MAYRA LEAL CHRISÓSTOMO BODART - UFRJ
126
UNIVERSIDADE FEDERAL DO RIO DE JANEIRO FACULDADE DE FARMÁCIA Programa de Pós-Graduação Stricto Sensu Mestrado Profissional em Ciência e Tecnologia Farmacêutica MAYRA LEAL CHRISÓSTOMO BODART Desenvolvimento e estudo de estabilidade de solução oral magistral de L-Carnitina para tratamento de doenças metabólicas RIO DE JANEIRO 2019
Transcript of MAYRA LEAL CHRISÓSTOMO BODART - UFRJ

UNIVERSIDADE FEDERAL DO RIO DE JANEIRO
FACULDADE DE FARMÁCIA
Programa de Pós-Graduação Stricto Sensu
Mestrado Profissional em Ciência e Tecnologia Farmacêutica
MAYRA LEAL CHRISÓSTOMO BODART Desenvolvimento e estudo de estabilidade de solução oral magistral de
L-Carnitina para tratamento de doenças metabólicas
RIO DE JANEIRO 2019

MAYRA LEAL CHRISÓSTOMO BODART
Desenvolvimento e estudo de estabilidade de solução oral magistral de L-Carnitina
para tratamento de doenças metabólicas
Dissertação de Mestrado Profissional apresentada ao Programa de Pós-graduação em Ciência e Tecnologia Farmacêutica, Faculdade de Farmácia, Universidade Federal do Rio de Janeiro, como parte dos requisitos necessários à obtenção do título de Mestre em Ciência e tecnologia Farmacêutica.
Orientadora: Profª. Drª. Elisabete Pereira dos Santos Coorientadora: Profª. Drª. Ana Lúcia Vazquez Villa
Rio de Janeiro
2019

CIP - Catalogação na Publicação
Elaborado pelo Sistema de Geração Automática da UFRJ com os dados fornecidospelo(a) autor(a), sob a responsabilidade de Miguel Romeu Amorim Neto - CRB-7/6283.
B666dBodart, Mayra Leal Chrisóstomo Desenvolvimento e estudo de estabilidade desolução oral magistral de L-Carnitina paratratamento de doenças metabólicas / Mayra LealChrisóstomo Bodart. -- Rio de Janeiro, 2019. 125 f.
Orientadora: Elisabete Pereira dos Santos. Coorientadora: Ana Lúcia Vazquez Villa. Dissertação (mestrado) - Universidade Federal doRio de Janeiro, Faculdade de Farmácia, Programa dePós-Graduação em Ciências Farmacêuticas, 2019.
1. Carnitina, . 2. Erro inato de metabolismo. 3.Formulação líquida. 4. Estabilidade. 5. FarmáciaMagistral. I. Santos, Elisabete Pereira dos,orient. II. Villa, Ana Lúcia Vazquez, coorient.III. Título.


Dedicatória
Dedico esse trabalho a todos os pacientes da Farmácia Universitária

AGRADECIMENTOS
Agradeço principalmente a Deus por me dar forças e por me impulsionar nos momentos mais difíceis. Eu senti Sua presença em vários momentos desse projeto ao colocar anjos no meu caminho a cada vez que uma dificuldade aparecia!
Ao meu marido que entendeu toda a minha ausência, que me mandava dormir quando eu virava noites estudando ou escrevendo, ou por me dar apoio e chocolate nas inúmeras vezes que eu chegava desesperada com as diversas dificuldades que encontrei no caminho. Sem você, eu não teria conseguido!
Aos meus pais que sempre acreditaram em mim, que me ensinaram a ser o que sou hoje e que comemoraram cada pequena vitória encarada nesse projeto, sem nem ao menos entender o que significava! Se eu consegui essa conquista hoje, é porque vocês me apoiaram desde o início! Obrigada por tudo! Essa conquista é nossa!
À minha irmã, afilhada, minha “filhinha” e amiga, que tanto me deu apoio durante esse período, que mandava mensagens carinhosas e fotos malucas tentando me animar quando eu mais precisava de um carinho!
À professora Elisabete Pereira dos Santos pela orientação e confiança para realização desse trabalho.
À professora Ana Lúcia Vazquez Villa por acreditar mais em mim do que eu, pela paciência, contribuições, incentivos, broncas e amizade.
Às professoras Mariana Sato de Souza de Bustamante Monteiro e Zaida Maria Freitas, pela contribuição e cuidado com a orientação na Banca de Acompanhamento.
À Banca examinadora por aceitar o convite e fornecer preciosas contribuições para o trabalho.
À professora Helena Keiko pela ajuda, contribuições e disponibilidade em realizar os testes microbiológicos da formulação.
A imensa ajuda que recebi de todos do Laboratório de Imunofarmacologia, chefiado pela Bartira Rossi Bergmann, que abriu as portas do laboratório para mim. Agradeço especialmente ao Douglas e Maria Paula, pela amizade, ajuda, por não me deixarem com fome quando eu não almoçava para ganhar tempo; à Ariane por todas as dicas, contribuições, avaliações e orientações; à Ariele, Felipe e Izabelle por toda torcida, ajuda e amizade! Eu me senti parte do laboratório de vocês! Vocês foram fundamentais à realização do projeto!
Aos queridos Aline e Jonatas da Central Analítica.

Ao LabTIF, especialmente à Letícia Coli, Paloma Wettler, Mariani Grillo e Fernanda Locatelli por me ajudarem com algumas análises.
Ao Marco Antonio que disponibilizou o HPLC e tempo para me ajudar.
À todos do LabCQ, especialmente ao Pedro e Luis que tanto me ajudaram.
Às grandes amigas Cláudia, Fernanda e Elaine. Vocês acompanham meu crescimento pessoal e profissional e me ajudaram e ajudam sempre! Eu não teria conseguido sem vocês! Obrigada pelos maravilhosos momentos de diversão, de ajuda e de incentivo!
À Érika, da Seção de Referência da Biblioteca Central do CCS, que tanto me ajudou com a busca de Referencial Bibliográfico.
À Sarah Noslien que desde o começo me ajudou, me incentivou e me ajudou!
À Larissa Rodrigues, minha amiga e aluna de iniciação científica, que me alavancou e fez com que o projeto desse certo. Você foi essencial nos momentos em que mais precisei!
À Ana, Fernanda Cardoso e Valery que me tanto me ajudaram.
Aos meus amigos da “Panela” Ana Elisa, Sarah, Felippe e Walter, que sempre escutaram minhas reclamações, medos e sempre me incentivaram!
Aos amigos feitos no Laboratório de Desenvolvimento Galênico (LADEG): Priscila, Ana Paula, Luciana, Fiammeta, Tatiele, Márcio, Raphaela, Mirella, Deise, Bruna, Leide. Vocês fizeram meus dias mais leves, felizes e animados!
À todos do meu setor que seguraram as pontas quando eu precisava resolver algum problema ou realizar alguma análise! Muito obrigada a todos! Em especial à amiga Tatiana Zanela que tanto me apoiou, me incentivou, me deu forças e segurou o setor quando eu mais precisava! Muito obrigada! Com certeza sem seu apoio e ajuda, eu não teria conseguido!
Às queridas Cleonice Marques e Letícia Dysarz que escutavam meus momentos de desespero, que me apoiaram, me incentivaram e muitas vezes me tranquilizaram!
À todos os amigos da Farmácia Universitária, que me apoiaram e me incentivaram tanto durante esses anos de mestrado.
À todos, os meus mais sinceros agradecimentos! Muito obrigada!

“A persistência é o caminho do êxito” (Charles Chaplin)

RESUMO
BODART, Mayra Leal Chrisóstomo. Desenvolvimento e estudo de estabilidade de solução oral magistral de L-Carnitina para tratamento de doenças metabólicas. Rio de Janeiro, 2019. Dissertação (Mestrado em Ciência e Tecnologia Farmacêutica) – Faculdade de Farmácia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, 2019. Erros inatos do metabolismo (EIM) são distúrbios genéticos que levam a falhas
enzimáticas causando interrupção de vias metabólicas, o que pode comprometer
diversos processos celulares. O rápido diagnóstico e tratamento das patologias
associadas aos EIM evitam danos irreversíveis neurológicos e cognitivos,
fornecendo uma melhor qualidade de vida aos pacientes. A administração de L-
carnitina é amplamente utilizada nesse perfil de tratamento e no Brasil, apenas
suplementos voltados para o público esportivo pode ser encontrada sob a fórmula
líquida, não existindo formulação medicamentosa de L-carnitina voltada para o
público alvo. Na Farmácia Universitária, é usualmente dispensada sob a forma de
sachê, porém apresenta alta higroscopicidade o que dificulta sua pesagem e
manipulação. A necessidade de multidoses diárias a pacientes pediátricos e com
problemas cognitivos evidenciou a necessidade do desenvolvimento de uma solução
magistral de L-carnitina. Todavia, a escolha dos excipientes a serem utilizados para
esse público alvo, que apresenta uma serie de restrições e alterações metabólicas,
se torna um desafio e, além disso, a L-carnitina é um excelente substrato para
microrganismos, apresentando problemas em sua estabilidade. Diante disso, esse
trabalho teve como objetivo desenvolver uma formulação líquida oral de L-Carnitina,
com excipientes que atendam ao público alvo garantindo boa estabilidade físico-
química e microbiológica. Foram desenvolvidas oito formulações com duas
concentrações de L-carnitina (10 e 20%) e diferentes combinações de agentes de
viscosidade, acidulante e conservantes. Duas dentre as oito formulações
apresentaram melhores características físico-químicas, organolépticas e
microbiológicas, sendo conduzidas ao estudo de estabilidade completo. O estudo de
estabilidade realizado foi do tipo “em uso”, onde alíquotas diárias foram retiradas das
embalagens multidoses para mimetizar as condições de uso. As amostras foram
armazenadas em condições controladas de temperatura (5ºC, 25ºC e 40ºC) e em
diferentes materiais de embalagem primárias (vidro âmbar e plástico PET leitoso).
As amostras foram analisadas nos tempos 0, 15, 30 e 45 dias após suas

manipulações. Elas foram analisadas em relação as suas características
organolépticas e físico-químicas como aspecto, odor, pH (Brasil, 2005) e teor do
princípio ativo (EUA, 2018), utilizando a cromatografia líquida de alta eficiência
(CLAE), essa metodologia passou por uma validação parcial e teve a adequabilidade
do sistema verificada. Para análise microbiológica, os testes foram determinados
pela farmacopeia brasileira para soluções orais não estéreis (contagem total de
bactérias aeróbias, contagem de leveduras e fungos e presença de Escherichia coli)
(Brasil, 2005). Para a observação de produtos de degradação, as formulações foram
expostas a condições de estresse como aquecimento, solução ácida, básica e
oxidativa e apenas apresentou degradação quando mantida sob meio fortemente
ácido e básico com aquecimento a 70ºC. A melhor formulação apresentou boa
estabilidade físico-química e microbiológica durante os 45 dias de estudo de
estabilidade quando mantida sob temperatura ambiente. Portanto, podemos concluir
que a estabilidade foi garantida mesmo com a abertura subsequente da embalagem
primária para mimetização das condições de uso pelo paciente, tendo essa
formulação um perfil seguro para o público alvo e será comercializada pela Farmácia
Universitária da UFRJ.
Palavras chaves: Carnitina, estabilidade, formulação líquida, erro inato de
metabolismo.

Resumo em Língua estrangeira BODART, Mayra Leal Christóstomo. DEVELOPMENT AND STABILITY STUDY OF MAGISTRAL ORAL SOLUTION OF L-CARNITIN FOR TREATMENT OF PATHOLOGIES ASSOCIATED TO INBORN ERRORS OF METABOLISM (IEM). Rio de Janeiro, 2019. Dissertation (MsC in Science and Pharmaceutical Technology) – Faculdade de Farmácia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, 2019.
Inborn errors of metabolism (IEM) are genetic disturbs witch can be correlated with
enzymatic defects; these defects are able to cause interruption of several metabolic
paths compromising many cellular processes. A fast diagnosis and treatment of
these pathologies associated to IEM allows to avoid irreversible neurological and
cognitive damages, therefore the patients could have an improvement in their quality
of life. The supplementation with L-carnitine is widely use in this treatment.
Commonly dispensed in sachet, it has a high hygroscopic that makes difficult the
process of weighing and manipulating. The requirement of multiple daily doses of
pediatric patients with cognitive issues manifested the haste to develop a magistral
solution of L-carnitine. However, the excipient selection to use it in this public, which
has an enormous series of restriction and metabolic alterations, became a challenge,
furthermore, L-carnitine is an excellent substrate for microorganisms that shows
stability problems. For that reason, this work aims to develop an oral solution
formulation of L-carnitine with excipients that fulfill the public needs, warranting a
good physics-chemistry and microbiological stability. To this end, eight formulations
were developed, containing two distinct concentration of L-carnitine (10% and 20%),
and different combinations of thickening agent, acidifiers and preservatives. Two of
them presented better organoleptic characteristics and were piloted to the fully
stability study. The stability study method applied was “in use”; in witch, daily
samples were taken from the multidose bottles to mime the use conditions of the
patient. To run the stability study, the samples were kept in dissimilar controlled
temperature levels (5ºC, 25ºC e 40ºC) and different sorts of primary packaging
(amber glass and plastic PET). The analysis were performed along 0, 15, 30 and 45
days after the manipulation. The samples were analyzed by their organoleptic and
physic-chemistry characteristic as aspect, odor, pH (Brazil, 2005) and content of
active (USA, 2018) using high performance liquid chromatography (HPLC) which had
an initial validation process to adequate to the system. The microbiological assay

was made as described in Brazilian Pharmacopoeia 5nd edition for oral solution non-
sterile (aerobic bacteria total count, yeast and fungus count and presence of
Escherichia coli). In order to evaluate the degradation products, the formulations
were exposed to stress conditions as heat, acid, basic and oxidative solutions. The
oral solution that exhibited the better stability physic-chemistry, microbiological and
organoleptic along 45 days was the stocked in shelf at ambient temperature. Thus,
we can conclude that even opening the primary packaging everyday to mime the use
condition of the patient, the oral solution maintained the stability condition, presenting
a safe profile to the patient and will be commercialized by the Farmácia Universitária
of UFRJ.
Key words: carnitine, stability, liquid formulation, Inborn errors of metabolism

LISTA DE FIGURAS
Figura 01. Metabolismo dos aminoácidos de cadeia ramificada e a sinalização das etapas interrompidas pelas acidemiaspropiônica, metilmalônica, isovalérica e glutárica.
34
Figura 02. Via principal do catabolismo da leucina e bloqueio da enzima isovaleril-CoAdesidrogenase.
35
Figura 03. Vias metabólicas alternativas do isovaleril-CoA em caso de interrupção da via principal.
36
Figura 04. Metabólitos secundários produzidos quando ocorre o bloqueio enzimático da PCC.
36
Figura 05. Transporte dos ácidos graxos do meio extramitocondrial para a matriz mitocondrial com auxílio de enzimas.
41
Figura 06. Estrutura da L-carnitina. 42
Figura 07. Estrutura química das isoformas de carnitina: levógira e dextrógira.
43
Figura 08. Estrutura do sal cloridrato de L-carnitina. 43
Figura 09. Ilustração do processo de manipulação durante o processo de mistura.
46
Figura 10. Representação esquemática de um sistema de cromatografia líquida.
58
Figura 11. Metabólitos possiveis da L-carnitina quando consumida por microrganismos
59
Figura 12. Espectro referente à análise de IV-TF do padrão de L-carnitina 79
Figura 13. Espectro referente à análise de IV-TF da matéria-prima de L-carnitina
79
Figura 14. Principais bandas presentes no espectro IV-TF representadas na estrutura da L-Carnitina.
80
Figura 15. Cromatograma do solvente (água ultrapurificada). 82

Figura 16. Cromatograma da Fase Móvel 82
Figura 17. Cromatograma da L-carnitina padrão a 2,0 mg/mL 83
Figura 18 Espectro UV/VIS do pico cromatográfico da L-carnitina padrão a 2,0 mg/mL
83
Figura 19. Cromatograma da solução com padrão de L-carnitina e padrão interno.
83
Figura 20. Cromatograma de solução com matéria-prima de L-carnitina e padrão interno.
84
Figura 21. Cromatograma dos excipientes usados no desenvolvimento da formulação em presença do padrão interno.
84
Figura 22. Cromatograma da L-carnitina em presença do padrão interno e excipientes usados no desenvolvimento da formulação: ácido cítrico e benzoato de sódio.
85
Figura 23. Comparação entre o espectro de varredura do pico cromatográfico correspondente à L-carnitina na solução padrão e na solução com os excipientes.
85
Figura 24. Curva padrão da L-carnitina a partir da média de valores encontradas no primeiro dia de análise
87
Figura 25. Curva padrão da L-carnitina a partir da média de valores encontradas no segundo dia de análise
87
Figura 26. Curva padrão da L-carnitina a partir da média de valores encontradas no terceiro dia de análise
87

LISTA DE QUADROS
Quadro 01. Níveis de variações aceitáveis de DPR para precisão 89
Quadro 02. Resultado das análises de pH no estudo de estabilidade completo das formulações a 20% (p/v)
102
Quadro 03 Resultado das análises de teor no estudo de estabilidade completo das formulações a 20% (p/v)
105
Quadro 04 Resultados do estudo de estabilidade microbiológico da formulação 6 acondicionada em vidro e em plástico
108
Quadro 05 Resultados do estudo de estabilidade microbiológico da formulação 7 acondicionada em vidro e em plástico
109
Quadro 06 Resultados do estudo de estabilidade microbiológico da
formulação controle acondicionada em vidro e em
plástico
110

LISTA DE TABELAS
Tabela 01. Principais características de identificação da Carnitina 42
Tabela 02. Principais características dos materiais comumente usados para embalagem
54
Tabela 03. Principais resistências dos materiais comumente usados para embalagem
54
Tabela 04. Descrição dos excipientes utilizados nas formulações testadas e suas proporções
68
Tabela 05. Densidade da água de 20 a 30°C 69
Tabela 06. Características de incubação dos testes microbiológicos 76
Tabela 07. Esquema das condições de exposição das amostras para avaliação da estabilidade
77
Tabela 08. Principais bandas encontradas em espectro de IV-FT de L-carnitina
80
Tabela 09. Parâmetros aplicados no teste de linearidade 86
Tabela 10. Valores obtidos para LD e LQ. 88
Tabela 11. Resultado encontrado para o teste de precisão. 89
Tabela 12. Resultados obtidos para o teste de exatidão. 90
Tabela 13. Composição das formulações testes a 10% (p/v) 94
Tabela 14. Resultado da análise FIQ inicial das formulações a 10% (p/v). 94
Tabela 15. Resultado das análises microbiológicas no estudo de estabilidade prévio
95
Tabela 16. Resultado das análises físico-químicas no estudo de estabilidade prévio
96
Tabela 17. Composição das formulações testes a 20% (p/v) 97
Tabela 18. Resultado FIQ inicial da estabilidade prévia das formulações a 20% (p/v)
97

Tabela 19. Resultado das análises FIQ das formulações a 20% (p/v) 98
Tabela 20. Resultado das análises MIC das formulações a 20% (p/v) 99
Tabela 21. Resumo do estudo de estabilidade completo em uso 101
Tabela 22. Análise estatística dos valores de pH avaliando cada formulação nas diferentes embalagens
104
Tabela 23. Resultado da análise estatística do teste de teor realizado em comparação com as embalagens para cada formulação
106
Tabela 24. Teor de L-carnitina da Formulação 6 sob soluções degradantes dez vezes menos concentrada
112
Tabela 25. Resumo do estudo de degradação forçada sob diferentes condições.
112
Tabela 26. Tempos de retenção dos picos encontrados no estudo de estabilidade.
113
Tabela 27. Áreas dos picos encontrados no estudo de degradação. 113

LISTA DE ABREVIATURAS E SIGLAS
3-MMM 3-Metilcrotonil glicinúria
ANOVA Análise de Variância
ANVISA Agencia Nacional de Vigilância Sanitária
BPF Boas Práticas de Fabricação
CACT Deficiência de Carnitina/AcilcarnitinaTranslocase
CG Cromatografia Gasosa
CLAE Cromatografia Líquida de Alta Eficiência
CMC Carboximetilcelulose
CoA Coenzima A
CPT-II Deficiência da Palmitoil Carnitina Transferase
CTBA Contagem total de bactérias aeróbicas
CTFL Contagem total de fungos/leveduras
DAD Detector de Arranjo de Diodos
DCP Deficiência de Carnitina Primária
DLE Deficiência de Beta-Cetotiolase
DP Desvio Padrão
DPR Desvio Padrão Relativo
EIM Erro Inato do Metabolismo
FB Farmacopeia Brasileira
FD&C Food, drug and Cosmetics administration
FIQ Físico-químico
FU Farmácia Universitária
GA I AcidemiaGlutária tipo 1
GA II AcidemiaGlutária tipo 2
HGM Acidúria 3-Hidroxi-3-Metilglutárica
HUCFF Hospital Universitário Clementino Fraga Filho

IC Inclinação da curva de calibração
IPPMG Instituto de Puericultura e Pediatria Martagão Gesteira
IVA AcidemiaIsovalérica
IV-TF Espectroscopia de Infravermelho com Transformada de Fourier
Lacmac Laboratório de Controle de Qualidade de Medicamentos, Alimentos e Cosméticos
LCHADD Deficiência de 3-hidroxiAcil-Coenzima A Desidrogenase de Cadeia Longa
LD Limite de Detecção
LQ Limite de Quantificação
MCAD Deficiência da Desidrogenase das Acil-Coa dos Ácidos Graxos de Cadeia Média
MCC Deficiência isolada de 3-metilcrotonil-CoA carboxilase
MCM Deficiência da enzima metilmalonil-CoAmutase
MIC Microbiológico
Milli-Q Água Ultrapurificada
MMA AcidemiaMetilmalônica
PA AcidemiaPropiônica
PAH Fenilcetonúria
PCC Deficiência da enzima Propionil-CoACarboxilase
PEAD Polietileno de alta densidade
PEBD Polietileno de baixa densidade
PET Polietilenotereftalato
pH Potencial hidrogeniônico
PP Polipropileno
PS Poliestireno
PVC Policloreto de vinila
qs Quantidade suficiente

qsp Quantidade suficiente para
RDC Resolução da Diretoria Colegiada
RE Resolução
TR Tempo de retenção
SCOT Deficiência de SCOT
TI Informe técnico
T2 Anomalia da Acetoacetil-CoATiolase mitocondrial
UFC Unidade Formadora de Colônia
UFC/mL Unidade Formadora de Colônia por mL
UFRJ Universidade Federal do Rio de Janeiro
USP Farmacopeia dos Estados Unidos
UV Ultravioleta
VIS Visível
VLCAD Deficiência da Acil-Coenzima A Desidrogenase de Cadeira Muito Longa

LISTA DE FÓRMULAS E SÍMBOLOS
% Porcentagem
® Registrado
°C Grau Celsius
°C Graus Celsius
µm Micrograma
a Coeficiente angular
b Coeficiente linear
bar Unidade de pressão
cm-¹ Número de onda
d2020 Densidade relativa
g grama
h hora
H2O Água
H3O+ Íon hidrônio
HCl Ácido clorídrico
HO- Íon hidroxila
KBR Brometo de potássio
L Litro
log Logarítmo
M Molar
mg Miligrama
min Minuto
mL Mililitro
mL/min Mililitro por minuto
mm Milimetro

NaOH Hidróxido de sódio
p/p Peso por peso
p/v Peso por volume
r Coeficiente de correlação
R² Coeficiente de determinação
λ Comprimento de onda
ρt Densidade de massa

SUMÁRIO
1. INTRODUÇÃO ...................................................................................................... 26
2. REVISÃO BIBLIOGRÁFICA ................................................................................ 29
2.1 Erros Inatos do Metabolismo (EIM) ............................................................... 29
2.1.1 Manifestações Clínicas .............................................................................. 29
2.1.2 Diagnóstico ................................................................................................ 30
2.1.3 Classificação dos EIM ................................................................................ 31
2.1.3.1 AMINOACIDOPATIAS ........................................................................ 33
2.1.3.2 ACIDEMIAS ORGÂNICAS .................................................................. 33
2.1.3.3 OUTRAS DOENÇAS DE EIM TRATADAS COM L-CARNITINA ....... 38
2.2 L-Carnitina ....................................................................................................... 40
2.2.1 Bioquímica ................................................................................................. 40
2.2.2 Características Gerais ............................................................................... 41
2.2.3 Efeitos adversos e precauções .................................................................. 43
2.2.4 Farmacocinética......................................................................................... 44
2.3 Farmácia Universitária da UFRJ ........................... Erro! Indicador não definido.
2.4 Desenvolvimento De Formulações Líquidas ................................................ 48
2.4.1 Forma Farmacêutica Líquida ..................................................................... 48
2.4.2 Excipientes Farmacêuticos Usados Para Soluções Líquidas Orais ........... 49
2.4.2.1 SOLVENTE: ÁGUA PURIFICADA ..................................................... 49
2.4.2.2 CONSERVANTES ............................................................................... 50
2.4.2.3 AGENTE PROMOTOR DE VISCOSIDADE ........................................ 51
2.4.2.4 ACIDULANTE ..................................................................................... 52
2.4.2.5 EDULCORANTES ............................................................................... 52
2.4.3 Embalagens Usadas Para Soluções Líquidas Orais.................................. 52
2.5 Controle De Qualidade De Soluções Orais Magistrais ................................ 55

2.5.1 Densidade de Massa ................................................................................. 55
2.5.2 Determinação potenciométrica do pH ........................................................ 56
2.5.3 Doseamento do princípio ativo .................................................................. 56
2.5.4 Controle microbiológico ............................................................................. 59
2.6 Estudo De Estabilidade .................................................................................. 60
3 JUSTIFICATIVA ..................................................................................................... 63
4 OBJETIVOS ........................................................................................................... 64
4.1 Objetivo Geral ................................................................................................. 64
4.2 Objetivos Específicos ..................................................................................... 64
5 MATERIAIS E MÉTODOS .................................................................................... 65
5.1 Materiais ........................................................................................................... 65
5.1.1 Matérias-primas e Reagentes .................................................................... 65
5.1.2 Equipamentos e acessórios ....................................................................... 66
5.2 Métodos ........................................................................................................... 66
5.2.1 Análise de identificação da L-Carnitina ...................................................... 66
5.2.2 Desenvolvimento das formulações ............................................................ 67
5.2.3 Metodologia De Análise Físico-Química .................................................... 68
5.2.3.1 Determinação Do Aspecto ................................................................ 68
5.2.3.2 Determinação do odor ....................................................................... 68
5.2.3.3 Determinação do pH .......................................................................... 68
5.2.3.4 Determinação da densidade de massa ............................................ 69
5.2.3.5 Determinação do teor do princípio ativo ......................................... 70
5.2.3.5.1 PREPARO DE CURVA PADRÃO ................................................. 70
5.2.3.5.2 PREPARO DE SOLUÇÃO DE PADRÃO INTERNO ..................... 70
5.2.3.5.3 PREPARO DE SOLUÇÃO PADRÃO ............................................ 70
5.2.3.5.4 PREPARO DE SOLUÇÃO AMOSTRA ESTOQUE ....................... 70

5.2.3.5.5 PREPARO DE SOLUÇÃO AMOSTRA .......................................... 71
5.2.3.5.6 INFORMAÇÕES DE SISTEMA CROMATOGRÁFICO ................. 71
5.2.3.5.7 ADEQUAÇÃO DO SISTEMA ........................................................ 71
5.2.4 Validação Parcial da Metodologia de Análise de Doseamento .................. 72
5.2.4.1 Precisão .............................................................................................. 72
5.2.4.2 Exatidão .............................................................................................. 73
5.2.4.3 Seletividade ........................................................................................ 73
5.2.4.4 Linearidade ........................................................................................ 74
5.2.4.5 Faixa de trabalho ............................................................................... 75
5.2.5 Metodologia de Análise Microbiológica ...................................................... 75
5.2.5.1 Análise microbiológica ..................................................................... 75
5.2.6 Estudo de Estabilidade .............................................................................. 76
5.2.6.1 Estudo Prévio de Estabilidade ......................................................... 76
5.2.6.2 Estudo de Estabilidade Completo Em Uso ..................................... 76
5.2.7 Estudo de Degradação Forçada ................................................................ 77
5.2.8 Análise de dados ....................................................................................... 78
6 RESULTADOS E DISCUSSÃO ............................................................................. 79
6.1 Análise de identificação da L-Carnitina ........................................................ 81
6.2 Validação Parcial da Metodologia de Doseamento ...................................... 81
6.2.1 Determinação da faixa de trabalho ............................................................ 81
6.2.2 Seletividade ............................................................................................... 81
6.2.3 Linearidade ................................................................................................ 86
6.2.4 Precisão ..................................................................................................... 88
6.2.5 Exatidão ..................................................................................................... 90
6.3 Desenvolvimento da metodologia de análise ............................................... 91
6.4 Desenvolvimento das formulações ............................................................... 91

6.5 Estabilidade prévia das formulações iniciais ............................................... 95
6.6 Estudo de estabilidade completo em uso ................................................... 100
7 CONCLUSÃO ...................................................................................................... 116
8 REFERÊNCIAS BIBLIOGRÁFICAS .................................................................... 117

26
1. INTRODUÇÃO
Os erros inatos do metabolismo (EIM) são distúrbios de natureza genética
que representam uma importante causa de morbidade e mortalidade entre a
população pediátrica (SHAWKY; ABD-ELKHALEK; ELAKHDAR, 2015). Resultam em
anormalidades na síntese de proteínas, normalmente enzimas, alterando suas
principais funções. Tal alteração pode levar a bloqueio de uma determinada via
metabólica com consequente acúmulo de substratos e derivados, bem como a
diminuição da síntese do produto esperado. Dependendo da rota afetada, esse
bloqueio repercute de maneira clínica variável, sendo geralmente grave e muitas
vezes letal (SCRIVER et al., 2002).
Embora sejam individualmente raros, eles possuem uma incidência, em
conjunto, estimada em 1/1.000 nascidos vivos (ARAÚJO, 2004). Em muitos casos,
as crianças parecem perfeitamente normais ao nascimento e podem demorar a
apresentar manifestações clínicas. O rápido diagnóstico é essencial para impedir o
agravamento e a irreversibilidade dos sintomas, podendo representar a
sobrevivência do paciente em alguns casos (JARDIM; ASHTON-PROLLA, 1996).
Mas, a variabilidade de sintomas clínicos, o enorme número de doenças de grande
complexidade e o fato de serem considerados extremamente raros pela maioria dos
profissionais, pode dificultar o correto diagnóstico clínico (EL HUSNY; CALDATO,
2006).
Exemplos de patologias associadas à EIM e que possuem alta incidência, as
aminoacidopatias e acidemias orgânicas são ocasionadas por deficiência severa da
atividade de enzimas específicas de determinadas vias metabólicas, podendo levar
ao acúmulo de substratos que, em concentrações mais elevadas que o normal, se
tornam tóxicas. Elas apresentam grande impacto no sistema gastrointestinal e
nervoso, podendo apresentar manifestações como: vômito, recusa alimentar,
apneia, convulsão, coma, lesão cerebral e retardo mental e cognitivo (BEHRMAN, K,
2014; DEODATO et al., 2006a; GARCÍA et al., 2002a; NADAI; PINHEIRO, 2006).
Para a maioria das aminoacidopatias e acidemias, o tratamento é feito a partir
de restrição alimentar, administração de cofatores das enzimas deficientes e,
principalmente, com a suplementação de L-carnitina. Esta substância é capaz de se
conjugar e aumentar a excreção dos ácidos orgânicos acumulados nessas doenças,

27
evitando as indesejáveis manifestações clínicas ou retardando o progresso da
doença (BERRY, 1998).Sendo o público alvo composto em sua grande maioria por
pacientes pediátricos e, por muitas vezes, com dificuldades de deglutição, a forma
farmacêutica mais indicada é a líquida. As soluções de uso oral podem ser
administradas a pacientes de diferentes idades e com diferentes necessidades,
conferindo maior flexibilidade na administração da dosagem correta (NUNN;
WILLIAMS, 2005).
O desenvolvimento de formulações orais líquidas possui vários fatores críticos
e requer cuidados para garantia da segurança e qualidade do produto. Essas
preparações apresentam, em sua maioria, alto teor de água o que pode propiciar a
degradação físico-química e contaminação microbiológica(BILLANY, 2005;
LACHMAN; LIEBERMAN; KANING, 2001). Além disso, a L-carnitina apresenta
estrutura semelhante a de aminoácidos e pode ser utilizada por bactérias como fonte
de carbono, nitrogênio e de energia, favorecendo também o crescimento microbiano
(MEADOWS & WARGO, 2015).Para isso, substâncias inertes denominadas
excipientes podem auxiliar no desenvolvimento de soluções mais estáveis, eficazes
e atraentes. Mas diferentes estudos apontam algumas dessas substâncias
consideradas inertes como sendo responsáveis por reações adversas relacionadas
a medicamentos. Além disso, pacientes com EIM apresentam uma série de
restrições e, desta forma, a escolha dos excipientes da formulação não pode ser
realizada ao acaso e deve ser feita de maneira criteriosa sendo o mais simples
possível, no intuito de diminuir a probabilidade de reações não esperadas, mas
garantindo a qualidade e estabilidade da formulação (ANSEL; POPOVICH; ALLEN
JUNIOR, 2000; GUERRA, 2012).
Para identificar o período de utilização e condições de estocagem, mantendo
as mesmas propriedades que possuía no momento de sua fabricação, são
realizados os testes de estabilidade onde são retiradas amostras em tempos
definidos e encaminhadas para análises. A realização do estudo de estabilidade é
projetada para verificar as características físicas, químicas e microbiológicas de um
produto farmacêutico durante seu prazo de validade. Além de identificar a
degradação química ou mudança física de um produto farmacêutico quando
expostos a condições forçadas de armazenamento. Esse resultado é utilizado para

28
estabelecer ou confirmar o prazo de validade e recomendar as condições de
armazenamento (BRASIL, 2005).
A Farmácia Universitária (FU) da Universidade Federal do Rio de Janeiro
(UFRJ) recebe diariamente muitos pacientes provenientes do Instituto de
Puericultura e Pediatria Martagão Gesteira (IPPMG) e de outros centros de saúde
que apresentam alguma patologia associada à EIM. Eles, em sua grande maioria,
fazem uso de suplementação de L-carnitina que, atualmente, não possui formulação
oral líquida medicamentosa disponível no mercado brasileiro. Iniciando o
atendimento desses pacientes, a L-carnitina era dispensada na FU sob a forma de
sachê contendo a dose individualizada. Mas a forte característica higroscópica do
ativo requer uma manipulação, pesagem e selagem rápidas, além da necessidade
de dois funcionários envolvidos para uma boa qualidade do produto final. Esse
processo é impraticável com a demanda recebida pela FU e, desta maneira, o
desenvolvimento de uma formulação líquida de L-carnitina que atenda as
necessidades dos pacientes, foi importante para atendimento de todos os pacientes
além de aumentar a adesão ao tratamento. A formulação líquida oral de L-carnitina
foi desenvolvida nas apresentações de 10% e 20%, sendo avaliadas quanto às suas
estabilidades físico-química e microbiológica e apresentando resultados favoráveis
durante 45 dias mantidos a temperatura ambiente.

29
2. REVISÃO BIBLIOGRÁFICA
2.1 Erros Inatos do Metabolismo (EIM)
Os erros inatos do metabolismo (EIM) são um grupo altamente heterogêneo
de distúrbios de natureza genética e representam uma importante causa de
morbidade e mortalidade entre a população pediátrica (SHAWKY; ABD-ELKHALEK;
ELAKHDAR, 2015). Geralmente, correspondem a defeitos enzimáticos capazes de
ocasionar a interrupção de uma via metabólica, podendo causar falhas de síntese,
degradação, armazenamento ou transporte de moléculas no organismo. Como
consequência, tem-se a ausência de um produto esperado, o acúmulo de substrato
da etapa anterior à interrompida ou o surgimento de uma rota metabólica alternativa,
o que podem levar ao comprometimento de diversos processos celulares (EL
HUSNY & FERNANDES-CALDATO, 2006).
O estudo dos EIM originou-se no início dos anos 1900, quando Archibald E.
Garrod descobriu e identificou as primeiras doenças metabólicas: alcaptonúria,
pentosúria, porfiria e albinismo(SHLOMI, CABILI & RUPPIN, 2009). Desde as
primeiras descobertas de Garrod, no início do século XX, diversos novos distúrbios
foram descritos, na medida em que novas ferramentas de diagnóstico bioquímico e
molecular tornaram-se disponíveis(SHAWKY, ABD-ELKHALEK & ELAKHDAR,
2015). Atualmente, existem cerca de 500 distúrbios conhecidos e, embora sejam
individualmente raros, eles possuem uma incidência, em conjunto, estimada em
1/1.000 nascidos vivos. Desta forma, cumulativamente, causam importante agravo à
saúde (ARAÚJO, 2004).
2.1.1 Manifestações Clínicas
Os sintomas e sinais dos EIM podem abranger diferentes sistemas e podem
se iniciar em diferentes faixas etárias, apesar de o distúrbio ter origem genética.
Neonatos com EIM são, geralmente, saudáveis ao nascimento, com os sintomas
podendo se iniciar desde as primeiras horas até as primeiras semanas de vida. Em
alguns casos, podem ser adiados por meses até que algum evento, de natureza
exógena, altere o equilíbrio bioquímico mantido pela criança até então (ARAÚJO,

30
2004; EL-HATTAB, 2015; JARDIM & ASHTON-PROLLA, 1996). Embora o quadro
clínico possa variar, os sintomas iniciais geralmente são inespecíficos, incluindo
letargia, redução da alimentação, vômitos, taquipneia e convulsões. Na medida em
que a doença metabólica progride, pode haver estupor progressivo ou coma,
associados com alterações de tônus (hipotonia, hipertonia), de postura (opistótono)
e dos movimentos (interposição lingual, estalar dos lábios, mioclonia), bem como
presença de ascite (acúmulo de fluido na cavidade peritoneal), síndromes
dismórficas (malformações da face) e apneia do sono (EL-HATTAB, 2015; NASSER
et al., 2012; RAGHUVEER, GARG & GRAF, 2006).
De uma maneira geral, os pacientes portadores de EIM apresentam
acometimento de diversos sistemas, sendo as manifestações neurológicas,
hepáticas e cardíacas as principais envolvidas. Os sintomas neurológicos incluem
quadros de retardo mental, disfunções no desenvolvimento psicomotor, regressão
psicomotora e convulsões. Sintomas psiquiátricos costumam estar presentes em
quase todos os EIM que acometem o sistema nervoso. Já as manifestações
hepáticas incluem hepatomegalia e hipoglicemia, icterícia colestática ou insuficiência
hepática com icterícia, transaminases elevadas, entre outros. O quadro cardíaco
envolve doenças como cardiomiopatia, falência cardíaca e arritmias(ARAÚJO, 2004;
EL-HATTAB, 2015).
2.1.2 Diagnóstico
A quantidade, complexidade e variedade de manifestações clínicas
relacionadas aos EIM dificultam o correto diagnóstico clínico dessas doenças, o que
pode trazer danos irreparáveis ao organismo da criança. Por outro lado, a detecção
e a intervenção precoces levam a medidas terapêuticas satisfatórias, seguidas de
evoluções clínicas favoráveis (BURTON, 1998; JARDIM & ASHTON-PROLLA,
1996).
Segundo RAO et al., 2009, quando existe a suspeita de um EIM, cinco
importantes aspectos devem ser considerados e seguidos:
1- Histórico familiar: observar a existência de consanguinidade entre os pais,
casos de familiares falecidos precocemente ou abortos sem causa definida,

31
ou a ocorrência de um irmão ou irmã previamente afetado por um quadro
clínico semelhante.
2- Exames físicos: avaliação de aspectos como dermatite, alopecia, dismorfia
facial, catarata, entre outros.
3- Testes iniciais de triagem: realização de exames laboratoriais, como
hemograma completo, dosagem de eletrólitos, glicose, amônia, lactato,
razão lactato/piruvato, substâncias redutoras, ácidos orgânicos,
aminoácidos e cetonas.
4- Testes avançados de triagem: realização de ensaios com base no contexto
clínico, que podem incluir ácidos graxos de cadeia longa, quantificação de
aminoácidos, ácidos orgânicos, carboidratos e outros metabólitos.
5- Testes definitivos de diagnóstico: para confirmar o distúrbio detectado,
ensaio enzimático específico em leucócitos, plasma ou células vermelhas,
imunoensaios e análises baseadas em mutações gênicas serão
realizados(RAO et al., 2009).
Cabe ao pediatra, portanto, se familiarizar com os sinais e sintomas
característicos dos EIM e com os exames laboratoriais necessários para se chegar a
um diagnóstico preciso. A clínica que alia a história da doença e um bom exame
físico é capaz de conduzir para a realização de exames confirmatórios mais
adequados, para a terapêutica precoce, bem como para o importante
aconselhamento genético (BURTON, 1998; EL HUSNY & FERNANDES-CALDATO,
2006).
2.1.3 Classificação dos EIM
Segundo Saudubray & Charpentier (1995), os EIM podem ser divididos em
duas categorias, conforme suas características fisiopatológicas e fenótipo clínico:
Categoria 1, que corresponde às alterações que afetam um único órgão ou sistema
orgânico, e que são normalmente mais facilmente diagnosticadas; e Categoria 2,
que engloba doenças onde a alteração bioquímica compromete vias metabólicas
comuns a diversos órgãos e, por isso, os relatos clínicos são muito diversificados,
tornando difícil o diagnóstico pelo profissional da saúde. A fim de facilitar o estudo

32
dessas doenças, a Categoria 2 foi dividida em três principais grupos: Grupo I, Grupo
II e Grupo III.
As doenças do Grupo I afetam a síntese ou o catabolismo de moléculas
complexas. Os sintomas são permanentes, progressivos com o passar do tempo,
podem apresentar-se em todas as idades, não estão relacionados com a ingesta
alimentar e também não possuem relação com eventos intercorrentes, como
infecções. Entre os distúrbios enquadrados neste grupo estão as doenças com
alterações de depósitos lisossômicos, doenças dos peroxissomos, alteração no
metabolismo de lipídios, alteração na síntese de purinas e pirimidinas e alteração no
transporte de metais. As principais manifestações clínicas desse grupo de doenças
são: hidropsia fetal, hepato e/ou esplenomegalia, alterações esqueléticas,
convulsões, hipotonia (não sustentar cabeça e membros), fácies grotesca,
neurodegeneração subaguda, mieloneuropatia subaguda, deficiência auditiva,
achados dismórficos, alterações oculares e da pele, limitações e deficiências
articulares (EL HUSNY & FERNANDES-CALDATO, 2006; SAUDUBRAY et al.,
2002).
As doenças enquadradas no Grupo III são doenças resultantes de deficiência
na produção ou utilização de energia por erros inatos do metabolismo intermediário
no fígado, miocárdio, músculo ou cérebro. Algumas manifestações clínicas
comumente encontradas são: hipoglicemia, miopatia, insuficiência cardíaca, retardo
de crescimento e até morte súbita (EL HUSNY & FERNANDES-CALDATO, 2006;
EL-HATTAB, 2015).
Já as doenças enquadradas no Grupo II são erros inatos do metabolismo
intermediário que culminam em intoxicação e a maioria das doenças são tratadas
com suplementação de L-carnitina. As intoxicações podem ser decorrentes do
acúmulo de substrato tóxico por conta do bloqueio de rotas do metabolismo
intermediário e podem se tornar agudas ou crônicas. Apresentam relação evidente
com o aporte alimentar e com as intercorrências (infecções), podendo permanecer
sem manifestação clínica por grandes intervalos de tempo. Substâncias exógenas
podem desestabilizar o equilíbrio e ocasionar uma descompensação metabólica,
levando a uma intoxicação aguda e recorrente ou crônica e progressiva. Grande
parte de patologias desse grupo apresentam como tratamento suplementação de L-
carnitina associada com cofatores enzimáticos. Os principais distúrbios

33
caracterizados neste grupo são as aminoacidopatias, acidemias orgânicas, defeitos
do ciclo da ureia e intolerância aos açúcares (SAUDUBRAY et al., 2002).
2.1.3.1 AMINOACIDOPATIAS
As aminoacidopatias são causadas por mutações de genes que codificam
proteínas específicas. Tais mutações podem alterar a estrutura primária de uma
proteína ou a quantidade de proteína sintetizada. Essa alteração pode comprometer
a capacidade funcional da proteína, podendo levar ao bloqueio de determinadas vias
metabólicas e ao acúmulo de substratos que, em concentrações mais elevadas que
o normal, podem ser tóxicas (BEHRMAN, KLIEGMAN & JENSON, 2013). O sistema
nervoso é, normalmente, muito impactado pelo acúmulo dos substratos, podendo
desencadear manifestações clínicas como coma (em fase aguda). Em algumas
aminoacidopatias, a intoxicação somente se manifesta depois de vários meses, até
mesmo anos (SAUDUBRAY et al., 2002).
A patogenia mais conhecida nesse grupo de EIM é a fenilcetonúria, que é
causada pela deficiência completa ou quase completa da fenilalanina hidroxilase
(PAH). Esta enzima atua sobre a conversão de fenilalanina a tirosina, e por conta da
deficiência enzimática, ocorre o acúmulo do substrato no organismo. Tal situação
perturba os processos essenciais no cérebro, como a mielinização e a síntese de
proteínas, resultando em lesão cerebral e retardo mental (GARCÍA et al., 2002b). As
crianças afetadas podem nascer livres de sintomas, mas se não tratadas no período
pós-natal, eles surgem tardiamente nos primeiros meses e o retardo se torna
progressivamente pior. O tratamento consiste em uma dieta pobre em fenilalanina e
suplementação com L-carnitina (NADAI et al., 2006).
2.1.3.2 ACIDEMIAS ORGÂNICAS
As acidúrias ou acidemias orgânicas são normalmente causadas por
deficiência severa da atividade de enzimas envolvidas no metabolismo de
aminoácidos ramificados, o que resulta em acúmulo tecidual de ácidos carboxílicos
(SCRIVER et al., 2002). As acidemias orgânicas mais comuns são as
acidemiaspropiônica (PA), metilmalônica (MMA), isovalérica (IVA) e glutárica Tipo 1
(GA I) e Tipo 2 (GA II). Elas são assim classificadas a partir dos ácidos orgânicos
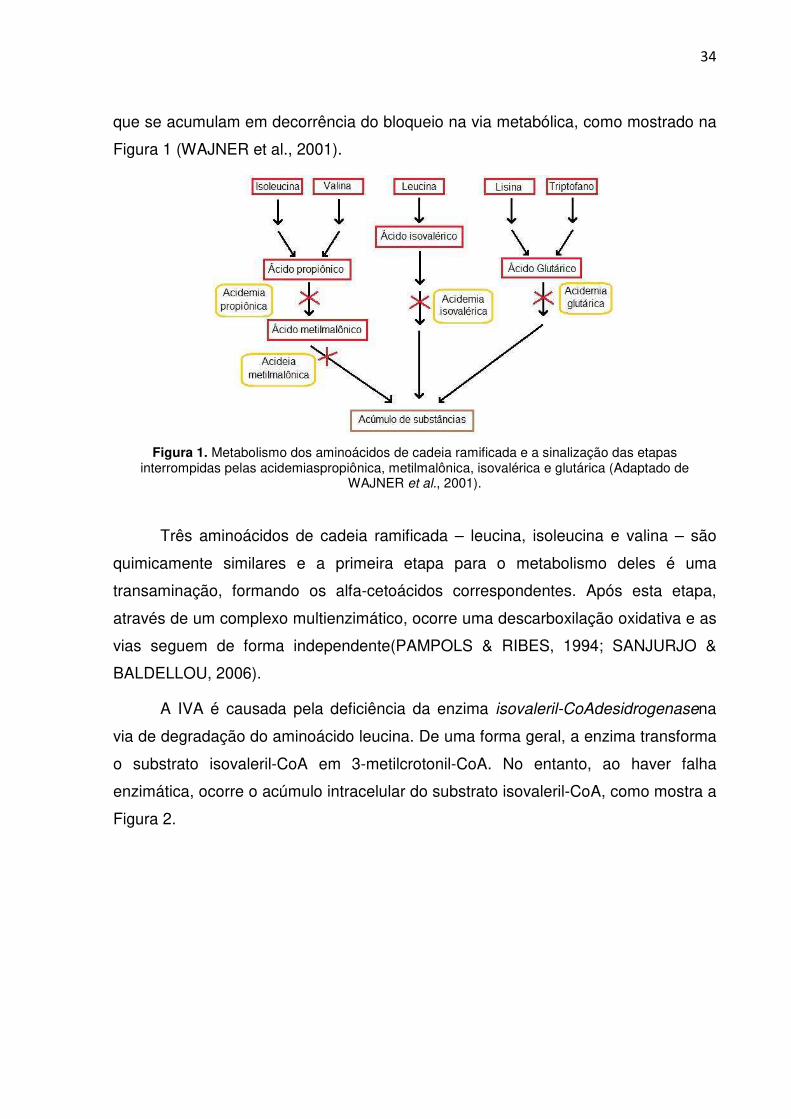
34
que se acumulam em decorrência do bloqueio na via metabólica, como mostrado na
Figura 1 (WAJNER et al., 2001).
Figura 1. Metabolismo dos aminoácidos de cadeia ramificada e a sinalização das etapas
interrompidas pelas acidemiaspropiônica, metilmalônica, isovalérica e glutárica (Adaptado de WAJNER et al., 2001).
Três aminoácidos de cadeia ramificada – leucina, isoleucina e valina – são
quimicamente similares e a primeira etapa para o metabolismo deles é uma
transaminação, formando os alfa-cetoácidos correspondentes. Após esta etapa,
através de um complexo multienzimático, ocorre uma descarboxilação oxidativa e as
vias seguem de forma independente(PAMPOLS & RIBES, 1994; SANJURJO &
BALDELLOU, 2006).
A IVA é causada pela deficiência da enzima isovaleril-CoAdesidrogenasena
via de degradação do aminoácido leucina. De uma forma geral, a enzima transforma
o substrato isovaleril-CoA em 3-metilcrotonil-CoA. No entanto, ao haver falha
enzimática, ocorre o acúmulo intracelular do substrato isovaleril-CoA, como mostra a
Figura 2.

35
Figura 2. Via principal do catabolismo da leucina e bloqueio da enzima isovaleril-CoAdesidrogenase
(1) (Adaptado de VARGAS & WAJNER, 2001)
O substrato isovaleril-CoA, quando descompensado, desencadeia a formação
de metabólitos alternativos (Figura 3), que podem ser encontrados no sangue e na
urina. Do mesmo modo, pode-se encontrar nos fluidos biológicos o próprio isovaleril-
CoA, que se encontra em excesso devido ao seu acúmulo. Essas alterações são
acompanhadas dos seguintes sintomas clínicos: acidose, cetose, acidose lática,
hiperamonemia, hiperglicinemia e níveis diminuídos de carnitina durante as crises
metabólicas (PAMPOLS & RIBES, 1994; SANJURJO & BALDELLOU, 2006).
Também pode ocorrer uma via secundária, que é a via endógena de excreção da
isovaleril-CoA, através da sua conjugação com a glicina. O produto formado é
excretado na urina e os níveis de isovaleril-CoA nos fluidos biológicos são
diminuídos. Esta via é a base dos principais tratamentos de suplementação com
glicina. Outra suplementação importante é a de L-carnitina, que remove o excesso
de ácido isovalérico. Com a suplementação destes dois componentes, associados a
uma dieta com baixa quantidade de proteína, a possibilidade de desenvolvimento de
retardo mental é baixa, porém, o déficit de atenção e de aprendizado ainda assim
podem aparecer (COWETT, 1998).

36
Figura 3. Vias metabólicas alternativas do isovaleril-CoA em caso de interrupção da via principal
(Adaptado de CAMPISTOL et al., 2007).
A PA é causada pela deficiência da enzima propionil-CoAcarboxilase (PCC),
enzima mitocondrial que possui a biotina como cofator do catabolismo dos
aminoácidos isoleucina, valina, treonina e metioninia, dos ácidos graxos de número
ímpar de carbonos e da cadeia lateral do colesterol. Este processo metabólico
fornece como produto o propionil-CoA, que é convertido pela enzima PCC em
metilmalonil-CoA, e seus produtos entram no ciclo de Krebs e na fosforilação
oxidativa, gerando energia. A deficiência da enzima PCC resulta em acúmulo
intramitocondrial de propionil-CoA, que passa a ser metabolizado por vias
secundárias e produz metabólitos secundários, como mostra a Figura 4 (FENTON,
GRAVEL & ROSENBLATT, 2001).
Figura 4. Metabólitos secundários produzidos quando ocorre o bloqueio enzimático da PCC (2)
(FENTON, GRAVEL & ROSENBLATT, 2001).

37
Existe um grande número de metabólitos decorrentes das rotas alternativas, e
a maioria deles são ácidos orgânicos. Eles encontram-se elevados em fluidos
fisiológicos e a identificação deles na urina pode levar ao diagnóstico da doença,
especialmente o metil-citrato e o 3-OH-propionato. O acúmulo de propionil-CoA
intracelular induz a inibição da enzima N-acetilglutamatosintetase e do sistema
mitocondrial de transporte de glicina, levando ao aumento dos níveis de amônia e de
glicina, comumente encontrados em pacientes com enzima deficiente. O propionil-
CoA também pode se conjugar com a carnitina, dando origem à propionil-carnitina. A
produção desse metabólito pode ter como consequência a deficiência secundária de
carnitina no plasma, sendo necessária a sua suplementação(FENTON, GRAVEL &
ROSENBLATT, 2001; SANJURJO & BALDELLOU, 2006).
A MMA é um grupo de EIM de aminoácidos que se caracteriza pelo acúmulo
de metilmalonil-CoA e de ácido metilmalônico nos fluidos biológicos (FENTON,
GRAVEL & ROSENBLATT, 2001; HOFFMAN, 1994). Esse acúmulo pode ser
causado pela deficiência da enzima metilmalonil-CoAmutase (MCM), ou pela
deficiência de seu cofator cobalamina, ambos essenciais para a produção do
substrato succinil-CoA (SANJURJO & BALDELLOU, 2006). Os metabólitos
característicos da PA também podem se acumular secundariamente na MMA, tais
como o ácido propiônico, o 3-OH-propiônico e o substrato metil-citrato e, por conta
disso, as manifestações clínicas de ambas as acidemias são muito semelhantes
(NYHAN, BARSHOP & OZAND, 2005). Normalmente, os sintomas clínicos são
percebidos nos primeiros dias de vida extra-uterina, já que ocorre a perda da função
dialisadora da placenta materna. As manifestações clínicas incluem sintomas
gastrointestinais, vômitos, recusa alimentar, desidratação, apneia, hepatomegalia,
manifestações neurológicas, hipotonia, letargia e convulsões. A demora no
diagnóstico e tratamento podem trazer danos irreparáveis ao sistema nervoso
central da criança, podendo progredir para o coma, danos neurológicos
permanentes ou morte (DEODATO et al., 2006b). Os achados laboratoriais mais
frequentes incluem cetoacidose metabólica, hiperglicemia, hiperamonemia, acidose
lática, anemia, trombocitopenia, deficiência de carnitina livre e aumento de
acilcarnitinas (GOSEN, 2008; LEHNERT et al., 1994).
O tratamento dos pacientes com PA ou MMA deve ser feito com restrição
proteica, administração de cofatores das enzimas deficientes (biotina para a PA e

38
hidroxicobalamina para a MMA) e, principalmente, com a administração de L-
carnitina. Esta substância é capaz de se conjugar e aumentar a excreção dos ácidos
orgânicos acumulados nessas doenças. A enzima carnitina aciltransferase catalisa a
formação de ésteres de carnitina como a propionilcarnitina e metilmalonilcarnitina.
Além disso, a L-carnitina também pode restaurar a razão acil-CoA/CoA livre
intramitocondrial e corrigir a deficiência secundária de carnitina (HOPPEL, 2003;
WALTER, 2003).
2.1.3.3 OUTRAS DOENÇAS DO GRUPO II
Além das doenças explicadas anteriormente, várias outras doenças
apresentam tratamento com L-carnitina. Alguns exemplos resumidos são:
3-Metilcrotonil glicinúria (3-MMM), a Deficiência Isolada de 3-metilcrotonil-CoA
Carboxilase (MCC), Acidúria 3-Hidroxi-3-Metilglutárica (HMG) são falhas no
catabolismo da leucina que causam hipoglicemia, ausência de corpos cetônicos e
acumulo de produtos tóxicos no plasma, urina e tecidos (GRÜNERT et al., 2012);
A Deficiência de Beta-cetotiolase (DLE) é uma anomalia da Acetoacetil-
CoATiolase mitocondrial (T2) envolvida no catabolismo da isoleucina, geralmente
grave, por vezes acompanhados por letargia/coma(GRÜNERT et al., 2012);
A Deficiência de Carnitina Primária (DCP) causa deficiência no transportador
de carnitina impedindo o transporte de ácidos graxos para o interior da mitocôndria,
podendo ocasionar encefalopatia hepática, hipoglicemia e cardiomiopatia
(MCGOVERN et al., 2004);
A Deficiência de Carnitina/AcilcarnitinaTranslocase (CACT) catalisa a troca da
carnitina interna com as acilcarnitinas externas, se apresentam no período neonatal
ou na primeira infância e pode causar morte súbita, além de anomalias neurológicas,
hipoglicemia, hiperamonemia, hepatomegalia e cardiomiopatia (VITORIA et al.,
2015);
A Deficiência da Acil-Coenzima A Desidrogenase de Cadeira Muito Longa
(VLCAD), enzima voltada na beta-oxidação de ácidos graxos de cadeia muito longa,
gera acúmulo dos intermediários reacionais e pode se manifestar ao nascer, na
infância ou na fase adulta. Manifestações metabólicas, diarreia, sonolência extrema,

39
mudança de comportamento, hiperglicemia e vômitos, cardiomegalia, hepatomegalia
e problemas musculares podem ser observadas(TENOPOULOU et al., 2015);
A Deficiência da Desidrogenase das Acil-Coa dos Ácidos Graxos de Cadeia
Média (MCAD) é responsável pela quebra de ácidos graxos para obtenção de
energia, também impede a formação de reservas energéticas. Os sintomas incluem
letargia, hipoglicemia, vômitos e, em casos extremos, convulsões, danos cerebrais,
coma e morte súbita (DRENDEL et al., 2015);
A Deficiência da Palmitoil Carnitina Transferase (CPT-II), que é responsável
pela entrada de ácidos graxos na mitocondria, podelevar à cardiomegalia com
batimento cardíaco irregular, hepatomegalia, fraqueza muscular, perda de apetite,
sonolência extrema, indícios de descompensação metabólica (MALIK et al., 2015);
A Deficiência de SCOT (SCOT)é uma doença recessiva do metabolismo dos
corpos cetônicos. A enzima SCOT é responsável pela ativação do acetoacetato,
transformando-o em acetoacetil-Coa para a utilização pela mitocôndria. Pode
compreender como sintomas a letargia, hipotonia, vômitos e até coma em casos
graves (FUKAO et al., 2011);
A Deficiência de 3-hidroxi-Acil-Coenzima-A-Desidrogenase de Cadeia Longa
(LCHADD), que é responsável pela oxidação dos ácidos graxos de cadeia longa (12
a 16 carbonos), pode desencadear uma cardiomegalia com batimento irregular,
hepatomegalia e fraqueza muscular. Costuma se manifestar na faixa dos 15 a 30
anos, em neonatos pode apresentar como sintomas catarata, imperfeições no
cérebro e óbito.
A escolha da terapêutica adequada dependerá do EIM responsável pela
doença e da substância acumulada ou deficiente que está levando ao desequilíbrio
bioquímico. Uma das principais abordagens de tratamento para esse perfil de
doenças apresentadas é a suplementação de L-carnitina, que evita o acúmulo do
metabólito que causa os sintomas apresentados nas patologias acima descritas (EL
HUSNY & FERNANDES-CALDATO, 2006; SCHWARTZ, SOUZA & GIUGLIANI,
2008).

40
2.2 L-Carnitina
2.2.1 Bioquímica
A L-carnitina é uma amina quaternária com importante função na oxidação de
ácidos graxos, principal fonte de energia das células. Ela é um componente de baixo
peso molecular, sendo sintetizada de forma endógena no fígado, rins e cérebro a
partir de dois aminoácidos essenciais: lisina e metionina. Para esse processo, é
necessária a presença de ferro, ácido ascórbico, niacina e piridoxina (HOPPEL,
2003).
Além da produção endógena, cerca de 75% da L-carnitina presente no
organismo é proveniente da alimentação, sendo as principais fontes a carne
vermelha e os laticínios (EVANS & FORNASINI, 2003). Ela é armazenada
principalmente nos músculos esqueléticos e cardíacos para fornecer rapidamente
energia para as atividades musculares (BREMER, 1983; REBOUCHE & ENGEL,
1984).
A L-carnitina atua no processo de produção de energia no organismo. Ela tem
importante função no transporte dos ácidos graxos para seu local de destino, onde
irá ocorrer a oxidação (produção de energia). Os ácidos graxos são a principal fonte
de energia do organismo e são moléculas orgânicas, de caráter lipofílico. Para
transitarem pelo sistema sanguíneo, são normalmente associados a proteínas
sanguíneas (albumina) para garantir solubilidade em meio aquoso. Entretanto,
quando ligados a proteínas, os ácidos graxos não conseguem penetrar na
mitocôndria, que é seu local de oxidação. Para atingirem a matriz mitocondrial, uma
série de reações enzimáticas deve ocorrer para realizar o transporte do meio extra
mitocondrial para o interior desta organela. Esse processo é de suma importância,
principalmente para ácidos graxos de cadeia longa (BROQUIST, 1994).
A primeira etapa envolve uma família de isoenzimas presentes na membrana
mitocondrial, denominadas acil-CoAsintetases. Nesta etapa, em quehá consumo de
ATP, o ácido graxo é ligado à coenzima A (CoA), formando acil-CoA. Com ação da
enzima carnitina palmitoiltransferase I, o acil-CoA e a carnitina formam o complexo
acil-carnitina, liberando uma CoA. Em seguida, a acil-carnitina é transferida pela
carnitina-acilcarnitinatranslocase para a matriz, ao mesmo tempo em que uma
molécula de carnitina é transportada para o meio extramitocondrial. Na matriz, a acil-

41
carnitina passa pela enzima carnitina palmitoiltransferase II e libera o grupo acil, que
se conjuga com a CoAintramitocondrial, liberando uma molécula de carnitina na
matriz intramitocondrial (Figura 5). Essas reações são importantes para manter
separadas a CoAextramitocondrial da intramitocondrial, já que estas possuem
funções diferentes. A CoA intramitocondrial atua na degradação oxidativa do
piruvato, dos ácidos graxos e de alguns aminoácidos, enquanto que a CoA citosólica
é normalmente utilizada para a biossíntese dos ácidos graxos. Esse processo
mediado pela carnitina regula a oxidação dos ácidos graxos (BROQUIST, 1994;
CHAMPE, HARVEY & FERRIER, 2006; NELSON & COX, 2014).
Figura 5. Transporte dos ácidos graxos do meio extramitocondrial para a matriz mitocondrial com
auxílio de enzimas (Adaptado de NELSON & COX, 2014).
2.2.2 Características Gerais
A L-carnitina (Figura 6) é um pó cristalino branco ou quase branco, com odor
característico, altamente higroscópico e muito solúvel em água. Por conta disso, é
indicado para formulações líquidas (MARTINDALE, 2009).

42
Figura 6. Estrutura da L-carnitina (HOPPEL, 2003).
É solúvel em álcool quente e praticamente insolúvel em acetona. Possui peso
molecular de 161,2g/mol, seu ponto de fusão é entre 196 e 197ºC, seu ponto
isoelétrico é 3,8 e possui densidade aparente de aproximadamente 0,64g/mL. Uma
solução a 5% em água apresenta pH entre 6,5 e 8,5 (SWEETMAN, 2009). As
principais características de identificação estão apresentadas na tabela 01:
Tabela 01. Principais características de identificação da Carnitina
Nome químico 3-hidroxi-4-N-trimetilamino-butirato
Sinônimos Carnitina,
Levocarnitina,
L-carnitina base,
Vitamina BT,
CAS 541-15-1
DCB/DCI 05242 – Levocarnitina
A L-carnitina é encontrada naturalmente nos animais, vegetais e
microrganismos. Em humanos, a sua biossíntese endógena não é suficiente para as
atividades do organismo, sendo necessário adquiri-la exógenamente. A dieta é o
principal meio de obtenção de L-carnitina e os alimentos que apresentam
abundância em sua composição são as carnes vermelhas, derivados lácteos, peixes
e ovos (BREMER, 1983; WALTER & SCHAFFHAUSER, 2000).
Na década de 80, a empresa suíça Lonza® desenvolveu e patenteou a técnica
para a síntese da L-carnitina em escala industrial, tornando-a acessível à população.
O processo de fabricação da L-carnitina era realizado por reações químicas na
presença de catalisadores quirais. Inevitavelmente, a síntese química conduzia à

43
mistura racêmica dos isômeros L-carnitina (Figura 7e) e D-carnitina (Figura 7d)
(BERNAL et al., 2007; CAVAZZA, 1981; WALTER & SCHAFFHAUSER, 2000).
L-Carnitina D-Carnitina
Figura 7. Estrutura química das isoformas de carnitina: (e) levógira e (d) dextrógira (HOPPEL, 2003).
Diversos estudos sugerem que a D-carnitina não seja biologicamente ativa.
Isso porque a substância pode se ligar ao sistema de transporte da L-carnitina,
diminuindo a concentração da isoforma levógira nas células e inibir reações
dependentes de levocarnitina, já que a concentração de L-carnitina livre nas células
é um importante regulador para a síntese e degradação de ácidos graxos. Diante
disso, a companhia suíça Lonza® mais uma vez desenvolveu e patenteou um
processo de biotransformação único, estereosseletivo, para a produção de L-
carnitina (livre da isoforma dextrógira) (SÁNCHEZ-HERNÁNDEZ et al., 2010;
WALTER & SCHAFFHAUSER, 2000).
Para contornar o forte odor característico e a alta higroscopicidade da L-
carnitina base, a companhia sintetizou o sal cloridrato de L-carnitina (Figura 8), que
não teve boa aceitação.
Figura 8. Estrutura do sal cloridrato de L-carnitina (C7H15NO3.HCl) (SWEETMAN, 2009).
2.2.3 Efeitos adversos e precauções
Após administração oral, alguns efeitos adversos podem ocorrer, tais como
náuseas, vômitos, diarreia e cólicas abdominais. Para evitar os distúrbios

44
gastrointestinais, a solução oral deve ser ingerida lentamente, com doses espaçadas
e uniformes ao longo do dia. Alguns pacientes podem exalar odor característico,
provavelmente devido à formação do metabólito trimetilamina, o que pode ser
reduzido ou eliminado com a redução da dose (PATRICK, HINCHCLIFFE &
JONATHAN, 2005; SWEETMAN, 2009).
Deve-se evitar administrar doses elevadas de L-carnitina oral por longos
períodos a pacientes com insuficiência renal grave, pois eles não conseguem
eliminar os metabólitos tóxicos trimetilamina e N-óxido de trimetilamina. Além disso,
deve-se monitorar a glicemia de pacientes diabéticos que fazem uso de insulina, a
fim de evitar crises de hipoglicemia (SWEETMAN, 2009).
2.2.4 Farmacocinética
A concentração plasmática de L-carnitina representa a soma do material
endógeno e exógeno. As doses orais de L-carnitina são absorvidas lentamente no
intestino delgado, através de um sistema de transporte dependente de sódio ou via
difusão passiva (LI et al., 1992). Após dose oral de 1 a 6 g de L-carnitina, a
biodisponibilidade encontrada é de 5 a 18% da dose administrada, enquanto que a
biodisponibilidade da L-carnitina proveniente da dieta é superior a 75%. A
concentração máxima é alcançada 3 a 4 horas após a administração oral. A porção
não absorvida é degradada pelos microrganismos presentes no intestino grosso,
conduzindo à formação de metabólitos (REBOUCHE & ENGEL, 1984). Alguns
metabólitos são recuperados na urina (óxido-N-trimetilamina) e nas fezes (γ-
butirobetaína). A L-carnitina possui alta eliminação renal, mas sofre intensa
reabsorção tubular (98-99%). Ela não se liga às proteínas plasmáticas e seu estado
de equilíbrio é alcançado após quatro dias de administração oral (EVANS &
FORNASINI, 2003).

45
2.3 Farmácia Universitária da UFRJ
A Farmácia Universitária (FU) da Universidade Federal do Rio de Janeiro
(UFRJ) atende cerca de 200 pacientes diários vindos de clínicas especializadas,
como as do Hospital Universitário Clementino Fraga Filho (HUCFF/UFRJ) e do
Instituto de Pediatria e Puericultura Martagão Gesteira (IPPMG/UFRJ). Eles são
atraídos por medicamentos de qualidade a preço de custo, se tornando um grande
benefício principalmente para pacientes de baixa renda (BARROS, 2016). Uma
grande parcela dos paciêntes provenientes do IPPMG apresentam alguma doença
relacionada a EIM e necessitam de suplementação de L-carnitina para seu
tratamento.
A FU é uma farmácia escola fundada em 1986 pela necessidade de haver um
local que oferecesse estágio em manipulação alopática para os alunos da Faculdade
de Farmácia (FF) da UFRJ. A FU recebe mais de 200 estudantes por ano, da
Faculdade de Farmácia da UFRJ, UFRJ Macaé e do Instituto Federal do Rio de
Janeiro (IFRJ) para realização do estágio curricular obrigatório de manipulação
alopática, que são orientados por farmacêuticos e docentes. A FU possibilita o
contato dos estudantes com a manipulação de formas farmacêuticas e orientação
aos pacientes sobre os principais efeitos adversos do medicamento, instruções de
administração, uso racional e cuidados no armazenamento (BARROS, 2016;
RODRIGUES et al., 2009).
Atuamente apresenta inúmeros projetos relevantes e foi classificada pela Pró-
reitoria de Extensão da UFRJ como um programa de extensão. Vale ressaltar o
caráter multidisciplinar das suas atividades que, ao longos dos anos, vem
desempenhando papel estratégico na formação de estudantes não só de graduação,
mas também de Pós-Graduação, em áreas carentes de recursos humanos, tais
como a busca de novas formulações de medicamentos, assistência farmacêutica,
farmacovigilância e projetos que consolidam a pesquisa na área de cosmetologia
(RODRIGUES et al., 2009).
Com uma grande parcela de pacientes em busca de suplementação de L-
carnitina e sem a existência de uma formulação de L-carnitina existente no mercado,

46
a FU buscou atender às prescrições na forma farmacêutica de pó dispensado em
sachês. O processo, relativamente simples, consiste em transferir a massa
equivalente à dose do ativo para o sachê, proceder com a expulsão do máximo de ar
do interior da embalagem e finalizar com completa selagem.
Porém, a L-carnitina base, sendo extremamente higroscópica, requer
associação com agente dessecante para reduzir a absorção de água durante o
processo de manipulação. Os constituintes da formulação devem ser tamisados
antes da homogeneização, que deve acontecer em gral e pistilo. A mistura deve ser
realizada em progressão geométrica até total homogeneização. O processo descrito,
a pesagem e a selagem do sachê, devem ocorrer o mais breve possível para evitar
grande exposição da formulação à umidade do ambiente. Com esse objetivo, no
máximo vinte sachês eram produzidos a cada manipulação, que contava com a
dedicação de dois funcionários dividindo as tarefas de pesar e selar no menor
intervalo de tempo possível. Mas, mesmo com esses cuidados, a mistura
apresentava grande absorção de água, tornando-se grumosa e, por muitas vezes,
líquida, tornando difícil sua pesagem e envase total da dose (Figura 09). Muitas
vezes não se conseguia manipular a quantidade de sachês planejada e uma
quantidade considerável de mistura tornava-se inutilizada e era descartada.
Figura 09. Ilustração da absorção de água durante o processo de manipulação da mistura (D), durante o processo de pesagem (M) e durante processo de envase (E).
Além dos problemas relativos à produção da formulação, a dispensação sob a
forma de pó leva a problemas na adesão do paciente. Para administração do
conteúdo do sache, é necessário que o pó seja disperso em líquidos ou em

47
alimentos durante as refeições. Este tipo de administração, no entanto, apresenta
alguns inconvenientes, principalmente pela dificuldade em fazer pacientes infantis ou
com dificuldade de deglutição em ingerirem a dose correta em todas as refeições. E
o forte odor característico desagradável da formulação pode dificultar ainda mais a
correta administração. Além disso, alguns sachês (normalmente os últimos
manipulados) podem apresentar conteúdo mais aglomerado (grumado) e apresentar
alteração no escoamento do pó, ficando retido na superfície interna da embalagem,
podendo levar a redução da dose a ser administrada.
Por conta das desvantagens da dispensação da L-carnitina em pó envasada
em sachê, o mais indicado para esse público alvo seria a disponibilização de uma
formulação líquida oral de L-carnitina. O mercado farmacêutico disponibiliza poucos
medicamentos na forma farmacêutica líquida e, até o momento, não existe registro
de formulação líquida oral de L-carnitina com finalidade medicamentosa, apenas
suplementos voltados para o público desportivo, normalmente associado a diversos
componentes vitamínicos e nutrientes energéticos. Por se tratarem de suplementos,
não são considerados como alternativa de tratamento, já que não são produzidos
com a qualidade e exigências necessárias para um medicamento, tornando a
qualidade e a quantidade declarada duvidosa. Em muitos casos, não necessitam
nem de registro em órgão regulatório (VENTURA, 2011).
As formas farmacêuticas líquidas são as mais indicadas para uso em
pediatria, já que, além de facilitarem a administração e contribuir para a adesão dos
doentes à terapêutica, apresentam grande flexibilidade, permitindo o ajuste simples
e rápido das doses que, em função da evolução da patologia e do desenvolvimento
da criança, podem sofrer ajustes. Estes aspectos são particularmente relevantes nos
casos de terapias prolongadas, como no tratamento das doenças relacionadas à
EIM (PINTO; BARBOSA, 2008). Esse público apresenta importantes diferenças e
alterações fisiológicas, além de apresentarem uma série de restrições. Dessa forma,
a formulação deve ser o mais simples possível, com o mínimo de excipientes, mas
sem deixar de apresentar a qualidade e segurança necessárias (EL HUSNY;
FERNANDES-CALDATO, 2006).

48
2.4 Desenvolvimento De Formulações Líquidas
2.4.1 Forma Farmacêutica Líquida
A forma farmacêutica é uma ferramenta fundamental na adesão ao
tratamento farmacoterapêutico, principalmente quando se trata de doenças crônicas,
onde o medicamento pode ser utilizado por longos períodos, além de assegurar a
eficácia e segurança do medicamento. Para pacientes pediátricos, idosos e que
apresentem alguma dificuldade de deglutição, a forma farmacêutica líquida é a mais
adequada, pois facilita a administração do medicamento e confere grande
flexibilidade quanto à dosagem, permitindo ajustes simples e rápidos que podem ser
necessários durante o tratamento (PINTO; BARBOSA, 2008; VENTURA, 2011).
No entanto, o desenvolvimento de formulações líquidas possui fatores críticos
e requer diferentes cuidados para garantia da segurança e qualidade do produto.
Por apresentarem elevado teor de água, estão passíveis de degradação físico-
química, podem apresentar menor estabilidade dos componentes da formulação e
apresentam maior susceptibilidade de proliferação de microrganismos (FERREIRA;
SOUZA, 2011a).
Além disso, a L-carnitina é sintetisada a partir dos aminácidos essenciais
lisina e metionina e, por apresentar estrutura semelhante a de um aminoácido, pode
ser usada como substrato para microrganismos. Bactérias Gram-positivas e Gram-
negativas podem consumir a L-carnitina como fonte de carbono, nitrogênio e de
energia podendo levar à formação de metabólitos indejesados (MEADOWS;
WARGO, 2015; SÁNCHEZ-HERNÁNDEZ et al., 2010). Caso o sistema conservante
não seja eficiente, a concentração da formulação pode ser comprometida, levando a
uma administração de dose subterapêutica, exposição do paciente a produtos de
degradação tóxicos ou ingestão de um número inaceitável de microrganismos
(BILLANY, 2005).
Na tentativa de impedir os problemas apontados, substâncias inertes
denominadas excipientes podem auxiliar no desenvolvimento de soluções mais
estáveis, eficazes e atraentes. Essas substâncias possuem diferentes funções,
sendo comumente usadas para solubilizar, espessar, diluir, estabilizar, conservar ou
flavorizar formulações farmacêuticas. As características organolépticas da

49
formulação são importantes fatores de adesão à terapêutica e diferentes opções de
excipientes são encontrados no mercado (GLASS et al., 2006).
Porém, estudos apontam que essas substâncias consideradas inertes, podem
ser responsáveis por algumas reações adversas relacionadas a medicamentos.
Além disso, alguns adjuvantes são reconhecidamente desaconselhados para uso
em crianças. Os edulcorantes, utilizados para tornar as formulações orais mais
palatáveis, podem conter açúcar sendo contra-indicado para pacientes diabéticos e
com erros inatos de metabolismo. Os corantes também são utilizados nessas
formulações e podem causar reações alérgicas em pacientes com hipersensibilidade
(VENTURA, 2011). Desta forma, a escolha dos excipientes da formulação não pode
ser realizada ao acaso e deve ser feita de maneira criteriosa sendo o mais simples
possível, no intuito de diminuir a probabilidade de reações adversas, mas garantindo
a qualidade e estabilidade da formulação (ANSEL; POPOVICH; ALLEN JUNIOR,
2000; GUERRA, 2012).
2.4.2 Excipientes Farmacêuticos Usados Para Soluções Líquidas Orais
Os excipientes são substâncias adicionadas aos compostos
farmacologiamente ativos para permitir a produção de formas farmacêuticas
estáveis, atraentes, eficazes e seguras e, normalmente, representam a maior parte
da forma farmacêutica. Os excipientes podem auxiliar na manutenção da
estabilidade física, química e microbiológica do produto, interferir na disponibilidade
do princípio ativo no organismo, melhorar a aceitabilidade do paciente e manter a
efetividade do produto durante a sua estocagem e uso.Cada excipiente tem uma
função definida no produto e podem funcionar como conservantes, solventes,
edulcorantes, aromatizantes, agentes doadores de viscosiade, veículo, entre outros.
Portanto, sua seleção vai depender das características desejadas do produto final
farmacêutico(AULTON, 2016; BRASIL, 2012a). Os principais tipos de excipientes
utilizados em soluções orais estão descritos abaixo:
2.4.2.1 SOLVENTE: ÁGUA PURIFICADA
A água é o solvente mais usado em formas farmacêuticas líquidas por
apresentar diversas vantagens como a ausência de toxicidade, compatibilidade

50
fisiológica e baixo custo. A água potável apresenta diferentes contaminantes que
podem interferir na qualidade da formulação final. Por isso, a água deve ser tratada
por sistemas de purificação como destilação, troca iônica ou osmose reversa, para
atender aos requisitos de pureza estabelecidos na monografia da Farmacopeia
Brasileira. A água purificada pode ser utilizada na fabricação de soluções não
estéreis de uso não parenteral e em formulações magistrais (BRASIL, 2010a).
2.4.2.2 CONSERVANTES
As formulações líquidas orais apresentam, em sua grande maioria, alto teor
de água em sua composição, o que favorece o crescimento microbiano durante sua
manipulação, armazenamento e uso. Os chamados conservantes são substâncias
usadas para inibir o crescimento microbiano, com o objetivo de prolongar a
estabilidade do produto, principalmente em formulações multidose. Um bom
conservante deve apresentar atividade de amplo espectro contra bactérias,
leveduras e mofos, ser efetivo em baixas concentrações, ser solúvel em água e, de
preferencia, ser incolor e inodoro (AULTON, 2016; PAULO et al., 2013).
Dentre os conservantes mais utilizados em formulações farmacêuticas,
destacam-se os parabenos, usados desde 1932, sendo o metilparabeno e
propilparabeno os mais conhecidos. Eles apresentam amplo espectro de atividade,
são ativos contra fungos e bactérias Gram positivas, mas fracos para as Gram
negativas. Apesar de só serem efetivos em fase aquosa, não apresentam alta
solubilidade em água necessitando de aquecimento prévio. Atuam em uma faixa de
pH de 3 a 8, sendo mais ativos em pH baixo e apresentam sabor não muito
convidativo. São normalmente utilizados entre 0,015 a 0,2% para soluções ou
suspensões orais (SONI et al., 2001).
O benzoato de sódio é o conservante mais antigo e utilizado. É um agente
bacteriostático e fungicida, bastante utilizado em formulações farmacêuticas orais e
considerado seguro quando usado em concentrações inferiores a 0,5%. Possui alta
solubilidade em água a 25°C e é pH depentende, apresentando maior atividade em
valores baixos de pH, quando é convertido em sua forma ativa, o ácido benzóico. É
possível também encontrar o ácido benzóico comercializado como conservante, mas
ele não apresenta boa solubilidade em meios aquosos (LACHMAN; LIEBERMAN;
KANING, 2001; MARTINDALE, 2009; VILAPLANA; ROMAGUERA, 2003).

51
O ácido cítrico é um ácido orgânico normalmente encontrado em frutas
cítricas, solúvel em água e com agradável sabor cítrico. Possue atividade
antimicrobiana devido à sua acidificação do meio e também por sua característica de
quelar íons metálicos, o que diminui o crescimento bacteriano, porém é menos
eficaz no controle de fungos. Não apresenta atividade em soluções com valores de
pH maiores do que 5,5 e sua atividade é potencializada com a diminuição do pH
(MAX et al., 2010).
O ácido sórbico e os sorbatos de sódio ou de potássio são potentes inibidores
de bolores e leveduras, possuindo pouca ou nenhuma efetividade na inibição de
bactérias. Também possuem melhor atividade em baixos valores de pH. É inodoro e
incolor, e utilizado em concentrações inferiores a 0,3% (FERREIRA, 2000b;
GUYNOT et al., 2005).
2.4.2.3 AGENTE PROMOTOR DE VISCOSIDADE
Usados para mudar a consistência de uma preparação, fornecem maior
resistência ao escoamento. São utilizados em formulações orais no intuito de
diminuir o contato do ativo com as papilas gustativas e facilitar a administração
(FERREIRA, 2000b). Alguns exemplos estão descritos abaixo:
A carboximetilcelulose (CMC) é um derivado hidrossolúvel de celulose,
normalmente encontrada na forma sódica (sal de sódio), é higroscópica, altamente
solúvel em água, tanto a frio quanto a quente, inodoro e insípido. É amplamente
utilizado em formulações de uso oral. Vários graus de viscosidade podem ser
comercializados (baixa, média e alta viscosidade) e proporcionam viscosidades
diferentes quando dispersadas em soluções. Apresenta boa estabilidade na faixa de
pH entre 4 e 10 (LUBI, 2002; PRISTA; ALVES; MORGADO, 1990).
A goma xantana é um polissacarídeo constituído de manose, glicose e ácido
glicurônico. A estrutura rígida do polímero e natureza específica da ramificação
fornece uma boa estabilidade da viscosidade de soluções aquosas, não sendo
afetadas por alteração de temperatura e pH(LUBI, 2002).
A hidroxietilcelulose é um polímero derivado da celulose, não iônico, solúvel
em água, estável em formulação de pH entre 2 e 12, entretanto, pode ocorrer
hidrólise em soluções com pH inferiores a 5. Em temperaturas elevadas, apresenta

52
diminuição de viscosidade, o que pode ser revertido com o resfriamento da
formulação. É amplamente usado em preparações de uso oftálmico e tópico, e
estudos sugerem sua administração em formulações orais na administração oral
(LUBI, 2002).
O alginato de sódio também é utilizado como agente suspensor em
formulações orais, com concentração usual entre 1 e 5%. É higroscópico, a solução
aquosa apresenta boa estabilidade quando está na faixa de pH entre 4 e 10. As
soluções de alginato de sódio são susceptíveis à contaminação microbiana, o que
pode alterar sua viscosidade. O mesmo pode acontecer caso a solução seja exposta
à temperaturas superiores a 70°C.
2.4.2.4 ACIDULANTE
É importante a prática de ajuste de pH das formulações manipuladas com o
objetivo de garantir a estabilidade físico-química e microbiológica. Muitos dos
excipientes adicionados à formulação necessitam de uma faixa de pH ideal para
garantir suas atividades na composição da formulação. Solução de ácido cítrico,
solução de ácido clorídrico e solução de ácido acético são importantes exemplos de
acidulantes descritos no formulário nacional 2ª Ed.(BRASIL, 2010a, 2012a;
FERREIRA, 2000b).
2.4.2.5 EDULCORANTES
Formulações líquidas orais podem apresentar sabor desagradável, sendo o
uso de edulcorantes uma forma de melhorar a palatabilidade da formulação,
favorecendo a adesão do paciente ao tratamento. São classificados em naturais,
semissintéticos e sintéticos (BALBANI; STELZER; MONTOVANI, 2006).Os principais
edulcorantes utilizados em preparações orais líquidas são: sacarose, sacarina
sódica, aspartame, ciclamato de sódio, sorbitol, manitol e frutose (FERREIRA;
SOUZA, 2011a)
2.4.3 Embalagens Usadas Para Soluções Líquidas Orais
A embalagem primária é destinada ao acondicionamento do produto acabado
e fica em contato direto com o seu conteúdo durante todo o tempo. Pode ser de

53
vidro ou de plástico e deve proteger o produto das condições ambientais
preservando sua estabilidade. Ela não deve interagir fisicamente ou quimicamente
com a preparação e deve preservar a concentração, qualidade e pureza da
formulação. (BRASIL, 2007b, 2010b).
O vidro utilizado na embalagem de produtos farmacêuticos necessita ter
qualidades protetoras superiores, ser quimicamente inerte, impermeável, forte, rígido
e não se deteriorar com o tempo. Para uso farmacêutico, são classificados de
acordo com a resistência ao ataque hidrolítico(ANSEL; POPOVICH; ALLEN
JUNIOR; FERREIRA, 2000c):
• Vidro tipo I – vidro neutro, de alta resistência térmica, mecânica e
hidrolítica. Destinado ao acondicionamento de medicamentos para
aplicação intravascular e uso parenteral.
• Vidro tipo II – vidro alcalino do tipo sódico/cálcico, de resistência hidrolítica
elevada. Destinado ao acondicionamento de soluções de uso parenteral,
neutras e ácidas, que não tenham seu pH alterado.
• Vidro tipo III – vidro alcalino do tipo sódico/cálcico, de resistência hidrolítica
média, porém com boa resistência mecânica, sem qualquer tratamento
superficial. Destinado ao acondicionamento de soluções de uso oral.
• Vidros tipo NP (Não parenteral) – vidro alcalino do tipo sódico/cálcico, de
resistência hidrolítica baixa e alta alcalinidade. Indicado ao
acondicionamento de produtos não parenterais, de uso tópico e oral.
Embora o vidro tenha muitas características adequadas, atualmente a maioria
dos produtos farmacêuticos são embalados em material plástico. Nesse grupo inclui-
se o polietileno de alta densidade (PEAD), polietileno de baixa densidade (PEBD),
polipropileno (PP), poliestireno (PS), policloreto de vinila (PVC), polietilenotereftalato
(PET) e vários outros. Algumas características de materiais plásticos podem ser
observadas na tabela abaixo:

54
Tabela 02. Principais características dos materiais comumente usados para embalagem
Característica PEAD PEBD PP PS PVC PET
Transparência Opaco
Transp.
Opavo
Transl. Transp. Transp. Transp. Transp.
Absorção de água B B B A B B
Permeabilidade ao vapor d’água B MB MB A B B
Permeabilidade ao oxigênio A A A A B B
Permeabilidade ao CO2 A A A A B B
Legenda: Transp. – Transparente; Transl. – Translúcido; MB – Muito Baixa; B – Baixa; A – Alta.
(adaptado de FERREIRA, 2000c).
Os materiais plásticos são mais leves quando comparados ao vidro,
apresentam maior resistência a impactos reduzindo custos de transporte e perdas,
maior versatilidade no design e cores de embalagens com baixo custo e maior
aceitação do consumidor. Porém, se pode encontrar diferentes problemas na
utilização de embalagens plásticas como: permeabilidade dos recipientes ao
oxigênio atmosférico e ao vapor de umidade, lixiviação dos constituintes do
recipiente para o conteúdo interno, absorção de fármacos do conteúdo para o
recipiente e possível alteração do recipiente após o armazenamento. Algumas
características devem ser levadas em consideração no momento da escolha da
composição do material de embalagem a ser utilizado. Algumas dessas
características podem ser observadas no tabela 03 (LACHMAN; LIEBERMAN;
KANING, 2001; ROCHA et al., 2014).
Tabela 03. Principais resistencias dos materiais comumente usados para embalagem
Característica PEAD PEBD PP PS PVC PET
Resistência a ácidos 4 4 4 3 4 4
Resistência a álcalis 3 3 4 3 3 3
Resistência ao calor 2 4 3 4 3 4
Resistência a alta umidade 5 5 5 5 5 5
Neutralidade (ser inerte) 5 5 3 a 4 1 2 4 Legenda: 1 – Muito baixa; 2 – baixa; 3 – boa; 4 – muito boa; 5 – excelente (adaptado de FERREIRA,
2000c).

55
2.5 Controle De Qualidade De Soluções Orais Magistrais
No Brasil, a atividade farmacêutica magistral é regulamentada pela RDC N°
67/2007 que dispõe sobre as boas práticas de manipulação de preparações
magistrais para uso humano em farmácias, para garantir a qualidade do produto
final. Essa resolução reforça a importância do estabelecimento em ter um laboratório
capacitado de controle de qualidade para realização dos ensaios determinados pela
farmacopeia brasileira 5ª ed.
De acordo com a RDC N°67/2007, para a realização do controle de qualidade
de preparações líquidas não estéreis, deve-se realizar no mínimo os ensaios de
descrição, aspecto, caracteres organolépticos, pH e peso ou volume antes do
envase. Devido a pequena escala de produção, testes destrutivos são
impossibilitados de serem realizados. Desta forma, os principais testes aplicáveis a
soluções orais magistrais produzidos rotineiramente são a determinação da
densidade de massa e determinação do pH. Além disso, podem ser realizados
periodicamente, testes microbiológicos para soluções orais e determinação do teor
do princípio ativo(BRASIL, 2007a, 2010a, 2012a).
2.5.1 Densidade de Massa
A densidade de massa (ρ) de uma substância é a razão de sua massa por
seu volume a 20°C. Ela é determinada considerando a temperatura da solução e é
calculada a partir da densidade relativa (dtt). A densidade relativa de uma substância
é a razão de sua massa pela massa de igual volume de água, ambas a 20°C (d2020).
Ela pode ser determinada com o auxílio de picnômetro.
Deve-se realizar a calibração do picnômetro antes da análise da amostra.
Para realizá-la, deve-se determinar a massa do picnômetro vazio e depois de seu
conteúdo preenchido com água na temperatura de 20°C, recentemente destilada e
fervida. Após a calibração, deve-se determinar a massa do picnômetro preenchido
com a amostra. A partir dessas informações, pode-se calcular a densidade relativa
(d2020) a partir da razão entre a massa da amostra líquida e a massa da água
(BRASIL, 2010b).

56
2.5.2 Determinação potenciométrica do pH
O valor de pH é definido como a medida da atividade do íon hidrogênio de
uma solução. Convencionalmente é usada a escala da concentração de íon
hidrogênio da solução. A água é um eletrólito extremamente fraco, cuja
autoionização produz íon hidrônio (hidrogênio hidratado) e íon hidróxido:
H2O+ H2O �� H3O+ + OH- (Equação III)
As concentrações do íon hidrônio nas soluções aquosas podem variar entre
limites amplos, que experimentalmente vão de 1 a 10-14 M, que é definida pela
relação:
pH = - log [H3O+] = log 1/[ H3O
+] (Equação IV)
Desta forma, a escala de pH é uma escala invertida em relação às
concentrações de íon hidrônio, ou seja, quanto menor a concentração de íon
hidrônio, maior o valor do pH.
A determinação potenciométrica do pH é feita pela medida da diferença de
potencial entre dois eletrodos adequados, imersos na solução em exame. Um destes
eletrodos é sensível aos íons hidrogênio e o outro é o eletrodo de referência, de
potencial constante. Os aparelhos comercialmente utilizados para a determinação de
pH são instrumentos potenciométricos, providos de amplificadores eletrônicos de
corrente com célula de vidro-calomelano, os quais são capazes de reproduzir
valores correspondentes a 0,02 de unidades de pH (BRASIL, 2010).
2.5.3 Doseamento do princípio ativo
O ensaio de doseamento visa quantificar o principio ativo e determinar seu
teor em preparações farmacêuticas. As monografias encontradas nos compêndios
farmacêuticos oficiais apresentam metodologia específica para cada ativo e suas
diferentes formas farmacêuticas. Diferentes técnicas podem ser encontradas e as
mais conhecidas e utilizadas são: métodos volumétricos (titulação),
espectrofotometria, Cromatografia Líquida de Alta Eficiência (CLAE) e a
Cromatografia Gasosa (CG). Essas metodologias de análise oficiais são

57
consideradas validadas, onde testes específicos são realizados para demonstrar que
o método analítico produz resultados confiáveis e adequados à finalidade a que se
destina. Métodos não oficiais precisam ser validados conforme disposto na RDC
N°166, de 24 de julho de 2017(BRASIL, 2017a; TERRA; ROSSI, 2005).
A análise volumétrica é efetuada pela determinação do volume de uma
solução, cuja concentração é exatamente conhecida (solução titulante), que reage
quantitativamente com um volume conhecido da solução amostra. A adição da
solução titulante é feita gradativamente para que ocorra a reação entre amostra e
solução titulante. Esse processo é denominado de titulação. Quando a reação entre
o titulante e o titulado é completa, obtém-se o ponto de equivalência ou ponto
estequiométrico, que é acompanhado de modificação física provocada pela própria
reação ou pela adição de uma solução indicadora. A concentração da amostra é
definida a partir do volume usado da solução titulante na reação. É considerado um
método de baixo custo e de fácil acesso(PAVIA; LAMPMAN; VYVYAN, 2013).
A espectrofotometria na região UV-VIS do espectro eletromagnético é uma
das técnicas analíticas mais empregadas, em função de sua robustez e custo
relativamente baixo. A incidência de uma radiação continua (feixe de luz) sobre uma
amostra resulta em absorção da radiação, onde átomos e moléculas passam de um
estado de energia mais baixa (estado fundamental) para um estado de energia
maior (excitado) e a diferença entre as energias encontradas (entre os estados
excitado e fundamental) pode ser quantificada. A absorção de energia depende da
estrutura eletrônica da molécula e, por isso, a espectroscopia de absorção na região
do ultravioleta-visível (UV-VIS) tem ampla aplicação na caracterização de espécies
orgânicas e inorgânicas. A absorção é influenciada pela concentração da amostra e
pela estrutura do analito analisado. De acordo com o intervalo de frequência da
energia eletromagnética aplicada, a espectrofotometria de absorção pode ser
dividida em ultravioleta, visível e infravermelho(BRASIL, 2010; PAVIA; LAMPMAN;
VYVYAN, 2013).
A cromatografia gasosa (CG) é uma técnica de separação cromatográfica
baseada na diferença de distribuição de espécies de uma mistura entre duas fases
não miscíveis, na qual a fase móvel é um gás de arraste que se move através da
fase estacionária contida em uma coluna. É baseada no mecanismo de adsorção,

58
distribuição de massa ou exclusão de tamanho. É aplicada a substâncias e seus
derivados que se volatilizam sob as temperaturas empregadas e é aplicada para
testes de identificação, de pureza e determinação quantitativa (BRASIL, 2010).
A cromatografia líquida de alta eficiência (CLAE) é uma técnica de separação
cromatográfica fundamentada na distribuição dos componentes de uma mistura
entre duas fases imiscíveis: a fase móvel (líquida) e a fase estacionária (sólida,
líquida ou super fluida) que se encontra retira na coluna cromatográfica. O
cromatógrafo é constituído por um sistema de bombeamento, injetor, coluna
cromatográfica (em alguns casos onde a metodologia de análise exigir temperaturas
superiores à ambiente, pode ser acoplado um forno), um detector (o mais utilizado é
o UV/VIS) e um sistema de aquisição de dados (ANTUNES, 2015; EUROPEAN
PHARMACOPOEIA, 2004). A representação dos componentes pode ser visualizada
na figura abaixo:
Figura 10. Representação esquemática de um sistema de cromatografia líquida. 1 –
Sistema de acondicionamento de Fase Móvel; 2 – Bomba; 3 – Injetor; 4 – Coluna cromatográfica; 5 – Detector; 6 – Sistema de aquisição de dados; 7 – Computador
(adaptado de Applies Porous Technologies Inc, 2004)
Os sistemas de bombas disponíveis atualmente podem oferecer sistemas de
eluição isocráticos, onde a fase móvel é formada por um único solvente ou por uma
mistura de solventes de composição constante, sistema de eluição por gradiente,
com composição variável ao longo da corrida. As fases estacionárias são
armazenadas em colunas cilíndricas de alta pressão denominadas de coluna
cromatográfica. Quando sistemas são compostos por fase estacionária polar e fase
móvel apolar, é definido como cromatografia de fase normal, enquanto o oposto

59
(fase estacionária apolar e fase móvel polar) é denominada de cromatografia de fase
reversa. A afinidade de uma substância pela fase estacionária e, consequentemente,
seu tempo de retenção na coluna é controlado pela polaridade da fase móvel em
relação à fase estacionária (BRASIL, 2010).
2.5.4 Controle microbiológico
A contaminação microbiana de um produto não estéril pode conduzir não
somente à sua deterioração, com perda da estabilidade físico-química, mas também
ao risco de infecção para o usuário. A L-carnitina é sintetisada a partir dos
aminácidos essenciais lisina e metionina e, por apresentar estrutura semelhante a
de um aminoácido, pode ser usada como substrato para microrganismos. Bactérias
Gram-positivas e Gram-negativas podem consumir a L-carnitina como fonte de
carbono, nitrogênio e de energia podendo levar à formação de metabólitos
indejesados (Figura 11) (MEADOWS; WARGO, 2015; SÁNCHEZ-HERNÁNDEZ et
al., 2010).
Figura 11 – Metabólitos possiveis da L-carnitina quando consumida por microrganismos
(MEADOWS & WARGO, 2015).
Por conta disso, produtos farmacêuticos orais não estéreis devem ser
submetidos aos controles de contaminação microbiana. Os ensaios microbiológicos
para preparações aquosas para uso oral não estéril são: contagem total de bactérias

60
aeróbias (máximo permitido de 10² UFC/mL), contagem total de fungos/ leveduras
(máximo permitido de 10 UFC/mL) e a pesquisa do patógeno específico Escherichia
coli (deve ser ausente em 1mL de formulação) (BRASIL, 2010b).
2.6 Estudo De Estabilidade
A estabilidade de preparações magistrais é o período no qual um produto
manipulado mantém, dentro dos limites especificados e nas condições de
armazenamento e uso, as mesmas características e propriedades que apresentava
ao final de sua manipulação. Formulações líquidas, principalmente as aquosas,
podem apresentar suscetibilidade à degradação físico-química e microbiológica
podendo implicar no comprometimento da estabilidade da formulação (BILLANY,
2005; LACHMAN; LIEBERMAN; KANING, 2001).
Os estudos de estabilidade têm como objetivo gerar evidências sobre as
variações que podem ocorrer com a formulação em função do tempo, diante de
fatores ambientais como temperatura, umidade e luz. As informações obtidas
também orientam quanto ao prazo de validade do medicamento e as condições de
armazenamento. Tais estudos se iniciam no desenvolvimento da formulação,
quando são avaliados fatores intrínsecos como a fórmula em si, a compatibilidade
entre seus componentes, pH e adequação da embalagem primária, entre outros
fatores. Uma vez definido, o produto farmacêutico segue por longo período de
armazenamento, em sua embalagem final, sob condições controladas de
temperatura e umidade, simulando aquelas em que o produto estará exposto, e
passa por avaliações físico-químicas e microbiológicas (ANSEL; POPOVICH; ALLEN
JUNIOR, 2000; BRASIL, 2012a).
Para as indústrias, atualmente são definidos três tipos de estudo de
estabilidade: acelerado, que é projetado para acelerar a degradação química e/ou
mudanças físicas de um produto farmacêutico em condições forçadas de
armazenamento, estudo de longa duração que é projetado para verificar as
características físicas, químicas, biológicas e microbiológicas de um produto
farmacêutico durante seu prazo de validade, e o estudo de acompanhamento que é
realizado para verificar se o produto farmacêutico mantêm suas características

61
encontradas no estudo de estabilidade de longa duração (BRASIL, 2012a; SILVA et
al., 2009).
Todos os estudos descritos acima são realizados para determinação do prazo
de validade considerando o produto em sua embalagem primária lacrada, ao abrir a
embalagem para uso, um medicamento multidose adquire a característica de um
medicamento extemporâneo, onde o manuseio e as condições de exposição
desconhecidas não foram avaliadas previamente em estudos de estabilidade(
BRASIL, 2012a).
O medicamento magistral é produzido de forma personalizada, para atender a
necessidade particular de um paciente para uso imediato, sendo considerada uma
preparação extemporânea. Dessa forma, não se atribui a ele um prazo de validade,
mas sim uma data limite para uso denominada de “Prazo de Uso”. A definição dessa
data limite tem sido um grande desafio, pois a estabilidade de produtos
extemporâneos pode variar de uma formulação para outra, dependendo do fármaco
utilizado, da composição da formulação, da forma farmacêutica do produto final, do
processo de manipulação, da embalagem e de condições de armazenamento, entre
outros diversos fatores que podem alterar a estabilidade da formulação (ANSEL;
POPOVICH; ALLEN JUNIOR; BRASIL, 2012a; FERREIRA, 2010).
Na tentativa de determinar o melhor prazo de uso de medicamentos
multidose, pode-se realizar o estudo de estabilidade em uso, que é projetado para
verificar a manutenção da qualidade do produto em situações semelhantes às de
uso, quando ocorre a abertura e subsequentes reaberturas da embalagem primária,
simulando as condições de conservação do produto pelo período de tempo
recomendado para o tratamento (BRASIL, 2010c).
O estudo de estabilidade deve ser conduzido em ambiente com condições de
temperatura e umidade controladas. Para determinação das condições de
armazenagem, deve-se levar em conta as características da Zona Climática em que
o Brasil se encontra, que é a zona IV com temperatura ambiente de 30°C ± 2°C e
umidade relativa de 70 ± 5%. E, para formulações que necessitem ser
acondicionadas sob baixas temperaturas, a temperatura de condicionamento
durante o teste deve ser de 5°C ± 3°C. Já para o estudo de estabilidade acelerado, a
temperatura deve ser de 40°C ± 2°C e umidade relativa de 75% ± 5%. Ao usar

62
embalagem de material impermeável a umidade não precisa ser controlada(BRASIL,
2012a).
Para avaliar a estabilidade contra fatores extrínsecos podem ser realizados
testes de degradação forçada, onde a formulação é avaliada sob efeitos da
temperatura, oxidação, luz e susceptibilidade à hidrólise em ampla faixa de pH. O
estudo permite a geração de produtos de degradação e, dessa forma, avaliar a
seletividade e especificidade do método analítico, bem como obter informações
acerca das possíveis rotas de degradação do produto (BRASIL, 2012b).

63
3 JUSTIFICATIVA
A relevância deste trabalho se baseia nos benefícios alcançados com a
suplementação de L-carnitina em pacientes portadores de erros inatos do
metabolismo. A reposição contínua desta substância pode proporcionar melhor
qualidade de vida aos pacientes fornecendo melhoria dos sintomas e prevenção de
graves sequelas.
A L-carnitina não possui preparação medicamentosa industrializada na forma
líquida no Brasil e é geralmente dispensada em farmácias de manipulação sob a
forma de pó (sachê) para mistura em alimentos ou líquidos durante refeições. Este
tipo de administração, no entanto, apresenta alguns inconvenientes, principalmente
pela dificuldade em fazer pacientes infantis ou com dificuldade de deglutição, que
necessitam de multidoses diárias, em ingerirem a dose correta em todas as
refeições. A apresentação em solução oral facilitaria também a administração do
medicamento por pacientes com sondas de gastrostomia.
Algumas características da formulação podem se tornar um desafio, como o
fato de formas farmacêuticas líquidas apresentarem alta proporção de veículo
aquoso, tornando propício o crescimento de microrganismos. Além disso, a L-
carnitina pode ser utilizada como fonte de carbono, nitrogênio e reserva energética,
sendo considerada um bom substrato para bactérias. A formulação também deve
apresentar boa textura satisfatória e a capacidade de agradar o paladar do público
alvo, que é formado em sua maioria por pacientes infantis e com alterações
metabólicas. Por conta disso, a formulação a ser desenvolvida deve ser o mais
simples possível, utilizando o mínimo de excipientes, mas apresentando boa
estabilidade físico-química e microbiológica, garantindo a qualidade e segurança da
formulação.

64
4 OBJETIVOS
4.1 Objetivo Geral
O presente trabalho tem como objetivo desenvolver uma solução aquosa
magistral de L-carnitina, avaliando sua estabilidade físico-química e microbiológica
em diferentes tempos e sob diferentes condições. Este estudo visa disponibilizar
essas soluções aos pacientes portadores de patologias associadas a Erros Inatos de
Metabolismo (EIM) atendidos pela Farmácia Universitária da UFRJ, com apelo
especial para os que realizam tratamento no Instituto de Puericultura e Pediatria
Martagão Gesteira (IPPMG), o hospital pediátrico da UFRJ.
4.2 Objetivos Específicos
1- Desenvolvimento e caracterização da formulação líquida de L-carnitina;
2- Desenvolvimento e validação de metodologia analítica para solução oral de
L-carnitina;
3- Estudo de estabilidade físico-química e microbiológico;
4- Estudo dedegradação forçada das formulações.

65
5 MATERIAIS E MÉTODOS
5.1 Materiais
As matérias-primas e reagentes utilizados foram de grau analítico, e os
solventes utilizados foram de grau HPLC. Todos os equipamentos e vidrarias
utilizados encontravam-se previamente calibrados. As soluções foram preparadas
com água destilada e água Milli-Q.
5.1.1Matérias-primas e Reagentes
• Ácido Cítrico (Infinity Pharma);
• Ácido p-aminobenzóico (Sigma);
• Ácido clorídrico (Vetec);
• Ácido fosfórico 85% (Tedia);
• Água destilada (FU);
• Água ultrapurificada (Milli-Q);
• Benzoato de sódio(Farmos);
• Brometo de potássio (Shimadzu);
• Carboximetilcelulose (Farmos);
• Filtro 0,45µm (Chromafil);
• Membrana de celulose regenerada 0,45µm para filtração de fase aquosa
(Merck);
• Heptanosulfonato de sódio (Sigma);
• Hidróxido de sódio (Vetec);
• L-carnitina Matéria-prima (Infinity Pharma);
• Padrão SQR de L-carnitina (Sigma);
• Metanol (Vetec);
• Metilparabeno (Farmos);
• Peróxido de hidrogênio 3% (Vetec);
• Solução tampão padrão pH 4,01 (Merck);
• Solução tampão padrão pH 7,01 (Merck);
• Sulfato de cobre (Vetec).

66
5.1.2 Equipamentos e acessórios
• Agitador mecânico com haste de hélice (Fisatom);
• Balança analítica (Sartorius);
• Banho de ultrassom (LGI);
• Caneco de aço inoxidável de 200mL, 500mL, 1000mL e 3000mL;
• Coluna cromatográfica C18 3,9 x 300mm - 10µm 125A (Waters);
• Cromatógrafo Líquido de Alta Eficiência (CLAE) (Shimadzu LC-20AT);
• Detector de arranjo de diodos (DAD – UV/VIS) (Shimadzu SPD-M20A);
• Espectrofotômetro de Transformada de Fourier (ShimadzuIRPrestige-21);
• Filtro de vidro, suporte e papel de filtro;
• Placa de aquecimento e agitação (IKA C-MAG HS7);
• Potenciômetro (Meter 922);
• Pipetador automático 5000µL.
5.2 Métodos
5.2.1 Análise de identificação da L-Carnitina
Para identificação e caracterização da L-Carnitina foi realizada a análise de
Espectroscopia de Infravermelho com Transformada de Fourier (FTIR). Essa técnica
fornece evidências da presença de grupos funcionais presentes na estrutura do
ativo, confirmando sua composição química. Para obter as medidas, a radiação no
infravermelho passa através da amostra e é comparada com aquela transmitida na
ausência de amostra. O espectrofotômetro registra o resultado na forma de bandas
de absorção. A região do espetro eletromagnético de maior interesse para essa
técnica se encontra entre 4000 a 400 cm-1 (FORATO et al., 2010).
As amostras foram preparadas com a mistura de 3mg de L-carnitina,
previamente seca à vácuo por 5 horas a temperatura de 50°C, com 297 mg de
brometo de potássio (KBr). A mistura homogeneizada foi compactada em prensa
hidráulica, sob pressão de 15 bar por 2 minutos. As pastilhas formadas foram
levadas ao espectrofotômetro para análise e a faixa de varredura dos espectros foi
feita entre 4000 a 400 cm-1, com resolução de 4 cm-1. O espectro é formado pela
intensidade das bandas de absorção dada em transmitância, e a posição em

67
número de onda (cm-1). As bandas observadas no espectro foram caracterizadas de
acordo com os grupos funcionais presentes na estrutura do ativo e comparada com
literatura (USP, 2016).
5.2.2 Desenvolvimento das formulações
Foram manipuladas 10 formulações diferentes de L-carnitina. Foram
produzidos lotes de bancada de 100mL de cada solução com o objetivo de
selecionar a melhor formulação. A técnica de preparo envolveu a dissolução da L-
carnitina em veículo aquoso (água destilada), seguida da adição do agente
conservante. Após total dissolução dos componentes, completou-se o volume final
com o veículo e filtrou-se a formulação em papel de filtro. Após essas etapas, as
soluções foram levadas ao agitador mecânico acoplado com haste em hélice e
polvilhou-se o agente de viscosidade, mantendo sempre agitação constante e
intensa até total incorporação nas formulações. Para ajuste do pH foi adicionada
quantidade suficiente de solução de ácido cítrico 50% até o pH indicado de cada
formulação (essa etapa contou com o auxílio do potenciômetro). Antes do envase,
realizou-se a conferencia do volume final. As proporções dos componentes
utilizados nas formulações estão descritas na Tabela 04.
As soluções foram acondicionadas em frascos de vidro neutro âmbar de 60
mL, com tampa branca de rosca e batoque e volume suficiente para preencher dois
terços da capacidade do frasco. Antes do envase, os frascos foram lavados,
higienizados com álcool 70% (p/p) e secos (ANVISA, 2004).

68
Tabela 04. Descrição dos excipientes utilizados nas formulações testadas e suas proporções.
Componentes F1 F2 F3 F4 F5 F6 F7 F8
L-carnitina 10% 10% 10% 10% 20% 20% 20% 20%
Benzoato de sódio 0,3% 0,4% - 0,3% 0,4% 0,3% - 0,2%
Metilparabeno - - 0,1% - - - - 0,1%
Ácido Cítrico q.s. q.s. q.s. 0,9% - 1,8% 1,8% q.s.
pH 5,5 5,5 5,5 5,0 5,0 5,0 5,0 5,5
Carboximetilcelulose 0,3% 0,3% 0,3% 0,3% 0,3% 0,3% 0,3% 0,3%
Água destilada q.s.p. q.s.p. q.s.p. q.s.p. q.s.p. q.s.p. q.s.p. q.s.p.
Legenda. F = Formulação; q.s.= “Quantidade suficiente” para pH 5,5; Q.s.p. = “Quantidade suficiente para”.
5.2.3 Metodologia De Análise Físico-Química
5.2.3.1 Determinação Do Aspecto
Após prévia agitação da solução, observou-se cor, homogeneidade, turvação e
presença precipitado. As avaliações foram realizadas no dia do preparo do produto e
após o(s) período(s) de exposição indicado(s) nos estudos de estabilidade (BRASIL,
2010; PRISTA; ALVES; MORGADO, 1990).
5.2.3.2 Determinação do odor
Para determinação do odor, foram realizados movimentos circulares com a
solução, próximo ao nariz para avaliação olfativa. As avaliações foram realizadas
após o preparo do produto e nas datas determinadas nos testes de estabilidade
(BRASIL, 2010; PRISTA; ALVES; MORGADO, 1990).
5.2.3.3 Determinação do pH
As determinações potenciométrica do pH foram realizadas no potenciômetro
digital, previamente calibrado com as soluções tampão padrões. A determinação do
pH foi efetuada diretamente na amostra, procedendo-se três leituras consecutivas e

69
obtendo-se como resultado a média das leituras (BRASIL, 2010b). A amostra deve
apresentar pH entre 4 e 6 (USP, 2016).
5.2.3.4 Determinação da densidade de massa
Para determinação da densidade de massa das amostras (ρt), utilizou-se
picnômetro para líquidos limpo e seco, com capacidade para 25mL. Inicialmente,
pesou-se o picnômetro e, em seguida transferiu-se água destilada, recentemente
destilada e fervida, a 20°C para o picnômetro e registrou-se a massa encontrada.
Obteve-se a massa de água pela diferença entre as massas obtidas. Após o
picnômetro estar limpo e seco, transferiu-se a amostra e removeu-se o excesso da
substância, caso necessário, e pesou-se. Obteve-se a massa da amostra pela
diferença da massa do picnômetro cheio e vazio. Registrou-se a temperatura
encontrada da solução amostra e calculou-se a densidade relativa (d2020) pela razão
entre a massa da amostra e a massa da água (expressa em g/mL ou kg/L). A partir
desse resultado, encontrou-se a densidade de massa da amostra (BRASIL,2010).
ρt = d (água) x dtt + 0,0012 (Equação I)
Onde: ρt = densidade de massa
dtt = densidade relativa (ver equação II)
d (água) = densidade da água na temperatura registrada (ver tabela 2)
Tabela 05.Densidade da água de 20 a 30°C (adaptado de BRASIL, 2010).
TEMPERATURA (°C) DENSIDADE (g/L) TEMPERATURA (°C) DENSIDADE (g/L)
20 0,99820 25 0,99704
21 0,99799 26 0,99678
22 0,99777 27 0,99651
23 0,99754 28 0,99623
24 0,99730 29 0,99594

70
5.2.3.5 Determinação do teor do princípio ativo
A análise das amostras foi realizada por Cromatografia Líquida de Alta
Eficiência (CLAE) conforme metodologia de análise para solução oral de L-carnitina
descrita na Farmacopeia Americana (USP,2017). Todos os padrões utilizados para
preparo das soluções descritas abaixo foram pesados em balança analítica com
quatro casas decimais, os balões volumétricos utilizados foram devidamente
calibrados, as soluções padrões e soluções amostra foram avolumadas com água
ultra purificada, homogeneizadas e filtradas com filtro de poro 0,45µm. Os ensaios
foram realizados em triplicata.
5.2.3.5.1 PREPARO DE CURVA PADRÃO
Foi preparada solução padrão de L-carnitina 04 mg/mL e, a partir desta, foram
feitas as soluções para a curva padrão nas concentrações de 1,6, 1,8, 2,0, 2,2, 2,4 e
2,6 mg/mL. As alíquotas foram transferidas para balão volumétrico, avolumadas com
água ultrapurificada.e filtradas em filtro 0,45µm.
5.2.3.5.2 PREPARO DE SOLUÇÃO DE PADRÃO INTERNO
Preparou-se uma solução de ácido p-aminobenzóico com concentração final
de 0,02 mg/mL, avolumando-se com água ultrapurificada.
5.2.3.5.3 PREPARO DE SOLUÇÃO PADRÃO
Para preparo da Solução Padrão, transferiu-se 10,0 mg de L-carnitina para
balão volumétrico de 5,0 mL, adicionou-se 1mL da Solução de Padrão Interno e
completou-se o volume com água ultra purificada .
5.2.3.5.4 PREPARO DE SOLUÇÃO AMOSTRA ESTOQUE
Para o preparo das Soluções Amostras Estoques, preparou-se soluções finais
com concentração de ativo de 10,0 mg/mL. Desse modo, para análise de
formulações de L-carnitina a 10%, transferiu-se alíquota de 5,0 mL de amostra para
balão volumétrico de 50,0 mL e avolumou-se com água ultrapurificada. Já para
análise de formulações de L-carnitina a 20%, transferiu-se alíquota de 5,0 mL de

71
amostra para balão volumétrico de 100,0 mL e completou-se o volume com água
ultra purificada.
5.2.3.5.5 PREPARO DE SOLUÇÃO AMOSTRA
Para preparo da solução amostra, foram transferidos 5,0 mL de Solução
Amostra Estoque para balão volumétrico de 25,0 mL. Adicionou-se 5,0 mL de
Solução de Padrão Interno e avolumou-se com água ultrapurificada. As soluções
finais foram filtradas em filtro de porosidade 0,45 µm.
5.2.3.5.6 INFORMAÇÕES DE SISTEMA CROMATOGRÁFICO
As análises foram realizadas em Cromatógrafo Líquido utilizando coluna
cromatográfica C18 (30 cm x 3,9 mm, 10 µm), com fase móvel composta por uma
mistura de 555mg de 1-heptanosulfonato de sódio, 980mL de Tampão Fosfato
0,05M pH 2,4 e 20mL de Metanol, que foi filtrada através de membrana de celulose
regenerada de porosidade 0,45µm, sob vácuo. O volume de injeção foi de 40 µL,
com fluxo de 1,5mL/min, sistema mantido a temperatura ambiente e tempo de
corrida de 15 min. A detecção foi realizada com detector DAD, com monitoramento
no comprimento de onda de 225 nm (USP, 2018).
De acordo com a Farmacopeia dos Estados Unidos (USP 40, 2017), o teor de
L-Carnitina em soluções orais deve estar entre 90 e 110% do valor declarado.
A partir dos resultados encontrados para a curva padrão, foi determinada a
equação da reta e os valores de coeficiente de correlação (R), coeficiente angular
(a) e coeficiente de determinação (R2). O critério mínimo aceitável de coeficiente de
correlação (R) é de 0,99. A partir da equação da reta, obteve-se a concentração
nominal de L-carnitina nas soluções amostras (BRASIL, 2017a).
5.2.3.5.7 ADEQUAÇÃO DO SISTEMA
O tempo de retenção relativo para L-carnitina e para o ácido p-aminobenzóico
é, respectivamente, 0,56 e 1,0. A resolução entre os picos de L-carnitina e do padrão
interno não deve ser menor que 1,5. E o desvio padrão relativo (DPR) não deve ser
maior que 2,0% (USP, 2016).

72
5.2.4 Validação Parcial da Metodologia de Análise de Doseamento
A Farmacopeia dos Estados Unidos (USP 40° Edição) contempla em seu
conteúdo, a monografia correspondente à solução oral de L-carnitina. Os métodos
analíticos compendiais devem ter sua adequabilidade demonstrada ao uso
pretendido, necessitando passar por um estudo de validação parcial. Esse
procedimento visa garantir que os resultados obtidos sejam confiáveis e
reprodutíveis. A validação parcial deve avaliar, pelo menos, os parâmetros de
precisão, exatidão e seletividade (BRASIL, 2017a).
Para a realização da validação parcial do método de doseamento da L-
carnitina em solução oral, foram usados critérios estabelecidos pela Resolução RDC
N° 166, de 24 de julho de 2017 da Agência Nacional de Vigilância Sanitária
(ANVISA).
5.2.4.1 Precisão
A precisão de um método analítico expressa a proximidade entre os
resultados obtidos de uma série de medidas obtidas de uma mesma amostra
homogênea em condições determinadas (BRASIL, 2017a). Em geral, a precisão
pode ser definida em níveis de repetibilidade (mesmas condições em um curto
intervalo de tempo) e precisão intermediária (diferentes dias e/ou diferentes
analistas) (VALDERRAMA; BRAGA; POPPI, 2009).
Foram analisadas seis diferentes soluções de L-carnitina com concentração
de 10mg/mL. As análises foram realizadas sob as mesmas condições de operação,
mesmo analista e mesma instrumentação, em uma única corrida. O procedimento foi
realizado em dois dias diferentes.
A precisão foi demonstrada calculando-se o desvio padrão relativo (DPR) da
série de medições, conforme Equação III (BRASIL, 2017a).
DPR = DP x 100 (Equação III) CMD
Onde: DPR = desvio padrão relativo
DP = Desvio padrão
CMD = Concentração média determinada

73
5.2.4.2 Exatidão
A exatidão de um método analítico é a proximidade dos resultados obtidos
pelo método avaliado em relação ao valor verdadeiro. É expressa pela relação entre
a concentração média determinada experimentalmente e a concentração teórica
(BRASIL, 2017a).
Foram preparadas soluções padrões de L-carnitina nas concentrações de 1,6,
2,0 e 2,4 mg/mL, representando os níveis de concentração baixo (80%), médio
(100%) e alto (120%). Foram realizadas análises em triplicata para cada nível
mencionado. O cálculo da exatidão pode ser calculado pela equação IV (BRASIL,
2017a).
Exatidão = CME x 100 (Equação IV) Cteórica
Onde: CME = Concentração Média Experimental
Cteórica = Concentração teórica
5.2.4.3 Seletividade
Seletividade é a capacidade que o método possui em identificar ou quantificar
o analito de interesse na presença de outros componentes como impurezas,
produtos de degradação, diluentes e componentes da matriz.
Foi preparada solução padrão de L-carnitina na concentração de 2,0 mg/mL,
levada para analise e comparada com:
• Amostra preparada com matéria prima do ativo na mesma concentração;
• com solução contendo todos os componentes da formulação
desenvolvida;
• com solução contendo apenas o veículo das formulações (branco);
• com fase móvel.
Esses testes foram realizados para comprovar que os picos de interesse não
sofrem interferência com a presença de nenhum componente utilizado (BRASIL,
2017a). Para demonstrar ausência de interferência de produtos de degradação, foi
realizado estudo de degradação forçado (item 5.2.7) para expor a amostra a
condições de degradação em ampla faixa de pH, de oxidação, de calor e de luz.

74
5.2.4.4 Linearidade
A linearidade de um método deve demonstrar sua capacidade de obter
respostas analíticas proporcionais à concentração do analito em uma amostra dentro
de um intervalo específico (BRASIL, 2017a).
Foi desenvolvida uma curva de calibração onde o sinal analítico denominado
variável dependente y é linearmente proporcional à sua concentração, denominada
variável independente x. Com os resultados obtidos obteve-se a equação da reta
com estrutura definida como (RIBEIRO et al., 2008):
y = ax + b (Equação V)
Onde: y = variável referente ao sinal analítico
a = Coeficiente angular
x = variável referente à concentração da amostra (mg/mL)
b = Coeficiente linear
Para preparo de três curvas de calibração, foram preparadas soluções
padrões de L-carnitina, em dias diferentes, nas concentrações de 1,6, 1,8, 2,0, 2,2,
2,4 e 2,6mg/mL. Os resultados foram tratados por métodos estatísticos para
determinação da equação da reta, do coeficiente de correlação (r) e do coeficiente
angular (a). O critério mínimo aceitável para o coeficiente de correlação (r) é de
0,990 e o coeficiente angular (a) deve ser significativamente diferente de zero. A
partir das curvas de calibração, estimou-se o limite de detecção (LD) e o limite de
quantificação (LQ) a aplicando as equações VI e VII (BRASIL, 2017a):
• Limite de detecção
LD = (3,3 x DP) (Equação VI) IC
• Limite de quantificação
LQ = (DP x 10) (Equação VII) IC

75
Onde: DP = desvio padrão do intercepto com o eixo Y das 3 curvas de calibração
IC = inclinação da curva de calibração
5.2.4.5 Faixa de trabalho
A faixa de trabalho foi estabelecida determinando o limite de quantificação
inferior e superior do método validado. Neste caso, o intervalo utilizado foi de 80 a
120% da concentração teórica do teste. A faixa foi estabelecida uma vez que o
método apresenta exatidão, precisão e linearidade adequada quando aplicados a
amostras de concentração dentro do intervalo especificado (BRASIL, 2017a).
5.2.5 Metodologia de Análise Microbiológica
As análises microbiológicas das formulações desenvolvidas foram realizadas
em parceria com a Professora Doutora Helena Keiko Toma, coordenadora do
Laboratório de Controle de Qualidade de Medicamentos, Alimentos e Cosméticos
(LACMAC) da Faculdade de Farmácia da UFRJ. As formulações foram submetidas
aos ensaios para soluções orais não estéreis, definidos pela Farmacopéia Brasileira
5° Edição, que conta com as análises de contagem total de bactérias aeróbicas,
contagem total de bolores e leveduras e pesquisa de patógenos (Escherichia coli).
5.2.5.1 Análise microbiológica
Higienizou-se a parte externa do frasco a ser analisado com álcool 70%. Em
fluxo laminar, transferiu-se 10 mL da amostra para 90 mL de solução de diluição
(tampão fosfato - pH 7,2) e ajustou-se o pH da solução final para 7 com HCl 0,1 M
ou NaOH 0,1 M (Solução 1:10). Repetiu-se o procedimento, transferindo 10 mL da
solução recém-preparada para 90 mL de solução de diluição, fazendo o ajuste
necessário para manter o pH final em 7 (solução 1:102). Repetiu-se o procedimento
para obter a solução 1:103. Esses meios foram mantidos por 24horas para
fortificação. Após esse período, transferiu-se 0,1 mL das soluções preparadas para
placas de Petri contendo 15mL de meio de cultura adequado para cada análise
(Tabela 06). Incubou-se as placas conforme temperatura e período indicado para

76
cada análise, apresentados na Tabela 06. Tomou-se a média aritmética das placas
de cada meio e calculou-se o número de UFC/mL do produto.
Tabela 06. Características de incubação dos testes microbiológicos
TESTE MEIO DE CULTURA TEMPERATURA TEMPO
Contagem total de bactérias aeróbias
Ágar caseína-soja 32,5ºC ± 2,5°C Até 7 dias
Ágar Sabouraud-dextrose 22,5°C ± 2,5°C Até 5 dias
Leveduras e fungos Ágar Sabouraud-dextrose 22,5ºC ± 2,5°C 5 a 7 dias
Patógeno específico (E.coli) Caldo MacConkey 32,5ºC ± 2,5°C 18 – 72 h
5.2.6 Estudo de Estabilidade
5.2.6.1 Estudo Prévio de Estabilidade
As amostras desenvolvidas no item “5.2.2 Desenvolvimento das formulações”
que não apresentaram incompatibilidades farmacotécnicas, foram encaminhadas
para estudo de estabilidade prévio. Nessa etapa elas foram avaliadas pelas análises
físico-químicas de aspecto, odor e pH, descritas no item “5.2.3 Metodologia de
Análise Físico-química”, e pelas análises microbiológicas descritas no item “5.2.5
Metodologia de Análise Microbiológica”. Os tempos de análises realizados foram:
após 15, 30 e 45 dias das formulações serem mantidas em estufa a temperatura
constante de 40°C. Como a estabilidade microbiológica era o maior desafio, esse
estudo prévio teve como objetivo identificar a formulação com melhor e maior tempo
de estabilidade microbiológica, que foi encaminhada para estudo de estabilidade
completo. Nessa etapa não foi avaliado o teor de L-carnitina nas formulações.
5.2.6.2 Estudo de Estabilidade Completo Em Uso
Lotes de bancada de 4 L foram produzidos de acordo com o modo de preparo
descrito no item “5.2.2 Desenvolvimento das formulações”. No momento do envase,
2.000 mL da formulação preparada foram envasados em frascos PET e os outros
2.000 mL foram envasados em frascos de vidro âmbar. As soluções foram divididas

77
e expostas em temperaturas controladas de 5°C, 25°C e 40°C e analisadas nos dias
0 (após a manipulação das formulações), 15, 30 e 45. Foram realizadas análises
físico-químicas e microbiológicas.
Por ser um produto de embalagem multidose, o estudo de estabilidade
realizado foi do tipo “em uso”, onde, a partir de um mesmo frasco alíquotas diárias
de 1,0 mL eram retiradas e desprezadas para simulação de uso do produto. Desse
frasco, foram retiradas alíquotas para realização das análises físico-químicas e
microbiológicas.
Foram produzidas também, soluções com a mesma composição da amostra
excetuando-se o princípio ativo, denominadas de “Amostras em Branco”, e elas
foram utilizadas apenas no teste de Teor. E ainda, foi produzida uma formulação
sem a presença de conservantes microbianos em sua composição, sendo
denominada de “Formulação Controle” para o estudo microbiológico.
As análises físico-químicas foram realizadas em triplicatas verdadeiras,
quando são retiradas três alíquotas a partir de um mesmo recipiente. Um resumo do
estudo de estabilidade está descrito na Tabela 7.
Tabela 07. Esquema das condições de exposição das amostras para avaliação da estabilidade.
Amostra Embalagem Temperatura Análise Tempo de
análise
Amostra 1
Branco 1¹
Amostra 2
Branco 2¹
Controle
PET
Vidro âmbar
5°C
25°C
40°C
Físico-Química
Microbiológica
0 dias ²
15 dias
30 dias
45 dias
Legenda.¹Amostras em Branco - apenas para análise de teor (FIQ); ² Após a manipulação das
formulações.
5.2.7 Estudo de Degradação Forçada
A RDC 53 de 2015 indica que os testes de degradação forçada devem
promover uma degradação maior que 10% da amostra e inferior àquela que
degradaria completamente a amostra. Baseado no Informe Técnico N° 01 de 2008,

78
as soluções foram testadas com igual volume de solução degradante. Para hidrólise
ácida foi adicionado 25mL de ácido clorídrico 0,1 N a 25mL de formulação. Para
hidrólise básica, 25mL de formulação foram misturadas com 25mL de hidróxido de
sódio 0,1N. Para avaliação da degradação térmica, a formulação foi mantida sob
aquecimento a 60°C. Todas as formulações foram mantidas sob agitação nas
condições especificadas por 24h. Em paralelo foi deixada uma solução padrão de L-
carnitina na mesma concentração da amostra sob agitação por 24 horas para
controle.
Por não apresentar degradação superior a 10%, foi realizada uma segunda
etapa do estudo, com base no trabalho de Khoshkam e Afshar (2014) onde 25,0 mL
da formulação e 25,0 mL de solução de ácido clorídrico 1M foram misturados e
aquecidos a 70°C por 24 horas com agitação constante e em sistema vedado. Ao
final do aquecimento, as amostras foram neutralizadas até o pH 7. O mesmo
procedimento foi realizado com 25 mL de amostra e 25 mL de solução de hidróxido
de sódio 1M para exposição à hidrólise básica. Para avaliação de degradação
oxidativa, 25 mL de solução amostra foram misturadas com 25 mL de solução de
peróxido de hidrogênio a 3% e foi mantida a temperatura ambiente por 4 horas,
protegidas da luz. Para avaliação da exposição ao calor, a amostra foi aquecida a
70°C por 12 e 24 horas sob agitação constante e em sistema vedado.
As soluções foram diluídas com água ultrapura até a concentração final de
10mg/mL, filtradas com filtro 0,45µm e analisadas em triplicata por CLAE para
determinação do teor de L-Carnitina e avaliação da existência de possíveis produtos
de degradação.
5.2.8 Análise de dados
Os resultados experimentais obtidos foram expressos como a média dos
resultados acompanhados do desvio padrão (DP). A análise estatística de dados foi
realizada usando o software Excel® (2010) aplicando-se o teste t de Student para
comparação de dois grupos ou ANOVA (AnalysisofVariance) para comparação de
três ou mais grupos, considerando-se o nível de significância de 0,05 (p < 0,05).

79
6 RESULTADOS E DISCUSSÃO
6.1 Análise de identificação da L-Carnitina
Foram realizadas análises por Espectroscopia de Infravermelho por
Transformada de Fourier (IV-TF) tanto do padrão, quanto da matéria-prima de L-
carnitina. Os espectros obtidos estão apresentados nas figuras 12 e 13, onde foram
sinalizadas as bandas características da molécula de L-carnitina (Figura 14).
Figura 12. Espectro referente à análise de IV-TF do padrão de L-carnitinacom a
demarcação das principais bandas (a -h).
Figura 13.Espectro referente à análise de IV-TF da matéria-prima de L-carnitina com a demarcação das principais bandas (a - h).
Pode-se observar que o perfil encontrado em ambos os espectros são
semelhantes e a identidade molecular da matéria-prima é a mesma que a do padrão,
e ambas estão coerentes com o encontrado na literatura (Tabela 08).

80
Figura 14. Principais bandas presentes no espectro IV-TF representadas na estrutura da L-
Carnitina.
Tabela 08. Principais bandas encontradas em espectro de IV-FT de L-carnitina
(adaptado de KIEFER; MAZZOLINI; STODDART, 2007 e 姜波et al, 2013).
IDENTIFICAÇÃO BANDA (cm-1) GRUPO CORRESPONDENTE
a 3461 O - H
b 1582 COOH
c 1481 CH2
d 1394 COOH
e 1147 C - COOH
f 1097 C - N+
g 975, 944, 903 C – N – (CH3)3
h 875 CH2
Com a identificação e caracterização da L-carnitina presente no padrão e na
matéria-prima que foi utilizada no desenvolvimento das formulações, as próximas
etapas puderam ser realizadas sabendo da integridade da amostra na etapa inicial.
Antes de iniciar o desenvolvimento das formulações, foi necessário desenvolver a
metodologia de análise e realizar o processo de validação parcial do doseamento do
ativo.
g h a c b,d
f
e

81
6.2 Validação Parcial da Metodologia de Doseamento
A quantificação da L-carnitina foi realizada por CLAE conforme descrito na
monografia de “L-carnitina solução oral” da Farmacopeia dos Estados Unidos
(USP,2017). E, de acordo com a RDC N°166, de 24 de Julho de 2017, da Agência
Nacional de Vigilância Sanitária (ANVISA), os métodos analíticos compendiais
devem ter sua adequabilidade comprovada por meio de validação parcial, que exige,
no mínimo, os parâmetros de precisão, exatidão e seletividade. Além desses, foram
avaliados também a especificidade, linearidade e faixa de trabalho, limite de
detecção e limite de quantificação.
6.2.1 Determinação da faixa de trabalho
A determinação da faixa de trabalho é estabelecida determinando-se a
concentração do analito de interesse em diferentes níveis de concentração. A partir
dos resultados obtidos, pode-se determinar a faixa de concentração na qual os
resultados apresentam nível de incerteza para o método empregado. Conforme
descrito na monografia para Solução Oral de L-carnitina (USP 40), a concentração
teórica do teste de teor é de 2mg/mL e, o intervalo utilizado para a validação foi de
80% a 120% da concentração teórica, sendo então a faixa de trabalho de 1,6 mg/mL
a 2,4mg/mL. A faixa de trabalho será confirmada a partir do estudo de linearidade.
6.2.2Seletividade
Seletividade é a capacidade que o método possui de identificar ou quantificar
o analito de interesse na presença de outros componentes. Para demonstrar a
ausência de interferentes, as análises foram realizadas com espectro de varredura
entre 190 e 800nm, com monitoramento no comprimento de onda de 225nm,
definido na monografia. Foram realizadas análises do solvente utilizado (água
ultrapurificada), da solução de L-carnitina a partir do padrão e da solução de L-
carnitina a partir da matéria-prima, de uma solução contendo componentes da
formulação a ser desenvolvida e de uma solução contendo apenas o veículo das
formulações (solução em branco).

82
Foi realizada a análise do solvente utilizado (água ultrapurificada) para diluir
as amostras do teste de doseamento e da fase móvel. A análise de ambos é
importante para comprovar que o solvente e a fase móvel utilizados não interferem
na resposta das análises, o que pode ser observado nos cromatograma abaixo
(Figura 15 e 16) onde nenhum pico de absorção é observado no comprimento de
onda de análise.
Figura 15. Cromatograma do solvente (água ultrapurificada).
Figura 16. Cromatograma da Fase móvel.
Uma solução de L-carnitina padrão foi analisada na concentração de 2,0
mg/mL utilizando água ultrapurificada como solvente. O tempo de retenção
encontrado foi de 2,938 minutos e não apresentou nenhum interferente em seu
cromatograma (Figura 17):

83
Figura 17. Cromatograma da L-carnitina padrão a 2,0 mg/mL (a).
O uso do detector de arranjo de diodo (DAD) permite mais uma forma de
avaliar a pureza do pico cromatográfico. Foi obtido então, o espectro de varredura
do pico cromatográfico correspondente à L-carnitina. Com essa avaliação pode-se
dizer que não há outra substância absorvendo nos comprimentos de onda. No
espectro (Figura 18), observa-se o mesmo perfil de espectros em todos os
comprimentos de onda, indicando não haver interferente no pico obtido.
Figura 18. Varredura de todos os comprimentos de onda do pico da L-carnitina padrão a 2,0 mg/mL.
No preparo das soluções para análise de teor, foi adicionado o ácido p-
aminobenzóico como padrão interno. As soluções de L-carnitina padrão e de L-
carnitina matéria-prima foram analisadas com a adição do padrão interno e seus
cromatogramas podem ser observados nas figuras 19 e 20.
Figura 19. Cromatograma da solução com padrão de L-carnitina (a) e padrão interno (b).

84
Figura 20. Cromatograma de solução com matéria-prima de L-carnitina (a) e padrão interno (b).
O tempo de retenção da L-carnitina na solução contendo padrão foi 2,926
minutos e o encontrado para o padrão interno foi 4,377 com resolução de 1,995. Já
no cromatograma com a L-carnitina matéria-prima, o tempo de retenção encontrado
foi de 2,927 minutos para a L-carnitina e 4,383 para o padrão interno, com resolução
de 2,004. A resolução entre os picos nos dois cromatogramas foi maior que 1,5,
mínimo aceitável especificado na monografia, indicando uma boa separação entre
os analitos. Não foram observados interferentes na análise cromatográfica.
Para avaliar o impacto da presença dos excipientes usados no
desenvolvimento da formulação, foi analisada uma solução contendo ácido cítrico a
40,0 µg/mL, padrão interno a 40,0 µg/mL, benzoato de sódio a 60,0 µg/mL e
carboximetilcelulose a 30,0 µg/mL. Ao filtrar a solução final, o CMC ficou retiro na
membrana e não é observado no cromatograma obtido, que pode ser visualizado na
figura 21.
Figura 21. Cromatograma dos excipientes usados no desenvolvimento da formulação (ácido cítrico
(a) e benzoato de sódio (d)), em presença do padrão interno (b).

85
A solução analisada acima foi contaminada com L-carnitina para avaliação da
interferência entre os componentes da formulação. O cromatograma obtido pode ser
visualizado na figura 22.
Figura 22. Cromatograma da L-carnitina (b) em presença do padrão interno (c) e excipientes usados
no desenvolvimento da formulação: ácido cítrico(a) e benzoato de sódio (d).
Observando os cromatogramas das figuras 21 e 22, pode-se observar que os
picos da L-carnitina e do padrão interno não sofreram interferência com a presença
dos excipientes adicionados. Os tempos de retenção encontrados também não
apresentaram alteração e houve boa separação entre os componentes da
formulação com valores de resolução maiores que 1,5 (R = 1,895, R = 1,992 e R =
4,803 respectivamente). Buscando avaliar a pureza do pico da L-carnitina foi obtido
seu espectro de varredura (figura 23) e comparado com o espectro do padrão de L-
carnitina apresentado na figura 18.
Figura 23. Comparação entre o espectro de varredura do pico cromatográfico correspondente à L-
carnitina na solução padrão(d) e na solução com os excipientes (e).
Com os resultados obtidos, pode-se afirmar que o método detectou o analito
de interesse na presença de outros componentes que podem estar presente nas
amostras, apresentando então especificidade e seletividade.

86
6.2.3 Linearidade
Foram feitas injeções em triplicata de soluções de L-carnitina de
concentrações: 1,6, 1,8, 2,0, 2,2, 2,4 e 2,6 mg/mL. A partir do resultado foi
construída curva padrão linear e obtida a equação da reta de regressão de y em x (y
= ax + b), estimada pelo método dos mínimos quadrados. A criação da curva de
calibração é importante para avaliar a associação linear entre as variáveis
apresentadas. Para isso, alguns coeficientes foram obtidos a partir da curva de
calibração: o coeficiente angular (a), o coeficiente linear (b), o coeficiente de
determinação (r²) e o coeficiente de correlação (r). Conforme preconizado na RDC
N° 166, de 24 de julho de 2017, o valor de r para uma correlação linear adequada
deve ser maior que 0,99.
As injeções foram repetidas em três dias diferentes, a partir de soluções
diferentes para cada dia, e então, foram obtidas três curvas que podem ser
visualizadas nas figuras 24, 25 e 26. O resumo dos resultados obtidos estão
descritos na tabela 09.
Tabela 09. Parâmetros aplicados no teste de linearidade
PARÂMETRO DIA 01 DIA 02 DIA 03 MÉDIA DP
Coeficiente Angular (a) 228078 233595 229904 230526 2811
Coeficiente Linear (b) -31949 -46294 -6030,1 -28091 20407
Coeficiente de correlação (r) 0,9998 0,9996 0,9997 0,9997 0,0001
Coeficiente de determinação (R²) 0,9995 0,9995 0,9996 0,9995 0,0001
Legenda: DP – Desvio padrão.

87
Figura 24. Curva padrão da L-carnitina a partir da média de valores
encontradas no primeiro dia de análise.
Figura 25. Curva padrão da L-carnitina a partir da média de valores
encontradas no segundo dia de análise.
Figura 26. Curva padrão da L-carnitina a partir da média de valores
encontradas no terceiro dia de análise.

88
A partir dos resultados obtidos, pode-se calcular o limite de detecção e o
limite de quantificação. Esses dados são importantes para definição da faixa de
trabalho. O limite de detecção (LD) demonstra a menor quantidade do analito que
pode ser detectada pelo método proposto, mas não necessariamente ser
quantificada. Por isso é calculado também o limite de quantificação (LQ), que
informa qual é a menor quantidade do analito que pode ser quantificada pelo
método, com precisão e exatidão aceitáveis. Os valores usados para o calculo e os
resultados obtidos foram apresentados na tabela 10.
Tabela 10. Valores obtidos para LD e LQ.
PARÂMETRO MÉDIA DOS VALORES
Desvio padrão dos valores de “b”(intercepto com o eixo Y)
20407
Inclinação da curva de calibração (IC) (média dos valores encontrados para “a”) 230526
Limite de Detecção 0,29 mg/mL
Limite de Quantificação 0,88 mg/mL
Desse modo, a faixa de trabalho proposta no item “6.2.1. Faixa de trabalho”
foi adequada por estar acima do limite de detecção e de quantificação, e o método
apresenta linearidade demonstrando sua capacidade de obter resposta analítica
diretamente proporcional à concentração do analito.
6.2.4 Precisão
A precisão do método analítico expressa a proximidade entre os resultados
obtidos na metodologia pretendida e pode ser considerada no nível de repetibilidade,
de precisão intermediária ou de reprodutibilidade. A repetibilidade avalia as
amostras sob as mesmas condições de operação, mesmo analista, mesmas
condições ambientais, em uma única corrida analítica (BRASIL, 2017a). Foram
então preparadas seis amostras de L-carnitina 20%, que foram diluídas até a
concentração 2,0 mg/mL e realizadas seis injeções em mesma corrida analítica.
Esse processo foi repetido no dia seguinte onde seis injeções foram realizadas em
uma mesma corrida, sob as mesmas condições ambientais e instrumentais, e com o
mesmo analista. Foi calculado o desvio padrão relativo dos resultados obtidos, que
expressam a capacidade de repetibilidade do método.
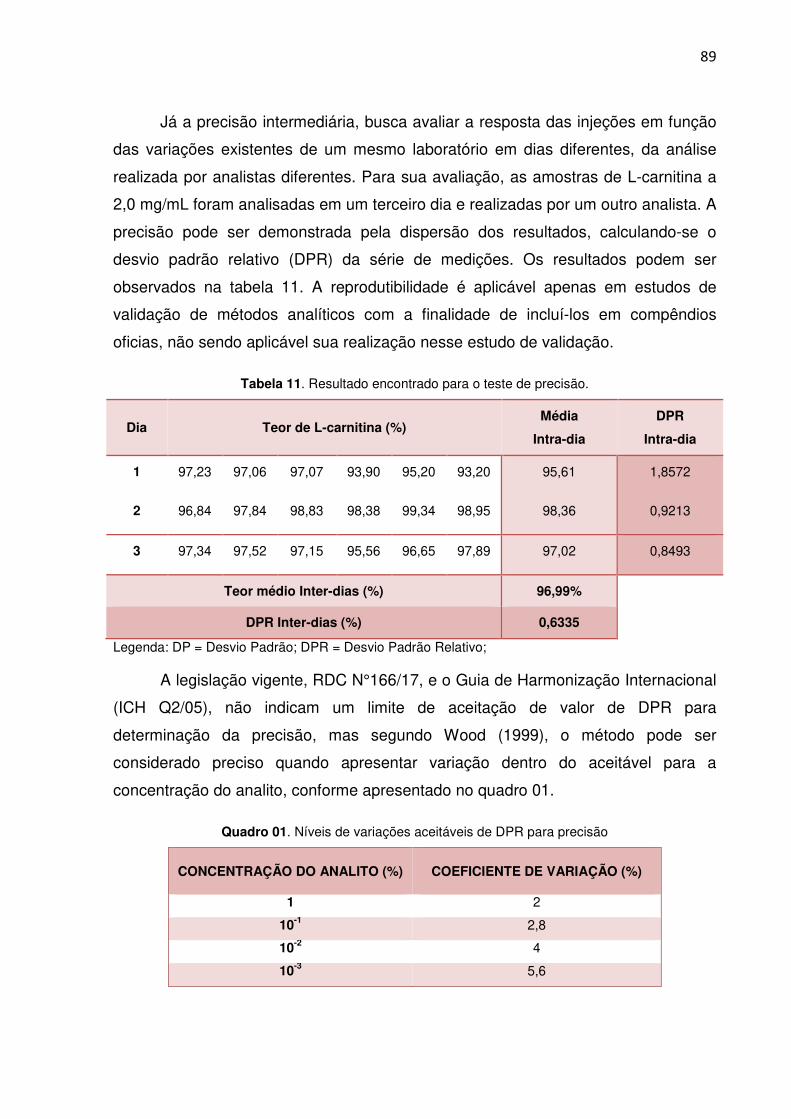
89
Já a precisão intermediária, busca avaliar a resposta das injeções em função
das variações existentes de um mesmo laboratório em dias diferentes, da análise
realizada por analistas diferentes. Para sua avaliação, as amostras de L-carnitina a
2,0 mg/mL foram analisadas em um terceiro dia e realizadas por um outro analista. A
precisão pode ser demonstrada pela dispersão dos resultados, calculando-se o
desvio padrão relativo (DPR) da série de medições. Os resultados podem ser
observados na tabela 11. A reprodutibilidade é aplicável apenas em estudos de
validação de métodos analíticos com a finalidade de incluí-los em compêndios
oficias, não sendo aplicável sua realização nesse estudo de validação.
Tabela 11. Resultado encontrado para o teste de precisão.
Dia Teor de L-carnitina (%) Média
Intra-dia
DPR
Intra-dia
1 97,23 97,06 97,07 93,90 95,20 93,20 95,61 1,8572
2 96,84 97,84 98,83 98,38 99,34 98,95 98,36 0,9213
3 97,34 97,52 97,15 95,56 96,65 97,89 97,02 0,8493
Teor médio Inter-dias (%) 96,99%
DPR Inter-dias (%) 0,6335
Legenda: DP = Desvio Padrão; DPR = Desvio Padrão Relativo;
A legislação vigente, RDC N°166/17, e o Guia de Harmonização Internacional
(ICH Q2/05), não indicam um limite de aceitação de valor de DPR para
determinação da precisão, mas segundo Wood (1999), o método pode ser
considerado preciso quando apresentar variação dentro do aceitável para a
concentração do analito, conforme apresentado no quadro 01.
Quadro 01. Níveis de variações aceitáveis de DPR para precisão
CONCENTRAÇÃO DO ANALITO (%) COEFICIENTE DE VARIAÇÃO (%)
1 2
10-1 2,8
10-2 4
10-3 5,6

90
As amostras analisadas apresentavam concentração equivalente a 2,0 x 10-
1g/100mL. Dessa forma, a partir dos resultados apresentados no teste de precisão,
com valores menores que 2,8%, pode-se considerar que o método é preciso
(WOOD, 1999).
6.2.5 Exatidão
A determinação da exatidão do método analítico foi obtida a partir da análise
de soluções com três diferentes concentrações contemplando o intervalo linear do
método analítico. As concentrações deveriam contemplar a faixa linear baixa, média
e alta e foram realizadas em triplicata. Foram usados os valores de 80% (baixa),
100% (média) e 120% (alta concentração) da faixa de trabalho, equivalente a 1,6
mg/mL, 2,0 mg/mL e 2,4mg/mL (BRASIL, 2017b).
As amostras foram preparadas de maneira independente, a partir uma mesma
solução mãe. O teor das amostras foi calculado, assim como o valor de DPR para
cada nível trabalhado (baixo, médio e alto). Os valores podem ser observados na
tabela 12.
Tabela 12. Resultados obtidos para o teste de exatidão
Nível
Concentração
média Teórica
(mg/mL)
Conc. média
Experimental
(mg/mL)
(%) Média
(%) DP DPR (%)
80%
(1,6 mg/mL)
1,6176 1,6072 99,36
99,77 1,08 1,08 1,6272 1,6101 98,95
1,6088 1,6247 100,99
100%
(2,0 mg/mL)
2,0220 2,0129 99,55
99,96 0,81 0,81 2,0340 2,0226 99,44
2,0110 2,0289 100,89
120%
(2,4 mg/mL)
2,4264 2,4179 99,65
99,32 0,29 0,29 2,4408 2,4205 99,17
2,4132 2,3924 99,14
A exatidão do método analítico busca refletir a proximidade entre o valor
verdadeiro ou de referência (obtido a partir de um padrão de pureza conhecida) e o

91
valor medido ou experimental (resultado da análise do método em processo de
validação), mostrando sua capacidade de obter o resultado mais próximo ao
verdadeiro. Observando os resultados obtidos, os valores médios de teor
encontram-se dentro da faixa de 98,0 a 102,0%, com baixo desvio padrão entre eles,
pequena amplitude e DPR menor do que 5,0%. Dessa maneira, considerou-se que o
método analítico apresenta exatidão adequada.
Com todos os parâmetros avaliados, pode-se concluir que o método analítico
apresenta desempenho adequado para as condições que foi aplicado, fornecendo
resultados confiáveis e não distorcidos, e atendendo às normas estabelecidas por
agências reguladoras. O método analítico em questão encontra-se validado e apto
para ser utilizado nas análises físico-químicas a seguir.
6.3 Desenvolvimento das formulações
Para desenvolvimento da formulação foi proposta a utilização do ativo,
conservante, agente de viscosidade e acidulante. Inicialmente selecionou-se o
metilparabeno como um dos conservantes a ser usado nas formulações testes. Ele é
o conservante com melhor aceitação no mercado farmacêutico e apresenta largo
espectro de ação, sendo eficiente contra bactérias gram positivas, bactérias gram
negativas, fungos e leveduras. É insípido, incolor, inodoro e é estável em uma ampla
faixa de pH, mas apresenta solubilidade relativa em meios aquosos necessitando de
aquecimento para sua solubilização em água. De acordo com SILVA et al., 2008,
numerosos casos de reações adversas foram associados à sua utilização e é
considerado um excipiente de risco quando utilizado em soluções orais. Ele será
testado e sua resposta será avaliada e comparada com a resposta dos outros
agentes conservantes. Dependendo de sua relação risco/benefício ele poderá ser
descartado (BALBANI; STELZER; MONTOVANI, 2006; DA SILVA et al., 2008;
FERREIRA; SOUZA, 2011a; SONI et al., 2001).
Outro agente conservante selecionado foi o benzoato de sódio, que apresenta
eficiente ação contra bolores, leveduras e bactérias gram positivas. Além disso,
apresenta boa solubilidade em água à temperatura ambiente (25°C), eliminando a
necessidade de aquecimento da formulação. Porém, sua efetividade é

92
potencializada em meio ácido, na faixa de pH de 2 a 5. Embora seja considerado
seguro quando usado em concentrações inferiores a 0,6%, também é apontado
como excipiente relacionado a reações adversas (VILAPLANA & ROMAGUERA,
2004).
O ácido sórbico e sorbato de potássio também são considerados potentes
inibidores de bolores e leveduras, mas possuem pouca ou nenhuma efetividade na
inibição de bactérias, importante microrganismo a ser inibido na formulação. O ácido
sórbico apresenta baixa solubilidade em meios aquosos enquanto que o sorbato de
potássio possui boa solubilidade em água. Quando administrados em longo prazo,
podem ocasionar reações alérgicas e sintomas como coceira, congestão nasal, dor
abdominal, diarreia, problemas gástricos, enxaqueca e vômito. Além disso, podem
causar deficiência nutricional e redução de absorção de vitaminas, minerais e
nutrientes, o que pode ser problemático para o público alvo.Com o perfil de
pacientes com patologias associadas à EIM e necessitando de multidoses diárias, a
formulação será de uso contínuo e esses fatores podem ser agravados. Dessa
forma, por não apresentar ação bactericida, o sorbato de potássio não foi testado
(NUNN; WILLIAMS, 2005; SENA et al., 2014; SWEETMAN, 2009).
A influência do pH também é um fator determinante para a estabilidade da
formulação. O ativo L-carnitina apresenta estabilidade em solução na faixa de pH 4 a
6 e os agentes conservantes escolhidos necessitam de pH acidificado para terem
sua ação potencializada e efetiva. Dessa forma, o ajuste de pH da formulação será
realizado pela adição de ácido cítrico e deve ser estabilizado em pH 5,0. O ácido
cítrico foi escolhido por apresentar atividade antimicrobiana devido à sua
acidificação do meio e por sua propriedade em quelar íons metálicos, o que diminui
o crescimento bacteriano. Apesar de apresentar baixa eficiência contra bolores e
leveduras, seu uso associado a outro sistema conservante é indicado, pois o poder
antimicrobiano é mais efetivo e ele potencializa a ação dos outros agentes
conservantes (MAX et al., 2010).
Muitos pacientes portadores de EIM apresentam dificuldades de deglutição,
principalmente quando as soluções possuem baixa viscosidade. Além disso, muitos
pacientes possuem sondas de gastrostomia, sendo necessária a administração do
medicamento pela sonda. O aumento da viscosidade da formulação ajuda na

93
administração da solução nestes dois casos. Além disso, quando a solução é
ingerida, apenas uma parte do fármaco dissolvido na formulação entra em contato
com as papilas gustativas do paciente, enquanto que o restante é carreado pela
solução viscosa, mascarando o sabor da formulação. Dessa forma, com a finalidade
de proporcionar um leve aumento da viscosidade da formulação, foi utilizado um
agente promotor de viscosidade (BILLANY, 2005).
A sacarose, amplamente utilizada no mercado farmacêutico, tem baixo custo,
não deixa gosto residual e, além de fornecer sabor adocicado e viscosidade a
medicamentos líquidos, também pode agir como conservante e antioxidante, porém
não pode ser utilizada para o público alvo que apresenta restrições ao seu consumo
(BALBANI; STELZER; MONTOVANI, 2006). Outra opção comumente utilizada é a
goma xantana, mas sua utilização não é recomendada em formulações voltadas
para crianças menores de um ano de idade, especialmente para crianças
prematuras. McCallum (2011) e Woods et.al (2019), acreditam que a goma xantana,
quando utilizada a longo prazo em crianças prematuras, pode alterar a microbiota no
intestino ainda prematuro dos bebês. Alguns relatos apontam casos fatais como a
enterocolitenecrosante. Portanto, por precaução, não é recomendado o uso da goma
para esse público e ela não foi testada(MCCALLUM, 2011; WOODS, 2019).
A Hidroxietilcelulose e a carboximetilcelulose são agentes de viscosidade que
não são convertidos em glicose após sua ingestão, tornando-se bons candidatos ao
uso. Normalmente utilizada em soluções tópicas ou oftálmicas, estudos apontam a
utilização de hidroxietilcelulose para formulações de uso oral. Ela apresenta boa
estabilidade em diferentes faixas de pH, no entanto, as soluções com pH abaixo de
5 são menos estáveis podendo intensificar reações de hidrólise. Além disso,
necessita de longo tempo para sua dispersão completa (em média ao menos uma
hora com aquecimento)(ROWE; SHESKEY; QUINN, 2009).Optou-se então pela
Carboximetilcelulose (CMC), um derivado hidrossolúvel de celulose, amplamente
utilizado em formulações orais, que apresenta boa estabilidade na faixa de pH entre
4 e 10. A faixa de concentração normalmente utilizada é de 0,50 a 2,0% (p/v), mas
usando a concentração de 0,3% (p/v) obteve-se o resultado esperado e, por estar
com concentração abaixo da usual, o aparecimento de reações indesejáveis aos
pacientes pode ser reduzido (ROWE; SHESKEY; QUINN, 2009).

94
Para determinação da concentração de L-carnitina a ser utilizada no
desenvolvimento da formulação, levou-se em consideração o fato da formulação ser
de uso contínuo, ter a dose calculada a partir do peso corporal do paciente e que
constantemente sofre ajustes, a concentração do ativo foi definida para ser flexível e
atender às diferentes dosagens apresentadas pelos pacientes. Com o objetivo de
fornecer uma concentração que atenda à maioria das prescrições recebidas na FU
foram desenvolvidas formulações de L-carnitina a 10% (p/v) e 20% (p/v), podendo
atender pacientes pediátricos, que necessitam de volumes menores, e pacientes
adultos, que necessitam de grandes volumes. As soluções iniciais desenvolvidas
para caracterização foram realizadas com a concentração de 10% (p/v) e estão
apresentadas na tabela 13:
Tabela 13. Composição das formulações testes a 10% (p/v)
COMPONENTES FORMULAÇÃO
1
FORMULAÇÃO
2
FORMULAÇÃO
3
FORMULAÇÃO
4
L-carnitina 10% 10% 10% 10%
Benzoato de sódio 0,3% 0,4% - 0,3%
Metilparabeno - - 0,1% -
Ácido Cítrico q.s. pH 5,5 q.s. pH 5,5 q.s. pH 5,5 0,9%
pH 5,5 5,5 5,5 5,0
Carboximetilcelulose 0,3% 0,3% 0,3% 0,3%
Água destilada q.s.p. q.s.p. q.s.p. q.s.p.
Legenda.q.s.= “Quantidade suficiente”; Q.s.p. = “Quantidade suficiente para”.
Todas as formulações foram analisadas inicialmente quanto às suas
características físicas. Analisou-se o aspecto visual, odor e pH das formulações e
buscou-se avaliar a existência de alguma incompatibilidade farmacotécnica após a
manipulação das formulações. O resultado dessas análises pode ser visualizado na
tabela 14.
Tabela 14. Resultado da análise FIQ inicial das formulações a 10% (p/v)
Análise Aspecto Odor pH
Formulação 1 Límpidas, incolores, sem a
presença de partículas e
sem grumos.
Aroma quase
imperceptível,
agradável.
5,60
Formulação 2 5,61
Formulação 3 5,74
Formulação 4 5,03

95
6.5 Estabilidade prévia das formulações iniciais
Após a análise inicial, observou-se que todas as formulações apresentavam
boa compatibilidade farmacotécnica e foram encaminhadas para estudo de
estabilidade prévio. As formulações foram armazenadas em estufa com temperatura
controlada a 40°C. Foram encaminhadas amostras para análise físico-química (FIQ)
(exceto a análise de teor) e microbiológica (MIC) após 15, 30 e 45 dias de
exposição. O resultado físico-químico pode ser observado na tabela 15 e o resultado
microbiológico, na tabela 16.
Tabela 15. Resultado das análises FIQ da estabilidade prévia das formulações a 10% (p/v)
Formulação 1 Teste 15 dias 30 dias 45 dias
Carnitina 10% Aspecto Límpido e incolor Límpido e incolor Límpido e incolor
Benzoato 0,3% Odor Característico Característico Característico
pH 5,5 pH 5,69 ± 0,07 5,74 ± 0,14 5,87± 0,11
Formulação 2 Teste 15 dias 30 dias 45 dias
Carnitina 10% Aspecto Límpido e incolor Límpido e incolor Límpido e incolor
Benzoato 0,4% Odor Característico Característico Característico
pH 5,5 pH 5,65± 0,13 5,69 ± 0,15 5,77± 0,09
Formulação 3 Teste 15 dias 30 dias 45 dias
Carnitina 10% Aspecto Límpido e incolor Límpido e incolor Límpido e incolor
Metilparabeno 0,1% Odor Característico Característico Característico
pH 5,5 pH 5,77± 0,11 5,77 ± 0,19 5,82 ± 0,15
Formulação 4 Teste 15 dias 30 dias 45 dias
Carnitina 10% Aspecto Límpido e incolor Límpido e incolor Límpido e incolor
Benzoato 0,3% Odor Característico Característico Característico
pH 5,0 pH 5,01± 0,15 5,08± 0,18 5,07± 0,06

96
Tabela 16. Resultado das análises MIC do estudo de estabilidade prévio das formulações a 10% (p/v)
Análise 15 dias 30 dias 45 dias
Formulação 1 Sem crescimento* Fora de especificação Fora de especificação
Formulação 2 Sem crescimento Fora de especificação Fora de especificação
Formulação 3 Sem crescimento Sem crescimento Fora de especificação
Formulação 4 Sem crescimento Sem crescimento Sem crescimento
*Crescimento de bactérias de 6UFC/mL. Máximo permitido 10 UFC/mL.
Conforme observado na tabela 15, todas as formulações apresentaram boa
estabilidade físico-química, sem alteração significativa de suas características
organolépticas e com pequenas variações de valores de pH não estatisticamente
significantes (p>0,05). Quanto a estabilidade microbiológica, apenas a Formulação 4
não apresentou crescimento bacteriano durante os 45 dias quando mantida sob alta
temperatura. Essa formulação é a única que apresentou pH final 5,0. Para esse
ajuste, foi necessário adicionar à formulação quantidade de ácido cítrico equivalente
à 0,9 % (p/v) da formulação. Como o ácido cítrico também tem poder antimicrobiano
e acidifica o meio, potencializando o efeito do benzoato de sódio, tornou o sistema
conservante da formulação eficiente (ANSEL; POPOVICH; ALLEN JUNIOR,2016).
A segunda formulação mais estável foi a Formulação 3, que apresentou
estabilidade por 30 dias. Essa formulação apresentou pH final de 5,74, mas
diferentemente das outras formulações, o agente conservante utilizado foi o
metilparabeno, que apresenta amplo espectro de atividade e é ativo contra fungos e
bactérias. A desvantagem do seu uso é sua baixa solubilidade em veículos aquosos,
o que implica na necessidade de aquecimento da formulação a fim de promover sua
solubilização. Além disso, é necessário aguardar o resfriamento da formulação para
prosseguir com a incorporação dos outros componentes. Além do tempo gasto
nessa etapa, o metilparabeno está fortemente relacionado a efeitos indesejáveis
após sua administração e apresentam sabor desagradável (PESSANHA et al., 2012;
SENA et al., 2014). E, em se tratar de formulação farmacêutica voltada,
principalmente, para crianças e neonatos, o potencial para toxicidade é maior.
Levando em consideração que a Formulação 4 apresentou melhor estabilidade
durante os 45 dias de exposição a altas temperaturas, ela será encaminhada para

97
estudo de estabilidade completo. Apesar do benzoato de sódio também estar
relacionado a efeitos indesejáveis como aparecimento de erupções cutâneas,
dermatites de contato e outros, sua utilização pode ser considerada positiva já que o
benzoato induz a excreção de produtos nitrogenados em pacientes com
determinadas patologias associadas à EIM e é amplamente usando nesse perfil de
pacientes (JARDIM; ASHTON-PROLLA, 1996).
Além da formulação a 10%, a formulação de concentração a 20% também é
importante para atender pacientes que utilizam um volume maior diariamente. Dessa
forma, foi realizado um segundo estudo de estabilidade prévio com soluções de L-
carnitina com concentração de 20% (p/v). A composição das formulações pode ser
observada na tabela 17.
Tabela 17. Composição das formulações testes a 20% (p/v)
COMPONENTES FORMULAÇÃO
5
FORMULAÇÃO
6
FORMULAÇÃO
7
FORMULAÇÃO
8
L-carnitina 20% 20% 20% 20%
Benzoato de sódio 0,4% 0,3% - 0,2%
Metilparabeno - - - 0,1%
Ácido Cítrico q.s. 1,8% 1,8% q.s.
pH 5,5 5,0 5,0 5,5
Carboximetilcelulose 0,3% 0,3% 0,3% 0,3%
Água destilada q.s.p. q.s.p. q.s.p. q.s.p.
Legenda. q.s.= “Quantidade suficiente” para pH 5,5; Q.s.p. = “Quantidade suficiente para”.
Após a manipulação, todas as soluções foram analisadas quanto às suas
características físico-químicas. Avaliou-se o aspecto das formulações, odor e pH, e
buscou-se observar incompatibilidade farmacotécnica. O resultado dessas análises
pode ser visualizado na tabela 18.
Tabela 18. Resultado FIQ inicial do estudo de estabilidade prévio das formulações a 20% (p/v)
Análise Aspecto Odor pH
Formulação 5 Límpidas, incolores, sem a
presença de partículas e
sem grumos.
Aroma quase
imperceptível,
agradável.
5,55
Formulação 6 5,02
Formulação 7 5,01
Formulação 8 5,53

98
Após a análise inicial, observou-se que todas as formulações apresentavam
boa compatibilidade farmacotécnica e foram encaminhadas para estudo de
estabilidade prévio, realizado sob as mesmas condições que o estudo anterior para
as soluções com 10% (p/v), onde as formulações foram armazenadas em estufa
com temperatura controlada a 40°C. Foram encaminhadas amostras para análise
físico-química (FIQ) (exceto a análise de teor) e microbiológica (MIC) após 15, 30 e
45 dias de exposição. O resultado físico-químico pode ser observado na tabela 19 e
o resultado microbiológico, na tabela 20.
Tabela 19. Resultado FIQ do Estudo de Estabilidade Prévio das formulações a 20% (p/v)
Formulação 5 Teste 15 dias 30 dias 45 dias
Carnitina 20% Aspecto Límpido e incolor Límpido e incolor Límpido e incolor
Benzoato 0,4% Odor Característico Característico Característico
pH 5,5 pH 5,57 ± 0,15 5, 61± 0,04 5,60 ± 0,13
Formulação 6 Teste 15 dias 30 dias 45 dias
Carnitina 20% Aspecto Límpido e incolor Límpido e incolor Límpido e incolor
Benzoato 0,3% Odor Característico Característico Característico
pH 5,0 pH 5,01± 0,07 5,03 ± 0,09 5,01± 0,16
Formulação 7 Teste 15 dias 30 dias 45 dias
Carnitina 20% Aspecto Límpido e incolor Límpido e incolor Límpido e incolor
Ácido cítrico 2% Odor Característico Característico Característico
pH 5,0 pH 5,02± 0,17 5,00 ± 0,11 5,01 ± 0,08
Formulação 8 Teste 15 dias 30 dias 45 dias
Carnitina 20% Aspecto Límpido e incolor Límpido e incolor Límpido e incolor
Benzoato 0,2%
Metilparabeno 0,1% Odor Característico Característico Característico
pH 5,5 pH 5,55± 0,15 5,59± 0,12 5,62± 0,06

99
Tabela 20. Resultado das análises MIC do estudo de estabilidade prévio das formulações a 20% (p/v)
Análise 15 dias 30 dias 45 dias
Formulação 5 Sem crescimento Fora de especificação Fora de especificação
Formulação 6 Sem crescimento Sem crescimento Sem crescimento
Formulação 7 Sem crescimento Sem crescimento Sem crescimento
Formulação 8 Sem crescimento Sem crescimento Fora de especificação
Especificação: Máximo permitido 10 UFC/mL.
A análise físico-química das formulações com concentração a 20% (p/v)
apresentou boa estabilidade, assim como observado nas formulações com
concentração de 10% (p/v). Todas as formulações apresentaram boa estabilidade
físico-química, sem alteração de suas características organolépticas e com
pequenas variações de valores de pH não estatisticamente significantes (p>0,05). A
Formulação 8 apresentou em sua composição a associação do benzoato de sódio
com o metilparabeno em concentrações menores, buscando potencializar o poder
antimicrobiano com o sinergismo dos dois conservantes. Porém, a conservação
microbiana foi mantida apenas por 30 dias, repetindo o resultado encontrado quando
o metilparabeno foi utilizado isoladamente. Isso pode ter acontecido pelo fato do
benzoato de sódio possuir maior ação em faixas mais ácidas e, nesse caso, como o
pH da formulação foi mantido em 5,5, seu uso não fez diferença no poder
conservante da formulação.
Já as formulações com valores de pH mais baixos apresentaram boa
estabilidade microbiológica durante os 45 dias de estudo, confirmando a importância
do ajuste do pH final para 5,0. A Formulação 7, inclusive, só contou com o ácido
cítrico como conservante e conseguiu preservar a formulação de forma eficiente.
Para estudo de estabilidade completo, foram encaminhadas as formulações 6 e 7
que apresentaram boa estabilidade físico-química e microbiológica. O estudo
completo contou com a retirada diária de alíquotas, mimetizando as condições de
uso em formulações multidoses onde há a abertura subsequente da formulação. A
Formulação 7 apresenta em sua composição apenas ácido cítrico como agente
conservante e, desta forma, apresentando pouca atividade contra bolores e

100
leveduras. O estudo mimetizando as condições de uso pode evidenciar a limitação
dos conservantes testados.
6.6 Estudo de estabilidade completo em uso
As formulações desenvolvidas visam, principalmente, o atendimento à
demanda dos pacientes da FU que apresentam alguma patologia associada à EIM.
Esse perfil de paciente é formado por diferentes faixas etárias e, dessa forma, por
diferentes massas corpóreas. A dose de carnitina é definida por quilograma de peso
do paciente e, nesse caso, existe uma enorme diferença nos volumes a serem
dispensados, podendo variar de 60 mL a 3 litros. As embalagens disponíveis para
esses volumes tão discrepantes são frascos de vidro (volume de 60 mL até 200 mL)
e de plástico PET (volume de 200mL até 1L).Um fator importante para garantia da
estabilidade do produto final é a determinação do material de embalagem primária a
ser utilizado, uma vez que os componentes da embalagem podem interagir com o
produto farmacêutico, promovendo uma possível instabilidade. Além disso, a
embalagem adequada garante ao paciente que o medicamento permaneça
completamente protegido contra reações externas adversas, sendo uma barreira
entre os meios internos e externos ao medicamento (ROCHA et al., 2014). Durante o
desenvolvimento de um produto é necessário conduzir estudo de estabilidade na
embalagem proposta para comercialização. Nesse caso, o estudo de estabilidade foi
realizado nos dois tipos de embalagem disponíveis.
Com base no estudo de estabilidade prévio realizado nas formulações iniciais,
o estudo de estabilidade completo foi realizado com as formulações6 e 7. Foi
incluída no estudo, formulação controle sem a presença de agente conservante,
contando apenas com L-carnitina a 20% (p/v), CMC 0,3% e água destilada. O
resumo do estudo pode ser observado na tabela 21.

101
Tabela 21. Resumo do estudo de estabilidade completo em uso
Formulação Embalagem Temperatura Análise Tempo de análise
Formulação 6
Formulação 7
Controle*¹
PET
(200mL)
Vidro âmbar (200mL)
5°C
25°C
40°C
Físico-Química
Microbiológica
0 dias *²
15 dias
30 dias
45 dias
Obs.: *¹ Formulação sem a presença de ativo; *²Análise realizada após a manipulação.
As formulações 6 e 7 apresentaram-se límpidas, transparentes, incolores, sem a
presença de partículas ou precipitações durante todo o estudo de estabilidade. A
Formulação Controle exposta em estufa apresentou alteração quanto à coloração e
ao odor após 30 dias de armazenamento, tornando-se amarelada e com odor mais
forte e característico. As soluções controle expostas em geladeira e temperatura
ambiente não apresentaram alteração nas características organolépticas. O
resultado encontrado nas análises de pH podem ser observados no quadro 02.

102
Quadro 02. Resultado das análises de pH no estudo de estabilidade completo das formulações a 20% (p/v)
Embalagem Composição Condição Análise inicial 15 dias 30 dias 45 dias p (dias) p (Temperatura)
Vidro
Formulação 6
Benzoato 0,4%
Ácido cítrico 2%
5°C
25°C
40°C
5,01 ± 0,05
5,01 ± 0,01
5,02 ± 0,01
5,02 ± 0,01
5,02 ± 0,01
5,03 ± 0,02
5,02 ± 0,01
5,01 ± 0,02
5,02 ± 0,04
5,02 ± 0,04
0,06 0,27
Formulação 7
Ácido Cítrico 2%
5°C
25°C
40°C
5,01 ± 0,03
5,00 ± 0,01
5,01 ± 0,01
5,01 ± 0,01
5,01 ± 0,01
5,02 ± 0,01
5,01 ± 0,02
5,01 ± 0,05
5,02 ± 0,02
5,02 ± 0,05
0,16 0,17
Controle
Sem conservante
5°C
25°C
40°C
5,91 ± 0,04
6,00 ± 0,01
6,07 ± 0,02
6,11 ± 0,02
5,92 ± 0,02
6,14 ± 0,04
6,23 ± 0,05
6,03 ± 0,06
6,31 ± 0,04
6,58 ± 0,06
0,09 0,25
Embalagem Composição Condição Análise inicial 15 dias 30 dias 45 dias p (dias) p (Temperatura)
PET
Formulação 6
Benzoato 0,4%
Ácido cítrico 2%
5°C
25°C
40°C
5,01 ± 0,05
5,01 ± 0,01
5,03 ± 0,01
5,02 ± 0,02
5,01 ± 0,02
5,02 ± 0,03
5,04 ± 0,02
5,00 ± 0,03
5,03 ± 0,04
5,03 ± 0,07
0,63 0,06
Formulação 7
Ácido Cítrico 2%
5°C
25°C
40°C
5,01 ± 0,03
5,00 ± 0,02
5,01 ± 0,02
5,01 ± 0,02
5,00 ± 0,01
5,02 ± 0,03
5,02 ± 0,05
5,00 ± 0,07
5,04 ± 0,03
5,05 ± 0,05
0,29 0,14
Controle
Sem conservante
5°C
25°C
40°C
5,91 ± 0,04
6,00 ± 0,04
6,07 ± 0,03
6,11 ± 0,04
6,01 ± 0,02
6,18 ± 0,04
6,44 ± 0,03
6,15 ± 0,01
6,15 ± 0,05
6,76 ± 0,02
0,13 0,23
Leg: o valor de “p (dias)” é a avaliação estatística entre os tempos analisados para cada formulação, o valor de p para temperatura avalia as diferenças
significativas entre os resultados encontrados para cada temperatura condicionada. (n = 3).

103
Inicialmente, foi realizada avaliação estatística comparando os resultados de
pH da formulação 6 em função do tempo, avaliando os resultados encontrados nos
dias 15, 30 e 45 dias (independente da temperatura de acondicionamento) onde p >
0,05 e, portanto, as alterações observadas ao longo desse período não foram
consideradas significativas. O mesmo foi realizado para a formulação 7 (p = 0,16) e
para a formulação controle (p = 0,09). Dessa forma, o valor de pH não foi alterado
para nenhuma das formulações (6, 7 e controle) ao longo dos 45 dias de estudo de
estabilidade.
Ao mesmo tempo, foi avaliado em cada formulação a influência da
temperatura sobre os resultados obtidos para o pH. Dessa forma, todos os valores
de pH da Formulação 6 exposta a 5°C foram comparadas com as de 25°C e 40°C e
não houve alteração significativa entre elas. Dessa forma, para a Formulação 6 os
valores de pH não são influenciados pela temperatura exposta. O mesmo foi
observado para a Formulação 7 e Formulação Controle.
As análises descritas acima foram realizadas para avaliar as alterações do
tempo e da temperatura de armazenamento da formulação para cada embalagem
de armazenamento avaliada. A tabela 22 une os valores de pH encontrados tanto na
embalagem de vidro quanto no frasco PET para verificar se há uma variação
significativa entre os materiais de embalagem comparados. Ao avaliar todos os
resultados obtidos (Vidro e PET) ao longo do 15, 30 e 45 dias foi obtido p = 0,09,
não sendo considerada então variação significativa ao longo do tempo mesmo em
embalagens diferentes.
Para avaliação da influência da temperatura nas formulações mantidas nas
diferentes embalagens, foi feita uma avaliação estatística entre os valores de pH sob
as diferentes temperaturas de exposição. Foi encontrado p = 0,06 e dessa forma não
há alteração significativa para os valores de pH encontrados quando há alteração de
temperatura de exposição das formulações, independente do material de
embalagem utilizado.

104
Tabela 22. Análise estatística dos valores de pH avaliando cada formulação nas diferentes embalagens
Não se observou diferença significativa nos valores de pH nas Formulações 6 e
7 para nenhuma das avaliações de tempo de armazenamento, temperatura de
armazenamento e tipo de material de embalagem. Não houve alteração significativa
após os 45 dias de exposição, entre as temperaturas e entre as embalagens
testadas. Mas observou-se diferença significativa na solução controle quando
avaliada em relação ao tempo com p < 0,05, indicando uma alteração de pH
significativa ao longo dos 45 dias.
O resultado encontrado no teste de teor pode ser observado no quadro 03. Todas as análises foram feitas em triplicatas (n = 3)
Formulação Embalagem Condição 0 15 30 45 Análise P (dias)
Análise P
(Temp.)
Formulação
6
VD
5°C 5,01 5,01 5,02 5,01
0,09 0,06
25°C 5,01 5,02 5,03 5,02
40°C 5,01 5,02 5,02 5,02
PET
5°C 5,01 5,01 5,01 5,00
25°C 5,01 5,03 5,02 5,03
40°C 5,01 5,02 5,04 5,03
Formulação
7
VD
5°C 5,01 5,00 5,01 5,01
0,06 0,12
25°C 5,01 5,01 5,02 5,02
40°C 5,01 5,01 5,01 5,02
PET
5°C 5,01 5,00 5,00 5,00
25°C 5,01 5,01 5,02 5,04
40°C 5,01 5,01 5,02 5,05
Controle
VD
5°C 5,91 6,00 5,92 6,03
0,003 0,28
25°C 5,91 6,07 6,14 6,31
40°C 5,91 6,11 6,23 6,58
PET
5°C 5,91 6,00 6,01 6,15
25°C 5,91 6,07 6,18 6,15
40°C 5,91 6,11 6,44 6,76

105
Quadro 03. Resultado das análises de teor no estudo de estabilidade completo das formulações a 20% (p/v)
Embalagem Composição Condição Análise inicial 15 dias 30 dias 45 dias Análise P (dias)
Análise P (Temp.)
Vidro
Formulação 6
Benzoato 0,4%
Ácido cítrico 2%
5°C
25°C
40°C
99,12%
99,32%
99,01%
98,75%
99,00%
99,89%
98,42%
99,96%
101,51%
99,77%
0,16 0,27
Formulação 7
Ácido Cítrico 2%
5°C
25°C
40°C
97,61%
96,82%
96,56%
97,82%
98,48%
96,63%
95,94%
97,28%
97,70%
96,67%
0,94 0,44
Controle
Sem conservante
5°C
25°C
40°C
98,01%
98,48%
96,92%
98,58%
97,98%
98,42%
99,00%
99,31%
98,64%
98,57%
0,47 0,44
Embalagem Composição Condição Análise inicial 15 dias 30 dias 45 dias Análise P (dias)
Análise P (Temp.)
PET
Formulação 6
Benzoato 0,4%
Ácido cítrico 2%
5°C
25°C
40°C
99,12%
100,52%
98,77% 101,02%
101,16%
101,18% 99,37%
99,72%
99,36% 100,99%
0,68 0,72
Formulação 7
Ácido Cítrico 2%
5°C
25°C
40°C
97,61%
98,30%
97,72%
96,96%
99,81%
98,55%
99,71%
99,49%
97,92%
98,45%
0,10 0,64
Controle
Sem conservante
5°C
25°C
40°C
98,01%
97,93%
98,34%
98,97%
96,16%
97,88%
98,44%
98,64%
98,67%
98,01%
0,46 0,40

106
Para verificar se houve alteração significativa dos resultados de teor, foram
utilizadas as mesmas análises estatísticas aplicadas na avaliação do pH. Avaliou-se
a existência de variação significativa ao longo do tempo de exposição (avaliação
estatística entre os resultados expostos em colunas) e encontrou-se valor de p maior
que 0,05 (p = 0,49), não havendo alteração significativa entre os resultados
encontrados ao longo do tempo de 15, 30 e 45 dias. O mesmo foi realizado em
relação às diferentes temperaturas de armazenamento (avaliação entre linhas) e em
relação aos dois tipos de embalagens utilizados (entre tabelas, por formulação).
Não se observou diferença significativa (p > 0,05) quanto aos valores de teor
para as formulações analisadas (formulação 6, 7 e controle) em nenhuma das
avaliações: ao longo dos 45 dias de exposição, entre as temperaturas de
armazenamento e entre as embalagens de vidro e de plástico (Tabela 23).
Tabela 23. Resultado da análise estatística do teste de teor realizado em comparação com as embalagens
para cada formulação
As formulações em branco (sem o ativo) também foram mantidas nas mesmas
condições de exposição das formulações e também foram analisadas por CLAE. O
preparo das soluções para injeção foi realizado no mesmo dia em que as soluções
amostras eram preparadas. Não foi observado nenhuma alteração nos
Formulação Embalagem Condição 0 15 30 45 Análise P (dias)
Análise P (Temp.)
Formulação 6
VD
5°C 99,32% 99,01% 98,75%
99,00% 99,89% 98,42%
99,96% 101,51% 99,77%
0,49 0,38
25°C 99,12%
40°C
PET
5°C 100,52% 98,77% 101,02%
101,16% 101,18% 99,37%
99,72% 99,36% 100,99%
25°C 99,12%
40°C
Formulação 7
VD
5°C 96,82% 96,56% 97,82%
98,48% 96,63% 95,94%
97,28% 97,70% 96,67%
0,62 0,07
25°C 97,61%
40°C
PET
5°C 98,30% 97,72% 96,96%
99,81% 98,55% 99,71%
99,49% 97,92% 98,45%
25°C 97,61%
40°C
Controle
VD
5°C
98,01%
97,93% 96,16% 98,64%
0,43 0,48
25°C 98,34% 97,88% 98,67%
40°C 98,97% 98,44% 98,01%
PET
5°C
98,01%
98,48% 97,98% 99,31%
25°C 96,92% 98,42% 98,64%
40°C 98,58% 99,00% 98,57%

107
cromatogramas e, dessa maneira, os excipientes utilizados não apresentaram
influência na estabilidade do produto.
Os estudos de estabilidade microbiológicos estão apresentados nos quadros
04 (Formulação 6), 05 (Formulação 7) e 06 (Formulação Controle). Pode-se
observar que as formulações 6 e 7 apresentaram-se estáveis durante todo o estudo
de estabilidade, independente da temperatura de armazenamento, do tempo de
exposição às condições e do tipo de material utilizado na embalagem primária,
mesmo após utilização subsequente das formulações.
Além de apresentar boa estabilidade quando mantida sob refrigeração, as
formulações também apresentaram boa estabilidade quando mantidas em
temperatura ambiente, dessa forma, a recomendação de acondicionamento pode
ser manutenção em temperatura na faixa de 15 a 30°C (temperatura ambiente).
Essa premissa é assegurada pelo estudo realizado em temperatura elevada que
visa potencializar o estresse da formulação e acelerar possível contaminação. E,
mesmo em temperatura elevada, as formulações não apresentaram contaminação
ao longo dos 45 dias de estudo. Esse resultado foi observado independente do
material utilizado como embalagem primária.
No preparo das amostras, planejou-se a manipulação de quantidade suficiente
para a realização das análises e para mais três amostras extras, para o caso de
perdas durante o estudo de estabilidade. Como o resultado microbiológico não
demonstrou crescimento microbiano em nenhuma das condições de exposição,
estendeu-se o estudo de estabilidade das formulações 6 e 7 por mais 15 dias nas
mesmas condições de temperatura, de embalagem e de retirada de alíquota diária.
As amostras extras de cada formulação foram encaminhadas ao LACMAC para
análise do tempo de 60 dias após a manipulação. Nenhuma das duas formulações
apresentou crescimento microbiano fora da especificação vigente, indicando boa
estabilidade microbiológica por 60 dias, inclusive na temperatura de 40°C.

108
Quadro 04. Resultados encontrados durante o estudo de estabilidade microbiológico da formulação 6 acondicionada em vidro e em plástico.
Embalagem Composição Condição Teste Análise
inicial 15 dias 30 dias 45 dias Especificação
Vidro
Formulação 6
Benzoato 0,4%
Ácido cítrico 2%
5°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
25°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
40°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
Embalagem Composição Condição Teste Análise
inicial 15 dias 30 dias 45 dias Especificação
PET
Formulação 6
Benzoato 0,4%
Ácido cítrico 2%
5°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
25°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
40°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente

109
Quadro 05. Resultados encontrados durante o estudo de estabilidade microbiológico da formulação 7 acondicionada em vidro e em plástico.
Embalagem Composição Condição Teste Análise
inicial 15 dias 30 dias 45 dias Especificação
Vidro Formulação 7
Ácido cítrico 2%
5°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
25°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
40°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
Embalagem Composição Condição Teste Análise
inicial 15 dias 30 dias 45 dias Especificação
PET Formulação 7
Ácido cítrico 2%
5°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
25°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
40°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente

110
Quadro 06. Resultados encontrados durante o estudo de estabilidade microbiológico da Formulação Controle acondicionada em vidro e em plástico.
Embalagem Composição Condição Teste Análise
inicial 15 dias 30 dias 45 dias Especificação
Vidro Controle
5°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
> 10 UFC/mL
Conforme
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
25°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
40°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
Embalagem Composição Condição Teste Análise
inicial 15 dias 30 dias 45 dias Especificação
PET Controle
5°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
> 10 UFC/mL
Conforme
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
25°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
Conforme
Conforme
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
40°C
Bactérias
Fungos
E.coli
Conforme
Conforme
Conforme
> 10 UFC/mL
Conforme
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
> 10 UFC/mL
> 10 UFC/mL
Conforme
< 10¹ UFC/mL
< 10² UFC/mL
Ausente
Legenda: Conforme – Conforme especificação, não houve crescimento; > 10 UFC/mL – Crescimento bacteriano não atendendo às especificações.

111
Já a formulação controle (sem conservante) apresentou crescimento
microbiano fora da especificação nas duas embalagens testadas. Durante 15 dias a
estabilidade microbiológica foi mantida, independente do material de embalagem
escolhido e independente da temperatura de armazenamento testada (ambiente ou
sob refrigeração), porém as análises após 30 e 45 dias de estudo apresentaram
grande contaminação, fora do permitido em especificação vigente. Observou-se, no
entanto, uma diferença quanto aos resultados do estudo de estabilidade acelerado,
onde a formulação mantida em temperatura elevada e acondicionada em
embalagem plástica apresentou crescimento microbiano com 15 dias de estudo,
enquanto que a formulação acondicionada em frasco de vidro só apresentou
contaminação após 30 dias.
Os valores encontrados nos testes de pH são coerentes com esses resultados,
a formulação controle quando envasada em embalagem plástica, apresenta um leve
aumento do pH (p < 0,05), o que pode ser relacionado a uma intensa contaminação
microbiana. E pode-se observar que as formulações ajustadas em pH 5,0
apresentaram-se estáveis, não havendo nenhum crescimento microbiano durante os
45 dias de estudo, mesmo quando foi realizada abertura e descarte diário de
alíquotas para mimetizar as condições de uso. Além disso, ambas as formulações
apresentaram boa estabilidade físico-química durante todo o estudo de estabilidade,
apresentando resultados de teor do ativo dentro dos limites especificados, sem
variações significativas.
Dessa forma, as formulações 6 e 7 foram consideradas estáveis por 45 dias
nos estudos de estabilidade físico-químico e microbiológico. A Formulação 7, apesar
de apresentar em sua composição apenas o ácido cítrico como conservante,
apresentou bons resultados para o estudo de estabilidade microbiológico, inclusive
quando mimetizadas as condições de uso e potencializadas com altas temperaturas.
Com o intuito de obter informações acerca de possíveis produtos de
degradação, a Formulação 7 foi exposta às condições de estresse conforme Informe
Técnico N° 01/2008 (IT 01/08). A formulação foi exposta à hidrólise ácida (HCl 0,1N),
básica (NaOH 0,1N) e ao aquecimento constante a temperatura de 60°C.Os
resultados podem ser observados na tabela 24.

112
Tabela 24. Teor de L-carnitina da Formulação 6 sob soluções degradantes dez vezes menos concentrada
Condição de estresse 24h
HCl 0,1N 91,45% ± 0,49
NaOH 0,1N 90,81% ± 0,25
Aquecimento (60°C) 105,01%± 0,25
Padrão 99,99% ± 0,31
Legenda: HCl: ácido clorídrico, NaOH: hidróxido de sódio
Como informado na RDC 53 de 2005, é indicado degradar mais que 10% do
ativo, o que não foi observado no estudo. Observou-se também que a formulação
submetida a aquecimento apresentou teor um pouco elevado, podendo indicar uma
falha no sistema de vedação do refluxo, tendo sofrido evaporação e concentração do
ativo.
Visando submeter a formulação à condições degradantes para avaliação dos
produtos formados, um novo estudo foi realizado com base na degradação forçada
de L-carnitina descrita por Khoshkam e Afshar (2014). A formulação foi exposta a
uma solução ácida, básica, solução oxidante e ao aquecimento extremo por 12 e 24
horas em sistema de vidraria vedado para evitar possível evaporação. O resultado
para o teste pode ser observado na tabela 25.
Tabela 25.Resumo do estudo de degradação forçada sob diferentes condições.
Condição de estresse Temperatura 4h 12h 24h Impureza detectada
Aquecimento 70°C - 99,00%± 0,82 98,36%± 0,93 0
HCl 1N 70°C - 48,92%± 0,49 22,01%± 0,57 1
NaOH 1N 70°C - 51,29%± 0,25 25,33%± 1,11 1
H2O2 (abrigo da luz) 25°C 89,19%± 0,25 - - 0
Padrão (sob agitação) 25°C - - 99,32% ± 0,29 -
Assim como demonstrado por Khoshkam e Afshar (2014), a L-carnitina
apresentou drástica degradação em meios ácidos e básicos aquecidos

113
permanecendo apenas 22,01%e 25,33% respectivamente após 24 horas de
exposição. Foi observado pico adicional na análise ácida e básica e ambos
apresentaram o mesmo tempo de retenção relativo conforme apresentado na tabela
26.
Tabela 26.Tempo de retenção dos picos encontrados no estudo de estabilidade.
Condição de estresse 4 horas 12 horas 24 horas Substância
equivalente
Aquecimento - 2,161 ± 0,29
3,193 ± 0,52
2,179 ± 0,32
3,196 ± 0,70
Ácido Cítrico
Carnitina
HCl 1N -
-
2,178 ± 0,28
3,193 ± 0,16
2,013 ± 0,75
2,182 ± 0,18
3,202 ± 0,28
Impureza
Ácido Cítrico
Carnitina
NaOH 1N -
1,898 ± 0,37
2,199 ± 0,62
3,081 ± 0,37
1,985 ± 0,20
2,209 ± 0,52
3,143 ± 0,81
Impureza
Ácido Cítrico
Carnitina
H2O2(abrigo da luz) 2,161 ± 0,78
3,133 ± 0,41 - -
Ácido Cítrico
Carnitina
As áreas encontradas para as amostras com impureza detectada estão
apresentadas na tabela 27.
Tabela 27. Áreas dos picos encontrados no estudo de degradação.
Condição de estresse 4 horas 12 horas 24 horas Substância
equivalente
Aquecimento - 21092701 20956535 Carnitina
HCl 1N - -
10437730
1922109
4712386
Impureza
Carnitina
NaOH 1N - 872551
10941969
1459959
5418745
Impureza
Carnitina
H2O2 (abrigo da luz) 19005535 - - Carnitina
Padrão - - 21160783 Carnitina

114
A formulação magistral é produzida de forma individualizada, sob prescrição
médica e preparada para uso em até 48h após sua manipulação, sendo considerada
uma preparação extemporânea (BRASIL, 2012c). Dessa forma, ele não possui um
prazo de validade, mas sim uma data limite para uso denominada de “Prazo de Uso”
e cabe ao farmacêutico a responsabilidade de definir esse prazo, de acordo com
material científico e referência disponíveis. No entanto, fatores externos não podem
ser controlados, mas a realização do estudo de degradação forçada visa mostrar
indícios do que pode ocorrer com a substância frente às condições extremas de
armazenamento.
Uma vez que a degradação encontrada possa ocorrer pela reação de um
excipiente, ou por impurezas presentes em sua embalagem primária, ou até mesmo
pela natureza da embalagem utilizada (como é o caso de vidro tipo I, II e III), é
necessário observar o perfil de degradação e tomar as medidas cabíveis para evitar
a perda da estabilidade ao longo do prazo de uso. Com esses testes, é mais fácil a
proteção do fármaco, realizando apenas ajustes. O farmacêutico ganha maior
controle e pode ser mais assertivo sobre o desenvolvimento da formulação e a
determinação do prazo de uso da formulação de forma segura.
O estudo de estabilidade completo realizado sob temperaturas elevadas
(estudo de estabilidade acelerado), corroborado com o estudo de degradação
forçada por aquecimento, indicam que a L-carnitina não apresenta problemas quanto
à sua estabilidade térmica e não necessita de acondicionamento da formulação sob
refrigeração. Foram realizados testes t de Student comparando resultados
encontrados para 5°C e para 25°C nos estudos de estabildiade apresentados e não
foi encontrada diferença significativa, reforçando a assertiva apresentada.
Por mais que a degradação em meio ácido e básico tenha ocorrido em
concentrações altas das soluções degradantes, a intensa degradação percebida
aponta a necessidade de cuidados para a manutenção do valor de pH. Como
observado no estudo de estabilidade completo realizado, a formulação controle
apresentou contaminação microbiana que foi acompanhada por aumento nos
valores de pH da formulação. Mas os sistemas conservantes avaliados nas duas
formulações mantiveram a estabilidade microbiana ao longo dos 60 dias de estudo,
mesmo quando condicionados a temperatura de 40°C, que visa acelerar os
processos degradantes. Vale lembrar que o estudo realizado foi feito com a abertura

115
subsequente da embalagem primária na tentativa de mimetizar as condições reais
de uso.
Dessa forma, as formulações 6 e 7 apresentaram boa estabilidade físico-
química e microbiológica, podendo ser acondicionadas sob temperatura ambiente
(sem a necessidade de refrigerador) durante o período de 45 dias. Além disso, o seu
envase pode ser realizado tanto em frasco de vidro quanto em frasco plástico leitoso
de PET dependendo apenas do volume final necessário pelo paciente.
A Formulação 7 apresentar menor quantidade de excipientes e, como o perfil
do público alvo possui diversas restrições por conta das alterações metabólicas,
indica-se a formulação com a menor quantidade de excipientes possíveis visando
evitar o aparecimento de efeitos indesejáveis atrelados ao uso do benzoato de sódio
(PIFFERI; RESTANI, 2003; SENA et al., 2014). Entretanto, o estudo de estabilidade
foi realizado em ambiente que possui boas práticas de fabricação e, dessa forma,
apresenta rigor quanto à limpeza, e é provável que o ambiente onde o paciente
realizará o manuseio do frasco e a administração da formulação apresente alguma
propensão maior à realizada quanto a contaminação microbiana. Dessa forma, para
assegurar a manutenção da estabilidade microbiana após dispensação, indica-se
utilizar a formulação 6 que apresenta em sua composição o benzoato de sódio,
podendo assegurar ainda mais a ação contra leveduras e bactérias e garantia da
segurança da formulação.

116
7 CONCLUSÃO
A Formulação 6 (com benzoato de sódio e ácido cítrico) e a Formulação 7
(apenas com ácido cítrico) apresentaram estabilidade físico-química e microbiológica
satisfatória durante os 45 dias de estudo de estabilidade propostos, mesmo quando
armazenadas em temperatura ambiente (15 a 30°C).
A estabilidade também foi mantida nos dois materiais de embalagem testados
e, dessa forma, a formulação pode ser envasada e dispensada em frasco de vidro
âmbar ou em frasco plástico PET leitoso, sem alteração da estabilidade e com
garantia de segurança para o paciente.
Apesar de ambas as formulações apresentarem boa estabilidade, a
Formulação 6 pode garantir uma maior segurança após a dispensação ao paciente.
Dessa forma, a Formulação 6 será incluída no Memento Terapêutico da
Farmácia Universitária da UFRJ e será disponibilizada para venda e atendimento
aos pacientes. Ela estará disponível nas concentrações de 10 e 20%, com prazo de
uso seguro de 45 dias, podendo ser armazenada sob temperatura ambiente (15 a
30°C) e envasada tanto em frasco de vidro âmbar como em frasco plástico PET
leitoso.

117
8 REFERÊNCIAS BIBLIOGRÁFICAS ALENCAR, T.O.S. A reforma sanitária brasileira e questão medicamentos/assitência farmacêutica.2016. Tese (Doutorado em Saúde Coletiva) - Instituto de Saúde Coletiva, Universidade Federal da Bahia. Salvador, Bahia, 2016.
ANSEL, H. C.; POPOVICH, N. G.; ALLEN JUNIOR, L. V. Ansel'sPharmaceutical Dosage Forms and Drug Delivery Systems. 10. ed. Philadelphia: Wolters Kluwer Health, 2014.
ANTUNES, D. J. M. Estudos de Estabilidade de Formas Farmacêuticas: HPLC e Controlo Microbiológico. 2015. Dissertação (Metrado em Engenharia Farmacêutica) - Faculdade de Farmácia, Técnico Lisboa, Lisboa, 2015.
ARAÚJO, A. P. DE Q. C. Doenças metabólicas com manifestações psiquiátricas. Rev. Psiq. Clín., Rio de Janeiro, v. 31, n. 6, p. 285–289, 2004.
AULTON, M. E. Delineamento de Formas Farmacêuticas. 4a ed. Rio de Janeiro: Elsevier, 2016. p.1713.
BALBANI, A.P.S.; STELZER, L. B.; MONTOVANI, J. C. Excipientes de medicamentos e as informações da bula. Revista Brasileira de Otorrinolaringologia, São Paulo, v. 72, n. 3, p. 400–406, 2006.
BARROS, R. C. A. Apresentação da Farmácia Universitária da UFRJ. In: WORKSHOP DA FARMÁCIA UNIVERSITÁRIA DA UFRJ, 2., 2016, Rio de Janeiro, Anais. Rio de Janeiro: UFRJ, 2016.
BEHRMAN, R.E.; KLIEGMAN, R.M.;JENSON, H.B.Tratado de pediatria de Nelson. 19ª ed. Rio de Janeiro: Guanabara Koogan, v. 1, p. 1–3033, 2014.
BERNAL, V.; SEVILLA, A.; CÁNOVAS, M.; IBORRA, J. Production of L-carnitine by secondary metabolism of bacteria. Microbial cell factories, Spain, v. 6, p. 31, 2007.
BERRY, G.T. Inborn errors of amino acid and organic acid metabolism. Principles of Perinatal - Neonatal Metabolism, New York: Springer, v. 2 p.799, 1998.
BILLANY, M. Soluções. Em: Delineamento de formas farmacêuticas. Porto Alegre, RS: Artmed, 2005. p. 318–329.
BRASIL. ANVISA. Agência Nacional de Vigilância Sanitária; Guia de estabilidade de produtos cosméticos. 1ª ed. Brasília: ANVISA, 2004.
BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RE N° 1, de 29 de julho de 2005. Guia para realização dos testes de estabilidade de produtos

118
farmacêuticos. Diário Oficial da República Federativa do Brasil, Poder Executivo, Brasília, DF, 01 de agosto de 2005.
BRASIL. ANVISA. Agência Nacional de Vigilância Sanitária. Resolução RDC nº 67, de 08 de outubro de 2007. Dispõe sobre Boas Práticas de Manipulação de Preparações Magistrais e Oficinais para Uso Humano em farmácias. Diário Oficial da República Federativa do Brasil, Poder Executivo, Brasília, DF, 08 outubro de 2007.
BRASIL. Agência Nacional de Vigilância Sanitária. Consulta Pública no 25, de 23 de março de 2010. Revisão, atualização e inclusão de Métodos Gerais da Farmacopéia Brasileira. Diário Oficial da República Federativa do Brasil, Poder Executivo, Brasília, DF, 25 de março de 2010.
BRASIL. Agência Nacional de Vigilância Sanitária. Resolução da Diretoria Colegiada - RDC N° 45, de 9 de agosto de 2012. Dispõe sobre a realização de estudos de estabilidade de insumos farmacêuticos ativos. Diário Oficial da República Federativa do Brasil, Poder Executivo, Brasília, DF, 10 de agosto de 2012.
BRASIL. Agência Nacional de Vigilância Sanitária. Resolução da Diretoria Colegiada - RDC N° 166, de 24 de julho de 2017. Dispõe critérios para validação de métodos analíticos. Diário Oficial da República Federativa do Brasil, Poder Executivo, Brasília, DF, 25 de julho de 2017.
BRASIL. ANVISA. Agência Nacional de Vigilância Sanitária. Farmacopeia Brasileira. 5.ed., 2010.
BRASIL. Agência Nacional de Vigilância Sanitária. Formulário Nacional Da Farmacopeia Brasileira. 2. ed. Brasília, Brasil: Ministério da Saúde, 2012b.
BREMER, J. Carnitine - metabolism and functions. Physiological reviews, v. 63, n. 4, p. 1420–1480, 1983.
BROQUIST, H. P. Modern nutrition in health and disease. 10. ed. Philadelphia: Lippincott Williams & Wilkins, 1994.
BURRESON, J.; COUTEUR, P. LE. Os botões de Napoleão: As 17 moléculas que mudaram a história. 1. ed. Zahar, 2006.
BURTON, B. K. Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics, v. 102, n. 6, p. 1–9, dez. 1998.
CAMPISTOL, J. et al. Protocolo de diagnóstico y tratamiento de las acidemias propiónica, metilmalónica e isovalérica. Protocolos de diagnóstico y tratamiento de los errores congénitos del metabolismo, p. 27–51, 2007.
CAVAZZA, M. D-camphorate of L-carnitinamide and D-camphorate of D-carnitinamide. BE patent.877609AI, 1981.

119
CHAMPE, P. C.; HARVEY, R. A.; FERRIER, D. R. Bioquímica Ilustrada. In: CARLA DALMAZ. Bioquímica Ilustada. 3. ed. Porto Alegre, RS: Artmed, 2006. p. 179–216.
DA SILVA, A. V. A. et al. Presença de excipientes com potencial para indução de reações adversas em medicamentos comercializados no Brasil. Revista Brasileira de Ciências Farmacêuticas, v. 44, n. 3, p. 397–405, 2008.
DEODATO, F. et al. Methylmalonic and Propionic Aciduria FEDERICA. American Journal of Medical Genetics Part C Semin Med Genet, v. 142C, n. 2, p. 104–112, 2006a.
DRENDEL, H. M. et al. Intermediate MCAD Deficiency Associated with a NovelMutation of the ACADM Gene: c.1052CT. Case Reports in Genetics, v. 2015, p. 4, 2015.
EL-HATTAB, A. W. Inborn Errors of Metabolism. Clinics in Perinatology, v. 42, n. 2, p. 413–439, 2015.
EL HUSNY, A. S.; CALDATO, M. C. F. Erros Inatos Do Metabolismo: Revisão De Literatura. Revista Paraense de Medicina, v. 20, n. 2, p. 41–45, 2006.
EL HUSNY, A. S.; FERNANDES-CALDATO, M. C. Erros inatos do metabolismo: revisão de literatura. Revista Paraense de Medicina, v. 20, n. 2, p. 41–45, 2006.
ESTEFAN, I.J.S. O ensino de Farmácia. Cad. Saúde Pública, v. 2, n. 4, 1986.
EVANS, A. M.; FORNASINI, G. Pharmacokinetics of L-Carnitine. Clin Pharmacokinet, v. 42, n. 11, p. 941–967, 2003.
FENTON, W. A; GRAVEL, R. A; ROSENBLATT, D. S. Disorders of Propionate and Methylmalonate Metabolism. The Online Metabolic and Molecular Bases of Inherited Diseases, p. 2165–2194, 2001.
FERREIRA, A. DE O. Manipulação. In:Guia Prático da Farmácia Magistral. 2a ed. Juiz de Fora: Pharmabooks Editora, 2000. p. 109–492.
FERREIRA, A. DE O.; SOUZA, G. F. Preparações orais líquidas. 3a ed. São Paulo: Pharmabooks Editora, 2011.
FORATO, L. A. et al. A Espectroscopia na região do Infravermelho e algumas aplicações.1. ed. São Paulo: Embrapa Instrumentação, 2010.
FUKAO, T. et al. Clinical and molecular characterization of five patients with succinyl-CoA:3-ketoacid CoA transferase (SCOT) deficiency. Biochim Biophys Acta, v. 1812, n. 5, p. 619–24, 2011.

120
GARCÍA, E. G. et al. Artículos originales CARACTERIZACIÓN MOLECULAR DE FENILCETONÚRICOS. Revista cubana de pediatría, v. 75, n. 2, p. 101–5, 2002.
GARCÍA, E. G. et al. Caracterización molecular de fenilcetonúricos cubanos. Rev Cubana de Pediatría, v. 75, n. 2, p. 101–5, 2002.
GOSEN, L.V. Organic Acidemias: A Methylmalonic and propionic focus.Journal of Pediatric Nursing. v. 23, n. 3, 2008.
GRÜNERT, S. et al. 3-methylcrotonyl-CoA carboxylase deficiency: clinical, biochemical, enzymatic and molecular studies in 88 individuals. Orphanet J Rare Dis, v. 7, n. 31, 2012.
GUERRA, M.P.S.B. Desenvolvimento e estudo da estabilidade de uma formulação líquida oral de amiodarona. 2012. Dissertação (Mestrado em Farmacotecnica Avançada) - Faculdade de Farmácia, Universidade de Lisboa, Lisboa, 2012.
GUYNOT, M. E. et al. Study of benzoate, propionate, and sorbate salts as mould spoilage inhibitors on intermediate moisture bakery products of low pH (4.5-5.5). International Journal of Food Microbiology, v. 101, n. 1–2, p. 161–168, 2005.
HOFFMAN, G. F. Selective screening for inborn erros of metabolism: past, present and future. Eur J Pedriatr, v. 153, p. 2–8, 1994.
HOPPEL, C. The Role of Carnitine in Normal and Altered Fatty Acid Metabolism.American Journal of Kidney Diseases, v. 41, n. 4, p. 4–12, 2003.
JAIN, N. K. et al. Sorbitol intolerance in adults. Am J Gastroenterol, v. 80, n. 9, p. 678–81, 1985.
JARDIM, L. B.; ASHTON-PROLLA, P. Erros inatos do metabolismo em crianças e recém-nascidos agudamente enfermos : guia para o seu diagnóstico e manejo. Jornal de Pediatria, v. 72, p. 63–70, 1996.
KHOSHKAM, R.; AFSHAR, M. Validation of a Stability-Indicating RP-HPLC Method for Determination of L-Carnitine in Tablets.International Scholarly Research Notices, v. 2014, p. 1-8, 2014.
KIEFER, W.; MAZZOLINI, A. P.; STODDART, P. R. Monitoring oxidation of multiwalled carbon nanotubes by Raman spectroscopy. Journal of Raman Spectroscopy, v. 38, p. 356-363, 2007.
LACHMAN, L.; LIEBERMAN, H. A.; KANING, J. L. Líquidos. In: Teoria e Prática na Indústria Farmacêutica. Fundação C ed. Lisboa: p. 783–818.

121
LEAL, L. B. Estudo de fármacos e medicamentos manipulados em farmácias magistrais utilizados no tratamento de doenças reumatológicas. 2007. Tese (Doutorado em Ciências Farmacêuticas) - Faculdade de Farmácia, Universidade Federal de Pernambuco Centro, 2007.
LEHNERT, W. et al. Propionic acidemia: clinical, biochemical and therapeutic aspects. Experience in 30 patients. European Journal of Pediatrics, v. 153, p. 68–80, 1994.
LEITE, F. et al. Medicamentos pediátricos e cáries dentárias - Percecões e atitudes de um grupo de tutores pediátricos em Vila Nova de Gaia. Revista Portuguesa de Estomatologia, Medicina Dentaria e Cirurgia Maxilofacial, v. 52, n. 4, p. 193–199, 2011.
LI, B. et al. The effect of enteral carnitine administration in humans. The American Journal of Clinical Nutrition, v. 55, p. 838–845, 1992.
LUBI, N. CRISTINA. Desenvolvimento de forma farmacêutica líquida de uso oral, isenta de açúcar com extrato fluído de guaco (Mikania glomerata Sprengel, Asteraceae), para afecções do aparelho respiratório. 2002. Dissertação (Mestrado em Ciências Farmacêuticas) - Faculdade de Farmácia, Universidade Federal do Paraná, 2002.
MALIK, S. et al. Neonatal Carnitine Palmitoyltransferase II Deficiency: A Lethal Entity. J Clin Diagn Res, v. 10, n. 9, p. SD01–SD02, 2015.
MARTINDALE, W. Martindale: The complete drug reference. 30. ed. London: Pharmaceutical Press, 2009.
MAX, B. et al. Biotechnological production of citric acid. Brazilian Journal of Microbiology, v. 41, n. 4, p. 862–875, 2010.
MCCALLUM, S. Addressing Nutrient Density in the Context of the Use of Thickened Liquids in Dysphagia Treatment. ICAN: Infant, Child, & Adolescent Nutrition, v. 3, n. 6, p. 351–360, 2011.
MCGOVERN, P. E. et al. Fermented beverages of pre- and proto-historic China. Proceedings of the National Academy of Sciences, v. 101, n. 51, p. 17593–17598, 2004.
MEADOWS, J. A.; WARGO, M. J. Carnitine in bacterial physiology and metabolism. Microbiology (United Kingdom), v. 161, n. 6, p. 1161–1174, 2015.
NADAI, C. P.; PINHEIRO, P. R. E. Aminoacidopatias: caraterísticas clínicas e genéticas. REVISTA BRASILEIRA DE MEDICINA, v. 64, n. 10, p. 465 à 473, 2006.
NASSER, M. et al. Carnitine supplementation for inborn errors of metabolism

122
(Review). Cochrane Database of Systematic Reviews, n. 2, p. 1981–90, 2012.
NELSON, D. L.; COX, M. M. A Oxidação dos Ácidos Graxos. In: AL, C. T. ET. Princípios de bioquímica de Lehninger. 6. ed. Porto Alegre, RS: Artmed, 2014. p. 465–485.
NUNN, T.; WILLIAMS, J. Formulation of medicines for children. British Journal of Clinical Pharmacology, v. 59, n. 6, p. 674–676, 2005.
NYHAN, W.L.; BARSHOP, B.A.; OZAND, P.T. Atlas of Metabolic Diseases. 2. ed. Hodder Arnold: London, 2005.
PAMPOLS, T.; RIBES, A. Bioquímica Clínica. In: FRANCESC GONZÁLEZ SASTRE. Bioquímica Clínica. Barcelona: Barcanova, 1994. p. 579–631.
PATRICK, F. N.; HINCHCLIFFE, C.; JONATHAN, W. V. L-carnitine. Journal of Alternative Medicine Review, v. 10, n. 1, p. 42–50, 2005.
PAULO, J. et al. Estudo Das Relações Entre Estrutura E Atividade De Parabenos: Uma Aula Prática. Quim. Nova, v. 36, n. 6, p. 890–893, 2013.
PAVIA, D. L.; LAMPMAN, G. S. K.; VYVYAN, J. R. Introdução à espectroscopia. 4a ed. Washington: Cengage Learning, 2013.
PESSANHA, A. F. V. et al. Influência dos excipientes multifuncionais no desempenho dos fármacos em formas farmacêuticas. Rev. Bras. Farm, v. 93, n. 2, p. 136–145, 2012.
PIFFERI, G.; RESTANI, P. The safety of pharmaceutical excipients. Il Farmaco, v. 58, n. 8, p. 541–550, 2003.
PINTO, S.; BARBOSA, C. M. Medicamentos Manipulados em Pediatria. Arquivos de Medicina, v. 22, n. 2/3, p. 75–84, 2008.
PRISTA, L.N.; ALVES, A.C.; MORGADO, R. Técnica farmacêutica e farmácia galênica. 3. ed. Lisboa: Fundação Calouste Gulbenkian, 1990.
RAGHUVEER, T. S.; GARG, U.; GRAF, W. D. Inborn errors of metabolism in infancy and early childhood: an update. American Family Physician, v. 73, n. 11, p. 1981–90, 2006.
RAO, A. N. et al. Inborn errors of metabolism: review and data from a tertiary care center. Indian Journal of Clinical Biochemistry, v. 24, n. 3, p. 215–22, jul. 2009.
REBOUCHE, C. J.; ENGEL, A. G. Kinetic Compartmental Analysis of Carnitine Metabolism in the Human Carnitine Deficiency Syndromes Carnitine Transport.

123
Academy of Sciences, v. 30, p. 857–867, 1984.
RIBEIRO, F. A. DE L. et al. Planilha De ValidaçãO: Uma Nova Ferramenta Para Estimar Figuras De MéRito Na ValidaçãO De MéTodos AnalíTicos Univariados. Quim. Nova, v. 31, n. 1, p. 164–171, 2008.
ROCHA, J. et al. Comparação da Estabilidade do Paracetamol Solução Oral Armazenada em Frasco Vidro. Cienc. Biol. Agrar. Saúde, v. 18, n. 3, p. 143–150, 2014.
RODRIGUES, C. R. et al. A Faculdade de Farmácia da UFRJ. In: QUEIROZ, A. C. DE B.; OLIVEIRA, A. J. B. (Eds.). . Universidade e lugares de memória II. Rio de Janeiro: Sistema de Bibliotecas e Informação,. p. 55–69, 2009.
ROWE, R. C.; SHESKEY, P. J.; QUINN, M. Handbook od pharmaceutical excipients. 6a ed. Grayslake: Pharmaceutical Press, 2009.
SÁNCHEZ-HERNÁNDEZ, L. et al. Sensitive determination of d-carnitine as enantiomeric impurity of levo-carnitine in pharmaceutical formulations by capillary electrophoresis-tandem mass spectrometry. Journal of Pharmaceutical and Biomedical Analysis, v. 53, n. 5, p. 1217–1223, 2010.
SANJURJO, P.; BALDELLOU, A. Diagnostico y Tratamiento de Las Enfermidades Metabólicas Hereditarias. 2. ed. 2006.
SAUDUBRAY, J. M. et al. Clinical approach to inherited metabolic disorders in neonates: An overview. Seminars in Neonatology, v. 7, n. 1, p. 3–15, 2002.
SCHWARTZ, I. V.; SOUZA, C. F. M. DE; GIUGLIANI, R. Treatment of inborn errors of metabolism. Jornal de pediatria, v. 84, n. 4 Suppl, p. S8–S19, 2008.
SCRIVER, C. R. et al. The metabolic and molecular bases of inherited disease. Biochemistry (Moscow), v. 67, n. 5, p. 611–612, 2002.
SENA, L. C. S. et al. Excipientes Farmacêuticos E Seus Riscos À Saúde: Uma Revisão Da Literatura Excipientes Farmacéuticos Y Sus Riesgos Para La Salud: Una Revisión De La Literatura Pharmaceutical Excipients and Its Risks for Health: a Literature Review. Rev. Bras. Farm. Hosp. Serv. Saúde São Paulo, v. 5 n.4, p. 25–34, 2014.
SHAWKY, R. M.; ABD-ELKHALEK, H. S.; ELAKHDAR, S. E. Selective screening in neonates suspected to have inborn errors of metabolism. Egyptian Journal of Medical Human Genetics, v. 16, n. 2, p. 165–171, 2015.
SHLOMI, T.; CABILI, M. N.; RUPPIN, E. Predicting metabolic biomarkers of human inborn errors of metabolism. Molecular Systems Biology, v. 5, n. 263, p. 263, jan. 2009.

124
SILVA, K. E. R. et al. Modelos de avaliação da estabilidade de fármacos e medicamentos para a indústria farmacêutica. Revista de Ciencias Farmaceuticas Basica e Aplicada, v. 30, n. 2, p. 129–135, 2009.
SONI, M. G. et al. Safety assessment of propyl paraben: A review of the published literature. Food and Chemical Toxicology, v. 39, n. 6, p. 513–532, 2001.
SWEETMAN, S. C. Martindale: The Complete Drug Reference. 37. ed. London: Pharmaceutical Press, 2009.
TENOPOULOU, M. et al. Strategies for Correcting Very Long Chain Acyl-CoA Dehydrogenase Deficiency. J Biol Chem, v. 290, n. 16, p. 10486–10494, 2015.
TERRA, J.; ROSSI, V. SOBRE O DESENVOLVIMENTO DA ANÁLISE VOLUMÉTRICA E ALGUMAS APLICAÇÕES ATUAIS Juliana Terra e Adriana Vitorino Rossi. Quim. Nova, v. 28, n. 1, p. 166–171, 2005.
USP. The United States Pharmacopeia Convention. 40. ed. p. 4823–24.
VALDERRAMA, P.; BRAGA, J. W. B.; POPPI, R. J. Estado da arte de figuras de mérito em calibração multivariada. Quimica Nova, v. 32, n. 5, p. 1278–1287, 2009.
VARGAS, C. R.; WAJNER, M. Cidúrias Orgânicas: Diagnóstico E Tratamento. Revista AMRIGS, v. 45, n. 1,2, p. 77–82, 2001.
VENTURA, D. M. Desenvolvimento farmacotécnico de formulações de suspensões de hidroclorotiazida obtidas por transformação de formas farmacêuticas.Quim. Nova, p. 15–24, 2011.
VILAPLANA, J.; ROMAGUERA, C. Fixed drug eruption from sodium benzoate. Contact Dermatitis, v. 49, n. 6, p. 290–291, 2003.
VITORIA, I. et al. Carnitine-Acylcarnitine Translocase Deficiency: Experience with Four Cases in Spain and Review of the Literature. JIMD Reports, v. 20, p. 11–20, 2015.
WAJNER, M. et al. Acidúrias orgânicas: diagnóstico em pacientes de alto risco no Brasil. Jornal de Pediatria, v. 77, n. 5, p. 401–406, 2001.
WALTER, J. H. L -Carnitine in inborn errors of metabolism : What is the evidence ? Journal of Inherited Metabolic Disease, v. 26, n. 2, p. 181–188, 2003.
WALTER, P.; SCHAFFHAUSER, A. O. ‘Vitamin-Like Substance’ for Functional Food. Annals of Nutrition & Metabolism, v. 44 (2), p. 75–96, 2000.
WOOD, R. How to validate analytical methods. TrAC Trends in Analytical

125
Chemistry, v. 18, n. 9, p. 624–632, 1999.
WOODS, C. et al. Development of necrotizing enterocolitis in premature infants receiving thickened feeds using SimplyThick. J Perinatol, v. 32, n. 2, p. 150–152, 2012.
WOODS, D. J. Veículos - Agentes Suspensores e Espessantes. In: FONSECA, S. G. C.Formulação na Prática Farmacêutica. 2a ed. Ceará, Fortaleza: PharmInfoTech, 2019. p. 3–4.
姜波, 刘东风, 王小雪, 张春苗. Levocarnitine compound and preparation method thereof. CN103910645A, 07 jan. 2013. Disponível em: <https://patents.google.com/ patent/CN103910645A/en>. Acesso em: 30 abr. 2019.