
Klinické a genetické - lf2.cuni.cz · MND s nástupem v dosp ě losti • Amyotrofická...
22
Nemoci motorického neuronu (amyotrofická laterální skleróza) Klinické a genetické souvislosti
Transcript of Klinické a genetické - lf2.cuni.cz · MND s nástupem v dosp ě losti • Amyotrofická...

Nemoci motorického neuronu (amyotrofická
laterální
skleróza)
Klinické
a genetické
souvislosti

Onemocnění
motoneuronůEtiologicky heterogenní
skupina chorob
Interakce genetických a získaných poruchs familiárním a sporadickým výskytem
Selektivní
degenerace periferního a/nebo centrálního motoneuronu
Progrese svalové
slabosti a/nebo spasticképarézy

Základní
formy MND
•
Časný nástup v dětství•
Pouze periferní
motoneuron
•
Proximální
příznaky•
Spinální
svalové
atrofie I-IV
•
Delece
7+8 exonu
SMN1 genu v lokusu
5q13
•
Tíže fenotypu
SMA záleží
na počtu kopií
SMN2 genu
•
Pozdní
nástup v dospělosti•
Kombinace periferního i centrálního motoneuronu
•
Fokální, později generalizované
příznaky
•
Amyotrofická
laterální skleróza –
ALS
•
Varianty MND

MND s nástupem v dospělosti
•
Amyotrofická
Laterální
Skleróza
(80%) sporadická
nebo familiární
forma
•
Izolované
formy MND progresivní
svalová
atrofie (PMA) -
jen dolní
MN (10%)
progresivní
bulbární
paralýza (PBP) -
jen bulbární
MN (8%)primární
laterální
skleróza (PLS) -
jen horní
MN (2%)
(El Escorial
kritéria –
poslední
revize 2008 –
www.alsa.org)

Epidemiologie ALS
ALS (Lou
Gehrig´s disease) Prevalence -
4-6 : 100 000
Incidence -
0.5-3 : 100 000
90-95% sporadické
formy ALS (SALS)•
absence pozitivní
rodinné
anamnézy
5-10% familiární
formy ALS (FALS)
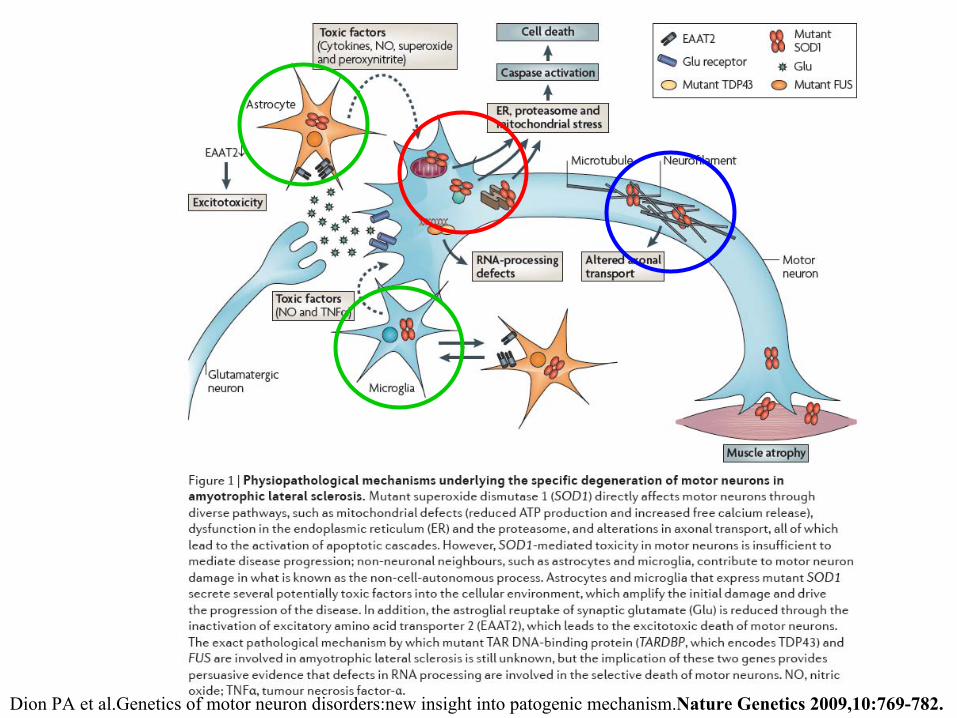
Dion
PA et
al.Genetics
of
motor neuron disorders:new
insight
into
patogenic
mechanism.Nature
Genetics
2009,10:769-782.

Klasická
ALS
Před klinickou manifestací
je 50 –
60 % motoneuronůdysfunkčních nebo zničeno (experimentální
práce)
První
symptomy :končetiny 75% bulbární
sy
25%
Horní
končetiny -
41%Dolní
končetiny -
34%

Klinický obraz ALS
•
Kombinace léze
centrálního a periferního motoneuronu•
Fascikulace
v kosterních svalech
•
Progresívní
průběh•
Nebolestivý vývoj
•
Normální
čití•
Bez okohybných a sfinkterových
poruch
•
Věk manifestace nejčastěji 45-65 let •
CAVE : fokální
začátek ( amyotrofie drobných svalů
na HK
jako n.ulnaris
nebo na DK jako n.peroneus)

Diagnostická
jistota ?
•
Možná
–
1 spinální
region s UMN + LMN nebo UMN ve 2 regionech nebo LMN je rostrálně
k UMN
•
Pravděpodobná
–
2 spinální
regiony s UMN + LMN
•
Jistá
–
bulbární
+ 2 spinální
regiony nebo 3 spinální regiony UMN + LMN
•
podrobně
website
www.alsa.org

Diagnóza ALS
•
Per exclusionem
–
neexistuje biomarker•
Klinický obraz
•
EMG vyš.•
MRI mozku a míchy
•
Likvorové
vyšetření
•
Někdy testy k potvrzení
paraneoplastické etiologie –
ALS like
syndromy

Léčba MND
•
Kauzální
terapie není
k dispozici•
Existuje léčba neuroprotektivní
riluzol
-
inhibitor glutamátu
(viz dále) antioxidanty –
koenzym Q10 + vitamin E
Zkoušelo se více než
24 molekul –
např.IGF-1, minocyklin, kreatin, vitamin E, tamoxifen…

ALS a Rilutek
•
jediný schválený FDA>> efekt
léčby
•
osoby
<
45 let•
rychle progresivní
průběh
•
bulbární
sy
na počátku•
hodnota FVC při 1.vyš.není
relevantní
pro
predikci efektu léčby ani progrese
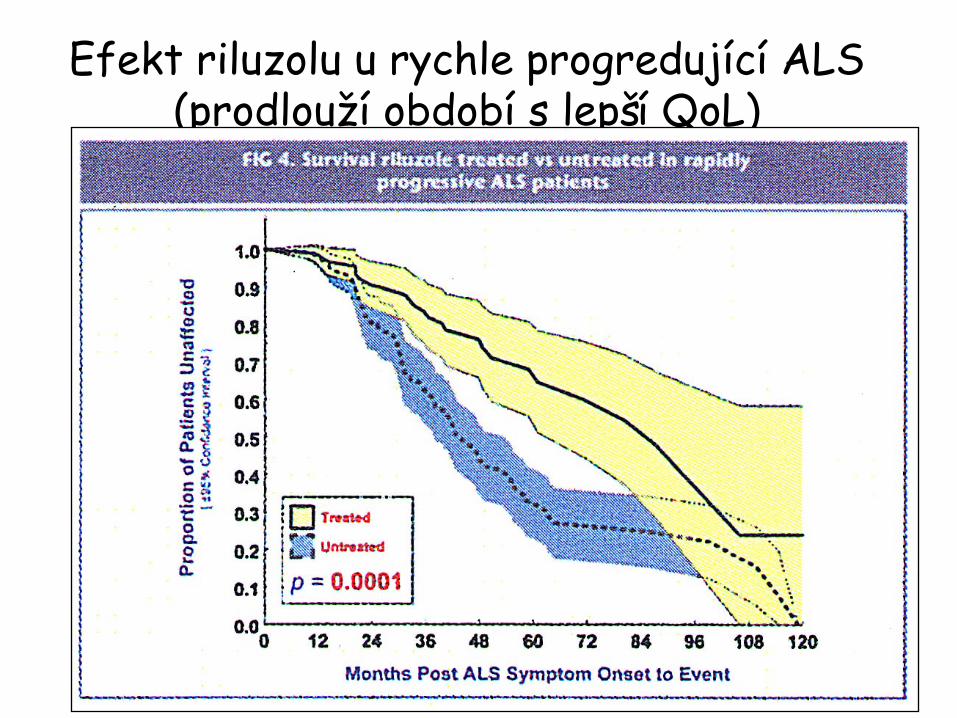
Efekt riluzolu
u rychle progredující
ALS (prodlouží
období
s lepší
QoL)

Léčba MND
Symptomatická
paliativní-
dnes zásadní1.
Mobilita
– pomůcky-hole, vozík, lůžko
2.
Nutrice
–
vážení, PEG + Nutrizon3.
Komunikace
–
tužka, písmena,tabulky, PC
4.
Psychika –
antideperesiva, psychoterapie5.
Respirace –
neinvazivní
nebo UPV
Důležitá
je komunikace s rodinou.

Algoritmus péče o ALS pacienty•
Obvykle 3 měsíční
kontroly-závisí
na stupni
postižení•
Biochemie-ALT-riluzol, spirometrie, vážení
•
Neurologické
+ svalové
testy –
hand
grip•
EMG –
pokud je dg.definitivní
nemá
smysl
opakovat•
Psychoterapie
–
na konzultace i rodina –
postupné
sdělení
diagnózy –
fáze paniky-vzteku- úzkosti-zmaru-smíření
•
Naděje na účinnou terapii

Klinické
studie•
Buněčná
terapie -
mesenchymální
kmenové
buňky
•
Systémová
aplikace i.v. •
Intrathekální
aplikace (Německo, Čína, ČR)
•
Kombinace i.v. + i.th.•
Intraspinální
aplikace –
nejprve L mícha, pak C
mícha
(USA, Itálie, Turecko)
•
Chybí
data, v ČR není
registr-
nevíme kolik pacientů
s ALS v ČR žije

ALS a dexpramipexol
–
ongoing
RCT
ALS Phase
3 Study Dexpramipexole
(KNS-760704) NCT01281189
is a randomized, double-blind, placebo-controlled, multi-center study of the safety and efficacy of dexpramipexole in subjects with amyotrophic lateral sclerosis (ALS). Dexpramipexole was previously known as KNS-760704. BIOGEN – 12týdnů
300 mg denně
–31% zpomalení
progrese

Roste význam molekulární
genetiky !
•
2012 –
známe > 30 genů, které
působí
MND
•
ALS –
familiární
ALS, ale i sporadická
ALS
•
Spinální
svalová
atrofie (SMA)
•
Hereditární
spastická
paraplegie (HSP)
•
Bulbospinální
svalová
atrofie (Kennedyho
choroba)
•
Distální
hereditární
motorická
neuropatie (dHMN)

Klinické
rozdíly FALS a SALS
•
Nejsou jasné
rozdíly v klinických symptomech
•
FALS časnější
nástup s průměrným věkem 46 let
•
FALS častěji postiženy DK
Zdroj:www.neuro.wustl.edu

ALS asociovaná
s genetickými defekty
Zdroj:www.neuro.wustl.edu

SOD1 –
Cu/Zn
superoxiddismutáza
•
Antioxidační
enzym•
Lokus
21q22.11,
> 130
mutací
•
20% všech FALS s AD typem dědičnosti
•
Objev u SALS ve 3%•
Variabilita v nástupu příznaků
a
délce trvání
nemoci•
p.Ala4Val –
50% v USA
(maligní
forma<12měsíců)•
p.Gly37Arg (pomalejší
průběh
>80měsíců)•
„gain
of
function“
s tvorbou
agregátů
a poruchy konformace proteinu
Zdroj:www.neuro.wustl.edu

Některé
zajímavé
genotyp-fenotyp
korelace
•
TARDBP
-
TAR-DNA binding
protein 43–
1p36 –
ALS 10•
5% FALS –
ALS10
+ FTD –
overlap
dvou agresivních
neurodegenerativních
jednotek
–
multisystémové kontinuum (15%FTD pacientů
vyvine ALS, 50%ALS
pacientů
vyvine frontální
dysexekutivní
syndrom vedoucí k FTD)
•
SETX –
senataxin
–
9q34.13 –
AD typ dědičnosti –
ALS 4•
Juvenilní
forma
s nástupem do 20 let a pomalá
progrese,
postižení
distálních
svalů
na HK i DK, kolem 50 let upoutání
na vozík


















