
Evaluación del Cambio de Tecnología y Mejora del ...
114
UNIVERSIDAD NACIONAL MAYOR DE SAN MARCOS FACULTAD DE FARMACIA Y BIOQUÍMICA E.A.P. DE FARMACIA Y BIOQUÍMICA Evaluación del Cambio de Tecnología y Mejora del Procedimiento de Fabricación de Tabletas Recubiertas de Paracetamol 500 mg + Diclofenaco Sódico 50 mg TESIS Para optar el Título Profesional de Químico Farmacéutico AUTOR Christian Joel Charri Prudencio ASESOR Norma Julia Ramos Cevallos LIMA – PERÚ 2014
Transcript of Evaluación del Cambio de Tecnología y Mejora del ...

UNIVERSIDAD NACIONAL MAYOR DE SAN MARCOS
FACULTAD DE FARMACIA Y BIOQUÍMICA
E.A.P. DE FARMACIA Y BIOQUÍMICA
Evaluación del Cambio de Tecnología y Mejora del
Procedimiento de Fabricación de Tabletas Recubiertas de
Paracetamol 500 mg + Diclofenaco Sódico 50 mg
TESIS
Para optar el Título Profesional de Químico Farmacéutico
AUTOR
Christian Joel Charri Prudencio
ASESOR
Norma Julia Ramos Cevallos
LIMA – PERÚ
2014

A mis padres quienes me dieron vida, educación, apoyo y consejos. A mis hermanos,
a mis maestros y amigos, quienes sin su ayuda nunca hubiera podido hacer esta tesis.
A todos ellos se los agradezco desde el fondo de mi alma. Para todos ellos hago esta
dedicatoria.

A la Facultad de Farmacia y Bioquímica, por hacer de mí un excelente profesional y
prepararme para los retos de la vida.
A mis padres que hicieron todo en la vida para que yo pudiera lograr mis sueños, por
motivarme y darme la mano cuando sentía que el camino se terminaba.
A Mis hermanos por ser un ejemplo y por su apoyo incondicional en cada momento de
mi vida.
A la Dra. Norma Ramos por ser el soporte y aliento para no desanimar y conseguir mis
metas, quién influyó con sus lecciones y experiencias en formarme como una persona
de bien y preparada para los retos que pone la vida.
Un agradecimiento especial a los Doctores: Juan Ponce, Jáuregui Maldonado, Julio
Díaz y Paul Gutiérrez, por sus sabios consejos, sus palabras de aliento y su gran apoyo
para el desarrollo del presente trabajo
A mi gran amigo y compañero de trabajo Frank Pérez por brindarme su apoyo en el
desarrollo de esta tesis y por sus grandes consejos.

ÍNDICE
RESUMEN
SUMMARY
I. INTRODUCCIÓN 1
a. Objetivos
1.1. Objetivo general 2
1.2. Objetivos específicos 2
II. GENERALIDADES 3
a. Conceptos generales 3
b. Método de fabricación de tabletas 5
c. Pruebas de caracterización sobre la mezcla pulverulenta 7
d. Pruebas de caracterización farmacocinética de tabletas 9
e. Características fisicoquímicas de los comprimidos 11
f. Características microbiológicas 12
g. Estabilidad 12
h. Tipos de estudio de estabilidad 13
III. PARTE EXPERIMENTAL 14
a. Equipos, materiales y reactivos 14
b. Metodología 15
i. Etapa preliminar 15
ii. Descripción del proceso de manufactura 20
c. Especificaciones de las tabletas recubiertas 31
IV. RESULTADOS 34
a. Resultados de caracterización de mezcla pulverulenta 34
i. Aspecto 34
ii. Humedad 34
iii. Densidad aparente 35

iv. Densidad aparente de asentamiento 36
v. Índice de Hausner 37
vi. Índice de carr (índice de compresibilidad) 38
vii. Distribución Granulométrica 39
b. Dosaje de Principios activos (mezcla sin lubricante) 40
c. Mezcla con lubricantes (mezcla con lubricantes) 45
d. Estudio de caracterización de comprimidos (núcleos) 47
4.4.1 Aspecto 47
4.4.2 Dimensiones 47
4.4.3 Humedad 46
4.4.4 Peso promedio 49
4.4.5 Dureza 51
4.4.6 Friabilidad 52
4.4.7 Desintegración 53
4.4.8 Uniformidad de contenido 54
e. Estudio de caracterización de tabletas recubiertas 57
f. Estudio de pruebas microbiológicas 74
g. Resultados de estabilidad tabletas recubiertas 76
h. Impacto económico y ahorro de tiempo 77
V. DISCUSIÓN 79
VI. CONCLUSIÓN 85
VII. GLOSARIO DE TÉRMINOS 86
VIII. REFERENCIAS BIBLIOGRÁFICAS 88
IX. ANEXOS

RESUMEN
El presente trabajo tuvo como objetivo evaluar el impacto del uso de tecnología
automatizada sobre la productividad y mejora del proceso de manufactura, en
operaciones unitarias de fabricación de Paracetamol 500 mg + Diclofenaco sódico 50
mg tabletas recubiertas, mediante la transferencia tecnológica en las etapas de
granulación, compresión y recubierta de las tabletas. La metodología consistió en
desarrollar las etapas del proceso para la obtención de tabletas recubiertas, se utilizó el
granulador de lecho fluido, amasador de alta velocidad, tableteadora rotativa y
recubridora de bombo cerrado, en reemplazo de equipos de tecnología convencional.
En el diseño del proceso para el cambio a esta nueva tecnología se tomó en cuenta las
características de los productos, tamaño de diseño, características de los equipos y los
controles a realizar durante el proceso. Los resultados físicos, fisicoquímicos y
microbiológicos obtenidos del producto bajo el nuevo diseño de proceso en los nuevos
equipos, fueron conformes a las especificaciones de calidad establecidas para las
tabletas recubiertas de Paracetamol 500mg + Diclofenaco sódico 50 mg. La
investigación se realizó en dos lotes industriales, un lote ingresó a estudio de
estabilidad mostrando resultados conformes. Del trabajo se concluye que el empleo de
tecnología automatizada y el incremento de tamaño de lote, bajo el diseño propuesto,
fue satisfactorio, asimismo se identificaron los parámetros críticos de proceso en estas
etapas y los atributos de calidad de estos productos fueron conformes, se reporta un
ahorro significativo de 40% por lote y un ahorro anual de 400% aproximadamente
(Horas-Hombre y Horas-Maquina) en tiempos, lo que permite incrementar la
operatividad para la planta farmacéutica.
Palabras clave: Tecnología automatizada, productividad, optimización, Paracetamol,
diclofenaco sódico.

SUMMARY
This work had as a main objective to assess the impact of using automated technology
on the process improvement and productivity, during the manufacturing, specifically on
the units operations of Paracetamol 500 mg + sodium diclofenaco 50 mg coating
tablets, through technological transfer during granulation, compressing and coating
phases. The methodology of working was based on develop the phases of the process
to obtain coating tablets. There were used a fluid bed granulator, a high shear kneader,
a rotative tablet press, and a coater closed hype, instead of using conventional
technology. In the design’s process were technology was changed for this new
machinery, I took into account the following considerations: product characteristics,
batch size, characteristics of the machinery and controls during process. The obtained
results (physical, physicochemical and microbiological) applying this new process which
performs new technology, complain according to the quality specifications for coating
tablets of Paracetamol 500mg + sodium diclofenac 50 mg. This work was perform in
two industrial batch, both of them were included for stability study, this one showed
results conforms. In conclusions, the employment of automated technology and the
increasing of batch size according to the proposed design was satisfactory, there were
identified critical parameters of the process in this phases, quality attributes of this
products were agree with the specifications. It is shown an important saving of 40% by
lot and an annual saving of 400% approximately (Hours-men y hours-Machine), which
allows to increase the operability of a pharmaceutical industry.
Key words: automated technology, productivity, optimization, Paracetamol, diclofenac.

1
I. INTRODUCCIÓN
Los comprimidos son una de las formas farmacéuticas de administración oral más
empleados; por ello, el desarrollo y generalización de los mismos ha crecido y se ha
multiplicado debido a su diseño, carácter compacto y tamaño reducido, convirtiéndola
en una de las formas farmacéuticas de más fácil administración y transporte, sumado a
la facilidad de su fabricación a gran escala.
Sin embargo, cabe señalar que los comprimidos requieren de una fabricación compleja
debido a las diferentes operaciones unitarias que se necesitan para tener el producto
final. Entre las mismas tenemos que el granulado debe tener buenas propiedades de
flujo, compactación y uniformidad de mezcla para poder comprimir; asimismo, los
comprimidos deben poseer una dureza, friabilidad, peso, entre otros atributos de
calidad que permitan asegurar que el producto es capaz de resistir las manipulaciones
y transportes posteriores sin alterar la liberación rápida de las sustancias y cumpliendo
eficazmente su acción.
Es cada vez más frecuente recurrir a la utilización de tecnologías con diseños
automatizados, con la finalidad de optimizar las características del producto y el
proceso de manufactura. El objetivo final de todo proceso productivo es el producto,
resultado de las actividades preliminares que se emplearon para su obtención, y los
indicadores de optimización y productividad representan el manejo adecuado de la
tecnología dentro de la organización; en este sentido, la transferencia tecnológica de
una empresa productora puede ser evaluado en función de diferentes capacidades:
mejora de procesos, capacidades de producción, conocimiento de los equipos que
participan en el proceso y vinculación necesaria para recibir y transmitir información
tecnológica de agentes externos, como proveedores. Toda organización debería estar
en condiciones de desarrollar sus propias capacidades tecnológicas, resultado de un
proceso de evolución.
En la actualidad, la tecnología farmacéutica viene desarrollándose constantemente con
el fin de optimizar los procesos de manufactura. Dentro de este contexto, la evaluación
y mejora de los procedimientos de fabricación aplicando nuevas tecnologías es de
suma importancia. En el presente estudio se desarrolló el proceso de manufactura

2
aplicando una nueva tecnología para la obtención de tabletas recubiertas de
Paracetamol 500 mg + Diclofenaco sódico 50 mg, cumpliendo con las especificaciones
técnicas de calidad previamente establecidas. Asimismo, logramos una reducción
significativa en tiempos y costos de producción de las tabletas recubiertas de
Paracetamol 500 mg + Diclofenaco sódico 50 mg. Por lo tanto con el presente estudio
consideramos contribuir a la industria farmacéutica nacional en la búsqueda de la
automatización de sus procesos productivos; los cuales traen un impacto positivo en
las operaciones que desarrollan como laboratorios manufactureros.
1.1 OBJETIVOS
1.1.1 OBJETIVO GENERAL
Desarrollar el proceso de manufactura aplicando una nueva tecnología de
fabricación para la obtención de tabletas recubiertas de Paracetamol
500mg + Diclofenaco sódico 50 mg.
1.1.2 OBJETIVOS ESPECÍFICOS
Mejorar las etapas de fabricación: granulación, compresión y
recubrimiento para la obtención de las tabletas recubiertas, aplicando
nueva tecnología.
Demostrar por medio de los estudios de estabilidad, que las tabletas
recubiertas obtenidas por cambio de tecnología, se mantienen estables
durante el tiempo.
Reducir tiempo y costo en el proceso de fabricación del producto por el
cambio de tecnología.

3
II. GENERALIDADES
2.1 Conceptos generales
a) Formas farmacéuticas sólidas
Los comprimidos son formas farmacéuticas sólidas, cada una de las cuales
contiene una unidad de dosificación de uno o más principios activos. Se obtiene
por compresión de un volumen uniforme de partículas a partir de polvos,
cristales o granulados y con adición de diversos excipientes. Las tabletas
pueden ser producidas en una amplia variedad de tamaños, formas,
dependiendo del diseño de los punzones. (1-3)
b) Composición de las tabletas
Los comprimidos están constituidos por uno o más principios activos y por
excipientes que conjuntamente con la tecnología aplicada garantizan el
cumplimiento del objetivo terapéutico. Los excipientes, que son sustancias
adyuvantes, materiales farmacológicamente inertes, que se asocian al principio
activo y que nos permiten obtener una forma farmacéutica; para esto deben
poseer ciertas características físicas y mecánicas como fluidez adecuada,
cohesividad y lubricación. No tan solo tienen la finalidad de vehiculizar el
principio activo contribuyendo a la obtención de la forma de dosificación que
responda a las especificaciones previamente establecidas, sino que el mismo le
compete el permitir la disgregación o disolución o la liberación secuencial del
principio activo en la cavidad bucal o en el tracto gastrointestinal. A continuación
se revisan brevemente los distintos tipos de excipientes empleados en la
elaboración de comprimidos. (2-5)
c) Diluyentes
Un gran número de fármacos se utilizan a dosis relativamente bajas, en estos
casos, para producir comprimidos de un tamaño razonable, se precisa la adición
de agentes diluyentes, por ello la sustancia inerte (diluyente) se agrega para
aumentar el volumen, con el propósito de que la tableta tenga un tamaño

4
adecuado para la compresión. Es fundamental que el diluyente presente
adecuadas características de compresión. (2-5)
d) Desintegrantes
Son sustancias o mezclas de estas, que promueven y aceleran la desintegración
del comprimido cuando se pone en contacto con medio de naturaleza acuosa o
jugos gástricos, su objetivo principal es provocar la rápida disgregación del
comprimido, así como incrementar el área superficial de los fragmentos del
mismo, con el fin de conseguir la rápida liberación del principio activo. Las
sustancias activas deben liberarse de la matriz del comprimido tan efectivamente
como sea posible, rompiéndose las uniones formadas durante la compresión
como: la fuerza de Van Der Waals, uniones capilares, puentes de hidrógeno y
uniones de fusión o disolución parcial. (2-5)
e) Deslizantes
Son sustancias que mejoran las características de flujo de una mezcla de polvos,
donde es importante optimizar el orden de la adición y el proceso de mezclado
de estos materiales, con el objetivo de maximizar su efecto. Los deslizantes
pueden actuar por interposición entre partículas del granulado y formar una capa
protectora que reduce de este modo la fricción interparticular y la tendencia a su
adhesión; además, al introducirse en las rugosidades de los gránulos, hacen su
forma más regular y facilitan el llenado homogéneo de la matriz. (2-5)
f) Lubricantes
Los lubricantes actúan en la interface gránulo – metal, por lo que facilitan el
deslizamiento del granulo, la carga de la matriz y evitan adherencias de los
comprimidos a punzones y matrices, deben incorporarse al final de la etapa de
mezclado, evitando un mezclado excesivo para que la máxima cantidad posible
de lubricante quede retenida en la superficie de las partículas. (2-5)

5
g) Aglutinantes
Los agentes cohesivos o aglutinantes imparten a la formulación de la tableta
cohesividad que asegura que la formulación se mantenga intacta después de
comprimirla y mejora las cualidades de fluidez mediante la formación de
gránulos, del tamaño y la dureza que deseen. (2-5)
2.2 Método de fabricación de tabletas (2,3,10,11)
La importancia del proceso tecnológico en la elaboración de comprimidos, hace
que estos puedan clasificarse según el método de obtención: en comprimidos
obtenidos por compresión directa y comprimidos obtenidos por compresión de un
granulado.
a) Comprimidos obtenidos por compresión directa:
Es el proceso por el cual los comprimidos son obtenidos directamente por
compresión de mezclas de polvos de la sustancia activa y excipientes
apropiados, los cuales fluyen uniformemente en la cavidad de la matriz formando
un compacto firme, no siendo necesario el apretamiento de las mezclas de los
polvos por granulación húmeda y seca. Para lograr la compresión directa se
debe cumplir perfectamente estas dos premisas:
- Propiedades de flujo y deslizamiento
- Compactibilidad
b) Comprimidos obtenidos por compresión de un granulado
La granulación tiene como objetivo la transformación de partículas de polvo
cristalizado o amorfo en agregados sólidos más o menos resistentes y porosos
denominados granulados. Las partículas se unen mediante enlaces
interatómicos e intermoleculares de diferente naturaleza: fuerza de Van Der
Waals, enlaces por puente de hidrógeno, etc. Las principales razones por la que
se recurre a la granulación son:
- Prevenir la segregación de los componentes en el mezclado de polvos.

6
- Mejorar las propiedades de flujo de la mezcla
- Aumentar las características de compresión de la mezcla
- Favorecer la expulsión de aire interpuesto
- Reducir la cantidad de polvo generado en el proceso
- Mejorar la velocidad de disolución
- Incrementar la densidad del producto que se va a comprimir
c) Granulación húmeda
Es el método más clásico de elaboración de comprimidos y está basado en
promover la unión entre partículas mediante una sustancia aglutinante, con el
objetivo de incrementar el tamaño de la partícula y mejorar las propiedades de
flujo. Todo ello se consigue gracias a la granulación, operación que permite
obtener un granulado, a partir de una mezcla homogénea de partículas. La
granulación húmeda pretende transformar partículas irregulares, de tamaño muy
variado, a veces pequeño y de composición heterogénea, en partículas de
tamaño más grande y bastante parecido, esféricas y de igual composición. Esta
transformación permite de una parte mejorar el deslizamiento y de otra, limitar
las consecuencias del fenómeno de separación. Los pasos principales de la
granulación húmeda son: Mezclado - Amasado - Tamizado – Secado -
Tamizado y Lubricación.
d) Granulación seca
Este método es una alternativa de la granulación húmeda y se emplea para
principios activos que son sensibles a la humedad o no soportan temperaturas
altas durante el secado y cuando los constituyentes de las tabletas poseen
suficientes propiedades cohesivas intrínsecas. Consiste en crear aglomerados a
través de una pre-compresión, que son posteriormente desmoronados con
objeto de obtener una forma granular que fluye mucho más uniformemente que
la mezcla pulverulenta inicial, los pasos principales para la granulación seca son:
Mezclado - Pre-compresión – Molienda – Tamizado – Lubricación.

7
e) Compresión
La técnica de la compresión se realiza mediante dos punzones, uno superior y
otro inferior, y una matriz. Los punzones ejercen una fuerza axial sobre el
granulado o el polvo. Se trata de piezas metálicas, casi siempre cilíndricas. La
matriz es una pieza metálica perforada. Puede tener uno o varios orificios con
una sección circular, triangular o de otro tipo. La operación se basa en la
compresión axial del granulado o del polvo dentro de la cavidad (cámara de
compresión) de la matriz. La forma de esta cavidad y la de las superficies de
contacto de los punzones determinan el aspecto del comprimido: de bordes
cóncavos, convexos, lisos, con bisel, con forma oblonga, etc. Con esta operación
se busca una forma farmacéutica (comprimidos) cuya dosificación resulte
precisa, tenga una estabilidad máxima y cuya biodisponibilidad propicie el mayor
efecto terapéutico posible.
f) Recubrimiento Pelicular
El recubrimiento pelicular consiste en la deposición, habitualmente por
atomización, de una fina película de polímero que rodea al núcleo del
comprimido. El polímero puede ir disuelto en solvente orgánico o bien
dispersado en agua y adicionado en otros componentes. En este método, la
formulación del líquido de recubrimiento incluye agente filmógeno, solventes,
plastificantes y colorantes. El agente filmógeno de naturaleza polimérica
constituyen el componente esencial del recubrimiento. Se aplican disueltos o
dispersos en gran variedad de solventes, incluida el agua, y tiene la ventaja de
formar películas finas, lisas y mecánicamente resistentes.
2.3 Pruebas de caracterización sobre la mezcla pulverulenta (3,4,6,7)
Las pruebas de caracterización se llevan a cabo para poder realizar los ajustes
necesarios del producto, al inicio y durante el proceso de fabricación, detectando a
tiempo cualquier parámetro que no se encuentre dentro de las especificaciones
establecidas. Dentro de esto tenemos:

8
a) Aspecto:
El aspecto general del granulado, su identificación visual es esencial para el
cumplimiento de las especificaciones. El control de la apariencia implica el
estudio de varios parámetros tales como: color, textura, tamaño, etc.
b) Humedad
El contenido de humedad de una mezcla pulverulenta influye sobre sus
propiedades reológicas, así como sobre la estabilidad de sus componentes.
Además juega un papel importante en la compresión ya que puede alterar la
fluidez del producto, obteniéndose comprimidos de elevada friabilidad.
c) Granulometría
La granulometría es un parámetro de gran repercusión en las propiedades
reológicas de la mezcla pulverulenta y en las características finales de los
comprimidos, ya que una granulometría poco adecuada puede provocar la
aparición de fenómenos de desmezclado, con la consiguiente alteración de la
dosificación de los comprimidos y de algunas de sus propiedades.
d) Densidad
Es un parámetro tecnológico con una gran repercusión sobre sus propiedades
reológicas. Podemos distinguir los siguientes:
- Densidad aparente (dap)
- Densidad aparente de asentamiento (das)
- Índice de Carr o compresibilidad (%C)
Determina la capacidad de un polvo de formar una masa compacta al estar
sometido a una presión.
Se calcula a partir de la siguiente expresión:
𝐶(%) =𝑑𝑎𝑠 − 𝑑𝑎𝑝
𝑑𝑎𝑠 𝑥 100

9
e) Índice de Hausner (I.H.)
Este parámetro relaciona ambas densidades según la razón siguiente:
𝐼. 𝐻. = 𝑑𝑎𝑠
𝑑𝑎𝑝
La escala de fluidez está dada en la siguiente tabla:
Tabla 1: Escala de fluidez de granulados (15, 16)
2.4 Pruebas de caracterización farmacocinética de tabletas (3,4,6,7)
Las pruebas de caracterización se llevan a cabo para poder realizar los ajustes
necesarios del producto, al inicio y durante el proceso de compresión, detectando a
tiempo cualquier parámetro que no se encuentre dentro de las especificaciones
establecidas. Dentro de esto tenemos:
a) Aspecto y dimensiones
En la apariencia general de un comprimido, su identificación visual con
predominio de un aspecto elegante, es esencial en cuanto a la aceptación por
parte del paciente, y como consecuencia de ello, de las propiedades
biofarmacéuticas que se deba tener. El control de la apariencia externa de un
comprimido implica el estudio de varios parámetros tales como: forma, color,
Índice de
compresibilidad (%)
Característica de flujo Índice de Hausner
< 10 Excelente 1,00 – 1,11
11 – 15 Bueno 1,12 – 1,18
16 – 20 Adecuado 1,19 – 1,25
21 – 25 Aceptable 1,26 – 1,34
26 – 31 Pobre 1,35 – 1,45
32 – 37 Muy pobre 1,46 – 1,59
>38 Muy, muy pobre >1,60

10
tamaño, textura superficial, brillo, existencia o no de fenómenos como agrietado,
etc.
b) Dureza
Este ensayo engloba junto con los de resistencia de abrasión o friabilidad, y
resistencia a la deformación local o dureza dentro del apartado referente a las
propiedades mecánicas de los comprimidos. El ensayo podemos definirlo como
la fuerza mínima que es necesaria ejercer sobre el eje mayor del comprimido
para fracturarlo, en un aparato denominado durómetro.
c) Friabilidad
Expresa la resistencia que opone la superficie de los comprimidos sin cubierta a
la pérdida de masa por erosión; se lleva a cabo con aparatos que reciben el
nombre de friabilizador, que se encuentra descrito en la USP. Los límites
aceptados de friabilidad en comprimidos varían según autores entre 0,5 y el 1%
según las distintas farmacopeas.
d) Desintegración
La desintegración es la fase previa a la liberación del principio activo. Es la
rotura del comprimido en pequeñas partículas semejantes a la mezcla previa a la
compresión. Los mecanismos que se han descrito para la disgregación de un
comprimido son: Hinchamiento, desarrollo de una red capilar, energía liberada
por hidratación y separación- disociación.
e) Uniformidad de peso
El cumplimiento del peso del comprimido entre límites establecidos es un
requisito de interés terapéutico puesto que la actividad del mismo radica en la
dosis del principio activo administrado, que a su vez depende del peso del
comprimido.

11
f) Velocidad de disolución
La disponibilidad del principio activo es comprimidos depende, además del
tiempo de disgregación, de la velocidad de disolución del mismo. Este parámetro
está ligado a la eficacia terapéutica y tiene interés particularmente en
comprimidos de disponibilidad rápida y en los de liberación modificada. El
análisis se lleva a cabo en un equipo de paleta y un equipo de cestillo en los que
a intervalos de tiempo prefijado se retiran volumen del medio de disolución y de
acuerdo con el método analítico propuesto permiten conocer las cantidades de
principio activo en función del tiempo.
2.5 Características fisicoquímicas de los comprimidos(3,4,6,7)
a) Dosaje
En contenido del principio activo supone implícitamente la actividad terapéutica.
El cumplimiento de esta especificación y de la posible presencia o no de
sustancias relacionadas está estrechamente ligado a la seguridad del
medicamento. La aplicación de la técnica analítica prescrita en la monografía del
producto de una farmacopea o bien mediante una técnica convenientemente
validada por el propio laboratorio, permite evaluar la riqueza que se adecuará a
sus límites prefijados.
b) Uniformidad de contenido (L1)
La homogeneidad inicial de la mezcla a comprimir no presupone que se
mantenga a lo largo del proceso de compresión. El método se basa en
determinar los contenidos individuales del principio activo de 10 comprimidos
escogidos al azar y confirmar si los mismos están dentro de los límites
establecidos con respecto al dosaje media de la muestra. Se cumple con los
requisitos de uniformidad de dosificación si el valor de aceptación de las 10
primeras unidades de dosificación es menor o igual a L1 (uniformidad de
contenido). Si el valor de aceptación es mayor que L1, se analizan las siguientes
20 unidades y se calcula el valor de aceptación. Se cumple con los requisitos si

12
el valor de aceptación final de las 30 unidades de dosificación es menor o igual a
L1.
2.6 Características microbiológicas
Deben tomarse medidas adecuadas que garanticen la calidad microbiana durante
la fabricación de las preparaciones farmacéuticas.
2.7 Estabilidad(7,9, 14,15)
Es la capacidad de un producto farmacéutico para conservar sus propiedades
químicas, físicas, microbiológicas y biofarmacéuticas, dentro de los límites
especificados, a lo largo de su tiempo de conservación. Depende, por una parte de
factores ambientales tales como: temperatura, humedad y luz ambiental; así como
los factores relacionados con el producto, por ejemplo: las propiedades químicas y
físicas de las sustancias activas y de los excipientes farmacéuticos, la forma
farmacéutica y su composición, el proceso de fabricación, la naturaleza del sistema
de cierre del envase y las propiedades de los materiales de envase.
Objetivos a cumplir
- Determinar causas de alteración
- Identificar productos de degradación
- Detectar incompatibilidades
- Determinar período de validez
Para los fines de las pruebas de estabilidad en todo el mundo se distinguen cuatro
zonas climáticas (tabla 1):
- Zona I: Templada
- Zona II: Subtropical
- Zona III: Cálida/seca
- Zona IV: Cálida/húmeda
Condiciones climáticas medias de las cuatro zonas a nivel mundial.

13
Tabla 2: Condiciones climáticas de estabilidad (8)
ZONA Temperatura media
cinética (Cº)
Procedimiento anual de la humedad
relativa (%)
Zona I 21 45
Zona II 25 60
Zona III 30 35
Zona IV A 30 65
Zona IV B 30 75
Siendo para el Perú, la zona IV A como condición climática, de acuerdo a la última
directiva técnica de estabilidad de medicamentos Nº 031 MINSA/DIGEMID-V01 (25
de noviembre del 2009).
2.8 Tipos de estudio de estabilidad (7,9, 14,15)
a) Estudio de estabilidad en tiempo real y a largo plazo
Experimentos relacionados con las características físicas, químicas, biológicas,
biofarmacéuticas y microbiológicas de un medicamento, durante y más allá del
tiempo de conservación, y el período de almacenamiento previstos. Las
condiciones experimentales de almacenamiento serían tan parecidas a las
condiciones reales de almacenamiento previstas en el sistema de distribución
como sea factible. Los estudios de tiempo real tendrán que continuar hasta el
final del tiempo de conservación.
b) Estudios de estabilidad acelerada
Estudios ideados para aumentar la tasa de degradación química y de alteración
física de un medicamento sometiéndolo a condiciones de almacenamiento
excesivas como parte del programa estructurado de pruebas de estabilidad.

14
III. PARTE EXPERIMENTAL
3.1 Equipos, materiales y reactivos
3.1.1 Equipos de fabricación
Granulador de lecho fluido modelo FL-120 A
Mezclador granulador de alta velocidad modelo HLSG 200
Molino granulador tornado
Tableteadora rotativa modelo ZP – 1100
Recubridora de tabletas modelo BG – 150 E
Mezclado en “V” marca Zuma (capacidad de 60 kg)
Mezclador en “V” marca PK (capacidad de 180 kg)
Mezclador Octogonal (capacidad de 500 kg)
3.1.2 Equipos de análisis
Balanza analítica
Balanza de Humedad Mettler Toledo
Cromatografía líquida de alta performance (HPLC)
Desintegrador
Durómetro
Friabilizador
Disolutor
3.1.3 Materiales de fabricación
Tamices
Recipientes de acero inoxidable
Agitador neumático
Agitador manual de acero inoxidable
3.1.4 Materiales de análisis
Tamices
Agitador de tamices

15
3.1.5 Reactivos de análisis
Agua destilada
Metanol HPLC
Hidróxido de sodio
Fosfato monobásico de potasio
Hidróxido de potasio
Sodio fosfato monobásico monohidratado
3.1.6 Estándar de análisis
Diclofenaco sódico
Paracetamol
3.2 Metodología
3.2.1. Etapa preliminar: Evaluación de la tecnología a utilizarse
Se evaluó las características y parámetros a controlar en los equipos en las
diferentes etapas del proceso de manufactura de las Tabletas Recubiertas
de Paracetamol 500 mg + Diclofenaco sódico 50 mg y se comparó frente a
los equipos convencionales.
3.2.1.1 Granulador Lecho Fluido
Tabla 3. Características del Granulador lecho fluido
Característica Especificación
Modelo FL 120 A
Volumen del recipiente 420 L
Diámetro de recipiente 1 200 mm
Capacidad máxima 120 kg
Presión de Ingreso de aire 950 mm/H2O altura min
Temperatura Ajustable
Filtro del sistema de ingreso de aire Pre filtro y filtro HEPA de 99,9 %

16
3.2.1.2 Mezclador Granulador de alta velocidad
Tabla 4. Características del granulador de alta velocidad
- rpm: revoluciones por minuto
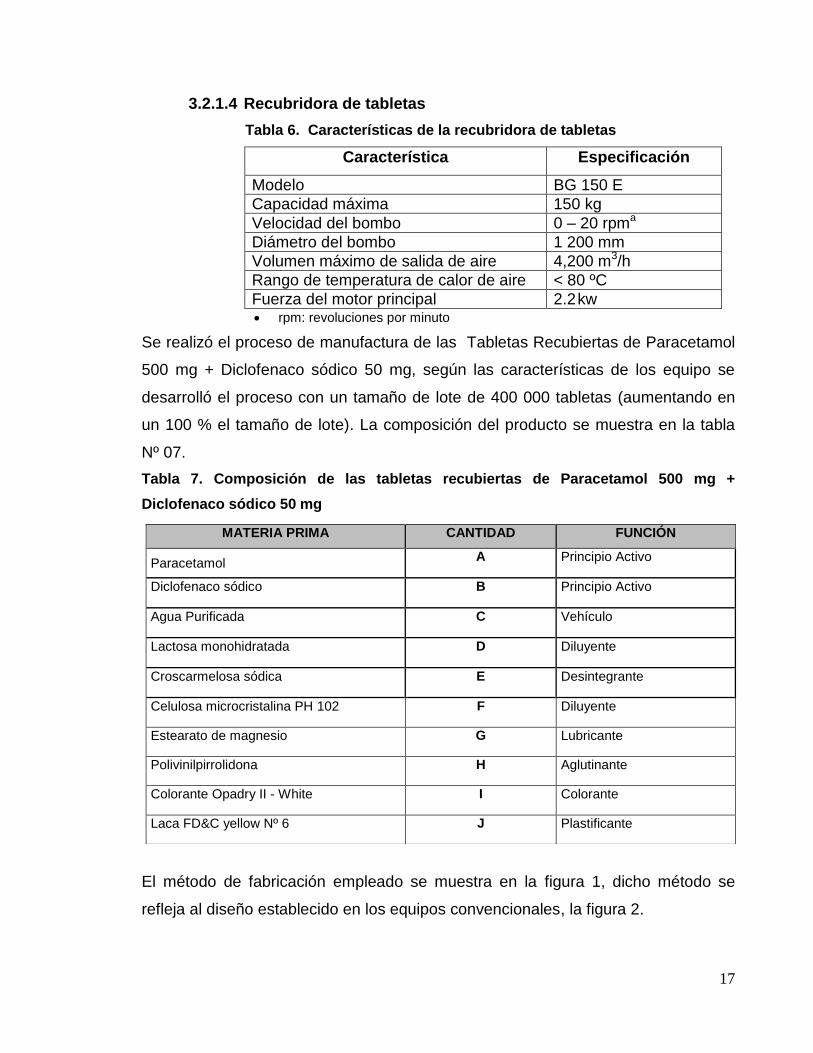
3.2.1.3 Tableteadora rotativa
Tabla 5. Características de la tableteadora rotativa
a) No aparecen unidades en el equipo, son marcaciones de posición.
b) N: Newton
c) rpm: revoluciones por minuto
Característica Especificación
Modelo Amasador HLSG 200
Dimensiones 2115 x 1650 x 2350 mm
Volumen de cámara 270 L
Volumen de trabajo 50 – 180 L
Velocidad de agitación de paleta 6 – 230rpma
Velocidad de cuchillas 300 – 3 000 rpma
Capacidad máxima 100 kg
Característica Especificación
Modelo ZP 1 100
Punzones 24
Tolva de alimentación (posición)a 3 -4
Marcación de durezaa 6 - 7
Marcación de llenado de matriza 10 - 11
Marcación de precompresióna 5 - 6
Límite de presión superior 270 – 280 Nb
Límite de presión inferior 30 – 40 Nb
Velocidad del motor principal 30 – 36 rpmc
17
3.2.1.4 Recubridora de tabletas
Tabla 6. Características de la recubridora de tabletas
Característica Especificación
Modelo BG 150 E
Capacidad máxima 150 kg
Velocidad del bombo 0 – 20 rpma
Diámetro del bombo 1 200 mm
Volumen máximo de salida de aire 4,200 m3/h
Rango de temperatura de calor de aire < 80 ºC
Fuerza del motor principal 2.2 kw rpm: revoluciones por minuto
Se realizó el proceso de manufactura de las Tabletas Recubiertas de Paracetamol
500 mg + Diclofenaco sódico 50 mg, según las características de los equipo se
desarrolló el proceso con un tamaño de lote de 400 000 tabletas (aumentando en
un 100 % el tamaño de lote). La composición del producto se muestra en la tabla
Nº 07.
Tabla 7. Composición de las tabletas recubiertas de Paracetamol 500 mg +
Diclofenaco sódico 50 mg
El método de fabricación empleado se muestra en la figura 1, dicho método se
refleja al diseño establecido en los equipos convencionales, la figura 2.
MATERIA PRIMA CANTIDAD FUNCIÓN
Paracetamol A Principio Activo
Diclofenaco sódico B Principio Activo
Agua Purificada C Vehículo
Lactosa monohidratada D Diluyente
Croscarmelosa sódica E Desintegrante
Celulosa microcristalina PH 102 F Diluyente
Estearato de magnesio G Lubricante
Polivinilpirrolidona H Aglutinante
Colorante Opadry II - White I Colorante
Laca FD&C yellow Nº 6 J Plastificante

18
a) Granulación
b) Tamizado
c) Mezclado (se realiza en dos parciales)
d) Compresión
e) Recubierta
f) Envasado
Figura 1: Diagrama de flujo del proceso de fabricación empleando nueva tecnología
(Equipos: amasador de alta velocidad, granulador de lecho fluido, tableteadora rotativa de
alta velocidad y recubridora de tabletas de bombo cerrado).
Lactosa monohidratada
Diclofenaco sódico Tamizar y Mezclar (Mezclador
en “V”)
Povidona
Agua purificada Dispersar Granular en lecho fluido
Secado en lecho fluido
Paracetamol
Celulosa microcristalina
Agua purificada
Amasar en amasador de alta
velocidad (3 parciales de amasado)
Molino tornado (malla
1,5 mm)
Povidona
Croscarmelosa sódica
Mezclador en “V”
Tamizar
Estearato de magnesio
Mezclador en “V”
Compresión
Laca FD&C yellow
Agua purificada
Colorante Opadry II
Homogenizado
de la suspensión
Recubrimiento y secado en
bombo cerrado
Envasado
Mezclador Octogonal (mezcla
de parcial 1 y 2)

19
a) Granulación
b) Tamizado
c) Mezclado
d) Compresión
e) Recubierta
f) Envasado
Figura 2: Diagrama de flujo del proceso de fabricación con tecnología convencional.
(Equipos: amasador planetario, secadora de lecho estático, tableteadora rotativa y
recubridora de tabletas de bombo abierto).
Lactosa monohidratada
Diclofenaco sódico Tamizar y Mezclar
Povidona
Agua purificada Dispersar Amasado
Secado en lecho estático
Celulosa microcristalina
Agua purificada Amasar
Molino Diaff (malla 1 mm)
Povidona
Croscarmelosa sódica
Mezclador en “V”
Tamizar
Estearato de magnesio Mezclador en “V”
Compresión
Laca FD&C yellow
Agua purificada
Colorante Opadry II
Homogenizado
de la suspensión
Recubrimiento y secado en
bombo abierto
Envasado
Paracetamol

20
3.2.2. Descripción del proceso de manufactura
Se estableció el proceso de manufactura de las tabletas recubiertas de
Paracetamol 500 mg + Diclofenaco 50 mg Tabletas, de acuerdo al siguiente
procedimiento de fabricación:
Tamizar por malla N° 16 y mezclar en el mezclador en “V”; las materias
primas: Diclofenaco sódico y Lactosa monohidratada.
Preparación de la solución granuladora: Dispersar la polivinilpirrolidona en
agua purificada hasta obtener una fase homogénea.
Granulación y secado: Colocar en el recipiente del granulador de lecho
fluido la mezcla e iniciar la granulación con la solución granuladora
preparada. Culminada la granulación iniciar el secado de acuerdo a la
programación ingresada al equipo, granulador lecho fluido.
Humectación (Amasado): Realizar la humectación en el mezclador-
granulador de alta velocidad e iniciar el funcionamiento del equipo de
acuerdo a la programación ingresada. La carga de las materias primas se
realiza como se indica a continuación: Paracetamol (1/2), celulosa
microcristalina, granulado seco Paracetamol (2/2).
Mezclado en el mezclador en “V” (se realiza en dos parciales iguales): La
mezcla se realiza con las siguientes materias primas: Granulado obtenido
en la humectación, croscamelosa sódica y polivinilpirrolidona.
Mezclado en el mezclador octogonal (mezcla de parcial 1 y 2). Se
procede a realizar la mezcla final de los parciales obtenidos y estearato
de magnesio, en el equipo mezclador octogonal.
Compresión: La compresión se realizó en la tableteadora rotativa modelo
ZP 1110, controlando los parámetros durante todo el proceso.
Recubrimiento: La etapa de recubrimiento se realizó en la recubridora de
tabletas modelo BG 150 E, controlando los parámetros durante todo el
proceso.

21
3.2.3. Estudio de caracterización de la mezcla pulverulenta (mezcla final)
Este estudio tiene como fin caracterizar el granulado obtenido para
determinar parámetros de aceptación que aseguren la reproducibilidad del
nuevo método de fabricación empleado. Para tener un mejor control del
proceso de manufactura se caracterizó el granulado obtenido, con la
finalidad de demostrar que el proceso se encontrará estable y con resultados
dentro de las especificaciones de calidad establecidas. A continuación se
detalla los tipos de análisis realizados:
a) Aspecto
En el presente estudio el granulado obtenido fue de color blanco libre de
partículas extrañas. Para la evaluación del aspecto se tomaron 3
muestras de granulado, divididos en tres niveles de muestreo, 1 superior,
1 mitad y 1 inferior del mezclador. Se realizó un análisis organoléptico
incidiendo en los siguientes puntos: uniformidad del color y ausencia de
partículas extrañas.
Criterio de aceptación:
- Conforme: Granulado de color blanco libre de partículas extrañas.
- No conforme: No cumple especificación.
b) Humedad
El contenido de humedad del granulado obtenido, bajo el nuevo diseño
del proceso de manufactura, se realizó por el método gravimétrico,
utilizando el equipo Mettler Toledo. Se tomaron tres muestras distribuidas
en tres niveles de muestreo: 1 superior, 1 medio y 1 inferior (tamaño de
muestra: 2 g).
Criterio de aceptación:
- Conforme: 3% - 6,5 %
- No conforme: Menor a 3% y mayor a 6,5 %.
c) Densidad aparente
Para la determinación de la densidad aparente del granulado obtenido se
utilizó el método de la probeta. Se tomaron tres muestras distribuidas en
tres niveles de muestreo: 1 superior, 1 medio y 1 inferior (tamaño de
muestra: 40 g). Se introdujo cuidadosamente la mezcla pulverulenta en

22
cada probeta seca de 100 mL, se leyó el volumen aparente de vertido (sin
sedimentar). En ensayo se realizó en cada nivel de muestreo.
d) Densidad aparente de asentamiento
Posteriormente obtenido los datos del polvo vertido en la probeta, de los
tres ensayos, se procedió a determinar la densidad por asentamiento,
mediante la secuencia de golpes por caídas. Luego de cada secuencia se
verificó el volumen obtenido, el ensayo se realizó en las tres muestras
obtenidas para la determinación de densidad aparente.
e) Índice de Hausner
Este parámetro se calculó mediante la relación de ambas densidades
según la siguiente razón:
𝐼. 𝐻. = 𝑑𝑎𝑠
𝑑𝑎𝑝
f) Índice de carr (índice de compresibilidad)
Determina la capacidad de un polvo para formar una masa compactada al
estar sometida a presión. Se calcula a partir de la siguiente expresión y en
función a las densidades obtenidas en cada nivel de muestreo (1 superior,
1 medio y 1 inferior).
𝐶(%) =𝑑𝑎𝑠 − 𝑑𝑎𝑝
𝑑𝑎𝑠 𝑥 100
g) Distribución granulométrica
Para la realización de la prueba el uso de tamices en cascada resultó
suficientemente útil para los rangos de tamaños de partículas que
utilizamos en nuestro estudio. El tamaño de apertura del tamiz es el
siguiente:
Tabla 8. Secuencia de tamices utilizados
TAMICES
Número de tamiz Tamaño de apertura (um)
20 850
40 425
60 250
80 180
100 150

23
Se taró los tamices y se anotó los pesos obtenidos, luego se procedió a
armar los tamices y se colocó la muestra (granulado sin compactar) de 20
g +5 g. Seguidamente, se procedió a ejecutar la prueba en el agitador de
tamices. La prueba se realizó por triplicado, muestras retiradas de tres
niveles de muestreo (1 superior, 1 medio y 1 inferior).
3.2.4. Estudio de caracterización de comprimidos (núcleos)
Para tener un mejor control del proceso de compresión se realizaron
pruebas al comprimido obtenido, con la finalidad de demostrar que el
proceso se encontrará estable y con resultados dentro de las
especificaciones de calidad establecidas. A continuación se detalla los tipos
de análisis realizados:
a) Aspecto
En el presente estudio los comprimidos obtenidos fueron de color blanco
con ligero moteado en forma oblonga y con ranura en una de sus caras.
Para el estudio de apariencia se tomaron 10 tabletas al inicio, 10 tabletas
a la mitad y 10 tabletas a la final del proceso de compresión. Se realizó un
análisis organoléptico incidiendo en los siguientes puntos: forma, textura
superficial (posible aparición de grietas, rugosidades, problemas de
laminado, etc.) y correcto biselado.
Criterio de aceptación:
- Conforme: Núcleos de color blanco con ligero moteado en forma
oblonga, con ranura en una de sus caras.
- No conforme: No cumple especificación
b) Dimensiones
En el presente trabajo los comprimidos presentan una altura de 5,7 – 6,10
mm. Para el estudio de las dimensiones se tomaron 10 tabletas al inicio,
10 tabletas a la mitad y 10 tabletas a la final del proceso de compresión.
Se realizó las mediciones con ayuda de un vernier calibrado.
Criterio de aceptación:
- Conforme: 5,70 - 6,10 mm

24
- No conforme: Menor a 5,70 mm y mayor a 6,10 mm
c) Humedad
El contenido de humedad de las muestras (núcleos), bajo el nuevo diseño
del proceso de manufactura, se realizó por el método gravimétrico,
utilizando el equipo Mettler Toledo. Se tomaron tres muestras en total
distribuidas en tres niveles de muestreo: 1 inicio, 1 medio y 1 final de la
operación de compresión (tamaño de muestra: 10 tabletas).
Criterio de aceptación:
- Conforme: 3% - 6%
- No conforme: Menor a 3% y mayor a 6%.
d) Peso promedio
La prueba se realizó según el método oficial descrito en la USP 35. Se
midió la variación de peso de 10 comprimidos cada 15 minutos, se
calcularon los valores medios y se realizó la prueba de capacidad de
proceso de los pesos obtenidos en la etapa de compresión.
Criterio de aceptación:
- Conforme: 760 mg/tab – 840 mg/tab.
- No conforme: Menor a 760 mg/tab y mayor a 840 mg/tab.
e) Dureza
Se tomaron muestras de 10 tabletas de la máquina tableteadora en
operación; con ayuda de una pinza metálica se colocaron en el durómetro
Erweka, se midió la fuerza en kilopond (kp) requerida para romper la
tableta, se calculó la media y registró. Se tomaron tres muestras en total
distribuidas en tres niveles de muestreo: 1 inicio, 1 medio y 1 final de la
operación de compresión (tamaño de muestra: 10 tabletas)
Criterio de aceptación:
- Conforme: 10 kp – 21 kp.
- No conforme: Menor a 10 kp y mayor a 21 kp.

25
f) Friabilidad
La friabilidad se evaluó a partir de la pérdida de peso porcentual de 10
comprimidos centrifugados en un friabilizador. Se tomaron tres muestras
en total distribuidas en tres niveles de muestreo: 1 inicio, 1 medio y 1 final
de la operación de compresión (tamaño de muestra: 10 tabletas).
Inicialmente se limpiaron los comprimidos y pesaron (P: peso de diez
comprimidos al inicio del ensayo), se introdujeron en el tambor de
friabilidad y ejecutaron las 100 rotaciones, se sacaron los comprimidos del
tambor, eliminando el polvo adherido a los mismos (P’: peso de los diez
comprimidos al final del ensayo). El porcentaje de pérdida de masa se
calculó mediante la siguiente expresión:
𝑓(%) = 𝑃 − 𝑃′
𝑃 𝑥 100
Criterio de aceptación:
- Conforme: Máximo 1%.
- No conforme: Diferente a lo especificado.
g) Desintegración
Para medir el tiempo requerido para la desintegración de seis
comprimidos, colocados cada uno en un tubo de un aparato de análisis de
desintegración USP (Erweka). Se tomaron tres muestras en total
distribuidas en tres niveles de muestreo: 1 inicio, 1 medio y 1 final de la
operación de compresión (tamaño de muestra: 6 tabletas).
Criterio de aceptación:
- Conforme: Máximo 30 minutos.
- No conforme: Diferente a lo especificado.
3.2.5. Estudio de caracterización de tabletas recubiertas
Para tener un mejor control del proceso de recubrimiento se realizaron
pruebas a las tabletas recubiertas, con la finalidad de demostrar que el
proceso se encontrará estable y con resultados dentro de las

26
especificaciones de calidad establecidas. A continuación se detalla las
pruebas realizadas:
a) Aspecto
En el presente trabajo las tabletas recubiertas Tabletas de forma de
oblonga con recubierta de color anaranjado (con ligero moteado propio
del colorante utilizado). Para el estudio de apariencia se tomaron 10
tabletas de cada parcial de recubierta (3 parciales de recubrimiento). Se
realizó un análisis organoléptico incidiendo sobre los distintos aspectos
como la uniformidad del color, forma, textura superficial (posible aparición
de grietas, rugosidades, etc.), brillo.
Criterio de aceptación:
- Conforme: Tabletas de forma de oblonga con recubierta de color
anaranjado (con ligero moteado propio del colorante utilizado).
- No conforme: Diferente a lo especificado.
b) Humedad
El contenido de humedad de las muestras de tabletas recubiertas, bajo el
nuevo diseño del proceso de manufactura, se realizó por el método
gravimétrico, utilizando el equipo Mettler Toledo. Se tomó tres muestras
en total distribuidas en tres niveles de muestreo: 1 muestra de cada
parcial de recubierta (3 parciales de recubrimiento), el análisis se realizó
con 10 tabletas, en cada punto de muestreo.
Criterio de aceptación:
- Conforme: 3% - 6%.
- No conforme: Menor a 3% y mayor a 6%.
c) Peso promedio
La prueba se realizó según el método oficial descrito en la USP 35. Se
midió la variación de peso de 20 tabletas recubiertas de cada parcial de

27
recubrimiento (3 parciales de recubrimiento) y se calcularon los valores
medios y la capacidad de proceso de las tabletas.
Criterio de aceptación:
- Conforme: 786,6 mg/tab – 869,4 mg/tab.
- No conforme: Menor a 786,6 mg/tab y mayor a 869,4 mg/tab.
d) Dureza
Se tomaron muestras de 10 tabletas de cada parcial de recubrimiento (3
parciales de recubrimiento); con ayuda de una pinza metálica se
colocaron en el durómetro Erweka, se midió la fuerza se midió la fuerza
en kilopond (kp) requerida para romper la tableta, se calculó la media y
registró.
Criterio de aceptación:
- Conforme: Mayor a 4 kp.
- No conforme: Diferente a lo especificado.
e) Friabilidad
La friabilidad se evaluó a partir de la pérdida de peso porcentual de 10
comprimidos centrifugados en un friabilizador, el análisis de realizo a cada
parcial de recubierta (3 parciales de recubrimiento). Inicialmente se
limpiaron los comprimidos y pesaron (P: peso de diez comprimidos al
inicio del ensayo), se introdujeron en el tambor de friabilidad y ejecutaron
las 100 rotaciones, se sacaron los comprimidos del tambor, eliminando el
polvo adherido a los mismos (P’: peso de los diez comprimidos al final del
ensayo). El porcentaje de pérdida de masa se calculó mediante la
siguiente expresión:
𝑓(%) = 𝑃 − 𝑃′
𝑃 𝑥 100
Criterio de aceptación:
- Conforme: Mayor a 1%.
- No conforme: Diferente a lo especificado.
28
f) Desintegración
Para medir el tiempo requerido para la desintegración de seis
comprimidos, en cada parcial de recubrimiento, colocados cada uno en un
tubo de un aparato de análisis de desintegración USP (Erweka).
Criterio de aceptación:
- Conforme: Máximo 30 minutos.
- No conforme: Diferente a lo especificado.
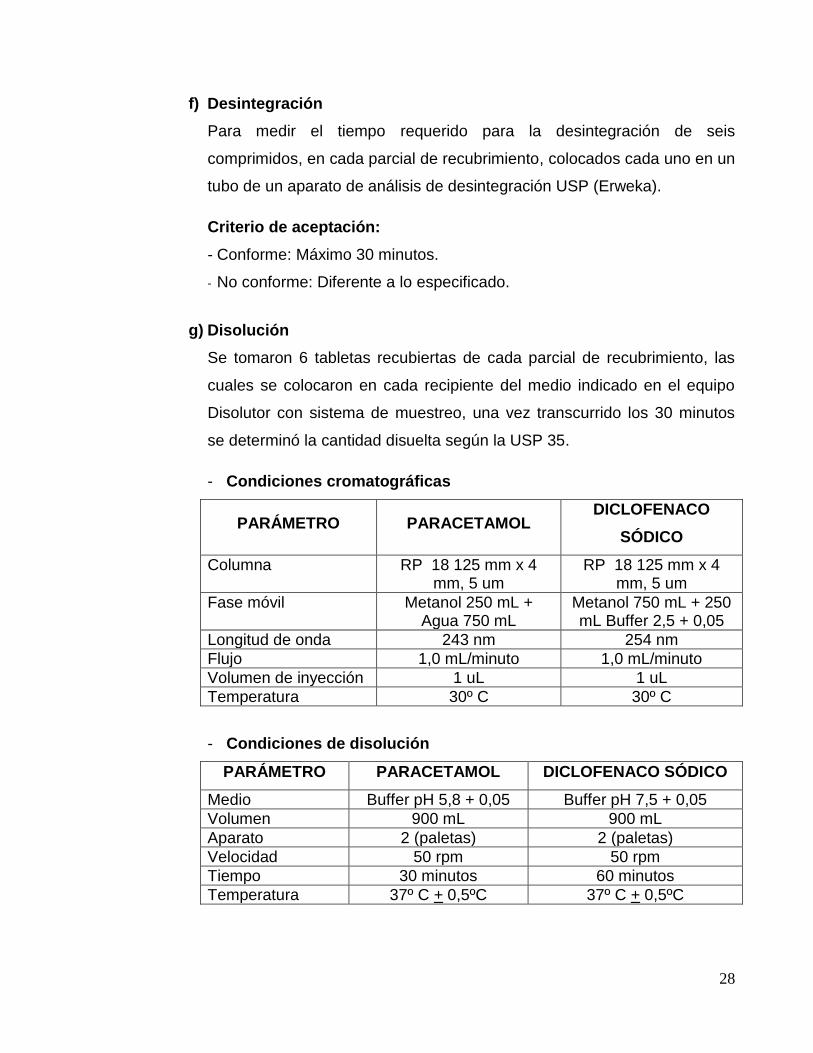
g) Disolución
Se tomaron 6 tabletas recubiertas de cada parcial de recubrimiento, las
cuales se colocaron en cada recipiente del medio indicado en el equipo
Disolutor con sistema de muestreo, una vez transcurrido los 30 minutos
se determinó la cantidad disuelta según la USP 35.
- Condiciones cromatográficas
PARÁMETRO PARACETAMOL DICLOFENACO
SÓDICO
Columna RP 18 125 mm x 4 mm, 5 um
RP 18 125 mm x 4 mm, 5 um
Fase móvil Metanol 250 mL + Agua 750 mL
Metanol 750 mL + 250 mL Buffer 2,5 + 0,05
Longitud de onda 243 nm 254 nm
Flujo 1,0 mL/minuto 1,0 mL/minuto
Volumen de inyección 1 uL 1 uL
Temperatura 30º C 30º C
- Condiciones de disolución
PARÁMETRO PARACETAMOL DICLOFENACO SÓDICO
Medio Buffer pH 5,8 + 0,05 Buffer pH 7,5 + 0,05
Volumen 900 mL 900 mL
Aparato 2 (paletas) 2 (paletas)
Velocidad 50 rpm 50 rpm
Tiempo 30 minutos 60 minutos
Temperatura 37º C + 0,5ºC 37º C + 0,5ºC

29
Criterio de aceptación:
- Conforme: Diclofenaco sódico: No menos de 80% (Q) en 60 minutos.
Paracetamol: No menos de 80% (Q) en 30 minutos.
- No conforme: Diferente a lo especificado.
3.2.6. Estudio de caracterizaciones fisicoquímicas
Se realizaron pruebas fisicoquímicas en las diferentes etapas del proceso,
con la finalidad de demostrar que el proceso es seguro, estable y produce
resultados dentro de las especificaciones de calidad establecidas. A
continuación se detalla los tipos de análisis realizados:
a) Dosaje de diclofenaco sódico
Para esto se analizaron muestras obtenidas de las etapas de granulación,
compresión y recubrimiento, cada muestra se analizó individualmente por
el método de cromatografía líquida de alta performance (HPLC), según lo
indicado en la técnica analítica.
CONDICIONES CROMATOGRÁFICAS DICLOFENACO SÓDICO
Columna RP 18 125 mm x 4 mm, 5 um
Fase móvil Metanol 750 mL + 250 mL Buffer 2,5 + 0,05
Longitud de onda 254 nm
Flujo 1,0 mL/minuto
Volumen de inyección 1 uL
Temperatura 30º C
b) Dosaje de paracetamol
Para esto se analizaron muestras obtenidas de las etapas de granulación,
compresión y recubrimiento, cada muestra se analizó individualmente por
el método de cromatografía líquida de alta performance (HPLC), según lo
indicado en la técnica analítica.

30
CONDICIONES CROMATOGRÁFICAS PARACETAMOL
Columna RP 18 125 mm x 4 mm, 5 um
Fase móvil Metanol 250 mL + Agua 750 mL
Longitud de onda 243 nm
Flujo 1,0 mL/minuto
Volumen de inyección 1 uL
Temperatura 30º C
c) Uniformidad de contenido
Para esto se analizaron muestras en las etapas de compresión y
recubierta, cada muestra se analizó individualmente por el método de
cromatografía líquida de alta performance (HPLC) y por variación de peso
para el Diclofenaco sódico y Paracetamol respectivamente, según lo
indicado en la técnica analítica.
3.2.7. Estudio de propiedades microbiológicas
En el presente estudio se realizaron pruebas microbiológicas en las
diferentes etapas del proceso (mezcla, compresión y recubrimiento), con la
finalidad de demostrar que el proceso es seguro y se mantiene las
condiciones microbiológicas dentro de los parámetros previamente
establecidos. A continuación se detalla los tipos de análisis realizados:
Tabla 9. Pruebas de análisis microbiológico
RECUENTO MICROBIANO ESPECIFICACIONES
Recuento total de microorganismos aerobios < 103 UFC/g
Recuento Total Combinado de Hongos Filamentosos y Levaduras
< 102 UFC/g
MICROORGANISMOS ESPECÍFICOS ESPECIFICACIONES
Escherichia coli AUSENCIA/g
Salmonella spp. AUSENCIA/10 g
Pseudomonas aeruginosa AUSENCIA/g
Staphylococcus aureus AUSENCIA/g

31
3.2.8. Estudio de estabilidad: El producto obtenido fue sometido a estabilidad
bajo las siguientes condiciones ambientales:
zona II (25°C +/- 2 °C y 60 % +/- 5%)
zona IVb (30 °C + 2 °C / 75% + 5%)
El estudio de estabilidad se realizará al tiempo inicial, sexto y noveno mes,
durante las cuales se realizaron las pruebas físicas y fisicoquímicas de
acuerdo a las especificaciones de calidad establecidas.
3.2.9. Estudio del impacto de la transferencia e incremento de tamaño de lote
en el tiempo y costo
Se realizará una evaluación comparativa de las horas hombre y horas
máquina, entre la nueva tecnología y la tecnología convencional, de igual
forma el ahorro generado por el incremento de tamaño de lote.
3.3 Especificaciones de las tabletas recubiertas
3.3.1 Etapa del proceso: Producto intermedio (granulado)
Tabla 10. Especificaciones del granulado
3.3.2 Etapa del proceso: Producto intermedio (Núcleo)
Tabla 11. Especificaciones de tabletas sin recubrir
DESCRIPCIÓN ESPECIFICACIONES
Aspecto Granulado de color blanco libre de partículas extrañas.
Humedad(Método OHAUS) 3 – 6,5 %
DESCRIPCIÓN ESPECIFICACIONES
Aspecto Núcleos de color blanco con ligero moteado en forma de oblonga de 20 mm x 7 mm con ranura en una de sus caras.
Altura 5,70 - 6,10 mm
Peso promedio 800 mg/tab + 5,0% 760 mg/tab - 840 mg/tab

32
3.3.3 Etapa del proceso: GRANEL (tabletas recubiertas)
Tabla 12. Especificaciones de tabletas recubiertas
Desintegración Máximo 30 minutos
Dureza 10 Kp - 21 Kp
Friabilidad Máximo 1%
DESCRIPCIÓN ESPECIFICACIONES
Aspecto Tabletas de forma de oblonga con recubierta de color anaranjado (con ligero moteado propio del colorante utilizado).
Peso promedio 828 mg/tab
786,6 mg/tab - 869,4 mg/tab 95% - 105%
Desintegración (Con discos en agua)
Máximo 30 minutos.
Dureza Mayor a 4 Kp
Friabilidad Máximo 1%
IDENTIFICACIÓN ESPECIFICACIONES
Diclofenaco sódico POSITIVO
Paracetamol POSITIVO
CUANTIFICACIÓN ESPECIFICACIONES
Diclofenaco sódico 50 mg/ tab.
(45 mg/tab. – 55 mg/tab.) 90% -110%
Paracetamol 500 mg/ tab.
(450 mg/tab. – 550 mg/tab.) 90% -110%
Uniformidad de unidades de dosificación Valor de Aceptación (V.A.) DICLOFENACO
L1 < 15
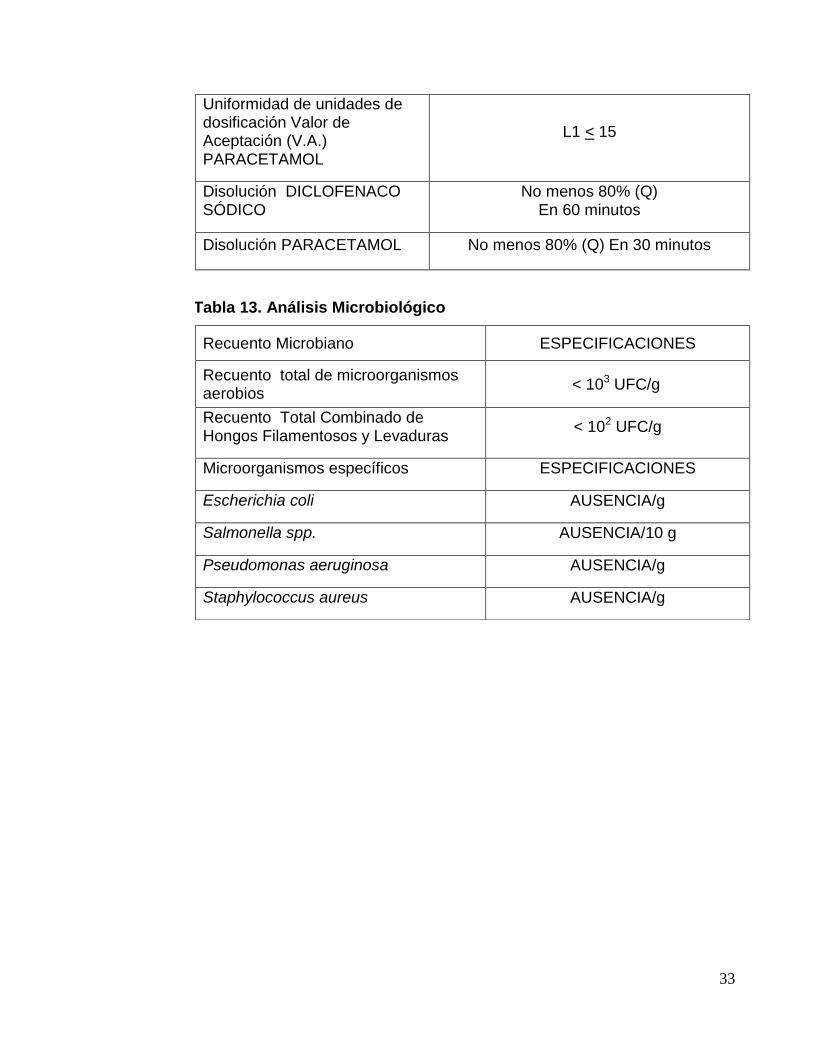
33
Uniformidad de unidades de dosificación Valor de Aceptación (V.A.) PARACETAMOL
L1 < 15
Disolución DICLOFENACO SÓDICO
No menos 80% (Q) En 60 minutos
Disolución PARACETAMOL No menos 80% (Q) En 30 minutos
Tabla 13. Análisis Microbiológico
Recuento Microbiano ESPECIFICACIONES
Recuento total de microorganismos aerobios
< 103 UFC/g
Recuento Total Combinado de Hongos Filamentosos y Levaduras
< 102 UFC/g
Microorganismos específicos ESPECIFICACIONES
Escherichia coli AUSENCIA/g
Salmonella spp. AUSENCIA/10 g
Pseudomonas aeruginosa AUSENCIA/g
Staphylococcus aureus AUSENCIA/g
34
IV. RESULTADOS
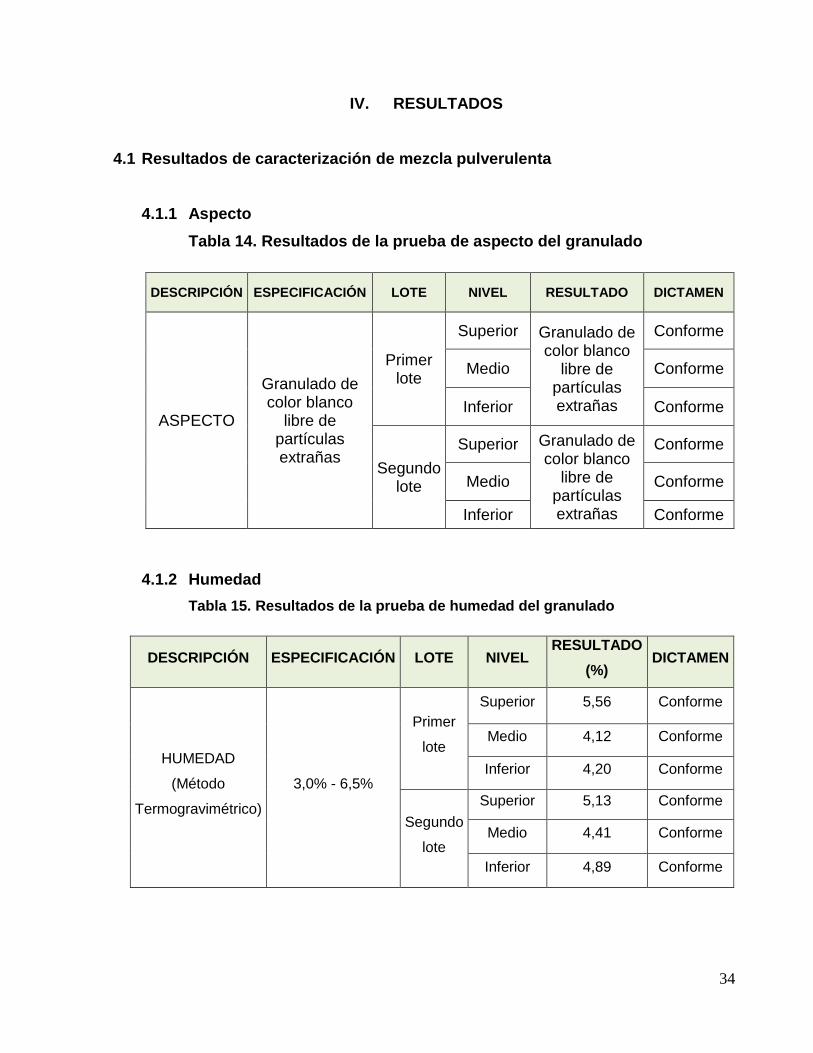
4.1 Resultados de caracterización de mezcla pulverulenta
4.1.1 Aspecto
Tabla 14. Resultados de la prueba de aspecto del granulado
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO DICTAMEN
ASPECTO
Granulado de color blanco
libre de partículas extrañas
Primer lote
Superior Granulado de color blanco
libre de partículas extrañas
Conforme
Medio Conforme
Inferior Conforme
Segundo lote
Superior Granulado de color blanco
libre de partículas extrañas
Conforme
Medio Conforme
Inferior Conforme
4.1.2 Humedad
Tabla 15. Resultados de la prueba de humedad del granulado
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
(%) DICTAMEN
HUMEDAD
(Método
Termogravimétrico)
3,0% - 6,5%
Primer
lote
Superior 5,56 Conforme
Medio 4,12 Conforme
Inferior 4,20 Conforme
Segundo
lote
Superior 5,13 Conforme
Medio 4,41 Conforme
Inferior 4,89 Conforme

35
Figura 3. Comparación de humedad en los lotes evaluados.
4.1.3 Densidad aparente
Tabla 16. Resultados de la prueba de densidad aparente del granulado
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
DENSIDAD
APARENTE Referencial
Primer
lote
Superior 0,555 g/mL
Medio 0,563 g/mL
Inferior 0,556 g/mL
Segundo
lote
Superior 0,500 g/mL
Medio 0,510 g/mL
Inferior 0,530 g/mL
0.00%
1.00%
2.00%
3.00%
4.00%
5.00%
6.00%
7.00%
Superior Medio Inferior
5.56%
4.12% 4.20%
5.13% 4.41% 4.89%
% d
e H
um
edad
Nivel de Muestreo
Primer lote
Segundo lote

36
Figura 4. Comparación de la densidad aparente en los lotes evaluados
4.1.4 Densidad aparente de asentamiento
Tabla 17. Resultados de la prueba de densidad compactada del granulado
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
DENSIDAD
COMPACTADA Referencial
Primer
lote
Superior 0,654 g/mL
Medio 0,655 g/mL
Inferior 0,657 g/mL
Segundo
lote
Superior 0,580 g/mL
Medio 0,600 g/mL
Inferior 0,610 g/mL
0.46
0.48
0.5
0.52
0.54
0.56
0.58
SUPERIOR MEDIO INFERIOR
DEN
SID
AD
AP
AR
ENTE
(g/
mL)
NIVEL DE MUESTREO
PRIMER LOTE
SEGUNDO LOTE

37
Figura 5. Comparación de la densidad compactada en los lotes evaluados.
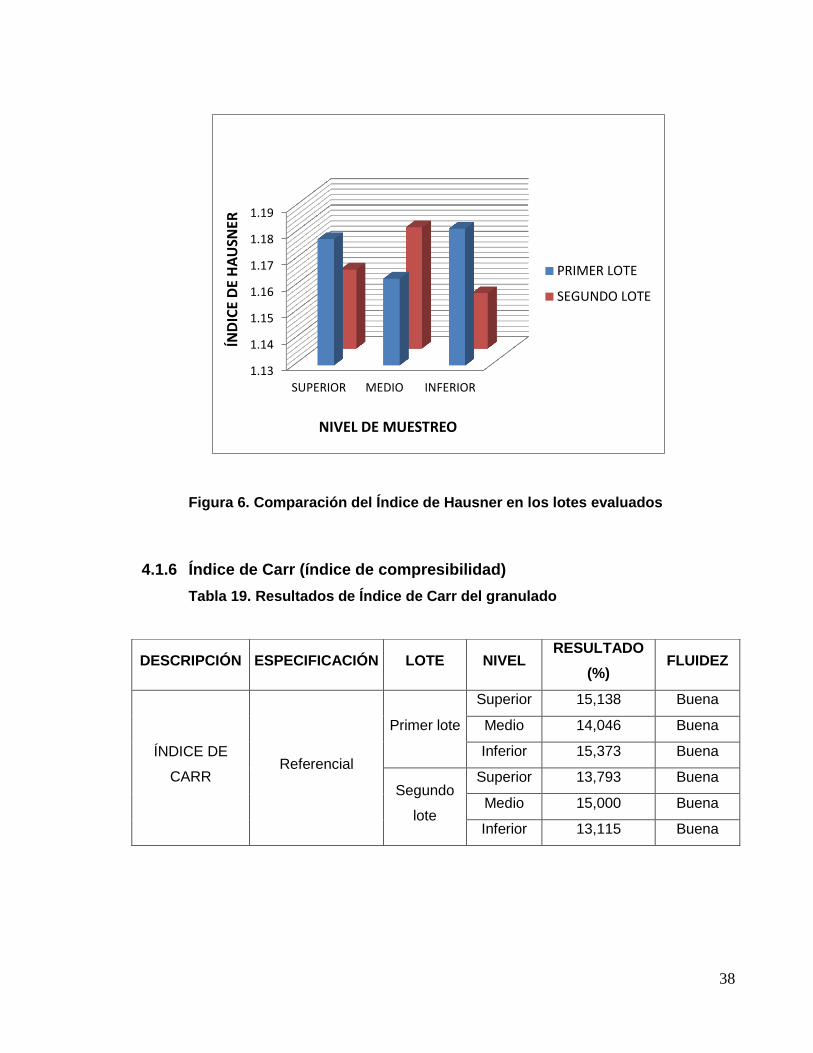
4.1.5 Índice de Hausner
Tabla 18. Resultados de Índice de Hausner del granulado.
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO FLUIDEZ
ÍNDICE DE
HAUSNER Referencial
Primer
lote
Superior 1,178 Buena
Medio 1,163 Buena
Inferior 1,182 Buena
Segundo
lote
Superior 1,160 Buena
Medio 1,176 Buena
Inferior 1,151 Buena
0.5
0.55
0.6
0.65
0.7
SUPERIOR MEDIO INFERIOR
DEN
SID
AD
CO
MP
AC
TAD
A (
g/m
L)
NIVEL DE MUESTREO
PRIMER LOTE
SEGUNDO LOTE
38
Figura 6. Comparación del Índice de Hausner en los lotes evaluados
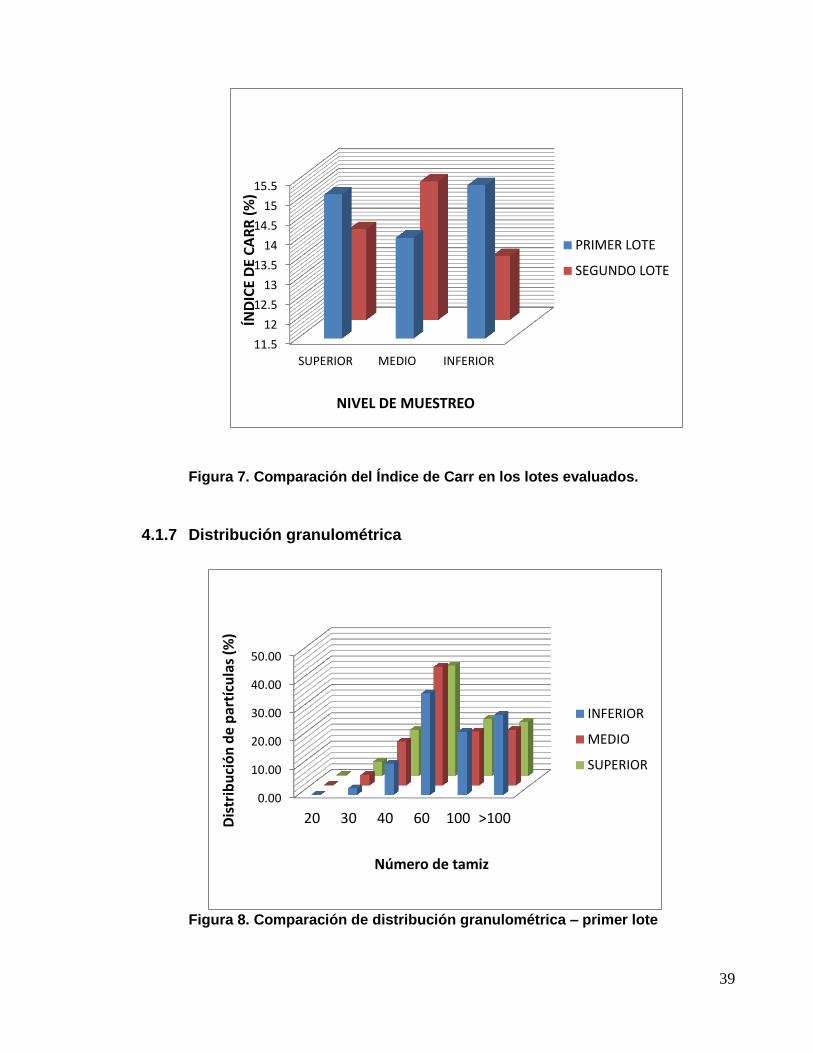
4.1.6 Índice de Carr (índice de compresibilidad)
Tabla 19. Resultados de Índice de Carr del granulado
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
(%) FLUIDEZ
ÍNDICE DE
CARR Referencial
Primer lote
Superior 15,138 Buena
Medio 14,046 Buena
Inferior 15,373 Buena
Segundo
lote
Superior 13,793 Buena
Medio 15,000 Buena
Inferior 13,115 Buena
1.13
1.14
1.15
1.16
1.17
1.18
1.19
SUPERIOR MEDIO INFERIOR
ÍND
ICE
DE
HA
USN
ER
NIVEL DE MUESTREO
PRIMER LOTE
SEGUNDO LOTE
39
Figura 7. Comparación del Índice de Carr en los lotes evaluados.
4.1.7 Distribución granulométrica
Figura 8. Comparación de distribución granulométrica – primer lote
0.00
10.00
20.00
30.00
40.00
50.00
20 30 40 60 100 >100Dis
trib
uci
ón
de
par
tícu
las
(%)
Número de tamiz
INFERIOR
MEDIO
SUPERIOR
11.5
12
12.5
13
13.5
14
14.5
15
15.5
SUPERIOR MEDIO INFERIOR
ÍND
ICE
DE
CA
RR
(%
)
NIVEL DE MUESTREO
PRIMER LOTE
SEGUNDO LOTE

40
Figura 9. Comparación de distribución granulométrica – segundo lote
4.2 Dosaje de principios activos (mezcla sin lubricante)
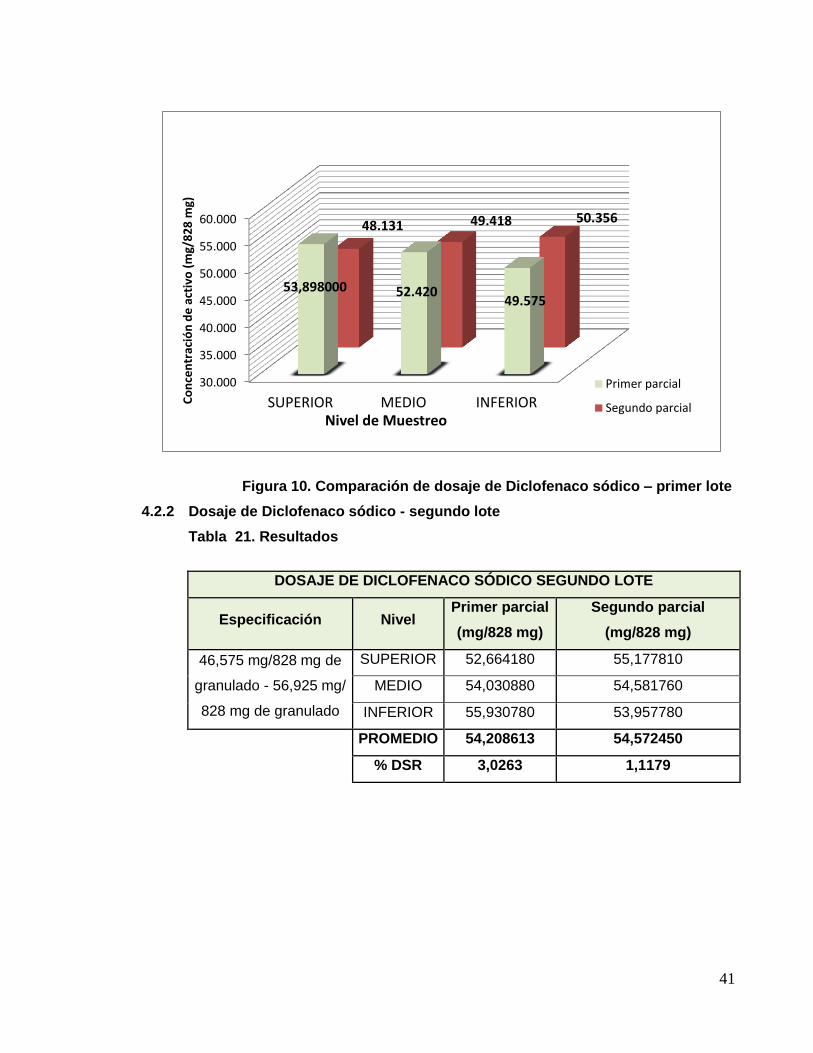
4.2.1 Dosaje de Diclofenaco sódico - primer lote
Tabla 20. Resultados de dosaje de Diclofenaco sódico – primer lote
DOSAJE DE DICLOFENACO SÓDICO PRIMER LOTE
Especificación Nivel Primer parcial
(mg/828 mg)
Segundo parcial
(mg/828 mg)
46,575 mg/828 mg de
granulado - 56,925 mg/ 828
mg de granulado
SUPERIOR 53,898000 48,130920
MEDIO 52,420320 49,418160
INFERIOR 49,575010 50,356240
PROMEDIO 51,964443 49,301773
% DSR 4,2284 2,2661
0.005.00
10.0015.0020.0025.0030.0035.0040.0045.00
20 30 40 60 100 >100
Dis
trib
uci
ón
de
par
tícu
las
(%)
Número de tamiz
Segundo lote
41
Figura 10. Comparación de dosaje de Diclofenaco sódico – primer lote
4.2.2 Dosaje de Diclofenaco sódico - segundo lote
Tabla 21. Resultados
DOSAJE DE DICLOFENACO SÓDICO SEGUNDO LOTE
Especificación Nivel Primer parcial
(mg/828 mg)
Segundo parcial
(mg/828 mg)
46,575 mg/828 mg de
granulado - 56,925 mg/
828 mg de granulado
SUPERIOR 52,664180 55,177810
MEDIO 54,030880 54,581760
INFERIOR 55,930780 53,957780
PROMEDIO 54,208613 54,572450
% DSR 3,0263 1,1179
30.000
35.000
40.000
45.000
50.000
55.000
60.000
SUPERIOR MEDIO INFERIOR
53,898000 52.420 49.575
48.131 49.418 50.356
Co
nce
ntr
ació
n d
e a
ctiv
o (
mg/
82
8 m
g)
Nivel de Muestreo
Primer parcial
Segundo parcial

42
Figura 11. Comparación de dosaje de Diclofenaco sódico – segundo lote.
4.2.3 Dosaje de Diclofenaco sódico en los lotes evaluados
Figura 12. Comparación de dosaje de Diclofenaco sódico en los lotes
evaluados.
30.000
35.000
40.000
45.000
50.000
55.000
60.000
SUPERIOR MEDIO INFERIOR
52.664 54.031 55.931
55.178 54.582 53.958
Co
nce
ntr
ació
n d
e a
ctiv
o (
mg/
82
8 m
g)
Nivel de muestreo
Primer parcial
Segundo parcial
30.000
35.000
40.000
45.000
50.000
55.000
60.000
Primer parcial Segundo parcial
51.964 49.302
54.209 54.572
Co
nce
ntr
ació
n d
e ac
tivo
(m
g/8
28
mg)
PACIALES DE MUESTREO
PRIMER LOTE
SEGUNDO LOTE
43
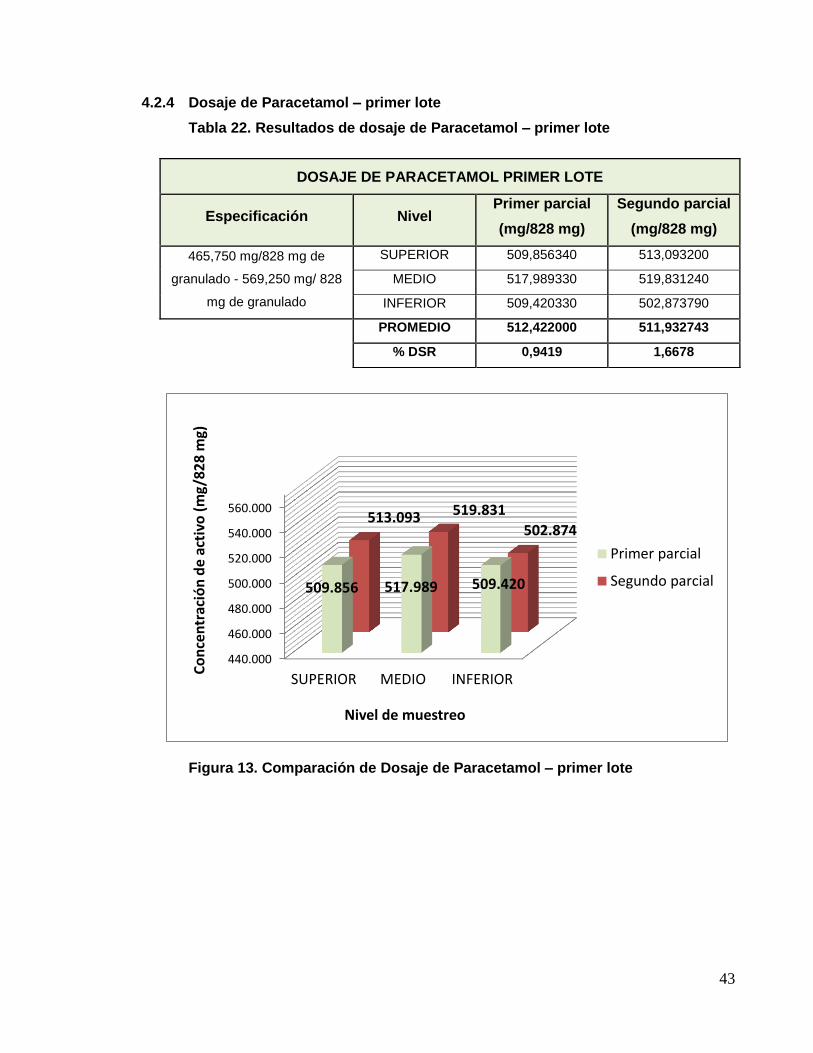
4.2.4 Dosaje de Paracetamol – primer lote
Tabla 22. Resultados de dosaje de Paracetamol – primer lote
Figura 13. Comparación de Dosaje de Paracetamol – primer lote
DOSAJE DE PARACETAMOL PRIMER LOTE
Especificación Nivel Primer parcial
(mg/828 mg)
Segundo parcial
(mg/828 mg)
465,750 mg/828 mg de
granulado - 569,250 mg/ 828
mg de granulado
SUPERIOR 509,856340 513,093200
MEDIO 517,989330 519,831240
INFERIOR 509,420330 502,873790
PROMEDIO 512,422000 511,932743
% DSR 0,9419 1,6678
440.000
460.000
480.000
500.000
520.000
540.000
560.000
SUPERIOR MEDIO INFERIOR
509.856 517.989 509.420
513.093 519.831
502.874
Co
nce
ntr
ació
n d
e ac
tivo
(m
g/8
28
mg)
Nivel de muestreo
Primer parcial
Segundo parcial

44
Tabla Nº 23: Resultados de dosaje de Paracetamol – segundo lote
Figura 14. Comparación de dosaje de Paracetamol en los lotes evaluados.
DOSAJE DE PARACETAMOL SEGUNDO LOTE
Especificación Nivel Primer parcial
(mg/828 mg)
Segundo parcial
(mg/828 mg)
465,750 mg/828 mg de
granulado - 569,250
mg/ 828 mg de
granulado
SUPERIOR 503,305990 501,370000
MEDIO 510,753480 502,072820
INFERIOR 479,758240 504,253160
PROMEDIO 497,939237 502,565327
% DSR 3,2493 0,2991
440.000
460.000
480.000
500.000
520.000
540.000
560.000
SUPERIOR MEDIO INFERIOR
503.306 510.753 479.758
501.370 502.073 504.253
Co
nce
ntr
ació
n d
e ac
tivo
(m
g/8
28
mg)
Nivel de muestreo
Primer parcial
Segundo parcial

45
4.3 Dosaje de principios activos (mezcla con lubricante)
Tabla 24. Resultados de dosaje de Diclofenaco sódico en los lotes evaluados
Figura 15. Comparación de dosaje de Diclofenaco sódico en los lotes evaluados
DOSAJE DE DICLOFENACO SÓDICO
Especificación Nivel Primer lote
(mg/828 mg) Segundo lote (mg/828 mg)
46,575 mg/828 mg de granulado -
56,925 mg/ 828 mg de granulado
INICO 53,42411 53,46516
MEDIO 53,98433 52,90049
FINAL 53,46819 53,72644
PROMEDIO 53,625542 53,364031
% DSR 0,5809 0,7911
30.000
35.000
40.000
45.000
50.000
55.000
60.000
INICO MEDIO FINAL
53.42411 53.98433 53.46819
53.46516 52.90049 53.72644
Co
nce
ntr
ació
n d
e ac
tivo
(m
g/8
28
mg)
Niveles de muestreo
Primer lote
Segundo lote
46
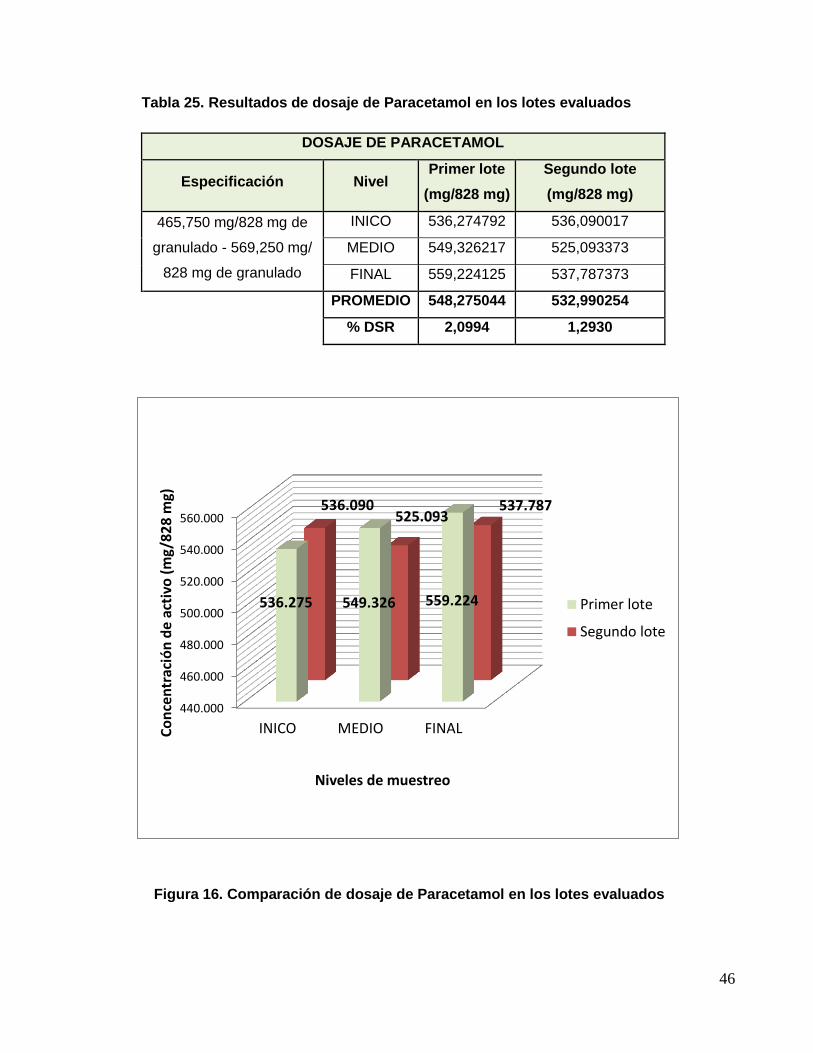
Tabla 25. Resultados de dosaje de Paracetamol en los lotes evaluados
DOSAJE DE PARACETAMOL
Especificación Nivel Primer lote
(mg/828 mg)
Segundo lote
(mg/828 mg)
465,750 mg/828 mg de
granulado - 569,250 mg/
828 mg de granulado
INICO 536,274792 536,090017
MEDIO 549,326217 525,093373
FINAL 559,224125 537,787373
PROMEDIO 548,275044 532,990254
% DSR 2,0994 1,2930
Figura 16. Comparación de dosaje de Paracetamol en los lotes evaluados
440.000
460.000
480.000
500.000
520.000
540.000
560.000
INICO MEDIO FINAL
536.275 549.326 559.224
536.090 525.093
537.787
Co
nce
ntr
ació
n d
e ac
tivo
(m
g/8
28
mg)
Niveles de muestreo
Primer lote
Segundo lote
47
4.4 Estudio de caracterización de comprimidos (núcleos)
4.4.1 Aspecto
Tabla 26. Resultados de aspecto de los núcleos de los lotes evaluados
4.4.2 Dimensiones
Tabla 27. Resultados de dimensiones de los núcleos de los lotes evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO DICTAMEN
ASPECTO
Núcleos de color blanco con ligero
moteado en forma de
oblonga de 20 mm x 7 mm con ranura en una de
sus caras
Primer lote
Inicio Núcleos de color blanco con ligero moteado en
forma de oblonga de 20 mm x 7 mm con ranura
en una de sus caras
Conforme
Medio Conforme
Final Conforme
Segundo lote
Inicio
Núcleos de color blanco con ligero moteado en
forma de oblonga de 20 mm x 7 mm con ranura
en una de sus caras
Conforme
Medio Conforme
Final Conforme
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
(mm) DICTAMEN
ALTURA 5,70 - 6,10 mm
Primer
lote
Inicio 6,01 Conforme
Medio 5,98 Conforme
Final 5,96 Conforme
Segundo
lote
Inicio 6,03 Conforme
Medio 6,02 Conforme
Final 6,06 Conforme

48
Figura 17. Comparación de la tendencia de las dimensiones en los lotes
evaluados
4.4.3 Humedad
Tabla Nº 28: Resultados de humedad de los núcleos de los lotes evaluados.
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO DICTAMEN
HUMEDAD
(Método
Termogravimétrico)
3,0% - 6,5%
Primer
lote
Inicio 4,57% Conforme
Medio 4,58% Conforme
Final 4,64% Conforme
Segundo
lote
Inicio 4,63% Conforme
Medio 4,58% Conforme
Final 4,70% Conforme
5.80
5.82
5.84
5.86
5.88
5.90
5.92
5.94
5.96
5.98
6.00
6.02
6.04
6.06
6.08
6.10
1 3 5 7 9 11 13 15 17 19 21 23 25 27 29
ALT
UR
A (
mm
)
Nùmero de muestra
PRIMER LOTE
SEGUNDO LOTE

49
Figura 18. Comparación de humedad en los lotes evaluado
4.4.4 Peso promedio
Figura 19. Gráfica de control de los pesos promedios – primer lote.
LES: límite de especificación superior; X: promedio de medias
LEI: límite de especificación inferior.
4.50%
4.55%
4.60%
4.65%
4.70%
Inicio Medio Final
4.57% 4.58% 4.64%
4.63%
4.58%
4.70% H
um
edad
Nivel de muestreo
Primer lote
Segundo lote
3128252219161310741
0,84
0,83
0,82
0,81
0,80
0,79
0,78
0,77
0,76
0,75
Muestra
Me
dia
de
la m
ue
stra
__X=0,80228
LES = 0,84 g
LEI = 0,76 g
655
6651
1
Gráfica de control de pesos promedios primer lote

50
Resultado: Ppk=2,11; proceso capaz
Figura 20. Prueba de capacidad de proceso – primer lote
LES: límite de especificación superior; X: promedio de medias; LEI: límite de
especificación inferior.
Figura21. Control de los pesos promedios – segundo lote
0,86
25
0,85
00
0,83
75
0,82
50
0,81
25
0,80
00
0,78
75
LEI LES
LEI 0,7866
O bjetiv o *
LES 0,8694
Media de la muestra 0,822626
Número de muestra 179
Desv .Est. (Dentro) 0,00554007
Desv .Est. (General) 0,00568258
Procesar datos
C p 2,49
C PL 2,17
C PU 2,81
C pk 2,17
Pp 2,43
PPL 2,11
PPU 2,74
Ppk 2,11
C pm *
C apacidad general
C apacidad (dentro) del potencial
PPM < LEI 0,00
PPM > LES 0,00
PPM Total 0,00
Desempeño observ ado
PPM < LEI 0,00
PPM > LES 0,00
PPM Total 0,00
Exp. Dentro del rendimiento
PPM < LEI 0,00
PPM > LES 0,00
PPM Total 0,00
Exp. Rendimiento general
Dentro de
General
Capacidad de proceso de pesos de tabletas recubiertas primer lote
343128252219161310741
0,84
0,83
0,82
0,81
0,80
0,79
0,78
0,77
0,76
0,75
Muestra
Med
ia d
e la
mue
stra
__X = 0,80107 g
LES = 0,84 g
LEI = 0,76 g
6
5
Gráfica de control de pesos promedio segundo lote

51
Figura 22. Prueba de capacidad de proceso – primer lote
Resultado: Ppk= 1,83; proceso capaz
4.4.5 Dureza
Tabla 29. Resultados de dureza de los núcleos de los lotes evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE RESULTADO
(Kp) DICTAMEN
Dureza 10 Kp – 21 Kp
Primer lote
13,70 Conforme 13,70
13,50
14,20 Conforme 13,30
14,20 Conforme 13,40
12,30
Segundo lote
12,80 Conforme 13,00
13,50 Conforme 12,95
13,40
12,75 Conforme 12,50
13,03
0,8360,8250,8140,8030,7920,7810,7700,759
LEI LES
LEI 0,76
O bjetiv o *
LES 0,84
Media de la muestra 0,801068
Número de muestra 340
Forma 216,724
Escala 0,803
Procesar datos
Pp 2,57
PPL 1,83
PPU 4,58
Ppk 1,83
C apacidad general
PPM < LEI 0,00
PPM > LES 0,00
PPM Total 0,00
Desempeño observ ado
PPM < LEI 6,60
PPM > LES 0,00
PPM Total 6,60
Exp. Rendimiento general
Capacidad de proceso de Pesos promedio segundo loteCálculos basados en el modelo de distribuciónWeibull

52
Figura 23: Comparación de dureza en los lotes evaluados
4.4.6 Friabilidad
Tabla 30. Resultados de friabilidad de los núcleos de los lotes evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
(%) DICTAMEN
FRIABILIDAD Máximo 1%
Primer lote
Inicio 0,17 Conforme
Medio 0,14 Conforme
Final 0,13 Conforme
Segundo
lote
Inicio 0,24 Conforme
Medio 0,21 Conforme
Final 0,15 Conforme
4.005.006.007.008.009.00
10.0011.0012.0013.0014.0015.00
1 2 3 4 5 6 7 8
13.7 13.7 13.5 14.2 13.3 14.2 13.4 12.3
12.8 13 13.5 12.95 13.4 12.75 12.5 13.03
Du
reza
(K
p)
Nùmero de Muestras
Primer lote
Segundo lote

53
Figura 24. Comparación de friabilidad en los lotes evaluados
4.4.7 Desintegración
Tabla 31. Resultados de desintegración de los núcleos de los lotes evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO DICTAMEN
DESINTEGRACIÓN Máximo 30
minutos
Primer lote
Inicio 9 min (2 tabletas)
10 min (4 tabletas) Conforme
Medio 11 min (6 tabletas) Conforme
Final 10 min (1 tabletas)
11 min (5 tabletas) Conforme
Segundo
lote
Inicio 10 min (6 tabletas) Conforme
Medio 9 min (6 tabletas) Conforme
Final 9 min (6 tabletas) Conforme
0.00%
0.05%
0.10%
0.15%
0.20%
0.25%
Inicio Medio Final
0.17% 0.14% 0.13%
0.24% 0.21%
0.15% Fr
iab
ilid
ad
Nivel de muestreo
Primer lote
Segundo lote

54
4.4.8 Uniformidad de contenido
Tabla 32. Resultados de uniformidad de contenido de Diclofenaco sódico de
los núcleos de los lotes evaluados
Figura 25. Comparación de uniformidad de contenido de Diclofenaco sódico
Tabla 33. Resultados de uniformidad de contenido de Paracetamol de los
núcleos de los lotes evaluados
UNIFORMIDAD DE UNIDADES DE DOSIFICACIÓN DE PARACETAMOL
Especificación Nivel Primer lote Segundo lote
L1 < 15
INICO 1,853 1,737
MEDIO 1,340 0,858
FINAL 1,024 1,539
UNIFORMIDAD DE UNIDADES DE DOSIFICACIÓN DE DICLOFENACO SÓDICO
Especificación Nivel Primer lote Segundo lote
L1 < 15
INICO 9,88 3,76
MEDIO 11,56 2,29
FINAL 7,23 3,12
0.00
2.00
4.00
6.00
8.00
10.00
12.00
INICO MEDIO FINAL
9.88 11.56 7.23
3.76 2.29
3.12
Un
ifo
rmid
ad d
e u
nid
ades
de
do
sifi
caci
ón
(%)
Nivel de muestreo
Primer lote
Segundo lote

55
Figura 26: Comparación de uniformidad de contenido de Paracetamol
4.4.9 Dosaje de principios activos
Tabla 34. Resultados de dosaje de Diclofenaco sódico de los núcleos de los
lotes evaluados
DOSAJE DE DICLOFENACO SÓDICO
Especificación Nivel Primer lote
(mg/828 mg)
Segundo lote
(mg/828 mg)
46,575 mg/828 mg de
núcleo - 56,925 mg/
828 mg de núcleo
INICO 52,665130 48,344010
MEDIO 49,879720 49,816430
FINAL 51,271090 50,553970
PROMEDIO 51,271980 49,571470
% DSR 2,7163 2,2698
0.000
0.500
1.000
1.500
2.000
INICO MEDIO FINAL
1.853
1.340 1.024
1.737
0.858
1.539 U
nif
orm
idad
de
un
idad
es d
e d
osi
fica
ció
n (
%)
Nivel de muestreo
Primer lote
Segundo lote
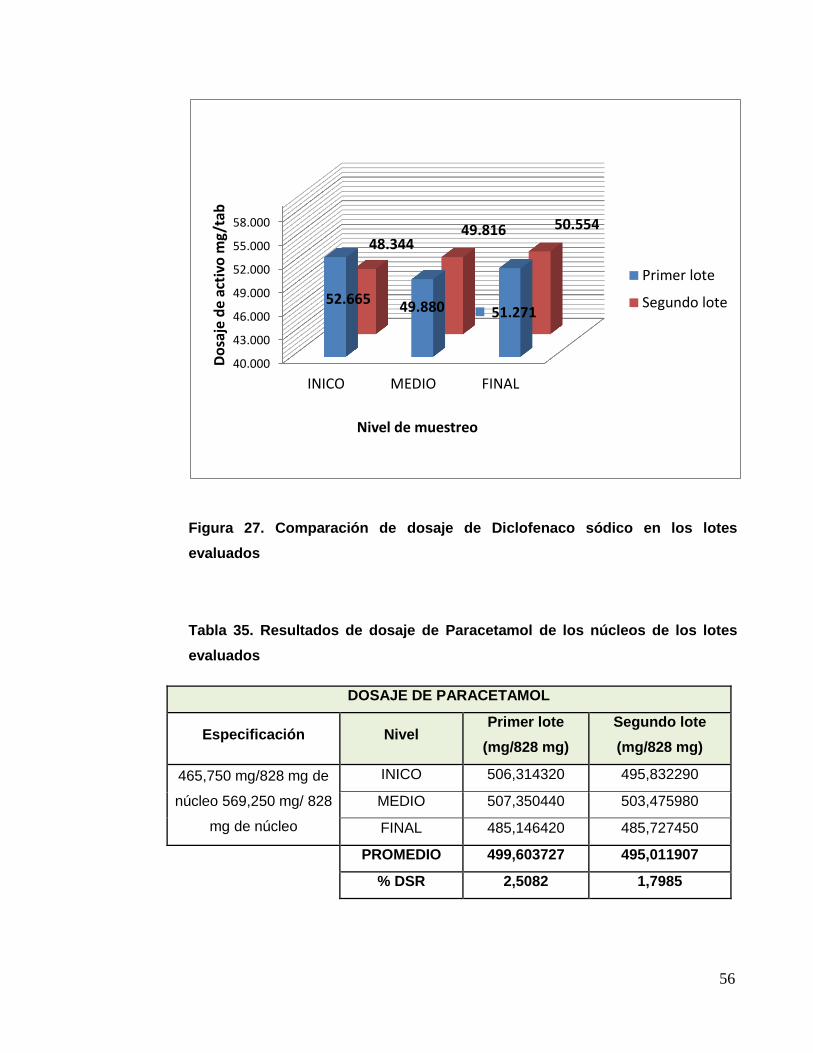
56
Figura 27. Comparación de dosaje de Diclofenaco sódico en los lotes
evaluados
Tabla 35. Resultados de dosaje de Paracetamol de los núcleos de los lotes
evaluados
DOSAJE DE PARACETAMOL
Especificación Nivel Primer lote
(mg/828 mg)
Segundo lote
(mg/828 mg)
465,750 mg/828 mg de
núcleo 569,250 mg/ 828
mg de núcleo
INICO 506,314320 495,832290
MEDIO 507,350440 503,475980
FINAL 485,146420 485,727450
PROMEDIO 499,603727 495,011907
% DSR 2,5082 1,7985
40.000
43.000
46.000
49.000
52.000
55.000
58.000
INICO MEDIO FINAL
52.665 49.880 51.271
48.344 49.816 50.554
Do
saje
de
acti
vo m
g/ta
b
Nivel de muestreo
Primer lote
Segundo lote

57
Figura 28. Comparación de dosaje de Paracetamol en los lotes evaluados.
4.5 Estudio de caracterización de tabletas recubiertas
4.5.1 Aspecto
Tabla 36. Resultados de aspecto de tabletas recubiertas de los lotes
evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO DICTAMEN
ASPECTO
Tabletas de forma de oblonga con
recubierta de color anaranjado (con ligero moteado
propio del colorante utilizado)
Primer lote
Primer parcial
Tabletas de forma de oblonga con recubierta de
color anaranjado (con ligero
moteado propio del colorante
utilizado)
Conforme
Segundo parcial
Conforme
Tercer parcial
Conforme
Segundo lote
Primer parcial
Tabletas de forma de oblonga con recubierta de
color anaranjado (con ligero
moteado propio del colorante
utilizado)
Conforme
Segundo parcial
Conforme
Tercer parcial
Conforme
430.000
450.000
470.000
490.000
510.000
530.000
550.000
570.000
INICO MEDIO FINAL
506.314 507.350 485.146
495.832 503.476
485.727
Do
saje
de
acti
vo (
mg/
tab
)
Nivel de muestreo
Primer lote
Segundo lote

58
4.5.2 Dimensiones
Tabla 37. Resultados de las dimensiones de tabletas recubiertas de los lotes
evaluados
Figura 29: Comparación de las dimensiones en los lotes evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
(mm) DICTAMEN
ALTURA 5,70 - 6,10 mm
Primer lote
Primer parcial
6,08 Conforme
6,07 Conforme
6,07 Conforme
Segundo parcial
6,07 Conforme
6,08 Conforme
6,09 Conforme
Tercer parcial
6,05 Conforme
6,05 Conforme
6,05 Conforme
Segundo lote
Primer parcial
6,06 Conforme
6,03 Conforme
6,05 Conforme
Segundo parcial
6,06 Conforme
6,06 Conforme
6,08 Conforme
Tercer parcial
6,07 Conforme
6,07 Conforme
6,08 Conforme
6.00
6.01
6.02
6.03
6.04
6.05
6.06
6.07
6.08
6.09
Primer parcial Segundo parcial Tercer parcial
6.08 6.07 6.07 6.07 6.08 6.09 6.05 6.05 6.05
6.06
6.03
6.05
6.06 6.06
6.08 6.07
6.07 6.08
Alt
ura
(m
m)
Nivel de muestreo
Primer lote
Segundo lote

59
2.00
2.50
3.00
3.50
4.00
4.50
5.00
5.50
6.00
Primer parcial Segundo parcial Tercer parcial
4.03 3.91 4.03 4.25 4.22 4.18 4.21 4.27 4.28
3.69 3.8 3.59 3.61 3.7 3.72 3.54 3.65 3.91
Hu
med
ad (
%)
Niveles de muestreo
Primer lote
Segundo lote
4.5.3 Humedad
Tabla 38. Resultados de humedad de tabletas recubiertas de los lotes
evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
(%) DICTAMEN
HUMEDAD 3,0% - 6,5%
Primer lote
Primer parcial
4,03 Conforme
3,91 Conforme
4,03 Conforme
Segundo parcial
4,25 Conforme
4,22 Conforme
4,18 Conforme
Tercer parcial
4,21 Conforme
4,27 Conforme
4,28 Conforme
Segundo lote
Primer parcial
3,69 Conforme
3,80 Conforme
3,59 Conforme
Segundo parcial
3,61 Conforme
3,70 Conforme
3,72 Conforme
Tercer parcial
3,54 Conforme
3,65 Conforme
3,91 Conforme
Figura 30. Comparación de humedad en los lotes evaluados

60
16314512710991735537191
0,87
0,86
0,85
0,84
0,83
0,82
0,81
0,80
0,79
0,78
Observación
Val
or in
divi
dual
_X = 0,82263 g
LES = 0,869 g
1
65
5
Gráfica de control de pesos de tabletas recubiertas primer lote
4.5.4 Peso promedio
Tabla 39. Resultados de pesos promedio de tabletas recubiertas de los lotes
evaluados
Figura 31. Gráfica de control de los pesos promedio de las tabletas
recubiertas – primer lote. LES: límite de especificación superior; X: promedio de
medias; LEI: límite de especificación inferior.
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
(mg/tab) DICTAMEN
PESO PROMEDIO
828 mg/tab
(786,6 - 869,4 mg/tab)
95% - 105%
Primer lote
Primer parcial
819,70 Conforme
820,80 Conforme
819,50 Conforme
Segundo parcial
821,00 Conforme
816,20 Conforme
819,80 Conforme
Tercer parcial
820,20 Conforme
822,20 Conforme
819,40 Conforme
Segundo lote
Primer parcial
814,10 Conforme
813,50 Conforme
813,60 Conforme
Segundo parcial
817,00 Conforme
817,60 Conforme
815,60 Conforme
Tercer parcial
814,80 Conforme
816,30 Conforme
815,70 Conforme
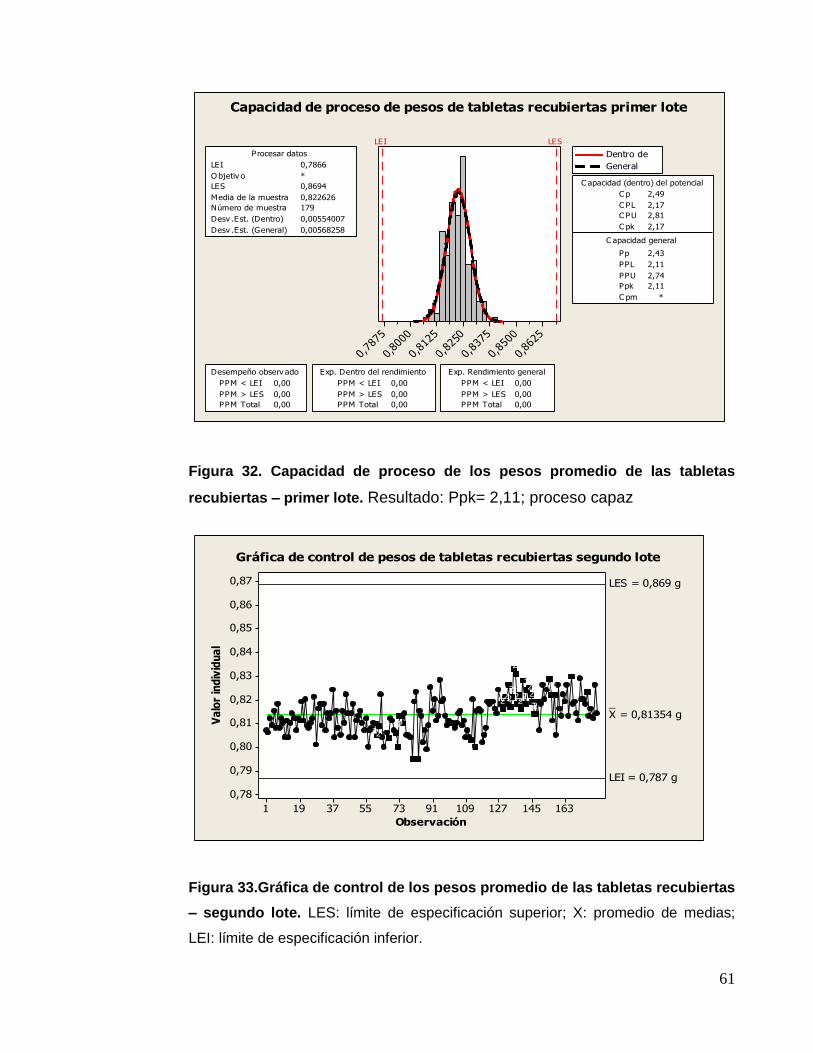
61
Figura 32. Capacidad de proceso de los pesos promedio de las tabletas
recubiertas – primer lote. Resultado: Ppk= 2,11; proceso capaz
Figura 33.Gráfica de control de los pesos promedio de las tabletas recubiertas
– segundo lote. LES: límite de especificación superior; X: promedio de medias;
LEI: límite de especificación inferior.
16314512710991735537191
0,87
0,86
0,85
0,84
0,83
0,82
0,81
0,80
0,79
0,78
Observación
Va
lor
ind
ivid
ua
l
_X = 0,81354 g
LES = 0,869 g
LEI = 0,787 g
6
5
66
6
2
22
2222
2
2
2
22
21
22
2
22
22
66
11
222
2
26
22
2
Gráfica de control de pesos de tabletas recubiertas segundo lote
0,86
25
0,85
00
0,83
75
0,82
50
0,81
25
0,80
00
0,78
75
LEI LES
LEI 0,7866
O bjetiv o *
LES 0,8694
Media de la muestra 0,822626
Número de muestra 179
Desv .Est. (Dentro) 0,00554007
Desv .Est. (General) 0,00568258
Procesar datos
C p 2,49
C PL 2,17
C PU 2,81
C pk 2,17
Pp 2,43
PPL 2,11
PPU 2,74
Ppk 2,11
C pm *
C apacidad general
C apacidad (dentro) del potencial
PPM < LEI 0,00
PPM > LES 0,00
PPM Total 0,00
Desempeño observ ado
PPM < LEI 0,00
PPM > LES 0,00
PPM Total 0,00
Exp. Dentro del rendimiento
PPM < LEI 0,00
PPM > LES 0,00
PPM Total 0,00
Exp. Rendimiento general
Dentro de
General
Capacidad de proceso de pesos de tabletas recubiertas primer lote

62
Figura 34. Capacidad de proceso de los pesos promedio de las tabletas
recubiertas – segundo lote. Resultado: Ppk= 1,22; proceso capaz
4.5.5 Dureza
Tabla 40. Resultados de dureza de tabletas recubiertas de los lotes
evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO
(Kp) DICTAMEN
DUREZA Mayor a 4 Kp
Primer lote
Primer parcial
22,70 Conforme
22,90 Conforme
23,00 Conforme
Segundo parcial
23,10 Conforme
24,30 Conforme
23,80 Conforme
Tercer parcial
24,00 Conforme
24,30 Conforme
24,30 Conforme
Segundo lote
Primer parcial
19,80 Conforme
20,00 Conforme
19,80 Conforme
Segundo parcial
20,50 Conforme
20,20 Conforme
20,10 Conforme
Tercer parcial
20,30 Conforme
19,60 Conforme
20,70 Conforme
0,86
4
0,85
2
0,84
0
0,82
8
0,81
6
0,80
4
0,79
2
LEI LES
LEI 0,7866
O bjetiv o *
LES 0,8694
Media de la muestra 0,813539
Número de muestra 180
Desv .Est. (Dentro) 0,00592986
Desv .Est. (General) 0,00737455
Procesar datos
C p 2,33
C PL 1,51
C PU 3,14
C pk 1,51
Pp 1,87
PPL 1,22
PPU 2,52
Ppk 1,22
C pm *
C apacidad general
C apacidad (dentro) del potencial
PPM < LEI 0,00
PPM > LES 0,00
PPM Total 0,00
Desempeño observ ado
PPM < LEI 2,77
PPM > LES 0,00
PPM Total 2,77
Exp. Dentro del rendimiento
PPM < LEI 129,62
PPM > LES 0,00
PPM Total 129,62
Exp. Rendimiento general
Dentro de
General
Capacidad de proceso de pesos de tabletas recubiertas segundo lote

63
Figura 35. Comparación de durezade tabletas recubiertas en los lotes
evaluados
4.5.6 Friabilidad
Tabla 41. Resultados de friabilidad de tabletas recubiertas de los lotes
evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO (%) DICTAMEN
FRIABILIDAD Máximo 1%
Primer lote
Primer parcial
0,01 Conforme
0,02 Conforme
0,00 Conforme
Segundo parcial
0,01 Conforme
0,00 Conforme
0,04 Conforme
Tercer parcial
0,01 Conforme
0,02 Conforme
0,01 Conforme
Segundo lote
Primer parcial
0,04 Conforme
0,02 Conforme
0,04 Conforme
Segundo parcial
0,01 Conforme
0,00 Conforme
0,00 Conforme
Tercer parcial
0,01 Conforme
0,00 Conforme
0,00 Conforme
0
5
10
15
20
25
Primer parcial Segundo parcial Tercer parcial
22.7 22.9 23 23.1 24.3 23.8 24 24.3 24.3
19.8 20 19.8 20.5 20.2 20.1 20.3 19.6 20.7 D
ure
za (
Kp
)
Niveles de muestreo
Primer lote
Segundo lote
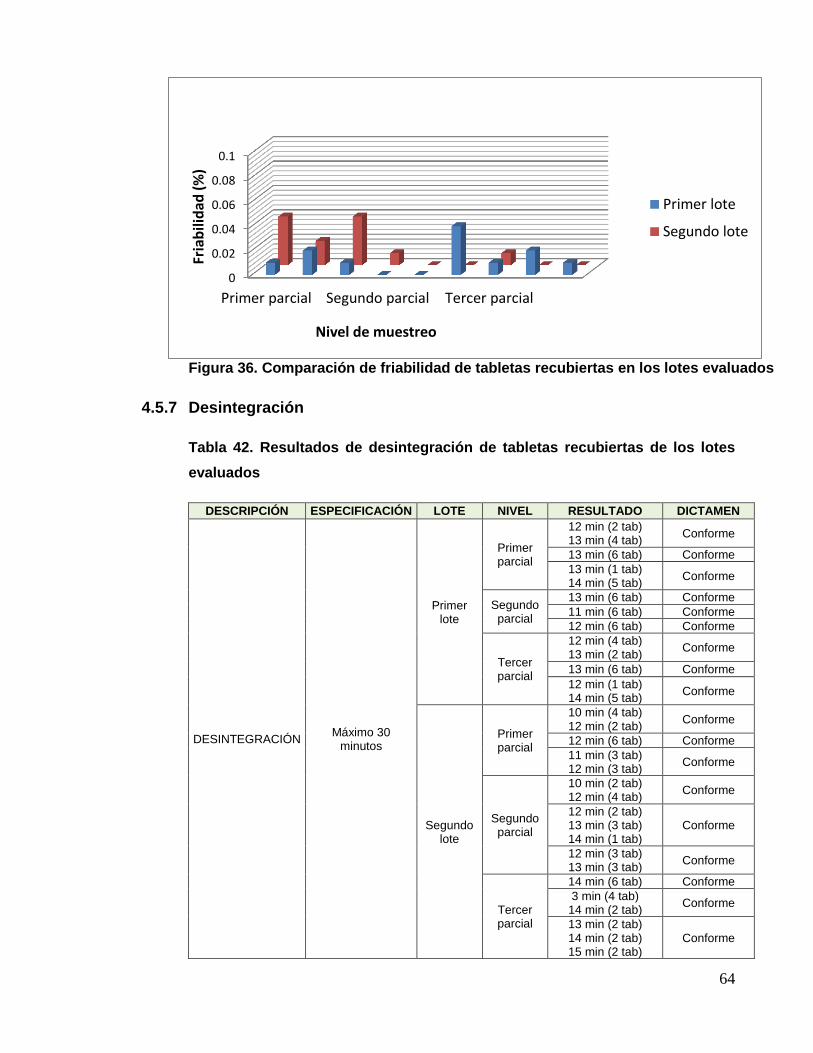
64
Figura 36. Comparación de friabilidad de tabletas recubiertas en los lotes evaluados
4.5.7 Desintegración
Tabla 42. Resultados de desintegración de tabletas recubiertas de los lotes
evaluados
DESCRIPCIÓN ESPECIFICACIÓN LOTE NIVEL RESULTADO DICTAMEN
DESINTEGRACIÓN Máximo 30
minutos
Primer lote
Primer parcial
12 min (2 tab) 13 min (4 tab)
Conforme
13 min (6 tab) Conforme
13 min (1 tab) 14 min (5 tab)
Conforme
Segundo parcial
13 min (6 tab) Conforme
11 min (6 tab) Conforme
12 min (6 tab) Conforme
Tercer parcial
12 min (4 tab) 13 min (2 tab)
Conforme
13 min (6 tab) Conforme
12 min (1 tab) 14 min (5 tab)
Conforme
Segundo lote
Primer parcial
10 min (4 tab) 12 min (2 tab)
Conforme
12 min (6 tab) Conforme
11 min (3 tab) 12 min (3 tab)
Conforme
Segundo parcial
10 min (2 tab) 12 min (4 tab)
Conforme
12 min (2 tab) 13 min (3 tab) 14 min (1 tab)
Conforme
12 min (3 tab) 13 min (3 tab)
Conforme
Tercer parcial
14 min (6 tab) Conforme
3 min (4 tab) 14 min (2 tab)
Conforme
13 min (2 tab) 14 min (2 tab) 15 min (2 tab)
Conforme
0
0.02
0.04
0.06
0.08
0.1
Primer parcial Segundo parcial Tercer parcial
Fria
bili
dad
(%
)
Nivel de muestreo
Primer lote
Segundo lote
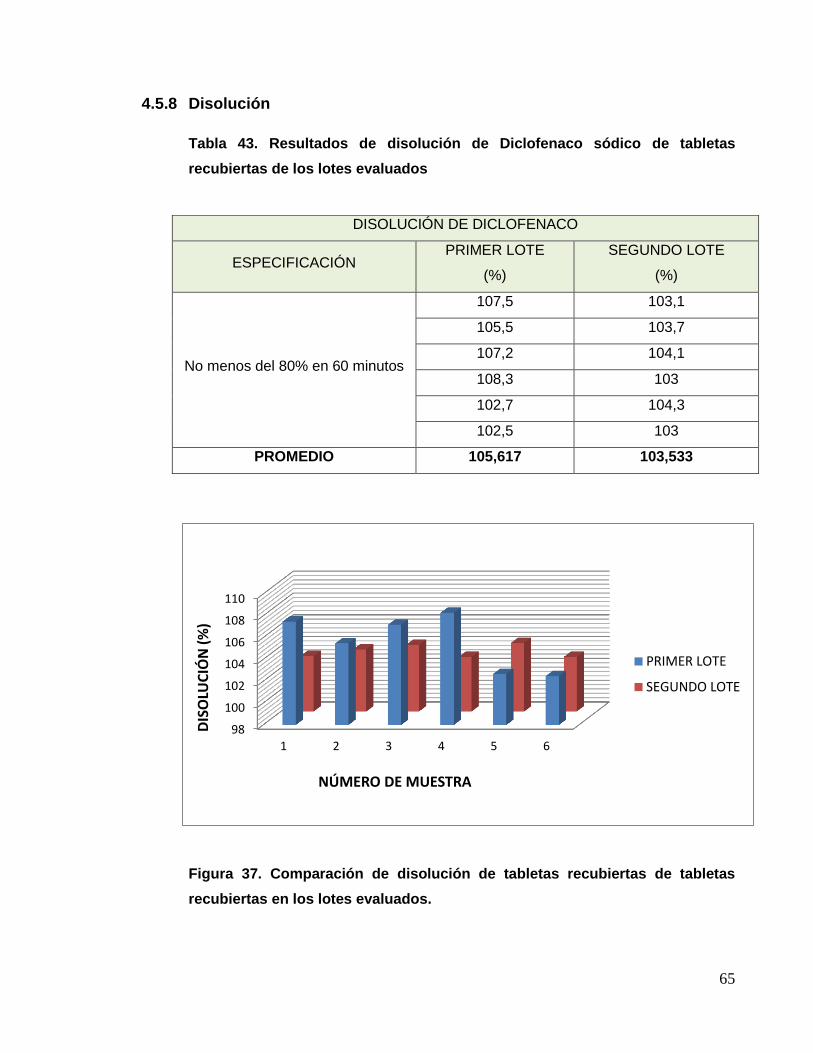
65
4.5.8 Disolución
Tabla 43. Resultados de disolución de Diclofenaco sódico de tabletas
recubiertas de los lotes evaluados
Figura 37. Comparación de disolución de tabletas recubiertas de tabletas
recubiertas en los lotes evaluados.
DISOLUCIÓN DE DICLOFENACO
ESPECIFICACIÓN PRIMER LOTE
(%)
SEGUNDO LOTE
(%)
No menos del 80% en 60 minutos
107,5 103,1
105,5 103,7
107,2 104,1
108,3 103
102,7 104,3
102,5 103
PROMEDIO 105,617 103,533
98
100
102
104
106
108
110
1 2 3 4 5 6
DIS
OLU
CIÓ
N (
%)
NÚMERO DE MUESTRA
PRIMER LOTE
SEGUNDO LOTE

66
Tabla 44. Resultados de disolución de Paracetamol de tabletas recubiertas de
los lotes evaluados
Figura 38. Comparación de disolución de tabletas recubiertas de tabletas
recubiertas en los lotes evaluados
DISOLUCIÓN DE PARACETAMOL
ESPECIFICACIÓN PRIMER LOTE
(%)
SEGUNDO LOTE
(%)
No menos del 80% en 30 minutos
97,100 98,300
97,500 98,000
96,400 99,500
97,600 96,700
97,300 97,500
96,700 98,700
PROMEDIO 97,100 98,117
94.000
95.000
96.000
97.000
98.000
99.000
100.000
1 2 3 4 5 6
DIS
OLU
CIÓ
N (
%)
NÚMERO DE MUESTRA
PRIMER LOTE
SEGUNDO LOTE

67
4.5.9 Uniformidad de contenido
Tabla 45. Resultados de Uniformidad de contenido de Diclofenaco sódico de
tabletas recubiertas de los lotes evaluados
Figura 39. Comparación de uniformidad de contenido de tabletas recubiertas
de tabletas recubiertas en los lotes evaluados
UNIFORMIDAD DE UNIDADES DE DOSIFICACIÓN DE DICLOFENACO SÓDICO
Especificación Nivel Primer
lote Segundo lote
L1 < 15
PRIMER PARCIAL 9,60 5,45
SEGUNDO PARCIAL 9,40 5,85
TERCER PARCIAL 5,63 4,52
PROMEDIO 8,210000 5,273333
0.00
2.00
4.00
6.00
8.00
10.00
PRIMERPARCIAL
SEGUNDOPARCIAL
TERCER PARCIAL
9.60 9.40
5.63
5.45 5.85 4.52
Un
ifo
rmid
ad d
e u
nid
ades
de
do
sifi
caci
ón
(%)
Nivel de muestreo
Primer lote
Segundo lote

68
Tabla 46. Resultados de Uniformidad de contenido de Paracetamol de tabletas
recubiertas de los lotes evaluados
Figura 40: Comparación de uniformidad de contenido de tabletas recubiertas
de tabletas recubiertas en los lotes evaluados
UNIFORMIDAD DE UNIDADES DE DOSIFICACIÓN DE PARACETAMOL
Especificación Nivel Primer lote Segundo lote
L1 < 15
PRIMER PARCIAL 1,927 6,394
SEGUNDO PARCIAL 2,084 1,256
TERCER PARCIAL 3,313 3,200
0.00
2.00
4.00
6.00
8.00
10.00
PRIMER PARCIAL SEGUNDOPARCIAL
TERCER PARCIAL
1.927 2.084 3.313
6.394
1.256
3.200
Un
ifo
rmid
ad d
e u
nid
ades
de
do
sifi
caci
ón
(%)
Nivel de muestreo
Primer lote
Segundo lote
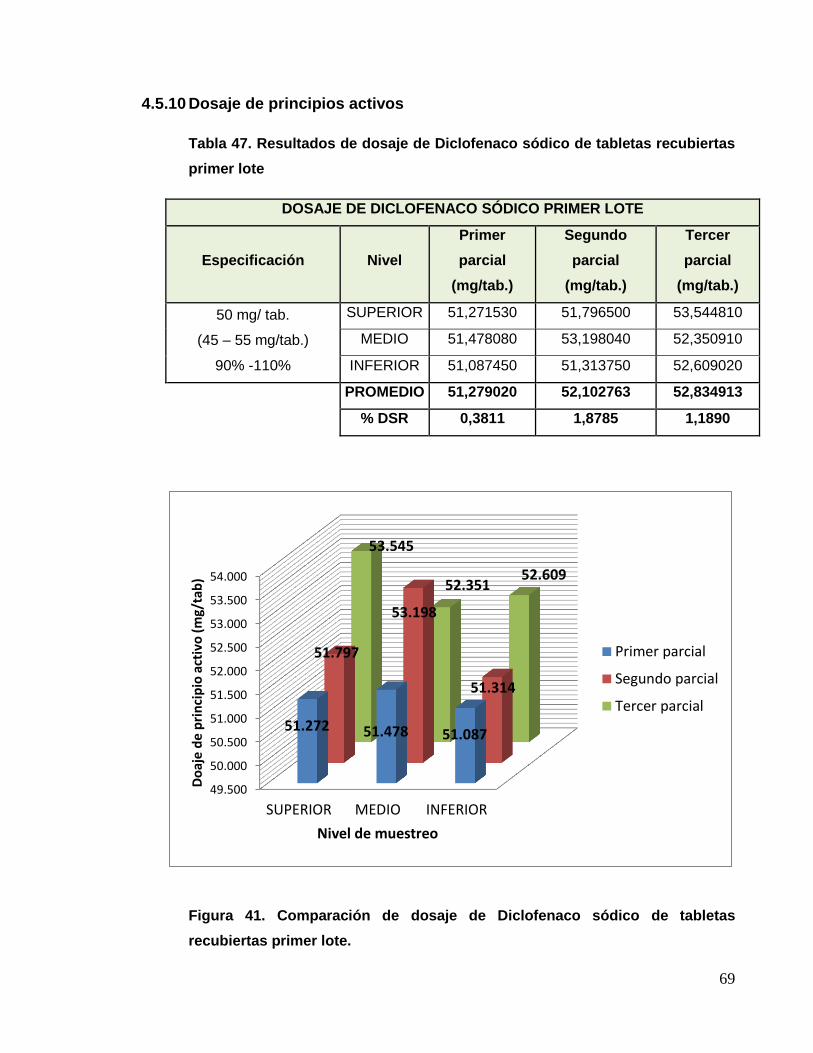
69
4.5.10 Dosaje de principios activos
Tabla 47. Resultados de dosaje de Diclofenaco sódico de tabletas recubiertas
primer lote
DOSAJE DE DICLOFENACO SÓDICO PRIMER LOTE
Especificación Nivel
Primer
parcial
(mg/tab.)
Segundo
parcial
(mg/tab.)
Tercer
parcial
(mg/tab.)
50 mg/ tab.
(45 – 55 mg/tab.)
90% -110%
SUPERIOR 51,271530 51,796500 53,544810
MEDIO 51,478080 53,198040 52,350910
INFERIOR 51,087450 51,313750 52,609020
PROMEDIO 51,279020 52,102763 52,834913
% DSR 0,3811 1,8785 1,1890
Figura 41. Comparación de dosaje de Diclofenaco sódico de tabletas
recubiertas primer lote.
49.500
50.000
50.500
51.000
51.500
52.000
52.500
53.000
53.500
54.000
SUPERIOR MEDIO INFERIOR
51.272 51.478 51.087
51.797
53.198
51.314
53.545
52.351 52.609
Do
aje
de
pri
nci
pio
act
ivo
(m
g/ta
b)
Nivel de muestreo
Primer parcial
Segundo parcial
Tercer parcial
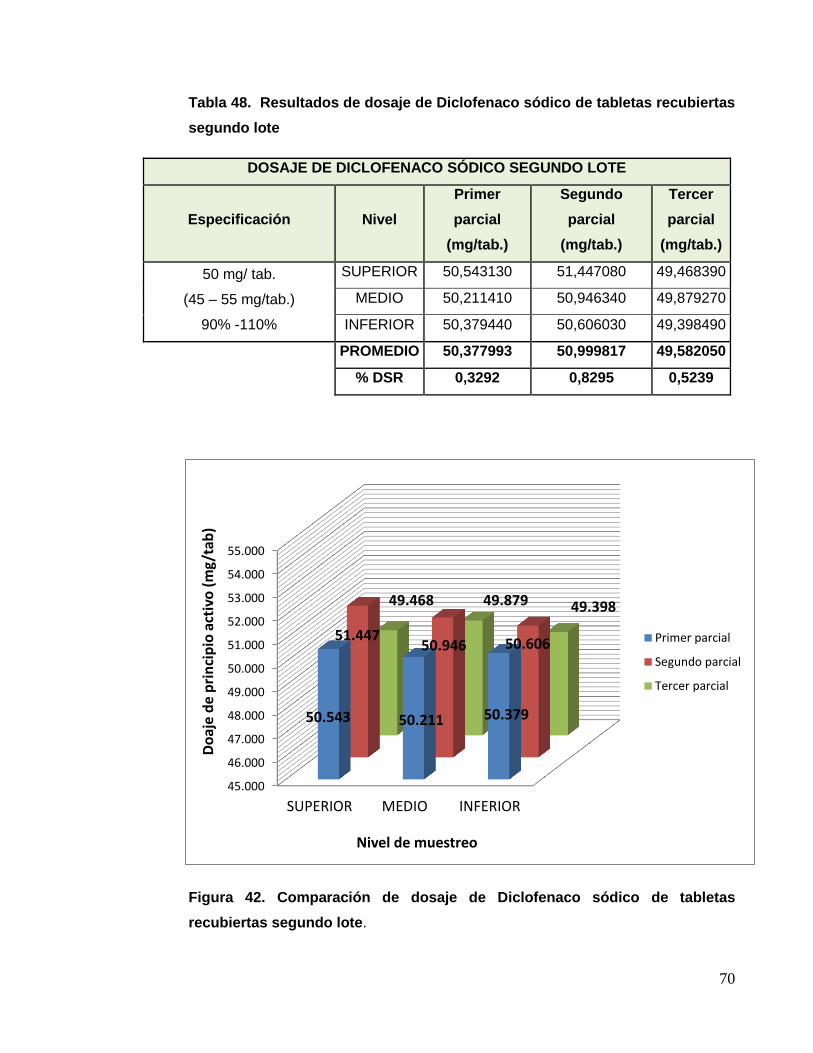
70
Tabla 48. Resultados de dosaje de Diclofenaco sódico de tabletas recubiertas
segundo lote
DOSAJE DE DICLOFENACO SÓDICO SEGUNDO LOTE
Especificación Nivel
Primer
parcial
(mg/tab.)
Segundo
parcial
(mg/tab.)
Tercer
parcial
(mg/tab.)
50 mg/ tab.
(45 – 55 mg/tab.)
90% -110%
SUPERIOR 50,543130 51,447080 49,468390
MEDIO 50,211410 50,946340 49,879270
INFERIOR 50,379440 50,606030 49,398490
PROMEDIO 50,377993 50,999817 49,582050
% DSR 0,3292 0,8295 0,5239
Figura 42. Comparación de dosaje de Diclofenaco sódico de tabletas
recubiertas segundo lote.
45.000
46.000
47.000
48.000
49.000
50.000
51.000
52.000
53.000
54.000
55.000
SUPERIOR MEDIO INFERIOR
50.543 50.211 50.379
51.447 50.946 50.606
49.468 49.879 49.398
Do
aje
de
pri
nci
pio
act
ivo
(m
g/ta
b)
Nivel de muestreo
Primer parcial
Segundo parcial
Tercer parcial
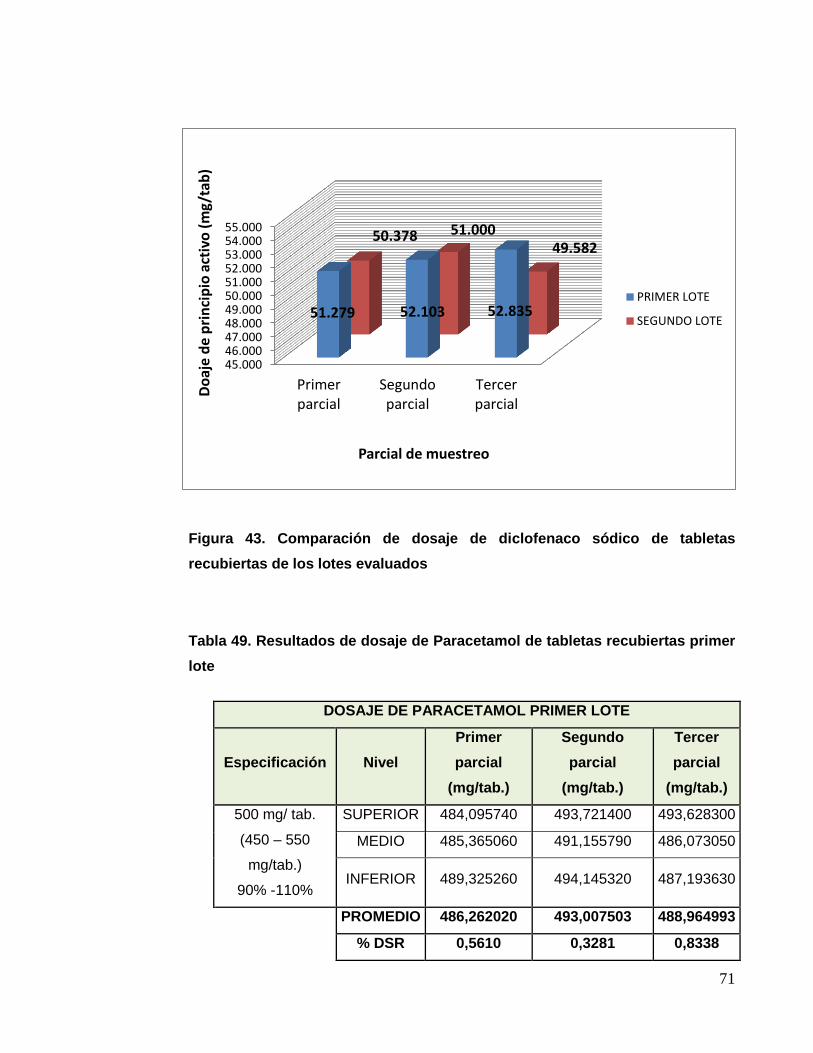
71
Figura 43. Comparación de dosaje de diclofenaco sódico de tabletas
recubiertas de los lotes evaluados
Tabla 49. Resultados de dosaje de Paracetamol de tabletas recubiertas primer
lote
DOSAJE DE PARACETAMOL PRIMER LOTE
Especificación Nivel
Primer
parcial
(mg/tab.)
Segundo
parcial
(mg/tab.)
Tercer
parcial
(mg/tab.)
500 mg/ tab.
(450 – 550
mg/tab.)
90% -110%
SUPERIOR 484,095740 493,721400 493,628300
MEDIO 485,365060 491,155790 486,073050
INFERIOR 489,325260 494,145320 487,193630
PROMEDIO 486,262020 493,007503 488,964993
% DSR 0,5610 0,3281 0,8338
45.00046.00047.00048.00049.00050.00051.00052.00053.00054.00055.000
Primerparcial
Segundoparcial
Tercerparcial
51.279 52.103 52.835
50.378 51.000 49.582
Do
aje
de
pri
nci
pio
act
ivo
(m
g/ta
b)
Parcial de muestreo
PRIMER LOTE
SEGUNDO LOTE
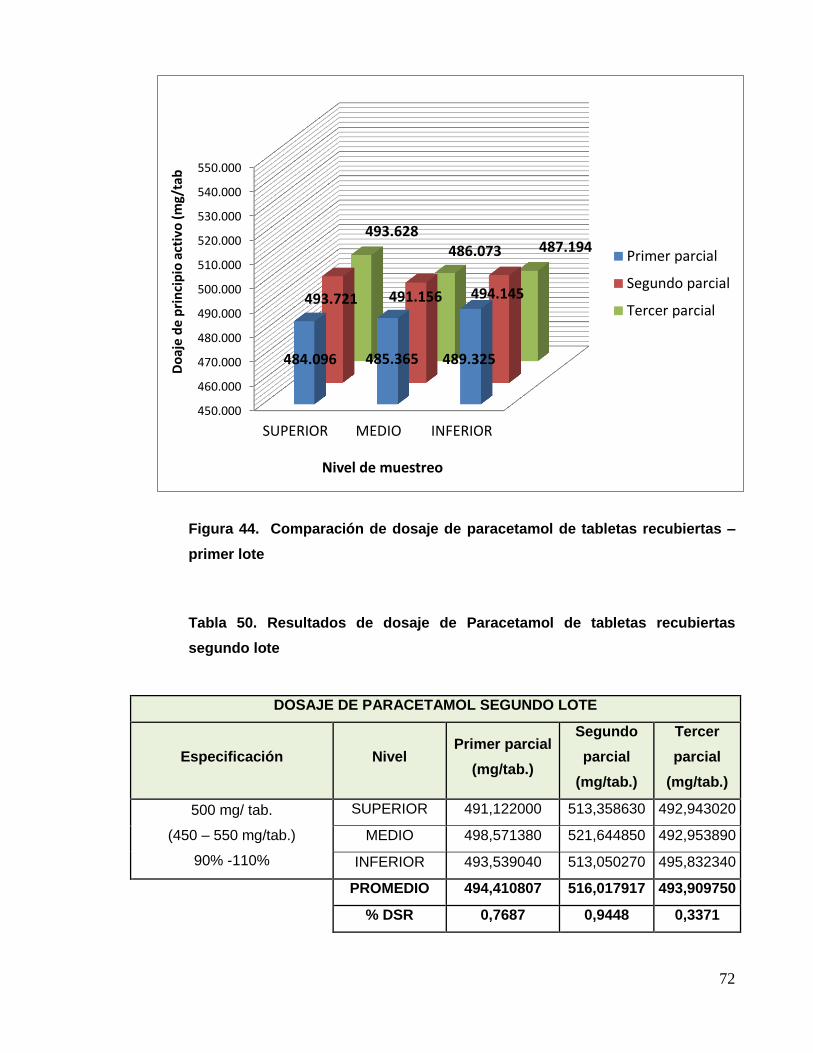
72
Figura 44. Comparación de dosaje de paracetamol de tabletas recubiertas –
primer lote
Tabla 50. Resultados de dosaje de Paracetamol de tabletas recubiertas
segundo lote
DOSAJE DE PARACETAMOL SEGUNDO LOTE
Especificación Nivel Primer parcial
(mg/tab.)
Segundo
parcial
(mg/tab.)
Tercer
parcial
(mg/tab.)
500 mg/ tab.
(450 – 550 mg/tab.)
90% -110%
SUPERIOR 491,122000 513,358630 492,943020
MEDIO 498,571380 521,644850 492,953890
INFERIOR 493,539040 513,050270 495,832340
PROMEDIO 494,410807 516,017917 493,909750
% DSR 0,7687 0,9448 0,3371
450.000
460.000
470.000
480.000
490.000
500.000
510.000
520.000
530.000
540.000
550.000
SUPERIOR MEDIO INFERIOR
484.096 485.365 489.325
493.721 491.156 494.145
493.628 486.073 487.194
Do
aje
de
pri
nci
pio
act
ivo
(m
g/ta
b
Nivel de muestreo
Primer parcial
Segundo parcial
Tercer parcial

73
Figura 45. Comparación de dosaje de Paracetamol de tabletas recubiertas –
segundo lote.
Figura 46. Comparación de dosaje de Paracetamol de tabletas recubiertas de
los lotes evaluados
450.000460.000470.000480.000490.000500.000510.000520.000530.000540.000550.000
Primerparcial
Segundoparcial
Tercerparcial
486.262 493.008 488.965
494.411 516.018
493.910
Do
aje
de
pri
nci
pio
act
ivo
(m
g/ta
b)
Parciales de muestreo
PRIMER LOTE
SEGUNDO LOTE
450.000
460.000
470.000
480.000
490.000
500.000
510.000
520.000
530.000
540.000
550.000
SUPERIOR MEDIO INFERIOR
491.122 498.571 493.539
513.359 521.645
513.050
492.943 492.954 495.832 D
oaj
e d
e p
rin
cip
io a
ctiv
o (
mg/
tab
)
Nivel de muestreo
Primer parcial
Segundo parcial
Tercer parcial
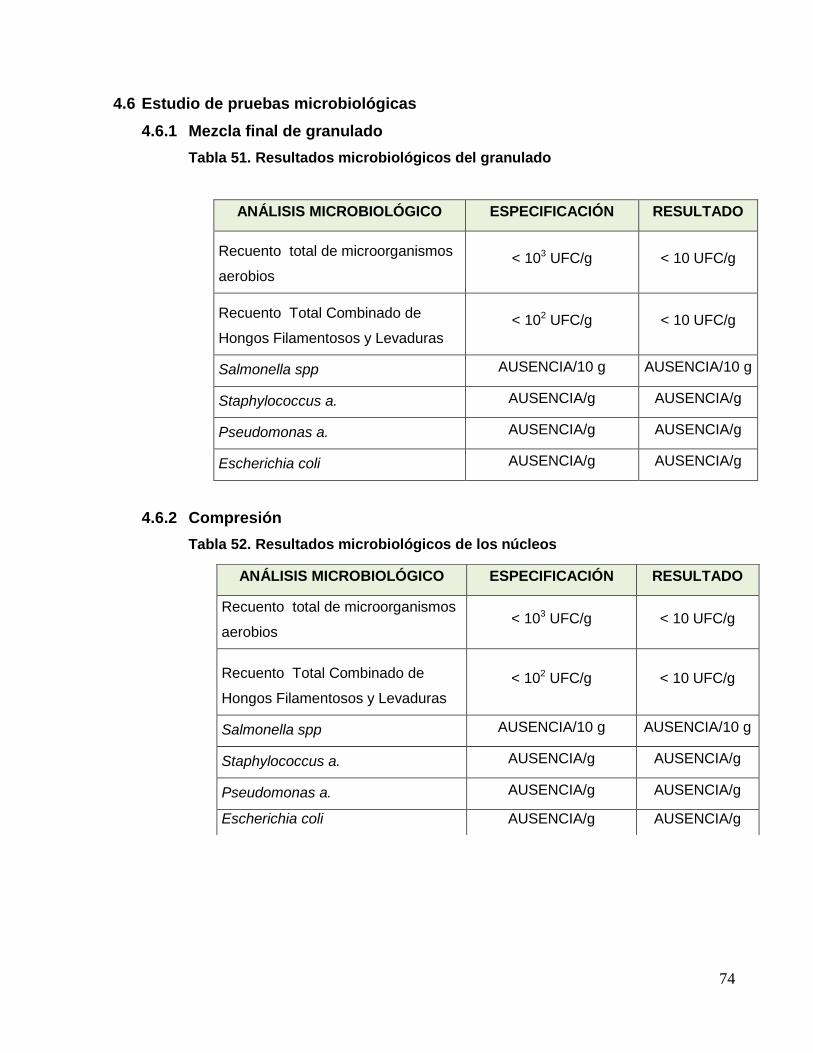
74
4.6 Estudio de pruebas microbiológicas
4.6.1 Mezcla final de granulado
Tabla 51. Resultados microbiológicos del granulado
ANÁLISIS MICROBIOLÓGICO ESPECIFICACIÓN RESULTADO
Recuento total de microorganismos
aerobios
< 103 UFC/g < 10 UFC/g
Recuento Total Combinado de
Hongos Filamentosos y Levaduras
< 102 UFC/g < 10 UFC/g
Salmonella spp AUSENCIA/10 g AUSENCIA/10 g
Staphylococcus a. AUSENCIA/g AUSENCIA/g
Pseudomonas a. AUSENCIA/g AUSENCIA/g
Escherichia coli AUSENCIA/g AUSENCIA/g
4.6.2 Compresión
Tabla 52. Resultados microbiológicos de los núcleos
ANÁLISIS MICROBIOLÓGICO ESPECIFICACIÓN RESULTADO
Recuento total de microorganismos
aerobios < 103 UFC/g < 10 UFC/g
Recuento Total Combinado de
Hongos Filamentosos y Levaduras
< 102 UFC/g < 10 UFC/g
Salmonella spp AUSENCIA/10 g AUSENCIA/10 g
Staphylococcus a. AUSENCIA/g AUSENCIA/g
Pseudomonas a. AUSENCIA/g AUSENCIA/g
Escherichia coli AUSENCIA/g AUSENCIA/g

75
4.6.3 Recubierta
Tabla 53. Resultados microbiológicos de tabletas recubiertas
ANÁLISIS MICROBIOLÓGICO ESPECIFICACIÓN RESULTADO
Recuento total de
microorganismos aerobios < 103 UFC/g < 10 UFC/g
Recuento Total Combinado de
Hongos Filamentosos y Levaduras < 102 UFC/g < 10 UFC/g
Salmonella spp AUSENCIA/10 g AUSENCIA/10 g
Staphylococcus a. AUSENCIA/g AUSENCIA/g
Pseudomonas a. AUSENCIA/g AUSENCIA/g
Escherichia coli AUSENCIA/g AUSENCIA/g
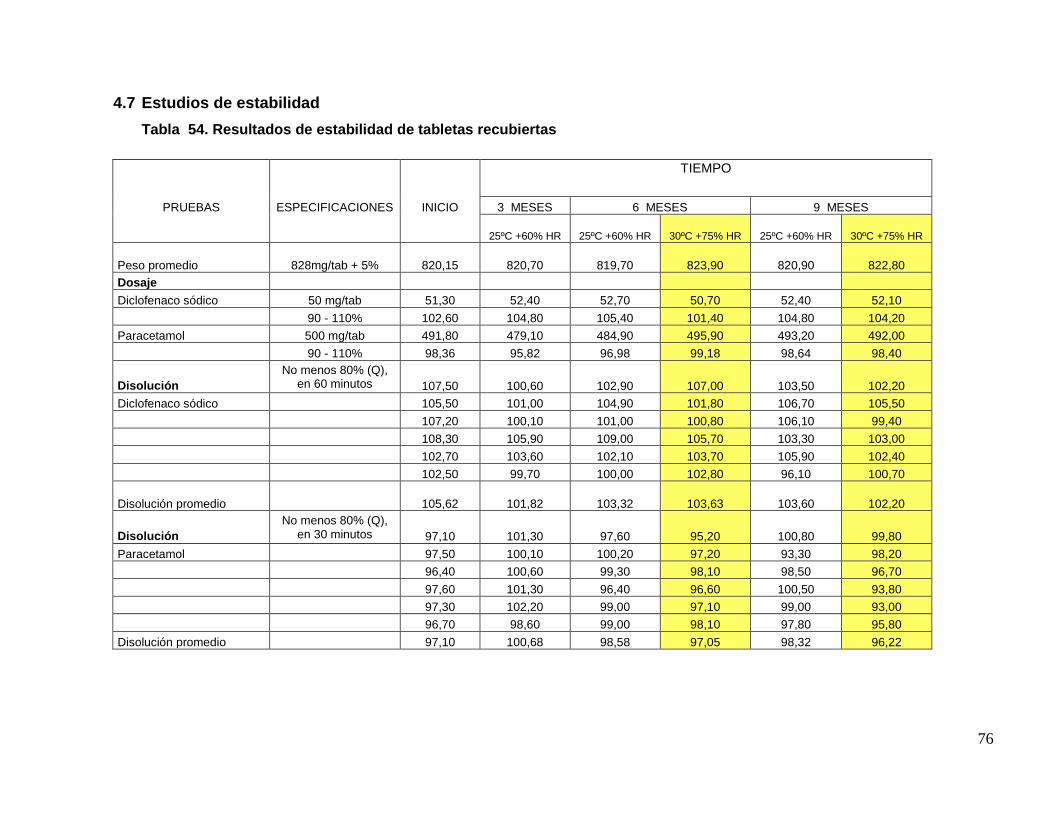
76
4.7 Estudios de estabilidad
Tabla 54. Resultados de estabilidad de tabletas recubiertas
TIEMPO
PRUEBAS ESPECIFICACIONES INICIO 3 MESES 6 MESES 9 MESES
25ºC +60% HR 25ºC +60% HR 30ºC +75% HR
25ºC +60% HR 30ºC +75% HR
Peso promedio 828mg/tab + 5% 820,15 820,70 819,70 823,90 820,90
822,80
Dosaje
Diclofenaco sódico 50 mg/tab 51,30 52,40 52,70 50,70 52,40 52,10
90 - 110% 102,60 104,80 105,40 101,40 104,80 104,20
Paracetamol 500 mg/tab 491,80 479,10 484,90 495,90 493,20 492,00
90 - 110% 98,36 95,82 96,98 99,18 98,64 98,40
Disolución
No menos 80% (Q), en 60 minutos 107,50 100,60 102,90 107,00 103,50 102,20
Diclofenaco sódico 105,50 101,00 104,90 101,80 106,70 105,50
107,20 100,10 101,00 100,80 106,10 99,40
108,30 105,90 109,00 105,70 103,30 103,00
102,70 103,60 102,10 103,70 105,90 102,40
102,50 99,70 100,00 102,80 96,10 100,70
Disolución promedio
105,62
101,82
103,32
103,63
103,60
102,20
Disolución
No menos 80% (Q), en 30 minutos 97,10 101,30 97,60 95,20 100,80 99,80
Paracetamol 97,50 100,10 100,20 97,20 93,30 98,20
96,40 100,60 99,30 98,10 98,50 96,70
97,60 101,30 96,40 96,60 100,50 93,80
97,30 102,20 99,00 97,10 99,00 93,00
96,70 98,60 99,00 98,10 97,80 95,80
Disolución promedio 97,10 100,68 98,58 97,05 98,32 96,22
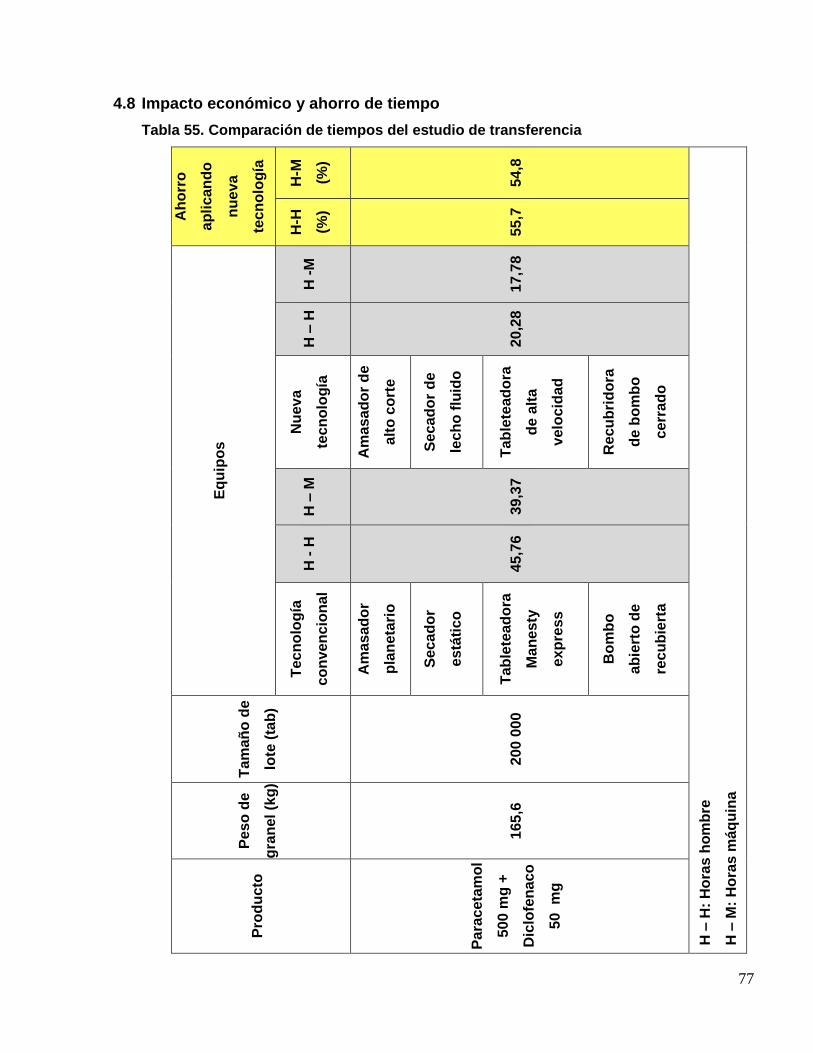
77
4.8 Impacto económico y ahorro de tiempo
Tabla 55. Comparación de tiempos del estudio de transferencia
Ah
orr
o
ap
lic
an
do
nu
eva
tecn
olo
gía
H-M
(%)
54,8
H –
H:
Ho
ras h
om
bre
H –
M:
Ho
ras m
áq
uin
a
H-H
(%)
55,7
Eq
uip
os
H -
M
17,7
8
H –
H
20,2
8
Nu
ev
a
tecn
olo
gía
Am
as
ad
or
de
alt
o c
ort
e
Se
ca
do
r d
e
lech
o f
luid
o
Tab
lete
ad
ora
de a
lta
velo
cid
ad
Re
cu
bri
do
ra
de b
om
bo
cerr
ad
o
H –
M
39,3
7
H -
H
45,7
6
Tec
no
log
ía
co
nven
cio
nal
Am
as
ad
or
pla
neta
rio
Se
ca
do
r
está
tic
o
Tab
lete
ad
ora
Ma
ne
sty
exp
ress
Bo
mb
o
ab
iert
o d
e
rec
ub
iert
a
Tam
añ
o d
e
lote
(ta
b)
200
00
0
Pe
so
de
gra
ne
l (k
g)
165
,6
Pro
du
cto
Pa
rac
eta
mo
l
500
mg
+
Dic
lofe
na
co
50 m
g

78
Tabla 56. Comparación de tiempo por incremento de tamaño de lote
Ah
orr
o
ap
lic
an
do
nu
eva
tecn
olo
gía
H –
M
(%)
34,3
3
H –
H:
Ho
ras h
om
bre
H –
M:
Ho
ras m
áq
uin
a
H –
H
(%)
42,2
8
Nu
ev
o t
am
añ
o
de l
ote
(400
0 0
00 t
ab
)
H -
M
29,8
6
H –
H
85,3
1
Tam
añ
o d
e
lote
ori
gin
al
(200
00
0 t
ab
)
H –
M
22,8
1
H -
H
73,9
0
Eq
uip
os
Am
as
ad
or
de
alt
o c
ort
e
Se
ca
do
r d
e
lech
o f
luid
o
Tab
lete
ad
ora
de a
lta
velo
cid
ad
Re
cu
bri
do
ra
de b
om
bo
cerr
ad
o
Pro
du
cto
Pa
rac
eta
mo
l
500
mg
+
Dic
lofe
na
co
50 m
g
Tabla 57. Ahorro anual por el estudio realizado

79
V. DISCUSIÓN
De acuerdo a los resultados obtenidos en el presente estudio, se observa que la
transferencia tecnológica contribuye significativamente a la mejora de los procesos
de manufactura (granulación, compresión y recubrimiento) del producto evaluado;
evidenciándose en el cumplimiento de las especificaciones de calidad establecidas
(pruebas físicas, fisicoquímicas y microbiológicas), obteniendo resultados
semejantes a los lotes fabricados en los equipos convencionales. Asimismo, la
mejora tecnológica asociado al incremento de tamaño de lote impacta positivamente
en la productividad de los procesos farmacéuticos, optimizando: tiempos, horas
máquina, horas hombre y análisis de producto; lo que genera una ventaja
competitiva para los laboratorios ya que se reduce el costo de manufactura y se
logra una mejor respuesta productiva frente a las necesidades del mercado.
Los equipos empleados en la transferencia tecnológica poseen parámetros de
medición automatizados los que se describen en las tablas N° 3, 4, 5 y 6. El control
y medición de estos parámetros a diferencia de los equipos convencionales
contribuyen a la estandarización y reproducibilidad del proceso de manufactura para
la obtención y cumplimiento de los atributos de calidad del producto.
La Organización Mundial de la Salud (OMS), mediante la guía de transferencia
tecnológica en la industria farmacéutica, define a la transferencia tecnológica como
"un procedimiento lógico que controla la transferencia de cualquier proceso, junto
con su documentación y experiencia profesional entre el desarrollo y la producción, o
entre los sitios de fabricación". Se trata de un procedimiento sistemático que se
sigue con el fin de transmitir los conocimientos y la experiencia adquiridos durante el
proceso (10); siguiendo esa línea, en el presente estudio se estableció la secuencia
del proceso de manufactura empleando nueva tecnología, los mismos que se
encuentran detallados en la figuras N° 1 y 2.
Los resultados obtenidos en la evaluación del desempeño de los equipos: amasador
de alta velocidad, granulador de lecho fluido, tableteadora rotativa y recubridora de

80
bombo cerrado; en las etapas de granulación, compresión y recubrimiento del
producto paracetamol 500 mg + diclofenaco 50 mg tabletas recubiertas, fueron
conformes al diseño establecido.
Los parámetros de amasado (amasador de alta velocidad): velocidad de la cuchilla,
velocidad de la paleta, velocidad de ingreso de la solución granuladora y tiempo de
amasado se controlan de manera automatizada; el control de estos parámetros en el
presente estudio, bajo el diseño establecido, nos ayuda a obtener un granulado con
características físicas adecuadas (aspecto y tamaño de partículas); en contraste al
amasado realizado en el equipo convencional (amasador planetario), donde no se
tiene un control automatizado de los parámetros mencionados anteriormente.
En el granulador de lecho fluido, los parámetros operativos: temperatura de aire de
ingreso, temperatura del lecho de secado, velocidad de ingreso del aire y tiempo de
secado de secado se controlan de manera automatizada; el control de estos
parámetros nos ayuda a obtener un granulado con características físicas adecuadas
(aspecto, humedad y tamaño de partículas); a diferencia del secado realizado en la
estufa estática.
La etapa de compresión, realizada en la tableteadora rotativa de alta velocidad,
presenta los siguientes parámetros: presión de tonelaje, dureza, llenado de matriz,
precompresión y velocidad de compresión, que se controlan de manera
automatizada; el control de estos parámetros nos ayuda a obtener tabletas que
cumplen los criterios de calidad previamente establecidos (aspecto, friabilidad, peso,
dureza, desintegración y dimensiones), esto difiere a la compresión realizada en la
tableteadora convencional donde no se tiene un control automatizado de los
parámetros.
En la etapa de recubrimiento, realizada en la recubridora de bombo cerrado, los
siguientes parámetros se controlan de manera automatizada: velocidad de bombo,
velocidad de ingreso de la suspensión de recubrimiento, temperatura de recubierta,

81
velocidad de aire de ingreso, temperatura de secado y distancia de pistolas; el
control de estos parámetros nos ayuda a obtener tabletas recubiertas que cumplen
con los criterios de calidad previamente establecidos (aspecto, peso, dureza,
desintegración, disolución y dimensiones); esto difiere al recubrimiento realizado en
la recubridora convencional (bombo abierto) donde no se tiene un control
automatizado de los parámetros.
Los resultados obtenidos de las pruebas físicas: aspecto y humedad del granulado
en los diferentes niveles de muestreo (superior, medio e inferior), que se observan en
las tablas N° 14 y 15, fueron conformes a las especificaciones de calidad
previamente establecidas para estos atributos; lo que indica que el cambio de
tecnología no afecta los atributos de calidad evaluados en comparación con los lotes
fabricados en los equipos convencionales, en adición el proceso se vuelve más
reproducible.
Con la finalidad de tener una mayor caracterización del granulado se realizaron
pruebas físicas adicionales: granulometría y densidad del granulado. En la prueba de
granulometría (figuras N° 8 y 9) de los lotes de evaluados en el presente estudio, se
puede observar que existe una distribución homogénea de los gránulos en los
diferentes tamices, esto es un indicativo que empleando nueva tecnología se obtiene
un granulado con mejores características reológicas, los cuales se refuerzan con los
resultados de densidad aparente y compactada de los lotes de transferencia (tablas
N° 16 y 17), que nos permite calcular los índices de Hausner y de Carr (tablas N° 18
y 19), que nos ayudan a determinar que el granulado obtenido tiene una buena
fluidez(15,16).
Las pruebas de dosaje de los principios activos: paracetamol y diclofenaco, en la
etapa de granulación (mezcla sin lubricación y mezcla con lubricación), fueron
conformes a las especificaciones de calidad establecidas. Se obtuvo resultados de
dosaje dentro del + 10 % para cada principio activo, según lo indicado en la USP (7).
Asimismo, se obtiene una distribución homogénea (uniformidad de mezcla) de los

82
activos en todos los puntos de muestreo (tablas N° 20 - 25). Por lo mencionado, la
fabricación del producto empleando los equipos: amasador de alta velocidad y
granulador de lecho fluido, no afecta el dosaje de activos (atributo crítico de calidad)
para esta etapa del producto y se logra un granulado homogéneo.
Los resultados obtenidos en la etapa de compresión cumplen con las
especificaciones previamente establecidas: aspecto, humedad, dimensiones,
friabilidad, peso, dureza y desintegración (tablas del N° 26 al N° 31); esto demuestra
que el cambio de tecnología no impacta en los atributos de calidad del producto para
esta etapa. Adicionalmente, se evaluó la capacidad de proceso (un proceso se
considera capaz si el índice Ppk es mayor a 1,00) del peso de las tabletas para la
tableteadora de alta rotación; obteniéndose un proceso capaz debido a que se
obtuvo un índice de Ppk igual a 2,11 y 1,83 (figura N° 20 y 22) para el primer y
segundo lote respectivamente. De esta manera, el proceso está centrado en el
objetivo y la tableteadora es capaz de producir tabletas con pesos conforme a
especificaciones. Asimismo, los pesos obtenidos después de la etapa de compresión
se encuentran en un estado de control; ya que los datos se mueven dentro de las
especificaciones inferior y superior (figura N° 19 y 20).
Los resultados de dosaje de los principios activos: paracetamol y diclofenaco, en la
etapa de compresión, fueron conformes a las especificaciones de calidad
establecidas. Se obtuvo resultados de dosaje dentro del + 10 % para cada principio
activo, según lo indicado en la USP(7). Asimismo, se obtiene una distribución
homogénea de los activos en las tabletas a lo largo del proceso de compresión
(tablas N° 34 - 35); en adición, se obtienen resultados conformes para la prueba de
uniformidad de unidades de dosificación (tablas N° 32 y 33). Por lo descrito, la
compresión del producto empleando el equipo: tableteadora de alta rotación, no
afecta la uniformidad de unidades de dosificación (atributo crítico de calidad) para
esta etapa del producto.

83
Los resultados obtenidos en la etapa de recubrimiento cumplen con las
especificaciones previamente establecidas: aspecto, humedad, dimensiones,
friabilidad, peso, dureza y desintegración (tablas N° 36 – 42); esto demuestra que el
cambio de tecnología no impacta en los atributos de calidad del producto para esta
etapa. Adicionalmente, se evaluó la capacidad de proceso (un proceso se considera
capaz si el índice Ppk es mayor a 1,00) del peso en las tabletas; obteniéndose un
proceso capaz debido a que se obtuvo un índice de Ppk igual a 2,11 y 1,22 (figura N°
32 y 34) para el primer y segundo lote respectivamente. De esta manera, el proceso
está centrado en el objetivo y en la etapa de recubrimiento es capaz de producir
tabletas recubiertas con pesos conforme a especificaciones. Asimismo, los pesos
obtenidos después de la etapa de compresión se encuentran en un estado de
control; ya que los datos se mueven dentro de las especificaciones inferior y superior
(figura N° 31 y 33).
Los resultados de disolución, dosaje y uniformidad de unidades de dosificación de
los principios activos: paracetamol y diclofenaco, en la etapa de recubrimiento,
fueron conformes a las especificaciones de calidad establecidas. Se obtuvieron
resultados de disolución (tablas N° 43 y 44) y resultados de dosaje para cada
principio activo, según lo indicado en la USP(7). Asimismo, se obtiene una distribución
homogénea de los activos en las tabletas recubiertas (tablas N° 47 - 50); en adición,
se obtienen resultados conformes para la prueba de uniformidad de unidades de
dosificación (tablas N° 45 y 46). Por lo descrito, el recubrimiento del producto
empleando el equipo: recubridora de tabletas cerrado, no afecta los atributos críticos
de calidad para esta etapa del producto.
La conformidad de los atributos de calidad (aspecto, peso promedio, desintegración,
dureza, friabilidad, dosaje de principios activos, uniformidad de unidades de
dosificación, disolución y análisis microbiológico) que han sido evaluados en las
diferentes etapas del proceso de manufactura del producto (granulación, compresión
y recubrimiento) muestran una correlación entre estas etapas; el dosaje de los
activos en las diferentes etas del proceso oscila para diclofenaco entre 95,80% -
108,08% y para paracetamol entre 92,71% - 108,06% (especificación entre 90% +

84
10%); esto indica que los atributos se mantienen estables entre una etapa y otra,
asegurando cumplir las especificaciones de calidad del producto a lo largo del ciclo
de manufactura.
La evaluación microbiológica realiza en las diferentes etapas del proceso de
manufactura fueron conformes a las especificaciones de calidad establecidas, los
mismo que asegura la obtención de productos de calidad (tablas N° 51, 52 y 53).
El producto Paracetamol 500 mg + diclofenaco 50 mg tabletas recubiertas, se
sometió a estudios de estabilidad, con la finalidad de garantizar que el producto
cumpla con las especificaciones de calidad, seguridad y eficacia durante su período
de vida útil, bajo las condiciones de almacenamiento establecidas (8). El presente
estudio muestra resultados conformes en los diferentes atributos de calidad, durante
el tiempo de estudio (tabla N° 54). Estos estudios se realizaron bajo las siguientes
condiciones climáticas: zona II (25°C +/- 2 °C y 60 % +/- 5%) y zona IVb (30 °C + 2
°C / 75% + 5%) (8), esta última es una condición más exigente para el producto,
debido que Perú se encuentra categorizado como zona IVa (30 °C + 2 °C / 65% +
5%). Este estudio de estabilidad natural a largo plazo se realizó para observar la
tendencia a la degradación química del principio activo, para proyectar el período de
vida útil sobre el límite de confianza de 95% (8).
El estudio de trasferencia contribuyó a reducir significativamente los tiempos del
proceso de manufactura (horas hombre y horas máquina), lo que genera un ahorro
por proceso que a largo plazo se evidenciará en un ahorro significativo de recursos
destinados para el proceso de obtención de este producto. Se obtuvo un ahorro del
42,28 % en horas hombre y 34,33% en horas máquina, en comparación al lote
manufacturado bajo los equipos convencionales (tablas N° 55, 56 y 57). Ello conlleva
a tener productos de calidad y optimizar las operaciones de las empresas
farmacéuticas.

85
VI. CONCLUSIONES
El estudio de transferencia tecnológica mediante el empleo de los equipos
automatizados para el desarrollo del proceso de fabricación de tabletas recubiertas
de Paracetamol 500mg + Diclofenaco sódico 50 mg son satisfactorios y cumplen
las especificaciones de calidad establecidas para el producto en relación a la
Farmacopea de los Estados Unidos (USP vigente).
Las etapas de fabricación, granulación, compresión y recubrimiento del producto
estudiado fueron optimizados por el cambio de tecnología, lo que permitió el
cumplimiento de las especificaciones de calidad previamente establecidas.
El producto de Paracetamol 500 mg + Diclofenaco sódico 50 mg tabletas
recubiertas, es estable a las condiciones de almacenamiento: zona II (25°C +/- 2 °C
y 60 % +/- 5%) y zona IVb (30 °C + 2 °C / 75% + 5%), lo que demuestra que el
cambio de tecnología para la obtención del producto no afecta sus atributos de
calidad en el tiempo.
El cambio de tecnología para la manufactura de Paracetamol 500mg + Diclofenaco
sódico 50 mg tabletas recubiertas, permitió un ahorro de 42,28% en horas hombre
y 34,33% en horas máquina, en comparación con la tecnología convencional.

86
VII. GLOSARIO DE TÉRMINOS
Capacidad de proceso: Es una propiedad medible de un proceso que puede
calcularse por medio del índice de capacidad del proceso, que permite calcular
cuántos componentes serán producidos fuera de los límites establecidos en la
especificación.
Cpk / Ppk: Capacidad de proceso por límites de especificación por ambos lados
Gráfica de control: Sirven para poder analizar el comportamiento de los
diferentes procesos y poder prever posibles fallos de producción mediante
métodos estadísticos.
L1: Máximo valor permitido para la prueba de uniformidad de unidades de
dosificación
Densidad aparente: Es la densidad del polvo que incluye los espacios que
existen entre las partículas y las burbujas de aire que hayan incrustadas en
estas.
Densidad compactada: Es la densidad aparente que se ha compactado o
asentado por vibración, dejando un volumen o peso específico del polvo en la
probeta.
Índice de carr: La relación entre las densidades aparente y compactada entre
más se compacte un polvo, más pobre serán sus propiedades de flujo.
Índice de Hausner: Relaciona el cociente entre las densidades aparente final
respecto a la inicial, y entre más alto sea éste menor será el flujo de los polvos.

87
Granulometría: Es la distribución de los tamaños de las partículas de un
agregado, tal como se determina por análisis de tamices.
Optimización: Es la acción que busca la mejor manera de realizar una actividad.
Automatización: Es aquella acción diseñada con el fin de usar la capacidad de
las máquinas para llevar a cabo determinadas tareas anteriormente efectuadas
por seres humanos, y controlar la secuencia de las operaciones sin intervención
humana.

88
VIII. REFERENCIAS BIBLIOGRÁFICAS
1. Fauli Trillo C. Tratado de Farmacia Galénica. 1ra Edición. Madrid. Editorial Lusan
5 ediciones, 1993: 59 -103, 405 – 12, 504 - 18.
2. Villa Jato JL. Tecnología Farmacéutica: Aspectos fundamentales de los
Sistemas Farmacéuticos y Operaciones Básicas. Madrid: Editorial Síntesis,
1997.
3. Salazar Macián R. Gestión de la Calidad en el Desarrollo y Fabricación Industrial
de Medicamentos. Barcelona: Editorial ROMARGRAF, 2001.
4. Gibson M. Pharmaceutical preformulation and formulation. New York: Editorial
Informa Healthcare, 2009.
5. Rowe R, Sheskey P, Owen S. 5th Ed. Handbook of pharmaceutical
excipients.2006.
6. Brittain H. Physical characterization of pharmaceutical solids. New York, 1995.
7. USP 35 – NF 30. The United States Pharmacopeial Convention, Rockville, MD,
2012.
8. MINSA/DIGEMID. Directiva Sanitaria Nº 31. Directiva Sanitaria que reglamenta
los estudios de estabilidad de medicamentos. Lima. 2009.
9. International Conference on Harmonization. Evaluation for Stability Data, ICH
Harmonised Tripatite Guideline, ICH Q1E. Febrero 2003.

89
10. Organización Mundial de la Salud (OMS). Who Guideline on Transfer of
Technology. 2009; 259 (1). (En línea), consultado el 15 de enero del 2013.
Disponible en:
http://www.who.int/medicines/areas/quality_safety/quality_assurance/TransferT
echnologyPharmaceuticalManufacturingTRS961Annex7.pdf.
11. Administración de Alimentos y medicamentos (FDA). Guidance for Industry:
Quality Systems Approach to Pharmaceutical Current Good Manufacturing
Practice Regulations. 2004.
12. Mark Gibson. Technology Transfer: An International Good Practice Guide for
Pharmaceuticals and Allied Industries. PDA Bethesda, MD, USA - DHI
Publishing, LLC River Grove, IL, USA 2005.
13. International Conference on Harmonization. Pharmaceutical Development, ICH
Harmonised Tripartite Guideline, ICH Q10. Junio, 2008.
14. Alva Bazalar, N. Evaluación del Cambio de Formulación y Mejora del
Procedimiento de Fabricación de Tabletas de Hioscina-N- Butil Bromuro de
10mg [Tesis para optar el título profesional de Químico Farmacéutico] Facultad
de Farmacia y Bioquímica, Universidad Nacional Mayor de San Marcos. Lima,
2002.
15. Castillo Guerra H., Panoca Alberto P. Diseño, Caracterización Farmacotécnica
y Evaluación de Nuevas Formulaciones de Comprimidos Recubiertos de
Nimodipino 30 mg [Tesis para optar el título profesional de Químico
Farmacéutico] Facultad de Farmacia y Bioquímica, Universidad Nacional Mayor
de San Marcos. Lima, 2011.
16. Ninamaco Sarmiento R., Quezada Balcázar D. Evaluación de la formulación
por cambio de excipientes y mejora en el proceso de fabricación de

90
comprimidos de espirolactona 100 mg [Tesis para optar el título profesional de
Químico Farmacéutico] Facultad de Farmacia y Bioquímica, Universidad
Nacional Mayor de San Marcos. Lima, 2011.
17. Rayat Institute of Pharmacy, Railmajra, District S.B.S. Nagar, Punjab, India.
Technology Transfer in Pharmaceutical Industry: A Discussion. International
Journal of Pharma and Bio Sciences. 2010; 001.
18. Ramon SM. Apuntes sobre tecnología farmacéutica. Fabricación y control de
formas farmacéuticas recubiertas. 1ra ed. Editorial Síntesis. Barcelona 2010. P.
414.

91
IX. ANEXOS
ANEXO 1: EQUIPOS DEL PROCESO DE MANUFACTURA / PRODUCTOS OBTENIDOS
DURANTE LE PROCESO
Tableteadora rotativa de alta velocidad Recubridora de bombo cerrado
Granulado de Paracetamol 500 mg +
Diclofenaco sódico 50 mg
Núcleos de Paracetamol 500 mg +
Diclofenaco sódico 50 mg
Tabletas recubiertas de Paracetamol
500 mg + Diclofenaco sódico 50 mg

92
ANEXO 2: RESULTADOS DE ASPECTO DE GRANULADO DE PARACETAMOL 500 mg
+ DICLOFENACO SÓDICO 50 mg

93
ANEXO 3: RESULTADOS DE HUMEDAD DE GRANULADO DE PARACETAMOL 500 mg
+ DICLOFENACO SÓDICO 50 mg

94
ANEXO 4: RESULTADOS DE GRANULOMETRÍA DE PARACETAMOL 500 mg +
DICLOFENACO SÓDICO 50 mg

95
ANEXO 4: RESULTADOS DE GRANULOMETRÍA DE PARACETAMOL 500 mg +
DICLOFENACO SÓDICO 50 mg

96
ANEXO 4: RESULTADOS GRANULOMETRÍA DE 500 mg + DICLOFENACO SÓDICO 50
mg

97
ANEXO 5: RESULTADOS DE DENSIDADES DE PARACETAMOL 500 mg +
DICLOFENACO SÓDICO 50 mg

98
ANEXO 6: RESULTADOS DE ASPECTO DE NÚCLEOS DE PARACETAMOL 500 mg +
DICLOFENACO SÓDICO 50 mg

99
ANEXO 7: RESULTADOS DE HUMEDAD DE NÚCLEOS DE PARACETAMOL 500 mg +
DICLOFENACO SÓDICO 50 mg

100
ANEXO 8: RESULTADOS DE FRIABILIDAD DE NÚCLEOS DE PARACETAMOL 500 mg
+ DICLOFENACO SÓDICO 50 mg

101
ANEXO 9: RESULTADOS DE DESINTEGRACIÓN DE NÚCLEOS DE PARACETAMOL
500 mg + DICLOFENACO SÓDICO 50 mg

102
ANEXO 10: RESULTADOS DIMENSIONES DE NÚCLEOS DE PARACETAMOL 500 mg
+ DICLOFENACO SÓDICO 50 mg

103
ANEXO 11: RESULTADOS DE DESINTEGRACIÓN DE TABLETAS RECUBIERTAS DE
PARACETAMOL 500 mg + DICLOFENACO SÓDICO 50 mg

104
ANEXO 12: RESULTADOS DE PESO PROMEDIO DE TABLETAS RECUBIERTAS DE
PARACETAMOL 500 mg + DICLOFENACO SÓDICO 50 mg

105
ANEXO 13: RESULTADOS DE DUREZA DE TABLETAS RECUBIERTAS DE
PARACETAMOL 500 mg + DICLOFENACO SÓDICO 50 mg

106
ANEXO 14: RESULTADOS DE DIMENSIONES DE TABLETAS RECUBIERTAS
DE PARACETAMOL 500 mg + DICLOFENACO SÓDICO 50 mg

107
ANEXO 14: RESULTADOS DE DIMENSIONES DE TABLETAS RECUBIERTAS DE
PARACETAMOL 500 mg + DICLOFENACO SÓDICO 50 mg


















