
Imaging of Enzymes in the Steroid Biosynthetic Pathway ...159489/FULLTEXT02.pdf · Imaging of...
56
ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2009 Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 607 Imaging of Enzymes in the Steroid Biosynthetic Pathway Synthesis of 18 F-Labelled Tracers MARIA ERLANDSSON ISSN 1651-6214 ISBN 978-91-554-7424-9 urn:nbn:se:uu:diva-89177
Transcript of Imaging of Enzymes in the Steroid Biosynthetic Pathway ...159489/FULLTEXT02.pdf · Imaging of...

ACTA UNIVERSITATIS
UPSALIENSISUPPSALA
2009
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 607
Imaging of Enzymes in the Steroid Biosynthetic Pathway
Synthesis of 18F-Labelled Tracers
MARIA ERLANDSSON
ISSN 1651-6214ISBN 978-91-554-7424-9urn:nbn:se:uu:diva-89177

������������������ ������������������������������������������ ���������������������������������� ���� ��������!��"���##$����"#%"&�'���!�� ������'������'(!����!�)�*!�����������+�������� ���� ���,����!)
��������
,��� �����)��##$)�-������'�,.��������!�� ���� ������!�����(��!+��)� ��!����
'�"/012������ �*������)�3���������������������������)����������� �������������� ������������������ �������� �������������������������������������4#5)�&����)��������)$5/1$"1&&�15���1$)
*!��� �!����� ����� +��!� �!�� ���!����� � � ��������� '� "/01������� � ��6��� ���� ���� � ��.������������� ��!������������������������������!��7(,*8���������'���!��������
'��!������� ��.�����""�1!� ��������� ���������)�*+����!�����"/01���������������!�����!������ ����� ���.�����������+���� ������ ��� ��!����������+������������ ���������������������)*!�� �+1����� ��������� ���! � +��� ��� � �� ���!������ ���� ���� �� '�� ��������
��������)�-��!��'�������������"/01������� ������� ��������� ���� �������������!������� ���!�����!���+������!����� �� ���� � �����������!��������6����������)�*!�� ����1������� 7 )�)8��� ��!����������� �+���!��!��������� ����!���6+��+1�����������������! �)9������������������ � ����!��������!���'�����������������������1�������������
���! �+�������������������)�:���!���'��� ������ ������! ����� �������������!���!�
������� ������ ��� �!�� � � '� �!���� ��6��� �!����� � � ��� � �!���� �������� �� �!�� "/01��������
���!����)�*!���1�����"/01������������!�������;���� ������������������� ��� ��� �!��!�������'����� ����������� � )�)��� ��!����������� ��!������+1��������!����)��:��!�����+���!��������!��������������+����� ��� ������ ��� ��!�� )�)��� ��!����������� �+����������!���!�������� �+��!���������!�����)�*!���1��������!�����������'�� ��!�����!����!� ����������+����!������������!����������������� ���!��*<3�,<2���0=0>)
���� ����(,*��1����� ����!������������� ��.������""?1!� �������������������)�)�)�����!�����'���������"/01�������)
� ���! �������"����� ��������#�������� ������$ ������������ �"�BMC, #%�&'("�������� ����� ����"��!)'&*+,��������"�������
@�������,��� ����##$
- >�"4&"14�"�- �>�$5/1$"1&&�15���1$��%�%��%��% ���1/$"55�7!���%AA��)6�)��A������B��C��%�%��%��% ���1/$"558

Till minne av Mormor


List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I. Synthesis of 11C-labelled metomidate analogues as adrenocortical imaging agents. Farhad Karimi, Maria Erlandsson, Örjan Lindhe, Bengt Långström, J. Label. Compd. Radiopharm. 2008, 51, 273-276.
II. 18F-Labelled metomidate analogues as adrenocortical imaging agents. Maria Erlandsson, Farhad Karimi, Örjan Lindhe, Bengt Lång-ström, Nucl. Med. Biol. 2009, Accepted.
III. Synthesis and in vitro evaluation of 18F-labelled di- and tri(ethylene glycol) metomidate esters. Maria Erlandsson, Håkan Hall, Bengt Långström, J. Label. Compd. Radiopharm. 2009, Accepted.
IV. 18F-Labelled vorozole analogues as PET tracer for aromatase. Maria Erlandsson, Farhad Karimi, Kayo Takahashi, Bengt Långström,
J. Label. Compd. Radiopharm. 2008, 51, 207-212. V. Pharmacological characterization of 18F-labeled vorozole ana-
logues. Håkan Hall, Kayo Takahashi, Maria Erlandsson, Sergio Estra-da, Elisabeth Bergström, Bengt Långström, Submitted.
VI. Automated synthesis of 18F-labelled analogues of etomidate, voro-
zole and harmine using commercial synthesizer TRACERLab FXFN. Obaidur Rahman, Maria Erlandsson, Elisabeth Blom, Bengt Långström, Manuscript.
Reprints were made with permission from the publishers: John Wiley & Sons Ltd (I, III & IV) and Elsevier Ltd (II).
Related patent application: Bengt Långström, Farhad Karimi, Elisabeth Blom, Maria Erlandsson. WIPO (World Intellectual Property Organization), WO/2007/144725, 2007.

Contribution Report
The author wishes to clarify her contributions to the papers presented in this thesis:
Paper I. Performed the synthetic work and characterization of the precursors. Responsible for the planning and performance of the metabolite analysis. Contributed significantly to the writing of the paper. Papers II, III & IV. Contributed to the planning of the research problems. Synthesised and char-acterized precursors and tracers. Planned and performed the metabolite analyses. Contributed significantly to the writing of the papers. Paper V. Responsible for planning the chemistry part. Performed all the synthetic work and labelling of precursors and tracers. Planned, performed and ana-lysed the metabolite study. Paper VI. Responsible for the synthetic work and characterization of the precursors and references. Contributed in the labelling procedure.

Contents
1. Introduction...............................................................................................11 1.1 Molecular imaging with PET .............................................................11 1.2 Aspects of tracer development ...........................................................12
1.2.1 Specific radioactivity ..................................................................12 1.2.2 Metabolism .................................................................................13 1.2.3 Lipophilicity ...............................................................................13 1.2.4 The time factor............................................................................14 1.2.5 Purification and identification of labelled compounds ...............14
1.3 Steroid biosynthesis pathway .............................................................15 1.3.1 Imaging of receptors and enzymes in the steroid pathway .........15 1.3.2 Imaging of the enzyme 11�-hydroxylase....................................16 1.3.3 Imaging of the enzyme aromatase ..............................................17
1.4 Strategies for 18F-labelling .................................................................18 1.4.1 The [18F]fluoride ion...................................................................18 1.4.2 Electrophilic radiofluorination....................................................19 1.4.3 Nucleophilic radiofluorination....................................................19 1.4.4 Synthetic labelling approaches ...................................................20
2. Aims of the Thesis ....................................................................................22
3. Results and Discussion .............................................................................23 3.1 Synthesis and biological evaluation of 11C-labelled MTO analogues (Paper I)....................................................................................................23
3.1.1 11C-Labelling synthesis...............................................................23 3.1.2 Biological evaluation ..................................................................24
3.2 Synthesis and biological evaluation of 18F-labelled ETO analogues (Paper II) ..................................................................................................25
3.2.1 Two-step 18F-labelling ................................................................26 3.2.2 Precursor synthesis .....................................................................27 3.2.3 One-step 18F-labelling.................................................................27 3.2.4 Biological evaluation ..................................................................28
3.3 Synthesis and biological evaluation of 18F-labelled di- and tri(ethylene glycol) esters of etomidate (Paper III)......................................................30
3.3.1 Precursor synthesis .....................................................................31 3.3.2 One-step 18F-labelling.................................................................31 3.3.3 Biological evaluation ..................................................................33

3.4 18F-Labelling and biological evaluation of vorozole (Papers IV & V) .......................................................................................34
3.4.1 Two-step 18F-labelling ................................................................35 3.4.2 One-step 18F-labelling.................................................................36 3.4.3 Product formation in the two- and one-step syntheses ...............37 3.4.4 Biological evaluation ..................................................................39
3.5 Technological developments ..............................................................41 3.5.1 Automation of 18F-labelling synthesis (Paper VI) ......................41 3.5.2 Radiolabelling with microwave heating (Papers II & III) ..........42
4. Conclusions...............................................................................................45
Summary in Swedish ....................................................................................47
Acknowledgments.........................................................................................49
References.....................................................................................................51

Abbreviations
�+ Positron Bq Becquerel Bu Butyl group, C4H9- d.c. Decay-corrected DMF N,N-Dimethylformamide DMSO Dimethyl sulfoxide ETO, Etomidate Ethyl 1-[(1R)-1-phenylethyl]-1H-
imidazole-5-carboxylate HPLC High performance liquid chromatog-
raphy LC-MS Liquid chromatography - mass spec-
trometry MeCN Acetonitrile, CH3CN MTO, Metomidate Methyl 1-[(1R)-1-phenylethyl]-1H-
imidazole-5-carboxylate n.c.a. Non-carrier-added NMR Nuclear magnetic resonance PET Positron emission tomography QMA Quaternary methylammonium r.t. Room temperature SN2 Bimolecular nucleophilic substitution SPE Solid phase extraction SRA Specific radioactivity T1/2 Half-life TBAF Tetrabutylammonium fluoride TBAH Tetrabutylammonium hydroxide Ts Tosyl group, CH3C6H4SO2- UV Ultraviolet VOZ, Vorozole 6-[(S)-(4-chlorophenyl)(1H-1,2,4-
triazol-1-yl)methyl]-1-methyl-1H-1,2,3-benzotriazole


11
1. Introduction
1.1 Molecular imaging with PET Molecular imaging enables the fulfilment of future demands to tailor medi-cal treatment not only to symptoms, but also to the biochemical profile of an individual’s disease state. Prescribing the right medicine for the right person at the right time will reduce side effects and increase effectiveness. Molecu-lar imaging can be used for, among other applications, early detection, char-acterization, “real-time” monitoring of diseases, and for investigating the function and maybe the efficacy of drugs.1
Positron emission tomography (PET) is a non-invasive in vivo molecular imaging technique used to detect �+-emitting tracers. An important attribute of the PET technique is its ability to explore relations between molecular events, biological functions and simultaneously observing molecular signal-ling; i.e. its correspondence to certain biological functions. Important aspects that can be studied using PET are receptor binding, enzyme inhibition, other possible protein enzymes as well as drug distribution. PET provides a pow-erful method to narrow the gap between molecular biology and targeted treatment.2,3
Investigations using PET require suitable tracers labelled with positron emitting radionuclides, e.g. 15O (t1/2 = 2 min), 13N (t1/2 = 10 min), 11C (t1/2 = 20 min), 68Ga (t1/2 = 68 min), 18F (t1/2 = 110 min), 76Br (t1/2 = 16.2 h), 124I (t1/2 = 4.2 d) etc.
A PET investigation is a complex procedure. The radionuclide has to be produced and incorporated into the tracer, which must be purified before it can be injected into a biological system. The short-lived radionuclide, e.g. 18F, decays by emitting a positron (�+ particle) and a neutrino. After travel-ling up to a few millimetres in a tissue, the positron annihilates with an elec-tron, producing a pair of annihilation photons that move in opposite direc-tions (Figure 1a). These are detected almost simultaneously by a scintillator material in the scanning device surrounding the subject (Figure 1b). The acquisition is followed by correction and reconstruction procedures to create PET images for further exploration.

12
Figure 1. (a) Radioactive decay of 18F. (b) Schematic picture of the detection of the annihilation photons (arrows) in the PET camera.4
1.2 Aspects of tracer development A radiotracer should be designed in such a way that it interacts with the sys-tem of interest without disturbing biological processes. It can be a tracer that competes with an endogenous molecule or a drug for a receptor, an enzyme or some other binding-protein structure. The tracer should have high speci-ficity in recognizing the biological target and selectivity over the other re-ceptors in the area of interest.
In order to develop a tracer for a defined task, several requirements must be explored:
1.2.1 Specific radioactivity Specific radioactivity (SRA) is defined as the concentration of a radioactive material in a sample, and is expressed as the radioactivity of the labelled compound divided by the molar amount of the compound (Bq/mol).
SRA is a parameter to be considered in the synthesis of PET tracers and its importance depends on the concentration of biological target. The use of tracers with high SRA is required in order to avoid perturbations such as the saturation of receptors, because unlabelled substances can compete for the same target as the labelled compound. Low SRA may lead to a marked oc-cupancy of the binding sites. A tracer having high SRA can be used in a lower amount for PET investigations.
The maximal theoretical SRA can be calculated from the following equa-tion: SRA = A/N = ln2/T1/2; (T1/2 = the half-life of the radionuclide, N = the number of atoms of the radioactive element, A = measured radioactivity). The maximal theoretical SRAs for 11C and 18F are 3.4 × 105 and 6.3 × 104 GBq/�mol, respectively.5,6 The SRA obtained in a labelling experiment is usually lower than the theoretical value due to isotopic dilution, which refers
(a) (b)

13
to the dilution of a given radionuclide by one or more of its other isotopes. To avoid this problem, the reaction conditions should be designed to mini-mise the use of contaminating chemicals, materials, and transport lines.
In 18F-labelling synthesis, it is difficult to eliminate the naturally occur-ring stable isotope 19F that originates from the 18O target water or the mobile phase used in HPLC-systems. The presence of anions other than fluoride may also compete in the labelling reaction.7 Unlabelled impurities with simi-lar biological properties as the labelled substance (pseudo carriers) can also be included in the product, as purification procedures are not a 100% effec-tive. With careful exclusion of non-radioactive fluoride in the production systems, SRA can be obtained in the range of 50–500 GBq/�mol.1 In the 11C-labelling synthesis, CO, CO2, CH4 and other sources of carbon contrib-ute to isotopic dilution.
1.2.2 Metabolism The position of the radiolabel in a molecule is a factor that has to be consid-ered during planning of the synthetic approach.
In a PET investigation, it is impossible to determine whether the radioac-tive uptake in a tissue is dominated by signals from the parent radiotracer or its radiolabelled metabolites. Therefore the radioisotope should be incorpo-rated into a metabolically stable position within the molecule so that it re-mains with that part of the molecule throughout the study.8 The loss of the radiolabel in a molecule by metabolic degradation will limit its usefulness as a PET ligand.
For example, 18F-labelling was hypothetically possible at two different positions in the etomidate precursor molecule: on the aromatic ring or by esterifying the carboxylic acid. It has been shown that the lead tracer is not too sensitive to changes in the ester part and that modifications in this part do not affect the biological properties of the tracer.9 We therefore decided to introduce the 18F-label via ester formation (Scheme 1).
When a fluorine atom is introduced into a drug molecule, both the rate and the route of its metabolism can be altered10 and consequently the meta-bolic stability of the drug analogue should be investigated. Blood samples are taken over time and analyzed by liquid chromatography (LC) in order to quantify the amount of intact tracer at different time points.
1.2.3 Lipophilicity Lipophilicity is an important physicochemical property that influences a drug’s absorption, distribution, metabolism and elimination.11 Lipophilicity is expressed as the log P value, or the octanol-water partition coefficient. The log P value can be calculated theoretically12 or measured experimen-tally.

14
Lipophilicity is an important factor when it comes to a tracer’s capacity to cross lipid membranes, which it must to reach targets in the CNS and intra-cellular targets (e.g. enzymes). It can also affect the extent of the tracer’s specific-to-non-specific binding ratios. For example, high lipophilicity leads to high protein binding, low free fraction and reduced delivery of the tracer to the specific sites. Tracers with low lipophilicity have higher water solubil-ity, faster clearance through the kidneys and limited blood-brain penetration. To cross the blood-brain barrier a recommended log P value is between 1 and 3.5.13
The incorporation of a fluorine atom in a molecule can have a profound effect on its lipophilicity and influence both the disposition of the drug and the interaction of the drug with its pharmacological target.10 The log P values are therefore important criteria in the evaluation and screening of PET-tracer candidates.
1.2.4 The time factor The length of a synthesis is another important parameter in labelling chemis-try. The relatively short physical half-lives of the radionuclides require the syntheses of PET tracers to be as short as possible. As a consequence of the time constraint, synthetic methods are often modified when used in tracer production. The time frame of a PET investigation depends on the half-life of the radionuclide used. The half-life of 11C allows for biological investiga-tions up to 1–1.5 hours, whereas tracers containing 18F can be studied for a longer period of time.14 The synthesis and purification time should be per-formed as fast as possible to minimise losses of radioactivity due to radioac-tive decay and therefore maximise the SRA.
Time optimization also influences the type of procedure used for synthe-sis. For example, one-pot syntheses reduce production time by simplifying the technical handling. Besides conventional heating, other techniques of reaction activation, e.g. microwave heating, are also used for radiochemical reactions.15,16 In fact, microwave heating has in many cases been shown to be more effective than conventional heating in terms of speed, purity and selec-tivity.1
1.2.5 Purification and identification of labelled compounds All PET tracers, whether for human or animal use, should show a high level of chemical and radiochemical purity (typically >95%). The purification of obtained labelled compounds is most frequently achieved by semi-preparative LC or solid phase extraction (SPE). The identity of the purified product is confirmed by analytical methods like HPLC analysis, which is performed in the presence of non-radioactive authentic reference compound. The analytical LC is equipped with radioactivity and UV-absorption detec-

15
tors. Comparing the retention times for the UV peak and the radiopeak al-lows the labelled compound to be identified. Non-radioactive reference sub-stances were synthesised for all labelled metomidate, etomidate and vorozole analogue compounds presented in this thesis, and were used to verify the identity of the labelled products. The reference substances were synthesised by one of the following three methods: a) appropriate one-step precursor was dissolved in a solvent and TBAF was added, b) appropriate two-step precur-sor was dissolved in a solvent and oxalyl chloride and 3-fluoropropanol were added, or c) appropriate two-step precursor was dissolved in a solvent and 1-bromo-2-fluoroethane was added. The reference compound’s structure was determined using LC-MS and 1H, 13C and 19F NMR spectroscopy.
The sensitivity of UV detection is not always enough for determining the SRA, especially in cases where the synthesised substances have a high SRA or show low UV-absorption. When lower detection limits are needed for compounds that can be protonated, the LC is connected to a mass spectrome-ter, which enables accurate chemical characterization of the product.17
1.3 Steroid biosynthesis pathway 1.3.1 Imaging of receptors and enzymes in the steroid pathway Different kinds of stress disturbances of the hypothalamic-pituitary-adrenal axis (HPA axis) can result in the accumulation of one steroid hormone and lack of another. This can, in the long term, produce changes in the limbic system and the HPA axis, like abnormal cortisol levels.18 Most steroid-hormone biosynthesis is conducted in the adrenal cortex and the gonads. Several tumours depend on steroid hormones to grow, and therefore have a high expression of steroid-hormone receptors.19
Two different categories of biosynthetic inhibitors exist; steroid and non-steroidal. The first is based on receptor-targeted antagonists, which bind to the receptor and block the normal agonist ligand from binding and activating the pathway.20 In many cases, these are structurally similar to the natural ligand, and can be regarded as antiestrogens. The other approach is to de-plete the ligand, with e.g. an enzyme inhibitor that blocks the synthesis of the naturally occurring agonist ligand (such as estradiol and estrone).20
The literature contains examples of 11C and 18F-labelled steroids used for the imaging of different kind of tumours.21,22,23 The first clinically successful adrenalcortical imaging agents based on cholesterol were 131I-19-iodocholesterol and 131I/75Se-6�-norcholesterol.24 More recently, adrenalcor-tical imaging with PET has focused on labelled enzyme inhibitors of 11�-hydroxylase, e.g. metomidate and etomidate (Figure 3).25,26,27,28,29 An example of a steroid PET tracer that images estrogen receptor-positive primary and metastatic breast tumours is 16�-[18F]fluoroestradiol.30,31 Imag-

16
ing of aromatase has previously been performed with the nonsteroidal aro-matase inhibitor 11C-labelled vorozole, a high-affinity aromatase-binding radiotracer.32,33,34,35,36
This thesis focuses on the radiolabelling of non-steroidal inhibitors of the enzymes 11�-hydroxylase and aromatase in the steroid biosynthesis (Figure 2).
Figure 2. The steroid biosynthesis pathway. 1) The enzyme 11�-hydroxylase, and 2) the enzyme aromatase.
1.3.2 Imaging of the enzyme 11�-hydroxylase The search for a suitable tracer for identifing tumours incidentally found in the adrenal cortex, so-called incidentalomas, has focused on the biosynthesis of the steroids cortisol and aldosterone.25 They are synthesised in the adrenal cortex, proceeding from cholesterol through several steroid hormones and cytochrome P-450 enzymes (Figure 2).
Etomidate and metomidate (Figure 3) interact selectively with the mito-chondrial cytochrome P-450 species in the adrenal cortex to inhibit the 11�-hydroxylase enzyme.37,38 The interaction with cytochrome P-450 are stereo-selective; the R enantiomers are more potent inhibitors than the S enanti-omers.37

17
N
N
OO
N
N
OO
(R)-Etomidate (R)-Metomidate
Figure 3. The structures of etomidate and metomidate.
11C-labelled etomidate and metomidate have previously been developed with the clinical goal of obtaining PET imaging agents for tumours of the adrenal cortex.25 PET is a specific and sensitive method for the visualisation and characterisation of lesions found incidentally around the adrenals during CT of the abdomen and can differentiate between malign and benign lesions found in incidentalomas.26 These PET tracers are alternatives to an invasive procedure for the pre-operative evaluation of incidentalomas.39
From the clinical perspective, the future goal is to select the most suitable 18F-labelled analogue, which must be selective for primary aldosteronism. To validate this method, microdosing the chosen tracer in a limited group of patients must provide proof of concept and then a clinical multi-centre study must be performed.39
1.3.3 Imaging of the enzyme aromatase Attempts to elucidate the biochemical basis for the gender difference have brought the sex-steroid pathways into focus. Aromatase is an enzyme re-sponsible for the conversion of C19 steroids (androgens) into C18 steroids (estrogens) (Figure 2). It is found on the endoplasmatic reticulum of the cells and consists of two proteins: cytocrome P450aromatase heamoprotein and an electron-donating flavoprotein, NADPH-cytocrome P450reductase.40
The S enantiomer of vorozole (Figure 4) is a very potent inhibitor of aro-matase but the R enantiomer is only a weak inhibitor.40,41,42 Although the molecular interactions behind this inhibition are not fully understood, the triazole derivative is believed to bind through the basic triazole N-4 atom to the iron atom in the haem group of the enzyme.40, 43

18
N
NN
NN
N
Cl
1
2
4
(S)-Vorozole
Figure 4. The structures of vorozole.
From a clinical perspective, vorozole was developed as a specific, high-affinity competitive aromatase inhibitor, designed to inhibit estrogen synthe-sis in patients with breast cancer, which is the most common cancer among women.41,44,45 Despite the promising therapeutic results obtained with voro-zole, it was withdrawn from testing because it performed no differently than the progestational agent megestrol acetate.46,47 Thus vorozole is an example of a compound arising from drug development that is unsuitable for thera-peutic applications, but that has the potential to be a good lead for the devel-opment of PET tracers. One reason for this is that PET tracers are given in-travenously at extremely low doses, so low that pharmacological side effects and toxicity are very rare.3
A PET tracer like vorozole can potentially be used for imaging areas where the enzyme aromatase exists. In the human body, aromatase is found in several different tissues such as placenta, granulosa cells of the ovary, skin, fat, the central nervous system, hair follicles, Leydig cells in the testi-cle, liver, muscle and various sites of the brain.48 The distribution of aroma-tase in the brain has been studied in several species, and the highest aroma-tase activity is found in the amygdala and the bed nucleus of the stria termi-nalis. Moderate levels are also detected in the preoptic area.49,50 Estrogens are believed to affect mood because women are more vulnerable than men to developing depression and anxiety disorders.51,52
There is still a need for a more long-lived and selective alternative to the 11C-labelled vorozole; therefore 18F-labelled vorozole analogues were syn-thesised and are described in this thesis.
1.4 Strategies for 18F-labelling 1.4.1 The [18F]fluoride ion Despite that fluorine is unusual in naturally occurring biological molecules, it has found a widespread use in pharmaceuticals.10
The introduction of a fluorine atom often induces modifications of the physical and biological properties of the initial molecule,53 although there

19
are a lot of examples where the fluorine atom has replaced either hydrogen or oxygen in a molecule that has subsequently retained comparable activi-ties.54 The fluorine atom has been considered to mimic these atoms with respect to steric requirements at binding sites of receptors and enzymes. Therefore it is used to synthesise fluorine analogues.
[18F]Fluoride is the most widely used �+-emitting radiohalogen in PET due to its convenient half-life (110 min), decay mode (97% �+ decay) and low positron energy (maximum 0.63 MeV). This allows, for example, low radiation doses and short ranges (maximum 2.4 mm) in the tissue. Moreover, the half-life of 110 min allows the commercial distribution of the radionu-clide to ‘‘satellite’’ hospitals that lack cyclotrons.
Both the electrophilic and nucleophilic radiofluorination methods are needed to satisfy the current need for 18F-labelled pharmaceuticals. How-ever, only nucleophilic 18F-fluorination was used in this thesis.
1.4.2 Electrophilic radiofluorination For electrophilic reactions, [18F]F2 gas is used. It can be produced via the nuclear reaction 20Ne(d,�)18F. The final 18F-labelled product has low SRA because unlabelled 19F2 gas is required to recover [18F]F2 from the surface of the target walls. The maximum achievable radiochemical yield is as low as 50% because only one of the fluorine atoms in [18F]F2 carries the 18F label.
Another approach that gives higher SRA [18F]F2 is to start from [18F]fluoride, converting it to [18F]methyl fluoride and then flush it with neon gas in a quartz discharged chamber containing a variable amount of F2.55 Generally, [18F]F2 is converted into less reactive and more selective 18F-fluorination agents.56,57,58
Despite drawbacks like lower radiochemical yield and lower SRA, elec-trophilic fluorination is still used in the production of some well-known PET tracers, e.g. [18F]FDOPA, due to its convenient chemistry.59
1.4.3 Nucleophilic radiofluorination To obtain 18F-labelled tracers with high SRA, the nucleophilic method is preferable. The n.c.a. [18F]fluoride ion is prepared by the nuclear reaction 18O(p,n)18F. Reactions involving [18F]fluoride require strict exclusion of water because the fluoride ion is a poor nucleophile in aqueous media and is thus inactivated for nucleophilic substitution reactions. Nucleophilic 18F-fluorination is also very sensitive to the presence of metal-ion traces formed by irradiation of the target.7 Therefore [18F]fluoride must be isolated and dried before starting a labelling synthesis. This can be done through azeotropic drying or with a QMA filter,60 which is a quaternary methylam-monium anion-exchange resin. To maintain [18F]fluoride solubility, a

20
counter ion and a phase-transfer catalyst is required. Potassium carbonate and Kryptofix 2.2.2 are commonly used.60
For aliphatic compounds, the reaction proceeds via an SN2 mechanism, in which a suitable leaving group is replaced by [18F]fluoride ion and a carbon-fluorine bond is formed. Different leaving groups, like halogens and sul-fonate esters (methanesulfonate, p-toluenesulfonate, trifluoromethanesul-fonate), can be used. The choice of leaving group depends mainly on the stability of the precursor, the ease of separation of the 18F-labelled product and the precursor, reagents and solvents, and the potential for formation of side products.1 Nucleophilic aromatic substitution of a nitro or trialkylam-monium group in activated aromatic systems can also be performed.61
1.4.4 Synthetic labelling approaches A two-step 18F-labelling synthesis of etomidate ([18F]FETO), has previously been reported.62 2-Bromo-[18F]fluoroethane was used as the [18F]fluoroalkylating agent (Scheme 1). Although the method to synthesise 2-bromo-[18F]fluoroethane63 has been established, the success of the total labelling synthesis of [18F]FETO depends on the purity of the labelled inter-mediate.62 Generally, the reproducibility of the synthesis of 2-bromo-[18F]fluoroethane was low; the decay-corrected (d.c.) radiochemical yield varied between 4–24%. After synthesis and purification, [18F]FETO was obtained with a d.c. radiochemical yield of 15% and a SRA of 42–214 GBq/�mol.62,64
We have developed an alternative two-step method to synthesise [18F]FETO (Scheme 1) and some new 18F-labelled etomidate analogues. The 18F-labelled tosylate intermediate was obtained with a d.c. radiochemical yield of 62–77% and, contrary to the prior two-step method, it could be used directly in the subsequent alkylation step. This two-step labelling method is a direct method to synthesise many compounds for use in early biological evaluation.
However, one-step synthesis is more valuable for clinical applications and our further work focused on reducing the synthesis time, simplifying the technical handling by automation and increasing the d.c. radiochemical yield and SRA. To perform the synthesis in one step (Scheme 1), some appropri-ate precursors were needed. These were synthesised by adding a leaving group to the end of the alkyl chain on the ester part of the metomidate struc-ture. These precursors could then be used directly in the 18F-labelling synthe-sis. The product from the one-step 18F-labelling synthesis had higher SRA and d.c. radiochemical yield than that from the two-step synthesis.
The usefulness of the two- and one-step approaches was further explored in the 18F-radiolabelling synthesis of vorozole.

21
N
N
O O NBu4
N
N
O OF18
N
N
O OOTs
BrF18
N
N
O OH
TsOF18
+
(a)
(b) (c)
[18F]FETO
Scheme 1. Labelling approaches. (a) Literature method,62 (b) our two-step method, (c) our one-step method using [K/K2.2.2]+18F-.

22
2. Aims of the Thesis
The objective of this thesis was to synthesise and further develop radio-labelled tracers for use in the PET imaging of two steroid enzymes, 11�-hydroxylase and aromatase.
The following specific aims were set:
♦ To synthesise metomidate analogue precursors, with halogen atoms at the para positions of the phenyl groups, to be used in 11C-labelling syn-thesis and biological evaluation of imaging agents for the enzyme 11�-hydroxylase.
♦ To synthesise and evaluate 18F-labelled etomidate analogues with differ-ent structural properties on the ester part of the molecule or with a halo-gen atom at the para position of the phenyl group. To develop various synthetic methods that met the requirements of a particular application, such as screening of a tracer library or biological/clinical evaluation of a lead tracer.
♦ To synthesise 18F-labelled vorozole analogues and evaluate their bio-logical properties as imaging agents for the enzyme aromatase.
♦ To automatize the one-step labelling synthesis of the synthesised etomi-date and vorozole precursors.
♦ To explore the microwave heating technique in the one-step 18F-labelling synthesis of some etomidate analogues.

23
3. Results and Discussion
3.1 Synthesis and biological evaluation of 11C-labelled MTO analogues (Paper I) Clinical findings with the radiotracer [11C]methyl 1-[(1R)-1-phenylethyl]-1H-imidazole-5-carboxylate (4, Scheme 2) have indicated high uptake in lesions of adrenocortical origin, including adenomas, but very low uptake in lesions of non-adrenocortical origin.25,26,27,28,29 A study using 4 in the diagno-sis of gliomas showed that tumours of all types had remarkable uptake of the tracer.25 A retrospective study showed that PET using 4 was useful in the imaging of adrenal incidentalomas and may be beneficial for the examina-tion of patients with primary aldosteronism or adrenocortical cancer.65 Al-though 4 has good biological properties for visualization of the adrenals and their tumours, there is still a need for tracers with higher adrenal to liver uptake ratios, because the assessment of the right adrenal is often hampered by the high uptake of 4 in the liver.
3.1.1 11C-Labelling synthesis We synthesised two potential 11C-labelled metomidate (MTO) analogues with halogens at the para position, [11C]methyl 1-[(1R)-1-(4-chlorophenyl)ethyl]-1H-imidazole-5-carboxylate (5) and [11C]methyl 1-[(1R)-1-(4-bromophenyl)ethyl]-1H-imidazole-5-carboxylate (6), and investi-gated how halogen introduction changed the biological properties. The pre-cursors 2 and 3 were synthesised from the corresponding arylethylamine by the modified literature procedure66 used to synthesise 1. Recently, a method to prepare enantiopure metomidate analogues with modifications at the para position on the phenyl ring was described in the literature9; this might be an alternative way to prepare 2 and 3 in the future.
The 11C-labelled MTO analogues were prepared as shown in Scheme 2. [11C]Methyl iodide was synthesised as previously described.67 The radio-chemical purity was >97% and the radiochemical yield of the compounds, determined by analytical HPLC, varied between 60–80%.

24
N
NX
O OH
N
NX
O O
C11 H3
(1) X = H
(2) X = Cl (3) X = Br
(4) X = H
(5) X = Cl (6) X = Br
(a)
Scheme 2. 11C-Labelling of MTO analogues. (a) i. TBAH, ii. [11C]methyl iodide, DMF, 130 ºC, 7 min.
3.1.2 Biological evaluation The MTO analogues 5 and 6 were evaluated by frozen-section autoradiogra-phy (Figure 5), organ distribution and metabolite analysis to investigate their binding properties as tracers, and whether they changed the liver uptake compared to the lead 4.25,68,69,70 Frozen-section autoradiography showed that the degrees of specific binding in rat adrenal were 83, 62 and 21% for 4, 5 and 6, respectively. In sections from the rhesus monkey adrenal, the specific binding was 76, 65 and 35%, respectively.
Organ distribution experiments indicated that varying the halogen group at the para position of the phenyl group could alter the biological properties of the MTO analogues, for instance by increasing the adrenal-to-liver uptake ratio. This may improve the characterization of incidentalomas because the right adrenal can be assessed more readily. The possibility of detecting adre-nal metastases in the liver was also improved with an increased adrenal-to-liver ratio.
The metabolite analysis revealed that tracer 5 was 62% and 16% intact 5 and 30 min after injection, respectively. Tracer 6 was 52% and 37% intact 5 and 30 min after injection, respectively. Thus analogues 5 and 6 have similar metabolism profiles to that of 4 in man.29
In conclusion, the adrenal-to-organ ratios indicate that analogue 6 has the most favourable properties with regard to liver uptake and stability in blood.

25
Figure 5. Frozen-section autoradiography of 4, 5 and 6. Total binding and non-specific binding (after blocking with 1 �M etomidate) are shown. Organs, from top: (a) rhesus monkey kidney, (b) rhesus monkey liver, (c) rhesus monkey adrenal, (d) pig kidney, (e) pig liver, (f) pig adrenal, (g) rat kidney, (h) rat liver and (i) rat adre-nal.
3.2 Synthesis and biological evaluation of 18F-labelled ETO analogues (Paper II) Although 4 and the previously synthesised 18F-labelled etomidate (ETO) analogue62,64 ([18F]FETO (11)) have good biological properties as tracers for adrenocortical imaging, we were interested in the further development of 18F-labelled analogues. The purpose was to find ETO analogues with im-proved properties as tracers for imaging adrenocortical tissues. The assess-ment of the right adrenal is often hampered by high uptake in the liver, which complicates the interpretation of the image.
We have developed two- and one-step methods to synthesise 11 and some 18F-labelled ETO analogues, which were tested in in vivo studies.

26
3.2.1 Two-step 18F-labelling The purpose of the two-step labelling synthesis was to have a method for synthesising many ETO analogues for biological evaluation relatively easily. Five 18F-labelled compounds, 2-[18F]fluoroethyl 1-[(1R)-1-phenylethyl]-1H-imidazole-5-carboxylate (11), 2-[18F]fluoroethyl 1-[(1R)-1-(4-chlorophenyl)ethyl]-1H-imidazole-5-carboxylate (12), 2-[18F]fluoroethyl 1-[(1R)-1-(4-bromophenyl)ethyl]-1H-imidazole-5-carboxylate (13), 3-[18F]fluoropropyl 1-[(1R)-1-(4-bromophenyl)ethyl]-1H-imidazole-5-carboxylate (14) and 3-[18F]fluoropropyl 1-[(1R)-1-phenylethyl]-1H-imidazole-5-carboxylate (15), were synthesised using a two-step approach (Scheme 3).
In the first step of the synthesis, 9 or 10 were prepared from 7 or 8, re-spectively, following modified literature procedures.71,72,73 There was no indication that unreacted 7 or 8 competed with the labelling reagent in the following alkylation step. The d.c. radiochemical yields of 9 and 10 were 62% and 77%, respectively, starting from [18F]fluoride after 47- to 68-min synthesis. The 18F-labelled tosylates were not isolated after the first step, but rather used directly to alkylate the appropriate precursor ammonium salt to obtain the product. The d.c. radiochemical yield, based upon [18F]fluoride at the start and the radioactivity of the HPLC-purified product, was in the range 10–29% for 11–15. The specific radioactivities after 80-min syntheses were 32, 50, 37, 218 and 3 GBq/�mol for compounds 11, 12, 13, 14 and 15, re-spectively. The SRAs depended on isotopic dilution with 19F from the reac-tion vial.
TsO F18
n
N
N
O OH
X
N
N
O O
X
NBu4
N
N
O O
X
F18
n
TsO OTsn
+
(11) X = H, n = 2(12) X = Cl, n = 2(13) X = Br, n = 2(14) X = Br, n = 3(15) X = H, n = 3
(9) n = 2(10) n = 3
(1) X = H(2) X = Cl(3) X = Br
(7) n = 2(8) n = 3
(a)
(b) (c)
Scheme 3. 18F-Labelled synthesis using a two-step method. (a) [K/K2.2.2]+18F-, DMF, MeCN, 70 ºC, 15 min, (b) TBAH, (c) DMF, 9 or 10, 150 ºC, 15 min.

27
3.2.2 Precursor synthesis Once a suitable analogue has been chosen for clinical evaluation, it will be advantageous to perform the [18F]fluoride incorporation reaction and the alkylation step with minimum delay, making a one-step 18F-labelling synthe-sis beneficial. To achieve the synthesis in one step, we synthesised the rele-vant precursors, 17, 19 and 20 (Scheme 4).
Precursor 17 was synthesised in two steps. In the first step, the alcohol 16 was synthesised in quantitative yield following a modified published proce-dure.74 In the second step, compound 17 was prepared in 46% yield by a reaction between p-toluenesulfonyl chloride and 16.75,76 To investigate whether the same synthetic route could be used with a longer alkyl chain, 19 was synthesised. Similar reaction conditions were used, except that the tem-perature was decreased to -75 ºC in the synthesis of compound 18, which polymerized at higher temperature. The yields were low: only 2% in the first step and 3% in the second step. However, compound 19 could be synthe-sised in slightly higher yield (12%), directly from 1, using cesium carbonate and 8. The same method produced compound 20 in 27% yield.
N
N
OOR 1
X
N
N
OOR 2
X
N
N
OHO
X
N
N
OOR 2
X
(16) X = H, R1 = (CH2)2OH
(18) X = H, R1 = (CH2)3OH(17) X = H, R2 = (CH2)2OTs(19) X = H, R2 = (CH2)3OTs
(19) X = H, R2 = (CH2)3OTs (20) X = Cl, R2 = (CH2)2OTs
(1) X = H(2) X = Cl
(a) or (b) (c)
(d)
Scheme 4. Synthesis of precursors. (a) i. TBAH, ii. 2-iodoethanol, MeCN, 80 ºC, 1 h, (b) i. TBAH, ii. 3-iodo-1-propanol, DMF, -75 ºC, 5 h (c) pyridine, p-toluenesulfonyl chloride, 0 ºC, 1 h, r.t., 2 h (d) i. cesium carbonate, DMF, 1-2 h, ii.7 or 8, 0 ºC.
3.2.3 One-step 18F-labelling Precursors 17, 19 and 20 were labelled by a one-step nucleophilic 18F-fluorination to give the radiolabelled compounds 11, 15 and 12, respectively (Scheme 5).

28
N
N
OO
X
F18
n
N
N
OO
X
OTsn
(17) X = H, n = 2 (19) X = H, n = 3 (20) X = Cl, n = 2
(11) X = H, n = 2(15) X = H, n = 3(12) X = Cl, n = 2
(a)
Scheme 5. One-step 18F-labelling synthesis. (a) [K/K2.2.2]+18F-, DMF, 150 ºC, 15 min.
The d.c. radiochemical yield for 11 was increased from 18 ± 5% in the two-step labelling to 27 ± 9% in the one-step procedure. The SRA was also in-creased from 32 GBq/�mol to 328 GBq/�mol. Furthermore, the SRA was higher compared to the reported two-step synthesis of 11.64
Switching from the two-step to the one-step synthesis produced the same improvements for compound 15, as for compound 11, i.e. increased d.c. radiochemical yield and SRA. The d.c. radiochemical yield increased from 10 ± 5% to 25 ± 10% and the SRA increased from 3 GBq/�mol to 85 GBq/�mol.
For compound 12, the d.c. radiochemical yield was increased from 29 ± 14% to 37 ± 4% upon implementing the one-step synthesis. However, the SRA decreased from 50 GBq/�mol, obtained from the two-step labelling synthesis, to 37 GBq/�mol.
The total synthesis time for the one-step synthesis was 40 min. The radio-chemical purities of the compounds, determined by analytical HPLC, were >98%.
3.2.4 Biological evaluation The analogues were biologically evaluated by frozen-section autoradiogra-phy, organ distribution and metabolite analysis to investigate their suitability for imaging of the adrenals and their tumours.
Analogues 11–14 were used in frozen-section autoradiography studies on various tissues of interest, e.g. liver, adrenal, conn adenoma and kidney (Figure 6). The incubation of frozen sections with 18F-labelled compounds (11–14) showed high levels of specific binding to the adrenal cortex, with a slightly higher degree of non-specific binding than with the 11C-labelled analogues. Compound 12 showed higher specific adrenal binding, 81% in normal human adrenal and 96% in human adenoma, than compound 13, which showed 72% and 68%, respectively.

29
Organ distribution studies indicated that the uptake ratios of adrenal to liver, kidney and blood favoured compound 12 over the other analogues.
In an earlier in vivo study of 11, rapid metabolism was observed for the first 10 min and was followed by a smooth decrease from 20 to 90 min.77 Metabolite studies were therefore performed on 12 and 13. These analogues were selected because they showed the best properties in the autoradiogra-phy and organ-distribution studies.
Metabolite analysis revealed that 35% and 6% of 12 was intact at 5 and 30 min after injection, respectively. Three hydrophilic metabolites were ob-served. For compound 13, 51% and 39% was intact 5 and 30 min after injec-tion, respectively and one hydrophilic metabolite was observed. Analogues 12 and 13 had similar metabolism profiles to 11. However, after 30 min compound 13 had metabolized less in the plasma than 11 had.
Both the 18F-labelling and the addition of a halogen at the para position contributed to improving the biological properties of the ETO analogues. Despite that compound 12 was metabolized slightly faster than compound 13, it will be selected for further clinical applications because it had the greatest adrenal to liver ratio and displayed the highest degree of specific binding.

30
Figure 6. Frozen-section autoradiography of (from left to right): [11C]MTO (4), 11, 12, 13, and 14. Organs, from top: (a) human liver, (b) human adrenal, (c) human conn adenoma, (d) rhesus liver, (e) rhesus adrenal, (f) guinea pig liver, (g) guinea pig adrenal, (h) rat liver, (i) rat adrenal and (j) rat kidney. In every second column from the left, parallel sections of the same tissue were used in the blocking experi-ment.
3.3 Synthesis and biological evaluation of 18F-labelled di- and tri(ethylene glycol) esters of etomidate (Paper III) To develop different 18F-labelled ETO analogues with modified ester parts, the focus was laid on ethoxy chains. The ethylene glycol chains of different lengths were expected to change the properties of the ETO analogues, e.g. decrease the lipophilicity, to combat the problem of high lipophilicity that can result from the addition of a fluoroalkyl group.78 We have developed two PET tracers that are based on the ETO structure and contain either di- or tri(ethylene glycol) substituents with [18F]fluorine atoms attached at the end of the chains (Scheme 7). These analogues were evaluated to find out how the chain modification altered the biological properties.

31
3.3.1 Precursor synthesis The precursors 21 and 22 were synthesised according to Scheme 6; both conventional and microwave heating were explored. The disadvantage of conventional heating using an oil bath is that the vessel walls are heated up first, causing a temperature gradient in the solution. However, very fast heat-ing can be obtained using microwave irradiation because the sample is heated more uniformly from the inside.79
Compound 21 could not be synthesised using either conventional or mi-crowave heating due to the formation of byproducts. Instead, the best yield of compound 21, 53%, was obtained when the reaction mixture was allowed to warm slowly from 0 �C to room temperature, then stirred at that tempera-ture for 20 h.
The synthesis of 22 using microwave heating gave a higher yield of the desired product and less byproduct formation than conventional heating did. The yield of compound 22 could be increased from 33% up to 56%, by switching heating methods.
N
N
OHO
N
N
OOR 1
(21) R1 = (CH2)2O(CH2)2Br (22) R1 = (CH2)2O(CH2)2O(CH2)2Cl
(1)
(a)
Scheme 6. Precursor synthesis. (a) i. TBAH, ii. 2-bromoethyl ether or 1,2-bis(2-chloroethoxy)ethane, MeCN.
3.3.2 One-step 18F-labelling Two 18F-labelled compounds, 2-(2-[18F]fluoroethoxy)ethyl 1-[(1R)-1-phenylethyl]-1H-imidazole-5-carboxylate (23) and 2-[2-(2-[18F]fluoroethoxy)ethoxy]ethyl 1-[(1R)-1-phenylethyl]-1H-imidazole-5-carboxylate (24) with chains of different lengths on the ester part, were syn-thesised from their respective precursors 21 and 22, respectively (Scheme 7).
Varying the chain length of the ethylene glycol group in compounds 23 and 24 adjusted their lipophilicity. The calculated log P values for the ETO analogues were 2.47 ± 0.59 (23) and 2.11 ± 0.69 (24), which are lower than the 11C-labelled MTO lead (2.73 ± 0.34).12,80
The specific activities for 23 and 24 were 20 ± 4 and 175 ± 18 GBq/�mol, respectively, after a 50–60 min synthesis time. The radiochemical purities of

32
both compounds, as determined by analytical HPLC, were >95% and the d.c. radiochemical yields were 26 ± 8% for 23 and 23 ± 8% for 24.
N
N
OOR 2
N
N
OOR 1
(21) R1 = (CH2)2O(CH2)2Br
(a)
(24) R2 = (CH2)2O(CH2)2O(CH2)218F
(23) R2 = (CH2)2O(CH2)218F
(22) R1 = (CH2)2O(CH2)2O(CH2)2Cl Scheme 7. 18F-Labelled compounds with ethoxy modifications. (a) [K/K2.2.2]+18F-, DMF, MeCN, 150 ºC, 15 min.
In the future it would be interesting to synthesise MTO analogue tracers with methylene glycol chains of different lengths and explore their biological properties.
In the process of synthesising the compound ([18F]fluoromethoxy)methyl 1-[(1R)-1-phenylethyl]-1H-imidazole-5-carboxylate (27) by a two-step label-ling synthesis, di(bromomethyl) ether (25) was 18F-labelled for use in the first step (Scheme 8). According to a modified published procedure,81 di(bromomethyl) ether was synthesised from thionyl bromide and parafor-maldehyde. 18F-Fluorination labelling experiments were performed with di(bromomethyl) ether using various temperatures and solvents. In those experiments, the labelled product 26 was obtained in a yield up to 18%. However, compound 26 was very light sensitive and it was difficult to iso-late the product due to its low boiling point. In the literature, a less hazard-ous solid bis(methylsulfonoxymethyl) ether has been synthesised and this compound might be a good alternative in future syntheses of 27.82,83

33
N
N
OHO
N
N
OO O F18
Br O Br Br O F18
(1) (27)
(26)(25)
Scheme 8. 18F-Labelled compounds with methoxy modification.
3.3.3 Biological evaluation The 18F-labelled compounds 23 and 24 were evaluated in metabolism stud-ies, frozen-section autoradiography (Figure 7) and organ distribution to find out whether the modifications conferred different biological properties.
Metabolite analyzes were performed for the two tracers 23 and 24 after waiting 5 and 30 min after injection in rat. Tracer 23 was 20 ± 6% and 2 ± 1% intact 5 and 30 min after injection, respectively. Tracer 24 was 27 ± 5% and 6 ± 4% intact 5 and 30 min after injection, respectively. Each tracer had one radioactive metabolite.
The biological evaluation of the compounds 23 and 24 was in accordance with previous results.25,26,64,77 Hence, strong accumulation was seen in nor-mal pig and human adrenal cortex in autoradiography and in rat adrenals in the organ distribution. The activity was slightly higher in bone, which indi-cates that defluorination occurred. The autoradiographic studies indicated that both compounds could be useful candidates for the detection of different types of adrenal carcinomas, as they showed low nonspecific binding (3% and 20–30% for compound 23 and 24, respectively). The metabolism, probably via cleavage of the ester, is rapid for both compounds, which might limit their usefulness as tracers for the in vivo visualization of adrenal tu-mours. Both compounds also accumulate in the lung, pancreas and liver, in line with earlier data that suggested rapid metabolism.77

34
Figure 7. Autoradiography showing total binding of 23 and 24 to various human (A-J) and pig (K) adrenal tissue. A, B: Normal adrenals; C, D: Adrenal cortex cancer; E, F: Aldosterone-producing adenoma; G, H: Cortisol-producing adenoma; I, J: Adrenal cortex hyperplasia; K: Pig adrenal; L: Standards on paper. The tissues to the left show the tracer binding and the tissues to the right are under blocking con-ditions.
3.4 18F-Labelling and biological evaluation of vorozole (Papers IV & V) Although 11C-labelled vorozole (VOZ) is a suitable PET tracer for the imag-ing of aromatase, the aim of our study was to synthesise a longer-lived and more selective alternative. In the 18F-labelling, the label was kept on the triazole structure, as in the 11C-labelled VOZ. However, the synthesis had to be designed with consideration for the possible formation of three products. This led to the development of a two- and a one-step synthesis of two 18F-labelled VOZ analogues and the evaluation of their biological properties relevant to in vivo imaging of aromatase.

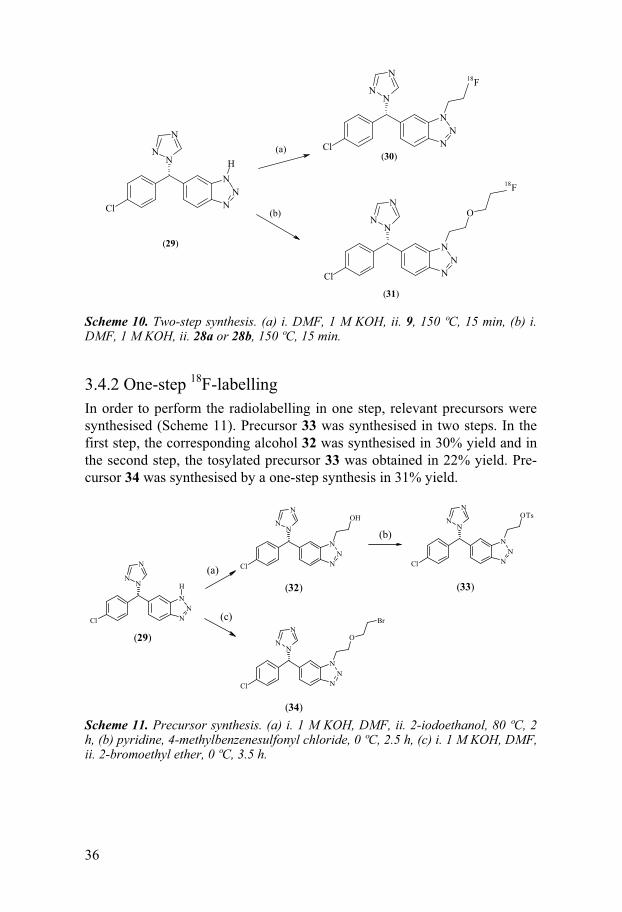
35
3.4.1 Two-step 18F-labelling A two-step labelling synthesis of the 18F-labelled VOZ analogues 6-[(S)-(4-chlorophenyl)(1H-1,2,4-triazol-1-yl)methyl]-1-(2-[18F]fluoroethyl)-1H-1,2,3-benzotriazole (30) and 6-[(S)-(4-chlorophenyl)(1H-1,2,4-triazol-1-yl)methyl]-1-[2-(2-[18F]fluoroethoxy)ethyl]-1H-1,2,3-benzotriazole (31) was developed, with a total synthesis time of 110- to 120 min (Schemes 9 and 10). Compound 30 was synthesised from the 18F-labelled intermediate 9. The d.c. radiochemical yield for 30 was 11 ± 2% and the SRA was 175 ± 7 GBq/�mol. Compound 31 could be synthesised from either of the 18F-labelled intermediates 28a or 28b. The d.c. radiochemical yield for 31 was 15 ± 3% using 28a as a starting material and 5 ± 1% using 28b. The varia-tion in results is probably due to the better leaving-group character of tosy-late. The SRA for compound 31 produced from the intermediate 28a was 56 GBq/�mol. The radiochemical purities of 30 and 31, determined by analyti-cal HPLC, were >96%.
OTsTsO
OTsTsOO
F18TsOO
BrBrO
F18BrO
F18TsO
(9)
(28a)
(28b)
(a)
(a)
(a)
Scheme 9. 18F-Labelled intermediates. (a) [K/K2.2.2]+18F-, DMF, MeCN, 70 ºC, 15 min.
36
NN
N
N
NN
Cl
H
NN
N
N
NN
Cl
F18
NN
N
N
NN
Cl
O
F18
(31)
(a)
(29)
(b)
(30)
Scheme 10. Two-step synthesis. (a) i. DMF, 1 M KOH, ii. 9, 150 ºC, 15 min, (b) i. DMF, 1 M KOH, ii. 28a or 28b, 150 ºC, 15 min.
3.4.2 One-step 18F-labelling In order to perform the radiolabelling in one step, relevant precursors were synthesised (Scheme 11). Precursor 33 was synthesised in two steps. In the first step, the corresponding alcohol 32 was synthesised in 30% yield and in the second step, the tosylated precursor 33 was obtained in 22% yield. Pre-cursor 34 was synthesised by a one-step synthesis in 31% yield.
NN
N
N
NN
Cl
H
NN
N
N
NN
Cl
O
Br
NN
N
N
NN
Cl
OH NN
N
N
NN
Cl
OTs
(34)
(33)(32)
(29)
(a)
(b)
(c)
Scheme 11. Precursor synthesis. (a) i. 1 M KOH, DMF, ii. 2-iodoethanol, 80 ºC, 2 h, (b) pyridine, 4-methylbenzenesulfonyl chloride, 0 ºC, 2.5 h, (c) i. 1 M KOH, DMF, ii. 2-bromoethyl ether, 0 ºC, 3.5 h.

37
The 18F-labelled VOZ analogues were prepared according to Scheme 12. The d.c. radiochemical yields for compounds 30 and 31 were 36% and >99%, respectively, after the 75-min one-step syntheses. The specific radio-activities were 100 GBq/μmol for compound 30 and 80 GBq/μmol for com-pound 31.
NN
N
N
NN
Cl
O
Br
NN
N
N
NN
Cl
O
F18
NN
N
N
NN
Cl
F18
NN
N
N
NN
Cl
OTs
(34)
(33) (30)
(31)
12
3
1
2
3
(a)
(a)
6
89
1011
6
89
1011
1
2
3
1
2
3
6
89
1011
6
89
1011
Scheme 12. One-step 18F-labelling. (a) [K/K2.2.2]+18F-, DMF, 150 ºC, 15 min.
3.4.3 Product formation in the two- and one-step syntheses The synthesis of [11C]VOZ via alkylation of 29 with [11C]methyl iodide was reported to result in a equimolar mixture of three product isotopomers.84 This may occur because three possible products can be synthesised, as the nega-tive charge of the base-generated anion can be delocalized in the triazole ring, which has the major electron densities on the first (N-1) and third (N-3) nitrogen atoms. It is also possible to obtain a product alkylated at N-2. The contamination of the 11C-labelled N-1 product with the N-3 product caused significant image degradation.84 Therefore, it was important to avoid that problem when developing the 18F-labelling synthesis of the VOZ analogues.
In our two-step [18F]fluoroalkylation reaction, the HPLC analysis of crude 30 showed two main products (fractions a and b) and unreacted 18F-labelled intermediate (fraction c) (Figure 8). LC-MS analysis showed that peaks a and b had the same molecular weight. The HPLC analysis of crude 31 showed two peaks. The first peak was the product (fraction d) and the sec-ond peak was unreacted 18F-labelled intermediate (fraction e) (Figure 8).

38
The peaks a and d contained the product of 30 and 31, respectively having the 18F-alkyl chain on N-1. This was verified by comparison with the corre-sponding reference compounds, whose structures were determined by NMR spectroscopic analysis.
However, despite that the products in fractions a and d were identified by comparison to their non-radioactive analogues, byproducts that are not sepa-rable under the HPLC conditions can still be present. The N-1 and N-3 prod-ucts have equal calculated log P values (Table 1), suggesting that they would elute close together in the HPLC analysis.
Our one-step 18F-labelling approach avoids the problem of different iso-topomers. The use of appropriate precursors provides the opportunity to determine which product that will be formed in the labelling synthesis.
The structures of the precursors 33 and 34 were identified by nuclear overhauser effect difference (NOE diff), heteronuclear single quantum co-herence (HSQC) and heteronuclear multiple bond correlation (HMBC) NMR analysis, and shown to have the alkyl chains on the first nitrogen atom in both cases. Their structural assignments were largely based on the NOE cor-relation of H-6 or H-9 to H-10 of the alkyl group (Scheme 12).
30 31
Figure 8. Analysis of the crude products in the syntheses of compounds 30 and 31.
Table 1. Calculated log P values.12
Isotopomer Log P* 18F-CH2CH2-N1 (30) 2.6 18F-CH2CH2-N2 (30) 3.5 18F-CH2CH2-N3 (30) 2.6 18F-(CH2)2O(CH2)2-N1 (31) 2.4 18F-(CH2)2O(CH2)2-N2 (31) 3.4 18F-(CH2)2O(CH2)2-N3 (31) 2.4

39
3.4.4 Biological evaluation The 18F-labelled analogues 30 and 31 were studied to determine their proper-ties as tracers for aromatase. Metabolite studies were performed for both product fractions of compound 30 to evaluate how much unmetabolized tracer remained after 40 min. Fractions a and b contained 28 ± 2% and 10 ± 1% unmetabolized tracer, respectively after this time. However, fraction b has not been analyzed by autoradiography/organ distribution. Hence, no statement is made regarding whether it binds to the same target as fraction a. For analogue 31, fraction d, 8 ± 1% unmetabolized tracer was present after 40 min. Thus both 18F-labelled analogues are metabolised faster than [11C]VOZ.33
In vitro autoradiography studies with the two analogues exhibited specific binding in the medial amygdala, the bed nucleus of stria terminals and the medial preoptic area; these results correspondent with those for [11C]VOZ (Figure 9). However, no binding was obtained with 31 after in vivo admini-stration of the ligand (ex vivo autoradiography).
A monkey PET study was performed with analogue 30, which appeared to have the better properties among the two tracers. Tracer 30 accumulated in the amygdala and hypothalamus, with no specific binding in the cortex (Figure 10). The specific binding of 30 was rapidly and totally displaced by intravenous administration of non-radioactive VOZ, indicating reversibility and rapid kinetics.
The initial biological results indicate that analogue 30 can be a suitable tracer for further studies in vitro and in vivo because it has the appropriate properties to reach the organ of interest, binds selectively to the receptors and penetrates the blood-brain barrier.

40
Figure 9. Autoradiograpic images of 30 and 31. a) The bed nucleus of stria termi-nalis and b) the amygdala.
Figure 10. A PET image of 30 in a monkey brain.
30
31
[11C]VOZ
a) b)

41
3.5 Technological developments Technological improvements are important because they can contribute to increasing SRA and the useful production of potential tracers.
3.5.1 Automation of 18F-labelling synthesis (Paper VI) Automation of labelling synthesis is important for the good manufacturing-practice production of a clinically useful tracer. Additionally, the implemen-tation of such a synthesis into a commercially available platform can make a tracer accessible for many clinical sites. The automatic syntheses of 18F-labelled ETO and VOZ analogues were explored.
The precursors for one-step 18F-fluorination were used to automate the synthesis of tracers 11 and 30 on the commercial platform TRACERLab FXFN (Figure 11). The one-step method was adapted for the synthesis (Scheme 13). The precursors 17 and 33 were used to synthesise compounds 11 and 30 in 30% and 25% d.c. radiochemical yields, respectively.
N
N
OO
OTs
N
N
OO
F18
N
NN
N
NN
OTs
Cl
N
NN
N
NN
F18
Cl
(a)
(b)
(17)
(33) (30)
(11)
Scheme 13. Reactions performed on TRACERLab FXFN. (a) [K/K2.2.2]+18F-, DMF, 110 ºC, 10 min, (b) [K/K2.2.2]+18F-, DMF, 140 ºC, 15 min.

42
Figure 11. Schematic diagram for TRACERLab FXFN. 1) and 3) Reservoirs for re-agents, 2) reservoir for crude product, 4) reactor, 5) reservoir for HPLC-purified product and 6) reservoir for final product.
3.5.2 Radiolabelling with microwave heating (Papers II & III) Microwave heating has been reported to enhance radiolabelling reactions, including one-step nucleophilic 18F-fluorination reactions, by making them faster, cleaner and higher-yielding.85,86
Experiments with microwave heating were performed to synthesise com-pounds 11, 12, 23 and 24 by one-step 18F-labelling methods. Different reac-tion conditions were screened to optimize the syntheses. The reactions were carried out in the microwave for times ranging from 2 to 120 s. Samples were collected from the reaction mixture at each time point and HPLC analysis was used to obtain the d.c. radiochemical yield. When the reaction conditions had been optimized, the syntheses were performed again and the final d.c. radiochemical yield and SRA were determined.
For the synthesis of compound 11, four different temperatures (120, 150, 170 and 210 �C) were tested, and in all cases the same solvent that had been used for conventional heating was used. Generally, the reaction time had no impact on the yield of the reaction. At 150 �C, the variation of the d.c. radio-chemical yields were large; the reaction produced yields in the range of 24 ± 21%. The best yields were obtained at 170 �C. The SRA of the product ob-tained at 170 �C was 313 GBq/�mol. The result was similar to that obtained

43
from conventional heating at 150�C for 15 min (Table 2). Thus the d.c. ra-diochemical yield of compound 11 could be increased from 27 ± 9% with 15 min conventional heating to 46 ± 3% with 2 s of microwave heating (Table 2).
Compound 12 was synthesised using the same reaction conditions as compound 11 (microwave, 170 �C, 2 s). Compared to conventional heating, the microwave synthesis gave higher d.c. radiochemical yield (79 ± 30% vs. 37 ± 4%) but lower SRA (19 GBq/�mol vs. 37 GBq/�mol, Table 2). The conditions for the synthesis of 12 require more optimization in order to give a higher SRA.
In the synthesis of compound 23, five different temperatures (60, 100, 150, 170 and 200 �C) and three different solvents (DMF, MeCN and DMSO) were explored. The microwave syntheses of analogue 23 indicated that the temperature was more important than the solvent in determining the product yield, as there was more variation in the d.c. radiochemical yield when the temperature was varied than when the solvent was changed. The best yield was obtained at 200 �C with 2 s of microwave heating in MeCN/DMF. Compared to our one-step synthesis with conventional heating, microwave heating increased the d.c. radiochemical yield of 23 from 26 ± 8% to 57 ± 12% (Table 3). It was also possible to increase the SRA for compound 23 to 46 ± 8 GBq/μmol with microwave heating, compared to 20 ± 4 GBq/μmol with conventional heating (Table 3).
In the synthesis of compound 24, both the solvent mixture and tempera-ture were important factors in determining the yield. The yield doubled upon increasing the temperature from 150 ºC to 200 ºC. To take advantage of the microwave-heating effect, the reaction was first tested in a solvent with high dielectric constant.87 However, the precursor decomposed in water and in DMF/water. When MeCN and DMF were used as solvents, the yield was lower than obtained with conventional heating. To find out if solvatization was a problem, the less polar solvent dioxane was added to DMF and DMF/DMSO. In these cases no products were obtained; only unreacted start-ing material and [18F]fluoride were recovered. A 3:1 mixture of DMF and DMSO gave the best yield; higher DMSO content decreased the yield. Mi-crowave heating at 200 ºC for 2 s increased the d.c. radiochemical yield for analogue 24 to 51 ± 18%, compared to 23 ± 8% with conventional heating (Table 3). However, the SRA decreased from 175 ± 18 GBq/μmol with con-ventional heating to 19 ± 7 GBq/μmol with microwave heating (Table 3).
When microwave heating was employed instead of conventional heating in the labelling synthesis of the ETO analogues, the reaction time decreased and the d.c. radiochemical yield increased.
This one-step, microwave-heated 18F-labelling syntheses have the poten-tial to be used in a fully automated system with an integrating microwave reactor, e.g. a TRACERLab FXFN or Synthia88 system.

44
Table 2. Radiochemical yield and SRA for compounds 11 and 12 using conventional and microwave heating.
11 12 Compound
Conventional
heating
Microwave
heatingc
Conventional
heating
Microwave
heatingc
Radiochemical
yielda (%)
27 ± 9
(n = 3)
46 ± 3
(n = 2)
37 ± 4
(n = 2)
79 ± 30
(n = 2)
SRAb (GBq/μmol) 328 313 37 19 aD.c., based upon [18F]fluoride at start and radioactivity of HPLC-purified product. n = number of experiments. bRatio of radioactivity to amount of substance, d.c. to time at start of synthesis. cOptimized reaction conditions were used in the synthesis.
Table 3. Radiochemical yield and SRA for compounds 23 and 24 using conventional and microwave heating.
23 24 Compound
Conventional
heating
Microwave
heatingc
Conventional
heating
Microwave
heatingc
Radiochemical
yielda (%)
26 ± 8
(n = 5)
57 ± 12
(n = 4)
23 ± 8
(n = 4)
51 ± 18
(n = 6)
SRAb
(GBq/μmol)
20 ± 4
(n = 2)
46 ± 8
(n = 2)
175 ± 18
(n = 2)
19 ± 7
(n = 4) aD.c., based upon [18F]fluoride at start and radioactivity of HPLC-purified product. n = number of experiments. bRatio of radioactivity to amount of substance, d.c. to time at start of synthesis. cOptimized reaction conditions were used in the synthesis.

45
4. Conclusions
♦ Two metomidate analogue precursors containing halogen atoms at the para position of the phenyl group were synthesised and labelled with 11C. The biological evaluation of these compounds using autoradiogra-phy, organ distribution and metabolite analysis indicated that the ana-logue containing a bromine atom had the best properties, particularly with respect to adrenal-to-liver ratio and stability in blood over time, for use as an imaging agent for the enzyme 11�-hydroxylase in the adrenal cortex.
♦ 18F-Labelled etomidate analogues with structurally different ester parts
or with halogen atoms at the para position of the phenyl groups have been synthesised using two- and one-step methods. The new two-step 18F-labelling synthesis gives higher yield than the previously published method, and no purification of the labelled intermediate is required after the first step. Suitable precursors were synthesised and used in a one-step 18F-labelling that gave faster and efficient labelling and higher ra-diochemical yields and specific radioactivities.
♦ The compound that best met clinical demands was the etomidate ana-
logue containing an 18F-labelled ethyl chain and a chloride in the para position on the phenyl group, because it had the greatest adrenal-to-liver ratio and highest degree of specific binding. This compound will be fur-ther investigated as a tracer for cancer in the adrenal cortex.
♦ The two- and one-step 18F-labelling methods were used to synthesise
two 18F-labelled vorozole analogues. The one-step synthesis provided higher reproducibility and radiochemical yield than the two-step synthe-sis did. The one-step synthesis made it possible to avoid the problem of isotopomer formations as appropriate precursors where synthesised. The precursors were characterized before the labelling synthesis to ensure that only the N-1 product was synthesised. The biological results indi-cated that the alkylated vorozole analogue had the best properties of the analogues as a tracer for the visualization of aromatase.

46
♦ The one-step synthesis of 18F-labelled etomidate and vorozole analogues was automated using TRACERLab FXFN. The implementation of these labelling syntheses into a commercially available platform makes the tracers more accessible for many clinical sites.
♦ When microwave heating was used in the one-step 18F-labelling synthe-
sis of some etomidate analogues, the reaction time was decreased to sec-onds, and increased radiochemical yields were obtained.

47
Summary in Swedish
Det övergripande målet med denna avhandling var att utveckla nya syntes-metoder för inmärkning av biologiskt aktiva molekyler för användning inom positronemissionstomografi (PET), med fokus på avbildandet av olika en-zymer i steroidsyntesen.
PET är en icke-invasiv avbildningsteknik som mäter strålning från en spårmolekyl märkt med en positronemitterande �+-radionuklid. Inom PET används ofta kortlivade radionuklider som 11C och 18F, med halveringstider på 20 och 110 min.
Genom att använda sig av skräddarsydda spårmolekyler kan information erhållas som underlättar diagnos av bl.a. cancertumörer och val av behand-ling för patienten. PET används allt mer inom läkemedelsforskning, då man kan följa läkemedelssubstanser eller andra biologiskt aktiva substansers transport, bindning och metabolism. En stor fördel med PET är att man an-vänder låga koncentrationer av spårmolekylen vilket möjliggör studier av biokemiska processer utan att dessa störs från normala mönster.
Vid en PET-undersökning injiceras spårmolekyler intravenöst i kroppen. Eftersom spårmolekylerna binder specifikt i de vävnader där målreceptormo-lekylerna finns, kan man med hjälp av en PET-kamera avbilda positronstrål-ningens utbredning och koncentrationen i vävnaden.
Kolatomer kan hittas i nästan alla biomolekyler, därför är det möjligt att med 11C syntetisera nästa identiska spårmolekyler som har liknande biolo-giska egenskaper.
Trots att fluoratomer är ovanliga i de flesta naturligt förekommande bio-logiska molekyler, har de en vida användning inom läkemedelsindustrin. Genom att introducera en fluoratom kan man ändra molekylens kemiska och biologiska egenskaper. Detta utnyttjas inom radiokemin och PET då man letar efter alternativa och förbättrade spårmolekyler.
Metomidat och etomidate är spårmolekyler som intragerar selektivt med enzymet 11�-hydroxylase som finns i adrenal cortex. I det första delarbetet utfördes 11C-inmärkning av olika metomidatanaloger. Våra experiment indi-kerar att man kan förbättra avbildningen genom att introducera en halogen-atom (d.v.s. brom eller klor) i spårmolekylen.
I det andra och tredje delarbetet studerades hur 18F-inmärkning och för-ändring av strukturen genom pegylering och alkylering påverkade etomidats förmåga att fungera som en spårmolekyl. Den korta halveringstiden ställer krav på att inmärkningen av spårmolekylen är snabb och effektiv. Det är

48
dessutom viktigt att erhålla maximal mängd produkt och minimalt med bi-produkt på kortast möjliga tid. Därför jämfördes två olika typer av inmärk-ningsvägar, en tvåstegsmetod och en enstegsmetod. Med enstegsmetoden ökade reproducerbarheten och på grund av undvikandet av sidoreaktioner ökade även utbytet. Inga radioaktiva intermediat behövde syntetiseras vilket bidrog till kortare reaktionstid. Vidare behövdes ingen preparering av start-materialet (prekursorn) före syntes. För att minska reaktionstiden ytterligare utfördes mikrovågskörningar. Detta bidrog till att syntestiden minskade till sekunder och att utbytet ökade ytterligare.
I det fjärde delarbetet utfördes 18F-inmärkningen genom en enstegs- och tvåstegssyntes av en spårmolekyl kallad vorozol. Detta är en spårmolekyl för aromatas, som är ett enzym som konverterar manliga könshormoner till kvinnliga hormoner. Syftet var att utveckla bättre eller andra typer av spår-molekyler för aromatas, än 11C-inmärkt vorozol. Två olika 18F-inmärkta vo-rozolanaloger utvecklades men p.g.a. snabb metabolism visade sig endast en analog vara ett alternativ till 11C-inmärkt vorozol. Tvåstegssyntesen gav förutom den önskade produkten ytterligare biprodukter. Detta kunde undvi-kas genom att använda en enstegssyntes. De två vorozolanalogernas biolo-giska egenskaper undersöktes i delarbete fyra och fem.
I det sjätte delarbete påvisades att enstegssyntesen av etomidat och voro-zole kan utföras i ett fullt automatiserat system som TRACERLab FXFN.

49
Acknowledgments
I would like to thank all of those who helped me directly or indirectly in the preparation of this thesis.
First of all I would like to express my sincere gratitude to my supervisor Prof. Bengt Långström for accepting me as a PhD student in your group and for your enthusiasm and encouragement during these years. For always be-ing available, no matter where in the world you were you always gave me scientific guidance and support. You have prepared me to meet new chal-lenges in my future scientific career. My assistant supervisors: Dr. Farhad Karimi for introducing me to the world of labelling chemistry and for your guidance during the first part of my PhD studies. Dr. Obaidur Rahman for supervision during the last two years, for chemistry related discussions, commenting on my thesis and your high spirit. Prof. Helena Grennberg for your support and for introducing me to the world of nanoparticles and its possibilities. My excellent co-workers: Prof. Håkan Hall for you nice personality and helpfulness with commenting on my thesis. For introducing me both to biol-ogy and the “latest” swimming fashion during the conference in Nice. Dr. Örjan Lindhe for fruitful collaboration on the MTO-projects. Elisabeth Bergström-Petterman for always taking time for assisting me with the bio-logical part of the project. Dr. Sergio Estrada and Dr. Kayo Takahashi for collaboration on the VOZ-projects. I would like to thank all past and present members of the group BLå, in par-ticular: Elisabeth Blom for you friendship during the graduate and PhD-years. For all the nice moments, from chemistry related work to “smör-gåspauser”. Ola Åberg for being a nice colleague. I am impressed that you have managed to survive during these years as “den enda tuppen i höns-gården”. Dr. Julien Barletta for bringing some French spirit into the lab. Dr. Jonas Eriksson for creating a nice working environment. Dr. Torben Ras-mussen for invaluable help with the computers. Dr. Oleksiy Itsenko for shar-ing my love for discussions no matter the subject and for commenting on my thesis. Dr Irina Velikyan for always welcoming me with a hug and for shar-

50
ing the funny “dress and shoe memory” in Nice, as well as for commenting on my thesis. All the people in the ASL group and the staff at Imanet are sincerely ac-knowledged for creating a friendly and stimulating atmosphere: Especially, Madeleine Svennebäck, Margareta Sprycha, Helena Wilking, Dr. Pernilla Johansen, Daniel Johansen, Eva-Lotta Nyberg and Anna Holmberg and Assoc. Prof. Tor Kihlberg. Assoc. Prof. Alf Thibblin, Sven-Åke Gustavsson, Assoc. Prof. Gunnar Antoni and Viktor Leander for the help with the 18F-Synthia in Hotlab 2. Joachim Schultz for keeping the cyclotron in good con-dition. Johan Ulin for your knowledge in the microwave technology. Pasha Razifar and Azita Monazzam for your kindness. Thanks to everyone at the department of biochemistry and organic chemis-try. Especially, Eva Pylvänen and Gunnar Svensson for technical and admin-istrative assistance. Prof. Adolf Gogoll for NMR support. Dr Tamara Church for linguistic corrections. Alla mina nära vänner som jag har haft glädjen att få lära känna under olika faser i mitt liv. Jag vill skänka ett varmt tack till min härliga släkt som under mina nio år i Uppsala har funnits till för mig och bevisat att det går in fler personer än man tror på en liten yta. Mina två fantastiska gudmödrar Erica och Helena, som bara är… Fredrik, mannen i mitt liv för din kärlek och ditt otroliga tålamod, som kan-ske har fått uthärda det mesta av min personlighet i synnerhet de sista åren, men nu blir det bara bättre och bättre. � Sist men inte minst vill jag tacka mina helt underbara föräldrar Börje och Birgitta, utan er kärlek och ert stöd under dessa år hade detta aldrig varit möjligt för mig. Ni har lärt mig att tro på mina drömmar, följa mitt hjärta och våga flyga. � Maria Uppsala, February 2009

51
References
1 Ametamey, S. M., Honer, M., Schubiger, P. A., Chem. Rev. 2008, 108,
1501. 2 Wagner, H. N., Szabo, Z., Buchanan, J. W., 1995, Principles of Nuclear
Medicine, W. B. Saunders Co.: Philadelphia, PA. 3 Pither, R., Expert Rev. Mol. Diagn. 2003, 3, 703. 4 Accessed January 20, 2009, at http://www-pet.umds.ac.uk/whatspet.htm 5 Bergman, J., Eskola, O., Lehikoinen, P., Solin, O., Appl. Radiat. Isot.
2001, 54, 927. 6 Långström, B., 1980, On the synthesis of 11C-compounds, Almqvist &
Wiksell International, LiberTryck, Stockholm. 7 Schlyer, D. J., Firouzbakht, M. L., Wolf, A. P., Appl. Radiat. Isot. 1993,
44, 1459. 8 Lappin, G., Temple, S., 2006, Radiotracers in drug development, Taylor
& Francis, p 44. 9 Zolle, I. M., Berger, M. L., Hammerschmidt, F., Hahner, S., Schirbel, A.,
Peric-Simov, B., J. Med. Chem. 2008, 51, 2244. 10 Park, K. B., Kitteringham, N. R., Drug Metabol. Rev. 1994, 26, 605. 11 Waterhouse, R. N., Mol. Imag. Biol. 2003, 5, 376. 12 www.acdlabs.com 13 Patel, S., Gibson, R., Mol. Imag. Biol. 2008, 35, 805. 14 Fowler, J. S., Wolf, A. P., Acc. Chem. Res. 1997, 30, 181. 15 Thorell, J. O., Stone-Elander, S., Elander, N., J. Label. Compd. Radi-
opharm. 1992, 31, 207. 16 Långström, B., Kihlberg, T., Bergström, M., Antoni, G., Björkman, M.,
Forngren, B. H., Forngren, T., Hartvig, P., Markides, K., Yngve, U., Ög-ren, M., Acta Chem. Scand. 1999, 53, 651.
17 Hyllbrant, B., Tyrefors, N., Markides, K. E., Långström, B., J. Pharm. Biomed. Anal. 1999, 20, 493.
18 Ulrich-Lai, Y. M., Xie, W., Meij, J. T. A., Dolgas, C. M., Yu, L., Herman J. P., Physiol. Behav. 2006, 88, 67.
19 Welch, M. J., Redvanly, C. S., 2003, Handbook of Radiopharmaceuticals, John Wiley & Sons, Ltd, p 715.
20 Mankoff, D. A., Link, J. M., Linden, H. M., Sundararajan, L., Krohn, K., J. Nucl. Med. 2008, 49, 149S.
21 Katzenellenbogen, J. A., J. Fluor. Chem. 2001, 109, 49.

52
22 Lidström, P., Neu, H., Långström, B., J. Label. Compd. Radiopharm.
1997, 39, 695. 23 Welch, M. J., Redvanly, C. S., 2003, Handbook of Radiopharmaceuticals,
John Wiley & Sons, Ltd, p 725. 24 Wagner, H. N., Szabo, Z., Buchanan, J. W., 1995, Principles of Nuclear
Medicine, W. B. Saunders Co.: Philadelphia, PA, p. 655. 25 Bergström, M., Bonasera, T. A., Lu, L., Bergström, E., Backlin, C., Juh-
lin, C., Långström, B., J. Nucl. Med. 1998, 39, 982. 26 Bergström, M., Juhlin, C., Bonasera, T. A., Sundin, A., Rastad, J., Åker-
ström, G., Långström, B., J. Nucl. Med. 2000, 41, 275. 27 Khan, T. S., Sundin, A., Juhlin, C., Långström, B., Bergström, M., Eriks-
son, B., Eur. J. Nucl. Med. 2003, 30, 403. 28 Zettinig, G., Mitterhauser, M., Wadsak, W., Becherer, A., Pirich, C.,
Vierhapper, H., Niederle, B., Dudczak, R., Kletter, K., Eur. J. Nucl. Med. 2004, 31, 1224.
29 Minn, H., Salonen, A., Friberg, J., Roivainen, A., Viljanen, T., Långsjö, J., Salmi, J., Välimäki, M., Någren, K., Nuutila, P., J. Nucl. Med. 2004, 45, 972.
30 McGuire, A. H., Dehdashti, F., Siegel, B. A., Lyss, A. O., Brodack, J. W., Mathias, C. J., Mintun, M. A., Katzenellenbogen, J. A., Welch, M. J., J. Nucl. Med. 1991, 32, 1526.
31 Mintun, M. A., Welch, M. J., Siegel, B. A., Mathias, C. J., Brodack, J. W., McGuire, A. H., Katzenellenbogen, J. A., Radiology. 1988, 169, 45.
32 Lidström, P., Bonasera, T. A., Kirilovas, D., Lindblom, B., Lu, L., Berg-ström, E., Bergström, M., Westlin, J. E., Långström, B., Nucl. Med. Biol. 1998, 25, 497.
33 Takahashi, K., Bergström, M., Frändberg, P., Vesström, E. L., Watanabe, Y., Långström, B., Nucl. Med. Biol. 2006, 33, 599.
34 Takahashi, K., Hallberg, M., Magnusson, K., Nyberg, F., Watanabe, Y., Långström, B., Bergström, M., NeuroReport, 2007, 18, 171.
35 Takahashi, K., Tamura, Y., Watanabe, Y., Långström, B., Bergström, M., NeuroReport. 2008, 19, 431.
36 Kirilovas, D., Naessen, T., Bergström, M., Bergström-Petterman, E., Carl-ström, K., Långström, B., Steroids. 2003, 68, 1139.
37 Vanden Bossche, H., Willemsens, G., Cools, W., Bellens, D., Biochem. Pharmacol. 1984, 33, 3861.
38 Weber, M. M., Lang, J., Abedinpour, F., Zeilberger, K., Adelmann, B., Engelhart, D., Clin. Investig. 1993, 71, 933.
39 Hellman, P., The Bengt Långström Symposium, 2008. 40 Bossche, H. V., Morereels, H., Koymans, L. M. H., Breast Cancer Res.
Treat. 1994, 30, 43. 41 Wounters, W., Snoeck, E., De Coster, R., Breast Cancer Res. Treat. 1994,
30, 89.

53
42 Wouters, W., De Coster, R., Dun, J., Krekels, M. D. W. G., Dillen, A.,
Raeymaekers, A., Freyne, E., Van Gelder, J., Sanz, G., Venet, M., Janssen, M., J. Steroid Biochem. Mol. Biol. 1990, 37, 1049.
43 Johnston, S. R. D., Dowsett, M., Nature Reviews Cancer. 2003, 3, 821. 44 Wouters, W., Van Ginckel, R., Krekels, M., Bowden, C., De Coster, R., J.
Steroid Biochem. Mol. Biol. 1993, 44, 617. 45 De Jong, P. C., Van de Ven, J., Nortier, H. W. R., Maitimu, I., Donker, T.
H., Thijssen, J. H. H., Slee, P. H. T. J., Blankenstein, R.A., Cancer Res. 1997, 57, 2109.
46 Santen, R. J., Harvey, H. A., Endocrine-Related Cancer. 1999, 6, 75. 47 Dowsett, M., Doody, D., Miall, S., Howes, A., English, J., Coombes, R.
C., Breast Cancer Res. Treat. 1999, 56, 25. 48 Simpson, E. R., Kilgore, M. W., Mahendroo, M. S., Means, G. D., Cor-
bin, C. J., Mendelson, C. R., J. Steroid Biochem. Mol. Biol. 1992, 43, 923. 49 Roselli, C. E., Stormshak, F., Resko, J. A., Brain Res. 1998, 811, 105. 50 Abdelgadir, S. E., Roselli, C. E., Choate, J. V. A., Resko, J. A., Biol. Re-
prod. 1997, 57, 772. 51 Kessler, R. C., McGonagle, K. A., Swartz, M., Blazer, D. G., Nelson, C.
B., J. Affect. Disord. 1993, 29, 85. 52 Nolen-Hoeksema, S., Psychol. Bull. 1987, 101, 259. 53 Le Bars, D., J. Fluorine Chem. 2006, 127, 1488. 54 Hagmann, W. K., J. Med. Chem. 2008, 51, 4359. 55 Bergman, J., Solin, O., Nucl. Med. Biol. 1997, 24, 677. 56 Fowler, J. S., Shiue, C. Y., Wolf, A. P., Salvadori, P. A., MacGregor, R.
R., J. Label. Compd. Radiopharm. 1982, 19, 1634. 57 Chirakal, R., Firnau, G., Schrobilgen, G. J., Mckay, J., Garnett, E. S.,
Appl. Radiat. Isot. 1984, 35, 401. 58 Satyamurthy, N., Bida, G. T., Phelps, M. E., Barrio, J. R., Appl. Radiat.
Isot. 1990, 41, 733. 59 Namavari, M., Bishop, A., Satyamurthy, N., Bida, G., Barrio, J. R., Appl.
Radiat. Isot. 1992, 43, 989. 60 Gomzina, N. A., Vasil’ev, D. A., Krasikova, R. N., Radiochemistry. 2002,
44, 403. 61 Angelini, G., Speranza, M., Wolf, A. P., Shiue, C. Y., J. Fluorine Chem.
1985, 27, 177. 62 Wadsak, W., Mitterhauser, M., J. Label. Compd. Radiopharm. 2003, 46, 379. 63 Wilson, A., Dasilva, J. N., Houle, S., Appl. Radiat. Isot. 1995, 46, 765. 64 Wadsak, W., Mitterhauser, M., Rendl, G., Schuetz, M., Mien, L. K., Ett-
linger, D. E., Dudczak, R., Kletter, K., Karanikas, G., Eur. J. Nucl. Med. Mol. Imaging. 2006, 33, 669.
65 Hennings, J., Lindhe, O., Bergström, M., Långström, B., Sundin, A., Hellman, P., J. Clin. Endocrin. Metab. 2006, 91, 1410.
66 Godefroi, E. F., Janssen, P. A. J., Van Der Eycken, C. A. M., Van Heertum, A. H. M. T., Niemegeers, C. J. E., J. Med. Chem. 1965, 8, 220.

54
67 Långström, B., Antoni, G., Gullberg, P., Halldin, C., Malmborg, P., Någ-
ren, K., Rimland, A., Svärd H., J. Nucl. Med. 1987, 28, 1037. 68 Bergström, M., Awad, R., Estrada, S., Mälman, J., Lu, L., Lendvai, G., Berg-
ström-Pettermann, E., Långström, B., Mol. Imaging. Biol. 2003, 5, 390. 69 Sihver, W., Sihver, S., Bergström, M., Murata, T., Matsumura, K., Onoe,
H., Andersson, Y., Bjurling, P., Fasth, K., Westerberg, G., Ögren, M., Ja-cobsson, G., Lundqvist, H., Oreland L., Watanabe, Y., Långström, B., Nucl. Med. Biol. 1997, 24, 723.
70 Sihver, S., Sihver, W., Bergström, M., Höglund, A. U., Sjöberg, P., Lång-ström, B., Watanabe, Y., J. Pharmacol. Exp. Ther. 1999, 290, 917.
71 Hara, T., Kosaka, N., Kishi, H., J. Nucl. Med. 2002, 43, 187. 72 Koivula, T., Perhola, O., Kämäräinen, E. L., Lipponen, T., Vepsäläinen,
J., Solin, O., J. Label. Compd. Radiopharm. 2005, 48, 463. 73 Kazumata, K., Dhawan, V., Chaly, T., Antonini, A., Margouleff, C., Be-
lakhlef, A., Neumeyer, J., Eidelberg, D., J. Nucl. Med. 1998, 39, 1521. 74 Hutson, L., Krstina, J., Moad, C. L., Moad, G., Morrow, G. R., Postma,
A., Rizzardo, E., Thang, S. H., Macromolecules. 2004, 37, 4441. 75 Lowary, T. L., Bundle, D. R., Tetrahedron Assymmetry. 1994, 5, 2397. 76 Bradshaw, J. S., Krakowiak, K. E., Lindh, G. C., Izatt, R. M., Tetrahe-
dron. 1987, 43, 4271. 77 Ettlinger, D. E., Wadsak, W., Mien, L. K., Machek, M., Wabnegger, L.,
Rendl, G., Karanikas, G., Viernstein, H., Kletter, K., Dudczak, R., Mitter-hauser, M., Eur. J. Nucl. Med. Molec. Imag. 2006, 33, 928.
78 Zang, W., Kung, M-P., Oya, S., Hou, C., Kung, H. F., Nucl. Med. Biol. 2007, 34, 89.
79 Tierney, J., Lindström, P., 2005, Microwave assisted organic synthesis, Blackwell Publishing Ltd.
80 Karimi, F., Erlandsson, M., Lindhe, Ö., Långström, B., J. Label. Compd. Radiopharm. 2008, 51, 273.
81 Head, F. S. H., J. Chem. Soc. 1963, 2963. 82 Amin, M., Twine, C. E., Kepler, J. A., J. Label. Compd. Radiopharm.
1997, 39, 875. 83 Hsiao, L. Y. Y., Musallam, H. A., U.S. Patent 5130438, 1992. 84 Kim, S. W., Biegon, A., Katsamanis, Z. E., Ehrlich, C. W, Hooker, J. M.,
Shea, C., Muenche, L., Xu, Y., King, P., Carter, P., Alexoff, D. L., Fowler, J. S., Nucl. Med. Biol. In press.
85 Jones, J. R., Lu, S. Y., 2002, Microwave-enhanced radiochemistry. In: Loupy A 1st edn. Microwaves in Organic Synthesis. Wiley-VCH Verlag GmbH & Co, Weinheim, p 456–459.
86 Elander, N., Jones, J. R., Lu, S-Y., Stone-Elander, S., Chem. Soc. Rev. 2000, 29, 239.
87 Leadbeater, N. E., Torenius, H. M., J. Org. Chem. 2002, 67, 3145. 88 Lazarova, N., Siméon, F. G., Musachio, J. L., Lu, S., Pike, V. W., J. La-
bel. Compd. Radiopharm. 2007, 50, 463.


ACTA UNIVERSITATIS
UPSALIENSISUPPSALA
2009
Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 607
Editor: The Dean of the Faculty of Science and Technology
A doctoral dissertation from the Faculty of Science and Technology, Uppsala University, is usually a summary of a number of papers. A few copies of the complete dissertation are kept at major Swedish research libraries, while the summary alone is distributed internationally through the series Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology. (Prior to January, 2005, the series was published under the title “Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology”.)
Distribution: publications.uu.se urn:nbn:se:uu:diva-89177


















