
MEK-ERK Signaling Dictates DNA-Repair Gene MGMT Expression and Temozolomide Resistance of Stem-Like...
10
CANCER STEM CELLS MEK-ERK Signaling Dictates DNA-Repair Gene MGMT Expression and Temozolomide Resistance of Stem-Like Glioblastoma Cells via the MDM2-p53 Axis ATSUSHI SATO, a,b JUN SUNAYAMA, a,c,d KEN-ICHIRO MATSUDA, a,b SHIZUKA SEINO, a,c,d KAORI SUZUKI, a,c,d ERIKO WATANABE, a,c,d KEN TACHIBANA, a ARATA TOMIYAMA, a TAKAMASA KAYAMA, b CHIFUMI KITANAKA a,c,d a Department of Molecular Cancer Science; and b Department of Neurosurgery, Yamagata University School of Medicine, Yamagata, Japan; c Oncology Research Center, Research Institute for Advanced Molecular Epidemiology, Yamagata University, Yamagata, Japan; d Global COE Program for Medical Sciences, Japan Society for the Promotion of Science, Tokyo, Japan Key Words. Glioma • Chemoresistance • Cancer stem cell • Mitogen-activated protein kinase • Combination therapy ABSTRACT Overcoming the resistance of glioblastoma cells against temo- zolomide, the first-line chemotherapeutic agent of choice for newly diagnosed glioblastoma, is a major therapeutic challenge in the management of this deadly brain tumor. The gene encoding O 6 -methylguanine DNA methyltransferase (MGMT), which removes the methyl group attached by temo- zolomide, is often silenced by promoter methylation in glio- blastoma but is nevertheless expressed in a significant fraction of cases and is therefore regarded as one of the most clinically relevant mechanisms of resistance against temozolomide. However, to date, signaling pathways regulating MGMT in MGMT-expressing glioblastoma cells have been poorly delineated. Here in this study, we provide lines of evidence that the mitogen-activated protein/extracellular signal-regu- lated kinase kinase (MEK)–extracellular signal-regulated ki- nase (ERK)–murine double minute 2 (MDM2)-p53 pathway plays a critical role in the regulation of MGMT expression, using stem-like glioblastoma cells directly derived from patient tumor samples and maintained in the absence of serum, which not only possess stem-like properties but are also known to phenocopy the characteristics of the original tumors from which they are derived. We show that, in stem-like glioblas- toma cells, MEK inhibition reduced MDM2 expression and that inhibition of either MEK or MDM2 resulted in p53 acti- vation accompanied by p53-dependent downregulation of MGMT expression. MEK inhibition rendered otherwise resist- ant stem-like glioblastoma cells sensitive to temozolomide, and combination of MEK inhibitor and temozolomide treatments effectively deprived stem-like glioblastoma cells of their tumorigenic potential. Our findings suggest that targeting of the MEK-ERK-MDM2-p53 pathway in combination with temozolomide could be a novel and promising therapeutic strategy in the treatment of glioblastoma. STEM CELLS 2011;29:1942–1951 Disclosure of potential conflicts of interest is found at the end of this article. INTRODUCTION Glioblastoma multiforme, the most common primary neoplasm of the brain in adults, is one of the most aggressive types of human cancer with a dismal prognosis, the median survival being less than 2 years despite multimodality treatment. The standard of care for glioblastoma is composed of maximal surgical resection fol- lowed by radiotherapy with concomitant and adjuvant chemother- apy [1, 2]. Temozolomide is a monofunctional alkylating agent currently used as the first-line chemotherapeutic agent against newly diagnosed glioblastoma and is also a drug of choice for recurrent disease [3, 4]. Sensitivity of tumor cells to temozolomide is therefore key to successful management of this intractable dis- ease; however, unfortunately glioblastoma cells often exhibit re- sistance against this alkylating agent. Among possible mecha- nisms, O 6 -methylguanine DNA methyltransferase (MGMT) expression has been well documented as the clinically most rele- vant mechanism of resistance against temozolomide-based glio- blastoma therapies [5]. MGMT is a repair enzyme that rapidly removes the methyl group attached by temozolomide at the O 6 position of the guanine residue and as such could theoretically counteract the antitumor effect of temozolomide. Indeed, accumu- lating evidence from correlative observations as well as from func- tional analyses, in either clinical settings or in vitro studies [5–9], now suggests that this is actually the case with glioblastoma, underscoring the absolute necessity for developing novel methods to inactivate MGMT in tumor cells to overcome temozolomide Author contributions: A.S.: concept and design, collection and assembly of data, data analysis and interpretation, and manuscript writing; J.S.: concept and design and data analysis and interpretation; K.-i.M., S.S., K.T., and A.T: data analysis and interpretation; K.S.: provision of study material or patients; E.W.: collection and assembly of data; T.K.: provision of study material or patients and data analysis and interpretation; C.K.: concept and design, data analysis and interpretation, manuscript writing, and final approval of manuscript. Correspondence: Chifumi Kitanaka, M.D., Ph.D., Department of Molecular Cancer Science, Yamagata University School of Medicine, Yamagata 990-9585, Japan. Telephone: þ81-23-628-5212; Fax: þ81-23-628-5215; e-mail: [email protected] Received June 29, 2011; accepted for publication September 11, 2011; first published online in STEM CELLS EXPRESS September 28, 2011. V C AlphaMed Press 1066-5099/2009/$30.00/0 doi: 10.1002/stem.753 STEM CELLS 2011;29:1942–1951 www.StemCells.com
-
Upload
atsushi-sato -
Category
Documents
-
view
213 -
download
0
Transcript of MEK-ERK Signaling Dictates DNA-Repair Gene MGMT Expression and Temozolomide Resistance of Stem-Like...

CANCER STEM CELLS
MEK-ERK Signaling Dictates DNA-Repair Gene MGMT Expression
and Temozolomide Resistance of Stem-Like Glioblastoma
Cells via the MDM2-p53 Axis
ATSUSHI SATO,a,b JUN SUNAYAMA,a,c,d KEN-ICHIRO MATSUDA,a,b SHIZUKA SEINO,a,c,d KAORI SUZUKI,a,c,d
ERIKO WATANABE,a,c,d
KEN TACHIBANA,aARATA TOMIYAMA,
aTAKAMASA KAYAMA,
bCHIFUMI KITANAKA
a,c,d
aDepartment of Molecular Cancer Science; and bDepartment of Neurosurgery, Yamagata University School of
Medicine, Yamagata, Japan; cOncology Research Center, Research Institute for Advanced Molecular
Epidemiology, Yamagata University, Yamagata, Japan; dGlobal COE Program for Medical Sciences, Japan
Society for the Promotion of Science, Tokyo, Japan
Key Words. Glioma • Chemoresistance • Cancer stem cell • Mitogen-activated protein kinase • Combination therapy
ABSTRACT
Overcoming the resistance of glioblastoma cells against temo-zolomide, the first-line chemotherapeutic agent of choicefor newly diagnosed glioblastoma, is a major therapeutic
challenge in the management of this deadly brain tumor. Thegene encoding O6-methylguanine DNA methyltransferase(MGMT), which removes the methyl group attached by temo-
zolomide, is often silenced by promoter methylation in glio-blastoma but is nevertheless expressed in a significant fraction
of cases and is therefore regarded as one of the most clinicallyrelevant mechanisms of resistance against temozolomide.However, to date, signaling pathways regulating MGMT in
MGMT-expressing glioblastoma cells have been poorlydelineated. Here in this study, we provide lines of evidence
that the mitogen-activated protein/extracellular signal-regu-lated kinase kinase (MEK)–extracellular signal-regulated ki-nase (ERK)–murine double minute 2 (MDM2)-p53 pathway
plays a critical role in the regulation of MGMT expression,
using stem-like glioblastoma cells directly derived from patienttumor samples and maintained in the absence of serum, whichnot only possess stem-like properties but are also known to
phenocopy the characteristics of the original tumors fromwhich they are derived. We show that, in stem-like glioblas-toma cells, MEK inhibition reduced MDM2 expression and
that inhibition of either MEK or MDM2 resulted in p53 acti-vation accompanied by p53-dependent downregulation of
MGMT expression. MEK inhibition rendered otherwise resist-ant stem-like glioblastoma cells sensitive to temozolomide, andcombination of MEK inhibitor and temozolomide treatments
effectively deprived stem-like glioblastoma cells of theirtumorigenic potential. Our findings suggest that targeting of
the MEK-ERK-MDM2-p53 pathway in combination withtemozolomide could be a novel and promising therapeuticstrategy in the treatment of glioblastoma. STEM CELLS
2011;29:1942–1951
Disclosure of potential conflicts of interest is found at the end of this article.
INTRODUCTION
Glioblastoma multiforme, the most common primary neoplasmof the brain in adults, is one of the most aggressive types of humancancer with a dismal prognosis, the median survival being lessthan 2 years despite multimodality treatment. The standard of carefor glioblastoma is composed of maximal surgical resection fol-lowed by radiotherapy with concomitant and adjuvant chemother-apy [1, 2]. Temozolomide is a monofunctional alkylating agentcurrently used as the first-line chemotherapeutic agent againstnewly diagnosed glioblastoma and is also a drug of choice forrecurrent disease [3, 4]. Sensitivity of tumor cells to temozolomideis therefore key to successful management of this intractable dis-
ease; however, unfortunately glioblastoma cells often exhibit re-sistance against this alkylating agent. Among possible mecha-nisms, O6-methylguanine DNA methyltransferase (MGMT)expression has been well documented as the clinically most rele-vant mechanism of resistance against temozolomide-based glio-blastoma therapies [5]. MGMT is a repair enzyme that rapidlyremoves the methyl group attached by temozolomide at the O6
position of the guanine residue and as such could theoreticallycounteract the antitumor effect of temozolomide. Indeed, accumu-lating evidence from correlative observations as well as from func-tional analyses, in either clinical settings or in vitro studies [5–9],now suggests that this is actually the case with glioblastoma,underscoring the absolute necessity for developing novel methodsto inactivate MGMT in tumor cells to overcome temozolomide
Author contributions: A.S.: concept and design, collection and assembly of data, data analysis and interpretation, and manuscriptwriting; J.S.: concept and design and data analysis and interpretation; K.-i.M., S.S., K.T., and A.T: data analysis and interpretation; K.S.:provision of study material or patients; E.W.: collection and assembly of data; T.K.: provision of study material or patients and dataanalysis and interpretation; C.K.: concept and design, data analysis and interpretation, manuscript writing, and final approval ofmanuscript.
Correspondence: Chifumi Kitanaka, M.D., Ph.D., Department of Molecular Cancer Science, Yamagata University School of Medicine,Yamagata 990-9585, Japan. Telephone: þ81-23-628-5212; Fax: þ81-23-628-5215; e-mail: [email protected] ReceivedJune 29, 2011; accepted for publication September 11, 2011; first published online in STEM CELLS EXPRESS September 28, 2011.VC AlphaMed Press 1066-5099/2009/$30.00/0 doi: 10.1002/stem.753
STEM CELLS 2011;29:1942–1951 www.StemCells.com

resistance. Understanding the molecular mechanism involved inthe regulation of MGMT expression/function is vital for identifica-tion of therapeutic targets; however, signaling pathways control-ling MGMT in glioblastoma cells remain poorly characterized.
In this study, we dissected the molecular pathway regulatingMGMT expression using stem-like glioblastoma cells as a clini-cally relevant in vitro model of glioblastoma. We used stem-likecells directly derived from glioblastoma patients because (1)stem-like glioblastoma cells more faithfully phenocopy and repre-sent the original tumors from which they are derived than their se-rum-cultured counterparts and conventional glioma cell lines [10,11], and because (2) stem-like cancer cells are presumed, due totheir inherent therapy resistance and high tumorigenic capacity, tobe the source of post-treatment recurrence and hence the most crit-ical target of therapy [12, 13]. Here, we provide lines of evidencethat, in stem-like glioblastoma cells, the MEK-extracellular sig-nal-regulated kinase (ERK) pathway dictates MGMT expressionand temozolomide resistance via the murine double minute 2(MDM2)-p53 axis. This study thus offers novel opportunities todevelop molecular targeting drugs for MGMT inactivation in glio-blastoma to be validated in future preclinical and clinical studies.
MATERIALS AND METHODS
Reagents and Antibodies
SL327 was purchased from Enzo Life Sciences (New York, NY,http://www.enzolifesciences.com), temozolomide was from LKTLaboratories (St. Paul, MN, http://www.lktlabs.com), Nutlin-3 wasfrom Calbiochem (Darmstadt, Germany, http://www.emdchemicals.com), and epidermal growth factor (EGF) and basic fibro-blast growth factor (bFGF) were from Peprotech (Princeton, NJ,http://www.peprotech.com). Anti-nestin (AB5922) and anti-p53 (OP-43) were from Chemicon (Billerica, MA, http://www.millipore.com). Anti-glial fibrillary acidic protein (AF2594), anti-bIII-tubulin(MAB1195), and anti-MDM2 (AF1244) were from R&D (Minneap-olis, MN, http://www.rndsystems.com). Anti-ERK1/2 (#4695), anti-phospho-ERK1/2 (#9106), and anti-p21 (#2947) were from Cell Sig-naling Technology (Danvers, MA, http://www.cellsignal.com). Anti-MGMT (ab212628) was from Abcam (Cambridge, MA, http://www.abcam.com). Anti-MEK1 (sc-219) and anti-MEK2 (sc-525)were from Santa Cruz Biotechnology (CA, http://www.scbt.com).Anti-actin (A1978) was from Sigma (St. Louis, MO, http://www.sigmaaldrich.com). Horseradish peroxidase (HRP)-conjugatedsecondary antibodies for immunoblotting were from JacksonImmuno Research (Carlsbad, CA, http://www.invitrogen.com).
Human Glioblastoma Tissues and Isolation, Culture,and Characterization of Patient-Derived Stem-LikeGlioblastoma Cells
Isolation and establishment of primary human stem-like glioblastomacells (GS-Y01 and GS-Y02) were carried out essentially as previouslydescribed [14] in accordance with a protocol approved by the Institu-tional Review Boards of Yamagata University School of Medicine,and the stem-like cells were maintained in the monolayer culture con-dition [15, 16]. The capacity for self-renewal, multipotency of differ-entiation, and tumorigenicity were tested and verified as in SupportingInformation Figure S1. To induce differentiation, cells were culturedin the differentiation culture condition (Dulbecco’s modified Eagle’smedium/F12 containing 10% fetal bovine serum). TGS01 and TGS04are generous gifts from the Department of Neurosurgery, Universityof Tokyo. Characterization of TGS01 and TGS04 has been describedelsewhere [17] and was also confirmed in this study (Supporting Infor-mation Fig. S1).
RNA Interference
Cells seeded in monolayer culture condition for stem-like cells onthe previous day were transfected with the short-interfering
RNAs (siRNAs) indicated below using Lipofectamin 2000 or Lip-ofectamin RNAi MAX (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) in accordance with the manufacturer’s protocols.
All the siRNAs used in this study were purchased from Invi-trogen and are described below.
Temozolomide Experiment and Cell Death Assay
Stem-like glioblastoma cells exposed to temozolomide for 4hours were cultured on fresh, temozolomide-free culture mediumfor 72 hours after washout of temozolomide and then subjected tocell death assay. Dead cells were detected on the basis of theirinability to exclude propidium iodide (PI). In brief, cells wereincubated with 5 lg/ml PI and, for nuclear staining, with 10 lg/mlHoechst 33342 for 10 minutes. The numbers of PI- and Hoechst-positive cells were scored under a fluorescence microscope(CKX41; Olympus, Tokyo, Japan; at least 500 cells per samplewere examined), and the percentage of PI-positive cells (deadcells) against Hoechst-positive cells (total cells) was determined.
Immunoblot Analysis
For immunoblot analysis, cells were lysed in lysis buffer (10 mMTris-HCl [pH 7.4], 0.1% SDS, 1% sodium deoxycholate, 0.15 MNaCl, 1 mM EDTA, 1% protease inhibitor cocktail set III [Calbio-chem]). After determination of protein concentrations using theBCA protein assay kit (Pierce, Rockford, IL, http://www.piercenet.com), cell lysates containing equal amounts of protein were sepa-rated by SDS-PAGE and transferred to PVDF membrane. Themembrane was probed with a primary antibody and then with anappropriate HRP-conjugated secondary antibody according to theprotocol recommended by the manufacturer of each antibody. Sig-nal was detected using Immobilon Western ChemiluminescentHRP Substrate (Millipore, Billerica, MA, http://www.millipore.com) or ECL Western Blotting Detection Reagents (GE Health-care, Amersham, UK, http://www.gelifesciences.com). Chemilumi-nescent images were captured using a CCD camera of Bio-Rad(Hercules, CA, http://www.bio-rad.com).
Animal Experiments
Stem-like glioblastoma cells (TGS01, 1 � 104) pretreated for 3 dayswith or without SL327 (10 lM) were subsequently exposed to temozo-lomide (50 lM) or vehicle control (dimethyl sulfoxide) in the absenceof SL327 for 4 hours. Then, after 3-day culture in temozolomide-freemedium, the cells were injected stereotactically into the right corpusstriatum (2.5 mm anterior and 2.5 mm lateral to the bregma, and 3.0mm deep) of 5-week-old male BALB/c nu/nu mice (CLEA Japan,Inc.). All animal experiments were performed under a protocolapproved by the Animal Research Committee of YamagataUniversity.
Statistical Analysis
The results of cell death analysis in Figures 1 and 2 are expressedas the means þ standard deviations of three independent experi-ments and were analyzed using the unpaired Student’s t test. InFigure 5, mouse survival was evaluated by the Kaplan-Meiermethod and analyzed using the log-rank test.
MGMT #1 MGMT-HSS106519MGMT #2 MGMT-HSS106520MGMT #3 MGMT-HSS106521MEK1 MAP2K1-HSS108560MEK2 MAP2K2-HSS108588p53 TP53-HSS110905MDM2 MDM2-HSS142909Nontargeting Control
siRNAStealth RNAi Negative
Control Medium GC Duplex #2
Sato, Sunayama, Matsuda et al. 1943
www.StemCells.com
RESULTS
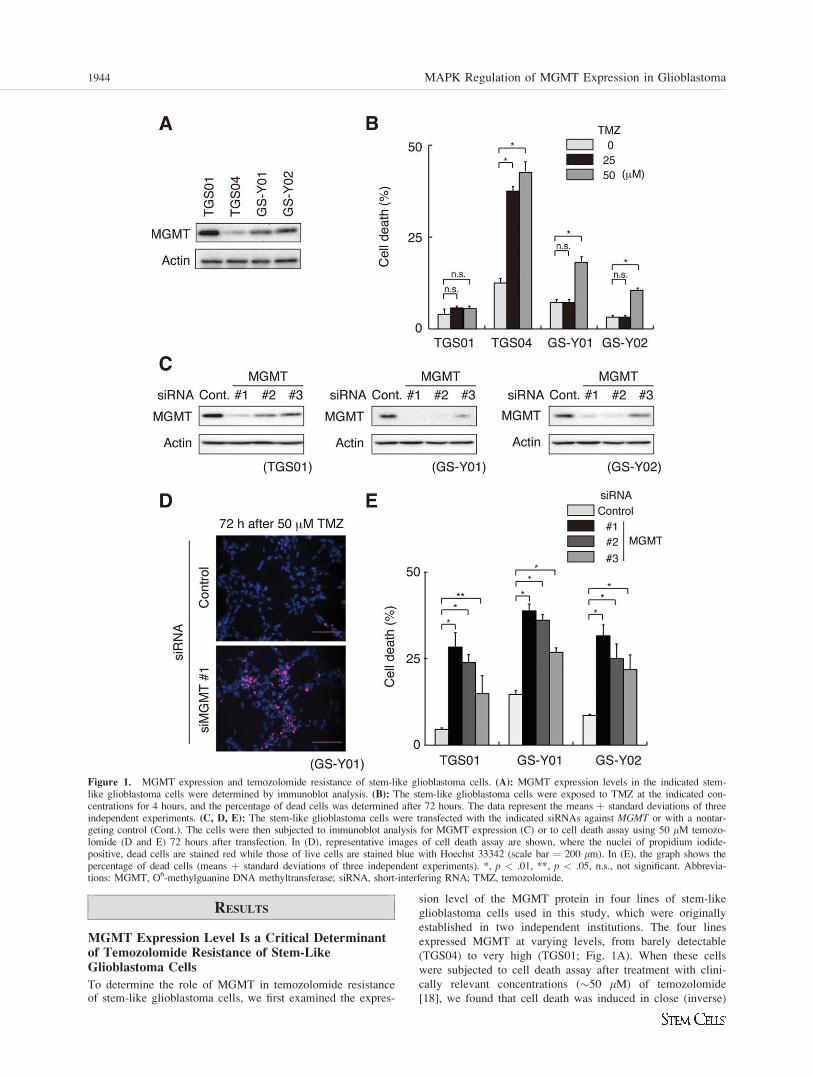
MGMT Expression Level Is a Critical Determinantof Temozolomide Resistance of Stem-LikeGlioblastoma Cells
To determine the role of MGMT in temozolomide resistanceof stem-like glioblastoma cells, we first examined the expres-
sion level of the MGMT protein in four lines of stem-like
glioblastoma cells used in this study, which were originally
established in two independent institutions. The four lines
expressed MGMT at varying levels, from barely detectable
(TGS04) to very high (TGS01; Fig. 1A). When these cells
were subjected to cell death assay after treatment with clini-
cally relevant concentrations (�50 lM) of temozolomide
[18], we found that cell death was induced in close (inverse)
Figure 1. MGMT expression and temozolomide resistance of stem-like glioblastoma cells. (A): MGMT expression levels in the indicated stem-like glioblastoma cells were determined by immunoblot analysis. (B): The stem-like glioblastoma cells were exposed to TMZ at the indicated con-centrations for 4 hours, and the percentage of dead cells was determined after 72 hours. The data represent the means þ standard deviations of threeindependent experiments. (C, D, E): The stem-like glioblastoma cells were transfected with the indicated siRNAs against MGMT or with a nontar-geting control (Cont.). The cells were then subjected to immunoblot analysis for MGMT expression (C) or to cell death assay using 50 lM temozo-lomide (D and E) 72 hours after transfection. In (D), representative images of cell death assay are shown, where the nuclei of propidium iodide-positive, dead cells are stained red while those of live cells are stained blue with Hoechst 33342 (scale bar ¼ 200 lm). In (E), the graph shows thepercentage of dead cells (means þ standard deviations of three independent experiments). *, p < .01, **, p < .05, n.s., not significant. Abbrevia-tions: MGMT, O6-methylguanine DNA methyltransferase; siRNA, short-interfering RNA; TMZ, temozolomide.
1944 MAPK Regulation of MGMT Expression in Glioblastoma
correlation with their MGMT expression levels. For instance,
whereas TGS04 expressing the lowest level of MGMT readily
underwent cell death when treated with temozolomide at a
lower concentration (25 lM), only minimal increase of cell
death was observed with the highest expressor TGS01 even
when treated at a higher concentration (50 lM) (Fig. 1B).
These results are consistent with a recent report showing that
nearly half of the brain tumor-initiating cell lines derived
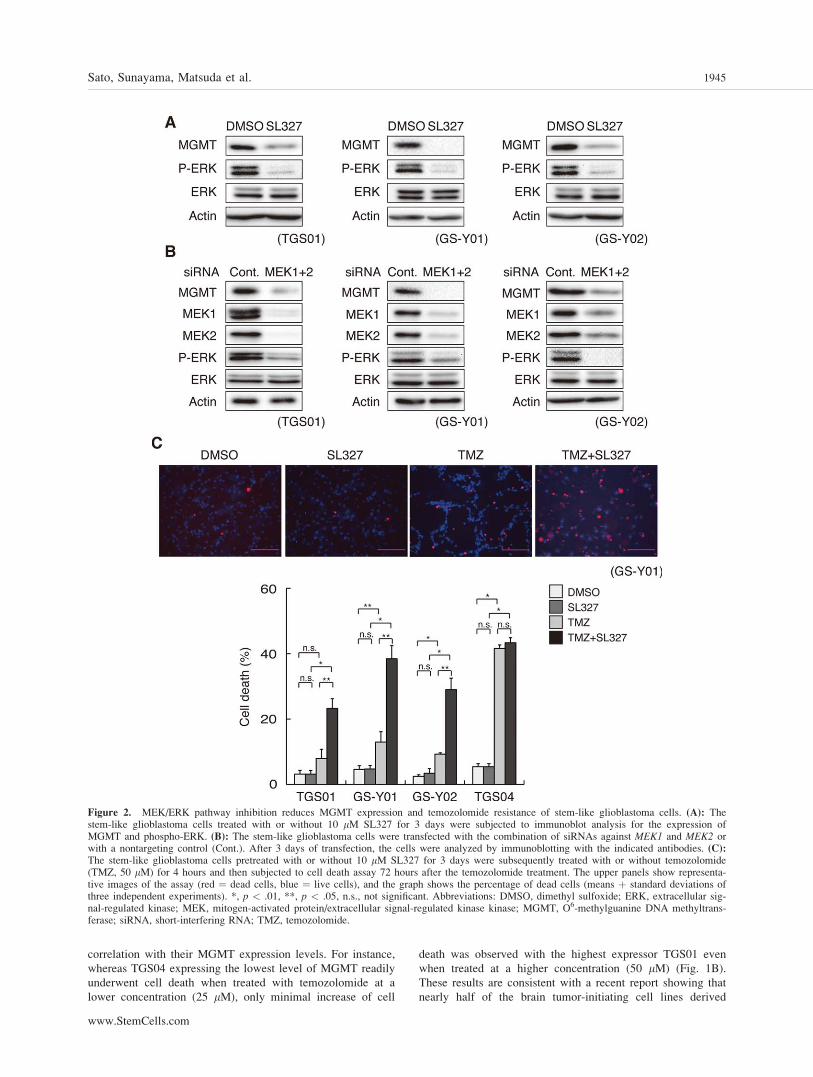
Figure 2. MEK/ERK pathway inhibition reduces MGMT expression and temozolomide resistance of stem-like glioblastoma cells. (A): Thestem-like glioblastoma cells treated with or without 10 lM SL327 for 3 days were subjected to immunoblot analysis for the expression ofMGMT and phospho-ERK. (B): The stem-like glioblastoma cells were transfected with the combination of siRNAs against MEK1 and MEK2 orwith a nontargeting control (Cont.). After 3 days of transfection, the cells were analyzed by immunoblotting with the indicated antibodies. (C):The stem-like glioblastoma cells pretreated with or without 10 lM SL327 for 3 days were subsequently treated with or without temozolomide(TMZ, 50 lM) for 4 hours and then subjected to cell death assay 72 hours after the temozolomide treatment. The upper panels show representa-tive images of the assay (red ¼ dead cells, blue ¼ live cells), and the graph shows the percentage of dead cells (means þ standard deviations ofthree independent experiments). *, p < .01, **, p < .05, n.s., not significant. Abbreviations: DMSO, dimethyl sulfoxide; ERK, extracellular sig-nal-regulated kinase; MEK, mitogen-activated protein/extracellular signal-regulated kinase kinase; MGMT, O6-methylguanine DNA methyltrans-ferase; siRNA, short-interfering RNA; TMZ, temozolomide.
Sato, Sunayama, Matsuda et al. 1945
www.StemCells.com

from glioblastoma expressed the MGMT protein and that
those with MGMT expression, but not those without, showed
resistance against temozolomide [7]. Such close association
between MGMT expression and temozolomide sensitivity
suggests a causal link between these two factors; however,
this has not yet been established in stem-like glioblastoma
cells, with only a single pilot study testing this point using
two glioma-initiating cell lines so far [9]. Therefore, to defini-
tively determine the role of MGMT expression in temozolo-
mide resistance of stem-like glioblastoma cells, we knocked
down MGMT expression in MGMT-expressing stem-like cells
and examined its impact on their temozolomide sensitivity.
We used three different sets of siRNA against MGMT, whichinhibited MGMT with varying efficiencies (Fig. 1C). Signifi-
cantly, cell death induced by temozolomide treatment directly
correlated with the efficiency of knockdown, namely, the
level of MGMT expression in all the three lines tested (Fig.
1D, 1E). Thus, the results indicate that the endogenous level
of MGMT expression confers temozolomide resistance on
stem-like glioblastoma cells in a quantitative, expression
level-dependent manner.
MEK-ERK Signaling Is Required for theMaintenance of MGMT Expression andTemozolomide Resistance of Stem-LikeGlioblastoma Cells
A previous study demonstrated that CD133-positive cancerstem cells derived from glioblastoma express much higherlevels of MGMT mRNA than autologous CD133-negativetumor cells, suggesting that MGMT expression may beassociated with stem-like state of glioblastoma cells [19].We therefore reasoned that inhibition of MEK in stem-likeglioblastoma cells, which we have recently shown to pro-mote their differentiation [14], could also reduce MGMTexpression along with other stem-like properties. We testedthis idea first by treating the stem-like glioblastoma cellswith chemical inhibitors of MEK, SL327 and U0126. Bothinhibitors induced differentiation (Supporting InformationFig. S3) under conditions in which they inhibited ERK phos-phorylation (Fig. 2A, Supporting Information Figs. S2 andS8), confirming that this is required for prevention of prema-ture differentiation of stem-like glioblastoma cells, as wehave shown using a different set of stem-like glioblastomacells [14]. Under identical experimental conditions, bothSL327 and U0126 suppressed MGMT expression, suggestingthat the kinase activity of MEK is required for MGMTexpression (Fig. 2A and Supporting Information Fig. S2).Knockdown experiments targeted against the MEK genesalso demonstrated MEK requirement for MGMT expression,corroborating the results of the chemical inhibitor study(Fig. 2B). We then examined the impact of MEK inhibition-mediated downregulation of MGMT expression on temozolo-mide sensitivity of the stem-like glioblastoma cells. Intrigu-ingly, when the cells were treated with SL327 for 3 days fol-lowed by washout of the inhibitor, MGMT expressionremained suppressed for at least 3 days after the washout,despite apparent recovery of ERK phosphorylation (Support-ing Information Fig. S4). We therefore treated stem-likeglioblastoma cells first with SL327 for 3 days, and afterwashout of SL327, then treated them with temozolomide inthe absence of the MEK inhibitor for 3 days to conduct celldeath assay. The result clearly indicated that MEK inhibitionpreceding temozolomide treatment, while having a minimaleffect on cellular viability by itself, remarkably promotescell death induction by temozolomide in cells expressing
MGMT (Fig. 2C, Supporting Information Fig. S5). In sharpcontrast, MEK inhibition failed to promote temozolomide-induced death of TGS04 cells that express only a low levelof MGMT and hence are originally sensitive to temozolo-mide (Figs. 1A, 1B, and 2C, Supporting Information Fig.S5), indicating that the cell death-promoting effect of MEKinhibition is dependent on MGMT expression. Together,these results suggest that MEK-ERK signaling contributes totemozolomide resistance of stem-like glioblastoma cellsthrough maintenance of MGMT expression.
Upregulation of p53 is Responsible for MEKInhibition-Mediated Suppression of MGMTExpression in Stem-Like Glioblastoma Cells
During our screening search for molecules differentiallyexpressed in stem-like glioblastoma cells before and afterdifferentiation, we noted remarkable upregulation of p53,together with its transcriptional target p21 (with the excep-tion of TGS04 cells), in cells undergoing differentiationafter SL327 treatment (Fig. 3A, Supporting InformationFigs. S6A and S8A). Subsequent knockdown experimentstargeted against the MEK genes confirmed that MEK inhibi-tion is indeed responsible for the observed p53 activation(Fig. 3B, Supporting Information Figs. S6B and S8B). Asprevious studies indicated that p53 is capable of inhibitingMGMT expression [20–23], we investigated the role of thisp53 upregulation in MEK inhibition-mediated suppressionof MGMT. Under conditions where p53 expression waseffectively knocked down by an siRNA against p53, sup-pression of MGMT expression either by SL327 (Fig. 3C,Supporting Information Fig. S6C) or by knockdown of theMEK genes (Fig. 3D, Supporting Information Fig. S6D)was abolished. Thus, suppression of MGMT expression ofstem-like glioblastoma cells by MEK inhibition is depend-ent on p53.
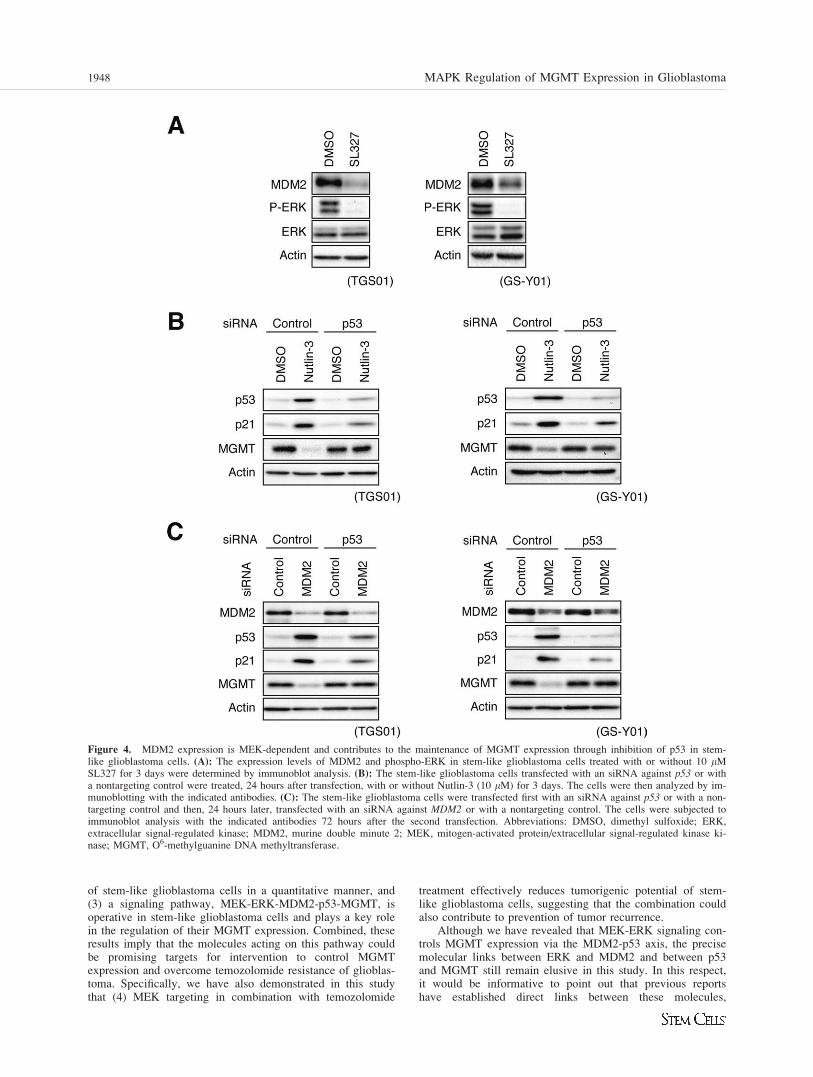
MEK-ERK Signaling is Required for MDM2Expression in Stem-Like Glioblastoma Cells, WhichPrevents p53 Activation and SubsequentSuppression of MGMT Expression
To further delineate the molecular pathway involved in MEKinhibition-mediated suppression of MGMT expression, wenext examined molecules that could modulate p53 expressionin response to MEK inhibition. Among other candidates, wefound in stem-like glioblastoma cells treated with SL327decreased expression of MDM2 (Fig. 4A, Supporting Infor-mation Figs. S7A and S8A), a negative regulator of p53expression and function [24], giving rise to the possibility thatMEK-ERK signaling maintains MDM2 expression to preventp53 activation and subsequent downregulation of MGMTexpression in stem-like glioblastoma cells. To test this possi-bility, we first attempted functional inactivation of MDM2using Nutlin-3, a chemical inhibitor of MDM2, which disruptsphysical interaction between MDM2 and p53 [25]. Nutlin-3treatment of stem-like glioblastoma cells increased the expres-sion of the p53 protein accompanied by increased anddecreased expressions of p21 and MGMT, respectively, andknockdown of p53 prevented the change of their expressionlevels (Fig. 4B, Supporting Information Fig. S7B). Similarresults were obtained when MDM2 was inactivated bysiRNA-mediated knockdown (Fig. 4C, Supporting InformationFig. S7C). In TGS04 cells, which originally express only alow level of MGMT, MDM2 inactivation led to increased p53expression but again no change in p21 or MGMT expressionlevel (Supporting Information Fig. S8C). Thus, inactivation ofMDM2, be it either chemical or genetic, resulted in p53
1946 MAPK Regulation of MGMT Expression in Glioblastoma

activation and p53-dependent suppression of MGMT expres-sion in stem-like glioblastoma cells with transcriptionallycompetent p53. Collectively, the results suggest that the MEKactivity is required for MDM2 expression, which in turn isrequired for the maintenance of MGMT expression throughnegative regulation of p53.
Combination of Temozolomide Treatment withMEK Inhibition Promotes Elimination ofTumorigenic Population from Stem-LikeGlioblastoma Cells
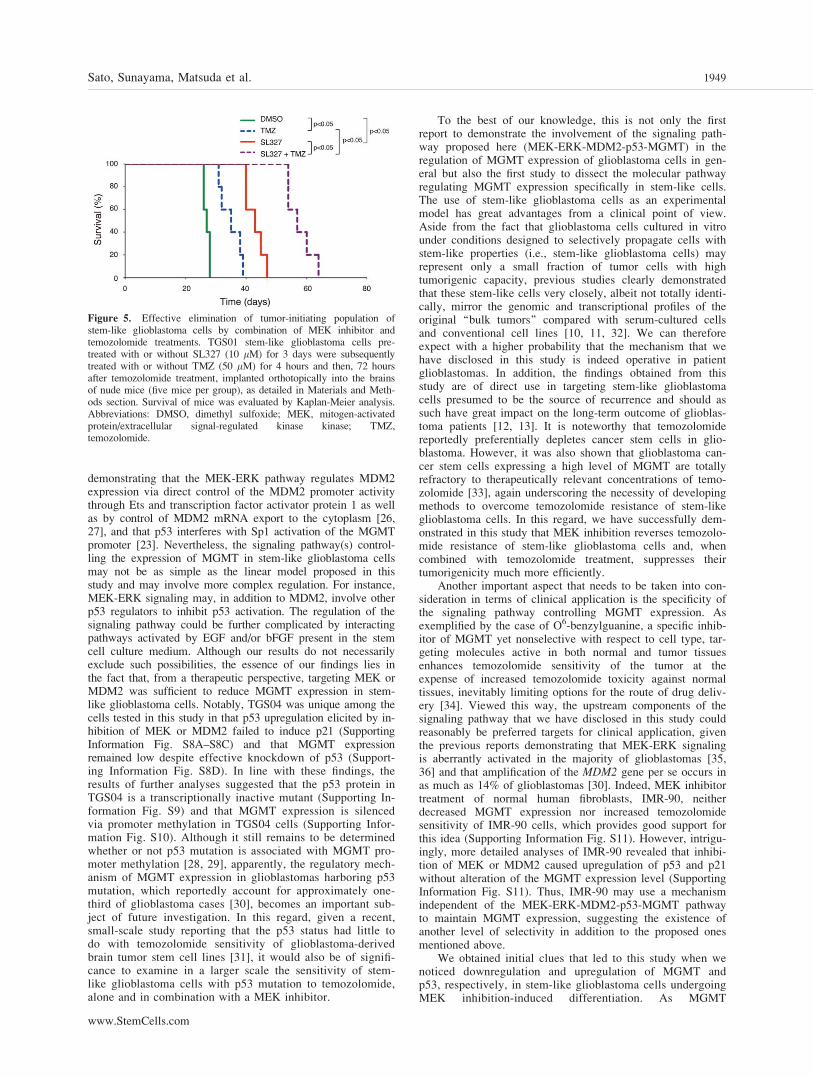
The results of this study thus far suggest that targeting of themolecule(s) in the MEK-ERK-MDM2-p53-MGMT pathwayin combination with temozolomide treatment would be arational therapeutic approach to glioblastoma. While stem-likeglioblastoma cells maintained in the absence of serum gener-ally closely phenocopy the original, bulk tumors from whichthey are derived, they are considered at the same time to spe-cifically represent a highly tumorigenic subpopulation withinthe original tumors [10–12]. Our recent study showed thatMEK inhibition promotes commitment of stem-like glioblas-toma cells to differentiation and thereby reduces their tumori-genic potential; however, the suppressive effect of SL327 as asingle-agent on stem-like glioblastoma cell tumorigenesiswas limited [14]. We therefore asked in this study whethertemozolomide treatment in combination with SL327, via thesynergistic mechanism observed in vitro earlier in this study(Fig. 2C), further eliminates a subpopulation of stem-like glio-blastoma cells that would have evaded commitment to differ-entiation and contributed to brain tumor formation if treated
singly with SL327 (Fig. 5). In line with our earlier reportusing a different set of stem-like cells [14], nude mice trans-planted intracranially with stem-like glioblastoma cells(TGS01) treated with SL327 survived significantly longerthan control mice receiving vehicle-treated cells (median sur-vival, 43 days for SL327 vs. 27 days for control). In contrast,temozolomide treatment (50 lM) alone extended survival ofthe mice significantly but modestly (median survival, 35days), consistent with the in vitro observation that TGS01cells are highly resistant against the cytotoxic effect of temo-zolomide unless pretreated with SL327. However, treatmentof stem-like glioblastoma cells with temozolomide in combi-nation with SL327 further extended mouse survival by morethan 2 weeks (median survival, 60 days) compared withSL327 treatment alone. Thus, the results suggest that combi-nation of temozolomide treatment with MEK inhibition issuperior to MEK inhibition alone in terms of depleting thetumorigenic population of glioblastoma cells, presumably dueto temozolomide-mediated killing of stem-like glioblastomacells that remained undifferentiated but had reduced MGMTexpression after MEK inhibition.
DISCUSSION
In this study, we have demonstrated that (1) a significant pro-portion (three out of four examined) of stem-like glioblastomacells directly derived from glioblastoma tissues (patient-derived stem-like glioblastoma cells) express MGMT,(2) MGMT expression contributes to temozolomide resistance
Figure 3. p53 mediates MEK inhibition-induced MGMT downregulation in stem-like glioblastoma cells. (A): The expression levels of p53 andp21 in stem-like glioblastoma cells treated with or without 10 lM SL327 for 3 days were determined by immunoblot analysis. (B): The stem-likeglioblastoma cells transfected with the combination of siRNAs against MEK1 and MEK2 or with a nontargeting control were subjected to immuno-blot analysis for p53 and p21 expression 72 hours after transfection. (C): The stem-like glioblastoma cells transfected with a siRNA against p53 orwith a nontargeting control were treated, 24 hours after transfection, with or without 10 lM SL327 for 3 days. The cells were then analyzed by im-munoblotting with the indicated antibodies. (D): The stem-like glioblastoma cells were transfected first with an siRNA against p53 or with a nontar-geting control and then, 24 hours later, transfected with the combination of siRNAs against MEK1 and MEK2 or with a nontargeting control. Thecells were subjected to immunoblot analysis with the indicated antibodies 72 hours after the second transfection. Abbreviations: DMSO, dimethylsulfoxide; ERK, extracellular signal-regulated kinase; MEK, mitogen-activated protein/extracellular signal-regulated kinase kinase; MGMT, O6-meth-ylguanine DNA methyltransferase; siRNA, short-interfering RNA.
Sato, Sunayama, Matsuda et al. 1947
www.StemCells.com
of stem-like glioblastoma cells in a quantitative manner, and(3) a signaling pathway, MEK-ERK-MDM2-p53-MGMT, isoperative in stem-like glioblastoma cells and plays a key rolein the regulation of their MGMT expression. Combined, theseresults imply that the molecules acting on this pathway couldbe promising targets for intervention to control MGMTexpression and overcome temozolomide resistance of glioblas-toma. Specifically, we have also demonstrated in this studythat (4) MEK targeting in combination with temozolomide
treatment effectively reduces tumorigenic potential of stem-like glioblastoma cells, suggesting that the combination couldalso contribute to prevention of tumor recurrence.
Although we have revealed that MEK-ERK signaling con-trols MGMT expression via the MDM2-p53 axis, the precisemolecular links between ERK and MDM2 and between p53and MGMT still remain elusive in this study. In this respect,it would be informative to point out that previous reportshave established direct links between these molecules,
Figure 4. MDM2 expression is MEK-dependent and contributes to the maintenance of MGMT expression through inhibition of p53 in stem-like glioblastoma cells. (A): The expression levels of MDM2 and phospho-ERK in stem-like glioblastoma cells treated with or without 10 lMSL327 for 3 days were determined by immunoblot analysis. (B): The stem-like glioblastoma cells transfected with an siRNA against p53 or witha nontargeting control were treated, 24 hours after transfection, with or without Nutlin-3 (10 lM) for 3 days. The cells were then analyzed by im-munoblotting with the indicated antibodies. (C): The stem-like glioblastoma cells were transfected first with an siRNA against p53 or with a non-targeting control and then, 24 hours later, transfected with an siRNA against MDM2 or with a nontargeting control. The cells were subjected toimmunoblot analysis with the indicated antibodies 72 hours after the second transfection. Abbreviations: DMSO, dimethyl sulfoxide; ERK,extracellular signal-regulated kinase; MDM2, murine double minute 2; MEK, mitogen-activated protein/extracellular signal-regulated kinase ki-nase; MGMT, O6-methylguanine DNA methyltransferase.
1948 MAPK Regulation of MGMT Expression in Glioblastoma
demonstrating that the MEK-ERK pathway regulates MDM2expression via direct control of the MDM2 promoter activitythrough Ets and transcription factor activator protein 1 as wellas by control of MDM2 mRNA export to the cytoplasm [26,27], and that p53 interferes with Sp1 activation of the MGMTpromoter [23]. Nevertheless, the signaling pathway(s) control-ling the expression of MGMT in stem-like glioblastoma cellsmay not be as simple as the linear model proposed in thisstudy and may involve more complex regulation. For instance,MEK-ERK signaling may, in addition to MDM2, involve otherp53 regulators to inhibit p53 activation. The regulation of thesignaling pathway could be further complicated by interactingpathways activated by EGF and/or bFGF present in the stemcell culture medium. Although our results do not necessarilyexclude such possibilities, the essence of our findings lies inthe fact that, from a therapeutic perspective, targeting MEK orMDM2 was sufficient to reduce MGMT expression in stem-like glioblastoma cells. Notably, TGS04 was unique among thecells tested in this study in that p53 upregulation elicited by in-hibition of MEK or MDM2 failed to induce p21 (SupportingInformation Fig. S8A–S8C) and that MGMT expressionremained low despite effective knockdown of p53 (Support-ing Information Fig. S8D). In line with these findings, theresults of further analyses suggested that the p53 protein inTGS04 is a transcriptionally inactive mutant (Supporting In-formation Fig. S9) and that MGMT expression is silencedvia promoter methylation in TGS04 cells (Supporting Infor-mation Fig. S10). Although it still remains to be determinedwhether or not p53 mutation is associated with MGMT pro-moter methylation [28, 29], apparently, the regulatory mech-anism of MGMT expression in glioblastomas harboring p53mutation, which reportedly account for approximately one-third of glioblastoma cases [30], becomes an important sub-ject of future investigation. In this regard, given a recent,small-scale study reporting that the p53 status had little todo with temozolomide sensitivity of glioblastoma-derivedbrain tumor stem cell lines [31], it would also be of signifi-cance to examine in a larger scale the sensitivity of stem-like glioblastoma cells with p53 mutation to temozolomide,alone and in combination with a MEK inhibitor.
To the best of our knowledge, this is not only the firstreport to demonstrate the involvement of the signaling path-way proposed here (MEK-ERK-MDM2-p53-MGMT) in theregulation of MGMT expression of glioblastoma cells in gen-eral but also the first study to dissect the molecular pathwayregulating MGMT expression specifically in stem-like cells.The use of stem-like glioblastoma cells as an experimentalmodel has great advantages from a clinical point of view.Aside from the fact that glioblastoma cells cultured in vitrounder conditions designed to selectively propagate cells withstem-like properties (i.e., stem-like glioblastoma cells) mayrepresent only a small fraction of tumor cells with hightumorigenic capacity, previous studies clearly demonstratedthat these stem-like cells very closely, albeit not totally identi-cally, mirror the genomic and transcriptional profiles of theoriginal ‘‘bulk tumors’’ compared with serum-cultured cellsand conventional cell lines [10, 11, 32]. We can thereforeexpect with a higher probability that the mechanism that wehave disclosed in this study is indeed operative in patientglioblastomas. In addition, the findings obtained from thisstudy are of direct use in targeting stem-like glioblastomacells presumed to be the source of recurrence and should assuch have great impact on the long-term outcome of glioblas-toma patients [12, 13]. It is noteworthy that temozolomidereportedly preferentially depletes cancer stem cells in glio-blastoma. However, it was also shown that glioblastoma can-cer stem cells expressing a high level of MGMT are totallyrefractory to therapeutically relevant concentrations of temo-zolomide [33], again underscoring the necessity of developingmethods to overcome temozolomide resistance of stem-likeglioblastoma cells. In this regard, we have successfully dem-onstrated in this study that MEK inhibition reverses temozolo-mide resistance of stem-like glioblastoma cells and, whencombined with temozolomide treatment, suppresses theirtumorigenicity much more efficiently.
Another important aspect that needs to be taken into con-sideration in terms of clinical application is the specificity ofthe signaling pathway controlling MGMT expression. Asexemplified by the case of O6-benzylguanine, a specific inhib-itor of MGMT yet nonselective with respect to cell type, tar-geting molecules active in both normal and tumor tissuesenhances temozolomide sensitivity of the tumor at theexpense of increased temozolomide toxicity against normaltissues, inevitably limiting options for the route of drug deliv-ery [34]. Viewed this way, the upstream components of thesignaling pathway that we have disclosed in this study couldreasonably be preferred targets for clinical application, giventhe previous reports demonstrating that MEK-ERK signalingis aberrantly activated in the majority of glioblastomas [35,36] and that amplification of the MDM2 gene per se occurs inas much as 14% of glioblastomas [30]. Indeed, MEK inhibitortreatment of normal human fibroblasts, IMR-90, neitherdecreased MGMT expression nor increased temozolomidesensitivity of IMR-90 cells, which provides good support forthis idea (Supporting Information Fig. S11). However, intrigu-ingly, more detailed analyses of IMR-90 revealed that inhibi-tion of MEK or MDM2 caused upregulation of p53 and p21without alteration of the MGMT expression level (SupportingInformation Fig. S11). Thus, IMR-90 may use a mechanismindependent of the MEK-ERK-MDM2-p53-MGMT pathwayto maintain MGMT expression, suggesting the existence ofanother level of selectivity in addition to the proposed onesmentioned above.
We obtained initial clues that led to this study when wenoticed downregulation and upregulation of MGMT andp53, respectively, in stem-like glioblastoma cells undergoingMEK inhibition-induced differentiation. As MGMT
Figure 5. Effective elimination of tumor-initiating population ofstem-like glioblastoma cells by combination of MEK inhibitor andtemozolomide treatments. TGS01 stem-like glioblastoma cells pre-treated with or without SL327 (10 lM) for 3 days were subsequentlytreated with or without TMZ (50 lM) for 4 hours and then, 72 hoursafter temozolomide treatment, implanted orthotopically into the brainsof nude mice (five mice per group), as detailed in Materials and Meth-ods section. Survival of mice was evaluated by Kaplan-Meier analysis.Abbreviations: DMSO, dimethyl sulfoxide; MEK, mitogen-activatedprotein/extracellular signal-regulated kinase kinase; TMZ,temozolomide.
Sato, Sunayama, Matsuda et al. 1949
www.StemCells.com

downregulation was also observed in stem-like glioblastomacells undergoing serum-induced differentiation (SupportingInformation Fig. S12), we initially assumed that MGMTexpression might be one of the stem-like properties of glio-blastoma cells and that p53 might also be a master regulatorof such stem-like properties including MGMT expression.However, our recent observation that p53 activation doesaccompany MEK inhibition-induced differentiation (thisstudy), but not serum-induced differentiation of stem-likeglioblastoma cells (Supporting Information Fig. S12), sug-gests that p53 activation may not always be essential fortheir differentiation and associated downregulation ofMGMT expression. Although the exact role of MDM2 andp53 in the maintenance/differentiation of stem-like glioblas-toma cells is still to be investigated, targeting moleculesinvolved in the maintenance of both stem-like state andMGMT expression (e.g., MEK) may be more reasonable inthat, by doing so, we can expect inhibition of tumorigenicpotential of stem-like glioblastoma cells as well in additionto MGMT inactivation.
CONCLUSION
To date, a number of methods have been described to inhibitMGMT; however, their significance in clinical use for thepurpose of overcoming temozolomide resistance is still lim-ited or unknown [3, 37, 38]. Here, we have shown that themolecular pathway linking MEK to MGMT expression could
offer novel and rational targets for MGMT inactivation, spe-cifically MEK (by SL327) and MDM2 (by Nutlin-3). Thisstudy thus constitutes a significant step forward in the battleagainst glioblastoma, by adding new arms to our arsenal tocombat this deadly disease.
ACKNOWLEDGMENTS
We thank Drs. Tomoki Todo and Nobuhito Saito at the Universityof Tokyo for generously providing us with the TGS cells; Dr.Motoo Nagane at Kyorin University for valuable advice on xeno-graft experiments; Keita Shibuya and Dr. Tomoko Kagawa of ourlaboratory for his expertise in flow cytometry and for her continu-ous support/encouragements, respectively. This work was sup-ported by Grants-in-Aid for Scientific Research, for ChallengingExploratory Research, and for Young Scientists from the Ministryof Education, Culture, Sports, Science and Technology of Japan,by a Grant-in-Aid from the Global COE Program of the Japan So-ciety for the Promotion of Science, by a Grant-in-Aid for CancerResearch from the Ministry of Health, Labor, and Welfare of Ja-pan, and by a grant from the Japan Brain Foundation.
DISCLOSURE OF POTENTIAL
CONFLICTS OF INTEREST
The authors indicate no conflicts of interest.
REFERENCES
1 Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008;359:492–507.
2 Khasraw M, Lassman AB. Advances in the treatment of malignantgliomas. Curr Oncol Rep 2010;12:26–33.
3 Villano JL, Seery TE, Bressler LR. Temozolomide in malignant glio-mas: Current use and future targets. Cancer Chemother Pharmacol2009;64:647–655.
4 Nishikawa R. Standard therapy for glioblastoma—A review of wherewe are. Neurol Med Chir (Tokyo) 2010;50:713–719.
5 Hegi ME, Liu L, Herman JG et al. Correlation of O6-methylguaninemethyltransferase (MGMT) promoter methylation with clinical out-comes in glioblastoma and clinical strategies to modulate MGMTactivity. J Clin Oncol 2008;26:4189–4199.
6 Hermisson M, Klumpp A, Wick W et al. O6-methylguanine DNAmethyltransferase and p53 status predict temozolomide sensitivity inhuman malignant glioma cells. J Neurochem 2006;96:766–776.
7 Blough MD, Westgate MR, Beauchamp D et al. Sensitivity totemozolomide in brain tumor initiating cells. Neuro Oncol 2010;12:756–760.
8 Spiegl-Kreinecker S, Pirker C, Filipits M et al. O6-methylguanineDNA methyltransferase protein expression in tumor cells predicts out-come of temozolomide therapy in glioblastoma patients. Neuro Oncol2010;12:28–36.
9 Kato T, Natsume A, Toda H et al. Efficient delivery of liposome-mediated MGMT-siRNA reinforces the cytotoxity of temozolomide inGBM-initiating cells. Gene Ther 2010;17:1363–1371.
10 Lee J, Kotliarova S, Kotliarov Y et al. Tumor stem cells derived fromglioblastomas cultured in bFGF and EGF more closely mirror the phe-notype and genotype of primary tumors than do serum-cultured celllines. Cancer Cell 2006;9:391–403.
11 Schulte A, Gunther HS, Phillips HS et al. A distinct subset of gliomacell lines with stem cell-like properties reflects the transcriptionalphenotype of glioblastomas and overexpresses CXCR4 as therapeutictarget. Glia 2011;59:590–602.
12 Neman J, Jandial R. Decreasing glioma recurrence through adjuvantcancer stem cell inhibition. Biologics 2010;4:157–162.
13 Clevers H. The cancer stem cell: Premises, promises and challenges.Nat Med 2011;17:313–319.
14 Sunayama J, Matsuda K, Sato A et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-
renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells2010;28:1930–1939.
15 Pollard SM, Yoshikawa K, Clarke ID et al. Glioma stem cell linesexpanded in adherent culture have tumor-specific phenotypes andare suitable for chemical and genetic screens. Cell Stem Cell 2009;4:568–580.
16 Fael Al-Mayhani TM, Ball SL, Zhao JW et al. An efficient methodfor derivation and propagation of glioblastoma cell lines that con-serves the molecular profile of their original tumours. J NeurosciMethods 2009;176:192–199.
17 Ikushima H, Todo T, Ino Y et al. Autocrine TGF-beta signaling main-tains tumorigenicity of glioma-initiating cells through Sry-relatedHMG-box factors. Cell Stem Cell 2009;5:504–514.
18 Ostermann S, Csajka C, Buclin T et al. Plasma and cerebrospinal fluidpopulation pharmacokinetics of temozolomide in malignant gliomapatients. Clin Cancer Res 2004;10:3728–3736.
19 Liu G, Yuan X, Zeng Z et al. Analysis of gene expression and chemo-resistance of CD133þ cancer stem cells in glioblastoma. Mol Cancer2006;5:67.
20 Harris LC, Remack JS, Houghton PJ et al. Wild-type p53 suppressestranscription of the human O6-methylguanine-DNA methyltransferasegene. Cancer Res 1996;56:2029–2032.
21 Grombacher T, Eichhorn U, Kaina B. p53 is involved in regulation ofthe DNA repair gene O6-methylguanine-DNA methyltransferase(MGMT) by DNA damaging agents. Oncogene 1998;17:845–851.
22 Srivenugopal KS, Shou J, Mullapudi SR et al. Enforced expressionof wild-type p53 curtails the transcription of the O(6)-methyl-guanine-DNA methyltransferase gene in human tumor cells andenhances their sensitivity to alkylating agents. Clin Cancer Res2001;7:1398–1409.
23 Bocangel D, Sengupta S, Mitra S et al. p53-mediated down-regulationof the human DNA repair gene O6-methylguanine-DNA methyltrans-ferase (MGMT) via interaction with Sp1 transcription factor. Anti-cancer Res 2009;29:3741–3750.
24 Hock A, Vousden KH. Regulation of the p53 pathway by ubiquitinand related proteins. Int J Biochem Cell Biol 2010;42:1618–1621.
25 Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53protein–protein interaction to reactivate p53 function: A novelapproach for cancer therapy. Annu Rev Pharmacol Toxicol 2009;49:223–241.
26 Ries S, Biederer C, Woods D et al. Opposing effects of Ras on p53:Transcriptional activation of mdm2 and induction of p19ARF. Cell2000;103:321–330.
1950 MAPK Regulation of MGMT Expression in Glioblastoma

27 Phelps M, Phillips A, Darley M et al. MEK-ERK signaling controlsHdm2 oncoprotein expression by regulating hdm2 mRNA export tothe cytoplasm. J Biol Chem 2005;280:16651–16658.
28 Shamsara J, Sharif S, Afsharnezhad S et al. Association betweenMGMT promoter hypermethylation and p53 mutation in glioblastoma.Cancer Invest 2009;27:825–829.
29 Groenendijk FH, Taal W, Dubbink HJ et al. MGMT promoter hyper-methylation is a frequent, early, and consistent event in astrocytomaprogression, and not correlated with TP53 mutation. J Neurooncol2011;101:405–417.
30 Network TCGAR. Comprehensive genomic characterization defineshuman glioblastoma genes and core pathways. Nature 2008;455:1061–1068.
31 Blough MD, Beauchamp DC, Westgate MR et al. Effect of aberrantp53 function on temozolomide sensitivity of glioma cell lines andbrain tumor initiating cells from glioblastoma. J Neurooncol 2011;102:1–7.
32 Ernst A, Hofmann S, Ahmadi R et al. Genomic and expression profiling ofglioblastoma stem cell-like spheroid cultures identifies novel tumor-rele-vant genes associated with survival. Clin Cancer Res 2009;15:6541–6550.
33 Beier D, Rohrl S, Pillai DR et al. Temozolomide preferentially depletescancer stem cells in glioblastoma. Cancer Res 2008;68:5706–5715.
34 Koch D, Hundsberger T, Boor S et al. Local intracerebral administra-tion of O(6)-benzylguanine combined with systemic chemotherapywith temozolomide of a patient suffering from a recurrent glioblas-toma. J Neurooncol 2007;82:85–89.
35 Feldkamp MM, Lala P, Lau N et al. Expression of activated epidermalgrowth factor receptors, Ras-guanosine triphosphate, and mitogen-activatedprotein kinase in human glioblastoma multiforme specimens. Neurosurgery1999;45:1442–1453.
36 Lopez-Gines C, Gil-Benso R, Benito R et al. The activation of ERK1/2 MAP kinases in glioblastoma pathobiology and its relationship withEGFR amplification. Neuropathology 2008;28:507–515.
37 Natsume A, Ishii D, Wakabayashi T et al. IFN-beta down-regulatesthe expression of DNA repair gene MGMT and sensitizes resistant gli-oma cells to temozolomide. Cancer Res 2005;65:7573–7579.
38 Bobustuc GC, Baker CH, Limaye A et al. Levetiracetam enhancesp53-mediated MGMT inhibition and sensitizes glioblastoma cells totemozolomide. Neuro Oncol 2010;12:917–927.
See www.StemCells.com for supporting information available online.
Sato, Sunayama, Matsuda et al. 1951











![The Answered Supplication · Ad-Du‘â’ according to the terminology of the Sharî‘ah [Islamic Law] is a request from a servant to His Lord. Supplication dictates that there](https://static.fdocument.pub/doc/165x107/5f2448b6da2b5035a94903bf/the-answered-supplication-ad-duaa-according-to-the-terminology-of-the-sharaah.jpg)






