
Hemoglobinopatías adams olivia
37
Instituto Politécnico Nacional Escuela Nacional de Medicina y Homeopatía “ Hemoglobinopatías” López Adams Dania Olivia Grupo. 6pm3 14-Marzo-16
-
Upload
olivia-adams -
Category
Health & Medicine
-
view
140 -
download
0
Transcript of Hemoglobinopatías adams olivia

Instituto Politécnico NacionalEscuela Nacional de Medicina y Homeopatía
“Hemoglobinopatías”
López Adams Dania Olivia Grupo. 6pm3 14-Marzo-16

Hemoglobinopatías
Defectos en la Hb
Heredados Adquiridos

Hemoglobinopatías Estructurales
• Alteración en las secuencia de aminoácidos de una de sus
cadenas globínicas.
• Alteración de las propiedades moleculares.
Son detectadas mediante electroforesis.
En su mayoría son asintomáticas
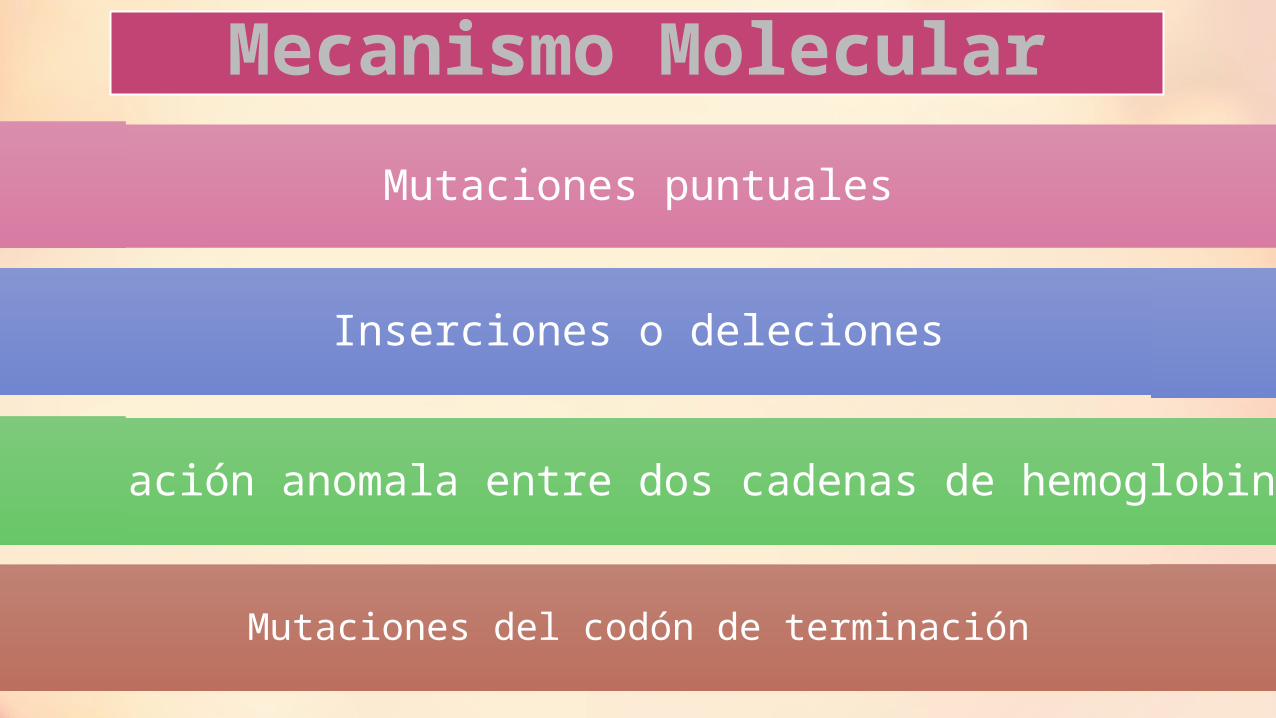
Mecanismo Molecular
Mutaciones puntuales
Inserciones o deleciones
Hibridación anomala entre dos cadenas de hemoglobina
Mutaciones del codón de terminación

Hemoglobinopatías Estructurales
Alteración de la carga
superficial
Inestables
Alteración de la afinidad por
el O2
Hemoglobino-patías M

Hemoglobinopatías con alteración de carga superficial
HbCHbS
• Hemoglobinopatías estructurales más
frecuentes.
• Fácil detección mediante electroforesis.

Hemoglobinopatía S (Hb S)
• Descrita por Pauling en 1949.
• Individuos de raza negra.
• En su forma homocigota es causante de la drepanocitosis.
• Hemoglobinopatía mas común y de mayor impacto sanitario.
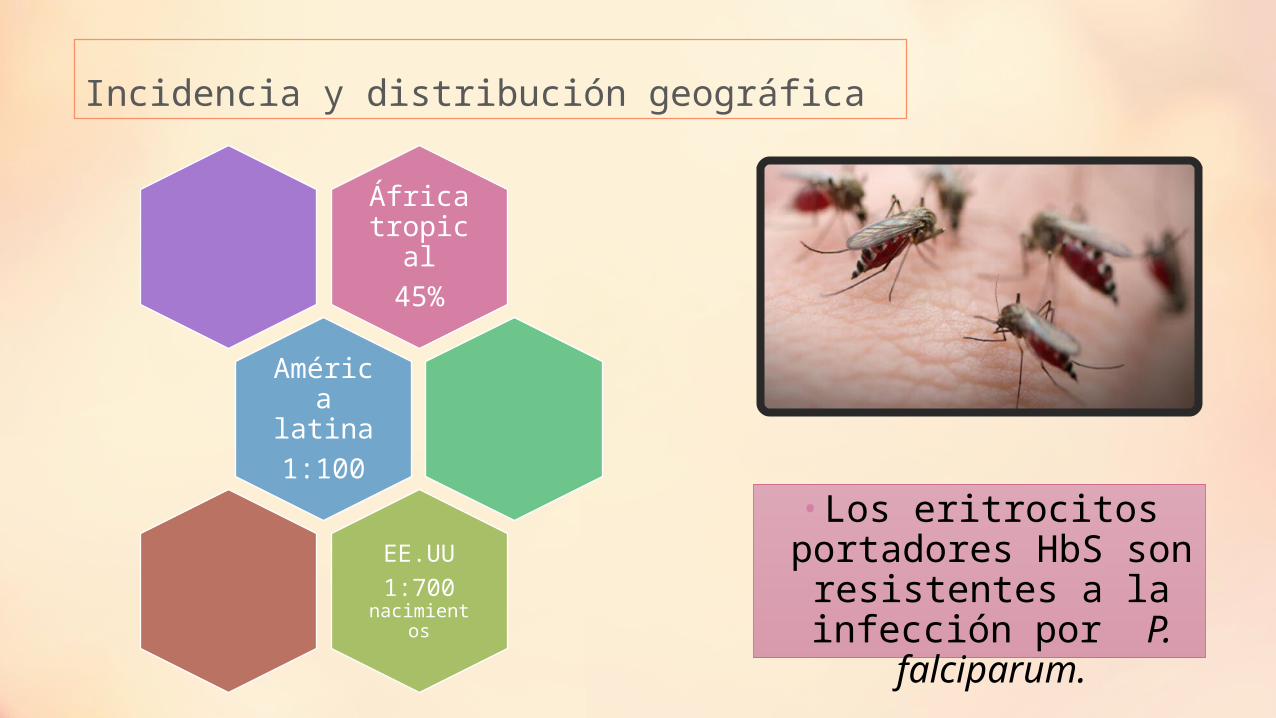
Incidencia y distribución geográfica
• Los eritrocitos portadores HbS son
resistentes a la infección por P.
falciparum.
África tropical
45%
América latina 1:100
EE.UU1:700
nacimientos
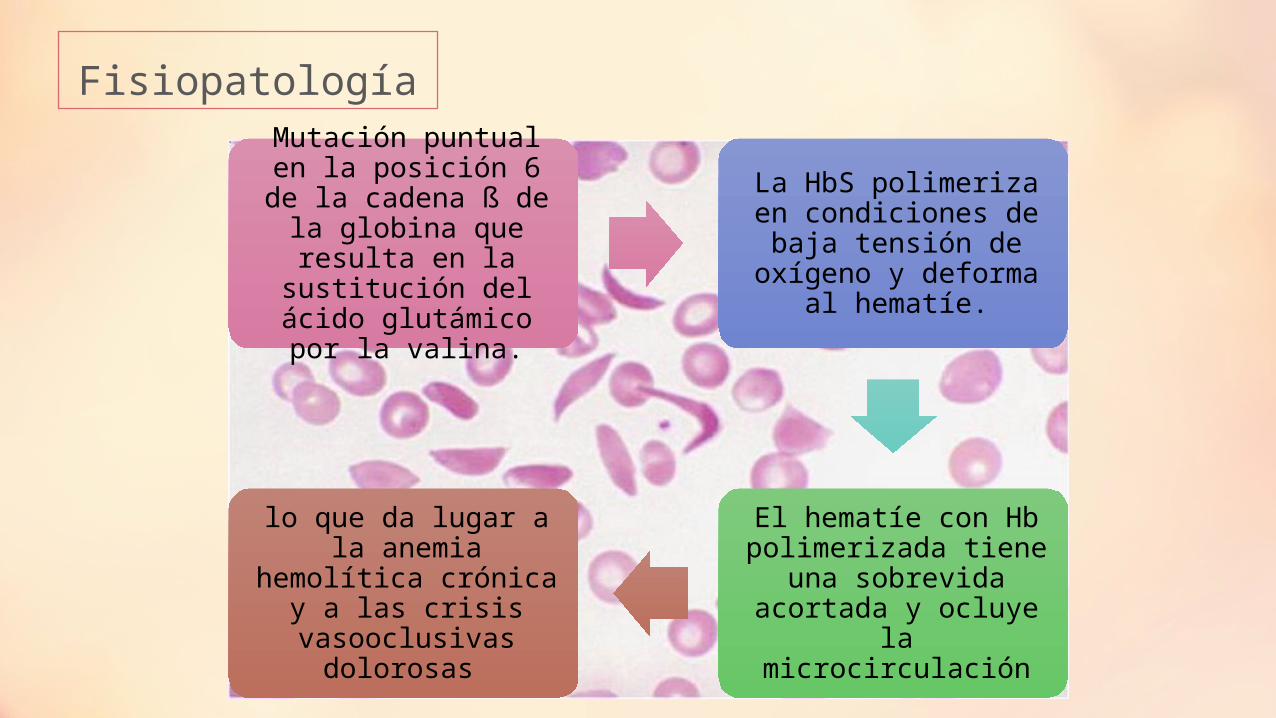
Fisiopatología
Mutación puntual en la posición 6 de la cadena ß de la globina que resulta
en la sustitución del ácido glutámico por la valina.
La HbS polimeriza en condiciones de baja tensión de oxígeno y deforma al hematíe.
El hematíe con Hb polimerizada tiene una
sobrevida acortada y ocluye la microcirculación
lo que da lugar a la anemia hemolítica crónica y a las crisis vasooclusivas
dolorosas

Fisiopatología

Manifestaciones Clínicas
•Rasgo Falciforme:• Ausencia de signos clínicos de la enfermedad.
• Examen morfológico de eritrocitos, sin alteraciones.
• VCM, HCM, CCMH normales.
• La enfermedad puede manifestarse en situaciones de estrés.

Manifestaciones Clínicas
•Anemia falciforme/ drepanocitosis:
• Pasados los primeros 4 a 6 meses de vida inicia el cuadro clínico.
1. Fase estacionaria2. Fase de expresividad aguda3. Fase de expresividad crónica
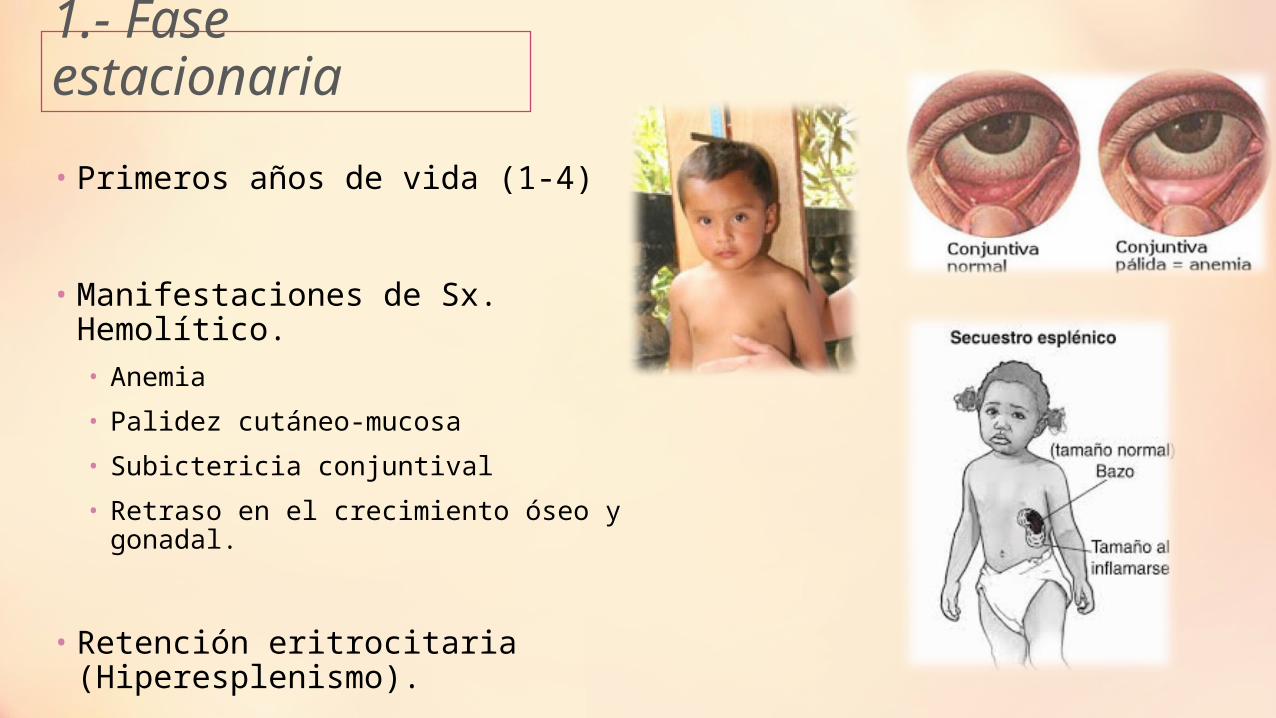
1.- Fase estacionaria
• Primeros años de vida (1-4)
• Manifestaciones de Sx. Hemolítico.• Anemia
• Palidez cutáneo-mucosa
• Subictericia conjuntival
• Retraso en el crecimiento óseo y gonadal.
• Retención eritrocitaria (Hiperesplenismo).

2.- Fase de expresividad aguda
• A partir de los 4 años de edad.
• Agravamiento del cuadro anémico. (Hb < 80g/L)
• Crisis vasooclusivas.• Ataques muy dolorosos y pasajeros que pueden durar
días o semanas.
• Desencadenados por situaciones diversas como hipoxia, fiebre, infecciones, acidosis, deshidrataciones, menstruación entre otros.
• Sx. Mano-pie : Dolor agudo con tumefacción en la superficie dorsal e impotencia funcional.
• Sx. Mesentérico: Dolor abdominal muy intenso y agudo.
• Oclusión de vasos cerebrales: Hemiplejia, monoplejia y convulsiones.
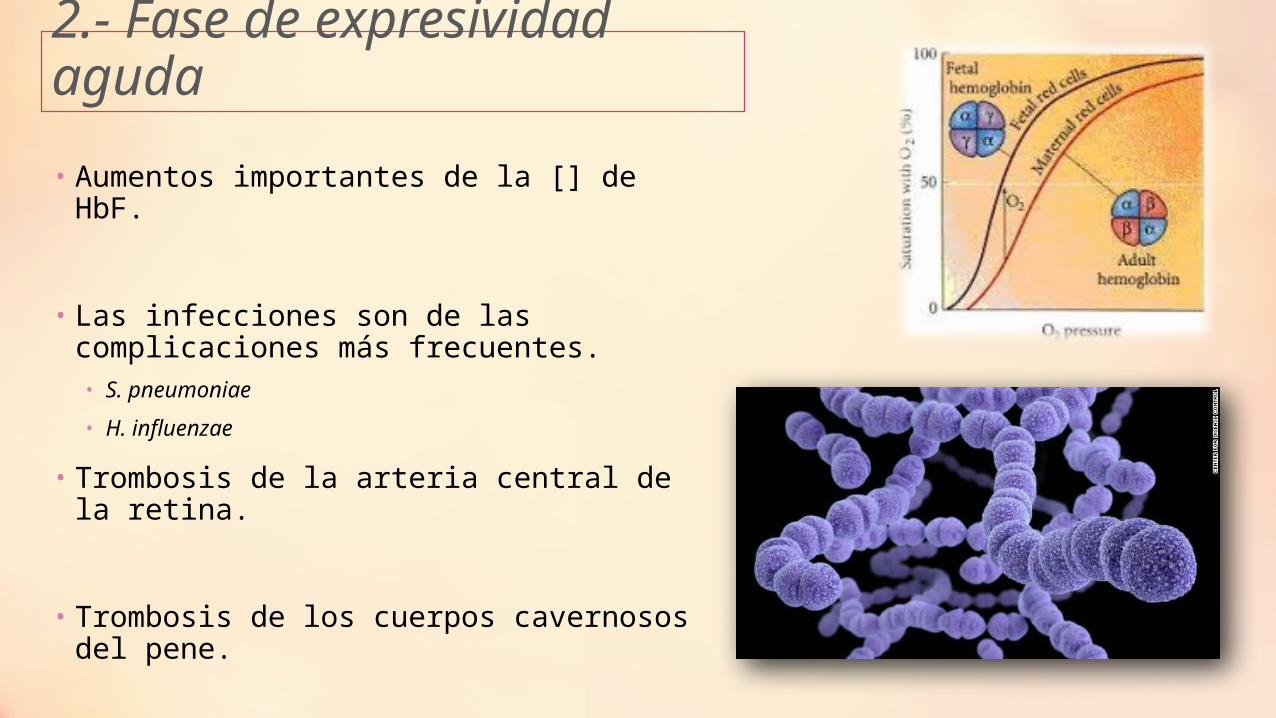
2.- Fase de expresividad aguda
• Aumentos importantes de la [] de HbF.
• Las infecciones son de las complicaciones más frecuentes. • S. pneumoniae
• H. influenzae
• Trombosis de la arteria central de la retina.
• Trombosis de los cuerpos cavernosos del pene.
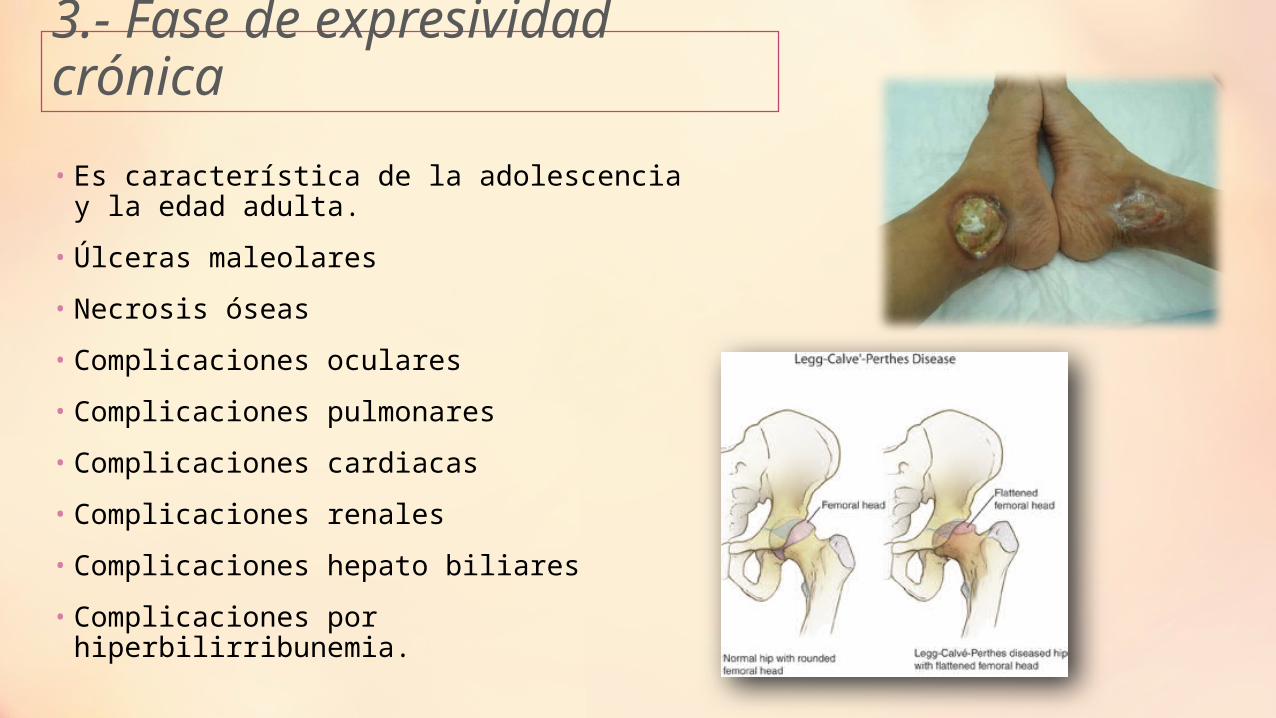
3.- Fase de expresividad crónica
• Es característica de la adolescencia y la edad adulta.
• Úlceras maleolares
• Necrosis óseas
• Complicaciones oculares
• Complicaciones pulmonares
• Complicaciones cardiacas
• Complicaciones renales
• Complicaciones hepato biliares
• Complicaciones por hiperbilirribunemia.

Diagnóstico
a) Hemograma con recuento de reticulocitos.
b) Examen de la morfología eritrocitaria.
c) Pruebas de solubilidad o falciformación.
d) Electroforesis de hemoglobinas.
e) Análisis del gen HbS mediante PCR.

Pronóstico y prevención
• El pronostico depende de la concentración de HbF. (mayor en mujeres), coexistencia de otras hemoglobinopatías.
• Medidas de prevención: Diagnostico precoz mediante programas de cribado neonatal.
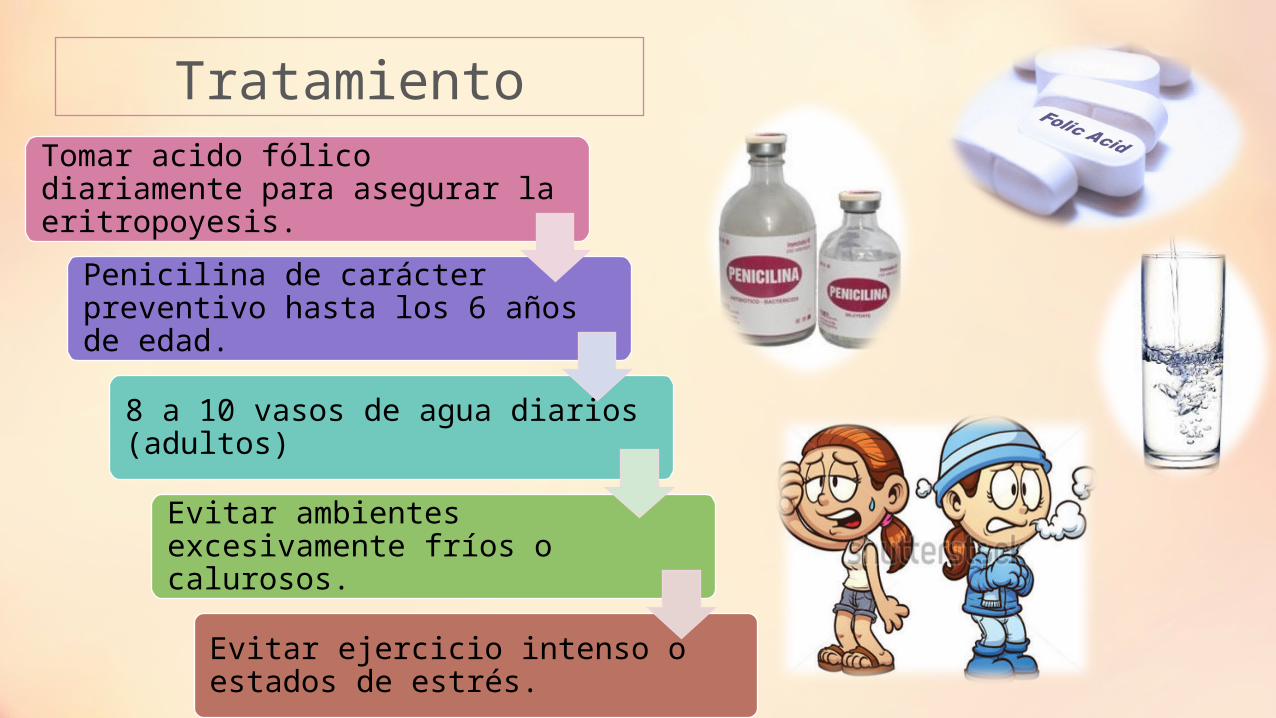
Tratamiento
Tomar acido fólico diariamente para asegurar la eritropoyesis.
Penicilina de carácter preventivo hasta los 6 años de edad.
8 a 10 vasos de agua diarios (adultos)
Evitar ambientes excesivamente fríos o calurosos.
Evitar ejercicio intenso o estados de estrés.

Hemoglobina C (HbC)• Prácticamente exclusiva de la raza negra.
• Mutación (G-A) del codón 6 del gen globina causando la sustitución del ácido glutámico por lisina.
• Alteración de la carga eléctrica superficial perdiendo la solubilidad y la Hb se cristaliza.
• Suele pasar desapercibida por su falta de expresividad clínica.

Manifestaciones Clínicas
HbC Homocigota• Sintomática• Anemia muy
moderada
HbC Heterocigota • Asintomática
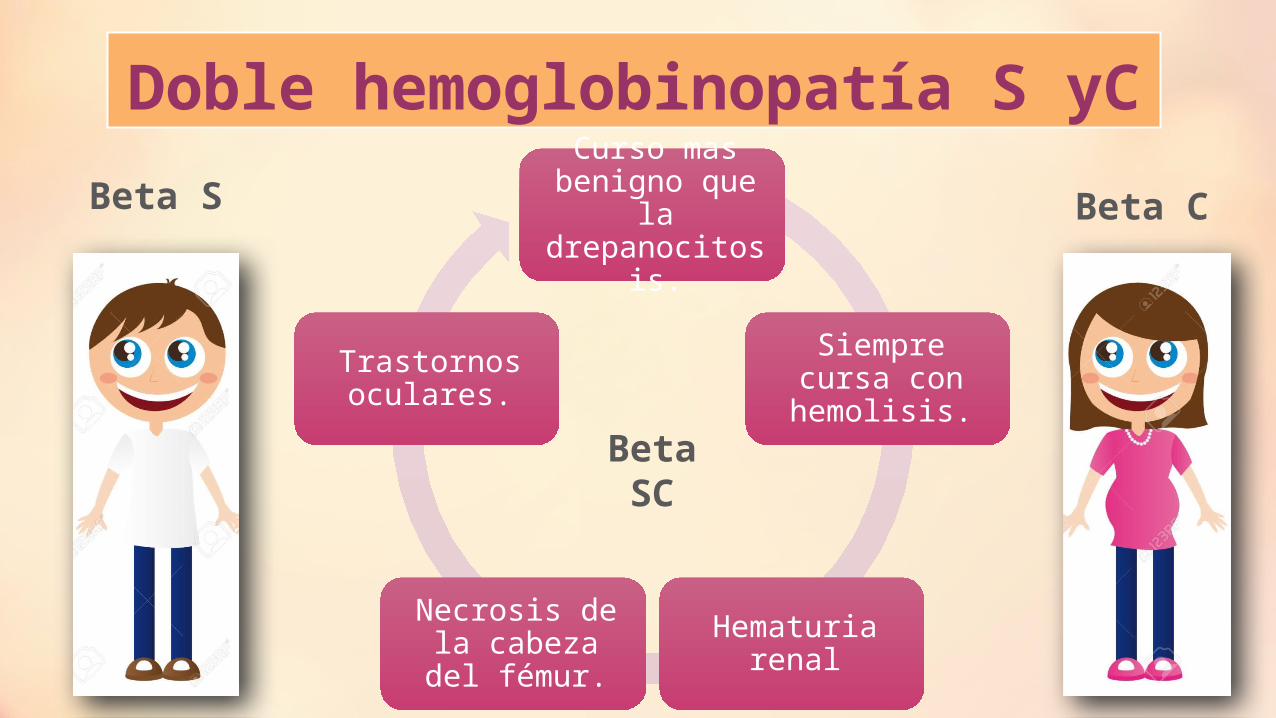
Doble hemoglobinopatía S yCCurso mas
benigno que la drepanocitosis.
Siempre cursa con hemolisis.
Hematuria renalNecrosis de la cabeza del fémur.
Trastornos oculares.
Beta S Beta C
Beta SC

Hemoglobinopatías Inestables
Homocigoto• Incompati-
blesHeterocigoto
• Sustituciones de aminoácidos en lugares críticos de la
molécula, baja de la solubilidad.
• Cuerpos de Heinz ( Hb precipitada y
desnaturalizada).
• Descritas mas de 150.

Mecanismo molecular y fisiopatología
Mutaciones en la cavidad hemo.
Sustitución de residuos internos con diferente grado de polaridad.
Mutaciones de la hélice a.
Alteraciones en las zonas de contacto alfa1 y beta1.
Precipitación de la Hb.

Mecanismo molecular y fisiopatología
Cuerpos de Heinz
Alteración morfológica
Dismin. La deformabilidad eritrocitaria
Hemólisis
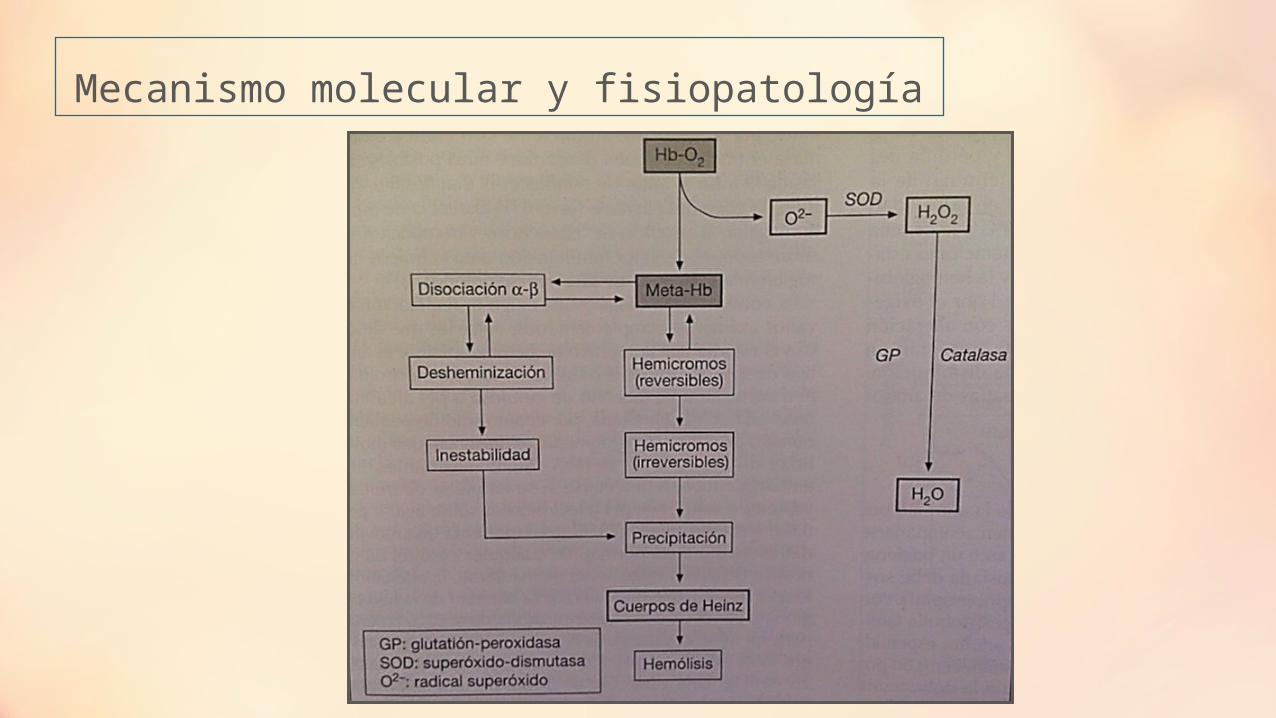
Mecanismo molecular y fisiopatología
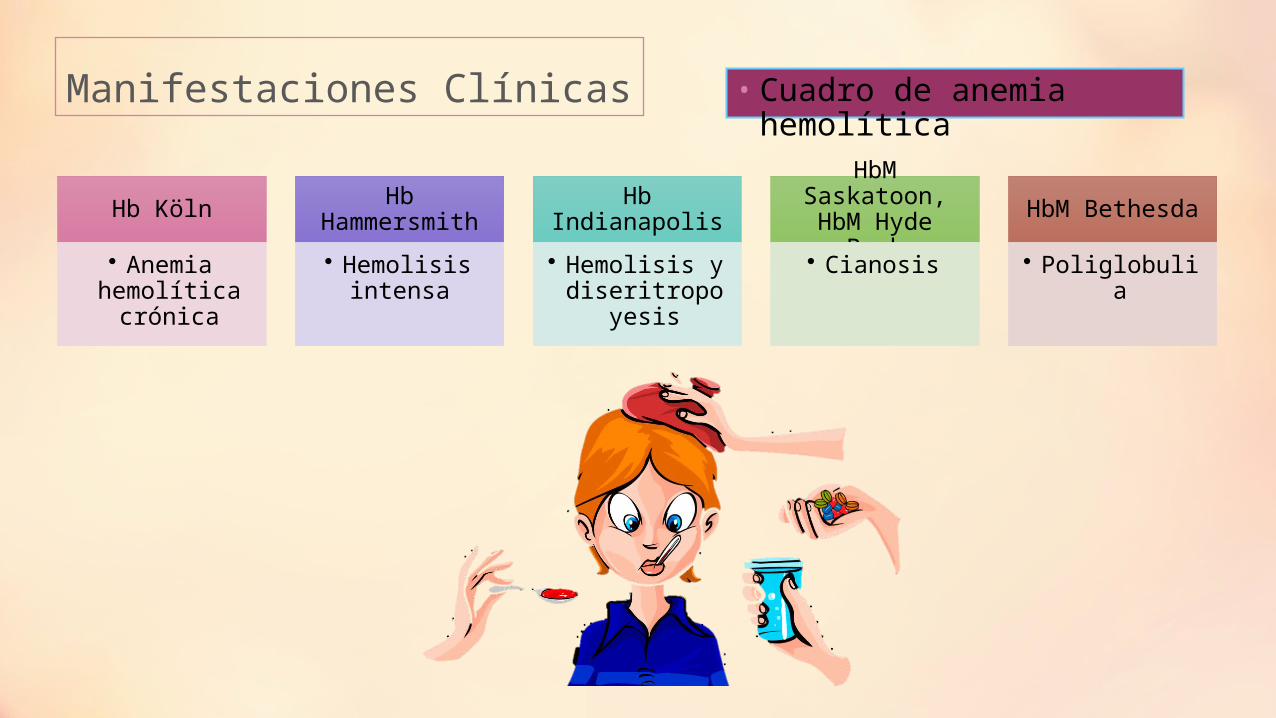
Manifestaciones Clínicas • Cuadro de anemia hemolítica
Hb Köln
• Anemia hemolítica
crónica
Hb Hammersmith
• Hemolisis intensa
Hb Indianapolis
• Hemolisis y diseritropoyesis
HbM Saskatoon, HbM Hyde Park
• Cianosis
HbM Bethesda
• Poliglobulia
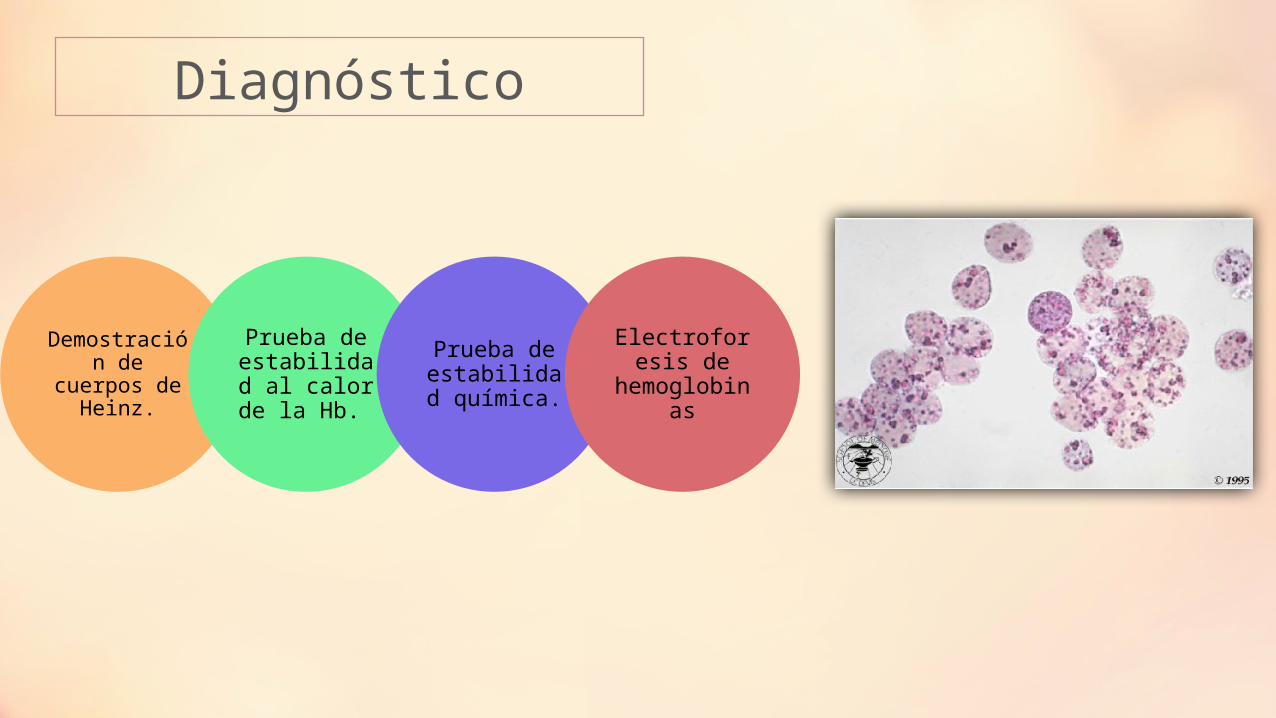
Diagnóstico
Demostración de cuerpos de
Heinz.
Prueba de estabilidad al calor de la Hb.
Prueba de estabilidad
química.
Electroforesis de
hemoglobinas
Tratamiento
La mayoría no requiere tratamiento.
Evitar medicamentos oxidantes.
Esplenectomía en anemia hemolítica intensa.

Hemoglobinopatías con alteración de la afinidad por el O2.
• Sólo se observan heterocigotos.
• Homocigotos son incompatibles con la vida.
• 1:1 HbA y patológica.
• Descritas mas de 118 variantes.
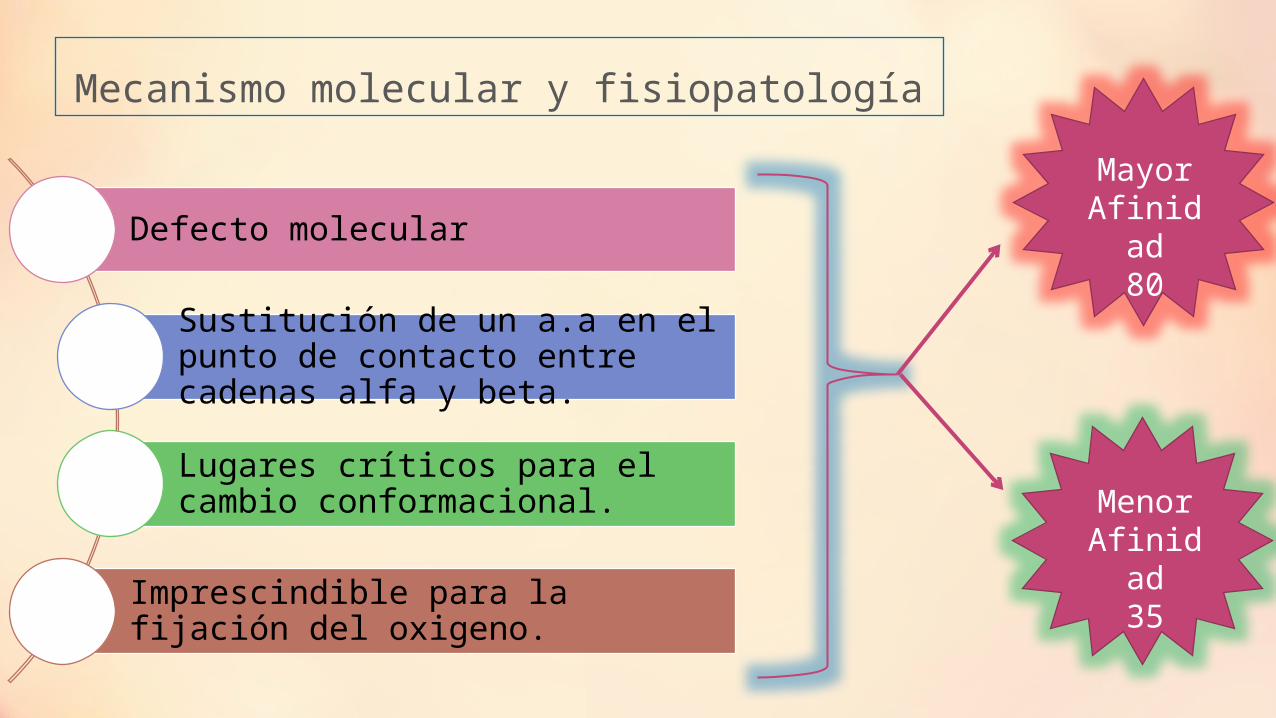
Mecanismo molecular y fisiopatología
Defecto molecular
Sustitución de un a.a en el punto de contacto entre cadenas alfa y beta.
Lugares críticos para el cambio conformacional.
Imprescindible para la fijación del oxigeno.
Mayor Afinidad
80
Menor Afinidad
35

Manifestaciones Clínicas
Afinidad por el O2
Aumento
Eritrocitosis
Diagnostico diferencial de
PV.
Disminución
Anemia
Cianosis
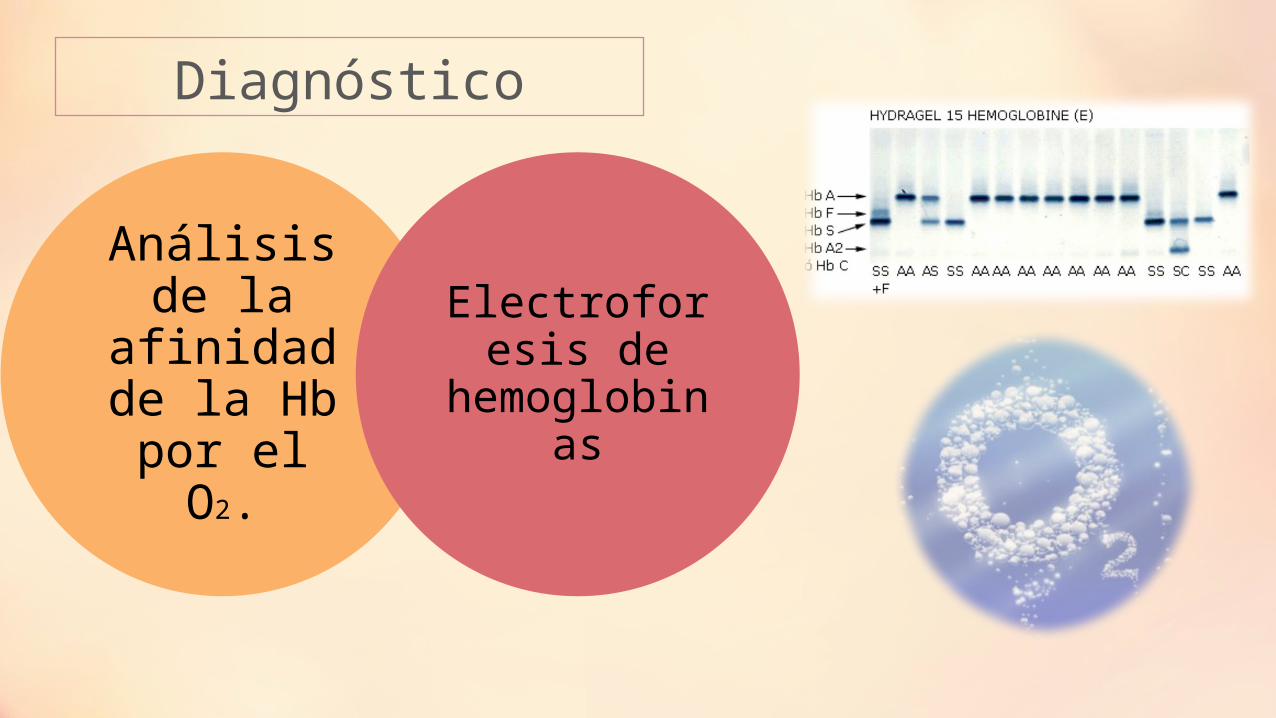
Diagnóstico
Análisis de la afinidad
de la Hb por el O2.
Electroforesis de
hemoglobinas

Tratamiento
Carácter benigno
En la mayoría de los casos no es necesario ningún tipo de tratamiento.
Sangrías sólo en caso de intensa poliglobulia que cause hiperviscosidad de la sangre.

Hemoglobinopatías M.
Puede afectar cadenas alfa o beta.
Preserva el hierro de 2 de los 4 gpos.
Hemo en estado férrico permanente.
Hb. Con oxigenación parcial
Parte de ellas se encuentra siempre
en estado desoxigenado.
Hb. Desoxigenada supera 50g/L la
sangre adquiere una tonalidad azulada.
Tx. Carece de tratamiento

Gracias.

Referencias
W. J. Williams: “Manual Williams de Hematología” 5 Ed. McGraw-Hill Interamericana. 2004.
ROSS. Histología, 4ª edición, Ed. Panamericana, 2005.
GENESER. Histología, 3ª edición, Ed. Médica Panamericana, 2000


















