
Enzymic Studies on t,he Biosynthesis of Serotonin in Mammalian Brain
12
THE JOURNAL OF BIOLOGWALCHEMISTRY Vol. 245, No. 7, Issue of April 10, pp. X99-1709.1970 Printed in U.S.A. Enzymic Studies on t,he Biosynthesis of Serotonin in Mammalian Brain* (Received for publication, December 3, 1969) ARATA ICHIYAMA, SHIGENOBU NAKAMURA,$ YASUTOMI NISHIZUKA,§ AND OSAMU HAYAISHI From the Department of Medical Chemistry, Kyoto University Faculty of Medicine, Kyoto, Japan SUMMARY A rapid, convenient and sensitive assay method for measur- ing tryptophan 5-monooxygenase in mammalian brain is developed with r.-tryptophan-carboxyl-r4C as substrate. The method is based on the differing afhnity of aromatic L-amino acid decarboxylase toward L-tryptophan and 5- hydroxy-L-tryptophan (K, values for L-tryptophan and 5-hydroxy-L-tryptophan are approximately 1.4 X 10V2 M and 5.4 X 1OV M, respectively). The reaction is carried out in the presence of an excess amount of the decarboxylase, and the radioactive COz preferentially liberated from 5-hydroxy- L-tryptophan, which is formed from L-tryptophanJ4C by the action of tryptophan S-monooxygenase, is measured. Tryptophan 5-monooxygenase activity is detected in both the soluble and particulate fractions from the guinea pig brain stem. When relatively low concentrations (below lo-+ M) of L-tryptophan-(side chain-1,2,3-14C) are used as substrate and incubated with the supernatant or the crude mitochondrial fractions in the presence of monoamine oxidase inhibitor, the radioactive COz evolved is almost entirely accounted for by the formation of serotoninJ4C. Only small amounts of tryptamineJ4C are produced and very little 5-hydroxy-L-tryptophan-r4C accumulates in the reaction mixture. Properties of tryptophan 5-monooxygen- ase in both the supernatant and crude mitochondrial fractions are examined and the appropriate conditions for the assay are described. It has been generally accepted that serotonin is synthesized from tryptophan through two consecutive reactions, the hy- * The data are taken from a dissertation which was submitted by A. Ichiyama to Kyoto University in partial fulfillment of the requirement for the degree of Doctor of Medical Science in May 1968. This investigation has been supported in part by Public Health Service Research Grants CA-04222, from the National Cancer Institute, and AM-10333, from the National Institute of Arthritis and Metabolic Diseases, and grants from the Jane Coffin Childs Memorial Fund for Medical Research, the Squibb Institute for Medical Research, and the Scientific Research Fund of the Ministry of Education of Japan. $ Present address, D,epartment of Geriatrics, Kyoto University Faculty of Medicine, Kyoto, Japan. $ Present address, Department of Biochemistry, Kobe Uni- versity School of Medicine, Kobe, Japan. droxylation of tryptophan to 5-hydroxytryptophan followed by decarboxylation of the latter to form serotonin (1). Tryptophan 5-monooxygenase in the brain was first shown by Grahame- Smith who showed that homogenates from dog and rabbit brain stems hydroxylate n-tryptophan to produce 5-hydroxy- n-tryptophan (2). Subsequent reports from several laboratories including our own have also described the hydroxylation of tryptophan in vitro with extracts from various mammalian brain tissues (3-12). This enzyme activity was also observed in intact cells of Chrcnnobacterium uioluceum (13), cell-free extracts of caroOinoid tumors (10, 14), and mouse neoplastic mast cells (15-18). In a preliminary report from this laboratory the partial purification and some properties of the monooxygenase from the rabbit brain stem were briefly described (7). However, the relatively low activity of this enzyme and the lack of a sensitive and convenient assay have hindered further studies on t,he purification and properties of the enzyme as well as the reaction mechanism involved. Assay methods so far available are based on the determination of radioactive serotonin or 5-hydroxy- tryptophan which are separated from a large excess of unreacted tryptophan by either paper chromatography, paper electrophore- sis, or ion exchange resins. Such methods are useful for mast cell tryptophan 5-monooxygenase which is exceptionally active and accumulates a sizable quantity of 5-hydroxytryptophan. For most tissues including brain, a more sensitive and convenient assay was highly desirable. The present study was undertaken to develop such a procedure and to study the detailed properties and mechanism of tryptophan 5-monooxygenase in mammalian brain. The enzyme responsible for the decarboxylation of 5-hydroxy- L-tryptophan to produce serotonin has been described in a variety of mammalian tissues (19-22). Lovenberg, Weissbach, and Udenfriend (20) showed that the decarboxylases obtained from guinea-pig kidney and dog brain react with various aro- matic n-amino acids including n-tryptophan and 5-hydroxy-L- tryptophan, and designated the enzyme as aromatic L-amino acid decarboxylase. The apparent K, value of the guinea pig kidney enzyme for tryptophan was far greater than that for 5- hydroxytryptophan (3 X 1O-3 M for tryptophan, 2 X 10V6 M for 5-hydroxytryptophan) (20). This difference is even greater in t,he case of enzymes obtained from guinea pig and bovine brains. The assay method for tryptophan 5-monooxygenase used in the present study has taken advantage of the relative affinity of the decarboxylase toward these two amino acids. The assay meas- ures the radioactive CO2 liberated from 5-hydroxy-n-t,ryptophan 1699 by guest on April 9, 2019 http://www.jbc.org/ Downloaded from
Transcript of Enzymic Studies on t,he Biosynthesis of Serotonin in Mammalian Brain

THE JOURNAL OF BIOLOGWAL CHEMISTRY Vol. 245, No. 7, Issue of April 10, pp. X99-1709.1970
Printed in U.S.A.
Enzymic Studies on t,he Biosynthesis of Serotonin in Mammalian Brain*
(Received for publication, December 3, 1969)
ARATA ICHIYAMA, SHIGENOBU NAKAMURA,$ YASUTOMI NISHIZUKA,§ AND OSAMU HAYAISHI
From the Department of Medical Chemistry, Kyoto University Faculty of Medicine, Kyoto, Japan
SUMMARY
A rapid, convenient and sensitive assay method for measur- ing tryptophan 5-monooxygenase in mammalian brain is developed with r.-tryptophan-carboxyl-r4C as substrate. The method is based on the differing afhnity of aromatic L-amino acid decarboxylase toward L-tryptophan and 5- hydroxy-L-tryptophan (K, values for L-tryptophan and 5-hydroxy-L-tryptophan are approximately 1.4 X 10V2 M and 5.4 X 1OV M, respectively). The reaction is carried out in the presence of an excess amount of the decarboxylase, and the radioactive COz preferentially liberated from 5-hydroxy- L-tryptophan, which is formed from L-tryptophanJ4C by the action of tryptophan S-monooxygenase, is measured.
Tryptophan 5-monooxygenase activity is detected in both the soluble and particulate fractions from the guinea pig brain stem. When relatively low concentrations (below lo-+ M) of L-tryptophan-(side chain-1,2,3-14C) are used as substrate and incubated with the supernatant or the crude mitochondrial fractions in the presence of monoamine oxidase inhibitor, the radioactive COz evolved is almost entirely accounted for by the formation of serotoninJ4C. Only small amounts of tryptamineJ4C are produced and very little 5-hydroxy-L-tryptophan-r4C accumulates in the reaction mixture. Properties of tryptophan 5-monooxygen- ase in both the supernatant and crude mitochondrial fractions are examined and the appropriate conditions for the assay are described.
It has been generally accepted that serotonin is synthesized from tryptophan through two consecutive reactions, the hy-
* The data are taken from a dissertation which was submitted by A. Ichiyama to Kyoto University in partial fulfillment of the requirement for the degree of Doctor of Medical Science in May 1968. This investigation has been supported in part by Public Health Service Research Grants CA-04222, from the National Cancer Institute, and AM-10333, from the National Institute of Arthritis and Metabolic Diseases, and grants from the Jane Coffin Childs Memorial Fund for Medical Research, the Squibb Institute for Medical Research, and the Scientific Research Fund of the Ministry of Education of Japan.
$ Present address, D,epartment of Geriatrics, Kyoto University Faculty of Medicine, Kyoto, Japan.
$ Present address, Department of Biochemistry, Kobe Uni- versity School of Medicine, Kobe, Japan.
droxylation of tryptophan to 5-hydroxytryptophan followed by decarboxylation of the latter to form serotonin (1). Tryptophan 5-monooxygenase in the brain was first shown by Grahame- Smith who showed that homogenates from dog and rabbit brain stems hydroxylate n-tryptophan to produce 5-hydroxy- n-tryptophan (2). Subsequent reports from several laboratories including our own have also described the hydroxylation of tryptophan in vitro with extracts from various mammalian brain tissues (3-12). This enzyme activity was also observed in intact cells of Chrcnnobacterium uioluceum (13), cell-free extracts of caroOinoid tumors (10, 14), and mouse neoplastic mast cells (15-18).
In a preliminary report from this laboratory the partial purification and some properties of the monooxygenase from the rabbit brain stem were briefly described (7). However, the relatively low activity of this enzyme and the lack of a sensitive and convenient assay have hindered further studies on t,he purification and properties of the enzyme as well as the reaction mechanism involved. Assay methods so far available are based on the determination of radioactive serotonin or 5-hydroxy- tryptophan which are separated from a large excess of unreacted tryptophan by either paper chromatography, paper electrophore- sis, or ion exchange resins. Such methods are useful for mast cell tryptophan 5-monooxygenase which is exceptionally active and accumulates a sizable quantity of 5-hydroxytryptophan. For most tissues including brain, a more sensitive and convenient assay was highly desirable. The present study was undertaken to develop such a procedure and to study the detailed properties and mechanism of tryptophan 5-monooxygenase in mammalian brain.
The enzyme responsible for the decarboxylation of 5-hydroxy- L-tryptophan to produce serotonin has been described in a variety of mammalian tissues (19-22). Lovenberg, Weissbach, and Udenfriend (20) showed that the decarboxylases obtained from guinea-pig kidney and dog brain react with various aro- matic n-amino acids including n-tryptophan and 5-hydroxy-L- tryptophan, and designated the enzyme as aromatic L-amino acid decarboxylase. The apparent K, value of the guinea pig kidney enzyme for tryptophan was far greater than that for 5- hydroxytryptophan (3 X 1O-3 M for tryptophan, 2 X 10V6 M for 5-hydroxytryptophan) (20). This difference is even greater in t,he case of enzymes obtained from guinea pig and bovine brains. The assay method for tryptophan 5-monooxygenase used in the present study has taken advantage of the relative affinity of the decarboxylase toward these two amino acids. The assay meas- ures the radioactive CO2 liberated from 5-hydroxy-n-t,ryptophan
1699
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

1700 Tryptophan 5-Mmooxygenase Vol. 245, No. 7
which is formed from L-tryptophan- (side chain-l ,2, 3J4C) or L-tryptophan-carboxyl-14C by the action of tryptophan 5-mono- oxygenase. With the new assay method, some basic properties of tryptophan 5-monooxygenase in mammalian brain stem have been re-examined and characterized.
A rapid and sensitive assay of the decarboxylase activity for 5-hydroxytryptophan was also developed by the use of 5-hydroxy-n-tryptophaa-(side chain-l ,2,3-l%) as substrate. A preliminary account of this work has been published (23).
EXPERIMENTAL PROCEDURE
Preparation of 5-Hydroxy-L-typtophan-(side chain-l ,2?,QJ4C)- L-SerineJ4C (uniformly labeled, 160 mCi per mmole) was ob- tained from the Radiochemical Centre, Amersham, England. The principle of the enzymic synthesis of 5-hydroxy-L-trypto- phan-(side chain-l ,2,3J4C) is based on the condensation of L-serine and 5-hydroxyindole to 5-hydroxy-n-tryptophan catalyzed by tryptophan synthetase from Escherichia coli, T3 mutant (24). The incubation mixture (1.0 ml) contained 0.1 mCi of uniformly labeled L-serine-‘4C (0.62 pmole), 3.0 pmoles of 5-hydroxyindole, 0.1 pmole of pyridoxal phosphate, 2 pmoles of reduced glutathione, 340 pmoles of NaCl, 120 pmoles of potassium phosphate (pH 7.8), and approximately 30 mg of tryptophan synthetase which was purified from the T3 mutant of E. co& as described below. The mixture was incubated for 3 hours at 37” with constant shaking. The reaction was stopped by the addition of 0.05 ml of 60% perchloric acid. An appropri- ate amount of 5-hydroxy-L-tryptophan was added to the mix- ture and the insoluble material was removed by centrifugation. The precipitate was washed five times with 1 ml each of 2% perchloric acid. The supernatant and washings were combined and pH was adjusted with 5 N KOH to approximately 6.0. Potassium perchlorate was removed by centrifugation at 4’ and the precipitate was washed three times with 0.5 ml each of cold water. To the combined supernatant and washings were added approximately 50 mg of charcoal which had been deac- tivated with 4% stearic acid (25). The suspension was stirred for 10 min at room temperature and was poured onto a glass filter. After the charcoal was washed with 50 ml of water, 5-hydroxy-n-tryptophan-*4C was eluted from the charcoal with 15 ml of 8% aqueous phenol solution (25). Phenol was then removed by extraction with excess ether, and the aqueous phase containing 5-hydroxy-L-tryptophan-l4C was passed through a column of Dowex 1-X8-formate (200 to 400 mesh; diameter 0.6 cm, length 2 cm). The column was washed with 30 ml of 0.01 N formic acid. The effluent and washings were combined and lyophilized. The dried residue was taken up with 3 ml of the upper layer of a mixture of 1-butanol-acetic acid-water (20:1:16). The material was then subjected to partition chromatography on a Sephadex G-25 column as described below. Fractions (5 ml each) were collected at a flow rate of 40 to 50 ml per hour and the radioactivity of each fraction was deter- mined. The fractions containing 5-hydroxy-n-tryptophanJ4C were combined and lyophilized. The dried residue was dis- solved in a small volume of 0.04 N acetic acid. The over-all yield of 5-hydroxy-L-tryptophan-14C from L-serine-14C was approximately 50%. The final product showed an ultraviolet absorption spectrum almost identical with that of authentic 5-hydroxytryptophan. A single radioactive peak was obtained which corresponded to 5-hydroxy-L-tryptophan upon paper
chromatography with the four different solvent systems described below, high voltage paper electrophoresis, thin layer chroma- tography, and partition chromatography. 5-Hydroxy-n-trypto- phan-(side chain-l ,2,3-l%) thus prepared was stable when stored frozen at -20”.
Preparation of z-Tryptophan-(side chain-l ,b,S-14C)-~-Trypto- phan-(side chain-l ,2,3J4C) was prepared and purified as described above except that 5-hydroxyindole was replaced by indole. The react.ion mixture was deproteinized and then neutralized with KOH as described above. After potassium perchlorate was removed by centrifugat,ion, the supernatant solution was put on a Whatman No. 3MM paper as a narrow band and chromatographed overnight at 4” with l-but.anol- acetic acid-water (12:3:5) as solvent. Rp values of L-trypto- phan and L-serine were 0.52 and 0.31, respectively. L-Trypto- phanJ4C was eluted from the paper strip at 4” with 0.04 N
acetic acid. To the eluate (usually 7 to 10 ml) were added 50 mg of the deactivated charcoal, and L-tryptophar+C adsorbed on the charcoal was eluted and further purified by partition chroma- tography as described above. The over-all yield of L-trypto- phan-14C from L-serineJ4C was approximately 50%. The final product showed an ultraviolet absorption spectrum almost iden- tical with that of authentic L-tryptophan. The radiochemical purity was shown to be more than 95% as judged by paper, thin layer, and partition chromatographies in the solvent systems described below and by paper electrophoresis. L-Tryptophan- 14C thus prepared was stored frozen at -20”.
Other Radioactive Materials-L-Tryptophan-(side chain-lJ4C) (9 mCi per mmole) was purchased from New England Nuclear and purified by partition chromatography on a Sephadex G-25 column. L-Tryptophan-(side chain-3-14C) was obtained from the Radiochemical Centre, Amersham, England.
Chemicals2-Amino-4-hydroxy-6,7 - dimethyl- 5,6,7,8 - tetra- hydropteridine and dithiothreitol were obtained from Calbio- them. or-Methylphenethylhydrazine (Catron) was a gift from Dr. M. Fujiwara, Department of Pharmacology, Kyoto Uni- versity Faculty of Medicine, and Dr. H. Matsumoto, Chugai Pharmaceutical Company, Ltd. Isonicotinic acid-2-isopropyl- hydrasine phosphate (iproniazid phosphate) was a product of Tokyo Chemical Industry Company, Ltd. 5-Hydroxyindole, 5-hydroxytryptamine creatinine sulfate, 5-hydroxy-nn-trypto- phan, tryptamine, 5-hydroxyindole-3-acetic acid, and indole-3- aldehyde were obtained from Sigma. Other chemicals were obtained from commercial sources. All reagents were prepared with twice distilled water.
Biological Materials--A tryptophan auxotroph of E. coli, strain T3, was supplied by Drs. T. Yura and M. Imai, the Virus Research Institute, Kyoto University. The bacteria were grown for 8 hours at 37” with constant shaking in Medium E of Vogel-Bonner (26) containing 0.2% glucose, 0.2y0 casamino acids, and 5 mg per liter of L-tryptophan. The cells were harvested by a Sharples centrifuge. Tryptophan synthetase was purified from the sonic extracts approximately 5-fold according to the method Crawford and Yanofsky (27); the extracts were subjected to heat treatment for 2 min at 52”, followed by streptomycin treatment (2’%) and ammonium sulfate fractionation (33 to 50%) at pH 6.2. The over-all yield of the enzyme was approximately 60%. The partially purified enzyme was dialyzed overnight against a large excess of 0.025 M potas- sium phosphate, pH 7.8, and stored at -20” until use. The
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Issue of April 10, 1970 A. Ichiyama, S. Nakamura, Y. Nishizuka, and 0. Hayaishi 1701
enzyme preparation thus obtained catalyzed the conversion of approximately 35 nmoles of indole to tryptophan per min per mg of protein under the standard assay conditions (28).
Guinea pig brain stem was mainly used as an enzyme source in the present study. Guinea pigs maintained on an ad libitum diet of Clea laboratory chow were killed by a blow on the neck followed by decapitation. Brains were removed immedi- ately and rinsed with cold-distilled water. The brain stem was separated by a spatula, weighed, and homogenized with an appropriate volume of medium (see below) in a Potter-Elvehjem type homogenizer with a Teflon pestle. Usually three to four passes of the pestle were made in an ice bath. All subsequent operations were carried out at 4” except where indicated. The crude mitochondrial fraction was prepared from the 10% homog- enate in 0.32 M sucrose by the method of Gray and Whittaker (29). The particulate fraction was pelleted by centrifugation for 20 min at 12,000 X g and suspended in 0.32 M sucrose to make 3 ml of suspension from 1 g of the starting tissue by the use of the homogenizer. Approximately 1 g of the starting tissue yielded 30 mg of protein of the crude mitochondrial fraction. The supernatant fraction was obtained from the 33 ‘% homogenate of the brain stem in 0.02 M Tris-acetate, pH 8.1, containing 10ea M dithiothreitol by centrifugation for 30 min at 105,000 X g. Usually 1 g of the starting tissue yielded 1.8 ml of the supernatant fluid containing approximately 15 mg of protein per ml. Trypto- phan 5-monooxygenase was partially purified from the super- natant fraction by ammonium sulfate fractionation (0 to 40%) followed by dialysis for 12 hours against 0.01 M Tris-acetate, pH 8.1, containing 2 X 10e3 M dithiothreitol, under NP at 4”.
Assay of Tryptophan 5-Monooxygenase-Tryptophan B-mono- oxygenase was assayed by measurement of the rate of 14C02 formation from either L-tryptophan-(side chain-l ,2,3J4C) or L-tryptophan-(side chain-lJ4C) as substrate. Two standard assay systems were used. The first (Assay System I) was used for the homogenate or crude mitochondrial fraction. The reaction mixture (0.5 ml) contained 50 pmoles of Tris-acetate (pH 8.1), 125 pmoles of sucrose, 30 to 60 nmoles of r,-tryptophanJ4C (approximately 100,000 cpm), and 0.15 ml of the enzyme preparation. The second (Assay System II) was used for the soluble tryptophan 5-monooxygenase. The reac- tion mixture (0.5 ml) contained 50 pmoles of Tris-acetate (pH S.l), 0.3 pmole of 2-amino-4-hydroxy-6,7-dimethyl-5,6,7,8- tetrahydropteridine, 1.2 pmoles of dithiothreitol, 100 nmoles of pyridoxal phosphate, 10 nmoles of ferrous ammonium sulfate, n-tryptophan-r4C as in Assay I, 30 pg of catalase, 5 to 10 units (see below) of aromatic L-amino acid decarboxylase which was purified from the bovine brain stem as described below, and 0.1 ml of the enzyme. The reaction was carried out for 60 min at 37” in a test tube (diameter 1.6 cm, length 10 cm) which was connected to a counting vial with a thick rubber tube. The vial contained a filter paper strip (Whatman No. 3MM, width 1.7 cm, length 8 cm) which was immersed with 0.3 ml of 20% 2- phenethylamine in methanol’ (30,31). At the end of incubation 0.8 ml of 4% perchloric acid containing 2 X 10-a M tiron was injected through the rubber tube. The acidified reaction mixture was incubated for additional 3 hours at 37” with gentle
1 The authors are grateful to Drs. K. Kawashima and K. Morita, Research and Development Division, Takeda Chemical Indus- tries, for making the basic experimental data on CO* measurement available.
shaking or allowed to stand for 12 hours at 4”. To each vial were then added 10 ml each of a scintillator solution consisting of 2.5 g of 2,5-diphenyloxazole (PPO) and 150 mg of 1,4-bis- [2-(5-phenyloxazolyl)]benzene (POPOP) in 1 lit.er of toluene (see “Determinations”) and the radioactivity was determined in a Beckman DPM-100 liquid scintillation spectrometer. Counting efficiency of 14COZ was approximately 85%. In all experiments a blank incubation was carried out which contained all ingredients except for a boiled enzyme preparation (loo”, 5 min). The nonenzymic evolution of 14C02 was subtracted from the value for the experimental incubations. The amount of CO2 formed was calculated from the specific activity of the 1 carbon atom which is expected to be one-third of that of L-tryptophan- (side chain-l ,2 ,3J4C) or the same as that of the L-tryptophan- (side chain-lJ4C) used as substrate.
Assay of 5-Hydroxy-z-typtophan Decarboxylase-The enzyme activity was determined by measurement of the rate of 14C02 evolution from 5-hydroxy-n-tryptophan-(side chain-l ,2, 3-r4C). The standard assay system contained 11 nmoles of 5-hydroxy- n-tryptophan-(side chain-l ,2,3J4C) (3,300 cpm per nmole), 50 nmoles of pyridoxal phosphate, 30 Mmoles of Tris-acetate (pH 8.8), and an enzyme preparation in a final volume of 0.3 ml. The reaction mixture was incubated for 15 min at 37“ with shaking. The reaction was stopped by the addition of per- chloric acid-tiron mixture, and the 14COa evolved was determined as described above. One unit of the decarboxylase activity was defined as the amount which catalyzed the decarboxylat.ion of 1 nmole of 5-hydroxy-n-tryptophan per hour under the standard assay conditions.
Chromatographic and Electrophoretic ProceduresPartition chromatography on a Sephadex G-25 column was carried out as follows.2 Sephadex G-25 was suspended in water and poured onto a column. The column (diameter, 0.8 cm; length 40 cm) was washed with approximately 280 ml of the lower layer of a two-phase solvent system of 1-butanol-acetic acid-water (20: 1: 16) and then with the upper layer until the top of the column became semitransparent. Samples were dissolved in 3 ml of the upper layer and put on the column. The elution was carried out with the upper layer at a flow rate of 40 to 50 ml per hour at room temperature.
Amberlite CG-50 column chromatography was carried out according to the method of Green and Sawyer (8). Ascending paper chromatography for analytical purposes was carried out on Whatman No. 1 filter paper at room temperature and for preparative purposes on Whatman No. 3MM paper at 4”. The following solvent systems were used: methanol-l-butanol- benzene-acetic acid-water (40:20:20: 1: 19) (32); l-butanol- acetic acid-water (12:3:5) (14); 1 M ammonium acetate, pH 5.0.ethanol (3 : 7) ; and isopropyl alcohol-ammonia-water (20: 1:2) (14). High voltage electrophoresis was carried out on Whatman No. 1 paper (width, 17 cm; length 55 cm) with a pyridine-acetic acid-water (5.0:3.4:90.0, pH 5.0) buffer, ac- cording to Ryle et al. (33). n-Hexane was used as a coolant and a potential of 4000 volts was applied for 60 min. Thin layer chromatography was carried out on a silica gel film with a solvent system of methyl acetate-isopropyl alcohol-ammonia (9:7:4) (34). Indole compounds were detected by spraying Ehrlich’s reagent followed by gentle heating for several minutes.
1 The method was developed by Dr. N. Itada in this laboratory.
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

1702 Tryptophan &Monooxygenase Vol. 245, No. 7
Eff lient 2N Formi’c acid eluate
Amberlite CGSO column
I
1-1
Effluent 4N Acetic acid eluate I 1
Partition chromatography on Sephadex G-25 column I
FIQ. 1. Outline of the method of separation of various indole derivatives. Details are described in the text.
Treatment of indole compounds with charcoal was performed according to the method of Asatoor and Dalgliesh (35).
Analysis of Reaction Products-The reaction products were analyzed as outlined in Fig. 1. The reaction was stopped by the addition of perchloric acid. Authentic samples of L-trypto- phan, 5-hydroxyl+tryptophan, serotonin, tryptamine, and 5-hydroxyindoleacetic acid were added as carriers, and denatured protein was removed by centrifugation. The residue was washed twice, each time with 3 ml of 2y0 perchloric acid. The supernatant and washings were combined and neutralized with 5 N KOH to approximately pH 6. After potassium perchlorate was removed by centrifugatjon at 4”, the mixture was passed through a small column of Dowex 1-formate (diameter 0.6 cm, length 4 cm). The column was washed with 20 ml of 0.01 N formic acid, and further with an additional 200 ml of 0.01 N formic acid. 5-Hydroxyindoleacetic acid was then eluted from the column with 50 ml of 2 N formic acid. The effluent and the first washing were combined, neutralized to pH 7, and applied to an Amberlite CG-50 column (H+ form, diameter 0.6 cm, length 5 cm). Tryptophan and 5-hydroxytryptophan were washed out from the column with 30 ml of water. After the column was washed with an additional 200 ml of water, trypta- mine and serotonin were eluted together with 15 ml of 4 N acetic acid. The effluent and eluate were lyophilized separately. The indole amino acids and amines were taken up separately in a small amount of the upper layer (organic layer) of a mixture of 1-butanol-acetic acid-water (20: 1: 16) and were subjected to partition chromatography on a Sephadex G-25 column as de- scribed above. By this procedure tryptophan and tryptamine were separated from the respective 5-hydroxylated compounds (see below). When complete separation of 5-hydroxytryptophan from tryptophan was required, the 5-hydroxytryptophan frac- tion was lyophilized and rechromatographed on a Sephadex G-25 column. Tryptamine thus obtained was lyophilized, dissolved in 0.1 N NaOH and purified further by extraction with benzene by vigorous shaking for 5 min at room temperature
(36). Determinations-Radioactivity was determined by the use of
either a Nuclear-Chicago gas flow counter as an infiniteIy thin sample or a Beckman DPM-100 or Packard Tri-Carb model 314A liquid scintillation spectrometer. Toluene containing
0.25% 2,5-diphenyloxazole (PPO) and 0.015% 1,4-bis[2’-(5’- phenyloxazolyl)]benzene (POPOP) or Bray’s fluorophor solu- tion (37) was used as scintilIa.tor. Radioactivity on paper chromatogram was determined by direct paper strip counting with a liquid scintillation spectrometer (38). Tryptophan and tryptamine were determined spectrophotometrically. 5-Hy- droxytryptophan and serotonin were determined in aqueous acidic solution either spectrophotometrically or fluorometrically (295 rnp activation, 550 rnp fluorescence) according to the method of Udenfriend (39). Extinction coefficients used for spectrophotometric determinations were: ens and tpgi for trypto- phan = 3.75 and 3.68, respectively (40,41); EU~ for tryptamine = 3.72 (40); ~276 for serotonin = 3.74 (40); ens for 5-hydroxytrypto- phan = 3.75 (42). Spectrophotometric and fluorometric measurements were carried out with a Shimadzu spectropho- tometer, type &R-50, and an Aminco-Bowman spectrofluorom- eter, respectively. Absorption spectra were measured with a Cary recording spectrometer, model 15. Protein was determined by the method of Lowry et al. (43) with bovine serum albumin as a standard. High speed centrifugation was carried out with a Spinco ultracentrifuge model L or a Hitachi preparative ultracentrifuge, type 40P. All other centrifugations were carried out with a Tominaga No. 90 UV refrigerated centri- fuge.
RESULTS
Enxymic Formation of Radioactive CO2 and Serotonin from 5-Hydroxy-L- tryptophan- (side chain-l ,d,S-14C)-When 5-hy- droxy-L-tryptophan-(side chain-l ,2,3-‘4C) was incubated with the supernatant fraction obtained from guinea pig brain stem as descrjbed under “Experimental Procedure,” radioactive COZ was evolved linearly with time and the amount of 14C02 pro- duced was proportional to the quantity of enzyme as shown in Fig. 2. The activity was maximal at pH 8.8. In order to identify the reaction product, guinea pigs were given an intra- peritoneal injection of Catron (10 mg per kg, body weight), a monoamine oxidase inhibitor, 2 hours prior to decapitation. The supernatant fraction was prepared from the brain stem as described under “Experimental Procedure,” and was incubated with 5-hydroxy-L-tryptophan-(side chain-l ,2,3-14C) in the presence of iproniazid phosphate. As shown in Table I, the evolution of radioactive COZ was accompanied by the stoichio- metric formation of serotonin and disappearance of 5-hydroxy- L-tryptophan-1%. Serotonin-YZ thus recovered was identified by paper chromatography with 1-butanol-acetic acid-water (12:3:5) and isopropyl alcohol-ammonia-water (20:1:2) as solvents. The results indicate that the radioactive COZ pro- duced from 5-hydroxy-L-tryptophan-(side chain-l ,2 ,3J4C) is entirely accounted for by the formation of serotonin from 5- hydroxy-L-tryptophan.
Purification and Properties of Aromatic L-Amino Acid De- carboxylase from Bovine Brain Stem-Fresh bovine brain was transported in ice from the slaughterhouse. All subsequent ma- nipulations were carried out at 4”. Brain stem (109 g) was ho- mogenized for 1 min in a Waring Blendor with 436 ml of 0.02 M
Tris-acetate, pH 8.1. The homogenate was centrifuged for 15 min at 9,OOOrpm. The precipitate was extracted again with 218 ml of 0.02 M Tris-acetate, pH 8.1, by the use of the Blendor as described above. To the combined supernatant solution (628 ml) were added 62.8 ml of 1 M phosphate buffer, pH 8.1. Then 38.7 g of solid ammonium sulfate were added slowly with stirring
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from
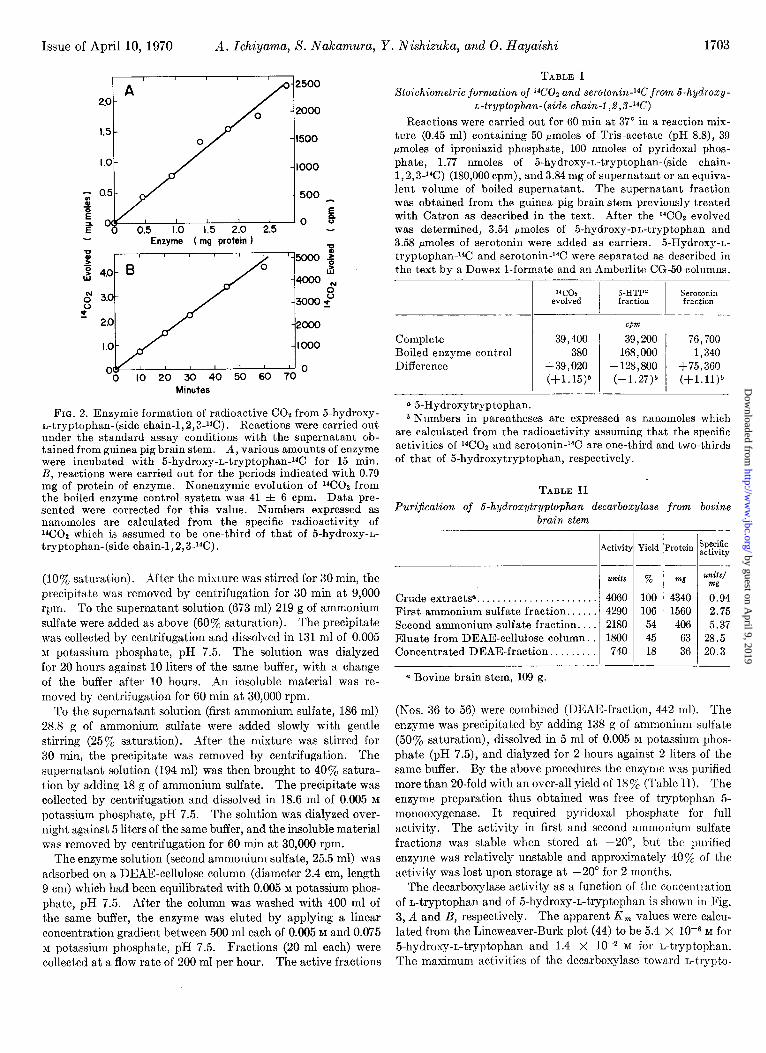
Issue of April 10, 1970 A. Ichiyama, S. Nakamura, Y. Nishixuka, and 0. Hayaishi 1703
Enzyme ( mg protein 1
o(y I i I I I 1 1’0
0 IO 20 30 40 50 60 70 Minutes
FIG. 2. Enzymic formation of radioactive Cot from B-hydroxy- L-tryptophan-(side chain-1,2,8-W). Reactions were carried out under the standard assay conditions with the supernatant ob- tained from guinea pig brain stem. A, various amounts of enzyme were incubated with 5-hydroxy-n-tryptophan-14C for 15 min. R, reactions were carried out for the periods indicated with 0.79 mg of protein of enzyme. Nonenzymic evolution of WOa from the boiled enzyme control system was 41 f 6 cpm. Data pre- sented were corrected for this value. Numbers expressed as nanomoles are calculated from the specific radioactivity of XJOt which is assumed to be one-third of that of 5-hydroxy-n- tryptophan-(side chain-l, 2, 3J4C).
(10% saturation). After the mixture was stirred for 30 min, the precipitate was removed by centrifugation for 30 min at 9,000 rpm. To the supernatant solution (673 ml) 219 g of ammonium sulfate were added as above (60% saturation). The precipitate was collected by centrifugation and dissolved in 131 ml of 0.005 M potassium phosphate, pH 7.5. The solution was dialyzed for 20 hours against 10 liters of the same buffer, with a change of the buffer after 10 hours. An insoluble material was re- moved by centrifugation for 60 min at 30,000 rpm.
To the supernatant solution (first ammonium sulfate, 186 ml) 28.8 g of ammonium sulfate were added slowly with gentle stirring (25% saturation). After the mixture was stirred for 30 min, the precipitate was removed by centrifugation. The supernatant solution (194 ml) was then brought to 40% satura- tion by adding 18 g of ammonium sulfate. The precipitate was collected by centrifugation and dissolved in 18.6 ml of 0.005 M
potassium phosphate, pH 7.5. The solution was dialyzed over- night against 5 liters of the same buffer, and the insoluble material was removed by centrifugation for 60 min at 30,000 rpm.
The enzyme solution (second ammonium sulfate, 25.5 ml) was adsorbed on a DEAE-cellulose column (diameter 2.4 cm, length 9 cm) which had been equilibrated with 0.005 M potassium phos- phate, pH 7.5. After the column was washed with 400 ml of t,he same buffer, the enzyme was eluted by applying a linear concentration gradient, between 500 ml each of 0.005 M and 0.075 M potassium phosphate, pH 7.5. Fractions (20 ml each) were collected at a flow rate of 200 ml per hour. The active fractions
TABLE I Stoichiometric formation of ‘4CO2 and serotonin-‘4C from 6-hydroxy-
L-tryptophan-(side chain-l ,.Z,S-‘4C)
Reactions were carried out for 60 min at 37” in a reaction mix- ture (0.45 ml) containing 50 pmoles of Tris-acetate (pH 8.8), 39 pmoles of iproniazid phosphate, 100 nmoles of pyridoxal phos- phate, 1.77 nmoles of 5-hydroxy-n-tryptophan-(side chain- 1,2, 3-14C) (180,000 cpm), and 3.84 mg of supernatant or an equiva- lent volume of boiled supernatant. The supernatant fraction was obtained from the guinea pig brain stem previously treated with Catron as described in the text. After the 14C02 evolved was determined, 3.54 rmoles of 5-hydroxy-nn-tryptophan and 3.58 pmoles of serotonin were added as carriers. 5-Hydroxy-n- tryptophan-14C and serotoninJ4C were separated as described in the text by a Dowex l-formate and an Amberlite CG-50 columns.
‘CO1 s-HTPL” Serotonin evolved fraction fraction
Complete Boiled enzyme control Difference
39,400 380
+39,020 (+1.15)6
CM
39,200 76,700 168,000 1,340
-128,800 +75,360 (-1.27)” (+l.ll)b
a5-Hydroxytryptophan. b Numbers in parentheses are expressed as nanomoles which
are calculated from the radioactivity assuming that the specific activities of i4C02 and serotonin-14C are one-third and two-thirds of that of 5-hydroxytryptophan, respectively.
TABLE II
Puri$cation of B-hydroxytryptophan decarboxylase from bovine brain stem
Activity Yield Protein
units 70 w
Crude extracts”. . . . . . . . . . 4060 100 4340 First ammonium sulfate fraction.. . 4290 106 1560 Second ammonium sulfate fraction.. 2180 54 406 Eluate from DEAE-cellulose column. 1800 45 63 Concentrated DEAE-fraction.. . 740 18 36
= Bovine brain stem, 109 g.
c a ipecific rctivity
units/ w 0.94 2.75 5.37
28.5 20.3
(Nos. 36 to 56) were combined (DEAE-fraction, 442 ml). The enzyme was precipitated by adding 138 g of ammonium sulfate (50% saturation), dissolved in 5 ml of 0.005 M potassium phos- phate (pH 7.5), and dialyzed for 2 hours against 2 liters of the same buffer. By the above procedures the enzyme was purified more than 20-fold with an over-all yield of 18% (Table II). The enzyme preparation thus obtained was free of tryptophan 5- monooxygenase. It required pyridoxal phosphate for full activity. The activity in first and second ammonium sulfate fractions was stable when stored at -2O”, but the purified enzyme was relatively unstable and approximately 40% of the activity was lost upon storage at -20” for 2 months.
The decarboxylase activity as a function of the concent,rat,ion of L-tryptophan and of 5-hydroxy-n-tryptophan is shown in Fig. 3, A and B, respectively. The apparent K, values were calcu- lated from the Lineweaver-Burk plot (44) to be 5.4 X IO+ M for 5-hydroxyn-tryptophan and 1.4 X lo-* M for L-tryptophan. The maximum activities of the decarboxylase toward L-trypto-
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from
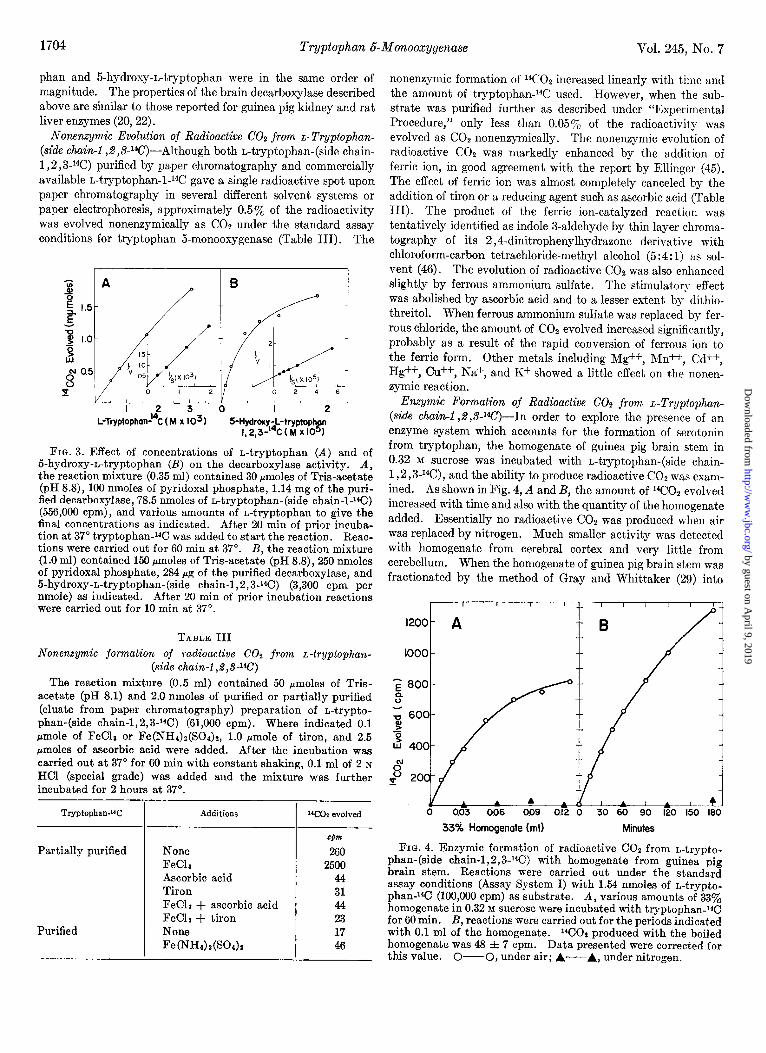
1704 Tryptophan 6-Monooxygenase Vol. 245, No. 7
phan and 5-hydroxy-L-tryptophan were in the same order of magnitude. The properties of the brain decarboxylase described above are similar to those reported for guinea pig kidney and rat liver enzymes (20,22).
Nonenzymic Evolution of Radioactive CO2 from L-Tryptophan- (side chain-l $,V*C)-Although both n-tryptophan-(side chain- 1,2 ,3J4C) purified by paper chromatography and commercially available n-tryptophan-l-*4C gave a single radioactive spot upon paper chromatography in several different solvent systems or paper electrophoresis, approximately 0.5% of the radioactivity was evolved nonenzymically as COZ under the standard assay conditions for tryptophan 5-monooxygenase (Table III). The
L-Tryptophad’C (M x IO31 5-Hydroxy-L-tryptophtn I 23-'4C(M~10 ) I 1
FIG. 3. Effect of concentrations of r.-tryptophan (A) and of 5-hydroxy-n-tryptophan (B) on the decarboxylase activity. A, the reaction mixture (0.35 ml) contained 30 pmoles of Tris-acetate (pH KS), 100 nmoles of pyridoxal phosphate, 1.14 mg of the puri- fied decarboxylase, 78.5 nmoles of n-tryptophan-(side chain-1-‘4C) (556,000 cpm), and various amounts of n-tryptophan to give the final concentrations as indicated. After 20 min of prior incuba- tion at 37” tryptophan-14C was added to start the reaction. Reac- tions were carried out for 60 min at 37”. B, the reaction mixture (1.0 ml) contained 150 amoles of Tris-acetate (pH KS), 250 nmoles of pyridoxal phosphate, 284 rg of the purified decarboxylase, and 5-hydroxy-n-tryptophan-(side chain-1,2,3-i4C) (3,300 cpm per nmole) as indicated. After 20 min of prior incubation reactions were carried out for 10 min at 37”.
TABLE III Nonenqmic formation of radioactive Cot from L-tryptophan-
(side chain-1$,3-W) The reaction mixture (0.5 ml) contained 50 rmoles of Tris-
acetate (pH 8.1) and 2.0 nmoles of purified or partially purified (eluate from paper chromatography) preparation of n-trypto- phan-(side chain-1,2,3J4C) (61,000 cpm). Where indicated 0.1 rmole of FeClp or Fe(NH&(SO.&, 1.0 bmole of tiron, and 2.5 pmoles of ascorbic acid were added. After the incubation was carried out at 37” for 60 min with constant shaking, 0.1 ml of 2 N
HCl (special grade) was added and the mixture was further incubated for 2 hours at 37”.
Tryptophan-W Additions “COP evolved
Partially purified
Purified
None FeC13 Ascorbic acid Tiron FeCla + ascorbic acid FeCla + tiron None Fe(NH4)z(SO&
CM
260 2500
44 31 44 23 17 46
nonenzymic formation of “CO* increased linearly with time and the amount of tryptophan-i4C used. However, when the sub- strate was purified further as described under “Experimental Procedure,” only less than 0.05% of the radioactivity was evolved as COP nonenzymically. The nonenzymic evolution of radioactive CO2 was markedly enhanced by the addition of ferric ion, in good agreement with the report by Ellinger (45). The effect of ferric ion was almost completely canceled by the addition of tiron or a reducing agent such as ascorbic acid (Table III). The product of the ferric ion-catalyzed reaction was tentatively identified as indole 3-aldehyde by thin layer chroma- tography of its 2,4-dinitrophenylhydrazone derivative with chloroform-carbon tetrachloride-methyl alcohol (5:4: 1) as sol- vent (46). The evolution of radioactive CO2 was also enhanced slightly by ferrous ammonium sulfate. The stimulatory effect was abolished by ascorbic acid and to a lesser extent by dithio- threitol. When ferrous ammonium sulfate was replaced by fer- rous chloride, the amount of CO2 evolved increased significantly, probably as a result of the rapid conversion of ferrous ion to the ferric form. Other metals including Mg++, Mn++, Cd++, Hg++, Cu++, Na+, and K+ showed a little effect on the nonen- zymic reaction.
Enzymic Form.ation of Radioactive COP from A-Tryptophan- (side chain-l ,8,SJ4C)-In order to explore the presence of an enzyme system which accounts for the formation of serotonin from tryptophan, the homogenate of guinea pig brain stem in 0.32 M sucrose was incubated with r,-tryptophan-(side chain- 1 ,2,3-14C), and the ability to produce radioactive CO* was exam- ined. As shown in Fig. 4, A and B, the amount of i4CO2 evolved increased with time and also with the quantity of the homogenate added. Essentially no radioactive CO2 was produced when air was replaced by nitrogen. Much smaller activity was detected with homogenate from cerebral cortex and very litt.le from cerebellum. When the homogenate of guinea pig brain stem was fractionated by the method of Gray and Whittaker (29) into
Y A . 4 * 1 . I . I 1 0 0.03 0.06 009 0.12 0 30 60 90 120 150 180
33% Homogenate (ml) Minutes
FIG. 4. Enzymic formation of radioactive COZ from L-trypto- phan-(side chain-1,2,3-14C) with homogenate from guinea pig brain stem. Reactions were carried out under the standard assay conditions (Assay System I) with 1.54 nmoles of n-trypto- phan-1% (100,000 cpm) as substrate. A, various amounts of 33% homogenate in 0.32 M sucrose were incubated with tryptophan-14C for 60 min. B, reactions were carried out for the periods indicated with 0.1 ml of the homogenate. r4COt produced with the boiled homogenate was 48 f 7 cpm. Data presented were corrected for this value. O-O, under air; A----A, under nitrogen.
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Issue of April 10, 1970 A. Ichiyama, S. Nakamura, Y. Nishixuka, and 0. Hayaishi 1705
nuclear, crude mitochondrial, microsomal, and supernatant frac- tions, approximately 4Ooj, of the enzyme activity was recovered in the crude mitochondrial fraction and 60% was detected in the supernatant fraction. The soluble enzyme required almost absolutely 2-amino-4-hydroxy-6,7-dimethyl-5,6,7,8-tetrahy- dropteridine and either NADPH or a sulfhydryl reagent such as dithiothreitol for full activity as described below. In contrast, neither homogenate nor crude mitochondrial fraction required any of these cofactors. A preliminary survey showed that the level of activity in brain stem homogenates from guinea pig, rat, mouse, hamster, rabbit, and cow is within the same order of magnitude.
Tryptophan 5-Monooxygenase in Soluble Fraction-The cofac- tors necessary for the enzymic formation of radioactive CO2 from L-tryptophanJ4C with the soluble enzyme system are shown in Table IV. Both 2-amino-4-hydroxy-6,7-dimethyl-5,6,7,8- tetrahydropteridine and a sulfhydryl compound, such as dithio- threitol, were required almost absolutely. Dithiothreitol was partially substituted by NADPH and to a lesser extent by NADH. But the reduced pyridine nucleotides did not stimulate further the activity in the presence of both reduced pteridine and dithiothreitol. Neither 2-amino-4-hydroxy- 6,7- dimethyl-5,6, 7,%tetrahydropteridine, NADPH, NADH, nor dithiothreitol alone affected the activity. The activity of a partially purified enzyme preparation was enhanced slightly by the addition of ferrous ion and was strongly inhibited by ar,cr’-dipyridyl or
TABLE IV Cofactor requirement of soluble tryptophan B-monooxygenase Tryptophan 5-monooxygenase was partially purified from the
supernatant fraction of guinea pig brain stem by ammonium sulfate fractionation (0 to 40%) and dialyzed overnight against 0.02 M Tris-acetate (pH 8.1) containing 10-s M p-mercaptoethanol under nitrogen. Reactions were carried out for 60 min at 37” in reaction mixture (0.5 ml) containing 50 Fmoles of Tris-acetate, (pH 8.1), 50 nmoles of pyridoxal phosphate, 5 units of aromatic n-amino acid decarboxylase purified from the bovine brain stem as described in the text, 3Opg of catalase, 10 nmoles of Fe(NH& (SO&, 0.1 ml of the enzyme preparation, and 3.225 nmoles of n-tryptophan-(side chain-1-i4C) (27,300 cpm). Where indicated 1.5 pmoles of dithiothreitol, 0.6 pmole of 2-amino-4-hydroxy-6,7- dimethyl-5,6,7,%tetrahydropteridine, 0.5 pmole of or,or’-di- pyridyl, and 0.5 pmole of o-phenanthroline were added. The 14COa evolved was trapped in phenethylamine and determined as described under “Experimental Procedure.”
Additions or omissions TOa evolved
None............................................... 3 Plus DTT*.......................................... 2 Plus DMPHbc.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10 Plus DMPH4, plus DTT. . . . . . . . . . . . . . . . . . . . . . . . . 36 Plus DMPH4, plus DTT, minus Fe(NH&(SO&. . . . . 24 Plus DMPH+ plus DTT, plus ol,a’-dipyridyl, minus
Fe(NH&(SO&. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 Plus DMPH4, plus DTT, plus o-phenanthroline, mi-
nus Fe(NH&(SO&. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
0 Values were calculated from the specific activity of n-trypto- phanJ4C.
* Dithiothreitol. c 2-Amino4-hydroxy6,7-dimethyl-5,6,7,8 -tetrahydropteri-
dine.
o-phenanthroline, indicating that ferrous ion may be involved in the enzymic reaction.
The optimum pH with the soluble enzyme was found to be around pH 8 to 8.5. The apparent I<, values for L-tlyptophan and for 2-amino-4-hydroxy-6,7-dimethyl-5,6,7,8-tetrahydro- pteridine were calculated to be in the order of 3 x 10M4 M and 6 X lop5 M, respectively.
Reaction ProductsIn order to identify the reaction products, guinea pigs weighing 380 to 430 g were given an intraperitoneal injection of Catron (15 mg per kg, body weight), 2 hours prior to decapitation. The supernatant fraction was prepared from the brain stem and was incubated under standard assay condi- tions (Assay System II) with n-tryptophan-(side chain-l ,3-14C) at t,hree different concentrations (6.1 X 10e6 M, 2.3 X 1O-6 M,
and 9.1 X lop5 M). Iproniazid phosphate was added in a final concentration of 7.8 X lop2 M as described under “Experimental Procedure.” After the radioactive CO2 was determined, the products were analyzed as described under “Experimental Procedure” (Fig. 1). When the indole amine fraction (acetic acid eluate from the Amberlite CG-50 column) was chromato-
5HTP b 2~
400 20 40 ’
800
Fmctions ( 5.3 ml )
FIG. 5. Partition chromatography of reaction products. Reac- tions were carried out under the standard assay conditions (Assay System II) except that iproniazid phosphate was added at a final concentration of 7.78 X KY2 M. The concentration of L-trypto- phan-1% and the volume of incubation were as follows. A, 9.1 X 10-s M L-tryptophan-(side chain-1,3-l%). Reaction mixture, 0.5 ml. B, n-Tryptophan-(side chain-1,3-14C), 2.3 X 10-b M. Reaction mixture, 2.0 ml. C and D, L-Tryptophan-(side chain- 1,3J4C), 6.1 X 10-e M. Reaction mixture, 4.0 ml. The super- natant fraction was prepared from the guinea pig brain stem previously treated with Catron as described under “Experimental Procedure.” L-Tryptophan-(side chain-l, 3J4C) was prepared by mixing L-tryptophan-(side chain-l-l%) and L-tryptophan- (side chain-3-14C). The specific radioactivities of carboxyl and methylene carbons were 10,600 and 1,960 cpm per nmole, respec- tively. After 14CO2 evolved was determined, indole amino acid and indole amine fractions were separated by Amberlite CG-50 column chromatography and subjected to partition chromatog- raphy on Sephadex G-25 column as described under “Experi- mental Procedure.” Fractions (5.3 ml) were collected and, after the optical density at 280 rnp (open bar) was determined, a l-ml portion of each fraction was mixed with 10 ml of Bray’s solution (37) and the radioactivity (solid bar) was determined for 10 min with a Packard Tri-Carb liquid scintillation spectrometer. A, B, C, partition chromatography of indole amine fraction. D, second partition chromatography of 5-hydroxytryptophan frac- tion. The radioactivity presented is corrected for the incubation volume. TryNHa, tryptamine; 6HT, 5-hydroxytryptamine (ser- otonin) ; 5HTP, 5-hydroxytryptophan.
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

1706 Tryptophan 5-Monooxygenase Vol. 245, No. 7
graphed on a Sephadex G-25 column, two radioactive products appeared in the tryptamine and serotonin fractions with a coincidence of i*C content and opt’ical density at 280 rnp (Fig. 5, A, B, and C). The radioactive serotonin was further identified as such by paper chromatography in four different solvent systems described under “Experimental Procedure.” Only very small amounts of tryptamine were produced when a relatively low concentrat,ion of tryptophanJ4C (6.1 X lo-6 M) was used (Fig. 5C). With higher concentrations of L-tryptophan (9.1 x lo+ M and 2.3 X lop5 M), however, an appreciable amount of ra- di0activit.y was recovered in the tryptamine fraction as shown in Fig. 5, A and B. The radioactive tryptamine thus produced was identified by paper chromatography with I-butanol-acetic acid-water (12:3:5) and methanol-l -butanol-benzene-acetic acid-water (4.0:20:20: 19: 1) as solvent. Although the nonen- zymic formation of serotoninfrom tryptophan has been suggested (47, 48), no serotonin-‘4C: was produced when a boiled enzyme preparation was used. Quantitative analysis showed that’, with a lower concentration of L-tryptophanJ4C (6.1 X 1OB6 M), the ra- dioactive CO2 evolved is almost entirely accounted for by the format,ion of serotonin (14C02; 71 pmoles, serotoninJ4C; 69 pmoles). However, with 2.3 x lop5 M and 9.1 X lop5 M L-
tiyptophan-14C, approximately 6% and IS%, respectively, of the total ‘4CO2 evolved were due to the direct decarboxylat8ion of tryptophan to tryptamine.
Only a little radioactivity was recovered in the 5-hydroxytryp- tophan fraction (Fig. 50), indicating that the hydroxylation of L-tryptophan is the rate-limiting step in the biosynt,hesis of serotonin under the above conditions.
5-Hydroxytryptophan as Intermediate-Guinea pigs were treated with Catron and the supernatant fraction was prepared from the brain stem as described above. A large scale incubation
TABLE V
Formation of 6-hydroxytryptophan-14C from L-tryptophan- (side chain-1,2,3-‘4C)
Reactions were carried out under the standard assay conditions (Assay System II) except that 1.0 ml of the reaction mixture was used and 7.78 X 10m3 M iproniaeid phosphate was added to the reaction mixture. Supernatant fraction was prepared from guinea pig brain stem previously treated with Catron as described in the text. Where indicated a-methyl-DL-m-tyrosine was added at a final concentration of 2 X 10e3 M. After the ‘4cO~ evolved was determined, 3.54 Imoles of 5-hydroxy-nn-tryptophan and 3.58 pmoles of serotonin were added as carriers and the reaction products were analyzed as described under “Experimental Pro- cedure.”
Additions co2 5-Hydroxy- tryptophan Serotcnin
None . . . . . . or-Methyl-m-tyrosine
3340 0
prides
Nil” 310.0
286b Nil
a Formation of COZ was calculated from the radioactivity assuming that the specific activity is one-third of that of L-
tryptophan-(side chain-l, 2, 3J4C). b Amounts of 5-hydroxytryptophan and serotonin produced
were calculated from the amount of respective carrier added before isolation and the specific radioactivity of the isolated compounds assuming that the specific activity of 5-hydroxy- tryptophan and serotonin is the same and two-thirds, respec- tively, of that of tryptophan-14C used as substrate.
(2.5 ml) was carried out with 10U5 M L-tryptophan-(side chain- 1,2, 3-14C) (40,000 cpm per nmole) as substrate under the stand- ard conditions (Assay System II), except that 2 pmoles of cu-methyl-nn-m-tyrosine, a decarbosylase inhibitor, were added to the reaction mixture. The control system contained all in- gredients except ar-methyl-nn-m-tyrosine. After ‘4CO2 was deter- mined, radioactive products were analyzed as outlined in Fig. 1. When the reaction was carried out in the absence of the decar- boxylase inhibitor practically no 5-hydroxytryptophan was pro- duced; the radioact,ive CO* evolved was almost entirely ac- counted for by the amount of serotonin-l4C formed. In contrast’, in the presence of a-methyl-nn-m-tyrosine, neither 14C02 nor sero- tonin 1% was produced, but 5-hydroxytryptophan% accumu- lated (Table V). 5-Hydroxytryptophan-14C thus produced was identified by paper chromatography with three different solvent systems: methanol-1-butanol-benzene-ammonia-water (40:20:- 20: 1: 19), I-butanol-acetic acid-water (4 : 1: l), and ammonium acetate, pH 5.1-ethanol (3 : 7).
Tryptophan 5-Monooxygenase in Particulate Fraction-U though the soluble tryptophan 5-monooxygenase required almost absolutely 2-amino-4-hydroxy-6,7-dimethyl-5,6,7 ,%tetrahy- dropteridine and either a sulfhydryl reagent or NADPH as described above, the enzyme activity in crude mitochondrial fraction did not respond at all to these cofactors. When the
TABLE VI
Cofactor requirement of tryptophan 5-monooxygenase in particulate fraction
Crude mitochondrial fraction was prepared from the guinea pig brain stem as described under “Experimental Procedure.” An aliquot of this fraction was placed on a layer of 0.88 M sucrose and centrifuged at 30,000 rpm for 120 min. The precipitate was suspended in 0.32 M sucrose by the use of a Potter-Elvehjem type homogenizer and again passed through a layer of 0.88 M sucrose as above. The precipitate from the second centrifugation was suspended in 0.05 M K~HPOI (crude mitochondrial fraction). The washed crude mitochondrial suspension was adjusted to pH 10.7 by 10 N KOH, and cold I-butanol (-30”) was added slowly with stirring to give a final butanol concentration of 4%. After stirring at 0” for additional 10 min the mixture was dialyzed at 4” for 6 hours against a large excess of 0.05 M KsHPO4. The mixture was then centrifuged at 40,000 rpm for 60 min and the precipitate was suspended in 0.05 M K~HPOI by the use of a Potter-Elvehjem type homogenizer. All enzyme preparations were prepared to make 2ml suspensions from 1 g of starting tissue. Reactions were carried out under the standard assay conditions (Assay System I) except that 0.5 pmole each of 2-amino-4-hydroxy- 6,7-dimethyl-5,6,7,8-tetrahydropteridine (reduced pteridine), NADH, and NADPH was added where indicated.
Crude mitochon-
drial Et%; fraction treatment
I I
p??des ‘CO2 evolvd~ None................................ 48.0 6.8 0.4 Reduced pteridine 26.0 7.5 2.7 NADPH . . . . . . . . . . . 30.6 5.1 1.0 NADH . . . . . . . . . . 21.5 0.2 Reduced pteridine + NADPH. 26.4 28.4 22.8 Reduced pteridine + NADH. . . . . . 23.5 17.0
a Values are calculated from the specific activity of 1 carbon atom which is expected to be one-third of n-tryptophan-(side chain-l, 2,3-l%).
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Issue of April 10, 1970 A. Ichiyama, S. Nakamura, Y. Nishizuka, and 0. Hayaishi 1707
TABLE VII
Enzymic formation of serotonin and tryptamine from tryptophan in crude mitochondrial fraction from guinea pig brain stem
Reactions were carried out in a total volume of 2.5 ml (Experi- ments 1 to 3) or 0.5 ml (Experiment 4) under the standard assay conditions (Assay System I) in the presence of 7.78 X 10e3 M ipro- niazid phosphate. The crude mitochondrial fraction was pre- pared from the guinea pig brain stem previously treated with Catron as described in the text. L-Tryptophan-(side chain-1,2,3- I%) (60,200 cpm per nmole) was used as substrate at concentra- tions as indicated. In Experiment 3, previous treatment of the animal with Catron was omitted and the reaction was carried out in the absence of iproniazid phosphate. After the radioactive CO2 evolved was determined, 4.24 pmoles of L-tryptophan, 4.25 pmoles of tryptamine, 3.92 pimoles of 5-hydroxy-nL-tryptophan, 4.32 pmoles of serotonin, and 4.05 @moles of 5-hydroxyindoleacetic acid were added as carriers. The radioactive products were analyzed as described under “Experimental Procedure.” The over-all recovery of carrier tryptamine, 5-hydroxy-DL-trypto- phan, and serotonin was 62.3,57.4, and 57.8oj,, respectively.
Experiment and substrate
a!
1. 2.7 X 1O-6
2. 2.7 X lo-”
3. 2.7 X 1O-6
4. 4 x 10-E
co2
208”
(4,180) 235
(4,710) 180
(3,610) 182
(3,660)
Serotonin
204b
(8,210)
(lOy!iO)
@?727) 145
(5,830)
_-
-
Tryptamine
3.3b
(136 )
2.5 (1’35)
(4ii)
S-Hydroxy- tryptophan
-b
<4.1 (<240)
~1 Numbers and those in parentheses are expressed as picomoles and radioactivity (counts per min), respectively. Nonenzymic evolution of WOS by the boiled enzyme (Experiment 1 and 2,150 cpm; Experiment 3, 84 cpm; Experiment 4, 92 cpm) is corrected.
b Formation of serotonin, tryptamine, and 5-hydroxytrypto- phan was calculated from the specific radioactivity of the isolated compounds and the amount of respective carrier added before isolation.
crude mitochondria were washed twice by passage through a layer of 0.88 M sucrose by centrifugation for 120 min at 30,000 rpm, the enzymic activity was enhanced by the addition of NAD or 2-amino-4-hydroxy-6,7-dimethyl-5,6,7, %tetrahydro- pteridine plus either NADPH or NADH (Table VI). After 1-butanol treatment at pH 10.7, the particulate enzyme required almost absolutely both 2-amino-4-hydroxy-6,7-dimethyl-5,6,7, %tetrahydropteridine and either NADPH or NADH. The apparent K, value for L-tryptophan of tryptophan 5-monooxy- genase in crude mitochondrial fraction was calculated to be 2 x lo- M.
Stoichiometry of Reaction with Particulate Fraction-Guinea pigs were given an intraperitoneal injection of Catron (10 mg per kg, body weight) 2 hours prior to decapitation, and the crude mitochondrial fraction was prepared from the brain stem as de- scribed under “Experimental Procedure.” A large scale incuba- tion (2.5 ml) was carried out with L-tryptophan-(side chain-l, 2,3-‘4C) (60,000 cpm per nmole) at two different concentrations (2.7 X lo-6 M and 4 X 1O-5 M) in the presence of iproniasid phosphate. The formation of 14C02 as well as of radioactive serotonin, tryptamine, and 5-hydroxytryptophan was deter- mined as described above. As shown in Table VII, when 2.7 x
10F6 M L-tryptophan was used as substrate, the radioactive CO2 evolved was almost entirely accounted for by the amount of serotonin formed (Experiment 1). However, wit.h 4 x lo+ M L-tryptophanJ4C, approximately 6 y0 of the total 14C02 evolved was due to the formation of tryptamine (Experiment 4). Essen- tially no radioactive 5-hydroxytryptophan was detected (Experi- ment 2), suggesting that the hydroxylation of tryptophan is the rate-limiting step in the biosynthesis of serotonin in the particu- late fraction of guinea pig brain stem under the conditions used. In the absence of the monoamine oxidase inhibitor, serotonin was not recovered in a stoichiometric quantity, probably a result of further degradation (Experiment 3).
DISCUSSION
Tryptophan 5-monooxygenase, the first enzyme in the bio- synthetic pathway of serotonin, has attracted much attention but the relatively low activity of this enzyme and the lack of a sensitive and convenient assay have hindered the studies on the purification and properties of this enzyme as well as the react’ion mechanisms involved. Assay methods so far available are based on the determination of radioactive serotonin or 5-hydroxy- tryptophan which are separated from a large excess of unreacted tryptophan by either paper chromatography, paper electrophore- sis, or ion exchange resins. In our earlier work, 5-hydroxytlyp- tophan formed from tryptophanJ4C was first converted to serotonin by the guinea pig kidney decarboxylase and t,hen separated from unreacted tryptophan-14C by high voltage paper electrophoresis at pH 5.0 (7). However, the method was not only tedious but also impracticable since serotonin fract,ion may be contaminated with other radioactive compounds unless the reaction mixture is first treated with Amberlite CG-50 column.
The advantages of the method described here are that it is extremely sensitive and can be performed rapidly. Since uni- formly labeled L-serine-14C, the specific radioactivity of which is as high as 160 mCi per mmole, is now available commercially, L-tryptophan-(side chain-l ,2 ,3J4C) (the specific radioactivity of 1 carbon atom is 53.3 mCi per mmole) can be prepared. Thus the formation of 1 pmole of serotonin is expected to be accom- panied by the evolution of 117 dpm of 14C02.
The disadvantages of the method are: first, this assay cannot be applied to crude fractions from tissues such as liver in which alternate pathways are known to exist for the catabolism of tryptophan. Second, in a reaction containing relatively high concentrations of L-tryptophan, an appreciable quantity of the CO:! evolved is due to the tryptoamine formation instead of serotonin formation. Therefore, substrate concentrations above 2 x 10m5 M should not be used for the assay.
The critical point of the assay procedure proposed in the present study is the purification of tryptophan-14C and the eliminationof ferric ion from the reaction mixture. Tryptophan- 14C purified as described under “Experimental Procedure” is rela- tively stable when stored frozen at -20’. Upon incubation under the standard assay conditions in the absence of enzyme, only less than 0.050/, of the radioactivity was evolved nonen- zymically as COZ. When the purification of tryptophan-‘4C was incomplete or a trace of ferric ion contaminated the re- action mixture, however, a considerable amount of radioactive CO2 was evolved which interfered with the assay of tryptophan 5-monooxygenase.
There have been apparent inconsistencies among the reports from several laboratories as to the subcellular distribution and
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Tryptophan 5-Monooxygenase Vol. 245, No. 7
the cofactor requirement of tryptophan 5-monooxygenase in brain. Lovenberg et al. reported that the activity in homogenate of rabbit brain stem was greatly stimulated by the addition of tetrahydropteridine and a high concentration of P-mercapto- ethanol (10) and that a greater part of the activity was either cytoplasmic or can be easily released from the mitochondrial fraction (49). Gal, Armstrong, and Ginsberg (9) and Green and Sawyer (S), however, have detected the enzyme activity in crude mitochondrial fraction and could find no stimulation by the addition of the pteridine cofactor. In a preliminary report from this laboratory (23), the tryptophan 5-monooxygenase activity was also shown to be localized in the crude mitochondrial fraction. When the crude mitochondrial fraction was further separated into fractions containing a preponderance of myelin, nerve endings, and mitochondria, according to the method of Grey and Whittaker (29), the enzyme activity was mostly concentrated into the nerve ending fraction. The activity in homogenate as well as in the particulate fraction did not respond to the addition of the pteridine cofactor (23). Recently Grahame-Smith (12) reported that the enzyme is localized in both the supernatant and a nerve ending fraction, and that the activity in homogenate responds to the pteridine cofactor only after hypo-osmotic treatment. Robinson, Lovenberg, and Sjoerdsma (50) showed, however, that tryptophan 5-monooxygenase in the mitochondrial fraction is inhibited by the presence of tetrahydropteridine and &mercaptoethanol. In confirmation of the reports by Loven- berg, Jequier, and Sjoerdsma (49) and Grahame-Smith (12), we found the enzyme activity in the supernatant fraction when 2- amino-4-hydroxy-6,7-dimethyl5,6,7, S-tetrahydropteridine and a high concentration of either ,&mercaptoethanol or dithiothreitol supplemented the reaction mixture. NADPH could substitute, at least in part, sulfhydryl reagent but not reduced pteridine. However, the enzyme activity in homogenate and crude mito- chondrial fraction was not stimulated at all by the addition of tetrahydropteridine, sulfhydryl reagents, NADPH, ascorbate, or their various combinations. The enzyme in crude mito- chondrial fraction required the cofactors only after 1-butanol treatment, suggesting that the reductant in this system was generated in situ by some endogenous mechanisms. Whether or not this mechanism is similar t.o that reported by Kaufman (51, 52) and his co-workers for phenylalanine-hydroxylating system in liver remains to be elucidated.
The apparent K, value of soluble tryptophan 5-monooxygen- ase for L-tryptophan was calculated to be in the order of 10m4 M
in good agreement with the report by Lovenberg et al. (49). The K, value of the particulate enzyme was approximately 2 x 1O-6 M, as has been reported previously (23), suggesting that either two different tryptophan 5-monooxygenases are present in brain or the structure of the particle affects the properties of a single enzyme. The latter explanation is more likely since subsequent studies have shown that, when a low concentration of L-tryptophan-14C is added to the crude mitochondrial fraction or nerve ending particles, txyptophan-14C is rapidly t’aken up by the particles and concentrated more than IO-fold. Available evidence indicated that the uptake of L-tryptophan by nerve ending particles described first by Grahame-Smith (53) is a common reaction to and hence inhibited competitively by many neutral L-amino acids including 5-hydroxy-L-tryptophan. The mechanism of the uptake reaction is apparently different from
that of so-called active transport process and is seemingly due to an exchange reaction.a
AcknowZecZgments-We are grateful to Dr. T. Nagatsu, Depart- ment of Biochemistry, Dental School, Aichi-Gakuin University, and to Dr. T. Deguchi, Department ot Psychiatry, Kyoto University Faculty of Medicine, for many stimulatory discus- sions and helpful suggestions. Thanks are also due Miss S. Okuno for her excellent technical assistance.
1.
2.
3. 4.
5.
;:
i:
10.
11.
12. 13.
14. 15.
16.
17.
18. 19.
20.
21. 22.
23.
24. 25. 26.
REFERENCES
UDENFRIEND, S., WEISSBACH, H., AND BOGDANSKI, D. F., in H. HOAGLAND (Editor), Hormones, brain function and be- havior, Academic Press, New York, 1957, p. 147.
GRAHAME-SMITH, D. G., Biochem. Biophys. Res. Commun., 16, 586 (19644).
GR.QHAME-&XI&, D. G., Biochem. J., 92,52P (1964). GRAHAME-SMITH, D. G., AND MOLONEY, L., Biochem. J., 96,
66P (1965). GAL, E. M., Fed. Proc., 24,580 (1965). GREEN, H., AND SAWYER, J. L., Fed. Proc., 24, 604 (1965). NAKAMURA, S., ICHIYAMA, A., AND HAYAISHI, O., Fed. Proc.,
24, 604 (1965). GREEN, H., AND SAWYER, J. L., Anal. Biochem., 16, 53 (1966). G.~L, E. M., ARMSTRONG, J. C., AND GINSBERG, B., J. Neuro-
them., 13, 643 (1966). LOVENBERG, W., JEQUIER, E., AND SJOERDSMA, A., Science,
166, 217 (1967). JEQUIER, E., LOVENBERG, W., AND SJOERDSMA, A., Mol. Phar-
macol., 3, 274 (1967). GRAHAME-SMITH, D. G., Biochem. J., 106,351 (1967). MITOMA, C., WEISSBACH, H., AND UDENFRIEND, S., Arch.
Biochem. Biophys., 63, 122 (1956). GRAHAME-SMITH, D. G., Biochim. Biophys. Acta, 86,176 (1964). LEVINE, R. J., LOVENBERG, W., AND SJOERDSMA, A., Biochem.
Pharmacol., 13, 1283 (1964). SATO, T., LOVENBERG, W., LEVINE, R. J., AND SJOERDSMA, A.,
Fed. Proc., 24, 580 (1965). LOVENBERG, W., LEVINE, R. J., AND SJOERDSMA, A., Biochem.
Pharmacoi., 14, 887 (lG65). ,
HOSODA, S., AND GLICK, D., J. Biol. Chem., 241, 192 (1966). CLARK, C. T., WEISSBACH, H., AND UDENFRIEND, S., J. Biol.
Chem., 210, 139 (1954). LOVENBERG. W.. WEISSBACH. H.. AND UDENFRIEND. S.. J.
Biol. Chek., 2$7, 89 (1962).’ ’ FELLMBN, J. H., Enzymologia, 20,366 (1959). AWAPARA, J., SANDMAN, R. P., AND HANLY, C., Arch. Biochem.
Biophys., 98, 520 (1962). ICHIYAMA, A., NAKAMURA, S., NISHIZUKA, Y., AND HAYAISHI,
O., in S. GARATTINI AND P. A. SHORE (Editors), Advances in pharmacology, ‘vol. 6A, Academic Press, New York, 1968, p. 5.
YdNOFSHY, C., J. Biol. Chem., 224,783 (1957). DALGLIESH. C. E.. J. Clin. Path.. 8. 73 (1955). VOGEL, H. ‘J., AN; BONNER, D. ‘MI, J. ‘Biol: Chem., 218, 97
(1956). 27. CRAWFORD, I. P., AND YANOFSKY, C., Proc. Nat. Acad. Sci.
U. S. A., 44, 1161 (1958). 28. YANOFSKY; C., in S. P. CO~OWICK AND N. 0. KAPLAN (Editors),
Methods in enzymology, vol. II, Academic Press, New York, 1955, p. 233.
29. GRAY, E. G., AND WHITTAKER, V. P., J. Anat., 96, 79 (1962). 30. GLIEMANN, J., Diabetes, 16, 643 (1967). 31. GLIEMANN, J., Diabetologica, 3, 382 (1967). 32. MASON, M., AND BERG, C. P., J. Biol. Chem., 188, 783 (1951). 33. RYLE, A. P., SANGER, F., SMITH, L. F., AND KITAI, R., Bio-
them. J., 60, 541 (1955).
* H. Fujisawa, A. Ichiyama, and 0. Hayaishi, unpublished data.
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Issue of April 10, 1970 A. Ichiyama, S. Nakamura, Y. Nishizuka, and 0. Hayaishi 1709
34. STAHL, E., AND HALDEWEY, H., Z. Physiol. Chem., 323, 182 (1961).
35. ASATOOR, A., AND DALGLIESH, C. E., J. Chem. Sot., 2291 (1956). 36. HESS, S. M., AND UDENFRIEND, S., J. Pharmacol. Exp. Ther.,
127, 175 (1959). 37. BRAY, G. A., Anal. Biochem., 1, 279 (1960). 38. WANG, C. H., AND JONES, D. E., Biochem. Biophys. Res. Com-
mm., 1, 203 (1959). 39. UDENFRIEND, S., in N. 0. KAPLAN AND H. A. SCHERAGA
(Editors), Fluorescence assay in biology and medicine, Aca- demic Press, New York, 1962, p. 170.
40. MCMENAMY, R. H., AND ONCLEY, J. L., J. Biol. Chem., 233, 1436 (1958).
41. EDWARDS, B. G., Arch. Biochem., 21, 103 (1949). 42. EK, A., AND WITKOP, B., J. Amer. Chem. Sot., 76, 500 (1953). 43. LOWRY, 0. H., ROSEBROUGH, N. J., FARR, A. L., AND RANDALL,
R. J., J. Biol. Chem., 193, 265 (1951).
44. LINEWEAVER, H., AND BURK, D., J. Amer. Chem. Sot., 66, 658 (1934).
45. BELLINGER, A., Berichte, 39, 2515 (1906). 46. BALLIN, G., J. Chromalogr.,
DALGLI~SH; C. 16, 152 (1964).
47. E., Arch: Biochem. Biophys., 68, 214 (1955). 48. DALPFNER. W.. AND CERLETTI. A.. Exverientia. 14. 376 (1968). 49. LOVENBER;, *., JEQUIER, 6., END- SJOERDSMA; A., ‘in s.
GARATTINI AND P. A. SHORE (Editors), Advances in phar- macology, Vol. 6A, Academic Press, New York, 1968, p. 21.
50. ROBINSON, D., LOVENBERG, W., AND SJOERDSMA, A., Arch. Biochem. Biophys., 123, 419 (1968).
51. KAUFMAN, S., in 0. H.AYAISHI (Editor), Oxygenases, Academic Press, New York, 1962, p. 129.
52. KAUFMAN, S., J. Biol. Chem., 239,332 (1964). 53. GRAHAME&M~TH, D. G., in-S. GARATTINI AND P. A. SHORE
(Editors), Advances in pharmacology, Vol. 6A, Academic Press, New York, 1968, p. 37.
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Arata Ichiyama, Shigenobu Nakamura, Yasutomi Nishizuka and Osamu HayaishiEnzymic Studies on the Biosynthesis of Serotonin in Mammalian Brain
1970, 245:1699-1709.J. Biol. Chem.
http://www.jbc.org/content/245/7/1699Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/245/7/1699.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on April 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from







![VEDLEGG I PREPARATOMTALE · serotonin-norepinefrin-reopptakshemmere (SNRIs), samt med legemidler som forringer metabolismen av serotonin (inkludert monoaminoksidasehemmere [MAO-hemmere]).](https://static.fdocument.pub/doc/165x107/5e52a2565bd51d52a71b872b/vedlegg-i-preparatomtale-serotonin-norepinefrin-reopptakshemmere-snris-samt-med.jpg)










