motor neuron disease
18
MOTOR NEURON DISEASES לללל לללללל, ללללל לללללללללל, לל"ל ללללל ללללל ללללל
-
Upload
eliran-jacobi -
Category
Health & Medicine
-
view
346 -
download
1
Transcript of motor neuron disease

MOTOR NEURON DISEASESמיכל הולצמן, מחלקת נוירולוגיה, בי"ח לגליל מערבי נהריה

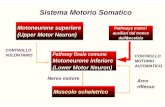
Upper motor neuron Characteristic of upper motor neurone
lesions: no wasting; increased tone of clasp-knife type; weakness most evident in anti-gravity
muscles; increased reflexes and clonus; extensor plantar responses.

Upper motor neuron Lesion situated peripherally in the cerebral
hemisphere produces weakness of part of the contralateral side of the body
Lesion of the internal capsule, are much more likely to produce weakness of the whole of the contralateral side of the body & visual loss
Lesion in the spinal cord in high cervical level will cause ipsilateral hemiparessis, below the neck level ipsilateral monoparessis

Lower motor neuroncharacteristics of lower motor neurone
lesions: wasting; fasciculation decreased tone (i.e. flaccidity) weakness decreased or absent reflexes flexor or absent plantar responses
Anterior horn

Bulbar weakness(CN 7-12)

Amyotrophic Lateral Sclerosis

Amyotrophic Lateral Sclerosis Degenerative disorder of upper and
lower motor neurons of the corticospinal tract
Anterior motor horn degeneration leads to lower motor neuron signs
Lateral corticospinal tract degeneration leads to upper motor neuron signs

Amyotrophic Lateral Sclerosis The process is remarkably selective, leaving
special senses, and cerebellar, sensory and autonomic functions intact
Tends to start either as a problem in the bulbar muscles, or as a problem in the limbs
Initially the involvement tends to be either lower motor neurone or upper motor neurone in nature
the coexistence of both, in the absence of sensory signs, is the hallmark of motor neurone disease

pathogenesis Most cases are sporadic, arising in
middle age adults. Zinc-copper superoxide dismutase
mutation (SOD1) is present in some familial cases; leads to free radical injury in neurons
Prior to their destruction, motor neurons develop protein-rich inclusions in their cell bodies and axons containing ubiquitin

Clinical presentationIn 75-80% of patients, symptoms begin with limb involvement. Initial complaints in patients with lower limb onset are often as
follows: Tripping, stumbling, or awkwardness when running Foot drop; patients may report a "slapping" gaitInitial complaints with upper limb onset include the following: Reduced finger dexterity, cramping, stiffness, and weakness
or wasting of intrinsic hand muscles Wrist drop interfering with work performanceWith bulbar onset (20-25%), initial complaints are as follows: Slurred speech, hoarseness, or decreased volume of speech Aspiration or choking during a meal

Clinical presentationEmotional and special cognitive difficulties in some
ALS patients are as follows: Involuntary laughing or crying Depression Impaired executive function Maladaptive social behaviorFeatures of more-advanced disease are as follows: Muscle atrophy becomes more apparent Spasticity may compromise gait and manual
dexterity Muscle cramps are common

Diagnosis

Treatment Riluzole, decreasing the release of
glutamate and modifies the rate of progression 50 mg orally twice daily,
Symptomatic treatment:Limb stiffness - antispasticity
agents(baclofen, tizanidine)Pain – NSAIDsCramps - Quinine sulfate,benzodiazepines

Prognosis Involvement of bulbar and respiratory
muscles accounts for most of the deaths in patients with motor neurone disease.
Median survival – 3 years 15% will live 5 years after diagnosis

נתונים שנאספו לראשונה בישראל מצאו כי הישרדות הישראלים שחולים ( ארוכה פי שניים עד ארבעה מזו שמדווחת ALSבמחלת ניוון שרירים )
מהחולים שורדים יותר מעשר 5%-10%בעולם. בספרות מדווח כי רק בקרב הישראלים שרדו יותר.20%שנים, בעוד למעלה מ־

Primary lateral sclerosis Disease of the voluntary muscle due to
UMN lesion
Symptoms: spasticity, weakness and stiffness of legs, drop foot.
occurs spontaneously after age 50 and progresses gradually over a number of year

Spinal Muscular Atrophy autosomal-recessive disorders, characterized by
progressive weakness of the lower motor neurons. Related to defect on SMN1 gene on chromosome 5q characterized by loss of anterior horn cells Patients usually presenting with muscle weakness and
wasting in the limbs, respiratory, and bulbar or brainstem muscles
The most common types are acute infantile (SMA type I, or Werdnig-Hoffman disease), chronic infantile (SMA type II), chronic juvenile (SMA type III or Kugelberg-Welander disease), and adult onset (SMA type IV) forms.

Thank you for listening…