
Dias nummer 1 - Dansk Patologiselskab · Grad af siderose Relateret vævsskade fibrose, cirrose,...
42
Metabolisk leversygdom Leverens og galdevejenes patologi Specialespecifikt kursus i patologisk anatomi og cytologi 2010
-
Upload
trinhthuan -
Category
Documents
-
view
216 -
download
0
Transcript of Dias nummer 1 - Dansk Patologiselskab · Grad af siderose Relateret vævsskade fibrose, cirrose,...

Metabolisk leversygdom
Leverens og galdevejenes patologiSpecialespecifikt kursus i patologisk anatomi og cytologi 2010

Metaboliske leversygdommeHereditærePorfyrinstofskiftet
• Porfyria cutanea tarda• Erytropoietisk protoporfyri
Sukkerstofskiftet• Glycogen storage-sygdomme (Type 0-
XI)• Galaktosæmi
Glykoproteiner og glykolipider• Mucopolysakkaridose (Type I-VII)• Mucolipidose (Type I-IV)
ER storage disease• α1-antitrypsinmangel• α1-antichymotrypsinmangel• Hypofibrinogenæmi
Aminosyrestofskiftet• Tyrosinæmi
Lipoproteiner og lipidstofskiftet• Abetalipoproteinæmi• Familiær hypobetalipoproteinæmi• Familiær hyperkolesterolæmi• Kolesterolester storage disease• Fabry, Farber, Gaucher, Niemann-Pick
Peroxisomale sygdomme• Zellweger, Refsum
Mitochondriesygdomme• Pearson, Navajo neurohepatopati
Kobberstofskiftet• Wilson
Jernstofskiftet• Hæmokromatose (Type 1-4), neonatal
HKBilirubinstofskiftet
• Benign recurrent intrahepatisk cholestase (BRIC), progressiv familiær intrahepatisk cholestase (PFIC)
• Crigler-Najjar, Gilbert, Dubin-JohnsonAndre
• Cystisk fibrose, albinisme
ErhvervedeAdipositasDiabetes mellitusMetabolisk syndrom

α1-antitrypsin
• AAT-gen på 14q31-32.3 (co-dominant)• Plasma-glykoprotein, 52 kD• Syntetiseres primært i hepatocytter • Kompetitiv hæmmer af leukocyt-elastase med
favorisering af antitrypsin/elastase-kompleks frem for elastase/elastin
• Akut fasereaktant
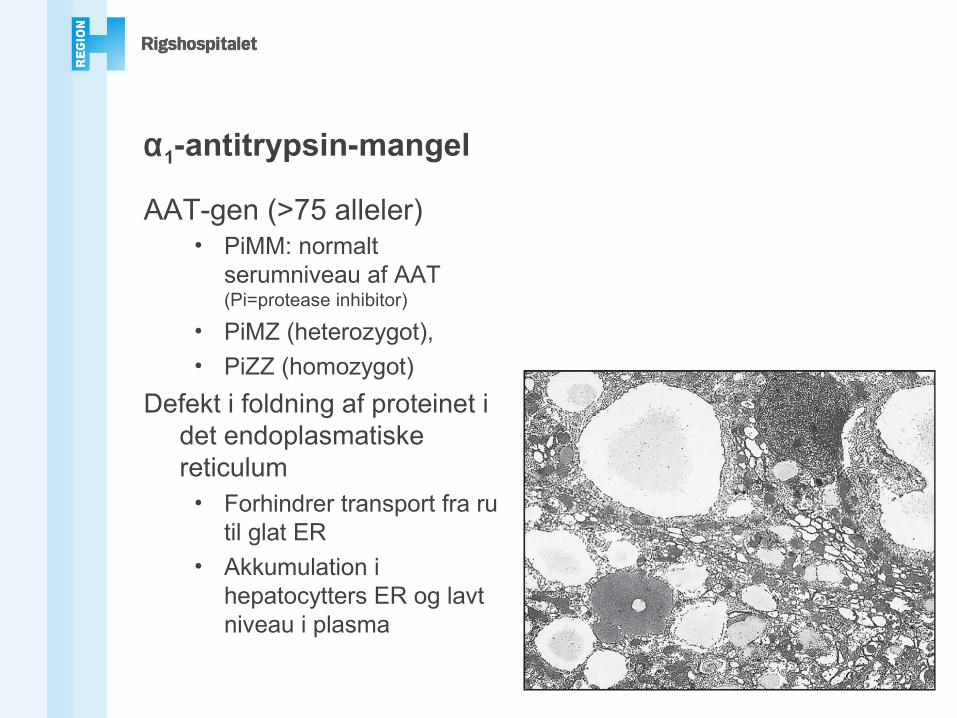
α1-antitrypsin-mangel
AAT-gen (>75 alleler)• PiMM: normalt
serumniveau af AAT (Pi=protease inhibitor)
• PiMZ (heterozygot),• PiZZ (homozygot)
Defekt i foldning af proteinet i det endoplasmatiske reticulum
• Forhindrer transport fra ru til glat ER
• Akkumulation i hepatocytters ER og lavt niveau i plasma
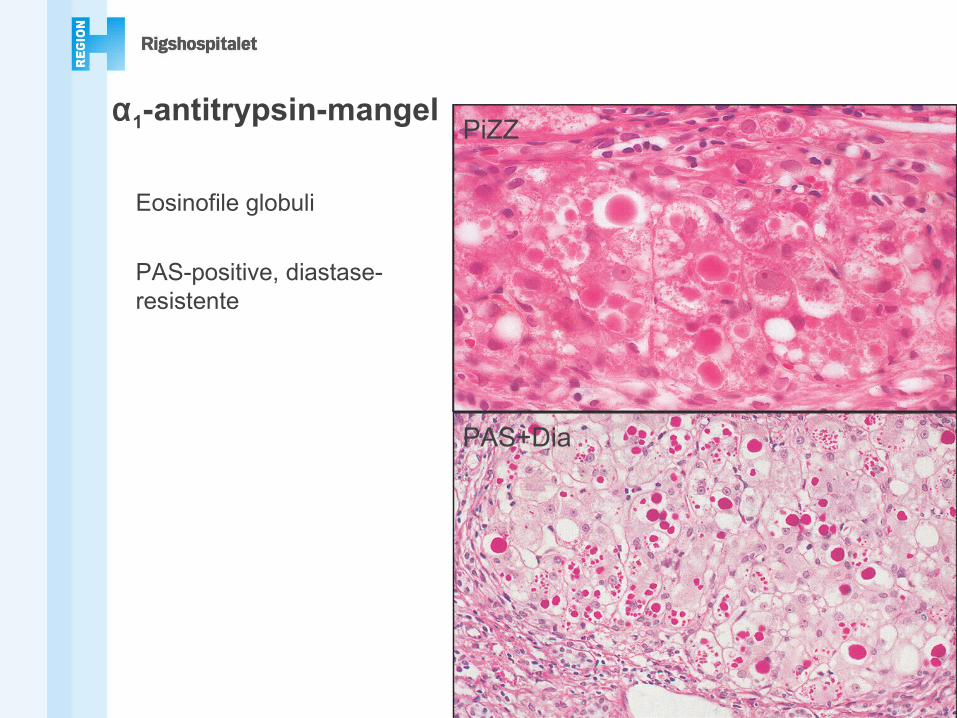
PiZZ
PAS+Dia
Eosinofile globuli
PAS-positive, diastase-resistente
α1-antitrypsin-mangel
DiagnoseLungesygdom (emfysem)Mistanke ved kolestase i barnealder og hos alle med udefineret leversygdom
• Serum α1-AT lav (OBS: akut fasereaktant, stiger ved inflammation – også hepatitis af anden årsag!!!)
• Typebestemmelse af protease inhibitor (Pi)
- ELISA- PCR (Z- og S-varianter; PiZ,
PiS)

LeverforandringerSes oftest hos prs. med Z-allel (og da kun hos få)
• 1-2 % af nordeuropæisk (kaukasisk) oprindelse, højst i Skandinavien
• Incidens 1:2000 – 1:5000
11 % udvikler symptomatisk leversygdom i barnealderen75 % har forhøjet ALAT
Serum α1-AT > 40 % af normal ⇒ normal ALAT
(Sveger, J Pediatr 1989; 104: 91-4)(Sveger, Acta Paediatr Scand 1998; 77: 847-51)

Debut af α1-AT-mangel
Neonatal debut: • Konjugeret hyperbilirubinæmi, svær kolestase• Kolestase forsvinder oftest inden 6 mdr., et mindre antal
dog kronisk leversygdom (bl.a. paucity of bile ducts)
Adult debut (prs. med mutation: % syge):• 41-50-årige: 5% (mand:kvinde 2:1)• 51-60-årige: 15% mænd, 0 % kvinder

Neonatal α1-antitrypsin-hepatitis
Minimal lobulær og portal inflammationPortalt ødemMinimal interfaseaktivitet
Ligner:• Neonatal hepatitis• Ekstrahepatisk biliær atresi
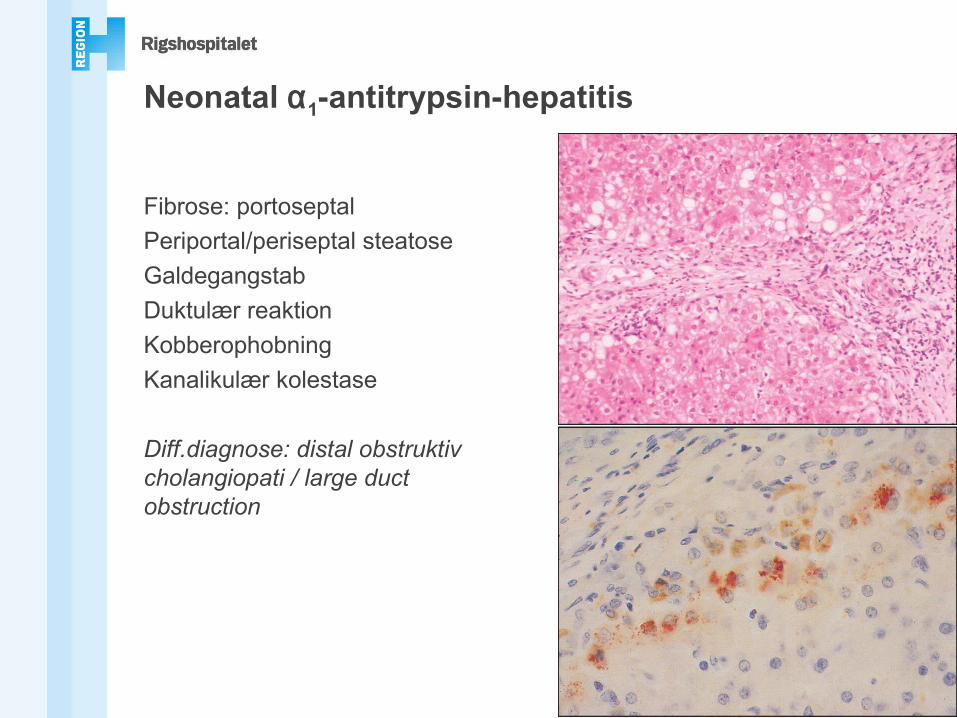
Neonatal α1-antitrypsin-hepatitis
Fibrose: portoseptalPeriportal/periseptal steatoseGaldegangstabDuktulær reaktion KobberophobningKanalikulær kolestase
Diff.diagnose: distal obstruktiv cholangiopati / large duct obstruction
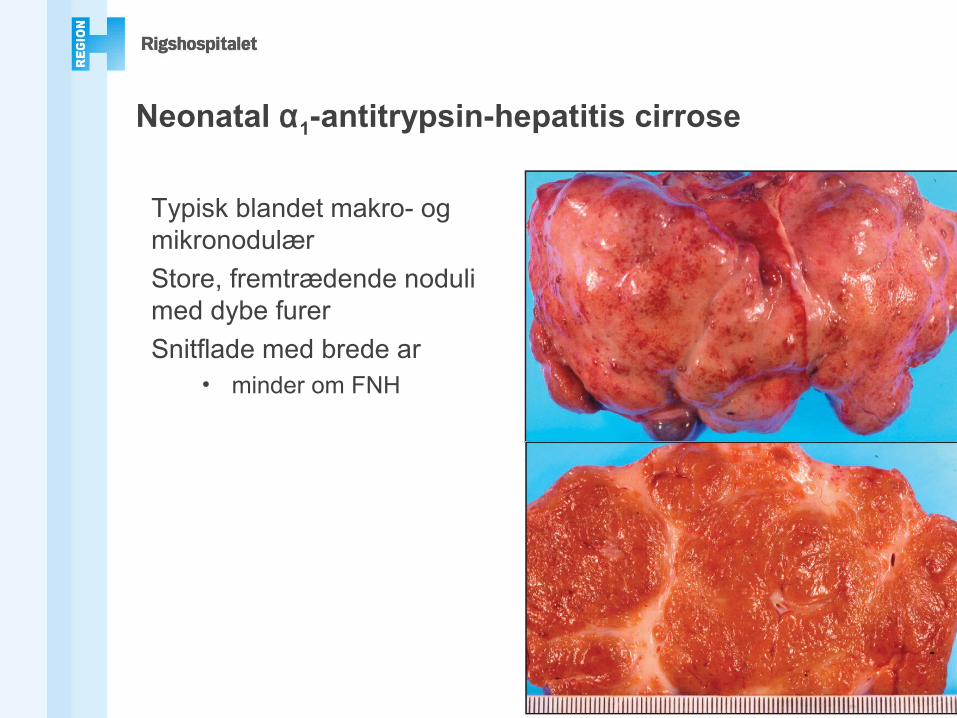
Neonatal α1-antitrypsin-hepatitis cirrose
Typisk blandet makro- og mikronodulærStore, fremtrædende noduli med dybe furerSnitflade med brede ar
• minder om FNH

Neonatal α1-antitrypsin-cirrose
Cirrotiske noduli• Omgivet af tæt kollagen• Adskilt af løst bindevæv
α1-antitrypsin-globuli• Pan-nodulært
IHC

Prognose af neonatal α1-AT-mangel
25 % kommer sig (3-10 år)25 % har persisterende forhøjet s-ALAT uden andre symptomer eller tegn25 % ikterus forsvinder, men vedvarende forhøjet ALAT med forstørret lever og milt 25 % dør pga. cirrose el. transplanteres (6 mdr.-17 år)
(Ibarguen, J Pediatr 1990; 117: 864-70)
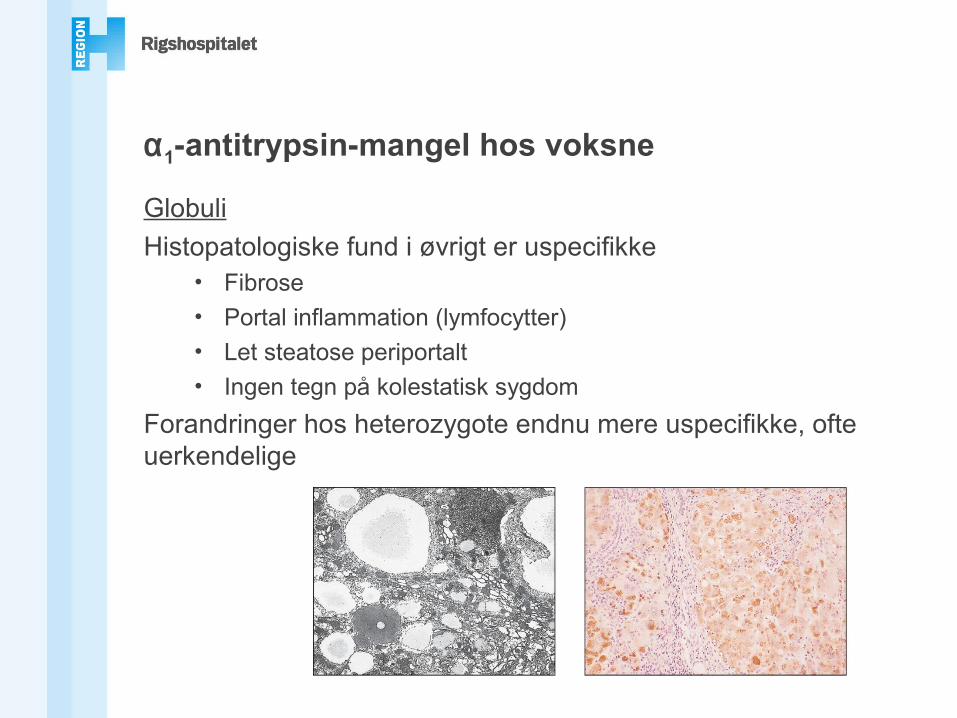
α1-antitrypsin-mangel hos voksne
GlobuliHistopatologiske fund i øvrigt er uspecifikke
• Fibrose• Portal inflammation (lymfocytter)• Let steatose periportalt• Ingen tegn på kolestatisk sygdom
Forandringer hos heterozygote endnu mere uspecifikke, ofte uerkendelige

Prognose, voksne
Lungesygdom (emfysem)
Cirrose er dødsårsag hos 10 % af ptt. med α1-AT-mangel
3 % udvikler HCC el. cholangiokarcinom- Ikke øget risiko hos heterozygote
(Berkowitz, Hepatology 1992; 15: 407-10)

Wilsons sygdomHepatolenticulær degeneration
Incidens 1:30.000
Gen ATP7B på 13q14.2-q21• P-type ATPase (Wilson ATPase), som
transporterer kobber- Funktioner: (1) inkorporering af Cu i
caeruloplasmin, (2) facilitering af ekskretion af Cu til galden
• Golgi-apparatetAutosomal recessiv
• 280 mutationer (Wilson Disease Mutation Database)
- http://www.wilsondisease.med.ualberta.ca/database.aspdatabase
• Ofte ”compound” heterozygote• Andre disponerende faktorer (forskelle i
præsentation hos tvillinger)- Apolipoprotein E
(Schiefermeier, Brain 2000; 123: 585-90)

Wilsons sygdom
Cu essentielt spormetalCuproproteiner
• Redox-reaktioner (elektronoverførsel) • Cellulær respiration, jernhomeostase, pigmentdannelse,
neurotransmitterproduktion, peptiddannelse, bindevævssyntese og antioxidation
Voksne: ca. 100 mg Cu• Diæt ca. 4-6 g Cu – 40 % absorberes• GI-absorption (ventrikel, duodenum)• Hurtig omsætning i lever
- efter 24 h kun 10% tilbage i cirkulation (bundet til histidin og andre a.a.)
• Biliær ekskretion – direkte proportional med konc. af Cu i leveren

Symptomer på mb. Wilson
Resultat af kobber-overload i div. væv og organer• Non-infektiøs kronisk hepatitis• Autoimmun-lignende billede
- Udslæt, artropati, øget gammaglobulin, pos. ANA el. ASMA• Sjældent debut med akut leversvigt
- Nær-normal ALAT

Symptomer på mb. WilsonØjensymptomer
• Kayser-Fleisher-ring- Cu-ophobning i Descemets
membran• Solsikke-katarakt
- Cu-ophobning i linsen
Andre:• Neuropsykiatriske symptomer er
hyppigste præsentationsform hos voksne (50 % af teenagere)
- Personlighedsforandringer, irritabilitet, psykose, depression
• Gynækomasti, • Amenorrhoea• Kardielle symptomer, nyresten,
nyreinsuff.

DiagnoseMistanke:
• Børn og unge med parenkymatøs leversygdom (↑ aminotransferaser) af ukendt årsag
• Sjældent symptomer før 3-årsalderen
Undersøgelser:S-caeruloplasmin
• <5 mg/dl: overensstemmende med Wilson• 5-20 mg/dl hos 95%: ikke diagnostisk
Kayser-Fleischer-ring• Diagnostisk (spaltelampeundersøgelse), dog ikke hos ptt. med kronisk kolestase
af anden årsag• Fravær hos 50 % hos voksne med sygdommen
Høj urin-kobber-udskillelse (24 timer)Cu-indhold i levervæv
• > 250 µg/g tørvægt (< 55 normal)

Behandling
Kelerende stoffer• Penicillamin
- Opløser de Cu-rige granula i hepatocytters lysosomer- Forhindrer udvikling af fulminant hepatitis
(Klein, J Hepatol 2000; 32: 193-201, rottemodel)
• Alt. triethylentetramin (Trientin) el. tetrathiomolybdat - øger udskillelsen af Cu
Zink (kompetitiv for optag af Cu)Diæt (lever, skalddyr, svampe, chokolade og nødder)

Wilson - histopatologi
Portal inflammation• Evt. interfaseaktivitet
Glykogenerede kerner periportalt
Fokal steatose
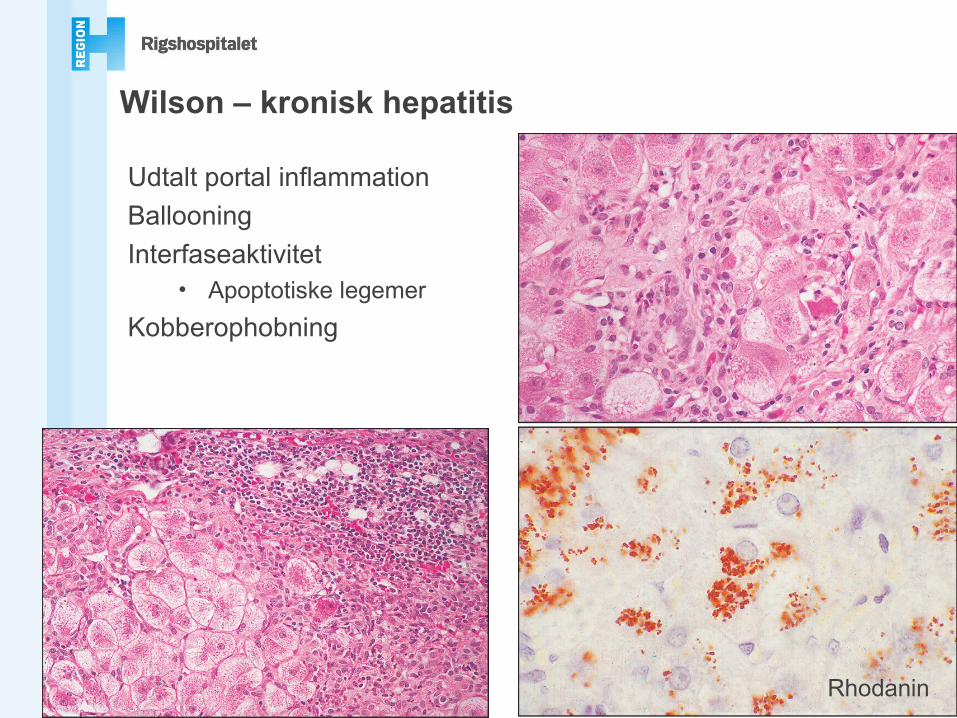
Wilson – kronisk hepatitis
Udtalt portal inflammationBallooningInterfaseaktivitet
• Apoptotiske legemerKobberophobning
Rhodanin
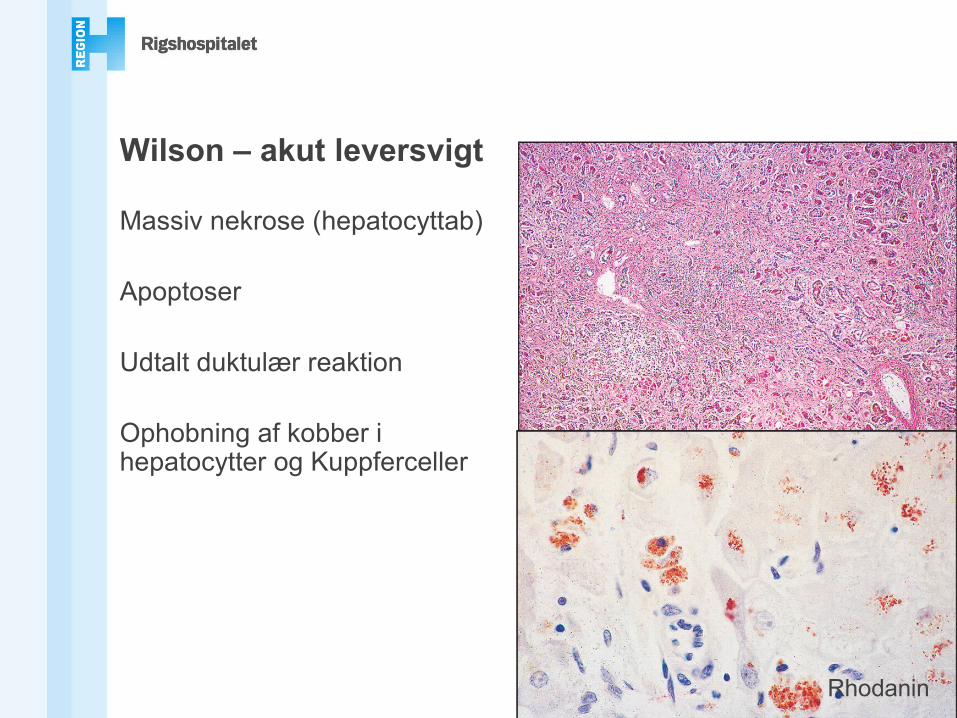
Wilson – akut leversvigt
Massiv nekrose (hepatocyttab)
Apoptoser
Udtalt duktulær reaktion
Ophobning af kobber i hepatocytter og Kuppferceller
Rhodanin
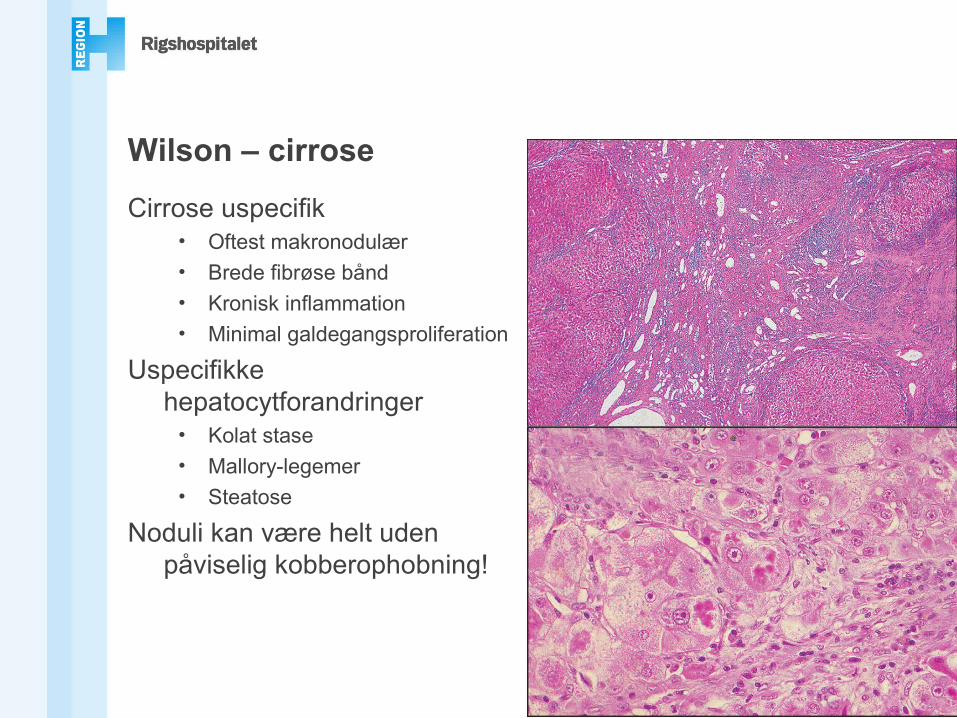
Wilson – cirrose
Cirrose uspecifik• Oftest makronodulær• Brede fibrøse bånd• Kronisk inflammation• Minimal galdegangsproliferation
Uspecifikke hepatocytforandringer
• Kolat stase• Mallory-legemer• Steatose
Noduli kan være helt uden påviselig kobberophobning!
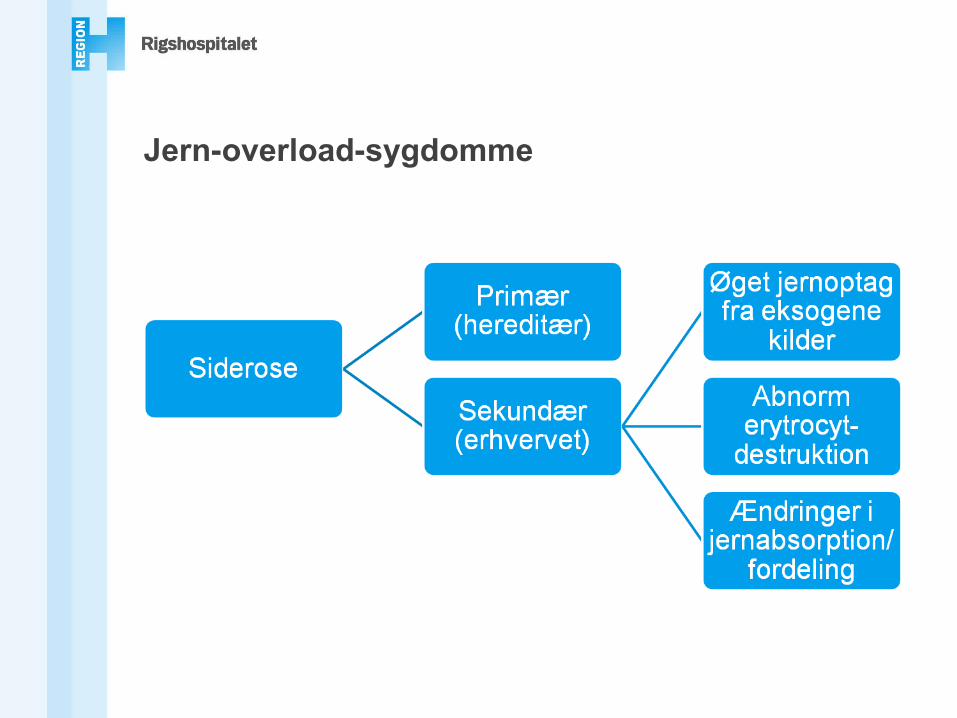
Jern-overload-sygdomme
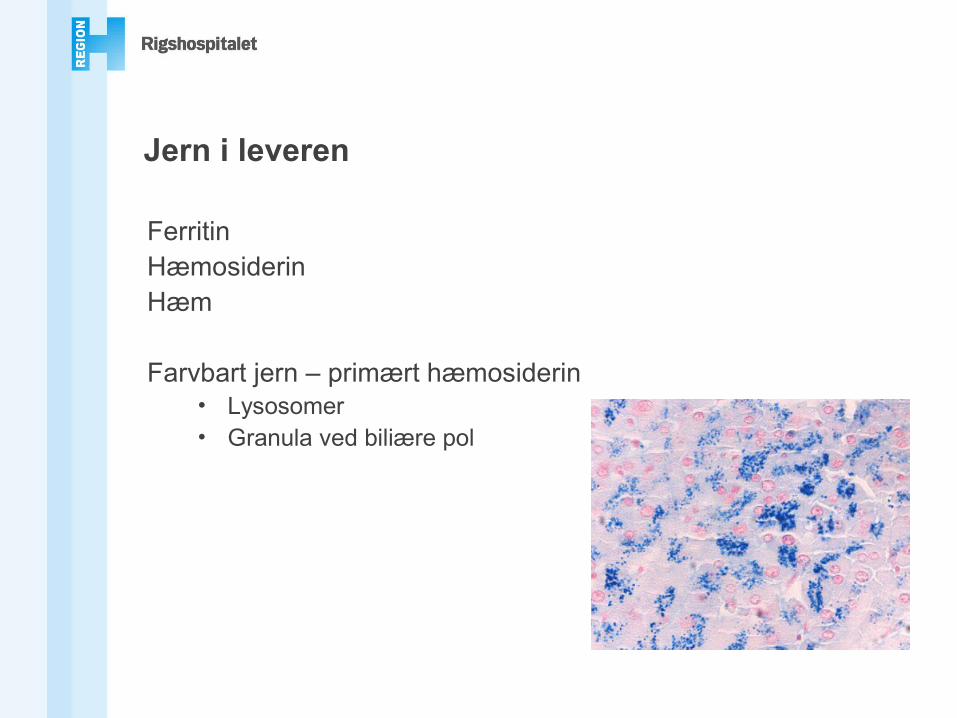
Jern i leveren
FerritinHæmosiderinHæm
Farvbart jern – primært hæmosiderin• Lysosomer• Granula ved biliære pol

Vurdering af siderotisk lever
Fordeling af farvbart jernhepatocytter, Kuppferceller, galdeepitel, endotel
Grad af siderose
Relateret vævsskadefibrose, cirrose, nekrose, HCC
Samtidig anden leversygdom

Primær / hereditær hæmokromatoseKlassisk HFE-relateret hæmokromatose (Type 1)
(HFE: High Iron Fe)Non-HFE hæmokromatose
• Juvenil hereditær hæmokromatose (Type 2)- Hæmojuvelin / HJV (HFE2)-relateret (Type 2A)- Hepcidin / HAMP-relateret (Type 2B)
• Transferrin receptor 2-relateret hæmokromatose (Type 3)• Ferroportin-relateret jern-overload (Type 4)
Andre, bl.a. atransferrinæmi, aceruloplasminæmi(sygdomme i jernbalance og distribution)

Sekundær siderose / hæmokromatoseSekundær/erhvervet siderose
• Jern-loading anæmier- Thalassaemia major- Sideroblastisk anæmi
• Porphyria cutanea tarda• Parenteral jern-overload• Anæmi pga. inflammation• Jern-overload ved kronisk leversygdom
- Hepatitis B og C- Alkoholisk siderose- Insulin-resistens (NAFLD/NASH)- End-stage cirrose

Hereditær hæmokromatose(von Recklinghausen 1889)
Triade:Diabetes, cirrose, melanin-
baseret pigmentering (Sheldon, Lancet 1934)
HFE-relateret 6p21.3Missense-mutation, hyppigst C282Y
alt. H63D eller S65C

PatogeneseMekanisme
• Teori 1: kryptprogrammeringshypotesen- HFE i duodenale kryptceller ”måler” jern i blodet og (ned-)regulerer
jernoptaget over duodenalslimhinden> Ved hæmokromatose har villusenterocytten relativ jernmangel og øger jernoptaget
ved opregulering af jerntransportgenerne DMT1 og ferroportin• Teori 2: hepcidinhypotesen
- HFE regulerer hepcidin, som regulerer jernoptaget (hæmning)> mutation i genet for hepcidin (HAMP) fører til mgl. hæmning af jernoptaget

KlinikFire kliniske stadier
• Genetisk prædisponering uden abnormiteter• Asymptomatisk jern-overload (2-5 g)• Jern-overload med tidlige symptomer• Jern-overload med organskade
Tidl. symptomer ofte uspecifikke og kræver høj grad af mistankeC282Y-homozygote
• Generel svaghed: 60 % • Artralgier/artritis: 30-40 %• Hepatomegali/cirrose: 13-60 %• Diabetes mellitus: 10-30 %• Seksuel dysfunktion: 10-40 %• Hjertearrhytmi: 20-29 %• Hjertesvigt: 15-35 %
(Adams, J Hepatol 2000; 33: 487-96)
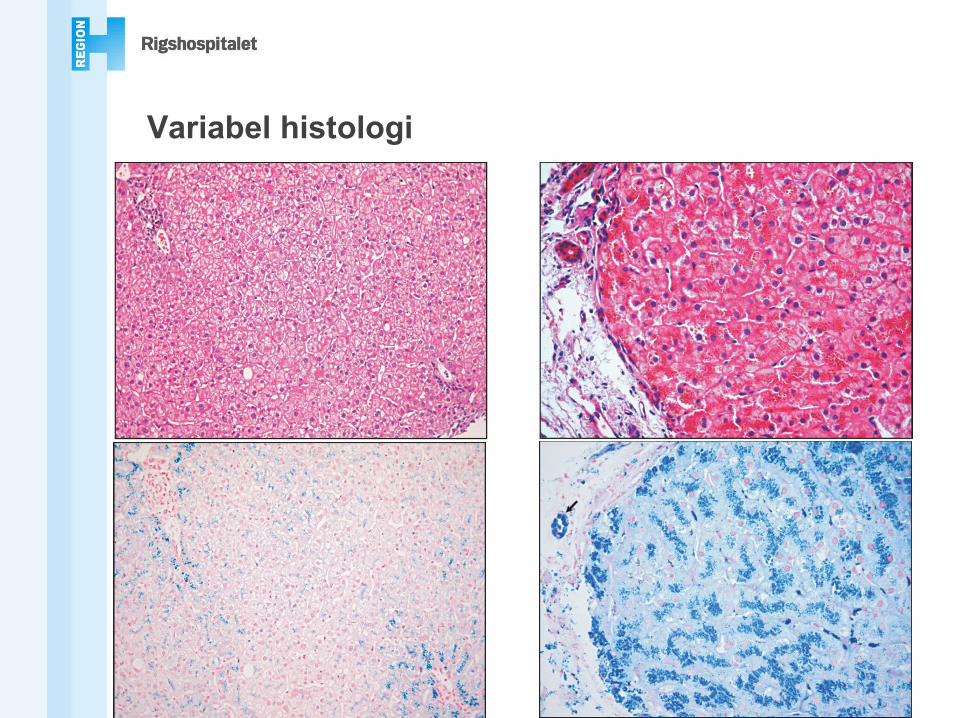
Variabel histologi
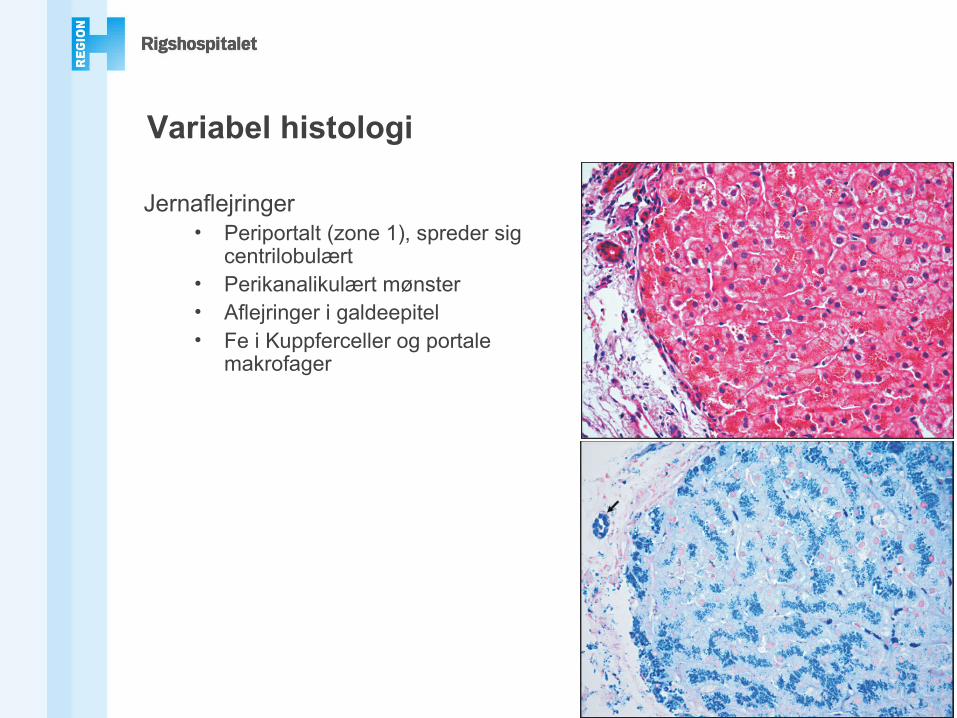
Variabel histologi
Jernaflejringer• Periportalt (zone 1), spreder sig
centrilobulært• Perikanalikulært mønster• Aflejringer i galdeepitel• Fe i Kuppferceller og portale
makrofager
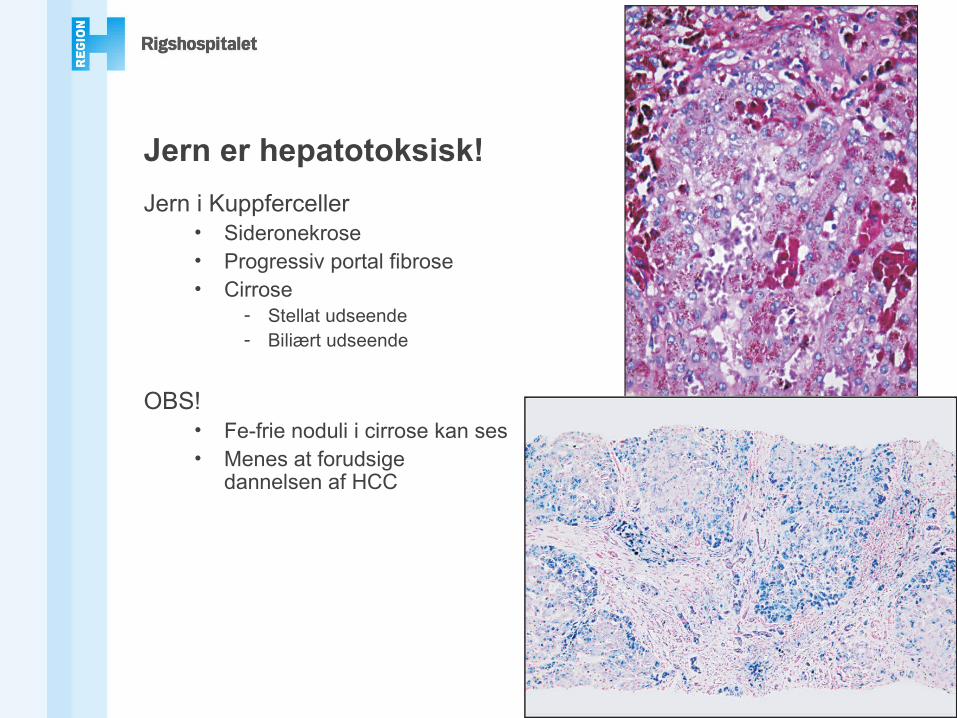
Jern er hepatotoksisk!
PAS
Jern i Kuppferceller• Sideronekrose• Progressiv portal fibrose• Cirrose
- Stellat udseende- Biliært udseende
OBS!• Fe-frie noduli i cirrose kan ses• Menes at forudsige
dannelsen af HCC

Gradering
Grad 4
Grad 0: Granula fraværende eller knap synlige ved x400
Grad 1: Granula knap synlige ved x200 og let erkendelige ved x400
Grad 2: Diskrete granula ved x100
Grad 3: Diskrete granula synlige ved x25
Grad 4: Jern synlige x10 eller med det blotte øje
(Subramaniam, World J Gastroenterol 2007; 13: 4755-60)
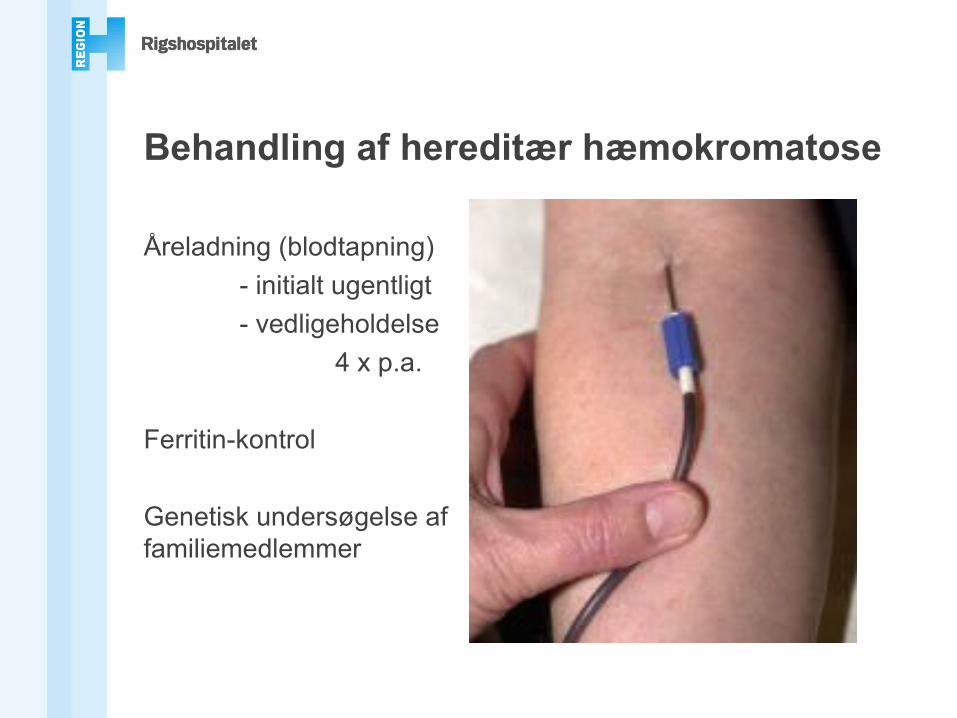
Behandling af hereditær hæmokromatose
Åreladning (blodtapning)- initialt ugentligt- vedligeholdelse
4 x p.a.
Ferritin-kontrol
Genetisk undersøgelse af familiemedlemmer

Kombination med alkoholisme
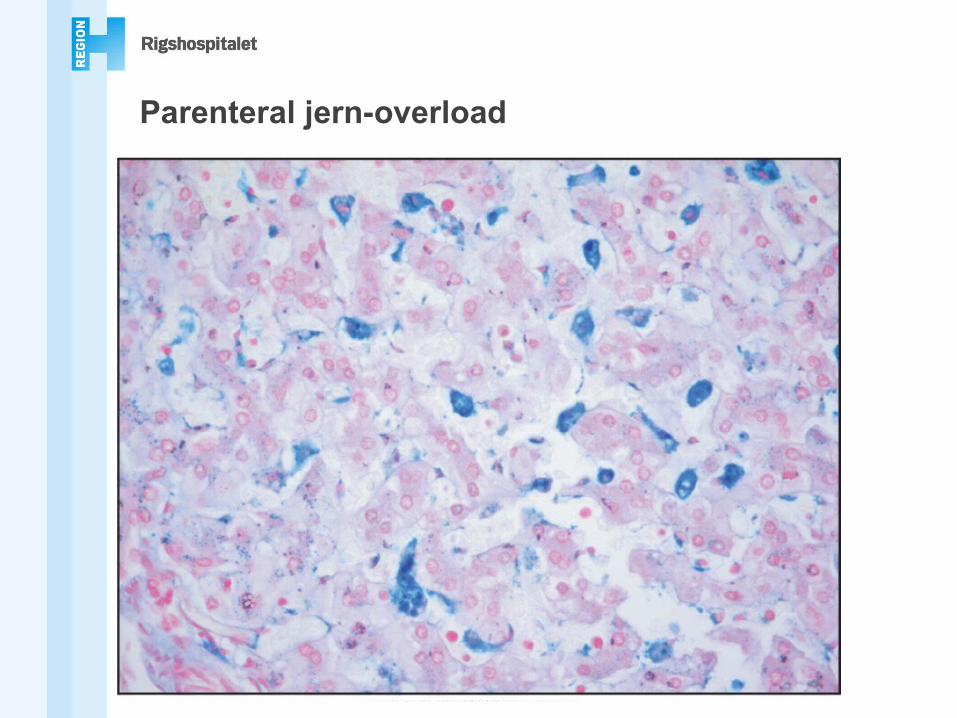
Parenteral jern-overload

Siderose ved viral hepatitis
Hepatitis B Hepatitis C
HBcAg med Perl-counterstaining
Jern er formentlig en selektiv fordel for virusreplikation(Kakizaki, Liver 2000; 20: 125-8)

Overvej altid ved kronisk leversygdom / leversymptomer! – essentielt for behandlingenHereditærePorfyrinstofskiftet
• Porfyria cutanea tarda• Erytropoietisk protoporfyri
Sukkerstofskiftet• Glycogen storage-sygdomme (Type 0-
XI)• Galaktosæmi
Glykoproteiner og glykolipider• Mucopolysakkaridose (Type I-VII)• Mucolipidose (Type I-IV)
ER storage disease• α1-antitrypsinmangel• α1-antichymotrypsinmangel• Hypofibrinogenæmi
Aminosyrestofskiftet• Tyrosinæmi
Lipoproteiner og lipidstofskiftet• Abetalipoproteinæmi• Familiær hypobetalipoproteinæmi• Familiær hyperkolesterolæmi• Kolesterolester storage disease• Fabry, Farber, Gaucher, Niemann-Pick
Peroxisomale sygdomme• Zellweger, Refsum
Mitochondriesygdomme• Pearson, Navajo neurohepatopati
Kobberstofskiftet• Wilson
Jernstofskiftet• Hæmokromatose (Type 1-4), neonatal
HKBilirubinstofskiftet
• Benign recurrent intrahepatisk cholestase (BRIC), progressiv familiær intrahepatisk cholestase (PFIC)
• Crigler-Najjar, Gilbert, Dubin-JohnsonAndre
• Cystisk fibrose, albinisme
ErhvervedeAdipositasDiabetes mellitusMetabolisk syndrom














