
Functional Domains of Murine Cytomegalovirus Nuclear ...jvi.asm.org/content/80/1/73.full.pdfwas able...
12
JOURNAL OF VIROLOGY, Jan. 2006, p. 73–84 Vol. 80, No. 1 0022-538X/06/$08.000 doi:10.1128/JVI.80.1.73–84.2006 Copyright © 2006, American Society for Microbiology. All Rights Reserved. Functional Domains of Murine Cytomegalovirus Nuclear Egress Protein M53/p38† Mark Lo ¨tzerich, Zsolt Ruzsics, and Ulrich H. Koszinowski* Max von Pettenkofer Institut, Ludwig-Maximilians-Universita ¨t Mu ¨nchen, Pettenkoferstrasse 9a, 80336 Munich, Germany Received 11 May 2005/Accepted 3 October 2005 Two conserved herpes simplex virus 1 proteins, UL31 and UL34, form a complex at the inner nuclear membrane which governs primary envelopment and nuclear egress of the herpesvirus nucleocapsids. In mouse cytomegalovirus, a member of the betaherpesvirus subfamily, the homologous proteins M53/p38 and M50/p35 form the nuclear egress complex (NEC). Since the interaction of these proteins is essential for functionality, the definition of the mutual binding sites is a prerequisite for further analysis. Using a comprehensive random mutagenesis procedure, we have mapped the M53/p38 binding site of M50/p35 (A. Bubeck, M. Wagner, Z. Ruzsics, M. Lo ¨tzerich, M. Iglesias, I. R. Singh, and U. H. Koszinowski, J. Virol. 78:8026–8035). Here we describe a corresponding analysis for the UL31 homolog M53/p38. A total of 72 individual mutants were reinserted into the genome to test the complementation of the lethal M53 null phenotype. The mutants were also studied for colocalization and for coprecipitation with M50/p35. The analysis revealed that the noncon- served N-terminal one-third of M53/p38 provides the nuclear localization signal as an essential function. The collective results for many mutants localized the binding site for M50/p35 to amino acids (aa) 112 to 137. No single aa exchange for alanine could destroy NEC formation, but virus attenuation revealed a major role for aa K128, Y129, and L130. The lethal phenotype of several insertion and stop mutants indicated the functional importance of the C terminus of the protein. Lytic replication of the herpesviruses gives rise to the pro- duction of infectious virus particles as a result of an assembly process that starts in the nucleus of the infected cell, where the viral genomes are packaged into the nucleocapsid. Nucleocap- sid formation is followed by a complex process of nucleocapsid transitions through cellular membranes. First, the nucleocap- sid buds from the inner nuclear membrane (INM) into the space between the INM and the outer nuclear membrane (ONM). This process is termed primary envelopment. By pri- mary envelope fusion with the ONM, the nucleocapsids reach the cytoplasm. A secondary envelopment occurs at a not yet clearly defined compartment of the cellular exocytotic path- way. Finally, enveloped virus particles are released into the extracellular space (17, 18). Virus maturation events are controlled by multiprotein assem- blies which require the interaction of a number of viral and cel- lular proteins. Studies of alphaherpesviruses have shown that two conserved viral proteins, the prototypic UL31 and UL34 gene products, play a major role during primary envelopment (7, 8, 10, 25–27). In the betaherpesvirus mouse cytomegalovirus (MCMV), the products of the M53 and M50 genes (3, 7, 10, 19) have this function, and in the gammaherpesvirus Epstein-Barr virus, the BFRF1 and BFLF2 gene products (7, 10, 12) have this function. UL34 and related proteins are type II membrane proteins which, upon isolated expression, circulate in the contiguous membranes of the endoplasmic reticulum and the INM (UL34, M50, and BFRF1) unless the nuclear protein UL31 and UL31-related pro- teins (M53 and BFLF2) arrest the viral membrane protein in the INM. Apparently, complex formation by the two proteins is piv- otal for the nuclear egress of herpesvirus nucleocapsids. The nu- clear egress complex (NEC) executes this task by interacting with cellular proteins which are already residing in or recruited to the INM and presumably recruits the viral capsids to the egress sites. These interactions lead to the displacement of the rigid nuclear lamina along with nucleocapsid budding (7, 10). In herpes simplex virus type 1 (HSV-1), the interaction be- tween the UL34 and UL31 gene products is required for function. To study the function of the betaherpesvirus NEC in more detail, we started with an analysis of M50/p35 (3). M50/p35, an essential protein, is localized at the nuclear envelope in MCMV-infected cells and recruits its viral interaction partner M53/p38 and cellular protein kinase C to the INM. We identified the M53/p38 binding region of M50/p35 and showed that this interaction is essential for productive MCMV infection (3). Here we report the first mutational analysis of a herpesvirus UL31 family member, the MCMV M53 gene. Deletion of the M53 gene from the MCMV bacterial artificial chromosome (BAC) abolishes virus replication, and reintroduction of the M53 open reading frame (ORF) at an ectopic position in the genome rescues the null phenotype. By an improved random Tn7-based linker-scanning mutagenesis method (2), a library of M53 inser- tion mutants was generated. A representative set of M53 mu- tants was reinserted into M53-MCMV-BAC for functional analysis in the genomic context. A nuclear localization signal embedded in nonessential sequences was mapped to the N terminus of the protein. The sequence required for M50/p35 binding was localized to amino acids (aa) 107 to 136 of M53/ p38. Lethal insertions accumulated within the C-terminal two- thirds of M53/p38. None of the C-terminal truncation mutants * Corresponding author. Mailing address: Max von Pettenkofer In- stitut, Pettenkoferstrasse 9a, 80336 Munich, Germany. Phone: 49-89- 5160-5203. Fax: 49-89-5160-5292. E-mail: [email protected] .med.uni-muenchen.de. † Supplemental material for this article may be found at http://jvi .asm.org/. 73 on May 21, 2018 by guest http://jvi.asm.org/ Downloaded from
Transcript of Functional Domains of Murine Cytomegalovirus Nuclear ...jvi.asm.org/content/80/1/73.full.pdfwas able...

JOURNAL OF VIROLOGY, Jan. 2006, p. 73–84 Vol. 80, No. 10022-538X/06/$08.00�0 doi:10.1128/JVI.80.1.73–84.2006Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Functional Domains of Murine Cytomegalovirus Nuclear EgressProtein M53/p38†
Mark Lotzerich, Zsolt Ruzsics, and Ulrich H. Koszinowski*
Max von Pettenkofer Institut, Ludwig-Maximilians-Universitat Munchen, Pettenkoferstrasse 9a, 80336 Munich, Germany
Received 11 May 2005/Accepted 3 October 2005
Two conserved herpes simplex virus 1 proteins, UL31 and UL34, form a complex at the inner nuclearmembrane which governs primary envelopment and nuclear egress of the herpesvirus nucleocapsids. In mousecytomegalovirus, a member of the betaherpesvirus subfamily, the homologous proteins M53/p38 and M50/p35form the nuclear egress complex (NEC). Since the interaction of these proteins is essential for functionality,the definition of the mutual binding sites is a prerequisite for further analysis. Using a comprehensive randommutagenesis procedure, we have mapped the M53/p38 binding site of M50/p35 (A. Bubeck, M. Wagner, Z.Ruzsics, M. Lotzerich, M. Iglesias, I. R. Singh, and U. H. Koszinowski, J. Virol. 78:8026–8035). Here wedescribe a corresponding analysis for the UL31 homolog M53/p38. A total of 72 individual mutants werereinserted into the genome to test the complementation of the lethal M53 null phenotype. The mutants werealso studied for colocalization and for coprecipitation with M50/p35. The analysis revealed that the noncon-served N-terminal one-third of M53/p38 provides the nuclear localization signal as an essential function. Thecollective results for many mutants localized the binding site for M50/p35 to amino acids (aa) 112 to 137. Nosingle aa exchange for alanine could destroy NEC formation, but virus attenuation revealed a major role foraa K128, Y129, and L130. The lethal phenotype of several insertion and stop mutants indicated the functionalimportance of the C terminus of the protein.
Lytic replication of the herpesviruses gives rise to the pro-duction of infectious virus particles as a result of an assemblyprocess that starts in the nucleus of the infected cell, where theviral genomes are packaged into the nucleocapsid. Nucleocap-sid formation is followed by a complex process of nucleocapsidtransitions through cellular membranes. First, the nucleocap-sid buds from the inner nuclear membrane (INM) into thespace between the INM and the outer nuclear membrane(ONM). This process is termed primary envelopment. By pri-mary envelope fusion with the ONM, the nucleocapsids reachthe cytoplasm. A secondary envelopment occurs at a not yetclearly defined compartment of the cellular exocytotic path-way. Finally, enveloped virus particles are released into theextracellular space (17, 18).
Virus maturation events are controlled by multiprotein assem-blies which require the interaction of a number of viral and cel-lular proteins. Studies of alphaherpesviruses have shown that twoconserved viral proteins, the prototypic UL31 and UL34 geneproducts, play a major role during primary envelopment (7, 8, 10,25–27). In the betaherpesvirus mouse cytomegalovirus (MCMV),the products of the M53 and M50 genes (3, 7, 10, 19) have thisfunction, and in the gammaherpesvirus Epstein-Barr virus, theBFRF1 and BFLF2 gene products (7, 10, 12) have this function.UL34 and related proteins are type II membrane proteins which,upon isolated expression, circulate in the contiguous membranesof the endoplasmic reticulum and the INM (UL34, M50, and
BFRF1) unless the nuclear protein UL31 and UL31-related pro-teins (M53 and BFLF2) arrest the viral membrane protein in theINM. Apparently, complex formation by the two proteins is piv-otal for the nuclear egress of herpesvirus nucleocapsids. The nu-clear egress complex (NEC) executes this task by interacting withcellular proteins which are already residing in or recruited to theINM and presumably recruits the viral capsids to the egress sites.These interactions lead to the displacement of the rigid nuclearlamina along with nucleocapsid budding (7, 10).
In herpes simplex virus type 1 (HSV-1), the interaction be-tween the UL34 and UL31 gene products is required for function.To study the function of the betaherpesvirus NEC in more detail,we started with an analysis of M50/p35 (3). M50/p35, an essentialprotein, is localized at the nuclear envelope in MCMV-infectedcells and recruits its viral interaction partner M53/p38 and cellularprotein kinase C to the INM. We identified the M53/p38 bindingregion of M50/p35 and showed that this interaction is essential forproductive MCMV infection (3).
Here we report the first mutational analysis of a herpesvirusUL31 family member, the MCMV M53 gene. Deletion of theM53 gene from the MCMV bacterial artificial chromosome(BAC) abolishes virus replication, and reintroduction of the M53open reading frame (ORF) at an ectopic position in the genomerescues the null phenotype. By an improved random Tn7-basedlinker-scanning mutagenesis method (2), a library of M53 inser-tion mutants was generated. A representative set of M53 mu-tants was reinserted into �M53-MCMV-BAC for functionalanalysis in the genomic context. A nuclear localization signalembedded in nonessential sequences was mapped to the Nterminus of the protein. The sequence required for M50/p35binding was localized to amino acids (aa) 107 to 136 of M53/p38. Lethal insertions accumulated within the C-terminal two-thirds of M53/p38. None of the C-terminal truncation mutants
* Corresponding author. Mailing address: Max von Pettenkofer In-stitut, Pettenkoferstrasse 9a, 80336 Munich, Germany. Phone: 49-89-5160-5203. Fax: 49-89-5160-5292. E-mail: [email protected].
† Supplemental material for this article may be found at http://jvi.asm.org/.
73
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

was able to rescue the M53 null phenotype, underscoring thefunctional importance of this part of the protein.
MATERIALS AND METHODS
Cells and viruses. NIH 3T3 murine fibroblasts (ATCC CRL 1658), M2-10B4cells (a bone marrow stromal cell line [ATCC CRL 1972]), human 293 cells(ATCC CRL 1573), and mouse embryonic fibroblasts (MEF) were propagated asdescribed previously (15). The wild-type (wt) MCMV Smith strain was recoveredfrom pSM3fr-BAC (32). All mutant viruses were derived from pSM3fr-16FRT17-BAC carrying an FRT site between the m16 and m17 genes. Thegrowth properties of 16FRT17 MCMV are identical to those of wt MCMV bothin vitro and in vivo (4). The wt and mutant MCMV BACs were reconstituted toviruses by the transfection of MEF, using Superfect transfection reagent (QIAGEN)according to the manufacturer’s instructions. Reconstituted viruses were prop-agated on M2-10B4 cells (15) and titrated by a plaque assay (23).
Virus infection. NIH 3T3 fibroblasts were infected with wt MCMV at a mul-tiplicity of infection (MOI) of 0.5 to determine the expression kinetics of M53/p38. To block DNA replication, phosphonoacetic acid (PAA; Sigma) was used ata concentration of 300 �g/ml as described previously (9). To compare the growthof wt and mutant viruses, NIH 3T3 fibroblasts were infected at a MOI of 0.1.Supernatants of the infected cultures were collected daily for 5 days, and thereleased viruses were quantified by a plaque assay.
Plasmids. The M53 ORF was cloned into the pOriR6K-zeo-ie rescue vector(3) from pCR3-M53 (3), using KpnI and NotI, resulting in pOriR6K-zeo-ie-M53,which was used as a wt M53 rescue and expression vector in this study. pOriR6K-zeo-ie-STM50, which was used for pull-down analysis, was constructed as follows.The synthetic oligonucleotides ieST1 and ieST2 were annealed and inserted intopOriR6K-zeo-ie by the use of ApaI and XhoI, resulting in pOriR6K-zeo-ie-ST.The M50 ORF was amplified with the AB6-02 and M50Strep primers and clonedinto pOriR6K-zeo-ie-ST, using KpnI and XhoI.
To generate N-terminal M53 deletion mutants lacking aa 16 to 106, pOriR6K-zeo-ie-M53 was amplified by inverse touchdown PCR (20) using the 5�-SapI-delN and 3�-Ndel-SapI primers, which carried a nuclear localization sequence(NLS) derived from the simian virus 40 (SV40) large T antigen. The PCRproduct was treated with SapI and religated, generating the pM53-�16-106NLSconstruct, in which the deleted sequence was replaced with the SV40 NLS. TheNLS sequence was removed from pM53-�16-106NLS by AgeI digestion, andreligation generated the pM53-�16-106 construct carrying only the 16-106 dele-tion. An N-terminal M53 deletion mutant lacking aa 16 to 136 was generated byinsertion of the NLS21 and NLS22 annealed synthetic oligonucleotides intoAgeI- and BspHI-treated pM53-�16-106NLS, resulting in pM53-�16-136NLS.
Point mutations were introduced into the M53 ORF by mutated overlappingprimers (see Table S1 in the supplemental material for a primer list), as follows.The M53 ORF was amplified by PCRs using AB6-02 and reverse mutagenesisprimers as well as AB7-02 and forward mutagenesis primers, thereby generating5� and 3� M53 fragments, respectively. The 5� fragments were digested withKpnI/SapI, and the 3� fragments were digested with SapI/MluI; the fragmentswere then inserted into KpnI- and MluI-treated pOriR6K-zeo-ie-M53 in a one-step reaction.
An M53-green fluorescent protein (GFP) fusion protein was generated asfollows. The mutant M53-s106 ORF was amplified by a PCR using primersAB6-SpAs and AB7-02. The PCR product was inserted into Litmus28 (NEB)with BamHI and SpeI. The subcloned wt and mutant M53-s106 fragment wasthen isolated by SpeI and PmeI digestion and inserted into NheI- and SmaI-treated pEGFPN1 (Clontech).
Linker-scanning mutagenesis of the M53 ORF. For the mutagenesis proce-dure, the M53 ORF was cloned from pCR3-M53 (3) into Litmus28 (NEB) byusing KpnI and XbaI, generating pL-M53, which was used as a target vector fortransposon mutagenesis. pST76K-S4, a new transposon donor vector with atemperature-sensitive origin of replication, was generated by inserting a mini-transposon carrying the KpnI/SacI fragment of pGPS-4 (NEB) into pST76K (21).pLit28-M53 was subjected to in vitro random transposon insertion mutagenesisusing TnsABC* transposase (NEB) and pST76K-S4 as the donor according tothe manufacturer’s instructions. Escherichia coli DH10B cells (Invitrogen) weretransformed with the transposition mixture, and insertion mutants were selectedby chloramphenicol and ampicillin at 43°C to remove both the intact acceptorand donor plasmids. Plasmid DNAs were prepared from this primary pool ofinsertion mutants, and the 1,084-bp KpnI/NsiI fragments containing the M53ORF and the randomly inserted mini-transposons were isolated and reclonedinto the KpnI/NsiI-treated pOriR6K-zeo-ie rescue plasmid. Recombinants con-taining the M53 ORF with the mini-transposon insertions were selected bychloramphenicol and Zeocin. Plasmid DNAs were prepared from this secondary
pool of insertion mutants, and the mini-transposon was removed by PmeI diges-tion and religation, generating a pOriR6K-zeo-ie-M53mut insertion librarywhich was maintained in E. coli PIR1 (Invitrogen). Insertion sites were identifiedfor selected clones by PCR screening and sequencing as described previously (3).
Generation and reconstitution of recombinant MCMV BACs. �M53-BAC wasgenerated on the basis of pSM3fr-16FRT17 (4). A linear recombination frag-ment carrying a kanamycin resistance marker was generated by PCR with apACYC177 template (NEB) by using the 5�-M53del and 3�-M53del primers. TheM53 ORF (nucleotide positions 78461 to 79459 of MCMV strain Smith, accord-ing to Rawlinson et al. [22]) was deleted from pSM3fr-16FRT17 by ET recom-bination in E. coli with this linear recombination fragment, as described previ-ously (33). The wt and mutant rescue plasmids were inserted into �M53-BAC atthe FRT site as described previously (3). For virus reconstitution, semiconfluentMEF in 6-cm dishes were transfected with 1.5 �g of purified DNA. Two inde-pendent BAC clones were always transfected in two replicates. Twenty-fourhours after transfection, cells were replated onto 10-cm dishes and then refedweekly. Cultures were inspected for 6 weeks after transfection. As a control,pM53E DNA was transfected, and a reconstitution experiment was consideredvalid when pM53E DNA-derived virus plaques occurred during the second weekposttransfection.
M53 transcomplementation. To create an M53/p38 complementing cell line,the M53 ORF from pOriR6K-zeo-ie-M53 was isolated by BsaI and SpeI andinserted into the PvuII- and NheI-cleaved pTRE2Hyg vector (Clontech), result-ing in pTRE-M53. Stable NIH 3T3 transfectants harboring pTRE-M53 wereselected according to the manufacturer’s instructions. Thirty-two independentcell clones were expanded and designated NT/M53-1 to -32.
An artificial transcription unit expressing tTA under the control of the HCMVimmediate-early promoter was cloned into pOriR6K-zeo-ie by using SpeI andNotI, generating pO6-tTA, which was inserted into pSM3fr-16FRT17 by the Flpsystem in E. coli (3), resulting in pm16-17FRT-tTA. Subsequently, the M53 genewas deleted from m16-17FRT-tTA by ET recombination using the recombina-tion cassette for M53 deletion described above, generating �M53-tTA-BAC. Toreconstitute the �M53-tTA virus, eight of the NT/M53 cell clones were trans-fected with �M53-tTA-BAC. After the appearance of a significant cytopathiceffect, the culture supernatants were harvested and pooled. The NT/M53 cellclones were tested for productivity, using the pooled supernatant for infection,and NT/M53-12 was chosen for the propagation of both m16-17FRT-tTA and�M53-tTA. The resulting virus stocks were titrated on NT/M53-12 by the tissueculture infective dose (TCID) method (15).
To determine virus production under multistep growth conditions, NIH 3T3cells were infected with either �M53-tTA or m16-17FRT-tTA at an MOI of 0.1.Supernatants of the infected cells were harvested at day 5 postinfection, and thereleased infectious units were determined by the 50% TCID (TCID50) method,using NT/M53-12 cells. To determine virus production under single-step growthconditions, NIH 3T3 cells were infected with either �M53-tTA or m16-17FRT-tTA at an MOI of 3. Supernatants of the infected cells were harvested at day 3postinfection, and the released infectious units were determined by the TCID50
method, using NT/M53-12 cells.Metabolic labeling and coimmunoprecipitation. Subconfluent 293 cells in
6-cm dishes were cotransfected with pOriR6K-zeo-ie-M53 or pOriR6k-zeo-ie-M53mut and with 3.5 �g of the pOriR6K-zeo-ie-M50 or pOriR6K-zeo-ie vector(3) by Ca2PO4 precipitation (29). Twenty-four hours after transfection, the newlysynthesized proteins were labeled with 300 �Ci/ml [35S]methionine-cysteine(Promix; Amersham Bioscience) for 1 hour. The cells were lysed, and coimmu-noprecipitation was carried out as described previously (3). An M50/p35-specificpolyclonal rabbit antiserum (19) and protein A-Sepharose (Amersham Bio-sciences) were used to precipitate the M50/p35-specific complexes. M53/p38 wasprecipitated on protein G-Sepharose beads (Amersham Biosciences) with aspecific rat polyclonal antiserum raised against a synthetic peptide representingthe 15 N-terminal amino acids of M53/p38 (3).
Strep tag pull-down assay. Subconfluent 293 cells in 6-cm dishes were cotrans-fected with 3.5 �g of the construct pOriR6K-zeo-ie-STM50, expressing Strep-tagged M50/p35, and 3.5 �g of plasmid pOriR6K-zeo-ie-M53 or pOriR6k-zeo-ie-M53mut by Ca2PO4 precipitation (29). Twenty-four hours after transfection,the cells were washed with phosphate-buffered saline, scratched from the plates,and resuspended in phosphate-buffered saline. Five percent of the cell suspen-sion was lysed directly in loading buffer (62.5 mM Tris, pH 6.8, 2% [vol/vol]sodium dodecyl sulfate [SDS], 10% [vol/vol] glycerol, 6 M urea, 5% [vol/vol]�-mercaptoethanol, 0.01% [wt/vol] bromophenol blue, 0.01% [wt/vol] phenolred), and the samples were separated by SDS-polyacrylamide gel electrophoresis(SDS-PAGE) to determine the total protein load. The rest of the cell suspensionwas collected by centrifugation, and pellets were lysed in hypotonic buffer (10mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2) by sonication. The nuclei were
74 LOTZERICH ET AL. J. VIROL.
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
collected by centrifugation and treated with M-Per mammalian protein extrac-tion reagent (Pierce). Strep-Tactin Sepharose (IBA) was used to pull downStrep-tagged M50/p35 complexes according to the manufacturer’s instructions.Desthiobiotin (IBA)-eluted samples were separated by SDS-PAGE. The pro-teins were transferred from the gels onto Hybond-P membranes (AmershamBiosciences) in the presence of blotting buffer (25 mM Tris, 192 mM glycine,20% [vol/vol] methanol, pH 8.3). Membranes were blocked in TBS-T (Tris-buffered saline, 0.05% Tween 20) containing 5% nonfat dry milk overnight at4°C. To detect M53/p38, the membrane was incubated at room temperature withTBS-T containing M53/p38-specific polyclonal rat antiserum (3). Membraneswere washed with TBS-T and incubated with the appropriate horseradish per-oxidase-conjugated secondary antibody (Dianova). The proteins were visualizedwith an ECL-Plus Western blot detection system (Amersham Biosciences).
Confocal laser scanning microscopy. Transfected NIH 3T3 cells were grownon glass coverslips and fixed as previously described (19). M53/p38 was visualizedwith a specific polyclonal rat antiserum as the primary antibody and fluoresceinisothiocyanate-conjugated goat anti-rat immunoglobulin G (Dianova) as thesecondary antibody. M50/p35 was visualized with a polyclonal rabbit antiserumas the primary antibody and Texas red-conjugated donkey anti-rabbit immuno-globulin G (Dianova) as the secondary antibody.
RESULTS
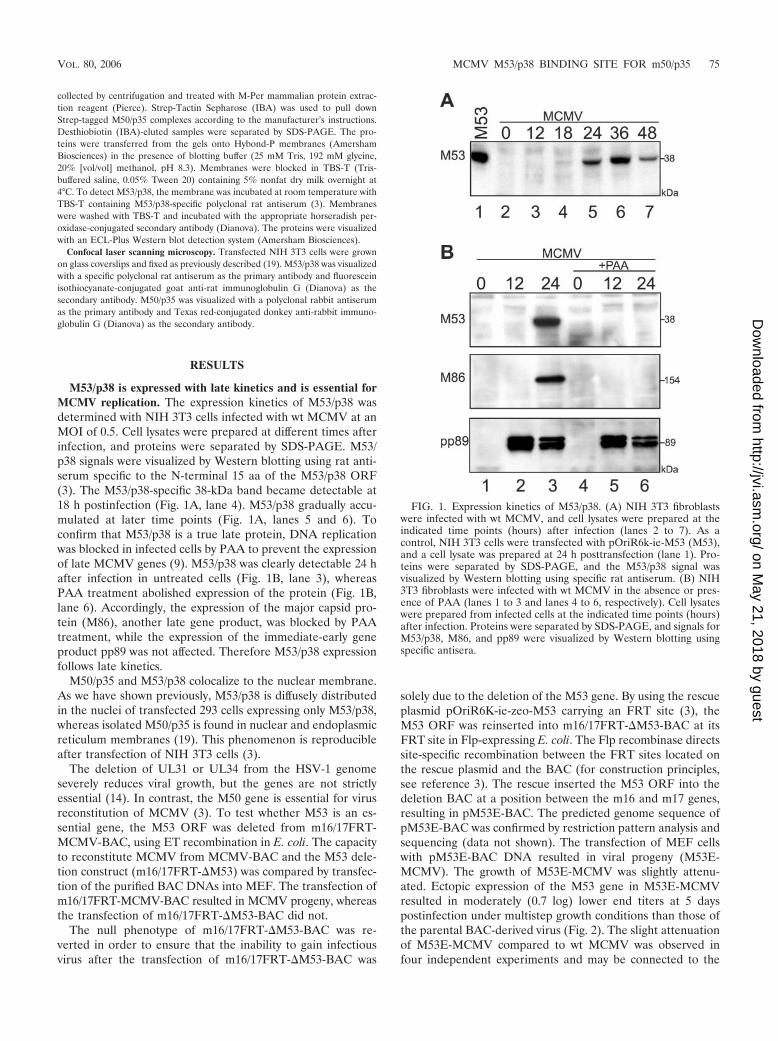
M53/p38 is expressed with late kinetics and is essential forMCMV replication. The expression kinetics of M53/p38 wasdetermined with NIH 3T3 cells infected with wt MCMV at anMOI of 0.5. Cell lysates were prepared at different times afterinfection, and proteins were separated by SDS-PAGE. M53/p38 signals were visualized by Western blotting using rat anti-serum specific to the N-terminal 15 aa of the M53/p38 ORF(3). The M53/p38-specific 38-kDa band became detectable at18 h postinfection (Fig. 1A, lane 4). M53/p38 gradually accu-mulated at later time points (Fig. 1A, lanes 5 and 6). Toconfirm that M53/p38 is a true late protein, DNA replicationwas blocked in infected cells by PAA to prevent the expressionof late MCMV genes (9). M53/p38 was clearly detectable 24 hafter infection in untreated cells (Fig. 1B, lane 3), whereasPAA treatment abolished expression of the protein (Fig. 1B,lane 6). Accordingly, the expression of the major capsid pro-tein (M86), another late gene product, was blocked by PAAtreatment, while the expression of the immediate-early geneproduct pp89 was not affected. Therefore M53/p38 expressionfollows late kinetics.
M50/p35 and M53/p38 colocalize to the nuclear membrane.As we have shown previously, M53/p38 is diffusely distributedin the nuclei of transfected 293 cells expressing only M53/p38,whereas isolated M50/p35 is found in nuclear and endoplasmicreticulum membranes (19). This phenomenon is reproducibleafter transfection of NIH 3T3 cells (3).
The deletion of UL31 or UL34 from the HSV-1 genomeseverely reduces viral growth, but the genes are not strictlyessential (14). In contrast, the M50 gene is essential for virusreconstitution of MCMV (3). To test whether M53 is an es-sential gene, the M53 ORF was deleted from m16/17FRT-MCMV-BAC, using ET recombination in E. coli. The capacityto reconstitute MCMV from MCMV-BAC and the M53 dele-tion construct (m16/17FRT-�M53) was compared by transfec-tion of the purified BAC DNAs into MEF. The transfection ofm16/17FRT-MCMV-BAC resulted in MCMV progeny, whereasthe transfection of m16/17FRT-�M53-BAC did not.
The null phenotype of m16/17FRT-�M53-BAC was re-verted in order to ensure that the inability to gain infectiousvirus after the transfection of m16/17FRT-�M53-BAC was
solely due to the deletion of the M53 gene. By using the rescueplasmid pOriR6K-ie-zeo-M53 carrying an FRT site (3), theM53 ORF was reinserted into m16/17FRT-�M53-BAC at itsFRT site in Flp-expressing E. coli. The Flp recombinase directssite-specific recombination between the FRT sites located onthe rescue plasmid and the BAC (for construction principles,see reference 3). The rescue inserted the M53 ORF into thedeletion BAC at a position between the m16 and m17 genes,resulting in pM53E-BAC. The predicted genome sequence ofpM53E-BAC was confirmed by restriction pattern analysis andsequencing (data not shown). The transfection of MEF cellswith pM53E-BAC DNA resulted in viral progeny (M53E-MCMV). The growth of M53E-MCMV was slightly attenu-ated. Ectopic expression of the M53 gene in M53E-MCMVresulted in moderately (0.7 log) lower end titers at 5 dayspostinfection under multistep growth conditions than those ofthe parental BAC-derived virus (Fig. 2). The slight attenuationof M53E-MCMV compared to wt MCMV was observed infour independent experiments and may be connected to the
FIG. 1. Expression kinetics of M53/p38. (A) NIH 3T3 fibroblastswere infected with wt MCMV, and cell lysates were prepared at theindicated time points (hours) after infection (lanes 2 to 7). As acontrol, NIH 3T3 cells were transfected with pOriR6k-ie-M53 (M53),and a cell lysate was prepared at 24 h posttransfection (lane 1). Pro-teins were separated by SDS-PAGE, and the M53/p38 signal wasvisualized by Western blotting using specific rat antiserum. (B) NIH3T3 fibroblasts were infected with wt MCMV in the absence or pres-ence of PAA (lanes 1 to 3 and lanes 4 to 6, respectively). Cell lysateswere prepared from infected cells at the indicated time points (hours)after infection. Proteins were separated by SDS-PAGE, and signals forM53/p38, M86, and pp89 were visualized by Western blotting usingspecific antisera.
VOL. 80, 2006 MCMV M53/p38 BINDING SITE FOR m50/p35 75
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

altered expression kinetics of ectopic M53/p38. Thus, the M53gene is essential for MCMV growth, and the M53 null pheno-type could be reverted by ectopic expression of the M53 ORF.
In order to trans-complement the M53 deletion, NIH 3T3-derived cell lines were generated that harbored the M53 ORFunder the control of the artificial TRE promoter (NT/M53clones). The TRE promoter is practically silent in mammaliancells in the absence of its fusion transactivator tTA, allowingthe establishment of stable cell lines even if the expression ofthe gene of interest would be deleterious for the host cell (11).A tTA expression cassette was introduced into m16/17FRT-MCMV-BAC, generating m16/17FRT-tTA, which possessesall essential functions of MCMV and expresses the tTA pro-tein constitutively. Subsequently, the M53 gene was deletedfrom this recombinant, as described above for m16/17FRT-�M53, resulting in �M53-tTA-BAC, which lacks the M53 genebut expresses tTA. m16/17FRT-tTA and �M53-tTA-BAC werereconstituted by transfecting different NT/M53 cell clones. Bothwere able to produce infectious viruses, which were then prop-agated on the complementing cell line NT/M53-12. �M53-tTAand m16/17FRT-tTA were tested for viability under bothmulti- and single-step growth conditions, using noncomple-menting NIH 3T3 and complementing NT/M53 cells. �M53-tTA was unable to produce progeny under noncomplementingconditions at both low and high MOIs (Table 1), confirmingthat M53 is an essential MCMV gene.
Construction of an insertion mutant library of M53 ORFsand analysis of the mutants in the context of the MCMVgenome. A library of M53 insertion mutants was generated.The M53 ORF was first subcloned into the Litmus28 vector,which served as the acceptor for Tn7-based transposon-medi-ated mutagenesis (2). We constructed a new transposon donorplasmid, which carries the Tn7-derived mini-transposon and aconditional origin of replication to allow donor plasmid elim-ination after the transposition reaction at a nonpermissive tem-perature (21). After in vitro transposition, acceptor plasmidscarrying the mini-transposon were selected and purified as apool. The M53 ORF, which was increased in size due to theinserted transposon, was isolated and inserted into the rescueplasmid pOriR6K-ie-zeo. PmeI digestion removed the mini-transposon, and subsequent religation resulted in the insertion
of 5 aa or a stop codon into the coding sequence. From thelibrary of 28,000 pOriR6K-ie-zeo-M53 mutants, 986 indepen-dent clones were screened by PCR, and 498 plasmids with aninsertion in the M53 ORF were sequenced. Depending on theprimary insertion site in the codon, M53 readthrough andtruncation mutants were found, some of which were mutatedat the same amino acid position. From this set, a total of 46in-frame insertion mutants, representing a distribution of aboutone insertion per 10 to 15 aa, and 8 stop mutants were selectedfor the complementation assays and were reinserted one byone into m16/17FRT-�M53-BAC.
Using the AlignX program (Invitrogen), the aa sequences ofa total of 36 members of the UL31 family (accession numberPF02718) (1; www.sanger.ac.uk/Software/Pfam) were aligned(Fig. 3). The average similarity between betaherpesvirus UL31members was 47.5%, with an average of 25.1% within thewhole family. However, the homology was not equally distrib-uted along the sequences. A variable N-terminal region and aconserved region including the central and C-terminal two-thirds of the protein were identified, with a similarity of 84.6%among the betaherpesviruses and of 44% for the whole family.Furthermore, when the entire UL31 family was aligned, thesimilarity plot indicated four peaks along the conserved region,which may reflect strictly conserved functional domains andwere designated conserved regions 1 to 4 (CR1 to CR4). Weassumed that, according to the sequence conservation, lethalmutations should accumulate in the conserved central andC-terminal regions (corresponding to aa 115 to 333 of M53/p38) rather than in the variable N-terminal region (corre-sponding to aa 1 to 114 of M53/p38). We expected to find thebinding site(s) for M50/p35 within one of the four conservedregions.
First, the ability of the selected mutant set to rescue theM53 null phenotype was tested. To this end, �M53-MCMV-BAC with reinserted M53 mutants was transfected intoMEF. The results of the virus reconstitution screen aresummarized in Fig. 3B. All in-frame insertions within theN-terminal part of the M53 ORF (aa 1 to 114) gave rise toviral progeny. In contrast, 32 of 37 insertion mutations in thecentral- and C-terminal domains caused a lethal phenotype.The clusters of lethal mutants are interspersed with singleviable insertion mutants. The 14 viable insertion mutantsshowed viral plaques comparable to those of the MCMV-M53E virus. A set of eight truncation mutants was chosen totest the essentiality of the four conserved sequence clusters.All truncation mutants turned out to be nonfunctional. Al-together, loss-of-function mutants accumulated in the con-
FIG. 2. Ectopic rescue of M53 deletion mutant. The growth kinet-ics of the viruses derived from pSMfr3 (wt) or pM53E (M53E) weredetermined on NIH 3T3 cells. Subconfluent cultures were infectedwith the respective viruses. Supernatants of the infected cells wereharvested on the indicated days, and the infectious progeny was quan-tified by a plaque assay.
TABLE 1. Growth of trans-complemented M53 deletion virusunder noncomplementing conditions
Virus Virus stocktiter a
Titer on NIH 3T3 cellsat 5 days postinfection
MOI of 0.1 MOI of 3.0
�M53-tTA 0.8 � 107 �101 �101
m16/17FRT-tTA 2.0 � 107 2.4 � 105 6.2 � 106
a The �M53-tTA and m16/17FRT-tTA virus stocks were titrated on the NT/M53-12 cell line by the TCID50 method. The titers of wt MCMV determined bythe TCID50 method are 0.5 to 1 log higher than the titers determined by thestandard plaque assay using MEF.
76 LOTZERICH ET AL. J. VIROL.
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
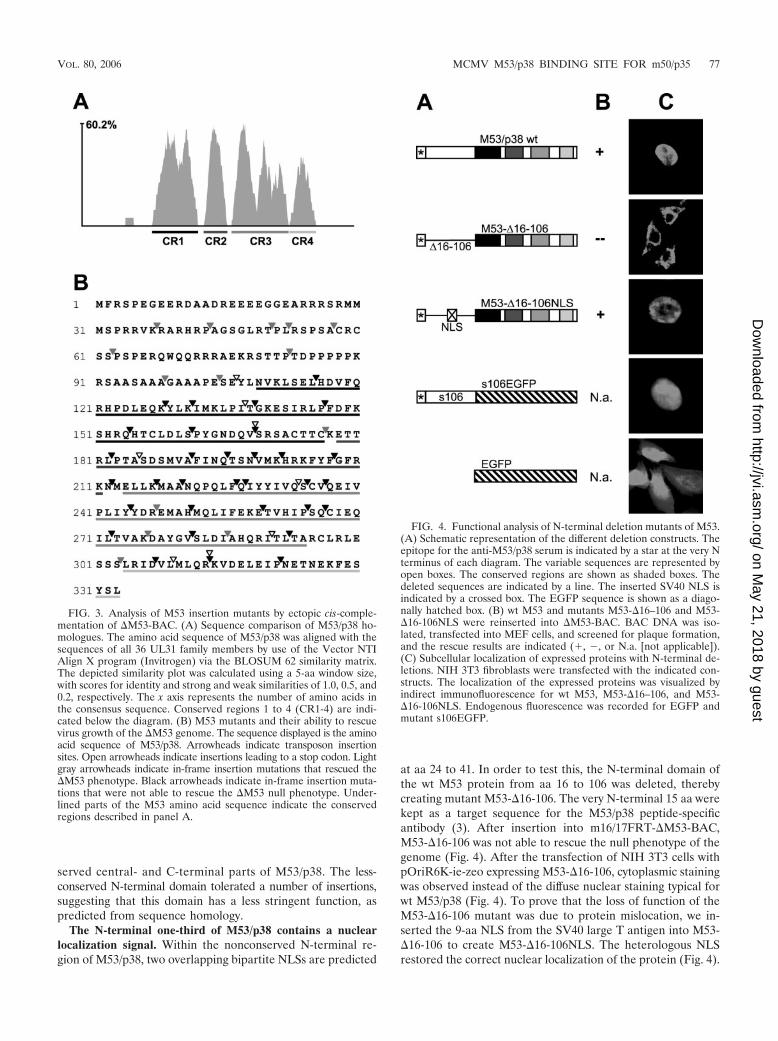
served central- and C-terminal parts of M53/p38. The less-conserved N-terminal domain tolerated a number of insertions,suggesting that this domain has a less stringent function, aspredicted from sequence homology.
The N-terminal one-third of M53/p38 contains a nuclearlocalization signal. Within the nonconserved N-terminal re-gion of M53/p38, two overlapping bipartite NLSs are predicted
at aa 24 to 41. In order to test this, the N-terminal domain ofthe wt M53 protein from aa 16 to 106 was deleted, therebycreating mutant M53-�16-106. The very N-terminal 15 aa werekept as a target sequence for the M53/p38 peptide-specificantibody (3). After insertion into m16/17FRT-�M53-BAC,M53-�16-106 was not able to rescue the null phenotype of thegenome (Fig. 4). After the transfection of NIH 3T3 cells withpOriR6K-ie-zeo expressing M53-�16-106, cytoplasmic stainingwas observed instead of the diffuse nuclear staining typical forwt M53/p38 (Fig. 4). To prove that the loss of function of theM53-�16-106 mutant was due to protein mislocation, we in-serted the 9-aa NLS from the SV40 large T antigen into M53-�16-106 to create M53-�16-106NLS. The heterologous NLSrestored the correct nuclear localization of the protein (Fig. 4).
FIG. 3. Analysis of M53 insertion mutants by ectopic cis-comple-mentation of �M53-BAC. (A) Sequence comparison of M53/p38 ho-mologues. The amino acid sequence of M53/p38 was aligned with thesequences of all 36 UL31 family members by use of the Vector NTIAlign X program (Invitrogen) via the BLOSUM 62 similarity matrix.The depicted similarity plot was calculated using a 5-aa window size,with scores for identity and strong and weak similarities of 1.0, 0.5, and0.2, respectively. The x axis represents the number of amino acids inthe consensus sequence. Conserved regions 1 to 4 (CR1-4) are indi-cated below the diagram. (B) M53 mutants and their ability to rescuevirus growth of the �M53 genome. The sequence displayed is the aminoacid sequence of M53/p38. Arrowheads indicate transposon insertionsites. Open arrowheads indicate insertions leading to a stop codon. Lightgray arrowheads indicate in-frame insertion mutations that rescued the�M53 phenotype. Black arrowheads indicate in-frame insertion muta-tions that were not able to rescue the �M53 null phenotype. Under-lined parts of the M53 amino acid sequence indicate the conservedregions described in panel A.
FIG. 4. Functional analysis of N-terminal deletion mutants of M53.(A) Schematic representation of the different deletion constructs. Theepitope for the anti-M53/p38 serum is indicated by a star at the very Nterminus of each diagram. The variable sequences are represented byopen boxes. The conserved regions are shown as shaded boxes. Thedeleted sequences are indicated by a line. The inserted SV40 NLS isindicated by a crossed box. The EGFP sequence is shown as a diago-nally hatched box. (B) wt M53 and mutants M53-�16–106 and M53-�16-106NLS were reinserted into �M53-BAC. BAC DNA was iso-lated, transfected into MEF cells, and screened for plaque formation,and the rescue results are indicated (�, �, or N.a. [not applicable]).(C) Subcellular localization of expressed proteins with N-terminal de-letions. NIH 3T3 fibroblasts were transfected with the indicated con-structs. The localization of the expressed proteins was visualized byindirect immunofluorescence for wt M53, M53-�16–106, and M53-�16-106NLS. Endogenous fluorescence was recorded for EGFP andmutant s106EGFP.
VOL. 80, 2006 MCMV M53/p38 BINDING SITE FOR m50/p35 77
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

The existence of an NLS within the N-terminal part of M53/p38 was tested by a fusion between aa 1 to 106 of M53/p38 andthe enhanced GFP (EGFP) ORF. NIH 3T3 cells expressingEGFP showed fluorescent protein in both the cytoplasm andthe nucleus, whereas the M53/1-106-EGFP fusion constructwas restricted to the nucleus. Furthermore, M53-�16-106NLS,but not M53-�16-106, colocalized with M50/p35 upon coex-pression (data not shown). Finally, the mutant M53-�16-106NLS,lacking aa 16 to 106 but carrying the SV40 NLS, but notM53-�16-106, rescued the null phenotype of m16/17FRT-�M53-BAC. Thus, nuclear targeting of the protein is the onlyessential function of the N-terminal part of M53/p38.
M50/p35 binding site of M53/p38. Recently, we showed thatM53/p38 binds directly to M50/p35 and that the binding regionof M50/p35 is essential for MCMV growth (3). We predictedthat the binding of M53/p38 to M50/p35 should also representan essential function. Therefore, the pool of lethal M53 mu-tants should contain mutants which have lost the ability to bindto M50/p35. For a first rough mapping of the binding region, theselected set of M53/p38 truncation mutants was analyzed. To thisend, NIH 3T3 fibroblasts were transfected with either wt M53/p38 or M53/p38 mutants in the presence or absence of M50/p35 coexpression. The subcellular localization of wt and mu-tated M53/p38 was analyzed by confocal microscopy afterindirect immunofluorescence staining with specific antisera(Fig. 5A). After isolated expression, all truncation mutants aswell as wt M53/p38 (M53) showed the typical diffuse nuclearstaining pattern (Fig. 5A, first column). After cotransfectionwith M50/p35, the same diffuse nuclear staining was observedonly for the stop mutant s106, which lacks all four conservedregions (Fig. 5A, second column). The other stop mutants, i.e.,s137, lacking CR2 to -4 and the C-terminal part of CR1, s168,lacking CR2 to -4 (Fig. 5A, third and fourth columns), s233,s290, and s309 (data not shown), colocalized with M50/p35 atthe nuclear rim.
These colocalization experiments indicated a critical role ofM53/p38 aa 1 to 137 for interaction with M50/p35. To confirmthe results obtained by subcellular localization of the M53truncation mutants, we performed coimmunoprecipitation ex-periments with the stop mutants. 293 cells were cotransfectedwith expression plasmids containing mutant M53 constructsand with pM50, expressing wt M50/p35. Controls were trans-fected with plasmids expressing wt M50, wt M53, or M53 trun-cation mutants alone. Cell lysates were precipitated by specificantisera. The wt M53/p38 protein was coprecipitated in thepresence of M50/p35 when M50-specific antiserum was used(Fig. 5B, lane 3). The M53s106 stop mutant was unable to bindto M50/p35 (lane 5). The other stop mutants, M53s137,M53s168, and M53s185, bound M50/p35, as shown by the spe-cific bands corresponding to the expected molecular massesafter coprecipitation with anti-M50/p35 (lanes 7, 9, and 11).The same result was found with the s233, s290, and s309 stop
FIG. 5. Analysis of interaction of M53 stop mutants with M50/p35.(A) Localization of M53 stop mutants. NIH 3T3 cells were transfectedwith M53 stop mutants s106, s137, and s168 alone (first column) ortogether with wt M50/p35 (second to fourth columns). Single trans-fected cells were stained with a specific rat antiserum against M53/p38.For detection, a fluorescein isothiocyanate-conjugated secondary an-tibody was used (green). Cotransfected cells were treated as describedabove and costained with an M50/p35-specific rabbit serum, which wasdetected by a Texas red-coupled secondary antibody (red). As a con-trol, wt M53/p38 was used (first row). (B) Coimmunoprecipitation ofM53 stop mutants and M50/p35. Plasmids expressing wt M53/p38 (lane1), wt M50/p35 (lane 2), and M53 stop mutants (lanes 4, 6, 8, and 10)were transfected into 293 cells. In parallel, wt M53/p38 and M53 stopmutants were cotransfected with wt M50/p35 (lanes 3, 5, 7, 9, and 11).Cells were radioactively labeled, and singly transfected probes wereprecipitated with anti-M53/p38-specific rat serum on protein G-Sepha-rose. Protein complexes with wt M50 were precipitated with proteinA-Sepharose, using anti-M50/p35-specific rabbit serum. Samples wereanalyzed by SDS-PAGE followed by autoradiography. (C) Pull-downassay of M53 stop mutants with Strep-tagged M50/p35. wt M53/p38and M53 stop mutants were cotransfected with M50ST into 293 cells.Total cell lysates were analyzed to test the protein expression byWestern blotting using a specific antiserum against M53/p38 (upper
panel, total protein [T]). Proteins in complex with M50ST were pre-cipitated by Strep-Tactin–Sepharose. Desthiobiotin eluates were ana-lyzed by SDS-PAGE and blotted onto membranes. wt M53/p38 andmutant proteins were detected by Western blotting with M53/p38-specific antiserum (lower panel, bound protein [B]).
78 LOTZERICH ET AL. J. VIROL.
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

mutants (data not shown). The binding pattern was also testedin pull-down assays after cotransfection of pM50ST, expressingStrep-II-tagged M50/p35, with wt and mutant M53 constructs.From the lysates of cotransfected 293 cells, proteins bound to
M50ST were recovered by Strep-Tactin–Sepharose beads. TheM50ST complexes were eluted from the beads by desthiobi-otin, separated by SDS-PAGE, and subjected to Western blotanalysis using M53/p38-specific antiserum. The data were infull agreement with the results obtained by coimmunoprecipi-tation with wt M50/p35. This convinced us that the taggedM50/p35 protein had kept the properties of wt M50/p35 withregard to M53/p38 binding (Fig. 5C).
These data, together with the functionality of the M53�16-106NLS mutant in the context of the virus, suggested that ashort sequence of M53/p38 CR1 is required for M50/p35 bind-ing. To describe the contribution of specific sequences involvedin the M50/p35-M53/p38 interaction in more detail, we ana-lyzed M53/p38 mutants with insertions in the region betweenaa 106 and aa 138 by confocal microscopy and by the M50ST-mediated protein pull-down assay. NIH 3T3 cells were trans-fected with different M53/p38 insertion mutants alone or to-gether with pM50. As expected, all analyzed M53 insertionmutants showed diffuse nuclear staining after isolated expres-sion (Fig. 6A, first column). After cotransfection with pM50,only mutant M53i128 was not able to colocalize with M50/p35at the nuclear rim (Fig. 6A, third column). All other insertionmutants tested, i.e., i104, i115, i131, and i138, tolerated 5-aainsertions inasmuch as they were recruited to the nuclear en-velope by M50/p35 coexpression. In addition, 293 cells werecotransfected with pM50ST and the same set of M53/p38 in-sertion mutants which was used for the colocalization experi-ments. In parallel, the total cell lysates were analyzed to de-termine the expression of the M53/p38 mutants by Westernblotting. Among the mutants tested, only the insertion mutantM53i128 was not pulled down by M50ST (Fig. 6B, lane 3),which is in line with the results of the colocalization studiesdescribed above.
Taken together, colocalization and protein-protein interac-tion assays using several C-terminal truncation and insertionmutants indicated that the region of M53/p38 which is neces-sary for M50/p35 binding is located at the beginning of CR1and that the most important residues are probably representedby aa 115 to 131 of the M53/p38 protein.
Characterization of the M50/p35 binding site of M53/p38.Insertions of five amino acids helped to identify a bindingmotif, but the contribution of the inserted amino acids thatdiffered between the different insertion mutants was difficult toevaluate. In order to define the relative roles of individualamino acids with respect to M50/p35 binding, the 12 aminoacids which are conserved in betaherpesviruses within the N-terminal half of CR1 (aa 112 to 137) were replaced one by onewith alanine. After the transfection of NIH 3T3 cells with theM53/p38 point mutants alone or cotransfection with M50/p35,the subcellular localization of the M53/p38 mutants was ana-lyzed. All M53/p38 point mutants showed diffuse nuclear stain-ing when expressed alone (Fig. 7, first column) (not all data areshown; see Fig. S1 in the supplemental material). After coex-pression with M50/p35, the intranuclear distribution of M53point mutants fell into three classes of phenotypes: (i) mutantsthat redistributed to the nuclear rim and recruited M50/p35 sim-ilar to wt M53/p38 (M53P123A, M53D124A, and M53E126A),(ii) mutants that had apparently lost the ability to redistribute tothe nuclear rim in the presence of M50/p35 (M53Y129A [Fig.7A], M53L130A, and M53I133A), and (iii) mutants with inter-
FIG. 6. Analysis of interaction of M53 insertion mutants with M50/p35. (A) Localization of M53 insertion mutants. NIH 3T3 cells weretransfected with M53 insertion mutants i104, i115, i128, i131, and i138alone (first column) or together with wt M50 (second to fourth col-umns). Single transfected cells were stained with a specific rat anti-serum against M53/p38. For detection, a fluorescein isothiocyanate-conjugated secondary antibody was used (green). Cotransfected cellswere treated as described above and costained with an M50/p35-spe-cific rabbit serum, which was detected by a Texas red-coupled second-ary antibody (red). (B) Pull-down analysis of M53 insertion mutantswith Strep-tagged M50/p35. 293 cells were cotransfected with M53insertion mutants and M50ST. Total cell lysates were analyzed bySDS-PAGE and Western blotting using an M53/p38-specific antiserum(upper panel, T). Proteins in complex with M50ST were precipitatedwith Strep-Tactin-Sepharose, and desthiobiotin-eluted proteins wereseparated by SDS-PAGE. Signals for M53/p38 insertion mutants werevisualized by Western blotting using a specific antiserum against M53/p38 (lower panel, B).
VOL. 80, 2006 MCMV M53/p38 BINDING SITE FOR m50/p35 79
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
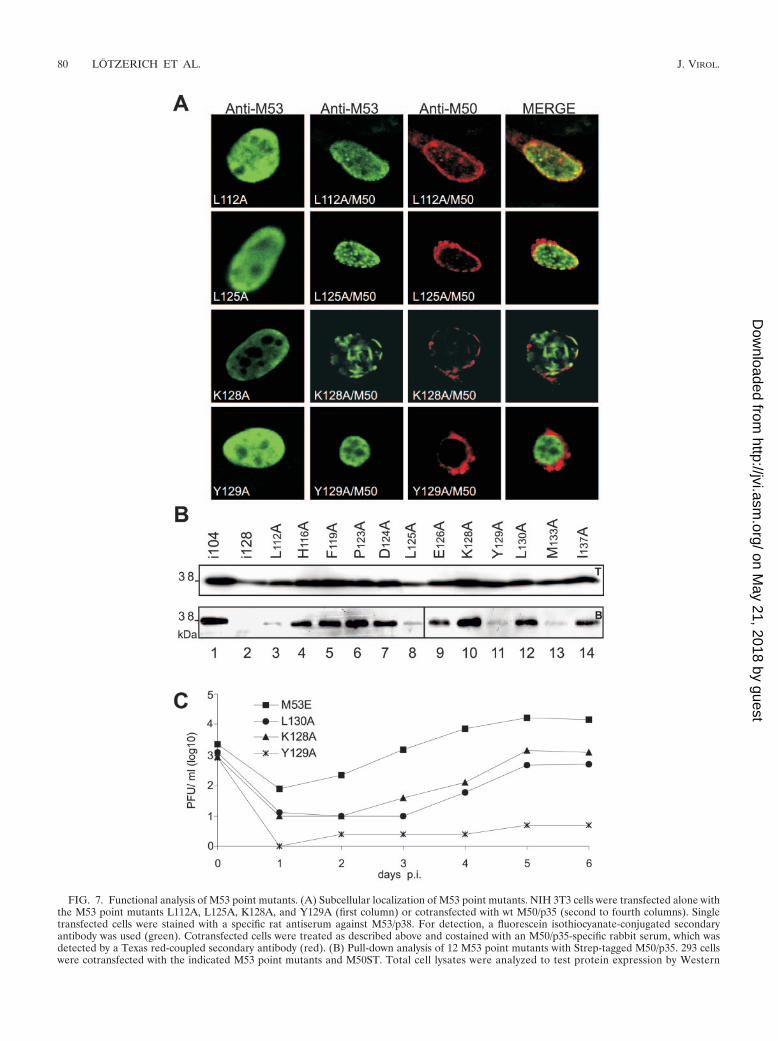
FIG. 7. Functional analysis of M53 point mutants. (A) Subcellular localization of M53 point mutants. NIH 3T3 cells were transfected alone withthe M53 point mutants L112A, L125A, K128A, and Y129A (first column) or cotransfected with wt M50/p35 (second to fourth columns). Singletransfected cells were stained with a specific rat antiserum against M53/p38. For detection, a fluorescein isothiocyanate-conjugated secondaryantibody was used (green). Cotransfected cells were treated as described above and costained with an M50/p35-specific rabbit serum, which wasdetected by a Texas red-coupled secondary antibody (red). (B) Pull-down analysis of 12 M53 point mutants with Strep-tagged M50/p35. 293 cellswere cotransfected with the indicated M53 point mutants and M50ST. Total cell lysates were analyzed to test protein expression by Western
80 LOTZERICH ET AL. J. VIROL.
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

mediate phenotypes. Several mutants fell into the last class,and their phenotypic appearance was variable. One subsetshowed abundant diffuse intranuclear staining in addition tocolocalization with M50/p35 at the nuclear rim (M53L112A[Fig. 7A], M53H116A, M53F119A, and M53I137A). The sec-ond type of mutants colocalized with M50/p35, at least to someextent. The remarkable phenotype, however, was a new distri-bution of M53/p38 mutants upon coexpression of M50/p35resulting in the formation of discrete intranuclear aggregates(M53L125A) and fibrous structures (M53K128A) (Fig. 7A)which did not colocalize with M50/p35. In the absence of M50/p35, the intranuclear distribution of the mutants was indistin-guishable from that of wt M53/p38.
Next, 293 cells were cotransfected with M50ST and the set ofalanine-scanning M53/p38 mutants. Strep-Tactin–Sepharosepull-down assays were performed to analyze the binding ca-pacity of the M53 point mutants to M50ST. Remarkably, de-spite the different distribution phenotypes, all M53 point mu-tants could be retrieved by M50ST pull-down assays, includingthose that did not show a detectable interaction in the colo-calization assay (Fig. 7B). Notably, binding was found to besignificantly reduced for the mutants M53L112A, M53L125A,M53Y129A, and M53M133A.
Since there was a substantial degree of variability in thecolocalization assay and a certain heterogeneity of interactionsdetectable by the pull-down assay, we tested the M53 pointmutants for functionality in the viral context. Interestingly, thenull phenotype of �M53FRT-BAC could be reverted by allM53 point mutants. About 11 days are usually required todetect the first virus plaques after transfection of the ectopi-cally complemented deletion genome. The virus reconstitutiontime and plaque formation were significantly delayed, to 3weeks, for mutants M53K128A and M53L130A. Virus reconsti-tution from the BAC carrying M53Y129A required 6 weeks.Accordingly, the growth of mutants M53K128A and M53L130Awas reduced by 1 order of magnitude, and M53Y129A wasattenuated 3 orders of magnitude, compared with a genomecomplemented by the wt M53 ORF (Fig. 7C). These data showthat already the exchange of a single aa within the bindingregion of M53/p38 can strongly affect M50/p35 binding butcannot completely abolish the M50/p35-M53/p38 interactionand thus can rescue the null phenotype.
To rigorously test the binding site, we replaced either twoadjacent critical amino acids (M53YL129-130A) or threeadjacent amino acids (M53KYL128-130A) by alanine. In ad-dition, we deleted the sequence of aa 108 to 136 of M53(M53�108-136). As expected, each of these mutants com-pletely failed to colocalize with M50/p35 (see Fig. S2 in thesupplemental material), failed to interact with M50ST in thepull-down assay (Fig. 8A), and also failed to rescue the M53
null phenotype. The N-terminal deletion mutant M53-�16-106NLS, carrying a deletion of aa 16 to 106 and an artificialNLS, rescued the M53 deletion after ectopic reinsertion intothe mutant MCMV-BAC, indicating that this mutant can in-teract with M50/p35. To confirm the M50/p35 interaction sitemapping data, the N-terminal deletion was increased to aa 16to aa 136 (M53-�16-136NLS). As expected, M53-�16-106NLSdid interact with M50ST in the pull-down assay, whereas M53-�16-136NLS did not (Fig. 8B). Also, in contrast to M53-�16-106NLS, the N-terminal deletion mutant M53-�16-136NLSwas not able to rescue the M53 null phenotype upon insertioninto the deletion BAC.
DISCUSSION
The comparative analysis of herpesvirus common proteinfunctions is an interesting area of research. Although the threesubgroups of herpesviruses diverged from a common ancestorprobably more than 50 million years ago, the amino acid se-quences of proteins involved in virus morphogenesis still indi-
blotting using a specific antiserum against M53/p38 (upper panel, T). Proteins in complex with M50ST were precipitated by Strep-Tactin–Sepharose. Desthiobiotin eluates were analyzed by SDS-PAGE and blotted onto membranes. Signals for M53 point mutants were visualized byWestern blotting with M53/p38-specific antiserum (lower panel, B). As a positive control for precipitation, the functional insertion mutant M53i104was used (lane 1), and the M53 insertion mutant M53i128 served as a negative control (lane 2). (C) Rescue of �M53-BAC by M53 point mutantsK128A, Y129A, and L130A. The growth kinetics of the viruses derived from pM53E, pM53K128AE, pM53Y129AE, and pM53L130AE are shown.NIH 3T3 cells were infected with the respective viruses. Supernatants of the infected cells were harvested on the indicated days, and virus titerswere determined by a plaque assay.
FIG. 8. Analysis of interaction of M50/p35 with M53 point andN-terminal deletion mutants. (A) Pull-down analysis of M53 pointmutants YL129,130A, KYL128-130A, and M53-�108-136 with Strep-tagged M50. 293 cells were cotransfected with M53 point mutants andM50ST. The expression of the constructs was tested by SDS-PAGEwith total cell lysates and by Western blotting using an M53/p38-specific antiserum (upper panel, T). Proteins in complex with M50STwere precipitated by Strep-Tactin–Sepharose. Desthiobiotin eluateswere analyzed by SDS-PAGE and blotted onto membranes. Signals forM53 point mutants were visualized by Western blotting with an M53/p38-specific antiserum (lower panel, B). As a positive control, a func-tional insertion mutant, M53i104, was used (lane 1), and M53i128,which failed to bind to M50/p35, served as a negative control (lane 2).(B) Pull-down analysis of N-terminal deletion mutants of M53/p38. 293cells were cotransfected with the M53 N-terminal deletion mutantM53-�16-106NLS or M53-�16-136NLS and M50ST. Analyses of totalcell lysates (upper panel, T) and protein complex formation (lowerpanel, B) were performed as described above.
VOL. 80, 2006 MCMV M53/p38 BINDING SITE FOR m50/p35 81
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

cate similar functions. However, sequence homology does notpredict function, and proteins with no detectable sequencehomology can still carry out similar functions. Since proteins ofviruses and hosts may change motifs and domains during evo-lution, the spectrum of activities executed by related proteinsmay differ considerably.
We owe our basic knowledge of the herpesvirus commonproteins governing the egress of the herpesvirus nucleocapsidfrom the cell nucleus to pioneering studies of alphaherpesvi-ruses. To achieve nucleocapsid egress, the UL31 and UL34proteins need to form a complex at the nuclear membrane.Deletion of either protein severely compromises virus replica-tion but still gives rise to progeny (5, 8, 14, 25, 26, 35). For beta-and gammaherpesviruses, the conditions are even more strin-gent. The lack of either protein abolishes virus replication (3,7, 10). Therefore, these studies could only be initiated aftercloning and mutagenesis of complete infectious herpesvirusgenomes as BACs in E. coli (16, 34). Published work and datapresented in this study converge to the conclusion that UL31and UL34 homologues of alpha-, beta-, and gammaherpesvi-ruses have to interact at the inner nuclear membrane to exe-cute their function as NECs (3, 5, 7, 10, 12, 14, 19, 24, 25, 31,35). This might involve contact with several unknown cellularand viral proteins. The mutual binding sites need to be deter-mined to address the question of which functions of theseproteins are conducted in isolation and which are executedsubsequent to NEC formation. In a previous paper, we re-ported on the binding domain in M50/p35, the UL34 homologof a betaherpesvirus (3). The M50 gene was subjected to ran-dom mutagenesis followed by reintroduction into the viral ge-nome (3). Due to frequent insertions into the rescue vectorbackbone, the efficiency of random insertion mutagenesis ofthe M50 ORF was �20% and required laborious screening.Here we excluded vector insertions by subcloning the primarypool of transposon-labeled M53 ORFs into the rescue vector,and the use of a temperature-sensitive transposon donor raisedthe successful insertion frequency to 50%. We created a libraryof about 28,000 insertion/stop mutants in the M53 ORF,screened 986 by PCR, and sequenced 498 PCR-positive clones.A total of 54 random mutants (46 M53 insertion mutants and8 truncation mutants) were tested both at the level of isolatedexpression and for complementation of the M53 null pheno-type in the viral context. In addition, a total of 18 targetedmutants were also tested in the genomic context. Altogether,we give the first report of the binding site of a UL31 proteinfamily member, M53/p38.
We report the following observations. (i) A sequence com-parison of 36 known members of the UL31 family of herpes-virus proteins revealed an N-terminal variable region and fourconserved regions (CR) located within the C-terminal two-thirds of the sequences. (ii) The only essential function of thenonconserved N-terminal end of M50/p35 is to provide anNLS. (iii) Within CR1 (aa 115 to 174 in MCMV M53/p38), wefound the binding motif for M50/p35. This motif is representedby aa 115 to 137, which are necessary for binding to M50/p35.(iv) Mutation of all conserved amino acids in this region intoalanines resulted in different phenotypes with regard to NECformation, revealing a prominent role for the residues K128,Y129, and L130. Nevertheless, all single point mutants wereable to bind to M50/p35 and to rescue the M53 null phenotype
of the genome. However, when two or three of the criticalresidues were replaced, the mutants failed to bind to M50/p35and did not rescue the M53 null phenotype. (v) The C-terminalconserved regions 2 to 4 (CR2-4) bear an as yet unidentifiedessential function(s). Insertion mutants in these regions couldbind to M50/p35 (data not shown) but lacked functionality.
An alignment of M53 with members of the UL31 familydemonstrates a conserved central and C-terminal part withinUL31 family homologues, whereas the N-terminal region isvariable. The results of the functional analysis of M53 insertionmutants were in line with the in silico predictions. Nonfunc-tional insertion mutants accumulated only within the con-served two-thirds of the M53 ORF. Within the N-terminalvariable region of M53/p38, two overlapping NLSs were pre-dicted between aa 24 and 42. The deletion mutant of M53/p38lacking the region from aa 16 to 106 failed to rescue virusgrowth in the absence of the wt M53 gene; however, the in-troduction of an artificial 9-aa NLS could restore the function-ality of this mutant, demonstrating that the variable N-terminaldomain of M53/p38 contains an NLS as a functional element.The consensus sequence for a nuclear targeting signal (6) ispresent in only 14 of 36 members of the UL31 family. How-ever, if an NLS is predicted, it is always located within theN-terminal variable region. All analyzed members of the UL31protein family are localized exclusively in the nucleus uponisolated expression, indicating that they have an active NLS,and we believe that this is the function of the N-terminalvariable domain.
A conserved feature in all herpesvirus subgroups is the in-teraction between M50/p35 homologues (UL34) and M53/p38homologues (UL31). A stop mutant suggested that the N-terminal 136 aa of M53/p38 are necessary and sufficient forM50/p35 binding. The N-terminal sequence of aa 16 to 106 wasreplaced without a loss of functionality and placed the M50/p35 binding region between aa 106 and 136. The nonfunctionalinsertion mutants M53i115 and i131 further localized se-quences necessary for M50/p35 binding within aa 115 to 131 ofM53/p38. Alanine-scanning mutagenesis for the 12 conservedaa of this essential region confirmed and extended these ob-servations. All point mutants showed wt nuclear distributionupon isolated expression. However, a variety of phenotypesoccurred in the presence of M50/p35, including mutants whichformed intranuclear aggregates (M53L125A) or filamentousstructures (M53K128A). Since these structures were only seenin the presence of M50/p35, we assume that a transient M50/p35-M53/p38 interaction is responsible for the observed phe-notypic changes. This transient interaction was apparently suf-ficient to cause the aggregation of M53/p38 alone or in complexeswith thus far unknown cellular partners. Surprisingly, all testedpoint mutants could bind to M50/p35 to some extent in pull-down assays, and more importantly, all rescued the M53 nullphenotype in the viral context, including those without appar-ent colocalization. This indicates that using only one assay tostudy a protein-protein interaction might not be sufficient. Co-localization studies show the major steady-state phenotype,and residual low-affinity interactions may be overlooked. Co-immunoprecipitation reveals the potential to bind.
However, the growth of reconstituted viruses was poor formutants M53K128A, M53Y129A, and M53L130A. These dataimply that wt M50/p35-M53/p38 complexes are required for
82 LOTZERICH ET AL. J. VIROL.
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

efficient productive infection and that a small number of tran-sient complexes may suffice for virus production. However, iftwo or more aa within the predicted binding region were ex-changed, these mutants neither colocalized with nor bound toM50/p35 and were not able to rescue the M53 null phenotype.Our data show that a loss of M53/p38 binding to M50/p35 isassociated with the inability to replicate. However, M50/p35binding might not be the only function of CR1. Notably, thereare M53 insertion mutants with mutations within the identifiedbinding motif (i115 and i131) which bind to M50/p35 but arenevertheless lethal.
Interestingly, the M50/p35 binding region of M53/p38 isstrictly conserved only in betaherpesviruses. The homology ofalpha- or gammaherpesvirus UL31 family members to the be-taherpesvirus sequence (aa 115 to 136) is lower than the aver-age similarity of the conserved regions. The sequence conser-vation is considerably high only within subfamilies. Therefore,we expect that sequences in the conserved regions, but notnecessarily the same sequences in CR1, functionally define thebinding region in alpha- and gammaherpesvirus homologues.This divergence is already experimentally proven for the bind-ing regions of UL34 members from the alpha- and betaher-pesvirus subfamilies. We showed that MCMV M50/p35 aa 53to 57 and aa 114 are important for M53/p38 binding (3). Usinga set of nine HSV-1 UL34 mutants, one of which was intro-duced into the genomic context, the binding region was local-ized to a different region, namely, aa 137 to 181 of UL34(which correspond to aa 129 to 173 of M50/p35) (13).
The C-terminal regions CR2 to CR4 are conserved in theUL31 family, and several insertion mutants have a null phenotypein the genomic context, although they bind to M50/p35, indicatingthat the C-terminal half of M53/p38 bears as yet unidentifiedessential functions. Under certain conditions, HSV-1 UL31 inter-acts with lamins A/C and is involved in chromatin reorganization(24, 30, 31). M50/p35 and BFRF1 have indirect effects on thenuclear lamina (7, 19), and in Epstein-Barr virus, the complex ofBFLF2 and BFRF1 interacts with lamin B (10). These features,and also capsid recruitment and regulation of the subsequentbudding event, may involve CR2-4 of the UL31 family proteins.Since the mutual binding sites have been defined for the M50/p35-M53/p38 interaction, the decisive features dependent oncomplex formation can now be addressed. Experimental ap-proaches such as testing mutants for dominant negative effects(28) or protein pull-down assays using a functional or nonfunc-tional NEC may pave the way to the elucidation of the herpesvi-rus NEC functions.
ACKNOWLEDGMENTS
We thank S. Boos for excellent technical assistance and L. Dolkenfor a critical reading of the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaftthrough SFB 455, “Viral functions and immune modulation.”
REFERENCES
1. Bateman, A., L. Coin, R. Durbin, R. D. Finn, V. Hollich, S. Griffiths-Jones,A. Khanna, M. Marshall, S. Moxon, E. L. Sonnhammer, D. J. Studholme, C.Yeats, and S. R. Eddy. 2004. The Pfam protein families database. NucleicAcids Res. 32:D138–D141.
2. Biery, M. C., F. J. Stewart, A. E. Stellwagen, E. A. Raleigh, and N. L. Craig.2000. A simple in vitro Tn7-based transposition system with low target siteselectivity for genome and gene analysis. Nucleic Acids Res. 28:1067–1077.
3. Bubeck, A., M. Wagner, Z. Ruzsics, M. Lotzerich, M. Iglesias, I. R. Singh,and U. H. Koszinowski. 2004. Comprehensive mutational analysis of a her-
pesvirus gene in the viral genome context reveals a region essential for virusreplication. J. Virol. 78:8026–8035.
4. Bubic, I., M. Wagner, A. Krmpotic, T. Saulig, S. Kim, W. M. Yokoyama, S.Jonjic, and U. H. Koszinowski. 2004. Gain of virulence caused by loss of agene in murine cytomegalovirus. J. Virol. 78:7536–7544.
5. Chang, Y. E., C. Van Sant, P. W. Krug, A. E. Sears, and B. Roizman. 1997.The null mutant of the U(L)31 gene of herpes simplex virus 1: constructionand phenotype in infected cells. J. Virol. 71:8307–8315.
6. Dingwall, C., and R. A. Laskey. 1991. Nuclear targeting sequences—a con-sensus? Trends Biochem. Sci. 16:478–481.
7. Farina, A., R. Feederle, S. Raffa, R. Gonnella, R. Santarelli, L. Frati, A.Angeloni, M. R. Torrisi, A. Faggioni, and H. J. Delecluse. 2005. BFRF1 ofEpstein-Barr virus is essential for efficient primary viral envelopment andegress. J. Virol. 79:3703–3712.
8. Fuchs, W., B. G. Klupp, H. Granzow, N. Osterrieder, and T. C. Mettenleiter.2002. The interacting UL31 and UL34 gene products of pseudorabies virusare involved in egress from the host-cell nucleus and represent componentsof primary enveloped but not mature virions. J. Virol. 76:364–378.
9. Gold, M. C., M. W. Munks, M. Wagner, U. H. Koszinowski, A. B. Hill, andS. P. Fling. 2002. The murine cytomegalovirus immunomodulatory genem152 prevents recognition of infected cells by M45-specific CTL but doesnot alter the immunodominance of the M45-specific CD8 T cell response invivo. J. Immunol. 169:359–365.
10. Gonnella, R., A. Farina, R. Santarelli, S. Raffa, R. Feederle, R. Bei, M.Granato, A. Modesti, L. Frati, H. J. Delecluse, M. R. Torrisi, A. Angeloni,and A. Faggioni. 2005. Characterization and intracellular localization of theEpstein-Barr virus protein BFLF2: interactions with BFRF1 and with thenuclear lamina. J. Virol. 79:3713–3727.
11. Gossen, M., and H. Bujard. 1992. Tight control of gene expression in mam-malian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci.USA 89:5547–5551.
12. Lake, C. M., and L. M. Hutt-Fletcher. 2004. The Epstein-Barr virus BFRF1and BFLF2 proteins interact and coexpression alters their cellular localiza-tion. Virology 320:99–106.
13. Liang, L., and J. D. Baines. 2005. Identification of an essential domain in theherpes simplex virus 1 UL34 protein that is necessary and sufficient tointeract with UL31 protein. J. Virol. 79:3797–3806.
14. Liang, L., M. Tanaka, Y. Kawaguchi, and J. D. Baines. 2004. Cell lines thatsupport replication of a novel herpes simplex virus 1 UL31 deletion mutantcan properly target UL34 protein to the nuclear rim in the absence of UL31.Virology 329:68–76.
15. Menard, C., M. Wagner, Z. Ruzsics, K. Holak, W. Brune, A. E. Campbell,and U. H. Koszinowski. 2003. Role of murine cytomegalovirus US22 genefamily members in replication in macrophages. J. Virol. 77:5557–5570.
16. Messerle, M., I. Crnkovic, W. Hammerschmidt, H. Ziegler, and U. H.Koszinowski. 1997. Cloning and mutagenesis of a herpesvirus genome as aninfectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 94:14759–14763.
17. Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537–1547.
18. Mettenleiter, T. C. 2004. Budding events in herpesvirus morphogenesis.Virus Res. 106:167–180.
19. Muranyi, W., J. Haas, M. Wagner, G. Krohne, and U. H. Koszinowski. 2002.Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lam-ina. Science 297:854–857.
20. Padgett, K. A., and J. A. Sorge. 1996. Creating seamless junctions indepen-dent of restriction sites in PCR cloning. Gene 168:31–35.
21. Posfai, G., M. D. Koob, H. A. Kirkpatrick, and F. R. Blattner. 1997. Versatileinsertion plasmids for targeted genome manipulations in bacteria: isolation,deletion, and rescue of the pathogenicity island LEE of the Escherichia coliO157:H7 genome. J. Bacteriol. 179:4426–4428.
22. Rawlinson, W. D., H. E. Farrell, and B. G. Barrell. 1996. Analysis of thecomplete DNA sequence of murine cytomegalovirus. J. Virol. 70:8833–8849.
23. Reddehase, M. J., F. Weiland, K. Munch, S. Jonjic, A. Luske, and U. H.Koszinowski. 1985. Interstitial murine cytomegalovirus pneumonia after ir-radiation: characterization of cells that limit viral replication during estab-lished infection of the lungs. J. Virol. 55:264–273.
24. Reynolds, A. E., L. Liang, and J. D. Baines. 2004. Conformational changes inthe nuclear lamina induced by herpes simplex virus type 1 require genesU(L)31 and U(L)34. J. Virol. 78:5564–5575.
25. Reynolds, A. E., B. J. Ryckman, J. D. Baines, Y. Zhou, L. Liang, and R. J.Roller. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1form a complex that accumulates at the nuclear rim and is required forenvelopment of nucleocapsids. J. Virol. 75:8803–8817.
26. Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines.2002. Ultrastructural localization of the herpes simplex virus type 1 UL31,UL34, and US3 proteins suggests specific roles in primary envelopment andegress of nucleocapsids. J. Virol. 76:8939–8952.
27. Roller, R. J., Y. Zhou, R. Schnetzer, J. Ferguson, and D. DeSalvo. 2000.Herpes simplex virus type 1 U(L)34 gene product is required for viralenvelopment. J. Virol. 74:117–129.
VOL. 80, 2006 MCMV M53/p38 BINDING SITE FOR m50/p35 83
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

28. Rupp, B., Z. Ruzsics, T. Sacher, and U. H. Koszinowski. 2005. Conditionalcytomegalovirus replication in vitro and in vivo. J. Virol. 79:486–494.
29. Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratorymanual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
30. Scott, E. S., and P. O’Hare. 2001. Fate of the inner nuclear membraneprotein lamin B receptor and nuclear lamins in herpes simplex virus type 1infection. J. Virol. 75:8818–8830.
31. Simpson-Holley, M., J. Baines, R. Roller, and D. M. Knipe. 2004. Herpessimplex virus 1 U(L)31 and U(L)34 gene products promote the late matu-ration of viral replication compartments to the nuclear periphery. J. Virol.78:5591–5600.
32. Wagner, M., S. Jonjic, U. H. Koszinowski, and M. Messerle. 1999. Systematicexcision of vector sequences from the BAC-cloned herpesvirus genomeduring virus reconstitution. J. Virol. 73:7056–7060.
33. Wagner, M., and U. H. Koszinowski. 2004. Mutagenesis of viral BACswith linear PCR fragments (ET recombination). Methods Mol. Biol.256:257–268.
34. Wagner, M., Z. Ruzsics, and U. H. Koszinowski. 2002. Herpesvirus geneticshas come of age. Trends Microbiol. 10:318–324.
35. Ye, G. J., and B. Roizman. 2000. The essential protein encoded by the UL31gene of herpes simplex virus 1 depends for its stability on the presence ofUL34 protein. Proc. Natl. Acad. Sci. USA 97:11002–11007.
84 LOTZERICH ET AL. J. VIROL.
on May 21, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from


















