

Médico Hematologista COI -...
34
Outras anemias hemolíticas Dr. Márcio Hori Médico Hematologista – COI
Transcript of Médico Hematologista COI -...

Outras anemias hemolíticas
Dr. Márcio Hori
Médico Hematologista – COI

Hemoglobina
• Ligação de oxigênio à Hb para transporte no corpo
• Transporte na hemácias para preservar a Hb por 120 dias
• Estrutura: – Tetrâmero com 2 pares de cadeias de globina (2 cadeias α e 2 cadeias
não-α) • Formação de dímeros de cadeia α + não α (β, γ ou δ) com atração de cargas
– Grupamento heme • protoporfirinas + ferro

Hemoglobina
• Tipos de Hemoglobina normais – HbA 2 cadeias α e 2 cadeias β
– HbA2 2 cadeias α e 2 cadeias δ
– HbF 2 cadeias α e 2 cadeias γ
• No período neonatal – Produção predominante de cadeias γ sobre cadeias β
– Maior produção de HbF
• Vida adulta – Produção muito pequena de cadeias γ
– Produção abundante de cadeia β
– Maior produção de HbA e pouca HbF

Hemoglobina
• Adulto normal – HbA 95%
– HbA2 2,5%
– HbF 2,5%
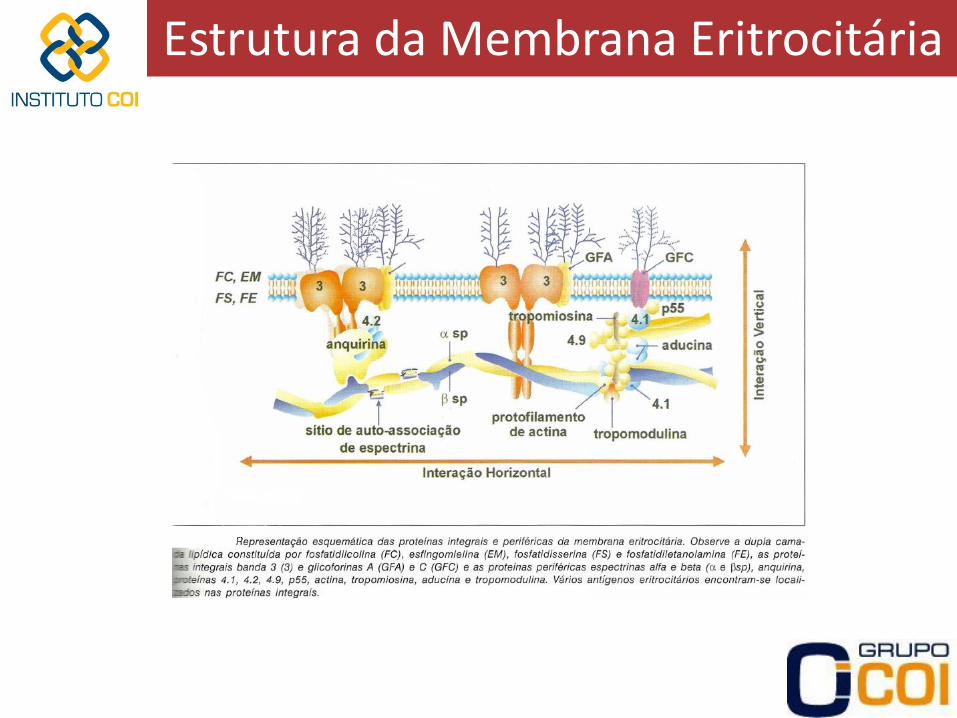
Estrutura da Membrana Eritrocitária

Esferocitose Hereditária
• Epidemiologia – Incidência de 1:2.000 a 5.000 nascimentos
– Mais frequentes em brancos do que negros
– Formas leves e portadores assintomáticos 1% da população
– Podem decorrer (60% dos casos) de deficiências de:
• Espectrina isolada
• Espectrina e anquirina
• Banda 3
• Proteína 4.2

Introdução
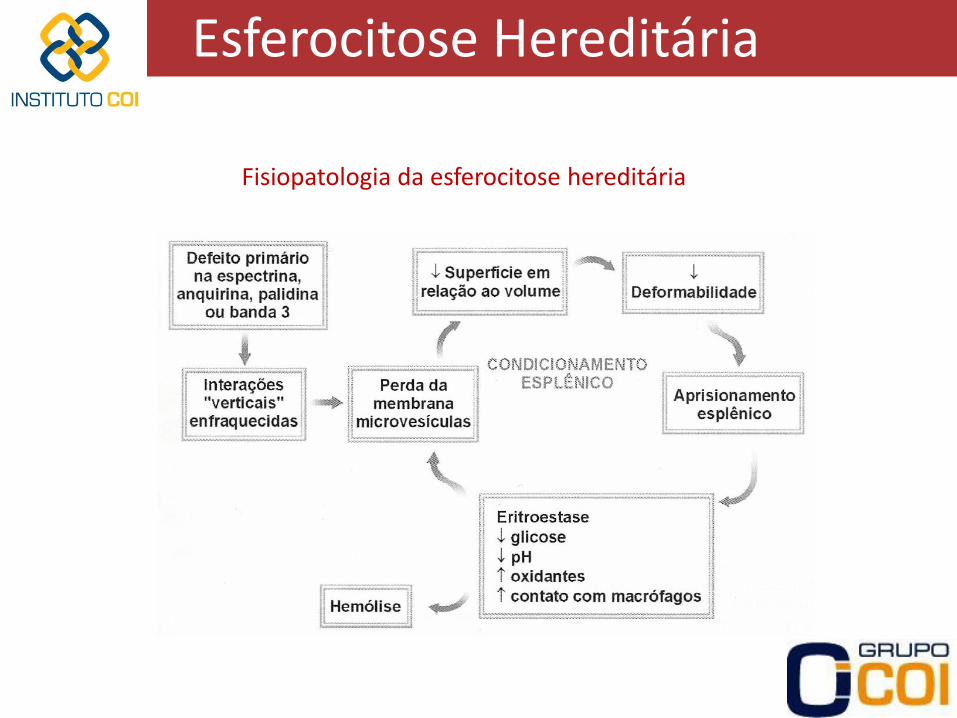
Esferocitose Hereditária
Fisiopatologia da esferocitose hereditária

Esferocitose Hereditária
• Manifestações clínicas – Forma típica
• Ictericia leve
• Esplenomegalia
• Anemia ausente (compensação medular, leve ou moderada)
– Pacientes com traço e formas leves
• Usualmente assintomáticos (sem anemia compensação medular)
• Reticulócitos normais ou levemente aumentados
• Sem padrão hemolítico
• Sangue periférico com discreta esferocitose
• Diagnóstico em investigação de familiar mais sintomático ou durante infecção viral transitória (aparecimento de sintomas) ou na 3ª idade por incapacidade de resposta medular adequada

Esferocitose Hereditária
– Formas graves
• Hemólise importante de aparecimento precoce, com necessidade transfusional
• Esplenomegalia
– Em neonatos
• 50% com icterícia neonatal fototerapia ou, raramente, exsanguineotransfusão
• Diagnóstico difícil pois hemácias com HbF mais resistentes à lise osmótica
• Maioria evolui sem sintomas
– Complicações
• Colelitíase por cálculos de bilirrubinato
• Crises hemolíticas em infecções
• Crises aplásticas infecções por parvovírus B19
• Crises megaloblásticas carência de folato (elevado turnover eritrocitário)

Esferocitose Hereditária
– Diagnóstico
Sangue periférico normal Esferocitose hereditária
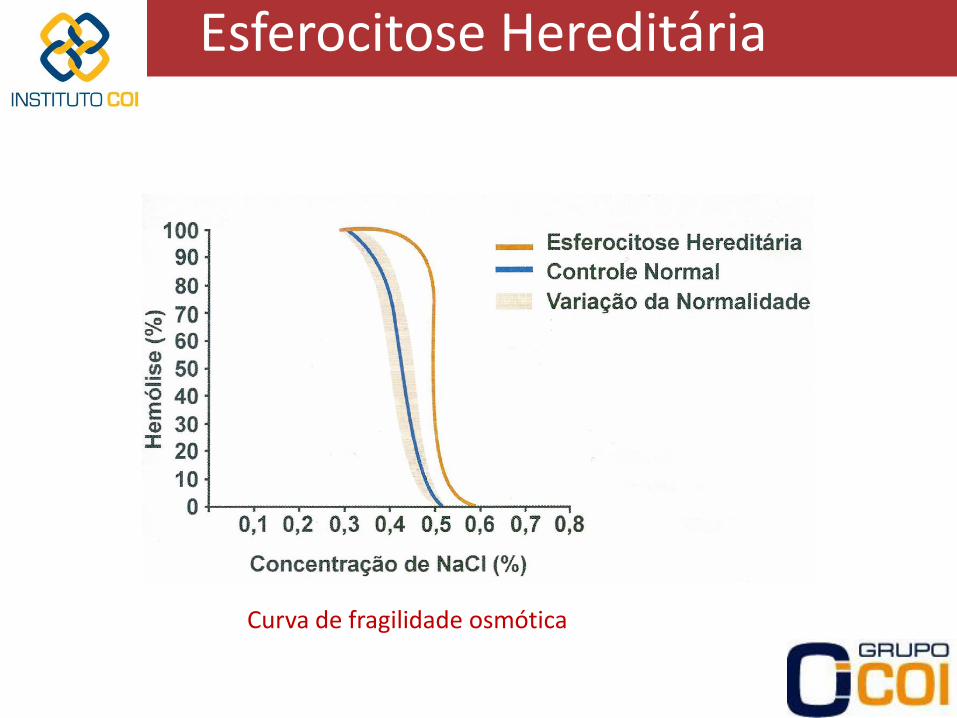
Esferocitose Hereditária
Curva de fragilidade osmótica

Tratamento (doenças de membrana)
• Suplementação de ácido fólico
• Esplenectomia – Cura de quase todas as doenças de membrana eritrocitária
– Formas graves não curáveis, mas com melhora da anemia
– Risco de sepse adiar até 5 anos de idade
– Vacinação antes do procedimento
– Antibioticoprofilaxia após o procedimento
– Indicações:
• Doença de membrana grave
• Doença de membrana moderada que compromete atividade
• Úlceras de perna
• Hematopoiese extramedular
– Falha baço acessório ou associação com outro defeito eritrocitário

Deficiência de G6PD
• Introdução – Gene de G6PD ligado ao cromossomo X
• Mulheres com sintomas se homozigotas
• Homens com sintomas se heterozigotos
– Deficiência em mais de 200 milhões de pessoas, manifestações clínicas apenas em uma parcela
– Maior frequência em negros africanos e povos mediterrâneos
• Vantagem seletiva contra a malária endêmica
• Negros americanos prevalência de 12-15%
• Italianos prevalência de 1,3-2% (14-48% na Sardenha)
– Hemácias não possuem mitocôndria via glicolítica apenas
– G6PD primeira enzima das vias glicolítica e das pentoses
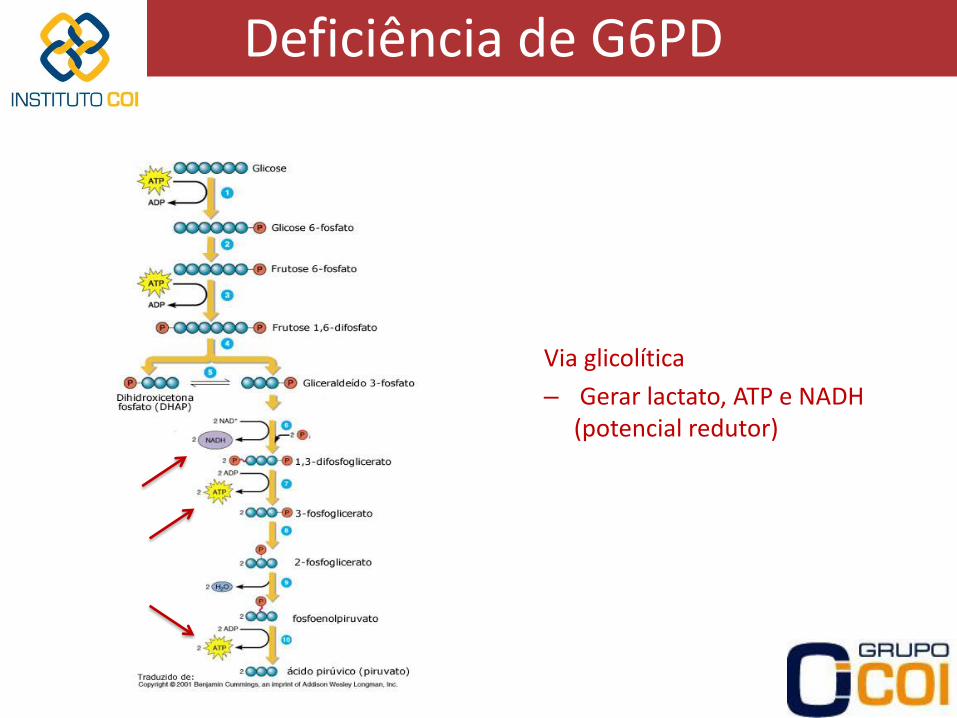
Deficiência de G6PD
Via glicolítica
– Gerar lactato, ATP e NADH (potencial redutor)

Deficiência de G6PD
– Via das pentoses geração de CO2 e NADPH (potencial redutor)
• Ativada em estresse oxidativo
• Se deficiência de G6PD não gera NADH (sem potencial redutor)
•Oxidação de grupamentos tióis na membrana •Formação de corpúsculos de Heinz •Aumento de cálcio intraeritrocitário
Hemólise

Deficiência de G6PD
• Manifestações clínicas – Depende da variante da enzima da qual o paciente é portador
• Forma normal B
• 3 variantes mais comuns
– A-
– A+
– Mediterrânea (ou B-)

Deficiência de G6PD
• Variante A-
– Pessoas de origem africana
– Atividade enzimática residual muito diminuída (3-7%), mas suficiente em condições normais assintomáticos
– Hemólise aguda se expostas a determinadas substâncias
• Variante A+
– Presentes em muitos negros
– Atividade enzimática residual de 80% sem manifestações clínicas
• Variante mediterrânea ou B-
– Atividade enzimática residual muito pequena (Menor que na A-)
– Hemólise mais grave se exposição a determinadas substâncias

Deficiência de G6PD
• Crise hemolítica aguda
– Após 2-3 dias de exposição hemoglobinúria, dor abdominal ou lombar, icterícia e sintomas de anemia
– Queda rápida de Hb e aumento rápido de reticulócitos
– Corpúsculos de Heinz nas hemácias nos primeiros dias
Corpúsculos de Heinz desnaturação oxidativa da Hb

Deficiência de G6PD
– Após 5-6 dias há parada da hemólise e melhora clínica, mesmo se houver continuação da ingesta Reticulócitos apresentam maior quantidade de enzima
Reticulocitose aumento do conteúdo enzimático redução da hemólise
– Crises também podem ser deflagradas por infecções
– Tratamento:
• Hidratação venosa e diuréticos (prevenção de insuficiência renal)
• Transfusão cautelosa de hemácias
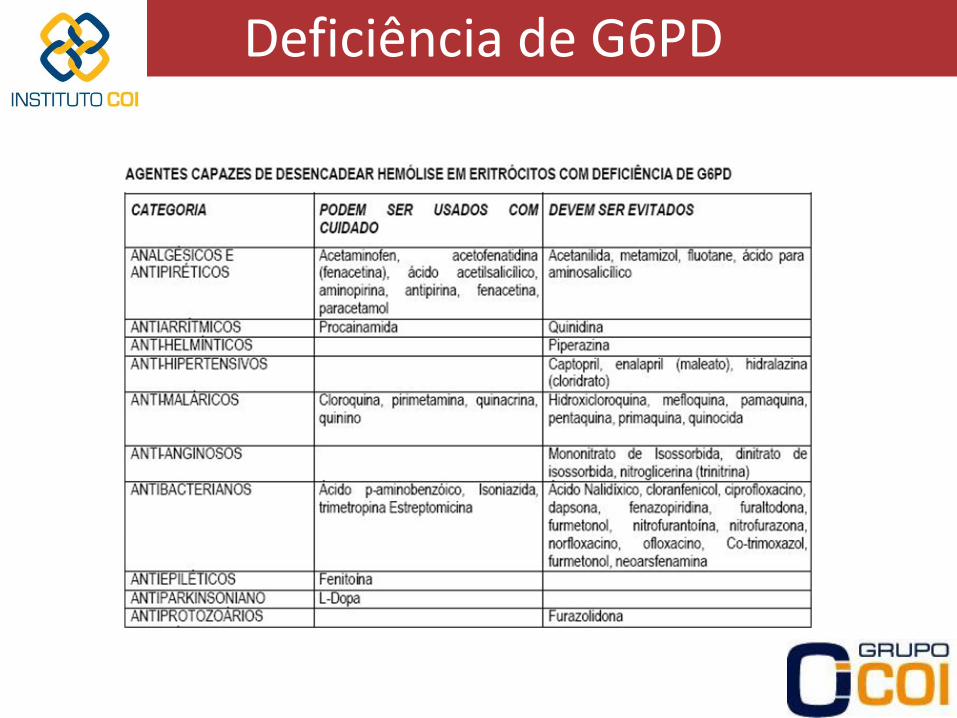
Deficiência de G6PD
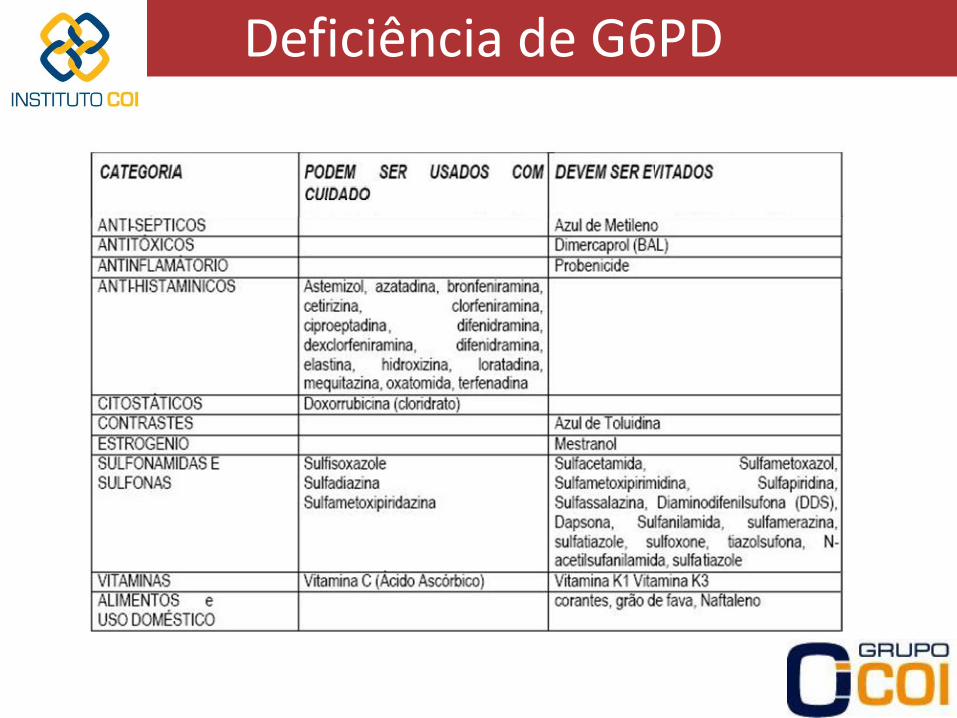
Deficiência de G6PD

Anemias hemolíticas extrínsecas não imunes
• Microangiopatia – Hemólise extravascular geralmente; intravascular se dano grave à
hemácia
– Redução leve ou moderada de haptoglobina
– Outras alterações de acordo com a causa (ex: trombocitopenia na PTT; consumo de fatores de coagulação na CIVD)
– Fragmentação eritrocitária no sangue periférico
Esquizócitos

Anemias hemolíticas extrínsecas não imunes
– Dano microvascular
• PTT/SHU
• Associados com gestação preeclampsia, eclampsia, Síndrome HELLP
• Associados a neoplasias CIVD, uso de mitomicina
• Vasculites poliarterites, glomerulonefrite aguda, granulomatose de Wegner, infeccções Rickettsia-like, esclerodermia
• Alterações da vasculatura renal hipertensão maligna, glomerulonefrite aguda, rejeição de enxerto, uso de ciclosporina
• CIVD
– Mal-formações atrioventriculares
• Síndrome de Kasabach-Merritt
• Hemangioendoteliomas
• Shunts atrioventriculares para condições congênitas ou adquiridas (stents, TIPS, shunts)

Anemias hemolíticas extrínsecas não imunes
– Alterações cardíacas
• Troca valvar
• Jatos de regurgitação ou estenose aórtica
– Drogas
• Ciclosporina
• Mitomicina
• Ticlopidina
• Tacrolimus
• Cocaína

Anemias hemolíticas extrínsecas não imunes
• Circulação extracorpórea
– Síndrome de reperfusão quadro inflamatório febril com ativação de complemento ao passar pelo oxigenador hemólise intravascular aguda e leucopenia
– Tratamento de suporte até resolução espontânea
• Doença renal – Lesão de pequenas arteríolas renais hemólise microangiopática
– Suscetibilidade a lesão oxidativa das hemácias hemólise
• Hemólise na doença hepática – Por hiperesplenismo secundário a hipertensão portal
– Em doença hepática grave spur cells remoção esplênica com esplenomegalia

Anemias hemolíticas extrínsecas não imunes
• Infecção – Infecções parasitárias
• Malária, bartonelose e babesiose
– Alteração da superfície eritrocitária por produtos bacterianos
• Polissacarídeo capsular de Haemophyllus influenzae (PRP) se liga à superfície da hemácia Ac contra PRP hemólise imune intra e extravascular
– Hemólise mediada diretamente por produtos bacterianos
• Liberação de enzimas por Clostridium que degradam os fosfolipídeos e PTNs de membrana hemólise intravascular súbita
– Colecistite aguda, cirurgia da árvore biliar e infecções obstétricas
– Avaliar CIVD
– Início imediato de antibióticos
Plasmodium

Anemias hemolíticas extrínsecas não imunes
• Hiperesplenismo – Sequestro de hemácias, com estase e maior contato com macrófagos maior destruição eritocitária
– Tamanho do baço determina o grau de sequestro eritrocitário
• Hemólise intravascular por picada de aranha marrom ou víbora
Aranha marrom Víbora

Hemoglobinúria paroxística noturna
• Características – Hemólise intravascular
– Tendência a trombose
– Falência da medula óssea
• Alteração no glicofosfatidilinositol – Proteína transmembrana
– Âncora para inúmeras proteínas da membrana
– Redução da expressão de CD55 e CD59 susceptibilidade à lise pelo sistema complemento

Hemoglobinúria paroxística noturna
• Manifestações clínicas – Anemia hemolítica paroxística
• Hemólise intravascular
• Sintomas de anemia
– Insuficiência renal
• Hemólise muito intensa hemoglobinúria maciça e dano renal
• Hemólise lenta e gradual depósito de ferro renal fibrose intersticial e dano tubular proximal
– Manifestações decorrentes de consumo de óxido nítrico por Hb livre
• Espasmos esofageanos
• Cólicas intestinais
• Disfunção erétil
– Astenia não relacionada ao grau de anemia

Hemoglobinúria paroxística noturna
– Tromboses venosas (arteriais menos comumente)
– Manifestações de pancitopenia (às vezes manifestação como aplasia de medula óssea)
• Diagnóstico – Quadro clínico suspeito
– Imunofenotipagem eritrocitária
• Redução de expressão de CD55 e CD 59

Hemoglobinúria paroxística noturna
• Tratamento – Hemotransfusão se necessário
– Reposição de ferro se não estiver recebendo tx
• Perda crônica de ferro por hemossiderinúria
– Reposição de ácido fólico
– Drogas para o tratamento da HPN
• Prednisona
• Danazol
• Eculizumab

Púrpura trombocitopênica trombótica
Fisiopatologia
Redução da atividade da enzima
ADAMTS13
Redução da clivagem do fator de von
Willebrand de alto peso molecular
Lesão endotelial
Consequências
• Anemia microangiopática
• Trombocitopenia
• Oclusão microvascular


















