
Dulce Marléne Martins Fontoura - …repositorium.sdum.uminho.pt/bitstream/1822/15966/1/Dulce...
127
Dulce Marléne Martins Fontoura Outubro de 2010 Universidade do Minho Escola de Ciências Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
-
Upload
duongkhanh -
Category
Documents
-
view
220 -
download
0
Transcript of Dulce Marléne Martins Fontoura - …repositorium.sdum.uminho.pt/bitstream/1822/15966/1/Dulce...

Dulce Marléne Martins Fontoura
Outubro de 2010
Universidade do Minho
Escola de Ciências
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar

Dissertação de Mestrado Mestrado em Genética Molecular
Dulce Marléne Martins Fontoura
Outubro de 2010
Universidade do Minho
Escola de Ciências
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
Trabalho efectuado sob a orientação doProfessor Doutor Adelino F. Leite Moreirae daProfessora Doutora Maria João Sousa

DE ACORDO COM A LEGISLAÇÃO EM VIGOR, NÃO É PERMITIDA A REPRODUÇÃO DE QUALQUER PARTE DESTA TESE
Universidade do Minho, ___/___/______
Assinatura: ________________________________________________

iii
AGRADECIMENTOS
Agradeço a todos aqueles que contribuíram para a realização desta tese e me
ajudaram no caminho exigente da investigação em Fisiopatologia Cardiovascular.
Ao Professor Doutor Adelino F. Leite Moreira, orientador desta tese, que de forma
incansável me ensinou os conteúdos práticos e teóricos da Fisiopatologia Cardiovascular.
Agradeço igualmente pela sua disponibilidade permanente e dedicação durante a minha
formação como investigadora, pelos seus conselhos e orientação profissional, por ter
garantido todas as condições necessárias para o desenvolvimento e realização do projecto
científico presente nesta tese, assim como, pela oportunidade e privilégio que me deu em
colaborar com a sua equipa de investigação. Desta forma, manifesto a minha sincera
gratidão e admiração.
À Professora Doutora Maria João Sousa pelo seu incansável apoio e orientação ao
longo do Mestrado em Genética Molecular.
A todos os elementos do Serviço de Fisiologia, da Faculdade de Medicina do Porto,
o meu profundo reconhecimento pelo apoio que sempre souberam prestar. À Professora
Doutora Carmen Brás Silva pela sua preocupação, apoio e amizade. À Professora Doutora
Inês Pires pela disponibilidade e recente colaboração científica. O meu reconhecido
agradecimento ao Professor Doutor Amândio Rocha, ao Professor Doutor Roberto Roncon
de Albuquerque, ao Professor Doutor Tiago Henriques Coelho e ao Professor Doutor Paulo
Chaves. Ao Doutor André Lourenço pelo seu profissionalismo, entusiasmo, rigor,
persistência, exigência e dedicação, pela preocupação constante com o decorrer do meu
trabalho, por acreditar e incentivar todas as minhas actividades como investigadora, assim
como pela amizade que me tem confiado. À Dra. Antónia Teles pela atenciosa
disponibilidade durante os seus ensinamentos e por ser uma pessoa encantadora. À Marta
Oliveira e à Maria José pelo apoio técnico prestado. À Daniela Miranda, ao Francisco
Vasques Nóvoa, ao Vasco Sequeira, ao Rui Cerqueira, ao José Pedro Pinto, ao Pedro
Ferreira e à Carolina Rocha pelo convívio pessoal e saudável espírito de equipa. À D.
Francelina Marques, à D. Rosa Gonçalves, à D. Margarida, ao Sr. Armando, ao Sr. Alberto
e ao Pedro Leitão o meu reconhecido agradecimento.

iv
À Professora Doutora Cândida Lucas pela sua disponibilidade e dedicação. Ao
Professor Doutor Rui Oliveira pelo convívio científico, amizade e estímulo ao longo do meu
percurso profissional.
Aos meus pais pelo incansável apoio, amor, compreensão e paciência ao longo do
meu percurso de vida e por me terem privilegiado com formação de qualidade. Ao meu
irmão e à minha cunhada pelos momentos bem passados, pelas palavras de apoio e
verdadeira amizade. Aos meus avós pelo ensinamento dos seus valores e sabedoria. À
Professora Doutora Hercília Guimarães e ao Professor Doutor Jorge Areias pelo fantástico
apoio e amizade, assim como à Doutora Cristina Areias e ao Doutor José Castro. Ao Luís
Pinto Leite e seus familiares pelo privilégio da sua amizade, paciência e atitude positiva. À
Isis Alonso, ao Augusto Silva, à Filipa Neiva, à Manuela Araújo e à Paula Brilhante Simões
pela amizade e apoio que sempre me confiaram.

v
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas
em Modelos Experimentais de Hipertensão Pulmonar (Resumo)
A hipertensão pulmonar (HP) é um síndrome de etiologia diversificada, que conduz à
hipertrofia e falência ventricular direita (VD), partilhando muitas características com a
insuficiência cardíaca (IC), que é um síndrome complexo caracterizado pelo
comprometimento da ejecção ou do preenchimento do miocárdio. HP e IC associam-se a
activação neuroendócrina e inflamatória e a manifestações sistémicas como a caquexia
cardíaca, que pode definir-se por perda ponderal superior a 6% num intervalo mínimo de 6
meses, e acompanha 50% dos casos de IC severa, constituindo um preditor de mau
prognóstico. Entre os mecanismos complexos subjacentes destacam-se alterações
metabólicas e activação imunitária e neuroendócrina. Regimes alimentares hipercalóricos
são um factor de risco cardiovascular estabelecido, no entanto, e paradoxalmente, a
obesidade tem sido associada a melhor prognóstico na IC e a suplementação nutricional,
na doença crónica, contraria a anorexia e a caquexia. Os efeitos de uma dieta hipercalórica
(DH) na HP com IC poderão ser, portanto, inteiramente distintos. Neste trabalho, avaliámos
as repercursões de uma DH, rica em gordura saturada e carbo-hidratos simples, na IC e
caquexia associadas à HP. Induzimos HP em ratos Wistar machos por injecção subcutânea
única de 60 mg.Kg-1 de monocrotalina (MCT), distribuindo aleatoriamente os animais por
regimes alimentares com DH (5,4 kcal.g-1) ou dieta normal (DN) (2,9 Kcal.g-1). A evolução
ponderal, ingestão calórica e mortalidade foram registadas. Às 5 semanas, procedeu-se à
avaliação hemodinâmica e morfométrica, à avaliação histológica pulmonar e miocárdica, da
apoptose miocárdica, da expressão génica da endotelina-1 (ET-1), interleucina-6 (IL-6),
factor de necrose tumoral-α (TNF-α) e de enzimas metabólicas, bem como à quantificação
sérica do TNF-α e miocárdica da actividade dos factores de transcrição nuclear -κB (NF-κB)
e do receptor activado pelo proliferador do peroxissoma-α (PPAR-α). Ratos injectados com
MCT alimentados com DN, desenvolveram HP e IC, com elevada mortalidade, bem como
remodelagem e apoptose miocárdica. Apresentaram menor ingestão calórica e ganho
ponderal. A HP foi acompanhada por maior actividade do factor de transcrição NF-κB,
sobre-expressão de ET-1, TNF-α e IL-6, e aumento de TNF-α circulante, com redução da
actividade do PPAR-α. A DH atenuou a IC e caquexia, melhorando a sobrevida, a função e
remodelagem miocárdicas e reduzindo a actividade inflamatória e neuroendócrina. Os
resultados incentivam a realização de ensaios clínicos com DH em pacientes com caquexia
cardíaca.

vi

vii
Neuroendocrine and Metabolic Gene Expression and Subcellular Pathway Changes in
Experimental Pulmonary Hypertension (Abstract)
Pulmonary hypertension (PH), a syndrome of diverse etiology that leads to right
ventricular (RV) hypertrophy and failure, shares many features with heart failure (HF), a
syndrome characterized by impaired ability of the myocardium to fill or eject. PH and HF are
associated with neuroendocrine and inflammatory activation and systemic manifestations
such as cardiac cachexia, defined by a weight loss of more than 6% over 6 months.
Cachexia accompanies HF in up to 50% of severe cases and independently determines a
poor prognosis. As part of the complex and still incompletely revealed mechanisms
underlying cachexia associated with HF and PH are abnormalities of general metabolism as
well as of the immune and neuroendocrine systems. Hypercaloric diet (HD) regimens are
well-established cardiovascular risk factors, but since obesity has paradoxically been shown
to predict a better prognosis in HF and nutritional and energetic support have been shown to
partly counteract anorexia and cachexia in chronic diseases, the effects of a HD regimen
may have quite distinct effects in severe PH and terminal HF. Our study was designed to
evaluate the repercussions of a HD, rich in saturated fat and simple carbohydrates in HF
and cachexia associated with PH. We induced PH in male Wistar rats by subcutaneous
injection of 60 mg.Kg-1 monocrotaline (MCT) and randomly allocated rats to be fed with
either a 5.4 kcal.g-1 HD or a normal 2.9 Kcal.g-1 diet (ND). Calorie intake, weight gain and
mortality were recorded daily. Five weeks after, we evaluated RV and left ventricular
hemodynamics and morphometry, lung and myocardial histology, myocardial expression of
endothelin-1 (ET-1), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α) and metabolic
enzymes, plasma TNF-α levels as well as myocardial apoptosis and nuclear factor-κB (NF-
κB) and peroxisome proliferator-activated receptor-α (PPAR-α) transcription factor activities.
ND fed MCT-injected rats showed PH and HF, associated with high mortality, myocardial
remodeling and apoptosis, reduced calorie intake, and loss of body weight, lean and fat
mass that were attenuated by the HD. PH was accompanied in ND fed MCT-injected rats by
increased myocardial NF-κB transcription factor activity and ET-1, TNF-α and IL-6
overexpression, reduced PPAR-α activity, and increased circulating TNF-α, which were
offset by the HD. HD attenuated experimental PH with HF and cachexia, improving survival,
myocardial function and remodeling, and reduced inflammatory and neuroendocrine
activities. Our results encourage the conduction of clinical trials with HD regimens in patients
with cardiac cachexia.

viii

ix
ÍNDICE
Agradecimentos.…………………………………………………………………...…………….…..iii
Resumo.………………………………………………………………………………………...….....v
Abstract.………………………………………………………………………………...………...….vii
Índice.……………………………………………………………………………………...………….ix
Lista de abreviaturas.……………………………………………………………………..……......xii
1. Introdução .......................................................................................................................... 1
1.1. Hipertensão Pulmonar ................................................................................................. 1
1.2. Diagnóstico, apresentação e avaliação clínica ............................................................ 2
1.3. Prognóstico .................................................................................................................. 3
1.4. Modelos experimentais ................................................................................................ 3
1.5. Fisiopatologia ............................................................................................................... 5
1.6. HPA como panvasculopatia ......................................................................................... 5
1.7. Mecanismos genéticos ................................................................................................ 6
1.8. Inflamação ................................................................................................................... 6
1.9. O miocárdio na HP ....................................................................................................... 7
1.10. Terapêutica ................................................................................................................ 8
1.11. IC e caquexia ............................................................................................................. 9
1.12. Fisiopatologia da caquexia cardíaca ........................................................................ 10
1.13. Remodelagem metabólica na progressão da IC ...................................................... 11
1.14. Nutrição na IC e caquexia ........................................................................................ 14
2. Objectivos………….. ........................................................................................................ 15
3. Metodologia ...................................................................................................................... 16
3.1. Modelos animais ........................................................................................................ 16
3.2. Estudos de biologia molecular ................................................................................... 17
3.2.1. Quantificação de DNA e conteúdo protéico do miocárdio do VD e VE ................ 17
3.2.2. Quantificação de mRNA ...................................................................................... 17
3.2.3. Quantificação das isoformas das cadeias pesadas de miosina ........................... 19
3.2.4. Quantificação do TNF-α no plasma ..................................................................... 20
3.2.5. Apoptose miocárdica ........................................................................................... 20
3.2.6. Quantificação do NF-κB e do PPAR-α ................................................................ 21
4. Resultados ....................................................................................................................... 23

x
A hipertensão pulmonar experimental associada à insuficiência cardíaca e caquexia é
atenuada pela dieta rica em gorduras e carbo-hidratos simples (trabalho original em
revisão na revista American Journal of Physiology Heart and Circulatory Physiology)....24
5. Discussão e Conclusões .................................................................................................. 52
Relevância Clínica e Perspectivas Futuras ...................................................................... 58
6. Anexo ............................................................................................................................... 59
Novos Conceitos Fisiopatológicos e Abordagem Terapêutica da Hipertensão Pulmonar
(trabalho de revisão submetido para a revista International Journal of Cardiology)… … 60
7. Referências ...................................................................................................................... 97

xi
LISTA DE ABREVIATURAS
ACSL1 Acil-CoA Ligase de Ácidos Gordos de Cadeia Longa tipo 1
AG Ácidos Gordos
BCC Bloqueadores dos Canais de Ca2+
BMP Proteínas de Morfogénese Óssea
BMPR2 Receptor das Proteínas de Morfogénese Óssea do tipo 2
BNP Peptídeo Natriurético do tipo B
CCD Cateterismo Cardíaco Direito
CoA Coenzima A
DC Débito Cardíaco
DPOC Doença Pulmonar Obstrutiva Crónica
ET-1 Endotelina-1
FO Fosforilação Oxidativa
GI Gastrointestinal
GLUT-4 Transportador de Glicose do tipo 4
HP Hipertensão Pulmonar
HPA HP Arterial
HPAf HPA familiar
HPAi HPA idiopática
HpnA HP não-Arterial
HRP Peroxidase de Rábano
IC Insuficiência Cardíaca
IL-6 Interleucina-6
I-κB Inibidor-κB
LCAD Desidrogenase de Acil-CoA de Cadeia Longa
LPS Lipopolissacarídeo
MCAD Desidrogenase de Acil-CoA de Cadeia Média
MCT Monocrotalina
NF-κB Factor de Transcrição Nuclear-κB
PAP Pressão(ões) da Artéria Pulmonar
PDH Desidrogénase do Piruvato
PDK Cínase da Desidrogénase do Piruvato
PDK4 PDK do tipo 4
PmAP Pressão(ões) médias da Artéria Pulmonar

xii
PPAR Receptor Activado pelo Proliferador do Peroxissoma
PPRE Elemento de Resposta do Proliferador do Peroxissoma
ROS Radicais Livres de Oxigénio
SDS-PAGE Electroforese em Gel de Poliacrilamida e Dodecil Sulfato de Sódio
TEP Tromboembolismo Pulmonar
TGF-β1 Factor de Transformação de Crescimento-β1
Tm Temperatura de fusão ou melting
TM6M Teste de Marcha durante 6 Minutos
TNF-α Factor de Necrose Tumoral-α
TUNEL Marcação dos Terminais Desoxiuridina Trifosfato pela Desoxinucleotidil
Transferase (terminal UDP nick-end labeling)
VD Ventrículo(ar) Direito(a)
VE Ventrículo(ar) Esquerdo(a)
VEGF Factor de Crescimento do Endotélio Vascular

xiii

xiv

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
1
1. Introdução
1.1. Hipertensão Pulmonar
A hipertensão pulmonar (HP) é um síndrome rapidamente progressivo e fatal, apesar
das alternativas terapêuticas actuais. É classicamente diagnosticada pela demonstração
hemodinâmica de pressões médias da artéria pulmonar (PmAP) superiores a 25 mmHg em
repouso, ou 30 mmHg durante o exercício (Fishman, 2004).
Na maioria dos casos de HP, esta acompanha doenças cardio-respiratórias comuns,
não existindo doença vascular pulmonar intrínseca. No entanto, a HP também se pode
manifestar, menos frequentemente, sob a forma de HP arterial (HPA), uma doença
vasoproliferativa que envolve as pequenas artérias pulmonares e que se define, em termos
hemodinâmicos, não só pela elevação das PmAP, mas também pela presença de pressões
de enchimento ventriculares esquerdas (VE) normais, ou seja pressões de encravamento
capilar pulmonar ou pressões de enchimento VE inferiores a 15 mmHg (Chin e Rubin,
2008). A HPA apresenta traços anatomopatológicos característicos, alguns dos quais,
nomeadamente as lesões plexiformes, não observáveis noutras formas de HP (Rabinovitch,
2008).
De modo a melhor traduzir os mecanismos fisiopatológicos e apresentação clínica, a
classificação etiológica da HP tem vindo a sofrer modificações. Resumidamente, a HP pode
ser classificada em HPA e HP não-arterial (HPnA), sendo esta última composta por vários
subtipos de doença, com causa estabelecida, indefinida ou multifactorial. A classificação
adoptada mais recentemente é sumariada na tabela 2 do trabalho de revisão anexado.
A incidência e prevalência estimadas da HPA, com base em vários estudos
populacionais, são cerca de 2,4 a 7,6 casos/milhão e 15 a 26 casos/milhão de habitantes
por ano, respectivamente (Humbert et al., 2006; Peacock et al., 2007). Com a excepção da
HPA idiopática (HPAi), não há estimativas precisas da incidência e prevalência da HP, mas,
uma vez que as manifestações clínicas são inespecíficas, qualquer previsão constituirá
uma subestimativa (Provencher et al., 2005). Nas últimas décadas, a HPnA tem sido
crescentemente reconhecida, nomeadamente a HP associada à doença pulmonar
obstrutiva crónica (DPOC), uma vez que, segundo a maioria das previsões, em 2020,
devido ao tabagismo, será a 3ª causa de morte (Murray e Lopez, 1997). De facto, a DPOC
é presentemente responsável por até 84% dos casos de cor pulmonale. A insuficiência
cardíaca (IC) direita é a sua complicação mais grave, sendo responsável por 10 a 30% dos
internamentos devido à IC descompensada (MacNee, 1994).

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
2
A HP e a IC direita subsequente constituem uma importante causa de mortalidade e
incapacidade a nível mundial (Humbert, 2009). No entanto, o impacto na saúde é
provavelmente maior do que o reconhecido, sobretudo pelas associações recentemente
estabelecidas com a hemodiálise (Fruchter e Yigla, 2008) e com o síndrome metabólico
(Robbins et al., 2009), assim como, com patologias infecciosas altamente prevalentes nos
países em desenvolvimento (Butrous et al., 2008).
As duas últimas décadas foram pródigas em estudos experimentais e clínicos que
permitiram desvendar um pouco dos seus complexos mecanismos fisiopatológicos e
tornaram disponíveis vários novos fármacos (Rabinovitch, 2008). No entanto, o prognóstico
HP continua a ser pouco satisfatório e muitos dos pacientes acabam por necessitar de
transplantação (Keogh et al., 2009).
1.2. Diagnóstico, apresentação e avaliação clínica
Os sintomas, como por exemplo a fadiga e a dispneia, são normalmente inespecíficos,
insidiosos e indefinidos, tornando o diagnóstico um desafio para os clínicos (Provencher et
al., 2005). A HP é sub-diagnosticada, sendo a maior parte dos doentes identificados apenas
após o aparecimento de manifestações como dor torácica, palpitações, edema, ascite e
síncope. Normalmente o diagnóstico é efectuado com 2 ou 3 anos de atraso, e muitas
vezes apenas após episódios de síncope, quando o débito cardíaco (DC) está já
comprometido e as pressões da artéria pulmonar (PAP) atingem níveis supra-sistémicos
(Nef et al., 2010; Rabinovitch, 2008). No entanto, o diagnóstico e tratamento precoce, em
estadios reversíveis, é fundamental para melhorar a resposta à terapêutica. Para facilitar o
diagnóstico, na definição mais recente foram incluídos novos critérios de diagnóstico, não-
invasivos e mais práticos, baseados na ecocardiográfica (Nef et al., 2010). Contudo, em
termos práticos, a ecocardiografia é normalmente utilizada para rastreio, uma vez que não
permite a quantificação do fluxo transpulmonar e das pressões venosas, sendo a
confirmação do diagnóstico feita pelo cateterismo cardíaco direito (CCD), que continua a
ser o gold standard (Chin e Rubin, 2008). O CCD também é essencial na selecção de
doentes que poderão beneficiar de terapia crónica com bloqueadores dos canais de Ca2+
(BCC) (McLaughlin et al., 2009; Sitbon et al., 2005). Vários exames auxiliares de
diagnóstico podem fornecer indicações úteis ao rastreio, diagnóstico definitivo e diferencial
(McLaughlin et al., 2009; Nef et al., 2010; van Wolferen et al., 2007). Outros exames
auxiliares são empregues para avaliar a capacidade funcional, como por exemplo a
espirometria, a difusão de monóxido de carbono e o teste de marcha durante 6 minutos
(TM6M) (Sun et al., 2003). A descrição detalhada destes não se encontra no âmbito da

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
3
presente tese, mas pode ser consultada no trabalho de revisão em anexo. O TM6M é um
indicador sensível e reprodutível no compromisso moderado do desempenho
cardiopulmonar, sendo vulgarmente empregue para quantificar a melhoria clínica após
intervenções terapêuticas em ensaios clínicos (Miyamoto et al., 2000). As orientações da
American Thoracic Society relativamente ao TM6M constituem uma perspectiva abrangente
sobre as suas indicações, contra-indicações, precauções de segurança, aspectos técnicos
e limitações (ATS statement, 2002).
1.3. Prognóstico
A raridade da HPA, a sua etiologia diversa e a constante evolução na abordagem
terapêutica tem dificultado as estimativas da taxa de mortalidade anual. Um registo
recolhido entre 1981 e 1985, estimou taxas de sobrevida de 68% e 48% aos 1 e 3 anos,
respectivamente na HPAi (McLaughlin et al., 2004). Contudo, registos mais recentes
apontam para uma melhoria no prognóstico, com sobrevidas de 83 a 88% após 1 ano e de
58 a 72% aos 3 anos (McLaughlin e Suissa, 2010). A progressão da HP nas formas de
HPnA é normalmente mais lenta e o prognóstico geralmente mais favorável. No entanto, o
desenvolvimento de HP tem um impacto importante na qualidade de vida e sobrevida
(Hoeper et al., 2009).
1.4. Modelos experimentais
Embora nenhum modelo animal recapitule completamente todas as características da
HPA humana, o estudo da fisiopatologia da HP tem sido levado a cabo essencialmente em
modelos animais de HPA. A combinação de diferentes agressões, de acordo com a
hipótese das agressões múltiplas, permitiu desenvolver fenótipos mais severos que melhor
mimetizam as características da HPA humana (Robbins, 2004).
O modelo experimental mais frequentemente utilizado pela sua reprodutibilidade e
facilidade de execução, é o modelo de HP induzida pela monocrotalina (MCT) no rato
(Brown et al., 1998; Jasmin et al., 2001; Lalich e Merkow, 1961; Lourenço et al., 2006). A
MCT é um alcalóide pirrolizidínico, isolado a partir da Crotalaria spectabilis, que após
desidrogenação pelo citocromo P450 hepático num pirrol reactivo, adquire acção tóxica no
endotélio, provocando uma endarterite obliterativa das arteríolas pulmonares. As lesões
histológicas pulmonares são semelhantes às da HP, sendo a MCT desprovida de acção
tóxica directa no miocárdio (Chen et al., 1998). Este modelo experimental contribuiu de
forma muito relevante para a evolução terapêutica recente na HP. Praticamente todos os

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
4
fármacos que tiveram sucesso na HPA humana também foram eficazes neste modelo, com
a excepção do beraprost (Stenmark et al., 2009). Adicionalmente, neste modelo a activação
neuro-humoral assemelha-se à da IC (Brunner, 1999; Leineweber et al., 2002), pelo que
tem sido empregue para estudar a hipertrofia miocárdica (Buermans et al., 2005) e
comparar alterações funcionais e moleculares entre os dois ventrículos (Kögler et al., 2003;
Leineweber et al., 2002; Schott et al., 2005; Seyfarth et al., 2000), o VE que é apenas
influenciado por mediadores neuroendócrinos e o ventrículo direito (VD) que é
simultaneamente sobrecarregado. Foi este modelo animal, que evolui rapidamente para IC
descompensada e fatal, com anorexia e caquexia acentuadas e perda de massa músculo-
esquelética (Ceconi et al., 1989; Hessel et al., 2006; Lourenço et al., 2006; Steffen et al.,
2008; Werchan et al., 1989) que seleccionamos para desenvolver o trabalho experimental
que levamos a cabo e que descrevemos nas secções de metodologia, resultados e
discussão.
No entanto, o modelo de HP induzida pela MCT apresenta algumas limitações
(Robbins, 2004). O espectro anatomopatológico mais amplo da HPAi só é reproduzido por
completo quando aos ratos tratados com MCT se associa a realização de pneumectomia.
Neste caso, pode observar-se a formação de neo-íntima e obliteração de pequenas
arteríolas pulmonares (Tanaka et al., 1996). A MCT também tem acção tóxica, embora
menos marcada, no endotélio renal e hepático, o que acarreta efeitos sistémicos. O modelo
é menos reprodutível no murganho, e mesmo estirpes diferentes de ratos apresentam
susceptibilidades diversas. Finalmente, há diferenças na gravidade do fenótipo atribuíveis
ao timming da injecção (Lalich e Merkow, 1961).
Outro modelo experimental utilizado frequentemente é o modelo de HP induzida pela
hipóxia. Este modelo experimental é mais relevante no estudo da HP associada à hipóxia e
doença respiratória e não tanto da HPA. Os animais são expostos a pressões alveolares de
oxigénio < 70 mmHg, de forma a desencadear vasoconstrição pulmonar. Na exposição
prolongada ocorre remodelagem dos ramos distais das artérias pulmonares, com
hiperplasia endotelial e miocitária (Fallon et al., 1998; Rhodes, 2005; Taraseviciene-Stewart
et al., 2001). Para além destes dois modelos principais, vários outros têm sido usados
ocasionalmente. Os ratos de capuz castanho, que têm um defeito plaquetário no
armazenamento de serotonina, desenvolvem HP espontaneamente, podendo o fenótipo ser
acelerado pela exposição à hipóxia (Stenmark et al., 2009). A administração de uma dose
única de semaxinib (SU5416), um antagonista do receptor do factor de crescimento do
endotélio vascular (VEGF) leva ao desenvolvimento de lesões oclusivas da neo-íntima em
ratos expostos à hipoxia (Taraseviciene-Stewart et al., 2001). O modelo de HP induzida

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
5
pela embolização com coágulos autólogos ou microsferas de diversos materiais (ex.
sefadex, polidextrano), possibilita o estudo das formas agudas e crónicas de HP associada
ao tromboembolismo pulmonar (TEP) (Shelub et al., 1984; Watts et al., 2006; Watts et al.,
2010). Adicionalmente, têm sido estudados também animais manipulados geneticamente
(Greenway et al., 2004; Ivy et al., 2005; Steiner et al., 2009).
1.5. Fisiopatologia
Como vimos, a HPA tem sido largamente estudada sobretudo recorrendo a modelos
animais. Paradoxalmente, poucos estudos têm sido realizados sobre a fisiopatologia das
formas mais comuns, a HPnA. No entanto, admite-se que a fisiopatologia destas formas
tem aspectos similares à HPA (Hoeper et al., 2009). De facto, sabe-se que na HP
associada a doenças respiratórias crónicas a vasoconstrição hipóxica não é um mecanismo
fundamental (Chaouat et al., 2005), sendo mais importantes a disfunção endotelial e a
activação neuroendócrina e inflamatória (Wright et al., 2005); que na HP associada ao TEP
crónico a elevação das resistências vasculares pulmonares não se deve apenas à oclusão
vascular pelos êmbolos, mas também a graus variáveis de arteriopatia de pequenos vasos
(Hoeper et al., 2006); e, finalmente, que na disfunção VE e patologia valvular, para além da
transmissão à circulação pulmonar de pressões de preenchimento VE elevadas também
ocorre disfunção endotelial e perturbação da produção de serotonina, tromboxano A2 e
angiotensina-II, que contribuem para o agravamento adicional da HP (Moraes et al., 2000).
1.6. HPA como panvasculopatia
A concepção mais prevalente nas últimas décadas admite que a HP resulta de um
desequilíbrio entre vasoconstritores e vasodilatadores pulmonares (Christman et al., 1992).
A síntese de prostaciclina e óxido nítrico, vasodilatadores e mediadores antiproliferativos,
está diminuída na HPA, enquanto a de endotelina-1 (ET-1), um vasoconstritor e mitógeneo
das células musculares lisas, está aumentada. Efectivamente, os prostanóides e outros
vasodilatadores pulmonares, têm constituído a base terapêutica na HPA. No entanto, sabe-
se actualmente que esta concepção é insuficiente para explicar a complexidade dos
mecanismos fisiopatológicos da HP. De facto, apesar dos avanços terapêuticos mais
recentes, o prognóstico continua a ser muito reservado (Rabinovitch, 2008). De acordo com
a maioria dos autores, a HPA é actualmente encarada como uma panvasculopatia, com
vários traços histológicos característicos identificáveis nas pequenas artérias e arteríolas
pulmonares (McLaughlin et al., 2009). Como hipótese fisiopatológica admite-se que após

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
6
apoptose endotelial persistente sejam seleccionados fenótipos endoteliais resistentes à
apoptose, que interagem com as células musculares lisas através da produção de factores
de crescimento, induzindo proliferação muscular lisa e transdiferenciação das células
endoteliais e dos fibroblastos em miócitos (Sakao et al., 2009). Adicionalmente, a activação
de metaloproteases conduz à perda de integridade da barreira da membrana basal, e ao
recrutamento de células inflamatórias que participam também na libertação de peptídeos
mitogénicos (Cowan et al., 2000).
Outra hipótese fisiopatológica sustenta que a HPA e o cancro partilham anomalias do
metabolismo mitocondrial, designadas por “fenótipo de Warburg”. Este consiste na
progressiva substituição da fosforilação oxidativa (FO) pela glicólise anaeróbia, mesmo em
condições de normóxia, que se associa a proliferação celular e inibição da apoptose e se
deve à alteração da sensibilidade mitocondrial ao O2 (Archer et al., 2008; Bonnet et al.,
2006). Uma descrição molecular detalhada das perspectivas fisiopatológicas mais recentes
sobre a HP é apresentada no trabalho de revisão em anexo.
1.7. Mecanismos genéticos
Em mais de 80% dos pacientes com HPA familiar (HPAf) é possível identificar
mutações inactivadoras do receptor das proteínas de morfogénese óssea do tipo 2
(BMPR2), um receptor activado pela superfamília do factor de transformação de
crescimento-β1 (TGF-β1), que inclui as proteínas de morfogénese óssea (BMP). Estas
mutações levam à inibição da via de sinalização da smad, e à menor diferenciação das
células musculares lisas, à proliferação celular e ao aumento do tono muscular (Morrell et
al., 2001; Yang et al., 2005), uma vez que a via das smads regula normalmente a
transcrição génica promovendo respostas citostáticas. Contudo, as mutações do BMPR2
constituem apenas 10 - 20% dos casos de HPA não familiar, e mesmo na HPAf a
penetrância é baixa (Newman et al., 2004), portanto, outros mecanismos genéticos deverão
estar envolvidos. Para alguns genes, foram identificados polimorfismos de nucleotídeos
singulares relacionados com predisposição para HPA (MacLean e Dempsie, 2009;
Provencher et al., 2005; Shapiro et al., 2007).
1.8. Inflamação
O estado pró-inflamatório dos vasos pulmonares tem, recentemente, sido apontado
como um evento primário e mecanismo de progressão da doença, alternativamente à visão
tradicional, que o encarava como mera consequência da doença. A presença de auto-
anticorpos circulantes e a infiltração de células inflamatórias nos pulmões são

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
7
características da HPA associada às doenças do tecido conjuntivo, mas também podem
surgir na HPAi (Dorfmuller et al., 2003). As citocinas, como o factor de necrose tumoral-α
(TNF-α) e a interleucina-6 (IL-6), que estão igualmente envolvidas na patogénese de várias
doenças crónicas, como neoplasias, IC e DPOC, parecem desempenhar uma função
importante na vasculopatia (Steiner et al., 2009).
1.9. O miocárdio na HP
A principal função do VD é conduzir sangue desoxigenado através da circulação
pulmonar, com baixa pressão de perfusão, até ao VE, possibilitando trocas gasosas
eficazes e adaptações rápidas de retorno venoso (Bogaard et al., 2009a). É mais
complacente e fino do que o VE e adapta-se facilmente quer a variações de pré-carga, quer
ao trabalho de ejecção de volume. Pelo contrário, não tolera elevações repentinas de pós-
carga, porque é incapaz de desenvolver agudamente pressões máximas superiores a 40
mmHg, quando uma sobrecarga de pressão repentina ultrapassa este valor, o volume de
ejecção diminui e o VD dilata (Blaise et al., 2003). Contudo, se a sobrecarga de pressão for
progressiva o VD desenvolve hipertrofia gradual e remodelagem que lhe permitem
desenvolver pressões mais elevadas, compensando a elevação de pós-carga (Ritchie et al.,
1993; van Wolferen et al., 2007). No entanto, tal como sucede na sobrecarga de pressão
VE, vários mecanismos acabam por levar à disfunção miocárdica (Bogaard et al., 2009a). A
activação neuroendócrina local contribui não só para a hipertrofia (Lourenço et al., 2006;
Miyauchi et al., 1993), mas também para fibrose, apoptose e rarefação capilar no
miocárdio. A sobrecarga prolongada associa-se igualmente a maior stress oxidativo,
activação de citoquinas inflamatórias e de factores de crescimento que também são
importantes determinantes de remodelagem adversa e disfunção (Bogaard et al., 2009a,
2009b). Adicionalmente, o miocárdio VD poderá adaptar-se, segundo alguns autores, a um
estado de hipoperfusão relativa e entrar num processo semelhante ao de hibernação
miocárdica, característico da doença isquémica, uma vez que, dadas a maior pressão e
massa do VD, é maior a resistência ao fluxo sistólico nas artérias coronárias (van Wolferen
et al., 2008). De facto, a sobrecarga e a hipertrofia VD reduzem a capacidade mitocondrial
de produção energética (Daicho et al., 2009) e tornam o miocárdio altamente dependente
do metabolismo anaeróbio da glicose (Piao et al., 2009).
As alterações morfofuncionais e a resposta miocárdica são determinantes fundamentais
da qualidade de vida e sobrevida dos pacientes com HP (van Wolferen et al., 2007). A
modulação da remodelagem e disfunção miocárdica constituirá um alvo terapêutico

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
8
promissor (Archer et al., 2010), o que é, ainda mais, reforçado pela reversibilidade potencial
da lesão miocárdica, como se verifica após transplante pulmonar (Ritchie et al., 1993).
1.10. Terapêutica
O tratamento da HPA evoluiu consideravelmente na última década, vários algoritmos
terapêuticos têm sido propostos. A terapêutica deve ser ajustada individualmente, em
função da gravidade da doença, das comorbilidades, dos efeitos laterais dos fármacos e da
experiência clínica (McLaughlin et al., 2009). O teste de vasoreactividade é efectuado em
pacientes com HPAi e hemodinâmica estável. Um teste positivo prevê a possibilidade de
resultados favoráveis com terapêutica crónica com BCC (Rich et al., 1992). Em caso
contrário, os antagonistas dos receptores da ET - 1 ou os inibidores da fosfodiestérase são
normalmente seleccionados como terapêutica de primeira linha, exceptuando os casos em
que a via oral não é praticável, os casos em classe IV da Organização Mundial de Saúde
ou que apresentam sinais clínicos evidentes de IC direita. Nestes, a primeira escolha são
os prostanóides administrados por via intravenosa (Barst et al., 1996). No entanto, com a
excepção dos doentes com respostas favoráveis aos BCC, a maior parte dos fármacos
apenas atinge reduções de 10 - 20% da PmAP e muitos dos doentes permanecem
sintomáticos. Neste caso, devem ponderar-se quer a terapêutica combinada,
particularmente quando surgem efeitos laterais, quer a inclusão em ensaios clínicos de
novos agentes farmacológicos. Todavia, antes que ocorra uma deterioração sistémica
irreversível, devem ponderar-se a septostomia interauricular (Reichenberger et al., 2003) e
o transplante. Nos casos refratários é crucial a referenciação precoce para transplante
(Keogh et al., 2009; McLaughlin et al., 2009). Apesar da mortalidade substancial associada
à transplantação, o resultado a longo prazo é favorável, sendo a sobrevida de 47% aos 5
anos (Trulock et al., 2007). No grupo de pacientes com HPnA é consensual que a
optimização da terapêutica dirigida à doença primária terá efeitos benéficos, mas apesar
desta abordagem, pode persistir HP e alguns pacientes têm mesmo HP desproporcionada,
isto é, um grau de HP que não é facilmente atribuível à condição médica subjacente. Em
muitos destes casos, os novos fármacos aprovados para a HPA têm sido utilizados com
algum sucesso (Hoeper et al., 2009). Para uma perspectiva mais completa sobre as
abordagens farmacológicas e cirúrgicas da HP, nas suas diversas etiologias, pode
consultar-se o trabalho de revisão em anexo.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
9
1.11. IC e caquexia
A IC pode definir-se clinicamente como um síndrome complexo resultante de uma
perturbação cardíaca funcional ou estrutural que compromete a capacidade do ventrículo
para se preencher ou ejectar (Hunt et al., 2009). De forma sumária, após uma lesão, a
capacidade de bombeamento diminui, desencadeando-se um processo lento e progressivo
de deterioração funcional, inicialmente compensado por sistemas homeostáticos, como o
sistema adrenérgico e os sistemas de retenção de sal e água, assim como pela activação
de moléculas vasodilatadoras, incluindo peptídeos natriuréticos, que contrariam a
vasoconstrição excessiva. Os doentes podem manter-se assintomáticos durante anos, mas
acabam por descompensar devido quer à activação excessiva de citocinas e sistemas
neuro-humonais, quer às alterações de remodelagem (Mann e Bristow, 2005). Os
cardiomiócitos de pacientes com IC sofrem alterações progressivas que comprometem a
função contráctil, nomeadamente o aumento da expressão das isoformas β das cadeias
pesadas de miosina, a perda de miofilamentos, alterações na composição do citoesqueleto,
perturbação do acoplamento excitação-contração e menor sensibilidade à sinalização
adrenérgica (Dhalla et al., 2009; Lehnart et al., 2009). A prevalência e a incidência da IC
continuam a aumentar, em grande parte devido ao envelhecimento populacional, ao
aumento da prevalência de hipertensão arterial e aterosclerose, bem como aos avanços
tecnológicos e modificações de estilos de vida que contribuíram para a prevenção e
tratamento da doença coronária (Young, 2004). Os inibidores da enzima de conversão da
angiotensina e os antagonistas β-adrenérgicos impedem a progressão da disfunção
miocárdica e previnem a remodelagem, mas apesar destes avanços terapêuticos a IC
continua a ser uma doença crónica de prognóstico reservado (Hunt et al., 2009).
Outro síndrome, a caquexia cardíaca, acompanha até 50% dos casos de IC grave
(Filippatos et al., 2000) e constitui um preditor independente e importante de mau
prognóstico (Anker et al., 1997). O termo caquexia (do Grego: kakós – má; hexis -
condição) corresponde à perda de massa corporal que surge nos estadios avançados de
várias doenças crónicas. A primeira definição do síndrome foi avançada por Hipócrates “A
carne é consumida e transforma-se em água, ... o abdómen preenche-se com água, os pés
e as pernas edemaciam, os ombros, as clavículas, o tórax e as coxas definham ... A doença
é fatal” (Doehner e Anker, 2002). Apenas deve ser considerada a perda ponderal não
edematosa, ou seja, relativa ao peso corporal excluindo edemas. O limiar mais consensual
para perda ponderal significativa derivou da reanálise do estudo SOLVD, nesta o limiar que
melhor previa a mortalidade era 6%, durante um período de pelo menos 6 meses (Anker et
al., 2003). Mais recentemente, numa reunião de consenso, definiu-se caquexia como a

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
10
complicação de doença crónica que surge quando os pacientes perdem pelo menos 5% do
peso corporal em 12 meses, ou menos, ou têm índice de massa corporal inferior a 20, e
cumprem pelo menos 3 de 5 critérios (diminuição da força muscular, fadiga, anorexia, baixo
índice de massa gorda livre ou inflamação, anemia ou diminuição da albumina sérica)
(Evans et al., 2008).
1.12. Fisiopatologia da caquexia cardíaca
Tanto a IC como a HP são síndromes multi-sistémicos que afectam os sistemas
músculo-esquelético, renal, neuroendócrino e imunológico (Dorfmuller et al., 2003; Francis,
2001). Como parte do envolvimento sistémico, muitos doentes desenvolvem caquexia. Os
mecanismos envolvidos são a elevação do metabolismo basal, a redução do apetite e
perturbações gastrointestinais (GI) que limitam a ingestão calórica, assim como um
comprometimento geral do metabolismo e dos sistemas imunológico e neuroendócrino (von
Haehling et al., 2007).
A activação inflamatória e as perturbações neuroendócrinas e imunológicas
correlacionam-se fortemente quer com a perda de massa corporal quer com a sobrevida,
na IC (Anker et al., 2003). O TNF-α, em particular, parece estar implicado nas vias que
conduzem à caquexia e poderá constituir a via final comum de todas as suas etiologias.
Inicialmente isolado a partir de murganhos infectados com soro do bacilo de Calmette-
Guérin após tratamento com lipopolissacarídeo (LPS), foi designado “caquexina” porque
mimetizava a acção de necrose tumoral do LPS e inibia a lipase das lipoproteínas,
pressumindo-se que desempenharia um papel na caquexia cardíaca (Beutler et al., 1985).
Sabe-se hoje que os seus efeitos integram as principais alterações que acompanham a
progressão da IC, tais como a disfunção ventricular, a remodelagem ventricular, a apoptose
dos cardiomiócitos, o desenvolvimento de anorexia e caquexia, a redução do fluxo
sanguíneo muscular, a disfunção endotelial e a insulino-resistência (Meldrum, 1998).
A anorexia é um elemento comum a todas as formas de caquexia. Na caquexia
cardíaca os pacientes apresentam redução de apetite e da ingestão alimentar, o que
poderá dever-se parcialmente ao aumento da actividade da melanocortina e à sensação de
saciedade induzidas por citocinas pró-inflamatórias (Inui, 1999). No caso da caquexia
cardíaca, o tracto GI parece desempenhar um papel importante. O edema e hipoperfusão
GI contribuem para a má-absorção (Witte e Clark, 2002) e perda de proteínas,
particularmente na IC direita. Nos casos mais extremos, os pacientes apresentam o
síndrome de enteropatia com perda de proteínas e hipoproteinémia (Thorne et al., 1998).

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
11
Por outro lado, o edema da parede GI também facilita a translocação bacteriana e,
portanto, a endotoxemia. De facto, níveis elevados de endotoxina normalizam após o
tratamento diurético (Niebauer et al., 1999). A translocação bacteriana também é agravada
como consequência da hipoperfusão e isquemia da mucosa GI, que aumentam a
permeabilidade da parede intestinal (Krack et al., 2005). O TNF-α e outras citocinas são
induzidas nos monócitos (Rauchhaus et al., 2000) e nos cardiomiócitos pelas endotoxinas
circulantes (Wagner et al., 1998), embora uma hipótese alternativa considere que a
activação pró-inflamatória possa surgir na sequência da menor perfusão e hipóxia tecidual
(Tsutamoto et al., 1998).
O principal factor de transcrição subjacente quer à actividade celular quer à indução da
síntese do TNF-α é o factor de transcrição nuclear-κB (NF-κB). A activação do NF-κB tem
sido associada a vários processos patológicos que acompanham a progressão da IC,
incluindo a libertação de citocinas pelos cardiomiócitos, o stress oxidativo, a hipertrofia e a
remodelagem do miocárdio (Valen et al., 2001). A família de factores de transcrição NF-κB
é responsável pela regulação de mais de 150 genes alvo. Na maioria das células, os
complexos NF-κB estão presentes no citoplasma sob a forma inactiva, complexados com o
inibidor-κB (I-κB). Vários estímulos, entre os quais o LPS e citocinas, resultam na
fosforilação, ubiquitinação e degradação subsequente do I-κB, libertando um conjunto de
aminoácidos no NF-κB que promove a translocação nuclear e a ligação a elementos de
resposta específicos dos genes alvo nos cardiomiócitos (Valen et al., 2001).
Para além das alterações inflamatórias, várias modificações endócrinas acompanham
também a progressão da caquexia (Cicoira et al., 2003; Doehner e Anker, 2002; Leyva et
al., 1998; Nagaya et al., 2001).
1.13. Remodelagem metabólica na progressão da IC
O miocárdio saudável depende de um fornecimento constante de O2 para manter o seu
metabolismo. O oxigénio é essencial para a FO, que é responsável por aproximadamente
90% da produção de ATP (Ventura-Clapier et al., 2004). Vários metabolitos, como ácidos
gordos (AG), glicose e lactato, podem ser utilizados como substrato pela FO no miocárdio
(Bing et al., 1954). O metabolismo energético miocárdico e a taxa de hidrólise de ATP
ajustam-se às exigências inerentes ao trabalho cardíaco. Em condições de esforço
máximas, as vias metabólicas são capazes de produzir quantidades elevadas de ATP,
aumentando a actividade mitocondrial para cerca de 80 - 90% da sua capacidade máxima
(Mootha et al., 1997). Numa situação de repouso, a β-oxidação de AG é responsável por 60

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
12
- 90% do total de energia produzida (van der Vusse et al., 2002), enquanto a oxidação do
piruvato e do lactato, provenientes da glicólise, representam apenas 10 - 40% (Wisneski et
al., 1985). A preferência por substratos energéticos é baseada a curto prazo no equilíbrio
entre as necessidades e o fornecimento, sendo ditada por factores mecânicos e
endócrinos, dependendo principalmente de uma efectiva reciprocidade entre os substratos.
De facto, em quantidade suficiente, cada substrato inibe automaticamente as restantes vias
metabólicas. A rápida adaptação aos substratos metabólicos disponíveis e às exigências
variáveis do trabalho cardíaco são características primordiais do miocárdio saudável
(Stanley et al., 2005).
Nas formas avançadas da IC, o miocárdio torna-se incapaz de converter eficientemente
a energia química em trabalho contráctil e o seu conteúdo de ATP encontra-se
substancialmente diminuído, em boa parte devido ao comprometimento do metabolismo
oxidativo (Ormerod et al., 2008). Para além de uma menor eficiência metabólica global, as
vias metabólicas sofrem adaptações, entre as quais se destaca a substituição progressiva
da oxidação de AG pela glicólise anaeróbia como principal mecanismo de produção de
energia (Davila-Roman et al., 2002). Apesar das consequências da disfunção metabólica na
IC serem ainda pouco compreendidas, um número crescente de evidências apoia um papel
causal na génese da disfunção contráctil e na remodelagem do miocárdio. As perturbações
do metabolismo miocárdico que acompanham a progressão da IC têm vindo a ser
exploradas como potencial alvo terapêutico. Estas adaptações a longo prazo, dependem de
alterações na expressão génica e na actividade de transportadores ou de enzimas chave,
da fosforilação de proteínas e dos níveis de co-fatores que possibilitam alterações
pronunciadas nas vias metabólicas (Stanley et al., 2005). O factor de transcrição do
receptor activado pelo proliferador do peroxissoma (PPAR) tem sido implicado como
potencial responsável por parte destas adaptações.
O PPAR, que apresenta três isoformas, todas elas activadas pelos AG, hetero-dimeriza
com o receptor retinóide X, sendo o complexo resultante translocado para o núcleo. No
núcleo, é activada a transcrição de genes após ligação ao elemento de resposta do
proliferador do peroxissoma (PPRE). O PPAR aumenta a expressão dos genes que
codificam as proteínas envolvidas na captação, transporte e β-oxidação dos AG (Finck e
Kelly, 2002) e diminui reciprocamente a utilização de glicose por modificação da expressão
génica de moduladores do metabolismo da glicose (Kodde et al., 2007). De facto, níveis
elevados de lípidos circulantes e de AG no citoplasma dos cardiomiócitos conduzem, por
activação transcricional mediada pelo PPAR-α, à sobre-expressão de cínase da
desidrogénase do piruvato (PDK). A PDK, por sua vez, fosforila e inactiva o complexo da

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
13
desidrogénase do piruvato (PDH), que é responsável pelo principal passo regulador da
oxidação da glicose. O complexo da PDH da membrana mitocondrial interna, transporta o
piruvato através da membrana e catalisa a transformação irreversível em acetil-Coenzima A
(CoA), sendo esta uma etapa fundamental da oxidação dos carbo-hidratos. Por outro lado,
a activação da oxidação de AG aumenta as razões acetil-CoA/CoA livre e NADH/NAD+ na
mitocôndria, activando a PDK. Reciprocamente, a redução da oxidação dos AG aumenta a
oxidação de glicose e de lactato pela desinibição da PDH (Stanley et al., 2005). A isoforma
α dos PPAR é a principal reguladora a nível cardíaco, mas a isoforma γ exerce uma
influência miocárdica indirecta por acção em adipócitos (Barak et al., 1999) e elevação dos
níveis circulantes de AG.
Na doença cardíaca prolongada, que conduz a hipertrofia e posteriormente a IC, a
expressão de PPAR-α diminui e, concomitantemente, o miocárdio diminui a sua capacidade
de metabolização de AG e aumenta a sua capacidade de utilização de glicose (Garnier et
al., 2003). O significado desta alteração metabólica não está completamente elucidado,
sendo ainda indefinido se terá uma acção adaptativa e benéfica ou desadaptativa e
prejudicial. Alguns estudos recentes verificaram que a activação do PPAR-α induz
disfunção contrátil moderada e acentua a hipertrofia em modelos experimentais de
hipertrofia induzida pela sobrecarga de pressão (Young et al., 2001) e de diabetes mellitus
(Kiec-Wilk et al., 2005), respectivamente, enquanto outros demonstraram efeitos benéficos
na hipertrofia cardíaca induzida pela sobrecarga de pressão em ratos após tratamento com
fenofibrato, que é um indutor dos PPAR (Ogata et al., 2002). Os resultados contraditórios
podem dever-se às diferenças existentes entre espécies, à duração das intervenções, à
idade dos animais e aos modelos experimentais.
Outra característica da remodelagem metabólica que acompanha a progressão da IC é
o desenvolvimento de insulino-resistência, mesmo em pacientes não diabéticos (Mamas et
al., 2010). O estudo RESOLV demonstrou que a insulino-resistência se correlaciona com a
severidade da IC (Suskin et al., 2000) e com os níveis plasmáticos de peptídeo natriurético
do tipo B (BNP) (Tassone et al., 2009). Para além disso, a insulino-resistência e as
alterações no metabolismo da glicose são preditores independentes de mortalidade
(Doehner et al., 2005). Contudo, ainda não estão definidos os mecanismos responsáveis
pela associação da insulino-resistência à IC ou o modo como cada uma destas condições
predispõe à outra. Nos cardiomiócitos, a insulina regula tanto o metabolismo dos carbo-
hidratos como o dos AG, na insulino-resistência, a entrada de glicose no miocárdio, através
do transportador de glicose do tipo 4 (GLUT-4), está comprometida e, consequentemente
também a sua via metabólica. Além do mais, os efeitos periféricos da insulino-resistência

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
14
levam ao aumento dos níveis de AG na circulação reforçando as vias metabólicas dos AG
e, suprimindo adicionalmente o metabolismo de glicose. Estas modificações parecem
associar-se a menor eficiência cardíaca e maior stress metabólico (Witteles e Fowler,
2008). De facto, o excesso de AG induz lipotoxicidade no miocárdio, maior formação de
radicais livres de oxigénio (ROS), contribuindo para o desenvolvimento da cardiomiopatia
(Pillutla et al., 2005).
1.14. Nutrição na IC e caquexia
O papel da nutrição como factor de risco cardiovascular está bem estabelecido. A
American Heart Association recomenda a redução do total de calorias ingeridas, a redução
da ingestão de gorduras, especialmente saturadas e colesterol, bem como o aumento da
ingestão de legumes, frutas e produtos lácteos com baixo teor em gordura (Azhar e Wei,
2006; Krauss et al., 2000). A avaliação clínica de períodos longos de restrição calórica em
pacientes não-obesos corrobora estas recomendações (Meyer et al., 2006). No entanto,
estas recomendações podem não se aplicar à IC grave, associada a perturbações do
metabolismo cardiomiocitário e caquexia.
Embora as dietas hipercalóricas, do tipo ocidental, sejam um factor de risco bem
estabelecido para o desenvolvimento de doença cardiovascular, paradoxalmente, estudos
epidemiológicos demonstraram que a obesidade é um factor de prognóstico favorável na IC
(Abel et al., 2008). Efectivamente, pacientes com IC que apresentem simultaneamente
excesso ponderal ou obesidade têm menor mortalidade (Oreopoulus et al., 2008) e o
suporte nutricional pode contrariar a anorexia das doenças crónicas e prevenir a caquexia
(Laviano et al., 2005). No entanto, a suplementação nutricional não pode reverter
completamente o processo de perda ponderal que acompanha a caquexia, porque a
anorexia, por si só, não explica toda a complexidade fisiopatológica da caquexia cardíaca.
A investigação experimental e clínica foca cada vez mais o papel que a nutrição pode
desempenhar na progressão da IC e caquexia (Akner e Cederholm, 2001; Sandek et al.,
2008). E as mais diversas abordagens têm sido testadas (Azhar e Wei, 2006).

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
15
2. Objectivos
O principal objectivo dos trabalhos levados a cabo foi testar os efeitos de um regime
alimentar hipercalórico, próximo da típica dieta Ocidental (rico em gordura animal e carbo-
hidratos simples) na sobrevida, no grau de HP, na função e remodelagem do miocárdio, na
IC e caquexia que acompanham o modelo experimental de HP severa, e rapidamente
progressiva, induzida pela MCT. Como objectivo adicional procuramos estudar os
mecanismos moleculares inerentes à remodelagem metabólica, às alterações
morfofuncionais do miocárdio e à caquexia que acompanham este modelo, bem como os
mecanismos pelos quais o regime alimentar hipercalórico modificou a sua evolução.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
16
3. Metodologia
3.1. Modelos animais
Ratos Wistar, machos, com pesos corporais compreendidos entre 180 - 200 g e cerca
de 6 - 7 semanas de idade (Charles-River Barcelona, Espanha; n = 192), receberam
aleatoriamente uma única injecção subcutânea de 60 mg.kg-1 de MCT (Sigma Chemical, St.
Louis, MO) ou igual volume de veículo (soro fisiológico a pH neutro). Quarenta e oito horas
depois, os dois grupos passaram, aleatoriamente, a ser alimentados com uma dieta
hipercalórica, rica em lípidos e carbo-hidratos simples (F2685; BioServe Frenchtown; 5,4
Kcal.g-1, 35% de gordura animal, 35% de carbo-hidratos simples, 20% de proteínas e 0,4%
Na+) ou mantiveram uma dieta normal, de composição padrão (A04; Scientific Animal
Food&Engineering; 2,9 Kcal.g-1, 3% de gordura não-animal, 60% de carbo-hidratos
complexos, 16% de proteínas e 0,25% Na+). Registou-se diariamente o peso corporal, a
ingestão alimentar e a mortalidade. A insulino-resistência e a tolerância à glicose oral foram
avaliadas 20 - 23 dias após injecção. Os ratos foram colocados em gaiolas metabólicas
durante 24 horas, 24 dias após injecção. A avaliação hemodinâmica e a colheita de
amostras foram realizadas após um período de 24 horas com ração regular, dos 28 aos 32
dias. Os animais foram alojados em grupos de 5 por caixa, em ambiente controlado (ciclo
luminosidade-obscuridade 12:12 horas, a 22 ºC e dieta ad libitum). Foram respeitadas a lei
Portuguesa para o bem-estar animal e o National Institutes of Health Guide for the Care and
Use of Laboratory Animals (NIH Pub. No. 85 - 23, revista em 1996).
A avaliação metabólica, hemodinâmica e histológica padrão podem ser consultadas no
trabalho original submetido para publicação, que consta na secção resultados desta tese. A
avaliação estatística foi efectuada com auxílio de software (SigmaStat 3.5, Systat software,
Inc.) e pode ser consultada na mesma secção. Durante a realização dos trabalhos
experimentais, tive a oportunidade de aprender e executar as técnicas de biologia
molecular que foram empregues neste trabalho, assim como aprender e contribuir para a
análise e interpretação de dados e redacção do mesmo. Em seguida será apresentada de
forma detalhada a metodologia de biologia molecular.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
17
3.2. Estudos de biologia molecular
3.2.1. Quantificação de DNA e conteúdo protéico do miocárdio do VD e VE
Foram analisados 10 mg das amostras de miocárdio do VD e do VE, provenientes de
sete animais de cada grupo, para quantificar DNA genómico e proteína total. As amostras
biológicas foram tratadas com uma solução tampão de lise composta por tiocianato de
guanidina, inactivando, deste modo, DNases, RNases e proteases e garantindo, portanto, o
isolamento de DNA, RNA e proteínas intactas. As amostras foram, então, homogeneizadas
com um rotor (PRO 200, Cat. No. 01 - 02200, Oxford, CT, USA), durante cerca de 30
segundos, para assegurar uma quantidade significativa de ácidos nucléicos livres. O
restante processo de extracção foi levado a cabo de acordo com as instruções do
fabricante por eluição sequencial em colunas (AllPrep® DNA/RNA/Protein Mini Kit, Cat. No.
80004, Qiagen, København, Dinamarca).
A concentração total de proteína foi testada pelo método do ácido bicinconínico
(Pierce® BCA Protein Assay Kit-Reducing Agent Compatible, Cat. No. 23250, Rockford, IL
USA). O ensaio consiste na redução de Cu+2 a Cu+ por proteínas, em meio alcalino,
apresentando elevada sensibilidade e especificidade na detecção e na quantificação
colorimétrica de proteínas. O efeito de redutores de cobre foi minimizado através de um
reagente de compatibilidade que altera os grupos dissulfito dos agentes redutores. A
absorvância das amostras foi quantificada por espectrofotometria com um comprimento de
onda de 562 nm e comparada com uma curva padrão obtida por diluição seriada de
padrões de albumina do soro bovino (µg/mL).
A concentração de DNA foi determinada por espectrofotometria, medindo a absorvância
a 260 nm (Eppendorf 6131000.012) após diluição das amostras numa solução tampão de
Tris-Cl com pH neutro. A pureza foi estimada pela razão das absorvâncias a 260 nm e 280
nm (A260/A280). O DNA puro tem uma razão A260/A280 de 1,7 - 1,9. Tanto o DNA como o
conteúdo protéico foram normalizados para a massa inicial do tecido do miocárdio.
3.2.2. Quantificação de mRNA
As amostras biológicas foram colhidas imediatamente após a eutanásia dos animais,
colocadas em reagente de estabilização de RNA (RNAlater) após fragmentação em
dimensões inferiores a 1 mm, e armazenadas, então, a -80 ºC. A extracção de RNA foi
efectuada a partir de 30 mg de tecido de cada amostra, por homogeneização em tampão de
lise com um rotor (PRO 200, Cat. No. 01 - 02200, Oxford, CT, USA), e eluição sequencial
em colunas utilizando diversos tampões de acordo com o método de guanidina-tiocianato e

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
18
ligação selectiva a uma membrana de gel de sílica seguindo as instruções do fabricante
(RNeasy® Mini Kit, Cat. No. 74124, Qiagen). Este método exclui RNA com extensão inferior
a 200 nucleótidos, como o rRNA e o tRNA, que em conjunto correspondem a 15 - 20% do
RNA total. A concentração e a pureza do RNA foram avaliadas por espectrofotometria
(Eppendorf 6131000.012), admitindo como RNA puro amostras com razões entre as
absorvâncias a 260 e 280 nm de 1,9 a 2,1, após diluição em tampão de Tris-Cl com pH de
7,5.
A quantificação de mRNA foi realizada, nas amostras de VD, VE e gordura mesentérica
provenientes de 7 animais de cada grupo, através da técnica RT-PCR em tempo real. No
miocárdio, avaliou-se a expressão relativa da ET-1, das citocinas TNF-α e IL-6, do PPAR-α
e das enzimas reguladoras do metabolismo, da PDK do tipo 4 (PDK4), da Acil-CoA ligase
de AG de cadeia longa tipo 1 (ACSL1), da desidrogenase de acil-CoA de cadeia média
(MCAD) e de cadeia longa (LCAD). A β-actina foi quantificada como gene de controlo
interno. Na gordura visceral quantificamos o TNF-α, a IL-6 e a adipocina adiponectina,
usando o desidrogenase do gliceraldeído-3-fosfato como gene de controlo interno, uma vez
que para a β-actina foram observadas diferenças significativas entre os grupos. Os pares
de primers específicos para o estudo destes genes foram desenhados com recurso a
software (DNAstar™, Lasergene) e são indicados no trabalho apresentado na secção
resultados, atendendo às seguintes características: comprimento dos primers até 18 - 21
nucleótidos; comprimento da sequência do produto de PCR com 100 - 150 pares de base;
conteúdo de guaninas e citosinas de 40 - 60%; temperatura de fusão, ou melting (Tm), do
transcrito 8 ºC superior à dos primers e temperaturas de fusão dos primers não diferindo
mais de 2 ºC; e, finalmente, escolha preferencial de sequências de primers intron spanning
para amplificação selectiva de RNA, ou, em alternativa, caso isto não seja possível, intron
flanking que possibilitam diferenciar amplificação de RNA da de DNA (caso seja transcrito
tem maior dimensão, uma vez que inclui uma sequência de um intrão).
A transcrição reversa foi realizada seguindo a metodologia de random primers. Num
volume total de reacção de 20 µL: 40 U/reacção (0,3 µL) de transcriptase reversa
(Superscript™ II, Invitrogen 18064 - 014), 20 U/reacção (0,5 µL) do inibidor de Rnases
(Promega N2515), 30 ng de random primes (Invitrogen 48190 - 011), solução tampão (50
mM Tris-HCl, 75 mM KCl; 3 mM MgCl2), 10 mM de ditiotreitol e 0,5 mM de
desoxiribonucleotídeos trifosfatados (MBI Fermentas R0192). A transcrição foi efectuada
num termociclador (Whatman Biometra 050 - 901) seguindo os seguintes passos: 10
minutos a 22 ºC para desnaturação do RNA, 50 minutos a 42 ºC para a transcrição reversa
e 10 minutos a 95 ºC para inactivação da enzima e destruição dos produtos de reacção. O

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
19
cDNA resultante foi armazenado a -20 ºC e posteriormente amplificado por PCR em tempo
real. A reacção de amplificação génica processou-se num volume final de 20 µL, com 500
nM de cada primer, mix com nucleotídeos, uma hot-start DNA polimerase e SYBR green
como marcador fluorescente de acordo com as instruções do fabricante
(QuantiTect™SYBR® Green PCR Kit, Cat. No. 204143, Qiagen). A reacção decorreu em
capilares de acordo com as instruções do fabricante (Lightcycler II, Roche), brevemente,
após 15 minutos a 95 ºC para activação da polimerase, sucederam-se 35 - 55 ciclos de 15
segundos a 94 ºC para a desnaturação, 20 - 30 segundos a 50 ºC para annealing entre
primers e DNA e 10 - 30 segundos a 72 ºC para extensão.
Criaram-se curvas padrão fazendo diluições seriadas (2,5; 5; 12,5; 25; 50; 100; 250 ng)
do RNA de uma amostra de um animal do grupo controlo. A transcrição reversa foi
efectuada, numa experiência única, incluindo a curva padrão e 25 ng de RNA de cada
amostra. Subsequentemente, os PCRs foram efectuados para cada gene, incluindo todas
as amostras e usando como referência a curva padrão. O SYBR green liga-se ao DNA de
dupla cadeia emitindo fluorescência, durante a amplificação exponencial para cada amostra
e padrão, o valor máximo da segunda derivada do traçado de fluorescência (início da fase
exponencial de amplificação) foi obtido automaticamente pelo software (Lightcycler II,
Roche). As experiências foram efectuadas em duplicado.
No final do PCR, para cada gene, o DNA foi desnaturado, por aquecimento lento de 65
a 95 ºC, e a fluorescência adquirida continuamente, por forma a obter traçados de melting,
assegurando a pureza do produto amplificado e a ausência de dímeros de primers. Para
uma amostra de cada gene correu-se uma electroforese com o DNA amplificado, num gel
de agarose a 2% corado com brometo de etídio (0,5 µg/mL) para reconfirmar a pureza do
produto de amplificação. A expressão génica foi normalizada para a expressão do gene de
controlo interno, que não variou entre os grupos experimentais. Os resultados são
apresentados relativamente ao valor médio do grupo injectado com veículo e alimentado
com dieta normal, que foi definido como unidade arbitrária.
3.2.3. Quantificação das isoformas das cadeias pesadas de miosina
A comutação de isoformas das cadeias pesadas de miosina do tipo α para o tipo β é
um traço característico da remodelagem do miocárdio que ocorre na progressão da IC
(Mann e Bristow, 2005). Em amostras de VD e VE, de seis animais de cada grupo
experimental, foram extraídas proteínas por lise tecidual. Posteriormente, 15 µg de proteína
total, diluídas 1:1 em tampão de Laemmli num volume final de 20 µL, e desnaturadas por
agitação durante 5 minutos a 95 ºC, foram separados por electroforese em gel de

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
20
poliacrilamida e dodecil sulfato de sódio (SDS-PAGE), juntamente com um marcador de
peso molecular. Resumidamente, prepararam-se dois géis, de separação (5% de
Acrilamida/Bisacrilamida) e de concentração (3% de Acrilamida/Bisacrilamida), preencheu-
se o sistema de electroforese com solução tampão de separação e a electroforese decorreu
a voltagem constante de 30 mA, durante 6 horas (PowerPac 200, Cat. No. 165 - 3301, BIO-
RAD). As isoformas das cadeias pesadas de miosina α e β visualizaram-se, no final, como
duas bandas distintas com um peso molecular aproximado de 200 KDa. Os géis foram
então corados, durante 15 - 20 minutos, com uma solução de azul brilhante de Coomassie
R-250. O gel foi digitalizado a 700 nm num sistema de aquisição de imagem (Odyssey, LI-
COR Biosciences, Lincoln, NE, USA). A percentagem da isoforma β, para cada amostra, foi
quantificada a partir da razão entre as absorvâncias da isoforma β e a total.
3.2.4. Quantificação do TNF-α no plasma
O plasma colhido no final da avaliação hemodinâmica em 7 animais por grupo, foi
utilizado para quantificar a concentração plasmática de TNF-α através de um imunoensaio
enzimático do tipo sandwich em fase sólida. Brevemente, neste ensaio altamente sensível,
que possibilita a detecção de concentrações mínimas de TNF-α (0,7 pg/mL), as amostras,
juntamente com a curva padrão e os controlos positivo e negativo, foram incubados com
anticorpos monoclonais específicos do TNF-α que revestem as microplacas (TNF-α (Rat)
Ultrasensitive EIA, 45-TNFRTU-E01, Alpco Diagnostics™, Salem, NH). Posteriormente,
adicionou-se sequencialmente anticorpo secundário biotinilado, estreptavidina-peroxidase
de rábano (HRP) e uma solução de substrato, o corante TMB, com passos de lavagem
intermédios. Após a reacção o substrato cromogénio amarelado muda de cor, para azul,
após oxidação, sendo adicionada solução de bloqueio. A intensidade da cor produzida é
directamente proporcional à concentração de TNF-α presente na amostra, de acordo com a
curva padrão. As absorvâncias no comprimento de onda de 450 nm (UVM-340
Monochromator based reader, ASYS Hitech GmbH, Eugendorf, Áustria) foram analisadas
(Microplate instrument analyser, versão 1.2.0.2). Todas as avaliações foram efectuadas em
duplicado.
3.2.5. Apoptose miocárdica
A morte celular programada, ou apoptose, é um dos mecanismos que acompanha a
remodelagem miocárdica na progressão da IC, determinando concomitantemente o
comprometimento funcional pela perda de unidades contrácteis. Na base desta, encontra-
se a degradação do DNA genómico por endonucleases específicas dependentes de cálcio.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
21
O grau de apoptose miocárdica VD e VE foi avaliada por histoquímica, em secções
histológicas de 7 animais de cada grupo. Empregou-se a metodologia enzimática de
marcação com fluoresceína dos terminais desoxiuridina trifosfato pela desoxinucleotidil
transferase (TUNEL, terminal UDP nick-end labeling), segundo as instruções do fabricante
(TACS™ TdT In Situ Apoptosis Detection Kit, R&D Systems, Minneapolis, MN USA).
Sumariamente, cinco secções histológicas intervaladas de 100 µm foram obtidas
aleatoriamente e desparafinadas, a 57 ºC, num aquecedor de placas. As amostras de
tecido foram então fixadas, para evitar a perda de fragmentos de DNA de baixo peso
molecular, procedeu-se à rehidratação das amostras e, finalmente, adicionou-se proteinase
K para permeabilizar os tecidos. As lâminas histológicas foram, então, imergidas em
solução supressora da actividade peroxidásica e marcadas com solução tampão de
marcação. Após bloqueio da reacção com uma solução tampão, as lâminas são coradas
com HRP e marcador azul TACS. O resultado final foi um precipitado escuro insolúvel que
assinala zonas em que ocorreu fragmentação de DNA. As lâminas são, então, contra-
coradas com hematoxilina. Numa das secções adicionou-se TACS-Nuclease™ por forma a
gerar fragmentação do DNA na maioria das células, confirmando, como controlo positivo,
se a permeabilização e marcação decorreram adequadamente. Enquanto que, noutra
secção, não foi adicionado tampão de marcação
Dois observadores independentes, fizeram a contagem dos núcleos de cardiomiócitos
marcados, em 50 campos escolhidos ao acaso em cada secção histológica (400 x). A taxa
de apoptose foi expressa como percentagem de núcleos de cardiomiócitos apoptóticos pelo
número total de cardiomiócitos.
3.2.6. Quantificação do NF-κB e do PPAR-α
Procedeu-se à extracção de proteínas nucleares do miocárdio VD e do VE de 7 animais
por cada grupo experimental, segundo as instruções do fabricante (Nuclear Extraction Kit,
Cat. No. 11906 - 100, Marligen Biosciences, Inc., Ijamsville, MD, USA). Os extractos
nucleares foram quantificados pelo método de Bradford, com leitura de absorvâncias a 595
nm por espectrofotometria (Eppendorf 6131000.012). Nos extractos quantificou-se a
actividade dos factores de transcrição NF-κB e PPAR-α, de acordo com as instruções do
fabricante (NF-κB (p65) Transcription Factor Assay Kit e PPAR-α Transcription Factor
Assay Kit, Cat. No. 1007889 e Cat. No. 10006915, Cayman Chemical Company, Ann
Harbor, MI, USA, respectivamente). Brevemente, 20 µL de proteínas nucleares de cada
amostra foram adicionados aos poços de microplacas revestidos com sequências
específicas de DNA de dupla cadeia, o elemento de resposta NF-κB e o PPRE,

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
22
adicionando-se, de seguida, um anticorpo primário específico do NF-κB e do PPAR-α,
respectivamente (Cat. No. 10007442, Cayman Chemical Company, Ann Harbor, MI, USA).
Após incubação a 4 ºC, durante a noite, adicionou-se um anticorpo secundário conjugado
com um marcador infravermelho (IRDye 800CW Goat Anti-Rabbit Labeled Secondary
Antibody, Cat. No. 926 - 32211, Li-COR biosciences, Lincoln, NE, USA), diluído em solução
de bloqueio (1:1000) (Odyssey Blocking Buffer, Cat. No. 927 - 40000, Li-COR biosciences,
Lincoln, NE, USA) com 0,2% de Tween-20. A absorvância no comprimento de onda de 800
nm foi, então, lida num sistema de aquisição de imagem (Odyssey, LI-COR Biosciences,
Lincoln, NE, USA). As amostras foram avaliadas em duplicado, em conjunto com controlos
positivo e negativo, e um controlo em que se adicionou DNA de dupla cadeia que competiu
com o factor de transcrição para a ligação, por forma a confirmar a especificidade da
ligação.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
23
4. Resultados
Experimental pulmonary hypertension with heart failure and cardiac cachexia
is attenuated by a high-fat high-simple carbohydrate diet
(A hipertensão pulmonar experimental associada à insuficiência cardíaca e
caquexia é atenuada pela dieta rica em gorduras e carbo-hidratos simples)
Artigo em revisão na revista American Journal of Physiology Heart and Circulatory Physiology,
submetido pelos autores Lourenço AP, Vasques-Nóvoa F, Fontoura D, Brás-Silva C, Roncon-
Albuquerque R, Jr., e Leite-Moreira AF, em Setembro de 2010.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
24
Experimental pulmonary hypertension with heart failure and cardiac cachexia
is attenuated by a high-fat high-simple carbohydrate diet
André P. Lourenço, Francisco Vasques-Nóvoa, Dulce Fontoura, Carmen Brás-Silva,
Roberto Roncon-Albuquerque Jr, Adelino F. Leite-Moreira
Department of Physiology, Faculty of Medicine, University of Porto, Porto, Portugal
Running Head: Hypercaloric diet attenuates cardiac cachexia
Keywords: hypercaloric diet; cytokines; inflammation; neuroendocrine; myocardial function
Correspondence to:
Adelino F. Leite-Moreira, MD PhD
Department of Physiology
Faculty of Medicine
Al Prof. Hernâni Monteiro
4200 - 319
Porto, Portugal
Tel: +351225513644
Fax: +351225513646
Email: [email protected]

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
25
Abstract
To evaluate the repercussions of a hypercaloric diet in cardiac cachexia (CC) and heart
failure (HF), male Wistar rats randomly underwent (i) subcutaneous injection of 60 mg.Kg-1
monocrotaline (MCT) or vehicle (Ctrl) and (ii) feeding with either a 5.4 kcal.g-1, 35% simple
carbohydrate and 35% animal fat (hypercaloric diet, HD), or a 2.9 Kcal.g-1, 60% complex
carbohydrate and 3% vegetable fat (normal diet, ND). Calorie intake, weight gain and
mortality were recorded. Metabolic studies, insulin resistance and oral glucose tolerance
testing were performed. Five weeks after injection, we evaluated right (RV) and left
ventricular (LV) haemodynamics under general anesthesia. Morphometry, lung and
myocardial histology, plasma levels of tumor necrosis factor-α (TNF-α) as well as
myocardial apoptosis, nuclear factor κ-B (NF-κB) and peroxisome proliferator-activated
receptor-α (PPAR-α) transcription factor activation, and gene expression of endothelin-1
(ET-1), interleukin-6 (IL-6), TNF-α and metabolic enzymes were also assessed. MCT ND
showed pulmonary hypertension (PH) and RV and LV failure, associated with high mortality,
myocardial remodeling and apoptosis, reduced calorie intake, and loss of body weight, lean
and fat mass that were all attenuated in MCT HD. PH was accompanied by increased
myocardial NF-κB transcription factor activity and by ET-1, TNF-α and IL-6 overexpression,
as well as by increased circulating TNF-α, which were offset in MCT HD. PPAR-α activity,
however, was decreased in MCT ND but not in MCT HD. The hypercaloric diet attenuated
experimental PH associated with CC and HF, improving survival, myocardial function and
remodeling, and reduced inflammatory and neuroendocrine activities.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
26
Introduction
Cardiac cachexia (CC), defined by a weight loss of more than 6% over 6 months (Anker et
al., 2003), accompanies heart failure (HF) in up to 50% of severe HF cases (Filippatos et al.,
2000) and independently determines a poor prognosis (Anker et al., 1997). As part of the
complex and still incompletely revealed mechanisms underlying CC are abnormalities of
general metabolism as well as of the immune and neuroendocrine systems (Anker and
Coats 1999). Besides increased basal metabolism, reduced appetite and gastrointestinal
derangements that compromise caloric intake (von Haehling et al., 2007), the failing heart
undergoes extensive and early metabolic changes, namely a myocardial shift from fatty acid
oxidation (FAO) to glycolysis and reduced mitochondrial oxidative capacity that partly
underlie functional disturbances and remodelling (Stanley et al., 2005). Caloric restriction
has been shown to reduce cardiovascular risk (Fontana et al., 2004) and improve overall
metabolism and organ function (Chou et al., 2010). American Heart Association nutritional
guidelines recommend avoiding simple carbohydrates and fat, particularly saturated, as
main energy sources in a balanced diet (Krauss et al., 2000). This may not apply however to
severe HF with disturbed cardiomyocyte metabolism and CC. Among the experimental
models of CC, monocrotaline (MCT) - induced pulmonary hypertension (PH) stands out by
rapidly progressive HF and cachexia (Dalla Libera et al., 2004, Steffen et al., 2008). Our
goal was to test the effects of a high-fat high-simple carbohydrate diet on survival, PH,
myocardial function and remodeling, neuroendocrine and inflammatory activities, and
cachexia, in severe MCT-induced PH.
Materials and Methods
Animal model
Seven-week old 180 - 200 g male Wistar rats (Charles-River, Barcelona, Spain; n = 192)
randomly received either a 60 mg.Kg-1 MCT (Sigma Chemical, St. Louis, MO) single
subcutaneous injection or an equal volume of vehicle. Both groups were randomly allocated,
48 h later, to be fed ad libitum with either a high-fat high-simple carbohydrate diet (F2685;
BioServe Frenchtown; 5.4 kcal.g-1, 35% animal fat, 35% simples carbohydrate, 20% protein,
and 0.4% Na+) or a regular diet (A04; Scientific Animal Food&Engineering; 2.9 Kcal.g-1, 3%
non-animal fat, 60% complex carbohydrate, 16% protein, and 0.25% Na+). Body weight
(BW), food ingestion and mortality were recorded daily (n = 136 for survival analysis). Insulin
resistance (IR) and oral glucose tolerance (OGT) were evaluated 20 - 23 days after

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
27
injection. Rats were then placed in metabolic cages for 24 h at the 24th day. Hemodynamic
evaluation and sample collection were carried out after 24 h of regular chow, from the 28th to
the 32nd day. Animals were housed in groups of 5 per cage with a controlled environment
under a 12:12 h light-dark cycle at a room temperature of 22 °C. Investigation conformed to
the Guide for the Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH Publication No. 85 - 23, revised 1996) and was approved by the
ethics committee of the Faculty of Medicine of the University of Porto. Methods are
presented in detail as supplemental data.
Metabolic studies
In 7 animals per group, IR and OGT were evaluated, after a 12 h fasting period, by
recording baseline, 15, 30, 45, 60, 90, and 120 min plasma glucose levels (Freestyle-Mini™)
after 0.5 U.Kg-1 intraperitoneal (ip) insulin injection and 1 g.Kg-1 glucose gavage,
respectively. Weight gain, diuresis, and calorie and water intake were measured during the
24 h metabolic cage stay (Techniplast, Buguggiate, Italy).
Haemodynamic evaluation
Briefly, 7 animals of each experimental group were sedated with 100 µg.Kg-1 ip fentanyl (50
µg.mL-1) and 5 mg.Kg-1 ip midazolam (5 mg.mL-1) and anaesthetized by inhalation of 8%
sevoflurane (Penlon Sigma Delta Anaesthetic Vaporizer). After endotracheal intubation and
mechanical ventilation, with respiratory rate set at 130.min-1, tidal volume adjusted for body
weight according to the manufacturer’s instructions (Harvard rodent ventilator model 683,
Harvard Apparatus, Holliston, MA) and positive end-expiratory pressure held at 4 cmH2O,
anaesthesia was maintained with 2.5 - 3% sevoflurane. Rats were placed on a heating pad.
The electrocardiogram was monitored, body temperature was kept at 38 ºC and 8 mL.Kg-
1.h-1 intravenous warm lactated Ringer solution was infused (Multi-Phaser, NE-1000, New
Era Pump Systems, Wantagh, NY). A left thoracotomy was performed under surgical
microscopy with animals in right lateral semidecubitus. Silk threads were passed around the
inferior vena cava (IVC). Pressure-volume (P-V) catheters were positioned along the long
axes of the left ventricle (LV) and right ventricle (RV) (models SPR-838, 2F, and PVR-1045,
1F, Millar Instruments, Houston, TX, respectively). Signals were continuously acquired
(MPVS 300, Millar Instruments, Houston, TX) and digitally recorded at 1000 Hz (ML880
PowerLab 16/30, ADinstruments Pty Ltd., Australia). A flow probe (200-367, Triton
Technology, San Diego, CA) positioned around the ascending aorta allowed cardiac output
(CO) measurement with an ultrasonic flowmeter (Active Redirection Transit Time
Flowmeter, System 6, Triton Technology, San Diego, CA). After stabilization for 15 minutes,

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
28
baseline and IVC occlusion recordings were obtained, with ventilation suspended at end-
expiration. Parallel conductance was calibrated by injection of 50 µL of 10% saline. Data
was analysed with the aid of software (PVAN 3.5, Millar Instruments, Houston, TX). Blood
was collected and stored at -80 ºC for subsequent analysis. After euthanasia with
intravenous pentobarbital 100 mg.Kg-1, heparinized blood was collected for volume
calibration with standard cuvettes (Part No. 910 - 1048, Millar Instr., Houston, TX), and the
lungs, heart and mesenteric fat were weighted. After careful dissection the RV free-wall and
the LV free-wall plus interventricular septum weights were determined. The right upper lung
lobe, RV and LV free-walls, and mesenteric fat were snap frozen in liquid nitrogen and
stored at -80 ºC for molecular biology studies. The weights of the liver, kidneys,
gastrocnemius muscle, perirenal and perigonadal fat, and the length of the tibia were
measured at necropsy.
Histology
In 7 additional animals per group, lungs were inflated and fixed with tracheal instillation of
ethanol. Lung and heart were immersed in 10% formalin. Transverse 4 µm-thick sections of
paraffin-embedded lung and myocardium (encompassing the RV, interventricular septum
and LV) were stained with H&E and Masson’s trichrome and photographed with a digital
camera (Leica DFC320). Sections were evaluated by two independent blinded observers.
For the myocardium, the shortest diameter of 50 transversally cut, randomly selected
cardiomyocytes from the RV and LV free wall myocardium (400 x) was measured at the
level of the nucleus in H&E sections, with image analyzer software (Leica IM-1000). We also
measured the percentage of blue in Masson’s trichrome staining as indicative of fibrosis in
10 randomly selected RV and LV myocardial fields (400 x) using the NIH Image Program
(Image J). For the lung, medial hypertrophy of pulmonary arterioles was assessed in 10
arterioles from randomly selected fields (400 x) as a percentage of the radius, by obtaining 8
orthogonal measurements of inner diameter (distance between internal elastic laminae) and
outer diameters (distance between external elastic laminae).
Quantification of LV and RV myocardial DNA and protein content
Total DNA and protein were assessed in 10 mg of RV and LV free-wall myocardial samples
from 7 animals of each group. Briefly, after tissue homogenation with a rotor stator (PRO
200, Cat. No. 01 - 02200, Oxford, CT, USA) samples were extracted according to the
manufacturer’s instructions (AllPrep® DNA/RNA/Protein Mini Kit, Cat. No. 80004, Qiagen,
København, Danmark). DNA concentration was assayed by spectrophotometry at 260 nm
(Eppendorf 6131000.012). Protein was dissolved in 5% Sodium-dodecyl sulphate and

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
29
concentration assayed by the bicinchoninic acid method (Pierce®, BCA Protein Asssay Kit-
Reducing Agent Compatible, Cat. No. 23250, Rockford, IL USA). DNA and protein contents
were normalized for the initial mass of myocardial tissue.
mRNA quantification
Briefly, two-step real-time reverse transcription (RT)-polymerase chain reaction (PCR) was
performed in mesenteric fat and LV and RV free-wall samples extracted from 7 animals per
group by the guanidium-thiocyanate selective silica-gel membrane-binding method (no.
74124; Qiagen). Concentration and purity were assayed by spectrophotometry (Eppendorf
6131000.012). After RT on a thermocycler (Whatman Biometra 050-901), with 40 U/reaction
reverse transcriptase (Invitrogen 18064-014), 20 U/reaction RNase inhibitor (Promega
N2515) and 30 ng/µl random primers (Invitrogen 48190-011), 0.5 mM nucleotide mix (MBI
Fermentas R0192) for a final volume of 20 µL, real-time PCR was performed using SYBR
green (Qiagen 204143) as fluorescent marker, according to the manufacturer’s instructions
(Lightcycler II, Roche). Standard curves were obtained with specific primer pairs for
endothelin-1 (ET-1; fw: 5’ – CGG GGC TCT GTA GTC AAT GTG - 3’ and rev: 5’ – CCA
TGC AGA AAG GCG TAA AAG - 3’), tumor necrosis factor-α (TNF-α; fw: 5’ – TGG GCT
ACG GGC TTG TCA CTC - 3’ and rev: 5’ – GGG GGC CAC CAC GCT CTT C - 3’),
interleukin-6 (IL-6; fw: 5’ – GAA GTT GGG GTA GGA AGG AC - 3’ and rev: 5’ – CCG TTT
CTA CCT GGA GTT TG - 3’), peroxisome proliferator activated receptor-α (PPAR-α; fw: 5’ –
TTC CAG CCC CTC CTC AGT CAG - 3’ and rev: 5’ – AGC CCT TGC AGC CTT CAC AT -
3’), pyruvate-dehidrogenase kinase type 4 (PDK4; fw: 5’ – GAG GCC ACC GTC GTC TTG -
3’ and rev: 5’ – ACA GGC GTT GGA GCA GTG G - 3’), long-chain-fatty-acid-CoA ligase 1
(ACSL1; fw: 5’ – CTA CAG GCA ACC CCA AAG GAG - 3’ and rev: 5’ – GGG CGA GAG
GCA AGA AAG ATA - 3’), medium chain acyl CoA dehydrogenase (MCAD; fw: 5’ – ACG
ATA AAA GCG GGG AAT ACC - 3’ and rev: 5’ – AGG CCA AGA CCA CCA CAA CTC - 3’),
long chain acyl CoA dehydrogenase (LCAD; fw: 5’ – CAT TTT CCG GGA GAG TGT AA - 3’
and rev: 5’ – CTT GCC AGC TTT TTC CCA GAG - 3’), adiponectin (fw: 5’ – GGG CTA CGG
GCT GCT CTG A - 3’ and rev: 5’ – TAT GGG GAA GGG GAC AAC AAT G - 3’), β-actin (fw:
5’ – ATC TGG GTC ATC TTT TCA CGG TTG G - 3’ and rev: 5’ – GAT TTG GCA CCA CAC
TTT CTA CA - 3’) and glyceraldehyde-3-phosphate dehydrogenase(GAPDH; fw: 5’ – CCG
CCT GCT TCA CCA CCT TCT - 3’ and rev: 5’ – TGG CCT TCC GTG TTC CTA CCC - 3’).
Equal amounts of mRNA from every sample underwent experiments in duplicate.
Thresholds for amplification were determined automatically by the second derivative
maximum method. Results are presented after normalization for β-actin and GAPDH internal

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
30
control genes, in the myocardium and adipose tissue, respectively, and setting the mean
value of the Ctrl ND group as arbitrary unit (AU).
Myosin-heavy chain (MHC) isoforms
In RV and LV samples from 6 animals of each experimental group, 15 µg of total protein
were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (3 and 5%
stacking and running gels, respectively) at 30 mA for 6 h (no. 165-3301, BIO-RAD), allowing
a clear separation of α and β myosin heavy-chain isoform bands (200 KDa). After staining
with Comassie Brilliant Blue R-250 gels were scanned at 700 nm by an image acquisition
system (Odyssey; LI-COR Biosciences, Lincoln, NE, USA). The percentage of β-isoform
was assessed as the ratio of β isoform absorbance to total absorbance.
Plasma TNF-α quantification
Plasma concentration of TNF-α was determined by solid phase sandwich quantitative
enzyme immunoassay with specific antibodies pre-coated onto microplates (45-TNFRTU-
E01, Alpco Diagnostics™, Salem, NH). Samples from 7 animals per group were run in
duplicates along with a standard curve and controls. A secondary biotinylated antibody,
streptavidin-peroxidase and a substrate solution were sequentially added with intermediate
washing steps and reading was done at a 450 nm wavelength (UVM-340 monochromator
based reader, ASYS Hitech GmbH, Eugendorf, Austria) and analysed (Microplate
instrument analyser, version 1.2.0.2).
Myocardial apoptosis
The extent of RV and LV myocardial apoptosis was assessed by immunohistochemical
terminal deoxynucleotidyl-transferase-mediated dUTP nick end-labeling (TUNEL) of
histological sections from 7 animals of each group. Briefly, 5 random sections from each
animal, obtained at 100 µm intervals, were deparaffinised, immersed in phosphate-buffered
saline, permeabilized with proteinase K, quenched for endogenous peroxidase activity with
5.0% hydrogen peroxide, and sequentially incubated in terminal deoxynucleotidyl
transferase (TdT) labeling mix, blocking buffer and streptavidin-HRP. Sections were then
developed with TACS Blue Label and counterstained with Nucler Fast Red according to the
manufacturer’s instructions (TdT In Situ Apoptosis Detection Kit, R&D Systems,
Minneapolis, MN USA). Positive and negative controls consisted of treatment with TACS-
Nuclease™ before labelling and a TdT-free labeling mix, respectively. TUNEL-positive
cardiomyocytes were counted by two independent blinded observers in 50 random fields

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
31
(400 x) of each section and apoptotic rate was expressed as percentage of apoptotic
cardiomyocytes nuclei per total number of cardiomyocytes.
Nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) and PPAR-α
transcription factor assays
Nuclear protein extracts were prepared from frozen RV and LV free-wall myocardium
according to the manufacturer’s instructions (Cat. No. 11906 - 100, Marligen Biosciences,
Inc., Ijamsville, MD USA). Briefly, nuclear factor κ-light-chain-enhancer of activated B cells
(NF-κB) and peroxisome proliferator activated receptor-α (PPAR-α) activation was assayed
by adding 20 µg of nuclear protein sample to microplate wells with the specific dsDNA
response elements for NF-κB (p65) and PPAR-α immobilized onto the bottom (Cat. No.
1007889 and Cat. No. 10006915, Cayman Chemical Company, Ann Harbor, MI, USA,
respectively). Samples were run in duplicate and positive and negative controls were
included. After an overnight 4 ºC incubation, transcription factor binding was detected by
addition of a specific primary antibody (Cat. No. 10007442, Cayman Chemical Company,
Ann Harbor, MI, USA). Finally, infrared labelling was performed with an IRDye 800CW-
conjugated secondary antibody (Cat. No. 926 - 32211, Li-COR biosciences, Lincoln, NE,
USA) diluted (1:1000) in blocking buffer (Cat. no. 927 - 40000, Li-COR biosciences, Lincoln,
NE, USA) containing 0.2% Tween-20. Absorbance at 800 nm was read with an image
acquisition system (Odyssey; LI-COR Biosciences, Lincoln, NE, USA).
Statistical analysis
Data were analyzed by Kaplan-Meier survival analysis with Gehan-Breslow statistic, by two-
way repeated measures ANOVA for BW and caloric ingestion progression, and by two-way
ANOVA for all other purposes. Results are presented as mean ± SEM. Two-tailed P < 0.05
was considered significant.
Results
Survival, weight gain, caloric intake, body composition and metabolism
Mortality was decreased in MCT HD compared with MCT ND (Figure 1, Panel A). MCT ND
rats presented signs of overt HF, namely laboured breathing, ruffled hair, lethargy, ascites
and pleural effusion, while MCT HD appeared more active.
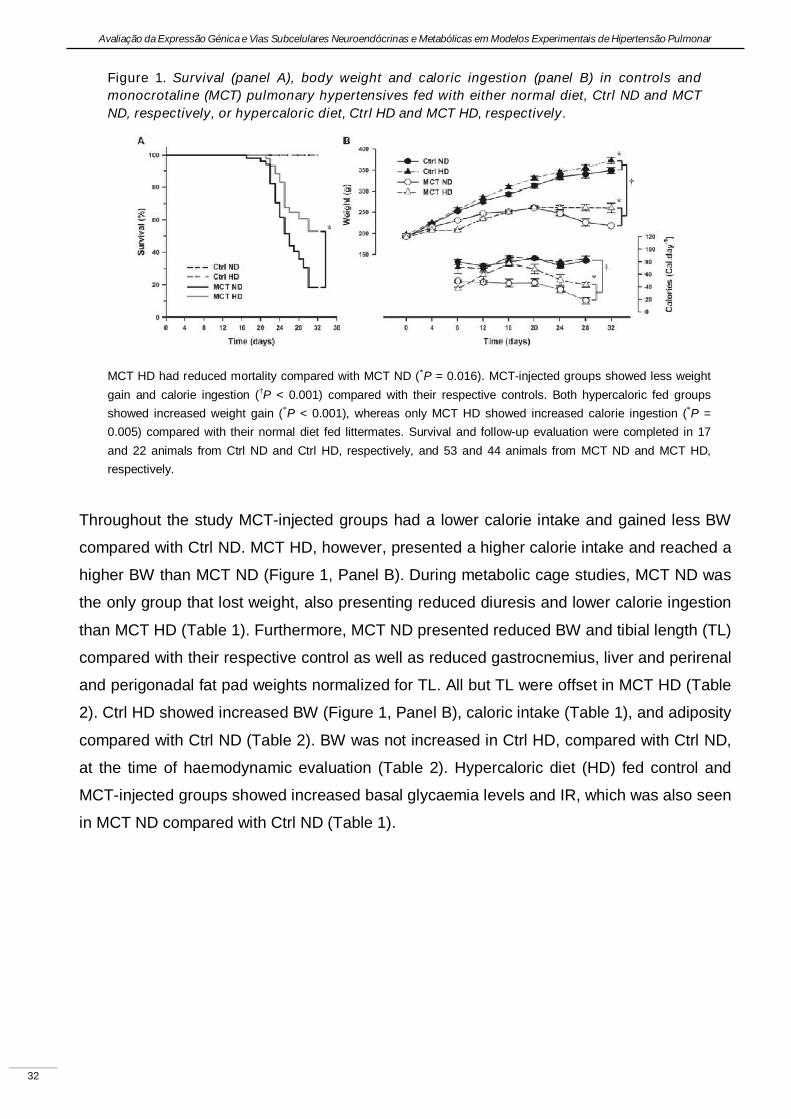
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
32
Figure 1. Survival (panel A), body weight and caloric ingestion (panel B) in controls and
monocrotaline (MCT) pulmonary hypertensives fed with either normal diet, Ctrl ND and MCT
ND, respectively, or hypercaloric diet, Ctrl HD and MCT HD, respectively.
MCT HD had reduced mortality compared with MCT ND (*P = 0.016). MCT-injected groups showed less weight
gain and calorie ingestion (†P < 0.001) compared with their respective controls. Both hypercaloric fed groups
showed increased weight gain (*P < 0.001), whereas only MCT HD showed increased calorie ingestion (*P =
0.005) compared with their normal diet fed littermates. Survival and follow-up evaluation were completed in 17
and 22 animals from Ctrl ND and Ctrl HD, respectively, and 53 and 44 animals from MCT ND and MCT HD,
respectively.
Throughout the study MCT-injected groups had a lower calorie intake and gained less BW
compared with Ctrl ND. MCT HD, however, presented a higher calorie intake and reached a
higher BW than MCT ND (Figure 1, Panel B). During metabolic cage studies, MCT ND was
the only group that lost weight, also presenting reduced diuresis and lower calorie ingestion
than MCT HD (Table 1). Furthermore, MCT ND presented reduced BW and tibial length (TL)
compared with their respective control as well as reduced gastrocnemius, liver and perirenal
and perigonadal fat pad weights normalized for TL. All but TL were offset in MCT HD (Table
2). Ctrl HD showed increased BW (Figure 1, Panel B), caloric intake (Table 1), and adiposity
compared with Ctrl ND (Table 2). BW was not increased in Ctrl HD, compared with Ctrl ND,
at the time of haemodynamic evaluation (Table 2). Hypercaloric diet (HD) fed control and
MCT-injected groups showed increased basal glycaemia levels and IR, which was also seen
in MCT ND compared with Ctrl ND (Table 1).

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
33
Table 1. Metabolic studies and glucose metabolism.
Ctrl ND Ctrl HD MCT ND MCT HD
Weight gain, % 2.48 ± 1.29 4.50 ± 1.18 -0.08 ± 1.36* 3.81 ± 0.73†
Caloric intake, cal.g-1 0.246 ± 0.009 0.370 ± 0.031† 0.224 ± 0.024 0.326 ± 0.036†
Diuresis, µL.g-1 35.0 ± 3.4 33.1 ± 4.8 19.5 ± 2.8* 24.3 ± 4.5
Glicemia, mg.dL-1 107 ± 4 121 ± 5† 121 ± 5* 134 ± 3*†
OGT AUC, mg.min 21009 ± 2232 22370 ± 2074 21117 ± 1146 23674 ± 1002
IR AUC, mg.min 9735 ± 798 16442 ± 568† 12519 ± 1240* 16232 ± 1123†
Data collected during 24 h metabolic cage studies, oral glucose tolerance (OGT) and insulin resistance tests
(IR) from monocrotaline-injected (MCT) and vehicle injected rats (Ctrl) fed a normal (ND) or a hypercaloric
diet (HD). AUC, area under curve. *P < 0.05 vs Ctrl; †P < 0.05 vs similar group fed ND; n = 7 in each group.
Table 2. Morphometry
Ctrl ND Ctrl HD MCT ND MCT HD
Weight, g 354 ± 6 361 ± 7 242 ± 7* 261 ± 7*†
Tibial length, cm 4.00 ± 0.03 3.93 ± 0.04 3.85 ± 0.02* 3.86 ± 0.04
Lung/tibia, mg.cm-1 383 ± 10 417 ± 12 650 ± 28* 520 ± 32*†
Gastrocnemius/tibia, mg.cm1 551 ± 14 513 ± 18 375 ± 21* 467 ± 10*†
Liver/tibia, mg.cm-1 3248 ± 186 3686 ± 328 2141 ± 125* 2844 ± 256*†
Perigonadal fat/tibia, mg.cm-1 804 ± 77 1155 ± 71† 473 ± 26* 633 ± 40*†
Perirenal fat/tibia, mg.cm-1 802 ± 77 1243 ± 158† 347 ± 45* 591 ± 36*†
Morphometric data from monocrotaline-injected rats fed either a normal (MCT ND) or a hypercaloric diet
(MCT HD) and their controls (Ctrl ND and Ctrl HD, respectively). *P < 0.05 vs Ctrl; †P < 0.05 vs ND; n = 7 in
each group.
Haemodynamics
Data are summarized in table 3.
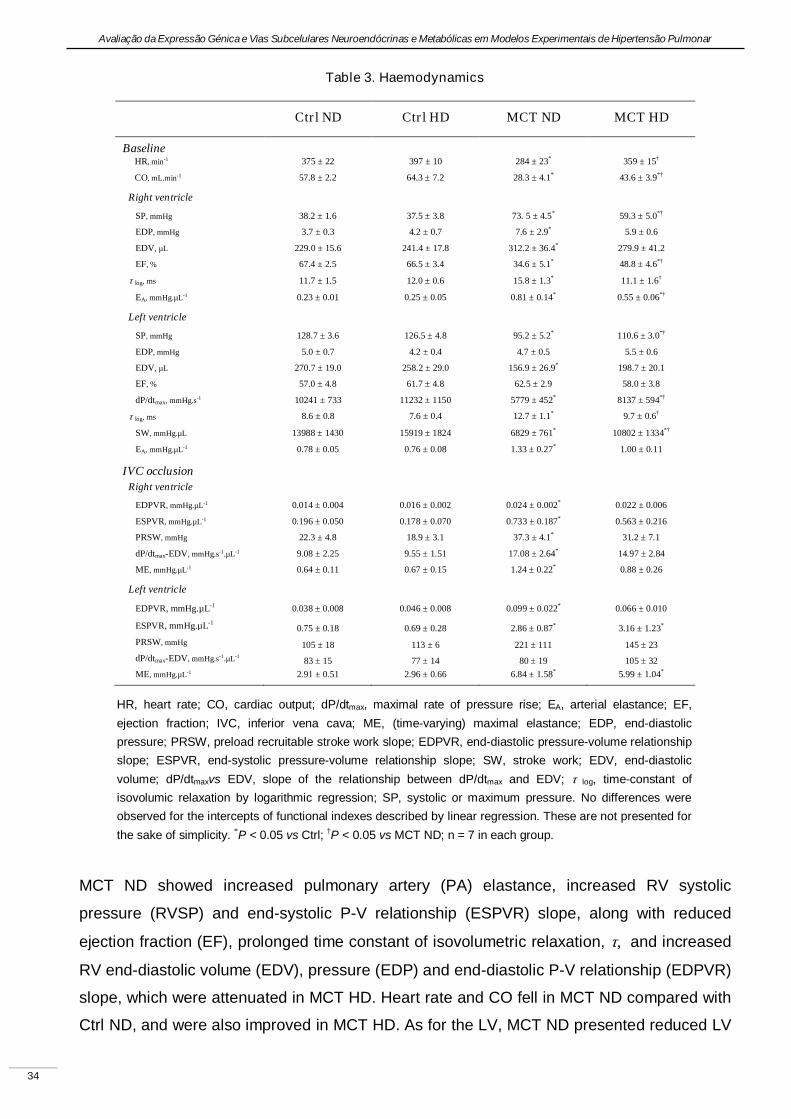
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
34
Table 3. Haemodynamics
Ctrl ND Ctrl HD MCT ND MCT HD
Baseline HR, min-1 375 ± 22 397 ± 10 284 ± 23* 359 ± 15†
CO, mL.min-1 57.8 ± 2.2 64.3 ± 7.2 28.3 ± 4.1* 43.6 ± 3.9*†
Right ventricle
SP, mmHg 38.2 ± 1.6 37.5 ± 3.8 73. 5 ± 4.5* 59.3 ± 5.0*†
EDP, mmHg 3.7 ± 0.3 4.2 ± 0.7 7.6 ± 2.9* 5.9 ± 0.6
EDV, µL 229.0 ± 15.6 241.4 ± 17.8 312.2 ± 36.4* 279.9 ± 41.2
EF, % 67.4 ± 2.5 66.5 ± 3.4 34.6 ± 5.1* 48.8 ± 4.6*†
τ log, ms 11.7 ± 1.5 12.0 ± 0.6 15.8 ± 1.3* 11.1 ± 1.6†
EA, mmHg.µL-1 0.23 ± 0.01 0.25 ± 0.05 0.81 ± 0.14* 0.55 ± 0.06*†
Left ventricle
SP, mmHg 128.7 ± 3.6 126.5 ± 4.8 95.2 ± 5.2* 110.6 ± 3.0*†
EDP, mmHg 5.0 ± 0.7 4.2 ± 0.4 4.7 ± 0.5 5.5 ± 0.6
EDV, µL 270.7 ± 19.0 258.2 ± 29.0 156.9 ± 26.9* 198.7 ± 20.1
EF, % 57.0 ± 4.8 61.7 ± 4.8 62.5 ± 2.9 58.0 ± 3.8
dP/dtmax, mmHg.s-1 10241 ± 733 11232 ± 1150 5779 ± 452* 8137 ± 594*†
τ log, ms 8.6 ± 0.8 7.6 ± 0.4 12.7 ± 1.1* 9.7 ± 0.6†
SW, mmHg.µL 13988 ± 1430 15919 ± 1824 6829 ± 761* 10802 ± 1334*†
EA, mmHg.µL-1 0.78 ± 0.05 0.76 ± 0.08 1.33 ± 0.27* 1.00 ± 0.11
IVC occlusion
Right ventricle
EDPVR, mmHg.µL-1 0.014 ± 0.004 0.016 ± 0.002 0.024 ± 0.002* 0.022 ± 0.006
ESPVR, mmHg.µL-1 0.196 ± 0.050 0.178 ± 0.070 0.733 ± 0.187* 0.563 ± 0.216
PRSW, mmHg 22.3 ± 4.8 18.9 ± 3.1 37.3 ± 4.1* 31.2 ± 7.1
dP/dtmax-EDV, mmHg.s-1.µL-1 9.08 ± 2.25 9.55 ± 1.51 17.08 ± 2.64* 14.97 ± 2.84
ME, mmHg.µL-1 0.64 ± 0.11 0.67 ± 0.15 1.24 ± 0.22* 0.88 ± 0.26
Left ventricle
EDPVR, mmHg.µL-1 0.038 ± 0.008 0.046 ± 0.008 0.099 ± 0.022* 0.066 ± 0.010
ESPVR, mmHg.µL-1 0.75 ± 0.18 0.69 ± 0.28 2.86 ± 0.87* 3.16 ± 1.23*
PRSW, mmHg 105 ± 18 113 ± 6 221 ± 111 145 ± 23
dP/dtmax-EDV, mmHg.s-1.µL-1 83 ± 15 77 ± 14 80 ± 19 105 ± 32
ME, mmHg.µL-1 2.91 ± 0.51 2.96 ± 0.66 6.84 ± 1.58* 5.99 ± 1.04*
HR, heart rate; CO, cardiac output; dP/dtmax, maximal rate of pressure rise; EA, arterial elastance; EF,
ejection fraction; IVC, inferior vena cava; ME, (time-varying) maximal elastance; EDP, end-diastolic
pressure; PRSW, preload recruitable stroke work slope; EDPVR, end-diastolic pressure-volume relationship
slope; ESPVR, end-systolic pressure-volume relationship slope; SW, stroke work; EDV, end-diastolic
volume; dP/dtmaxvs EDV, slope of the relationship between dP/dtmax and EDV; τ log, time-constant of
isovolumic relaxation by logarithmic regression; SP, systolic or maximum pressure. No differences were
observed for the intercepts of functional indexes described by linear regression. These are not presented for
the sake of simplicity. *P < 0.05 vs Ctrl; †P < 0.05 vs MCT ND; n = 7 in each group.
MCT ND showed increased pulmonary artery (PA) elastance, increased RV systolic
pressure (RVSP) and end-systolic P-V relationship (ESPVR) slope, along with reduced
ejection fraction (EF), prolonged time constant of isovolumetric relaxation, τ, and increased
RV end-diastolic volume (EDV), pressure (EDP) and end-diastolic P-V relationship (EDPVR)
slope, which were attenuated in MCT HD. Heart rate and CO fell in MCT ND compared with
Ctrl ND, and were also improved in MCT HD. As for the LV, MCT ND presented reduced LV
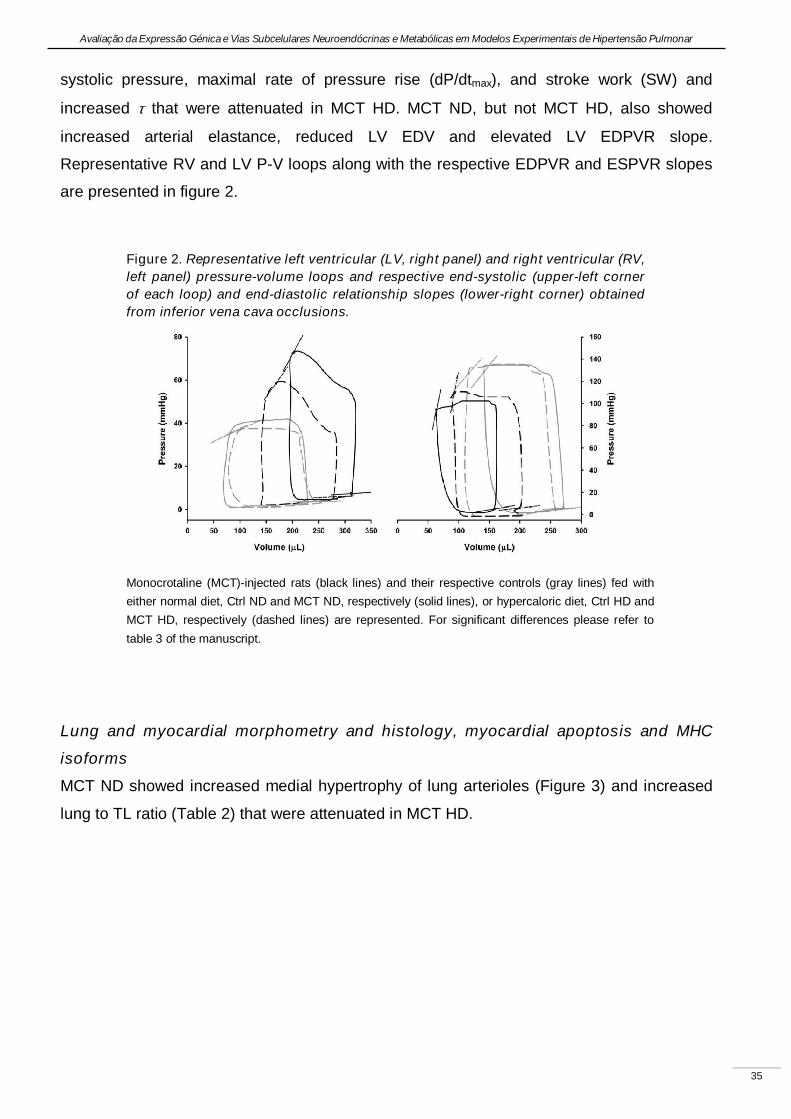
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
35
systolic pressure, maximal rate of pressure rise (dP/dtmax), and stroke work (SW) and
increased τ that were attenuated in MCT HD. MCT ND, but not MCT HD, also showed
increased arterial elastance, reduced LV EDV and elevated LV EDPVR slope.
Representative RV and LV P-V loops along with the respective EDPVR and ESPVR slopes
are presented in figure 2.
Figure 2. Representative left ventricular (LV, right panel) and right ventricular (RV,
left panel) pressure-volume loops and respective end-systolic (upper-left corner
of each loop) and end-diastolic relationship slopes (lower-right corner) obtained
from inferior vena cava occlusions.
Monocrotaline (MCT)-injected rats (black lines) and their respective controls (gray lines) fed with
either normal diet, Ctrl ND and MCT ND, respectively (solid lines), or hypercaloric diet, Ctrl HD and
MCT HD, respectively (dashed lines) are represented. For significant differences please refer to
table 3 of the manuscript.
Lung and myocardial morphometry and histology, myocardial apoptosis and MHC
isoforms
MCT ND showed increased medial hypertrophy of lung arterioles (Figure 3) and increased
lung to TL ratio (Table 2) that were attenuated in MCT HD.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
36
Figure 3. Lung histology (200x) of control and monocrotaline (MCT)-injected rats fed
with either normal diet, Ctrl ND and MCT ND, respectively, or hypercaloric diet, Ctrl HD
and MCT HD, respectively (upper panel) and media layer thickness of pulmonary
arterioles (lower panel).
MCT ND showed increased media thickness (*P = 0.003 vs Ctrl ND) that was abrogated in their
pulmonary hypertensive MCT HD littermates (†P = 0.002 vs MCT ND). Histological sections from 7
animals per group were analysed.
As for the myocardium, both MCT groups presented increased RV cardiomyocyte diameter
whereas no changes were seen in LV free-wall myocytes (Table 4). Nevertheless, while RV
weight normalized for TL increased in both MCT groups, TL-normalized weight of LV +
interventricular septum (IVS) decreased only in MCT ND (Table 4).
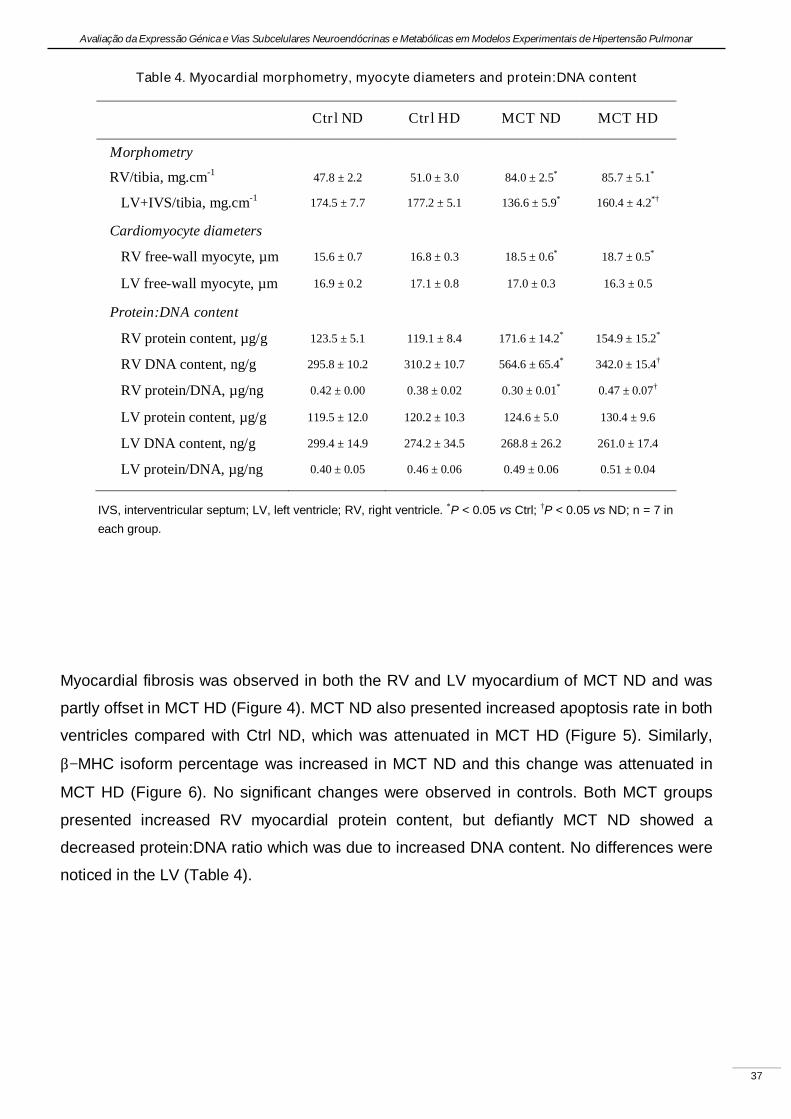
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
37
Table 4. Myocardial morphometry, myocyte diameters and protein:DNA content
Ctrl ND Ctrl HD MCT ND MCT HD
Morphometry
RV/tibia, mg.cm-1 47.8 ± 2.2 51.0 ± 3.0 84.0 ± 2.5* 85.7 ± 5.1*
LV+IVS/tibia, mg.cm-1 174.5 ± 7.7 177.2 ± 5.1 136.6 ± 5.9* 160.4 ± 4.2*†
Cardiomyocyte diameters
RV free-wall myocyte, µm 15.6 ± 0.7 16.8 ± 0.3 18.5 ± 0.6* 18.7 ± 0.5*
LV free-wall myocyte, µm 16.9 ± 0.2 17.1 ± 0.8 17.0 ± 0.3 16.3 ± 0.5
Protein:DNA content
RV protein content, µg/g 123.5 ± 5.1 119.1 ± 8.4 171.6 ± 14.2* 154.9 ± 15.2*
RV DNA content, ng/g 295.8 ± 10.2 310.2 ± 10.7 564.6 ± 65.4* 342.0 ± 15.4†
RV protein/DNA, µg/ng 0.42 ± 0.00 0.38 ± 0.02 0.30 ± 0.01* 0.47 ± 0.07†
LV protein content, µg/g 119.5 ± 12.0 120.2 ± 10.3 124.6 ± 5.0 130.4 ± 9.6
LV DNA content, ng/g 299.4 ± 14.9 274.2 ± 34.5 268.8 ± 26.2 261.0 ± 17.4
LV protein/DNA, µg/ng 0.40 ± 0.05 0.46 ± 0.06 0.49 ± 0.06 0.51 ± 0.04
IVS, interventricular septum; LV, left ventricle; RV, right ventricle. *P < 0.05 vs Ctrl; †P < 0.05 vs ND; n = 7 in
each group.
Myocardial fibrosis was observed in both the RV and LV myocardium of MCT ND and was
partly offset in MCT HD (Figure 4). MCT ND also presented increased apoptosis rate in both
ventricles compared with Ctrl ND, which was attenuated in MCT HD (Figure 5). Similarly,
β−MHC isoform percentage was increased in MCT ND and this change was attenuated in
MCT HD (Figure 6). No significant changes were observed in controls. Both MCT groups
presented increased RV myocardial protein content, but defiantly MCT ND showed a
decreased protein:DNA ratio which was due to increased DNA content. No differences were
noticed in the LV (Table 4).
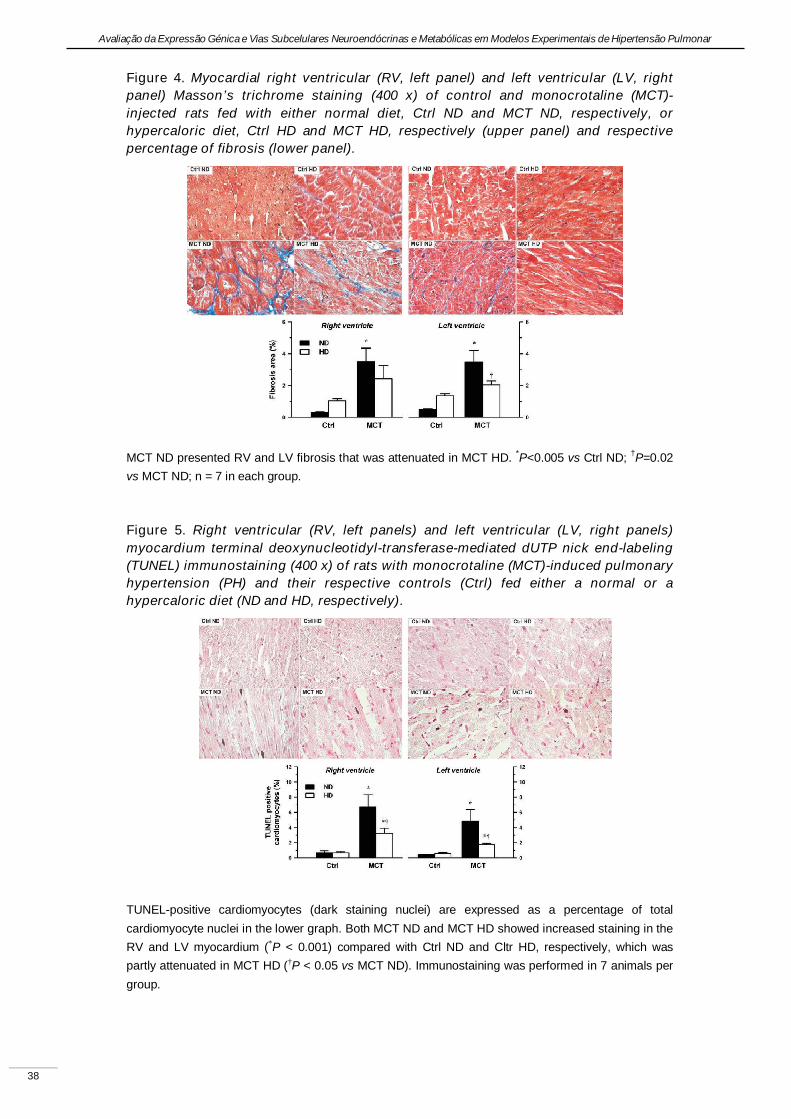
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
38
Figure 4. Myocardial right ventricular (RV, left panel) and left ventricular (LV, right
panel) Masson’s trichrome staining (400 x) of control and monocrotaline (MCT)-
injected rats fed with either normal diet, Ctrl ND and MCT ND, respectively, or
hypercaloric diet, Ctrl HD and MCT HD, respectively (upper panel) and respective
percentage of fibrosis (lower panel).
MCT ND presented RV and LV fibrosis that was attenuated in MCT HD. *P<0.005 vs Ctrl ND;
†P=0.02
vs MCT ND; n = 7 in each group.
Figure 5. Right ventricular (RV, left panels) and left ventricular (LV, right panels)
myocardium terminal deoxynucleotidyl-transferase-mediated dUTP nick end-labeling
(TUNEL) immunostaining (400 x) of rats with monocrotaline (MCT)-induced pulmonary
hypertension (PH) and their respective controls (Ctrl) fed either a normal or a
hypercaloric diet (ND and HD, respectively).
TUNEL-positive cardiomyocytes (dark staining nuclei) are expressed as a percentage of total
cardiomyocyte nuclei in the lower graph. Both MCT ND and MCT HD showed increased staining in the
RV and LV myocardium (*P < 0.001) compared with Ctrl ND and Cltr HD, respectively, which was
partly attenuated in MCT HD (†P < 0.05 vs MCT ND). Immunostaining was performed in 7 animals per
group.
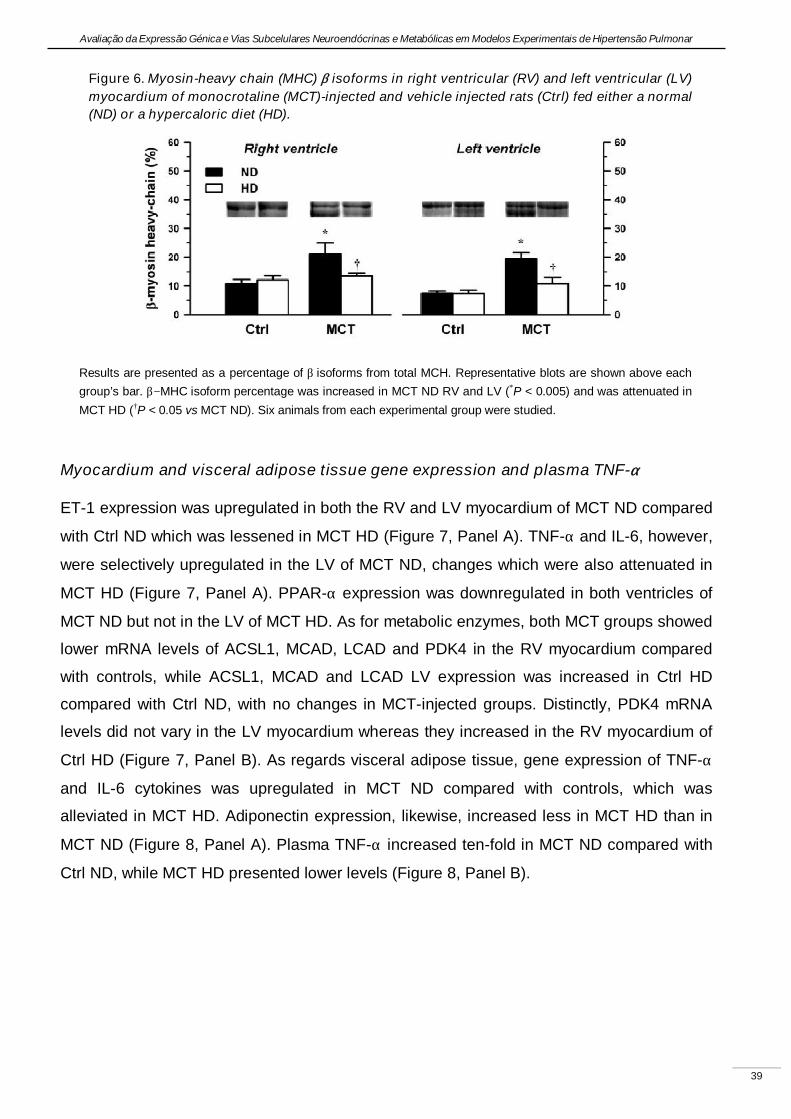
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
39
Figure 6. Myosin-heavy chain (MHC) β isoforms in right ventricular (RV) and left ventricular (LV)
myocardium of monocrotaline (MCT)-injected and vehicle injected rats (Ctrl) fed either a normal
(ND) or a hypercaloric diet (HD).
Results are presented as a percentage of β isoforms from total MCH. Representative blots are shown above each
group’s bar. β−MHC isoform percentage was increased in MCT ND RV and LV (*P < 0.005) and was attenuated in
MCT HD (†P < 0.05 vs MCT ND). Six animals from each experimental group were studied.
Myocardium and visceral adipose tissue gene expression and plasma TNF-α
ET-1 expression was upregulated in both the RV and LV myocardium of MCT ND compared
with Ctrl ND which was lessened in MCT HD (Figure 7, Panel A). TNF-α and IL-6, however,
were selectively upregulated in the LV of MCT ND, changes which were also attenuated in
MCT HD (Figure 7, Panel A). PPAR-α expression was downregulated in both ventricles of
MCT ND but not in the LV of MCT HD. As for metabolic enzymes, both MCT groups showed
lower mRNA levels of ACSL1, MCAD, LCAD and PDK4 in the RV myocardium compared
with controls, while ACSL1, MCAD and LCAD LV expression was increased in Ctrl HD
compared with Ctrl ND, with no changes in MCT-injected groups. Distinctly, PDK4 mRNA
levels did not vary in the LV myocardium whereas they increased in the RV myocardium of
Ctrl HD (Figure 7, Panel B). As regards visceral adipose tissue, gene expression of TNF-α
and IL-6 cytokines was upregulated in MCT ND compared with controls, which was
alleviated in MCT HD. Adiponectin expression, likewise, increased less in MCT HD than in
MCT ND (Figure 8, Panel A). Plasma TNF-α increased ten-fold in MCT ND compared with
Ctrl ND, while MCT HD presented lower levels (Figure 8, Panel B).
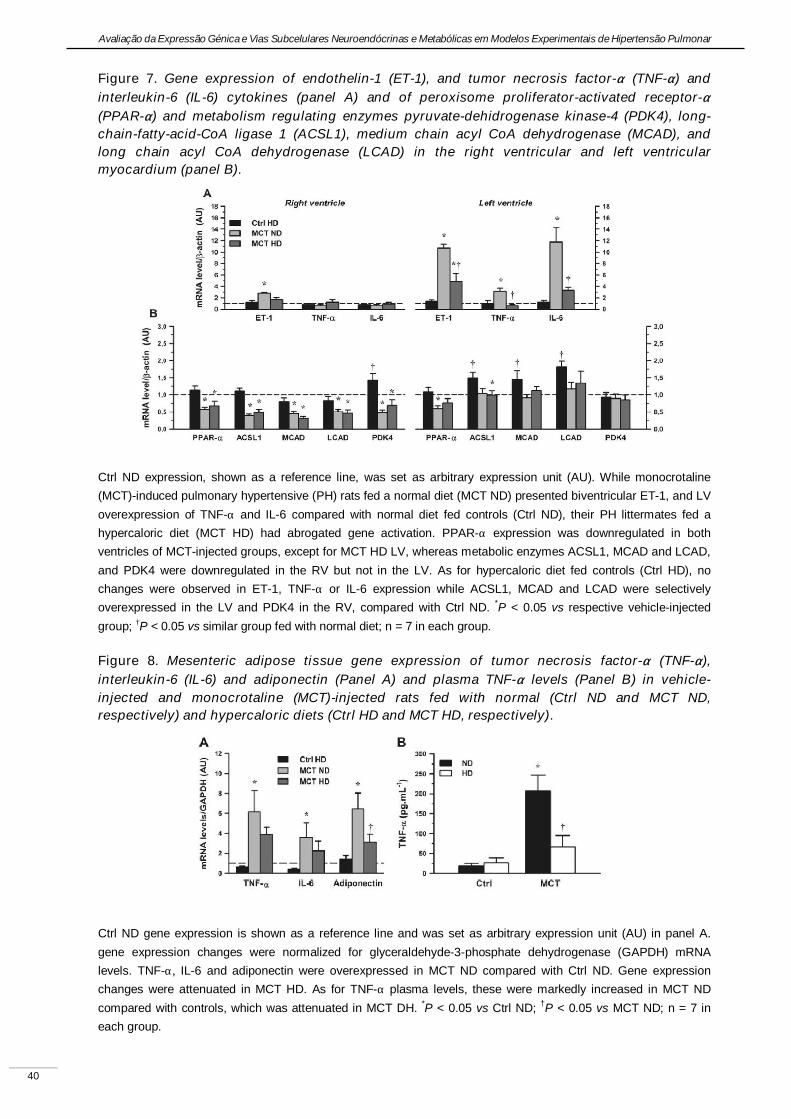
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
40
Figure 7. Gene expression of endothelin-1 (ET-1), and tumor necrosis factor-α (TNF-α) and
interleukin-6 (IL-6) cytokines (panel A) and of peroxisome proliferator-activated receptor-α
(PPAR-α) and metabolism regulating enzymes pyruvate-dehidrogenase kinase-4 (PDK4), long-
chain-fatty-acid-CoA ligase 1 (ACSL1), medium chain acyl CoA dehydrogenase (MCAD), and
long chain acyl CoA dehydrogenase (LCAD) in the right ventricular and left ventricular
myocardium (panel B).
Ctrl ND expression, shown as a reference line, was set as arbitrary expression unit (AU). While monocrotaline
(MCT)-induced pulmonary hypertensive (PH) rats fed a normal diet (MCT ND) presented biventricular ET-1, and LV
overexpression of TNF-α and IL-6 compared with normal diet fed controls (Ctrl ND), their PH littermates fed a
hypercaloric diet (MCT HD) had abrogated gene activation. PPAR-α expression was downregulated in both
ventricles of MCT-injected groups, except for MCT HD LV, whereas metabolic enzymes ACSL1, MCAD and LCAD,
and PDK4 were downregulated in the RV but not in the LV. As for hypercaloric diet fed controls (Ctrl HD), no
changes were observed in ET-1, TNF-α or IL-6 expression while ACSL1, MCAD and LCAD were selectively
overexpressed in the LV and PDK4 in the RV, compared with Ctrl ND. *P < 0.05 vs respective vehicle-injected
group; †P < 0.05 vs similar group fed with normal diet; n = 7 in each group.
Figure 8. Mesenteric adipose tissue gene expression of tumor necrosis factor-α (TNF-α),
interleukin-6 (IL-6) and adiponectin (Panel A) and plasma TNF-α levels (Panel B) in vehicle-
injected and monocrotaline (MCT)-injected rats fed with normal (Ctrl ND and MCT ND,
respectively) and hypercaloric diets (Ctrl HD and MCT HD, respectively).
Ctrl ND gene expression is shown as a reference line and was set as arbitrary expression unit (AU) in panel A.
gene expression changes were normalized for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA
levels. TNF-α, IL-6 and adiponectin were overexpressed in MCT ND compared with Ctrl ND. Gene expression
changes were attenuated in MCT HD. As for TNF-α plasma levels, these were markedly increased in MCT ND
compared with controls, which was attenuated in MCT DH. *P < 0.05 vs Ctrl ND; †P < 0.05 vs MCT ND; n = 7 in
each group.
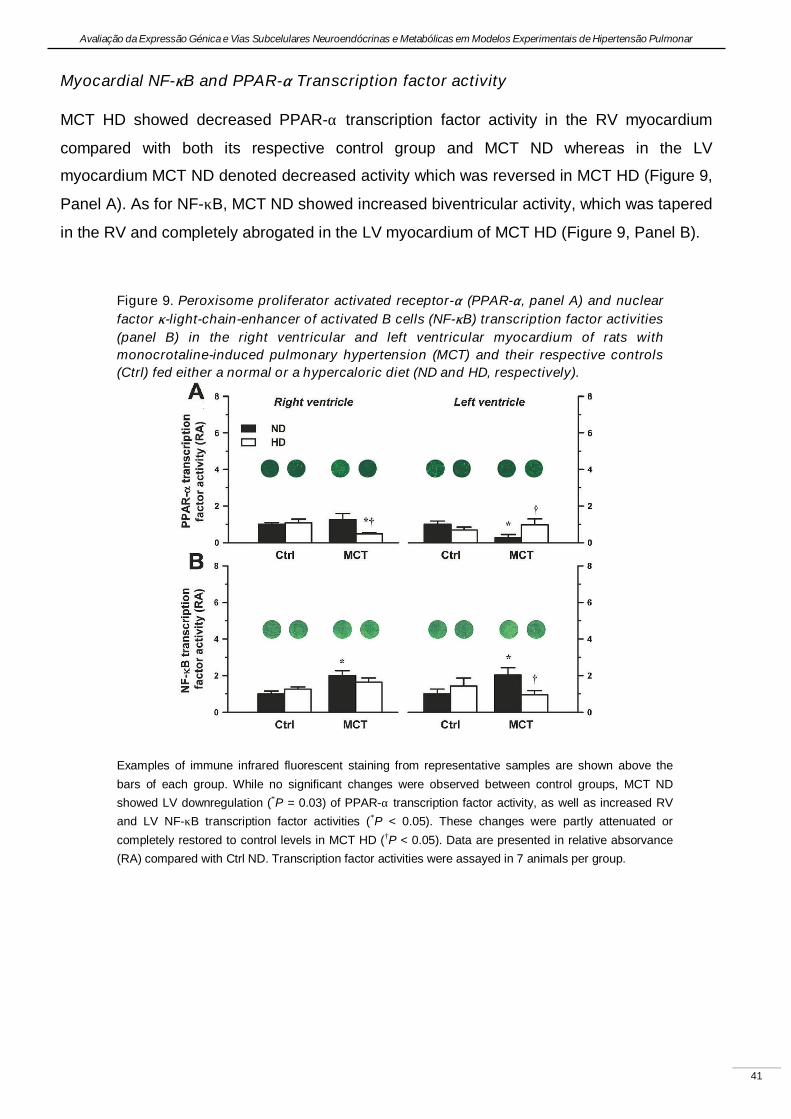
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
41
Myocardial NF-κB and PPAR-α Transcription factor activity
MCT HD showed decreased PPAR-α transcription factor activity in the RV myocardium
compared with both its respective control group and MCT ND whereas in the LV
myocardium MCT ND denoted decreased activity which was reversed in MCT HD (Figure 9,
Panel A). As for NF-κB, MCT ND showed increased biventricular activity, which was tapered
in the RV and completely abrogated in the LV myocardium of MCT HD (Figure 9, Panel B).
Figure 9. Peroxisome proliferator activated receptor-α (PPAR-α, panel A) and nuclear
factor κ-light-chain-enhancer of activated B cells (NF-κB) transcription factor activities
(panel B) in the right ventricular and left ventricular myocardium of rats with
monocrotaline-induced pulmonary hypertension (MCT) and their respective controls
(Ctrl) fed either a normal or a hypercaloric diet (ND and HD, respectively).
Examples of immune infrared fluorescent staining from representative samples are shown above the
bars of each group. While no significant changes were observed between control groups, MCT ND
showed LV downregulation (*P = 0.03) of PPAR-α transcription factor activity, as well as increased RV
and LV NF-κB transcription factor activities (*P < 0.05). These changes were partly attenuated or
completely restored to control levels in MCT HD (†P < 0.05). Data are presented in relative absorvance
(RA) compared with Ctrl ND. Transcription factor activities were assayed in 7 animals per group.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
42
Discussion
A HD rich in animal fat and simple carbohydrates improved survival and attenuated PH and
CC in experimental MCT-induced PH. Both RV and LV dysfunction and remodeling were
attenuated, as well as neuroendocrine and inflammatory activities.
MCT-induced PH extensively activates neuroendocrine systems and rapidly evolves
towards fatal decompensated HF (Ceconi et al., 1989, Hessel et al., 2006, Lourenço et al.,
2006). Accordingly, MCT ND presented medial PA thickening, increased PA elastance, an
index of pulmonary vascular load, and PH, as assessed by RVSP. The overloaded RV
showed increased EDP and EDV, hypertrophy, fibrosis and increased apoptotic rate, along
with an increase in both DNA and protein content with lower protein:DNA ratio. EF and CO
were decreased, whereas τ and EDPVR slope were increased, denoting compromised
relaxation and diastolic dysfunction, respectively. Regression towards the fetal β-MHC
isoform and local overexpression of ET-1 were also observed, as previously reported
(Correia-Pinto et al., 2009, Lourenço, Roncon-Albuquerque, Bras-Silva, Faria, Wieland,
Henriques-Coelho, Correia-Pinto and Leite-Moreira 2006).
Besides overload and neuroendocrine activation, other mechanisms, namely mitochondrial
dysfunction, reactive oxygen species (ROS), apoptosis and extracellular matrix
modifications underlie RV myocardial compromise (Bogaard et al., 2009, Redout et al.,
2007). Not surprisingly, anti-inflammatory agents such as statins have been effectively
employed as experimental therapeutics (Sun and Ku 2008). Another successful anti-
inflammatory approach has been caloric restriction, it favourably alters energy metabolism
while reducing ROS and neuroendocrine activity (Chou, Lee, Huang, Wang, Yu and Lau
2010). Indeed, caloric restriction attenuated RV hypertrophy and polyamine synthesis 21
days after MCT injection (Hacker 1993). It also diminished inflammation and cachexia in the
slow developing salt-sensitive Dahl rat arterial hypertension model (Seymour et al., 2006).
MCT-induced PH, however, is a severe and rapidly progressive model that usually evolves
from compensated RV hypertrophy, up to 4 weeks after injection (Correia-Pinto, Henriques-
Coelho, Roncon-Albuquerque, Lourenço, Melo-Rocha, Vasques-Novoa, Gillebert and Leite-
Moreira 2009, Hessel, Steendijk, den Adel, Schutte and van der Laarse 2006), to
decompensated HF, accompanied by marked anorexia and cachexia, beyond 21 days after
injection (Dalla Libera, Ravara, Volterrani, Gobbo, Della Barbera, Angelini, Danieli Betto,
Germinario and Vescovo 2004, Steffen, Lees and Booth 2008). Our study was intentionally
conducted when rats presented overt HF, reduced diuresis and a high mortality rate, as well
as decreased calorie ingestion and extensive body wasting, encompassing both lean and fat
masses. At this point, animals also had decreased heart rate, SW and LVSP along with

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
43
decreased EDV and compromised LV diastolic function indexes, consonant with ventricular
interdependence.
Although no changes were observed in LV cardiomyocyte diameters we found decreased
LV+IVS mass, increased apoptosis rate and fibrosis, along with marked overexpression not
only of ET-1 but also of IL-6 and TNF-α. We have previously suggested that systemic
neuroendocrine activity could trigger intrinsic LV myocardial dysfunction and neuroendocrine
activation, namely of ET-1 (Lourenço, Roncon-Albuquerque, Bras-Silva, Faria, Wieland,
Henriques-Coelho, Correia-Pinto and Leite-Moreira 2006). Based on current results we
propose that LV myocardial cytokine activation might also be explained by cachexia and
unloading. Indeed we have previously described lower LV mass and TNF-α overexpression
in cachectic rats with nephrotic syndrome (Moreira-Rodrigues et al., 2007). Inflammatory
cytokines, that may have been partly responsible for LV dysfunction and remodeling
(Feldman et al., 2000), were also overexpressed in visceral adipose tissue, alongside
adiponectin, an adipokine marker of worse prognosis in cachexia (Kistorp et al., 2005).
Compared with MCT ND, MCT HD presented a higher calorie intake and was able to
preserve BW, lean and fat mass. Although rats were not force-fed by gavaging, the typical
“western” HD is highly palatable and it provided additional calorie content per mass
ingested. Substrate and energy supply are fundamental in critical illness (Martindale et al.,
2009). Although nutritional supplementation in HF is constrained by fluid and salt restriction
and cannot circumvent anorexia, early satiety, reduced nutrient absorption and increased
catabolism, some small studies have shown benefits in BW gain and muscle function after
energy supplementation, mainly by nasoenteric route, with few cases of decompensation
due to a refeeding-like syndrome (Akner and Cederholm 2001). Interestingly, RV
hypertrophy was not attenuated in MCT HD compared with MCT ND despite reduced PH.
Still, myocardial DNA content did not increase, whilst myocardial apoptosis rate, fibrosis and
β-MHC isoform switch were attenuated, compared with MCT ND. We speculate the normal
DNA content compared with MCT ND could be due to a lower apoptosis rate, as suggested
for afterload-induced hypertrophy (Paquette et al., 2008). The higher substrate supply may
have accounted for differences in RV myocardial remodeling and hypertrophy. Additionally,
RV and LV myocardial function as well as CO were improved in MCT HD, along with
improved survival and attenuated inflammatory and neuroendocrine activities. These
findings are somewhat surprising but can be supported by several lines of evidence. Firstly,
observational studies suggest obesity and hypercholesterolemia improve survival in HF
(Curtis et al., 2005). The reverse epidemiology of cardiovascular risk factors (“obesity
paradox”) has been attributed not only to the fact that long term cardiovascular risk factors

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
44
may not behave as such in a poor prognosis condition such as severe HF (Kalantar-Zadeh
et al., 2004), but also to the lipoprotein-endotoxin hypothesis, which states lipoproteins,
which are increased in obesity and high-fat-feeding, neutralize bacterial lipopolysaccharides
(LPS) therefore protecting against inflammation (Rauchhaus et al., 2000). Chronic
inflammation accompanying HF partly results from intestinal bacterial translocation and
activation of cytokine production mostly from mononuclear cells but also from
cardiomyocytes (Anker and Coats 1999). Accordingly, the higher myocardial NF-κB activity,
and increased cytokine expression and plasma levels of TNF-α seen in MCT ND , were
mitigated in MCT HD. NF-κB signalling has been linked to cytokine release, oxidative stress,
hypertrophy, and remodeling in the failing myocardium (Valen et al., 2001). LPS or cytokines
activate inhibitor-κB (I-κB) kinase-mediated phosphorylation and degradation of I-κB,
unmasking a sequence within NF-κB that promotes nuclear translocation and binding to
response elements in target genes of failing cardiomyocytes (Valen, Yan and Hansson
2001). Some of these genes are cytokines, such as IL-6 and TNF-α, that underlie not only
many of the HF progression mechanisms (Feldman, Combes, Wagner, Kadakomi, Kubota,
Li and McTiernan 2000) but also IR, muscle wasting and cachexia (Sharma and Anker
2002), due to their pronounced effects in both cellular metabolism and PPAR-α function (Ye
2008). Decreased expression and activity of PPAR-α in the non-overloaded LV of MCT ND
might partly be attributed to TNF-α overexpression (Ye 2008).
Substantial metabolic adaptations accompany HF progression, triggering, and sustaining
functional and structural remodeling. PPAR plays a pivotal role in the transcriptional
regulation of genes involved in both FAO and glucose metabolism (Gulick et al., 1994).
Features such as impaired conversion of chemical energy into mechanical work, decline in
high-energy-phosphate content, mitochondrial dysfunction and loss of metabolic flexibility,
with a switch of energy source from FAO to anaerobic glycolysis are typically seen in the
failing myocardium (Ingwall 2009). Inactivation of PPAR-α during cardiac hypertrophy might
partly underlie this switch by transcriptional inactivation of FAO enzymes and PDK4 (Barger
et al., 2000). PDK4 phosphorylates and inhibits pyruvate dehydrogenase, an enzyme in
control of glycolysis-derived pyruvate oxidation (Stanley, Recchia and Lopaschuk 2005).
Although glucose metabolism increases the ATP production to O2 consumption ratio thereby
increasing energy efficiency (Stanley, Recchia and Lopaschuk 2005), this may not be
entirely adaptive, since more ATP is produced per mole of fatty acids (FA) (Barger, Brandt,
Leone, Weinheimer and Kelly 2000, van Bilsen et al., 2009). Indeed, cardiac work and
efficiency in human HF are definitely very dependent on FA (Tuunanen et al., 2006). The
combination of FA and carbohydrates in the HD regimen might have contributed to a better

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
45
preservation of myocardial function, as seen in MCT HD. MCT ND showed decreased RV
expression of PPARα, FAO enzymes and PDK4. LV PPAR-α expression and activity were
also decreased in MCT ND but not in MCT HD, which could be partly explained by PPAR-α
induction by the lipids in the HD (Gulick, Cresci, Caira, Moore and Kelly 1994). PPAR
activation normalizes cardiac substrate metabolism in RV hypertrophy and HF (Jucker et al.,
2007). Albeit we did not find LV FAO or PDK4 expression changes, these are not solely
regulated at the transcriptional level and expression levels do not translate enzymatic
activity (Stanley, Recchia and Lopaschuk 2005). Indeed, a high fat diet did not worsen
cardiac hypertrophy, in experimental LV pressure overload, and noticeably prevented the
decline in FAO enzyme activity and mitochondrial oxidative capacity (Chess et al., 2009).
Additionally, medium chain triglyceride diet enrichment in spontaneously hypertensive rats
preserved PPAR-α transcriptional activity, prevented FAO enzyme downregulation and
improved myocardial function (Iemitsu et al., 2008). Mitochondrial dysfunction, reduced
energy producing ability and increased production of ROS are known to accompany the
progression towards RV dysfunction in MCT-induced PH (Daicho et al., 2009, Redout,
Wagner, Zuidwijk, Boer, Musters, van Hardeveld, Paulus and Simonides 2007). Finally, we
must point out that recent evidence suggests PPAR agonists can attenuate PH (Hansmann
et al., 2007) and thus the high lipid content in the HD might have induced PPAR and
attenuated MCT-induced PH.
In summary, in a rapidly evolving experimental model of PH, HF and CC a HD regiment rich
in saturated fat and simple carbohydrates, which is a well-established nutritional
cardiovascular risk factor, attenuated PH, improved survival and myocardial function with
concomitant attenuation of neuroendocrine and inflammatory activities, possibly due to
beneficial changes in myocardial metabolism and transcription factor activity. Nevertheless,
we must acknowledge that a longer period of high-fat feeding could have led to cardiac
triglyceride accumulation and a decline in ventricular function (Akki and Seymour 2009).
Whether patients with CC and decompensated HF may profit from HD regimens remains to
be settled.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
46
Acknowledgments
The authors would like to thank Dra. Antónia Teles for her excellent technical support.
Carmen Brás-Silva’s current affiliation in Faculty of Nutrition and Food Sciences, University
of Porto, Porto, Portugal.
Grants
This work was supported by the Portuguese Foundation for Science and Technology (grant
PTDC/SAU/65793/2006).
Disclosures
None to declare.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
47
References
1. Akki A, and Seymour AM. Western diet impairs metabolic remodelling and
contractile efficiency in cardiac hypertrophy. Cardiovasc Res 81: 610-617. Epub 2008 Nov
2021., 2009.
2. Akner G, and Cederholm T. Treatment of protein-energy malnutrition in chronic
nonmalignant disorders. Am J Clin Nutr 74: 6-24, 2001.
3. Anker SD, and Coats AJ. Cardiac cachexia: a syndrome with impaired survival and
immune and neuroendocrine activation. Chest 115: 836-847, 1999.
4. Anker SD, Negassa A, Coats AJ, Afzal R, Poole-Wilson PA, Cohn JN, and Yusuf
S. Prognostic importance of weight loss in chronic heart failure and the effect of treatment
with angiotensin-converting-enzyme inhibitors: an observational study. Lancet 361: 1077-
1083, 2003.
5. Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM,
Harrington D, Kox WJ, Poole-Wilson PA, and Coats AJ. Wasting as independent risk
factor for mortality in chronic heart failure. Lancet 349: 1050-1053, 1997.
6. Barger PM, Brandt JM, Leone TC, Weinheimer CJ, and Kelly DP. Deactivation of
peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin
Invest 105: 1723-1730., 2000.
7. Bogaard HJ, Abe K, Vonk Noordegraaf A, and Voelkel NF. The right ventricle
under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary
hypertension. Chest 135: 794-804., 2009.
8. Ceconi C, Condorelli E, Quinzanini M, Rodella A, Ferrari R, and Harris P.
Noradrenaline, atrial natriuretic peptide, bombesin and neurotensin in myocardium and
blood of rats in congestive cardiac failure. Cardiovasc Res 23: 674-682., 1989.
9. Chess DJ, Khairallah RJ, O'Shea KM, Xu W, and Stanley WC. A High Fat Diet
Increases Adiposity but Maintains Mitochondrial Oxidative Enzymes without Affecting
Development of Heart Failure with Pressure Overload. Am J Physiol Heart Circ Physiol 18:
18, 2009.
10. Chou SH, Lee YC, Huang CF, Wang YR, Yu HP, and Lau YT. Gender-specific
effects of caloric restriction on the balance of vascular nitric oxide and superoxide radical.
Cardiovasc Res 87: 751-759. Epub 2010 Mar 2025., 2010.
11. Correia-Pinto J, Henriques-Coelho T, Roncon-Albuquerque R, Jr., Lourenço AP,
Melo-Rocha G, Vasques-Novoa F, Gillebert TC, and Leite-Moreira AF.Time course and
mechanisms of left ventricular systolic and diastolic dysfunction in monocrotaline-induced
pulmonary hypertension. Basic Res Cardiol 104: 535-545. Epub 2009 Mar 2014., 2009.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
48
12. Curtis JP, Selter JG, Wang Y, Rathore SS, Jovin IS, Jadbabaie F, Kosiborod M,
Portnay EL, Sokol SI, Bader F, and Krumholz HM. The obesity paradox: body mass index
and outcomes in patients with heart failure. Arch Intern Med 165: 55-61., 2005.
13. Daicho T, Yagi T, Abe Y, Ohara M, Marunouchi T, Takeo S, and Tanonaka K.
Possible involvement of mitochondrial energy-producing ability in the development of right
ventricular failure in monocrotaline-induced pulmonary hypertensive rats. J Pharmacol Sci
111: 33-43., 2009.
14. Dalla Libera L, Ravara B, Volterrani M, Gobbo V, Della Barbera M, Angelini A,
Danieli Betto D, Germinario E, and Vescovo G. Beneficial effects of GH/IGF-1 on skeletal
muscle atrophy and function in experimental heart failure. Am J Physiol Cell Physiol 286:
C138-144. Epub 2003 Sep 2017., 2004.
15. Feldman AM, Combes A, Wagner D, Kadakomi T, Kubota T, Li YY, and
McTiernan C. The role of tumor necrosis factor in the pathophysiology of heart failure. J Am
Coll Cardiol 35: 537-544, 2000.
16. Filippatos GS, Tsilias K, Venetsanou K, Karambinos E, Manolatos D, Kranidis
A, Antonellis J, Kardaras F, Anthopoulos L, and Baltopoulos G. Leptin serum levels in
cachectic heart failure patients. Relationship with tumor necrosis factor-alpha system. Int J
Cardiol 76: 117-122, 2000.
17. Fontana L, Meyer TE, Klein S, and Holloszy JO. Long-term calorie restriction is
highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci U S A
101: 6659-6663. Epub 2004 Apr 6619., 2004.
18. Gulick T, Cresci S, Caira T, Moore DD, and Kelly DP. The peroxisome proliferator-
activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression.
Proc Natl Acad Sci U S A 91: 11012-11016., 1994.
19. Hacker AD. Dietary restriction, polyamines and monocrotaline-induced pulmonary
hypertension. Biochem Pharmacol 45: 2475-2481., 1993.
20. Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh
AY, Suen RS, Stewart DJ, and Rabinovitch M. Pulmonary arterial hypertension is linked
to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma
activation. Circulation 115: 1275-1284, 2007.
21. Hessel MH, Steendijk P, den Adel B, Schutte CI, and van der Laarse A.
Characterization of right ventricular function after monocrotaline-induced pulmonary
hypertension in the intact rat. Am J Physiol Heart Circ Physiol 291: H2424-2430. Epub 2006
May 2426., 2006.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
49
22. Iemitsu M, Shimojo N, Maeda S, Irukayama-Tomobe Y, Sakai S, Ohkubo T,
Tanaka Y, and Miyauchi T. The benefit of medium-chain triglyceride therapy on the cardiac
function of SHRs is associated with a reversal of metabolic and signaling alterations. Am J
Physiol Heart Circ Physiol 295: H136-144. Epub 2008 May 2002., 2008.
23. Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res 81:
412-419, 2009.
24. Jucker BM, Doe CP, Schnackenberg CG, Olzinski AR, Maniscalco K, Williams C,
Hu TC, Lenhard SC, Costell M, Bernard R, Sarov-Blat L, Steplewski K, and Willette RN.
PPARdelta activation normalizes cardiac substrate metabolism and reduces right ventricular
hypertrophy in congestive heart failure. J Cardiovasc Pharmacol 50: 25-34, 2007.
25. Kalantar-Zadeh K, Block G, Horwich T, and Fonarow GC. Reverse epidemiology
of conventional cardiovascular risk factors in patients with chronic heart failure. J Am Coll
Cardiol 43: 1439-1444., 2004.
26. Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, and
Hildebrandt P. Plasma adiponectin, body mass index, and mortality in patients with chronic
heart failure. Circulation 112: 1756-1762, 2005.
27. Krauss RM, Eckel RH, Howard B, Appel LJ, Daniels SR, Deckelbaum RJ,
Erdman JW, Jr., Kris-Etherton P, Goldberg IJ, Kotchen TA, Lichtenstein AH, Mitch
WE, Mullis R, Robinson K, Wylie-Rosett J, St Jeor S, Suttie J, Tribble DL, and
Bazzarre TL. AHA Dietary Guidelines: revision 2000: A statement for healthcare
professionals from the Nutrition Committee of the American Heart Association. Circulation
102: 2284-2299., 2000.
28. Lourenço AP, Roncon-Albuquerque R, Jr., Bras-Silva C, Faria B, Wieland J,
Henriques-Coelho T, Correia-Pinto J, and Leite-Moreira AF. Myocardial dysfunction and
neurohumoral activation without remodeling in left ventricle of monocrotaline-induced
pulmonary hypertensive rats. Am J Physiol Heart Circ Physiol 291: H1587-1594. Epub 2006
May 1585., 2006.
29. Martindale RG, McClave SA, Vanek VW, McCarthy M, Roberts P, Taylor B,
Ochoa JB, Napolitano L, and Cresci G. Guidelines for the provision and assessment of
nutrition support therapy in the adult critically ill patient: Society of Critical Care Medicine
and American Society for Parenteral and Enteral Nutrition: Executive Summary. Crit Care
Med 37: 1757-1761., 2009.
30. Moreira-Rodrigues M, Roncon-Albuquerque R, Henriques-Coelho T, Lourenço
AP, Sampaio-Maia B, Santos J, Pestana M, and Leite-Moreira AF.Cardiac remodeling
and dysfunction in nephrotic syndrome. Kidney International 71: 1240-1248, 2007.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
50
31. Paquette PA, Duguay D, El-Ayoubi R, Menaouar A, Danalache B, Gutkowska J,
DeBlois D, and Mukaddam-Daher S. Control of left ventricular mass by moxonidine
involves reduced DNA synthesis and enhanced DNA fragmentation. Br J Pharmacol 153:
459-467, 2008.
32. Rauchhaus M, Coats AJ, and Anker SD. The endotoxin-lipoprotein hypothesis.
Lancet 356: 930-933., 2000.
33. Redout EM, Wagner MJ, Zuidwijk MJ, Boer C, Musters RJ, van Hardeveld C,
Paulus WJ, and Simonides WS. Right-ventricular failure is associated with increased
mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc Res
75: 770-781. Epub 2007 May 2017., 2007.
34. Seymour EM, Parikh RV, Singer AA, and Bolling SF. Moderate calorie restriction
improves cardiac remodeling and diastolic dysfunction in the Dahl-SS rat. J Mol Cell Cardiol
41: 661-668, 2006.
35. Sharma R, and Anker SD. Cytokines, apoptosis and cachexia: the potential for TNF
antagonism. Int J Cardiol 85: 161-171, 2002.
36. Stanley WC, Recchia FA, and Lopaschuk GD. Myocardial substrate metabolism in
the normal and failing heart. Physiol Rev 85: 1093-1129., 2005.
37. Steffen BT, Lees SJ, and Booth FW. Anti-TNF treatment reduces rat skeletal
muscle wasting in monocrotaline-induced cardiac cachexia. J Appl Physiol 105: 1950-1958.
Epub 2008 Sep 1918., 2008.
38. Sun X, and Ku DD. Rosuvastatin provides pleiotropic protection against pulmonary
hypertension, right ventricular hypertrophy, and coronary endothelial dysfunction in rats. Am
J Physiol Heart Circ Physiol 294: H801-809. Epub 2007 Nov 2030., 2008.
39. Tuunanen H, Engblom E, Naum A, Nagren K, Hesse B, Airaksinen KE, Nuutila P,
Iozzo P, Ukkonen H, Opie LH, and Knuuti J. Free fatty acid depletion acutely decreases
cardiac work and efficiency in cardiomyopathic heart failure. Circulation 114: 2130-2137.
Epub 2006 Nov 2136., 2006.
40. Valen G, Yan ZQ, and Hansson GK. Nuclear factor kappa-B and the heart. J Am
Coll Cardiol 38: 307-314, 2001.
41. van Bilsen M, van Nieuwenhoven FA, and van der Vusse GJ. Metabolic
remodelling of the failing heart: beneficial or detrimental? Cardiovasc Res 81: 420-428.
Epub 2008 Oct 2014., 2009.
42. von Haehling S, Doehner W, and Anker SD. Nutrition, metabolism, and the
complex pathophysiology of cachexia in chronic heart failure. Cardiovasc Res 73: 298-309,
2007.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
51
43. Ye J. Regulation of PPARgamma function by TNF-alpha. Biochem Biophys Res
Commun 374: 405-408. Epub 2008 Jul 2023., 2008.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
52
5. Discussão e Conclusões
A dieta hipercalórica, do tipo ocidental, rica em gordura animal e carbo-hidratos
simples, melhorou a sobrevida e atenuou a HP e a caquexia cardíaca no modelo
experimental de HP severa e rapidamente progressiva, induzida pela MCT. A disfunção e a
remodelagem foram significativamente atenuadas no VD e no VE, assim como as
actividades neuroendócrina e inflamatória.
No grupo de animais injectados com MCT e alimentados com dieta normal, de acordo
com o esperado neste modelo experimental, observámos espessamento da camada média
das arteríolas pulmonares, HP, disfunção VD, com redução da fracção de ejecção, do DC e
elevação do volume e pressão telediastólicas, e remodelagem miocárdica, incluindo
hipertrofia, fibrose e apoptose. Acompanhando estas alterações, observámos igualmente
alterações moleculares típicas da regressão para o fenótipo cardiomiocitário fetal, que
usualmente acompanha a progressão da IC, nomeadamente a comutação para as
isoformas do tipo β, com actividade ATPásica lenta, das cadeias pesadas de miosina e a
maior expressão local de ET-1, como relatámos anteriormente (Correia-Pinto et al., 2009;
Lourenço et al., 2006). Para além da sobrecarga e da activação neuroendócrina, outros
mecanismos, designadamente a disfunção mitocondrial, a maior produção de ROS, a
apoptose e as modificações da matriz extracelular, podem contribuir para a disfunção do
miocárdio VD na HP (Bogaard et al., 2009a, 2009b; Redout et al., 2007). Não constitui
surpresa, portanto, que agentes anti-inflamatórios, como as estatinas, e o enriquecimento
alimentar com óleo de peixe (Baybutt et al., 2002) tenham sido bem sucedidos como terapia
experimental (Sun e Ku, 2008). Outra abordagem anti-inflamatória bem sucedida é a
restrição calórica, que modifica favoravelmente o metabolismo energético, reduz a
formação de ROS, a actividade neuroendócrina (Chou et al., 2010) e a degradação proteíca
(Redman et al., 2008; Weindruch et al., 2001). De facto, a restrição calórica atenuou a
hipertrofia e a síntese de poliaminas, 21 dias após a injecção de MCT (Hacker, 1993) e
atenuou a inflamação e a caquexia no modelo de hipertensão arterial sensível ao sal em
ratos Dahl, caracterizado pelo desenvolvimento lento de hipertensão arterial, IC e caquexia
(Seymour et al., 2006). No entanto, na avaliação do efeito da restrição calórica na HP
induzida pela MCT só foi estudada a hipertrofia ventricular direita e a síntese de poliaminas
(Hacker, 1993). Além disso, os animais foram estudados precocemente após a injecção,
quando geralmente apresentam um fenótipo de hipertrofia VD compensada sem sinais de
IC ou caquexia (Hessel et al., 2006; Werchan et al., 1989). Por outro lado, o modelo de
hipertensão arterial em ratos Dahl tem uma evolução muito lenta, de várias semanas, até à
ocorrência de caquexia e IC. O nosso estudo foi realizado 5 semanas após a injecção com

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
53
MCT, quando os animais apresentavam já mortalidade elevada e sinais clínicos evidentes
de IC, como a diminuição da diurese e edemas generalizados, e deterioração sistémica,
incluindo anorexia e caquexia. De facto, na avaliação dos animais do grupo injectado com
MCT e alimentado com dieta normal observamos para além de diminuição de ingestão
calórica, perda ponderal marcada, incluindo a massa magra e a massa gorda, mas também
bradicardia, diminuição do trabalho de ejecção e da pressão sistólica VE. No VE
observámos igualmente diminuição do volume telediastólico e comprometimento dos
índices de função diastólica, em consonância com a perturbação do preenchimento
decorrente da interdependência ventricular (Menzel et al., 2000). Contudo, não foram
observadas alterações nos diâmetros dos cardiomiócitos VE, apesar de se ter registado
uma diminuição da massa do conjunto constituído pelo VE e pelo septo interventricular e
aumento quer da taxa de apoptose quer da fibrose miocárdicas. No miocárdio VE,
observamos ainda sobre-expressão acentuada não só da ET-1, mas também de citocinas
inflamatórias, como a IL-6 e o TNF-α. Num trabalho anterior, demonstramos que a
actividade neuroendócrina sistémica poderia desencadear activação neuroendócrina VE,
nomeadamente de ET-1, e disfunção miocárdica intrínseca (Lourenço et al., 2006). Com
base nos resultados deste trabalho experimental propomos que a activação de citocinas no
miocárdio possa também dever-se à caquexia e à redução da pré-carga (unloading) VE. De
facto, descrevemos anteriormente num modelo experimental de caquexia associada ao
síndrome nefrótico diminuição de massa VE e sobre-expressão de TNF-α (Moreira-
Rodrigues et al., 2007). A expressão de citocinas inflamatórias, cuja acção miocárdica pode
ter sido parcialmente responsável pela disfunção e remodelagem (Feldman et al., 2000),
não aumentou apenas no VE, mas também no tecido adiposo visceral, e os níveis
circulantes destas encontram-se seguramente elevados, como confirmamos para o TNF-α.
No tecido adiposo observamos ainda maior expressão de adiponectina, uma adipocina que,
apesar de desempenhar funções cardiovasculares benéficas, é, paradoxalmente, um
marcador de mau prognóstico na caquexia cardíaca (Kistorp et al., 2005).
Quanto ao grupo injectado com MCT e alimentado com dieta hipercalórica, rica em
gordura animal e carbo-hidratos simples, apesar dos ratos não terem sido alimentados por
gavagem, e, portanto, terem ingerido calorias ad libitum, observamos um maior consumo
calórico, com preservação do peso corporal, e das massas magra e gorda. Este resultado
está de acordo com a elevada palatabilidade deste tipo de ração e com a maior composição
em calorias e substratos metabólicos energéticos por massa ingerida. Parte das melhorias
encontradas na evolução ponderal pode ter-se devido ao maior aporte energético e
calórico, visto que a manutenção do equilíbrio energético e metabólico é essencial no

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
54
doente crítico (Martindale et al., 2009). Curiosamente, apesar destas diferenças e da
redução no nível de HP, a hipertrofia VD não foi atenuada comparativamente com o regime
alimentar normal. De qualquer modo, a taxa de apoptose miocárdica VD e VE, a fibrose e a
comutação para a isoforma β das cadeias pesadas de miosina foram atenuadas. No grupo
injectado com MCT e alimentado com dieta hipercalórica observamos também melhor
sobrevida e melhoria do perfil hemodinâmico. Assim, os animais deste grupo,
comparativamente com os alimentados com dieta normal, não apresentaram dilatação VD,
e registaram melhorias significativas no DC e nos índices de função VD e VE.
Paralelamente, registamos uma atenuação da actividade inflamatória e neuroendócrina. Os
nossos resultados são originais e algo surpreendentes, face aos efeitos bem estabelecidos
deste tipo de regime alimentar como factor de risco cardiovascular. No entanto, podem ser
interpretados atendendo a várias linhas de evidência. Com efeito, apesar da suplementação
nutricional na caquexia ser limitada pela necessidade de restrição de fluídos e sal e de não
ser possível contornar a anorexia, saciedade precoce, redução da absorção de nutrientes e
o aumento do metabolismo que acompanham a IC (Azhar e Wei, 2006), pequenos estudos
têm relatado benefícios na manutenção do peso corporal e da função muscular após a
suplementação energética, principalmente por via nasoentérica, com um número reduzido
de casos de descompensação associada a um síndrome semelhante ao síndrome de
realimentação (Akner e Cederholm, 2001). Para além destes pequenos estudos clínicos,
diversos estudos observacionais de larga escala constituem evidência epidemiológica de
que a obesidade e a hipercolesterolémia são factores de bom prognóstico e melhoram a
sobrevida na IC (Curtis et al., 2005). Esta epidemiologia reversa dos factores de risco
cardiovascular, denominada por “paradoxo da obesidade”, tem sido atribuída não só à
possibilidade de que factores de risco a longo prazo possam não se comportar como tal
numa situação de rápida evolução e mau prognóstico, como a IC terminal (Kalantar-Zadeh
et al., 2004), mas também à hipótese lipoproteína-endotoxina. Segundo esta hipótese, as
lipoproteínas ligam-se e neutralizam os LPS circulantes, protegendo o organismo da
resposta inflamatória (Rauchhaus et al., 2000). A IC é uma patologia multissistémica que se
faz acompanhar cronicamente de activação inflamatória, esta activação parece ser devida
parcialmente à translocação bacteriana a nível intestinal. Os produtos bacterianos
circulantes serão activadores fundamentais da produção de citocinas (Anker e Coats,
1999). Esta hipótese é concordante com os resultados obtidos. Com efeito, no grupo de
ratos injectado com MCT e posteriormente alimentado com dieta normal, verificamos maior
actividade miocárdica do NF-κB, maior expressão de citocinas no miocárdio VE e no tecido
adiposo visceral, e níveis circulantes aumentados de TNF-α. Estas alterações foram

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
55
atenuadas pelo regime alimentar hipercalórico. As citocinas, como o TNF-α, para além de
contribuírem para mecanismos de progressão da IC como a remodelagem, apoptose e
disfunção do miocárdio (Feldman et al., 2000), também induzem insulino-resistência, sub-
nutrição energético-protéica, perda de massa muscular e caquexia (Sharma e Anker, 2002).
Estes efeitos dever-se-ão essencialmente à sua influência marcada no metabolismo celular
e na função do PPAR-α. A diminuição da expressão e da actividade do PPAR-α observada
no VE dos ratos injectados com MCT e alimentados com dieta normal, pode ser
parcialmente atribuída à maior expressão e actividade do TNF-α (Ye, 2008). Em larga
medida, a inflamação crónica parece ser o elo principal entre IC e caquexia (von Haehling
et al., 2007). Para além dos pontos anteriormente apontados, a progressão da IC também
se acompanha de adaptações metabólicas substanciais, que poderão desencadear ou
facilitar a remodelagem estrutural e funcional. O PPAR-α parece desempenhar um papel
fundamental neste contexto. É um dos principais reguladores da transcrição de genes que
codificam enzimas envolvidas na oxidação dos AG e no metabolismo da glicose (Gulick et
al., 1994). No miocárdio insuficiente são típicas alterações metabólico-energéticas como
uma conversão ineficiente de energia química em trabalho mecânico, uma diminuição do
conteúdo de grupos fosfato armazenadores de energia química, a disfunção mitocondrial e
uma perda da flexibilidade metabólica com maior dependência energética da glicólise
anaeróbia em detrimento da oxidação de AG (Ingwall, 2009). A inactivação do PPAR-α
durante a hipertrofia cardíaca pode, em parte, ser responsável pela inactivação da
transcrição das enzimas responsáveis pela oxidação dos AG e da isoforma 4 da PDK
(Barger et al., 2000). A PDK4 fosforila e inibe a PDH, a principal enzima reguladora da
capacidade oxidativa de carbo-hidratos, que converte o produto final da glicólise, o piruvato,
em acetil-CoA, possibilitando a sua metabolização no ciclo de Krebs (Stanley et al., 2005).
Admite-se que a preferência metabólica pelos carbo-hidratos possa ter um papel adaptativo
nas circunstâncias em que as necessidades energéticas do miocárdio estão aumentadas,
como é o caso da hipertrofia e da IC, porque a contractilidade miocárdica melhora quando o
miocárdio oxida preferencialmente glicose e lactato, em detrimento dos AG (Burkhoff et al.,
1991), ao passo que concentrações plasmáticas elevadas de AG livres diminuem a
eficiência mecânica (Hutter et al., 1984), provavelmente por desacoplamento da FO
(Schrauwen et al., 2003). Mais, o metabolismo da glicose tem uma maior razão entre a
produção de ATP e o consumo de O2, o que lhe confere maior eficiência energética
comparativamente com os AG (Stanley et al., 2005), e as enzimas glicolíticas
compartimentalizam-se nas imediações do retículo sarcoplasmático e do sarcolema (Pierce
e Philipson, 1985), sendo fundamentais à captação de Ca2+ pelo retículo endoplasmático

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
56
(Entman et al., 1977) e ao relaxamento do miocárdio durante situações de stress, como a
isquemia (Kusuoka e Marban, 1994). Contudo, o consumo preferencial de glicose pode não
ser completamente adaptativo, uma vez que por cada mole de AG é gerado mais ATP
(Barger et al., 2000; van Bilsen et al., 2009). De facto, nos doentes com IC, o trabalho e a
eficiência cardíaca são muito dependentes dos AG (Tuunanen et al., 2006). Nos animais
alimentados com dieta hipercalórica a conjugação de AG e carbo-hidratos poderá, deste
modo, ter contribuído para uma melhor preservação da função miocárdica
comparativamente com o grupo alimentado com dieta normal. Em consonância com as
observações anteriormente mencionadas, que relatam diminuição da actividade no PPAR-α
na progressão da IC (Barger et al., 2000), a expressão e actividade do PPAR-α diminuiu no
miocárdio VE dos animais do grupo injectado com MCT que manteve o regime alimentar
normal. O mesmo não se verificou, no entanto, no grupo alimentado com dieta
hipercalórica, o que poderá ser parcialmente explicado pela indução do PPAR-α pelos
lípidos oriundos da dieta (Gulick et al., 1994). Em concordância com os nossos resultados,
a activação de PPAR-α normalizou o metabolismo na hipertrofia e falência VD experimental
(Jucker et al., 2007). Apesar de não termos verificado alterações da expressão génica das
enzimas envolvidas na oxidação de AG e da isoforma 4 da PDK, devemos salvaguardar
que estas não são apenas reguladas a nível transcricional e que os seus níveis de
expressão nem sempre traduzem a actividade enzimática (Stanley et al., 2005). Contudo,
podemos pressupor que as alterações na expressão e actividade do PPAR-α induzidas
pela dieta hipercalórica tiveram muito provavelmente um efeito metabólico benéfico, uma
vez que dietas ricas em lípidos preveniram o declínio da actividade das enzimas
responsáveis pela oxidação dos AG e da actividade oxidativa mitocondrial, sem agravarem
a hipertrofia, quer num modelo experimental de sobrecarga de pressão VE (Chess et al.,
2009), quer em ratos espontaneamente hipertensos. Concomitantemente, a actividade de
transcrição do PPAR-α e a função mitocondrial e miocárdica foram preservadas (Iemitsu et
al., 2008). A disfunção mitocondrial, maior produção de ROS e a menor capacidade de
produção energética acompanham a progressão da disfunção do VD na HP induzida pela
MCT (Daicho et al., 2009; Redout et al., 2007). A atenuação da HP pela dieta hipercalórica
é aparentemente um resultado surpreendedor, no entanto, evidências recentes sugerem
que os agonistas do PPAR podem de facto atenuar a HP (Hansmann et al., 2007) e,
portanto, o elevado conteúdo lipídico da dieta hipercalórica ingerida pelos ratos injectados
com MCT poderá ter atenuado a HP simplesmente por indução do PPAR.
Concluindo, num modelo experimental de HP severa, com rápida progressão para IC e
caquexia, demonstramos que um regime alimentar hipercalórico, rico em gorduras

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
57
saturadas e carbo-hidratos simples, que normalmente é tido como um factor nutricional de
risco cardiovascular, atenuou a HP, melhorou a sobrevida e a função miocárdica,
possivelmente devido a alterações benéficas no metabolismo miocárdico e à atenuação
concomitante da actividade neuroendócrina e inflamatória, o que poderá ter-se devido, em
parte, à modificação da actividade de factores de transcrição. No entanto, devemos
salvaguardar que um período mais prolongado de alimentação hipercalórica poderia ter
induzido acumulação cardíaca de triglicerídeos e comprometido a função miocárdica (Akki e
Seymour, 2009).

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
58
Relevância Clínica e Perspectivas Futuras
Neste trabalho produzimos evidências experimentais e fisiopatológicas que apoiam os
achados epidemiológicos paradoxais de melhor prognóstico nos insuficientes cardíacos
mais obesos (“paradoxo da obesidade”).
Os resultados apresentados sugerem que pacientes com caquexia cardíaca e IC
descompensada poderão beneficiar de regimes alimentares hipercalóricos.
Para confirmar esta hipótese, será importante reproduzir estes achados em modelos
adicionais de caquexia cardíaca e, posteriormente, através de ensaios clínicos
randomizados, avaliar o efeito de regimes alimentares similares em pacientes com
caquexia cardíaca.
Se as hipóteses levantadas forem confirmadas, a atitude médica perante a nutrição dos
pacientes com IC terminal e caquexia poderá sofrer uma evolução importante, com
potencial melhoria dos cuidados de saúde prestados.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
59
6. Anexo
Current Pathophysiological Concepts and
Management of Pulmonary Hypertension
(Novos Conceitos Fisiopatológicos e
Abordagem Terapêutica da Hipertensão Pulmonar)
Artigo de revisão submetido para peer review na revista International Journal of Cardiology, pelos
autores Lourenço AP, Fontoura D, Henriques-Coelho T, e Leite-Moreira AF, em Setembro de 2010.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
60
Current Pathophysiological Concepts and Management of Pulmonary Hypertension
Authors
André P. Lourenço1, Dulce Fontoura1, Tiago Henriques-Coelho1, Adelino F. Leite-Moreira1
1Department of Physiology, Faculty of Medicine, University of Porto, Porto, Portugal
Word count: 4998 (abstract+manuscript+figure legends)
Address for correspondence:
Prof. Dr. Adelino F. Leite-Moreira
Departments of Physiology and Cardio-Thoracic Surgery
Faculty of Medicine, University Hospital São João
Alameda Professor Hernâni Monteiro
4200 - 319 Porto, PORTUGAL
Tel: +351225513644; Fax: +351225513646
Email: [email protected]

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
61
Abstract
Pulmonary hypertension (PH), increasingly recognized as a major health burden, remains
underdiagnosed due to unspecific symptoms and the need for right-heart catheterization.
Pulmonary artery hypertension (PAH) has been extensively investigated. Pathophysiological
knowledge derives mostly from experimental models. Paradoxically, common non-PAH PH
forms remain largely unexplored. Drugs targeting lung vascular tonus became available
during the last two decades, notwithstanding disease progresses in many patients. The aim
of this review is to summon recent advances in epidemiology, pathophysiology and
management with particular focus on associated myocardial and systemic compromise and
experimental therapeutic possibilities. PAH, currently viewed as a panvasculopathy, is due
to a crosstalk between endothelial and smooth muscle cells, inflammatory activation and
altered subcellular pathways. Cardiac cachexia and right ventricular compromise are
fundamental determinants of PH prognosis. Combined vasodilator therapy is already
mainstay for refractory cases, but drugs directed at these new pathophysiological avenues
may constitute a significant advance.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
62
Abbreviations
5-HT, serotonin
5-HT2A, serotonin type 2A receptor
6MWT, 6-minute walk test
AC, adenylate cyclase
AS, atrial septostomy
BMP, bone morphogenetic protein
BMPR1, bone morphogenetic protein receptor 1
BMPR2, bone morphogenetic protein receptor 2
BNP, type B natriuretic peptide
CaL, L-type Ca2+
-channel
CC, cardiac cachexia
CCB, Ca2+
-channel blocker
CCR2, chemokine receptor 2
CCR5, chemokine receptor 5
CCT, chest computerized tomography
CDK, cyclin-dependent kinase
cGMP, cyclic guanosine monophosphate
CHD, congenital heart disease
CHLT, combined heart-lung transplantation
CO, cardiac output
CO-A/R, co-repressors or activators
COPD, chronic obstructive pulmonary disease
CTD, connective tissue disease
CTEPH, chronic thromboembolic pulmonary hypertension
CVC, central venous catheter
CX3CR1, chemokine receptor 1
CXCR4, α-chemokine receptor
DCA, dichloroacetate
DLCO, carbon monoxide diffusion
e-, electron
ECM, extracellular matrix
EF, ejection fraction
EGFR, epidermal growth factor receptor
EPC, endothelial progenitor cells
ERA, endothelin-1 receptor antagonists
ET-1, endothelin-1
ETA, endothelin-1 type A receptor
ETC, electron transport chain
FDA, Food and Drug Administration
fPAH, familial pulmonary arterial hypertension
GC, guanylate cyclase
Gq, protein Gq
HF, heart failure
HIF-1α, hypoxia-inducible factor-1α
HIV, human immunodeficiency virus
Id, inhibitor of DNA binding proteins
IL-6, Interleukin-6
IP1, prostaglandin receptor
IP3, inositol 3-phosphate
iPAH, idiopathic pulmonary arterial hypertension
iv, intravenous
Kv1.5, O2-sensitive K+-channels
LHD, left-heart disease
LV, left ventricular
LT, lung transplantation
MCP-1, monocyte chemotactic protein-1
MLC, myosin light-chain
MLCK (-P), myosin light-chain kinase, and respective phosphorylated
form
MMP, matrix metalloproteinases
mPAP, mean pulmonary artery pressure
NFAT, nuclear factor of activated T lymphocytes
NIH, National Institutes of Health
NO, nitric oxide
NRCT, non-randomized clinical trial
O2
.-, superoxide anion
PA, pulmonary arterial
PAP, pulmonary artery pressure
PAH, pulmonary arterial hypertension
PASMC, pulmonary artery smooth muscle cell
PEA, pulmonary endarterectomy
PCH, pulmonary capillary haemangiomatosis
PDE, phosphodiesterases
PDE5, type 5 phosphodiesterase
PDEi, phosphodiesterase inhibitors

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
63
PDGF, platelet derived growth factor
PDGFR, platelet derived growth factor receptor
PDH (-P), pyruvate dehydrogenase, and respective phosphorylated form
PDK, pyruvate dehydrogenase kinase
PGI2, prostacyclin
PH, pulmonary hypertension
PKA, protein-kinase A
PKG, protein-kinase G
PTE, pulmonary thromboembolism
PVOD, pulmonary veno-occlusive disease
PVR, pulmonary vascular resistance
QOL, quality of life
RANTES, regulated upon activation, normal T expressed and secreted
RCT, randomized clinical trials
RHC, right-heart catheterisation
ROS, reactive oxygen species
RV, right ventricular
RVAD, right ventricular assist device
sc, subcutaneous
SDF-1, stromal cell-derived factor-1
SLE, systemic lupus erythematosus
SOD, superoxyde dismutase
SPAP, systolic pulmonary artery pressure
SR, sarcoendoplasmic reticulum
TCW, time to clinical worsening
TGF-β, transforming growth factor -β
TGF-βR, transforming growth factor -β receptor
TP, ThromboxaneA2 receptor
TnC, tenascin C
TNF-α, tumor necrosis factor-α
Trp, transient receptor potential
TxA2, thromboxane A2
US, United States
VEGF, vascular endothelial growth factor
VEGR, vascular endothelial growth factor receptor
WHO, World Health Organization

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
64
Introduction
Pulmonary hypertension (PH), classically diagnosed by mean pulmonary arterial (PA)
pressure (mPAP) elevation above 25 mmHg at rest.1 In most cases, PH accompanies
cardio-respiratory conditions and does not involve the pulmonary vasculature. However,
more rarely it may present itself as pulmonary arterial hypertension (PAH), defined
additionally by normal left ventricular (LV) filling pressure.2 PAH is viewed as a
vasoproliferative disease with characteristic pathological abnormalities, such as arteriolar
plexiform lesions. Initial symptoms, mainly fatigue and dyspnea, are usually vague and
insidious, thus most cases are diagnosed when cardiac output (CO) is already low.3 Right
ventricular (RV) failure due to PH is an important cause of death4 whose complex
pathophysiological mechanisms are just beginning to be understood. The last two decades
have been fertile in experimental and clinical studies. Several new drugs have become
available.3 Nevertheless, the prognosis remains poor, and many patients require
transplantation.5 The present review aims to summarize much of what has recently been
published on the epidemiology, pathophysiology, diagnosis and management of PH.6,7
Aetiology and classification
Several conferences on PH have been fostered by the World Health Organization (WHO). A
classification was proposed in 1973 and then modified at Evian in 1988 to better reproduce
pathophysiology and clinical presentation. At Venice in 2003, the term primary PH was
substituted for idiopathic PAH (iPAH) and pulmonary veno-occlusive disease (PVOD) and
pulmonary capillary haemangiomatosis (PCH) were grouped under a single PAH
subcategory. In 2008, the 4th World Symposium held in Dana Point (Table 1) endorsed the
expression “non-PAH PH” to address categories other than PAH.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
65
Table 1. New classification for pulmonary hypertension (PH) from the 4th World Symposium on
PH (Dana Point, 2008).
Pulmonary arterial
hypertension (PAH)
Non-PAH pulmonary hypertension (PH)
Well defined cause Unclear or
multifactorial
PAH (1)
Idiopathic
Hereditary
Drug/toxin induced
Disease associated:
CTD
HIV infection
Portal hypertension
Systemic-pulmonary
shunts
Schistosomiasis
Chronic haemolytic
anaemia
Subclass of PAH (1’)
PVOD and PCH
Left-heart disease (2)
Systolic dysfunction
Diastolic dysfunction
Valvular Disease
Lung diseases /hypoxia (3)
COPD
Interstitial lung disease
Sleep-disordered breathing
Chronic exposure to high
altitude
Broncho pulmonary dysplasia
Developmental abnormalities
CTEPH (4)
Unclear/multifactorial
mechanisms (5)
Haematologic disorders
Myeloproliferative disorders,
etc.
Systemic disorders
Vasculitis, sarcoidosis,
neurofibromatosis, etc.
Metabolic disorders
Glycogen storage disease,
thyroid disorders, etc.
Congenital heart disease (Other than systemic-pulmonary shunt)
Other
Fibrosing mediastinitis,
Chronic renal failure on
dialysis, etc.
Classes are presented between parenthesis. CTD, connective tissue disease; HIV, human
immunodeficiency virus; PVOD, pulmonary veno-occlusive disease; PCH, pulmonary capillary angiomatosis;
COPD, chronic obstructive pulmonary disease; CTEPH, chronic thromboembolic PH.
Diagnosis
During the 4th Conference exercise values were excluded as a criteria for diagnosis since
the increase in mPAP during exercise frequently exceeds 30 mmHg among the elderly.8
Additionally, non-invasive echocardiographic criteria of systolic tricuspid regurgitant velocity
were contemplated (Table 2).9 Nevertheless, transpulmonary flow and pulmonary venous
pressure are not reliably measured by echocardiography thus right-heart catheterisation
(RHC) remains the gold standard while echocardiography is usually a screening exam. RHC
is mandatory for the selection of patients that may benefit from Ca2+-channel blockers
(CCB), the positive responders during vasoreactivity test, those in whom mPAP drops more
than 10 mmHg or to values bellow 40 mmHg with normal or increased CO, after

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
66
administration of a short-acting vasodilator, such as nitric oxide (NO),10 epoprostenol or
adenosine.7
Table 2. New diagnostic criteria for pulmonary hypertension (PH) from the 4th World Symposium
on PH (Dana Point, 2008)
Method Normal Borderline Clear
mPAP (mmHg) < 21 21 - 25 > 25
systolic tricuspid regurgitation (m.s-1) < 2.5 2.5 - 2.8 > 2.8
mPAP, mean pulmonary artery pressure.
Epidemiology
The incidence and prevalence of PAH were estimated to be 2.4 - 7.6 cases/million/year and
15 - 26 cases/million, respectively, in large population studies.11, 12 Worldwide prevalence is
hard to appraise, but it is surely underdiagnosed13 and its onus is likely greater than
recognized, given the newly revealed associations with haemodialysis,14 the metabolic
syndrome,15 and developing world diseases, such as human immunodeficiency virus (HIV)
infection, schistosomiasis, and sickle cell disease.16 Apart from iPAH no precise estimates
of incidence or prevalence are available. Nevertheless, non-PAH PH is increasingly
recognized as a major health burden. Not only up to 60% of patients with severe LV systolic
dysfunction but also 70% of those with heart failure (HF) and normal ejection fraction (EF)17
develop PH.18, 19 Moreover, PH afflicts 70% of patients with rheumatic heart disease.20 Many
patients develop chronic thromboembolic PH (CTEPH) after pulmonary thromboembolism
(PTE)20 or PH during the progression of chronic obstructive pulmonary disease (COPD).
Prevalence ranges from 35 to 90% according to stage,21, 22 mostly limited to systolic PAP
(SPAP) values of 25 - 35 mmHg, though severe PH is not uncommon in advanced COPD.23
Some patients develop disproportionate PH and warrant particular attention,21 but even
modest PH has a strong impact on quality of life (QOL) and survival.22 Right HF, its most
severe complication, is responsible for 10 - 30% of admissions due to decompensated HF.24
Presently COPD is already responsible for 84% of cor pulmonale cases and, due to
smoking, will be the 3rd cause of death by 2020.23

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
67
Clinical Presentation and Workup
Severe disease may present with chest pain, palpitation, oedema, ascites, and syncope9 but
earlier treatment, at reversible stages, is fundamental. Diagnosis is challenging, a delay of 2
to 3 years is common and a high suspicion level is needed.13 The clinician may find RV
hypertrophy on the electrocardiogram and hilar PA prominence on the chest X-ray.
Echocardiography, generally undertaken after a suspicion, may show increased SPAP,
estimated by the velocity of tricuspid regurgitation jet, and/or increased RV outflow tract
acceleration time. It is fundamental to evaluate valve or primary myocardial disease, as well
as the degree of RV hypertrophy and dysfunction.9 Magnetic resonance imaging techniques
have also been used.25 Regarding differential diagnosis, patients with suspicion of PTE
should undergo the highly sensitive ventilation-perfusion (V-Q) scan. Nevertheless, final
staging and surgical indication rely on chest computerized tomography (CCT) and/or
angiography with RHC. High-resolution CCT is useful to assess PVOD or PCH and to
diagnose interstitial lung or connective tissue disease (CTD).7, 9 Finally antinuclear
antibodies, autoimmune disease markers, HIV and viral hepatitis screening, coagulation
disorder markers (eg, protein S and C, lupus anticoagulants, von Willebrand factor) and type
B natriuretic peptide (BNP) may be carried out for differential diagnosis.7, 9 Functional
respiratory evaluation relies on spirometry and carbon monoxide diffusion (DLCO).
Spirometry may be markedly altered in lung disease, whereas minor changes are found in
iPAH. DLCO impairment correlates with lung vascular surface area and PAH severity.26 The
6-minute walk test (6MWT), a common clinical trial end-point, evaluates moderate to severe
heart or lung disease (Table 3). It correlates well with CO, PVR, O2 consumption, QOL, and
predicts mortality in PAH.27 Nevertheless, since it depends on many individual variables, it is
not a reliable marker of disease progression.7 Additionally, its validity has been questioned
for CTD.28

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
68
Table 3. The 6-minute walk test (6MWT) in pulmonary hypertension (PH).
Features
Submaximal exercise test
Correlates well with activities of daily living (useful for moderately severe functional impairment)
Non-specific (evaluates the response of all systems)
Well tolerated (nevertheless, appropriate response to an emergency should be available)
Measurements
Dyspnoea and fatigue self-rating at the beginning and end (according to the Borg scale, see legend)
Distance walked
Demographic and anthropometric determinants
Gender, age and ethnicity
Height and weight
Advantages
Practical and inexpensive to perform (no equipment or specially trained technicians needed)
Reproducible (estimated coefficient of variability of 8%)
Ongoing monitoring of cardiopulmonary disease progression
Evaluation of response to therapy
Limitations
Merely a rough estimate of the general functional status (does not discard specific assessment tools)
Lack of validation for connective tissue disease associated PAH (musculoskeletal involvement)
“Ceiling effect” for patients with better baseline capacity
Biases: disturbed cognition, motivational factors, test repetition, musculoskeletal limitations, etc.
A comprehensive perspective on the 6MWT including indications, contraindications, safety precautions,
technical aspects, biases, can be found in the guidelines from the American Thoracic Society.119
The 6MWT
measures the distance that a patient can walk on a flat surface in a period of 6 minutes, patients are allowed
to stop and rest. The normal walked distance for healthy 60 year-old men and women of average constitution
is approximately 630 and 550 m, respectively,120 whereas idiopathic PAH patients on World Health
Organization functional class IV usually walk less than 200 m.27 A clinically important improvement in walking
distance for the PAH patient is generally 44 - 76 m.89
Borg scale: (0) nothing at all, (0.5) just noticeable, (1) very slight, (2) slight,(3) moderate, (4) somewhat
severe, (5) severe, (7) very severe, and, finally, (10) maximal.121

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
69
Pathophysiology
Although no animal model completely recapitulates human PAH, combining multiple insults,
according to the multiple-hit hypothesis, yielded severe phenotypes that closely mimic it.29
Pathophysiological knowledge, derived mostly from these animal studies, once viewed PH
as an imbalance between pulmonary vasoconstrictors and vasodilators.30 While prostacyclin
(PGI2) and NO normally govern vascular tone, endothelin-1 (ET-1), thromboxane A2 (TxA2)
and serotonin (5-HT) take over in PH. Not surprisingly, lung vasodilators have been the
mainstay of therapy (Figure 1).3 Nevertheless, recent research showed this view to be highly
incomplete.
PAH as panvasculopathy
PAH is currently viewed as a panvasculopathy, accompanied by histological features as
intimal hyperplasia, medial hypertrophy, and arteriolar occlusion by thrombosis, infiltration
by inflammatory cells or angioproliferative plexiform lesions (Figure 1).7 It is hypothesized
that apoptosis generates apoptosis-resistant endothelial cell phenotypes that cross-talk with
PA smooth muscle cell (PASMC) through growth factors such as transforming growth factor-
β (TGF-β), that are involved in endothelial cell and fibroblast transdifferentiation and PASMC
proliferation.31 Metalloproteinase activation leads to the disruption of the basement
membrane enabling inflammatory cell recruitment and further generation of mitogenic
peptides.32 The main mechanisms involved in inflammation, endothelial progenitor cell
(EPC) recruitment, growth factor activity and extracellular matrix remodeling are
summarized in figure 1. PAH shares a mitochondrial-metabolic abnormality with cancer, the
“Warburg phenotype”, a shift from oxidative phosphorylation to glycolysis (despite adequate
O2 supply) that enhances proliferation and prevents apoptosis (Figure 2). Hyperpolarization
of the mitochondrial membrane, reduced production of reactive oxygen species (ROS),
normoxic-activation of hypoxia inducible factor-1α, overexpression of pyruvate
dehydrogenase kinase (PDK) and decreased expression of O2-sensitive K+ channels
(Kv1.5) have been postulated to underlie changes in mitochondrial O2 sensing.33 PDK
activation suppresses aerobic glucose metabolism and decreased Kv1.5 conductance
depolarizes the membrane. Dichloroacetate (DCA), a mitochondrial PDK inhibitor and Kv1.5
channel opener, improved PAH33 both by activating pyruvate dehydrogenase (PDH) and
aerobic metabolism and by restoring membrane potential and ROS production.34

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
70
Figure 1. Pulmonary artery smooth muscle cell (PASMC) constriction and proliferation mechanisms.
The major sites of action of lung vasodilator drug classes are shown
in the upper panel, namely Ca2+
-channel blockers (CCB),
endothelin-1 (ET-1) receptor antagonists (ERA), phosphodiesterase
inhibitors (PDEi) and prostanoids. Myosin light-chain (MLC) kinase
(MLCK) is inactivated upon phosphorylation (MLCK-P). Other
mechanisms are presented in the lower panel. Several cytokines,
beyond interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α),
mostly produced by fibroblasts, such as stromal cell-derived factor-1
(SDF-1), monocyte chemotactic protein-1 (MCP-1), fractalkine,
RANTES (regulated upon activation, normal T expressed and
secreted), and vascular endothelial growth factor (VEGF) are
upregulated and induce PASMC proliferation and monocyte
recruitment, while monocytes upregulate the α-chemokine receptor
(CXCR4) and chemokine receptors 1, 2 and 5 (CX3CR1, CCR2
and CCR5, respectively).40 Elastase, early activated in PH, triggers
growth factors release from the extracellular matrix (ECM) and
induces tenascin C (TnC) through the activation of matrix
metalloproteinases (MMP). When TnC binds surface integrins on
PASMCs cell-survival signals are generated and growth factor
receptors are further activated. Serotonin (5-HT) induces
proliferation of PASMCs by stimulation of S100A4/Mts1, a S100
Ca2+-binding protein family member with metastasis-inducing
ability.3 Endothelial progenitor cells (EPC) may participate in vessel
repair, but on the other hand also take part in plexiform lesions.6
Other abbreviations: TxA2, thromboxane A2; Gq, protein Gq; IP3,
inositol 3-phosphate; SR, sarcoendoplasmic reticulum; ETA, ET-1
type A receptor; TP, TxA2 receptor; 5-HT2A, 5-HT type 2A receptor;
CaL, Type L Ca2+-channel; GC, guanylate cyclase; AC, adenylate
cyclase; IP1, prostacyclin receptor; PDE5, phosphodiesterase type
5; PKG, protein-kinase G; PKA, protein-kinase A; VEGFR, VEGF
receptor; PDGF, platelet derived growth factor; TGF-β, transforming
growth factor-β; PDGFR, PDGF receptor; TGFβR, TGF-β receptor;
EGFR, epidermal growth factor receptor.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
71
Figure 2. Reactive oxygen species (ROS), disturbed O2 sensing, and mitochondrial dysfunction
in pulmonary arterial hypertension (PAH).
During oxidative phosphorylation, electrons (e-) are conveyed by the electron transport chain (ETC) from donors
(NADH and FAH) to O2, but minor side reactions also generate by-products, as superoxide anion (O
2
.-) that must be
detoxified to H2O2 by superoxyde dismutase (SOD).133
Under normoxia H2O2 constitutively opens plasma membrane O2-
sensitive K+-channels (Kv1.5) and inhibits hypoxia-inducible factor-1α (HIF-1α) activity, whereas during hypoxic
vasoconstriction, ROS and H2O2 production are decreased, Kv1.5 channels close, the plasma membrane depolarizes, Ca2+
enters the cell and myocytes contract. In PAH, mitochondrial abnormalities, most notably pyruvate dehydrogenase kinase
(PDK) activation, shift metabolism toward anaerobic glycolysis and impair the ETC. Reduced ROS production, nuclear
translocation of HIF-1α, and decreased expression of Kv1.5 ultimately lead to sustained membrane depolarization, L-type
Ca2+
-channel (CaL) activation and hypertrophy by Ca2+
-calcineurin-dependent activation of the nuclear factor of activated T
lymphocytes (NFAT).6 PDH, pyruvate dehydrogenase, and respective phosphorylated form (-P); ET-1, endothelin-1; VEGF,
vascular endothelial growth factor.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
72
Genetics of PAH
Mutations in bone morphogenetic protein (BMP) receptor-2 (BMPR2), a constitutively active
receptor responsive to TGF-β superfamily (including BMP), are seen in more than 80% of
familial PAH (fPAH) cases, leading to loss of smad signaling (Figure 3) and therefore to
increased proliferation and decreased differentiation of PASMC 35, 36. Still, penetrance is low
and the mutation is seen only in 10 to 20% of non-fPAH 37. Other genetic mechanisms
predispose to PAH, namely single-nucleotide polymorphisms of Kv1.5,18 transient receptor
potential (trp) channels,13 and serotonin transporters.38 Trp channels regulate contractility
and cell proliferation by intracellular Ca2+.39 Elevated 5-HT levels and 5-HT transport have
been implicated in PAH pathogenesis.38
Figure 3. Growth-promoting pathways in bone morphogenetic protein (BMP) receptor type 2 (BMPR-2)
mutations.
Bone morphogenetic protein (BMP) receptor type 1 (BMPR-1)
and -2 dimerize upon activation by BMP and initiate a cytosolic
receptor-activated Smad protein-signaling cascade. Smads
(homology with drosophila’s mothers against decapentaplegic –
MAD - and Caenorhabditis elegans’ small phenotype - sma -
proteins) ultimately complex with common partner Smad4 and
translocate to the nucleus. The weak Smad-DNA interaction
requires co-repressors or activators (Co-A/R).134 Signal
disrupting mutations in BMPRII can be found in the ligand-
binding domain, in the kinase domain or in the cytoplasmic tail.
Supression of Smad signalling partly underlies the hypertrophic
and proliferative phenotype of pulmonary artery smooth muscle
cells (PASMC).135 In normal PASMC, BMP stimulates
transcriptional activation of cyclin-dependent kinase (CDK)
inhibitors and repression of c-myc. CDK inhibitor prevents
progression in cell cycle, while c-myc encodes a transcriptional
activator responsible for growth and proliferation.136 Inhibitor of
DNA (Id) binding proteins, a family deprived of DNA-binding
domain that acts by inhibition of transcription factors, are also
major targets. Failure to induce Id genes makes PASMC
unresponsive to the growth suppressive effects of BMPs.137
Prostanoids, by cyclic adenosine monophosphate and a direct
effect on the Id promoter, drive the expression of Id proteins.134

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
73
Inflammation
The inflammatory state of the vessel wall has recently gained interestas primary event,
rather than mere consequence of the disease.40 Autoantibodies and infiltration by
inflammatory cells are common in PAH associated with CTD but are also seen in iPAH.40
Increased levels of cytokines and their receptors have been demonstrated, particularly in
iPAH patients,41 who also present heightened expression of inflammatory cell-associated
nuclear factor of activated T lymphocytes (NFAT).42 Cytokines involved in the pathogenesis
of chronic inflammatory diseases and cancer, such as tumor necrosis factor-α (TNF-α) and
IL-6, may play a role in PAH vasculopathy.43 Our group has tested an anti-inflammatory
approach in experimental models of PH with variable success44, 45. Inflammatory activation
may also underlie systemic manifestations, for instance cardiac cachexia (CC). CC is
characterized not only by neuroendocrine and inflammatory activation but also by
suppressed appetite and nutritional derangements46 and poses a significant prognostic
burden on HF patients.47 CC accompanies the progression of PH, indeed, patients with
severe PH have exaggerated and early post-prandial satiety hormone response.48
The myocardium in PH
The RV effectively serves as a thin, compliant reservoir for blood returning to the LV whose
primary function of is to deliver deoxygenated blood to the lungs, while maintaining low-
pressure perfusion.49 It is thus best suited for volume work and unable to suddenly generate
mPAP > 40mmHg. Sudden afterload decreases stroke volume and dilates the RV50,
whereas progressive overload allows gradual hypertrophy, remodeling and substantial
increases in mPAP. Curiously, although RV response partly determines the outcome,25, 51
despite the fact that the RV can be targeted therapeutically in PH,52 and even though RV
remodeling is potentially reversible, as seen after lung transplantation (LT),53 little is known
about the mechanisms underlying RV dysfunction.49 Many have shown neuroendocrine
activation can contribute to RV hypertrophy,54, 55 fibrosis and apoptosis, as well as to
oxidative stress, and activation of inflammatory cytokines and growth factors.49, 56 A state of
myocardial hibernation has been proposed based on systolic flow impediment to coronary
arteries which is proportional to RV pressure and mass.57 In contrast to the normal flexible
metabolism, in RV hypertrophy the myocardium relies solely on anaerobic glucose
metabolism partly due to PDK activation58 and possibly impaired mitochondrial energy-
producing ability.59 Moreover, changes in cardiomyocyte redox state can underlie

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
74
electrophysiological instability and remodeling, by mechanisms similar to those already
described for pulmonary vessels.60 Experimental findings and clinical observations suggest
that elevated mPAP cannot be the single driver for RV failure,56 therefore, targeting the RV
may be a promising approach.6 As for the LV myocardium, echocardiography shows
compromised LV function in various aetiologies of PH,61 mainly due to ventricular
interdependence and impaired filling.62 Nevertheless, myocardial abnormalities partly
underlie LV dysfunction. Indeed, despite immediate restoration of LV geometry and RV
function, LV filling is only normalized 1 year after single-LT in severe PH,63 and combined
heart-lung transplantation (CHLT) is favored if LV function is impaired because the LV may
not recover after LT alone.64 We have confirmed intrinsic LV myocardial dysfunction and
neuroendocrine activation experimentally.55, 65
Pathophysiology of non-PAH PH
Contrarily to PAH, and paradoxically, few data are available on the pathophysiology of the
far more common non-PAH PH. Regarding chronic pulmonary disease, several mechanisms
are potentially responsible. Hypoxia, such as is found at high altitude, is known to induce
PH, but low arterial O2 is not an independent predictor of mPAP, therefore after the Evian
Conference COPD-associated PH was no longer classified as ‘associated with
hypoxemia’.26 Pulmonary vessels in COPD consistently develop intimal fibro-elastic
thickening and overall muscularisation but this does also not provide a consistent
explanation.66 Endothelial dysfunction and inflammation, are currently viewed as the key to
vascular remodeling.67 Findings strongly suggest an involvement of vasoactive mediators
and cytokines.66 Plasma IL-6 correlates with mPAP and certain IL-6 genotypes are
associated with PH development in COPD.68 Symptomatic CTEPH affects 3.8% of patients
within 2 years of initial PTE accompanied by variable degrees of PAH-like vasculopathy.69
As for left-heart disease (LHD), increased filling pressures are transmitted to the pulmonary
circulation and generate, initially, pulmonary venous hypertension, but, later on, also PVR
increase, which leads to endothelial dysfunction and disturbances of 5-HT, TxA2 and
angiotensin-II production that further aggravate PH.70

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
75
Prognosis
Although PAH has been most extensively studied, its rarity, diverse etiology and changing
therapeutics preclude an estimation of yearly mortality rates. An early registry followed 194
patients with iPAH from 1981 to 1985 and estimated a median survival of 2.8 years, with 1-,
3-, and 5-year survival rates of 68, 48, and 34%, respectively.71 Present-day registries,
however, reveal a better prognosis, with 1 year survival ranging 83 to 88% and 3 year
survival 58 to 72%.72 A risk-prediction equation could be derived from multivariate analysis,
including gender, 6MWT, and CO at diagnosis as covariates.73 Four variables were
associated with increased 1-year survival: WHO functional class I, 6MWT ≥ 440 m, BNP <
50 pg/mL, and DLCO ≥ 80% of predicted.73 The progression in non-PAH PH is generally
slower and the overall prognosis is better. Still, there is a substantial impact on QOL and
survival.22, 74 The level of PAP is a good indicator of prognosis in COPD and a 50% 5-year
survival rate has been reported with PH.75 Regarding CTEPH, survival changed
dramatically. Before the advent of pulmonary endarterectomy (PEA) patients who had
mPAP higher than 30 mmHg steadily progressed to PH and 2 year-survival was lower than
20% after it reached 50 mmHg.76 Currently, in experienced centers and carefully selected
patients, PEA provides remarkable haemodynamic and clinical improvement with low
procedural mortality rate.5 In severe HF, the EF of the RV is the most important determinant
of short-term prognosis among hemodynamic variables.77 Although increased mPAP is
frequently coupled with reduced RV function, exceptions must be taken in account during
prognostic stratification.19
Therapeutics
Treatment of PAH has evolved considerably over the past decade, many treatment
algorithms have been proposed, mainly based on studies conducted in patients with iPAH
and PAH associated with CTDs. In Table 4 we summarize the major therapeutic studies on
PAH.
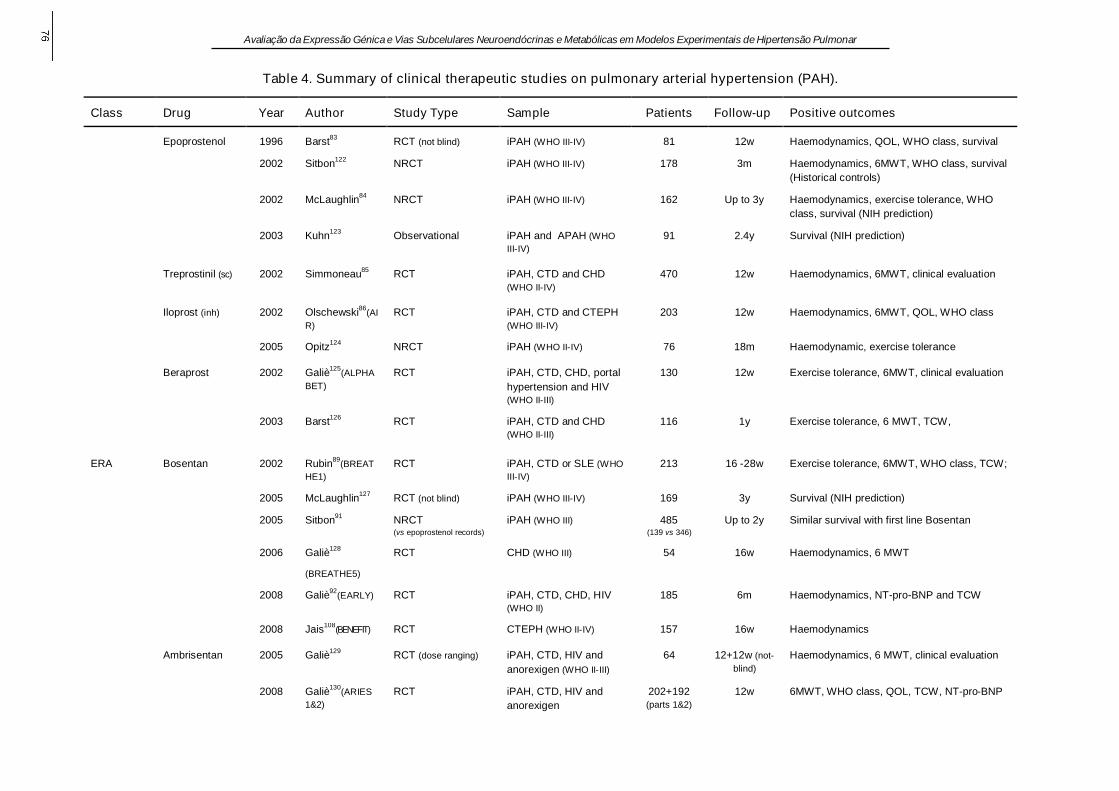
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
Table 4. Summary of clinical therapeutic studies on pulmonary arterial hypertension (PAH).
Class Drug Year Author Study Type Sample Patients Follow-up Positive outcomes
Epoprostenol 1996 Barst83
RCT (not blind) iPAH (WHO III-IV) 81 12w Haemodynamics, QOL, WHO class, survival
2002 Sitbon122
NRCT iPAH (WHO III-IV) 178 3m Haemodynamics, 6MWT, WHO class, survival
(Historical controls)
2002 McLaughlin84
NRCT iPAH (WHO III-IV) 162 Up to 3y Haemodynamics, exercise tolerance, WHO
class, survival (NIH prediction)
2003 Kuhn123
Observational iPAH and APAH (WHO
III-IV) 91 2.4y Survival (NIH prediction)
Treprostinil (sc) 2002 Simmoneau85
RCT iPAH, CTD and CHD
(WHO II-IV) 470 12w Haemodynamics, 6MWT, clinical evaluation
Iloprost (inh) 2002 Olschewski86
(AI
R) RCT iPAH, CTD and CTEPH
(WHO III-IV) 203 12w Haemodynamics, 6MWT, QOL, WHO class
2005 Opitz124
NRCT iPAH (WHO II-IV) 76 18m Haemodynamic, exercise tolerance
Beraprost 2002 Galiè125
(ALPHA
BET) RCT iPAH, CTD, CHD, portal
hypertension and HIV (WHO II-III)
130 12w Exercise tolerance, 6MWT, clinical evaluation
2003 Barst126
RCT iPAH, CTD and CHD (WHO II-III)
116 1y Exercise tolerance, 6 MWT, TCW,
ERA Bosentan 2002 Rubin89
(BREAT
HE1) RCT iPAH, CTD or SLE (WHO
III-IV) 213 16 -28w Exercise tolerance, 6MWT, WHO class, TCW;
2005 McLaughlin127
RCT (not blind) iPAH (WHO III-IV) 169 3y Survival (NIH prediction)
2005 Sitbon91
NRCT (vs epoprostenol records)
iPAH (WHO III) 485 (139 vs 346)
Up to 2y Similar survival with first line Bosentan
2006 Galiè128
(BREATHE5)
RCT CHD (WHO III) 54 16w Haemodynamics, 6 MWT
2008 Galiè92
(EARLY) RCT iPAH, CTD, CHD, HIV
(WHO II) 185 6m Haemodynamics, NT-pro-BNP and TCW
2008 Jais108
(BENEFIT) RCT CTEPH (WHO II-IV) 157 16w Haemodynamics
Ambrisentan 2005 Galiè129
RCT (dose ranging) iPAH, CTD, HIV and
anorexigen (WHO II-III)
64 12+12w (not-
blind) Haemodynamics, 6 MWT, clinical evaluation
2008 Galiè130
(ARIES
1&2) RCT iPAH, CTD, HIV and
anorexigen
202+192 (parts 1&2)
12w 6MWT, WHO class, QOL, TCW, NT-pro-BNP
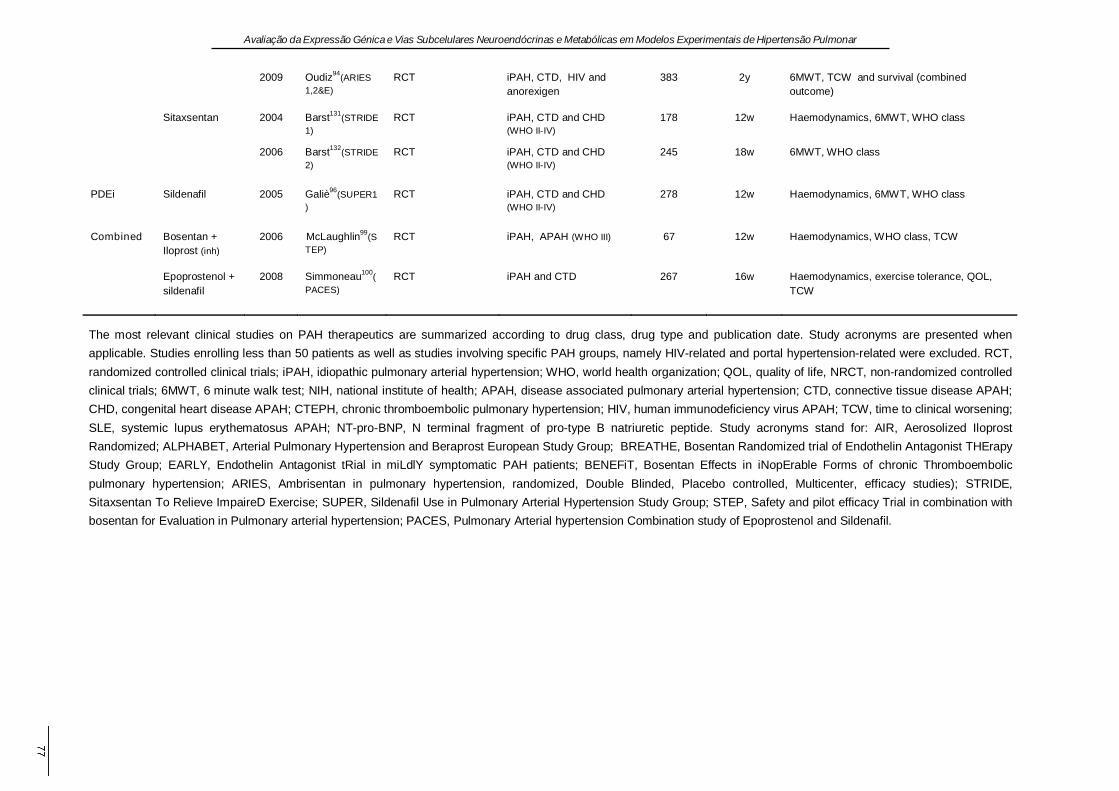
Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
2009 Oudiz94
(ARIES
1,2&E) RCT iPAH, CTD, HIV and
anorexigen
383 2y 6MWT, TCW and survival (combined
outcome)
Sitaxsentan 2004 Barst131
(STRIDE
1) RCT iPAH, CTD and CHD
(WHO II-IV) 178 12w Haemodynamics, 6MWT, WHO class
2006 Barst132
(STRIDE
2) RCT iPAH, CTD and CHD
(WHO II-IV) 245 18w 6MWT, WHO class
PDEi Sildenafil 2005 Galiè96
(SUPER1
) RCT iPAH, CTD and CHD
(WHO II-IV) 278 12w Haemodynamics, 6MWT, WHO class
Combined Bosentan +
Iloprost (inh)
2006 McLaughlin99
(S
TEP) RCT iPAH, APAH (WHO III) 67 12w Haemodynamics, WHO class, TCW
Epoprostenol +
sildenafil
2008 Simmoneau100
(
PACES) RCT iPAH and CTD 267 16w Haemodynamics, exercise tolerance, QOL,
TCW
The most relevant clinical studies on PAH therapeutics are summarized according to drug class, drug type and publication date. Study acronyms are presented when
applicable. Studies enrolling less than 50 patients as well as studies involving specific PAH groups, namely HIV-related and portal hypertension-related were excluded. RCT,
randomized controlled clinical trials; iPAH, idiopathic pulmonary arterial hypertension; WHO, world health organization; QOL, quality of life, NRCT, non-randomized controlled
clinical trials; 6MWT, 6 minute walk test; NIH, national institute of health; APAH, disease associated pulmonary arterial hypertension; CTD, connective tissue disease APAH;
CHD, congenital heart disease APAH; CTEPH, chronic thromboembolic pulmonary hypertension; HIV, human immunodeficiency virus APAH; TCW, time to clinical worsening;
SLE, systemic lupus erythematosus APAH; NT-pro-BNP, N terminal fragment of pro-type B natriuretic peptide. Study acronyms stand for: AIR, Aerosolized Iloprost
Randomized; ALPHABET, Arterial Pulmonary Hypertension and Beraprost European Study Group; BREATHE, Bosentan Randomized trial of Endothelin Antagonist THErapy
Study Group; EARLY, Endothelin Antagonist tRial in miLdlY symptomatic PAH patients; BENEFiT, Bosentan Effects in iNopErable Forms of chronic Thromboembolic
pulmonary hypertension; ARIES, Ambrisentan in pulmonary hypertension, randomized, Double Blinded, Placebo controlled, Multicenter, efficacy studies); STRIDE,
Sitaxsentan To Relieve ImpaireD Exercise; SUPER, Sildenafil Use in Pulmonary Arterial Hypertension Study Group; STEP, Safety and pilot efficacy Trial in combination with
bosentan for Evaluation in Pulmonary arterial hypertension; PACES, Pulmonary Arterial hypertension Combination study of Epoprostenol and Sildenafil.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
78
Several general measures can be recommended. Regarding exercise practice, patients
should practice low level aerobic exercise, such as walking, whereas strenuous efforts
should be avoided. Some patients may not tolerate high altitudes, for instance during
airplane flights, and therefore require O2. Dietary sodium restriction is advised (≤ 2.4 g.d-1)
particularly in RV failure. Immunization against common respiratory pathogens is
recommended.7 Pregnancy should be avoided or early terminated due to the high mortality
rate.78
Despite the lack of randomized controlled trials (RCT), the initial therapeutic, and largely
supportive, approaches to the treatment of PAH were anticoagulation, diuretics, O2 therapy
and digoxin. Observational studies suggested improved survival after anticoagulation in
patients with iPAH, therefore most experts recommend anticoagulation (titrated to
international normalized ratio of 1.5 - 2.5). This is only advised for severe non-iPAH.7
Diuretics are used to manage HF symptoms. O2 therapy in hypoxemia is employed strictly to
avoid vasoconstriction. Based on a short-term effect study, digoxin may be used in patients
with low CO particularly with supraventricular arrhythmias.79 During the last two decades
substantial RCT and pharmacological research have yielded several new and more effective
alternatives to treat PH. The main pharmacological classes will be briefly presented. Most of
the studies are small scaled and short-termed, not suitable for survival analysis, but a recent
meta-analysis found an overall benefit in mortality.80 Nevertheless, most therapies reduce
mPAP by only 10 - 20%, with the exception of strong responders to CCB. Despite all the
advancement, many patients still remain symptomatic, with a suboptimal QOL and warrant
combined therapy or even invasive or surgical procedures.
Ca2+ channel blockers
Rich et al.81 have shown a marked improvement in survival rates, for patients with iPAH and
a positive vasoreactivity test, after long-term high-dose CCB therapy. Long acting nifedipine,
diltiazem, or amlodipine are more commonly used. If there is no recovery to functional
classes I or II patients are deemed as non-responders and should discontinue CCB. True
responders are rare in non-iPAH.82 Indiscriminate use is not recommended, due to systemic
vasodilation and negative inotropic effects.9

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
79
Prostanoids
There are presently several commercially available prostanoid formulations. Intravenous (iv)
epoprostenol was the first shown to improve functional class, hemodynamics and survival in
a 12 week follow-up period in patients with iPAH of classes III and IV.83 These beneficial
effects were reproduced in long-term observational comparisons with historical controls.84
Moreover, epoprostenol was also evaluated in CTD associated PAH and other forms of non
iPAH with favourable outcomes. Presently, because of the complex administration and
cumbersome follow-up, epoprostenol use is mainly confined to highly experienced centers.
Patients must keep a central venous catheter (CVC) and handle drug preparation and
infusion. Dosing must be carefully titrated. Most patients do well with an initial in-hospital
dose of 2 ng/kg/min and a dose range between 25 and 40 ng.kg-1.min-1. Unfortunately,
substantial side-effects have been reported, namely flushing, headache, and sudden death
after abrupt discontinuation, as well as risk of infection related to CVC.7 Treprostinil, a longer
half-life prostanoid, amenable to administration by subcutaneous (sc) route, circumventing
the need for CVC, showed minor beneficial effects in patients with functional classes II - IV
of idiopathic, CTD and (CHD) associated PAH.85 The United States (US) Food and Drug
Administration (FDA) approved it for functional classes II - IV also by iv route, when the sc
route is not tolerated due to pain or erythema. On another attempt to facilitate
administration, iloprost was developed for inhalation by aerosol device. After a 12-weeks
administration, iloprost improved the 6MWT and functional class in a multicenter RCT
enrolling patients with PAH of different aetiologies.86
Endothelin receptor antagonists
We have reviewed the role of ET-1 and its antagonists (ERA) in cardiovascular
pathophysiology.87 Briefly, after a small magnitude RCT had shown improvement in the
6MWT, mPAP and CO with the non-selective ERA bosentan,88 a larger scale study
conducted in patients with idiopathic or CTD associated PAH, reproduced these findings
and reported improvement in time to clinical worsening (TCW), a secondary endpoint
defined as a composite of mortality, LT, hospitalization, discontinuation due to lack of
recovery or need for epoprostenol or atrial septostomy (AS).89 As an important side-effect,
bosentan dose-dependently altered hepatic function. Anemia can also occur and the US
FDA therefore recommends liver function test and haematocrit surveillance.7 Long-term

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
80
evaluation as a first-line drug in functional class III patients also revealed good results,
although many patients demanded prostanoids.90 In fact, improved survival was only
demonstrated by comparison with historical data from epoprostenol treated iPAH WHO
class III patients, and unfortunately the two cohorts were not comparable.91 By now,
bosentan has also been tested in CHD, HIV-associated PAH and CTEPH with favourable
results. Moreover, it has been successfully used in a large sample of mildly symptomatic,
class II, multiple cause-PAH patients improving haemodynamics and TCW.92 Sixtasentan a
selective ETA ERA initially raised concerns due to fatal hepatitis but was later shown to have
comparable effects to bosentan in iPAH and PAH associated with CTD or CHD, with a
similar side-effect profile, with lower doses 93. It is currently approved in the European Union
but not in the US. Ambrisentan, another selective ETA ERA, also improved the 6MWT and
TCW, which was reproducible in long-term studies.94 It is approved by the FDA since 2007
for PAH patients in functional classes II and III.
Phosphodiesterase inhibitors
Phosphodiesterases (PDE) degrade cyclic guanosine monophosphate (cGMP) therefore
PDE inhibitors (PDEi) potentiate the effects of cGMP generated by NO activation of
guanylate cyclase. NO and NO donors have been extensively used as a rescue therapy to
mitigate mPAP in the perioperative period and in the critically ill patient, particularly in
children.95 Sildenafill, the first used PDEi, was shown to improve 6MWT, WHO functional
class and mPAP in idiopathic, CTD or CHD associated PAH, but there were no differences
in TCW.96 The FDA approved sildenafil in low doses for patients with PAH although there is
some debate as to whether higher doses might confer additional benefits.97 Other PDEi are
currently under study.
Combination therapy
The possibility to combine distinct drug classes that target different molecular pathways in
order to improve clinical efficacy and minimize side-effects is an attractive perspective. After
an initial attempt to combine bosentan and epoprostenol in a small scale and underpowered
trial conducted on patients with either iPAH or PAH associated to CTD that proved
unsuccessful,98 another trial that combined inhaled iloprost with bosentan in patients who

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
81
remained symptomatic showed improvement in functional class, TCW and
haemodynamics.99 More recently, the addition of sildenafil to PAH patients who remained
symptomatic on a stable dose of iv epoprostenol improved the 6MWT, as well as mPAP,
CO, and TCW.100
Invasive and surgical strategies in PAH
These include AS and LT or CHLT. Other possibilities, such as the RV mechanical assist
devices (RVAD) are still poorly investigated. AS creates a right-to-left shunt that unloads the
RV, decreases mPAP, and improves LV filling. The increase in CO offsets the shunting of
deoxygenated blood and ameliorates O2 delivery. Increased CO allows bridging to
transplantation in up to 40% of patients.101 Nevertheless, it is merely palliative and
procedural mortality is still high therefore it is just a last resort for patients on maximal
medical therapy and inotropic support. Improved techniques are being currently explored to
reduce procedural risk.5 Currently, PAH is responsible for approximately 4% of LT and
CHLT, and although there is a substantial procedural related mortality, the long-term
outcome is favourable, with a 47% survival after 5 years.102 The type of transplant is still a
matter of debate and highly related to the experience of each centre. Generally CHLT is
preferred either when patients have intractable HF or are dependent on inotropic support or
if PH is secondary to CHD or LHD.64
Therapeutic algorithm
Management must be tailored to each patient according to disease severity, comorbid
conditions, drug side-effects and each centre’s experience. Vasoreactivity test is usually
reserved for iPAH patients with stable hemodynamics, otherwise first line therapy should
consist of ERA or PDEi, unless the oral route is not available, patients are in functional class
IV or present overt RV failure. In these cases, the first choice is an iv prostanoid. Moreover,
combination therapy should always be kept in mind, particularly when side-effects arise or
patients are not responding. Enrolment in clinical trials with newer pharmacological agents
may be an option but AS and transplantation should be considered before systemic
deterioration. Early referral for transplantation is crucial particularly for refractory cases.7 A
simplified therapeutic algorithm is suggested in Figure 4.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
82
Figure 4. Algorithm for pulmonary arterial hypertension (PAH) management.
CCB, calcium channel blockers; WHO, World Health Organization; ERA, endothelin-1
receptor antagonists; PDEi, phosphodiesterase inhibitors; RCT, randomized clinical
trials; iv, intravenous; sc, subcutaneous.
Non-PAH PH
Patients will benefit from medical optimization of their primary disease, but significant PH
may persist. Some patients actually present disproportionate PH not easily attributable to
the underlying condition. Particularly in these cases new PAH drugs have been used with
success.
In left-heart disease prostanoids, with the exception of inhaled route, are usually
contraindicated due to systemic vasodilation.103 ERA trials have been interrupted
prematurely due mainly to side-effects and absence of clinical benefit, even with reduced
dose,104 but selected cases may benefit from short trials as a bridge to transplantation.105 As
for PDEi short-term hemodynamic benefits, as well as long-term improvements have been
documented.106
Mild levels of PH are amenable to optimization of medical therapy in COPD. If PH is
disproportionate, and other PH causes have been ruled out, many centres are routinely
employing vasodilators despite V-Q mismatch.107 CTEPH is potentially curable with PEA
(usually with concomitant inferior vena cava filter placement).5 Yet, many patients are not

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
83
candidates so they remain anticoagulated and on diuretics. Many centres are promptly using
new PAH drugs off-label if there is associated vasculopathy.108
Recent progresses and future targets in PH
Based upon the most recent experimental findings, clinical trials targeting altered metabolic
and signalling pathways are warranted. DCA and Kv1.5 channel gene transfer have been
successful in experimental studies,33 as well as trp channel inhibitors, growth factor receptor
inhibitors and intracellular kinase inhibitors.3, 109 Inflammatory response modulation has also
been a major research topic. After several animal studies demonstrating beneficial effects of
statins,109 possibly due to pleiotropic effects, a human study disappointingly showed no
long-lasting improvement.110 Other immunomodulatory agents have been successful in
animal experiments,44, 111 but beneficial effects are mainly confined to CTD associated
PAH.112 We have also reported disturbances in endogenous endocrine and paracrine
systems113, 114 that may be targeted. Another tempting possibility is the recruitment or
infusion of EPC. The number and function of EPCs predicts prognosis, and most currently
used drugs increase circulating EPC numbers.115 Circulating EPCs home to sites of
endothelial injury, promote revascularization and improve vascular homeostasis,116
endothelial dysfunction may be related to the lack of EPCs.115 Finally, we must bear in mind
that RV failure is the final and most severe complication of PH. Agents such as
levosimendan that vasodilate lung vessels but are also positive inotropes are predictably
good therapeutic tools. Still, the clinical efficacy of these drugs has only just started to be
evaluated.117, 118
Acknowledgements
The authors would like to thank Daniela Silva, José Pinto, Francisco Vasques-Nóvoa, Rui
Cerqueira and Duarte Pinto for their contribution to the manuscript.
Conflict of Interest
None declared.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
84
References
1. Fishman AP. A century of pulmonary hemodynamics. Am J Respir Crit Care Med
2004;170:109-113.
2. Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol
2008;51:1527-1538.
3. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin
Invest 2008;118:2372-2379.
4. Humbert M. Update in pulmonary hypertension 2008. Am J Respir Crit Care Med
2009;179:650-656.
5. Keogh AM, Mayer E, Benza RL, Corris P, Dartevelle PG, Frost AE, et al.
Interventional and surgical modalities of treatment in pulmonary hypertension. J Am
Coll Cardiol 2009;54:S67-77.
6. Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for
clinicians: new concepts and experimental therapies. Circulation;121:2045-2066.
7. McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al.
ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of
the American College of Cardiology Foundation Task Force on Expert Consensus
Documents and the American Heart Association developed in collaboration with the
American College of Chest Physicians; American Thoracic Society, Inc.; and the
Pulmonary Hypertension Association. J Am Coll Cardiol 2009;53:1573-1619.
8. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during
rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009;34:888-
894. Epub 2009 Mar 2026.
9. Nef HM, Mollmann H, Hamm C, Grimminger F, Ghofrani HA. Pulmonary
hypertension: updated classification and management of pulmonary hypertension.
Heart;96:552-559.
10. Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, et al. Long-term
response to calcium channel blockers in idiopathic pulmonary arterial hypertension.
Circulation 2005;111:3105-3111. Epub 2005 Jun 3106.
11. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary
arterial hypertension in France: results from a national registry. Am J Respir Crit Care
Med 2006;173:1023-1030. Epub 2006 Feb 1022.
12. Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological
study of pulmonary arterial hypertension. Eur Respir J 2007;30:104-109. Epub 2007
Mar 2014.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
85
13. Provencher S, Jais X, Yaici A, Sitbon O, Humbert M, Simonneau G. Clinical
challenges in pulmonary hypertension: Roger S. Mitchell lecture. Chest
2005;128:622S-628S.
14. Fruchter O, Yigla M. Underlying aetiology of pulmonary hypertension in 191 patients:
a single centre experience. Respirology 2008;13:825-831.
15. Robbins IM, Newman JH, Johnson RF, Hemnes AR, Fremont RD, Piana RN, et al.
Association of the metabolic syndrome with pulmonary venous hypertension. Chest
2009;136:31-36. Epub 2009 Feb 2002.
16. Butrous G, Ghofrani HA, Grimminger F. Pulmonary vascular disease in the
developing world. Circulation 2008;118:1758-1766.
17. Leite-Moreira AF. Current perspectives in diastolic dysfunction and diastolic heart
failure. Heart 2006;92:712-718.
18. Shapiro BP, McGoon MD, Redfield MM. Unexplained pulmonary hypertension in
elderly patients. Chest 2007;131:94-100.
19. Ghio S, Gavazzi A, Campana C, Inserra C, Klersy C, Sebastiani R, et al. Independent
and additive prognostic value of right ventricular systolic function and pulmonary
artery pressure in patients with chronic heart failure. J Am Coll Cardiol 2001;37:183-
188.
20. Elliott CG, Barst RJ, Seeger W, Porres-Aguilar M, Brown LM, Zamanian RT, et al.
Worldwide physician education and training in pulmonary hypertension: pulmonary
vascular disease: the global perspective. Chest;137:85S-94S.
21. Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducolone A, et al. Severe
pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit
Care Med 2005;172:189-194. Epub 2005 Apr 2014.
22. Weitzenblum E, Hirth C, Ducolone A, Mirhom R, Rasaholinjanahary J, Ehrhart M.
Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary
disease. Thorax 1981;36:752-758.
23. Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause
1990-2020: Global Burden of Disease Study. Lancet 1997;349:1498-1504.
24. MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary
disease. Part One. Am J Respir Crit Care Med 1994;150:833-852.
25. van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG,
Spreeuwenberg MD, et al. Prognostic value of right ventricular mass, volume, and
function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007;28:1250-
1257. Epub 2007 Jan 1222.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
86
26. Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Pulmonary function in primary
pulmonary hypertension. J Am Coll Cardiol 2003;41:1028-1035.
27. Miyamoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita M, et al. Clinical
correlates and prognostic significance of six-minute walk test in patients with primary
pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J
Respir Crit Care Med 2000;161:487-492.
28. Pamidi S, Mehta S. Six-minute walk test in scleroderma-associated pulmonary
arterial hypertension: are we counting what counts? J Rheumatol 2009;36:216-218.
29. Robbins IM. Advancing therapy for pulmonary arterial hypertension: can animal
models help? Am J Respir Crit Care Med 2004;169:5-6.
30. Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, et
al. An imbalance between the excretion of thromboxane and prostacyclin metabolites
in pulmonary hypertension. N Engl J Med 1992;327:70-75.
31. Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial
hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res
2009;10:95.
32. Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase
inhibitors induce regression, and tenascin-C antisense prevents progression, of
vascular disease. J Clin Invest 2000;105:21-34.
33. Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Haromy A, et
al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway
disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded
rats: similarities to human pulmonary arterial hypertension. Circulation
2006;113:2630-2641. Epub 2006 May 2630.
34. Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK.
Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-
1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and
cancer. Am J Physiol Heart Circ Physiol 2008;294:H570-578. Epub 2007 Dec 2014.
35. Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, et al.
Dysfunctional Smad signaling contributes to abnormal smooth muscle cell
proliferation in familial pulmonary arterial hypertension. Circ Res 2005;96:1053-1063.
Epub 2005 Apr 1021.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
87
36. Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, et al. Altered
growth responses of pulmonary artery smooth muscle cells from patients with primary
pulmonary hypertension to transforming growth factor-beta(1) and bone
morphogenetic proteins. Circulation 2001;104:790-795.
37. Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, et al. Genetic
basis of pulmonary arterial hypertension: current understanding and future directions.
J Am Coll Cardiol 2004;43:33S-39S.
38. MacLean MR, Dempsie Y. Serotonin and pulmonary hypertension--from bench to
bedside? Curr Opin Pharmacol 2009;9:281-286. Epub 2009 Mar 2013.
39. Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth
muscle cell proliferation. News Physiol Sci 2004;19:44-50.
40. Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial
hypertension. Eur Respir J 2003;22:358-363.
41. Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, et al.
Increased interleukin-1 and interleukin-6 serum concentrations in severe primary
pulmonary hypertension. Am J Respir Crit Care Med 1995;151:1628-1631.
42. Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, et al. The
nuclear factor of activated T cells in pulmonary arterial hypertension can be
therapeutically targeted. Proc Natl Acad Sci U S A 2007;104:11418-11423. Epub
12007 Jun 11427.
43. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6
Overexpression Induces Pulmonary Hypertension. Circ Res 2009;104:236-244.
44. Henriques-Coelho T, Oliveira SM, Moura RS, Roncon-Albuquerque R, Jr., Neves AL,
Santos M, et al.Thymulin inhibits monocrotaline-induced pulmonary hypertension
modulating interleukin-6 expression and suppressing p38 pathway. Endocrinology
2008;149:4367-4373. Epub 2008 May 4329.
45. Henriques-Coelho T, Brandao-Nogueira A, Moreira-Goncalves D, Correia-Pinto J,
Leite-Moreira AF. Effects of TNF-alpha blockade in monocrotaline-induced pulmonary
hypertension. Rev Port Cardiol 2008;27:341-348.
46. von Haehling S, Doehner W, Anker SD. Nutrition, metabolism, and the complex
pathophysiology of cachexia in chronic heart failure. Cardiovasc Res 2007;73:298-
309.
47. Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al.
Wasting as independent risk factor for mortality in chronic heart failure. Lancet
1997;349:1050-1053.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
88
48. le Roux CW, Ghatei MA, Gibbs JS, Bloom SR. The putative satiety hormone PYY is
raised in cardiac cachexia associated with primary pulmonary hypertension. Heart
2005;91:241-242.
49. Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under
pressure: cellular and molecular mechanisms of right-heart failure in pulmonary
hypertension. Chest 2009;135:794-804.
50. Blaise G, Langleben D, Hubert B. Pulmonary arterial hypertension: pathophysiology
and anesthetic approach. Anesthesiology 2003;99:1415-1432.
51. D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al.
Survival in patients with primary pulmonary hypertension. Results from a national
prospective registry. Ann Intern Med 1991;115:343-349.
52. Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, et al.
Phosphodiesterase type 5 is highly expressed in the hypertrophied human right
ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility.
Circulation 2007;116:238-248. Epub 2007 Jul 2002.
53. Ritchie M, Waggoner AD, Davila-Roman VG, Barzilai B, Trulock EP, Eisenberg PR.
Echocardiographic characterization of the improvement in right ventricular function in
patients with severe pulmonary hypertension after single-lung transplantation. J Am
Coll Cardiol 1993;22:1170-1174.
54. Miyauchi T, Yorikane R, Sakai S, Sakurai T, Okada M, Nishikibe M, et al.
Contribution of endogenous endothelin-1 to the progression of cardiopulmonary
alterations in rats with monocrotaline-induced pulmonary hypertension. Circ Res
1993;73:887-897.
55. Lourenço AP, Roncon-Albuquerque R, Jr., Bras-Silva C, Faria B, Wieland J,
Henriques-Coelho T, et al.Myocardial dysfunction and neurohumoral activation
without remodeling in left ventricle of monocrotaline-induced pulmonary hypertensive
rats. Am J Physiol Heart Circ Physiol 2006;291:H1587-1594. Epub 2006 May 1585.
56. Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, et
al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart
failure. Circulation 2009;120:1951-1960. Epub 2009 Nov 1952.
57. van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM,
Bronzwaer JG, et al. Right coronary artery flow impairment in patients with pulmonary
hypertension. Eur Heart J 2008;29:120-127. Epub 2007 Dec 2008.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
89
58. Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, et al. The inhibition
of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical
remodeling in two models of right ventricular hypertrophy: resuscitating the
hibernating right ventricle. J Mol Med 2009;88:47-60.
59. Daicho T, Yagi T, Abe Y, Ohara M, Marunouchi T, Takeo S, et al. Possible
involvement of mitochondrial energy-producing ability in the development of right
ventricular failure in monocrotaline-induced pulmonary hypertensive rats. J
Pharmacol Sci 2009;111:33-43.
60. Hool LC. The L-type Ca(2+) channel as a potential mediator of pathology during
alterations in cellular redox state. Heart Lung Circ 2009;18:3-10. Epub 2008 Dec
2031.
61. Chang SM, Lin CC, Hsiao SH, Lee CY, Yang SH, Lin SK, et al. Pulmonary
hypertension and left heart function: insights from tissue Doppler imaging and
myocardial performance index. Echocardiography 2007;24:366-373.
62. Dong SJ, Crawley AP, MacGregor JH, Petrank YF, Bergman DW, Belenkie I, et al.
Regional left ventricular systolic function in relation to the cavity geometry in patients
with chronic right ventricular pressure overload. A three-dimensional tagged magnetic
resonance imaging study. Circulation 1995;91:2359-2370.
63. Xie GY, Lin CS, Preston HM, Taylor CG, Kearney K, Sapin PM, et al. Assessment of
left ventricular diastolic function after single lung transplantation in patients with
severe pulmonary hypertension. Chest 1998;114:477-481.
64. Pielsticker EJ, Martinez FJ, Rubenfire M. Lung and heart-lung transplant practice
patterns in pulmonary hypertension centers. J Heart Lung Transplant 2001;20:1297-
1304.
65. Correia-Pinto J, Henriques-Coelho T, Roncon-Albuquerque R, Jr., Lourenço AP,
Melo-Rocha G, Vasques-Novoa F, et al.Time course and mechanisms of left
ventricular systolic and diastolic dysfunction in monocrotaline-induced pulmonary
hypertension. Basic Res Cardiol 2009;104:535-545. Epub 2009 Mar 2014.
66. Wright JL, Levy RD, Churg A. Pulmonary hypertension in chronic obstructive
pulmonary disease: current theories of pathogenesis and their implications for
treatment. Thorax 2005;60:605-609.
67. Agusti A. Systemic effects of chronic obstructive pulmonary disease: what we know
and what we don't know (but should). Proc Am Thorac Soc 2007;4:522-525.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
90
68. Chaouat A, Savale L, Chouaid C, Tu L, Sztrymf B, Canuet M, et al. Role for
interleukin-6 in COPD-related pulmonary hypertension. Chest 2009;136:678-687.
Epub 2009 Apr 2006.
69. Hoeper MM, Mayer E, Simonneau G, Rubin LJ. Chronic thromboembolic pulmonary
hypertension. Circulation 2006;113:2011-2020.
70. Moraes DL, Colucci WS, Givertz MM. Secondary pulmonary hypertension in chronic
heart failure: the role of the endothelium in pathophysiology and management.
Circulation 2000;102:1718-1723.
71. McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, et al.
Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice
guidelines. Chest 2004;126:78S-92S.
72. McLaughlin VV, Suissa S. Prognosis of Pulmonary Arterial Hypertension. The Power
of Clinical Registries of Rare Diseases. Circulation 2010.
73. Humbert M, Sitbon O, Yaici A, Montani D, O'Callaghan DS, Jais X, et al. Survival in
incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur
Respir J 2010.
74. Hoeper MM, Barbera JA, Channick RN, Hassoun PM, Lang IM, Manes A, et al.
Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension
pulmonary hypertension. J Am Coll Cardiol 2009;54:S85-96.
75. Bishop JM, Cross KW. Physiological variables and mortality in patients with various
categories of chronic respiratory disease. Bull Eur Physiopathol Respir 1984;20:495-
500.
76. Riedel M, Stanek V, Widimsky J, Prerovsky I. Longterm follow-up of patients with
pulmonary thromboembolism. Late prognosis and evolution of hemodynamic and
respiratory data. Chest 1982;81:151-158.
77. Gavazzi A, Berzuini C, Campana C, Inserra C, Ponzetta M, Sebastiani R, et al.Value
of right ventricular ejection fraction in predicting short-term prognosis of patients with
severe chronic heart failure. J Heart Lung Transplant 1997;16:774-785.
78. Weiss BM, Zemp L, Seifert B, Hess OM. Outcome of pulmonary vascular disease in
pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol
1998;31:1650-1657.
79. Rich S, Seidlitz M, Dodin E, Osimani D, Judd D, Genthner D, et al. The short-term
effects of digoxin in patients with right ventricular dysfunction from pulmonary
hypertension. Chest 1998;114:787-792.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
91
80. Galie N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-
analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart
J 2009;30:394-403. Epub 2009 Jan 2020.
81. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers
on survival in primary pulmonary hypertension. N Engl J Med 1992;327:76-81.
82. Montani D, Savale L, Natali D, Jais X, Herve P, Garcia G, et al.Long-term response
to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur
Heart J 2010;31:1898-1907.
83. Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A
comparison of continuous intravenous epoprostenol (prostacyclin) with conventional
therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension
Study Group. N Engl J Med 1996;334:296-302.
84. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension:
the impact of epoprostenol therapy. Circulation 2002;106:1477-1482.
85. Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, et al. Continuous
subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with
pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial.
Am J Respir Crit Care Med 2002;165:800-804.
86. Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, et al.
Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002;347:322-329.
87. Brunner F, Bras-Silva C, Cerdeira AS, Leite-Moreira AF. Cardiovascular endothelins:
essential regulators of cardiovascular homeostasis. Pharmacol Ther 2006;111:508-
531. Epub 2006 Feb 2002.
88. Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al.
Effects of the dual endothelin-receptor antagonist bosentan in patients with
pulmonary hypertension: a randomised placebo-controlled study. Lancet
2001;358:1119-1123.
89. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan
therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896-903.
90. Provencher S, Sitbon O, Humbert M, Cabrol S, Jais X, Simonneau G. Long-term
outcome with first-line bosentan therapy in idiopathic pulmonary arterial hypertension.
Eur Heart J 2006;27:589-595.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
92
91. Sitbon O, McLaughlin VV, Badesch DB, Barst RJ, Black C, Galie N, et al. Survival in
patients with class III idiopathic pulmonary arterial hypertension treated with first line
oral bosentan compared with an historical cohort of patients started on intravenous
epoprostenol. Thorax 2005;60:1025-1030.
92. Galie N, Rubin L, Hoeper M, Jansa P, Al-Hiti H, Meyer G, et al. Treatment of patients
with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY
study): a double-blind, randomised controlled trial. Lancet 2008;371:2093-2100.
93. Barst RJ, Rich S, Widlitz A, Horn EM, McLaughlin V, McFarlin J. Clinical efficacy of
sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial
hypertension: open-label pilot study. Chest 2002;121:1860-1868.
94. Oudiz RJ, Galie N, Olschewski H, Torres F, Frost A, Ghofrani HA, et al. Long-term
ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll
Cardiol 2009;54:1971-1981.
95. Germann P, Braschi A, Della Rocca G, Dinh-Xuan AT, Falke K, Frostell C, et al.
Inhaled nitric oxide therapy in adults: European expert recommendations. Intensive
Care Med 2005;31:1029-1041. Epub 2005 Jun 1023.
96. Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil
citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005;353:2148-
2157.
97. Hoeper MM, Welte T. Sildenafil citrate therapy for pulmonary arterial hypertension. N
Engl J Med 2006;354:1091-1093; author reply 1091-1093.
98. Humbert M, Barst RJ, Robbins IM, Channick RN, Galie N, Boonstra A, et al.
Combination of bosentan with epoprostenol in pulmonary arterial hypertension:
BREATHE-2. Eur Respir J 2004;24:353-359.
99. McLaughlin VV, Oudiz RJ, Frost A, Tapson VF, Murali S, Channick RN, et al.
Randomized study of adding inhaled iloprost to existing bosentan in pulmonary
arterial hypertension. Am J Respir Crit Care Med 2006;174:1257-1263. Epub 2006
Aug 1231.
100. Simonneau G, Rubin LJ, Galie N, Barst RJ, Fleming TR, Frost AE, et al. Addition of
sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary
arterial hypertension: a randomized trial. Ann Intern Med 2008;149:521-530.
101. Reichenberger F, Pepke-Zaba J, McNeil K, Parameshwar J, Shapiro LM. Atrial
septostomy in the treatment of severe pulmonary arterial hypertension. Thorax
2003;58:797-800.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
93
102. Trulock EP, Christie JD, Edwards LB, Boucek MM, Aurora P, Taylor DO, et al.
Registry of the International Society for Heart and Lung Transplantation: twenty-
fourth official adult lung and heart-lung transplantation report-2007. J Heart Lung
Transplant 2007;26:782-795.
103. Sablotzki A, Czeslick E, Gruenig E, Friedrich I, Schubert S, Borgermann J, et al. First
experiences with the stable prostacyclin analog iloprost in the evaluation of heart
transplant candidates with increased pulmonary vascular resistance. J Thorac
Cardiovasc Surg 2003;125:960-962.
104. Teerlink JR. Recent heart failure trials of neurohormonal modulation (OVERTURE
and ENABLE): approaching the asymptote of efficacy? J Card Fail 2002;8:124-127.
105. Perez-Villa F, Cuppoletti A, Rossel V, Vallejos I, Roig E. Initial experience with
bosentan therapy in patients considered ineligible for heart transplantation because
of severe pulmonary hypertension. Clin Transplant 2006;20:239-244.
106. Lewis GD, Shah R, Shahzad K, Camuso JM, Pappagianopoulos PP, Hung J, et al.
Sildenafil improves exercise capacity and quality of life in patients with systolic heart
failure and secondary pulmonary hypertension. Circulation 2007;116:1555-1562.
Epub 2007 Sep 1554.
107. Maloney JP. Advances in the treatment of secondary pulmonary hypertension. Curr
Opin Pulm Med 2003;9:139-143.
108. Jais X, D'Armini AM, Jansa P, Torbicki A, Delcroix M, Ghofrani HA, et al. Bosentan
for treatment of inoperable chronic thromboembolic pulmonary hypertension:
BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic
pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol
2008;52:2127-2134.
109. Rhodes CJ, Davidson A, Gibbs JS, Wharton J, Wilkins MR. Therapeutic targets in
pulmonary arterial hypertension. Pharmacol Ther 2009;121:69-88. Epub 2008 Oct
2028.
110. Wilkins MR, Ali O, Bradlow W, Wharton J, Taegtmeyer A, Rhodes CJ, et al.
Simvastatin as a treatment for pulmonary hypertension trial. Am J Respir Crit Care
Med 2010;181:1106-1113.
111. Suzuki C, Takahashi M, Morimoto H, Izawa A, Ise H, Hongo M, et al. Mycophenolate
mofetil attenuates pulmonary arterial hypertension in rats. Biochem Biophys Res
Commun 2006;349:781-788. Epub 2006 Aug 2028.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
94
112. Sanchez O, Sitbon O, Jais X, Simonneau G, Humbert M. Immunosuppressive
therapy in connective tissue diseases-associated pulmonary arterial hypertension.
Chest 2006;130:182-189.
113. Henriques-Coelho T, Correia-Pinto J, Roncon-Albuquerque R, Jr., Baptista MJ,
Lourenço AP, Oliveira SM, et al.Endogenous production of ghrelin and beneficial
effects of its exogenous administration in monocrotaline-induced pulmonary
hypertension. Am J Physiol Heart Circ Physiol 2004;287:H2885-2890. Epub 2004
Aug 2826.
114. Falcao-Pires I, Goncalves N, Henriques-Coelho T, Moreira-Goncalves D, Roncon-
Albuquerque R, Jr., Leite-Moreira AF. Apelin decreases myocardial injury and
improves right ventricular function in monocrotaline-induced pulmonary hypertension.
Am J Physiol Heart Circ Physiol 2009;296:H2007-2014. Epub 2009 Apr 2003.
115. Diller GP, van Eijl S, Okonko DO, Howard LS, Ali O, Thum T, et al. Circulating
endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic
pulmonary arterial hypertension. Circulation 2008;117:3020-3030. Epub 2008 Jun
3022.
116. Povsic TJ, Goldschmidt-Clermont PJ. Endothelial progenitor cells: markers of
vascular reparative capacity. Ther Adv Cardiovasc Dis 2008;2:199-213.
117. Cavusoglu Y, Beyaztas A, Birdane A, Ata N. Levosimendan is not effective in
reducing pulmonary pressures in patients with idiopathic pulmonary arterial
hypertension: report of two cases. J Cardiovasc Med (Hagerstown) 2009;10:503-507.
118. Morais RJ. Levosimendan in severe right ventricular failure following mitral valve
replacement. J Cardiothorac Vasc Anesth 2006;20:82-84. Epub 2005 Dec 2015.
119. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med
2002;166:111-117.
120. Rasekaba T, Lee AL, Naughton MT, Williams TJ, Holland AE. The six-minute walk
test: a useful metric for the cardiopulmonary patient. Intern Med J 2009;39:495-501.
121. Borg GA. Psychophysical bases of perceived exertion. Med Sci Sports Exerc
1982;14:377-381.
122. Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Herve P, et al. Long-term
intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic
factors and survival. J Am Coll Cardiol 2002;40:780-788.
123. Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd JE, Robbins IM. Outcome in 91
consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am
J Respir Crit Care Med 2003;167:580-586.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
95
124. Opitz CF, Wensel R, Winkler J, Halank M, Bruch L, Kleber FX, et al. Clinical efficacy
and survival with first-line inhaled iloprost therapy in patients with idiopathic
pulmonary arterial hypertension. Eur Heart J 2005;26:1895-1902.
125. Galie N, Humbert M, Vachiery JL, Vizza CD, Kneussl M, Manes A, et al. Effects of
beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial
hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol
2002;39:1496-1502.
126. Barst RJ, McGoon M, McLaughlin V, Tapson V, Rich S, Rubin L, et al. Beraprost
therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2003;41:2119-2125.
127. McLaughlin VV, Sitbon O, Badesch DB, Barst RJ, Black C, Galie N, et al. Survival
with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J
2005;25:244-249.
128. Galie N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, et al. Bosentan
therapy in patients with Eisenmenger syndrome: a multicenter, double-blind,
randomized, placebo-controlled study. Circulation 2006;114:48-54.
129. Galie N, Badesch D, Oudiz R, Simonneau G, McGoon MD, Keogh AM, et al.
Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol
2005;46:529-535.
130. Galie N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al.Ambrisentan
for the treatment of pulmonary arterial hypertension: results of the ambrisentan in
pulmonary arterial hypertension, randomized, double-blind, placebo-controlled,
multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117:3010-3019.
131. Barst RJ, Langleben D, Frost A, Horn EM, Oudiz R, Shapiro S, et al. Sitaxsentan
therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med
2004;169:441-447.
132. Barst RJ, Langleben D, Badesch D, Frost A, Lawrence EC, Shapiro S, et al.
Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor
antagonist sitaxsentan. J Am Coll Cardiol 2006;47:2049-2056.
133. Nagaya N, Uematsu M, Kojima M, Ikeda Y, Yoshihara F, Shimizu W, et al. Chronic
administration of ghrelin improves left ventricular dysfunction and attenuates
development of cardiac cachexia in rats with heart failure. Circulation 2001;104:1430-
1435.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
96
134. Yang J, Li X, Al-Lamki RS, Southwood M, Zhao J, Lever AM, et al. Smad-dependent
and smad-independent induction of id1 by prostacyclin analogues inhibits
proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ Res
2010;107:252-262. Epub 2010 Jun 2013.
135. West J. Cross talk between Smad, MAPK, and actin in the etiology of pulmonary
arterial hypertension. Adv Exp Med Biol 2010;661:265-278.
136. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the
nucleus. Cell 2003;113:685-700.
137. Yang J, Davies RJ, Southwood M, Long L, Yang X, Sobolewski A, et al. Mutations in
bone morphogenetic protein type II receptor cause dysregulation of Id gene
expression in pulmonary artery smooth muscle cells: implications for familial
pulmonary arterial hypertension. Circ Res 2008;102:1212-1221. Epub 2008 Apr
1224.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
97
7. Referências
Abel ED, Litwin SE, and Sweeney G. Cardiac remodeling in obesity. Physiol Rev 88: 389-
419, 2008.
Akki A, and Seymour AM. Western diet impairs metabolic remodelling and contractile
efficiency in cardiac hypertrophy. Cardiovasc Res 81: 610-617, 2009.
Akner G, and Cederholm T. Treatment of protein-energy malnutrition in chronic
nonmalignant disorders. Am J Clin Nutr 74: 6-24, 2001.
Anker SD, and Coats AJ. Cardiac cachexia: a syndrome with impaired survival and
immune and neuroendocrine activation. Chest 115: 836-847, 1999.
Anker SD, Negassa A, Coats AJ, Afzal R, Poole-Wilson PA, Cohn JN, and Yusuf S.
Prognostic importance of weight loss in chronic heart failure and the effect of treatment with
angiotensin-converting-enzyme inhibitors: an observational study. Lancet 361: 1077-1083,
2003.
Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, Harrington
D, Kox WJ, Poole-Wilson PA, and Coats AJ. Wasting as independent risk factor for
mortality in chronic heart failure. Lancet 349: 1050-1053, 1997.
Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, and Weir EK.
Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-
Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J
Physiol Heart Circ Physiol 294: H570-578, 2008.
Archer SL, Weir EK, and Wilkins MR. Basic science of pulmonary arterial hypertension for
clinicians: new concepts and experimental therapies. Circulation 121:2045-66, 2010.
ATS statement. Guidelines for the six-minute walk test. Am J Respir Crit Care Med 166:
111-117, 2002.
Azhar G, and Wei JY. Nutrition and cardiac cachexia. Curr Opin Clin Nutr Metab Care 9:
18-23, 2006.
Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, and
Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue
development. Mol Cell 4: 585-595, 1999.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
98
Barger PM, Brandt JM, Leone TC, Weinheimer CJ, and Kelly DP. Deactivation of
peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin
Invest 105: 1723-1730, 2000.
Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, Tapson
VF, Bourge RC, Brundage BH, and et al. A comparison of continuous intravenous
epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension.
The Primary Pulmonary Hypertension Study Group. N Engl J Med 334: 296-302, 1996.
Baybutt RC, Rosales C, Brady H, and Molteni A. Dietary fish oil protects against lung and
liver inflammation and fibrosis in monocrotaline treated rats. Toxicology 175: 1-13, 2002.
Beutler B, Mahoney J, Le Trang N, Pekala P, and Cerami A. Purification of cachectin, a
lipoprotein lipase-suppressing hormone secreted by endotoxin-induced RAW 264.7 cells. J
Exp Med 161: 984-995, 1985.
Bing RJ, Siegel A, Ungar I, and Gilbert M. Metabolism of the human heart. II. Studies on
fat, ketone and amino acid metabolism. Am J Med 16: 504-515, 1954.
Blaise G, Langleben D, and Hubert B. Pulmonary arterial hypertension: pathophysiology
and anesthetic approach. Anesthesiology 99: 1415-1432, 2003.
Bogaard HJ, Abe K, Vonk Noordegraaf A, and Voelkel NF. The right ventricle under
pressure: cellular and molecular mechanisms of right-heart failure in pulmonary
hypertension. Chest 135: 794-804, 2009.
Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L,
Ockaili R, McCord JM, and Voelkel NF. Chronic pulmonary artery pressure elevation is
insufficient to explain right heart failure. Circulation 120: 1951-1960, 2009.
Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Haromy A,
Harry G, Moudgil R, McMurtry MS, Weir EK, and Archer SL. An abnormal mitochondrial-
hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers
pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary
arterial hypertension. Circulation 113: 2630-2641, 2006.
Brown L, Miller J, Dagger A, and Sernia C. Cardiac and vascular responses after
monocrotaline-induced hypertrophy in rats. J Cardiovasc Pharmacol 31: 108-115, 1998.
Brunner F. Cardiac endothelin and big endothelin in right-heart hypertrophy due to
monocrotaline-induced pulmonary hypertension in rat. Cardiovasc Res 44: 197-206, 1999.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
99
Buermans HP, Redout EM, Schiel AE, Musters RJ, Zuidwijk M, Eijk PP, van Hardeveld
C, Kasanmoentalib S, Visser FC, Ylstra B, and Simonides WS. Microarray analysis
reveals pivotal divergent mRNA expression profiles early in the development of either
compensated ventricular hypertrophy or heart failure. Physiol Genomics 21: 314-323, 2005.
Burkhoff D, Weiss RG, Schulman SP, Kalil-Filho R, Wannenburg T, and Gerstenblith
G. Influence of metabolic substrate on rat heart function and metabolism at different
coronary flows. Am J Physiol 261: H741-750, 1991.
Butrous G, Ghofrani HA, and Grimminger F. Pulmonary vascular disease in the
developing world. Circulation 118: 1758-1766, 2008.
Ceconi C, Condorelli E, Quinzanini M, Rodella A, Ferrari R, and Harris P.
Noradrenaline, atrial natriuretic peptide, bombesin and neurotensin in myocardium and
blood of rats in congestive cardiac failure. Cardiovasc Res 23: 674-682, 1989.
Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducolone A, Ehrhart M,
Kessler R, and Weitzenblum E. Severe pulmonary hypertension and chronic obstructive
pulmonary disease. Am J Respir Crit Care Med 172: 189-194, 2005.
Chen EP, Craig DM, Bittner HB, Davis RD, and Van Trigt P. Pharmacological strategies
for improving diastolic dysfunction in the setting of chronic pulmonary hypertension.
Circulation 97: 1606-1612, 1998.
Chess DJ, Khairallah RJ, O'Shea KM, Xu W, and Stanley WC. A High Fat Diet Increases
Adiposity but Maintains Mitochondrial Oxidative Enzymes without Affecting Development of
Heart Failure with Pressure Overload. Am J Physiol Heart Circ Physiol 297: H1585–H1593,
2009.
Chin KM, and Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol 51: 1527-
1538, 2008.
Chou SH, Lee YC, Huang CF, Wang YR, Yu HP, and Lau YT. Gender-specific effects of
caloric restriction on the balance of vascular nitric oxide and superoxide radical. Cardiovasc
Res 87: 751-759, 2010.
Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, and
Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites
in pulmonary hypertension. N Engl J Med 327: 70-75, 1992.
Cicoira M, Kalra PR, and Anker SD. Growth hormone resistance in chronic heart failure
and its therapeutic implications. J Card Fail 9: 219-226, 2003.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
100
Correia-Pinto J, Henriques-Coelho T, Roncon-Albuquerque R, Jr., Lourenço AP, Melo-
Rocha G, Vasques-Novoa F, Gillebert TC, and Leite-Moreira AF. Time course and
mechanisms of left ventricular systolic and diastolic dysfunction in monocrotaline-induced
pulmonary hypertension. Basic Res Cardiol 104: 535-545, 2009.
Cowan KN, Jones PL, and Rabinovitch M. Elastase and matrix metalloproteinase
inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular
disease. J Clin Invest 105: 21-34, 2000.
Curtis JP, Selter JG, Wang Y, Rathore SS, Jovin IS, Jadbabaie F, Kosiborod M,
Portnay EL, Sokol SI, Bader F, and Krumholz HM. The obesity paradox: body mass index
and outcomes in patients with heart failure. Arch Intern Med 165: 55-61, 2005.
Daicho T, Yagi T, Abe Y, Ohara M, Marunouchi T, Takeo S, and Tanonaka K. Possible
involvement of mitochondrial energy-producing ability in the development of right ventricular
failure in monocrotaline-induced pulmonary hypertensive rats. J Pharmacol Sci 111: 33-43,
2009.
Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, and
Gropler RJ. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated
cardiomyopathy. J Am Coll Cardiol 40: 271-277, 2002.
Dhalla NS, Saini-Chohan HK, Rodriguez-Leyva D, Elimban V, Dent MR, and Tappia PS.
Subcellular remodelling may induce cardiac dysfunction in congestive heart failure.
Cardiovasc Res 81: 429-438, 2009.
Doehner W, and Anker SD. Cardiac cachexia in early literature: a review of research prior
to Medline. Int J Cardiol 85: 7-14, 2002.
Doehner W, Rauchhaus M, Ponikowski P, Godsland IF, von Haehling S, Okonko DO,
Leyva F, Proudler AJ, Coats AJ, and Anker SD. Impaired insulin sensitivity as an
independent risk factor for mortality in patients with stable chronic heart failure. J Am Coll
Cardiol 46: 1019-1026, 2005.
Dorfmuller P, Perros F, Balabanian K, and Humbert M. Inflammation in pulmonary
arterial hypertension. Eur Respir J 22: 358-363, 2003.
Entman ML, Bornet EP, Van Winkle WB, Goldstein MA, and Schwartz A. Association of
glycogenolysis with cardiac sarcoplasmic reticulum: II. Effect of glycogen depletion,
deoxycholate solubilization and cardiac ischemia: evidence for a phorphorylase kinase
membrane complex. J Mol Cell Cardiol 9: 515-528, 1977.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
101
Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-
Zacdeh K, Lochs H, Mantovani G, Marks D, Mitch WE, Muscaritoli M, Najand A,
Ponikowski P, Rossi Fanelli F, Schambelan M, Schols A, Schuster M, Thomas D,
Wolfe R, and Anker SD. Cachexia: a new definition. Clin Nutr 27: 793-799, 2008.
Fallon MB, Abrams GA, Abdel-Razek TT, Dai J, Chen SJ, Chen YF, Luo B, Oparil S,
and Ku DD. Garlic prevents hypoxic pulmonary hypertension in rats. Am J Physiol 275:
L283-287, 1998.
Feldman AM, Combes A, Wagner D, Kadakomi T, Kubota T, Li YY, and McTiernan C.
The role of tumor necrosis factor in the pathophysiology of heart failure. J Am Coll Cardiol
35: 537-544, 2000.
Filippatos GS, Tsilias K, Venetsanou K, Karambinos E, Manolatos D, Kranidis A,
Antonellis J, Kardaras F, Anthopoulos L, and Baltopoulos G. Leptin serum levels in
cachectic heart failure patients. Relationship with tumor necrosis factor-alpha system. Int J
Cardiol 76: 117-122, 2000.
Finck BN, and Kelly DP. Peroxisome proliferator-activated receptor alpha (PPARalpha)
signaling in the gene regulatory control of energy metabolism in the normal and diseased
heart. J Mol Cell Cardiol 34: 1249-1257, 2002.
Fishman AP. A century of pulmonary hemodynamics. Am J Respir Crit Care Med 170: 109-
113, 2004.
Francis GS. Pathophysiology of chronic heart failure. Am J Med 110 Suppl 7A: 37S-46S,
2001.
Fruchter O, and Yigla M. Underlying aetiology of pulmonary hypertension in 191 patients: a
single centre experience. Respirology 13: 825-831, 2008.
Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, and Ventura-Clapier R.
Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac
and skeletal muscles. J Physiol 551: 491-501, 2003.
Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambartsumian N,
Lukanidin E, and Rabinovitch M. S100A4/Mts1 produces murine pulmonary artery
changes resembling plexogenic arteriopathy and is increased in human plexogenic
arteriopathy. Am J Pathol 164: 253-262, 2004.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
102
Gulick T, Cresci S, Caira T, Moore DD, and Kelly DP. The peroxisome proliferator-
activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression.
Proc Natl Acad Sci U S A 91: 11012-11016, 1994.
Hacker AD. Dietary restriction, polyamines and monocrotaline-induced pulmonary
hypertension. Biochem Pharmacol 45: 2475-2481, 1993.
Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY,
Suen RS, Stewart DJ, and Rabinovitch M. Pulmonary arterial hypertension is linked to
insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma
activation. Circulation 115: 1275-1284, 2007.
Hessel MH, Steendijk P, den Adel B, Schutte CI, and van der Laarse A. Characterization
of right ventricular function after monocrotaline-induced pulmonary hypertension in the intact
rat. Am J Physiol Heart Circ Physiol 291: H2424-2430, 2006.
Hoeper MM, Barbera JA, Channick RN, Hassoun PM, Lang IM, Manes A, Martinez FJ,
Naeije R, Olschewski H, Pepke-Zaba J, Redfield MM, Robbins IM, Souza R, Torbicki A,
and McGoon M. Diagnosis, assessment, and treatment of non-pulmonary arterial
hypertension pulmonary hypertension. J Am Coll Cardiol 54: S85-96, 2009.
Hoeper MM, Mayer E, Simonneau G, and Rubin LJ. Chronic thromboembolic pulmonary
hypertension. Circulation 113: 2011-2020, 2006.
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A,
Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M,
Haloun A, Laurent M, Hachulla E, and Simonneau G. Pulmonary arterial hypertension in
France: results from a national registry. Am J Respir Crit Care Med 173: 1023-1030, 2006.
Humbert M. Update in pulmonary hypertension 2008. Am J Respir Crit Care Med 179: 650-
656, 2009.
Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M,
Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW,
and Yancy CW. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for
the Diagnosis and Management of Heart Failure in Adults: a report of the American College
of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines:
developed in collaboration with the International Society for Heart and Lung Transplantation.
Circulation 119: e391-479, 2009.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
103
Hutter JF, Schweickhardt C, Piper HM, and Spieckermann PG. Inhibition of fatty acid
oxidation and decrease of oxygen consumption of working rat heart by 4-bromocrotonic
acid. J Mol Cell Cardiol 16: 105-108, 1984.
Iemitsu M, Shimojo N, Maeda S, Irukayama-Tomobe Y, Sakai S, Ohkubo T, Tanaka Y,
and Miyauchi T. The benefit of medium-chain triglyceride therapy on the cardiac function of
SHRs is associated with a reversal of metabolic and signaling alterations. Am J Physiol
Heart Circ Physiol 295: H136-144, 2008.
Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res 81: 412-
419, 2009.
Inui A. Cancer anorexia-cachexia syndrome: are neuropeptides the key? Cancer Res 59:
4493-4501, 1999.
Ivy DD, McMurtry IF, Colvin K, Imamura M, Oka M, Lee DS, Gebb S, and Jones PL.
Development of occlusive neointimal lesions in distal pulmonary arteries of endothelin B
receptor-deficient rats: a new model of severe pulmonary arterial hypertension. Circulation
111: 2988-2996, 2005.
Jasmin JF, Lucas M, Cernacek P, and Dupuis J. Effectiveness of a nonselective ET(A/B)
and a selective ET(A) antagonist in rats with monocrotaline-induced pulmonary
hypertension. Circulation 103: 314-318, 2001.
Jucker BM, Doe CP, Schnackenberg CG, Olzinski AR, Maniscalco K, Williams C, Hu
TC, Lenhard SC, Costell M, Bernard R, Sarov-Blat L, Steplewski K, and Willette RN.
PPARdelta activation normalizes cardiac substrate metabolism and reduces right ventricular
hypertrophy in congestive heart failure. J Cardiovasc Pharmacol 50: 25-34, 2007.
Kalantar-Zadeh K, Block G, Horwich T, and Fonarow GC. Reverse epidemiology of
conventional cardiovascular risk factors in patients with chronic heart failure. J Am Coll
Cardiol 43: 1439-1444, 2004.
Keogh AM, Mayer E, Benza RL, Corris P, Dartevelle PG, Frost AE, Kim NH, Lang IM,
Pepke-Zaba J, and Sandoval J. Interventional and surgical modalities of treatment in
pulmonary hypertension. J Am Coll Cardiol 54: S67-77, 2009.
Kiec-Wilk B, Dembinska-Kiec A, Olszanecka A, Bodzioch M, and Kawecka-Jaszcz K.
The selected pathophysiological aspects of PPARs activation. J Physiol Pharmacol 56: 149-
162, 2005.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
104
Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, and Hildebrandt
P. Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure.
Circulation 112: 1756-1762, 2005.
Kodde IF, van der Stok J, Smolenski RT, and de Jong JW. Metabolic and genetic
regulation of cardiac energy substrate preference. Comp Biochem Physiol A Mol Integr
Physiol 146: 26-39, 2007.
Kögler H, Hartmann O, Leineweber K, Nguyen van P, Schott P, Brodde OE, and
Hasenfuss G. Mechanical load-dependent regulation of gene expression in monocrotaline-
induced right ventricular hypertrophy in the rat. Circ Res 93: 230-237, 2003.
Krack A, Sharma R, Figulla HR, and Anker SD. The importance of the gastrointestinal
system in the pathogenesis of heart failure. Eur Heart J 26: 2368-2374, 2005.
Krauss RM, Eckel RH, Howard B, Appel LJ, Daniels SR, Deckelbaum RJ, Erdman JW,
Jr., Kris-Etherton P, Goldberg IJ, Kotchen TA, Lichtenstein AH, Mitch WE, Mullis R,
Robinson K, Wylie-Rosett J, St Jeor S, Suttie J, Tribble DL, and Bazzarre TL. AHA
Dietary Guidelines: revision 2000: A statement for healthcare professionals from the
Nutrition Committee of the American Heart Association. Circulation 102: 2284-2299, 2000.
Kusuoka H, and Marban E. Mechanism of the diastolic dysfunction induced by glycolytic
inhibition. Does adenosine triphosphate derived from glycolysis play a favored role in
cellular Ca2+ homeostasis in ferret myocardium? J Clin Invest 93: 1216-1223, 1994.
Lalich JJ, and Merkow L. Pulmonary arteritis produced in rat by feeding Crotalaria
spectabilis. Lab Invest 10: 744-750, 1961.
Laviano A, Muscaritoli M, Cascino A, Preziosa I, Inui A, Mantovani G, and Rossi-
Fanelli F.Branched-chain amino acids: the best compromise to achieve anabolism? Curr
Opin Clin Nutr Metab Care 8: 408-14, 2005.
Lehnart SE, Maier LS, and Hasenfuss G. Abnormalities of calcium metabolism and
myocardial contractility depression in the failing heart. Heart Fail Rev 14: 213-224, 2009.
Leineweber K, Brandt K, Wludyka B, Beilfuss A, Pönicke K, Heinroth-Hoffmann I, and
Brodde OE. Ventricular hypertrophy plus neurohumoral activation is necessary to alter the
cardiac beta-adrenoceptor system in experimental heart failure. Circ Res 91: 1056-1062,
2002.
Leyva F, Anker SD, Egerer K, Stevenson JC, Kox WJ, and Coats AJ. Hyperleptinaemia
in chronic heart failure. Relationships with insulin. Eur Heart J 19: 1547-1551, 1998.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
105
Lourenço AP, Roncon-Albuquerque R, Jr., Bras-Silva C, Faria B, Wieland J,
Henriques-Coelho T, Correia-Pinto J, and Leite-Moreira AF. Myocardial dysfunction and
neurohumoral activation without remodeling in left ventricle of monocrotaline-induced
pulmonary hypertensive rats. Am J Physiol Heart Circ Physiol 291: H1587-1594, 2006.
MacLean MR, and Dempsie Y. Serotonin and pulmonary hypertension-from bench to
bedside? Curr Opin Pharmacol 9: 281-286, 2009.
MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease.
Part One. Am J Respir Crit Care Med 150: 833-852, 1994.
Mamas MA, Deaton C, Rutter MK, Yuille M, Williams SG, Ray SG, New J, Gibson JM,
and Neyses L. Impaired glucose tolerance and insulin resistance in heart failure:
underrecognized and undertreated? J Card Fail 16: 761-768, 2010.
Mann DL, and Bristow MR. Mechanisms and models in heart failure: the biomechanical
model and beyond. Circulation 111: 2837-2849, 2005.
Martindale RG, McClave SA, Vanek VW, McCarthy M, Roberts P, Taylor B, Ochoa JB,
Napolitano L, and Cresci G. Guidelines for the provision and assessment of nutrition
support therapy in the adult critically ill patient: Society of Critical Care Medicine and
American Society for Parenteral and Enteral Nutrition: Executive Summary. Crit Care Med
37: 1757-1761, 2009.
McLaughlin VV, and Suissa S. Prognosis of Pulmonary Arterial Hypertension: the power of
clinical registries of rare diseases. Circulation122:106-108, 2010.
McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier
MA, McGoon MD, Park MH, Rosenson RS, Rubin LJ, Tapson VF, and Varga J.
ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the
American College of Cardiology Foundation Task Force on Expert Consensus Documents
and the American Heart Association developed in collaboration with the American College of
Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension
Association. J Am Coll Cardiol 53: 1573-1619, 2009.
McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, and
Ahearn G. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical
practice guidelines. Chest 126: 78S-92S, 2004.
Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol 274: R577-595, 1998.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
106
Menzel T,Wagner S, Kramm T, Mohr-Kahaly S, Mayer E, Braeuninger S, and Meyer J.
Pathophysiology of impaired right and left ventricular function in chronic embolic pulmonary
hypertension: changes after pulmonary thromboendarterectomy. Chest 118: 897–903, 2000.
Meyer TE, Kovács SJ, Ehsani AA, Klein S, Holloszy JO, and Fontana L. Long-term
caloric restriction ameliorates the decline in diastolic function in humans. J Am Coll Cardiol
47: 398-402, 2006.
Miyamoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita M, Nakanishi N, and
Miyatake K. Clinical correlates and prognostic significance of six-minute walk test in
patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise
testing. Am J Respir Crit Care Med 161: 487-492, 2000.
Miyauchi T, Yorikane R, Sakai S, Sakurai T, Okada M, Nishikibe M, Yano M,
Yamaguchi I, Sugishita Y, and Goto K. Contribution of endogenous endothelin-1 to the
progression of cardiopulmonary alterations in rats with monocrotaline-induced pulmonary
hypertension. Circ Res 73: 887-897, 1993.
Mootha VK, Arai AE, and Balaban RS. Maximum oxidative phosphorylation capacity of the
mammalian heart. Am J Physiol 272: H769-775, 1997.
Moraes DL, Colucci WS, and Givertz MM. Secondary pulmonary hypertension in chronic
heart failure: the role of the endothelium in pathophysiology and management. Circulation
102: 1718-1723, 2000.
Moreira-Rodrigues M, Roncon-Albuquerque R, Henriques-Coelho T, Lourenço AP,
Sampaio-Maia B, Santos J, Pestana M, and Leite-Moreira AF. Cardiac remodeling and
dysfunction in nephrotic syndrome. Kidney International 71: 1240-1248, 2007.
Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, and Trembath
RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with
primary pulmonary hypertension to transforming growth factor-beta(1) and bone
morphogenetic proteins. Circulation 104: 790-795, 2001.
Murray CJ, and Lopez AD. Alternative projections of mortality and disability by cause 1990-
2020: Global Burden of Disease Study. Lancet 349: 1498-1504, 1997.
Nagaya N, Uematsu M, Kojima M, Date Y, Nakazato M, Okumura H, Hosoda H, Shimizu
W, Yamagishi M, Oya H, Koh H, Yutani C, and Kangawa K. Elevated circulating level of
ghrelin in cachexia associated with chronic heart failure: relationships between ghrelin and
anabolic/catabolic factors. Circulation 104: 2034-2038, 2001.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
107
Nef HM, Mollmann H, Hamm C, Grimminger F, and Ghofrani HA. Pulmonary
hypertension: updated classification and management of pulmonary hypertension. Heart 96:
552-559, 2010.
Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, Coccolo F, Ventura
C, Phillips JA, 3rd, Knowles JA, Janssen B, Eickelberg O, Eddahibi S, Herve P,
Nichols WC, and Elliott G. Genetic basis of pulmonary arterial hypertension: current
understanding and future directions. J Am Coll Cardiol 43: 33S-39S, 2004.
Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, Poole-
Wilson PA, Coats AJ, and Anker SD. Endotoxin and immune activation in chronic heart
failure: a prospective cohort study. Lancet 353: 1838-1842, 1999.
Ogata T, Miyauchi T, Sakai S, Irukayama-Tomobe Y, Goto K, and Yamaguchi I.
Stimulation of peroxisome-proliferator-activated receptor alpha (PPAR alpha) attenuates
cardiac fibrosis and endothelin-1 production in pressure-overloaded rat hearts. Clin Sci
(Lond) 103: 284S-288S, 2002.
Oreopoulos A, Padwal R, Kalantar-Zadeh K, Fonarow GC, Norris CM, and McAlister
FA. Body mass index and mortality in heart failure: a meta-analysis. Am Heart J 156:13-22,
2008.
Ormerod JO, Ashrafian H, and Frenneaux MP. Impaired energetics in heart failure - a
new therapeutic target. Pharmacol Ther 119: 264-274, 2008.
Peacock AJ, Murphy NF, McMurray JJ, Caballero L, and Stewart S. An epidemiological
study of pulmonary arterial hypertension. Eur Respir J 30: 104-109, 2007.
Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang
HJ, Haber I, Rehman J, Lopaschuk GD, and Archer SL. The inhibition of pyruvate
dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two
models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol
Med 88: 47-60, 2009.
Pierce GN, and Philipson KD. Binding of glycolytic enzymes to cardiac sarcolemmal and
sarcoplasmic reticular membranes. J Biol Chem 260: 6862-6870, 1985.
Pillutla P, Hwang YC, Augustus A, Yokoyama M, Yagyu H, Johnston TP, Kaneko M,
Ramasamy R, and Goldberg IJ. Perfusion of hearts with triglyceride-rich particles
reproduces the metabolic abnormalities in lipotoxic cardiomyopathy. Am J Physiol
Endocrinol Metab 288: E1229-1235, 2005.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
108
Provencher S, Jais X, Yaici A, Sitbon O, Humbert M, and Simonneau G. Clinical
challenges in pulmonary hypertension: Roger S. Mitchell lecture. Chest 128: 622S-628S,
2005.
Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest
118: 2372-2379, 2008.
Rauchhaus M, Coats AJ, and Anker SD. The endotoxin-lipoprotein hypothesis. Lancet
356: 930-933, 2000.
Redman LM, Martin CK, Williamson DA, and Ravussin E. Effect of caloric restriction in
non-obese humans on physiological, psychological and behavioral outcomes. Physiol Behav
94: 643-648, 2008.
Redout EM, Wagner MJ, Zuidwijk MJ, Boer C, Musters RJ, van Hardeveld C, Paulus
WJ, and Simonides WS. Right-ventricular failure is associated with increased mitochondrial
complex II activity and production of reactive oxygen species. Cardiovasc Res 75: 770-781,
2007.
Reichenberger F, Pepke-Zaba J, McNeil K, Parameshwar J, and Shapiro LM. Atrial
septostomy in the treatment of severe pulmonary arterial hypertension. Thorax 58: 797-800,
2003.
Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: historical clues from
brisket disease. J Appl Physiol 98: 1092-1100, 2005.
Rich S, Kaufmann E, and Levy PS. The effect of high doses of calcium-channel blockers
on survival in primary pulmonary hypertension. N Engl J Med 327:76-81, 1992.
Ritchie M, Waggoner AD, Davila-Roman VG, Barzilai B, Trulock EP, and Eisenberg PR.
Echocardiographic characterization of the improvement in right ventricular function in
patients with severe pulmonary hypertension after single-lung transplantation. J Am Coll
Cardiol 22: 1170-1174, 1993.
Robbins IM, Newman JH, Johnson RF, Hemnes AR, Fremont RD, Piana RN, Zhao DX,
and Byrne DW. Association of the metabolic syndrome with pulmonary venous
hypertension. Chest 136: 31-36, 2009.
Robbins IM. Advancing therapy for pulmonary arterial hypertension: can animal models
help? Am J Respir Crit Care Med 169: 5-6, 2004.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
109
Sakao S, Tatsumi K, and Voelkel NF. Endothelial cells and pulmonary arterial
hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res 10: 95,
2009.
Sandek A, Rauchhaus M, Anker SD, and von Haehling S. The emerging role of the gut in
chronic heart failure. Curr Opin Clin Nutr Metab Care 11: 632-639, 2008.
Schott P, Singer SS, Kögler H, Neddermeier D, Leineweber K, Brodde OE, Regitz-
Zagrosek V, Schmidt B, Dihazi H, and Hasenfuss G. Pressure overload and
neurohumoral activation differentially affect the myocardial proteome. Proteomics 5: 1372-
1381, 2005.
Schrauwen P, Hoeks J, Schaart G, Kornips E, Binas B, Van De Vusse GJ, Van Bilsen
M, Luiken JJ, Coort SL, Glatz JF, Saris WH, and Hesselink MK. Uncoupling protein 3 as
a mitochondrial fatty acid anion exporter. Faseb J 17: 2272-2274, 2003.
Seyfarth T, Gerbershagen HP, Giessler C, Leineweber K, Heinroth-Hoffmann I,
Pönicke K, and Brodde OE. The cardiac beta-adrenoceptor-G-protein(s)-adenylyl cyclase
system in monocrotaline-treated rats. J Mol Cell Cardiol 32: 2315-2326, 2000.
Seymour EM, Parikh RV, Singer AA, and Bolling SF. Moderate calorie restriction
improves cardiac remodeling and diastolic dysfunction in the Dahl-SS rat. J Mol Cell Cardiol
41: 661-668, 2006.
Shapiro BP, McGoon MD, and Redfield MM. Unexplained pulmonary hypertension in
elderly patients. Chest 131: 94-100, 2007.
Sharma R, and Anker SD. Cytokines, apoptosis and cachexia: the potential for TNF
antagonism. Int J Cardiol 85: 161-171, 2002.
Shelub I, van Grondelle A, McCullough R, Hofmeister S, and Reeves JT. A model of
embolic chronic pulmonary hypertension in the dog. J Appl Physiol 56: 810-815, 1984.
Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F,
Herve P, and Simonneau G. Long-term response to calcium channel blockers in idiopathic
pulmonary arterial hypertension. Circulation 111: 3105-3111, 2005.
Stanley WC, Recchia FA, and Lopaschuk GD. Myocardial substrate metabolism in the
normal and failing heart. Physiol Rev 85: 1093-1129, 2005.
Steffen BT, Lees SJ, and Booth FW. Anti-TNF treatment reduces rat skeletal muscle
wasting in monocrotaline-induced cardiac cachexia. J Appl Physiol 105: 1950-1958, 2008.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
110
Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, and Waxman AB. Interleukin-6
overexpression induces pulmonary hypertension. Circ Res 104: 236-244, 2009.
Stenmark KR, Meyrick B, Galie N, Mooi WJ, and McMurtry IF. Animal models of
pulmonary arterial hypertension: the hope for etiological discovery and pharmacological
cure. Am J Physiol Lung Cell Mol Physiol 297: L1013-1032, 2009.
Sun X, and Ku DD. Rosuvastatin provides pleiotropic protection against pulmonary
hypertension, right ventricular hypertrophy, and coronary endothelial dysfunction in rats. Am
J Physiol Heart Circ Physiol 294: H801-809, 2008.
Sun XG, Hansen JE, Oudiz RJ, and Wasserman K. Pulmonary function in primary
pulmonary hypertension. J Am Coll Cardiol 41: 1028-1035, 2003.
Suskin N, McKelvie RS, Burns RJ, Latini R, Pericak D, Probstfield J, Rouleau JL,
Sigouin C, Solymoss CB, Tsuyuki R, White M, and Yusuf S. Glucose and insulin
abnormalities relate to functional capacity in patients with congestive heart failure. Eur Heart
J 21: 1368-1375, 2000.
Tanaka Y, Schuster DP, Davis EC, Patterson GA, and Botney MD. The role of vascular
injury and hemodynamics in rat pulmonary artery remodeling. J Clin Invest 98: 434-442,
1996.
Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J,
Voelkel NF, and Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic
hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe
pulmonary hypertension. Faseb J 15: 427-438, 2001.
Tassone F, Gianotti L, Rolfo F, Visconti G, Borretta G, and Feola M. B-type natriuretic
peptide levels and insulin resistance in patients with severe ischemic myocardial
dysfunction. J Endocrinol Invest 32: 805-809, 2009.
Thorne SA, Hooper J, Kemp M, and Somerville J. Gastro-intestinal protein loss in late
survivors of Fontan surgery and other congenital heart disease. Eur Heart J 19: 514-520,
1998.
Trulock EP, Christie JD, Edwards LB, Boucek MM, Aurora P, Taylor DO, Dobbels F,
Rahmel AO, Keck BM, and Hertz MI. Registry of the International Society for Heart and
Lung Transplantation: twenty-fourth official adult lung and heart-lung transplantation report-
2007. J Heart Lung Transplant 26: 782-795, 2007.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
111
Tsutamoto T, Hisanaga T, Wada A, Maeda K, Ohnishi M, Fukai D, Mabuchi N, Sawaki
M, and Kinoshita M. Interleukin-6 spillover in the peripheral circulation increases with the
severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic
predictor in patients with congestive heart failure. J Am Coll Cardiol 31: 391-398, 1998.
Tuunanen H, Engblom E, Naum A, Nagren K, Hesse B, Airaksinen KE, Nuutila P, Iozzo
P, Ukkonen H, Opie LH, and Knuuti J. Free fatty acid depletion acutely decreases cardiac
work and efficiency in cardiomyopathic heart failure. Circulation 114: 2130-2137, 2006.
Valen G, Yan ZQ, and Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll
Cardiol 38: 307-314, 2001.
van Bilsen M, van Nieuwenhoven FA, and van der Vusse GJ. Metabolic remodelling of
the failing heart: beneficial or detrimental? Cardiovasc Res 81: 420-428, 2009.
van der Vusse GJ, van Bilsen M, Glatz JF, Hasselbaink DM, and Luiken JJ. Critical
steps in cellular fatty acid uptake and utilization. Mol Cell Biochem 239: 9-15, 2002.
van Wolferen SA, Marcus JT, Boonstra A, Marques KM, Bronzwaer JG,
Spreeuwenberg MD, Postmus PE, and Vonk-Noordegraaf A. Prognostic value of right
ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur
Heart J 28: 1250-1257, 2007.
van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM,
Bronzwaer JG, Henkens IR, Gan CT, Boonstra A, Postmus PE, and Vonk-Noordegraaf
A. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart
J 29: 120-127, 2008.
Ventura-Clapier R, Garnier A, and Veksler V. Energy metabolism in heart failure. J
Physiol 555: 1-13, 2004.
von Haehling S, Doehner W, and Anker SD. Nutrition, metabolism, and the complex
pathophysiology of cachexia in chronic heart failure. Cardiovasc Res 73: 298-309, 2007.
Wagner DR, McTiernan C, Sanders VJ, and Feldman AM. Adenosine inhibits
lipopolysaccharide-induced secretion of tumor necrosis factor-alpha in the failing human
heart. Circulation 97: 521-524, 1998.
Watts JA, Marchick MR, and Kline JA. Right ventricular heart failure from pulmonary
embolism: key distinctions from chronic pulmonary hypertension. J Card Fail 16: 250-259,
2010.

Avaliação da Expressão Génica e Vias Subcelulares Neuroendócrinas e Metabólicas em Modelos Experimentais de Hipertensão Pulmonar
112
Watts JA, Zagorski J, Gellar MA, Stevinson BG, and Kline JA. Cardiac inflammation
contributes to right ventricular dysfunction following experimental pulmonary embolism in
rats. J Mol Cell Cardiol 41: 296-307, 2006.
Weindruch R, Kayo T, Lee CK, and Prolla TA. Microarray profiling of gene expression in
aging and its alteration by caloric restriction in mice. J Nutr 131: 918S-923S, 2001.
Werchan PM, Summer WR, Gerdes AM, and McDonough KH. Right ventricular
performance after monocrotaline-induced pulmonary hypertension. Am J Physiol 256:
H1328-1336, 1989.
Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Morris DL, and Craig JC. Metabolic
fate of extracted glucose in normal human myocardium. J Clin Invest 76: 1819-1827, 1985.
Witte KK, and Clark AL. Nutritional abnormalities contributing to cachexia in chronic illness.
Int J Cardiol 85: 23-31, 2002.
Witteles RM, and Fowler MB. Insulin-resistant cardiomyopathy clinical evidence,
mechanisms, and treatment options. J Am Coll Cardiol 51: 93-102, 2008.
Wright JL, Levy RD, and Churg A. Pulmonary hypertension in chronic obstructive
pulmonary disease: current theories of pathogenesis and their implications for treatment.
Thorax 60: 605-609, 2005.
Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C,
Chen H, Trembath RC, and Morrell NW. Dysfunctional Smad signaling contributes to
abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ
Res 96: 1053-1063, 2005.
Ye J. Regulation of PPARgamma function by TNF-alpha. Biochem Biophys Res Commun
374: 405-408, 2008.
Young JB. The global epidemiology of heart failure. Med Clin North Am 88: 1135-1143,
2004.
Young ME, Laws FA, Goodwin GW, and Taegtmeyer H. Reactivation of peroxisome
proliferator-activated receptor alpha is associated with contractile dysfunction in
hypertrophied rat heart. J Biol Chem 276: 44390-44395, 2001.


















