chains is more susceptible to endoprote-olysis than type IV collagen isolated from normalkidney containing �1 to �6 chains. This observationsuggests that the Alport’s syndrome GBM could bemore vulnerable to deterioration than the normalGBM.20 It is likely that the GBM in H-JEB could alsobe more susceptible to many insults to the patient,such as sepsis, disseminated intravascular coagula-tion, anemia, malnutrition, hypoxia, and antibiotics,compared with that of normal kidney. Throughoutthe patient’s clinical course, blood urea nitrogen andcreatinine levels, as well as creatinine clearance, re-mained at normal levels despite massive albumin-uria. It is of interest to note that compositionalchanges in laminin isoforms and collagen isoforms inthe GBM cause different types of nephropathy (ie,nephrosis and nephritis, respectively), but the pre-cise mechanisms remain to be explored. Studies ofmutant mice that genetically lack laminin-5 couldelucidate the underlying mechanism.21
Proteinuria and mild hypoalbuminemia (not in thenephrotic range), secondary to focal segmental glo-merulosclerosis, have been reported in epidermoly-sis bullosa with pyloric atresia, an autosomal reces-sive disease caused by mutations in the genesencoding either 1 of the 2 subunits of �6�4 integrin,which are expressed in the hemidesmosomes of avariety of epithelial tissues, including human skinand the gastrointestinal tract.22 In the case of epider-molysis bullosa with pyloric atresia, reduced �4 in-tegrin expression was demonstrated in glomerularpodocytes.22 The finding that �6�4 integrin functionsas a receptor for laminin-523 supports the functionalimportance of laminin-5 in human kidney. It hasbeen shown that both laminin-5 and laminin-bindingintegrins play a role in kidney development andureteric bud branching morphogenesis in embryonicrats.24
From the therapeutic point of view, the observa-tions for our patient strongly suggest that H-JEBshould be considered not only a hereditary skin dis-ease but also one of the congenital nephrotic syn-dromes. Currently, H-JEB among affected infants isinvariably lethal within the first 1 year of life. Apotential approach for treatment may be combina-tion therapy with meticulous skin care and the treat-ment protocol proposed for infants with congenitalnephritic syndromes, ie, massive intravenous albu-min supplementation and nutritional support (tomake normal growth and development possible),followed by bilateral nephrectomy and peritonealdialysis (to stop protein loss from the kidney) andfinally by renal transplantation. Because keratinocytegene therapy for H-JEB is now under investiga-tion,25,26 combination therapy with gene therapy forskin lesions and the aforementioned treatment pro-tocol for congenital nephrotic syndromes might beworth investigating for H-JEB in the near future.
ACKNOWLEDGMENTSWe thank Yutaka Hamazaki for providing control renal tissue,
Professor Tatsutoshi Nakahata, Department of Pediatrics, KyotoUniversity, for critical reading of the manuscript, and SatoruMutuo for assistance.
REFERENCES1. Fine JD, Eady RA, Bauer EA, et al. Revised classification system for
inherited epidermolysis bullosa: report of the Second International Con-sensus Meeting on diagnosis and classification of epidermolysis bul-losa. J Am Acad Dermatol. 2000;42:1051–1066
2. Verrando P, Blanchet-Bardon C, Pisani A, et al. Monoclonal antibodyGB3 defines a widespread defect of several basement membranes and akeratinocyte dysfunction in patients with lethal junctional epidermoly-sis bullosa. Lab Invest. 1991;64:85–92
3. Pulkkinen L, Uitto J, Christiano AM. The molecular basis of the junc-tional forms of epidermolysis bullosa. In: Fine JD, Bauer EA, McGuire J,Moshell A, eds. Epidermolysis Bullosa: Clinical, Epidemiologic, and Labora-tory Advances, and the Findings of the National Epidermolysis Bullosa Reg-istry. Baltimore, MD: Johns Hopkins University Press; 1999:300–325
4. Colognato H, Yurchenco PD. Form and function: the laminin family ofheterotrimers. Dev Dyn. 2000;218:213–234
5. Miner JH. Renal basement membrane components. Kidney Int. 1999;56:2016–2024
6. Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP.The renal glomerulus of mice lacking S-laminin/laminin �2: nephrosisdespite molecular compensation by laminin �1. Nat Genet. 1995;10:400–406
e606 NEPHROTIC SYNDROME AND RENAL LAMININ ISOFORMS by guest on June 12, 2018www.aappublications.org/newsDownloaded from

7. Sugiyama S, Utani A, Yamada S, Kozak CA, Yamada Y. Cloning andexpression of the mouse laminin �2 (B2t) chain, a subunit of epithelialcell laminin. Eur J Biochem. 1995;228:120–128
8. Sterk LMT, deMelker AA, Kramer D, et al. Glomerular extracellularmatrix components and integrins. Cell Adhes Commun. 1998;5:177–192
9. Mizushima H, Miyata Y, Kikkawa Y, et al. Differential expression oflaminin-5/ladsin subunits in human tissues and cancer cell lines andtheir induction by tumor promoter and growth factors. J Biochem. 1996;120:1196–1202
10. Mizushima H, Koshikawa N, Moriyama K, et al. Wide distribution oflaminin-5 �2 chain in basement membranes of various human tissues.Horm Res. 1998;50:7–14
11. Airenne T, Haakana H, Sainio K, et al. Structure of the human laminin�2 chain gene (LAMC2): alternative splicing with different tissue distri-bution of two transcripts. Genomics. 1996;32:54–64
12. Pulkkinen L, McGrath JA, Christiano AM, Uitto J. Detection of sequencevariants in the gene encoding the �3 chain of laminin 5 (LAMB3). HumMutat. 1995;6:77–84
13. Nakano A, Pfendner E, Pulkkinen L, Hashimoto I, Uitto J. Herlitzjunctional epidermolysis bullosa: novel and recurrent mutations in theLAMB3 gene and the population carrier frequency. J Invest Dermatol.2000;115:493–498
14. Koshikawa N, Moriyama K, Takamura H, et al. Overexpression of �2chain monomer in invading gastric carcinoma cells. Cancer Res. 1999;59:5596–5601
15. Akaogi K, Okabe Y, Sato J, et al. Specific accumulation of tumor-derivedadhesion factor in tumor blood vessels and in capillary tube-like struc-tures of cultured vascular endothelial cells. Proc Natl Acad Sci USA.1996;93:8384–8389
16. Miner HM. Developmental biology of glomerular basement membranecomponents. Curr Opin Nephrol Hypertens. 1998;7:13–19
17. Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function oflaminins in the neuromuscular system of developing, adult, and mutantmice. J Cell Biol. 1997;139:1507–1521
18. Miner JH, Li C. Defective glomerulogenesis in the absence of laminin �5demonstrates a developmental role for the kidney glomerular basementmembrane. Dev Biol. 2000;217:278–289
19. Zenker M, Aigner T, Wendler O, et al. Human laminin �2 deficiencycauses congenital nephrosis with mesangeal sclerosis and distinct eyeabnormalities. Hum Mol Genet. 2004;13:2625–2632
20. Kalluri R, Shield CF, Todd P, Hudson BG, Neilson EG. Isoform switch-ing of type IV collagen is developmentally arrested in X-linked Alportsyndrome leading to increased susceptibility of renal basement mem-branes to endoproteolysis. J Clin Invest. 1997;99:2470–2474
21. Kuster JE, Guarnieri MH, Ault JG, Flaherty L, Swiatek PJ. IAP insertionin the murine LamB3 gene results in junctional epidermolysis bullosa.Mamm Genome. 1997;8:673–681
22. Kambham N, Tanji N, Seigle RL, et al. Congenital focal segmentalglomerulosclerosis associated with �4 integrin mutation and epider-molysis bullosa. Am J Kidney Dis. 2000;36:190–196
23. Xia Y, Gill SG, Carter WG. Anchorage mediated by integrin �6�4 tolaminin 5 (epiligrin) regulates tyrosine phosphorylation of a membrane-associated 80-kD protein. J Cell Biol. 1996;132:727–740
24. Zent R, Bush KT, Pohl ML, et al. Involvement of laminin bindingintegrins and laminin-5 in branching morphogenesis of the ureteric budduring kidney development. Dev Biol. 2001;238:289–302
25. Masunaga T, Shimizu H, Matsui C, et al. LAMB3 gene transfection intoSV40-transformed keratinocytes from patient with Herlitz junctionalepidermolysis bullosa. Arch Dermatol Res. 2000;292:195–197
26. Robbins PB, Lin Q, Goodnough JB, Tian H, Chen X, Khavari PA.In vivo restoration of laminin 5 �3 expression and function injunctional epidermolysis bullosa. Proc Natl Acad Sci USA. 2001;98:5193–5198
www.pediatrics.org/cgi/doi/10.1542/peds.2005-0160 e607 by guest on June 12, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2005-0160 originally published online September 1, 2005; 2005;116;e601Pediatrics
MiyazakiNakano, Katsuto Tamai, Jouni Uitto, Michio Nagata, Kayano Moriyama and Kaoru
Daisuke Hata, Maki Miyazaki, Shiro Seto, Eiji Kadota, Eri Muso, Kosho Takasu, AoiEpidermolysis Bullosa
Glomerular Basement Membranes for an Infant With Herlitz Junctional Nephrotic Syndrome and Aberrant Expression of Laminin Isoforms in
ServicesUpdated Information &
http://pediatrics.aappublications.org/content/116/4/e601including high resolution figures, can be found at:
Referenceshttp://pediatrics.aappublications.org/content/116/4/e601#BIBLThis article cites 24 articles, 5 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/urology_subUrologyfollowing collection(s): This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtmlin its entirety can be found online at: Information about reproducing this article in parts (figures, tables) or
Reprintshttp://www.aappublications.org/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
by guest on June 12, 2018www.aappublications.org/newsDownloaded from

DOI: 10.1542/peds.2005-0160 originally published online September 1, 2005; 2005;116;e601Pediatrics
MiyazakiNakano, Katsuto Tamai, Jouni Uitto, Michio Nagata, Kayano Moriyama and Kaoru
Daisuke Hata, Maki Miyazaki, Shiro Seto, Eiji Kadota, Eri Muso, Kosho Takasu, AoiEpidermolysis Bullosa
Glomerular Basement Membranes for an Infant With Herlitz Junctional Nephrotic Syndrome and Aberrant Expression of Laminin Isoforms in
http://pediatrics.aappublications.org/content/116/4/e601located on the World Wide Web at:
The online version of this article, along with updated information and services, is
1073-0397. ISSN:60007. Copyright © 2005 by the American Academy of Pediatrics. All rights reserved. Print
the American Academy of Pediatrics, 141 Northwest Point Boulevard, Elk Grove Village, Illinois,has been published continuously since 1948. Pediatrics is owned, published, and trademarked by Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
by guest on June 12, 2018www.aappublications.org/newsDownloaded from


















