
Kobe University Repository : ThesispreS2 preS1 Reverse transcriptase Lipid bilayer HBcAg DNA...
139
Kobe University Repository : Thesis 学位論文題目 Title Hepatitis B virus derived carriers to effectively express pharmaceutical activity in target cancer cells(標的癌細胞において効果的な薬剤活性を 発現するB型肝炎ウイルス由来キャリア) 氏名 Author Nishimura, Yuya 専攻分野 Degree 博士(工学) 学位授与の日付 Date of Degree 2013-03-25 資源タイプ Resource Type Thesis or Dissertation / 学位論文 報告番号 Report Number 甲5767 権利 Rights JaLCDOI URL http://www.lib.kobe-u.ac.jp/handle_kernel/D1005767 ※当コンテンツは神戸大学の学術成果です。無断複製・不正使用等を禁じます。著作権法で認められている範囲内で、適切にご利用ください。 PDF issue: 2020-09-06
Transcript of Kobe University Repository : ThesispreS2 preS1 Reverse transcriptase Lipid bilayer HBcAg DNA...

Kobe University Repository : Thesis
学位論文題目Tit le
Hepat it is B virus derived carriers to effect ively express pharmaceut icalact ivity in target cancer cells(標的癌細胞において効果的な薬剤活性を発現するB型肝炎ウイルス由来キャリア)
氏名Author Nishimura, Yuya
専攻分野Degree 博士(工学)
学位授与の日付Date of Degree 2013-03-25
資源タイプResource Type Thesis or Dissertat ion / 学位論文
報告番号Report Number 甲5767
権利Rights
JaLCDOI
URL http://www.lib.kobe-u.ac.jp/handle_kernel/D1005767※当コンテンツは神戸大学の学術成果です。無断複製・不正使用等を禁じます。著作権法で認められている範囲内で、適切にご利用ください。
PDF issue: 2020-09-06

博士論文
Hepatitis B virus derived carriers to effectively express
pharmaceutical activity in target cancer cells
標的癌細胞において効果的な薬剤活性を発現する
B 型肝炎ウイルス由来キャリア
平成 25 年 1 月
神戸大学大学院
工学研究科 応用化学専攻
西 村 勇 哉

1
CONTENTS
Introduction
Synopsis
Part I.
Protein-encapsulated bio-nanocapsules production with ER
membrane localization sequences
Part II.
Complex carriers of affibody-displaying bio-nanocapsules and
composition-varied liposomes for HER2-expressing breast
cancer cell-specific protein delivery
Part III.
Targeting cancer cell-specific RNA interference by siRNA
delivery using a complex carrier of affibody-displaying
bio-nanocapsules and liposomes
Part IV.
An affinity chromatography method used to purify
His-tag-displaying bio-nanocapsules
Part V.
Granting specificity for breast cancer cells using a Hepatitis B
core particle with a HER2-targeted affibody molecule
General conclusion
Ackowledgments
Publication lists
1
16
21
44
74
95
109
131
134
136

1
INTRODUCTION
[Cancer treatment]
The cancer is the world’s third-biggest killer after heart and infectious
diseases, and about 7.5 million people die every year (Kievit, 2011). Although the
mechanism and treatments of the cancer have been studied over a long period, there
remain many unclear points due to its system complexity. In the drug treatment of the
cancer, major problems are described below (Kievit, 2011).
1) inability to bypass biological barriers
2) non-specific delivery and poor biological distribution of drug
3) ineffectiveness against metastatic disease
4) drug resistance of cancer
5) lack of an effective modality for treatment monitoring
To solve some of these problems, it is expected to apply nanotechnology as the
innovative treatment of the cancer (Galvin, 2012).
[Nanotechnology]
A nanotechnology is the multidisciplinary field to design nanodevices based
on the principles of chemistry, biology, physics and engineering. In the medicine field, it
is expected to benefit tremendously from a nanotechnology, and the study has already
progressed and is most progressive in oncology area. The benefits include the advances
of detection, imaging and treatment for diseases. In the nanotechnology fields,
nanoparticles (NPs) have the great potential to lead to paradigm shift of detection,

2
treatment and prevention especially for the treatment of cancer. Since NPs enable to
deliver drugs to the cancer stably and safely, it is expected to drastically improve the
effect of cancer treatment. Therefore, both universities and companies have focused on
the development of a novel NPs toward medical anti-cancer application (Siddiqui, 2012;
Tiwari, 2012).
NPs are the vesicular system to encapsulate drugs, and their sizes are 10
-1000 nm. However, >200 nm NPs are too heavy as the carrier, <200 nm NPs have been
frequently utilized. These <200 nm NPs are also expected to accumulate in tumor and
inflamed area selectively by enhanced permeability and retention (EPR) effect.
Therefore, this system can be available to deliver the drugs into specific tumor, improve
bioavailability and enable to the sustained release. Accordingly, the advantages of NPs
for drug delivery are provided through the characters of small sized and biodegradable
materials (Singh, 2009; Galvin, 2012; Tiwari, 2012).
[Drug delivery system]
It is important to optimize effects of medication and to reduce side effects for
the cancer in drug delivery system (DDS). Therefore, NPs have been developed as
carriers to deliver drugs effectively in a target-cell-specific manner. To use NPs as drug
carriers, mainly three functions are required as below (Fig. 1) (Nagai, 2005; Tabata,
2006).
1) Stabilization of drugs
To prevent the degradation and to keep the activity of drugs, various drugs

3
including low-molecular compounds, genes and proteins are incorporated into the
carriers.
2) Specificity to target cells
To minimize side effects and to maximize the effects of drugs, the carriers should
specifically recognize target tumors.
3) Expression of pharmaceutical activity
To express the activity of drugs effectively, it is required that the carriers arrive
in cell interior and release the drugs into cytoplasm or localize at nucleus.
The carrier with these functions is thought to be essential for effective cancer treatment.
[Bio-nanocapsule]
To establish efficient carrier system for drug delivery, a novel carrier based on
a hepatitis B surface antigen (HBsAg) derived from a hepatitis B virus (HBV) had been
developed. The HBV is an enveloped DNA virus of Hepadnaviridae. The HBV genome
encodes three envelope proteins: the S protein, the major constituent (226 amino acid
residues) of the HBV envelope protein and empty surface antigen (HBsAg) particles;
the M protein, containing 55 additional amino acid residues (pre-S2 peptide) at the
N-terminus of the S protein; and the L protein, containing 108 (subtype y) or 119
(subtype d) additional amino acid residues (pre-S1 peptide) at the N-terminus of the M
protein (Heermann, 1984; Tiollais, 1985; Neurath, 1998). The HBV specifically infects
only human and chimpanzee hepatocytes. It was reported that an attachment site of
HBV to the hepatocyte is located within the pre-S region (Fig. 2) (Neurath, 1989;
Pontisso, 1989; De Meyer, 1997).

4
Fig. 1. The main functions required for carriers of drug delivery system.
Release of drug
Endosome
1) Stabilization of drugs
drug
2) Specificity to target cells
ligand Receptor
3) Expression of pharmaceutical activity
Nucleus
Cellular uptake
cytoplasm
Release of drug
Endosome
1) Stabilization of drugs
drug
2) Specificity to target cells
ligand Receptor
3) Expression of pharmaceutical activity
Nucleus
Cellular uptake
cytoplasm

5
Fig. 2. Diagramatic illustration of Hepatitis B virus and Bio-nanocapsule. HBV is a
human liver-specific DNA virus, the 3.2-kbp genome of which harbors three
overlapping envelope genes in a single open reading frame. Depending on the three
translation initiation codons, three related transmembrane proteins are produced,
designated small (S), middle (M), and large (L). Bio-nanocapsule is hollow nano-sized
particle consisting of the L protein and a lipid bilayer. The L protein contains pre-S1,
pre-S2, and S regions. The S region is a transmembrane protein indispensable for the
formation of the particles. The pre-S1 region on the surface of an L particle is
responsible for the specific infection of human hepatocytes.
S
preS2
preS1
Reverse transcriptase
Lipid bilayerHBcAg
DNA
Hepatitis B Virus (HBV)
HBsAg
Bio-nanocapsule (BNC)
L protein
Lipid bilayer
PreS1 PreS2 SL protein
M protein
S protein
1 108 163 389 aa
S
preS2
preS1
Reverse transcriptase
Lipid bilayerHBcAg
DNA
Hepatitis B Virus (HBV)
HBsAg
Bio-nanocapsule (BNC)
L protein
Lipid bilayer
PreS1 PreS2 SL protein
M protein
S protein
1 108 163 389 aa

6
Bio-nanocapsules (BNCs) are hollow nanoparticles consisting of the L protein
and phospholipids derived from endoplasmic reticulum (ER) membrane with an average
diameter of approximately 100 nm (Fig. 2). BNCs can be formed in yeast, insect and
mammal cells through aggregation of L protein to ER membrane and the following
budding process (Kuroda, 1992; Yamada, 2001). We previously reported a novel
efficient gene and drug delivery using the BNC (Yamada, 2003; Yu, 2005; Iwasaki,
2007). The BNC has many advantages compared with conventional carriers. First, the
BNC shows a high transfection efficiency consistent with an original HBV. Second, the
BNC is very safe because it is free from viral genome. Third, relatively large materials
can be enclosed efficiently by using complex of BNC and liposome; therefore, BNCs
can deliver low-weight chemical compounds as well as 100 nm fluorescent beads and >
30 kbp plasmids (Jung, 2008). Furthermore, BNC possesses the specificity for human
hepatocytes. The in vitro and in vivo transfection experiments have demonstrated that
genes and drugs were specifically transferred into human hepatocytes with BNC.
To target other types of cells, further approaches to engineer specificity of
BNC have been developed. The pre-S region which has specific affinity for human
hepatocytes was genetically eliminated from the L-protein region. Then, the ZZ domain
derived from protein A, which has the affinity for the Fc region of immunoglobulin G
(IgG), was inserted and the ZZ domain-displaying BNC (ZZ-BNC) was prepared.
Because the antibodies can be immobilized on the BNC through binding to the ZZ
domain, the produced antibody-fusing BNC successfully presented the specific
recognition ability to the variety of target cells (Tsutsui, 2007). Furthermore, pre-S
region was genetically substituted with affibody molecule which is an altered Z domain

7
derived from Staphylococcal protein A (Fig. 3). The ZHER2, a type of affibody, has high
specificity to HER2 receptor (Nygren, 2008), which is a member of human epidermal
growth factor tyrosine kinase receptor family. Because HER2 is associated with
resistance to therapy and poor prognosis, it has been an attractive target for molecular
therapy (Witton, 2003; Chen, 2003). Therefore, the ZHER2-displaying BNC (ZHER2-BNC)
is expected to be available for in vivo and in vitro medical application.
[Research objective]
In this study, we developed the new types of engineered BNCs and the
methodologies to realize an oncoming generation anti-cancer therapy. For this, we
mainly focused on the specific delivery of the proteins and nucleotides to the target
cancer cells, and the expression of their functions inside the cells.
In Part 1, we tried to incorporate proteins into BNCs. Although it has
previously succeeded to incorporate low-molecular drugs and genes into BNCs, it is still
difficult to incorporate proteins into BNCs after particle formation due to their
higher-order structures. By focusing attention on the mechanism of particle formation
during the budding processes in the fungus body, we tried to incorporate target proteins
into BNCs. To do this, we used the green fluorescent protein (GFP) as model protein
and fused it to membrane localization sequence (MLS) of N-Ras, which can localize
protein on endoplasmic reticulum (ER) membrane. The MLS-fused GFP was
co-expressed with HBsAg in insect cells to establish the method for producing
protein-encapsulated BNCs.

8
Fig. 3. Genetic alteration of specificity for human hepatocytes on Bio-nanocapsule. The
engineered BNC was prepared by genetically substituted pre-S region with Affibody
(Affibody-BNC).
Affibody-displaying BNC
S
Affibody
S
49 160
Affibody
S
preS2
preS1
L protein
Affibody-displaying BNC
S
Affibody
S
49 160
Affibody
S
49 160
Affibody
S
preS2
preS1
L protein

9
While it is attractive to develop the method for encapsulating proteins into
BNCs, it is also important to demonstrate the abilities of BNCs for specifically
delivering various types of drugs and expressing their functions in target cells.
Therefore, we tried to deliver proteins (Part 2) and nucleic acids (Part 3) with
pharmaceutical activity and express their function in the target cells. To target breast
cancer carcinoma, which expresses excess amount of HER2 receptors, we chose
ZHER2-BNC as the carrier. Furthermore, we used the BNC/liposome (LP) conjugation
method that has been developed by Jung et al to incorporate not only proteins but also
nucleic acids into BNC effectively (Fig. 4).
In Part 2, we tried to deliver the cell cytotoxic exotoxin A as protein drugs to
HER2-expressing breast cancer cells and kill them. First, we evaluated the influence of
electric charge of LPs on this method. Next, we tried to escape ZHER2-BNC/LP from
endosome by mixing helper lipid, which is pH-sensitive phospholipid with the ability to
endosomal escape, to LP. And then, we demonstrated that the ZHER2-BNC/LP complex
carrier permitted cell-specific delivery and effective pharmaceutical activity.
Although the therapeutic effect of small interfering RNA (siRNA) as nucleic
acid medicine is greatly expected, inability to specifically reach target cells and to cross
the cell membrane limits its in vivo applications. In Part 3, therefore, we tried to lead to
the cell-specific RNA interference (RNAi) by delivering siRNA with the ZHER2-BNC/LP
complex carrier to HER2-expressing breast cancer cells. To facilitate evaluation of
RNAi, the siRNA for inhibiting the GFP expression was selected.

10
Fig. 4. Diagramatic illustration of Bio-nanocapsule/Liposome conjugation method. In
this method, target materials are pre-encapsulated to liposome, and then BNCs are fused
to the surface of the LP.
Complex carrier (BNC/LP)
Liposome formulationDrug solution injection
Dried lipid
materials
Liposome incorporating materials
Freeze-dried BNCs
Conjugation
Liposome incorporating materials
Complex carrier (BNC/LP)
Liposome formulationDrug solution injection
Dried lipid
materials
Liposome incorporating materials
Freeze-dried BNCs
Conjugation
Liposome incorporating materials

11
Furthermore, we improved the purification method of BNCs. The purification
of BNC is laborious, and the yield and degree of purification are often not high enough
for commercial applications. Actually, although the conventional ultracentrifugation
method is available to purify various types of BNC, it has a serious problem with
moderate yield for cumbersome protocol. On the other hand, the affinity
chromatography method generally can offer high-yield purification but lacks versatility
to purifying BNCs, because it requires the optimal column suited to the antigens. In Part
4, therefore, we tried to establish the purification method of BNCs with affinity
chromatography by using histidine-tag (His-tag). We evaluated to permit simply and
high-yield purification of ZHER2-BNC by genetically fusing His-tag to ZHER2-BNC.
Finally, we developed a new type of carrier particle, which allows the
large-scale production in Escherichia coli and the purification with His-tag affinity
chromatography. The developed carrier is based on the capsid-like particle consisting of
a hepatitis B core protein (HBc). Generally, the hollow HBc particle, which is formed
by the self-assembly of HBc, has the ability to bind to various cells non-specifically via
the action of an arginine-rich domain. In Part 5, we therefore developed an engineered
HBc particle that specifically recognizes and targets HER2-expressing breast cancer
cells by despoiling the non-specific binding property and granting the target-cell
specific recognition ability to the HBc particle with ZHER2 affibody. By adapting a
variety of useful approaches in the establishment of engineered BNCs, the newly
engineered capsid-like HBc particle would become to more highly-sophisticated carrier
for DDS in near future.

12
REFERENCES
Chen, J.S., Lan, K., Hung, M.C., 2003. Strategies of target HER2/neu overexpression
for cancer therapy, Drug Resist Updat. 6, 129–136.
De Meyer, S., Gong, Z.J., Suwandhi, W., van Pelt, J., Soumillion, A., Yap, S.H., 1997.
Organ and species specificity of hepatitis B virus (HBV) infection: a review of literature
with a special reference to preferential attachment of HBV to human hepatocytes. J
Viral Hepat. 4, 145-153.
Galvin, P., Thompson, D., Ryan, K.B., McCarthy, A., Moore, A.C., Burke, C.S., Dyson,
M., Maccraith, B.D., Gun'ko, Y.K., Byrne, M.T., Volkov, Y., Keely, C., Keehan, E.,
Howe, M., Duffy, C., MacLoughlin, R., 2012. Nanoparticle-based drug delivery: case
studies for cancer and cardiovascular applications. Cell Mol Life Sci. 69(3), 389-404.
Heermann, K.H., Goldmann, U., Schwartz, W., Seyffarth, T., Baumgarten, H., Gerlich,
W.H., 1984. Large surface proteins of hepatitis B virus containing the pre-S sequence. J
Virol. 52, 396-402.
Iwasaki, Y., Ueda, M., Yamada, T., Kondo, A., Seno, M., Tanizawa, K., Kuroda, S.,
Sakamoto, M., Kitajima, M., 2007. Gene therapy of liver tumors with human
liver-specific nanoparticles. Cancer Gene Ther. 14, 74-81.
Jung, J., Matsuzaki, T., Tatematsu, K., Okajima, T., Tanizawa, K., Kuroda, S., 2008.

13
Bio-nanocapsule conjugated with liposomes for in vivo pinpoint delivery of various
materials. J Control Release. 126, 255-264.
Kievit, F.M., Zhang, M., 2011. Cancer nanotheranostics: improving imaging and
therapy by targeted delivery across biological barriers. Adv Mater. 23(36), 217-247.
Kuroda, S., Otaka, S., Miyazaki, T., Nakao, M., Fujisawa, Y., 1992. Hepatitis B virus
envelope L protein particles. Synthesis and assembly in Saccharomyces cerevisiae,
purification and characterization. J Biol Chem. 267, 1953-1961.
Nagai, T., 2005. Drug discovery and innovative drug delivery research in new drug
development. Pharm Tech Japan. 21, 1949-1951.
Neurath, A.R., Kent, S.B., Stick, N., Parker, K., 1986. Identification and chemical
synthesis of a host cell receptor binding site on hepatitis B virus. Cell. 46, 429-436.
Neurath, A.R., Kent, S.B., 1998. The pre-S region of hepadnavirus envelope proteins.
Adv Virus Res. 34, 65-142.
Nygren, P.A., 2008. Alternative binding proteins: affibody binding proteins developed
from a small three-helix bundle scaffold. FEBS J. 275, 2668–2676.
Pontisso, P., Ruvoletto, M.G., Gerlich, W.H., Heermann, K.H., Bardini, R., Alberti, A.,
1989. Identification of an attachment site for human liver plasma membranes on

14
hepatitis B virus particles. Virology. 173, 522-530.
Siddiqui, I.A., Adhami, V.M., Chamcheu, J.C., Mukhtar, H., 2012. Impact of
nanotechnology in cancer: emphasis on nanochemoprevention. Int J Nanomedicine. 7,
591-605.
Singh, R., Lillard, J.W. Jr., 2009. Nanoparticle-based targeted drug delivery. Exp Mol
Pathol. 86(3), 215-223.
Tabata, T., 2006. Drug delivery system: Basic technology for biomedical research,
medical treatment and health care. Biotechnology-Journal. 6, 553-555.
Tiollais, P., Pourcel, C., Dejean, A., 1985. The hepatitis B virus. Nature. 317, 489-495.
Tiwari, M., 2012. Nano cancer therapy strategies. J Cancer Res Ther. 8(1), 19-22.
Tsutsui, Y., Tomizawa, K., Nagita, M., Michiue, H., Nishiki, T., Ohmori, I., Seno, M.,
Matsui, H., 2007. Development of bionanocapsules targeting brain tumors. J Control
Release. 122, 159-164.
Witton, C.J., Reeves, J.R., Going, J.J., Cooke, T.G., Bartlett, J.M., 2003. Expression of
the HER1-4 familiy of receptor tyrosin kinase in breast cancer. J Pathol. 200, 290-297.
Yamada, T., Iwabuki, H., Kannno, T., Tanaka, H., Kawai, T., Fukuda, H., Kondo, A.,

15
Seno, M., Tanizawa, K., Kuroda, S., 2001. Physicochemical and immunological
characterization of hepatitis B virus envelope particles exclusively consisting of the
entire L (pre-S1 + pre-S2 + S) protein. Vaccine. 19, 3154-3163.
Yamada, T., Iwasaki, Y., Tada, H., Iwabuki, H., Chuah, M.K., VandenDriessche, T.,
Fukuda, H., Kondo, A., Ueda, M., Seno, M., Tanizawa, K., Kuroda, S., 2003.
Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat Biotechnol.
21, 885-890.
Yu, D., Amano, C., Fukuda, T., Yamada, T., Kuroda, S., Tanizawa, K., Kondo, A., Ueda,
M., Yamada, H., Tada, H., Seno, M., 2005. The specific delivery of proteins to human
liver cells by engineered bio-nanocapsules. FEBS J. 272, 3651-3660.

16
SYNOPSIS
PART I.
Protein-encapsulated bio-nanocapsules production with ER membrane localization
sequences
Bio-nanocapsules (BNCs) are hollow nanoparticles composed of the L
protein of hepatitis B virus (HBV) surface antigen (HBsAg), which can specifically
introduce genes and drugs into various kinds of target cells. Although the classic
electroporation method has typically been used to introduce highly charged molecules
such as DNA, it is rarely adopted for proteins due to its very low efficiency. In this study,
a novel approach to the preparation of BNC was established whereby a target protein
was pre-encapsulated during the course of nanoparticle formation. Briefly, because of
the process of BNC formation in a budding manner on the endoplasmic reticulum (ER)
membrane, the association of target proteins to the ER membrane with lipidation
sequences (ER membrane localization sequences) could directly generate
protein-encapsulating BNC in collaboration with co-expression of the L proteins. Since
the membrane-localized proteins are automatically enveloped into BNCs during the
budding event, this method can be protect the proteins and BNCs from damage caused
by electroporation and obviate the need for laborious consideration to study the optimal
conditions for protein encapsulation. This approach would be a useful method for
encapsulating therapeutic candidate proteins into BNCs.

17
PART II.
Complex carriers of affibody-displaying bio-nanocapsules and composition-varied
liposomes for HER2-expressing breast cancer cell-specific protein delivery
A bio-nanocapsule (BNC), a hollow particle composed of hepatitis B virus
(HBV) surface antigen (HBsAg), and liposome (LP) conjugation method (BNC/LP) has
been recently developed by Jung et al. (2008). The BNC/LP complex carrier could
successfully deliver fluorescence-labeled beads (100 nm) into liver cells. In this study,
we report the promising delivery of proteins incorporated in the complex carriers, which
were prepared by the BNC/LP conjugation method with specificity-altered BNC and
composition-varied LPs. The specificity-altered BNC, ZHER2-BNC was developed by
replacing the hepatocyte recognition site of BNC with ZHER2 binding to HER2 receptor
specifically. Using green fluorescent protein (GFP; 27 kDa) and cellular cytotoxic
protein (exotoxin A; 66 kDa) for the delivery, we herein present the impact of different
charges attributed to the composition of the LP on specific cell targeting and cellular
uptake of the complex carriers. In addition, we demonstrate that the mixture prepared by
mixing LPs with helper lipid possessing endosomal escaping ability boosts the
functional expression of the cellular cytotoxic exotoxin A activity specifically. Finally,
we further show the blending ratio of the LP mixture and ZHER2-BNC is a critical factor
in determining the highly-efficient expression of the cytotoxic activity of exotoxin A.

18
PART III.
Targeting cancer cell-specific RNA interference by siRNA delivery using a complex
carrier of affibody-displaying bio-nanocapsules and liposomes
Small interfering RNA (siRNA) has attracted attention in the field of nucleic
acid medicine as a RNA interference (RNAi) application that leads to gene silencing
due to specific messenger RNA (mRNA) destruction. However, since siRNA is unstable
in blood and unable to cross the cell membrane, encapsulation of siRNA into a carrier is
required. In this study, we used a carrier that combined ZHER2-displaying
bio-nanocapsule (derived from hepatitis B virus surface antigen) and liposomes in a
complex in order to investigate the feasibility of effective and target-cell-specific RNAi
applications. As a result, by observing RNAi only in HER2-expressing breast cancer
cells, using our proposed methodology, we successfully demonstrated
target-cell-specific delivery and effective function expression of siRNA.

19
PART IV.
An affinity chromatography method used to purify His-tag-displaying
bio-nanocapsules
A bio-nanocapsule (BNC) derived from hepatitis B virus (HBV) is expected
to be useful as a drug delivery system (DDS) carrier. Because various types of BNCs
have been developed, a simple and versatile purification method for BNCs would be
useful. Therefore, we planned to establish a simple purification method using affinity
chromatography by inserting a histidine tag (His-tag) into a BNC. The method achieved
a simple, one-step purification with a yield that was 2.5-fold higher than conventional
ultracentrifugation, and thus would be a desirable alternative method for BNC
purification.

20
PART V.
Granting specificity for breast cancer cells using a Hepatitis B core particle with a
HER2-targeted affibody molecule
Capsid-like particles consisting of a hepatitis B core protein (HBc) have
been studied for their potential as carriers for drug delivery systems (DDS). The hollow
HBc particle, which is formed by the self-assembly of core proteins comprising 183
amino acid residues, has the ability to bind to various cells non-specifically via the
action of an arginine-rich domain. In this study, we developed an engineered HBc
particle that specifically recognizes and targets HER2-expressing breast cancer cells. To
despoil the non-specific binding property of an HBc particle, we genetically deleted the
C-terminal 150-183 aa part of the core protein that encodes the arginine-rich domain (Δ
HBc). Then, we genetically inserted a ZHER2 affibody molecule into the 78-81 aa
position of the core protein to confer the ability of target-cell specific recognition. The
constructed a ZHER2-displaying HBc (ZHER2-ΔHBc) particle that specifically recognized
HER2-expressing SKBR3 and MCF-7 breast cancer cells. In addition, the ZHER2-ΔHBc
particle exhibited different binding amounts in accordance with the HER2 expression
levels of cancer cells. These results show that the display of other types of affibody
molecules on HBc particles would be an expandable strategy for targeting several kinds
of cancer cells that would help enable a pinpoint drug delivery system.

21
PART I.
Protein-encapsulated bio-nanocapsules production
with ER membrane localization sequences

22
INTRODUCTION
Over the past couple of decades, drug delivery systems (DDS) have been
intensively studied in order to improve the efficacy of chemotherapy and reduce its
adverse effects. The delivery of bioactive molecules such as genes, chemical
compounds and proteins to target cells is very significant for medical and biological
applications (Nagai, 2005; Tabata, 2006). For this reason, it is necessary to establish an
efficient carrier that ensures the internal stability of bioactive molecules, as well as their
delivery into the targeted cells.
The bio-nanocapsule (BNC) is an attractive carrier for the delivery of
bioactive molecules (Yamada et al., 2003). BNCs are hollow particles composed of the
L protein of the hepatitis B virus (HBV), surface antigen (HBsAg), and the lipid bilayer
derived from host cells (Kuroda et al., 1992). As carriers for drug delivery, these
virus-like particles have many advantages, as follows: high specificity for human
hepatocytes; high transfection efficiency, equivalent to the original HBV; reliable safety
arising from the absence of the viral genome; high stability in the blood; and, a high
capacity for encapsulation of genes and drugs (Yamada et al., 2003; Iwasaki et al., 2007;
Jung et al., 2008).
To target cells other than hepatocytes, the specificity of BNC can be altered
by genetic modifications. Varieties of specificity-altered BNCs have been produced by
deleting the preS region having specificity for hepatocytes in the L protein, and
inserting binding molecules targeting other cells (Kasuya et al., 2008; Kasuya et al.,
2009). Antibodies and peptides have often been selected as such affinity molecules. To
confer specificity for various kinds of cell surface receptors, antibody-mediated

23
targeting with the ZZ domain (derived from protein A) or with biotin, which binds to the
Fc region of immunoglobulin G (IgG) or streptavidin, has been developed as a practical
and versatile technique (Iijima et al., 2011; Shishido et al., 2009a). Similarly, affibody
molecules, which comprise a new class of affinity ligands derived from the Z domain
and bind a range of different proteins, e.g. insulin, HER2 and EGFR, were used as a
substitute for antibodies, while an arginine-rich peptide was displayed on BNC to
permit the delivery into various types of cells (Nygren, 2008; Shishido et al., 2009b).
As described above, BNCs are useful carriers to deliver drugs specifically to
different cell types. However, methods to encapsulate drugs into BNC have not been
studied extensively. Therefore, the classic electroporation method is commonly used for
this purpose (Yamada et al., 2003). Besides expensive equipment, this method requires
consideration of the appropriate conditions that affect the encapsulation efficiency
through various factors such as the intensity of electric voltage and pulse, temperature,
concentration of particles and drugs, and composition of buffers (Yamada et al., 2003).
Although electroporation has typically been used to introduce highly charged molecules
such as DNA, it is rarely adopted for proteins due to its very low efficiency.
Furthermore, many proteins, including pharmaceutical proteins, might suffer serious
damage from high voltage, because they have a tendency to be denatured and
agglutinated under severe conditions such as pH, heat and concentration (Chi et al.,
2003). Thus, a simple and effective method for encapsulating proteins into BNC without
using electroporation is needed.
In the present study, a novel approach to the preparation of BNC was
established, in which a target protein is pre-encapsulated in the course of particle
formation. We focused on the following mechanism for the formation of BNC (Fig.

24
1A): 1) L proteins localize and accumulate on the ER membrane; 2) aggregation of the
L proteins is initiated by the accumulated L proteins on the ER; 3) intermolecular
interactions trigger budding of the L particles; and, 4) hollow particles are formed
within the ER lumen by a nucleocapsid-independent extrusion process and then
exported from the cells via the vesicular transport pathway (Kuroda et al., 1992). BNC
is thus produced when budding forms on the ER membrane. Therefore, the working
assumption in the present study was that co-expression of the target proteins with the L
proteins that associate with the outer leaflet of the ER membrane (cytoplasm side) by
lipid modification could encapsulate the target proteins into the BNC, and would be
accompanied by the formation of particles (Fig. 1B). As a means for this approach,
lipidation sequences (membrane localization sequences; MLSs) derived from N-Ras,
which cause prenylation in the CAAX motif (Choy et al., 1999), were added to the
C-terminal of the target proteins. Since the ER membrane-localized target proteins were
automatically embedded in the BNC during the formation process, this approach never
required laborious consideration of the electroporation conditions after the preparation
of hollow BNC particles, despite procedures identical to the previous process for the
production and purification of BNC. We verified the feasibility of this strategy to
encapsulate the target proteins into the BNC with lipidation motifs.

25
Fig. 1. Schematic illustration for the process of BNC formation in insect cells. (A) A
common process of BNC formation. Translated L proteins are accumulated on the ER
membrane and aggregated by intermolecular interaction. Hollow particles are released
via budding events by self-assembly into the side of the ER lumen. (B) A strategy for
direct production of protein-encapsulating BNC. Since target proteins are localized on
the ER membrane by lipid modification, they are easily encapsulated inside BNC
through the same process of common particle formation.

26
MATERIALS AND METHODS
Construction of plasmids for the expression of membrane-localized proteins in insect
cells
MLS1 and MLS2 derived from N-Ras were selected as the lipidation
sequences (Sato et al., 2006). The plasmids for expression of the enhanced green
fluorescent protein (EGFP), attached with MLS1 or MLS2 in insect cells, were
constructed as described below (Fig. 2A). The fragments encoding the EGFP-MLS1 or
EGFP-MLS2 fusion gene were amplified by polymerase chain reaction (PCR) from
pEGFP (Takara Bio, Shiga, Japan) with the following primers: EGFP-MLS1
(5’-GGGGGATCCATGGTGAGCAAGGGCGAGGA-3’ and 5’-
GGGCCGCGGTTACATCACCACGCAGGGCAGGCCCATGCAGCCCTGCTTGTAC
AGCTCGTCCATGC-3’) and EGFP-MLS2
(5’-GGGGGATCCATGGTGAGCAAGGGCGAGGA-3’ and 5’-
GGGCCGCGGTTACATCACCACGCAGGGCAGGCCCATGGAGCCCTGCTTGTAC
AGCTCGTCCATGC-3’). The amplified fragments were digested with BamHI/SacII
and ligated into the pXIHAbla (Shishido et al., 2009c) (Fig. 2B). The resulting plasmids
were designated as pXIHAbla-EGFP-MLS1 and pXIHAbla-EGFP-MLS2. The
previously constructed plasmid pXIHAbla-EGFP (Shishido et al., 2009c) was used for
the expression of cytosolic EGFP in a comparative expression manner. In contrast,
plasmid pX-ML (Shishido et al., 2006) was used for the co-expression of BNC with
these plasmids in insect cells (Fig. 2C).

27
Fig. 2. Schematic representation of constructs to localize target proteins on the ER
membrane of insect cells. ER membrane-localized proteins would be easily
encapsulated into BNCs. (A) EGFP was used as a model for the target proteins. MLS1
and MLS2 derived from N-Ras were reported to localize on plasma or on the ER
membrane in mammalian cells. Gray characters indicate the amino acid residues
involved in lipid modifications. (B) Insect cell shuttle vector for expression of EGFP,
EGFP-MLS1 and EGFP-MLS2. (C) Expression vector for secretion of BNC in insect
cells.

28
Transfection of plasmids for the expression of EGFP-MLSs and/or BNC
A Trichoplusia ni BTI-TN-5B1-4 insect cell line (High Five) (Invitrogen,
Carlsbad, CA, USA) was maintained in a serum-free medium (Express Five SFM)
(Invitrogen) supplemented with 0.26 g/L L-glutamine and 10 mg/L gentamicin
(Invitrogen) at 27 °C. High Five cells were seeded on a 35 mm dish at a density of
2×105 cells/ml for 24 h before transfection, and the cells were then used for transfection.
For observation by confocal laser scanning microscopy, the EGFP expression
plasmid (pXIHAbla-EGFP, pXIHAbla-EGFP-MLS1 or pXIHAbla-EGFP-MLS2) was
transfected into the High Five cells using FuGENE HD transfection reagent (Roche,
Basel, Switzerland), following the manufacturer's procedure.
For purification of BNCs, pX-ML and EGFP expression plasmid
(pXIHAbla-EGFP, pXIHAbla-EGFP-MLS1 or pXIHAbla-EGFP-MLS2) were
co-transfected into High Five cells using FuGENE HD transfection reagent.
Confocal laser scanning microscopy observation of EGFP localization in insect cells
At 72 h after transfection, the cells were observed with a laser-scanning
confocal microscope (Carl Zeiss, Oberkochen, Germany), following the manufacturer's
procedure. Fluorescence images were acquired using the 488 nm line of an Ar laser for
excitation and a 505 nm band pass filter for emission. The specimens were viewed using
a 63-fold oil immersion objective.
Expression and Purification of BNCs co-expressed with EGFP-MLSs
At 72 h after transfection, the culture supernatant (20 ml) of transfected insect
cells was collected and mixed with polyethylene glycol (PEG) 6000 solution (33%, w/v).

29
After 2 h incubation, the mixture was centrifuged at 10,000 g for 30 min at 4 °C and the
precipitate was dissolved in 2.8 ml of phosphate-buffered saline (PBS). The solution
was layered onto a discontinuous cesium chloride (CsCl) gradient (11 ml, concentration:
10-40% (w/v) in buffer A [0.1 M sodium phosphate, 15 mM ethylene diamine
tetraacetic acid (EDTA)]) and centrifuged at 24,000 rpm for 16 h at 15 °C in a himac
CP70MXX centrifuge equipped with swing roter P40ST (Hitachi, Tokyo, Japan). The
amount of BNC in each fraction was analyzed using an IMx enzyme immunoassay
(EIA) kit (Abbott Laboratories, Abbott Park, IL, USA), following the manufacturer's
procedure, and BNC was dialyzed against PBS. After dialysis, the BNC solution was
layered onto a discontinuous sucrose gradient (11 ml, concentration: 10-50% (w/v) in
buffer A) and centrifuged at 24,000 rpm for 10 h at 4 °C. The amount of BNC in each
fraction was determined using the IMx EIA kit, and the expression of EGFP was
confirmed by western blotting. Fractions containing BNC were dialyzed against PBS
and stored at 4 °C.
SDS-PAGE and western blotting
The expression of EGFP in each fraction was confirmed by western blotting.
The supernatant was fractionated by sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE) and electrotransferred onto a polyvinilidene fluoride
(PVDF) membrane. Rabbit anti-EGFP antibodies (Medical Biological Laboratories,
Nagoya, Japan) were used for immunoblotting, followed by anti-rabbit antibodies
conjugated with alkaline phosphatase (AP) (Promega, Madison, WI, USA). The
membrane was stained with 5-bromo-4-chloro-3-indolyl phosphate (BCIP) and nitro
blue tetrazolium (NBT) (Promega).

30
Dynamic Light Scattering Analysis of Purified BNCs co-expressed with EGFP-MLSs
The size of the purified BNCs co-expressed with EGFP-MLSs was determined
by dynamic light scattering (DLS) using a Zetasizer Nano particle size analyzer
(Malvern Instruments Ltd., Worcestershire, UK), following the manufacturer's
procedure.
RESULTS AND DISCUSSION
Strategy for direct production of protein-encapsulating BNC
The aim of the present study was to establish a novel approach that would
enable the simple preparation of protein-encapsulating BNC. Because BNC is produced
by a bioprocess, we hypothesized that BNC that inherently encapsulated the protein
drug candidates could be prepared with genetic modifications. If protein-encapsulating
BNC could be produced by the same process that is commonly used for preparing
hollow BNC particles, this would permit the protection of BNC and proteins from
damage caused by electroporation and obviate the need for laborious efforts to study the
optimal conditions for protein encapsulation.
For these reasons, we focused on the formation mechanism of BNC, that is,
budding on the ER membrane, as shown in Fig. 1A (Kuroda et al., 1992). We assumed
that the co-expression of target proteins on the ER membrane might directly generate
protein-encapsulating BNC by enveloping the membrane-localized proteins during the
budding event (Fig. 1B). The strategy used to test the feasibility of this approach was to

31
introduce MLSs into the C-terminus of the target proteins. Two types of peptide motifs,
11-amino-acid sequences derived from N-Ras including the CAAX motif, were selected
as the MLSs for the lipidation. MLS1 (QGCMGLPCVVM) is lipidated through both
prenylation at the cysteine residue on the CAAX motif and palmitoylation at the
upstream cysteine residue (Choy et al., 1999) (Fig. 2A). However, MLS2
(QGSMGLPCVVM) is lipidated by only prenylation at the cysteine residue on the
CAAX motif, since the Cys3 of MLS1 is replaced with a serine residue (Choy et al.,
1999) (Fig. 2A). According to the literature, MLS1 was localized to the plasma
membrane, and MLS2 was localized to the ER membrane and golgi membrane
apparatus in mammal cells (Sato et al., 2006). For the present study, an insect cell
allowing secretory production of BNC (Shishido et al., 2006) was used as the host cell,
and EGFP was used as the model target protein, which facilitated the evaluation of both
localization and encapsulation of BNC.
Localization of target proteins with membrane localization sequences (MLSs) in
insect cells
To confirm whether MLSs have ER membrane localization abilities in insect
cells, plasmids were constructed expressing EGFP, EGFP-MLS1, and EGFP-MLS2 (Fig.
2A). These three types of plasmids were transfected into insect cells (High Five)
without the plasmid producing BNC, and their localization was observed with a
confocal laser-scanning microscope (Fig. 3).
Because of its lack of membrane localization ability, EGFP without MLS was
observed in the cytoplasm of insect cells. EGFP-MLS1 was evenly localized on the
plasma and ER membranes in insect cells, although MLS1 reportedly locates on the

32
Fig. 3. Fluorescence images for observation of the localization of EGFP in insect cells
with confocal microscopy. ER-tracker (Invitrogen) was used as the localization marker.
The upper images are the cells transfected with EGFP expression plasmid. The middle
images are the cells transfected with EGFP-MLS1 expression plasmid. The lower
images are the cells transfected with EGFP-MLS2 expression plasmid. Scale bars; 20
m.

33
plasma membrane in mammal cells. In contrast, EGFP-MLS2 was strongly but partially
localized to the ER membrane. Thus, in the present study, both MLS1 and MLS2
functioned as membrane localization sequences in insect cells and had the capacity to
localize EGFP on ER membranes, even though they varied in their ER localization
ability. This result indicates that both MLS1 and MLS2 are capable of localizing target
proteins on an ER membrane as therapeutic candidates in a similar fashion.
Production and purification of EGFP-encapsulating BNC
To investigate the validity of our approach, the three types of plasmids (for
expression of EGFP, EGFP-MLS1 and EGFP-MLS2) were co-transfected, with the
plasmid producing BNC, into insect cells (High Five). After 72 h of cultivation, the
supernatants were harvested and the BNCs were purified by gradient ultracentrifugation,
as described in materials and methods. The resultant fractions were analyzed by EIA to
measure the amount of BNC and by western blotting to evaluate whether the BNCs
encapsulated EGFP (Fig. 4). After dialysis, about 25 g of purified EGFP-MLS1/BNC
and EGFP-MLS2/BNC were obtained from 20 ml of culture medium supernatant (Table
1).
First, in the case of co-transfection of EGFP and BNC, although the main
peaks of BNCs appeared in 10-12 fractions, the bands of EGFP were not detected in the
same fractions (Fig. 4A). This result indicates that EGFP was not encapsulated in BNC,
although BNC was produced uneventfully in the insect cells. Second, in the case of
co-transfection of EGFP-MLS1 and BNC, the thick bands of EGFP were detected in
9-12 fractions, which displayed the main peaks of BNC (Fig. 4B). This suggests that
EGFP-encapsulating BNC was successfully produced by introduction of the MLS1

34
Fig. 4. Examination for encapsulation of EGFP into the purified BNCs. After sucrose
gradient centrifugation, the amount of BNC including each fraction was measured with
a IMx EIA kit (S/N value of EIA). The same fractions were tested for the presence of
EGFP by western blotting with anti-EGFP antibody. Co-expression of (A) BNC and
EGFP, (B) BNC and EGFP-MLS1, and (C) BNC and EGFP-MLS2.

35
24.9 35.5 0.7 after concentration
38.7 6.5 6.0 after Sucrose ultracentrifugal method
174.3 58.1 3.0 after CsCl ultracentrifugal method
2033.3 726.2 2.8 pellet after PEG settling method
53672.3 2683.6 20.0 culture medium supernatant
EGFP-MLS2/BNC
25.8 51.6 0.5 after concentration
19.4 3.2 6.0 after Sucrose ultracentrifugal method
164.6 54.9 3.0 after CsCl ultracentrifugal method
3416.0 1220.0 2.8 pellet after PEG settling method
55296.6 2764.8 20.0 culture medium supernatant
EGFP-MLS1/BNC
mass (g)concentration (g/ml)
volume (ml)stepSample name
24.9 35.5 0.7 after concentration
38.7 6.5 6.0 after Sucrose ultracentrifugal method
174.3 58.1 3.0 after CsCl ultracentrifugal method
2033.3 726.2 2.8 pellet after PEG settling method
53672.3 2683.6 20.0 culture medium supernatant
EGFP-MLS2/BNC
25.8 51.6 0.5 after concentration
19.4 3.2 6.0 after Sucrose ultracentrifugal method
164.6 54.9 3.0 after CsCl ultracentrifugal method
3416.0 1220.0 2.8 pellet after PEG settling method
55296.6 2764.8 20.0 culture medium supernatant
EGFP-MLS1/BNC
mass (g)concentration (g/ml)
volume (ml)stepSample name
Table 1. Purification summary of EGFP-MLS1/BNC and EGFP-MLS2/BNC.

36
motif. In the third case, co-transfection of EGFP-MLS2 and BNC displayed a result
similar to the case of EGFP-MLS1 and BNC (Fig. 4C), suggesting that the introduction
of MLS2 also allowed the production of EGFP-encapsulating BNC. The smaller
amounts of EGFP in the BNC with MLS2 might be attributed to partial localization on
the ER. However, since the EGFP-encapsulating BNC with MLS2 produced almost
twice the amount of particles as that with MLS1, this suggests that MLS2 might be a
better expression system for protein-encapsulating BNC (Fig. 4B and 4C). These
differences might be due to the presence or absence of the palmitoylation site between
MLS1 and MLS2.
Finally, the diameters of the BNC particles were evaluated using the DLS
method (Fig. 5). The diameters of the three types of particles were almost equivalent, at
150 nm, indicating that the diameter of EGFP-encapsulating BNCs was similar to that
of hollow BNC particles produced in insect cells (Kurata et al., 2008). In addition, it
was also confirmed that the EGFP-encapsulating BNCs kept the targeting abilities to
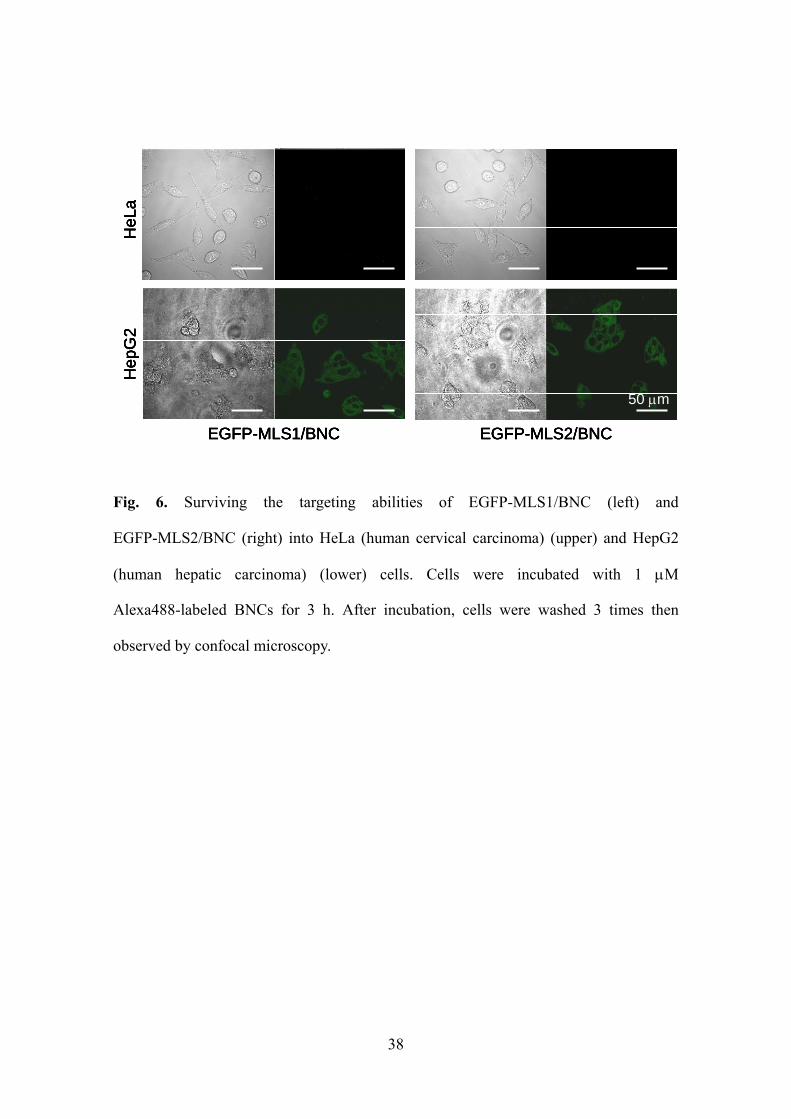
human hepatocytes (Fig. 6).

37
Fig. 5. DLS analyses of purified BNCs. Co-expression of (A) BNC and EGFP, (B) BNC
and EGFP-MLS1, and (C) BNC and EGFP-MLS2.
38
Fig. 6. Surviving the targeting abilities of EGFP-MLS1/BNC (left) and
EGFP-MLS2/BNC (right) into HeLa (human cervical carcinoma) (upper) and HepG2
(human hepatic carcinoma) (lower) cells. Cells were incubated with 1 M
Alexa488-labeled BNCs for 3 h. After incubation, cells were washed 3 times then
observed by confocal microscopy.
EGFP-MLS1/BNC EGFP-MLS2/BNC
HeL
aH
epG
2
EGFP-MLS1/BNC EGFP-MLS2/BNC
HeL
aH
epG
2
50 m
EGFP-MLS1/BNC EGFP-MLS2/BNC
HeL
aH
epG
2
EGFP-MLS1/BNC EGFP-MLS2/BNC
HeL
aH
epG
2
50 m

39
CONCLUSION
The feasibility of this approach to the direct production of
protein-encapsulating BNC by localizing the target proteins on the ER membrane was
successfully demonstrated. In this study, MLS1 and MLS2 of N-Ras were used to
localize the target proteins on the ER membrane either by prenylation or by
palmitoylation. While MLS1 and MLS2 could incorporate our approach, other ER
membrane localization sequences with different modification mechanisms might also be
utilized to produce protein-encapsulating BNCs. In addition, whereas therapeutic
candidate proteins might be encapsulated in BNC in the same manner as EGFP, this
should be demonstrated in the near future. This approach would be a useful tool for
encapsulating target proteins into BNCs.
ABBREVIATIONS
drug delivery system, DDS; bio-nanocapsule, BNC; hepatitis B virus, HBV; hepatitis B
virus surface antigen, HBsAg; endoplasmic reticulum, ER; immunoglobulin G, IgG;
human EGFR-related 2, HER2; epidermal growth factor receptor, EGFR; membrane
localization sequence, MLS; enhanced green fluorescent protein, EGFP; polymerase
chain reaction, PCR; polyethylene glycol, PEG; phosphate-buffered saline, PBS;
discontinuous cesium chloride, CsCl; ethylene diamine tetraacetic acid, EDTA; enzyme
immunoassay, EIA; sodium dodecyl sulphate-polyacrylamide gel electrophoresis,

40
SDS-PAGE; electrotransferred onto a polyvinilidene fluoride, PVDF; alkaline
phosphatase, AP; 5-bromo-4-chloro-3-indolyl phosphate, BCIP; nitro blue tetrazolium,
NBT; dynamic light scattering, DLS
REFERENCES
Chi, E.Y., Krishnan, S., Randolph, T.W., Carpenter, J.F., 2003. Physical stability of
proteins in aqueous solution: mechanism and driving forces in nonnative protein
aggregation. Pharm Res. 20, 1325-1336.
Choy, E., Chiu, V.K., Silletti, J., Feoktistov, M., Morimoto, T., Michaelson, D., Ivanov,
I.E., Philips, M.R., 1999. Endomembrane trafficking of ras: the CAAX motif targets
proteins to the ER and Golgi. Cell. 98, 69–80.
Iijima, M., Kadoya, H., Hatahira, S., Hiramatsu, S., Jung, G., Martin, A., Quinn, J., Jung,
J., Jeong, S.Y., Choi, E.K., Arakawa, T., Hinako, F., Kusunoki, M., Yoshimoto, N.,
Niimi, T., Tanizawa, K., Kuroda, S., 2011. Nanocapsules incorporating IgG Fc-binding
domain derived from Staphylococcus aureus protein A for displaying IgGs on
immunosensor chips. Biomaterials. 32, 1455-1464.
Iwasaki, Y., Ueda, M., Yamada, T., Kondo, A., Seno, M., Tanizawa, K., Kuroda, S.,
Sakamoto, M., Kitajima, M., 2007. Gene therapy of liver tumors with human

41
liver-specific nanoparticles. Cancer Gene Ther. 14, 74-81.
Jung, J., Matsuzaki, T., Tatematsu, K., Okajima, T., Tanizawa, K., Kuroda, S., 2008.
Bio-nanocapsule conjugated with liposomes for in vivo pinpoint delivery of various
materials. J Control Release. 126, 255-264.
Kasuya, T., Jung, J., Kadoya, H., Matsuzaki, T., Tatematsu, K., Okajima, T., Miyoshi,
E., Tanizawa, K., Kuroda, S., 2008. In Vivo Delivery of Bionanocapsules
Displaying Phaseolus vulgarisAgglutinin-L4 Isolectin to Malignant Tumors
Overexpressing N-Acetylglucosaminyltransferase V. Human Gene Therapy. 887-895.
Kasuya, T., Jung, J., Kinoshita, R., Goh, Y., Matsuzaki, T., Iijima, M., Yoshimoto, N.,
Tanizawa, K., Kuroda, S., 2009. Bio-Nanocapsule–Liposome Conjugates for In Vivo
Pinpoint Drug and Gene Delivery. Methods in Enzymology. 464, 147-166.
Kurata, N., Shishido, T., Muraoka, M., Tanaka, T., Ogino, C., Fukuda, H., Kondo, A.,
2008. Specific protein delivery to target cells by antibody-displaying bionanocapsules. J
Biochem. 144, 701-707.
Kuroda, S., Otaka, S., Miyazaki, T., Nakao, M., Fujisawa, Y., 1992. Hepatitis B virus
envelope L protein particles. Synthesis and assembly in Saccharomyces cerevisiae,
purification and characterization. J Biol Chem. 267, 1953-1961.
Nagai, T., 2005. Drug discovery and innovative drug delivery research in new drug

42
development. Pharm Tech Japan. 21, 1949-1951.
Nygren, P.A., 2008. Alternative binding proteins: affibody binding proteins developed
from a small three-helix bundle scaffold. FEBS J. 275, 2668-2676.
Sato, M., Ueda, Y., Umezawa, Y., 2006. Imaging diacylglycerol dynamics at organelle
membranes. Nat Methods. 3, 797-799.
Shishido, T., Muraoka, M., Ueda, M., Seno, M., Tanizawa, K., Kuroda, S., Fukuda, H.,
Kondo, A., 2006. Secretory production system of bionanocapsules using a stably
transfected insect cell line. Appl Microbiol Biotechnol. 73, 505-511.
Shishido, T., Azumi, Y., Nakanishi, T., Umetsu, M., Tanaka, T., Ogino, C., Fukuda, H.,
Kondo, A., 2009a. Biotinylated bionanocapsules for displaying diverse ligands toward
cell-specific delivery. J Biochem. 146, 867-874.
Shishido, T., Yonezawa, D., Iwata, K., Tanaka, T., Ogino, C., Fukuda, H., Kondo, A.,
2009b. Construction of arginine-rich peptide displaying bionanocapsules. Bioorg Med
Chem Lett. 19, 1473-1476.
Shishido, T., Kurata, N., Yoon, M.E., Tanaka, T., Yamaji, H., Fukuda, H., Kondo, A.,
2009c. A high-level expression vector containing selectable marker for continuous
production of recombinant protein in insect cells. Biotechnol Lett. 31, 623-627.
Tabata, T., 2006. Drug delivery system: Basic technology for biomedical research,

43
medical treatment and health care. Biotechnol J. 6, 553-555.
Yamada, T., Iwasaki, Y., Tada, H., Iwabuki, H., Chuah, M.K., VandenDriessche, T.,
Fukuda, H., Kondo, A., Ueda, M., Seno, M., Tanizawa, K., Kuroda, S., 2003.
Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat Biotechnol.
21, 885-890.

44
PART II.
Complex carriers of affibody-displaying bio-nanocapsules and
composition-varied liposomes for HER2-expressing
breast cancer cell-specific protein delivery

45
INTRODUCTION
A drug delivery system (DDS) is a technology that enables control of drug
distributions in the body on the basis of quantitative, spatial and temporal aspects. If the
delivery of biological active molecules (ex. DNA, RNA, medicinal chemicals and
pharmaceutical proteins) universally becomes available, improvements in therapeutic
effects and reductions in side effects should follow (Nagai, 2005; Tabata, 2006). The
development of a variety of tools and carriers for DDS is an area of emerging research.
As the carrier, a bio-nanocapsule (BNC) that is composed of the L protein of
the hepatitis B virus (HBV) surface antigen (HBsAg) and the lipid bilayer has many
attractive features (Kuroda, 1992). The original BNC shows high specificity for human
hepatocytes and high transfection efficiency equivalent to the original HBV. Moreover,
BNC exhibits a reliable safety profile and can incorporate drugs and genes by an
electroporation method since it is a viral-genome-free hollow nanoparticle (Yamada,
2003).
Previously, we and other researchers succeeded in altering the cell-specificity
of BNC by genetic modifications (Kasuya, 2008; Shishido, 2009a; Shishido, 2009b).
Several varieties of specificity-altered BNCs could be generated by deleting the
hepatocyte-specific recognition site (located in the preS region) in the L protein and
inserting binding molecules with the ability to target other cells. Using this technique
and an affibody molecule, a new class of affinity ligands derived from the Z domain of
staphylococcal protein A (Orlova, 2006; Lee, 2008), we have constructed the ZHER2
affibody molecule displaying BNC on its surface (ZHER2-BNC) whose specificity was
successfully altered from hepatocytes to HER2 receptor expressing cells such as breast

46
cancer and ovarian cancer cells (Shishido, 2010). In our previous study, we reported the
specificity alteration of BNC by using the small and easily detectable molecule
fluorescein, although further characterizations and applications of ZHER2-BNC are still
needed. For example, since the original HBV possesses the unique infectious entry
mechanism of hepadnaviruses via receptor-mediated endocytosis followed by
processing of a surface protein including the preS region in endosomes (Stoeckl, 2006),
the specificity-altered ZHER2-BNC in which the preS region is partly deleted, might
result in the problematic trapping of medicinal agents within the endosomes.
Alternatively, a new method to conjugate BNCs with the liposome (LP) by
first incorporating the materials together (BNC/LP conjugation method) was recently
developed by Jung et al (Jung, 2008) as an alternative to the conventional
electroporation method. They successfully demonstrated that the conjugated BNC/LP
complex could incorporate large materials including fluorescence-labeled beads (100
nm). They also succeeded in delivering a GFP expression plasmid (>30 kbp) and
specifically imparting green fluorescence to human hepatocytes both ex vivo and in vivo
using the original BNC. This suggested that a new type of complex carrier based on the
original BNC could release a gene into the cytoplasm by escaping from the endocytic
pathway because of the unique endocytosis mechanism derived from original HBV
(Jung, 2008). However, complex carriers prepared by conjugating the specificity-altered
BNCs with LPs, in addition to preparation of complexes incorporating proteins that are
comparatively large biomolecules have not been reported. Furthermore, the
characteristics of LPs have not been reported as the features of the lipids used for the
BNC/LP conjugation have never been evaluated closely.
In this study, we attempted for the first time to incorporate comparatively

47
large proteins into the complex carriers prepared by the BNC/LP conjugation method
with the specificity-altered BNC and also aimed to determine the impacts of
characteristic lipids on the protein delivery. To confer the specificity for HER2
expressing cells on the complex carriers, we selected ZHER2-BNC (ZHER2-displaying
BNC) for the conjugation with LPs. Moreover, we investigated the impact of LPs with
different charges on the cell targeting specificities of the complexes and cellular uptake
of the proteins when using three types of LPs, anionic-LP (ALP), nonionic-LP (NLP)
and cationic-LP (CLP) for conjugating ZHER2-BNC. Based on the obtained results, we
investigated boosting the expression efficiency of the incorporated protein activity by
using helper lipids with endosomal escaping abilities.
MATERIALS AND METHODS
Materials
BNCs were prepared from Saccharomyces cerevisiae AH22R- harboring the
plasmid pGLDsLd50-ZHER2 (Shishido, 2010) as described previously (Kuroda, 1992).
Briefly, yeast cells transformed with pGLDsLd50-ZHER2 by the spheroplast method were
cultured and disrupted with glass beads, the crude extract was precipitated with
polyethylene glycol (PEG) 6000 and subjected to cesium chloride (CsCl) isopycnic
ultracentrifugation and sucrose density gradient ultracentrifugation, and then the
purified ZHER2-BNC was obtained after freeze-drying in the presence of 5% sucrose.
Green fluorescent protein (GFP) was obtained from One Shot® TOP10 ElectrocompTM

48
Escherichia coli (Invitrogen Life Technologies, Carlsbad, CA, USA) harboring the
plasmid to express the enhanced GFP containing His tag (pBAD, unpublished plasmid)
by purifying the soluble fraction of the lysate using TALON metal affinity resins
(Clontech Laboratories / Takara Bio, Shiga, Japan). Liposomes (LPs) were purchased
from NOF (Tokyo, Japan). COATSOME EL-01-A (dipalmitoyl-phosphatidylcholine
(DPPC) : cholesterol (CHOL) : dipalmitoyl-phosphatidylglycerol (DPPG) = 30 : 40 : 30
(mol/vial)), COATSOME EL-01-N (DPPC : CHOL : DPPG = 54 : 40 : 6 (mol/vial))
and COATSOME EL-01-C (DPPC : CHOL : stearyl-amine = 52 : 40 : 8 (mol/vial))
were respectively selected as ALP, NLP and CLP. COATSOME EL-01-D
(1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) : CHOL :
O,O’-ditetradecanoyl-N-(-trimethylammonioacetyl) diethanolamine chloride (DC6-14)
= 0.75 : 0.75 : 1.00 (mol/vial)) was selected as the helper lipid. Pseudomonas exotoxin
A from Pseudomonas aeruginosa was purchased from Sigma-Aldrich (St. Louis, MO,
USA). Gibco® Fetal bovine serum (FBS), L-glutamine and Molecular Probes®
LIVE/DEAD® viability/cytotoxicity assay kit were purchased from Invitrogen Life
Technologies. RPMI 1640 medium and Dulbecco’s modified Eagle medium (DMEM)
were purchased from Nacalai Tesque (Kyoto, Japan). Leibovitz L-15 medium was
purchased from MP Biomedicals (Irvine, CA, USA). Sephacryl™ S-500 HR column
was purchased from GE Healthcare (Buckinghamshire, England).
Preparation of ZHER2-BNC/ALP, NLP and CLP complexes incorporating GFP or
exotoxin A
Complex carriers of ZHER2-BNC and LPs (ALP, NLP and CLP), in which GFP
or exotoxin A was incorporated, were prepared by referring to the previously described

49
BNC/LP conjugation method with some modifications (Jung, 2008). Freeze-dried LPs
(COATSOME EL-01-A, 61 mg; EL-01-N, 61 mg; and EL-01-C, 57 mg) were dissolved
in distilled water (2 ml) containing 2 mg/ml of GFP or 100 g/ml of exotoxin A. After
incubation for 1 h at room temperature, gel-filtration chromatography was performed
only for the LPs containing GFP using a Sephacryl™ S-500 HR column with an AKTA
system. The obtained LPs incorporating GFP or exotoxin A (100 l) were added to
freeze-dried ZHER2-BNC (100 g as protein) and incubated at room temperature for 1 h
to form BNC/LP complexes incorporating GFP or exotoxin A. The resultant complex
carriers were named ZHER2-BNC/ALP, ZHER2-BNC/NLP and ZHER2-BNC/CLP.
Preparation of ZHER2-BNC/DLP (DOPE-containing LP) complexes incorporating
exotoxin A
Complex carriers of ZHER2-BNC and DOPE-containing LP mixtures, in which
exotoxin A was incorporated, were prepared according to the above-described method
with the following modifications. To generate DOPE-containing LP mixtures (DLPs;
ADLP or NDLP), 2.2 mg of COATSOME EL-01-A (ALP) or COATSOME EL-01-N
(NLP) was added to 1 vial (1.5 mg) of COATSOME EL-01-D (DOPE-containing
cationic helper lipid). By mixing various amounts of COATSOME EL-01-A (ALP) into
a certain amount (1.5 mg) of COATSOME EL-01-D (DLP), the mixture ratio was
determined to give the negative zeta potential (Table 1). The generated LP mixture
(ADLP or NDLP; 3.7 mg) was used as a substitute for the freeze-dried LPs in the
previous section and dissolved in distilled water (1 ml). The amount of ZHER2-BNC was
varied from 0 g to 100 g (in terms of protein). The resultant complex carriers were
named as ZHER2-BNC/ADLP and ZHER2-BNC/NDLP.

50
Table 1. The sizes and zeta potentials of DOPE-containing LP mixtures.

51
Cell culture
SKBR3 cells (human breast carcinoma, approximately 106 HER2 molecules
expressed per cell (McLarty, 2009)) were maintained in RPMI 1640 medium
supplemented with 10% (v/v) FBS at 37°C in 5% CO2. MDA-MB-231 cells (human
breast carcinoma) were maintained in Leibovitz L-15 medium supplemented with 15%
FBS and 2 mM L-glutamine at 37°C without CO2. HeLa cells (human cervical
carcinoma) and MCF-7 cells (human breast carcinoma) were maintained in DMEM
medium supplemented with 10% FBS at 37°C in 5% CO2.
Microscopic observation of GFP delivery
Approximately 5×104 SKBR3 or MDA-MB-231 cells were seeded in 35 mm
glass bottom dishes. After washing with serum-free medium, 20 l of the complex
carriers and LPs containing GFP were added to 980 l of the medium and then the cells
were cultured for 1 h. After washing with serum-free medium twice, cells were
incubated with FBS-containing medium for 2 h. Cells were observed by a LSM 5
PASCAL laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany) using
a 63-fold oil immersion objective lens with excitation using the 488-nm line of an argon
laser and emission collection using a 505-nm long pass filter.
Microscopic observation of exotoxin A delivery
Approximately 2×105 SKBR3, MCF-7 or HeLa cells were seeded in 12-well
plates. After washing with serum-free medium, the required volumes of the complex
carriers and LPs containing exotoxin A were added to the medium and the volume was
adjusted to 1 ml and then cells were cultured for 1 h. After washing with serum-free

52
medium twice, the cells were incubated with FBS-containing medium for 47 h. Dead
cells were stained with ethidium homodimer-1 (EthD-1) from the LIVE/DEAD®
viability/cytotoxicity assay kit according to the manufacturer’s instructions. Cells were
observed by laser scanning confocal microscope using the same procedure described in
the previous section except for employing excitation using the 543-nm line of an He-Ne
laser and emission collection using a 560-nm long pass filter.
Flow cytometric evaluation of exotoxin A delivery
The cells were treated with the complex carriers and LPs containing exotoxin A
in the same manner as described in the previous section. Live cells were stained with
calcein AM from the LIVE/DEAD® viability/cytotoxicity assay kit according to the
manufacturer’s instructions. Cells were suspended into sheath solution and subjected to
a BD FACSCanto II flow cytometer equipped with a 488-nm blue laser (BD
Biosciences, San Jose, CA, USA). The green fluorescence signals were collected
through a 530/30-nm band-pass filter. The data were analyzed using the BD FACSDiva
software v5.0 (BD Biosciences). Dead cell numbers were estimated by subtracting the
viable cell counts from total cell counts.
Measuring the sizes and zeta potentials of particles.
The sizes and zeta potentials of the LPs and BNC-LP complexes were
determined by a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK),
following the manufacturer's procedure.

53
RESULTS AND DISCUSSION
The main purpose of this study was to investigate how cancer cell-specific
drug delivery is affected by the type of LP which is used to prepare the complex carriers
that incorporate the medicinal agents by the BNC/LP conjugation method (Jung, 2008).
Additionally, we also investigated target-cell-specific protein delivery since there have
been no reports regarding complex carriers using specificity-altered BNCs and the
incorporation of proteins. By conjugating various kinds of LPs with ZHER2-displaying
BNC (ZHER2-BNC) in which the hepatocyte-specific recognition site of original BNC
was genetically altered to the HER2-specific binding molecule ZHER2 (Shishido, 2010),
we evaluated the specificity of the complexes to target HER2-expressing SKBR3 breast
cancer cells and the expression efficiency of the incorporated protein activity. At the
same time, we used three types of HER2-negative cells (MDA-MB-231, HeLa and
MCF-7) to show clearly HER2-specific binding ability of the complexes. First, we
visually observed the targeting specificities and cellular uptakes of the BNC/LP
complexes incorporating the fluorescent protein GFP (27 kDa). Subsequently, we
examined the expression efficiencies of cell cytotoxicity with the variety of
composition-altered BNC/LP complexes, which incorporate the cytotoxic protein,
Pseudomonas exotoxin A (66 kDa), that can kill cells by inhibiting protein synthesis via
the ADP ribosylation of elongation factor 2 (Allured, 1986).
Influence of different charges of LPs with varied lipid compositions on cell targeting
specificities and cellular uptakes of ZHER2-displaying BNC/LP complexes
To examine the influence of different charges of LPs with varied lipid

54
compositions on the cell targeting specificities and cellular uptakes of ZHER2-displaying
BNC/LP complexes, we conjugated ZHER2-BNC with three kinds of LPs: anionic ALP,
nonionic NLP and cationic CLP (COATSOME EL-01-A, EL-01-N and EL-01-C),
respectively. The resultant complex carriers were named ZHER2-BNC/ALP,
ZHER2-BNC/NLP and ZHER2-BNC/CLP. Through the conjugations, GFP was
incorporated into each ZHER2-BNC/LP complex so that its cellular localization could be
visualized. All three types of LPs incorporating GFP without the conjugation to the
ZHER2-BNC were also used for comparison purposes. After 1 h of incubation with the
LPs or the ZHER2-BNC/LP complexes, HER2-positive SKBR3 cells (Davison, 2011) and
HER2-negative MDA-MB-231 cells (Davison, 2011) were washed twice and
additionally incubated for 2 h, and then observed by the confocal laser scanning
microscope (Fig. 1).
Both types of cells treated with the ALP and the NLP lacking the ZHER2-BNC
conjugations did not show fluorescence (Fig. 1). MDA-MB-231 cells also exhibited no
fluorescence after treatment with the ZHER2-BNC/ALP and ZHER2-BNC/NLP complexes,
whereas after treatment, SKBR3 cells exhibited the green fluorescence inside the cells
(Fig. 1). These results indicate that the LPs with an anionic or nonionic charge could
obtain the HER2-specific targeting ability by conjugation with the ZHER2-displaying
BNC, thereby permitting cellular uptake of the BNC/LP complexes incorporating GFP.
Both SKBR3 and MDA-MB-231 cells treated with the CLP and the
ZHER2-BNC/CLP complex showed green fluorescence on the periphery of the cell
membranes (Fig. 1). Due to the cationic charge of CLP, it would be presumed that even
the conjugated complex with ZHER2-BNC has non-specifically bound to the
negatively-charged cell membrane through electrostatic interactions. Indeed, the

55
Fig. 1. Fluorescence images of HER2-positive SKBR3 and HER2-negative
MDA-MB-231 cells treated with ZHER2-BNC/LP complexes incorporating GFP. ALP,
NLP and CLP with different (anionic, nonionic and cationic) charges were dissolved in
distilled water containing 2 mg/ml of GFP, and then were used to prepare the
ZHER2-BNC/LP complexes as described in materials and methods. All three types of LPs
incorporating GFP without conjugation to the ZHER2-BNC were also used as the carriers
for comparison. Cells were incubated for 1 h in the media adjusted to 1 ml by adding 20
l of the complex carriers and LPs, respectively. After washing twice, cells were
additionally incubated for 2 h and then observed by a confocal laser scanning
microscope. Scale bar, 50 m.

56
ZHER2-BNC/CLP complex showed the positively-charged zeta potential (Table 2). These
results suggest that the LPs with anionic or nonionic charges are favorable for the
preparation of the BNC/LP complex by conjugation because the cell targeting
specificity of BNC can be maintained.
Incorporation of cytotoxic protein, exotoxin A, into ZHER2-displaying BNC/LP
complexes prepared by using LPs with different charges
To investigate whether the ZHER2-BNC/LP complexes prepared by using the
LPs with different charges are able to express the incorporated protein activity in
keeping with the specificity towards HER2-expressing target cells, we prepared the LPs
and ZHER2-BNC/LP complexes incorporating the cell cytotoxic exotoxin A with ALP,
NLP and CLP. After 1 h of incubation with the obtained LPs or ZHER2-BNC/LP
complexes, HER2-positive SKBR3 cells and HER2-negative MCF-7 cells (Davison,
2011) were washed twice and additionally incubated for 47 h, and then stained with
EthD-1, which displays red fluorescence via a dead-cell-specific uptake mechanism.
Then, the stained cells were observed by confocal laser scanning microscopy to check
the expression of cell killing activity resulting from the effectiveness of exotoxin A (Fig.
2).
Both the SKBR3 (target cells) and MCF-7 (control cells) treated with the CLP
and ZHER2-BNC/CLP complexes incorporating exotoxin A exhibited red fluorescence,
showing that both carriers non-specifically caused cell death regardless of whether or
not the cells were expressing the HER2 receptor (Fig. 2). It is thought that the carriers
containing the LPs with cationic charges bound to the cell surface in a non-specific
fashion and then released the exotoxin A into the cytoplasm by membrane fusion in both

57
Table 2. The sizes and zeta potentials of LPs, LP mixtures and ZHER2-BNC/LP
complexes.

58
Fig. 2. Fluorescence images of HER2-positive SKBR3 and HER2-negative MCF-7 cells
treated with ZHER2-BNC/LP complexes incorporating exotoxin A. ALP, NLP and CLP
with different (anionic, nonionic and cationic) charges were dissolved in distilled water
containing 100 g/ml of exotoxin A, and then were used to prepare the ZHER2-BNC/LP
complexes as described in materials and methods. All three types of LPs incorporating
exotoxin A without conjugation to the ZHER2-BNC were also used as the carriers for
comparison. Cells were incubated for 1 h in the media adjusted to 1 ml by adding 20 l
of the complex carriers and LPs, respectively. After washing twice, cells were
additionally incubated for 47 h and then stained with EthD-1. Then, the stained cells
were observed by a confocal laser scanning microscope. Scale bar, 50 m.

59
target and non-target cells.
Both cells treated with the ZHER2-BNC/ALP and ZHER2-BNC/NLP complexes
never displayed red fluorescence (Fig. 2). As expected, both cells treated with the ALP
and NLP lacking the ZHER2-BNC also showed nearly no fluorescence (Fig. 2). These
results indicated that the ZHER2-BNC/LP complexes composed of the LPs with anionic
and nonionic charges did not lead to the death of MCF-7 control cells or SKBR3 target
cells since they were probably unable to effectively express the cytotoxic activity of
exotoxin A inside the cells. This was also supported by the results of quantitative FACS
analysis used to measure the fatality rates of target cells (SKBR3) and non-target cells
(HeLa) (Jia, 2003) with the same complexes containing exotoxin A (Fig. 3A and 3B;
white bars). Because SKBR3 cells treated with the ZHER2-dispalying BNC/LP
complexes incorporating GFP by the conjugation with ALP and NLP showed locally
punctate fluorescence patterns (Fig. 1), it was expected that the exotoxin A would be
introduced into HER2-expressing SKBR3 cells via receptor-mediated endocytosis but
remain within the endosome without being released into cytoplasm, resulting in no
expression of cell killing activity. This seemed to be different from the result obtained
for the plasmid incorporated complex carrier prepared by using the original BNC (Jung,
2008). Previously it had been elucidated that the endosomal escape of original HBV is
induced by the unmasked cell-permeable peptide [translocation motif (TLM); located in
the preS region] that is exposed on the surface of mature viral particles across the
conformational changes in surface proteins arising from processing by endosomal
proteases (Stoeckl, 2006). However, since the TLM sequence was deleted when
constructing the ZHER2-BNC with the aim to minimize the preS region in order to reduce
the antigenic region and the possibility of protease degradation (Shishido, 2009a;

60
Fig. 3. Fatality rates of HER2-positive SKBR3 and HER2-negative HeLa cells treated
by the exotoxin A containing ZHER2-BNC/LP complexes with and without helper lipid.
DOPE-containing cationic LP was selected as the helper lipid and mixed with ALP or
NLP to generate DOPE-containing LP mixtures (ADLP or NDLP). The LP mixtures
(ADLP or NDLP) or LPs (ALP or NLP) were dissolved in distilled water containing
100g/ml of exotoxin A, and then used to prepare the ZHER2-BNC/LP complexes as
described in materials and methods. Cells were incubated for 1 h in the media adjusted
to 1 ml by adding the indicated volumes of the complex carriers. After washing twice,
cells were additionally incubated for 47 h and then stained with calcein AM. Then, the
stained cells were analyzed using a flow cytometer. Dead cell numbers were estimated
by subtracting the viable cell counts from total cell counts.

61
Shishido, 2009b), the unique endocytosis mechanism of the original BNC might be
attenuated (Oess, 2000).
These results suggest that the target-cell-specific expression of the cell-killing
activity of exotoxin A with the ZHER2-displaying BNC/LP complex containing anionic or
nonionic LP requires a mechanism to release the incorporated proteins from the
endosome into the cytoplasm.
Expression of exotoxin A activity specifically in HER2-expressing cells with
ZHER2-BNC/LP complex prepared by mixing helper lipid
In order to improve the ability of the ZHER2-BNC/ALP or ZHER2-BNC/NLP
complex to escape the endosome, we conceived the use of a helper lipid. We chose a
pH-sensitive DOPE contained cationic LP (COATSOME EL-01-D) as the helper lipid to
assist the escape from the endosome through the destabilization of the endosomal
membrane via a conformational change in acidic conditions. We incorporated exotoxin
A into the LP mixtures which were prepared by mixing the DOPE contained cationic LP
with the anionic and nonionic ALP and NLP, respectively. The obtained LP mixtures
(ADLP and NDLP) were conjugated with ZHER2-BNC to produce the DOPE and
exotoxin A contained complex carriers (ZHER2-BNC/ADLP and ZHER2-BNC/NDLP).
After 1 h of incubation with the various amounts of ZHER2-BNC/ADLP and
ZHER2-BNC/NDLP complexes containing exotoxin A, HER2-positive SKBR3 cells and
HER2-negative HeLa cells were washed twice and incubated for an additional 47 h, and
then stained with calcein AM, which displays green fluorescence via live-cell-specific
uptake. Then the fatality rates of the cells were measured by subtracting the
live-cell-counts from the total-cell-counts obtained from the quantitative FACS analysis

62
(Fig. 3A and 3B; gray bars).
HER2-positive SKBR3 target cells added to more than 20 l of the
DOPE-containing ZHER2-BNC/ADLP complex (Fig. 3A; gray bars) showed higher
fatality rates than the DOPE-free ZHER2-BNC/ALP (Fig. 3A; white bars). As the
addition of 50 l of the ZHER2-BNC/ADLP complex rarely led to the death of non-target
cells (HeLa; Fig. 3A), it confirmed that displaying ZHER2 on the BNC/ADLP complex
functionally provided the HER2 positive SKBR3-specific cytotoxic effect. These results
suggest that the DOPE-containing LP in the ZHER2-BNC/ADLP complex successfully
functioned as a helper lipid to assist the endosomal escape and express the cell cytotoxic
activity of the incorporated exotoxin A, although the fatality rates of SKBR3 cells were
still not very high.
The DOPE and exotoxin A contained ZHER2-BNC/NDLP complex produced
high fatality rates in target SKBR3 cells (Fig. 3B; gray bars). However, the complex
also produced a high fatality rate even in non-target HeLa cells (Fig. 3B; HeLa, gray
bar). The ZHER2-BNC/NDLP complex, which was prepared with the mixture of the
nonionic LP (NLP) and the DOPE contained cationic LP was introduced into both target
and non-target cells non-specifically perhaps because of a shift of surface charge in the
complex to cationic. Indeed, the zeta potential was a negative value for the
ZHER2-BNC/ADLP complex and a positive value for the ZHER2-BNC/NDLP complex
(Table 2). The LP mixtures (ADLP and NDLP) also showed the same tendencies (Table
2). In addition, the sizes of ZHER2-BNC/ADLP and ZHER2-BNC/NDLP complexes were
almost unchanged (Table 2). These results demonstrated that the conjugation of
ZHER2-displaying BNC with the LP mixture (ADLP) obtained by mixing the anionic LP
(ALP; COATSOME EL-01-A) and the DOPE containing cationic LP (COATSOME

63
EL-01-D) could functionally confer the ability to enable the endosomal escape of the
incorporated protein as well as the ability to specifically target the HER2-expressing
cells.
Effective expression of exotoxin A activity using the complex carrier with the
optimized blended ratio of ZHER2-BNC and DOPE-containing anionic LP mixture
(ADLP)
Next, we considered an approach to enrich the expression efficiency of the
cytotoxic effect of the ZHER2-BNC/ADLP complex incorporating exotoxin A. We
assumed that the reason for the low fatality rates of the target SKBR3 cells (Fig. 3A)
was that the ratio of the ADLP to the ZHER2-BNC was linked to the expression of the
cell-killing activity of the incorporated exotoxin A. A relatively higher amount of
ZHER2-BNC conjugated to the ADLP might disturb the expression of the endosomal
escaping function by obscuring the efficacy of DOPE.
On the basis of this idea, we examined the cell-killing activity by modifying
the amounts of ZHER2-BNC available to conjugate with a certain amount of ADLP
incorporating exotoxin A (Fig. 4A). The sizes and zeta potentials of the complexes were
measured and shown in Table 3. The amounts of ZHER2-BNC had a large impact on the
fatality rates of SKBR3 cells as was expected. Surprisingly, the fatality rates fluctuated
dramatically depending on the amounts of ZHER2-BNC, and reached approximately
100% when the additive ZHER2-BNC was reduced to half of the original amount (from
100 g to 50 g as protein against 100 l-ADLP (3.7 mg/ml)) (Fig. 4A). It was inferred
that the dramatic decrease of the fatality rates was probably caused by attenuation of the
HER2-specific binding ability when the amount of additive

64
Table 3. The sizes and zeta potentials of ZHER2-BNC/ADLP complex carriers.

65
Fig. 4. Modification of the amounts of ZHER2-BNC for conjugation with the DOPE
contained LP mixture (ADLP). The LP mixture was dissolved in distilled water
containing 100g/ml of exotoxin A, and then used to prepare the complex carriers with
varied amounts of ZHER2-BNC as described in materials and methods. Cells were
incubated for 1 h in the media adjusted to 1 ml by adding the indicated volumes of the
complex carriers. After washing twice, cells were additionally incubated for 47 h. (A)
Fatality rates of HER2-positive SKBR3 cells treated with the complex carriers
containing varied amounts of ZHER2-BNC. The abscissa axis shows relative amounts of
the added ZHER2-BNC against 100 l-ADLP (3.7 mg/ml); For 0, x1, x1/2, x1/3, x1/4 and
x1/6, 0, 100, 50, 33, 25 and 16 g of ZHER2-BNC (as protein) were used, respectively.
Cells were stained with calcein AM and then analyzed using a flow cytometer. Dead cell
numbers were estimated by subtracting the viable cell counts from total cell counts. (B)
Fluorescence images of HER2-positive SKBR3 and HER2-negative HeLa cells treated
with LP mixture (ADLP) or the optimized blended ZHER2-BNC/ADLP complex (x1/2).
Cells were stained with EthD-1 and then observed by a confocal laser scanning
microscope. Scale bar, 50 m.

66
ZHER2-BNC was reduced to less than 1/3. When the ZHER2-BNC-halved complex was
added to SKBR3 and HeLa, cell death was specific to only the target SKBR3 cells (Fig.
4B). The anionic LP mixture (ADLP) containing exotoxin A never showed the
cell-killing activity in both cells (Fig. 4B). These results indicated that we could express
the function of the incorporated protein into the ZHER2-BNC/ADLP complex by
determining the optimized blending ratio of ZHER2-BNC and anionic LP mixture
(ZHER2-BNC : ADLP = 50 g as protein : 100 l (3.7 mg/ml)). Thus, we succeeded in
developing the specificity-altered BNC/LP complex containing the bifunctional
properties of cell-specificity and endosomal escape.
Evaluation of cell-killing activity when adding various amounts of the optimized
blended ZHER2-BNC/ADLP complexes containing exotoxin A
Finally, we evaluated the cell-killing activities when adding various amounts
of the optimized blended complexes containing exotoxin A (Fig. 5). Even when adding
5 l of the optimized blended ZHER2-BNC/ADLP complex, the fatality rate of SKBR3
cells was over 50%. The fatality rates were enriched in a dose-dependent manner,
producing approximately 100% cell death when 20 l of the complex was added. The
ADLP lacking ZHER2-BNC containing exotoxin A could not kill the SKBR3 cells
efficiently despite increasing the addition volume up to 50 l. Direct addition of 50 l
of exotoxin A (100 g/ml) without any carriers also never produced the death of SKBR3
cells. These results indicate that cellular uptake and release into the cytoplasm of
exotoxin A were surely attributed to the ZHER2-BNC/ADLP complex carrier. In addition,
the ZHER2-BNC/ADLP complex without exotoxin A rarely affected the fatality rate of
SKBR3 cells, suggesting that the complex itself displayed low cellular toxicity.

67
Fig. 5. Fatality rates of HER2-positive SKBR3 and HER2-negative HeLa cells treated
by the indicated carriers with and without exotoxin A. The DOPE-containing LP
mixture (ADLP) was dissolved in distilled water containing 100g/ml of exotoxin A,
and then was used to prepare the optimized blended ZHER2-BNC/ADLP complex shown
in Fig. 4. Cells were incubated for 1 h in the media adjusted to 1 ml by adding the
indicated volumes of carriers. After washing twice, cells were additionally incubated for
47 h and then stained with calcein AM. Then, the stained cells were analyzed using a
flow cytometer. Dead cell numbers were estimated by subtracting the viable cell counts
from total cell counts.

68
Furthermore, high specificity to HER2-expressing cells was exhibited by the low
fatality rate of non-target HeLa cells even when adding 50 l of the ZHER2-BNC/ADLP
complex. The complex carrier prepared with the BNC displaying wild-type Z domain
(ZWT-BNC/ADLP) never exhibited cellular toxicity to HER2-expressing SKBR3 cells
(Fig. 6). These results successfully demonstrate that the optimized blended
ZHER2-BNC/ADLP complex could be a potential carrier to provide protein therapy for
HER2 positive breast cancer cells.
CONCLUSION
As previously reported, the results presented herein confirm the
HER2-specific targeting ability of ZHER2-BNC. However, the delivery capacity of
ZHER2-BNC for protein therapy has remained obscure. We report here the design of an
advanced complex carrier for protein delivery by blending the DOPE contained cationic
helper lipid with the anionic LP that was used for the conjugation to the ZHER2-BNC.
The DOPE contained helper lipid successfully assisted the endosomal escape of the
incorporated protein, exotoxin A. The charge of LP was a critical factor in designing the
DOPE-containing BNC/LP complex in order to maintain the specificity to HER2
positive breast cancer cells. The optimized blending ratio of the ZHER2-BNC to the
DOPE-containing LP mixture dramatically improved the cell-killing activity of the
exotoxin A incorporated complex carrier. The conjugation of the LP mixture with the
helper lipid should prove to be a useful method for also incorporating virus-like

69
Fig. 6. Fatality rates of HER2-positive SKBR3 treated with the ZWT-BNC/ADLP
complex (optimized blended ratio, x1/2). Wild-type Z domain (ZWT)-displaying BNC
(ZWT-BNC) was conjugated with ADLP incorporating exotoxine A (ZWT-BNC/ADLP).
The LP mixture (ADLP) was dissolved in distilled water containing 100g/ml of
exotoxin A, and then used to prepare the ZWT-BNC/ADLP complexes along with the
materials and methods of the optimized blended ZHER2-BNC/ADLP complex (x1/2).
Cells were incubated for 1 h in the media adjusted to 1 ml by adding the indicated
volumes of the complex carriers. After washing twice, cells were additionally incubated
for 47 h and then stained with calcein AM. Then, the stained cells were analyzed using a
flow cytometer. Dead cell numbers were estimated by subtracting the viable cell counts
from total cell counts.

70
particles to take advantage of their various functions.
ABBREVIATIONS
DDS, drug delivery system; BNC, bio-nanocapsule; HBV, hepatitis B virus; HBsAg, hepatitis B
virus surface antigen; HER2, human EGFR-related 2; EGFR, epidermal growth factor receptor;
LP, liposome; GFP, green fluorescent protein; PEG, polyethylene glycol; CsCl, cesium chloride;
DPPC, dipalmitoyl-phosphatidylcholine; CHOL, cholesterol; DPPG,
dipalmitoyl-phosphatidylglycerol; DOPE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine;
DC6-14, O,O’-ditetradecanoyl-N-(-trimethylammonioacetyl) diethanolamine chloride; FBS,
Fetal bovine serum; DMEM, Dulbecco’s modified Eagle medium; LSM, laser scanning
microscope; EthD-1, ethidium homodimer-1; TLM, translocation motif;
REFERENCES
Allured, V.S., Collier, R.J., Carroll, S.F., McKay, D.B., 1986. Structure of exotoxin A
of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc Natl Acad Sci USA. 83
(5), 1320-1324.
Davison, Z., Blacquiere, G.E., Westley, B.R., May, F.E.B., 2011. Insulin-like Growth

71
Factor-Dependent Proliferation and Survival of Triple-Negative Breast Cancer Cells:
Implication for Therapy. Neoplasia. 13, 504-515.
Jia, L.T., Zhang, L.H., Yu, C.J., Zhao, J., Xu, Y.M., Gui, J.H., Jin, M., Ji, Z.L., Wen,
W.H., Wang, C.J., Chen, S.Y., Yang, A.G., 2003. Specific Tumoricidal Activity of a
Secreted Proapototic Protein Consisting of HER2 Antibody and Constitutively Active
Caspase-3. Cancer Res. 63, 3257-3262.
Jung, J., Matsuzaki, T., Tatematsu, K., Okajima, T., Tanizawa, K., Kuroda, S., 2008.
Bio-nanocapsule conjugated with liposomes for in vivo pinpoint delivery of various
materials. J Control Release. 126, 255-264.
Kasuya, T., Jung, J., Kadoya, H., Matsuzaki, T., Tatematsu, K., Okajima, T., Miyoshi,
E., Tanizawa, K., Kuroda, S., 2008. In Vivo Delivery of Bionanocapsules
Displaying Phaseolus vulgarisAgglutinin-L4 Isolectin to Malignant Tumors
Overexpressing N-Acetylglucosaminyltransferase V. Human Gene Therapy. 887-895.
Kuroda, S., Otaka, S., Miyazaki, T., Nakao, M., Fujisawa, Y., 1992. Hepatitis B virus
envelope L protein particles. Synthesis and assembly in Saccharomyces cerevisiae,
purification and characterization. J Biol Chem. 267, 1953-1961.
Lee, S.B., Hassan, M., Fisher, R., Chertov, O., Chernomordik, V., Kramer-Marek, G.,
Gandjbakhche, A., Capala, J., 2008. Affibody molecules for in vivo characterization of
HER2-positive tumors by near-infrared imaging. Clin Cancer Res. 14, 3840-3849.

72
McLarty, K., Cornelissen, B., Scollard, D.A., Done, S.J., Chun, K., Reilly, R.M., 2009.
Associations between the uptake of 111In-DTPA-trastuzumab, HER2 density and
response to trastuzumab (Herceptin) in athymic mice bearing subcutaneous human
tumour xenografts. Eur J Nucl Med Mol Imaging. 36, 81-93.
Nagai, T., 2005. Drug discovery and innovative drug delivery research in new drug
development. Pharm Tech Japan. 21, 1949-1951.
Oess, S., Hidt, E., 2000. Novel cell permeable motif derived from the PreS2-domain of
hepatitis-B virus surface antigens. Gene Therapy. 7, 750-758.
Orlova, A., Magnusson, M., Eriksson, T.L., Nilsson, M., Larsson, B.,
Hoiden-Guthenberg, I., Widstrom, C., Carlsson, J., Tolmachev, V., Stahl, S., Nilsson,
F.Y., 2006. Tumor imaging using a picomolar affinity HER2 binding affibody molecule.
Cancer Res. 66, 4339–4348.
Shishido, T., Azumi, Y., Nakanishi, T., Umetsu, M., Tanaka, T., Ogino, C., Fukuda, H.,
Kondo, A., 2009a. Biotinylated bionanocapsules for displaying diverse ligands toward
cell-specific delivery. J Biochem. 146, 867-874.
Shishido, T., Yonezawa, D., Iwata, K., Tanaka, T., Ogino, C., Fukuda, H., Kondo, A.,
2009b. Construction of arginine-rich peptide displaying bionanocapsules. Bioorg Med
Chem Lett. 19, 1473-1476.

73
Shishido, T., Mieda, H., Hwang S.Y., Nishimura, Y., Tanaka, T., Ogino, C., Fukuda, H.,
Kondo, A., 2010. Affibody-displaying bionanocapsules for specific drug delivery to
HER2-expressing cancer cells. Bioorg Med Chem Lett. 20, 5726-5731.
Stoeckl, L., Funk, A., Kopitzki, A., Brandenburg, B., Oess, S., Will, H., Sirma, H., Hildt,
E., 2006. Identification of a structural motif crucial for infectivity of hepatitis B viruses.
PNAS. 103 (17), 6730-6734.
Tabata, T., 2006. Drug delivery system: Basic technology for biomedical research,
medical treatment and health care. Biotechnol J. 6, 553-555.
Yamada, T., Iwasaki, Y., Tada, H., Iwabuki, H., Chuah, M.K., VandenDriessche, T.,
Fukuda, H., Kondo, A., Ueda, M., Seno, M., Tanizawa, K., Kuroda, S., 2003.
Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat Biotechnol.
21, 885-890.

74
PART III.
Targeting cancer cell-specific RNA interference
by siRNA delivery using a complex carrier of
affibody-displaying bio-nanocapsules and liposomes

75
INTRODUCTION
RNA interference (RNAi) is expected to become a new approach in treating a
variety of diseases, such as virus infection, cancer and neurodegenerative diseases,
owing to specific and effective gene silencing (Cardoso, 2009; Tseng, 2009). The
mechanism of RNAi involves double-stranded RNA injected into cells that are first cut
into short RNA (small interfering RNA (siRNA)), 21–23 bp long, using ribonuclease
(RNase) III enzyme that is referred to as the Dicer. The duplex siRNA forms a
RNA-induced silencing complex (RISC), which contains an endonuclease and an
Argonaute protein. The siRNA duplex is dissociated into unwound single-stranded RNA
using ATP-dependent helicase; therefore, the RISC with antisense strand against target
mRNA leads to RNA destruction and results in a downregulation of gene expression
(Lundberg, 2007; Chang, 2011; Miele, 2012; Spagnou, 2004). Although the use of
siRNA is a promising approach for nucleic acid medicine, several problems remain with
respect to in vivo use, such as an inability to cross membranes, an instability in the
blood, and a lack of ability to specifically target abnormal cells (Cardoso, 2009; Wirth,
2011).
A bio-nanocapsule (BNC) is a hollow nano particle composed of the L protein
of the hepatitis B virus (HBV), surface antigen (HBsAg), and a lipid bilayer. The BNC
exhibits a reliable safety profile due to being viral-genome-free and shows high
specificity for human hepatocytes and a high transfection efficiency that is equivalent to
the original HBV (Kuroda, 1992). Therefore, the BNC has been studied as a carrier for
the delivery of drugs and genes (Yamada, 2003).

76
Previously, we and other researchers succeeded in altering the cell-specificity
of BNCs by deleting the hepatocyte-specific recognition site (located in the preS region)
in the L protein and inserting binding molecules with the ability to target other cells
(Kasuya, 2008). For example, among an affibody molecule which is a new class of
binding proteins derived from the Z domain of staphylococcal protein A (Orlova, 2006),
we displayed the ZHER2 affibody molecule on the surface of BNC (ZHER2-BNC). Thus,
we succeeded in altering the specificity of a BNC from hepatocytes to HER2 receptor
expressing cells such as those found in breast and ovarian cancer (Shishido, 2010).
Additionally, the fusion of medicinal proteins (Kurata, 2008) and an
electroporation (Yamada, 2003) and a BNC/liposome (BNC/LP) conjugation (Jung,
2008) have been previously reported as methods used for encapsulating drugs and genes
into BNCs. In particular, the BNC/LP conjugation method has succeeded in
encapsulating various-sized materials including low-molecular compounds, genes and
proteins into BNC/LP complex carriers (Kasuya, 2009; Nishimura, 2012). The complex
carriers are formed by fusing BNCs to the surface of LPs, in which target materials have
been pre-encapsulated. By changing the phospholipid composition of LPs or the types
of BNCs, a variety of characteristic features are granted to the BNC/LP complex
carriers. For example, anionic phospholipid can avoid non-specific binding to non-target
cells (Nishimura, 2012); pH-responsive phospholipids
(1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DOPE) provide the ability for
endosomal escape (Nishimura, 2012); and, affibody-displaying BNCs can alter
cell-specificity (Shishido, 2010). By using this method, we previously constructed
ZHER2-BNC/LP complex carriers and succeeded in the specific and functional delivery
of proteins for HER2-expressing breast cancer cells (Nishimura, 2012).

77
In the present study, to overcome the problems in siRNA therapy, we tried the
specific delivery of siRNA into target cancer cells by using the BNC/LP complex as the
carrier. To facilitate the evaluation of RNAi, an siRNA that would inhibit GFP
expression was selected. We describe how the ZHER2-BNC/LP complex can specifically
deliver siRNA into HER2-expressing breast cancer cells and effectively lead to the
cell-specific targeting of RNAi.
MATERIALS AND METHODS
Materials
BNCs were prepared from Saccharomyces cerevisiae AH22R- harboring the
plasmid pGLDsLd50-ZHER2 or pGLDsLd50-ZWT (Shishido, 2010) as described
previously (Kuroda, 1992). Briefly, yeast cells transformed with pGLDsLd50-ZHER2 or
pGLDsLd50-ZWT by the spheroplast method were cultured and disrupted with glass
beads, the crude extract was precipitated with polyethylene glycol (PEG) 6000 and
subjected to cesium chloride (CsCl) isopycnic ultracentrifugation and sucrose density
gradient ultracentrifugation, and then the purified BNCs were obtained after
freeze-drying in the presence of 5% sucrose. Liposome (LP), COATSOME EL-01-D
(1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) : CHOL :
O,O’-ditetradecanoyl-N-(-trimethylammonioacetyl) diethanolamine chloride
(DC6-14) = 0.75 : 0.75 : 1.00 (mol/vial)) was purchased from NOF (Tokyo, Japan).
Silencer® GFP (eGFP) siRNA and LipofectaminTM RNAiMAX Reagent was purchased

78
from Invitrogen Life Technologies (Carlsbad, CA, USA). Gibco® Fetal bovine serum
(FBS) and an L-glutamine and Molecular Probes® LIVE/DEAD® viability/cytotoxicity
assay kit were purchased from Invitrogen Life Technologies. RPMI 1640 medium and
Dulbecco’s modified Eagle medium (DMEM) were purchased from Nacalai Tesque
(Kyoto, Japan). Blasticidin was purchased from InvivoGen (San Diego, CA, USA).
Preparation of BNC/LP complex and incorporation of siRNA
Complex carriers of ZHER2-BNC and ZWT-BNC and LP, in which siRNA was
incorporated, were prepared by referring to the previously described BNC/LP
conjugation method with some modifications (Jung, 2008). Freeze-dried LP
(COATSOME EL-01-D, 1.5 mg) were dissolved in distilled water (1 ml) containing 200
nM of siRNA. After incubation for 1 h at room temperature, LP-mixing siRNA (100 l)
was added to freeze-dried ZHER2-BNC or ZWT-BNC (50 g as protein) and incubated at
room temperature for 1 h to form BNC/LP complexes. The resultant complex carriers
were named ZHER2-BNC/LP and ZWT-BNC/LP.
Cell culture
To evaluate and quantify the RNAi efficacy, we used the cells constantly
expressing the chromosomally-integrated GFP. SKBR3 cells (human breast carcinoma)
were maintained in RPMI 1640 medium supplemented with 10% (v/v) FBS and 5 g/ml
Blasticidin at 37°C in 5% CO2. HeLa cells (human cervical carcinoma) were maintained
in DMEM medium supplemented with 10% FBS and 5 g/ml Blasticidin at 37°C in 5%
CO2.

79
Measuring the zeta potential and diameter of particles
The zeta potentials of the LPs and a BNC/LP complex were determined using
a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK), following the
manufacturer's procedure.
Flow cytometric evaluation of RNAi and cell viability
Approximately 1×105 SKBR3 and HeLa cells were seeded in 12-well plates
and incubated 37°C for 24 h. After washing with serum-free medium, the required
volumes of the complex carriers and LPs containing siRNA were added to the medium
and the volume was adjusted to 1 ml. Cells were incubated for 4 h, and then were
washed twice with serum-free medium and cultured with FBS-containing medium for
44 h.
siRNA was also directly transfected to cells with RNAiMAX, following the
manufacturer's procedure. Briefly, the required volumes of 50 mM siRNA and 2 l of
RNAiMAX were added to 200 l of serum-free medium in 12-well plates, and the
medium was incubated for 20 min at room temperature. Then, 1 ml of serum-free
medium containing cells (1×105) was added to the 12-well plates. Cells were incubated
for 4 h, washed twice with serum-free medium and cultured with FBS-containing
medium for 44 h.
To quantify RNAi efficacy, green fluorescence was detected and the
decrement of GFP-expressing cells was counted. To quantify cell viability, red
fluorescence was detected and the dead cells stained with EthD-1 were counted. The
EthD-1 staining was performed using a LIVE/DEAD® viability/cytotoxicity assay kit
according to the manufacturer’s instructions. Cells were suspended into a sheath

80
solution and subjected to a BD FACSCanto II flow cytometer equipped with a 488-nm
blue laser (BD Biosciences, San Jose, CA, USA). The green and red fluorescence
signals were collected through 530/30 and 585/42-nm band-pass filter, respectively. The
data were analyzed using BD FACSDiva software v5.0 (BD Biosciences).
Microscopic observation of GFP-expressing cells treated with siRNA
The introduction of siRNA basically followed the above-described procedure
with some modifications as follows: 12-well plates were changed to 35 mm glass
bottom dishes; the final concentration of siRNA in the medium was fixed to 25 nM; and
the total volume of medium was adjusted to 2 ml.
Cells were observed using a LSM 5 PASCAL laser scanning confocal
microscope (Carl Zeiss, Oberkochen, Germany) equipped with a 63-fold oil immersion
objective lens with excitation using the 488-nm line of an argon laser and emission
collection using a 505-nm long pass filter.
RESULTS AND DISCUSSION
Target cell-specific RNAi with ZHER2-BNC/LP
First, to examine the specific delivery of siRNA, we used HER2-expresing
SKBR3 cells (human breast carcinoma) as target cells (Davison, 2011).
HER2-non-expressing HeLa cells (human cervical carcinoma) were used as the
non-target cells (Jia, 2003). To evaluate and quantify the RNAi efficacy, we used the

81
cells constantly expressing the chromosomally-integrated GFP. RNAiMAX, LP and
ZHER2-BNC/LP were tested to deliver siRNA. After 48 h of incubation, the efficacies of
RNAi depending on the additive concentration of siRNA were determined by measuring
the cell population rates missing GFP fluorescence.
The efficacies of RNAi for HER2-expressing SKBR3 cells and
HER2-non-expressing HeLa cells are shown in Fig. 1A and 1B, respectively. In the
case of using RNAiMAX (white bars), RNAi was observed even at lower concentration
of siRNA (1 nM~) in both SKBR3 and HeLa cells. This indicated that the transfection
reagent never showed the specificity to the target cells although it has the ability for
high transfection efficiency. The LPs without ZHER2-BNC (gray bars) also triggered
RNAi in both cells as similar to the case of RNAiMAX. The zeta potential of the LPs
encapsulating siRNA showed a positive charge (Table 1), suggesting that it was bound
to cells non-specifically due to an electrostatic interaction. However, the ZHER2-BNC/LP
complex displayed the specific effect of RNAi only for HER2-expressing SKBR3 cells
(Fig. 1A and 1B; black bars). Furthermore, the RNAi efficacies of ZHER2-BNC/LP that
were >10 nM were equal to that of RNAiMAX. This result indicates that the siRNA
delivery with ZHER2-BNC/LP was HER2-expressing breast cancer cell-specific siRNA
delivery and that it led to an effective expression of the RNAi function.
Viability of cells treated with ZHER2-BNC/LP
To evaluate the biocompatibility of each carrier containing siRNA, we
measured cell survival rates with a type of EthD-1 that permits the detection of dead
cells under the progression of RNAi. The cell viabilities of SKBR3 (Fig. 2A) and HeLa
(Fig. 2B) were similar, but slightly different in each carrier. The slight decrease in

82
Fig. 1. Quantification of RNAi in HER2-positive SKBR3 (A) and HER2-negative HeLa
(B) cells treated by siRNA combined with RNAiMAX (white bars), LPs (gray bars) and
ZHER2-BNC/LP complex (black bars). The GFP expressions of the cells were analyzed
using a flow cytometer and results are expressed as a percentage of the GFP-expressing
cellular quantity in untreated controls. The x-axis represents the final concentration of
siRNA in the medium adjusted to 2 ml.
0
20
40
60
80
100
1 2.5 5 10 12.5 15 20 25
0
20
40
60
80
100
1 2.5 5 10 12.5 15 20 25
Concentrations of additive siRNA [nM]
% o
f R
NA
inte
rfer
ence
Concentrations of additive siRNA [nM]
% o
f R
NA
inte
rfer
ence
A. SKBR3 cell
B. HeLa cell
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
0
20
40
60
80
100
1 2.5 5 10 12.5 15 20 25
0
20
40
60
80
100
1 2.5 5 10 12.5 15 20 25
Concentrations of additive siRNA [nM]
% o
f R
NA
inte
rfer
ence
Concentrations of additive siRNA [nM]
% o
f R
NA
inte
rfer
ence
A. SKBR3 cell
B. HeLa cell
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP

83
Table. 1. The sizes and zeta potentials of LPs only, LPs with siRNA and ZHER2-BNC/LP
complex with siRNA.
-5.1 ± 1.2 229 ± 27.0ZHER2-BNC/LP containing siRNA
1.4 ± 2.9 167 ± 14.2LP containing siRNA
3.9 ± 1.8 187 ± 3.8LP
Zeta potential [mV]Diameter [nm]carrier
-5.1 ± 1.2 229 ± 27.0ZHER2-BNC/LP containing siRNA
1.4 ± 2.9 167 ± 14.2LP containing siRNA
3.9 ± 1.8 187 ± 3.8LP
Zeta potential [mV]Diameter [nm]carrier
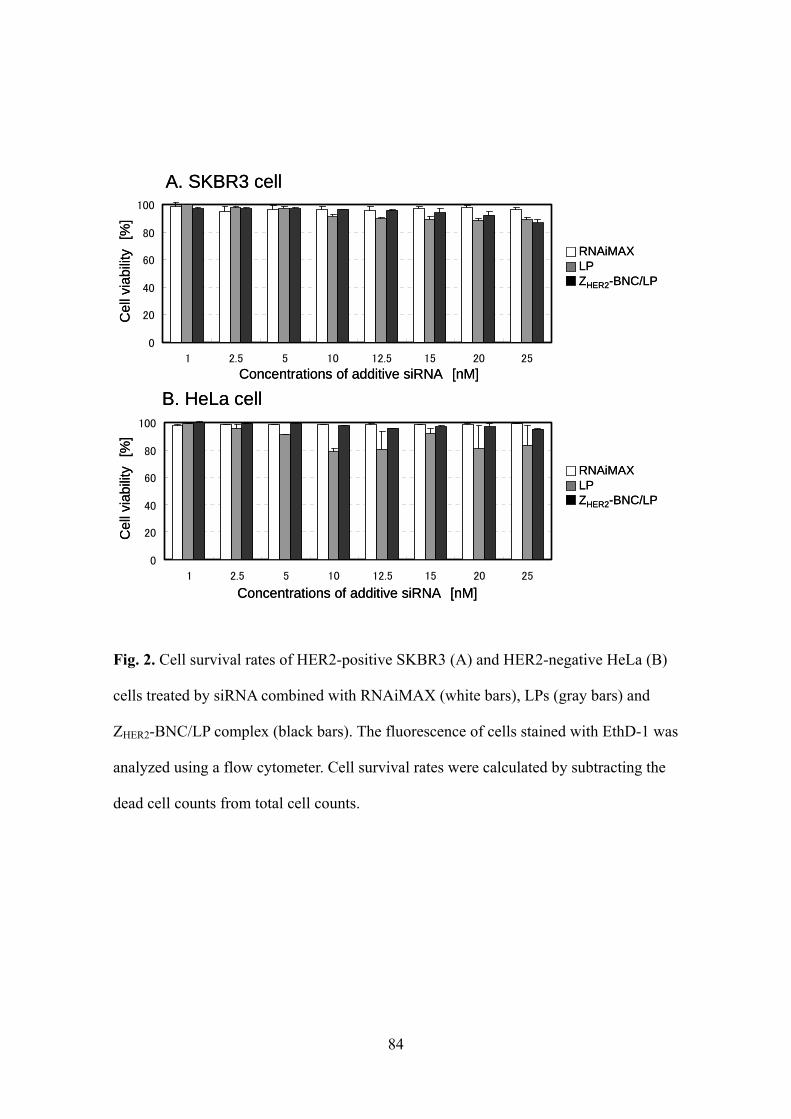
84
Fig. 2. Cell survival rates of HER2-positive SKBR3 (A) and HER2-negative HeLa (B)
cells treated by siRNA combined with RNAiMAX (white bars), LPs (gray bars) and
ZHER2-BNC/LP complex (black bars). The fluorescence of cells stained with EthD-1 was
analyzed using a flow cytometer. Cell survival rates were calculated by subtracting the
dead cell counts from total cell counts.
0
20
40
60
80
100
1 2.5 5 10 12.5 15 20 25
0
20
40
60
80
100
1 2.5 5 10 12.5 15 20 25
Cel
l via
bilit
y[%
]
Concentrations of additive siRNA [nM]
Cel
l via
bilit
y[%
]
Concentrations of additive siRNA [nM]
A. SKBR3 cell
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
B. HeLa cell
0
20
40
60
80
100
1 2.5 5 10 12.5 15 20 25
0
20
40
60
80
100
1 2.5 5 10 12.5 15 20 25
Cel
l via
bilit
y[%
]
Concentrations of additive siRNA [nM]
Cel
l via
bilit
y[%
]
Concentrations of additive siRNA [nM]
A. SKBR3 cell
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
RNAiMAXLPZHER2-BNC/LP
B. HeLa cell

85
viability in the case of LPs (gray bars) might have been due to the excess non-specific
binding of phospholipids to the cell membrane. A significant decrease in viability was
not observed in the case of ZHER2-BNC/LP (black bars). This result suggests that
ZHER2-BNC/LP was non-toxic to cells.
Confirmation of the occurrence of siRNA-specific and affibody-dependent RNAi
To confirm whether the inhibition of protein synthesis was really led by the
action of siRNA, we used the siRNA that never inhibits GFP expression (negative
siRNA) (Fig. 3). We added 25 nM carriers containing negative siRNA to SKBR3 and
HeLa cells. As a result, RNAi was never detected in the presence of any of these carriers
(Fig. 3A and 3B). This result clearly shows that the decrement of GFP-expressing cells
in Fig. 1 was surely guided by siRNA-specific action.
To establish the validity of using ZHER2-BNC to grant cell-specificity, we
compared the ZHER2-BNC/LP with ZWT-BNC/LP using the ZWT (Z domain)-displaying
BNC without HER2 recognition ability to form the BNC/LP complex (Fig. 4). Each
complex carrier with siRNA was added to SKBR3 and HeLa cells (final conc. 25 nM as
siRNA), and the rates of RNAi and cell viability were evaluated after 48 h of incubation.
As a result, ZHER2-BNC/LP triggered SKBR3-specific RNAi, whereas ZWT-BNC/LP did
not invoke RNAi in either cell. Thus, the importance of the affibody-displaying BNC
for specific siRNA delivery was confirmed.
Microscopic observation of GFP interference
To visually confirm the inhibition of GFP synthesis by RNAi, we treated
SKBR3 and HeLa cells with RNAiMAX, LPs and ZHER2-BNC/LP containing siRNA

86
Fig. 3. Quantification of RNAi (black bars) and cell survival rates (gray bars) of
HER2-positive SKBR3 (A) and HER2-negative HeLa (B) cells treated by negative
siRNA combined with RNAiMAX, LPs and ZHER2-BNC/LP complex (final conc. 25 nM
as siRNA).
0
20
40
60
80
100
0
20
40
60
80
100
ZHER2-BNC/LP
[%]
[%]A. SKBR3 cell
B. HeLa cell
RNAiCell viability
RNAiCell viability
RNAiMAX LP
ZHER2-BNC/LPRNAiMAX LP0
20
40
60
80
100
0
20
40
60
80
100
ZHER2-BNC/LP
[%]
[%]A. SKBR3 cell
B. HeLa cell
RNAiCell viability
RNAiCell viability
RNAiMAX LP
ZHER2-BNC/LPRNAiMAX LP

87
Fig. 4. Quantification of RNAi (black bars) and cell survival rates (gray bars) of
HER2-positive SKBR3 and HER2-negative HeLa cells treated by siRNA combined
with ZHER2-BNC/LP (left side) and ZWT-BNC/LP (right side) (final conc. 25 nM as
siRNA).
0
20
40
60
80
100[%]
ZHER2-BNC/LP ZWT-BNC/LP
SKBR3 HeLa SKBR3 HeLa
RNAi
Cell viability
0
20
40
60
80
100[%]
ZHER2-BNC/LP ZWT-BNC/LP
SKBR3 HeLa SKBR3 HeLa
RNAi
Cell viability

88
(final conc. 25 nM) and observed the cells using a confocal laser scanning microscope
(CLSM) following 24 and 48 h of incubation (Fig. 5A and Fig. 5B). In the case of
RNAiMAX, green fluorescence was rarely observed in either cell, and the non-specific
inhibition of GFP synthesis was confirmed. In the case of LP, inhibition of GFP
synthesis was scarcely provoked after 24 h but was confirmed after 48 h in both cells.
This indicated that LPs would bind to cells in a non-specific manner, and it took longer
to induce RNAi than with the transfection reagent. However, ZHER2-BNC/LP had no
impact on the expression of GFP in HeLa cells, while the inhibition of GFP synthesis
was clearly confirmed in SKBR3 cells during 48 h of incubation. Furthermore,
diminished GFP fluorescence was observed even after 24 h, indicating that
ZHER2-BNC/LP had fast-acting properties that were equivalent to with the transfection
reagent. These results demonstrated that ZHER2-BNC/LP can stabilize siRNA via the
formulation of a complex carrier to efficiently deliver siRNA inside specific
HER2-expressing cells through endosomal escape, which would allow RNAi to
effectively inhibit protein expression.
CONCLUSION
Although the therapeutic effect of siRNA has been highly anticipated, its
inability to specifically target cells and to cross the cell membrane has limited its in vivo
application (Cardoso, 2009). In this study, we succeeded in delivering and introducing
siRNA into targeted breast carcinoma cells, which led to the effective use of RNAi by

89
24 h 48 h
control
RNAiMAX
LP
ZHER2-BNC/LP
A. SKBR3 cell 24 h 48 h
control
RNAiMAX
LP
ZHER2-BNC/LP
A. SKBR3 cell

90
Fig. 5. Fluorescence images of HER2-positive SKBR3 (A) and HER2-negative HeLa
(B) cells treated by siRNA combined with RNAiMAX, LPs and ZHER2-BNC/LP
complex (final conc. 25 nM as siRNA). The cells were incubated for 24 and 48 h, and
then observed using a confocal laser scanning microscope. Scale bars, 50 m.
control
RNAiMAX
LP
ZHER2-BNC/LP
24 h 48 hB. HeLa cell
control
RNAiMAX
LP
ZHER2-BNC/LP
24 h 48 hB. HeLa cell

91
using ZHER2-BNC/LP as the carrier. Thus, in the field of nucleic acid medicine,
ZHER2-BNC/LP can be a useful carrier for siRNA delivery, and could also become a
useful tool for gene silencing and to accomplish protein knock-down.
REFERENCES
Cardoso, A., Trabulo, S., Moreira, J.N., Düzgüneş, N., de Lima, M.C., 2009. Targeted
lipoplexes for siRNA delivery. Methods Enzymol. 465, 267-287.
Chang, C.I., Kim, H.A., Dua, P., Kim, S., Li, C.J., Lee, D.K., 2011. Structural diversity
repertoire of gene silencing small interfering RNAs. Nucleic Acid Ther. 21(3), 125-131.
Davison, Z., Blacquiere, G.E., Westley, B.R., May, F.E.B., 2011. Insulin-like Growth
Factor-Dependent Proliferation and Survival of Triple-Negative Breast Cancer Cells:
Implication for Therapy. Neoplasia. 13, 504-515.
Jia, L.T., Zhang, L.H., Yu, C.J., Zhao, J., Xu, Y.M., Gui, J.H., Jin, M., Ji, Z.L., Wen,
W.H., Wang, C.J., Chen, S.Y., Yang, A.G., 2003. Specific Tumoricidal Activity of a
Secreted Proapototic Protein Consisting of HER2 Antibody and Constitutively Active
Caspase-3. Cancer Res. 63, 3257-3262.
Jung, J., Matsuzaki, T., Tatematsu, K., Okajima, T., Tanizawa, K., Kuroda, S., 2008.

92
Bio-nanocapsule conjugated with liposomes for in vivo pinpoint delivery of various
materials. J Control Release. 126, 255-264.
Kasuya, T., Jung, J., Kadoya, H., Matsuzaki, T., Tatematsu, K., Okajima, T., Miyoshi,
E., Tanizawa, K., Kuroda, S., 2008. In Vivo Delivery of Bionanocapsules Displaying Phaseolus
vulgarisAgglutinin-L4 Isolectin to Malignant Tumors Overexpressing
N-Acetylglucosaminyltransferase V. Human Gene Therapy. 887-895.
Kasuya, T., Jung. J., Kinoshita. R., Goh. Y., Matsuzaki. T., Iijima. M., Yoshimoto. N.,
Tanizawa. K., Kuroda. S., 2009. Bio-nanocapsule-liposome conjugates for in vivo
pinpoint drug and gene delivery. Methods Enzymol. 464, 147-166.
Kurata, N., Shishido, T., Muraoka, M., Tanaka, T., Ogino, C., Fukuda, H., Kondo, A.,
2008. Specific protein delivery to target cells by antibody-displaying bionanocapsules. J
Biochem. 144(6), 701-707.
Kuroda, S., Otaka, S., Miyazaki, T., Nakao, M., Fujisawa, Y., 1992. Hepatitis B virus
envelope L protein particles. Synthesis and assembly in Saccharomyces cerevisiae,
purification and characterization. J Biol Chem. 267, 1953-1961.
Lundberg, P., El-Andaloussi, S., Sütlü, T., Johansson, H., Langel, U., 2007. Delivery of
short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 21(11),
2664-2671.

93
Miele, E., Spinelli, G.P., Miele, E., Di, Fabrizio, E., Ferretti, E., Tomao, S., Gulino, A.,
2012. Nanoparticle-based delivery of small interfering RNA: challenges for cancer
therapy. Int J Nanomedicine. 7, 3637-3657.
Nishimura, Y., Ishii, J., Okazaki, F., Ogino, C., Kondo, A., 2012. Complex carriers of
affibody-displaying bio-nanocapsules and composition-varied liposomes for
HER2-expressing breast cancer cell-specific protein delivery. J Drug Target. 20(10),
897-905.
Orlova, A., Magnusson, M., Eriksson, T.L., Nilsson, M., Larsson, B.,
Hoiden-Guthenberg, I., Widstrom, C., Carlsson, J., Tolmachev, V., Stahl, S., Nilsson,
F.Y., 2006. Tumor imaging using a picomolar affinity HER2 binding affibody molecule.
Cancer Res. 66, 4339–4348.
Shishido, T., Mieda, H., Hwang S.Y., Nishimura, Y., Tanaka, T., Ogino, C., Fukuda, H.,
Kondo, A., 2010. Affibody-displaying bionanocapsules for specific drug delivery to
HER2-expressing cancer cells. Bioorg Med Chem Lett. 20, 5726-5731.
Spagnou, S., Miller, A.D., Keller, M., 2004. Lipidic carriers of siRNA: differences in
the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry. 43(42),
13348-13356.
Tseng, Y.C., Mozumdar, S., Huang, L., 2009. Lipid-based systemic delivery of siRNA.
Adv Drug Deliv Rev. 61(9), 721-731.

94
Wirth, M., Fritsche, P., Stojanovic, N., Brandl, M., Jaeckel, S., Schmid, R.M., Saur, D.,
Schneider, G., 2011. A simple and cost-effective method to transfect small interfering
RNAs into pancreatic cancer cell lines using polyethylenimine. Pancreas. 40(1),
144-150.
Yamada, T., Iwasaki, Y., Tada, H., Iwabuki, H., Chuah, M.K., VandenDriessche, T.,
Fukuda, H., Kondo, A., Ueda, M., Seno, M., Tanizawa, K., Kuroda, S., 2003.
Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat Biotechnol.
21, 885-890.

95
PART IV.
An affinity chromatography method
used to purify His-tag-displaying bio-nanocapsules

96
INTRODUCTION
A bio-nanocapsule (BNC) consisting of a hepatitis B surface antigen (HBsAg)
and a lipid bilayer is a hollow virus-like particle (Kuroda et al., 1992) that has been
studied as a carrier for drug delivery systems (DDS) (Yamada et al., 2003). Since wild
type (WT) BNCs specifically recognize hepatic cells, they are used as carriers to deliver
drugs to hepatocarcinomas (Yamada et al., 2003). In the past, WT BNCs have also been
altered into modified BNCs that recognize other types of cells by replacing the
hepatocyte recognition site in the pre-S region with other targeting molecules (Kasuya
et al., 2008; Shishido et al., 2009a, 2009b). For example, a ZHER2-displaying BNC
(ZHER2-BNC) specifically recognizes HER2-expressing cells that include breast cancer
cells and ovarian cancer cells (Shishido et al., 2010). The ZHER2 is one of a type of
affibodies that are the mutant proteins derived from the Z domain of Staphylococcal
protein A and function as affinity ligands (Orlova et al., 2006; Lee et al., 2008). These
WT and altered BNCs can be mass-produced in yeast cells (Kuroda et al., 1992;
Shishido et al., 2010). Approaches using ultracentrifugation (Kuroda et al., 1992) or
affinity chromatography combined with gel filtration (Kasuya et al., 2009) have been
reported as methods for purification of the BNCs from crude yeast extract. The
advantage of the ultracentrifugation method is a general versatility, which is available to
purify both WT BNC and altered BNCs; and, therefore, it is used conventionally for the
purification of BNCs. However, the yield and degree of purification are often not high
enough because the density gradient methods are laborious for the complete removal of
foreign substances, and relatively many purification steps lead to a loss in target
proteins and time consumption. However, the affinity chromatography method can

97
provide high-yield, high-degree purification using procedures that are simple and brief.
However, these methods have not been frequently used to purify BNCs, because it was
believed they were not applicable. This procedure commonly requires appropriate
purification columns that depend on the types of produced BNCs, such as a sulfated
cellulofine column (for WT BNC; Kasuya et al., 2009) or a porcine IgG column (for Z
domain-displaying BNC; Kasuya et al., 2009), and often there is a failure to find a
suitable affinity column. Thus, since both methods have advantages and disadvantages,
a simple, versatile and high-recovery purification method that would be applicable to
any BNC would be useful. Therefore, we attempted to establish an affinity
chromatography method to purify BNCs using the His-tag that is a gold standard for
protein purification (Sakamoto et al., 2010). In the present study, we tried to develop
affinity purification by genetically fusing hexahistidine sequences (His6) to a targeting
molecule substituted for the pre-S region within the BNC. This simple method of only
inserting an His6-tag is expected to apply only to WT and altered BNCs. Therefore, we
investigated whether we could successfully purify ZHER2-BNCs using an His6-tag and
compared the effectiveness with the conventional ultracentrifugation purification
method.
RESULTS AND DISCUSSION
To purify BNCs by His6 affinity chromatography, we constructed a plasmid
that would express the His6-tagged ZHER2-BNC in which His6 was fused to the

98
N-terminus of a ZHER2 fragment (Fig. 1). The plasmid for expression of the His6-tagged
ZHER2-BNC was constructed to replace the pre-S region in the L protein with a
His6-ZHER2 molecule, as described below. The fragment encoding His6-fused ZHER2
(His6-ZHER2) was amplified by polymerase chain reaction (PCR) using
pGLDsLd50-ZHER2 (Shishido et al., 2010) as a template with the following primers: (5’-
GGG GGA TCC CAC CAC CAC CAC CAC CAC GCG CAA CAC GAT GAA GCC
GTA GAC AAC AAA TTC AAC AA -3’ and 5’- GGG GCG GCC GCC TTT CGG
CGC CTA AGC ATC AT -3’). The amplified fragment was digested with BamHI/NotI
and ligated into the BamHI/NotI sites of pGLDsLd50-ZHER2. The resultant plasmid was
designated as a pGLDsLd50-His6-ZHER2. A Saccharomyces cerevisiae AH22R- yeast
strain was transformed with the constructed plasmid using the spheroplast method, and
was cultured and disrupted with glass beads (Kuroda et al., 1992). Then we examined
whether the crude extract contained the His6-ZHER2 fusion proteins by western blotting
using anti-His6 antibody and anti-protein A antibody (data not shown). We detected the
same sized bands in both lanes, which indicated that the produced BNCs contained His6
and ZHER2 as a fusion protein.
A crude yeast extract was purified by each of the purification methods listed
below. We evaluated the degree of the purified samples collected in each step of
purification via silver staining using Sil-Best stain One (Nacalai Tesque, Kyoto, Japan)
(Fig. 2). For the ultracentrifugation purification method, we followed the previous
specified procedures (Kuroda et al., 1992). The objective bands were shown through
five steps of purification, which contained polyethylene glycol (PEG) precipitation for 2
h (lane 4), dialysis for 6 h after CsCl ultracentrifugation for 16 h (lane 5), dialysis for 6

99
Fig. 1. Schematic representations of constructed BNCs (WT BNC, ZHER2-BNC and
His6-ZHER2-BNC).

100
Fig. 2. Analysis of the degree of purification by silver staining. Lanes 1–2, His6 affinity
chromatography purification; lanes 3–8, ultracentrifugation purification. Sample was
subjected to SDS-PAGE followed by crude yeast extract (lane 1 and lane 3) into an
affinity column and dialyzed for 6 h (lane 2), PEG precipitation for 2 h (lane 4), dialysis
for 6 h after CsCl ultracentrifugation for 16 h (lane 5), dialysis for 6 h after CsCl
ultracentrifugation for 14 h (lane 6), dialysis for 6 h after sucrose ultracentrifugation for
10 h (lane 7) and condensation (lane 8).

101
h after CsCl ultracentrifugation for 14 h (lane 6), dialysis for 6 h after sucrose
ultracentrifugation for 10 h (lane 7), and condensation (lane 8). However, it was thought
that the purified samples contained unnecessary proteins, because other thin bands and a
smear band were also detected throughout the lanes.
Next, a His6 affinity chromatography method was performed as described
below. The crude extract was prepared by disrupting the cultured yeast cells with glass
beads in 75 ml of dissociation buffer [7.5 M Urea, 170 mM Na2HPO4, 40 mM NaH2PO4,
15 mM disodium ethylene diamine tetraacetic acid (EDTA/2Na), 2 mM
phenylmethylsulfonyl fluoride (PMSF), and 0.01% sorbitan polyoxyethylene
monooleate (Tween 80)]. The pellet was removed by centrifugation at 14,000×g and
4 °C for 10 min. The His6-ZHER2-BNC was separated from the crude extract using Ni2+
-chelate affinity chromatography. A column with 5 ml of Ni2+-chelate agarose (Nacalai
Tesque) was pre-equilibrated with a 5-fold volume of dissociation buffer. The crude
extract was loaded into the column and washed with 15 ml of dissociation buffer. Then,
bound proteins were eluted with 15 ml of elution buffer (dissociation buffer with 1 M
imidazole). The eluate was fractionated in 1 ml aliquots. The aliquots of each fraction
were analyzed by silver staining to select the fractions containing proteins. The proteins
of purified fractions were finally dialyzed in phosphate-buffered saline (PBS) to remove
the urea. The method was very simple, and amounted to simply applying crude yeast
extract into the affinity column and dialyzing it for 6 h (lane 2). Two objective, albeit
different, bands were observed near 44 kDa that were produced by the presence and
absence of N-glycosylation, as previously reported (Shishido et al., 2010). The thin
band observed at near 25 kDa was a degraded His6-fused protein. This useless protein

102
should be omitted by optimizing the process (e.g., changing the cultivation and/or
extraction conditions) when the BNCs are used for commercial purposes. Nevertheless,
clear, objective bands were obtained without the smear bands produced by
ultracentrifugation purification, demonstrating a high purity for the His6 affinity
chromatography.
We measured the collateral concentrations of proteins contained within each
step of purification using a Lowry protein assay and then calculated the yields. In the
case of the ultracentrifugation method (Table 1B), we eventually obtained 1.3 mg
protein (yield, 0.61%). When combined with the results of silver staining, however, the
actual yield would be less than 0.61%. In the case of the His6 affinity chromatography
method, we eventually obtained 2.9 mg protein (yield, 1.52%). From the results of the
silver staining, these values would be almost the same as the actual amount and yield of
the His6-tagged target protein, because the useless proteins were rarely contaminated.
As a result, the His6 affinity chromatography method achieved a yield that was almost
2.5-fold higher than that achieved with ultracentrifugation, despite being a simple
one-step purification (Table 1A).
To evaluate the quality of purified His6-ZHER2-BNCs, we measured the
diameter by DLS using a Zetasizer Nano particle size analyzer (Malvern Instruments
Ltd., Worcestershire, UK). The His6-ZHER2-BNCs purified by His6 affinity
chromatography were about 100 nm in diameter, and were similar to the
His6-ZHER2-BNCs that were purified via the ultracentrifugation method. Since both
diameters were almost equal to that of the ZHER2-BNCs (without His6-tag) purified by

103
Table. 1. The yield of purified protein for each step of (A) affinity chromatography
method and (B) ultracentrifugation method. The number of steps was compatible with
each lane of Fig. 2.
100.00191.162.52940.11. Crude extract
1.522.915.0189.92. After affinity purification
(A) Step of affinitychromatography method
Concentration[g/ml]
Liquid measure[ml]
Mass[mg]
Yield[%]
100.00191.162.52940.11. Crude extract
1.522.915.0189.92. After affinity purification
(A) Step of affinitychromatography method
Concentration[g/ml]
Liquid measure[ml]
Mass[mg]
Yield[%]
Yield[%]
Mass[mg]
Liquid measure[ml]
Concentration[g/ml]
(B) Step of ultracentrifugation method
0.611.310.0133.18. After condensation
1.362.9103.828.07. After ultracentrifugation 3
3.417.347.5154.26. After ultracentrifugation 2
13.4128.755.0522.1 5. After ultracentrifugation 1
43.6993.527.03461.84. After PEG precipitation
100.00214.060.03566.93. Crude extract
Yield[%]
Mass[mg]
Liquid measure[ml]
Concentration[g/ml]
(B) Step of ultracentrifugation method
0.611.310.0133.18. After condensation
1.362.9103.828.07. After ultracentrifugation 3
3.417.347.5154.26. After ultracentrifugation 2
13.4128.755.0522.1 5. After ultracentrifugation 1
43.6993.527.03461.84. After PEG precipitation
100.00214.060.03566.93. Crude extract

104
the ultracentrifugation method (Shishido et al., 2010), this indicated that the insertion of
a His6-tag into the ZHER2-BNC had no influence on particle formation.
Finally, to determine the ability of cell specificity, we reacted the purified
BNCs with Alexa Fluor 488 succinimidyl esters (Invitrogen Life Technologies,
Carlsbad, CA, USA) (2.6 mol equivalent) in PBS for 1 h at room temperature and
dialyzed the mixture against PBS overnight to remove the free Alexa Fluor 488. Then,
we added them to HER2-positive SKBR3 cells (human breast carcinoma) (Davison et
al., 2011) and HER2-negative HeLa cells (human cervical carcinoma) (Jia et al., 2003).
The fluorescence of the cells was observed using a confocal laser scanning microscope
(CLSM) (Fig. 3). We observed fluorescence in both SKBR3 cells treated with
His6-ZHER2-BNCs purified by ultracentrifugation and the His6 affinity chromatography
also with ZHER2-BNCs (without a His6-tag) purified by ultracentrifugation, which
indicated that the His6-fused ZHER2-BNCs had no adverse effect on cell specificity. This
result showed that the insertion of a His6-tag into the BNCs is a successful alternate for
the previously reported BNC purification techniques.
CONCLUSION
We successfully developed a simple, versatile and high-recovery purification
method using His6 affinity chromatography. This method permits the basic purification
of altered BNCs, and would be useful for large-scale purification in commercial

105
Fig. 3. Fluorescence images of HER2-positive SKBR3 and HER2-negative HeLa cells
treated with Alexa Fluor 488 labeled His6-ZHER2-BNC purified by ultracentrifugation or
His6 affinity chromatography and ZHER2-BNC (without His6) purified by
ultracentrifugation. Cells were incubated for 1 h in the media adjusted to 10 g/ml as
protein. After washing twice, cells were additionally incubated for 2 h and then
observed by a confocal laser scanning microscope. Scale bar, 50 m.

106
applications.
REFERENCES
Davison, Z., Blacquiere, G.E., Westley, B.R., May, F.E.B., 2011. Insulin-like Growth
Factor-Dependent Proliferation and Survival of Triple-Negative Breast Cancer Cells:
Implication for Therapy. Neoplasia. 13, 504-515.
Jia, L.T., Zhang, L.H., Yu, C.J., Zhao, J., Xu, Y.M., Gui, J.H., Jin, M., Ji, Z.L., Wen,
W.H., Wang, C.J., Chen, S.Y., Yang, A.G., 2003. Specific Tumoricidal Activity of a
Secreted Proapototic Protein Consisting of HER2 Antibody and Constitutively Active
Caspase-3. Cancer Res. 63, 3257-3262.
Kasuya, T., Jung, J., Kadoya, H., Matsuzaki, T., Tatematsu, K., Okajima, T., Miyoshi,
E., Tanizawa, K., Kuroda, S., 2008. In Vivo Delivery of Bionanocapsules
Displaying Phaseolus vulgarisAgglutinin-L4 Isolectin to Malignant Tumors
Overexpressing N-Acetylglucosaminyltransferase V. Human Gene Therapy. 887-895.
Kasuya, T., Jung. J., Kinoshita. R., Goh. Y., Matsuzaki. T., Iijima. M., Yoshimoto. N.,
Tanizawa. K., Kuroda. S., 2009. Bio-nanocapsule-liposome conjugates for in vivo
pinpoint drug and gene delivery. Methods Enzymol. 464, 147-166.

107
Kuroda, S., Otaka, S., Miyazaki, T., Nakao, M., Fujisawa, Y., 1992. Hepatitis B virus
envelope L protein particles. Synthesis and assembly in Saccharomyces cerevisiae,
purification and characterization. J Biol Chem. 267, 1953-1961.
Lee, S.B., Hassan, M., Fisher, R., Chertov, O., Chernomordik, V., Kramer-Marek, G.,
Gandjbakhche, A., Capala, J., 2008. Affibody molecules for in vivo characterization of
HER2-positive tumors by near-infrared imaging. Clin Cancer Res. 14, 3840-3849.
Orlova, A., Magnusson, M., Eriksson, T.L., Nilsson, M., Larsson, B.,
Hoiden-Guthenberg, I., Widstrom, C., Carlsson, J., Tolmachev, V., Stahl, S., Nilsson,
F.Y., 2006. Tumor imaging using a picomolar affinity HER2 binding affibody molecule.
Cancer Res. 66, 4339–4348.
Sakamoto. T., Sawamoto. S., Tanaka. T., Fukuda. H., Kondo. A., 2010.
Enzyme-mediated site-specific antibody-protein modification using a ZZ domain as a
linker. Bioconjug Chem. 21 (12), 2227-2233.
Shishido, T., Azumi, Y., Nakanishi, T., Umetsu, M., Tanaka, T., Ogino, C., Fukuda, H.,
Kondo, A., 2009a. Biotinylated bionanocapsules for displaying diverse ligands toward
cell-specific delivery. J Biochem. 146, 867-874.
Shishido, T., Yonezawa, D., Iwata, K., Tanaka, T., Ogino, C., Fukuda, H., Kondo, A.,
2009b. Construction of arginine-rich peptide displaying bionanocapsules. Bioorg Med
Chem Lett. 19, 1473-1476.

108
Shishido, T., Mieda, H., Hwang S.Y., Nishimura, Y., Tanaka, T., Ogino, C., Fukuda, H.,
Kondo, A., 2010. Affibody-displaying bionanocapsules for specific drug delivery to
HER2-expressing cancer cells. Bioorg Med Chem Lett. 20, 5726-5731
Yamada, T., Iwasaki, Y., Tada, H., Iwabuki, H., Chuah, M.K., VandenDriessche, T.,
Fukuda, H., Kondo, A., Ueda, M., Seno, M., Tanizawa, K., Kuroda, S., 2003.
Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat Biotechnol.
21, 885-890.

109
PART V.
Granting specificity for breast cancer cells using a Hepatitis B
core particle with a HER2-targeted affibody molecule

110
INTRODUCTION
Anticancer drugs act against abnormal proteins in cancer cells and present a
large treatment effect. However, they are often limited by their systemic toxicities and
side effects (Wang, 2012). Therefore, targeting ability is an important factor for the
development of drug delivery systems (DDS). To attain pinpoint delivery to target cells,
studies have extensively focused on fusing targeting molecules with the drug itself
(Lorberboum-Galski, 2011) or on modifying the surface of the DDS carrier (Liong,
2008; Nie, 2007). As the targeting molecules, binding molecules such as antibodies (Ma,
2011), peptides (Accardo, 2012) and aptamers (Zhang, 2012) are often used.
As a binding protein, the affibody is an attractive molecule. An affibody is a
small molecule that is based on the Z domain derived from Staphylococcus aureus
protein A (Nygren, 2008). As a type of affibody, ZHER2 has the ability to bind to HER2
that is a type 2 epidermal growth factor receptor (EGFR) and is expressed on the surface
of breast cancer cells and ovarian cancer cells (Orlova, 2006; Lee, 2008). Since natural
ligands against HER2 have yet to be found in nature (Shojaei, 2012), ZHER2 has been
used as an alternative molecular probe to diagnose (Gao, 2011) or target HER2
expressing cells (Puri, 2008). In addition, various other types of affibodies such as ZWT,
Z440 and Z955 can be used as the binding molecules to the Fc regions of immunoglobulin
G, IGF1R and EGFR, respectively (Nygren, 2008; Li, 2010; Nordberga, 2007).
Hepatitis B virus (HBV) core protein (HBc) has been studied for developing
viral genome-free particles as DDS carriers. The HBc is a 183 amino acid (aa) protein
and assembles spontaneously into icosahedral capsid-like particles comprising 180~240
subunits (Cooper, 2005). The important feature of HBc is to transiently dissociate and

111
re-associate in the presence or absence of denaturants, thereby enabling it to enclose
molecules such as drugs (Leea, 2008). In addition, the HBc can be produced in large
quantities, because it can be expressed in Escherichia coli (Wizemann, 1999). The
original HBc has been used as a permeable particle because it has the ability to bind to
every cell (Cooper, 2005), which is caused by an arginine-rich domain (150~183 aa)
that recognizes the cell surface heparan sulfate proteoglycan with an electrostatic
interaction (Cooper, 2006). Additionally, foreign molecules (e.g. GFP) have been
successfully displayed on the surface of a HBc particle without drastically altering its
structure via insertion into the 78~81 aa position of the original HBc core protein (Kratz,
1999).
ΔHBc consisting of the first 149 aa residues of a core protein was developed
as a deletion mutant lacking a non-specific binding ability (Birnbaum, 1990). The ΔHBc
particle is far more suitable for a DDS capsule than the original HBc because of the
particle’s capacity to incorporate drugs (Beteramsa, 2000) and the avoidance of
host-derived RNA/DNA binding functions (Birnbaum, 1990). However, despite the
successful development of the ΔHBc particle, there have been no reports of a
binding-molecule-fused ΔHBc.
In the present study, we developed a concept for constructing a DDS carrier
that is based on the ΔHBc particle and that can specifically recognize target cancer cells.
By genetically inserting a ZHER2 affibody between the 78~81 aa of a ΔHBc core protein,
the engineered particle (ZHER2-ΔHBc) specifically binded to HER2-expressing breast
cancer cells.

112
MATERIALS AND METHODS
Construction of plasmids for the expression of core particles
The plasmids for expression of HBc, ΔHBc and ZHER2-ΔHBc were
constructed as described below. Fragment 1 and fragment 2 encoding HBc were
amplified by polymerase chain reaction (PCR) with the following primers: fragment 1
(5’- GGG GCT AGC AAT AAT TTT GTT TAA CTT TAA GAA GGA GAT ATA CAT
ATG ATG GAC ATT GAC CCG TAT AA -3’ and 5’- ATT CTC TAG ACT CGA GAT
TAC TTC CCA CCC AGG TGG -3’) and fragment 2 (5’- TAA TCT CGA GTC TAG
AGA ATT AGT AGT CAG CTA TGT -3’ and 5’- CCC GTC GAC TTA GTG GTG
GTG GTG GTG GTG ACA TTG AGA TTC CCG AGA TT -3’). Then, the whole-length
fragment encoding of HBc was amplified from fragment 1 and fragment 2 with the
following primers: (5’- GGG GCT AGC AAT AAT TTT GTT TAA CTT TAA GAA
GGA GAT ATA CAT ATG ATG GAC ATT GAC CCG TAT AA -3’ and 5’- CCC GTC
GAC TTA GTG GTG GTG GTG GTG GTG ACA TTG AGA TTC CCG AGA TT -3’).
The amplified fragment was digested with NheI/SalI and ligated into the XbaI/SalI sites
of pET-22b (+) (Novagen). The resultant plasmid was designated as pET-22b-HBc.
Fragment encoding of ΔHBc was amplified from pET-22b-HBc with the following
primers: (5’- TAA TCT CGA GTC TAG AGA ATT AGT AGT CAG CTA TGT -3’ and
5’- GGG GTC GAC AAG CTT TTA GTG GTG GTG GTG GTG GTG AAC AAC AGT
AGT TTC CGG AA -3’). The amplified fragment was digested with XbaI/SalI and
ligated into the same sites of pET-22b-HBc. The resultant plasmid was designated as
pET-22b-ΔHBc. A fragment encoding ZHER2 was amplified from pGLDsLd50-ZHER2

113
(Shishido, 2010) with the following primers: (5’- GGG CTC GAG GAC GGT GGT
GGT GGT TCT GCG CAA CAC GAT GAA GCC GT -3’ and 5’- GGG TCT AGA ACC
ACC ACC ACC TTT CGG CGC CTG AGC ATC AT -3’). The amplified fragment was
digested with XhoI/XbaI and ligated into the same sites of pET-22b-ΔHBc. The resultant
plasmid was designated as pET-22b-ZHER2-ΔHBc.
Expression of core particles in E. coli
The plasmids for expression of HBc, ΔHBc and ZHER2-ΔHBc were transformed
into E. coli BL21. The culture of E. coli BL21 carrying each plasmid was diluted with 1
L of fresh LB-medium (1% tryptone, 0.5% yeast extract, 0.5% NaCl) in the presence of
100 g/ml ampicillin and grown to OD600 = 0.7 at 37 °C and a shaking speed of 150
rpm. The culture was induced by adding isopropyl-β-thiogalactopyranoside (IPTG) to a
final concentration of 0.1 mM at 25 °C overnight. Cells were then collected at 3,000
rpm and 4 °C for 15 min.
Purification of core particles
To purify core particles fused with a His6-tag, we followed the procedure
described by H. Wizemann and A. von Brunn (1999) with minor modifications. Briefly,
the cell pellet was resuspended in 50 ml of lysis buffer (pH 8.0)(50 mM Tris-HCl, 100
mM NaCl, 5 mM EDTA, 0.2% Triton X-100, 10 mM β-mercaptethanol). The cells were
lysed on ice by 3 cycles of sonication for 1 min each with 2 min intervals to avoid
heating of the material. The supernatant was removed by centrifugation at 15,000 rpm
and 4 °C for 30 min. The core particles in the pellet were washed in 50 ml of lysis
buffer and collected by centrifugation at 12,000 rpm and 4 °C for 15 min twice. The

114
pellet containing E. coli proteins was dissolved in 50 ml of dissociation buffer (pH
9.5)(4 M Urea, 200 mM NaCl, 50 mM Sodium Carbonate, 10 mM β-mercaptethanol) by
overnight incubation in an ice-cold water bath. Then, the soluble fraction was separated
by centrifugation at 15,000 rpm and 4 °C for 20 min.
Contaminating proteins were separated from the core particle proteins using
Ni2+-chelate affinity chromatography. A column with 10 ml of Ni2+-chelate agarose
(Nacalai Tesque, Kyoto, Japan) was pre-equilibrated with a 5-fold volume of
dissociation buffer. The prepared sample was loaded into a column and washed with 30
ml of dissociation buffer. Then, bound proteins were eluted with 30 ml of elution buffer
(pH 9.5) (4 M Urea, 200 mM NaCl, 50 mM sodium Carbonate, 10 mM
β-mercaptethanol, 1 M Imidazole). The eluate was fractionated in 1 ml aliquots. The
aliquots of each fraction were subjected to sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE) and stained with Coomassie Brilliant Blue (CBB) to
analyze their purity. The proteins of purified fractions were polymerized to core
particles by the removal of the urea in a polymerization buffer (pH 7.0)(500 mM NaCl,
50 mM Tris-HCl, 0.5 mM EDTA).
SDS-PAGE and western blotting
The expression of each core monomer was confirmed by western blotting. The
purified core particles were analyzed by SDS-PAGE and electrotransferred onto a
polyvinilidene fluoride (PVDF) membrane. For the detection of the His6 tag, Rabbit
anti-6-His antibodies (Bethyl Laboratories, Montgomery, TX, USA) were used as a
primary antibody for immunoblotting, followed by anti-rabbit antibodies conjugated
with alkaline phosphatase (AP) (Promega, Madison, WI, USA) used as a second

115
antibody. For the detection of Z protein, Goat anti-protein A antibodies (Rockland
Immunochemicals Inc, Gilbertsville, PA, USA) were used as the primary antibody for
immunoblotting, followed by anti-goat antibodies conjugated with alkaline phosphatase
(AP) (Promega) used as the second antibody. The membrane was stained with
5-bromo-4-chloro-3-indolyl phosphate (BCIP) and nitro blue tetrazolium (NBT)
(Promega).
AFM Analysis of Purified core particles
A gold chip (100 nm thickness of Au wafer, KST world, Fukui, Japan) was
covered with 200 l of solution containing core particles at room temperature for 1 h.
The gold chip was then washed with 10 ml of polymerization buffer. After washing, the
core particles adsorbed onto the surface of the gold chip were measured using an
SPA400-Nanonavi atomic force microscopy (AFM) unit (SII Nanotechnology Inc,
Chiba, Japan) with a cantilever (BL-RC150VB-C1 from Olympus; Tokyo, Japan) at 0.8
kHz scan speed according to the manufacturer's procedure.
Dynamic light scattering analysis of purified core particles
The size of the purified core particles was determined by dynamic light
scattering (DLS) using a Zetasizer Nano ZS (Malvern Instruments Ltd., Worcestershire,
UK), following the manufacturer's procedure.
Cell culture
SKBR3 cells (human breast carcinoma) were maintained in RPMI 1640
medium supplemented with 10% (v/v) FBS at 37 °C in 5% CO2. MCF-7 cells (human

116
breast carcinoma) and HeLa cells (human cervical carcinoma) were maintained in
DMEM medium supplemented with 10% FBS at 37 °C in 5% CO2.
Flow cytometric evaluation
Purified core particles were reacted with Alexa Fluor 488 Succinimidyl Esters
(Invitrogen Life Technologies, Carlsbad, CA, USA) (2.6 mol equiv) in PBS for 1 h at
room temperature. The mixture then was dialyzed against polymerization buffer
overnight to remove free Alexa Fluor 488. Approximately 2×105 of SKBR3, MCF-7 and
HeLa cells were seeded in individual 12-well plates. After washing with serum-free
medium, indicated volumes of Alexa Fluore 488-labeled core particles were added to
the medium, each of which was adjusted to a volume of 1 ml, followed by culturing of
the cells for 1 h. After washing with serum-free medium twice, the cells were incubated
with FBS-containing medium for 2 h. Cells were suspended into a sheath solution and
subjected to a BD FACSCanto II flow cytometer equipped with a 488-nm blue laser
(BD Biosciences, San Jose, CA, USA). The green fluorescence signal was collected
through a 530/30-nm band-pass filter. The data were analyzed using the BD FACSDiva
software v5.0 (BD Biosciences).
Confocal laser scanning microscopy observation
Approximately 5×104 of SKBR3, MCF-7 and HeLa cells were seeded in
individual 35 mm glass-bottom dishes. After washing with serum-free medium, Alexa
Fluore 488-labeled core particles (10 g/ml) were added and then cells were cultured
for 1 h. After washing with serum-free medium twice, the cells were incubated with
FBS-containing medium for 2 h. The cells were observed using a LSM 5 PASCAL laser

117
scanning confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with a
63-fold oil immersion objective lens with excitation by the 488-nm line of an argon
laser and emission collection by a 505-nm long pass filter.
RESULTS AND DISCUSSION
Expression of core proteins and formation of particles
As shown in Fig. 1, we first constructed the plasmid to express the His6 tag
fused HBc, which consisted of 183 amino acid residues of full-length core proteins. To
eliminate the non-specific binding property of a core protein, we constructed the
plasmid to express a ΔHBc consisting of 149 amino acid residues by deleting the
corresponding sequence to the C-terminal arginine-rich domain (Fig. 1). Finally, we
constructed the expression plasmid for ZHER2-ΔHBc, in which a ZHER2 affibody
molecule was inserted between the 78~81 amino acid positions of the ΔHBc (Fig. 1). As
described in the materials and methods section, we introduced these plasmids into E.
coli and produced each kind of HBc particle.
First, to check the expression and purification of each core protein, we
performed western blot analysis with anti-His6 antibody. Since three His6-specific
bands appeared at each desired position (HBc, 21 kDa; ΔHBc, 17 kDa; and,
ZHER2-ΔHBc, 24 kDa, respectively), successful expression and purification of the core
proteins was confirmed (Fig. 2, left). When using anti-protein A antibody that can
specifically bind to the Z-derived affibodies, we detected the single band only for

118
Fig. 1. Schematic representations of constructed core proteins (HBc, ΔHBc and
ZHER2-ΔHBc). Wild-type HBc core protein consisted of an assembly domain (gray
prismatic body) and an arginine-rich domain (white prismatic body). For ΔHBc core
protein, the arginine-rich domain (150-183 aa) was deleted. For the ZHER2-ΔHBc core
protein, ZHER2 affibody (diagonal prismatic body) was inserted at the XhoI and XbaI
sites between 78 and 81 aa. For all constructs, His6-tag (black prismatic body) was
fused to the C-termini.

119
ZHER2-ΔHBc (24 kDa), as expected (Fig. 2, right). Furthermore, secondary structural
formation of each core protein was confirmed by circular dichroism (CD) spectra
analysis (data not shown).
Second, to examine whether each core protein-formed particle could be
attributed to the capsid-like structure, we analyzed the purified core proteins by DLS
(Fig. 3) and AFM (Fig. 4). Two measurement analysis suggested similar results, and it
was strongly supported that every core protein showed same particle-like structure that
was approximately 50 nm in diameter. According to previously reported for wild-type
HBc particles analysis [20, 21], it was assumed that the deletion of the arginine-rich
domain and the insertion of the ZHER2 affibody did not affect the self-assembly of the
engineered core proteins.
Examination of the ability of a ZHER2-ΔHBc particle to recognize HER2-expressing
breast cancer cells
Next, to evaluate the binding ability to HER2-expressing breast cancer cells,
we labeled the particles with Alexa Fluor 488. Then, each kind of particle was added to
two types of HER2 positive human breast cancer cells (SKBR3, expressing an abundant
amount of HER2; and, MCF-7, expressing a tiny amount of HER2) and a HER2
negative human cervical cancer cell (HeLa) at several concentrations. After 3 hours of
incubation, we measured the fluorescence intensity of these cells by FACS.
As shown in Fig. 5A, the addition of a wild-type HBc particle caused a
dose-dependent increase in fluorescence for all three kinds of cancer cells (SKBR3,
MCF-7 and HeLa), indicating that the original HBc particle binds to the cells in a
non-specific manner. The addition of a ΔHBc particle didn’t exhibit fluorescence for all

120
Fig. 2. Western blotting analyses of purified core particles. Purified samples (HBc,
ΔHBc and ZHER2-ΔHBc) were subjected to SDS-PAGE followed by immune blotting
using anti-His6 antibody (for His6 tag; left image) and anti-protein A antibody (for
ZHER2 affibody; right image).

121
Fig. 3. Particle size distribution analysis by DLS. The average sizes of (A) HBc, (B)
ΔHBc and (C) ZHER2-ΔHBc are 55.38±10.84 nm, 47.41±10.07 nm and 45.05±1.50
nm, respectively.
0
10
20
30
40
0.1 1 10 100 1000 10000
0
10
20
30
40
0.1 1 10 100 1000 10000
0
10
20
30
40
0.1 1 10 100 1000 10000
A. HBc
B. ΔHBc
C. ZHER2-ΔHBc
Inte
nsity
Inte
nsity
Inte
nsity
Diameter
Diameter
Diameter
0
10
20
30
40
0.1 1 10 100 1000 10000
0
10
20
30
40
0.1 1 10 100 1000 10000
0
10
20
30
40
0.1 1 10 100 1000 10000
A. HBc
B. ΔHBc
C. ZHER2-ΔHBc
Inte
nsity
Inte
nsity
Inte
nsity
Diameter
Diameter
Diameter

122
Fig. 4. AFM analyses of purified core particles. The macro and micro photographs
show 2D and 3D images, respectively. HBc core particles were analyzed on the surface
of the gold chip. (A) HBc, (B) ΔHBc and (C) ZHER2-ΔHBc. Scale bars, 50 m.
A. HBc B. ΔHBc
C. ZHER2-ΔHBc
A. HBc B. ΔHBc
C. ZHER2-ΔHBc

123
three cells, supporting the theory that the deletion of the arginine-rich domain can
cancel the non-specific binding ability of the original HBc (Fig. 5B). By contrast, the
addition of a ZHER2-ΔHBc particle promoted an apparent fluorescence for SKBR3 cells
in a dose-dependent manner (white bars), whereas it never exhibited fluorescence for
the HeLa cells (black bars) (Fig. 5C). Additionally, the addition of ZHER2-ΔHBc to
MCF-7 showed a weaker fluorescence than in the case of SKBR3 (Fig. 5C, gray bars).
These results indicate that ZHER2-ΔHBc specifically recognized HER2-expressing breast
cancer cells, and that the binding amount of ZHER2-ΔHBc differed in accordance with
the HER2 expression levels in the cells.
Finally, to visually observe the binding ability of these particles to the cells,
Alexa Fluor 488-labeled core particles were added into the cell cultures (SKBR3,
MCF-7 and HeLa) to give a final concentration of 10 g/ml. After 3 hours of incubation,
the cells were observed using a confocal laser scanning microscope (CLSM) (Fig. 6).
These results were consistent with the results of the FACS analyses (Fig. 5),
demonstrating that the ZHER2-ΔHBc particle has the ability to specifically bind to
HER2-expressing breast cancer cells.
CONCLUSION
We developed a HBc particle displaying a ZHER2 affibody that specifically
recognizes HER2-expressing breast cancer cells. It would be possible to incorporate
drugs into ZHER2-HBc particles by using the dissociation and association mechanism

124
Fig. 5. Relative fluorescence units (RFU) of HER2 positive and negative cells
treated with several concentrations of Alexa Fluor 488 labeled HBc core particles.
RFUs were determined by FACS measurement. (A) HBc, (B) ΔHBc and (C)
ZHER2-ΔHBc. White bars, SKBR3 (HER2, +++); gray bars, MCF-7 (HER2, +); and,
black bars, HeLa (HER2, –).
Concentration of core particles [g/ml]
0 1 2.5 5.0 7.5 10
0 1 2.5 5.0 7.5 10
0 1 2.5 5.0 7.5 10
6
5
4
3
2
1
0
16
14
12
10
8
6
0
4
2
RF
U [
-]
Concentration of core particles [g/ml]
RF
U [
-]
Concentration of core particles [g/ml]
RF
U [
-]
A. HBc
B. ΔHBc
C. ZHER2-ΔHBc
6
5
4
3
2
1
0
Concentration of core particles [g/ml]
0 1 2.5 5.0 7.5 10
0 1 2.5 5.0 7.5 10
0 1 2.5 5.0 7.5 10
6
5
4
3
2
1
0
16
14
12
10
8
6
0
4
2
RF
U [
-]
Concentration of core particles [g/ml]
RF
U [
-]
Concentration of core particles [g/ml]
RF
U [
-]
A. HBc
B. ΔHBc
C. ZHER2-ΔHBc
6
5
4
3
2
1
0

125
Fig. 6. Fluorescence images of SKBR3 (HER2, +++), MCF-7 (HER2, +) and HeLa
(HER2, –) treated with Alexa Fluor 488 labeled HBc core particles (10 g/ml). Cells
were observed on a confocal laser scanning microscope. Scale bars, 50 m.
MCF-7 HeLaSKBR3
HB
cΔ
HB
cZ
HE
R2-Δ
HB
c
MCF-7 HeLaSKBR3
HB
cΔ
HB
cZ
HE
R2-Δ
HB
c

126
regulated by salt concentration (Kann, 1994) or urea denaturant (Wizemann, 1999), or
by fusing peptidic drugs to C-terminal tail of core protein (Beteramsa, 2000). By
inserting other types of affibody molecules to the ΔHBc, these engineered HBc core
particles could be used as pinpoint carriers to target various kinds of cancer cells.
ABBREVIATIONS
DDS, drug delivery system; HER2, human EGFR-related 2; EGFR, epidermal growth
factor receptor; IGF1R, insulin-like growth factor-1 receptor ; HBV, hepatitis B virus;
HBc, hepatitis B core; GFP, green fluorescence protein; OD, overdose; IPTG,
isopropyl-β-thiogalactopyranoside; SDS-PAFE, sodium dodecyl
sulphate-polyacrylamide gel electrophoresis; CBB, Coomassie Brilliant Blue; RVDF,
polyvinilidene fluoride; AP, phosphatase; BCIP, 5-bromo-4-chloro-3-indolyl phosphate;
NBT, nitro blue tetrazolium; AFM, atomic force microscopy; FBS, Fetal bovine serum;
DMEM, Dulbecco’s modified Eagle medium; CLSM, confocal laser scanning
microscope; CD, circular dichroism; ΔHBc, HBc deletion mutant lacking arginine-rich
domain
REFERENCES

127
Accardo, A., Salsano, G., Morisco, A., Aurilio, M., Parisi, A., Maione, F., Cicala, C.,
Tesauro, D., Aloj, L., Rosa, G.D., Morelli, G., 2012. Peptide-modified liposomes for
selective targeting of bombesin receptors overexpressed by cancer cells: a potential
theranostic agent. Int J Nanomed. 7, 2007-2017.
Beteramsa, G., Bottcher, B., Nassal, M., 2000. Packaging of up to 240 subunits of a 17
kDa nuclease into the interior of recombinant hepatitis B virus capsids. FEBS Letters.
481, 169-176.
Birnbaum, F., Nassal, M., 1990. Hepatitis B Virus Nucleocapsid Assembly: Primary
Structure Requirements in the Core Protein. J Virol. 64 (7), 3319-3330.
Cooper, A., Shaul, Y., 2005. Recombinant viral capsids as an efficient vehicle of
oligonucleotide delivery into cells. BBRC. 327, 1094-1099.
Cooper, A., Shaul1, Y., 2006. Clathrin-mediated Endocytosis and Lysosomal Cleavage
of Hepatitis B Virus Capsid-like Core Particles. JBC. 281 (24), 16563-16569.
Gao, J., Chen, K., Miao, Z., Ren, G., Chen, X., Gambhir, S.S., Cheng, Z., 2011.
Affibody-based nanoprobes for HER2-expressing cell and tumor imaging. Biomaterials.
32 (8), 2141-2148.
Kann, M., Gerlich, W., 1994. Effect of Core Protein Phosphorylation by Protein Kinase
C on Encapsidation of RNA within Core Particles of Hepatitis B Virus. J Virol. 68 (12),

128
7993-8000.
Kratz, P.A., Bottcher, B., Nassal, M., 1999. Native display of complete foreign protein
domains on the surface of hepatitis B virus capsids. PNAS. 96, 1915-1920.
Lee, S.B., Hassan, M., Fisher, R., Chertov, O., Chernomordik, V., Kramer-Marek, G.,
Gandjbakhche, A., Capala, J., 2008. Affibody molecules for in vivo characterization of
HER2-positive tumors by near-infrared imaging. Clin Cancer Res. 14, 3840-3849.
Leea, K.W., Tana, W.S., 2008. Recombinant hepatitis B virus core particles: Association,
dissociation and encapsidation of green fluorescent protein. J Virol Meth. 151, 172-180.
Li, J., Lundberg, E., Vernet, E., Larsson, B., Hoiden-Guthenberg, I., Graslund, T., 2010.
Selection of affibody molecules to the ligand-binding site of the insulin-like growth
factor-1 receptor. Biotechnol Appl Biochem. 55, 99-109.
Liong, M., Lu, J., Kovochich, M., Xia, T., Ruehm, S.G., Nel, A.E., Tamanoi, F., Zink,
J.I., 2008. Multifunctional Inorganic Nanoparticles for Imaging, Targeting, and Drug
Delivery. ACS Nano. 2 (5), 889-896.
Lorberboum-Galski, H., 2011. Human toxin-based recombinant immunotoxins/chimeric
proteins as a drug delivery system for targeted treatment of human diseases. Epert Opin
Drug Deliv. 5, 605-621.

129
Ma, L.L., Tam, J.O., Willsey, B.W., Rigdon, D., Ramesh, R., Sokolov, K., Johnston,
K.P., 2011. Selective Targeting of Antibody Conjugated Multifunctional Nanoclusters
(Nanoroses) to Epidermal Growth Factor Receptors in Cancer Cells. Langmuir. 27 (12),
7681-7690.
Nie, S., Xing, Y., Kim, G.J., Simons, J.W., 2007. Nanotechnology applications in cancer.
Annu Rev Biomed Eng. 9, 257-288.
Nordberga, E., Friedmanb, M., Gostringa, L., Adamsd, G.P., Brismare, H., Nilssona, F.Y.,
Stahlb, S., Glimeliusf, B., Carlssona, J., 2007. Cellular studies of binding,
internalization and retention of a radiolabeled EGFR-binding affibody molecule. Nuc
Med Bio. 34, 609-618.
Nygren, P.-A., 2008. Alternative binding proteins: Affibody binding proteins developed
from a small three-helix bundle scaffold. FEBS J. 275, 2668-2676.
Orlova, A., Magnusson, M., Eriksson, T.L.J., Nilsson, M., Larsson, B.,
Hoiden-Guthenberg, I., Widstrom, C., Carlsson, J., Tolmachev, V., Stahl, S., Nilsson,
F.Y., 2006. Tumor Imaging Using a Picomolar Affinity HER2 Binding Affibody
Molecule. Cancer Res. 66, 4339-4348.
Puri, A., Kramer-Marek, G., Campbell-Massa, R., Yavlovich, A., Tele, S.C., Lee, S.B.,
Clogston, J.D., Patri, A.K., Blumenthal, R., Capala, J., 2008. HER2-specific
affibody-conjugated thermosensitive liposomes (Affisomes) for improved delivery of

130
anticancer agents. J Liposome Res. 18 (4), 293-307.
Shishido, T., Mieda, H., Hwang, S.Y., Nishimura, Y., Tanaka, T., Ogino, C., Fukuda,
H., Kondo, A., 2010. Affibody-displaying bionanocapsules for specific drug delivery to
HER2-expressing cancer cells. Bioorg Med Chem Lett. 20, 5726-5731.
Shojaei, S., Gardaneh, M., Shamabadi, A.R., 2012. Target Points in Trastuzumab
Resistance. Int J Breast Cancer.
Wang, S., Placzek, W.J., Stebbins, J.L., Mitra, S., Noberini, R., Koolpe, M., Zhang, Z.,
Dahl, R., Pasquale, E.B., Pellecchia, M., 2012. Novel Targeted System To Deliver
Chemotherapeutic Drugs to EphA2-Expressing Cancer Cells. J Med Chem. 55,
2427-2436.
Wizemann, H., von Brunn, A., 1999. Purification of E. coli-expressed HIS-tagged
hepatitis B core antigen by Ni2+-chelate affinity chromatography. J Virol Meth. 77,
189-197.
Zhang, K., Sefah, K., Tang, L., Zhao, Z., Zhu, G., Ye, M., Sun, W., Goodison, S., Tan,
W., 2012. A Novel Aptamer Developed for Breast Cancer Cell Internalization, Chem
Med Chem. 7, 79-84.

131
GENERAL CONCLUSION
In the cancer therapy, effective medicinal treatments without significant side
effects are required. Since nanoparticles (NPs) are attracted attention as a mean of
achieving the goal, various types of drug carriers have been developed. However,
optimal carriers have not yet been established, because a variety of characteristics
including safety, stability, specificity, high transfection efficiency and controlled release
of drugs are needed. Hence, we have used hollow NPs, bio-nanocapsules (BNCs) that
are composed of lipids bilayer and L proteins derived from Hepatitis B virus (HBV)
surface antigen (HBsAg). BNCs are very safe without viral genome and show the
ability for high transfection and the specificity to hepatocyte due to the action of HBsAg.
It was demonstrated that BNCs encapsulating drugs and genes could be delivered to
target hepatocyte cancer effectively; therefore, BNCs would be expected to become
effective carriers for drug delivery. In this study, we evaluated the in vitro delivery of
protein and siRNA with BNCs and the large scale purification of them toward in vivo
applications.
First, to apply BNCs for protein therapy, we established the method for
encapsulating proteins into BNCs. In this method, we tried to encapsulate proteins at the
same time as BNC formation, since it is difficult to encapsulate proteins into BNCs after
particle formation. We succeeded to encapsulate proteins into BNCs by fusing
membrane localization sequence (MLS) to target proteins and co-expressing the
proteins with L protein in insect cells. This result indicated that we could encapsulate
pharmaceutical proteins into BNCs by using this method.

132
Next, to develop more efficient protein delivery, we used the BNC/LP
conjugation method to fuse BNCs on the surface of LP encapsulating proteins. We used
Affibody; ZHER2, which binds to HER2 specifically, displaying BNC (ZHER2-BNC) and
tried to target HER2-expressing breast cancer cells. It has been indicated that the
conjugation of BNC with anionic LP permitted target-cell-specific delivery and the
mixing of helper lipids possessing the endosome escaping ability with anionic LP
allowed to express effective activity of pharmaceutical protein. These results
successfully showed target-cell-specific protein therapy in vitro. Thus, ZHER2-BNC/LP
complex carrier would be a useful tool for protein delivery in vivo.
Additionally, we tried to deliver siRNA which has now drawn attention as
nucleic acid medicine. Although siRNA is greatly specific and effective, there is a
problem for instability in the blood and inability to specifically reach target cells and to
cross the cell membrane. Therefore, we used ZHER2-BNC/LP to deliver siRNA,
confirming effective RNAi specifically in target breast cancer cells. This result indicated
that ZHER2-BNC/LP complex would be permitted as a carrier for nucleic acid medicine
as well as protein therapy.
Although the availability of BNC has been shown as described above,
large-scale production is required for in vivo application. Therefore, we established the
new purification method alternative to conventional ultracentrifugation method. We
genetically fused His-tag to ZHER2-BNC to carry out affinity chromatography. As a
result, we succeeded to simplify laborious purification process and to achieve 2.5-fold
higher yields than ultracentrifugation method. Since this method could purify BNCs in

133
large scale in one-step, it would be able to apply not only to in vivo application but also
to commercial production.
Finally, to develop a new type of carrier particle, we used a hepatitis B core
protein (HBc) which allows the large-scale production and the purification with His-tag
affinity chromatography. Since HBc has abilities to self-assembly formation and bind to
various cells non-specifically, we tried to develop an engineered HBc particle that
specifically recognizes and targets HER2-expressing breast cancer cells by despoiling
the non-specific binding property and granting the target-cell specific recognition ability
to the HBc particle with ZHER2 affibody. As a result, we could add the ability of
specificity for HER2-expressing cells to HBc.
In summary, BNCs have a variety of attractive abilities such as, safety,
specificity to target cells and high transfection efficiency. Furthermore, since stability
and controlled release are improved by conjugating BNCs with LP, it would be
indicated that BNCs acquire the abilities required for drug delivery carrier. Therefore, it
is expected to apply BNCs as nanotechnology for innovative cancer treatment in vivo.
Additionally, HBc particle would become to more highly-sophisticated carrier for DDS
in near future.

134
ACKOWLEDGMENTS
This is a thesis submitted by the author to Kobe University for the degree of
Doctor of Engineering. The studies reported here were carried out between from 2007 to
2013 under the direction of Professor Akihiko Kondo in the Laboratory of Biochemical
Engineering, Department of Chemical Science and Engineering, Graduate School of
Engineering, Kobe University.
First of all, the author would like to express his sincerest gratitude to his
research advisor, Professor Akihiko Kondo, for continuous guidance and invaluable
suggestions during the course of his studies. Next, the author would like to express his
hearty gratitude to Professor Hideki Fukuda for invaluable discussion and kind
support throughout this research. The author is also deeply grateful to Associate
Professor Chiaki Ogino and Toshinobu Fujiwara, Assistant Professor Fumiyoshi
Okazaki, Kazunori Nakashima and Tomohisa Hasunuma for their valuable advice
and hearty encouragement.
The author further wishes to acknowledge the contributions of Professor
Shun-ichi Kuroda (Nagoya University) for invaluable strategic and technical advice on
Bio-nanocapsules.
I wish to express my gratitude to the official reviewer, Professor Hideki
Yamaji and Atsunori Mori, who had time to take interest in the manuscript and to give
constructive criticism at the final stage of preparation.

135
The helpful discussions and advice of Assistant Professor Jun Ishii and Dr.
Takuya Shishido (Nitto Denko Co., Ltd.) and the technical assistance and
encouragement of, Mr. Naoya Kurata (Chugai Pharmaceutical Co., Ltd.), Mr. Daisaku
Yonezawa (Suntory Co., Ltd), Ms. Yuki Azumi (Nissha Printing Co., Ltd.), Mr.
Hiroaki Mieda (Canon Co., Ltd.), Ms. Wakiko Mimura (Canon Co., Ltd.), Mr.
Koichi Takeda, Mr. Izzat fahimuddin Mohamed sufflan, Mr. Ryosuke Ezawa, Mr.
Takayuki Sakamoto and all the members of Professor Kondo’s laboratory are also
sincerely acknowledged.
This work was, in partially, supported by Grants-in-aid for Research on
Advanced Medical Technology from the Ministry of Health, Labour and Welfare, Japan.
Last but not least, the author expresses my deep appreciation to my parents,
Hidetaka and Chieko Nishimura for continuous moral and financial support.
Yuya Nishimura
Department of Chemical Science and Engineering
Graduate School of Engineering
Kobe University

136
PUBLICATION LISTS
PART I.
Nishimura, Y., Shishido, T., Ishii, J., Tanaka, T., Ogino, C., Kondo, A., 2012.
Protein-encapsulated bio-nanocapsules production with ER membrane localization
sequences. J Biotechnol. 157(1), 124-129.
PART II.
Nishimura, Y., Ishii, J., Okazaki, F., Ogino, C., Kondo, A., 2012. Complex carriers of
affibody-displaying bio-nanocapsules and composition-varied liposomes for
HER2-expressing breast cancer cell-specific protein delivery. J Drug Target. 20(10),
897-905.
PART III.
Nishimura, Y., Mieda, H., Ishii, J., Ogino, C., Fujiwara, T., Kondo, A., Targeting cancer
cell-specific RNA interference by siRNA delivery using a complex carrier of
affibody-displaying bio-nanocapsules and liposomes. In revision.
PART IV.
Nishimura, Y., Takeda, K., Ishii, J., Ogino, C., Kondo, A., 2013. An affinity
chromatography method used to purify His-tag-displaying bio-nanocapsules. J Virol
Methods. 189(2), 393-396.
PART V.
Nishimura, Y., Mimura, W., Mohamed suffian, I.F., Amino, T., Ishii, J., Ogino, C.,
Kondo, A., 2013. Granting specificity for breast cancer cells using a Hepatitis B core
particle with a HER2-targeted affibody molecule. J Biochem. 153(3), 251-256.
OTHER PUBLICATIONS
Shishido, T., Mieda, H., Hwang S.Y., Nishimura, Y., Tanaka, T., Ogino, C., Fukuda, H.,
Kondo, A., 2010. Affibody-displaying bionanocapsules for specific drug delivery to
HER2-expressing cancer cells. Bioorg Med Chem Lett. 20, 5726-5731.


![Journal of Allergy & Therapy · ISSN:2155-6121 JAT an open access journal Volume 5 Issue 5 1000187 ... aged skin, xerosis, rosacea, and acne [1–5]. Many of these ... lipid bilayer,](https://static.fdocument.pub/doc/165x107/5cacbed588c993d4278cc386/journal-of-allergy-therapy-issn2155-6121-jat-an-open-access-journal-volume.jpg)















