
Der Elongationsfaktor PAF1 aus S. cerevisiae Aufbau ...
157
Der Elongationsfaktor PAF1 aus S. cerevisiae: Aufbau, Funktion und Interaktionen Analyse der Protein-Protein Interaktionen innerhalb des Hefe PAF1-Komplexes und Untersuchung der Wechselwirkung des Elongationsfaktors mit yFACT, Casein-Kinase II und Bdf1 als Mittel zur funktionellen Charakterisierung Den Naturwissenschaftlichen Fakultäten der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades vorgelegt von Alexander B. Schwahn aus Coburg
Transcript of Der Elongationsfaktor PAF1 aus S. cerevisiae Aufbau ...

Der Elongationsfaktor PAF1 aus S. cerevisiae:
Aufbau, Funktion und Interaktionen
Analyse der Protein-Protein Interaktionen innerhalb des Hefe PAF1-Komplexes und Untersuchung der Wechselwirkung des
Elongationsfaktors mit yFACT, Casein-Kinase II und Bdf1 als Mittel zur funktionellen Charakterisierung
Den Naturwissenschaftlichen Fakultäten
der Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades
vorgelegt von
Alexander B. Schwahn
aus Coburg

Als Dissertation genehmigt von den Naturwissenschaftlichen Fakultäten der
Universität Erlangen-Nürnberg
Tag der mündlichen Prüfung: 19. April 2007
Vorsitzender der Promotionskommission: Prof. Dr. E. Bänsch
Erstberichterstatter: Prof. Dr. C. Koch
Zweitberichterstatter: Prof. Dr. G. Kreimer

für meine Eltern


Zusammenfassung
V
Zusammenfassung
An der Modulation der Chromatinstruktur und der Regulation der Transkription ist die reversible posttranslationale Modifikation der in den Nukleosomen enthaltenen Core-Histone maßgeblich beteiligt. Besonders die Funktion der co-transkriptionellen Histon-Methylierung als regulatorisches Signal für das Durchlaufen des Transkriptionszyklus konnte innerhalb der letzten Jahre immer genauer aufgeklärt werden. Der Elongationsfaktor PAF1 erweist sich durch seinen regulatorischen Einfluss auf alle drei bekannten Histon-Methyltransferasen der Hefe als entscheidender Faktor für die co-transkriptionelle Methylierung von Histon H3 und besitzt zudem eine Funktion bei der auf den Transkriptionsverlauf abgestimmten Prozessierung des 3'-Endes der naszierenden mRNA.
In der vorliegenden Arbeit wird die Zusammensetzung des Elongationsfaktors PAF1 sowie die Protein-Protein Interaktionen zwischen den Komponenten des Komplexes eingehend untersucht und anhand der Daten ein Modell für den Aufbau des PAF1-Komplexes vorgeschlagen. Außerdem wird der Einfluss der einzelnen Bestandteile des PAF1-Komplexes auf die Elongation und das telomere Silencing phänotypisch und funktionell charakterisiert. Zudem werden die Interaktionen des Komplexes mit dem Elongationsfaktor yFACT sowie der Casein-Kinase II untersucht und eine physikalische Wechselwirkung mit dem Bromodomainfactor 1 aufgeklärt.
Der Elongationsfaktor PAF1 erweist sich dabei als pentamerer Proteinkomplex, der
homogen aus den Proteinen Ctr9, Rtf1, Leo1, Paf1 und Cdc73 aufgebaut ist. Dabei zeigt sich, dass die direkte Assoziation von Paf1 und Ctr9 für die Stabilität des PAF1-Komplexes maßgeblich ist. Besonders Ctr9 wirkt aufgrund seiner seriellen Aneinanderreihung von insgesamt mindestens 10 TPR-Motiven als Bindungspartner für die übrigen Komponenten, wobei die Position der Bindungsdomänen für Paf1 und Cdc73 auf diskrete Anordnungen aus jeweils 2-3 N- bzw. C-terminal gelegenen TPR-Motiven lokalisiert werden kann. Die Voraussetzung der Paf1-Ctr9-Bindung für die Stabilität des Elongationsfaktors spiegelt sich unmittelbar in dem Befund wider, dass beide Proteine die Funktion des PAF1-Komplexes während der Elongation und auch den Einfluss auf das Telomersilencing entscheidend vermitteln. Besonders anhand der phänotypischen Untersuchung der Elongationsdefekte von Deletionsmutanten wird jedoch deutlich, dass Rtf1 und Cdc73 teilweise antagonistisch zu Paf1 wirken, was auf eine regulatorische Selbstinhibierung des Elongationsfaktors hinweisen könnte. Auch die Interaktion zwischen dem Elongationsfaktor PAF1 und yFACT sowie der Casein-Kinase II (CKII) wird durch Paf1 und Ctr9 bestimmt. Zudem konnte in vitro die CKII-abhängige Phosphorylierung von Ctr9, Rtf1, Leo1 und Paf1 gezeigt und die Lokalisation der Phosphorylierung von Ctr9 auf dessen C-Terminus begrenzt werden. Daneben konnte erstmalig die physikalische Interaktion zwischen dem Elongationsfaktor PAF1 und dem Bromo-domainfactor 1 demonstriert werden.
Aufgrund der beobachteten Interaktion zwischen den Elongationsfaktoren PAF1 und yFACT sowie der Casein-Kinase II und des Bromodomainfactor 1 und anhand von Literaturdaten wird ein Modell für eine ggf. gemeinsame Beteiligung aller vier Faktoren am Capping-Kontrollpunkt vorgeschlagen und diskutiert.

Summary
VI
Summary
Modulation of chromatin structure and regulation of transcription are strongly dependent on posttranslational modifications of core histones. Especially, the importance of cotranscriptional histone methylation as a regulatory signal for progression through the transcription cycle has been realised over the past years. All three histone methyltransferases in yeast are functionally dependent on elongation factor PAF1. Additionally, the PAF1 complex has a role in coupling 3'-end processing of premature mRNA with transcription elongation and termination.
This work investigates the composition of the elongation factor PAF1 in yeast and the protein-protein interactions between members within the complex. On the basis of these data a model for the assembly of PAF1 is suggested. In addition, the individual effects of its components on elongation and telomeric silencing are analysed genetically and physiologically. Furthermore, the interactions between PAF1 complex, elongation factor yFACT, and Casein Kinase II are analyzed and a physical interaction between PAF1 complex and Bromodomainfactor 1 is decribed.
The data demonstrate that the elongation factor PAF1 is a homogeneously composed pentameric complex of the proteins Ctr9, Rtf1, Leo1, Paf1 and Cdc73. The association between Paf1 and Ctr9 is crucial for stability of the complex. Ctr9 acts as a binding partner for most of the other components due to its arrangement of at least 10 serial TPR motives. The location of binding domains for Paf1 and Cdc73 can be mapped to 2-3 consecutive TPR motives on the N- or C-terminal end of Ctr9, respectively. The importance of Paf1 and Ctr9 for the stability of the PAF1 complex is reflected in their specific importance for transcriptional elongation and their influence on telomeric silencing. The study of individual yeast mutants suggested that both Rtf1 and Cdc73 negatively regulate the activity of Paf1. This could indicate that the function of elongation factor PAF1 is subject to negative autoregulation. The interaction between PAF1 complex, yFACT, and Casein Kinase II (CKII) are also determined by Paf1 and Ctr9. Ctr9, Rtf1, Leo1, and Paf1 were found to be phosphorylated by Casein Kinase II in vitro and the phosphorylation site(s) in Ctr9 could be mapped to its the C-terminal end. Furthermore, this work demonstrates a physical interaction between PAF1 complex and Bromodomainfactor 1 (Bdf1) in yeast.
On the basis of the observed interactions between the elongation factors PAF1 and yFACT as well as CKII a model for a combined participation of all four factors on the transcriptional capping checkpoint is suggested and discussed.

Inhaltsverzeichnis
VII
Inhaltsverzeichnis
Zusammenfassung V
Summary VI
1. Einleitung 1
1.1 Der Transkriptionszyklus und die C-terminale Domäne der RNA- Polymerase II
2
1.2 DNA-Elemente eukaryotischer Gene, Transkriptionsaktivierung und (Prä)-Initiation
3
1.3 Promotorfreigabe (Clearance) und Capping-Kontrollpunkt 3
1.4 Elongation und co-transkriptionelle Modifizierung von Histonen 4
1.5 Termination und Polyadenylierung 6
1.6 Der Elongationsfaktor PAF1 7
1.7 Zielsetzung und Inhalte dieser Arbeit 8
2. Ergebnisse 9
2.1 Der Elongationsfaktor PAF1: Zusammensetzung und Aufbau 9
2.1.1 Der PAF1-Komplex ist aus den Proteinen Ctr9, Rtf1, Leo1, Paf1 und Cdc73 aufgebaut
9
2.1.2 Ctr9 liegt im PAF1-Komplex als Monomer vor 122.1.3 Paf1 und Ctr9 sind für den Komplexzusammenhalt entscheidend, während
Rtf1, Leo1 und Cdc73 ohne Einfluss auf die Stabilität entfernt werden können
14
2.1.4 Ctr9 ist aus einer seriellen Anordnung von TPR-Motiven zusammengesetzt 182.1.5 Die C-terminalen TPR-Motive 9 & 10 in Ctr9 bilden die Bindungsdomäne
für Cdc73, während der N-terminale Bereich für die Interaktion mit Rtf1, Leo1 und Paf1 notwendig ist
22
2.2 Der Elongationsfaktor PAF1: Funktion 30
2.2.1 Die gleichzeitige Deletion von CDC73 und RTF1 induziert einen dramatischen Wachstumsdefekt
30
2.2.2 Der Wachstumsdefekt einer cdc73Δ rtf1Δ Doppelmutante kann durch die zusätzliche Deletion von PAF1 oder CTR9 zum Teil aufgehoben werden
33
2.2.3 Das Netzwerk synthetisch letaler Interaktionen zwischen dem PAF1- Komplex, Rad6/Bre1 und den Histon-Methyltransferasen Set1, Set2 und Dot1
35
2.2.4 Die Sensitivität gegen 6-Azauracil offenbart unterschiedliche Einflüsse der einzelnen Komponenten des PAF1-Komplexes auf die Elongationsfähigkeit der RNA-Polymerase II
39
2.2.5 Der PAF1-Komplex ist für das Silencing der Genexpression an Telomeren notwendig
43

Inhaltsverzeichnis
VIII
2.2.6 Der Dot1-abhängige Methylierungsstatus von Histon H3 an Lysin 79 wird von den Komponenten des PAF1-Komplexes unterschiedlich beeinflusst
47
2.2.7 Die Assoziation mit der RNA-Polymerase II kann nur von einem intakten PAF1-Komplex ausgebildet werden
49
2.3 Der Elongationsfaktor PAF1: Interaktionen 57
2.3.1 Interaktionen zwischen yFACT und dem PAF1-Komplex 572.3.2 Interaktionen zwischen Casein-Kinase II und dem PAF1-Komplex 602.3.3 Interaktionen zwischen dem Bromodomainfactor 1 (Bdf1) und dem
PAF1-Komplex 74
3. Diskussion 83
3.1 Protein-Protein-Interaktionen innerhalb und außerhalb des PAF1- Komplexverbundes
83
3.2 Die Silencingdefekte in Mutanten des PAF1-Komplexes können nicht alleine auf den Ausfall der Dot1-abhängigen Methylierung von Histon H3Lys79 zurückgeführt werden
89
3.3 Hinweise für eine mögliche regulatorische Selbstinhibierung des PAF1-Komplexes
92
3.4 Die Interaktion zwischen den Elongationsfaktoren PAF1 und yFACT im Kontext der Promotorfreigabe und des Capping-Kontrollpunkts
94
3.5 Die Interaktion zwischen PAF1-Komplex, Bdf1 und Casein-Kinase II 96
3.6 Der PAF1-Komplex in höheren Eukaryoten 99
4. Material & Methoden 100
4.1 Organismen und Reagenzien 100
4.1.1 Saccharomyces cerevisiae Stämme 1004.1.2 Escherichia coli Stämme 1054.1.3 Vektoren, Plasmide und Oligonukleotide 1054.1.4 Enzyme, Substrate und spezielle Feinchemikalien 1084.1.5 Antikörper und Affinitätsmatrizes 1094.1.6 Protein- und DNA-Größenstandards 1104.1.7 Verwendete Kits 1104.1.8 Kultur- und Selektionsmedien 111
4.2 Mikro- und Molekularbiologische Methoden 113
4.2.1 Kultivierung von S. cerevisiae und E. coli 1134.2.2 Amplifikation von DNA-Sequenzen durch Polymerase-Kettenreaktion
(PCR) 113
4.2.3 Subklonierung von Sequenzbereichen des CTR9-Gens und molekular- biologische Klonierung von Plasmiden
114
4.2.4 Transformation von S. cerevisiae mit Lithium-Acetat 1154.2.5 Kreuzung von Hefestämmen und Tetradenanalyse 1154.2.6 Kolonie-Färbetest zur Ermittlung der Carboxypeptidase-Aktivität
116

Inhaltsverzeichnis
IX
4.2.7 Untersuchung von Hefestämmen hinsichtlich einer auftretenden Temperatursensitivität sowie Elongations- und Silencingdefekten
117
4.3 Spezielle biochemische Methoden 117
4.3.1 Tandem-Affinitätsreinigung 1174.3.2 Immunpräzipitation von Protein-TAP-tag Fusionen über magnetische
Dynabeads 120
4.3.3 Chromatin-Immunpräzipitation 1214.3.4 In vitro Bindestudien von heterolog in E. coli exprimierten GST-Ctr9
Fragmenten mit Cdc73-HA6 bzw. Paf1-HA3 aus Hefe 124
4.3.5 In vitro Phosphorylierung und Kinaseaktivitätstest 1254.3.6 TCA-Extraktion und Westerntransfer von Histonen 126
4.4 Bioanalytische Verfahren 127
4.4.1 Elektrophoretische Trennung von DNA-Fragmenten im Agarosegel 1274.4.2 Proteinbestimmung nach Bradford 1274.4.3 Trennung von Proteinen durch SDS-Polyacrylamidgelelektrophorese 1284.4.4 Färbung von Polyacrylamidgelen zum Nachweis von Proteinen 1294.4.5 Immunologischer Nachweis von Proteinen durch Westernblot-Analyse 129
Danksagungen 131
Abkürzungsverzeichnis 132
Literaturverzeichnis 134
Anhang: SL-Datensatz 143


Einleitung
1
1. Einleitung
Kurz aber prägnant, fast nebensächlich weisen Watson und Crick in ihrem Artikel zur Struktur der DNA auf das fundamentale Prinzip der Selbstduplikation hin, welches der Vervielfältigung des genetischen Materials zugrunde liegt#. Da die beiden DNA-Stränge komplementär zueinander sind, beinhaltet jeder Strang die Information, die zum Aufbau des anderen Strangs benötigt wird (Watson and Crick, 1953).
Das Prinzip der spezifischen Basenpaarung ist nicht nur die Grundlage für die Replikation, sondern stellt ebenso die Informationsquelle für die Synthese der RNA durch die DNA-abhängigen RNA-Polymerasen dar (Kornberg, 1996; Myer and Young, 1998; Weinmann and Roeder, 1974; Wittekind et al., 1988). So einfach und brillant dieses grundlegende Prinzip auch erscheint, bei genauerer Betrachtung erweist sich insbesondere die Transkription proteinkodierender Gene durch die RNA-Polymerase II in Eukaryoten als höchst komplexer Prozess, der die konzertierte, koordinierte und exakt regulierte Interaktion zahlreicher weiterer Faktoren benötigt (Übersicht in: Orphanides and Reinberg, 2002). Diese betreffen nicht nur die unmittelbare Synthese der mRNA, genauer gesagt der prä-mRNA, sondern berücksichtigen von Anfang an alle nachfolgenden Schritte, wie die Prozessierung der prä-mRNA zur reifen mRNA und deren Export aus dem Kern. Capping, Splicing und Polyadenylierung laufen dabei ineinandergreifend und parallel zur Transkription und nicht getrennt und nacheinander ab. Sogar der mRNA Export wird bereits zu Beginn der Transkription vorbereitet und ist zudem funktionell an das Splicing gekoppelt (Rodriguez-Navarro et al., 2004; Strasser et al., 2002). Diese Verknüpfung der mRNA-Prozessierung mit der Transkription ermöglicht der Zelle eine stetige Qualitätskontrolle der reifenden mRNA und stellt sicher, dass diese nur vollständig prozessiert den Zellkern verlässt und als Matrize zur Proteinbiosynthese eingesetzt wird. Dazu kommt, dass für eine erfolgreiche Initiation der Transkription das Hindernis der dicht gepackten Chromatinstruktur mit Hilfe von Strukturmodulatoren überwunden werden muss, um die DNA-Matrize für die RNA-Polymerase überhaupt erst zugänglich zu machen (Biswas et al., 2004; Cosma et al., 1999; Richards and Elgin, 2002). Auch für die Elongation entlang der mit Nukleosomen besetzten DNA werden Chromatin-Strukturmodulatoren und spezielle Elongationsfaktoren benötigt (Rhoades et al., 2004; Simic et al., 2003), welche die Zahl der am Transkriptionsgeschehen beteiligten Faktoren noch einmal erhöht.
Diese Arbeit beschäftigt sich mit dem Aufbau, der Funktion und den Interaktionen des Elongationsfaktor PAF1 (Koch et al., 1999; Shi et al., 1997), dessen Funktion sich innerhalb der letzten 10 Jahre als ein wichtiges Elemente für die Regulation der Transkription und die Synchronisation der co-transkriptionellen RNA-Prozessierung herausgestellt hat (Übersicht in: Rosonina and Manley, 2005)
# “If only specific pairs of bases can be formed, it follows that if the sequence of bases on one chain is given, then the sequence on the other chain is automatically determined … It has not escaped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material.” (Watson and Crick, 1953)

Einleitung
2
1.1 Der Transkriptionszyklus und die C-terminale Domäne der RNA-Polymerase II
Die Transkription proteinkodierender Gene durch die RNA-Polymerase II kann in fünf
Phasen unterteilt werden: Prä-Initiation, Initiation, Promotorfreigabe (Clearance), Elongation und Termination. Die serielle Abfolge dieser Phasen bildet den Transkriptionszyklus (Übersicht in: Sims et al., 2004). Die mit der RNA-Polymerase II assoziierten Faktoren variieren mit den Phasen des Transkriptionszyklus und sind unmittelbar am Ablauf des Transkriptionsprozesses beteiligt. Daher kann das zu Beginn der Transkription am Promotor ausgebildete und als Prä-Initiationskomplex (PIC) bezeichnete Holoenzym der RNA-Polymerase II von der elongationskompetenten RNA-Polymerase II unterschieden werden, die auch als transkriptioneller Elongationskomplex bezeichnet wird (Kornberg, 1996; Shilatifard et al., 2003).
Eine Besonderheit, die ausschließlich bei der RNA-Polymerase II auftritt, findet sich in der C-terminalen Domäne (CTD) ihrer größten Untereinheit, in Hefe dem Protein Rpb1. Die CTD von Rpb1 bzw. vereinfachend, die CTD der RNA-Polymerase II, besteht aus einer vielfachen Aneinanderreihung der Heptapeptidsequenz YS2PTS5PS. Die Häufigkeit der Sequenzwiederholung nimmt dabei phylogenetisch betrachtet von der Hefe (26-fach) zum Säuger (52-fach) stetig zu (Allison et al., 1988). Die Heptapeptidsequenzen der CTD werden im Laufe des Transkriptionszyklus u. a. an Serin 2 (S2) und Serin 5 (S5) durch eine Reihe spezifischer Kinasen und Phosphatasen reversibel phosphoryliert bzw. dephosphoryliert (Komarnitsky et al., 2000; O'Brien et al., 1994). Das Durchlaufen des Transkriptionszyklus und das synchrone Ablaufen der co-transkriptionellen RNA-Prozessierung wird dabei entscheidend durch den Phosphorylierungsstatus der CTD reguliert, da sie je nach Phosphorylierung als spezifische Bindestelle für bestimmte Faktoren der Transkription bzw. der RNA-Prozessierung wirkt (Übersicht in: Orphanides and Reinberg, 2002; Proudfoot et al., 2002). Die CTD des Prä-Initiationskomplexes ist zunächst unphosphoryliert (Laybourn and Dahmus, 1989). Der Übergang zur Elongation wird durch exzessive Phosphorylierung der CTD begleitet, die auch im transkriptionellen Elongationskomplex erhalten bleibt. Die Phosphorylierung von Serin 5 bleibt auf die promotornahen Sequenzbereiche aktiv transkribierter Gene beschränkt und kann an deren 3'-Enden nicht mehr nachgewiesen werden. Die Phosphorylierung von Serin 5 ist daher für die Initiation und Promotorfreigabe charakteristisch. Die Phosphorylierung von Serin 2 kann dagegen mit der Elongations- und Terminationsphase korreliert werden, da sie typischerweise bei Polymerasen beobachtet wird, die gerade mit dem Leserahmen oder der 3'-untranslatierten Region assoziiert sind (Komarnitsky et al., 2000; O'Brien et al., 1994). Die CTD wird schließlich für das erneute Durchlaufen des Transkriptionszyklus durch die CTD-spezifische Phosphatase FCP1 dephosphoryliert (Cho et al., 2001; Mandal et al., 2002). Die CTD-Phosphatase FCP1 wird dabei ihrerseits durch die Initiationsfaktoren TFIIF, TFIIB und die Casein-Kinase II reguliert (Abbott et al., 2005; Cho et al., 2001; Palancade et al., 2002). Auch die co-transkriptionelle Modifikation der Histone ist an den Phosphorylierungsstatus der CTD gekoppelt (Xiao et al., 2003).

Einleitung
3
1.2 DNA-Elemente eukaryotischer Gene, Transkriptionsaktivierung und (Prä)-Initiation
Eukaryotische Gene sind durch diskrete DNA-Elemente mit konservierter Sequenz
gekennzeichnet, die der spezifischen Bindung regulatorischer Proteine - den generellen und genspezifischen Transkriptionsfaktoren - und der Rekrutierung der Transkriptionsmaschinerie dienen (Guarente, 1984; Struhl, 1987). Die Positionierung der RNA-Polymerasen erfolgt über ein als Promotor bezeichnetes DNA-Element. Der Promotor proteinkodierender Gene besitzt zu diesem Zweck mindestens zwei diskrete Sequenzbereiche, die als TATA-Box und transkriptioneller Startpunkt (transcriptional start site) bezeichnet werden. Die TATA-Box dient der Positionierung der RNA-Polymerase II. Das erste transkribierte Nukleotid definiert den transkriptionellen Startpunkt. Daneben treten weitere regulatorische Sequenzen auf, die zur Aktivierung oder Repression der Transkription und zur Regulation der Transkriptionsrate dienen. Zu diesen DNA-Elementen zählen u. a. Upstream Activating Sequences (UAS), Enhancer-Sequenzen, Upstream Repressing Sequences (URS) und Silencer-Sequenzen (Übersicht in: Lee and Young, 2000).
Die genspezifische Aktivierung der Transkription wird u. a. durch die Bindung transkriptioneller Aktivatoren an UAS-Elemente des betreffenden Promotors vermittelt. Die gebundenen Transkriptionsaktivatoren führen dann zur Rekrutierung von Chromatin-Strukturmodulatoren, wie etwa dem Swi/Snf- oder SAGA-Komplex (Cosma et al., 1999; Johnson et al., 2005; Timmers and Tora, 2005). Durch die Reorganisation der Chromatinstruktur über diese Komplexe wird die Bindung weiterer Transkriptionsfaktoren an den Promotor des aktivierten Gens und damit die Initiation der Transkription ermöglicht.
Die Prä-Initiation ist durch die Ausbildung des Prä-Initiationskomplexes (PIC) an dem Promotor des transkribierten Gens gekennzeichnet. Der Prä-Initiationskomplex umfasst u. a. die generellen Transkriptionsfaktoren (GTFs) TFIID, TFIIB, TFIIE, TFIIF und TFIIH sowie den aus 12 Komponenten bestehenden RNA-Polymerase II Core-Komplex (Orphanides et al., 1996). Der Prä-Initiationskomplex wird dabei schrittweise aufgebaut, beginnend mit der Bindung des TATA-binding protein (TBP) und der assoziierten Faktoren (TAFs) des generellen Transkriptionsfaktors TFIID an die TATA-Box des Promotors. Die Formierung des Prä-Initiationskomplexes ermöglicht schließlich die Ausbildung eines „offenen Komplexes“ zwischen RNA-Polymerase II und der DNA-Matrize. Der DNA-Doppelstrang wird hierbei in einem ATP-abhängigen Prozess aufgeschmolzen, so dass der kodierende Einzelstrang als Matrize für die Synthese der mRNA verwendet werden kann (Asturias et al., 1997; Kim et al., 2000; Kornberg, 1996; Sims et al., 2004). Durch die Verknüpfung der ersten zwei Ribonukleotide über eine Phosphodiesterbindung wird die Initiation abgeschlossen und der Eintritt in die Elongationsphase beginnt über den Prozess der Promotorfreigabe.
1.3 Promotorfreigabe (Clearance) und Capping-Kontrollpunkt
Die Promotorfreigabe kennzeichnet den Eintritt in die Elongationsphase und damit den eigentlichen Beginn der prozessiven Biosynthese des mRNA-Stranges (Goodrich and Tjian,

Einleitung
4
1994; Maxon et al., 1994). Während der Promotorfreigabe wird der Prä-Initiationskomplex teilweise aufgelöst bzw. umarrangiert. Ein Teil der GEFs verbleibt dabei an dem Promotor um die Re-Initiation der Transkription, also den Aufbau eines neuen Prä-Initiationskomplexes zu beschleunigen. Von den GEFs bleibt einzig TFIIF auch während der Elongation mit der RNA-Polymerase II assoziiert, die während der Promotorfreigabe durch die Assoziation mit den Elongationsfaktoren in ihre elongationskompetente Form, den transkriptionellen Elongationskomplex (TEC), umstrukturiert wird. Der Übergang zwischen Initiation und Elongation wird gleichzeitig als transkriptioneller Kontrollpunkt genutzt und stellt eine der wichtigsten Phasen für die Regulation der Transkription dar (Sims et al., 2004). Dieser als Capping-Kontrollpunkt bezeichnete Prozess synchronisiert die Modifizierung des 5’-Endes der entstehenden mRNA mit dem 5’-Methylguanosin-Cap und stellt sicher, dass der Eintritt in die
2 25 5
2 25 5
2 25 5
P P
2 25 5
2 25 5
PP PP
TBP
RNAPII (PIC)
RNAPII
2 25 5
P P
2 25 5
2 25 5
PP PP
RNAPII
DSIFNELFNELF
5‘-
5‘-
NELF
DSIF
NELFNELF
DSIF
2 25 5
P P
2 25 5
2 25 5
PP PP
RNAPII
DSIF NELFNELF
Ceg1
GpppXCeg1
2 25 5
P P
2 25 5
2 25 5
PP PP
RNAPII
GpppX
DSIFDSIF
p-TEFbp-TEFbPP
PP
RNAPII
GpppX
2 25 5
PP PP
NELFNELF
NELFNELF
RNAPII (TEC)
PPPP
C-terminale Domäne
a)
b)
c)
d)
e)
f)
5‘-Ende dernasziernden mRNA
Abb. 1.1 Vereinfachtes Modell der Abläufe am Capping-Kontrollpunkt (verändert nach Orphanides und Reinberg (2002))
(a → b) Während der Promotorfreigabe wird die CTD durch TFIIH an Serin 5 phosphoryliert. (b → c) Die Bindung von DSIF an die CTD führt zur Rekrutierung von NELF, der das Pausieren der RNA-Polymerase II verursacht. (c → d) Die Capping-Maschinerie (repräsentiert durch Ceg1) wird durch die Interaktion mit DSIF und der an Serin 5 phosphorylierten CTD rekrutiert und modifiziert das 5'-Ende der naszierenden mRNA mit dem 5'-Methylguanosin-Cap. (d → e) Die Rekrutierung von p-TEFb nach erfolgreicher Modifizierung des 5'-Endes mit der Cap-Struktur führt zur Phosphorylierung von DSIF und der CTD an Serin 2 (e → f) Die repressive Wirkung von NELF wird dadurch aufgehoben und die RNA-Polymerase II tritt in die Phase der transkriptionellen Elongation ein.

Einleitung
5
Elongationsphase der Transkription nur bei erfolgreicher Ausbildung der 5’-Cap-Struktur erfolgt (Orphanides and Reinberg, 2002; Rodriguez et al., 2000). Die Abläufe des Capping-Kontrollpunkts sind vereinfacht in Abb. 1.1 dargestellt. Im Rahmen der Promotorfreigabe wird die CTD der RNA-Polymerase II durch die Kin28-Kinase des generellen Transkriptionsfaktors TFIIH an Serin 5 phosphoryliert, was zur Rekrutierung des Transkriptionsfaktors DSIF führt. Die Bindung von DSIF an die RNA-Polymerase II vermittelt die Assoziation mit dem negativen Elongationsfaktor NELF, der ein Pausieren der Polymerase bewirkt. Die zur Ausbildung des 5’-Methylguanosin-Caps benötigten Enzyme werden durch die Interaktion mit DSIF und der an Serin 5 phosphorylierten CTD zur RNA-Polymerase II rekrutiert. Nach erfolgreicher Modifizierung des freien 5’-Endes der entstehenden mRNA mit dem 5’-Methylguanosin-Cap wird der positive Elongationsfaktor p-TEFb durch die Interaktion mit der Capping-Maschinerie zur RNA-Polymerase II rekrutiert, was zur Phosphorylierung von DSIF und der CTD an Serin 2 durch p-TEFb führt (Orphanides and Reinberg, 2002). Diese Phosphorylierungsereignisse besitzen Signalwirkung und im Zusammenspiel mit den Elongationsfaktoren Spt6 und FACT, die ebenfalls zur RNA-Polymerase II rekrutiert werden, wird die repressive Wirkung des negativen Elongationsfaktors NELF aufgehoben (Endoh et al., 2004; Lindstrom et al., 2003; Wada et al., 2000). Der Eintritt in die Elongationsphase beginnt.
1.4 Elongation und co-transkriptionelle Modifizierung von Histonen
Als Elongation wird die aktive Transkriptionsphase der RNA-Polymerase II bezeichnet, während der die DNA-Matrize fortwährend in die mRNA umgeschrieben wird (Orphanides and Reinberg, 2000). Für die Elongation der RNA-Polymerase II wird eine Vielzahl spezifischer Elongationsfaktoren benötigt, die unter anderem durch ihre Wirkung als Chromatin-Strukturmodulator bzw. als Histon-Chaperone die Passage der Polymerase entlang des mit Nukleosomen besetzten DNA-Stranges ermöglichen (Übersicht in: Shilatifard et al., 2003). Zu diesen Faktoren zählen unter anderem die auch am Capping-Kontrollpunkt beteiligten Elongationsfaktoren FACT und Spt6 (Belotserkovskaya and Reinberg, 2004).
Parallel zur Elongation erfahren die Histone eine Reihe reversibler kovalenter Modifikationen, die maßgeblich an der Regulation des Transkriptionszyklus und auch an der Modulation der Chromatinstruktur beteiligt sind. Hierbei besitzen vor allem die Mono-Ubiquitinierung von Histon H2B, die Methylierung von Histon H3, sowie die abgestimmte Acetylierung und Deacetylierung von Histon H4 Signalwirkung für das kontrollierte Durchlaufen des Transkriptionszyklus (Carrozza et al., 2005; Joshi and Struhl, 2005; Keogh et al., 2005; Morillon et al., 2005). Auch die Ausbildung des Heterochromatins und das transkriptionelle Silencing werden durch diese Modifikation der Histone reguliert, welche dabei einen direkten Einfluss auf die lokale Packungsdichte des Chromatins ausüben können oder als Signal für die Rekrutierung spezifischer Chromatin-Strukturmodulatoren wirken, ein Aspekt, dem in der so genannten Histon-Code-Hypothese Rechnung getragen wird (Lee and Young, 2000; Strahl and Allis, 2000).
Im Zusammenhang mit dem PAF1-Komplex stehen vor allem die genannten Modifikationen der Histone H2B und H3. In Hefe sind bislang drei Histon-Methyltransferasen

Einleitung
6
(HMTs) bekannt, die jeweils einen spezifischen Lysin-Rest von Histon H3 methylieren. Die ε-Aminogruppe des Lysin-Restes kann dabei mit bis zu drei Methylgruppen bis zum quartären Amin modifiziert werden, wobei die resultierende Signalwirkung von dem Grad der Methylierung abhängig ist (Seol et al., 2006; Shahbazian et al., 2005). Die Histon-Methyltransferase Set1 ist Bestandteil des Multiproteinkomplexes COMPASS und katalysiert spezifisch die Methylierung von Histon H3 an Lysin 4 (Briggs et al., 2001). Set2 ist spezifisch für die Methylierung von Histon H3 an Lysin 36 (Li et al., 2002; Strahl et al., 2002). Die Methylierung von Lysin 79 wird schließlich durch die Histon H3-spezifische Methyltransferase Dot1 übernommen (Feng et al., 2002; Lacoste et al., 2002). Die beiden Histon-Methyltransferasen Set1 und Dot1 benötigen für ihre Funktionalität die vorausgehende Mono-Ubiquitinierung von Histon H2B an Lysin 123 (Ng et al., 2002; Sun and Allis, 2002). Auch die Aktivität der Histon-Methyltransferase Set2 wird indirekt durch die Mono-Ubiquitinierung an H2B beeinflusst, da sie an die Funktion von Set1 gekoppelt ist (Morillon et al., 2005). Maßgeblich für diese Histon-Modifizierung ist das Ubiquitin-konjugierende Enzym Rad6 in Verbindung mit der H2B-spezifischen Ubiquitin-Ligase Bre1 (Robzyk et al., 2000; Wood et al., 2003a).
Auch die Phosphorylierung der CTD beeinflusst den Methylierungsstatus von Histon H3 (Xiao et al., 2003). Besonders die Rekrutierung bzw. Assoziation von Set2 mit der RNA-Polymerase II ist direkt von der Phosphorylierung an Serin 2 der CTD anhängig, da diese die Bindestelle für die Histon-Methyltransferase bereitstellt (Li et al., 2003). Dagegen ist die Assoziation von Set1 mit der an Serin 5 phosphorylierten, nicht aber mit der an Serin 2 phosphorylierten CTD der RNA-Polymerase II korreliert. Dies spiegelt sich auch in dem Methylierungsprofil von Histon H3 entlang des aktiv transkribierten Gens wider. Die Set1-abhängige Tri-Methylierung von Histon H3Lys4 bleibt auf die promotornahen Bereiche aktiv transkribierter Gene beschränkt, während die Di-Methylierung von Histon H3Lys4 genomweit und transkriptionsunabhängig beobachtet werden kann (Ng et al., 2003b). Die Set2-abhängige Methylierung von Histon H3Lys36 erfolgt dagegen nahezu komplementär zur Tri-Methylierung von Histon H3Lys4 über den gesamten transkribierten Genbereich (Krogan et al., 2003c). Die Dot1-abhängige Methylierung von Histon H3Lys79 kann bei 90% aller Nukleosomen beobachtet werden (van Leeuwen et al., 2002).
1.5 Termination und Polyadenylierung
Die Elongation der Transkription wird nach dem Überschreiten des DNA-kodierten Polyadenylierungssignals durch den Prozess der Termination gestoppt, was letztlich zu dem Ablösen der RNA-Polymerase II von der DNA-Matrize führt. Die Termination ist dabei eng an die enzymatische Abspaltung (Cleavage) des gerade synthetisierten RNA-Stranges von der RNA-Polymerase II und die unmittelbar daran anschließende Polyadenylierung des neuen 3'-Endes gekoppelt (Proudfoot et al., 2002). Auch für die Termination sowie die Abspaltung und Polyadenylierung der mRNA werden eine Reihe spezifischer Faktoren benötigt, die bereits gegen Ende der Elongation zur RNA-Polymerase II rekrutiert werden. Gleichzeitig lösen sich nicht länger benötigte Elongationsfaktoren von dem transkriptionellen Elongationskomplex, so

Einleitung
7
dass auch der Übergang der Elongation zur Termination, wie bereits der Übergang zwischen Initiation und Elongation, von einer Restrukturierung des RNA-Polymerase II Holoenzyms begleitet wird (Kim et al., 2004).
1.6 Der Elongationsfaktor PAF1
Das Protein Paf1 wird durch die Isolierung und Charakterisierung der RNA-Polymerase II bereits ursprünglich und dem Namen nach als mit der RNA-Polymerase II assoziierter Faktor beschrieben (Shi et al., 1996; Wade et al., 1996). Cdc73 wird in derselben Studie ebenfalls als assoziierter Faktor der RNA-Polymerase II aufgeklärt und wenig später als Interaktionspartner von Paf1 identifiziert (Shi et al., 1997). Unabhängig davon führt die Suche nach Regulatoren der Expression von G1-Cyclinen zur Isolierung von Ctr9 als Bestandteil eines Multiproteinkomplexes, an dem auch die Proteine Paf1 und Cdc73 beteiligt sind (Koch et al., 1999). Die Studie zeigt, dass die Expression von CLN2 in paf1Δ und ctr9Δ Mutanten, nicht aber in cdc73Δ Mutanten, signifikant reduziert ist, so dass die beiden Proteine an der Regulation der Swi4-abhängigen Genexpression (Hadwiger et al., 1989; Koch et al., 1993; Koch et al., 1996; Nasmyth and Dirick, 1991) beteiligt sein müssen, zumal sich sowohl CTR9 als auch PAF1 in cln3Δ Mutanten als essentiell erweisen (Di Como et al., 1995; Koch et al., 1999; Richardson et al., 1989). Diese Befunde und die weitere Charakterisierung von CDC73 zeigten zudem, dass es sich bei dem Gen – entgegen den Erwartungen und der irreführenden Genbezeichnung – nicht um ein „Cell Division Cycle“ Protein handelt (C. Koch, persönliche Mitteilung).
Aufgrund der beobachteten Assoziation mit der RNA-Polymerase II und der Tatsache, dass neben der Expression von CLN1 und CLN2 auch die Expression zahlreicher weiterer Gene von der Funktionalität des PAF1-Komplexes abhängig ist, wurde dem PAF1-Komplex zunächst eine Funktion während der Transkriptionsinitiation als alternativer Mediatorkomplex zugewiesen (Shi et al., 1997). Der Einfluss des PAF1-Komplexes auf die transkriptionelle Elongation wurde erst später durch seine Interaktion mit den Elongationsfaktoren yDSIF und yFACT auf genetischer und physikalischer Ebene deutlich (Squazzo et al., 2002) und eindrucksvoll anhand von Chromatin-Immunpräzipitations-Studien demonstriert, die erstmals zeigen konnten, dass der PAF1-Komplex auch während der Elongationsphase mit der aktiv transkribierenden RNA-Polymerase II stabil assoziiert bleibt (Pokholok et al., 2002).
Die Suche nach Faktoren, die die Modifikation von Histonen beeinflussen können sowie die darauf aufbauenden Studien zeigen schließlich, dass der jetzt als Elongationsfaktor geltende PAF1-Komplex am Anfang einer Signalkaskade steht, die über die Mono-Ubiquitinierung von Histon H2B durch den Rad6/Bre1-Komplex zur Methylierung von Histon H3 durch die drei bekannten Histon-Methyltransferasen Set1, Set2 und Dot1 führt (Krogan et al., 2003a; Krogan et al., 2003c; Ng et al., 2003a; Ng et al., 2003b; Wood et al., 2003b). Insbesondere wird dabei deutlich, dass sowohl die Funktionalität als auch die Assoziation von Rad6/Bre1 mit der elongierenden RNA-Polymerase II unmittelbar von dem Elongationsfaktor PAF1 abhängt (Wood et al., 2003b).

Einleitung
8
Zusätzlich zu dieser co-transkriptionellen Funktion übernimmt der Elongationsfaktor PAF1 eine Rolle bei der Polyadenylierung der mRNA sowie der Termination bzw. der Ausbildung des 3’-Endes von nicht-polyadenylierten Transkripten der RNA-Polymerase II (Mueller et al., 2004; Sheldon et al., 2005).
1.7 Zielsetzung und Inhalte dieser Arbeit
In der vorliegenden Arbeit wurde ein besonderes Augenmerk auf die Untersuchung der Zusammensetzung und der Protein-Protein-Interaktionen innerhalb des Elongationsfaktors PAF1 gerichtet, um anhand der Daten ein Modell für den Aufbau des PAF1-Komplexes zu erstellen. Aufgrund des Modells und der phänotypischen und funktionellen Charakterisierung des Komplexes bezüglich seines Einflusses auf die Elongation und das Telomersilencing sollten neue Einsichten in die Funktionsweise des Elongationsfaktors gewonnen werden. Daneben wurden auch die physikalischen und funktionellen Interaktionen des PAF1-Komplexes mit dem Elongationsfaktor und Chromatin-Strukturmodulator yFACT und der Casein-Kinase II untersucht, sowie erstmalig eine Interaktion zwischen dem PAF1-Komplex und dem Bromodomainfactor 1 beschrieben.

Ergebnisse
9
2. Ergebnisse 2.1 Der Elongationsfaktor PAF1: Zusammensetzung und Aufbau
Die Assoziation der Proteine Paf1, Ctr9 und Cdc73 zu einem Multiproteinkomplex konnte erstmals durch eine Untersuchung von Koch et al. (1999) gezeigt werden. Aufgrund der ermittelten apparenten Masse dieses Komplexes wurde dabei bereits auf die Existenz weiterer Komplexbestandteile geschlossen. Ausgehend von dieser Studie war die eingehende Charakterisierung der Zusammensetzung und des Aufbaus des PAF1-Komplexes eine Zielsetzung dieser Arbeit.
2.1.1 Der PAF1-Komplex ist aus den Proteinen Ctr9, Rtf1, Leo1, Paf1 und Cdc73 aufgebaut
Der PAF1-Komplex wurde zur Charakterisierung seiner Zusammensetzung durch
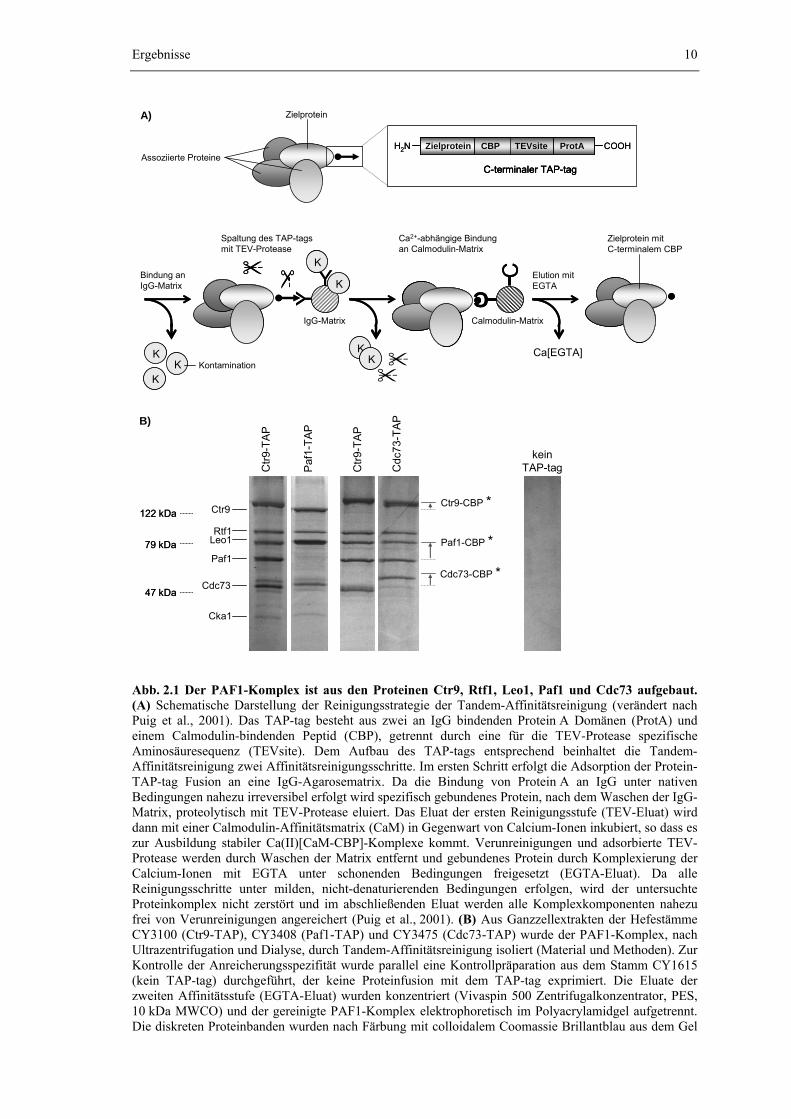
Tandem-Affinitätsreinigung über das gleichnamige Affinitätstag isoliert (Puig et al., 2001). Hierzu wurden jeweils C-terminale Protein-TAP-tag Fusionen (vgl. Abb. 2.1 A) von Ctr9, Paf1 oder Cdc73 eingesetzt. Die Einführung des TAP-tags erfolgte dabei auf chromosomaler Ebene durch homologe Rekombination eines genspezifischen TAP-tag Moduls in den Leserahmen des Zielgens (Knop et al., 1999; Puig et al., 2001). Damit verbleibt der Zelle nur das affinitätsmarkierte „TAP-Allel“ als Quelle für das entsprechende Protein. Eine Kompetition mit der endogenen Wildtypform des Proteins um den Einbau in den PAF1-Komplex wird so von vorneherein unterbunden. Zudem erfolgt die Expression der Protein-TAP-tag Fusion weiterhin mit der natürlichen Expressionsrate unter Kontrolle des eigenen Promotors. Damit kann auch eine Beeinflussung der Komplexstöchiometrie durch eine veränderte Proteinkonzentration weitestgehend ausgeschlossen werden. Genetische und phänotypische Untersuchungen zeigten, dass die Einführung des TAP-tags zu keiner erkennbaren Beeinträchtigung der physiologischen Funktion des PAF1-Komplexes führt (Daten nicht gezeigt). Die Strategie der Tandem-Affinitätsreinigung wird genauer in Abb. 2.1 A erklärt. Um die Ausbeute an gereinigtem PAF1-Komplex zu erhöhen, wurde zusätzlich eine Deletion des PEP4-Gens eingeführt, die zu einem Ausfall der vakuolären Peptidasen A, B und C führt (Jones, 1991).
Die Identifizierung der im gereinigten PAF1-Komplex enthaltenen Proteine erfolgte durch massenspektrometrische Analyse, die in Zusammenarbeit mit der Universität Manchester von Dr. Guido Sauer (Department of Biomolecular Sciences, BmS) durchgeführt wurde. Der gereinigte PAF1-Komplex wurde dazu elektrophoretisch im Polyacrylamidgel (10%) getrennt und die einzelnen Proteinbanden aus dem Gel ausgeschnitten. Die in den Gelfragmenten enthaltenen Proteine wurden mit Trypsin verdaut, die resultierenden Peptidfragmente eluiert und über eine C18 Reversed Phase Matrix (Millipore Zip Tip C18) angereichert und gereinigt. Die Fragmente wurden durch Matrix-unterstützte Laserdesorption/Ionisation (MALDI) mit α-Cyano-Zimtsäure ionisiert und durch ein "Time Of Flight"-Massenspektrometer (Micromass,
Ergebnisse
10
A)
B)
122 kDa
79 kDa
47 kDa
122 kDa
79 kDa
47 kDa
Ctr9
-TAP
Ctr9
-TAP
Paf
1-TA
P
Cdc
73-T
AP
Cdc73-CBP *
Paf1-CBP *
Ctr9-CBP *Ctr9
Rtf1Leo1
Cdc73
Cka1
Paf1
keinTAP-tag
Assoziierte Proteine
Zielprotein
KKKK
YY
K
K
YY
KK
KK
KKKK
C CC C
Ca[EGTA]
IgG-Matrix Calmodulin-Matrix
Bindung anIgG-Matrix
Spaltung des TAP-tagsmit TEV-Protease
Ca2+-abhängige Bindungan Calmodulin-Matrix
Elution mitEGTA
KKKontamination
Zielprotein mit C-terminalem CBP
C-terminaler TAP-tag
H2N Zielprotein CBP TEVsite ProtA COOH
C-terminaler TAP-tagC-terminaler TAP-tag
H2N Zielprotein CBP TEVsite ProtA COOH
Abb. 2.1 Der PAF1-Komplex ist aus den Proteinen Ctr9, Rtf1, Leo1, Paf1 und Cdc73 aufgebaut. (A) Schematische Darstellung der Reinigungsstrategie der Tandem-Affinitätsreinigung (verändert nach Puig et al., 2001). Das TAP-tag besteht aus zwei an IgG bindenden Protein A Domänen (ProtA) und einem Calmodulin-bindenden Peptid (CBP), getrennt durch eine für die TEV-Protease spezifische Aminosäuresequenz (TEVsite). Dem Aufbau des TAP-tags entsprechend beinhaltet die Tandem-Affinitätsreinigung zwei Affinitätsreinigungsschritte. Im ersten Schritt erfolgt die Adsorption der Protein-TAP-tag Fusion an eine IgG-Agarosematrix. Da die Bindung von Protein A an IgG unter nativen Bedingungen nahezu irreversibel erfolgt wird spezifisch gebundenes Protein, nach dem Waschen der IgG-Matrix, proteolytisch mit TEV-Protease eluiert. Das Eluat der ersten Reinigungsstufe (TEV-Eluat) wird dann mit einer Calmodulin-Affinitätsmatrix (CaM) in Gegenwart von Calcium-Ionen inkubiert, so dass es zur Ausbildung stabiler Ca(II)[CaM-CBP]-Komplexe kommt. Verunreinigungen und adsorbierte TEV-Protease werden durch Waschen der Matrix entfernt und gebundenes Protein durch Komplexierung der Calcium-Ionen mit EGTA unter schonenden Bedingungen freigesetzt (EGTA-Eluat). Da alle Reinigungsschritte unter milden, nicht-denaturierenden Bedingungen erfolgen, wird der untersuchte Proteinkomplex nicht zerstört und im abschließenden Eluat werden alle Komplexkomponenten nahezu frei von Verunreinigungen angereichert (Puig et al., 2001). (B) Aus Ganzzellextrakten der Hefestämme CY3100 (Ctr9-TAP), CY3408 (Paf1-TAP) und CY3475 (Cdc73-TAP) wurde der PAF1-Komplex, nach Ultrazentrifugation und Dialyse, durch Tandem-Affinitätsreinigung isoliert (Material und Methoden). Zur Kontrolle der Anreicherungsspezifität wurde parallel eine Kontrollpräparation aus dem Stamm CY1615 (kein TAP-tag) durchgeführt, der keine Proteinfusion mit dem TAP-tag exprimiert. Die Eluate der zweiten Affinitätsstufe (EGTA-Eluat) wurden konzentriert (Vivaspin 500 Zentrifugalkonzentrator, PES, 10 kDa MWCO) und der gereinigte PAF1-Komplex elektrophoretisch im Polyacrylamidgel aufgetrennt. Die diskreten Proteinbanden wurden nach Färbung mit colloidalem Coomassie Brillantblau aus dem Gel

Ergebnisse
11
ausgeschnitten und die Identität der Proteine anhand ihres „Peptide Mass Fingerprints“ durch massenspektrometrischen Analyse bestimmt. Nach der Reinigung verbleibt das Calmodulin Binding Peptide (CBP, in der Abb. mit einem Stern markiert) am Fusionspartner des TAP-tags. Das veränderte Molekulargewicht führt daher zur Verschiebung der entsprechenden Proteinbande (Stern).
Mathilda) analysiert. Die Identifizierung der Komplexbestandteile erfolgte anhand ihres „Peptide Mass Fingerprints“ durch Abgleich mit div. Proteindatenbanken unter Zuhilfenahme des Programms „Mascot“ (Helsens et al., 2007).
Wie Abb. 2.1 B zeigt, offenbart die elektrophoretische Auftrennung des gereinigten PAF1-Komplexes fünf prominente Proteinbanden. In einer Kontrollpräparation aus einem Hefestamm, der keine Proteinfusion mit dem TAP-tag exprimiert, treten diese Banden dagegen nicht auf, was die Spezifität der Methode und die hohe Reinheit des isolierten Komplexes dokumentiert. Durch die massenspektrometrische Analyse des gereinigten Komplexes wurden Ctr9, Paf1 und Cdc73 als Komponenten des PAF1-Komplexes bestätigt und zudem die Proteine Rtf1 und Leo1 als weitere Komponenten des PAF1-Komplexes identifiziert (A. Schwahn, Diplomarbeit, 2002 und unveröffentlichte Daten, nicht gezeigt). Diese fünf Proteine werden im Folgenden als Kernbestandteile des PAF1-Komplexes bezeichnet.
Neben den fünf Kernbestandteilen treten weitere schwache Banden auf, die von Interaktionspartnern des PAF1-Komplexes herrühren, zum Teil jedoch sicher auf Verunreinigungen während der Präparation zurückzuführen sind (A. Schwahn, Diplomarbeit, 2002 und unveröffentlichte Daten, nicht gezeigt). Durch die Massenspektrometrie wurde dabei die Assoziation der Spt16-Komponente des Elongationsfaktors yFACT (im Gel überlagert durch Ctr9) und der α-Kette der Casein-Kinase II (Cka1) mit dem gereinigten PAF1-Komplex aufgedeckt, was einen ersten Hinweis auf eine potentielle Interaktion zwischen dem PAF1-Komplex und yFACT bzw. der Casein-Kinase II lieferte (Kapitel 2.3).
Aus Abb. 2.1 B wird auch deutlich, dass die Reinigung des PAF1-Komplexes unabhängig davon, ob Paf1, Ctr9 oder Cdc73 als Fusionspartner des TAP-tags eingesetzt werden, immer zu dem Auftreten von fünf markanten Proteinbanden im elektrophoretisch aufgetrennten Komplex führt. Ein Vergleich der Laufweite dieser Banden zeigt, dass durch die Variation der zur Reinigung ausgenutzten Protein-TAP-tag Fusion jeweils eine Bande zu höheren Molekulargewichten verschoben wird. Dabei handelt es sich immer um das mit dem TAP-tag fusionierte Protein selbst, da seine Masse aufgrund der verbleibende Fusion mit dem Calmodulin-bindenden Peptid (CBP) des TAP-tags erhöht wird. Die Laufhöhe aller anderen Banden bleibt dagegen unverändert. Zudem treten durch die Variation des mit dem TAP-tag fusionierten Proteins keine zusätzlichen Banden auf. Die Tandem-Affinitätsreinigung des PAF1-Komplexes resultiert also immer in der Isolierung der fünf Kernbestandteile Ctr9, Rtf1, Leo1, Paf1 und Cdc73. Zusätzliche, vom Fusionspartner des TAP-tags abhängige Banden bzw. Komplexkomponenten treten dagegen nicht auf. Die fünf Kernkomponenten bilden damit eine homogene, strukturelle und vermutlich auch funktionelle Einheit ohne erkennbare Überlappung mit anderen Proteinkomplexen. Übereinstimmend mit dieser Interpretation konnte für alle fünf Komponenten durch indirekte Immunfluoreszenz eine subzelluläre und ausschließliche Lokalisation im Zellkern beobachtet werden (Daten nicht gezeigt).

Ergebnisse
12
Diese Ergebnisse werden von mehreren Studien bestätigt, die voneinander unabhängig zur Aufklärung der Proteine Ctr9, Rtf1, Leo1, Paf1 und Cdc73 als Kernbestandteile des PAF1-Komplexes führen und zudem eine Interaktion mit yFACT und der Casein-Kinase II andeuten (Krogan et al., 2002b; Mueller and Jaehning, 2002; Squazzo et al., 2002).
2.1.2 Ctr9 liegt im PAF1-Komplex als Monomer vor Für den PAF1-Komplex wurde durch Gelfiltrationschromatographie ein apparentes
Molekulargewicht von ca. 670 kDa ermittelt (Koch et al., 1999). Ein pentamerer Komplex aus den Komponenten Ctr9, Rtf1, Leo1, Paf1 und Cdc73 würde, rein rechnerisch, aber nur ein Molekulargewicht von 340,6 kDa (bzw. ca. 410 kDa bei Zugrundlage der durch die SDS-Polyacrylamidgelelektrophorese ermittelten apparenten Massen) aufweisen. Mehrere unabhängige massenspektrometrische Analysen des gereinigten PAF1-Komplexes geben zwar Rückschlüsse auf einige Interaktionspartner des Komplexes, lassen aber keine zusätzlichen, stöchiometrischen Komponenten erwarten, welche die Differenz aus apparenter und berechneter Masse erklären könnten (Krogan et al., 2002b; Mueller and Jaehning, 2002; Squazzo et al., 2002; eigene Beobachtung, vgl. Kapitel 2.1.1).
Eine mögliche Interpretation für das beobachtete Elutionsverhalten des PAF1-Komplexes bei der Gelfiltrationschromatographie wäre eine gestreckte Geometrie des Komplexes, oder auch die Ausbildung eines dimeren PAF1-Komplexes, was jeweils zu einer beschleunigten Elution und damit in einer Überschätzung der tatsächlichen Masse resultieren würde. Die Differenz zwischen apparenter und berechneter Komplexmasse könnte aber auch durch eine Abweichung von einer einfachen 1:1 Stöchiometrie der einzelnen Komponenten zueinander verursacht werden. Besonders Ctr9 fällt im gereinigten Komplex durch seine im Vergleich zu den Banden der anderen Bestandteile deutlich intensivere Proteinbande auf, die, unabhängig von der Wahl des TAP-tag Fusionspartners und auch unabhängig von der Färbemethode, die Stärke der Banden für Rtf1, Leo1, Paf1 und Cdc73 deutlich übertrifft (Abb. 2.1). Dies könnte zum einen durch eine mögliche mehrfache Anwesenheit von Ctr9 im PAF1-Komplex verursacht werden, zum anderen aber auch schlicht auf das hohe Molekulargewicht von Ctr9 und eine damit einhergehende intensivere Färbung der korrespondierenden Bande oder die Überlagerung mit Spt16 zurückgeführt werden (Kapitel 2.1.1). Um ein mögliches mehrfaches Vorkommen von Ctr9 im PAF1-Komplex zu überprüfen, wurde durch Kreuzung ein diploider Hefestamm erzeugt, der sowohl eine Ctr9-TAP, als auch eine Ctr9-Myc6 Proteinfusion exprimiert. Die Isolierung des PAF1-Komplexes aus diesem Stamm sollte bei mehrfacher Präsenz von Ctr9 auch zur immunologisch nachweisbaren Anreicherung der Ctr9-Myc6 Variante im durch Tandem-Affinitätsreinigung über Ctr9-TAP Fusionen gereinigten Komplex führen.
Wie Abb. 2.2 B zeigt, kann der PAF1-Komplex auch aus dem diploiden Stamm (CTR9-TAP/CTR9-MYC6) mit guter Effizienz gereinigt werden. Die immunologische Untersuchung des zur Reinigung eingesetzten Dialysats des Zellextraktes bestätigt zudem die Expression beider Varianten von Ctr9 als Fusion mit dem TAP- bzw. dem Myc6-tag

Ergebnisse
13
**
Ctr9-TAPCtr9-CBP
Ctr9-Myc6
Dialysat TEV-Eluat EGTA-Eluat
(nur
Myc
6-ta
g)
(nur
TAP
-tag)
(TAP
-tag
& M
yc6-
tag
)
(nur
Myc
6-ta
g)
(nur
TAP
-tag)
(TAP
-tag
& M
yc6-
tag
)
(nur
Myc
6-ta
g)
(nur
TAP
-tag)
(TAP
-tag
& M
yc6-
tag
)
anti-CBP
anti-Myc
2n 1n 1n 1n 2n 1n 1n 2n 1nCtr9-CBP /Ctr9-Myc6
Rtf1Leo1Paf1
Cdc73
(nur
Myc
6-ta
g)
(nur
TAP
-tag)
(TAP
-tag
& M
yc6-
tag
)
1n 2n 1n
EGTA-Eluat
Ctr9-CBP /Ctr9-Myc6
Rtf1Leo1Paf1
Cdc73
(nur
Myc
6-ta
g)
(nur
TAP
-tag)
(TAP
-tag
& M
yc6-
tag
)
1n 2n 1n
EGTA-EluatA) B)
Abb. 2.2 Ctr9 liegt im PAF1-Komplex als Monomer vor. Der PAF1-Komplex wurde durch Tandem-Affinitätsreinigung (Material & Methoden) aus den haploiden Hefestämmen CY3100 (1n; CTR9-TAP) und CY1616 (1n, CTR9-MYC6), sowie aus dem diploiden Hefestamm CY4551 (2n; CTR9-TAP/CTR9-MYC6) isoliert. Die Präparation aus dem Ctr9-Myc6 Stamm CY1616, der kein TAP-tag exprimiert, dient dabei zur Kontrolle der Anreicherungsspezifität. (A) Der gereinigte PAF1-Komplex wurde elektrophoretisch im Polyacrylamidgel (10%) aufgetrennt und immunologisch im Westernblot mit Antikörpern gegen das CBP des TAP-tags bzw. mit Antikörpern gegen das Myc-Epitop analysiert (beide aus Kaninchen). Die Sterne markieren N-terminale Abbauprodukte von Ctr9, die typischerweise für Ctr9-TAP und Ctr9-Myc6 beobachtet werden. (B) Der gereinigte PAF1-Komplex wurde elektrophoretisch im Polyacrylamidgel (10%) aufgetrennt und die isolierten Komplexbestandteile durch Reduktion von Silbernitrat gefärbt.
(Abb. 2.2 A). Die Banden von Ctr9-Myc6 und Ctr9-TAP werden aufgrund ihrer sehr ähnlichen Masse im Gel nicht aufgelöst. Da der verwendete anti-Myc Antikörper aus Kaninchen gewonnen wurde und deswegen durch die Protein A Domänen des TAP-tags gebunden wird, tritt in allen drei Dialysaten ein Bande auf Höhe von Ctr9-Myc6 auf. Weder im Eluat der ersten (TEV-Eluat), noch im Eluat der zweiten Reinigungsstufe (EGTA-Eluat) lässt sich dagegen Ctr9-Myc6 immunologisch nachweisen und wird daher nicht mit Ctr9-TAP angereichert. Ctr9 muss also monomer im PAF1-Komplex vorliegen und die beobachtete Diskrepanz zwischen apparenter und berechneter Masse wird weder durch ein mehrfaches Vorkommen von Ctr9 im PAF1-Komplex, noch durch eine Dimerisierung des gesamten Komplexes verursacht, die jeweils in der parallelen Anreicherung von Ctr9-Myc6 neben Ctr9-CBP resultieren müsste. Der Einfluss der Komplexgeometrie auf das Elutionsverhalten des PAF1-Komplexes während der Gelfiltrationschromatographie wurde nicht eingehender untersucht. Obwohl die Massenspektrometrie keinen Hinweis auf eine stöchiometrische Assoziation von yFACT und der Casein-Kinase II mit dem PAF1-Komplex liefert, würde ein Superkomplex aus diesen drei Subkomplexen rechnerisch dagegen ein Molekulargewicht von 668,4 kDa besitzen, das perfekt mit der durch die Gelfiltration ermittelten Masse übereinstimmt.

Ergebnisse
14
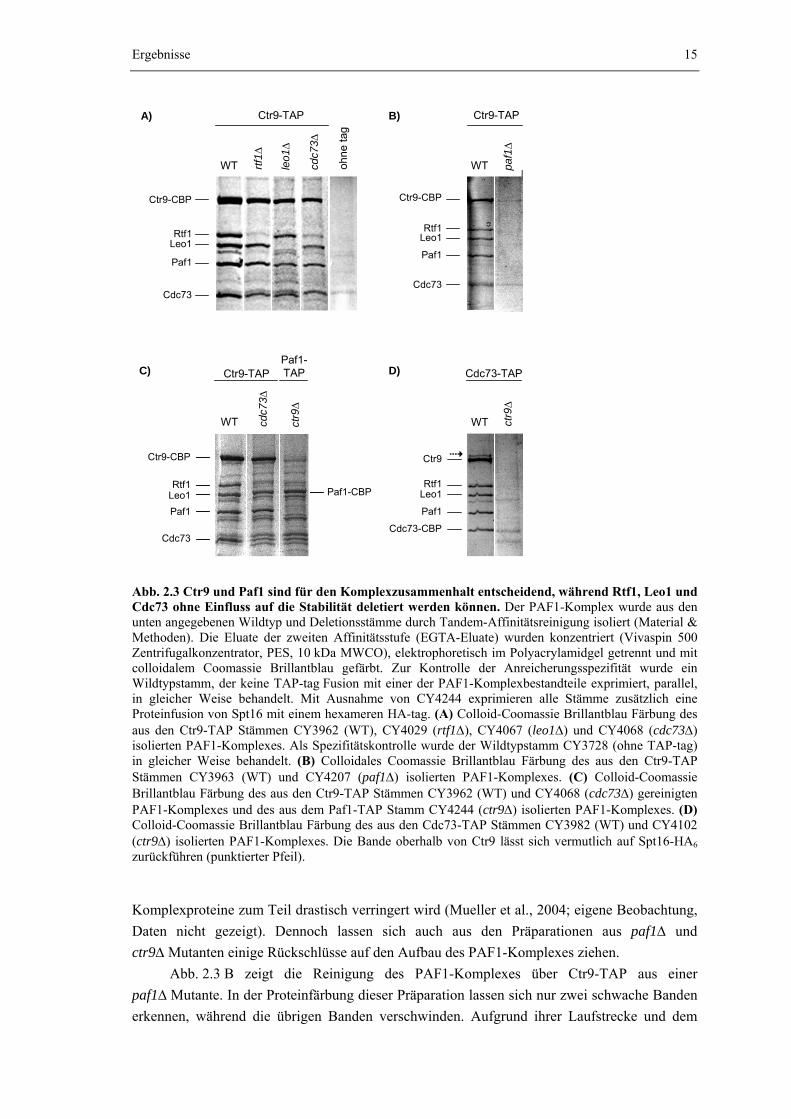
2.1.3 Paf1 und Ctr9 sind für den Komplexzusammenhalt entscheidend, während Rtf1, Leo1 und Cdc73 ohne Einfluss auf die Stabilität entfernt werden können Um einen genaueren Einblick in den Aufbau des PAF1-Komplexes zu erhalten, wurden
die fünf Kernbestandteile auf chromosomaler Ebene deletiert und die Zusammensetzung des aus der jeweiligen Deletionsmutante isolierten PAF1-Komplexes analysiert.
Die eingesetzten paf1Δ, ctr9Δ sowie cdc73Δ Stämme standen bereits aus einer früheren Studie zur Verfügung (Koch et al., 1999). Die Deletion von RTF1 und LEO1 erfolgte durch Austausch des jeweiligen Leserahmens mit einer KanMX Deletionskassette, wodurch als Selektionsmarker ein Resistenzgen gegen das Antibiotikum Geneticin eingeführt wird (Material & Methoden). Um die immunologische Detektion im Westernblot zu ermöglichen, wurden zusätzlich Stämme erzeugt, die jeweils Proteinfusionen mit C-terminalem HA6-tag exprimieren (Knop et al., 1999). Die Kombination unterschiedlicher Genotypen wurde durch die anschließende Kreuzung geeigneter Hefestämme erreicht. Die resultierenden diploiden Zellen wurden sporuliert und haploide Nachkommen mit dem gewünschten Genotyp für die weiteren Experimente eingesetzt (Material & Methoden). Alle hieraus resultierenden Hefestämme exprimieren neben dem TAP-tag zusätzlich eine Spt16-Variante mit C-terminal fusioniertem HA6-tag, die zur Untersuchung der Wechselwirkung zwischen yFACT und dem PAF1-Komplex eingesetzt wurde (siehe Kapitel 2.3.1).
Abb. 2.3 A zeigt die Auswirkung der Deletion von RTF1, LEO1 bzw. CDC73 auf die Zusammensetzung des PAF1-Komplexes. Wie die Proteinfärbung verdeutlicht, kann der PAF1-Komplex aus allen drei Deletionsmutanten mit etwa gleicher Ausbeute isoliert werden. Ein Vergleich der auftretenden Banden zeigt zudem, dass, mit Ausnahme der Komponente, deren Gen deletiert wurde, jeweils alle vier verbleibenden Kernbestandteile des PAF1-Komplexes angereichert werden. Rtf1, Leo1 oder Cdc73 können daher aus dem Komplexverbund entfernt werden ohne die Assoziation der jeweils verbleibenden vier Bestandteile zu unterbinden. Auch das Verhältnis der Bandenintensitäten innerhalb einer Präparation wird durch die Deletion von RTF1 oder LEO1 nicht beeinflusst. Wie auch bei der Präparation aus dem Wildtyp, tritt unter den fünf Kernbestandteilen die Ctr9-Bande jeweils am stärksten in Erscheinung und die durch Rtf1, Leo1, Paf1 und Cdc73 verursachten Banden sind in etwa gleichstark ausgeprägt. Daher wird auch die Ausbeute der einzelnen Komponenten nicht durch die Deletion von RTF1 oder LEO1 beeinträchtigt. Aus der cdc73Δ Mutante wird Rtf1 dagegen mit verminderter Ausbeute angereichert, da die Rtf1-Bande im Vergleich zu Paf1 und Leo1 deutlich schwächer ausfällt. Der Verlust von Cdc73 scheint daher zu einer Schwächung der Assoziation von Rtf1 mit den verbleibenden drei Kernkomponenten zu führen (Abb. 2.3.A und C).
Im Gegensatz zu rtf1Δ, leo1Δ und cdc73Δ konnte der PAF1-Komplex im gewählten Präparationsmaßstab weder aus paf1Δ noch aus ctr9Δ Mutanten mit befriedigender Ausbeute isoliert werden. Wie vor allem die Abb. 2.3 B und D zeigen, führt die Proteinfärbung des aus diesen Mutanten isolierten Komplexes zum Auftreten nur schwacher Banden und auch das mit dem TAP-tag fusionierte Protein wird nur schlecht angereichert. Die geringe Komplexausbeute passt zu dem Befund, dass durch Deletion von PAF1 oder CTR9 die Menge der übrigen
Ergebnisse
15
Ctr9-CBP
Rtf1Leo1
Paf1
Cdc73
Ctr9-CBP
Rtf1Leo1
Paf1
Cdc73
Ctr9-TAP
WT rtf1Δ
leo1
Δ
cdc7
3Δ
ohne
tag
WT paf1
Δ
Ctr9-TAP
Ctr9-CBP
Rtf1Leo1Paf1
Cdc73
Paf1-CBP
Ctr9-TAPPaf1-TAP
ctr9
Δ
cdc7
3Δ
WT
A)
C)
B)
Cdc73-TAP
Ctr9
Rtf1Leo1
Paf1
Cdc73-CBP
ctr9
Δ
WT
D)
Abb. 2.3 Ctr9 und Paf1 sind für den Komplexzusammenhalt entscheidend, während Rtf1, Leo1 und Cdc73 ohne Einfluss auf die Stabilität deletiert werden können. Der PAF1-Komplex wurde aus den unten angegebenen Wildtyp und Deletionsstämme durch Tandem-Affinitätsreinigung isoliert (Material & Methoden). Die Eluate der zweiten Affinitätsstufe (EGTA-Eluate) wurden konzentriert (Vivaspin 500 Zentrifugalkonzentrator, PES, 10 kDa MWCO), elektrophoretisch im Polyacrylamidgel getrennt und mit colloidalem Coomassie Brillantblau gefärbt. Zur Kontrolle der Anreicherungsspezifität wurde ein Wildtypstamm, der keine TAP-tag Fusion mit einer der PAF1-Komplexbestandteile exprimiert, parallel, in gleicher Weise behandelt. Mit Ausnahme von CY4244 exprimieren alle Stämme zusätzlich eine Proteinfusion von Spt16 mit einem hexameren HA-tag. (A) Colloid-Coomassie Brillantblau Färbung des aus den Ctr9-TAP Stämmen CY3962 (WT), CY4029 (rtf1Δ), CY4067 (leo1Δ) und CY4068 (cdc73Δ) isolierten PAF1-Komplexes. Als Spezifitätskontrolle wurde der Wildtypstamm CY3728 (ohne TAP-tag) in gleicher Weise behandelt. (B) Colloidales Coomassie Brillantblau Färbung des aus den Ctr9-TAP Stämmen CY3963 (WT) und CY4207 (paf1Δ) isolierten PAF1-Komplexes. (C) Colloid-Coomassie Brillantblau Färbung des aus den Ctr9-TAP Stämmen CY3962 (WT) und CY4068 (cdc73Δ) gereinigten PAF1-Komplexes und des aus dem Paf1-TAP Stamm CY4244 (ctr9Δ) isolierten PAF1-Komplexes. (D) Colloid-Coomassie Brillantblau Färbung des aus den Cdc73-TAP Stämmen CY3982 (WT) und CY4102 (ctr9Δ) isolierten PAF1-Komplexes. Die Bande oberhalb von Ctr9 lässt sich vermutlich auf Spt16-HA6 zurückführen (punktierter Pfeil).
Komplexproteine zum Teil drastisch verringert wird (Mueller et al., 2004; eigene Beobachtung, Daten nicht gezeigt). Dennoch lassen sich auch aus den Präparationen aus paf1Δ und ctr9Δ Mutanten einige Rückschlüsse auf den Aufbau des PAF1-Komplexes ziehen.
Abb. 2.3 B zeigt die Reinigung des PAF1-Komplexes über Ctr9-TAP aus einer paf1Δ Mutante. In der Proteinfärbung dieser Präparation lassen sich nur zwei schwache Banden erkennen, während die übrigen Banden verschwinden. Aufgrund ihrer Laufstrecke und dem

Ergebnisse
16
Vergleich mit den Banden einer Wildtyppräparation können die Banden sehr wahrscheinlich der Ctr9-CBP Fusion und vermutlich Cdc73 zugeordnet werden. Ctr9 und Cdc73 scheinen also miteinander interagieren zu können. Allerdings führte die Tandem-Affinitätsreinigung reproduzierbar zu einer auch in der Negativkontrolle auftretenden Bande oberhalb von Cdc73, was die eindeutige Zuordnung der Bande zu Cdc73 in der paf1Δ Mutante verhindert (Abb. 2.3 A, ohne tag).
Aus einer ctr9Δ Mutante konnten über eine Cdc73-TAP Fusion, mit Ausnahme von Cdc73-CBP, keine weiteren Proteine angereichert werden. Wie die Proteinfärbung der entsprechenden Präparation des PAF1-Komplexes in Abb. 2.3 D zeigt, tritt ausschließlich auf der charakteristischen Höhe von Cdc73-CBP eine Bande auf. Daher scheint Cdc73 nicht mit Paf1, Rtf1 oder Leo1 zu interagieren und seine Assoziation mit dem PAF1-Komplex ist daher von Ctr9 abhängig, was indirekt wiederum eine Bindung zwischen Cdc73 und Ctr9 erwarten ließ.
Auch zwischen Ctr9 und Paf1 muss eine direkte Bindung bestehen, da Rtf1, Leo1 und Cdc73 jeweils aus dem Komplex entfernt werden können ohne dabei die Assoziation von Paf1 mit Ctr9 zu beeinträchtigen (Abb. 2.3 A). Da Rtf1 und Leo1 jedoch nicht aus einer paf1Δ Mutante mit Ctr9-TAP angereichert werden (Abb. 2.3 B), scheinen die beiden Proteine eine direkte Bindung mit Paf1 einzugehen. Um dies zu überprüfen wurde der PAF1-Komplex über Paf1-TAP aus einer ctr9Δ Mutante gereinigt. Die gelelektrophoretische Analyse einer entsprechenden Präparation ist in Abb. 2.3 C gezeigt. Da die Fusion mit dem CBP des TAP-tags das Molekulargewicht von Paf1 vergrößert, wird die Paf1-Bande auf die Höhe der durch Leo1 verursachten Bande verschoben (vgl. Abb. 2.1). Daher kann keine Aussage darüber getroffen werden, ob Leo1 aus der ctr9Δ Mutante mit Paf1-TAP angereichert wurde. Auf Höhe von Rtf1 lässt sich keine eindeutige Bande erkennen, so dass die Assoziation von Rtf1 mit den verbleibenden Komponenten des PAF1-Komplexes sowohl durch die Deletion von PAF1 (Abb. 2.3 B) als auch durch die Deletion von CTR9 verloren geht (Abb. 2.3 C). Rtf1 scheint daher sowohl direkt mit Paf1 als auch mit Ctr9 assoziiert zu sein. Auch Cdc73 kann aus der ctr9Δ Mutante, übereinstimmend mit Ergebnissen aus Abb. 2.3 B und D, nicht mit der Paf1-TAP Fusion angereichert werden. Bei den auch in der Präparation aus der ctr9Δ Mutante auf Höhe von Cdc73 auftretenden Banden handelt es sich in diesem Fall jedoch um Verunreinigungen, die auch in der Vergleichspräparation aus einer cdc73Δ Mutante auftreten. Ein Vergleich der Bandenintensitäten zwischen den Präparationen aus Wildtyp, cdc73Δ und ctr9Δ zeigt jedoch, dass Cdc73 in der ctr9Δ Mutante nicht angereichert wird.
Diese Ergebnisse verdeutlichen, dass die Stabilität des PAF1-Komplexes entscheidend durch die Assoziation zwischen Paf1 und Ctr9 determiniert wird, so dass ein Verlust dieser Bindung vermutlich auch in vivo zur Dissoziation des Komplexes führt.
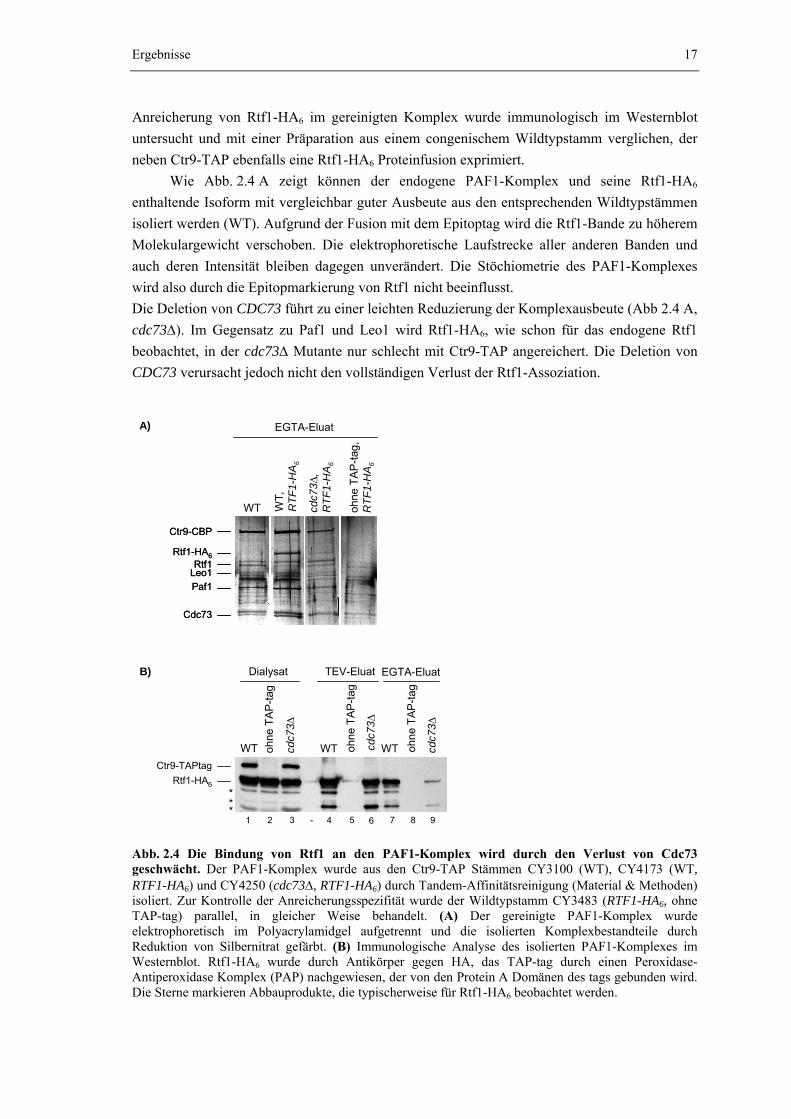
Die Bindung von Rtf1 an den PAF1-Komplex wird durch den Verlust von Cdc73 geschwächt
Um den Einfluss von Cdc73 auf die Assoziationsfähigkeit von Rtf1 genauer zu
untersuchen, wurde der PAF1-Komplex aus einer cdc73Δ Mutante isoliert, die neben einer Ctr9-TAP Fusion zusätzlich eine Fusion von Rtf1 mit einem HA6-tag exprimiert. Die
Ergebnisse
17
Anreicherung von Rtf1-HA6 im gereinigten Komplex wurde immunologisch im Westernblot untersucht und mit einer Präparation aus einem congenischem Wildtypstamm verglichen, der neben Ctr9-TAP ebenfalls eine Rtf1-HA6 Proteinfusion exprimiert.
Wie Abb. 2.4 A zeigt können der endogene PAF1-Komplex und seine Rtf1-HA6 enthaltende Isoform mit vergleichbar guter Ausbeute aus den entsprechenden Wildtypstämmen isoliert werden (WT). Aufgrund der Fusion mit dem Epitoptag wird die Rtf1-Bande zu höherem Molekulargewicht verschoben. Die elektrophoretische Laufstrecke aller anderen Banden und auch deren Intensität bleiben dagegen unverändert. Die Stöchiometrie des PAF1-Komplexes wird also durch die Epitopmarkierung von Rtf1 nicht beeinflusst. Die Deletion von CDC73 führt zu einer leichten Reduzierung der Komplexausbeute (Abb 2.4 A, cdc73Δ). Im Gegensatz zu Paf1 und Leo1 wird Rtf1-HA6, wie schon für das endogene Rtf1 beobachtet, in der cdc73Δ Mutante nur schlecht mit Ctr9-TAP angereichert. Die Deletion von CDC73 verursacht jedoch nicht den vollständigen Verlust der Rtf1-Assoziation.
Ctr9-TAPtagRtf1-HA6
Ctr9-CBP
Rtf1-HA6Rtf1
Leo1Paf1
Cdc73
Ctr9-CBP
Rtf1-HA6Rtf1
Leo1Paf1
Cdc73
WT WT,
RTF
1-H
A 6cd
c73Δ
,R
TF1-
HA 6
ohne
TA
P-ta
g,R
TF1-
HA 6
WTWT WTohne
TA
P-ta
g
ohne
TAP
-tag
ohne
TAP
-tag
cdc7
3Δ
cdc7
3Δ
cdc7
3Δ
Dialysat TEV-Eluat EGTA-EluatB)
A) EGTA-Eluat
***
1 2 3 4 5 6 7 8 9- Abb. 2.4 Die Bindung von Rtf1 an den PAF1-Komplex wird durch den Verlust von Cdc73 geschwächt. Der PAF1-Komplex wurde aus den Ctr9-TAP Stämmen CY3100 (WT), CY4173 (WT, RTF1-HA6) und CY4250 (cdc73Δ, RTF1-HA6) durch Tandem-Affinitätsreinigung (Material & Methoden) isoliert. Zur Kontrolle der Anreicherungsspezifität wurde der Wildtypstamm CY3483 (RTF1-HA6, ohne TAP-tag) parallel, in gleicher Weise behandelt. (A) Der gereinigte PAF1-Komplex wurde elektrophoretisch im Polyacrylamidgel aufgetrennt und die isolierten Komplexbestandteile durch Reduktion von Silbernitrat gefärbt. (B) Immunologische Analyse des isolierten PAF1-Komplexes im Westernblot. Rtf1-HA6 wurde durch Antikörper gegen HA, das TAP-tag durch einen Peroxidase-Antiperoxidase Komplex (PAP) nachgewiesen, der von den Protein A Domänen des tags gebunden wird. Die Sterne markieren Abbauprodukte, die typischerweise für Rtf1-HA6 beobachtet werden.

Ergebnisse
18
Wie der Westernblot in Abb. 2.4 B zeigt, lässt sich Rtf1-HA6 auch in der Präparation des isolierten PAF1-Komplexes der cdc73Δ Mutante immunologisch klar nachweisen (Spur 6 und 9). Die im Vergleich zur Präparation aus dem Wildtyp (Spur 4 und 7) verringerte Intensität der Bande bestätigt jedoch die signifikant reduzierte Ausbeute an Rtf1-HA6 in der cdc73Δ Mutante. Bereits im Eluat der ersten Reinigungsstufe der Tandem-Affinitätsreinigung (TEV-Eluat) des aus der cdc73Δ Mutante isolierten Komplexes wird Rtf1-HA6 im Vergleich zum Wildtyp weniger stark angereichert (vgl. Spur 4 mit 6). Im Eluat der zweiten Reinigungsstufe (EGTA-Eluat) ist Rtf1-HA6 jedoch nur noch mit einer drastisch verringerten Konzentration vorhanden (Spur 9). Zwar wird Rtf1-HA6 auch im Wildtyp in der zweiten Reinigungsstufe mit verringerter Ausbeute angereichert, um gut 50% gegenüber der ersten Reinigungsstufe (vgl. Spur 4 mit 7), im Paf1-Komplex, der aus der cdc73Δ Mutante isoliert wurde, erreicht Rtf1-HA6 jedoch in der zweiten Reinigungsstufe bestenfalls ein Zehntel bis ein Zwanzigstel der in der ersten Reinigungsstufe erzielten Ausbeute (vgl. Spur 6 mit 9). Cdc73 besitzt demnach tatsächlich einen Einfluss auf die Stabilität der Rtf1-Bindung.
2.1.4 Ctr9 ist aus einer seriellen Anordnung von TPR-Motiven zusammengesetzt Tetratricopeptide Repeat- (TPR-) Motive bilden eine gut charakterisierte
Faltungsdomäne, die durch spezifische Bindung von Peptidliganden bzw. Proteindomänen die Interaktion zwischen Proteinen und den Zusammenhalt von Multiproteinkomplexen vermitteln können (Cortajarena and Regan, 2006; D'Andrea and Regan, 2003). Das Strukturmotiv wird aus einer 34 Aminosäuren umfassenden Sequenz gebildet, die zwar generell wenig konserviert erscheint, hinsichtlich der physikalischen Eigenschaften der Aminosäuren an einigen Positionen jedoch strengen Regeln folgt (Tabelle 2.1). Insbesondere die Aminosäuren an den Positionen 4 (W/L/F), 7 (L/I/M), 8 (G/A/S), 11 (Y/L/F), 20 (A/S/E), 24 (F/Y/L), 27 (A/S/L) und 32 (P/K/E) sind hoch konserviert und können daher zur Definition einer Konsensussequenz für TPR-Motive herangezogen werden (Blatch and Lassle, 1999; Sikorski et al., 1990).
TPR-Motive sind häufig in serieller Anordnung von zumeist drei oder mehr Motiven arrangiert, können aber auch außerhalb solcher Gruppierungen auftreten (Blatch and Lassle, 1999; Cliff et al., 2005). Ein einzelnes TPR-Motiv bildet dabei unabhängig zwei α-Helices aus, die antiparallel zueinander orientiert sind (Helic-Turn-Helix-Motiv). Aufeinanderfolgende TPR-Motive falten sich weiter zu einer rechtsgängigen helikalen Superstruktur, wobei die α-Helices aneinander angrenzender TPR-Motive wiederum antiparallel angeordnet sind (Blatch and Lassle, 1999). Die Röntgenstruktur des synthetischen TPR-consensus Peptids 2FO7 (T. Kajander et al., 2006; RCSB-Datenbank, structureId=2FO7, DOI 10.2210/pdb2fo7/pdb; http://www.rcsb.org/) ist als Strukturbeispiel in Abb. 2.5 C gezeigt. Während die Ausbildung der gut untersuchten TPR-Struktur der Proteinphosphatase 5 (PP5) nur in Anwesenheit des Proteinliganden erfolgt, scheint die Faltung des TPR-Motivs in den meisten Fällen jedoch ligandenunabhängig zu erfolgen und auch die Bindung des Interaktionspartners führt nur zu unwesentlichen oder gar keinen strukturellen Veränderungen der TPR-Konformation (Cliff et al., 2005; Cortajarena and Regan, 2006).
Ergebnisse
19
1 2 3 4 5 6 7 8 910 ccN C
A)
B)
TPR-Motiv (TPR score > 0,3) TPR-Motiv (TPR score ≤ 0,3) cc Coiled coil
score = 0,3
0,0
0,2
0,4
0,6
0,8
1,0
0 100 200 300 400 500 600 700 800 900 1000
Aminosäure
scor
eTPR
Coiled coil
1 2 3 4 5 6 7 8 910 ccN C1 2 3 4 5 6 7 8 910 ccN C
A)
B)
TPR-Motiv (TPR score > 0,3) TPR-Motiv (TPR score ≤ 0,3) cc Coiled coil
score = 0,3
0,0
0,2
0,4
0,6
0,8
1,0
0 100 200 300 400 500 600 700 800 900 1000
Aminosäure
scor
eTPR
Coiled coil
CLUSTAL FORMAT for T-COFFEE Version_4.85 [http://www.tcoffee.org]; Nseq=2, Len=143
2FO7 AEAWYNLGNAYYKQGDYDEAIEYYQKALELDPRSAEAWYNLGNAYYKQGDYDEAIEYYQK Ctr9 model ------PKEQGKSKHSYLKAIQLYQKVLQVDPFNIFAAQGLAIIFAESKRLGPALEILRK : .: .* :**: ***.*::** . * .*. : :. . *:* :*
2FO7 ALELDPRSAEAWYNLGNAYYKQGDYDEAIEYYQKALEL--DPRSAEAWYN-LGNAYYKQG Ctr9 model VRD-SLDNEDVQLNLAHCYLEMREYGKAIENYELVLKKFDNEKTRPHILNLLGRAWYARA . : . . :. **.:.* : :*.:*** *: .*: : :: * **.*:* :.
2FO7 DYDEAIEYYQKALELDPR----S Ctr9 model IKETSVNFYQKALENAKTALDLF : ::::******
Synthetic Consensus TPR Protein 2FO7:A
HomologiemodellCtr9AS[671-806]
C)
D) CLUSTAL FORMAT for T-COFFEE Version_4.85 [http://www.tcoffee.org]; Nseq=2, Len=143
2FO7 AEAWYNLGNAYYKQGDYDEAIEYYQKALELDPRSAEAWYNLGNAYYKQGDYDEAIEYYQK Ctr9 model ------PKEQGKSKHSYLKAIQLYQKVLQVDPFNIFAAQGLAIIFAESKRLGPALEILRK : .: .* :**: ***.*::** . * .*. : :. . *:* :*
2FO7 ALELDPRSAEAWYNLGNAYYKQGDYDEAIEYYQKALEL--DPRSAEAWYN-LGNAYYKQG Ctr9 model VRD-SLDNEDVQLNLAHCYLEMREYGKAIENYELVLKKFDNEKTRPHILNLLGRAWYARA . : . . :. **.:.* : :*.:*** *: .*: : :: * **.*:* :.
2FO7 DYDEAIEYYQKALELDPR----S Ctr9 model IKETSVNFYQKALENAKTALDLF : ::::******
Synthetic Consensus TPR Protein 2FO7:A
HomologiemodellCtr9AS[671-806]
C)
D)
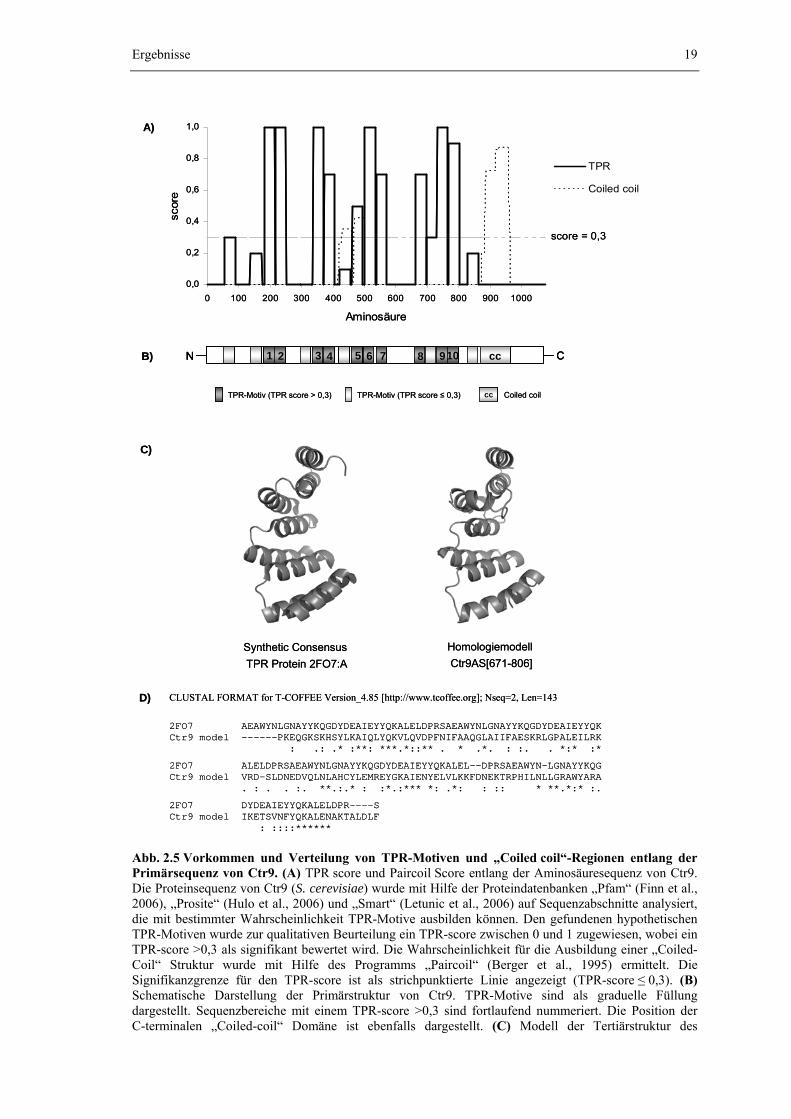
Abb. 2.5 Vorkommen und Verteilung von TPR-Motiven und „Coiled coil“-Regionen entlang der Primärsequenz von Ctr9. (A) TPR score und Paircoil Score entlang der Aminosäuresequenz von Ctr9. Die Proteinsequenz von Ctr9 (S. cerevisiae) wurde mit Hilfe der Proteindatenbanken „Pfam“ (Finn et al., 2006), „Prosite“ (Hulo et al., 2006) und „Smart“ (Letunic et al., 2006) auf Sequenzabschnitte analysiert, die mit bestimmter Wahrscheinlichkeit TPR-Motive ausbilden können. Den gefundenen hypothetischen TPR-Motiven wurde zur qualitativen Beurteilung ein TPR-score zwischen 0 und 1 zugewiesen, wobei ein TPR-score >0,3 als signifikant bewertet wird. Die Wahrscheinlichkeit für die Ausbildung einer „Coiled-Coil“ Struktur wurde mit Hilfe des Programms „Paircoil“ (Berger et al., 1995) ermittelt. Die Signifikanzgrenze für den TPR-score ist als strichpunktierte Linie angezeigt (TPR-score ≤ 0,3). (B) Schematische Darstellung der Primärstruktur von Ctr9. TPR-Motive sind als graduelle Füllung dargestellt. Sequenzbereiche mit einem TPR-score >0,3 sind fortlaufend nummeriert. Die Position der C-terminalen „Coiled-coil“ Domäne ist ebenfalls dargestellt. (C) Modell der Tertiärstruktur des

Ergebnisse
20
C-terminalen Ctr9-Bereichs von Aminosäure 671-806. Die Homologiemodellierung erfolgte mit Hilfe des Programms 3D-JIGSAW (Bates et al., 2001) aufgrund der bekannten Kristallstruktur des synthetischen TPR-Consensus Polypeptids 2FO7 (T. Kajander et al., 2006; RCSB-Datenbank, structureId=2FO7, DOI 10.2210/pdb2fo7/pdb; http://www.rcsb.org/) (D) Alignement der für die Homologiemodellierung benutzten Aminosäuresequenzen von Ctr9 und 2FO7.
Das Protein Ctr9 enthält eine serielle Anordnung dieser TPR-Motive und ist für die Stabilität des PAF1-Komplexes von entscheidender Bedeutung (vgl. Kapitel 2.1.3). Daher liegt die Vermutung nahe, dass der Zusammenhalt des PAF1-Komplexes wesentlich durch die TPR-Motive in Ctr9 determiniert wird. Um die Anzahl und Position der TPR-Motive in Ctr9 genauer zu charakterisieren, wurde dessen Aminosäuresequenz mit Hilfe der Proteindatenbanken bzw. der darin integrierten Suchmaschinen „Pfam“, „Prosite“ und „Smart“ analysiert (Finn et al., 2006; Hulo et al., 2006; Letunic et al., 2006), welche die Vorhersage von Proteindomänen anhand der Aminosäuresequenz durch Vergleich mit der annotierten Domänenstruktur bekannter Proteine ermöglichen. Die Analysen wurden in Zusammenarbeit mit dem Institut für Biochemie der Universität Erlangen durchgeführt (AG Bioinformatik, Prof. Dr. H. Sticht). Zur qualitativen Bewertung wurde den durch die drei Methoden ermittelten, hypothetischen TPR-Motiven eine Kenngröße (TPR-score) zwischen 0 und 1 zugewiesen, welche die Zuverlässigkeit eines gefunden TPR-Motivs charakterisiert. Die Wahrscheinlichkeit, dass die entsprechende Aminosäuresequenz tatsächlich eine TPR-Struktur ausbildet, ist dabei umso größer, je höher ihr korrespondierender TPR score ist (H. Sticht, persönliche Mitteilung). Um die Zuweisung der gefundenen Sequenzbereiche als TPR-Motiv weiter abzusichern und um ein Aussage über die evolutionäre Konservierung dieser Sequenzabschnitte treffen zu können, wurde die Analyse für Ctr9 und die jeweils orthologen Proteine aus vier weiteren Organismen (Magnaporthe grisea, Schizosaccharomyces pombe, Caenorhabditis elegans und Drosophila melanogaster) durchgeführt. Für die weitere Auswertung wurde ein TPR-score > 0,3 als signifikant bewertet. Zudem wurde aufgrund der Röntgenstruktur des synthetischen TPR-consensus Peptids 2FO7 (T. Kajander et al., 2006; RCSB-Datenbank, structureId = 2FO7, DOI 10.2210/pdb2fo7/pdb; http://www.rcsb.org/) durch „comparative modeling“ mit Hilfe des Programms 3D-JIGSAW (Bates et al., 2001) ein Homolgiemodell des C-terminalen Proteinbereichs von Ctr9 (Aminosäure 671-801) erstellt. Die Homologiemodellierung erfolgte in Zusammenarbeit mit dem Lehrstuhl für Biotechnik und wurde freundlicherweise von Martin Stiebritz zur Verfügung gestellt. Die Ergebnisse zur Verteilung der TPR-Motive in Ctr9 sind in Abb. 2.5 und in Tabelle 2.1 zusammengefasst.
In der Aminosäuresequenz von Ctr9 treten 16 Abschnitte auf, die potentiell in der Lage sind TPR-Motive auszubilden. Von diesen 16 hypothetischen TPR-Motiven erreichen 10 einen TPR-score oberhalb der Signifikanzgrenze (score > 0,3), vier liegen im Bereich zwischen 0,2 ≤ score ≤ 0,3 und zwei Abschnitte können vermutlich als TPR-Motive ausgeschlossen werden (score < 0,2). Die zehn signifikanten Sequenzabschnitte werden im Folgenden als TPR 1 bis TPR 10 bezeichnet. Mit Ausnahme von TPR 5 sind alle zehn TPR-Motive mit signifikantem Score evolutionär konserviert und werden in allen vier untersuchten Ctr9-Orthologen gefunden. Die TPR-Motive sind über das gesamte Ctr9-Protein verteilt und als
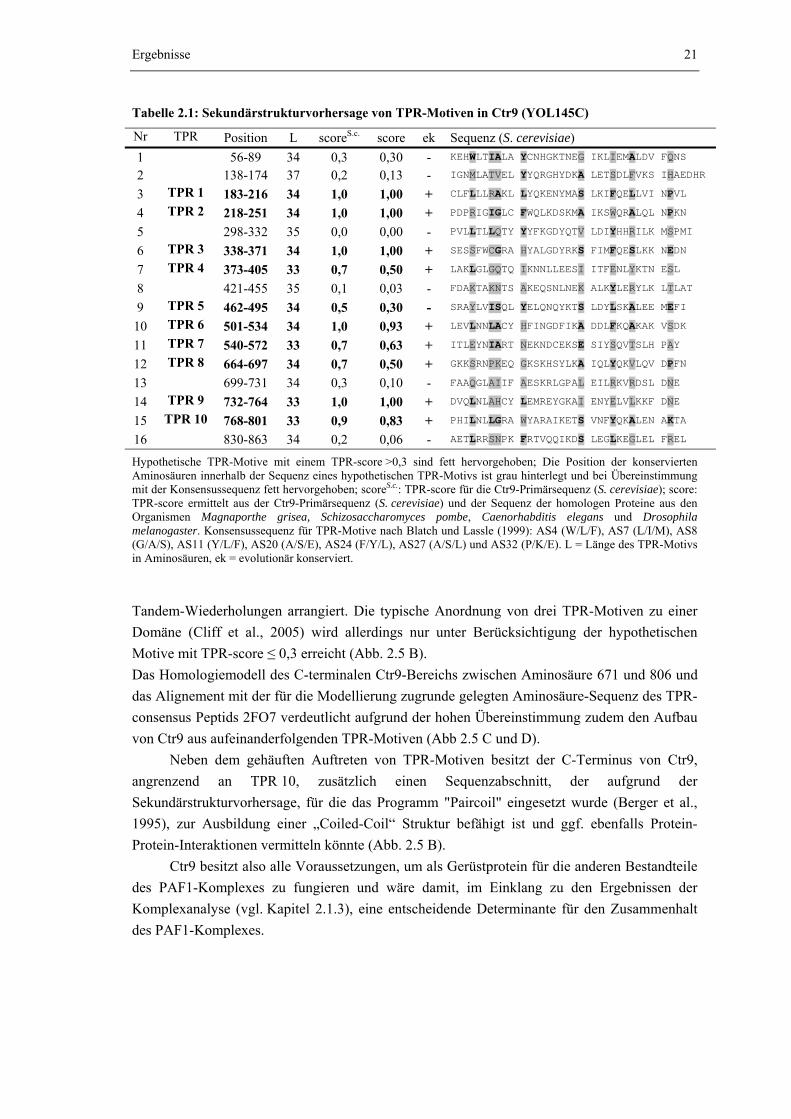
Ergebnisse
21
Tabelle 2.1: Sekundärstrukturvorhersage von TPR-Motiven in Ctr9 (YOL145C)
Nr TPR Position L scoreS.c. score ek Sequenz (S. cerevisiae) 1 56-89 34 0,3 0,30 - KEHWLTIALA YCNHGKTNEG IKLIEMALDV FQNS
2 138-174 37 0,2 0,13 - IGNMLATVEL YYQRGHYDKA LETSDLFVKS IHAEDHR
3 TPR 1 183-216 34 1,0 1,00 + CLFLLLRAKL LYQKENYMAS LKIFQELLVI NPVL
4 TPR 2 218-251 34 1,0 1,00 + PDPRIGIGLC FWQLKDSKMA IKSWQRALQL NPKN
5 298-332 35 0,0 0,00 - PVLLTLLQTY YYFKGDYQTV LDIYHHRILK MSPMI
6 TPR 3 338-371 34 1,0 1,00 + SESSFWCGRA HYALGDYRKS FIMFQESLKK NEDN
7 TPR 4 373-405 33 0,7 0,50 + LAKLGLGQTQ IKNNLLEESI ITFENLYKTN ESL
8 421-455 35 0,1 0,03 - FDAKTAKNTS AKEQSNLNEK ALKYLERYLK LTLAT
9 TPR 5 462-495 34 0,5 0,30 - SRAYLVISQL YELQNQYKTS LDYLSKALEE MEFI
10 TPR 6 501-534 34 1,0 0,93 + LEVLNNLACY HFINGDFIKA DDLFKQAKAK VSDK
11 TPR 7 540-572 33 0,7 0,63 + ITLEYNIART NEKNDCEKSE SIYSQVTSLH PAY
12 TPR 8 664-697 34 0,7 0,50 + GKKSRNPKEQ GKSKHSYLKA IQLYQKVLQV DPFN
13 699-731 34 0,3 0,10 - FAAQGLAIIF AESKRLGPAL EILRKVRDSL DNE
14 TPR 9 732-764 33 1,0 1,00 + DVQLNLAHCY LEMREYGKAI ENYELVLKKF DNE
15 TPR 10 768-801 33 0,9 0,83 + PHILNLLGRA WYARAIKETS VNFYQKALEN AKTA
16 830-863 34 0,2 0,06 - AETLRRSNPK FRTVQQIKDS LEGLKEGLEL FREL
Hypothetische TPR-Motive mit einem TPR-score >0,3 sind fett hervorgehoben; Die Position der konservierten Aminosäuren innerhalb der Sequenz eines hypothetischen TPR-Motivs ist grau hinterlegt und bei Übereinstimmung mit der Konsensussequenz fett hervorgehoben; scoreS.c.: TPR-score für die Ctr9-Primärsequenz (S. cerevisiae); score: TPR-score ermittelt aus der Ctr9-Primärsequenz (S. cerevisiae) und der Sequenz der homologen Proteine aus den Organismen Magnaporthe grisea, Schizosaccharomyces pombe, Caenorhabditis elegans und Drosophila melanogaster. Konsensussequenz für TPR-Motive nach Blatch und Lassle (1999): AS4 (W/L/F), AS7 (L/I/M), AS8 (G/A/S), AS11 (Y/L/F), AS20 (A/S/E), AS24 (F/Y/L), AS27 (A/S/L) und AS32 (P/K/E). L = Länge des TPR-Motivs in Aminosäuren, ek = evolutionär konserviert.
Tandem-Wiederholungen arrangiert. Die typische Anordnung von drei TPR-Motiven zu einer Domäne (Cliff et al., 2005) wird allerdings nur unter Berücksichtigung der hypothetischen Motive mit TPR-score ≤ 0,3 erreicht (Abb. 2.5 B). Das Homologiemodell des C-terminalen Ctr9-Bereichs zwischen Aminosäure 671 und 806 und das Alignement mit der für die Modellierung zugrunde gelegten Aminosäure-Sequenz des TPR-consensus Peptids 2FO7 verdeutlicht aufgrund der hohen Übereinstimmung zudem den Aufbau von Ctr9 aus aufeinanderfolgenden TPR-Motiven (Abb 2.5 C und D).
Neben dem gehäuften Auftreten von TPR-Motiven besitzt der C-Terminus von Ctr9, angrenzend an TPR 10, zusätzlich einen Sequenzabschnitt, der aufgrund der Sekundärstrukturvorhersage, für die das Programm "Paircoil" eingesetzt wurde (Berger et al., 1995), zur Ausbildung einer „Coiled-Coil“ Struktur befähigt ist und ggf. ebenfalls Protein-Protein-Interaktionen vermitteln könnte (Abb. 2.5 B).
Ctr9 besitzt also alle Voraussetzungen, um als Gerüstprotein für die anderen Bestandteile des PAF1-Komplexes zu fungieren und wäre damit, im Einklang zu den Ergebnissen der Komplexanalyse (vgl. Kapitel 2.1.3), eine entscheidende Determinante für den Zusammenhalt des PAF1-Komplexes.

Ergebnisse
22
2.1.5 Die C-terminalen TPR-Motive 9 & 10 in Ctr9 bilden die Bindungsdomäne für Cdc73, während der N-terminale Bereich für die Interaktion mit Rtf1, Leo1 und Paf1 notwendig ist Um den Einfluss der TPR-Motive in Ctr9 auf den Zusammenhalt des PAF1-Komplexes
genauer zu untersuchen, wurden im Folgenden zwei Strategien angewendet. Zum einen wurden definierte Proteinbereiche und TPR-Motive aus Ctr9 durch sukzessive C-terminale Verkürzung des Proteins deletiert und die Auswirkung der Entfernung dieser Domänen auf die Zusammensetzung des PAF1-Komplexes untersucht. Zum anderen wurden diskrete Bereiche von Ctr9 heterolog in E. coli exprimiert und getestet, ob die so erzeugten Ctr9-Fragmente in vitro direkt mit dem PAF1-Komplex bzw. mit dessen Bestandteilen interagieren können.
Der N-terminale Bereich von Ctr9 ist für die Interaktion mit Rtf1, Leo1 und Paf1 erforderlich
Zur Expression C-terminal verkürzter Ctr9-Varianten wurde der CTR9-Leserahmen durch
zielgerichtete homologe Rekombination mit dem TAP-tag Moduls partiell deletiert (Knop et al., 1999; Puig et al., 2001). Die Ctr9-Verkürzungen werden dadurch als Fusion mit einem C-terminalen TAP-tag exprimiert, so dass die Isolierung des PAF1-Komplexes durch Tandem-Affinitätsreinigung direkt ermöglicht wird. Auf diese Weise wurden vier graduell verkürzte Ctr9-Varianten hergestellt (Material & Methoden), deren Struktur schematisch in Abb. 2.6 A dargestellt ist. Dabei wurde zunächst der direkt an das TPR-Motiv 10 anschließende Proteinbereich entfernt, der ein mögliches TPR-Motiv mit einem nicht-signifikanten scoreS.c. von 0,2 und die „Coiled-Coil“-Domäne enthält (Abb. 2.5). Die Verkürzung wurde dann in den nächsten Ctr9-Varianten um die TPR-Motive 9 & 10, 7 & 8 und schließlich um die TPR-Motive 5 & 6 ausgeweitet, wodurch das Protein schließlich um mehr als 50% seiner ursprünglichen Länge verkürzt wurde. Die über die Ctr9-Verkürzungen gereinigten Proteinkomplexe wurden elektrophoretisch in ihre Bestandteile aufgetrennt und die Anwesenheit der Komponenten des PAF1-Komplexes durch Proteinfärbung untersucht. Da sich die einzelnen Bestandteile des PAF1-Komplexes in ihrem Färbeverhalten je nach angewandter Methode leicht unterschiedlich verhalten und sich insbesondere Leo1 nur bedingt durch Reduktion von Silbernitrat anfärben lässt, wurden die isolierten Proteine jeweils durch colloidales Coomassie Brillantblau und durch Reduktion mit Silbernitrat gefärbt. Die Proteinfärbungen der einzelnen Präparationen sind in Abb. 2.6 wiedergegeben.
Wie der Westernblot in Abb. 2.6 B zeigt, wurden alle Ctr9-Varianten exprimiert. Die zunehmende C-terminale Verkürzung bewirkt dabei eine immer ausgeprägtere Verschiebung der elektrophoretischen Laufweite zu geringeren Molekulargewichten.
Dieser Befund wird auch durch die Proteinfärbungen der über die Ctr9-Verkürzungen isolierten Komplexe deutlich (Abb. 2.6 C). Zur leichteren Orientierung ist die Bande der jeweiligen Ctr9-TAP Fusion durch einen Stern gekennzeichnet. Die Intensität der Banden nimmt im Vergleich zur Präparation aus dem Wildtyp ab, was eine verringerte Ausbeute der Reinigungen über die verkürzten Ctr9-Varianten anzeigt. Die Ctr9-Verkürzung um die Aminosäuren 459-1077 fällt dabei durch einen beträchtlichen Einbruch der Ausbeute auf.
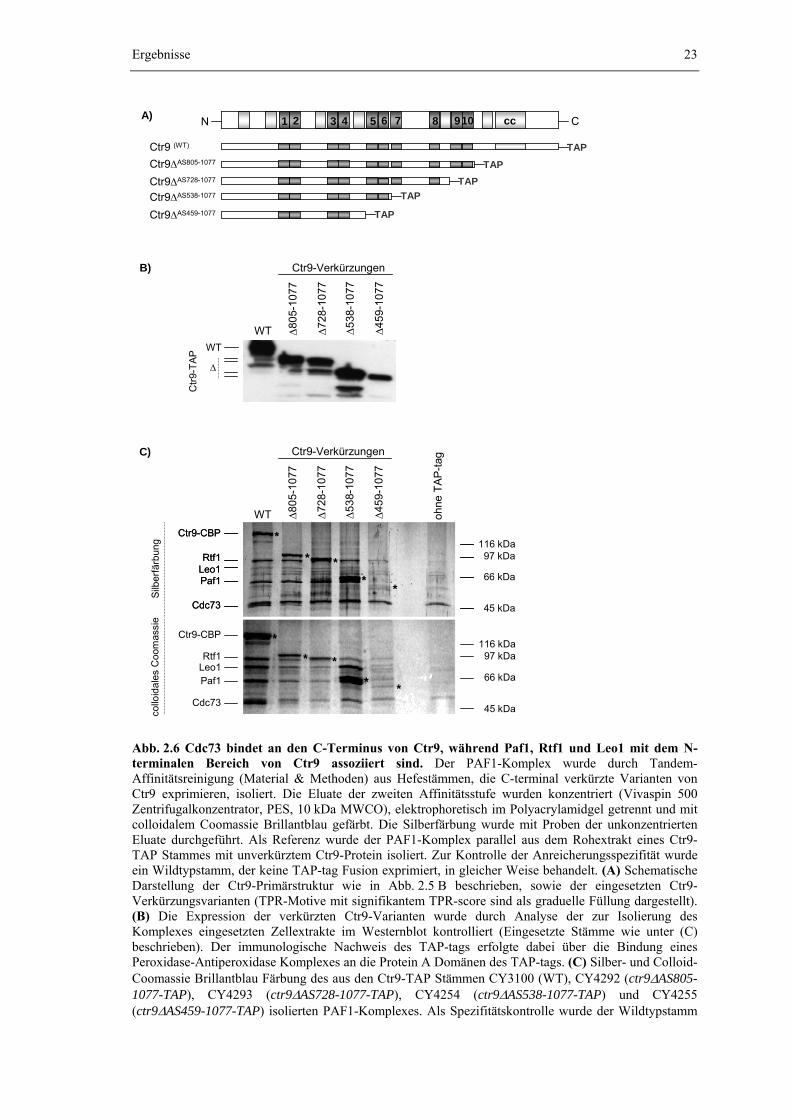
Ergebnisse
23
1 2 3 4 5 6 7 8 910 ccN C
TAP
TAPTAP
TAPCtr9 (WT)
Ctr9ΔAS805-1077
Ctr9ΔAS728-1077
Ctr9ΔAS459-1077
A)
Ctr9-CBP
Rtf1Leo1Paf1
Cdc73
Ctr9-CBP
Rtf1Leo1Paf1
Cdc73
Ctr9-CBP
Rtf1Leo1Paf1
Cdc73
116 kDa97 kDa
66 kDa
45 kDa
116 kDa97 kDa
66 kDa
45 kDa
Silb
erfä
rbun
gco
lloid
ales
Coo
mas
sie
WT Δ805
-107
7
Δ728
-107
7
Δ538
-107
7
Δ459
-107
7
ohne
TA
P-ta
gCtr9-Verkürzungen
B)
*
*
* **
*
* ** *
TAPCtr9ΔAS538-1077C
tr9-T
AP
WT
Δ
WT Δ80
5-10
77
Δ728
-107
7
Δ538
-107
7
Δ459
-107
7
Ctr9-Verkürzungen
C)
Abb. 2.6 Cdc73 bindet an den C-Terminus von Ctr9, während Paf1, Rtf1 und Leo1 mit dem N-terminalen Bereich von Ctr9 assoziiert sind. Der PAF1-Komplex wurde durch Tandem-Affinitätsreinigung (Material & Methoden) aus Hefestämmen, die C-terminal verkürzte Varianten von Ctr9 exprimieren, isoliert. Die Eluate der zweiten Affinitätsstufe wurden konzentriert (Vivaspin 500 Zentrifugalkonzentrator, PES, 10 kDa MWCO), elektrophoretisch im Polyacrylamidgel getrennt und mit colloidalem Coomassie Brillantblau gefärbt. Die Silberfärbung wurde mit Proben der unkonzentrierten Eluate durchgeführt. Als Referenz wurde der PAF1-Komplex parallel aus dem Rohextrakt eines Ctr9-TAP Stammes mit unverkürztem Ctr9-Protein isoliert. Zur Kontrolle der Anreicherungsspezifität wurde ein Wildtypstamm, der keine TAP-tag Fusion exprimiert, in gleicher Weise behandelt. (A) Schematische Darstellung der Ctr9-Primärstruktur wie in Abb. 2.5 B beschrieben, sowie der eingesetzten Ctr9-Verkürzungsvarianten (TPR-Motive mit signifikantem TPR-score sind als graduelle Füllung dargestellt). (B) Die Expression der verkürzten Ctr9-Varianten wurde durch Analyse der zur Isolierung des Komplexes eingesetzten Zellextrakte im Westernblot kontrolliert (Eingesetzte Stämme wie unter (C) beschrieben). Der immunologische Nachweis des TAP-tags erfolgte dabei über die Bindung eines Peroxidase-Antiperoxidase Komplexes an die Protein A Domänen des TAP-tags. (C) Silber- und Colloid-Coomassie Brillantblau Färbung des aus den Ctr9-TAP Stämmen CY3100 (WT), CY4292 (ctr9ΔAS805-1077-TAP), CY4293 (ctr9ΔAS728-1077-TAP), CY4254 (ctr9ΔAS538-1077-TAP) und CY4255 (ctr9ΔAS459-1077-TAP) isolierten PAF1-Komplexes. Als Spezifitätskontrolle wurde der Wildtypstamm

Ergebnisse
24
CY1615 (ohne TAP-tag) in gleicher Weise behandelt. Die Laufhöhe der Ctr9-Verkürzungen ist in den Proteinfärbungen durch Sterne markiert.
Ein ähnlicher Effekt wurde auch für die Präparation des PAF1-Komplexes aus einer ctr9Δ Mutante beobachtet (Abb. 2.3). Die Verkürzung von Ctr9 um die Aminosäuren 459-1077 verursacht daher, in Analogie zu ctr9Δ, vermutlich bereits eine erhebliche Beeinträchtigung der Stabilität bzw. die teilweise Dissoziation des PAF1-Komplexes.
Ein Vergleich der Banden in Abb. 2.6 C ermöglicht weitere Einblicke in den Aufbau des PAF1-Komplexes. In den drei Präparationen über die Ctr9-Verkürzungen Ctr9ΔAS805-1077-TAP, Ctr9ΔAS728-1077-TAP und Ctr9ΔAS538-1077-TAP lassen sich jeweils Banden auf der charakteristischen Höhe von Rtf1, Leo1 und Paf1 nachweisen. Sogar in der Colloid-Commassie-Färbung der Präparation über Ctr9ΔAS459-1077-TAP treten, wenn auch sehr schwach, Banden auf den entsprechenden Höhen auf. Die Assoziation von Rtf1, Leo1 und Paf1 mit dem PAF1-Komplex wird daher durch die Entfernung des C-terminalen Ctr9-Bereichs zwischen den Aminosäuren 538-1077 nicht beeinträchtigt. Obwohl die Stabilität des Komplexes durch die weitere Verkürzung von Ctr9 bis zu Aminosäure 459 bereits erheblich geschwächt wird, scheint auch die Ctr9ΔAS459-1077-TAP Verkürzung zumindest noch eingeschränkt zur Interaktion mit Paf1, Rtf1 und Leo1 befähigt zu sein. Da die Assoziation von Rtf1 die Interaktion zwischen Paf1 und Ctr9 voraussetzt (Kapitel 2.1.3, Abb. 2.3), kann die Lokalisation der mit Paf1 interagierenden Domäne von Ctr9 auf den N-terminalen Proteinbereich bis zu Aminosäure 459 eingeschränkt werden. Diese Interpretation steht im Einklang mit der Beobachtung, dass auch eine um die Aminosäuren 396-974 verkürzte Ctr9-Variante noch zur Co-Immunpräzipitation mit Paf1-HA3 befähigt ist (C. Koch, persönliche Mitteilung). Der N-terminale Ctr9-Bereich weist in dem Abschnitt bis Aminosäure 396 zwei als Tandem arrangierte TPR-Motive mit signifikantem TPR score auf (Tabelle 2.1). Diese beiden TPR-Motive stellen daher die aussichtsreichsten Kandidaten für die Paf1-bindende Domäne in Ctr9 dar.
Im Gegensatz zu Paf1, Rtf1 und Leo1 scheint die Assoziation von Cdc73 bereits durch die Entfernung der C-terminalen Aminosäuren 805-1077 beeinträchtigt zu werden. Die Intensität der auf der für Cdc73 charakteristischen Höhe verlaufenden Bande ist in der Präparation des PAF1-Komplexes über diese Ctr9-Verkürzung, im Vergleich zur Präparation aus dem Wildtyp, deutlich geschwächt. Die weitere Ausdehnung der C-terminalen Verkürzung bis zu Aminosäure 728 bzw. darüber hinaus führt schließlich zu dem vollständigen Verschwinden dieser Bande. Cdc73 scheint daher direkt mit dem C-terminalen Bereich von Ctr9 zwischen Aminosäure 728 und 1077 zu interagieren. Diese Interpretation wird jedoch durch das Auftreten einer zusätzlichen Bande unmittelbar oberhalb von Cdc73 erschwert. Da diese Bande auch in der Kontrollpräparation (kein TAP-tag) auftritt, kann sie jedoch nicht durch eine spezifische Assoziation mit dem PAF1-Komplex verursacht werden.
Die Position der Cdc73-bindenden Domäne in Ctr9 kann auf den C-Terminus zwischen Aminosäure 728 und 1077 eingeschränkt werden
Um die Abhängigkeit der Wechselwirkung zwischen Cdc73 und dem C-Terminus von
Ctr9 unbeeinflusst zu verifizieren, wurde der PAF1-Komplex durch Immunpräzipitation der
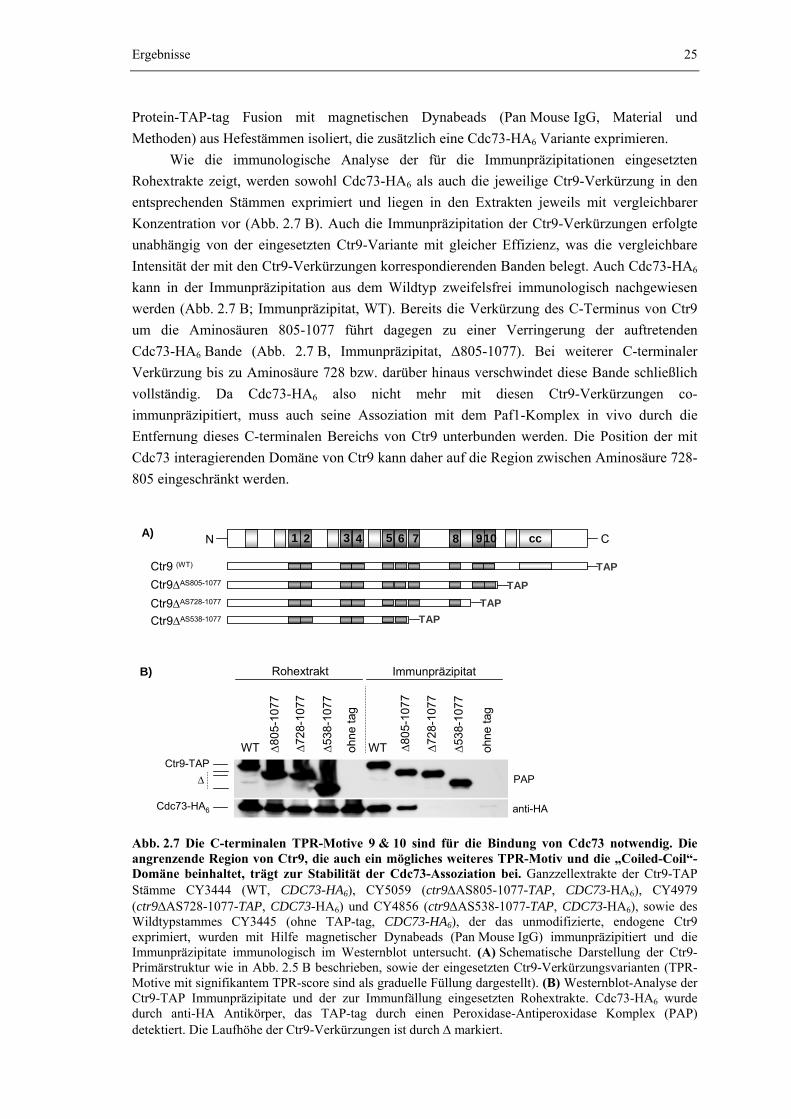
Ergebnisse
25
Protein-TAP-tag Fusion mit magnetischen Dynabeads (Pan Mouse IgG, Material und Methoden) aus Hefestämmen isoliert, die zusätzlich eine Cdc73-HA6 Variante exprimieren.
Wie die immunologische Analyse der für die Immunpräzipitationen eingesetzten Rohextrakte zeigt, werden sowohl Cdc73-HA6 als auch die jeweilige Ctr9-Verkürzung in den entsprechenden Stämmen exprimiert und liegen in den Extrakten jeweils mit vergleichbarer Konzentration vor (Abb. 2.7 B). Auch die Immunpräzipitation der Ctr9-Verkürzungen erfolgte unabhängig von der eingesetzten Ctr9-Variante mit gleicher Effizienz, was die vergleichbare Intensität der mit den Ctr9-Verkürzungen korrespondierenden Banden belegt. Auch Cdc73-HA6 kann in der Immunpräzipitation aus dem Wildtyp zweifelsfrei immunologisch nachgewiesen werden (Abb. 2.7 B; Immunpräzipitat, WT). Bereits die Verkürzung des C-Terminus von Ctr9 um die Aminosäuren 805-1077 führt dagegen zu einer Verringerung der auftretenden Cdc73-HA6 Bande (Abb. 2.7 B, Immunpräzipitat, Δ805-1077). Bei weiterer C-terminaler Verkürzung bis zu Aminosäure 728 bzw. darüber hinaus verschwindet diese Bande schließlich vollständig. Da Cdc73-HA6 also nicht mehr mit diesen Ctr9-Verkürzungen co-immunpräzipitiert, muss auch seine Assoziation mit dem Paf1-Komplex in vivo durch die Entfernung dieses C-terminalen Bereichs von Ctr9 unterbunden werden. Die Position der mit Cdc73 interagierenden Domäne von Ctr9 kann daher auf die Region zwischen Aminosäure 728-805 eingeschränkt werden.
1 2 3 4 5 6 7 8 910 ccN C
TAPTAP
TAPCtr9 (WT)
Ctr9ΔAS805-1077
Ctr9ΔAS728-1077
A)
Cdc73-HA6
Ctr9-TAPΔ
WT Δ805
-107
7
Δ728
-107
7
Δ538
-107
7
ohne
tag
WT Δ805
-107
7
Δ728
-107
7
Δ538
-107
7
ohne
tag
anti-HA
PAP
Rohextrakt ImmunpräzipitatB)
TAPCtr9ΔAS538-1077
Abb. 2.7 Die C-terminalen TPR-Motive 9 & 10 sind für die Bindung von Cdc73 notwendig. Die angrenzende Region von Ctr9, die auch ein mögliches weiteres TPR-Motiv und die „Coiled-Coil“-Domäne beinhaltet, trägt zur Stabilität der Cdc73-Assoziation bei. Ganzzellextrakte der Ctr9-TAP Stämme CY3444 (WT, CDC73-HA6), CY5059 (ctr9ΔAS805-1077-TAP, CDC73-HA6), CY4979 (ctr9ΔAS728-1077-TAP, CDC73-HA6) und CY4856 (ctr9ΔAS538-1077-TAP, CDC73-HA6), sowie des Wildtypstammes CY3445 (ohne TAP-tag, CDC73-HA6), der das unmodifizierte, endogene Ctr9 exprimiert, wurden mit Hilfe magnetischer Dynabeads (Pan Mouse IgG) immunpräzipitiert und die Immunpräzipitate immunologisch im Westernblot untersucht. (A) Schematische Darstellung der Ctr9-Primärstruktur wie in Abb. 2.5 B beschrieben, sowie der eingesetzten Ctr9-Verkürzungsvarianten (TPR-Motive mit signifikantem TPR-score sind als graduelle Füllung dargestellt). (B) Westernblot-Analyse der Ctr9-TAP Immunpräzipitate und der zur Immunfällung eingesetzten Rohextrakte. Cdc73-HA6 wurde durch anti-HA Antikörper, das TAP-tag durch einen Peroxidase-Antiperoxidase Komplex (PAP) detektiert. Die Laufhöhe der Ctr9-Verkürzungen ist durch Δ markiert.

Ergebnisse
26
Die heterolog exprimierten Ctr9-TPR-Motive 9 & 10 sind für die Bindung von Cdc73 hinreichend und der heterolog exprimierte N-terminale Ctr9-Bereich kann die Bindung von Paf1 vermitteln
Um die Bindung von Paf1 und Cdc73 an diskrete Bereiche von Ctr9 zu überprüfen
wurden ausgewählte Proteinabschnitte von Ctr9 heterolog in E. coli exprimiert. Die so erzeugten Ctr9-Fragmente wurden dann in Bindungsstudien auf ihre Fähigkeit hin überprüft, die Wechselwirkung mit Cdc73 oder Paf1 aus Hefe zu vermitteln. Zur Expression in E. coli wurde das T7-System (Rosenberg et al., 1987) unter Verwendung des Vektors pGEX-4T-3 ausgenutzt, der die Expression von Polypeptiden als Fusion mit N-terminaler Glutathion-S-Transferase (GST) ermöglicht (Smith and Johnson, 1988; GenBank Accession No. U13855). Definierte Bereiche von Ctr9 wurden durch PCR aus genomischer Hefe-DNA amplifiziert, wobei durch Wahl geeigneter Primer, die notwendigen Schnittstellen für die zur Klonierung verwendeten Restriktionsendonukleasen BamHI und XhoI eingeführt wurden (Material & Methoden). Zur heterologen Expression wurde der E. coli Stamm Rosetta(DE3) eingesetzt und die exprimierten GST-Protein Fusionen nach Aufschluss der Zellen an eine Glutathion-Sepharose-Matrix (GSH-Matrix) gebunden (Material & Methoden). Die beladene GSH-Matrix wurde anschließend mit Zellextrakten aus Hefestämmen inkubiert, die eine Fusion von Cdc73 oder Paf1 mit einem HA6- bzw. HA3-tag exprimieren. Zur Untersuchung der Wechselwirkung zwischen den Ctr9-Fragmenten und Cdc73 bzw. Paf1 wurden die GST-Fusionen von der GSH-Matrix eluiert und die Anwesenheit von Cdc73-HA6 bzw. Paf1-HA3 in den Eluaten immunologisch im Westernblot untersucht. Um die Bindung an bzw. die Elution der GST-Ctr9-Fragmente von der GSH-Matrix sicher zu stellen, wurden die Eluate zusätzlich immunologisch mit Antikörpern gegen das GST-tag untersucht (Abb. 2.8 B und Abb. 2.9 B). Die eingesetzten Ctr9-Fragmente und die Position der darin enthaltenen TPR-Motive sind schematisch in Abb. 2.8 A bzw. in Abb. 2.9 A dargestellt. Zunächst wurde die Affinität der heterolog exprimierten Ctr9-TPR-Motive 9 & 10 und die Affinität von Ctr9-Fragmenten, welche zusätzlich die N- bzw. C-terminal an TPR 9 bzw. TPR 10 angrenzenden Proteinbereiche enthalten, zu Cdc73-HA6 untersucht, das im Verbund mit dem intakten PAF1-Komplex vorliegt.
Die heterolog exprimierten Ctr9-Fragmente, welche die TPR-Motive 9 & 10 enthalten, können selektiv, endogenes Cdc73 binden, auch wenn dieses im Kontext des intakten PAF1-Komplexes vorliegt. Wie die auftretenden Banden in dem in Abb. 2.8 wiedergegebenen Westernblot (anti-HA) zeigen, ließ sich Cdc73-HA6 in den Eluaten der mit diesen Fragmenten beladenen GSH-Matrix immunologisch eindeutig nachweisen (Abb. 2.8 B, [AS550-816], [AS716-816] und [AS716-1077]). Dagegen führte weder die Inkubation der mit der Glutathion-S-Transferase (GST) beladenen noch die Inkubation der unbeladenen GSH-Matrix mit dem Hefezellextrakt zu einer nachweisbaren Anreicherung von Cdc73-HA6. Die Präzipitation von Cdc73-HA6 wird daher spezifisch durch die an die GSH-Matrix gebundenen Ctr9-Fragmente vermittelt. Dabei erweist sich der Ctr9-Bereich zwischen Aminosäure 715 und 816, der nur die Tandemanordnung aus den beiden TPR-Motiven 9 & 10 beinhaltet, als hinreichend für die Bindung von Cdc73-HA6 an den PAF1-Komplex. Dies bestärkt die vorangegangene Interpretation, dass diese beiden TPR-Motive eine spezifische Bindedomäne für Cdc73 bilden.

Ergebnisse
27
Cdc73-HA6
[AS
716-
1077
]
[AS
550-
816]
[AS7
16-8
16]
GST
GSH
-Mat
rix
Hef
e-R
ohex
trakt
anti-HA
anti-GST
GST-Ctr9-Fragmente
1 2 3 4 5 6 7 8 910 ccN C
GSTGST
GST
GST
GST-Ctr9[AS716-1077]:GST-Ctr9[AS716-816]GST-Ctr9[AS550-816]
Ctr9:
::
A)
B)
*
* *
*+
Abb. 2.8 Die TPR-Motive 9 & 10 aus Ctr9 bilden die Bindungsdomäne für Cdc73 (Abbildung entnommen und verändert nach A. Alemany Schmidt, Diplomarbeit 2005) Definierte Fragmente von Ctr9 wurden als Fusion mit einem N-terminalen GST-tag, heterolog in den E. coli Rosetta(DE3) Stämmen CE76 (GST-CTR9[AS550-816]), CE77 (GST-CTR9[AS716-816]) und CE79 (GST-CTR9[AS716-1077]) exprimiert und an eine Glutathion-Sepharosematrix (GSH-Matrix) gebunden. Die beladene GSH-Matrix wurde mit einem Hefe-Rohextrakt inkubiert, der aus dem Cdc73-HA6 Stamm CY3445 gewonnen wurde, anschließend mehrfach gewaschen und gebundenes Protein durch Erhitzen in 3x SDS/2ME-Puffer eluiert (Material & Methoden). Die Anwesenheit von Cdc73-HA6 in den Eluaten wurde immunologisch im Westernblot untersucht. Um die Spezifität der Interaktion zwischen Cdc73-HA6 und den Ctr9-Fragmenten zu überprüfen wurde sowohl eine nur mit dem GST-tag beladene, als auch die unbeladene GSH-Matrix mit dem Heferohextrakt inkubiert und anschließend in gleicher Weise analysiert. Der unfusionierte GST-tag wurde aus dem E. coli Rosetta(DE3) Stamm CE83 gewonnen. (A) Schematische Darstellung der Ctr9-Primärstruktur wie in Abb. 2.5 B beschrieben, sowie der heterolog exprimierten GST-Ctr9-Fragmente (TPR-Motive mit signifikantem TPR-score sind als graduelle Füllung dargestellt). (B) Westernblot-Analyse der GSH-Präzipitate. Die Anwesenheit von Cdc73-HA6 wurde mit Antikörpern gegen HA, das GST-tag und die Ctr9-Fragmente durch einen gegen das GST-tag gerichteten Antikörper nachgewiesen. Die Laufhöhe der GST-Ctr9-Fragmente bzw. des GST-tags, ist durch Pfeile hervorgehoben. Sterne markieren Abbauprodukte der GST-Ctr9-Fragmente, auftretende GST-Dimere sind durch „+“ gekennzeichnet.
Da die Quelle für Cdc73 jedoch ein Extrakt aus Hefezellen bildet, in denen der intakte PAF1-Komplex enthalten ist, kann zunächst dennoch nicht völlig ausgeschlossen werden, dass die Präzipitation von Cdc73-HA6 mit den Ctr9-Fragmenten die Folge einer indirekten Interaktion darstellt, die durch eine der weiteren Komponenten des Komplexes vermittelt wird. Die Deletion von PAF1 oder CTR9 resultiert jedoch in einer signifikanten Destabilisierung des PAF1-Komplexes und es kann davon ausgegangen werden, dass Cdc73 in einer ctr9Δ paf1Δ Doppelmutante als freies Protein vorliegt (vgl. Kapitel 2.1.3). Um entscheiden zu können, ob Cdc73 direkt mit den TPR-Motiven 9 &10 aus Ctr9 interagiert, wurde daher die Bindestudie mit dem Zellextrakt einer ctr9Δ paf1Δ Doppelmutante wiederholt, die eine Cdc73-HA6 Fusion exprimiert.

Ergebnisse
28
1 2 3 4 5 6 7 8 910 ccN C
GSTGSTGST
GSTGST
GST-Ctr9[AS716-1077]GST-Ctr9[AS716-816]
Ctr9:A)
GST-Ctr9[AS2-549]
Cdc73-HA6
Paf1-HA3
[AS7
16-8
16]
[AS7
16-1
077]
[AS2
-549
]
GS
H-M
atrix
GST
GST-Ctr9-FragmenteB)
anti-GST+
*
* *
[AS7
16-8
16]
[AS7
16-1
077]
[AS2
-549
]
GS
H-M
atrix
GS
T
GST-Ctr9-FragmenteH
efe-
Roh
extra
kt
anti-HA
anti-HA
C)
D)
Abb. 2.9 Cdc73 kann direkt mit den C-terminalen TPR-Motiven 9 und 10 in Ctr9 interagieren, bindet wie Paf1 aber auch an eine heterolog exprimierte, N-terminale Ctr9-Region. Definierte Fragmente von Ctr9 wurden als Fusion mit einem N-terminalen GST-tag heterolog in den E. coli Rosetta(DE3) Stämmen CE77 (GST-CTR9[AS716-816]), CE79 (GST-CTR9[AS716-1077]) und CE90 (GST-CTR9[AS2-549]) exprimiert und an eine Glutathion-Sepharosematrix (GSH-Matrix) gebunden. Die beladene GSH-Matrix wurde mit einem Hefe-Rohextrakt inkubiert, der aus dem Cdc73-HA6 Stamm CY5224 (ctr9Δ, paf1Δ) bzw. dem Paf1-HA3 Stamm CY5221 (ctr9Δ, cdc73Δ) gewonnen wurde, so dass der PAF1-Komplex aufgrund der jeweiligen Deletion von CTR9 weitestgehend dissoziiert oder zumindest destabilisiert vorliegt. Die GSH-Matrix wurde anschließend mehrfach gewaschen und gebundenes Protein durch Erhitzen in 3x SDS/2ME-Puffer eluiert (Material & Methoden). Die Anwesenheit von Cdc73-HA6 bzw. Paf1-HA3 in den Eluaten wurde immunologisch im Westernblot untersucht. Um die Spezifität der Interaktion zwischen den Ctr9-Fragmenten und Cdc73-HA6 bzw. Paf1-HA6 zu überprüfen, wurde sowohl eine nur mit dem GST-tag beladene, als auch die unbeladene GSH-Matrix mit den Heferohextrakten inkubiert und anschließend in gleicher Weise analysiert. Der unfusionierte GST-tag wurde aus dem E. coli Rosetta(DE3) Stamm CE83 gewonnen. (A) Schematische Darstellung der Ctr9-Primärstruktur wie in Abb. 2.5 B beschrieben, sowie der heterolog exprimierten GST-Ctr9-Fragmente (TPR-Motive mit signifikantem TPR-score sind als graduelle Füllung dargestellt). (B) Westernblot-Analyse der GST-Präzipitate mit anti-GST Antikörpern. Die Laufhöhe der GST-Ctr9-Fragmente, sowie des GST-tags alleine, ist durch Pfeile hervorgehoben. Sterne markieren Abbauprodukte der GST-Ctr9-Fragmente. Auftretende GST-Dimere sind durch „+“ gekennzeichnet. (C) Inkubation der mit GST-Ctr9 Fragmenten beladenen GSH-Matrix mit dem Heferohextrakt aus CY5224. Die Anwesenheit von Cdc73-HA6 in den GST-Präzipitaten wurde mit Antikörpern gegen das HA-tag nachgewiesen. (D) Inkubation der mit GST-Ctr9 Fragmenten beladenen GSH-Matrix mit dem Heferohextrakt aus CY5221. Die Anwesenheit von Paf1-HA6 in den GST-Präzipitaten durch Antikörper gegen das HA-tag nachgewiesen.
Wie der Westernblot in Abb. 2.9 C zeigt, wurde Cdc73-HA6 auch aus dem Zellextrakt der ctr9Δ paf1Δ Doppelmutante effektiv mit den Ctr9-Fragmenten präzipitiert, die den Ctr9-Bereich zwischen Aminosäure 715 und 816 enthalten. Auch das außerhalb des PAF1-Komplexes frei

Ergebnisse
29
vorliegende Cdc73-HA6 kann daher direkt und ohne Vermittlung durch etwaige „Brückenproteine“ an die heterolog exprimierten TPR-Motiven 9 & 10 aus Ctr9 binden. Unerwarteter Weise konnte Cdc73-HA6 aber auch im Eluat der mit dem bislang ungetesteten, N-terminalen Ctr9-Fragment GST-Ctr9[AS2-549] beladenen Matrix nachgewiesen werden. Cdc73 besitzt also zumindest in vitro auch eine Affinität zu diesem Ctr9-Bereich. Dieselbe N-terminale Ctr9-Region ist in vivo jedoch für die Bindung von Cdc73 weder notwendig noch hinreichend, wie die Studien mit den C-terminal verkürzten Ctr9-Varianten zeigen (vgl. Abb. 2.7). Es bleibt daher unklar, ob diese Bindung an den N-Terminus spezifisch erfolgt, oder ob es sich dabei um ein in vitro Artefakt handelt, das von einer intrinsischen Affinität von Cdc73 zu den TPR-Motiven in Ctr9 herrühren könnte. Im Gegensatz dazu erweist sich die Tandemanordnung aus den beiden TPR-Motiven 9 & 10 in vitro als hinreichend und zudem in vivo als notwendig für die Bindung von Cdc73. Die TPR-Motive 9&10 in Ctr9 können daher als spezifische Bindedomäne für Cdc73 aufgefasst werden.
Zudem wurde in einer weiteren Bindestudie die Wechselwirkung zwischen endogenem Paf1 und den in Abb. 2.9 A schematisch dargestellten Ctr9-Fragmenten untersucht. Speziell für diese Studie wurde ein Zellextrakt aus einer ctr9Δ cdc73Δ Doppelmutante eingesetzt, die eine Paf1-HA3 Fusion exprimiert, da auch die gleichzeitige Deletion von CTR9 und CDC73 zu einem frei vorliegendem Paf1-Protein, oder einem trimeren Komplex aus Paf1, Rtf1 und Leo1 führen sollte (vgl. Kapitel 2.1.3). Da Rtf1 und Leo1 keinen offensichtlichen Einfluss auf die Assoziationsfähigkeit der weiteren Untereinheiten besitzen, kann auch die Interaktion zwischen einem trimeren Komplex aus Paf1, Rtf1 und Leo1 mit den heterolog exprimierten Ctr9-Fragmenten auf eine direkte Wechselwirkung mit Paf1 zurückgeführt werden. Die Ergebnisse dieser Bindungsstudien sind ebenfalls in Abb. 2.9 wiedergegeben. Auch die bereits bei den Studien mit den verkürzten Ctr9-Varianten beobachtete Wechselwirkung zwischen Paf1 und dem N-terminalen Ctr9-Bereich (vgl. Abb. 2.6) kann durch Bindungsstudien mit heterolog exprimierten Ctr9-Fragmenten bestätigt werden. Wie Abb. 2.9 D zeigt, kann das Paf1-HA3 Protein selektiv mit dem N-terminalen Ctr9-Fragment, das die Aminosäuren 2-549 umfasst, präzipitiert werden. Die beiden getesteten, C-terminalen Ctr9-Fragmente führten dagegen nicht zur Präzipitation von Paf1-HA3 und auch die unbeladene bzw. mit GST beladene GSH-Matrix führte nach Inkubation mit dem Hefezellextrakt nicht zu einer Anreicherung von Paf1-HA3 in den entsprechenden Eluaten. Die Bindung von Paf1 an Ctr9 erfolgt also durch spezifische Interaktion mit dem N-Terminus von Ctr9. Dabei erweisen sich die TPR-Motive 1 bis 6 durch die in vitro Interaktionsstudien als hinreichend zur Bindung von Paf1. Da auch eine um die Aminosäuren 396-974 verkürzte Ctr9-Variante zur Co-Immunpräzipitation mit Paf1-HA3 in der Lage ist (C. Koch, persönliche Mitteilung), kann die in Frage kommende Paf1-Interaktionsdomäne vermutlich noch weiter auf die TPR-Motive 1-3 eingeschränkt werden.
Ctr9 besitzt damit mindestens zwei separate Interaktionsdomänen, über die die direkte Wechselwirkung mit den Bestandteilen Paf1 und Cdc73 des PAF1-Komplexes erfolgt. Die Assoziation von Rtf1 und Leo1 mit dem PAF1-Komplex setzt zudem die Interaktion zwischen Paf1 und Ctr9 voraus (vgl. Kapitel 2.1.3). Ctr9 kann daher legitim als Strukturgeber des PAF1-Komplexes betrachtet werden, wobei sich die TPR-Motive tatsächlich als essentiell für die Stabilität des PAF1-Komplexes erweisen.

Ergebnisse
30
2.2 Der Elongationsfaktor PAF1: Funktion Die Analyse des PAF1-Komplexes zeigt, dass vor allem die beiden Proteine Paf1 und
Ctr9 für die Stabilität des PAF1-Komplexes von Bedeutung sind, wohingegen Cdc73, Rtf1 und Leo1 ohne nennenswerten Einfluss auf die strukturelle Integrität des Komplexes bleiben. Eine ähnliche Hierarchie lässt sich aus dem Phänotyp der einzelnen Mutanten ableiten. Hierbei fallen paf1Δ und ctr9Δ Mutanten durch die schwerwiegendere Ausprägung des beobachteten Wachstumsdefekts und der auftretenden Temperatursensitivität auf, die in cdc73Δ, rtf1Δ und leo1Δ Mutanten wesentlich milder in Erscheinung treten (Koch et al., 1999; Magdolen et al., 1994; Mueller and Jaehning, 2002). Sowohl die Deletion von PAF1, als auch die Deletion von CTR9 resultiert in einer drastisch verlängerten Generationszeit, die mit einer Ansammlung stark in ihrem Volumen vergrößerter Zellen ohne Knospe einhergeht (Koch et al., 1999). Dieser Phänotyp weist bereits auf eine Störung im Durchlaufen des Zellzyklusses hin, da der Eintritt in die S-Phase verzögert erfolgt. Tatsächlich ist die Expression der für den Übergang von der G1- zur S-Phase benötigten G1-Zykline Cln1 und Cln2 sowohl in paf1Δ, als auch in ctr9Δ Mutanten signifikant verringert (Koch et al., 1999; Mueller and Jaehning, 2002). Interessanterweise scheint die Deletion von RTF1 oder LEO1 zumindest teilweise den Wachstumsdefekt von paf1Δ Mutanten unterdrücken zu können und auch die CLN1-Expression erreicht in der paf1Δ rtf1Δ Doppelmutante wieder das Niveau des Wildtyps (Mueller and Jaehning, 2002).
2.2.1 Die gleichzeitige Deletion von CDC73 und RTF1 induziert einen dramatischen Wachstumsdefekt Der PAF1-Komplex ist im Gegensatz zu den Elongationsfaktoren yFACT und yDSIF
nicht essentiell, da paf1Δ, ctr9Δ wie auch cdc73Δ, rtf1Δ und leo1Δ Mutante lebensfähig sind (Koch et al., 1999; Mueller and Jaehning, 2002; Shi et al., 1996). Die systematische Untersuchung von Hefedoppelmutanten durch die Methode der „Synthetic Genetic Array“ Analyse (SGA; Tong et al., 2001) wies jedoch in zwei unabhängigen Studien auf eine synthetisch letale Interaktion zwischen CDC73 und RTF1 bzw. CDC73 und LEO1 auf genetischer Ebene hin (Krogan et al., 2003b; Tong et al., 2004). Dies wäre interessant, da die Beobachtung entsprechend des Konzepts der synthetischen Letalität auf zusätzliche, ggf. von ihrer Beteiligung an dem PAF1-Komplex unabhängige Funktionen für Cdc73, Rtf1 und Leo1 hinweisen würde.
Das Konzept der synthetischen Letalität Synthetische Letalität bedeutet, dass die gleichzeitige Deletion zweier bestimmter Gene
zur Letalität der betroffenen Zelle führt, während sich Zellen mit den entsprechenden Einzeldeletionen dagegen als lebensfähig erweisen. In Analogie dazu wird die Potenzierung eines Defektphänotyps durch die gleichzeitige Deletion zweier Gene als synthetischer Wachstumsdefekt bezeichnet (Ye et al., 2005).
Ergebnisse
31
Tabelle 2.2 Tetradenstatistik der Kreuzungen zur Ermittlung einer synthetischen Letalität
Parentalstämme TSpo TSI NT nletal ΔΔletal ΔΔvital Δ3vital
cdc73Δ X leo1Δ CY2003 X CY3916 30°C 30°C 11 12 7 0 --- cdc73Δ X rtf1Δ CY2003 X CY3919 25°C 25°C 12 2 1 7 --- cdc73Δ X rtf1Δ CY2003 X CY3919 30°C 30°C 24 51 13 0 --- leo1Δ X rtf1Δ CY3916 X CY4654 30°C 30°C 4 0 0 5 --- rtf1Δ X leo1Δ CY3919 X CY4657 30°C 30°C 4 0 0 4 --- paf1Δ X cdc73Δ CY1994 X CY2002 30°C 30°C 12 3 1 12 --- paf1Δ X ctr9Δ CY1994 X CY2239 30°C 30°C 28 7 1 24 --- paf1Δ X leo1Δ CY1994 X CY3916 30°C 30°C 28 7 0 25 --- paf1Δ X rtf1Δ CY1994 X CY3919 30°C 30°C 8 3 0 8 --- ctr9Δ X cdc73Δ CY2239 X CY2003 30°C 30°C 14 5 0 12 --- ctr9Δ X rtf1Δ CY2239 X CY4654 30°C 30°C 19 12 2 10 --- rtf1Δ X cdc73Δ paf1Δ CY3919 X CY4686 25°C 25°C 44 8 2 14 20 rtf1Δ X cdc73Δ paf1Δ CY3919 X CY4686 25°C 30°C 28 21 7 7 6 rtf1Δ X cdc73Δ paf1Δ CY3919 X CY4686 30°C 30°C 14 10 2 6 7 rtf1Δ X cdc73Δ paf1Δ CY3919 X CY4686 37°C 30°C 14 33 ? 2 2 rtf1Δ X cdc73Δ ctr9Δ CY3919 X CY4690 25°C 25°C 40 20 3 21 6 rtf1Δ X cdc73Δ ctr9Δ CY3919 X CY4690 25°C 30°C 28 15 0 10 8 rtf1Δ X cdc73Δ ctr9Δ CY3919 X CY4692 25°C 25°C 14 2 0 6 4
TSpo … Sporulationstemperatur TSI … Inkubationstemperatur der Sporen NT … Anzahl der ausgewerteten Tetraden (Anzahl der vitalen Sporen = 4*NT-nletal)
nletal … Anzahl der nicht angewachsenen Sporen ΔΔletal … Anzahl der letalen Sporen mit nachweislich beiden Gendeletionen (cdc73Δ & rtf1Δ bei drei Deletionen) ΔΔvital … Anzahl der vitalen Sporen mit beiden Gendeletionen (cdc73Δ & rtf1Δ bei drei Deletionen ) Δ3
vital … Anzahl der vitalen Sporen mit allen drei Gendeletionen
Für das Auftreten eines synthetisch letalen Phänotyps bzw. eines synthetischen Wachstumsdefekts gibt es zwei prinzipielle Interpretationsmöglichkeiten, die entweder auf der redundanten Funktion beider Gene bzw. Allele im selben Prozessweg (z. B. der Synthese einer essentiellen Aminosäure) beruhen oder auf eine parallele Beeinflussung der gleichen zellulären Funktion über zwei unterschiedliche jedoch funktionell konvergierende Prozesswege (z. B. zwei konvergierende Signaltransduktionskaskaden) schließen lassen (Ye et al., 2005). Im ersten Fall tritt der Funktionsverlust stufenweise auf und führt erst durch die Doppeldeletion zum vollständigen Erliegen des Prozesses. Im zweiten Fall fallen die funktionell konvergierenden Reaktionswege jeweils bereits durch die Einzeldeletion aus. Der Ausfall wird aber durch die verbleibende Funktion des parallelen Reaktionswegs abgefangen. In beiden Fällen führt die Doppeldeletion jedoch zur Letalität, wenn eine für die Zelle essentielle Funktion betroffen ist. Das Auftreten einer synthetischen Letalität oder eines synthetischen Wachstumsdefekts kann daher zur Aufklärung einer funktionellen Interaktion zweier Gene eingesetzt werden, worauf auch die SGA-Anaylse aufbaut (Tong et al., 2001).
Verifizierung der synthetischen Letalität zwischen CDC73 und RTF1 bzw. LEO1 Um die Ergebnisse dieser SGA-Analysen (Krogan et al., 2003b; Tong et al., 2004) zu
verifizieren, wurden gezielt Doppelmutanten aller Kombinationen, der für die Proteine des PAF1-Komplexs kodierenden Gene, hergestellt sowie zusätzlich Tripelmutanten mit

Ergebnisse
32
paf1Δ cdc73Δ rtf1Δ oder ctr9Δ cdc73Δ rtf1Δ Genotyp. Dazu wurden Hefestämme mit Deletionen der entsprechenden Gene untereinander gekreuzt und der Genotyp der resultierenden Sporen durch Replikaplattierung anhand ihres selektierbaren Markergens und/oder durch PCR bestimmt. Die auftretenden Genotypkombinationen der überlebenden Sporen lassen dabei Rückschlüsse auf den Genotyp der nicht angewachsenen Sporen und damit auf eine eventuell auftretende synthetische Letalität zu (Copenhaver et al., 2000; Material & Methoden). Die aus den Sporen der Kreuzungen zwischen cdc73Δ und rtf1Δ bzw. cdc73Δ und leo1Δ Mutanten resultierenden Kolonien sind exemplarisch in Abb. 2.10 gezeigt.
Wie Abb. 2.10 und Tabelle 2.2 zeigen, konnte die synthetische Letalität zwischen CDC73 und RTF1 bzw. CDC73 und LEO1 weitestgehend bestätigt werden, da eine signifikante Anzahl der Sporen mit cdc73Δ rtf1Δ bzw. cdc73Δ leo1Δ nicht anwuchsen. Vor allem bei Erniedrigung der Inkubationstemperatur der Sporen von 30°C auf 25°C konnten zwar dennoch Zellkolonien mit cdc73Δ rtf1Δ Genotyp beobachtet werden (die cdc73Δ leo1Δ Kombination wurde nicht eingehender untersucht), die aufgrund ihrer geringen Koloniegröße und ihres Wachstumsverhaltens jedoch bereits auf einen schwerwiegenden synthetischen Wachstumsdefekt cdc73Δ rtf1Δ Doppelmutante hinwiesen (Abb. 2.10 C). Keine der weiteren in Tabelle 2.2 angegebenen Kreuzungen lieferte dagegen Hinweise für das Auftreten eines weiteren synthetisch letalen Phänotyps zwischen zwei Komponenten des PAF1-Komplexes. Zudem konnten auch die Tripelmutanten mit paf1Δ cdc73Δ rtf1Δ oder ctr9Δ cdc73Δ rtf1Δ Genotyp im Rahmen dieser Kreuzungsanalysen hergestellt werden (Tabelle 2.2), wobei vor allem die Kolonien mit paf1Δ cdc73Δ rtf1Δ Genotyp erste Hinweise dafür lieferten, dass der Wachstumsdefekt einer cdc73Δ rtf1Δ Mutante durch die zusätzliche Deletion von PAF1 unterdrückt werden kann.
A) cdc73Δ x leo1Δ
Sporulation: Sporenwachstum:
30°C30°C
A) cdc73Δ x leo1Δ
Sporulation: Sporenwachstum:
30°C30°C
B) cdc73Δ x rtf1Δ
Sporulation: Sporenwachstum:
30°C30°C
B) cdc73Δ x rtf1Δ
Sporulation: Sporenwachstum:
30°C30°C
C) cdc73Δ x rtf1Δ
Sporulation: Sporenwachstum:
25°C25°C
C) cdc73Δ x rtf1Δ
Sporulation: Sporenwachstum:
25°C25°C
-/+
-/-
+/-
+/+
+/-
+/-
-/+
-/+
-/-
+/+
-/+
+/-
+/-
-/-
-/+
+/+
-/-
-/+
+/-
+/+
+/-
+/+
-/-
-/+
-/-
-/+
+/-
+/+
+/+
+/-
-/+
-/-
-/-
+/-
-/+
+/+
-/+
+/+
+/-
-/-
?
?
+/+
?
-/+
+/+
-/-
+/-
-/-
-/-
+/+
+/+
+/+
-/+
-/-
+/-
+/-
+/+
-/+
-/-
+/-
-/+
-/+
+/-
-/-
-/+
+/+
+/-
Abb. 2.10 Die Tetradenanalyse offenbart einen drastischen synthetischen Wachstumsdefekt zwischen CDC73 und RTF1 bzw. CDC73 und LEO1 Deletionsmutanten. Haploide Parentalstämme mit einer Gendeletion von CDC73, RTF1 oder LEO1 wurden gekreuzt und anschließend bei 25°C oder 30°C sporuliert. Die Sporen der resultierenden Tetraden wurden vereinzelt und ebenfalls bei 25°C oder 30°C über einen Zeitraum von sechs Tagen auf YEPD-Medium inkubiert. Der Genotyp einzelner haploider Nachkommen wurde durch Replikaplattierung und PCR ermittelt. (A) cdc73Δ (CY2003) × leo1Δ (CY3916); Sporulation und Sporenwachstum bei 30°C. (B) cdc73Δ (CY2003) × rtf1Δ (CY3919); Sporulation und Sporenwachstum bei 30°C. (C) Kreuzung wie unter (B) beschrieben, jedoch Sporulation und Sporenwachstum bei 25°C. (A-C) lebensfähige und letale Doppeldeletionsmutanten sind durch Kreise hervorgehoben; Genotyptabellen: jeweils CDC73/LEO1 bzw. CDC73/RTF1; Wildtyp-Allel: +, Deletionsallel: -

Ergebnisse
33
2.2.2 Der Wachstumsdefekt einer cdc73Δ rtf1Δ Doppelmutante kann durch die zusätzliche Deletion von PAF1 oder CTR9 zum Teil aufgehoben werden Zur weiteren Charakterisierung der in Kapitel 2.2.1 beschriebenen Mutanten des PAF1-
Komplexes wurde das Wachstumsverhalten und die Temperatursensitivität der entsprechenden Stämme auf YEPD-Festmedium oder in Flüssigkultur ermittelt und die Zellmorphologie mikroskopisch im Hellfeld mit Phasenkontrast untersucht. Die Ergebnisse dieser Untersuchungen sind in Abb. 2.11 dargestellt.
Übereinstimmend mit früheren Beobachtungen (Koch et al., 1999; Magdolen et al., 1994; Mueller and Jaehning, 2002) fallen die Zellen, die Träger der ctr9Δ oder der pafΔ Deletion sind, durch ihr im Vergleich zum Wildtyp stark vergrößertes Zellvolumen auf, das mit einer signifikanten Verlängerung der Generationszeit und einer Temperatursensitivität bei 37°C einhergeht (Abb. 2.11 C und D). Dieser Effekt tritt bei der rtf1Δ und bei der cdc73Δ Mutante nur wesentlich milder in Erscheinung und die Zellen sind zudem zum Wachstum bei 37°C in der Lage (Abb. 2.11 E und F). Diese milde Ausprägung des Wachstumsdefekts wird bei gleichzeitiger Deletion von RTF1 und CDC73 jedoch gravierend verstärkt und übertrifft, mit Ausnahme der Generationszeitverlängerung, sogar die Auswirkungen einer ctr9Δ bzw. paf1Δ Deletion. Zudem erweist sich die rtf1Δ cdc73Δ Doppelmutante als stark temperatursensitiv.
Interessanterweise kann die cdc73Δ Deletion den Wachstumsdefekt der paf1Δ Mutante zum Teil unterdrücken, was sich in einer verringerten Zellgröße und verkürzten Generationszeit äußert (Abb. 2.11 H). Dieser Effekt tritt dagegen bei der Kombination der cdc73Δ Deletion mit ctr9Δ wesentlich weniger deutlich in Erscheinung und auch die Generationszeit bleibt unverändert (Abb. 2.11 G). Dass CDC73 und RTF1 ggf. antagonistisch zu PAF1 und weniger stark auch zu CTR9 wirken könnten wird spätestens im Phänotyp der entsprechenden Tripelmutanten klar (Abb. 2.11 K und L). Obwohl beide Tripelmutanten nach wie vor temperatursensitiv für das Wachstum bei 37°C sind, zeigen sie jedoch eine deutliche Reduzierung des Zellvolumens, das im Falle der paf1Δ Kombination mit der cdc73Δ und rtf1Δ Deletion nahezu wieder das normale Niveau des Wildtyps erreicht. Auch die Generationszeit nähert sich in der paf1Δ Tripelmutante wieder der für den Wildtypstamm charakteristischen Zeitspanne.
Cdc73, Rtf1 und vermutlich auch Leo1, für das keine Tripelmutante mit paf1Δ cdc73Δ bzw. ctr9Δ cdc73Δ charakterisiert wurde, wirken demnach anscheinend „antagonistisch“ zu ihren Komplexpartnern Paf1 und Ctr9. Dies lässt eine Hyperaktivität von Paf1 und/oder Ctr9 vermuten, die ohne den regulierenden Einfluss von Cdc73 und Rtf1 schädlich für die Zelle wirkt. Die Funktion des PAF1-Komplexes könnte daher einer Art regulatorischer Selbstinhibierung unterliegen. Andererseits könnte der schwerwiegende Wachstumsdefekt der cdc73Δ rtf1Δ Doppelmutante auf eine möglicherweise redundante, aber getrennte Funktion von Cdc73 und Rtf1 innerhalb des PAF1-Komplexes hinweisen. Um diese Möglichkeit zu untersuchen, wurde im Folgenden das Netzwerk aus synthetisch letalen Interaktionen, welche die einzelnen Bestandteile des PAF1-Komplexes eingehen, analysiert und überprüft, ob sich dadurch Rückschlüsse auf funktionelle Unterschiede in den Aktivitäten von Paf1, Ctr9, Rtf1, Leo1 und Cdc73 ziehen lassen.

Ergebnisse
34
WT (W303)
GT28°C: 1,8 hts37°C: nein
pep4::URA3
GT28°C: 1,8 hts37°C: nein
paf1::URA3
GT28°C: 3,3 hts37°C: ja
cdc73::HIS3
GT28°C: 2,3 hts37°C: nein
rtf1::KanMX4
GT28°C: 3,1 hts37°C: nein
paf1::URA3cdc73::HIS3
GT28°C: 2,5 hts37°C: ja
rtf1::KanMX4cdc73::HIS3
GT28°C: 2,5 hts37°C: ja
ctr9::LEU2rtf1::KanMX4cdc73::HIS3
GT28°C: 2,9 hts37°C: ja
paf1::URA3rtf1::KanMX4cdc73::HIS3
GT28°C: 2,1 hts37°C: ja
A) B)
C) D)
E) F)
G) H)
I) J)
K) L)
ctr9::LEU2
GT28°C: 4,7 hts37°C: ja
ctr9::LEU2cdc73::HIS3
GT28°C: 4,7ts37°C: ja
rtf1::KanMX4cdc73::HIS3
GT28°C: 2,9 hts37°C: ja
Abb. 2.11 Die gleichzeitige Deletion von CDC73 und RTF1 resultiert in einem synthetischen Wachstumsdefekt, der durch eine zusätzliche Deletion von PAF1 oder CTR9 zum Teil aufgehoben werden kann. Das Wachstum von Zellen der angegebenen Deletionsstämme wurde in YEPD-Flüssigkultur bei 28°C verfolgt und die Zellmorphologie mikroskopisch im Phasenkontrast untersucht. Die Generationszeit (GT28°C) wurde anhand der durch photometrische Trübungsmessung ermittelten Wachstumskurve bestimmt. Zur Ermittlung der Temperatursensitivität (ts37°C) wurden Zellen der entsprechenden Deletionsstämme auf YEPD-Festmedium vereinzelt und die Fähigkeit zur Koloniebildung bei 37°C untersucht (ts37°C = „nein“ bedeutet dabei, dass der jeweilige Stamm zur Koloniebildung bei 37°C in der Lage und damit nicht temperatursensitiv ist.). (A) W303 Wildtyp, CY1078. (B) CY1615 (pep4::URA3). (C) CY5455 (ctr9::LEU2). (D) CY1994 (paf1::URA3). (E) CY2003 (cdc73::HIS3). (F) CY3919 (rtf1::KanMX4). (G) CY4690 (ctr9::LEU2,

Ergebnisse
35
cdc73::HIS3). (H) CY4686 (paf1::URA3, cdc73::LEU2). (I) CY5452 (cdc73::HIS3, rtf1::KanMX4). (J) CY5447 (cdc73::HIS3, rtf1::KanMX4). (K) CY5453 (ctr9::LEU2, cdc73::HIS3, rtf1::KanMX4). (L) CY5445 (paf1::URA3, cdc73::HIS3, rtf1::KanMX4).
2.2.3 Das Netzwerk synthetisch letaler Interaktionen zwischen dem PAF1-Komplex, Rad6/Bre1 und den Histon-Methyltransferasens Set1, Set2 und Dot1 Die vollständige Sequenzierung des Hefegenoms (Goffeau et al., 1996) und die
Herstellung einer nahezu vollständigen Kollektion von Gendeletionsmutanten der Hefe (Giaever et al., 2002) ermöglicht die systematische Erfassung aller Doppelmutantenphänotypen innerhalb des Hefegenoms in einem kompletten Datensatz. Dieser Datensatz ist frei verfügbar und unter anderem Bestandteil der BioGRID-Datenbank (Reguly et al., 2006; www.thebiogrid.com). Entsprechend des Konzepts der SGA-Analyse (Kapitel 2.2.1) lässt die systematische Auswertung der Genpaare, die zum Auftreten eines synthetisch letalen Phänotyps führen, Rückschlüsse auf eine mögliche funktionelle Verbindung der durch diese Gene kodierten Proteine zu (Tong et al., 2001; Tong et al., 2004; Ye et al., 2005).
Erstellung eines SL-Datensatzes der synthetisch letalen Phänotypen für die Komponenten des Elongationsfaktors PAF1
Um weitere Hinweise auf gesonderte Funktionen einzelner Komponenten des PAF1-
Komplexes zu erhalten, wurden anhand der BioGRID-Datenbank alle Gene, deren Doppelmutantenphänotyp mit mindestens einer der fünf Komponenten des PAF1-Komplexes als synthetisch letal eingestuft wurde, zu einem Datensatz (SL-Datensatz) zusammengefasst und entsprechend ihrer Funktion bzw. ihrer Beteiligung an definierten Proteinkomplexen klassifiziert (Anhang A1). Alle Gene mit der gleichen funktionalen Klassifizierung bzw. Komplexzugehörigkeit wurden dann zu einer Funktionsgruppe zusammengefasst, wobei einzelne Gene auch Bestandteil mehrerer Funktionsgruppen sein können (Tabelle 2.3 und Anhang A1). Beispielweise bilden alle Gene des SL-Datensatzes, die für Komponenten des SWR1-Komplexes kodieren, die gleichnamige Funktionsgruppe „SWR1-Komplex“. Aufgrund der Funktion des SWR1-Komplexes sind die entsprechenden Gene aber ebenfalls in der übergeordneten Funktionsgruppe „Chromatinstrukturmodulation“ enthalten. Die Anzahl der Gene einer Funktionsgruppe, die einen synthetisch letalen Phänotyp mit einer oder mehrerern Komponenten des PAF1-Komplexes eingehen, wurde ausgewertet und ist in Tabelle 2.3 wiedergegeben. Die Beteiligung des PAF1-Komplexes an einem gegebenen Prozess ist dabei umso wahrscheinlicher, je mehr Komponenten des PAF1-Komplexes mit der korrespondierenden Funktionsgruppe synthetisch letale Doppelmutantenphänotypen zeigen. Im Umkehrschluss kann aber auch das gehäufte Auftreten synthetisch letaler Phänotypen nahezu aller Gene einer Funktionsgruppe mit nur einer Komponente des PAF1-Komplexes als Hinweis auf eine spezielle Funktion dieser bestimmten Komponente des PAF1-Komplexes gewertet werden. Dabei muss jedoch beachtet werden, dass die synthetisch letalen Interaktionen von

Ergebnisse
36
CTR9 in der Datenbank nicht vollständig erfasst werden und daher unterrepräsentiert sind, da der zugehörige Leserahmen YOL145C in den zugrunde gelegten SGA-Analysen (Giaever et al., 2002; Tong et al., 2004) nicht berücksichtigt wurde.
Um die bekannten Funktionen des PAF1-Komplexes bei der Auswertung zu berücksichtigen und von möglichen zusätzlichen Funktionen unterscheiden zu können, wurde der SL-Datensatz um die synthetisch letalen Interaktionen von SET1, SET2, DOT1, RAD6 und BRE1 erweitert. Im Falle von RAD6 und BRE1 wurden nur Gene berücksichtigt, die jeweils mit beiden Genen zur Ausbildung eines synthetisch letalen Phänotyps führen, da Rad6 außerhalb seiner gemeinsamen Aktivität mit Bre1, als spezifische Histon H2B Protein-Ubiquitinligase, weitere Funktionen besitzt, die bei der Auswertung unberücksichtigt bleiben sollen (Broomfield et al., 2001; Wood et al., 2003a).
Funktionelle Charakterisierung des Elongationsfaktors PAF1 anhand seiner auftretenden synthetisch letalen Phänotypen
Die Auswertung des erweiterten SL-Datensatzes ist in Tabelle 2.3 wiedergegeben. Die
Komponenten des PAF1-Komplexes, die die jeweils häufigsten synthetisch letalen Interaktionen zu den Genen einer Funktionsgruppe zeigen, sind grau hinterlegt hervorgehoben. Auffällig ist das geballte Auftreten von synthetisch letalen Interaktionen mit CDC73, das mit 117 Genen des Hefegenoms einen dokumentierten synthetisch letalen Phänotyp zeigt (thebiogrid.com, Mehrfachnennungen manuell korrigiert), was 53% der insgesamt 220 Gene des zugrunde gelegten SL-Datensatzes entspricht (Tabelle 2.3). Genomweit betrachtet zeigt CDC73 damit überdurchschnittlich viele synthetisch letale Interaktionen; in einer repräsentativen Studie, die ~4000 Interaktionen unter ~1000 Genen umfasst, wurden im Mittel 34 synthetisch letale Phänotypen pro untersuchtem Gen beobachtet (Tong et al., 2004). Für die restlichen Komponenten des PAF1-Komplexes werden dagegen deutlich weniger synthetisch letale Phänotypen beschrieben: RTF1 58, PAF1 31, LEO1 20 und CTR9 8 (thebiogrid.com, Mehrfachnennungen manuell korrigiert, eine mögliche Unterrepräsentierung einzelner Gene, vor allem von CTR9, bleibt unberücksichtigt). Dies könnte eine funktionelle Sonderstellung von CDC73 innerhalb des PAF1-Komplexes andeuten. Mit Ausnahme von SET2 zeigen die Gene der Histon-Methyltransferasen dagegen signifikant weniger synthetisch letale Interaktionen als der genomweite Durchschnitt und vor allen Dingen auch weniger als RAD6, BRE1 oder die Komponenten des PAF1-Komplexes (SET1: 4, SET2: 76 DOT1: 9, RAD6: 84, BRE1: 65; thebiogrid.com, Mehrfachnennungen manuell korrigiert). Dies ist ein deutlicher Hinweis darauf, dass sowohl der PAF1-Komplex, als auch Rad6 und Bre1, neben ihrem Einfluss auf den Methylierungsstatus von Histon H3, weitere Funktionen besitzen müssen. Unter den Funktionsgruppen, die mit mehreren Komponenten des PAF1-Komplexes synthetisch letale Doppeldeletionskombinationen aufweisen, fallen vor allem der Chromatinremodulator SWR1-Komplex (Krogan et al., 2003b), die Histon-Methyltransferase Set2 (Li et al., 2002; Strahl et al., 2002) und Gene auf, die an der CTD-Modifizierung bzw. am Mediator-, Elongator-, NuA4- oder SAGA-Komplex beteiligt sind (Allard et al., 1999; Cho et al., 2001; Grant et al., 1997; Jona et al., 2001; Kim et al., 1994; Winkler et al., 2002). Diese

Ergebnisse
37
Tabelle 2.3: Funktionelle Clusterung synthetisch letaler Interaktionen der Gene des PAF1- und RAD6/BRE1-Komplexes, sowie der HMTs Set1, Set2 und Dot1
Funktionsgruppe Na)
PAF1
CTR
9
CD
C73
RTF
1
LEO
1
RA
D6
& B
RE1
b)
SET2
SET1
DO
T1
Synthetisch letale Interaktionen gesamt 220 31 8 117 53 20 41 76 4 9
Chromatinstrukturmodulation 16 5 … 12 10 8 10 8 … …INO80-Komplex 4 … … 4 … … 2 … … …ISW1-Rekrutierung 1 … … … 1 … … … … …SWR1-Komplex 8 4 … 8 8 8 8 8 … …H2A.Z 1 … … 1 1 1 1 1 … …yFACT 2 1 … … 1 … … … … …
DNA-Schadensantwort und Reparatur 9 … … 4 3 … … 3 … …HDAC 11 … … 5 1 1 … 7 … …
SET3-Komplex 6 … … 1 … … … 6 … …SIN3-Komplex 5 … … 4 1 1 1 1 … …
HMT 2 1 1 2 1 1 … … … …DOT1 1 … … 1 … … … 1 … …SET2 1 1 1 1 1 1 … … … …
Mitochondrielle Struktur und Funktion 18 … … 9 … … 8 … … …mRNA-Prozessierung und Export 20 2 … 17 … … 2 1 … …
mRNA-Degradation 4 … … 4 … … … … … …mRNA-Export 8 1 … 6 … … 2 … … …Splicing 4 … … 3 … … … 1 … …Termination / Polyadenylierung 3 1 … 3 … … … … … …Translation 1 … … 1 … … … … … …
Proteasom und Proteindegradation 12 6 … 7 6 … 4 … 2 2Replikation 4 … … 1 3 1 1 … … …Sekretation und Vesikeltransport 22 … … 16 2 … 1 6 … 1sonstige 37 … … 15 3 2 2 15 … 3Transkription 48 14 6 22 23 7 16 21 … 1
CTD-Modifizierung 6 2 … 3 2 … 3 1 … …Elongator 5 5 4 4 5 … 5 … … …Mediator 4 1 … 1 2 … … 1 … …NuA4 (HAT) 5 … … 4 4 3 3 3 … …SAGA 5 4 2 4 3 1 2 … … …
Zellzyklus, Mitose und Maiose 27 3 1 9 5 … 4 8 2 2SBF 3 2 … 3 … … 2 … … …Schwester-Chromatid Kohäsion 3 … … … 3 … … 1 … …Expression von Histongenen 3 … … 3 … … … … … …
a) Anzahl der Gene im zugrundegelegten Datensatz (siehe Anhang A1)b) Berücksichtigt wurden nur Gene, die mit RAD6 und BRE1 synthetisch letale Phänotypen zeigen
Funktionsgruppen zeigen zudem ein gehäuftes Auftreten synthetisch letaler Interaktionen mit RAD6/BRE1 und der Histon-Methyltransferase Set2, das deutlich die enge Verzahnung zwischen Chromatinstrukturmodulation und transkriptioneller Aktivierung, Initiation und Elongation widerspiegelt (Babiarz et al., 2006; Keogh et al., 2006; Kobor et al., 2004; Krogan et al., 2004)
Daneben treten aber auch Funktionsgruppen auf, deren Gene nahezu exklusiv synthetisch letale Phänotypen mit CDC73 oder RTF1 zeigen, was ggf. auf eine spezifische Funktion der entsprechenden Komponente des PAF1-Komplexes hindeuten könnte. RTF1 zeigt beispielsweise als einzige Komponente des PAF1-Komplexes eine synthetische Letalität mit allen Genen der Funktionsgruppe Schwester-Chromatid-Kohäsion. Die Gruppe umfasst die Gene CTF4, CTF8 und DCC1, die allesamt für den Zusammenhalt der Schwesterchromatiden

Ergebnisse
38
erforderlich sind (Hanna et al., 2001; Mayer et al., 2001) und zudem an der Signaltransmission des DNA-Replikationskontrollpunkts (Kontrollpunkt der S-Phase) beteiligt zu sein scheinen (Osborn and Elledge, 2003; Pan et al., 2006). Dagegen zeigt nur CDC73 einen synthetisch letalen Phänotyp mit den Genen RAD52, RAD53, MET18 und XRS2, die an der DNA-Schadensantwort und Reparatur beteiligt sind. RAD53 ist als Effektorkinase ebenfalls unmittelbar an der DNA-Replikationskontrolle und gleichzeitig auch an dem DNA-Schadenskontrollpunkt beteiligt (Allen et al., 1994). RAD52 und XRS2 kodieren für Komponenten der DNA-Doppelstrangreparatur und der homologen Rekombination und MET18 ist Bestandteil der Nukleotid-Exzisionsreparatur (Bressan et al., 1999; Mortensen et al., 1996; Queimado et al., 2001). Die Annahme, dass Rtf1 und Cdc73 jeweils einen unterschiedlichen Zweig des S Phase-Kontrollpunkts, der Replikationskontrolle und der DNA-Reparaturmaschinerie beeinflussen, bietet daher einen möglichen Ansatzpunkt um den erheblichen synthetischen Wachstumsdefekt der cdc73Δ rtf1Δ Doppelmutante zu beschreiben (vgl. Kapitel 2.2.2, Abb.2.11), zumal auch Rad6 und Dot1 unmittelbar an der Adaption des DNA-Schadenskontrollpunkts und der Post-Replikationsreparatur beteiligt sind (Broomfield et al., 2001; Game et al., 2006a; Giannattasio et al., 2005b; Pan et al., 2006; Toh et al., 2006; Wysocki et al., 2005). Eine weitere, exklusiv bei CDC73 auftretende synthetisch letale Interaktion besteht zu den Genen HIR2, HIR3 und HPC, die alle an der zellzyklusregulierten Transkription der Histongene beteiligt sind (Spector et al., 1997; Xu et al., 1992). Interessant ist dabei auch ihre Verbindung zu dem Elongationsfaktor yFACT (Formosa et al., 2002), der seinerseits physikalisch mit dem PAF1-Komplex interagiert. Für CDC73 fällt aber auch das gehäufte Auftreten von synthetisch letalen Phänotypen mit Genen auf, die an der Prozessierung und dem Export, sowie der Degradation der mRNA beteiligt sind (Tabelle 2.3). Dies könnte eine spezielle Beteiligung von Cdc73 an diesen Prozessen andeuten.
Das Netzwerk synthetisch letaler Interaktionen zwischen PAF1-Komplex, RAD6/BRE1 und der Histon-Methyltransferase Set2 (Tabelle 2.3) unterstreicht damit die zentrale Funktion des PAF1-Komplexes als Bestandteil der Transkriptionsmaschinerie. Zudem wird aber auch eine mögliche zusätzliche funktionelle Verbindung des PAF1-Komplexes mit den Prozessen der DNA-Reparatur und der Ausbildung der Replikations- und Schadenskontrollpunkte deutlich. Das auffällig häufige Auftreten synthetisch letaler Phänotypen von CDC73 mit Genen, die an verschiedenen Aspekten der mRNA-Prozessierung beteiligt sind, lassen außerdem eine mögliche weitere Funktion des PAF1-Komplexes neben seiner bekannten Rolle bei der Polyadenylierung und Termination erwarten, da gerade diese zwar von PAF1, CTR9 und RTF1 aber nicht von CDC73 abhängig ist (Sheldon et al., 2005).

Ergebnisse
39
2.2.4 Die Sensitivität gegen 6-Azauracil offenbart unterschiedliche Einflüsse der einzelnen Komponenten des PAF1-Komplexes auf die Elongations-fähigkeit der RNA-Polymerase II Elongationsdefekte der RNA-Polymerase II äußern sich häufig in einer Sensitivität gegen
das Urazil-analoge 6-Azauracil (6AU; Riles et al., 2004). 6AU wirkt dabei u. a. als Inhibitor der IMP-Dehydrogenase und der Orotidylat-Decarboxylase. Dies führt zu einer Reduzierung des zellulären Nukleotidpools und verursacht eine reduzierte Elongationsrate und Prozessivität der RNA-Polymerase II, was sich letztlich in der 6AU-Sensitivität vieler Elongationsfaktormutanten widerspiegelt (Exinger and Lacroute, 1992; Mason and Struhl, 2005).
Ein fehlerhafter PAF1-Komplex führt ebenfalls zu einem Elongationsdefekt der RNA-Polymerase II (Krogan et al., 2002b; Squazzo et al., 2002). Zur phänotypischen Charakterisierung dieses Defekts wurde im Folgenden daher das Wachstumsvermögen von Hefestämmen mit definierten Deletionen oder Deletionskombinationen der Komponenten des PAF1-Komplexes in Gegenwart von 6-Azauracil untersucht. Dabei wurde auch die Auswirkung der CTR9-Verkürzung (siehe Kapitel 2.1.5) auf die Sensitivität gegen 6AU miteinbezogen. Die für diesen Test notwendige Uracil-Prototrophie der untersuchten Stämme wurde entweder durch eine pep4::URA3 oder durch ein paf1::URA3 Gendisruption eingeführt. Die entsprechenden Hefestämme wurden zur Ermittlung der 6AU-Sensitivität als serielle Verdünnung einer Zellsuspension auf Uracil-Mangelmedium (Medium auf Casaminoacid-Basis, Material & Methoden) mit oder ohne den Zusatz von 50, 75 oder 100 mg·L-1 6-Azauracil aufgetropft und über einen Zeitraum von bis zu zehn Tagen bei 30°C inkubiert. Daneben wurde ein Uracil-Mangelmedium auf SCD-Basis (Material & Methoden) mit oder ohne Zusatz von 60 mg·L-1 6AU eingesetzt.
6AU-Sensitivität von Mutanten mit einfachen und multiplen Deletionen der Komponenten des PAF1-Komplexes
Wie Abb. 2.12 A zeigt, wächst der Wildtypstamm auch in Gegenwart von 100 mg·L-1
6AU bei allen sechs seriell verdünnten Zellsuspensionen. Dabei bilden unabhängig von der Verdünnung ca. 90% der aufgetropften Zellen eine Kolonie. Die Abbildung zeigt auch, dass, übereinstimmend mit früheren Beobachtungen (Squazzo et al., 2002), alle Deletionsstämme von Komponenten des PAF1-Komplexes eine mehr oder weniger stark ausgebildete Sensitivität gegen 6-Azauracil besitzen. Dabei zeigen paf1Δ und ctr9Δ Mutanten die höchste Sensitivität gegen 6AU, deren Wachstum im Konzentrationsbereich von 50-100 mg·L-1 durch 6AU nahezu vollständig unterbunden wird (Abb. 2.12 A und B, sowie Daten nicht gezeigt). Dieser Effekt tritt bei Deletion von CDC73 weniger deutlich in Erscheinung. Die Deletion von LEO1 oder RTF1 führt dagegen nur zu einer milden Sensitivität gegen 6-Azauracil. Alle untersuchten Hefestämme, inklusive der im Folgenden beschriebenen, wachsen problemlos auf dem jeweiligen Uracil-Mangelmedium ohne den Zusatz von 6-Azauracil (Abb. 2.12 A und Daten nicht gezeigt).
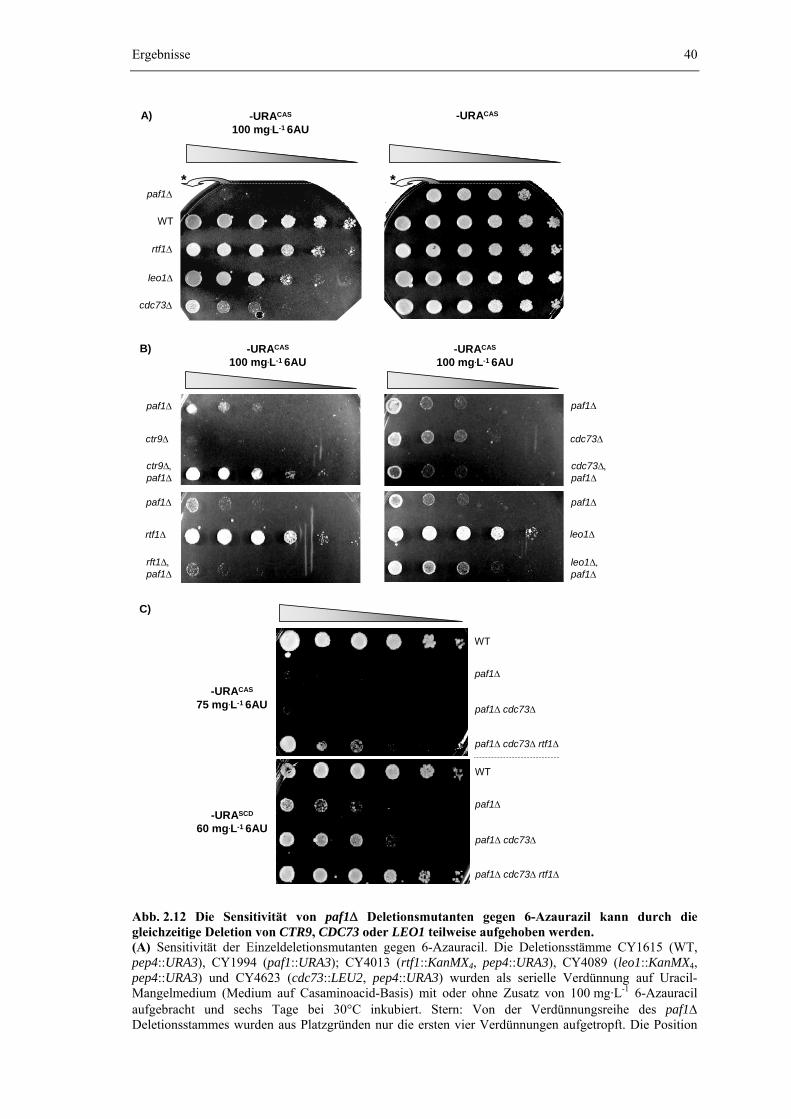
Ergebnisse
40
paf1Δ
WT
rtf1Δ
leo1Δ
cdc73Δ
-URACAS
100 mg.L-1 6AU-URACAS
paf1Δ paf1Δ
paf1Δ paf1Δ
ctr9Δ cdc73Δ
rtf1Δ leo1Δ
ctr9Δ,paf1Δ
cdc73Δ,paf1Δ
rft1Δ,paf1Δ
leo1Δ,paf1Δ
-URACAS
100 mg.L-1 6AU-URACAS
100 mg.L-1 6AU
WT
paf1Δ
paf1Δ cdc73Δ
paf1Δ cdc73Δ rtf1Δ
WT
paf1Δ
paf1Δ cdc73Δ
paf1Δ cdc73Δ rtf1Δ
-URACAS
75 mg.L-1 6AU
-URASCD
60 mg.L-1 6AU
B)
C)
A)
* *
Abb. 2.12 Die Sensitivität von paf1Δ Deletionsmutanten gegen 6-Azaurazil kann durch die gleichzeitige Deletion von CTR9, CDC73 oder LEO1 teilweise aufgehoben werden. (A) Sensitivität der Einzeldeletionsmutanten gegen 6-Azauracil. Die Deletionsstämme CY1615 (WT, pep4::URA3), CY1994 (paf1::URA3); CY4013 (rtf1::KanMX4, pep4::URA3), CY4089 (leo1::KanMX4, pep4::URA3) und CY4623 (cdc73::LEU2, pep4::URA3) wurden als serielle Verdünnung auf Uracil-Mangelmedium (Medium auf Casaminoacid-Basis) mit oder ohne Zusatz von 100 mg·L-1 6-Azauracil aufgebracht und sechs Tage bei 30°C inkubiert. Stern: Von der Verdünnungsreihe des paf1Δ Deletionsstammes wurden aus Platzgründen nur die ersten vier Verdünnungen aufgetropft. Die Position

Ergebnisse
41
der Verdünnungspunkte, bezogen auf die übrigen Verdünnungsreihen, wird dadurch jeweils um einen Schritt in Richtung des Pfeils versetzt. (B) Sensitivität der paf1Δ Doppeldeletionsmutanten gegen 6-Azauracil. Das Wachstum serieller Verdünnungen der angegebenen paf1Δ Doppeldeletionsmutanten in Gegenwart von 100 mg·L-1 6-Azauracil (Uracil-Mangelmedium auf Casaminoacid-Basis) wurde im Vergleich zu den entsprechenden Einzeldeletionsmutanten des PAF1-Komplexes über 6 Tage bei 30°C verfolgt. Die Uracil-Auxotrophie wird bei den Doppeldeletionsmutanten durch die paf1::URA3 Deletion, bei den Einzeldeletionsmutanten durch pep4::URA3 komplementiert. paf1Δ (CY1994), ctr9Δ (CY4627), ctr9Δ paf1Δ (CY4660), cdc73Δ (CY4623), cdc73Δ paf1Δ (CY4685), rtf1Δ (CY4013), rtf1Δ paf1Δ (CY4704), leo1Δ (CY4089), leo1Δ paf1Δ (CY4663). (C) Sensitivität der paf1Δ rtf1Δ cdc73Δ Tripelmutante gegen 6-Azauracil. Die Deletionsstämme CY1615 (WT, pep4::URA3), CY1994 (paf1::URA3), CY4686 (paf1::URA3, cdc73::HIS3) und CY5445 (paf1::URA3, cdc73::HIS3, rtf1::KanMX4) wurden als serielle Verdünnungsreihe auf Uracil-Mangelmedium mit 75 mg·L-1 bzw. 60 mg·L-1 6-Azauracil aufgetropft und über einen Zeitraum von 6 Tagen bei 30°C inkubiert. -URACAS … Uracil-Mangelmedium auf Casaminoacid-Basis, -URASCD … Uracil-Mangelmedium auf SCD-Basis
Überraschend war dagegen das Verhalten der paf1Δ ctr9Δ Doppelmutante: Während für die paf1Δ und ctr9Δ Mutante eine Hypersensitivität gegen 6-Azauracil beobachtet wurde, führte die gleichzeitige Deletion von PAF1 und CTR9 jedoch zu einer teilweisen Wiederherstellung des Wachstumsvermögens auf 6AU-haltigem Medium (Abb. 2.12 B). Ein ähnliches, aber weniger deutliches Bild, zeichnet sich unter den gewählten Versuchsbedingungen auch für die paf1Δ leo1Δ Doppeldeletionsmutante ab und wurde für eine paf1Δ rtf1Δ Doppelmutante bereits in ähnlicher Weise beschrieben (Mueller and Jaehning, 2002). Die Umstellung des verwendeten Uracil-Mangelmediums von Casaminoacid- auf SCD-Basis (Abb. 2.12 C) zeigte zudem, dass dieser Effekt auch bei gleichzeitiger Deletion von PAF1 und CDC73 auftritt und in einer paf1Δ cdc73Δ rtf1Δ Tripeldeletionsmutante noch wesentlich stärker zum Tragen kommt. Dieser Befund steht im Einklang mit der Beobachtung, dass eine paf1Δ cdc73Δ rtf1Δ Tripeldeletionsmutante wieder eine nahezu normale Zellmorphologie und Generationszeit besitzt (vgl. Kapitel 2.2.2). Allerdings ist keine der untersuchten paf1Δ Deletionskombinationen, und auch nicht die Tripelmutante, zum Wachstum bei 37°C in der Lage (Abb. 11 und Daten nicht gezeigt).
Einfluss des C-Terminus von Ctr9 auf die Sensitivität gegen 6-Azauracil Abb. 2.13 zeigt, dass auch die Stämme mit CTR9-Verkürzungsallelen mit Ausweitung der
C-terminalen Verkürzung von Ctr9 zunächst eine Zunahme der Sensitivität gegen 6-Azauracil erleiden, die mit dem Verlust der Cdc73-Bindungsdomäne im Bereich der TPR-Motive 9&10 korreliert (Kapitel 2.1.5). Die Ausweitung der Ctr9-Verkürzung auf die Bereiche vor AS 728 bis hin zu AS 459 führt jedoch wieder zu einer auffällig verringerten Empfindlichkeit gegen 6AU, die zuvor schon für die paf1Δ ctr9Δ Doppelmutante beobachtet wurde. Dieser Effekt lässt sich jedoch nicht auf einen Verlust der Paf1-Bindung zurückführen, da Paf1 auch zur Interaktion mit noch stärker verkürzten Ctr9-Fragmenten in der Lage ist (Kapitel 2.1.5). Die vollständige Deletion des CTR9-Leserahmens führt im Gegensatz dazu jedoch reproduzierbar wieder zu einer Hypersensitivität gegen 6AU und zeigt damit den gravierenden Elongationsdefekt der ctr9Δ Mutante an (Abb. 2.13 und Daten nicht gezeigt).

Ergebnisse
42
1 2 3 4 5 6 7 8 910 ccN C
TAP
TAPTAP
Ctr9 (WT)
Ctr9ΔAS805-1077
Ctr9ΔAS728-1077
Ctr9ΔAS459-1077
TAPCtr9ΔAS538-1077
WT
ctr9Δ
Ctr9ΔAS805-1077
Ctr9ΔAS728-1077
Ctr9ΔAS459-1077
Ctr9ΔAS538-1077
A)
B) -URACAS
100 mg.L-1 6AUYPD37°C
* *
* * Abb. 2.13 Der C-terminus von Ctr9 vermittelt und moduliert die Sensitivität gegen 6-Azauracil (A) Schematische Darstellung der Ctr9-Primärstruktur und der eingesetzten Ctr9-Verkürzungsvarianten (TPR-Motive mit signifikantem TPR-score sind als graduelle Füllung dargestellt). (B) Serielle Verdünnungen der Hefestämme CY4292, CY4293, CY4254 und CY4255, die graduell verkürzte Varianten von Ctr9 exprimieren, wurden im Vergleich zum Referenzstamm CY1615 (WT) und der ctr9Δ Deletionsmutante CY4627 auf Uracil-Mangelmedium (Medium auf Casaminoacid-Basis) mit oder ohne Zusatz von 100 mg·L-1 6-Azauracil aufgetropft und das Wachstum bei 30°C über einen Zeitraum von 6 Tagen verfolgt. Parallel dazu wurde dieselbe Verdünnungsreihe auf YEPD-Medium aufgetropft und zwei Tage bei 37°C inkubiert. Die Identität und Expression der jeweiligen Ctr9-Verkürzung wurde durch immunologischen Nachweis ihrer C-terminalen TAP-tag Fusion im Westernblot verifiziert (Daten nicht gezeigt). Die Expression des unverkürzten Ctr9-Proteins als C-terminale TAP-tag Fusion führt zu keiner beobachtbaren Sensitivität gegen 6-AU (Daten nicht gezeigt). Stern: Von der Verdünnungsreihe des Wildtypstammes und des ctr9Δ Deletionsstammes wurden aus Platzgründen jeweils nur die letzten vier Verdünnungen (WT) bzw. die ersten vier Verdünnungen (ctr9Δ) verwendet. Die Position der Verdünnungspunkte, bezogen auf die übrigen Verdünnungsreihen, wird dadurch jeweils um einen Schritt in Richtung des Pfeils versetzt.
Im Gegensatz zur Sensitivität gegen 6-Azauracil führt die Entfernung des C-Terminus von Ctr9 ab AS 805 bereits zu einem deutlichen Wachstumsdefekt bei 37°C und die Ausweitung der C-terminalen Verkürzung über AS 728 hinaus unterbindet das Wachstum schließlich vollständig (Abb. 2.13 B). Diese Temperatursensitivität kann aber im Gegensatz zur Sensitivität gegen 6AU nicht oder zumindest nicht im selben Ausmaß durch fortschreitende Verkürzung des C-Terminus kompensiert werden. Obwohl die untersuchten ctr9ΔAS538-1077 Mutanten bereits eine gemilderte Sensitivität gegen 6AU besitzen, ist keine der Mutanten in der Lage bei 37°C zu wachsen (Abb. 2.13 und Daten nicht gezeigt). Allein die ctr9ΔAS459-1077 Mutante scheint den Wachstumsdefekt bei 37°C ansatzweise ausgleichen zu können. Dieser Effekt tritt jedoch nicht auf, wenn ein vergleichbarer Hefestamm eingesetzt wird, der zwar das gleiche ctr9ΔAS459-1077 Allel aufweist, aber an Stelle der pep4::URA3 Deletion ein

Ergebnisse
43
telomergekoppeltes URA3 Auxotrophiemarkergen trägt (Kapitel 2.2.5, Abb. 2.15). Die vollständige Deletion des CTR9-Leserahmens führt in allen untersuchten Fällen ebenfalls zum vollständigen Verlust der Wachstumsfähigkeit bei 37°C.
2.2.5 Der PAF1-Komplex ist für das telomere Silencing notwendig Die Expression telomernaher Gene unterliegt einem positionsabhängigen Silencingeffekt,
der von der heterochromatischen Telomerregion ausgeht und die Expression dieser Gene unterdrückt (Gottschling et al., 1990). Der Ausfall von Faktoren, die das Silencing beeinflussen, ermöglicht dagegen die Genexpression auch nahe der telomeren Heterochromatingrenzen. Auch der Methylierungsstatus von Histon H3 beeinflusst die Silencingeffizienz an Telomeren.(Dhillon and Kamakaka, 2002). Entsprechend zeigen set1Δ und dot1Δ Mutanten erhebliche Defekte im telomeren Silencing, die aufgrund der funktionellen Abhängigkeit der beiden HMTs von der Mono-Ubiquitinierung an Histon H2B auch für rad6Δ und bre1Δ Mutanten beobachtet werden (Briggs et al., 2001; Krogan et al., 2002a; Sun and Allis, 2002; van Leeuwen et al., 2002). Da der Elongationsfaktor PAF1 am Anfang der Methylierungskaskade von Histon H3 (Kapitel 1.4) steht, wird verständlich, dass auch paf1Δ und rtf1Δ Mutanten beträchtliche Silencingdefekte aufweisen (Krogan et al., 2003a; Ng et al., 2003a).
Um die übrigen Komponenten des PAF1-Komplexes, Ctr9, Leo1 und Cdc73 sowie die hergestellten CTR9-Verkürzungsallele hinsichtlich eines auftretenden Silencingdefekts zu charakterisieren, wurde die Expression eines telomernahen URA3-Reportergens in den entsprechenden Mutanten des PAF1-Komplexes untersucht. 5-Fluororotsäure (5FOA) wirkt in Hefestämmen mit aktiver Orotidin-5’-phosphate Decarboxylase, die durch das URA3-Gen kodiert wird, toxisch auf die Zelle (Boeke et al., 1984). Das URA3-Reportergen wird in Stämmen mit funktionierendem Telomersilencing aufgrund der epigenetischen Variabilität der Heterochromatinbildung am Telomer nur in einem Teil der Zellpopulation abgelesen. Die übrigen Zellen können trotz der Anwesenheit des URA3-Reportergens auch in Gegenwart von 5FOA wachsen. Ein Silencingdefekt führt dagegen zum Verlust der 5FOA-Resistenz. Das Wachstum der untersuchten Stämme in Gegenwart von 5-Fluororotsäure kann daher als Indikator für auftretende Silencingdefekte genutzt werden (Craven and Petes, 2000).
Die für diese Versuchsreihe notwendigen Hefestämme wurden jeweils durch Kreuzung mit einem Hefestamm erhalten, der das oben beschriebene URA3-Reportergen als Insertion im Bereich des Telomers von Chromosom XII besitzt (TELVIIL::URA3; Craven and Petes, 2000). Die so hergestellten Hefestämme wurden als serielle Verdünnung einer Zellsuspension auf SCD-Medium (Material & Methoden) mit oder ohne den Zusatz von 1,0 mg·mL-1 5-Fluororotsäure aufgetropft und über einen Zeitraum sechs Tagen bei 30°C inkubiert.
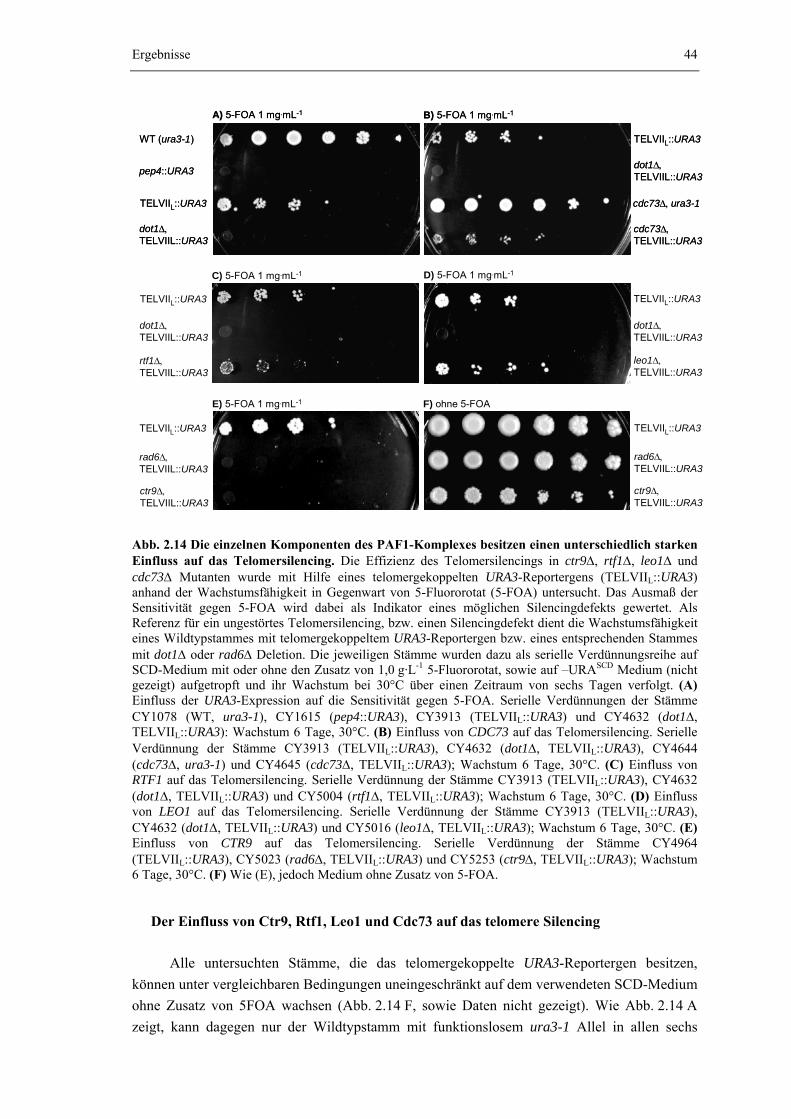
Ergebnisse
44
TELVIIL::URA3 TELVIIL::URA3
dot1Δ,TELVIIL::URA3
dot1Δ,TELVIIL::URA3
rtf1Δ,TELVIIL::URA3
leo1Δ,TELVIIL::URA3
WT (ura3-1)
pep4::URA3
TELVIIL::URA3
dot1Δ,TELVIIL::URA3
TELVIIL::URA3
dot1Δ,TELVIIL::URA3
cdc73Δ,TELVIIL::URA3
cdc73Δ, ura3-1
A) 5-FOA 1 mg.mL-1 B) 5-FOA 1 mg.mL-1
WT (ura3-1)
pep4::URA3
TELVIIL::URA3
dot1Δ,TELVIIL::URA3
TELVIIL::URA3
dot1Δ,TELVIIL::URA3
cdc73Δ,TELVIIL::URA3
cdc73Δ, ura3-1
A) 5-FOA 1 mg.mL-1 B) 5-FOA 1 mg.mL-1
C) 5-FOA 1 mg.mL-1 D) 5-FOA 1 mg.mL-1
TELVIIL::URA3
rad6Δ,TELVIIL::URA3
ctr9Δ,TELVIIL::URA3
E) 5-FOA 1 mg.mL-1 F) ohne 5-FOA
TELVIIL::URA3
rad6Δ,TELVIIL::URA3
ctr9Δ,TELVIIL::URA3
Abb. 2.14 Die einzelnen Komponenten des PAF1-Komplexes besitzen einen unterschiedlich starken Einfluss auf das Telomersilencing. Die Effizienz des Telomersilencings in ctr9Δ, rtf1Δ, leo1Δ und cdc73Δ Mutanten wurde mit Hilfe eines telomergekoppelten URA3-Reportergens (TELVIIL::URA3) anhand der Wachstumsfähigkeit in Gegenwart von 5-Fluororotat (5-FOA) untersucht. Das Ausmaß der Sensitivität gegen 5-FOA wird dabei als Indikator eines möglichen Silencingdefekts gewertet. Als Referenz für ein ungestörtes Telomersilencing, bzw. einen Silencingdefekt dient die Wachstumsfähigkeit eines Wildtypstammes mit telomergekoppeltem URA3-Reportergen bzw. eines entsprechenden Stammes mit dot1Δ oder rad6Δ Deletion. Die jeweiligen Stämme wurden dazu als serielle Verdünnungsreihe auf SCD-Medium mit oder ohne den Zusatz von 1,0 g·L-1 5-Fluororotat, sowie auf –URASCD Medium (nicht gezeigt) aufgetropft und ihr Wachstum bei 30°C über einen Zeitraum von sechs Tagen verfolgt. (A) Einfluss der URA3-Expression auf die Sensitivität gegen 5-FOA. Serielle Verdünnungen der Stämme CY1078 (WT, ura3-1), CY1615 (pep4::URA3), CY3913 (TELVIIL::URA3) und CY4632 (dot1Δ, TELVIIL::URA3): Wachstum 6 Tage, 30°C. (B) Einfluss von CDC73 auf das Telomersilencing. Serielle Verdünnung der Stämme CY3913 (TELVIIL::URA3), CY4632 (dot1Δ, TELVIIL::URA3), CY4644 (cdc73Δ, ura3-1) und CY4645 (cdc73Δ, TELVIIL::URA3); Wachstum 6 Tage, 30°C. (C) Einfluss von RTF1 auf das Telomersilencing. Serielle Verdünnung der Stämme CY3913 (TELVIIL::URA3), CY4632 (dot1Δ, TELVIIL::URA3) und CY5004 (rtf1Δ, TELVIIL::URA3); Wachstum 6 Tage, 30°C. (D) Einfluss von LEO1 auf das Telomersilencing. Serielle Verdünnung der Stämme CY3913 (TELVIIL::URA3), CY4632 (dot1Δ, TELVIIL::URA3) und CY5016 (leo1Δ, TELVIIL::URA3); Wachstum 6 Tage, 30°C. (E) Einfluss von CTR9 auf das Telomersilencing. Serielle Verdünnung der Stämme CY4964 (TELVIIL::URA3), CY5023 (rad6Δ, TELVIIL::URA3) und CY5253 (ctr9Δ, TELVIIL::URA3); Wachstum 6 Tage, 30°C. (F) Wie (E), jedoch Medium ohne Zusatz von 5-FOA.
Der Einfluss von Ctr9, Rtf1, Leo1 und Cdc73 auf das telomere Silencing Alle untersuchten Stämme, die das telomergekoppelte URA3-Reportergen besitzen,
können unter vergleichbaren Bedingungen uneingeschränkt auf dem verwendeten SCD-Medium ohne Zusatz von 5FOA wachsen (Abb. 2.14 F, sowie Daten nicht gezeigt). Wie Abb. 2.14 A zeigt, kann dagegen nur der Wildtypstamm mit funktionslosem ura3-1 Allel in allen sechs

Ergebnisse
45
Verdünnungsstufen auf 5FOA-haltigem Medium Kolonien bilden. In Gegenwart von 5FOA wird das Wachstum eines ansonsten isogenen Stammes durch die konstitutive Expression des URA3-Gens (pep4::URA3) jedoch vollständig unterbunden. Bereits der zum Wildtyp isogene Teststamm mit telomergekoppeltem URA3-Reportergen (TELVIIL::URA3) zeigt eine deutliche Wachstumsbeeinträchtigung durch 5FOA. Dies ist darauf zurückzuführen, dass auch in der Zellpopulation dieses Stammes bereits Zellen auftreten, welche die Orotidin-5’-phosphat-Decarboxylase (URA3) exprimieren (Abb. 2.14 A-E). Der bekannte Silencingdefekt der dot1Δ bzw. rad6Δ Mutante führt dagegen zur Transkription des telomergekoppelten URA3-Reportergens in allen Zellen und damit zur Sensitivität gegen 5-Fluororotat (Abb. 2.14 A-E).
Die cdc73Δ Mutante wird im Vergleich zu dem TELVIIL::URA3-Teststamm deutlich stärker durch 5FOA in ihrem Wachstum gehemmt. Wie Abb. 2.14 B zeigt, treten zwar auch bei der cdc73Δ Mutante, die das telomergekoppelte URA3-Reportergen besitzt, bis zur 4-ten Verdünnungsstufe Kolonien auf, die Kolonien sind jedoch deutlich kleiner. Die isogene cdc73Δ Mutante mit funktionslosem ura3-1 Allel kann dagegen bis zur 6-ten Verdünnungsstufe Kolonien ausbilden und wird in ihrem Wachstum nicht durch 5FOA beeinträchtigt. Das verringerte Ausmaß der Koloniebildung der cdc73Δ-Mutante mit dem Reportergen kann daher auf eine Beeinträchtigung der Silencingeffizienz durch die Deletion von CDC73 zurückgeführt werden. Mit derselben Argumentation lassen sich die Wachstumsdefekte aller untersuchten Deletionsmutanten auf eine Beeinträchtigung des telomeren Silencings zurückführen (Daten nicht gezeigt).
Die Deletion von RTF1 führt zu einem mit der cdc73Δ Mutante vergleichbarem Silencingphänotyp (Abb. 2.14 C). Die leo1Δ Mutante kann dagegen ähnlich gut wie der TELVIIL::URA3-Teststamm in Gegenwart von 5FOA wachsen, so dass die Deletion von LEO1 kaum zur Beeinträchtigung des Telomersilencings führt (Abb. 2.14 D). Im Gegensatz dazu werden durch die Deletion von CTR9 erhebliche Silencingdefekte verursacht, die das Wachstum der ctr9Δ Mutante in Gegenwart von 5FOA in allen getesteten Verdünnungsstufen verhindert (Abb. 2.14 E). Das Telomersilencing ist daher in einer ctr9Δ Mutante im selben Ausmaß beeinträchtigt wie in einer rad6Δ bzw. dot1Δ Mutante.
Der Einfluss der C-terminalen Region von Ctr9 auf das telomere Silencing Die Untersuchung der in Kapitel 2.1.5 beschriebenen CTR9-Verkürzungsallele in
Abb. 2.15 zeigt, dass ein der ctr9Δ-Mutante vergleichbarer Silencingdefekt bereits ab Verkürzung des C-Terminus von Ctr9 um AS 728-1077 auftritt (Abb. 2.15 B). Durch diese Verkürzung wird auch die vermutete Bindedomäne für Cdc73 entfernt (Kapitel 2.1.5). Der beobachtete Silencingdefekt des entsprechenden Hefestammes übersteigt jedoch die der cdc73Δ Mutante, so dass der Silencingdefekt nicht auf den Verlust der Cdc73-Bindung zurückgeführt werden kann. Alle getesteten Hefestämme mit noch stärker verkürztem Ctr9-Protein zeigen einen vergleichbaren Silencingdefekt in Gegenwart von 5FOA, können jedoch auf dem gleichen Medium ohne 5FOA problemlos wachsen (Abb. 2.15 B und C). Der C-Terminus von Ctr9 muss daher eine für das telomere Silencing relevante Funktion besitzen.
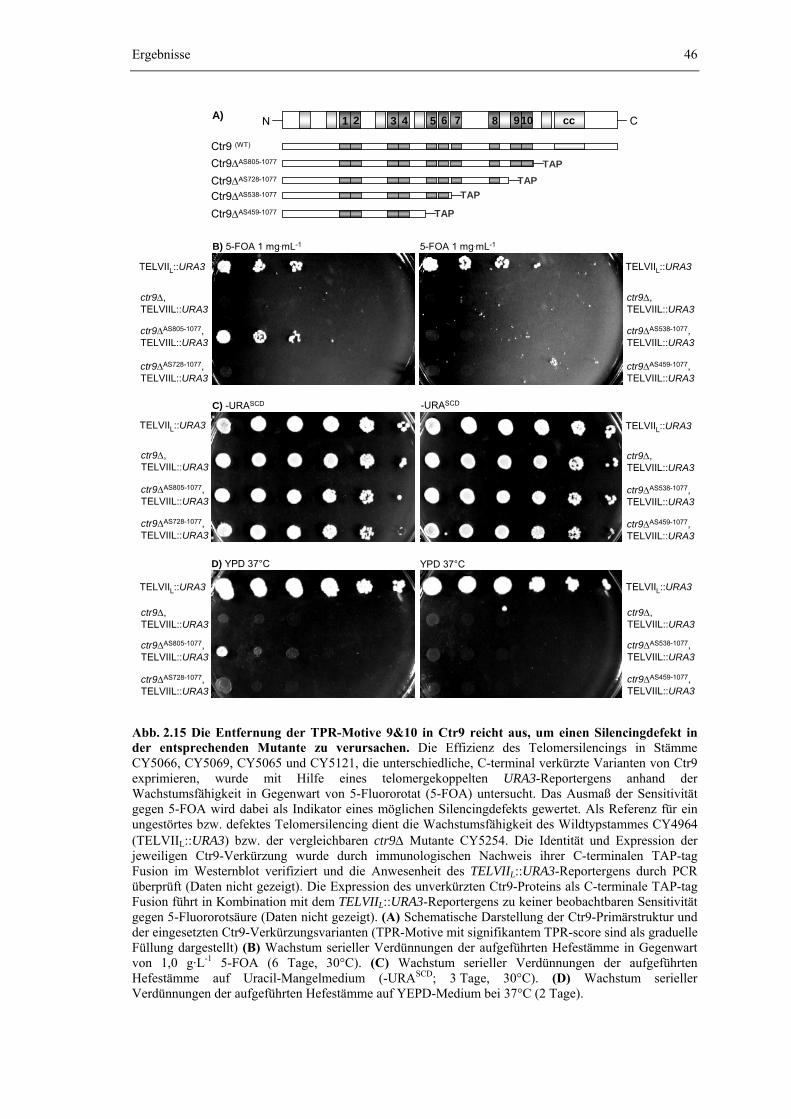
Ergebnisse
46
TELVIIL::URA3
ctr9Δ,TELVIIL::URA3
ctr9ΔAS805-1077,TELVIIL::URA3
ctr9ΔAS728-1077,TELVIIL::URA3
TELVIIL::URA3
ctr9Δ,TELVIIL::URA3
ctr9ΔAS805-1077,TELVIIL::URA3
ctr9ΔAS728-1077,TELVIIL::URA3
TELVIIL::URA3
ctr9Δ,TELVIIL::URA3
ctr9ΔAS805-1077,TELVIIL::URA3
ctr9ΔAS728-1077,TELVIIL::URA3
TELVIIL::URA3
ctr9Δ,TELVIIL::URA3
ctr9ΔAS538-1077,TELVIIL::URA3
ctr9ΔAS459-1077,TELVIIL::URA3
TELVIIL::URA3
ctr9Δ,TELVIIL::URA3
ctr9ΔAS538-1077,TELVIIL::URA3
ctr9ΔAS459-1077,TELVIIL::URA3
TELVIIL::URA3
ctr9Δ,TELVIIL::URA3
ctr9ΔAS538-1077,TELVIIL::URA3
ctr9ΔAS459-1077,TELVIIL::URA3
B) 5-FOA 1 mg.mL-1 5-FOA 1 mg.mL-1
C) -URASCD -URASCD
D) YPD 37°C YPD 37°C
1 2 3 4 5 6 7 8 910 ccN C
TAP
TAPTAP
Ctr9 (WT)
Ctr9ΔAS805-1077
Ctr9ΔAS728-1077
Ctr9ΔAS459-1077
TAPCtr9ΔAS538-1077
A)
Abb. 2.15 Die Entfernung der TPR-Motive 9&10 in Ctr9 reicht aus, um einen Silencingdefekt in der entsprechenden Mutante zu verursachen. Die Effizienz des Telomersilencings in Stämme CY5066, CY5069, CY5065 und CY5121, die unterschiedliche, C-terminal verkürzte Varianten von Ctr9 exprimieren, wurde mit Hilfe eines telomergekoppelten URA3-Reportergens anhand der Wachstumsfähigkeit in Gegenwart von 5-Fluororotat (5-FOA) untersucht. Das Ausmaß der Sensitivität gegen 5-FOA wird dabei als Indikator eines möglichen Silencingdefekts gewertet. Als Referenz für ein ungestörtes bzw. defektes Telomersilencing dient die Wachstumsfähigkeit des Wildtypstammes CY4964 (TELVIIL::URA3) bzw. der vergleichbaren ctr9Δ Mutante CY5254. Die Identität und Expression der jeweiligen Ctr9-Verkürzung wurde durch immunologischen Nachweis ihrer C-terminalen TAP-tag Fusion im Westernblot verifiziert und die Anwesenheit des TELVIIL::URA3-Reportergens durch PCR überprüft (Daten nicht gezeigt). Die Expression des unverkürzten Ctr9-Proteins als C-terminale TAP-tag Fusion führt in Kombination mit dem TELVIIL::URA3-Reportergens zu keiner beobachtbaren Sensitivität gegen 5-Fluororotsäure (Daten nicht gezeigt). (A) Schematische Darstellung der Ctr9-Primärstruktur und der eingesetzten Ctr9-Verkürzungsvarianten (TPR-Motive mit signifikantem TPR-score sind als graduelle Füllung dargestellt) (B) Wachstum serieller Verdünnungen der aufgeführten Hefestämme in Gegenwart von 1,0 g·L-1 5-FOA (6 Tage, 30°C). (C) Wachstum serieller Verdünnungen der aufgeführten Hefestämme auf Uracil-Mangelmedium (-URASCD; 3 Tage, 30°C). (D) Wachstum serieller Verdünnungen der aufgeführten Hefestämme auf YEPD-Medium bei 37°C (2 Tage).

Ergebnisse
47
Wie Abb. 2.15 D zeigt, entspricht die beobachtete Temperatursensitivität dieser Stämme den Befunden aus in Kapitel 2.2.4 (Abb. 2.13), mit der Ausnahme, dass die Ctr9-Verkürzung Ctr9ΔAS479-1077 in keiner der aufgetropften Zellverdünnungen anwächst. Für den TELVIIL::URA3-Teststamm wurde dagegen keine Temperatursensitivität beobachtet (Abb. 2.15 D).
2.2.6 Der Dot1-abhängige Methylierungsstatus von Histon H3 an Lysin 79 wird von den Komponenten des PAF1-Komplexes unterschiedlich beeinflusst Um zu untersuchen, ob die beobachteten Silencingdefekte von Mutanten des PAF1-
Komplexes mit einem Methylierungsdefekt von Histon H3 korrelieren, wurde im Folgenden exemplarisch die Dot1-abhängige Methylierung von Histon H3 an Lysin 79 in diesen Mutanten analysiert. Dazu wurde das Gesamtprotein der jeweiligen Hefestämme durch Fällung mit Trichloressigsäure (TCA) isoliert und die erhaltenen TCA-Extrakte nach elektrophoretische Trennung in einem 16,6% High Tris-Polyacrylamidgel mit einem Vernetzungsgrad von 5,1% immunologisch im Westernblot mit spezifischen Antikörpern gegen Di-Methyl-Histon H3Lys79 bzw. gegen Tri-Methyl-Histon H3Lys79 untersucht (Material und Methoden).
Wie die Westernblots in Abb. 2.16 A und B zeigen, lässt sich die Di- und Tri-Methylierung von Histon H3Lys79 in den Extrakten der Wildtypstämme (WT) jeweils durch eine Bande bei ca. 16 kDa spezifisch durch die verwendeten Antikörper nachweisen. Die dot1Δ Mutante weist dagegen eine vollständige Defizienz der Di- und Tri-Methylierung von Histon H3Lys79 auf, was durch das Verschwinden der jeweiligen Bande in den mit Antikörpern gegen Di-Methyl-Histon H3Lys79 (Abb. 2.16 A) bzw. Tri-Methyl-Histon H3Lys79 (Abb. 2.16 B) analysierten Westernblots deutlich wird. Auch die Deletion der Ubiquitin-Konjugase RAD6 verursacht einen Verslust der Di-Methylierung (Abb. 2.16 A und B, rad6Δ), was die funktionelle Abhängigkeit zwischen Dot1 und Rad6 demonstriert (Ng et al., 2002).
Die Analyse der Mutanten des PAF1-Komplexes zeigte, dass ausschließlich die Deletion von RTF1 zu einer Beeinträchtigung der Di-Methylierung von Histon H3Lys79 führt (Abb. 2.16 A, rtf1Δ). Dagegen konnte im selben Westernblot in der cdc73Δ, leo1Δ, ctr9Δ und auch in der paf1Δ Mutante die Di-Methylierung immunologisch detektiert werden. Die Untersuchung der Tri-Methylierung von Histon H3Lys79 in Abb. 2.16 B zeigt auch, dass diese wiederum durch die Deletion von RTF1 und zusätzlich durch die Deletion von PAF1 oder CTR9 Unterbunden wird. Die Deletion von LEO1 oder CDC73 führte dagegen zu keinem erkennbaren Einfluss auf die Tri-Methylierung von Histon H3Lys79. Der Befund, dass die Di-Methylierung von Histon H3Lys79 durch die Deletion von PAF1 nicht beeinflusst wird, steht im Widerspruch zu den Ergebnissen einer von dieser Arbeit unabhängigen Studie (Krogan et al., 2003a; vgl. Kapitel 3.2). Zur Verifizierung der Befunde wurden daher jeweils zwei voneinander unabhängige, zu den Laborstämmen W303 bzw. BY4741 (Material und Methoden) congene Stämme mit Deletionen in den Komponenten des PAF1-Komplexes untersucht. Wie Abb. 2.16 A und B zeigt, konnte dabei keine Abhängigkeit

Ergebnisse
48
Di-Methyl-Histon H3Lys79
Tri-Methyl-Histon H3Lys79
Di-Methyl-Histon H3Lys79
dot1
Δa)
WTa
)
paf1
Δa)
rtf1Δ
a)
leo1
Δa)
cdc7
3Δa)
rad6
Δa)
dot1
Δa)
WTa
)
paf1
Δa)
rtf1Δ
a)
leo1
Δa)
cdc7
3Δa)
rad6
Δa)
WTa
)
ctr9
Δb)
paf1
Δa)
paf1
Δb)
rtf1Δ
a)
rtf1Δ
b)
cdc7
3Δb)
dot1
Δa)
rad6
Δa)
WTa
)
ctr9
Δb)
paf1
Δa)
paf1
Δb)
rtf1Δ
a)
rtf1Δ
b)
cdc7
3Δb)
dot1
Δa)
rad6
Δa)
WTb
)
ctr9
Δb)
paf1
Δb)
rtf1Δ
b)
leo1
Δb)
dot1
Δa)
cdc7
3Δb)
*
*a) Stamm congen zu BY4741b) Stamm congen zu W303
A)
B)
Abb. 2.16 Nur Rtf1 ist für die Di-Methylierung von Histon H3Lys79 notwendig, die Tri-Methylierung ist dagegen zusätzlich von Paf1 und Ctr9 abhängig. Der Methylierungsstatus von Histon H3 an Lysin 79 in verschiedenen Mutanten mit Gendeletionen der Komponenten des PAF1-Komplexes wurde immunologisch im Westernblot mit spezifischen Antikörpern gegen Di-Methyl- bzw. Tri-Methyl-Histon H3Lys79 untersucht. TCA-Extrakte des Gesamtproteins der angegebenen Hefestämme wurden elektrophoretisch im Polyacrylamidgel (T = 16,6%, C = 5,1%) getrennt und zur Immunodetektion auf eine PVDF-Membran (Porenweite 0,2 µm) übertragen. (Material & Methoden). Soweit vorhanden wurde die gleiche Gendeletion jeweils in zwei unabhängigen Stämmen untersucht, deren genetischer Hintergrund entweder congen zum Wildtypstamm BY4741 (a) oder zum Wildtypstamm W303 (b) ist. BY4741 Derivate: CY4196 (WTa)), CY4070 (rtf1Δa)), CY4071 (leo1Δa)), CY4072 (paf1Δa)), CY4073 (cdc73Δa)), CY4074 (dot1Δa)), CY4077 (rad6Δa)); W303-Derivate: CY1615 (WTb)), CY1994 (paf1Δb)), CY2003 (cdc73Δb)), CY3916 (leo1Δb)), CY3919 (rtf1Δb)), CY5247 (ctr9Δb)). (A) Westernblot mit anti-Dimethyl-H3Lys79 Antikörpern (Upstate, Cat.-No.: 05-835). Der Stern markiert N-terminale Abbauprodukte von Histon H3 (Xiao et al., 2005). (B) Westernblot mit anti-Trimethyl-H3Lys79 Antikörpern (Abcam, AB2621). der Histonmethylierung vom genetischen Hintergrund der Stämme beobachtet werden. Insbesondere wurde für beide paf1Δ Mutanten keine Beeinträchtigung der Di-Methylierung von Histon H3Lys79 festgestellt.
Die beobachteten Silencingdefekte der Mutanten des PAF1-Komplexes(Kapitel 2.2.5) lassen sich daher nicht alleine durch die Beeinflussung der Dot1-abhängigen Methylierung von Histon H3Lys79 erklären, da das Telomersilencing in paf1Δ und ctr9Δ Mutanten im Vergleich zu rtf1Δ Mutanten deutlich stärker beeinträchtigt ist, aber nur Rtf1 sowohl die Di- als auch die Tri-Methylierung von Histon H3Lys79 beeinflusst. Bei den in paf1Δ und ctr9Δ Mutanten auftretenden Silencingdefekten handelt es sich daher um vermutlich um einen additiven Effekt, der durch den Ausfall mehrerer zellulärer Funktionen verursacht wird, die von dem Elongationsfaktor PAF1 abhängen.

Ergebnisse
49
2.2.7 Die Assoziation mit der RNA-Polymerase II kann nur von einem intakten PAF1-Komplex ausgebildet werden Um den Einfluss der einzelnen Bestandteile des PAF1-Komplexes auf die Assoziation
mit der RNA-Polymerase II zu untersuchen, wurde im Folgenden die Methode der Chromatin-Immunpräzipitation (ChIP; Dedon et al., 1991; Solomon and Varshavsky, 1985) eingesetzt, die erfolgreich zur Aufklärung einer Assoziation zwischen dem Elongationsfaktor PAF1 und der elongierenden RNA-Polymerase II eingesetzt wurde (Pokholok et al., 2002).
Das Prinzip der Chromatin-Immunpräzipitation Die Chromatin-Immunpräzipitation basiert auf der reversiblen Quervernetzung von
Proteinen und Nukleinsäuren mit Formaldehyd in vivo (Kuo and Allis, 1999; Solomon and Varshavsky, 1985; Tanaka et al., 1997). Auf diese Weise quervernetzte Protein-Protein- und Protein-DNA-Komplexe können nach Scherung der DNA-Stränge, z. B. durch Sonifizierung mittels Ultraschall, mit Hilfe spezifischer Antikörper oder geeigneter Affinitätstags gezielt präzipitiert und damit angereichert werden. Nach Lösung (Revertierung) der Quervernetzung lassen sich die mitgefällten DNA-Fragmente durch Extraktion mit Phenol/Chloroform isolieren und können als Template für eine PCR-Analyse eingesetzt werden. Die Assoziation eines untersuchten Proteins mit definierten Bereichen des Chromatins lässt sich dann durch ausgewählte Primerpaare ermitteln. Die durch Sonifizierung erzeugten DNA-Fragmente besitzen, abhängig von den gewählten Sonifizierungsbedingungen, eine mittlere Fragmentlänge von ca. 0,5 kb (Kuo and Allis, 1999; eigene Beobachtung, Daten nicht gezeigt). Die Auflösung, mit der sich zwei Sequenzabschnitte unabhängig voneinander untersuchen lassen, liegt daher ebenfalls in der Größenordnung zwischen 0,5 und 1 kb. Dies reicht aus, um die verschiedenen Phasen der Transkription voneinander abgrenzen zu können. Die RNA-Polymerase II selbst ist statistisch betrachtet über die gesamte Länge eines transkribierten Genbereichs mit dem Chromatin assoziiert. Die Bindung von Initiationsfaktoren bleibt dagegen auf den Promotor-Bereich des Gens beschränkt und vermittelt bei der Chromatin-Immunpräzipitation daher ausschließlich die Anreicherung promotornaher DNA-Fragmente. Proteine, die an der Elongation beteiligt sind, bleiben dagegen auch während dieser Transkriptionsphase mit der RNA-Polymerase II assoziiert und führen daher zusätzlich zur Präzipitation von DNA-Fragmenten, die für die 5’- nichttranslatierte Region (5’-UTR), den Leserahmen (ORF) und die 3’- nichttranslatierte Region (3’-UTR) des untersuchten Gens spezifisch sind. Faktoren der Termination und Polyadenylierung bleiben bis zum Ende eines Transkriptionszyklus mit der RNA-Polymerase II assoziiert und präzipitieren deswegen verstärkt auch Fragmente der 3’-UTR. Die Charakterisierung der jeweils untersuchten Proteine als Faktoren der Initiation, Elongation oder Termination ist daher mittels Chromatin-Immunpräzipitation anhand der Verteilung der co-präzipitierten DNA-Fragmente möglich.

Ergebnisse
50
Der PAF1-Komplex ist mit dem Promotor-, ORF- und 3’-UTR-Bereich proteinkodierender Gene assoziiert
Zur Chromatin-Immunpräzipitation wurde ein modifiziertes Protokoll nach Tanaka et al.
(1997) eingesetzt. Die Präzipitation erfolgte dabei entweder über die Adsorption von Proteinfusionen mit einem Myc6-Epitoptag an immobilisierte anti-Myc Antikörper (Maus, 9E11) oder durch die Bindung von Proteinfusionen mit dem TAP-tag an immobilisiertes IgG aus Kaninchen (IgG-Agarose) bzw. Pan-Maus-IgG Dynabeads (Material und Methoden).
Die Assoziation des PAF1-Komplexes mit den unterschiedlichen Genbereichen wurde zunächst am Beispiel des Cyclingens CLN2 untersucht. Die Positionen der bei der PCR-Analyse untersuchten Regionen des CLN2-Gens sind schematisch in Abb. 2.17 A wiedergegeben. Abb. 2.17 B zeigt die spezifische Amplifikation der getesteten Sequenzbereiche des CLN2-Gens aus den Chromatin-Immunpräzipitationen des PAF1-Komplexes über Ctr9-Myc6 bzw. Paf1-TAP Proteinfusionen mittels PCR (Material & Methoden).
Sowohl aus den durch Chromatin-Immunpräzipitation über Ctr9-Myc6 angereicherten DNA-Fragmenten, als auch aus den Eluaten der Paf1-TAP Immunpräzipitation lassen sich spezifische Sequenzbereiche des Promotors, des Leserahmens und der 3’-UTR durch PCR amplifizieren (Abb. 2.17 B). Die ChIP-Eluate der Negativkontrollen (ChIP aus dem Paf1-HA3 Stamm CY3099, der keine Protein-TAP Fusion exprimiert, mit 9E11-beladenen Dynabeads™ bzw. IgG-Agarose) führen unter den gleichen Bedingungen dagegen nicht zur Bildung eines PCR-Produkts (Abb. 2.17 B, Spuren N). Der Erfolg der Chromatin-Immunpräzipitation wird weder durch die Art des zur Fällung ausgenutzten Proteintags (Myc-tag bzw. TAP-tag) noch durch die verwendete Affinitätsmatrix (Dynabeads™ oder IgG-Agarose) beeinflusst und aus beiden Ansätzen (Ctr9-Myc6 bzw. Paf1-TAP) werden vergleichbar starke Signale der mit Ethidiumbromid gefärbten PCR-Produkte erhalten (Abb. 2.17 B, Spuren CE). Der PAF1-Komplex ist daher in vivo mit allen drei Bereichen des CLN2-Gens – Promotor, ORF und 3’-UTR – assoziiert, d. h. die Bindung an die RNA-Polymerase II bleibt auch während der Elongationsphase der Transkription bestehen. Dieser Befund wird durch die Untersuchung der PAF1-Assoziation mit dem Promotor, ORF und der 3’UTR des CLN3-Gens bestätigt (vgl. Abb. 2.25 und 2.27).
Die Ausweitung der PCR-Analyse auf zusätzliche Gene durch den Einsatz weiterer genspezifischer Primerpaare zeigte zudem, dass der PAF1-Komplex auch mit dem Leserahmen der Gene assoziiert ist, die für seine eigenen Bestandteile, die drei HMTs Dot1, Set1 und Set2, die Histon H2B spezifische Ubiquitin-Ligase Bre1 und weitere direkt an der Transkription beteiligte Proteine kodieren (Abb. 2.17 C). Diese Beobachtung ist übereinstimmend mit der Auffassung, dass der PAF1-Komplex während der aktiven Transkription aller proteinkodierender Gene mit der RNA-Polymerase II assoziiert ist (Mueller et al., 2004; Penheiter et al., 2005).

Ergebnisse
51
CLN2
Promotor
ORF
3'-UTR
R CE N R CE N
Paf1-TAP
Ctr9-Myc6
ATG STOPORF 3'-UTR
CLN21 1638-752 -408 715 1159 1866 2150
PromotorATG STOP
ORF 3'-UTR
CLN21 1638-752 -408 715 1159 1866 2150
Promotor
R …CE …
N …
Rohextrakt, sonifiziertChIP-EluatNegativkontrolle
A)
B)
R CE R R R R RCE CE CE CE CE
keinTAP-tag
keinTAP-tag
keinTAP-tag
Paf1-TAP
Paf1-TAP
Paf1-TAP
CLN1 PAF1 RTF1
SPT16 CKA1 BRE1
SET1 SET2 DOT1
C)
OR
F
Abb. 2.17 Der PAF1-Komplex ist mit der Promotor-, ORF- und 3’-UTR-Region von CLN2 assoziiert. Die Promotor-, ORF- und 3’UTR-Assoziation von Paf1 und Ctr9 wurde durch Chromatin-Immunpräzipitation (ChIP) untersucht. Ganzzellextrakte aus fixierten Zellen der angegebenen Stämme wurden nach Scherung der DNA durch Sonifizierung immunpräzipitiert und die mitgefällte DNA durch PCR analysiert. (A) Schematische Darstellung der durch PCR amplifizierten Regionen von CLN2 (Abbildung nicht maßstäblich). (B) Chromatin-Immunpräzipitation von Ctr9-Myc6 (CY3364) über an Dynabeads gekoppelte anti-Myc Antikörper (9E11) bzw. von Paf1-TAP (CY3408) durch Adsorption an eine IgG-Matrix (Kaninchen). Als Negativkontrolle wurde die ChIP aus dem Paf1-HA3 Stamm CY3099 jeweils in gleicher Weise durchgeführt. Von den ChIP-Eluaten bzw. dem sonifizierten Rohextrakt wurde je 1 µL als Template für die PCR-Analyse eingesetzt. Die Proteinassoziation mit der Promotor-, ORF- bzw. 3’-UTR-Region wurde am Beispiel des CLN2-Gens mit spezifischen Primerpaaren für die entsprechende Region untersucht (Promotorprimer: CK1165/CK1166, ORF-Primer CK1724/1725, 3’-UTR-Primer CK1738/CK1739). Der Erfolg der Immunpräzipitation wurde durch Westernblot Analyse und immunologischen Nachweis des Myc-tags bzw. des TAP-tags kontrolliert (nicht gezeigt). (C) Die aus der Chromatin-Immunpräzipitation über Paf1-TAP bzw. der Negativkontrolle (kein TAP-tag) erhaltenen und in (B) beschriebenen ChIP-Eluate wurden durch PCR mit spezifischen Primerpaaren (Material & Methoden) für den Leserahmen (ORF) der Gene CLN1, PAF1, RTF1, SPT16, CKA1, BRE1, SET1, SET2 und DOT1 untersucht. R: Rohextrakt, sonifiziert; CE: ChIP-Eluat
Prinzip der semi-quantitativen Auswertung von PCR-Amplifikaten Die Menge des amplifizierten PCR-Produkts hängt direkt von der Konzentration der
eingesetzten Template-DNA ab und kann daher auch zur Quantifizierung des Vorkommens einer bestimmten DNA-Sequenz in den durch Chromatin-Immunpräzipitation isolierten DNA-Fragmenten herangezogen werden. Damit wird gleichzeitig eine quantitative Beschreibung der Assoziation des untersuchten Proteins mit diesem Chromatinbereich möglich. Die exakte

Ergebnisse
52
quantitative Analyse durch PCR setzt jedoch die Messung der Amplifikationskinetik des entstehenden PCR-Produkts, z. B. durch „Real-time PCR“, voraus und stellt daher besondere apparative Anforderungen, die durch herkömmliche Thermocycler nicht erfüllt werden, da sie nur eine Endpunktbestimmung der amplifizierten DNA-Menge ermöglichen. Im begrenzteren Umfang ist aber auch diese Endpunktbestimmung zur relativen Quantifizierung einer bestimmten Sequenz aus den durch Chromatin-Immunpräzipitation angereicherten DNA-Fragmenten einsetzbar. Die relative Häufigkeit einer bestimmten Sequenz in einem ChIP-Eluat lässt sich z. B. über eine serielle Verdünnungsreihe der für die PCR eingesetzten Templatemenge ableiten. Die Signalintensität einer DNA-Bande ist in einem bestimmten Konzentrationsbereich proportional zur DNA-Menge. Damit können die erhaltene Intensitäten der DNA-Banden zu den Banden einer analogen Verdünnungsreihe aus den ChIP-Eluaten eines Referenzansatzes ins Verhältnis gesetzt werden, was letztlich auch eine Semi-Quantifizierung der Proteinassoziation mit einem bestimmten Chromatinbereich erlaubt.
Im Rahmen dieser Arbeit wurde diese semi-quantitative Methode zur Bewertung der Assoziationsfähigkeit des PAF1-Komplexes mit dem Leserahmen des CLN3-Gens angewendet. Die Signalintensität wurde als relatives Volumen durch Integration der digital dokumentierten DNA-Banden mit Hilfe der „BioCapt“-Software (Vilber Lourmat, V 12.3) bestimmt.
Die stabilisierenden Komponenten Paf1 und Ctr9 aber auch Rtf1 und Cdc73 sind für die Assoziation des PAF1-Komplexes mit der RNA-Polymerase II notwendig
Welche der fünf Komplexbestandteile letztlich die Interaktion mit der RNA-
Polymerase II vermitteln, wurde im Folgenden anhand der Assoziation des PAF1-Komplexes mit dem Leserahmen des Cyclingens CLN3 untersucht. Dazu wurde wiederum die Präzipitation von mit dem PAF1-Komplex quervernetzten Chromatinfragmenten durch ChIP untersucht, in diesem Fall jedoch aus Deletionsstämmen der einzelnen Komponenten des Komplexes bzw. aus Stämmen, die C-terminal verkürzten Ctr9-Varianten exprimieren (vgl. Kapitel 2.1.5). Ist die jeweils deletierte Komponente des PAF1-Komplexes an der Interaktion mit der RNA-Polymerase II während der Elongation beteiligt, sollte sich dies in einer reduzierten Assoziation mit dem CLN3-ORF widerspiegeln.
Um die Spezifität der Chromatin-Immunpräzipitationen sicher zu stellen, wurde bei jeder PCR-Analyse ein zusätzliches Primerpaar als interne Kontrolle eingesetzt. Dieses Primerpaar ist für eine ORF-freie Region auf Chromosom V spezifisch, mit der weder die RNA-Polymerase II noch der PAF1-Komplex assoziiert ist (Krogan et al., 2002b). Die Amplifikation dieses Sequenzbereichs kann daher zusätzlich - neben der Negativkontrolle durch einen Hefestamm, der keine Proteinfusion mit dem TAP-tag exprimiert - als Maß für die unspezifische Fällung co-präzipitierter DNA-Fragmente herangezogen werden.
Die in Abb. 2.18 A dargestellten PCR-Analysen zeigen, dass die isolierten DNA-Fragmente aus den Chromatin-Immunpräzipitationen der paf1Δ (Spur 4), ctr9Δ (Spur 3), rtf1Δ (Spur 6) und cdc73Δ (Spur 7) Mutanten im Vergleich zur Chromatin-Immunpräzipitationen aus dem Wildtyp (Spur 2 bzw. 5) zu einer deutlich verringerten Menge der amplifizierten ORF-Sequenz des CLN3-Gens führen. Die Assoziationsfähigkeit des PAF1-Komplexes mit dem
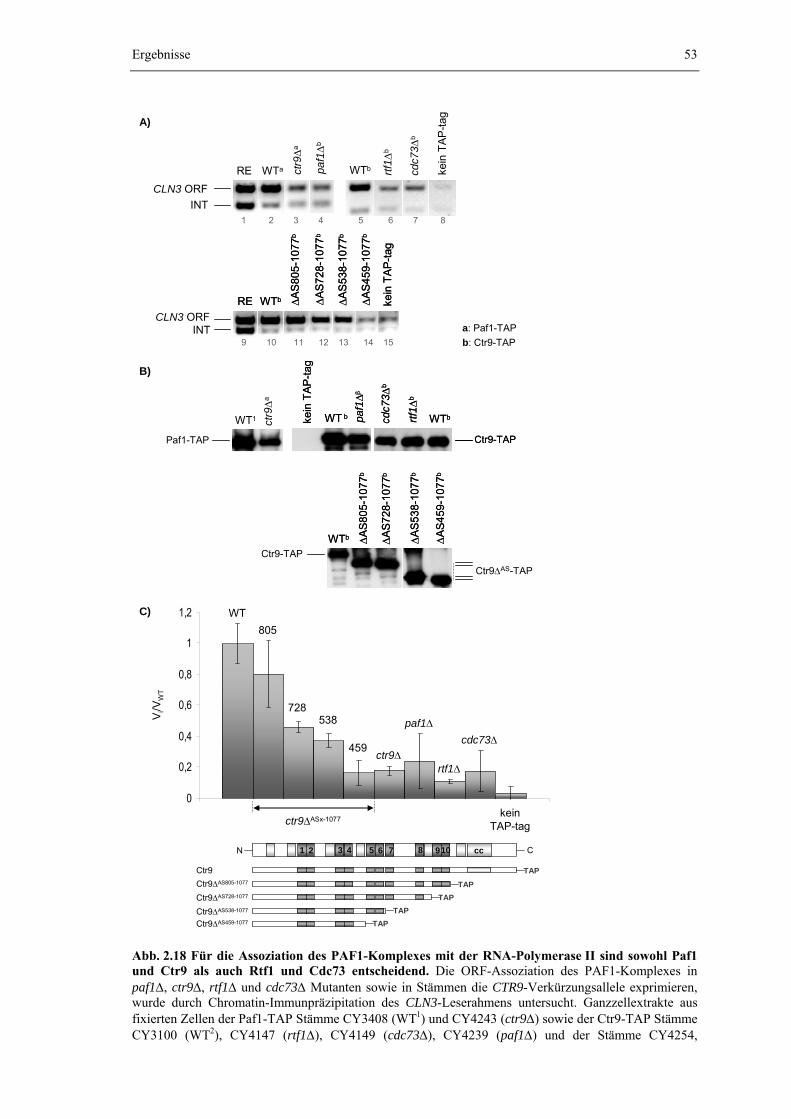
Ergebnisse
53
0
0,2
0,4
0,6
0,8
1
1,2
1
RE WTa ctr9
Δa
paf1
Δb
WTb rtf1Δ
b
cdc7
3Δb
kein
TAP
-tag
CLN3 ORFINT
Ctr9-TAP
kein
TAP
-tag
WT b WTbrtf1Δ
b
paf1
Δβ
cdc7
3Δb
Ctr9-TAP
kein
TAP
-tag
WT b WTbrtf1Δ
b
paf1
Δβ
cdc7
3Δb
Paf1-TAP
WT1 ctr9
Δa
ΔAS8
05-1
077b
ΔAS7
28-1
077b
ΔAS5
38-1
077b
ΔAS4
59-1
077b
WTb ΔAS8
05-1
077b
ΔAS7
28-1
077b
ΔAS5
38-1
077b
ΔAS4
59-1
077b
WTb
Ctr9-TAP
Ctr9ΔAS-TAP
RE WTb ΔAS
805-
1077
b
ΔAS7
28-1
077b
ΔAS
538-
1077
b
ΔAS4
59-1
077b
kein
TAP
-tag
RE WTb ΔAS
805-
1077
b
ΔAS7
28-1
077b
ΔAS
538-
1077
b
ΔAS4
59-1
077b
kein
TAP
-tag
CLN3 ORFINT
WT
ctr9Δ
805
728538
459
paf1Δ
ctr9ΔASx-1077
rtf1Δ
cdc73Δ
keinTAP-tag
V i/V
WT
C)
B)
A)
1 2 3 4 5 6 7 8 910 ccN C
TAPTAP
TAPTAP
TAPCtr9Ctr9ΔAS805-1077
Ctr9ΔAS728-1077
Ctr9ΔAS538-1077
Ctr9ΔAS459-1077
1 2 3 4 5 6 7 8
9 10 11 12 13 14 15a: Paf1-TAPb: Ctr9-TAP
Abb. 2.18 Für die Assoziation des PAF1-Komplexes mit der RNA-Polymerase II sind sowohl Paf1 und Ctr9 als auch Rtf1 und Cdc73 entscheidend. Die ORF-Assoziation des PAF1-Komplexes in paf1Δ, ctr9Δ, rtf1Δ und cdc73Δ Mutanten sowie in Stämmen die CTR9-Verkürzungsallele exprimieren, wurde durch Chromatin-Immunpräzipitation des CLN3-Leserahmens untersucht. Ganzzellextrakte aus fixierten Zellen der Paf1-TAP Stämme CY3408 (WT1) und CY4243 (ctr9Δ) sowie der Ctr9-TAP Stämme CY3100 (WT2), CY4147 (rtf1Δ), CY4149 (cdc73Δ), CY4239 (paf1Δ) und der Stämme CY4254,

Ergebnisse
54
CY4255, CY4292 und CY4293, die graduell verkürzte, C-terminale Ctr9-Varianten exprimieren, wurden nach Scherung der DNA durch Sonifizierung mit Hilfe einer IgG-Sepharosematrix (IgG aus Kaninchen) immunpräzipitiert und die mitgefällte DNA durch PCR analysiert. Als Negativkontrolle wurde der Wildtypstamm CY1615 in gleicher Weise behandelt (kein TAP-tag). Die Zellkulturen wurden nach der Fixierung in zwei bzw. drei getrennte Fällungsansätze aufgeteilt und unabhängig voneinander als parallele Doppelbestimmung behandelt (Dreifachansätze bei rtf1Δ, cdc73Δ und der Negativkontrolle). 5 µL der 1:25 verdünnten ChIP-Eluate wurden als Template für die PCR-Analyse eingesetzt und die Proteinassoziation mit der ORF-Region von CLN3 mit spezifischen Primerpaaren (CK1740/CK1741) untersucht. Die PCR wurde jeweils als Multiplex-PCR mit einem zusätzlichen Primerpaar (CK1744/1745) als interne Kontrolle (INT) durchgeführt, welches zur spezifischen Amplifikation einer ORF-freien Region auf Chromosom V führt. Als PCR-Referenz wurde der sonifizierte Rohextrakt (RE) der Negativkontrolle CY1615 als Template für die PCR eingesetzt. Alle PCR-Reaktionen wurden als Doppelbestimmung durchgeführt. (A) Die resultierenden PCR-Produkte einer Multiplex-PCR für die ORF-Region des CLN3 Gens bzw. die interne Kontrolle sind exemplarisch für alle Reaktionen gezeigt. Die PCR-Produkte wurden im 2%-Agarosegel elektrophoretisch getrennt und mit Ethidiumbromid gefärbt. (B) Der Erfolg der Immunpräzipitation wurde durch Westernblot und immunologischen Nachweis des TAP-tags mittels eines Peroxidase-Antiperoxidase Komplexes und anschließender ECL-Reaktion kontrolliert. (C) Graphische Darstellung der verringerten ORF-Assoziation des PAF1-Komplexes in unterschiedlichen Komplexmutanten. Die DNA-Produkte der PCR-Analysen wurden elektrophoretisch im 2% Agarosegel getrennt und durch Ethidiumbromid gefärbt. Die erhaltenen DNA-Banden wurden digital dokumentiert. Zur semi-quantitativen Auswertung wurde das Programm Photo-Capt (Version 12.3, Vilber Lourmat) eingesetzt, welches die Integration der aus der Photodokumentation ausgelesenen PCR-Signale ermöglicht. Pro Agarosegel wurden die Signale aus min. zwei Aufnahmen mit unterschiedlichen Belichtungszeiten ausgelesen. Fehlerbalken repräsentieren die Standardabweichung über alle Messungen eines Ansatzes. Die verkürzten Ctr9-Varianten sind zur Verdeutlichung schematisch analog zu Abb.: 2.6 A dargestellt. (A und B) a: Paf1-TAP; b: Ctr9-TAP
CLN3-Leserahmen wird daher sowohl durch die Deletion von PAF1 oder CTR9 als auch durch die Deletion von RTF1 oder CDC73 drastisch beeinträchtigt. Wie die semi-quantitative Auswertung verdeutlicht (Abb. 2.19 C), wird die Assoziation mit dem CLN3-Leserahmen in allen vier Deletionsmutanten – im Rahmen der Messgenauigkeit - um ca. 80% verringert. Die verwendeten Ctr9-TAP bzw. Paf1-TAP Fusionen lassen sich dagegen unabhängig von der jeweiligen Deletion einer weiteren Komponente des PAF1-Komplexes mit vergleichbarer Effizienz immunpräzipitieren (Abb. 2.19 B). Die verringerte Häufigkeit der untersuchten ORF-Sequenz in den durch ChIP isolierten Chromatinfragmenten kann daher nicht auf eine verringerte Proteinausbeute bei der Immunpräzipitation zurückgeführt werden und ist daher unmittelbar auf den Verlust der Assoziation mit der RNA-Polymerase II zurückführbar. Diese Befunde bestätigen zudem eine unabhängige Studie, in der die Abhängigkeit der Assoziation des PAF1-Komplexes mit dem TEF1-ORF von den vier Komponenten Paf1, Ctr9, Rtf1 und Cdc73 beschrieben wird (Mueller et al., 2004). Die Studie von Mueller et al. zeigt zudem anhand der Co-Immunpräzipitierbarkeit von Paf1 mit der Rpb3-Komponente der RNA-Polymerase II, dass die physikalische Interaktion zwischen Paf1 und der RNA-Polymerase II in der cdc73Δ und rtf1Δ Mutanten verloren geht, was die Auffassung untermauert, dass die ORF-Assoziation direkt die Interaktion mit der RNA-Polymerase II widerspiegelt. Abb. 2.18 A (Spuren 11-14) und Abb. 2.18 C zeigen, dass auch durch die stufenweise C-terminale Verkürzung des Ctr9-Proteins die Interaktionsfähigkeit des PAF1-Komplexes mit der RNA-Polymerase II in zunehmendem Maße beeinträchtigt wird und schließlich ab Verkürzung um den Proteinbereich zwischen Aminosäure 459-1077 das Ausmaß der

Ergebnisse
55
vollständigen CTR9-Deletion erreicht. Die verkürzten Ctr9-Varianten lassen sich aber wiederum mit vergleichbarer Ausbeute wie das unverkürzte Protein präzipitieren (Abb. 2.18 B).
Rtf1 und Cdc73 besitzen keinen nennenswerten Einfluss auf die Interaktion der jeweils verbleibenden vier Komplexbestandteile untereinander, ihre Deletion verhindert jedoch die Bindung des PAF1-Komplexes an die RNA-Polymerase II, so dass beide Proteine vermutlich für die spezifische Bindung des Elongationsfaktors an die Polymerase verantwortlich sind und diese ggf. kooperativ vermitteln. Dagegen wird die Gesamtstabilität des PAF1-Komplexes maßgeblich durch Ctr9 und Paf1 bestimmt (vgl. Kapitel 2.1.3), so dass der Verlust der Assoziation mit der RNA-Polymerase II in paf1Δ und ctr9Δ Mutanten vermutlich auf den Zerfall des PAF1-Komplexes zurückzuführen ist. Das Assoziationsverhalten der PAF1-Komplexe, die verkürzte Ctr9-Varianten beinhalten, lässt sich allerdings nicht nahtlos in ein Modell der kooperativen Bindung von Rtf1 und Cdc73 an die RNA-Polymerase II einfügen. Insbesondere wird die Assoziation mit der RNA-Polymerase II durch die Entfernung der Cdc73-Bindungsdomäne in Ctr9 nicht im gleichen Ausmaß wie durch die Deletion von CDC73 beeinträchtigt (Abb. 2.18 A, Spuren 7 und 12, sowie Abb. 2.18 C). Daher sollte im Folgenden überprüft werden, ob Cdc73 ggf. auch losgelöst aus dem PAF1-Komplexverbund mit der RNA-Polymerase II interagieren kann. Zu diesem Zweck wurde gezielt die Assoziation einer Cdc73-TAP Fusion mit dem CLN3-Leserahmen in einer ctr9Δ Mutante untersucht.
Cdc73 ist nur im Verbund des PAF1-Komplexes mit dem Leserahmen von CLN3 assoziiert
Die Auswertung der Chromatin-Immunpräzipitation über die Cdc73-TAP Proteinfusion
ist in Abb. 2.19 wiedergegeben. Wie ein Vergleich der Bandenintensitäten und die Semi-Quantifizierung der präzipitierten DNA-Fragmente verdeutlicht, wird die Assoziation von Cdc73 mit dem CLN3-Leserahmen in der ctr9Δ Mutante nahezu vollständig unterbunden; die Signalintensität der PCR-Produkte fällt in der ctr9Δ Mutante drastisch bis auf das Niveau der Negativkontrolle ab und erreicht nur noch ca. 10% des Wildtyps (Abb. 2.19 A). Cdc73 lässt sich aber auch aus der ctr9Δ Mutante mit vergleichbarer Ausbeute wie aus dem Wildtyp immunpräzipitieren (Abb. 2.19 B). Die Assoziation von Cdc73 mit dem CLN3-Leserahmen ist also, wie seine Bindung an den PAF1-Komplex, direkt von Ctr9 abhängig. Cdc73 scheint daher außerhalb des PAF1-Komplexverbundes nicht mit dem Leserahmen proteinkodierender Gene und damit sehr wahrscheinlich auch nicht mit der RNA-Polymerase II assoziiert zu sein. Damit bleibt das Modell einer kooperativen Vermittlung der Assoziation des PAF1-Komplexes mit der RNA-Polymerase II durch die beiden Komponenten Cdc73 und Rtf1 weiterhin im Bereich des Möglichen. Unterstützt wird diese Auffassung durch die Beobachtung, dass auch Rtf1 in Abwesenheit von Cdc73 nicht mehr mit dem Leserahmen des TEF1-Gens assoziiert ist und auch Paf1, Ctr9 und Leo1 benötigen für ihre ORF-Assoziation jeweils die gleichzeitige Anwesenheit von Cdc73 und Rtf1 (Mueller et al., 2004).
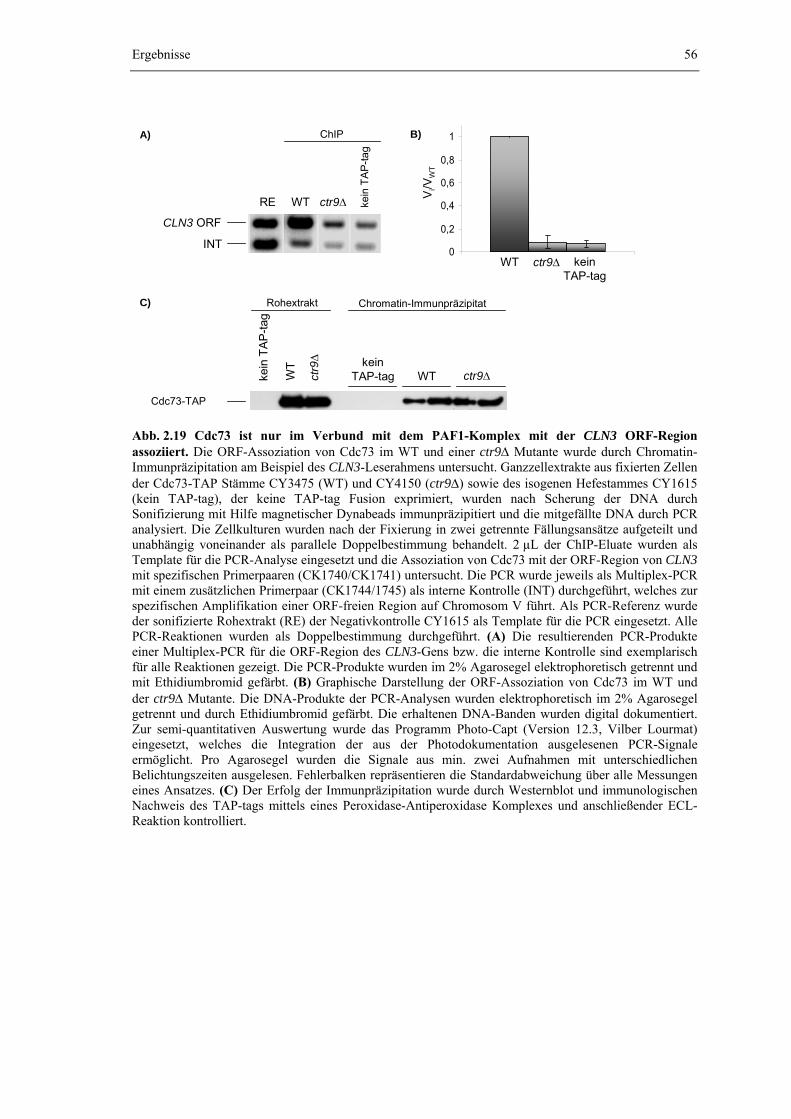
Ergebnisse
56
0
0,2
0,4
0,6
0,8
1
1
Cdc73-TAP
kein
TAP
-tag
WT
ctr9
Δ keinTAP-tag WT ctr9Δ
Rohextrakt Chromatin-Immunpräzipitat
CLN3 ORF
INT
RE WT
ctr9Δ
kein
TAP
-tag
ChIPA)
C)
WT keinTAP-tag
Vi/V
WT
B)
ctr9Δ
Abb. 2.19 Cdc73 ist nur im Verbund mit dem PAF1-Komplex mit der CLN3 ORF-Region assoziiert. Die ORF-Assoziation von Cdc73 im WT und einer ctr9Δ Mutante wurde durch Chromatin-Immunpräzipitation am Beispiel des CLN3-Leserahmens untersucht. Ganzzellextrakte aus fixierten Zellen der Cdc73-TAP Stämme CY3475 (WT) und CY4150 (ctr9Δ) sowie des isogenen Hefestammes CY1615 (kein TAP-tag), der keine TAP-tag Fusion exprimiert, wurden nach Scherung der DNA durch Sonifizierung mit Hilfe magnetischer Dynabeads immunpräzipitiert und die mitgefällte DNA durch PCR analysiert. Die Zellkulturen wurden nach der Fixierung in zwei getrennte Fällungsansätze aufgeteilt und unabhängig voneinander als parallele Doppelbestimmung behandelt. 2 µL der ChIP-Eluate wurden als Template für die PCR-Analyse eingesetzt und die Assoziation von Cdc73 mit der ORF-Region von CLN3 mit spezifischen Primerpaaren (CK1740/CK1741) untersucht. Die PCR wurde jeweils als Multiplex-PCR mit einem zusätzlichen Primerpaar (CK1744/1745) als interne Kontrolle (INT) durchgeführt, welches zur spezifischen Amplifikation einer ORF-freien Region auf Chromosom V führt. Als PCR-Referenz wurde der sonifizierte Rohextrakt (RE) der Negativkontrolle CY1615 als Template für die PCR eingesetzt. Alle PCR-Reaktionen wurden als Doppelbestimmung durchgeführt. (A) Die resultierenden PCR-Produkte einer Multiplex-PCR für die ORF-Region des CLN3-Gens bzw. die interne Kontrolle sind exemplarisch für alle Reaktionen gezeigt. Die PCR-Produkte wurden im 2% Agarosegel elektrophoretisch getrennt und mit Ethidiumbromid gefärbt. (B) Graphische Darstellung der ORF-Assoziation von Cdc73 im WT und der ctr9Δ Mutante. Die DNA-Produkte der PCR-Analysen wurden elektrophoretisch im 2% Agarosegel getrennt und durch Ethidiumbromid gefärbt. Die erhaltenen DNA-Banden wurden digital dokumentiert. Zur semi-quantitativen Auswertung wurde das Programm Photo-Capt (Version 12.3, Vilber Lourmat) eingesetzt, welches die Integration der aus der Photodokumentation ausgelesenen PCR-Signale ermöglicht. Pro Agarosegel wurden die Signale aus min. zwei Aufnahmen mit unterschiedlichen Belichtungszeiten ausgelesen. Fehlerbalken repräsentieren die Standardabweichung über alle Messungen eines Ansatzes. (C) Der Erfolg der Immunpräzipitation wurde durch Westernblot und immunologischen Nachweis des TAP-tags mittels eines Peroxidase-Antiperoxidase Komplexes und anschließender ECL-Reaktion kontrolliert.

Ergebnisse
57
2.3 Der Elongationsfaktor PAF1: Interaktionen Die massenspektrometrische Analyse des durch Tandem-Affinitätsreinigung isolierten
PAF1-Komplexes lieferte bereits erste Hinweise auf mögliche Interaktionspartner. Darunter wurden, als vielversprechendste Kandidaten im Rahmen dieser Arbeit, die Proteine Spt16 und Cka1 anhand ihres „Peptide-Mass-Fingerprints“ identifiziert (Kapitel 2.1.1). Spt16 bildet zusammen mit dem Protein Pob3 den Elongationsfaktor yFACT (Orphanides et al., 1999) und Cka1 ist eine der vier Untereinheiten der Casein-Kinase II (CKII; Chen-Wu et al., 1988). Durch den Befund der massenspektrometischen Analyse werden die Ergebnisse einer unabhängigen Studie bestätigt, die zudem die jeweils wechselseitigen Interaktionen zwischen PAF1-Komplex, Casein-Kinase II und dem Elongationsfaktor yFACT verdeutlicht (Krogan et al., 2002b). Gegenstand dieses Kapitels ist daher die genauere Untersuchung der Interaktion zwischen PAF1-Komplex, yFACT und Casein-Kinase II. Daneben wird die Interaktion zwischen PAF1- und SWR1-Komplex bzw. dem Bromodomainfactor 1 (Bdf1) eingehender beleuchtet, deren Verbindung ebenfalls über ihre gemeinsame Interaktion mit der Casein-Kinase II geschlossen und zudem durch die ausgeprägten genetischen Interaktionen zwischen den Bestandteilen des PAF1- und SWR1-Komplexes untermauert wird (Kapitel 2.2.3).
2.3.1 Interaktionen zwischen yFACT und dem PAF1-Komplex Die Proteine Spt16 und Pob3 bilden den heterodimeren Elongationsfaktor yFACT, der im
Zusammenspiel mit dem HMG-Box Protein Nhp6 den orthologen Komplex der Hefe zum humanen FACT-Komplex („facilitates chromatin transcription“) darstellt (Orphanides et al., 1999). Der FACT-Komplex wirkt sowohl in Hefe als auch im Menschen als Modulator der Chromatinstruktur und ermöglicht der RNA-Polymerase II die Passage durch die mit Nukleosomen besetzte DNA-Matrix (Belotserkovskaya et al., 2003; Orphanides et al., 1999; Singer and Johnston, 2004). Aufgrund seiner Aktivität als Histon-Chaperon bleibt die Funktion von yFACT nicht nur auf die Elongation beschränkt. Der Faktor übernimmt ebenfalls eine Rolle bei der Initiation der Transkription sowie der DNA-Replikation (Biswas et al., 2005b; O'Donnell et al., 2004; VanDemark et al., 2006). Die Komponenten des yFACT- bzw. des PAF1-Komplexes werden wechselseitig in den Präparationen des jeweils anderen Komplexes angereichert, interagieren also auf physikalischer Ebene direkt miteinander (Krogan et al., 2002b; eigene Beobachtung, vgl. Kapitel 2.1.1).
Um die physikalische Interaktion zwischen PAF1-Komplex und yFACT genauer zu untersuchen und insbesondere Rückschlüsse darauf zu erhalten, welche PAF1-Komponenten für diese Wechselwirkung notwendig sind, wurde auf die bereits in Kapitel 2.1.3 angewendete Strategie der Analyse des aus Deletionsmutanten isolierten Komplexes zurückgegriffen. Dazu wurde der PAF1-Komplex durch Tandem-Affinitätsreinigung aus paf1Δ, rtf1Δ, leo1Δ und cdc73Δ Mutanten isoliert, die neben der Fusion von Ctr9 bzw. Cdc73 mit dem TAP-tag zusätzlich eine Spt16-Variante mit C-terminal fusioniertem HA6-tag exprimieren. Aufgrund der physikalischen Wechselwirkung zwischen yFACT und PAF1-Komplex wird Spt16-HA6 in
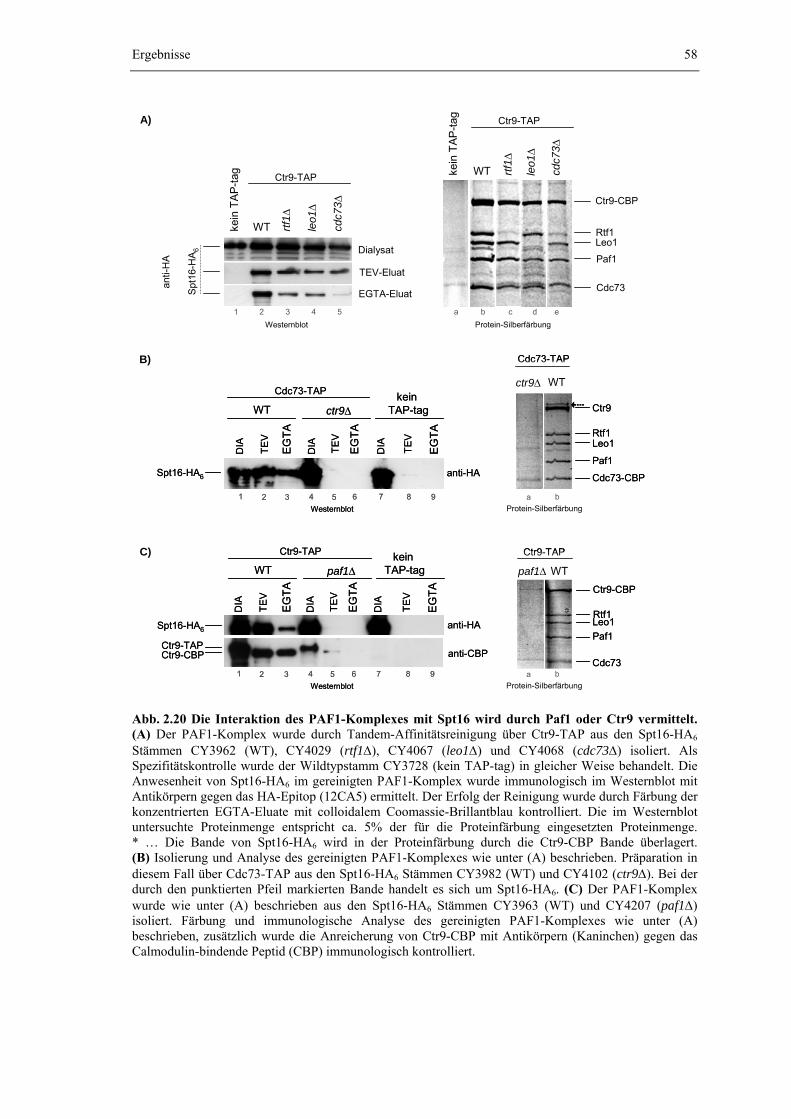
Ergebnisse
58
Dialysat
TEV-Eluat
EGTA-Eluat
kein
TAP
-tag
WT rtf1Δ
leo1
Δ
cdc7
3Δ Ctr9-CBP
Rtf1Leo1
Paf1
Cdc73Spt1
6-H
A 6
Ctr9-TAP kein
TAP
-tag
WT rtf1Δ
leo1
Δ
cdc7
3Δ
Ctr9-TAPA)
B)
C)
anti-
HA
1 2 3 4 5 a b c d eWesternblot Protein-Silberfärbung
DIA
DIA
DIA
TEV
TEV
TEV
EG
TA
EG
TA
EGTA
WT paf1Δkein
TAP-tag
Ctr9-TAP
Spt16-HA6 anti-HA
anti-CBPCtr9-TAPCtr9-CBP
Ctr9-CBP
WTpaf1Δ
Ctr9-TAP
Rtf1Leo1Paf1
Cdc731 2 3 4 5
Westernblot6 7 8 9 a
Protein-Silberfärbungb
DIA
DIA
DIA
TEV
TEV
TEV
EG
TA
EG
TA
EGTA
WT paf1Δkein
TAP-tag
Ctr9-TAP
Spt16-HA6 anti-HA
anti-CBPCtr9-TAPCtr9-CBP
Ctr9-CBP
WTpaf1Δ
Ctr9-TAP
Rtf1Leo1Paf1
Cdc731 2 3 4 5
Westernblot6 7 8 9 a
Protein-Silberfärbungb
Spt16-HA6
DIA
DIA
DIATE
V
TEV
TEV
EGTA
EGTA
EGTA
WT ctr9Δkein
TAP-tag
anti-HA
Cdc73-TAP
Ctr9
Rtf1Leo1
Paf1
Cdc73-CBP
ctr9Δ WT
Cdc73-TAP
1 2 3 4 5Westernblot
6 7 8 9 aProtein-Silberfärbung
b
Spt16-HA6
DIA
DIA
DIATE
V
TEV
TEV
EGTA
EGTA
EGTA
WT ctr9Δkein
TAP-tag
anti-HA
Cdc73-TAP
Ctr9
Rtf1Leo1
Paf1
Cdc73-CBP
ctr9Δ WT
Cdc73-TAP
1 2 3 4 5Westernblot
6 7 8 9 aProtein-Silberfärbung
b
Abb. 2.20 Die Interaktion des PAF1-Komplexes mit Spt16 wird durch Paf1 oder Ctr9 vermittelt. (A) Der PAF1-Komplex wurde durch Tandem-Affinitätsreinigung über Ctr9-TAP aus den Spt16-HA6 Stämmen CY3962 (WT), CY4029 (rtf1Δ), CY4067 (leo1Δ) und CY4068 (cdc73Δ) isoliert. Als Spezifitätskontrolle wurde der Wildtypstamm CY3728 (kein TAP-tag) in gleicher Weise behandelt. Die Anwesenheit von Spt16-HA6 im gereinigten PAF1-Komplex wurde immunologisch im Westernblot mit Antikörpern gegen das HA-Epitop (12CA5) ermittelt. Der Erfolg der Reinigung wurde durch Färbung der konzentrierten EGTA-Eluate mit colloidalem Coomassie-Brillantblau kontrolliert. Die im Westernblot untersuchte Proteinmenge entspricht ca. 5% der für die Proteinfärbung eingesetzten Proteinmenge. * … Die Bande von Spt16-HA6 wird in der Proteinfärbung durch die Ctr9-CBP Bande überlagert. (B) Isolierung und Analyse des gereinigten PAF1-Komplexes wie unter (A) beschrieben. Präparation in diesem Fall über Cdc73-TAP aus den Spt16-HA6 Stämmen CY3982 (WT) und CY4102 (ctr9Δ). Bei der durch den punktierten Pfeil markierten Bande handelt es sich um Spt16-HA6. (C) Der PAF1-Komplex wurde wie unter (A) beschrieben aus den Spt16-HA6 Stämmen CY3963 (WT) und CY4207 (paf1Δ) isoliert. Färbung und immunologische Analyse des gereinigten PAF1-Komplexes wie unter (A) beschrieben, zusätzlich wurde die Anreicherung von Ctr9-CBP mit Antikörpern (Kaninchen) gegen das Calmodulin-bindende Peptid (CBP) immunologisch kontrolliert.

Ergebnisse
59
Präparationen des intakten PAF1-Komplexes angereichert und kann über das HA6-tag immunologisch in den Eluaten der beiden Reinigungsstufen (TEV-Eluat bzw. EGTA-Eluat) nachgewiesen werden. Welche Komponente des PAF1-Komplexes mit yFACT interagiert, kann daher durch den Verlust der Anreicherung von Spt16-HA6 in dem aus der entsprechenden Mutante isolierten PAF1-Komplex ermittelt werden. Die Ergebnisse dieser Versuchsreihen sind in Abb. 2.20 wiedergegeben.
Zunächst wurde die Auswirkung der Deletion von RTF1, LEO1 und CDC73 auf die Assoziation von Spt16-HA6 mit dem PAF1-Komplex untersucht (Abb. 2.20 A). Sowohl im Eluat der ersten Reinigungsstufe (TEV-Eluat) als auch im Eluat der zweiten Reinigungsstufe (EGTA-Eluat) lässt sich Spt16-HA6, wie auch im dialysierten Zellextrakt (Dialysat), immunologisch eindeutig nachweisen (Spur 2). Spt16 wird also mit dem aus dem Wildtypstamm durch Tandem-Affinitätsreinigung isolierten PAF1-Komplex angereichert. In den Eluaten der Kontrollpräparation aus einem Spt16-HA6 Stamm ohne Ctr9-TAP Fusion konnte dagegen keine Bande detektiert werden (Spur 1). Auch die Reinigung des PAF1-Komplexes aus der rft1Δ, leo1Δ bzw. cdc73Δ Mutante führt zur Anreicherung von Spt16 (Spur 3-5). In allen drei Fällen lässt sich Spt16-HA6 eindeutig in TEV-Eluat und EGTA-Eluat nachweisen, im Vergleich zum Wildtyp jedoch in verminderter Menge und im EGTA-Eluat der Präparation aus der cdc73Δ Mutante nur noch in Spuren. Dennoch wird die Interaktion zwischen yFACT – repräsentiert durch Spt16 - und PAF1-Komplex durch die Deletion von RTF1, LEO1 oder CDC73 nicht unterbunden, so dass keine der drei Komponenten unmittelbar und ausschließlich die Assoziation zwischen yFACT und PAF1-Komplex vermittelt. Die Proteinfärbung der gereinigten Komplexe zeigt, dass die Ausbeute des isolierten Komplexes bereits in den drei Mutanten reduziert wird (Spur b-e), so dass die verringerte Anreicherung von Spt16-HA6 zum Teil auf diesen Effekt zurückzuführen ist. Die Bandenstärke von Spt16-HA6 im Westernblot verringert sich von der ersten zur zweiten Reinigungsstufe (TEV- bzw. EGTA-Eluat) in der rtf1Δ, der leo1Δ und vor allem der cdc73Δ Mutante und dies deutlich stärker (Spur 3-5) als es bei der Präparation aus dem Wildtyp (Spur 2) der Fall ist. Dies könnte darauf hindeuten, dass Rtf1, Leo1 oder Cdc73 zwar nicht unmittelbar die Assoziation von yFACT mit dem PAF1-Komplex vermitteln, aber dennoch einen Einfluss auf die Stabilität der Assoziation besitzen.
Durch die Deletion von CDC73 wird die Interaktion zwischen yFACT und PAF1-Komplex also zumindest beeinträchtigt. Um zu überprüfen, ob dieser Effekt auf eine direkte Interaktion zwischen Cdc73 und Spt16 zurückgeführt werden kann, wurde die Anreicherung von Spt16-HA6 mit einer aus einer ctr9Δ Mutante isolierten Cdc73-TAP Fusion in gleicher Weise untersucht (Abb. 2.20 B), wobei Cdc73 aufgrund der Deletion von CTR9 nicht mehr mit dem PAF1-Komplex assoziiert ist (vgl. Kapitel 2.1.3). Wie der Westernblot in Abb. 2.20 B zeigt, wurde Spt16-HA6 auch bei Präparation des PAF1-Komplexes über die Cdc73-TAP Fusion aus dem Wildtyp angereichert (Spur 2 und 3). Dagegen ließ sich Spt16-HA6 in der Präparation aus der ctr9Δ Mutante in keiner Reinigungsfraktion von Cdc73-TAP immunologisch nachweisen (Spur 5 und 6). Dies spricht gegen eine direkte Interaktion von Spt16 mit Cdc73. Die zugehörige Proteinfärbung zeigt jedoch, dass sich die Ccd73-TAP Fusion selbst nur mit minimaler Ausbeute aus einer ctr9Δ Mutante isolieren ließ (Spur a;), so dass Spt16 möglicherweise – wenn überhaupt – unterhalb der immunologischen Nachweisgrenze angereichert wird.

Ergebnisse
60
Im Gegensatz zur Situation in rtf1Δ, leo1Δ und cdc73Δ Mutanten kann Spt16-HA6 nicht durch die Isolierung der Ctr9-TAP Fusion aus einer paf1Δ Mutante angereichert werden. Wie der Westernblot mit anti-HA Antikörpern in Abb. 2.20 C zeigt, war Spt16-HA6 immunologisch weder im TEV-Eluat der ersten Reinigungsstufe, noch im EGTA-Eluat der zweiten Reinigungsstufe nachweisbar (Spur 5 und 6). Allerdings muss dabei berücksichtigt werden, dass bereits die Isolierung der Ctr9-TAP Fusion aus der paf1Δ Mutante, ähnlich wie im Falle der Cdc73-TAP Präparation aus der ctr9Δ Mutante, nur in Spuren gelang, wie die schwache Ctr9-CBP Bande sowohl in der Proteinfärbung (Spur a) als auch im Westernblot mit Antikörpern gegen das CBP (Spur 5 und 6) zeigt. Ursächlich hierfür ist, dass die Deletion von PAF1, sowie die Deletion von CTR9, die Dissoziation des PAF1-Komplexes (Kapitel 2.3.1) bewirkt. Da die Dissoziation des Komplexes natürlich auch die Interaktion mit yFACT unterbinden würde, kann keine Entscheidung darüber getroffen werden, ob Paf1 als Bindungspartner für yFACT fungiert. Rtf1, Leo1 und Cdc73 können jedoch als Bindungspartner für Spt16 ausgeschlossen werden. Daher muss die Assoziation von yFACT durch Paf1 und/oder durch Ctr9 vermittelt werden.
2.3.2 Interaktionen zwischen Casein-Kinase II und dem PAF1-Komplex Casein-Kinase II (CKII) ist eine in Eukaryoten evolutionär hoch konservierte
Serin/Threonin-Kinase. Das Holoenzym besteht aus den zwei katalytischen α-Untereinheiten Cka1 (α) und Cka2 (α’) sowie den beiden regulatorischen β-Untereinheiten Ckb1 (β) und Ckb2 (β’), die αα'ββ', α2ββ' und α'2ββ' Tetrameren ausbilden (Bidwai et al., 1994; Kubinski et al., 2006). CKII ist essentiell für die Lebensfähigkeit der Zelle. Zwar kann der einzelne Ausfall von Cka1 oder Cka2 toleriert werden, der gleichzeitige Funktionsverlust beider α-Untereinheiten ist jedoch letal (Padmanabha et al., 1990). Die beiden Isoformen der α-Untereinheiten vermitteln auch die Substratspezifität der Kinase (Ackermann et al., 2001; Berkey and Carlson, 2006).
Das Wirkungsspektrum der Kinase ist breit gefächert, die CKII-abhängige Proteinphosphorylierung beeinflusst als regulierendes Signal dabei unter anderem alle Phasen des Transkriptionszyklusses sowie die Modulation der Chromatinstruktur. Beispielsweise wird die Initiation bzw. Reinitiation der Transkription unter anderem durch die abwechselnde CKII-abhängige Phosphorylierung des generellen Transkriptionsfaktors TFIIF und der assoziierten CTD-Phosphatase Fcp1 kontrolliert (Abbott et al., 2005; Friedl et al., 2003; Palancade et al., 2002). Casein-Kinase II ist zudem sowohl mit dem Elongationsfaktor PAF1 als auch mit yFACT assoziiert (Krogan et al., 2002b; eigene Beobachtung, Kapitel 2.1.1), was die Frage nach einer möglichen funktionellen Interaktion dieser drei an der Transkription beteiligten Faktoren aufwirft.
Casein-Kinase II wird mit dem isolierten PAF1-Komplex angereichert und auch die isolierte Casein-Kinase II enthält Spt16 sowie Untereinheiten des PAF1-Komplexes
Um die wechselseitigen Interaktionen zwischen Casein-Kinase II und den beiden
Elongationsfaktoren PAF1 und yFACT zu verifizieren, wurde die Casein-Kinase II durch Tandem-Affinitätsreinigung aus Hefestämmen isoliert, die Varianten von Paf1 oder Spt16 als

Ergebnisse
61
C-terminale Fusion mit einem HA-tag exprimieren. Anschließend wurde immunologisch überprüft, ob auch die Komponenten des PAF1-Komplexes bzw. des Elongationsfaktors yFACT mit der Casein-Kinase II angereichert werden. Parallel dazu wurde der PAF1-Komplex aus einem Hefestamm gereinigt, der eine C-terminale HA-tag Fusion der CKII α-Untereinheit Cka1 exprimiert.
Die Färbung der isolierten Proteinkomplexe in Abb. 2.21 A zeigt, dass sowohl der PAF1-Komplex als auch die Casein-Kinase II mit guter Ausbeute durch die Tandem-Affinitätsreinigung isoliert werden konnten. In der Silberfärbung der über Cka1-TAP gereinigten Casein-Kinase II treten vier Banden auf (Spur 3 und 4), die aufgrund ihrer Laufstrecke im Polyacrylamidgel den vier Komponenten der Kinase zugeordnet werden können. Auffällig ist, dass Cka1 – als Fusion mit dem CBP des TAP-tags – und Ckb1 offenbar stärker angereichert wurden als ihre entsprechenden Isoformen Cka2 und Ckb2. Eine mögliche Ursache hierfür könnte das Vorkommen unterschiedlicher Zusammensetzungen der Casein-Kinase II in der Zelle sein. Da die Reinigung über die katalytische α-Untereinheit als Cka1-TAP erfolgt, werden ausschließlich CKII-Komplexe mit αα'ββ' und α2ββ' Konstitution bzw. die freie α-Untereinheit Cka1 angereichert. Komplexe mit α'2ββ' Konstitution oder die freie α'-Untereinheit Cka2 können dagegen nicht isoliert werden, so dass Cka2 (α') in der Präparation ggf. unterrepräsentiert wird. Unter der Voraussetzung, dass die regulatorischen Untereinheiten immer mit ββ' Konstitution in den aktiven CKII-Komplexen vorliegen (Kubinski et al., 2006), könnte dieser Befund jedoch auch auf eine teilweise Dissoziation der Casein-Kinase II oder zumindest der β'-Komponente Ckb2 während der Reinigung zurückzuführen sein.
Der Westernblot in Abb. 2.21 B zeigt, dass die katalytische α-Untereinheit Cka1 der Casein-Kinase II immunologisch klar in den Eluaten des über Paf1-TAP isolierten PAF1-Komplexes nachgewiesen werden konnte. Sowohl im Eluat der ersten Reinigungsstufe (TEV), als auch im Eluat der zweiten Reinigungsstufe (EGTA) tritt eine eindeutige, für die Cka1-HA6 Fusion spezifische Bande auf. Der Kontrollansatz ohne TAP-tag führt dagegen nicht zu einer Anreicherung von Cka1-HA6. Die Ergebnisse der massenspektrometrischen Analyse werden damit bestätigt. Casein-Kinase II ist mit dem gereinigten PAF1-Komplex assoziiert. Die Menge ist jedoch gering, da die Komponenten von CKII nicht durch Silberfärbung des gereinigten PAF1-Komplexes sichtbar werden (Abb. 2.21 A, Spur 1). Daher ist es unwahrscheinlich, dass die Kinase in vivo eine stöchiometrische Komponente des PAF1-Komplexes ist und die beobachtete Interaktion ist vermutlich zeitlich oder räumlich beschränkt bzw. wird indirekt über weitere Proteine vermittelt.
Die immunologische Untersuchung der Casein-Kinase II Präparationen aus den beiden Stämmen, die HA-tag Fusionen von Paf1 bzw. Spt16 exprimieren, zeigen jedoch eindeutig die Anreicherung von Paf1 und Spt16 mit der isolierten Casein-Kinase II (Abb. 2.21 C bzw. D). In beiden Fällen zeigt der Westernblot eindeutige und spezifische Signale für Paf1-HA3 (Abb. 2.21 C) bzw. für Spt16-HA6 (Abb. 2.21 D) sowohl im Eluat der ersten (TEV) als auch im Eluat der zweiten Reinigungsstufe (EGTA) der CKII Tandem-Affinitätsreinigung. Die Negativkontrolle aus Präparationen von Paf1-HA3 Stämmen bzw. Spt16-HA6 Stämmen, die keine TAP-tag Proteinfusion exprimieren, führte dagegen nicht zu einer Anreicherung der jeweiligen HA-epitopmarkierten Proteine. Einzig im TEV-Eluat des Spt16-HA6 Stammes tritt
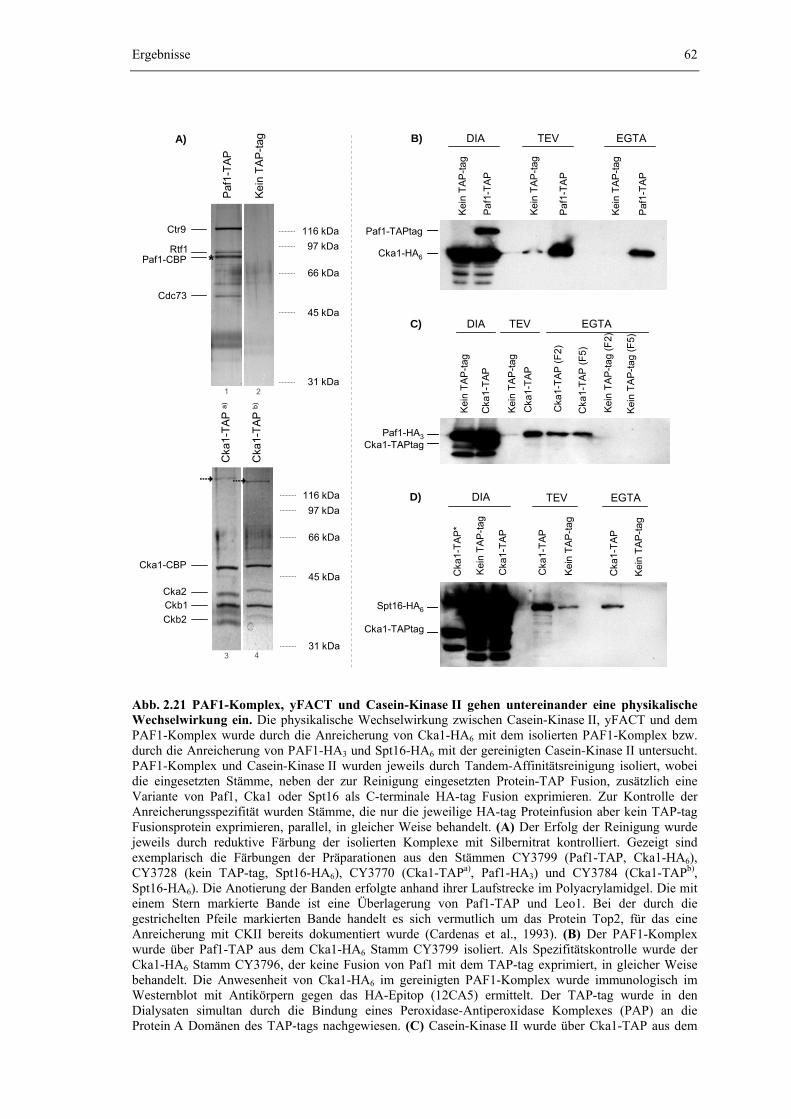
Ergebnisse
62
Paf1-TAPtag
Paf1-HA3Cka1-TAPtag
Spt16-HA6
Cka1-HA6
Kein
TAP
-tag
Paf1
-TA
P
Kein
TAP
-tag
Kein
TAP
-tag
Paf
1-TA
P
Paf1
-TA
P
DIA TEV EGTA
Kein
TA
P-ta
g
Kein
TA
P-ta
g
Cka
1-TA
P
Cka
1-TA
P
Cka
1-TA
P (F
2)
Kein
TA
P-ta
g (F
2)
Kein
TA
P-ta
g (F
5)
DIA TEV EGTA
Cka
1-TA
P (F
5)
Cka1-TAPtag
Kein
TA
P-ta
g
Cka
1-TA
P*
Cka
1-TA
P
Cka
1-TA
P
Cka
1-TA
P
Kein
TAP
-tag
Kein
TAP
-tag
DIA TEV EGTA
B)
C)
D)
Cka1-CBP
Cka2Ckb1Ckb2
116 kDa97 kDa
66 kDa
45 kDa
31 kDa
Ctr9
Rtf1Paf1-CBP
Cdc73
Paf
1-TA
P
Kein
TA
P-ta
g
116 kDa97 kDa
66 kDa
45 kDa
31 kDa
Cka
1-TA
P a)
Cka
1-TA
P b)
A)
*
1 2
3 4
Abb. 2.21 PAF1-Komplex, yFACT und Casein-Kinase II gehen untereinander eine physikalische Wechselwirkung ein. Die physikalische Wechselwirkung zwischen Casein-Kinase II, yFACT und dem PAF1-Komplex wurde durch die Anreicherung von Cka1-HA6 mit dem isolierten PAF1-Komplex bzw. durch die Anreicherung von PAF1-HA3 und Spt16-HA6 mit der gereinigten Casein-Kinase II untersucht. PAF1-Komplex und Casein-Kinase II wurden jeweils durch Tandem-Affinitätsreinigung isoliert, wobei die eingesetzten Stämme, neben der zur Reinigung eingesetzten Protein-TAP Fusion, zusätzlich eine Variante von Paf1, Cka1 oder Spt16 als C-terminale HA-tag Fusion exprimieren. Zur Kontrolle der Anreicherungsspezifität wurden Stämme, die nur die jeweilige HA-tag Proteinfusion aber kein TAP-tag Fusionsprotein exprimieren, parallel, in gleicher Weise behandelt. (A) Der Erfolg der Reinigung wurde jeweils durch reduktive Färbung der isolierten Komplexe mit Silbernitrat kontrolliert. Gezeigt sind exemplarisch die Färbungen der Präparationen aus den Stämmen CY3799 (Paf1-TAP, Cka1-HA6), CY3728 (kein TAP-tag, Spt16-HA6), CY3770 (Cka1-TAPa), Paf1-HA3) und CY3784 (Cka1-TAPb), Spt16-HA6). Die Anotierung der Banden erfolgte anhand ihrer Laufstrecke im Polyacrylamidgel. Die mit einem Stern markierte Bande ist eine Überlagerung von Paf1-TAP und Leo1. Bei der durch die gestrichelten Pfeile markierten Bande handelt es sich vermutlich um das Protein Top2, für das eine Anreicherung mit CKII bereits dokumentiert wurde (Cardenas et al., 1993). (B) Der PAF1-Komplex wurde über Paf1-TAP aus dem Cka1-HA6 Stamm CY3799 isoliert. Als Spezifitätskontrolle wurde der Cka1-HA6 Stamm CY3796, der keine Fusion von Paf1 mit dem TAP-tag exprimiert, in gleicher Weise behandelt. Die Anwesenheit von Cka1-HA6 im gereinigten PAF1-Komplex wurde immunologisch im Westernblot mit Antikörpern gegen das HA-Epitop (12CA5) ermittelt. Der TAP-tag wurde in den Dialysaten simultan durch die Bindung eines Peroxidase-Antiperoxidase Komplexes (PAP) an die Protein A Domänen des TAP-tags nachgewiesen. (C) Casein-Kinase II wurde über Cka1-TAP aus dem

Ergebnisse
63
Paf1-HA3 Stamm CY3770 isoliert. Als Spezifitätskontrolle wurde der Paf1-HA3 Stamm CY3718, der keine Fusion von Cka1 mit dem TAP-tag exprimiert, in gleicher Weise behandelt. Der immunologische Nachweis des HA- bzw. TAP-tags erfolgte wie unter (A) beschrieben. F2 bzw. F5 bezeichnet die Fraktionsnummer bei der fraktionierten Elution der Casein-Kinase II von Calmodulin-Affinitätsmatrix mit EGTA. (D) Reinigung und Analyse der Casein-Kinase II wie unter (B) beschrieben, jedoch aus den Spt16-HA6 Stämmen CY3784 (Cka1-TAP) und CY3728 (kein TAP-tag). Als Referenz für Cka1-TAP wurde im Westernblot das Dialysat des Stammes CY3785 (Cka1-TAP*) eingesetzt, der keine zusätzliche HA6-Proteinfusion exprimiert.
eine schwache Bande auf, die auf unspezifische Adsorption von Spt16-HA6 an die IgG-Matrix zurückzuführen ist. Sowohl die Anreicherung von Paf1-HA3 als auch die Anreicherung von Spt16-HA6 mit der isolierten Casein-Kinase II erfolgt daher durch eine spezifische Interaktion mit der Kinase. Wie schon im Falle des PAF1-Komplexes treten allerdings auch in der Silberfärbung der isolierten Casein-Kinase II keine zusätzlichen Banden auf, die eine stöchiometrische Anreicherung des PAF1-Komplexes bzw. des Elongationsfaktors yFACT erwarten lassen (Spur 3 und 4).
Diese wechselseitige Anreicherung von Komponenten der Elongationsfaktoren PAF1 oder yFACT mit der isolierten Casein-Kinase II bestätigt dennoch die Hypothese, dass PAF1-Komplex, yFACT und Casein-Kinase II auch in vivo auf physikalischer Ebene miteinander assoziiert sind.
Der gereinigte PAF1-Komplex enthält eine Kinaseaktivität, die auf die Assoziation mit der Casein-Kinase II zurückgeführt werden kann
Casein-Kinase II und PAF1-Komplex zeigen in vivo also eine physikalische Interaktion.
Zur weiteren Untersuchung dieser Wechselwirkung wurde getestet, ob der isolierte PAF1-Komplex tatsächlich eine CKII-abhängige Kinaseaktivität aufweist. Zu diesem Zweck wurde der isolierte PAF1-Komplex zunächst mit radioaktiv markiertem γ-32P-ATP in Gegenwart unterschiedlicher Mengen unmarkierten ATPs, mit und ohne Zusatz von Histon H1 aus Kalbsthymus (Roche) als potentielles Phosphorylierungssubstrat inkubiert. Der gesamte Reaktionsansatz wurde im Anschluss elektrophoretisch im Polyacrylamidgel aufgetrennt. Nach Trocknung des Gels wurden die radioaktiv phosphorylierten Polypeptide autoradiographisch analysiert. Eine Merkmal der Casein-Kinase II ist, dass sie sowohl ATP als auch GTP als Substrat nutzen kann, mit Km-Werten von 7,5 µmol·L-1 für ATP bzw. 55 µmol·L-1 für GTP bezogen auf das Hefeenzym (Tuazon and Traugh, 1991). Diese Besonderheit unterscheidet CKII von den meisten anderen Kinasen, und auch von Casein-Kinase I, die ausschließlich ATP als Phosphatdonor akzeptieren können (Tuazon and Traugh, 1991). Um eine mögliche Kinaseaktivität des isolierten PAF1-Komplexes auf die Anwesenheit der Casein-Kinase II zurückführen zu können, wurden alle Phosphorylierungsreaktionen daher zusätzlich mit γ-32P-GTP durchgeführt.
Die Autoradiographie in Abb. 2.22 A zeigt, dass bereits die Inkubation des PAF1-Komplexes alleine mit γ-32P-ATP (Spur 2) zum Auftreten von radioaktiv markierten Banden führt, die vermutlich auf die Phosphorylierung der Komplexbestandteile zurückzuführen ist. In
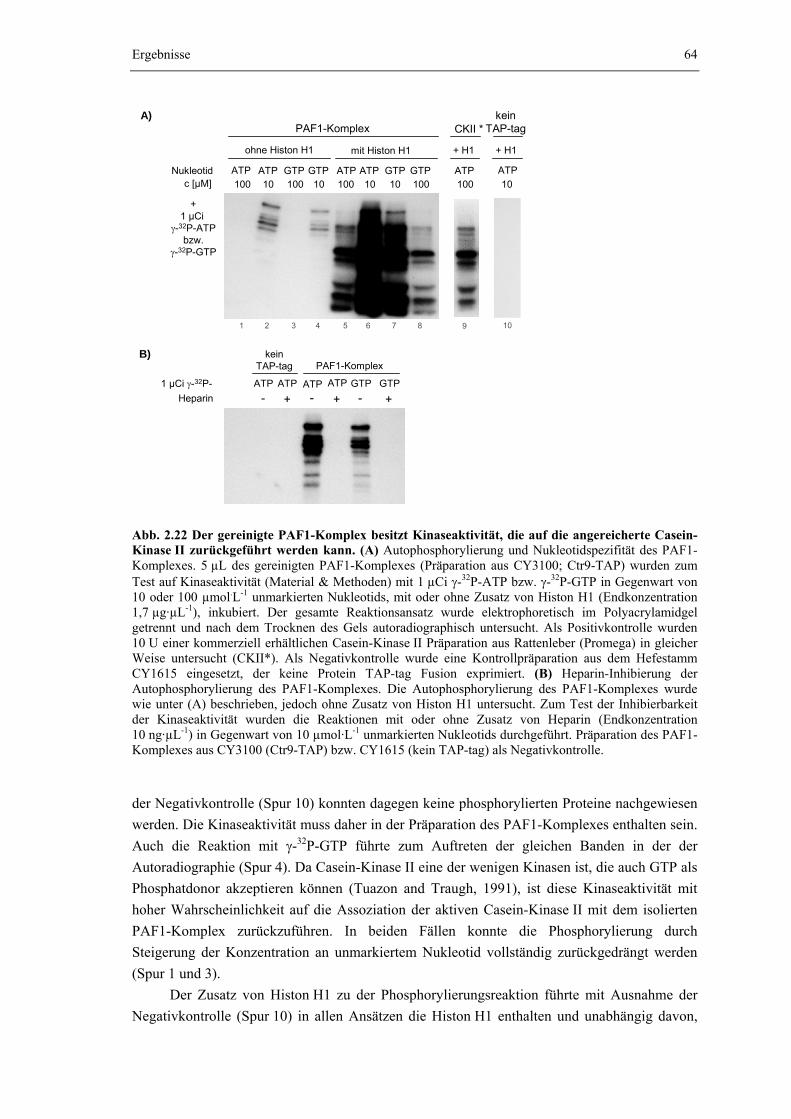
Ergebnisse
64
100 100 100 10 10010 10 10 100c [µM]Nukleotid ATP ATP ATP ATP ATPGTP GTP GTP GTP
ohne Histon H1 mit Histon H1
PAF1-Komplex
+ H1
CKII *
ATPATPATPATP GTP GTP
+1 µCi
γ-32P-ATPbzw.
γ-32P-GTP
Heparin1 µCi γ-32P-
keinTAP-tag PAF1-Komplex
+ ++ ---
A)
B)
ATP10
+ H1
keinTAP-tag
1 2 3 4 5 6 7 8 9 10
Abb. 2.22 Der gereinigte PAF1-Komplex besitzt Kinaseaktivität, die auf die angereicherte Casein-Kinase II zurückgeführt werden kann. (A) Autophosphorylierung und Nukleotidspezifität des PAF1-Komplexes. 5 µL des gereinigten PAF1-Komplexes (Präparation aus CY3100; Ctr9-TAP) wurden zum Test auf Kinaseaktivität (Material & Methoden) mit 1 µCi γ-32P-ATP bzw. γ-32P-GTP in Gegenwart von 10 oder 100 µmol.L-1 unmarkierten Nukleotids, mit oder ohne Zusatz von Histon H1 (Endkonzentration 1,7 µg·µL-1), inkubiert. Der gesamte Reaktionsansatz wurde elektrophoretisch im Polyacrylamidgel getrennt und nach dem Trocknen des Gels autoradiographisch untersucht. Als Positivkontrolle wurden 10 U einer kommerziell erhältlichen Casein-Kinase II Präparation aus Rattenleber (Promega) in gleicher Weise untersucht (CKII*). Als Negativkontrolle wurde eine Kontrollpräparation aus dem Hefestamm CY1615 eingesetzt, der keine Protein TAP-tag Fusion exprimiert. (B) Heparin-Inhibierung der Autophosphorylierung des PAF1-Komplexes. Die Autophosphorylierung des PAF1-Komplexes wurde wie unter (A) beschrieben, jedoch ohne Zusatz von Histon H1 untersucht. Zum Test der Inhibierbarkeit der Kinaseaktivität wurden die Reaktionen mit oder ohne Zusatz von Heparin (Endkonzentration 10 ng·µL-1) in Gegenwart von 10 µmol·L-1 unmarkierten Nukleotids durchgeführt. Präparation des PAF1-Komplexes aus CY3100 (Ctr9-TAP) bzw. CY1615 (kein TAP-tag) als Negativkontrolle.
der Negativkontrolle (Spur 10) konnten dagegen keine phosphorylierten Proteine nachgewiesen werden. Die Kinaseaktivität muss daher in der Präparation des PAF1-Komplexes enthalten sein. Auch die Reaktion mit γ-32P-GTP führte zum Auftreten der gleichen Banden in der der Autoradiographie (Spur 4). Da Casein-Kinase II eine der wenigen Kinasen ist, die auch GTP als Phosphatdonor akzeptieren können (Tuazon and Traugh, 1991), ist diese Kinaseaktivität mit hoher Wahrscheinlichkeit auf die Assoziation der aktiven Casein-Kinase II mit dem isolierten PAF1-Komplex zurückzuführen. In beiden Fällen konnte die Phosphorylierung durch Steigerung der Konzentration an unmarkiertem Nukleotid vollständig zurückgedrängt werden (Spur 1 und 3).
Der Zusatz von Histon H1 zu der Phosphorylierungsreaktion führte mit Ausnahme der Negativkontrolle (Spur 10) in allen Ansätzen die Histon H1 enthalten und unabhängig davon,

Ergebnisse
65
ob ATP oder GTP als Phosphatdonor verwendet wurde, zum Auftreten weiterer Banden in der Autoradiographie (Spur 5-8). Ein Vergleichsansatz mit einer Casein-Kinase II Präparation aus Rattenleber (Spur 9) führte in der Autoradiographie zudem zu einem identischen Bandenmuster (Spur 9). Die in der Histon H1 Präparation enthaltenen Polypeptide können also prinzipiell durch CKII und auch durch die mit dem gereinigten PAF1-Komplex assoziierte Casein-Kinase II der Hefe phosphoryliert werden. Die Identität der in der H1-Präparation auftretenden Banden wurde nicht genauer untersucht.
Um zu überprüfen, ob die Casein-Kinase II die alleinige Quelle der Kinaseaktivität des isolierten PAF1-Komplexes darstellt, oder ob ggf. eine weitere Kinase mit dem Komplex angereichert wird, wurde ein weiteres Charakteristikum der Casein-Kinase II ausgenutzt. Heparin wirkt als spezifischer Inhibitor der Casein-Kinase II ohne dabei die Aktivität anderer Kinasen zu beeinträchtigen, darunter u. a. Casein-Kinase I und die cAMP-abhängigen Kinasen I und II (Hathaway et al., 1980). Die I50 Konzentration, bei der 50% der Enzymaktivität inhibiert werden, wird für Heparin bezogen auf CKII im Bereich zwischen 0,13 – 0,3 µg·mL-1 angegeben (Hathaway et al., 1980; Tuazon and Traugh, 1991). Eine Konzentration von bis zu 15 µg·mL-1 Heparin bleibt dagegen ohne Einfluss auf die Aktivität anderer Proteinkinasen (Hathaway et al., 1980). Daher wurde die Autophosphorylierung des isolierten PAF1-Komplexes in parallelen Reaktionsansätzen mit oder ohne Zusatz von Heparin erneut durch Autoradiographie untersucht. Wie Abb. 2.22 B zeigt, konnte die Autophosphorylierung des PAF1-Komplexes sowohl bei Verwendung von γ-32P-ATP als auch bei Verwendung von γ-32P-GTP in allen Ansätzen vollständig durch Heparin gehemmt werden. Die Casein-Kinase II stellt daher die alleinige Quelle für die Kinaseaktivität des isolierten PAF1-Komplexes dar.
Ctr9, Rtf1, Leo1 und Paf1 können in vitro durch die assoziierte Casein-Kinase II phosphoryliert werden
Die Assoziation der Casein-Kinase II mit dem gereinigten PAF1-Komplex kann in vitro
zur Phosphorylierung der isolierten Proteine ausgenutzt werden. Da der PAF1-Komplex durch die Tandem-Affinitätsreinigung in hoher Reinheit angereichert wird, handelt es sich hierbei sehr wahrscheinlich um die Komponenten des Komplexes. Um diese Annahme zu überprüfen wurden die Präparationen des PAF1-Komplexes aus den rtf1Δ, leo1Δ , paf1Δ und cdc73Δ Mutanten sowie die Präparationen über verkürzte Ctr9-TAP Varianten (Kapitel 2.1.5), wie oben beschrieben, in einem Phosphorylierungstest eingesetzt. Falls die entsprechende Komponente des PAF1-Komplexes in vitro phosphoryliert werden kann, sollte ihre Deletion auch in der Autoradiographie zum Wegfall der korrespondierenden Bande führen. Ein vollständiger Verlust der Kinaseaktivität würde zudem den Interaktionspartner der Casein-Kinase II innerhalb des PAF1-Komplexes aufdecken. Die Autoradiographien dieser Phosphorylierungstests sind in Abb. 2.23 wiedergegeben.
Wie die Autoradiographie in Abb. 2.23 A zeigt, werden mindestens vier der Proteine, des aus einem Wildtypstamm (WT) über Ctr9-TAP isolierten PAF1-Komplexes, durch die Inkubation der Komplexpräparation mit γ-32P-ATP phosphoryliert. In der Autoradiographie der Kontrollpräparation (kein TAP-tag) traten dagegen keine Banden auf, was noch einmal die Kausalität zwischen PAF1-Komplex und Kinaseaktivität unterstreicht.
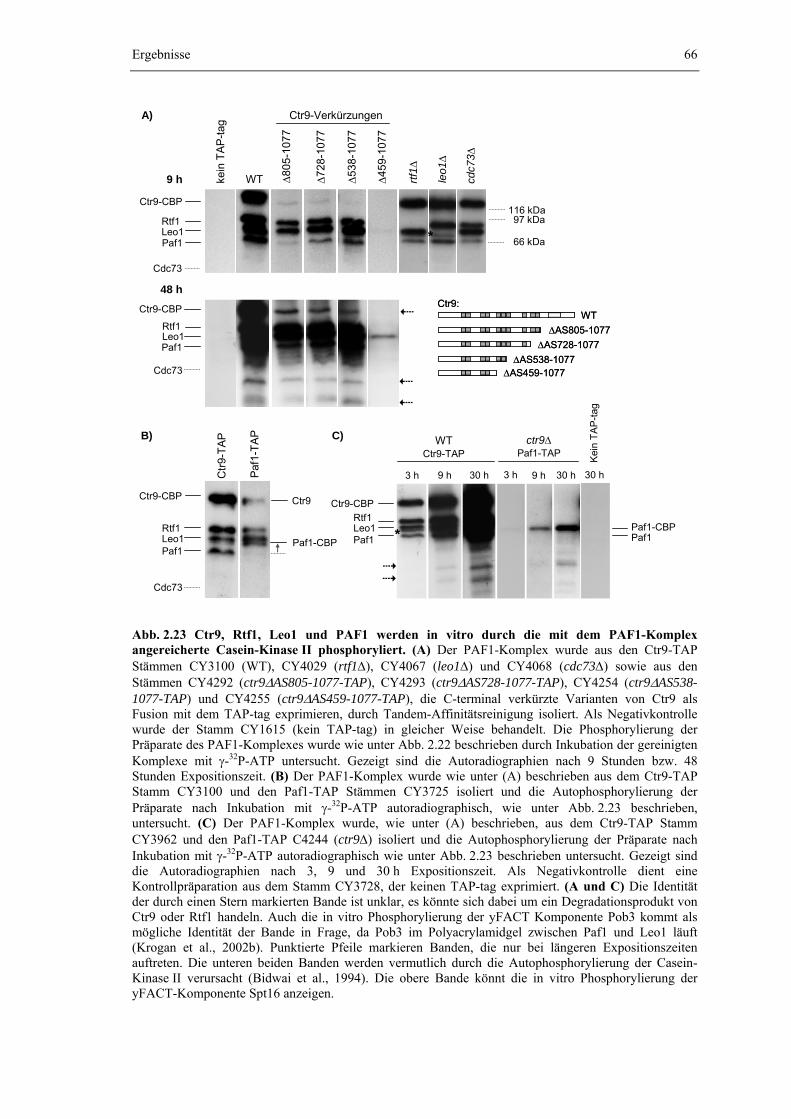
Ergebnisse
66
Ctr9-CBP
Rtf1Leo1Paf1
kein
TAP
-tag
WT Δ805
-107
7
Δ728
-107
7
Δ538
-107
7
Δ459
-107
7
Ctr9-Verkürzungen
rtf1Δ
Cdc73
leo1
Δ
cdc7
3Δ
*
A)
Ctr9-CBP
Rtf1Leo1Paf1
Paf1-CBP
Ctr9
-TAP
Paf1
-TAP
Ctr9
Cdc73
B) C)
Ctr9:WT
ΔAS805-1077ΔAS728-1077
ΔAS538-1077ΔAS459-1077
Ctr9:WT
ΔAS805-1077ΔAS728-1077
ΔAS538-1077ΔAS459-1077
Paf1-CBPPaf1*
Ctr9-CBPRtf1Leo1Paf1
3 h 3 h9 h 9 h30 h 30 h 30 h
WTCtr9-TAP
ctr9ΔPaf1-TAP Ke
in T
AP-ta
g
116 kDa97 kDa
66 kDa
Ctr9-CBP
Rtf1Leo1Paf1
Cdc73
9 h
48 h
Abb. 2.23 Ctr9, Rtf1, Leo1 und PAF1 werden in vitro durch die mit dem PAF1-Komplex angereicherte Casein-Kinase II phosphoryliert. (A) Der PAF1-Komplex wurde aus den Ctr9-TAP Stämmen CY3100 (WT), CY4029 (rtf1Δ), CY4067 (leo1Δ) und CY4068 (cdc73Δ) sowie aus den Stämmen CY4292 (ctr9ΔAS805-1077-TAP), CY4293 (ctr9ΔAS728-1077-TAP), CY4254 (ctr9ΔAS538-1077-TAP) und CY4255 (ctr9ΔAS459-1077-TAP), die C-terminal verkürzte Varianten von Ctr9 als Fusion mit dem TAP-tag exprimieren, durch Tandem-Affinitätsreinigung isoliert. Als Negativkontrolle wurde der Stamm CY1615 (kein TAP-tag) in gleicher Weise behandelt. Die Phosphorylierung der Präparate des PAF1-Komplexes wurde wie unter Abb. 2.22 beschrieben durch Inkubation der gereinigten Komplexe mit γ-32P-ATP untersucht. Gezeigt sind die Autoradiographien nach 9 Stunden bzw. 48 Stunden Expositionszeit. (B) Der PAF1-Komplex wurde wie unter (A) beschrieben aus dem Ctr9-TAP Stamm CY3100 und den Paf1-TAP Stämmen CY3725 isoliert und die Autophosphorylierung der Präparate nach Inkubation mit γ-32P-ATP autoradiographisch, wie unter Abb. 2.23 beschrieben, untersucht. (C) Der PAF1-Komplex wurde, wie unter (A) beschrieben, aus dem Ctr9-TAP Stamm CY3962 und den Paf1-TAP C4244 (ctr9Δ) isoliert und die Autophosphorylierung der Präparate nach Inkubation mit γ-32P-ATP autoradiographisch wie unter Abb. 2.23 beschrieben untersucht. Gezeigt sind die Autoradiographien nach 3, 9 und 30 h Expositionszeit. Als Negativkontrolle dient eine Kontrollpräparation aus dem Stamm CY3728, der keinen TAP-tag exprimiert. (A und C) Die Identität der durch einen Stern markierten Bande ist unklar, es könnte sich dabei um ein Degradationsprodukt von Ctr9 oder Rtf1 handeln. Auch die in vitro Phosphorylierung der yFACT Komponente Pob3 kommt als mögliche Identität der Bande in Frage, da Pob3 im Polyacrylamidgel zwischen Paf1 und Leo1 läuft (Krogan et al., 2002b). Punktierte Pfeile markieren Banden, die nur bei längeren Expositionszeiten auftreten. Die unteren beiden Banden werden vermutlich durch die Autophosphorylierung der Casein-Kinase II verursacht (Bidwai et al., 1994). Die obere Bande könnt die in vitro Phosphorylierung der yFACT-Komponente Spt16 anzeigen.

Ergebnisse
67
Mit Ausnahme dieses Kontrollansatzes ließen sich durch die Autoradiographie in allen Ansätzen phosphorylierte Proteine nachweisen. Die Assoziation der Casein-Kinase II mit dem isolierten PAF1-Komplex wird daher nicht durch die Deletion von RTF1, LEO1 oder CDC73 beeinträchtigt bzw. nicht durch diese Proteine vermittelt. Von den vier Präparationen des PAF1-Komplexes über C-terminal verkürzte Ctr9-TAP Varianten führte ausschließlich die gravierendste Verkürzung des Proteins durch die Entfernung der Aminosäuren 459-1077 zu einem scheinbaren Verlust der Kinaseaktivität, da sich in der Autoradiographie der entsprechenden Präparation zunächst keinerlei Banden nachweisen ließen. Durch Verlängerung der Expositionszeit von 9 h auf 48 h ließ sich jedoch auch in diesem Fall noch eine schwache Bande auf der Höhe von Leo1 nachweisen (Abb. 2.23 A, Δ459-1077, 48 h). Das drastisch verringerte Vorkommen von phosphorylierten Proteinen spiegelt vermutlich den dramatischen Einbruch der Komplexausbeute wider (vgl. Kapitel 2.1.5). Die Banden in den Autoradiographien der verbleibenden drei Ansätze mit Ctr9-Verkürzungen zeigen jedoch eindeutig, dass die Assoziation der Casein-Kinase II mit dem gereinigten PAF1-Komplex nicht durch die Entfernung des C-terminalen Ctr9-Bereichs bis zu Aminosäure 538 beeinträchtigt wird und vermutlich auch bei Ausweitung der Verkürzung bis Aminosäure 459 noch erhalten bleibt.
Alle nach 9 Stunden Expositionszeit in den Autoradiographien auftretenden Banden liegen oberhalb eines Molekulargewichts von 60 kDa. Damit kann die Phosphorylierung von Cdc73 (apparente Masse ca. 50 kDa) durch die assoziierte Casein-Kinase II in vitro ausgeschlossen werden, zumal auch die Verlängerung der Expositionszeit auf 48 h keine Bande auf der Höhe von ca. 50 kDa aufweist. Die Deletion von Rtf1 bzw. Leo1 führt zu dem Verschwinden der Banden bei ca. 90 kDa (rtf1Δ) bzw. 80 kDa (leo1Δ). Alle anderen auch in der Wildtyppräparation (WT) auftretenden Banden bleiben indessen erhalten. Die Banden bei 90 und 80 kDa können daher eindeutig auf Rtf1 bzw. Leo1 zurückgeführt werden. Beide Proteine können in vitro also durch die assoziierte Casein-Kinase II phosphoryliert werden. Durch die Deletion von LEO1 wurde zudem sichtbar, dass direkt unterhalb von Leo1 eine weitere Bande auftritt (Abb. 2.23 A, Stern), bei der es sich um ein Degradationsprodukt von Ctr9 oder Rtf1 handeln könnte. Aufgrund der teilweisen Überlagerung mit der durch Leo1 verursachten Bande kann dies jedoch nicht zweifelsfrei entschieden werden. Anhand der apparenten Masse dieses Proteins könnte es sich jedoch ggf. auch um die Pob3-Komponente des Elongationsfaktors yFACT handeln, die bei der Elektrophorese im Polyacrylamidgel zwischen den Proteinen Paf1 und Leo1 läuft (Krogan et al., 2002b).
Im Falle der Ctr9-Verkürzungen führt interessanterweise bereits die Entfernung des Proteinbereichs zwischen den Aminosäuren 805 und 1077 zu dem Verschwinden der Bande oberhalb von 116 kDa. Die Silberfärbung des isolierten Komplexes zeigt jedoch, dass die Ctr9-Bande aufgrund des geringeren Molekulargewichts dieser Verkürzung zwar nach unten verschoben wird, jedoch noch immer oberhalb der durch Rtf1 verursachten Bande verläuft (Abb. 2.6). In der Autoradiographie ist auf dieser Höhe jedoch keine Bande nachweisbar. Die markante Bande oberhalb von 116 kDa kann daher zweifelsfrei Ctr9 zugesprochen werden. Zudem scheint der entfernte Proteinbereich zwischen Aminosäure 805 und 1077 auch die durch CKII phosphorylierten Aminosäuren zu beinhalten. Auch die weitere Verkürzung von Ctr9 um den Proteinbereich zwischen Aminosäure 728 und 1077 bzw. 538 und 1077 führt zu einem

Ergebnisse
68
Verschwinden der durch die Phosphorylierung von Ctr9 verursachten Bande. In beiden Fällen tritt zudem keine zusätzliche Bande auf, die der apparenten Masse der jeweiligen Ctr9-Verkürzung entsprechen würde (Abb. 2.6). Die Verlängerung der Expositionszeit zeigte jedoch, dass sich unmittelbar unterhalb von Ctr9-CBP eine weitere, wenn auch schwache Bande befindet (Abb. 2.23 A, punktierter Pfeil). Eine Sequenzanalyse über die Datenbank "Prosite" (Gattiker et al., 2002; Hulo et al., 2006) ergibt, dass sowohl Spt16 als auch Pob3 in ihrer Primärstruktur mehrere Bereiche besitzen, die zumindest theoretisch durch die Casein-Kinase II phosphoryliert werden können (Daten nicht gezeigt). Die massenspektrometrische Analyse des PAF1-Komplexes zeigte zudem, dass die Ctr9-CBP Bande durch die Spt16-Komponente des Elongationsfaktors yFACT überlagert wird (Kapitel 2.1.1 und 2.3.1). Daher liegt die Vermutung nahe, dass diese Bande durch die in vitro Phosphorylierung von Spt16 hervorgerufen wird. Die unteren beiden Banden, die ebenfalls durch punktierte Pfeile markiert werden, sind sehr wahrscheinlich auf die Autophosphorylierung der Casein-Kinase II Untereinheiten zurückzuführen (Bidwai et al., 1994).
Ctr9, Rtf1 und Leo1 werden also in vitro durch CKII phosphoryliert. Um zu überprüfen, ob auch Paf1 durch CKII phosphoryliert werden kann und dadurch in der Autoradiographie die Bande auf Höhe von ca. 66 kDa verursacht, wurde das Phosphorylierungsexperiment mit Paf1-TAP Präparationen des PAF1-Komplexes wiederholt. Wie Abb. 2.23 B zeigt, tritt auch in dieser Präparation eine Autophosphorylierung auf, wobei die untere Bande bei 66 kDa in etwa auf die Höhe der Leo1-Bande verschoben wird. Eine vergleichbare Verschiebung der Paf1-Bande durch die verbleibende Fusion mit dem CBP des TAP-tags tritt auch in der Silberfärbung des über Paf1-TAP isolierten Komplexes auf (Abb. 2.1). Damit ist klar, dass auch Paf1 in vitro als Substrat der Casein-Kinase II genutzt wird.
Die in vitro Phosphorylierung der Komponenten des PAF1-Komplexes durch Casein-Kinase II bestätigt die Untersuchungen zum Aufbau des Elongationsfaktors
Mit Ausnahme von Cdc73 werden also alle Bestandteile des PAF1-Komplexes in vitro durch die assoziierte Casein-Kinase II phosphoryliert. Dies bot die willkommene Möglichkeit die Untersuchungen bezüglich des Aufbaus des PAF1-Komplexes zu verifizieren (Kapitel 2.1). Die Isolierung der Paf1-TAP Fusion aus einer ctr9Δ Mutante gelang nur in Spuren (vgl. Abb. 2.3). Die Autoradiographie dieser Präparation in Abb. 2.23 C zeigt jedoch mit steigender Expositionszeit deutlich das Auftreten einer Bande auf Höhe der Paf1-CBP Fusion bzw. auf der Höhe von Leo1 (Abb. 2.23 C, 9 und 30 h Exposition). Dagegen ließen sich keine Banden nachweisen, die Hinweise auf die Anwesenheit von Ctr9 oder Rtf1 in der Präparation geben könnten. Da in der Negativkontrolle nach einer Expositionszeit von 30 h (Abb. 2.23 C, kein TAP-tag, 30 h) ebenfalls keine Banden nachweisbar sind, kann die in der Präparation aus der ctr9Δ Mutante auftretende Bande jedoch spezifisch auf die Anreicherung von Paf1-CBP zurückgeführt werden. Dies unterstützt die Auffassung, dass die Stabilität des PAF1-Komplexes durch die Deletion von CTR9 weitestgehend verloren geht und liefert zudem einen Hinweis darauf, dass Paf1 vermutlich unmittelbar an der Assoziation der Casein-Kinase II beteiligt ist oder diese zumindest vermittelt.

Ergebnisse
69
Die Autophosphorylierung des PAF1-Komplexes bestätigt zudem, dass Rtf1, Leo1 und Cdc73 aus dem Komplexverbund gelöst werden können, ohne die Assoziation der verbleibenden Bestandteile des Komplexes zu stören (Kapitel 2.1.3). Gleiches gilt für die Verkürzung von Ctr9 um die Aminosäuren 538-1077 (Kapitel 2.1.5). Allerdings kann die Autoradiographie keine Aussage über die Assoziation von Cdc73 liefern, da das Protein in vitro nicht durch CKII phosphoryliert wird. Auch die Interpretation der Zusammensetzung des über die Ctr9-Variante mit um die Aminosäuren 459-1077 verkürztem C-Terminus isolierten Komplexes gestaltet sich problematisch. In der Silberfärbung dieser Präparation lassen sich schwache Banden erkennen, die möglicherweise auf die Anreicherung von Rtf1, Leo1 und Paf1 mit dieser Verkürzung hinweisen (Abb. 2.6). In der entsprechenden Autoradiographie tritt aber nur eine Bande auf Höhe von Leo1 auf (Abb. 2.23 A, Δ459-1077, 9 h). Die Annahme, dass Paf1 zwingend für die Assoziation mit der Casein-Kinase II verantwortlich ist und die Beobachtung, dass die Ctr9-Verkürzung selbst nicht mehr phosphoryliert wird, ermöglicht jedoch die Hypothese, dass neben der Ctr9-Verkürzung selbst zumindest noch Paf1, Leo1 und Spuren der Casein-Kinase II in der Präparation enthalten sein müssten.
Der C-Terminus von Ctr9 ist ein Substrat für Casein-Kinase II
Für die in vitro Bindestudien zwischen Ctr9 und Cdc73 wurden bereits erfolgreich heterolog in E. coli exprimierte Proteinfragmente von Ctr9 eingesetzt (vgl. Kapitel 2.1.5). Die Verwendung dieser Ctr9-Fragmente als Substrat für die isolierte Casein-Kinase II bot daher die Möglichkeit zu überprüfen, ob die Phosphorylierung von Ctr9 tatsächlich auf den C-terminalen Proteinbereich zwischen Aminosäure 805 und 1077 eingeschränkt werden kann. Die Ctr9-Fragmente wurden dazu in den jeweiligen E. coli Stämmen exprimiert und durch die Adsorption an eine Glutathion-Sepharosematrix (GSH-Matrix) isoliert. Die mit den Ctr9-Fragmenten beladene GSH-Matrix wurde dann als Substrat in einem in vitro Phosphorylierungstest eingesetzt. Als Kinase wurde die durch Tandem-Affinitätsreinigung aus der Hefe präparierte Casein-Kinase II verwendet. Der Nachweis einer Phosphorylierung erfolgte im Anschluss wiederum durch die Autoradiographie des elektrophoretisch im Polyacrylamidgel aufgetrennten Reaktionsansatzes. Ein Teil der als Kinasesubstrat eingesetzten, beladenen GSH-Matrix wurde zudem im Westernblot mit Antikörpern gegen GST untersucht, um die Bindung der Ctr9-Fragmente an die GSH-Matrix sicher zu stellen (Abb. 2.24 C). Die Autoradiographie dieser Phosphorylierungstests ist in Abb. 2.24 B wiedergegeben. Zur leichteren Orientierung ist in Abb. 2.24 A eine schematische Übersicht der eingesetzten Ctr9-Fragmente dargestellt.
Wie die Autoradiographie in Abb. 2.24 B zeigt, ließ sich ausschließlich und reproduzierbar in den Phosphorylierungsansätzen, die das Ctr9-Fragment Ctr9[AS716-1077] enthalten, eine deutliche Bande nachweisen. Das Fragment, das die Aminosäuren 716-816 umfasst, wurde dagegen nicht durch die zugesetzte Casein-Kinase II phosphoryliert. Die phosphorylierte Region in Ctr9 muss daher C-terminal, oberhalb von Aminosäure 816 gelegen sein. In Übereinstimmung dazu kann das Ctr9-Fragment Ctr9[AS550-816] und die beiden Fragmente Ctr9[AS386-549] und Ctr9[AS2-549] von CKII nicht phosphoryliert werden.
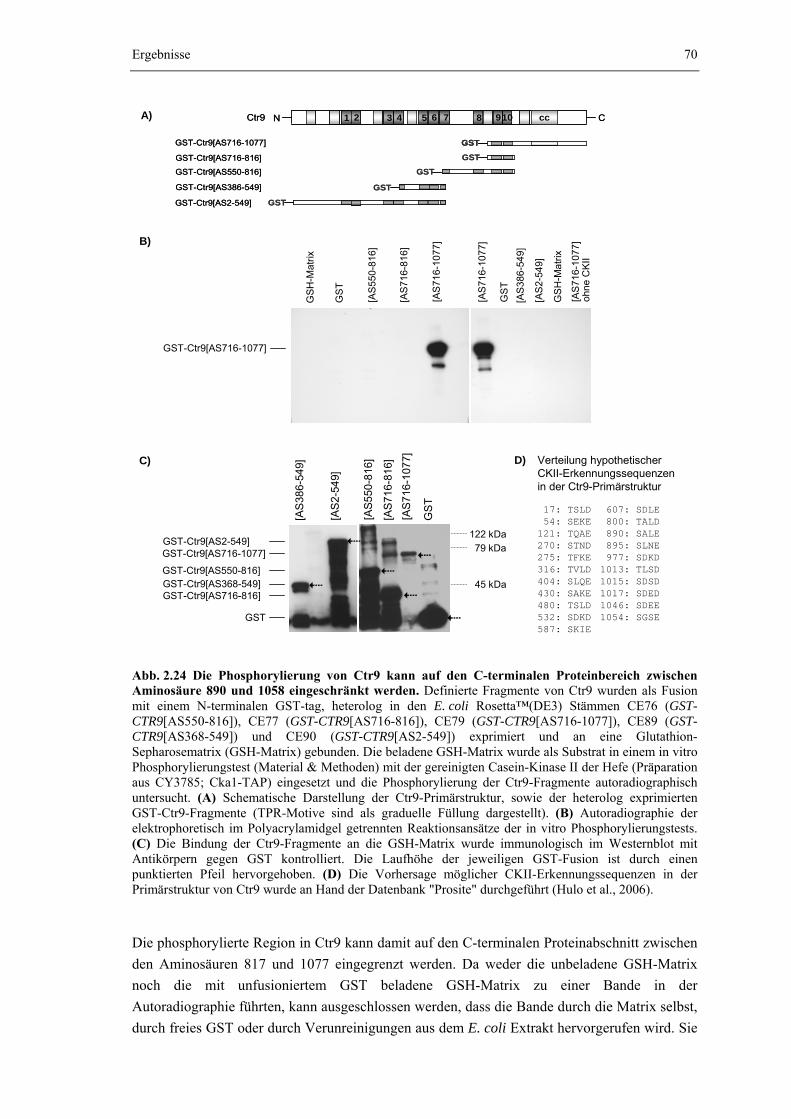
Ergebnisse
70
GSH
-Mat
rix
GST
[AS5
50-8
16]
[AS7
16-8
16]
[AS7
16-1
077]
[AS7
16-1
077]
[AS3
86-5
49]
[AS2
-549
]
GST
GS
H-M
atrix
[AS7
16-1
077]
oh
ne C
KII
1 2 3 4 5 6 7 8 910 ccN C
GST
GST
GST
GST-Ctr9[AS716-1077]
GST-Ctr9[AS716-816]
GST-Ctr9[AS550-816]
Ctr9
GSTGST-Ctr9[AS2-549]
GSTGST-Ctr9[AS386-549]
1 2 3 4 5 6 7 8 910 ccN C
GSTGST
GST
GST
GST-Ctr9[AS716-1077]
GST-Ctr9[AS716-816]
GST-Ctr9[AS550-816]
Ctr9
GSTGST-Ctr9[AS2-549]
GSTGST-Ctr9[AS386-549]
GST-Ctr9[AS716-1077]
B)
A)
[AS5
50-8
16]
[AS
716-
816]
[AS7
16-1
077]
GS
T
[AS
386-
549]
[AS
2-54
9]
122 kDa79 kDa
45 kDa
GST-Ctr9[AS2-549]GST-Ctr9[AS716-1077]
GST-Ctr9[AS550-816]GST-Ctr9[AS368-549]GST-Ctr9[AS716-816]
GST
C)
17: TSLD54: SEKE
121: TQAE270: STND275: TFKE316: TVLD 404: SLQE430: SAKE480: TSLD532: SDKD587: SKIE
607: SDLE800: TALD890: SALE895: SLNE977: SDKD
1013: TLSD1015: SDSD1017: SDED1046: SDEE1054: SGSE
D) Verteilung hypothetischerCKII-Erkennungssequenzenin der Ctr9-Primärstruktur
Abb. 2.24 Die Phosphorylierung von Ctr9 kann auf den C-terminalen Proteinbereich zwischen Aminosäure 890 und 1058 eingeschränkt werden. Definierte Fragmente von Ctr9 wurden als Fusion mit einem N-terminalen GST-tag, heterolog in den E. coli Rosetta™(DE3) Stämmen CE76 (GST-CTR9[AS550-816]), CE77 (GST-CTR9[AS716-816]), CE79 (GST-CTR9[AS716-1077]), CE89 (GST-CTR9[AS368-549]) und CE90 (GST-CTR9[AS2-549]) exprimiert und an eine Glutathion-Sepharosematrix (GSH-Matrix) gebunden. Die beladene GSH-Matrix wurde als Substrat in einem in vitro Phosphorylierungstest (Material & Methoden) mit der gereinigten Casein-Kinase II der Hefe (Präparation aus CY3785; Cka1-TAP) eingesetzt und die Phosphorylierung der Ctr9-Fragmente autoradiographisch untersucht. (A) Schematische Darstellung der Ctr9-Primärstruktur, sowie der heterolog exprimierten GST-Ctr9-Fragmente (TPR-Motive sind als graduelle Füllung dargestellt). (B) Autoradiographie der elektrophoretisch im Polyacrylamidgel getrennten Reaktionsansätze der in vitro Phosphorylierungstests. (C) Die Bindung der Ctr9-Fragmente an die GSH-Matrix wurde immunologisch im Westernblot mit Antikörpern gegen GST kontrolliert. Die Laufhöhe der jeweiligen GST-Fusion ist durch einen punktierten Pfeil hervorgehoben. (D) Die Vorhersage möglicher CKII-Erkennungssequenzen in der Primärstruktur von Ctr9 wurde an Hand der Datenbank "Prosite" durchgeführt (Hulo et al., 2006).
Die phosphorylierte Region in Ctr9 kann damit auf den C-terminalen Proteinabschnitt zwischen den Aminosäuren 817 und 1077 eingegrenzt werden. Da weder die unbeladene GSH-Matrix noch die mit unfusioniertem GST beladene GSH-Matrix zu einer Bande in der Autoradiographie führten, kann ausgeschlossen werden, dass die Bande durch die Matrix selbst, durch freies GST oder durch Verunreinigungen aus dem E. coli Extrakt hervorgerufen wird. Sie

Ergebnisse
71
ist daher eindeutig auf die Phosphorylierung des Fragments Ctr9[AS716-1077] zurückzuführen. Zudem führt die Inkubation dieses Fragments mit γ-32P-ATP ohne Zusatz der Casein-Kinase II in der Autoradiographie ebenfalls nicht zu dem Auftreten einer Bande. Eine Verunreinigung mit einer Kinase aus dem E. coli Extrakt kann damit erneut ausgeschlossen werden. CKII stellt somit die einzige Quelle der Kinaseaktivität dar.
Die Ergebnisse der Phosphorylierungstests mit den C-terminal verkürzten Ctr9-Varianten konnten also bestätigt werden, da bereits die um die C-terminalen Aminosäuren 805-1077 verkürzte Ctr9-Variante nicht mehr durch die Casein-Kinase II phosphoryliert wird (Abb. 2.24 A). Die Analyse der Ctr9-Sequenz anhand der "Prosite" Datenbank (Gattiker et al., 2002; Hulo et al., 2006) ergibt, dass die Primärstruktur von Ctr9 21 potentiell durch die Casein-Kinase II phosphorylierbare Sequenzabschnitte enthält (Abb. 2.24. D). Davon liegen acht in dem in Frage kommenden Bereich zwischen Aminosäure 816 und 1077. Dadurch kann die phosphorylierte Region in Ctr9 weiter auf den C-terminalen Proteinabschnitt zwischen den Aminosäuren 890 und 1054 eingeschränkt werden. Interessanterweise beinhaltet dieser Proteinabschnitt keine TPR-Motive, umfasst jedoch größten Teils die Region der möglichen "Coiled-Coil" Domäne.
Die Casein-Kinase II bleibt auch während der Elongationsphase der Transkription mit dem PAF1-Komplex bzw. mit der RNA-Polymerase II assoziiert
Die Methode der Chromatin-Immunpräzipitation kann zur Unterscheidung der
verschiedenen Phasen der Transkription eingesetzt werden (Kapitel 2.2.7). Diese Methode wurde im Folgenden auch zur genaueren Charakterisierung der physikalischen Interaktion zwischen Casein-Kinase II und PAF1-Komplex (bzw. RNA-Polymerase II) verwendet. Insbesondere sollte dadurch ermittelt werden, ob die Assoziation zwischen PAF1-Komplex und CKII zeitlich bzw. räumlich begrenzt ist. Zu diesem Zweck wurden Chromatin-Immunpräzipitationen des PAF1-Komplexes über Ctr9-TAP bzw. der Casein-Kinase II über Cka1-TAP durchgeführt und anschließend durch PCR überprüft, ob auch die Casein-Kinase II zur Präzipitation Promotor-, ORF- oder 3'-UTR-spezifischer DNA-Fragmente des CLN3-Gens führt. Die Resultate dieser ChIP-Studien sind in Abb. 2.25 wiedergegeben. Alle Chromatin-Immunpräzipitationen wurden als parallele Doppelbestimmung durchgeführt.
Abb. 2.25 A zeigt, dass sich durch PCR aus den ChIP-Eluaten des PAF1-Komplexes (Ctr9-TAP) spezifische Sequenzen für den Promotor, den Leserahmen und auch die 3'-nichttranslatierte Region amplifizieren lassen. In allen drei Fällen wurden starke PCR-Signale erhalten, die die Assoziation des Elongationsfaktors mit der RNA-Polymerase II über den gesamten transkribierten Abschnitt des CLN3-Gens widerspiegeln (Abb. 2.25 A, Ctr9-TAP; Promotor, ORF, 3'-UTR). Wie bereits in Kapitel 2.2.7 für CLN2 gezeigt, bleibt der PAF1-Komplex während der transkriptionellen Elongation mit der RNA-Polymerase II assoziiert.
Die Abbildung zeigt zudem, dass sich auch aus der Chromatin-Immunpräzipitation der Casein-Kinase II (Cka1-TAP) spezifische Sequenzbereiche für Promotor, ORF und 3'-UTR amplifizieren lassen. Die Signalintensität bleibt allerdings weit hinter den PCR-Signalen der Chromatin-Immunpräzipitation des PAF1-Komplexes zurück, übertrifft aber dennoch die
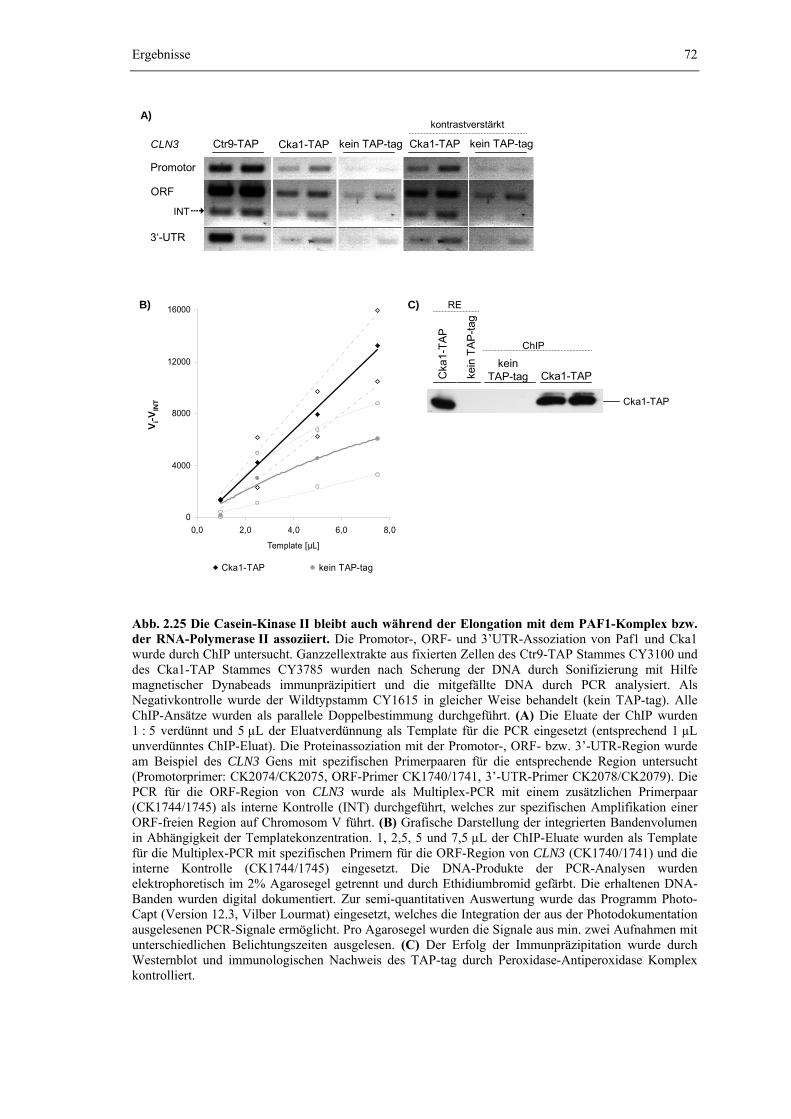
Ergebnisse
72
Ctr9-TAP Cka1-TAP Cka1-TAPkein TAP-tag kein TAP-tag
kontrastverstärkt
Promotor
ORF
3‘-UTR
INT
CLN3
0
4000
8000
12000
16000
0,0 2,0 4,0 6,0 8,0
Template [µL]
Vi
Cka1-TAP kein TAP-tag
A)
B) C)
Cka1-TAPkein
TAP-tagCka
1-TA
P
kein
TAP
-tag
ChIP
RE
Cka1-TAP
V i-V
INT
Abb. 2.25 Die Casein-Kinase II bleibt auch während der Elongation mit dem PAF1-Komplex bzw. der RNA-Polymerase II assoziiert. Die Promotor-, ORF- und 3’UTR-Assoziation von Paf1 und Cka1 wurde durch ChIP untersucht. Ganzzellextrakte aus fixierten Zellen des Ctr9-TAP Stammes CY3100 und des Cka1-TAP Stammes CY3785 wurden nach Scherung der DNA durch Sonifizierung mit Hilfe magnetischer Dynabeads immunpräzipitiert und die mitgefällte DNA durch PCR analysiert. Als Negativkontrolle wurde der Wildtypstamm CY1615 in gleicher Weise behandelt (kein TAP-tag). Alle ChIP-Ansätze wurden als parallele Doppelbestimmung durchgeführt. (A) Die Eluate der ChIP wurden 1 : 5 verdünnt und 5 µL der Eluatverdünnung als Template für die PCR eingesetzt (entsprechend 1 µL unverdünntes ChIP-Eluat). Die Proteinassoziation mit der Promotor-, ORF- bzw. 3’-UTR-Region wurde am Beispiel des CLN3 Gens mit spezifischen Primerpaaren für die entsprechende Region untersucht (Promotorprimer: CK2074/CK2075, ORF-Primer CK1740/1741, 3’-UTR-Primer CK2078/CK2079). Die PCR für die ORF-Region von CLN3 wurde als Multiplex-PCR mit einem zusätzlichen Primerpaar (CK1744/1745) als interne Kontrolle (INT) durchgeführt, welches zur spezifischen Amplifikation einer ORF-freien Region auf Chromosom V führt. (B) Grafische Darstellung der integrierten Bandenvolumen in Abhängigkeit der Templatekonzentration. 1, 2,5, 5 und 7,5 µL der ChIP-Eluate wurden als Template für die Multiplex-PCR mit spezifischen Primern für die ORF-Region von CLN3 (CK1740/1741) und die interne Kontrolle (CK1744/1745) eingesetzt. Die DNA-Produkte der PCR-Analysen wurden elektrophoretisch im 2% Agarosegel getrennt und durch Ethidiumbromid gefärbt. Die erhaltenen DNA-Banden wurden digital dokumentiert. Zur semi-quantitativen Auswertung wurde das Programm Photo-Capt (Version 12.3, Vilber Lourmat) eingesetzt, welches die Integration der aus der Photodokumentation ausgelesenen PCR-Signale ermöglicht. Pro Agarosegel wurden die Signale aus min. zwei Aufnahmen mit unterschiedlichen Belichtungszeiten ausgelesen. (C) Der Erfolg der Immunpräzipitation wurde durch Westernblot und immunologischen Nachweis des TAP-tag durch Peroxidase-Antiperoxidase Komplex kontrolliert.

Ergebnisse
73
Intensität der Signale aus der Negativkontrolle (kein TAP-tag). Dies wird besonders deutlich, wenn der Bildkontrast für die Signale der CKII-ChIP optimiert wird (Abb. 2.25 A, kontrastverstärkt). Die geringe Signalstärke kann jedoch nicht auf die Immunpräzipitation von Cka1-TAP zurückgeführt werden, da die Analyse der ChIP-Eluate im Westernblot zeigt, dass die Casein-Kinase II auch nach Fixierung mit Formaldehyd mit hoher Ausbeute präzipitiert werden kann (Abb. 2.25 C). Auch die Casein-Kinase II ist demnach sowohl mit dem Promotor, dem Leserahmen und der 3'-UTR des CLN3-Gens assoziiert. Ihre Assoziation mit dem PAF1-Komplex bzw. der RNA-Polymerase II ist nicht nur auf die Initiation beschränkt, sondern bleibt auch während der Elongation der RNA-Polymerase II bestehen. CKII verhält sich also wie ein Elongationsfaktor. Problematisch für die Auswertung ist jedoch die geringe Intensität der PCR-Signale, da auch die Amplifikation der ORF-freien Region (INT) zu, wenn auch deutlich schwächeren, Signalen bei der PCR führt.
Die geringere Signalintensität der PCR-Produkte bei der Chromatin-Immunpräzipitation der Casein-Kinase II ist jedoch nicht überraschend, da die physiologische Funktion der Casein-Kinase II nicht auf die Transkription beschränkt ist, so dass nur ein Bruchteil aller in der Zelle vorkommenden CKII-Komplexe zum Zeitpunkt der Formaldehydfixierung mit dem PAF1-Komplex bzw. der RNA-Polymerase II assoziiert sind. Da die Kinase nicht quantitativ präzipitiert wurde (Daten nicht gezeigt), führt die Chromatin-Immunpräzipitation zur statistischen Anreicherung aller CKII-Komplexe, unabhängig von ihrer augenblicklichen Funktion bzw. Lokalisation in der Zelle.
Um zu überprüfen, ob die Intensität der PCR-Signale aus der CKII Chromatin-Immunpräzipitation tatsächlich signifikant über den Signalintensitäten der Negativkontrolle liegt, wurden daher jeweils die Abhängigkeit der Signalintensität von der als Template eingesetzten Menge an ChIP-Eluat, semi-quantitativ durch Integration der resultierenden DNA-Banden der PCR-Produkte ermittelt. Die Untersuchung wurde als Multiplex-PCR mit Primerpaaren für den CLN3-ORF und die interne Kontrolle (INT) durchgeführt. Zur Auswertung wurden die integrierten Bandenvolumen der internen Kontrolle von den Volumina der CLN3-Signale abgezogen. Der Einfluss der Länge des amplifizierten PCR-Produkts auf die Signalintensität bleibt dabei unberücksichtigt. Die Abhängigkeit der Signalintensität von der eingesetzten Menge an ChIP-Eluat als Template für die PCR ist grafisch in Abb. 2.25 B wiedergegeben. Die durchgezogenen Linien geben die aus den Mittelwerten errechnete Regressionskurve für die CKII-ChIP bzw. die Negativkontrolle (kein TAP-tag) an. Die beiden gestrichelten Linien zeigen jeweils die Regressionskurven für die Werte der Einzelmessungen aus den zwei unabhängigen Eluaten der parallelen Fällungsansätze mit CKII (Cka1-TAP) bzw. der Negativkontrolle (kein TAP-tag).
Wie Abb. 2.25 B zeigt, nimmt die Signalintensität bzw. das Bandenvolumen im Fall der CKII-ChIP linear zum eingesetzten Templatevolumen zu und liegt immer deutlich über den Mittelwerten der Negativkontrolle. Die Steigung der Regressionskurve (lineare Regression 2. Ordnung) für die Werte der Negativkontrolle verläuft dementsprechend immer flacher als die Steigung der Regressionskurve für Cka1-TAP. Die Anreicherung von charakteristischen Sequenzen für den CLN3-ORF durch die Chromatin-Immunpräzipitation der Casein-Kinase II erfolgt daher spezifisch und kann damit auf die Assoziation zwischen CKII und der RNA-Polymerase II zurückgeführt werden. CKII verhält sich also tatsächlich wie ein

Ergebnisse
74
Elongationsfaktor und die Assoziation zwischen CKII und dem PAF1-Komplex bzw. der RNA-Polymerase II bleibt auch während der Elongation erhalten.
2.3.3 Interaktionen zwischen dem Bromodomainfactor 1 (Bdf1) und dem PAF1-Komplex Neben Präparationen von yFACT- und dem PAF1-Komplex wird Casein-Kinase II auch
in Reinigungen des Bromodomainfactor 1 (Bdf1) angereichert. Zudem ist die Phosphorylierung von Bdf1 durch Casein-Kinase II für dessen Funktionalität in vivo maßgeblich, da eine bdf1Δ bdf2Δ Mutante nicht durch eine Bdf1-Variante, deren durch CKII phosphorylierte Bereiche entfernt wurden, bzw. das entsprechende Allel komplementiert werden kann (Sawa et al., 2004). Bdf1 wirkt u. a. als Transkriptionsaktivator, ist mit dem generellen Transkriptionsfaktor TFIID assoziiert und stellt strukturell und funktionell die Hefeentsprechung zur C-terminalen Region des TAF1 aus höheren Eukaryoten dar (Martinez-Campa et al., 2004; Matangkasombut et al., 2000). yFACT und der PAF1-Komplex sind wie TFIID am Prozess der RNA-Polymerase II Initiation beteiligt (Biswas et al., 2005a; Stolinski et al., 1997), was die drei Proteinkomplexe sowohl in räumliche, als auch in funktionelle Nähe stellt. Bdf1 ist neben seiner Assoziation mit TFIID ebenfalls ein Bestandteil des SWR1-Komplexes, der zum Einbau der Histon H2A-Variante H2A.Z (Htz1) in Nukleosomen benötigt wird (Kobor et al., 2004; Krogan et al., 2004). H2A.Z ist an der Aufrechterhaltung der Heterochromatingrenzen beteiligt (Meneghini et al., 2003) und besitzt zudem eine Funktion bei der Transkriptionsregulation (Adam et al., 2001; Guillemette et al., 2005).
Bdf1 zeigt zudem eine zumindest indirekte genetische Wechselwirkung mit einzelnen Komponenten des PAF1-Komplexes (vgl. Kapitel 2.2.3, Tabelle 2.3). Daher sollte im Folgenden die Frage untersucht werden, ob auch Bdf1 und der PAF1-Komplex miteinander interagieren können und sich ggf. gegenseitig in ihrer Funktion beeinflussen.
Bdf1 und Paf1 stehen in physikalischer Wechselwirkung zueinander Um die mögliche Interaktion zwischen Bdf1 und dem PAF1-Komplex zu untersuchen,
wurde sowohl der PAF1-Komplex, als auch der SWR1-Komplex bzw. Bdf1 durch Tandem-Affinitätsreinigung isoliert und die Anwesenheit von Bdf1 im gereinigten PAF1-Komplex bzw. das Vorhandensein von Paf1 im isolierten SWR1-Komplex immunologisch im Westernblot untersucht (Abb. 2.26).
Wie die Silberfärbungen der gereinigten Komplexe in Abb. 2.26 zeigen, konnte sowohl der PAF1-Komplex als auch Bdf1 durch Tandem-Affinitätsreinigung mit guter Ausbeute isoliert werden. In der Präparation von Bdf1 sind zusätzliche Proteinbanden zu erkennen (Abb. 2.26 B; punktierte Pfeile), die vermutlich auf weitere Bestandteile des SWR1-Komplexes zurückzuführen sind, aber nicht genauer charakterisiert wurden. Die immunologische Untersuchung der gereinigten Komplexe im Westernblot zeigt zudem, dass Bdf1 spezifisch im isolierten PAF1-Komplex angereichert wurde (Abb. 2.26 A, Paf1-TAP)
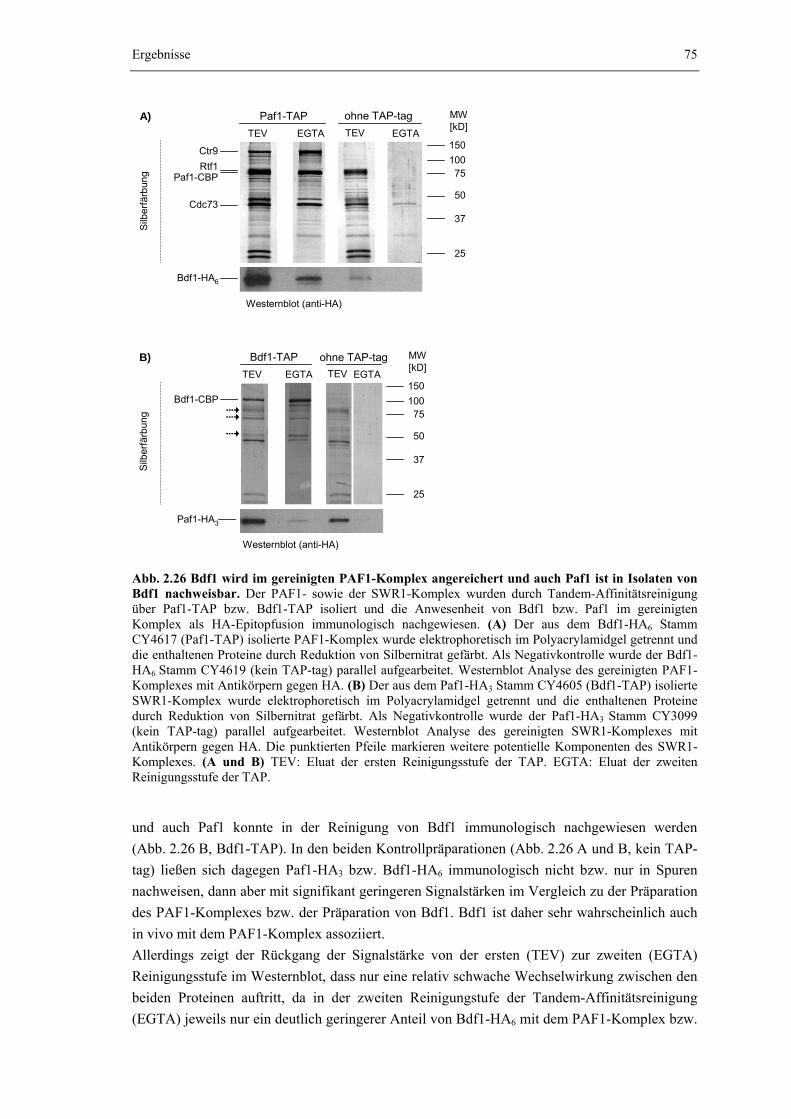
Ergebnisse
75
15010075
50
37
25
MW[kD]
Ctr9Rtf1
Paf1-CBP
Cdc73
Bdf1-HA6
TEV TEVEGTA EGTA
Paf1-TAP ohne TAP-tag
Westernblot (anti-HA)
A)
15010075
50
37
25
MW[kD]
Bdf1-CBP
Paf1-HA3
TEV EGTA TEV EGTA
Bdf1-TAP ohne TAP-tag
Westernblot (anti-HA)
B)
Silb
erfä
rbun
gSi
lber
färb
ung
Abb. 2.26 Bdf1 wird im gereinigten PAF1-Komplex angereichert und auch Paf1 ist in Isolaten von Bdf1 nachweisbar. Der PAF1- sowie der SWR1-Komplex wurden durch Tandem-Affinitätsreinigung über Paf1-TAP bzw. Bdf1-TAP isoliert und die Anwesenheit von Bdf1 bzw. Paf1 im gereinigten Komplex als HA-Epitopfusion immunologisch nachgewiesen. (A) Der aus dem Bdf1-HA6 Stamm CY4617 (Paf1-TAP) isolierte PAF1-Komplex wurde elektrophoretisch im Polyacrylamidgel getrennt und die enthaltenen Proteine durch Reduktion von Silbernitrat gefärbt. Als Negativkontrolle wurde der Bdf1-HA6 Stamm CY4619 (kein TAP-tag) parallel aufgearbeitet. Westernblot Analyse des gereinigten PAF1-Komplexes mit Antikörpern gegen HA. (B) Der aus dem Paf1-HA3 Stamm CY4605 (Bdf1-TAP) isolierte SWR1-Komplex wurde elektrophoretisch im Polyacrylamidgel getrennt und die enthaltenen Proteine durch Reduktion von Silbernitrat gefärbt. Als Negativkontrolle wurde der Paf1-HA3 Stamm CY3099 (kein TAP-tag) parallel aufgearbeitet. Westernblot Analyse des gereinigten SWR1-Komplexes mit Antikörpern gegen HA. Die punktierten Pfeile markieren weitere potentielle Komponenten des SWR1-Komplexes. (A und B) TEV: Eluat der ersten Reinigungsstufe der TAP. EGTA: Eluat der zweiten Reinigungsstufe der TAP.
und auch Paf1 konnte in der Reinigung von Bdf1 immunologisch nachgewiesen werden (Abb. 2.26 B, Bdf1-TAP). In den beiden Kontrollpräparationen (Abb. 2.26 A und B, kein TAP-tag) ließen sich dagegen Paf1-HA3 bzw. Bdf1-HA6 immunologisch nicht bzw. nur in Spuren nachweisen, dann aber mit signifikant geringeren Signalstärken im Vergleich zu der Präparation des PAF1-Komplexes bzw. der Präparation von Bdf1. Bdf1 ist daher sehr wahrscheinlich auch in vivo mit dem PAF1-Komplex assoziiert. Allerdings zeigt der Rückgang der Signalstärke von der ersten (TEV) zur zweiten (EGTA) Reinigungsstufe im Westernblot, dass nur eine relativ schwache Wechselwirkung zwischen den beiden Proteinen auftritt, da in der zweiten Reinigungstufe der Tandem-Affinitätsreinigung (EGTA) jeweils nur ein deutlich geringerer Anteil von Bdf1-HA6 mit dem PAF1-Komplex bzw.

Ergebnisse
76
von Paf1-HA3 mit Bdf1-TAP angereichert wird. Die Anreicherung von Bdf1 im PAF1-Komplex gelingt zudem mit höherer Ausbeute als im umgekehrten Fall, was darauf hindeutet, dass die Interaktion zwischen den beiden Komplexen im nicht-stöchiometrischen Verhältnis oder nur innerhalb eines eingeschränkten Zeitraums erfolgt, z. B. ausschließlich während der Initiation der Transkription. Um diese Hypothese zu testen, wurde das Assoziationsvermögen von Bdf1 mit aktiv transkribierten Genen und dessen Einfluss auf die Rekrutierung des PAF1-Komplexes zur RNA-Polymerase II untersucht.
Bdf1 ist ausschließlich mit der Promotorregion transkribierter Gene assoziiert Das Assoziationsvermögen von Bdf1 mit aktiv transkribierten Genen wurde durch
Chromatin-Immunpräzipitation über Bdf1-TAP Fusionsproteine ermittelt und die Präzipitation von DNA-Fragmenten des CLN3 Promotor-, ORF- und 3'-UTR-Bereichs durch PCR verfolgt. Wie Abb. 2.27 A zeigt, lassen sich aus dem Eluat der ChIP über Bdf1-TAP ausschließlich promotorspezifische Sequenzen amplifizieren. Primerpaare für den Leserahmen und die 3'-UTR von CLN3 führen dagegen nicht zu dem signifikanten Auftreten von PCR-Produkten. Die schwachen Banden sind mit der Signalstärke der Banden in der Negativkontrolle (ohne TAP-tag) identisch und werden daher durch unspezifische Fällung der DNA-Sequenz verursacht.
CLN3 Promotor
CLN3 ORF
INT
CLN3 3'-UTR
Paf1
-TAP
Bdf1
-TAP
ohne
TAP
-tag
RE ChIPA)
ohne
TA
P-ta
g
ohne
TA
P-ta
g
Paf1
-TAP
Paf1
-TAP
Bdf1
-TAP
Bdf1
-TAP
RE IPB)
Abb. 2.27 Bdf1 ist ausschließlich mit der Promotorregion transkribierter Gene assoziiert. Die Promotor-, ORF- und 3’UTR-Assoziation von Paf1 und Bdf1 wurde durch ChIP untersucht. Ganzzellextrakte aus fixierten Zellen des Paf1-TAP Stammes CY3408 und des Bdf1-TAP Stammes CY4604 wurden nach Scherung der DNA durch Sonifizierung mit Hilfe magnetischer Dynabeads immunpräzipitiert und die mitgefällte DNA durch PCR analysiert. Als Negativkontrolle wurde der Wildtypstamm CY1615 in gleicher Weise behandelt (ohne TAP-tag). (A) Die Eluate der ChIP wurden 1 : 5 verdünnt und 5 µL der Eluatverdünnung als Template für die PCR eingesetzt (entsprechend 1 µL unverdünntes ChIP-Eluat; Material & Methoden). Die Proteinassoziation mit der Promotor-, ORF- bzw. 3’-UTR-Region wurde am Beispiel des CLN3-Gens mit spezifischen Primerpaaren für die entsprechende Region untersucht (Promotorprimer: CK2074/CK2075, ORF-Primer CK1740/1741, 3’-UTR-Primer CK2078/CK2079). Die PCR für die ORF-Region von CLN3 wurde als Multiplex-PCR mit einem zusätzlichen Primerpaar (CK1744/1745) als interne Kontrolle (INT) durchgeführt, welches zur spezifischen Amplifikation einer ORF-freien Region auf Chromosom V führt. Als Referenz wurden die PCR-Reaktionen parallel mit dem sonifizierten Rohextrakt (RE) des Wildtypstammes CY1615 in gleicher Weise durchgeführt. (B) Der Erfolg der Immunpräzipitation wurde durch Westernblot Analyse und immunologischen Nachweis des TAP-tags durch den PAP-Komplex kontrolliert.

Ergebnisse
77
Bdf1 ist daher ausschließlich mit der Promotorregion, nicht aber mit dem Leserahmen oder der 3'-UTR des CLN3 Gens assoziiert. Interessanterweise lassen sich aber Sequenzen der ORF-freien Region auf Chromosom V im ChIP-Eluat von Bdf1 nachweisen (Abb. 2.27 A, INT). Bdf1 scheint daher auch mit dieser Region assoziiert zu sein. Dies steht im Einklang mit dem Befund einer Co-Lokalisierung zwischen Bdf1 und H2A.Z, da H2A.Z auch in intergenischen Regionen, die von einem 5’-Genende flankiert werden, verstärkt angereichert ist (Raisner et al., 2005; Zhang et al., 2005).
Im Gegensatz zu Bdf1 lässt sich die Assoziation des PAF1-Komplexes mit allen drei Genbereichen, nicht aber mit der ORF-freien Region auf Chromosom V, nachweisen. Die immunologische Analyse der ChIP-Eluate im Westernblot zeigt zudem, dass sowohl Paf1-TAP als auch Bdf1-TAP durch die Chromatin-Immunpräzipitation mit guter Ausbeute angereichert werden (Abb. 2.27 B).
Die Ergebnisse belegen also die Hypothese, dass die Wechselwirkung zwischen Bdf1 und dem PAF1-Komplex räumlich und zeitlich auf die Initiationsphase der Transkription beschränkt ist.
Die verringerte Assoziation des PAF1-Komplexes mit transkribierten Genen in bdf1Δ Mutanten kann auf eine generell reduzierte Transkriptionsrate zurückgeführt werden
Bdf1 besitzt wie der PAF1-Komplex einen Einfluss auf die Transkriptionsrate mancher,
aber nicht aller Gene (Ladurner et al., 2003). Die Interaktion zwischen Bdf1 und dem PAF1-Komplex könnte daher eine Abhängigkeit der Rekrutierung und/oder der Aktivität des PAF1-Komplexes von Bdf1 widerspiegeln. Bdf1 ist ausschließlich mit der Promotorregion transkribierter Gene assoziiert. Daher sollte nachfolgend die Frage beantwortet werden, ob Bdf1 für die Rekrutierung des PAF1-Komplexes zur RNA-Polymerase II notwendig ist oder diese zumindest beeinflussen kann.
Dazu wurde der BDF1-Leserahmen innerhalb eines Stammes, der eine Paf1-TAP Fusion exprimiert, durch Insertion einer KanMX4 Kassette deletiert und der Einfluss des Funktionsverlustes von Bdf1 auf die RNA-Polymerase II Assoziation des PAF1-Komplexes mittels Chromatin-Immunpräzipitation untersucht. Um die Möglichkeit einer generell reduzierten Transkriptionsrate in bdf1Δ Mutanten von einem direkten Einfluss auf die Rekrutierung des PAF1-Komplexes unterscheiden zu können, wurde die Rekrutierung der RNA-Polymerase II zum Promotor und ORF des CLN3 Gens im WT und in der bdf1Δ Mutante in gleicher Weise mittels ChIP untersucht. Dazu wurde die Rpb3 Untereinheit der RNA-Polymerase II auf genetischer Ebene ebenfalls mit einem C-terminalen TAP-tag fusioniert und die Deletion des BDF1-Leserahmens durch Kreuzung mit einem bdf1Δ Stamm eingeführt. Die Anwesenheit des TAP-tags führt dabei zu keiner beobachtbaren Beeinflussung der Polymerasefunktion (Borggrefe et al., 2001) oder phänotypischen Auffälligkeiten, wie Untersuchungen zum Wachstumsverhalten bei erhöhter Temperatur oder in Gegenwart von 6-Azauracil bzw. 5-Fluororotat zeigen (eigene Beobachtung, Daten nicht gezeigt).
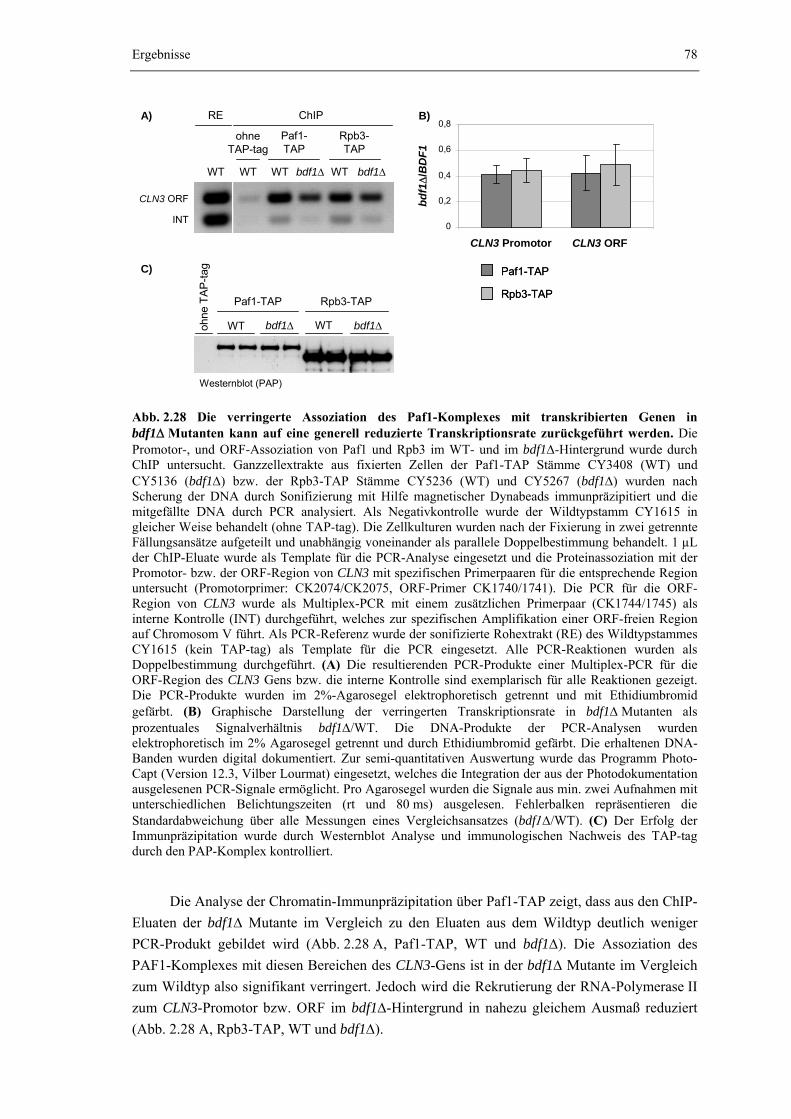
Ergebnisse
78
0
0,2
0,4
0,6
0,8
1CLN3 Promotor CLN3 ORF
bdf1
Δ/B
DF1
Paf1-TAP
Rpb3-TAP
Paf1-TAP
Rpb3-TAP
WT WT WT WTbdf1Δ bdf1Δ
ohneTAP-tag
Paf1-TAP
Rpb3-TAP
RE ChIP
CLN3 ORF
INToh
ne T
AP-
tag
WT WTbdf1Δ bdf1Δ
Paf1-TAP Rpb3-TAP
A)
C)
B)
Westernblot (PAP) Abb. 2.28 Die verringerte Assoziation des Paf1-Komplexes mit transkribierten Genen in bdf1Δ Mutanten kann auf eine generell reduzierte Transkriptionsrate zurückgeführt werden. Die Promotor-, und ORF-Assoziation von Paf1 und Rpb3 im WT- und im bdf1Δ-Hintergrund wurde durch ChIP untersucht. Ganzzellextrakte aus fixierten Zellen der Paf1-TAP Stämme CY3408 (WT) und CY5136 (bdf1Δ) bzw. der Rpb3-TAP Stämme CY5236 (WT) und CY5267 (bdf1Δ) wurden nach Scherung der DNA durch Sonifizierung mit Hilfe magnetischer Dynabeads immunpräzipitiert und die mitgefällte DNA durch PCR analysiert. Als Negativkontrolle wurde der Wildtypstamm CY1615 in gleicher Weise behandelt (ohne TAP-tag). Die Zellkulturen wurden nach der Fixierung in zwei getrennte Fällungsansätze aufgeteilt und unabhängig voneinander als parallele Doppelbestimmung behandelt. 1 µL der ChIP-Eluate wurde als Template für die PCR-Analyse eingesetzt und die Proteinassoziation mit der Promotor- bzw. der ORF-Region von CLN3 mit spezifischen Primerpaaren für die entsprechende Region untersucht (Promotorprimer: CK2074/CK2075, ORF-Primer CK1740/1741). Die PCR für die ORF-Region von CLN3 wurde als Multiplex-PCR mit einem zusätzlichen Primerpaar (CK1744/1745) als interne Kontrolle (INT) durchgeführt, welches zur spezifischen Amplifikation einer ORF-freien Region auf Chromosom V führt. Als PCR-Referenz wurde der sonifizierte Rohextrakt (RE) des Wildtypstammes CY1615 (kein TAP-tag) als Template für die PCR eingesetzt. Alle PCR-Reaktionen wurden als Doppelbestimmung durchgeführt. (A) Die resultierenden PCR-Produkte einer Multiplex-PCR für die ORF-Region des CLN3 Gens bzw. die interne Kontrolle sind exemplarisch für alle Reaktionen gezeigt. Die PCR-Produkte wurden im 2%-Agarosegel elektrophoretisch getrennt und mit Ethidiumbromid gefärbt. (B) Graphische Darstellung der verringerten Transkriptionsrate in bdf1Δ Mutanten als prozentuales Signalverhältnis bdf1Δ/WT. Die DNA-Produkte der PCR-Analysen wurden elektrophoretisch im 2% Agarosegel getrennt und durch Ethidiumbromid gefärbt. Die erhaltenen DNA-Banden wurden digital dokumentiert. Zur semi-quantitativen Auswertung wurde das Programm Photo-Capt (Version 12.3, Vilber Lourmat) eingesetzt, welches die Integration der aus der Photodokumentation ausgelesenen PCR-Signale ermöglicht. Pro Agarosegel wurden die Signale aus min. zwei Aufnahmen mit unterschiedlichen Belichtungszeiten (rt und 80 ms) ausgelesen. Fehlerbalken repräsentieren die Standardabweichung über alle Messungen eines Vergleichsansatzes (bdf1Δ/WT). (C) Der Erfolg der Immunpräzipitation wurde durch Westernblot Analyse und immunologischen Nachweis des TAP-tag durch den PAP-Komplex kontrolliert.
Die Analyse der Chromatin-Immunpräzipitation über Paf1-TAP zeigt, dass aus den ChIP-Eluaten der bdf1Δ Mutante im Vergleich zu den Eluaten aus dem Wildtyp deutlich weniger PCR-Produkt gebildet wird (Abb. 2.28 A, Paf1-TAP, WT und bdf1Δ). Die Assoziation des PAF1-Komplexes mit diesen Bereichen des CLN3-Gens ist in der bdf1Δ Mutante im Vergleich zum Wildtyp also signifikant verringert. Jedoch wird die Rekrutierung der RNA-Polymerase II zum CLN3-Promotor bzw. ORF im bdf1Δ-Hintergrund in nahezu gleichem Ausmaß reduziert (Abb. 2.28 A, Rpb3-TAP, WT und bdf1Δ).

Ergebnisse
79
Die semi-quantitative Auswertung der DNA-Mengen, die durch PCR aus den entsprechenden ChIP-Eluaten erhalten werden, zeigt, dass durch Verlust der Bdf1-Funktion sowohl die Assoziation von Paf1 als auch die Assoziation der RNA-Polymerase II mit der Promotor- bzw. der ORF-Region von CLN3 um ca. 50% im Vergleich zum Wildtyp verringert wird (Abb. 2.28 B). Die jeweilige Proteinfusion mit dem TAP-tag kann dabei aus der bdf1Δ Mutante im Vergleich zum Wildtyp jeweils mit der gleichen Effizienz und Ausbeute immunpräzipitiert werden (Abb. 2.28 C).
Paf1-Komplex und RNA-Polymerase II werden also durch Deletion von BDF1 in gleicher Weise in ihrer Promotor- und ORF-Rekrutierung beeinträchtigt. Bdf1 kann durch seine Bindung an acetyliertes Histon H4 die Rekrutierung des generellen Transkriptionsfaktors TFIID vermitteln und besitzt somit einen direkten Einfluss auf die Transkriptionsinitiation einiger Gene (Durant and Pugh, 2006). Hinzu kommt, dass der Austausch von Histon H2A durch H2A.Z direkt von Bdf1 abhängig ist und auch H2A.Z einen unmittelbaren Einfluss auf die Transkription besitzt. Daher liegt der Schluss nahe, dass die generelle Transkriptionsrate bezogen auf die Transkriptionsaktivierung durch den Funktionsverlust von Bdf1 reduziert wird. Aufgrund dieses generellen Effekts bleibt unklar, ob speziell die Assoziation des Paf1-Komplexes mit der RNA-Polymerase II durch Bdf1 beeinflusst wird. Die Assoziationsstudien durch die Chromatin-Immunpräzipitation lassen zwar einen leichten Trend für eine zusätzliche Reduzierung der Promotor- bzw. ORF-Assoziation des PAF1-Komplexes durch Deletion von BDF1 erkennen (Abb. 2.28 B), durch die Schwankung der Messwerte kann aber nicht entschieden werden, ob dieser Einfluss als signifikant bewertet werden kann.
bdf1Δ Mutanten zeigen keinen Elongationsdefekt bei Exposition mit 6-Azauracil, weisen aber eine deutliche Sensitivität gegen 5-Fluororotat in Gegenwart eines telomergekoppelten URA3-Reportergens auf
Bdf1 zeigt also keinen signifikanten Einfluss auf die Rekrutierung des Elongationsfaktors
PAF1 zur RNA-Polymerase II. Dennoch bleibt die Möglichkeit offen, dass die Aktivität des PAF1-Komplexes oder anderer Elongationsfaktoren (z. B. yFACT) durch die Interaktion mit Bdf1 beeinflusst bzw. reguliert wird. Ob die Deletion von BDF1 eine Auswirkung auf die transkriptionelle Elongation besitzt, sollte daher, entsprechend den Untersuchungen in Kapitel 2.2.4, durch das Wachstumsverhalten der bdf1Δ Mutante in Gegenwart von 6-Azauracil untersucht werden.
In Vorversuchen zeigte sich allerdings, dass die bdf1Δ Mutante nicht in der Lage ist auf dem standardmäßig eingesetzten Uracil-Mangelmedium auf Casaminoacid-Basis (Material & Methoden) zu wachsen. Dieser Wachstumsdefekt kann durch nachträgliche Zugabe von Uracil aufgehoben werden (Daten nicht gezeigt). Die Ursache hierfür wurde nicht genauer untersucht. Im Gegensatz dazu ist die bdf1Δ Mutante problemlos zum Wachstum auf SCD und SCD-URA Medium in der Lage. Das Wachstumsverhalten in Gegenwart von 6-Azauracil wurde daher auf SCD-URA Medium mit oder ohne Zusatz von 50, 75 und 100 mg·L-1 6-Azauracil untersucht.

Ergebnisse
80
WT
paf1Δ
cdc73Δ
bdf1Δ
6AU 100 mg.L-1
(Tag 6, 30°C)SCD-URA
(Tag 6, 30°C)
WT
rad6Δ
bdf1Δ
5FOA 1,0 mg.mL-1
(Tag 4, 30°C)SCD
(Tag 4, 30°C)
WT
paf1Δ
cdc73Δ
bdf1Δ
YPD 37°C(Tag 2)
A)
B)
C)
WT
paf1Δ
cdc73Δ
bdf1Δ
6AU 100 mg.L-1
(Tag 6, 30°C)SCD-URA
(Tag 6, 30°C)
WT
rad6Δ
bdf1Δ
5FOA 1,0 mg.mL-1
(Tag 4, 30°C)SCD
(Tag 4, 30°C)
WT
rad6Δ
bdf1Δ
5FOA 1,0 mg.mL-1
(Tag 4, 30°C)SCD
(Tag 4, 30°C)
WT
paf1Δ
cdc73Δ
bdf1Δ
YPD 37°C(Tag 2)
A)
B)
C)
Abb. 2.29 bdf1Δ Mutanten zeigen keinen Elongationsdefekt in Gegenwart von 6-Azauracil, weisen aber eine deutliche Sensitivität gegen 5-Fluororotat in Gegenwart eines telomergekoppelten URA3-Reportergens auf. (A) Sensitivität gegen 6-Azauracil. Zellen des Wildtypstamms CY1615 (pep4::URA3) und der Deletionsstämme CY1994 (paf1::URA3), CY4623 (cdc73Δ, pep4::URA3) und CY5134 (bdf1Δ, pep4::URA3) wurden in serieller Verdünnung auf SCD-URA Medium mit oder ohne 100 mg.L-1 6-Azaurazil aufgetropft und das Wachstum bei 30°C über einen Zeitraum von 8 Tagen verfolgt. (B) Sensitivität gegen 5-Fluororotsäure. Zellen des Wildtypstammes CY4964 (TELVIIL::URA3) und der Deletionsstämme CY5023 (rad6Δ, TELVIIL::URA3) und CY5263 (bdf1Δ, TELVIIL::URA3) wurden in serieller Verdünnung auf SCD-Medium mit und ohne 1,0 mg.mL-1 5-Fluororotsäure aufgetropft und die Expression des telomergekoppelten URA3-Gens anhand des Zellwachstums über einen Zeitraum von 8 Tagen verfolgt. (C) Wachstum bei 37°C. Zellen der Hefestämme aus (A) wurden in serieller Verdünnung auf YPD Medium aufgetropft und ihr Wachstum bei 37°C über einen Zeitraum von 3 Tagen verfolgt.
Die Deletion von BDF1 führt zu keiner signifikanten Beeinträchtigung des Zellwachstums in Gegenwart von 6-Azauracil. Wie Abb. 2.29 A zeigt, wächst die bdf1Δ Mutante auch bei einer 6-Azauracilkonzentration von 100 µg.L-1 annähernd gleich gut wie auf entsprechendem Medium ohne 6-Azauracil. Die Deletion von PAF1 führt dagegen wiederholt zu einer drastischen Sensitivität gegen 6-Azauracil und auch das Wachstum der cdc73Δ Mutante wird durch 6-Azauracil im Vergleich zur bdf1Δ Mutante stärker beeinträchtigt. Bdf1 scheint daher keinen direkten Einfluss auf die Elongationsfähigkeit der RNA-Polymerase II auszuüben. In diesem Zusammenhang ist allerdings interessant, dass die Deletion von H2A.Z zu einer Sensitivität gegen Mycophenolsäure und 6-Azauracil führt, was einen Einfluss von H2A.Z auf die Elongationsfähigkeit der RNA-Polymerase II andeuten würde (Desmoucelles et al., 2002).

Ergebnisse
81
Da der Einbau der Histonvariante H2A.Z in Nukleosomen zur Inhibierung der HMTs Set2 und Dot1 führt (Li et al., 2005) und die dot1Δ Mutante einen drastischen Defekt im Telomersilencing aufweist (Singer et al., 1998), wurde ein möglicher Effekt von Bdf1 auf das Telomersilencing mit Hilfe eines telomergekoppelten URA3-Reportergens und der Wachstumsfähigkeit in Gegenwart von 5-Fluororotsäure wie in Kapitel 2.2.5 beschrieben untersucht. Zur allgemeinen Charakterisierung der bdf1Δ Mutante unter Stressbedingungen wurde zusätzlich ihr Wachstumsverhalten unter Hitzestress von 37°C untersucht.
Im Gegensatz zur Resistenz gegen 6-Azauracil wird das Wachstum der bdf1Δ Mutante mit dem telomergekoppelten URA3-Reportergen durch 5-Fluororotsäure komplett eliminiert (Abb. 2.29 B). Wie auch im Falle der Deletion von RAD6, die unmittelbar zu einem Aktivitätsverlust von Dot1 und damit zu einer Unterbindung des Telomersilencings führt, scheint das URA3-Reportergen in der bdf1Δ Mutante nahezu uneingeschränkt exprimierbar zu sein. Der beobachtete Wachstumsdefekt ist dabei an die Anwesenheit des telomergekoppelten URA3-Reportergens geknüpft, da sich die Deletion von BDF1 alleine nicht in einer erhöhten Sensitivität gegen 5-Fluororotsäure äußert (Daten nicht gezeigt). Der Verlust der Bdf1-Aktivität führt also zu einer Aufhebung des Telomersilencings. Dies war nicht unbedingt zu erwarten, da die Histonvariante H2A.Z, deren Einbau in Nukleosomen direkt von Bdf1 abhängt, im Gegensatz dazu als Anti-Silencingfaktor wirkt und für die Aufrechterhaltung der Heterochromatingrenzen benötigt wird (Meneghini et al., 2003). Und auch Bdf1 wurde bereits als Anti-Silencingfaktor beschrieben, der die Ausbreitung der Sir-Proteine und damit die Ausspreitung von Heterochromatin in euchromatische Regionen unterbindet (Ladurner et al., 2003). Obwohl widersprüchlich zu der nachgewiesenen Anti-Silencing Funktion, führt aber auch der Verlust von H2A.Z durch Deletion des HTZ1 Gens zu einer Sensitivität der htz1Δ Mutante in Gegenwart eines telomergekoppelten URA3-Reportergens (Dhillon and Kamakaka, 2000).
bdf1Δ- und paf1Δ Mutanten teilen neben der Hypersensitivität gegen 5-Fluororotsäure in Anwesenheit eines telomergekoppelten URA3-Reportergens auch die Temperatursensitivität und sind nicht zum Wachstum bei 37°C befähigt (Abb. 2.29 C). Der Verlust von H2A.Z äußert sich dagegen nicht in einem temperatursensitiven Wachstumsverhalten (Daten nicht gezeigt), führt aber zu einer Sensitivität gegen 6-Azauracil und Mycophenolsäure (Desmoucelles et al., 2002), die für die bdf1Δ Mutante nicht beobachtet wird. Aufgrund dieser phänotypischen Diskrepanz zwischen bdf1Δ- und htz1Δ Mutanten (H2A.Z Mutante) wird deutlich, dass Bdf1 neben seiner Rolle als Austauschfaktor für H2A.Z noch weitere Funktionen besitzen muss, die auch durch seinen Einfluss auf die Rekrutierung von TFIID bereits dokumentiert werden (Durant and Pugh, 2006).
Die Di- und Tri-Methylierung von Histon H3Lys79 ist in bdf1Δ- Mutanten nicht beeinträchtigt
Da Bdf1 bereits als Anti-Silencingfaktor charakterisiert wurde (Ladurner et al., 2003), die
Sensitivität gegen 5-Fluororotsäure in Anwesenheit eines telomergekoppelten URA3-Reportergens jedoch einen Defekt im Telomersilencing andeutet, sollte als nächstes der

Ergebnisse
82
Methylierungsstatus von Histon H3 in der bdf1Δ Mutante untersucht werden. Da sowohl die Deletion der Histon-Methyltransferase SET1 als auch die Deletion von DOT1 zu einem Verlust des Telomersilencings führt und beide HMTs funktionell von Rad6 abhängig sind (Fingerman et al., 2005; Lacoste et al., 2002; Shahbazian et al., 2005), wurde exemplarisch die Dot1-abhängige Di- und Tri-Methylierung von Histon H3 an Lysin 79 in bdf1Δ, htz1Δ und rad6Δ Mutanten untersucht. Die Histone aus den entsprechenden Deletionsstämmen wurden, wie in Kapitel 2.2.6 beschrieben, durch Proteinextraktion mit Trichloressigsäure gewonnen und nach elektrophoretischer Auftrennung im High-Tris Polyacrylamidgel immunologisch im Westernblot mit Antikörpern gegen Di- bzw. Tri-methyliertes Histon H3Lys79 untersucht.
Weder Bdf1 noch H2A.Z (Htz1) sind für die Di- bzw. Tri-Methylierung von Histon H3Lys79 notwendig. Wie Abb. 2.33 zeigt, lässt sich die jeweilige Histonmodifikation in Proteinextrakten von bdf1Δ- bzw. htz1Δ-Stämmen immunologisch nachweisen. Die Deletion RAD6 führt dagegen klar zu einer signifikanten Beeinträchtigung bzw. zum vollständigen Verlust der Di- bzw. Tri-Methylierung von Histon H3Lys79.
Dieser Befund ermöglicht die Hypothese, dass die Aktivierung der Transkription, aber vermutlich nicht die Rekrutierung des PAF1-Komplexes zur RNA-Polymerase II in bdf1Δ Mutanten beeinträchtigt ist und stimmt mit der Beobachtung überein, dass bdf1Δ Mutanten keine ausgeprägten Elongationsdefekte zeigen (Abb.2.29 A). Zudem bleibt auch die Funktion des PAF1-Komplexes als Regulator des Rad6/Bre1-Komplexes (Xiao et al., 2005) von Bdf1 unbeeinflusst, da im Gegensatz zur rad6Δ Mutante keine Beeinträchtigung der Methylierung von Histon H3Lys79 in der bdf1Δ Mutante auftritt. Aus dem gleichen Grund kann auch die beobachtete Störung des telomeren Silencings in der bdf1D Mutante nicht auf den Ausfall der beiden Histon-Methyltransferasen Set1 und Dot1 zurückgeführt werden. Ob Bdf1 die posttranskriptionale Funktion des PAF1-Komplexes bei der mRNA 3’-Endprozessierung (Sheldon et al., 2005) beeinflusst, kann dagegen nicht entschieden werden.
Die Beobachtung, dass die Tri-Methylierung von H3Lys79 auch in der htz1Δ Mutante funktional ist, steht dabei im Einklang mit dem Befund, dass gerade der Einbau von H2A.Z in Nukleosomen zu einer Inhibierung der HMTs Set2 und Dot1 führt (Li et al., 2005).
htz1
Δ
Tri-Methyl H3Lys79
WTbdf1
Δ
Di-Methyl H3Lys79
*
rad6
Δ
htz1
Δ
Tri-Methyl H3Lys79
WTbdf1
Δ
Di-Methyl H3Lys79
*
rad6
Δ
Abb. 2.30 Die tri-Methylierung von Histon H3Lys79 ist in bdf1Δ- und htz1Δ-Mutanten nicht beeinträchtigt. Aus Zellen des Wildtypstammes CY1915 und der Deletionsstämme CY4077 (rad6Δ), CY5133 (bdf1Δ) und CY5379 (htz1Δ) wurde Gesamtprotein durch Extraktion mit Trichloressigsäure gewonnen. Die TCA-Extrakte wurden elektrophoretisch im 16,6% High-Tris Polyacrylamidgel (Vernetzungsgrad 5,1%) getrennt und immunologisch im Westernblot mit Antikörpern gegen di-methyliertes bzw. tri-methyliertes Histon H3Lys79 untersucht. Der Stern markiert N-terminale Abbauprodukte von Histon H3 (Xiao et al., 2005).

Diskussion
83
3. Diskussion
Der Einfluss des Elongationsfaktors PAF1 auf die co-transkriptionelle Methylierung von Histon H3 und seine Funktion bei der auf den Transkriptionsverlauf abgestimmten Prozessierung des 3'-Endes der naszierenden mRNA konnten innerhalb der letzten Jahre aufgeklärt werden (Krogan et al., 2003a; Krogan et al., 2003c; Ng et al., 2003a; Penheiter et al., 2005; Sheldon et al., 2005; Wood et al., 2003b). Die genaue Funktionsweise des Elongationsfaktors bleibt dabei bislang jedoch unverstanden. Um den Einfluss der einzelnen Untereinheiten des PAF1-Komplexes für dessen Funktion besser zu verstehen, wurde in dieser Arbeit die Protein-Protein-Interaktion zwischen den Komponenten des Komplexes und die Wechselwirkung zu Interaktionspartnern eingehend untersucht. Ausgehend von der Isolierung und Identifizierung der fünf Komponenten des Elongationsfaktors PAF1 konnte dabei ein genauerer Einblick in Zusammensetzung und Aufbau des pentameren Komplexes gewonnen werden, der sich auch in der funktionellen Charakterisierung des Elongationsfaktors bzw. seiner einzelnen Komponenten widerspiegelt. Zudem wurde die starke Interaktion zwischen den Elongationsfaktoren PAF1, yFACT und der Casein-Kinase II deutlich und zusätzlich eine Wechselwirkung zwischen PAF1-Komplex und dem Chromatin-Strukturmodulator SWR1 bzw. dessen Bdf1-Komponente aufgedeckt. 3.1 Protein-Protein-Interaktionen innerhalb und außerhalb des
PAF1-Komplexverbundes
Zusammensetzung und Aufbau des PAF1-Komplexes
Die verschiedenen Präparationen des Elongationsfaktors PAF1 durch Tandem-Affinitätsreinigung (Kapitel 2.1, Abb. 2.1-2.3) führten reproduzierbar und unabhängig davon, über welche Untereinheit die Reinigung erfolgte, zur selektiven Anreicherung von fünf Proteinen, die durch die massenspektrometrische bzw. immunologische Analyse als Ctr9, Paf1, Rtf1, Leo1 und Cdc73 identifiziert werden konnten (Kapitel 2.1.1). Der Elongationsfaktor PAF1 erweist sich daher als homogen aufgebauter Komplex mit pentamerer Struktur. Dieses Ergebnis stimmt mit den Befunden mehrerer unabhängiger Studien überein (Krogan et al., 2002b; Mueller and Jaehning, 2002; Squazzo et al., 2002).
Wie die in Kapitel 2.1.3 beschriebenen Untersuchungen zeigen, lassen sich durch die gezielte Deletion einzelner Komponenten auf genetischer Ebene weitere Einblicke in den Aufbau und die Protein-Protein-Interaktionen innerhalb des PAF1-Komplexes gewinnen. Dabei zeigt sich, dass die Interaktion zwischen Paf1 und Ctr9 von entscheidender Bedeutung für den Zusammenhalt des Komplexes ist (Abb. 2.3). Aus einer paf1Δ Mutante wurde über Ctr9-TAP nur Ctr9 selbst und Cdc73 angereichert. Die Interaktion mit Leo1 und Rtf1 geht durch die Deletion von PAF1 dagegen verloren. Auch aus einer ctr9Δ Mutante konnte Rtf1 nicht über Paf1-TAP angereichert werden. Ebenso ist Cdc73 in ctr9Δ Stämmen nicht mit Paf1 assoziiert,

Diskussion
84
was die Bindung von Cdc73 an Ctr9 zusätzlich demonstriert. Ob die Assoziation zwischen Leo1 und Paf1 in einer ctr9Δ Mutante erhalten bleibt, ist unklar, da die entsprechenden Banden im verwendeten elektrophoretischen System nicht aufgelöst wurden.
Im Gegensatz zu Paf1 und Ctr9 können Rtf1 und Leo1 aus dem Komplexverbund entfernt werden, ohne die Interaktion der verbleibenden vier Komponenten zu beeinflussen. Auch die Deletion von CDC73 führt nicht zu dem Verlust der Interaktion zwischen Paf1, Ctr9, Rtf1 und Leo1. Allerdings wird die Bindung von Rtf1 durch die Deletion von CDC73 geschwächt (Abb. 2.4). Damit muss Ctr9 also sowohl mit Paf1 als auch mit Cdc73 und vermutlich auch mit Rtf1 eine direkte Bindung eingehen, wobei die Bindung von Rtf1 nur bei gleichzeitiger Interaktion mit Paf1 erfolgt. Auch Paf1 scheint demzufolge unmittelbar mit Rtf1 assoziiert zu sein. Paf1 und Ctr9 sind also entscheidend für den Zusammenhalt des PAF1-Komplexes und der Verlust ihrer Interaktion führt aller Wahrscheinlichkeit nach zur Dissoziation des Elongationsfaktors. Die in Kapitel 2.1.3 gewonnenen Daten und die daraus abgeleiteten Schlussfolgerungen zum Aufbau des PAF1-Komplexes konnten zudem durch die in vitro Phosphorylierung von Ctr9, Paf1, Rtf1 und Leo1 durch die Casein-Kinase II untermauert und bestätigt werden (vgl. Kapitel 2.3.2), da die Proteinfärbung des isolierten PAF1-Komplexes sowie die Autoradiographie der radioaktiv phosphorylierten Komponenten zu einem sich entsprechenden Bandenmuster führt bzw. die Deletion bestimmter Untereinheiten den Verlust von Banden gleicher Identität in der Autoradiographie und der Proteinfärbung verursacht (Abb. 2.3 und 2.23).
Die zelluläre Konzentration bzw. Menge der jeweils verbleibenden Bestandteile des PAF1-Komplexes wird durch die Deletion von PAF1 oder CTR9 signifikant verringert (Mueller et al., 2004; eigene Beobachtung, Kapitel 2.1.3). Dies könnte die Folge einer verstärkten Proteindegradation der aufgrund der Destabilisierung des Komplexes frei vorliegenden Komponenten des PAF1-Komplexes sein und auch die drastisch verminderte Reinigungsausbeute des PAF1-Komplexes aus ctr9Δ bzw. paf1Δ Mutanten erklären (Abb. 2.3). Die Degradation frei vorliegender Untereinheiten des PAF1-Komplexes wäre dabei kein Sonderfall. Vielmehr kann dieser Effekt bei Multiproteinkomplexen sehr häufig beobachtet werden, wie die Instabilität der ribosomalen Untereinheiten außerhalb des Ribosomenverbundes oder der Abbau überschüssiger α- und β-Globin Untereinheiten außerhalb des tetrameren Hämoglobins zeigt (Übersicht in: Goldberg, 2003; Goldberg and Dice, 1974). Eine weitere Ursache für die reduzierte Proteinmenge könnte aber auch darin begründet sein, dass der Elongationsfaktor für die Expression seiner eigenen Komponenten benötigt wird. Die in Kapitel 2.2.7 beschriebenen Untersuchungen der Rekrutierung des PAF1-Komplexes zu bestimmten Genen würden diese Hypothese unterstützen, da der Komplex mit den Leserahmen der Gene seiner fünf Komponenten assoziiert ist (Abb. 2.17 und Daten nicht gezeigt). Allerdings liefert eine Studie, die den Einfluss des PAF1-Komplexes auf die genomweite Genexpression in Hefe untersucht, kein Indiz dafür, dass die Expression dieser Gene in paf1Δ oder ctr9Δ Mutanten beeinträchtigt ist (Penheiter et al., 2005), so dass eine Autoregulation der Expression der Komponenten des PAF1-Komplexes unwahrscheinlich ist.

Diskussion
85
TPR-Motive in Ctr9 bilden spezifische Bindedomänen für Paf1 und Cdc73
In der Primärstruktur von Ctr9 sind mindestens zehn Abschnitte enthalten, die anhand der Sequenzanalyse als Tetratricopeptide Repeat-Motive identifiziert werden können (vgl. Kapitel 2.1.4, Abb. 2.5). Diese Strukturmotive vermitteln die spezifische Interaktion zwischen Proteinen und ermöglichen die Ausbildung von Multiproteinkomplexen (Cortajarena and Regan, 2006; D'Andrea and Regan, 2003). Da Ctr9 eine entscheidende Determinante für die Stabilität des PAF1-Komplexes darstellt und mindestens Paf1 und Cdc73 direkt an Ctr9 binden, lag die Vermutung nahe, dass die TPR-Motive in Ctr9 die Bindung zu den übrigen Bestandteilen des Komplexes vermitteln. Um dies zu überprüfen, wurde die Auswirkung der gezielten Verkürzung des C-Terminus von Ctr9 um diskrete TPR-Motive auf die Zusammensetzung des über diese Ctr9-Varianten isolierten PAF1-Komplexes ermittelt. Außerdem wurde die Affinität von heterolog exprimierten Ctr9-Proteinfragmenten, die definierte TPR-Motive enthalten, zu Paf1 und Cdc73 in vitro untersucht. Die Ergebnisse dieser Untersuchungen (Kapitel 2.1.5) bestätigen die Vermutung, dass sich die TPR-Motive in Ctr9 zu spezifischen Bindedomänen falten und entscheidend am Zusammenhalt des PAF1-Komplexes beteiligt sind. Insbesondere konnte die Lokalisation der Bindedomäne für Paf1 bzw. Cdc73 ermittelt werden (Abb. 2.6-2.9). Die Interaktion zwischen Paf1 und Ctr9 geht in vivo auch bei Verkürzung der C-terminalen Aminosäuren 538-1077 von Ctr9 nicht verloren und bleibt vermutlich auch bei Ausweitung der Verkürzung bis zu Aminosäure 459-1077 erhalten. Zudem konnte gezeigt werden, dass Paf1 in vitro an den N-terminalen Ctr9-Bereich von Aminosäure 2-549 binden kann (Abb. 2.9). Auch eine um die Aminosäuren 396-974 verkürzte Ctr9-Variante ist noch zur Co-Immunpräzipitation mit Paf1-HA3 in der Lage (C. Koch, persönliche Mitteilung). Die Paf1-Interaktionsdomäne von Ctr9 muss also im Bereich der N-terminalen Aminosäuren 2-395 lokalisiert sein. In diesen Bereich fallen die TPR-Motive 1-3. TPR-Motiv 4 wird durch die partielle Deletion von Ctr9 bereits zum Teil zerstört. Jede einzelne TPR-Struktur besteht aus einem charakteristischen Helix-Turn-Helix-Motiv (D'Andrea and Regan, 2003). Erst die höhergeordnete Faltung aufeinander folgender TPR-Motive führt jedoch zur Ausbildung der Protein-Bindungsdomänen, wobei ein Modul aus 1,5 TPR-Motiven als Minimalvoraussetzung für die Ausbildung einer stabilen Bindungsdomäne beschrieben wurde (Blatch and Lassle, 1999; Main et al., 2003). Da das TPR-Motiv 3 durch 86 Aminosäuren von der Tandemanordnung der TPR-Motive 1 & 2 getrennt ist, wird die Bindedomäne für Paf1 daher vermutlich von den beiden TPR-Motiven 1 & 2 ausgebildet (Abb. 2.5 und 3.1). Ein direkter Beleg hierfür steht jedoch noch aus und könnte beispielsweise durch die in vitro Bindung eines synthetischen Peptids, das aus diesen beiden TPR-Motiven besteht, an freies und in reiner Form vorliegendes Paf1-Protein gezeigt werden. Auch für die Bindung von Cdc73 ist wahrscheinlich eine Anordnung aus zwei TPR-Motiven ausreichend. Wie im Falle der Paf1-Bindung bleibt die direkte Demonstration der Wechselwirkung dieser TPR-Motive mit isoliertem und in reiner Form vorliegendem Cdc73 jedoch offen. Allerdings konnte durch die sukzessive C-terminale Verkürzung von Ctr9 gezeigt werden, dass der C-terminale Ctr9-Bereich zwischen den Aminosäuren 728-1077 in vivo für die Bindung von Cdc73 erforderlich ist (Abb. 2.7). Durch die Untersuchung der Bindungsaffinität in vitro konnte die für die Bindung von Cdc73 erforderliche Region zudem weiter auf die C-terminalen Aminosäuren 716-816 eingeschränkt werden (Abb. 2.8). Dieser Bereich besteht

Diskussion
86
im Wesentlichen aus der Tandem-Anordnung der beiden TPR-Motive 9 & 10, flankiert von den 16 bzw. 15 anschließenden Aminosäuren (Abb. 2.5 und Tabelle 2.1). Die beiden TPR-Motive 9 & 10 bilden daher sehr wahrscheinlich die spezifische Bindedomäne für Cdc73 oder sind zumindest an ihr beteiligt, da sie sich in vitro als hinreichend und zudem in vivo als notwendig für die Bindung von Cdc73 erweisen. Dabei zeigt sich, dass auch der an das TPR-Motiv 10 angrenzende Bereich, welcher auch eine "Coiled-Coil" Domäne beinhaltet, Einfluss auf die Stärke der Cdc73-Bindung besitzt (Abb. 2.6 und 2.7). Dieser Bereich scheint daher stabilisierend auf die durch die TPR-Motive 9 & 10 gebildete Cdc73-Bindungsdomäne zu wirken.
Die Hypothese, dass die TPR-Motive 1 & 2 bzw. 9 & 10 spezifische Bindungsdomänen für Paf1 bzw. Cdc73 darstellen oder zumindest maßgeblich an diesen Domänen beteiligt sind, wird durch die in Kapitel 2.1.4 beschriebene Analyse der TPR-Motive in den zu Ctr9 orthologen Proteinen anderer Organismen gestützt. Die vier TPR-Motive werden auch in den Ctr9 Orthologen von M. grisea, S. pombe, C. elegans und D. melanogaster gefunden und sind daher wahrscheinlich evolutionär konserviert. Zudem zeigt ein Sequenzvergleich zwischen dem Ctr9-Protein der Hefe und dem humanen Ctr9-Protein, dass die evolutionäre Konservierung der Hefe TPR-Motive 1 & 2 bzw. 9 & 10 bis hin zum Menschen reicht und diese vier Domänen den in der Proteindatenbank Expasy ausgewiesenen N-terminalen TPR-Motiven 3 & 4 bzw. den C-terminalen TPR-Motiven 15 & 16 des humanen Proteins entsprechen (Daten nicht gezeigt und http://www.expasy.org/uniprot/Q6PD62).
Der Einfluss der einzelnen Untereinheiten des PAF1-Komplexes auf die Wechselwirkungen mit Interaktionspartnern
Die massenspektrometrische und immunologische Analyse des isolierten PAF1-Komplexes zeigt, dass der Komplex eine physikalische Wechselwirkung mit dem Elongationsfaktor yFACT und der Casein-Kinase II eingeht (Kapitel 2.3). yFACT und die Casein-Kinase II stellen daher sehr wahrscheinlich in vivo physiologisch relevante Interaktionspartner des PAF1-Komplexes dar. Die starke Wechselwirkung zwischen den beiden Elongationsfaktoren PAF1 und yFACT sowie der Casein-Kinase II wird dadurch deutlich, dass die Reinigung eines Komplexes jeweils zur Anreicherung der beiden anderen Proteinkomplexe führt (Kapitel 2.3, Abb. 2.20 und 2.21). Zudem konnte demonstriert werden, dass die Assoziation der Casein-Kinase II mit der RNA-Polymerase II auch während der Elongation bestehen bleibt (Kapitel 2.3, Abb. 2.25). Diese Befunde stehen im Einklang mit einer Studie von Krogan et al. (2002), die unabhängig von dieser Arbeit ebenfalls zeigt, dass die Elongationsfaktoren PAF und yFACT sowie die Casein-Kinase II jeweils in den Präparationen der anderen Faktoren enthalten sind.
Die Interaktion des PAF1-Komplexes mit yFACT und der Casein-Kinase II erfolgt unabhängig von den Komponenten Rtf1, Leo1 und Cdc73, da diese aus dem Komplexverbund entfernt werden können, ohne die Interaktion zwischen PAF1-Komplex und yFACT bzw. der Casein-Kinase II zu unterbinden (Abb. 2.20 bzw. 2.23). Durch die Untersuchung des aus verschiedenen Mutanten isolierten Elongationsfaktors PAF1 wird zudem unmittelbar deutlich, dass die Wechselwirkung zwischen yFACT und PAF1-Komplex an die Interaktion zwischen

Diskussion
87
Paf1 und Ctr9 gebunden ist, da Spt16 in einer ctr9Δ Mutante nicht mit Paf1 bzw. in einer paf1Δ Mutante nicht mit Ctr9 assoziiert ist (Abb. 2.20). Die Ergebnisse zeigen auch, dass Rtf1, Leo1 und vor allem Cdc73 einen stabilisierenden Einfluss auf die Assoziation zwischen yFACT und PAF1-Komplex besitzen (Abb. 2.20).
Die Inkubation des gereinigten PAF1-Komplexes mit γ-32P-ATP zeigt, dass die Assoziation des Komplexes mit der Casein-Kinase II zur in vitro Phosphorylierung der Komplexbestandteile Ctr9, Rtf1, Leo1 und Paf1 ausgenutzt werden kann (Abb. 2.23). Aufgrund der autoradiographischen Analyse wurde dabei deutlich, dass auch eine Präparation von Paf1 aus einer ctr9Δ Mutante noch CKII-abhängige Kinaseaktivität besitzt (Abb. 2.23). Da die Deletion von CTR9 aber die Interaktion von Paf1 mit Cdc73, Rtf1 und vermutlich auch mit Leo1 unterbindet (Abb. 2.3 und 2.23), muss die Interaktion zwischen Casein-Kinase II und dem Elongationsfaktor PAF1 direkt durch seine Paf1-Komponente vermittelt werden. Ob die Phosphorylierung des PAF1-Komplexes auch in vivo für die Funktion des Elongationsfaktors relevant ist, wurde anhand der physiologischen Auswirkungen untersucht, die die Entfernung des durch CKII-phosphorylierten Ctr9-Bereichs nach sich zieht. Die Position der CKII-abhängigen Phosphorylierung von Ctr9 konnte dabei auf die C-terminale Region zwischen Aminosäure 890 und 1054 eingegrenzt werden (Abb. 2.24). Die Verkürzung des C-Terminus von Aminosäure 805-1077 führte nur zu einer geringfügigen Schwächung der Stabilität des PAF1-Komplexes, da sie eine leichte Beeinträchtigung der Assoziation von Cdc73 verursacht (Abb. 2.7). Die entsprechende Mutante zeigt jedoch bereits eine gewisse Temperatursensitivität (Abb. 2.13 und 2.15). Dagegen wurde noch keine Sensitivität der Mutante gegen 6-Azauracil festgestellt, die erst bei weiterer C-terminaler Verkürzung von Ctr9 auftrat, so dass keine Hinweise auf eine mögliche Beeinträchtigung der Elongation durch den Wegfall der durch CKII-phosphorylierten Ctr9-Region erhalten wurden (Abb. 2.13). Zudem wurde auch keine Beeinträchtigung des telomeren Silencings in dieser Mutante beobachtet (Abb. 2.15). Daher bleibt unklar, ob der CKII-abhängigen Phosphorylierung von Ctr9 in vivo eine Funktion zukommt. Aber auch Rtf1, Leo1 und Paf1 können in vitro durch Casein-Kinase II phosphoryliert werden, so dass eine Regulation des PAF1-Komplexes durch die reversible, CKII-abhängige Phosphorylierung nach wie vor denkbar ist (siehe auch Kapitel 3.5).
Die Methode der Chromatin-Immunpräzipitation erwies sich als geeignetes Mittel zur Untersuchung der Interaktion zwischen PAF1-Komplex und RNA-Polymerase II (Kapitel 2.2.7). Dabei zeigt sich, dass die Deletion von PAF1, CTR9, RTF1 oder CDC73 jeweils die Assoziation zwischen PAF1-Komplex und RNA-Polymerase II verhindert (Abb. 2.18). Sowohl paf1Δ als auch ctr9Δ Mutanten können keinen funktionsfähigen PAF1-Komplex ausbilden, da die Ausbildung des Komplexes die Assoziation von Paf1 und Ctr9 voraussetzt (siehe oben). Damit kann auch der Verlust der Assoziation zwischen PAF1-Komplex und RNA-Polymerase II in diesen Mutanten auf den Zerfall des Elongationsfaktors zurückgeführt werden. Im Gegensatz dazu sind Rtf1 und Cdc73 nicht oder nur geringfügig für den Zusammenhalt des Komplexes von Bedeutung (Abb. 2.3). Dennoch führte die Deletion von RTF1 oder CDC73, in Übereinstimmung mit einer Studie von Mueller et al. (2004), zu einem quasi-vollständigen Verlust der Assoziation mit der RNA-Polymerase II. Rtf1 und Cdc73 sind daher unmittelbar für die Bindung des PAF1-Komplexes an die RNA-Polymerase II erforderlich. Interessanterweise ist dabei weder Rtf1 noch Cdc73 alleine ausreichend für die Ausbildung der Interaktion mit der RNA-Polymerase II (Abb. 2.18). Ähnlich einer Zange kommt die Assoziation nur durch die
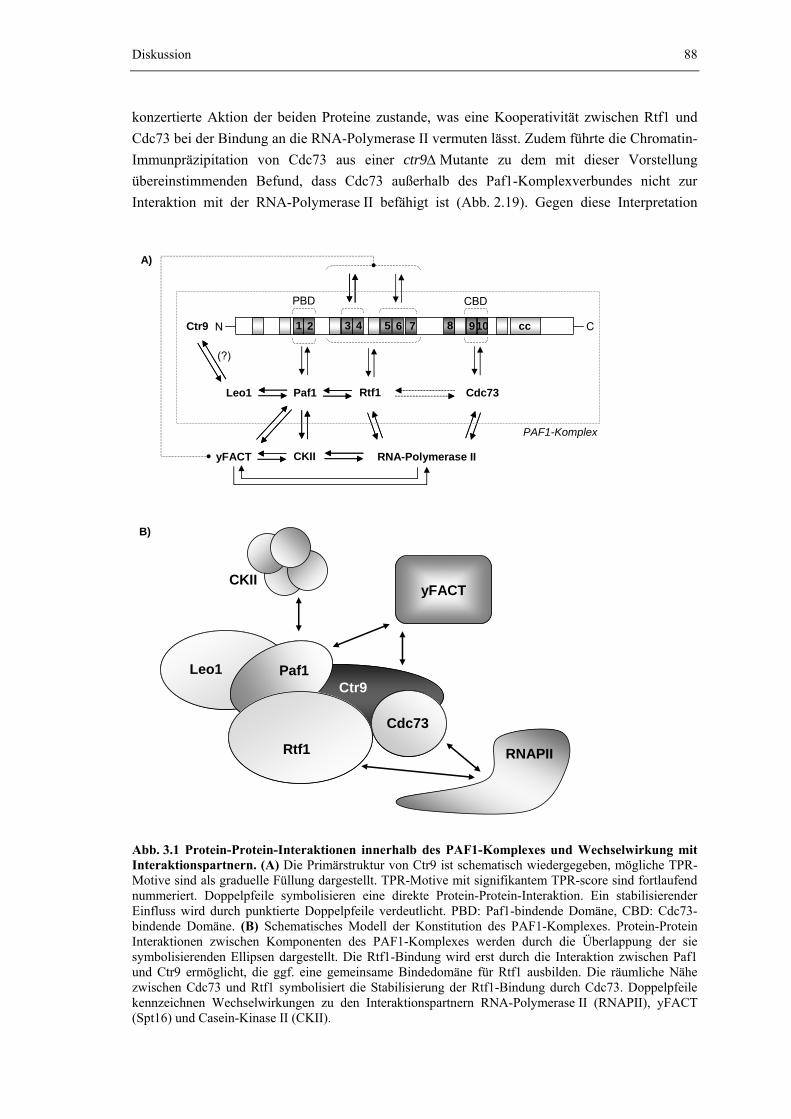
Diskussion
88
konzertierte Aktion der beiden Proteine zustande, was eine Kooperativität zwischen Rtf1 und Cdc73 bei der Bindung an die RNA-Polymerase II vermuten lässt. Zudem führte die Chromatin-Immunpräzipitation von Cdc73 aus einer ctr9Δ Mutante zu dem mit dieser Vorstellung übereinstimmenden Befund, dass Cdc73 außerhalb des Paf1-Komplexverbundes nicht zur Interaktion mit der RNA-Polymerase II befähigt ist (Abb. 2.19). Gegen diese Interpretation
yFACT
Paf1Ctr9
Cdc73
Rtf1
Leo1 Paf1Ctr9
Cdc73Cdc73
Rtf1
Leo1
CKII
RNAPII
1 2 3 4 5 6 7 8 910 ccN CCtr9
Paf1 Cdc73Rtf1Leo1
PBD CBD
CKII RNA-Polymerase II
(?)
yFACT
PAF1-Komplex
A)
B)
Abb. 3.1 Protein-Protein-Interaktionen innerhalb des PAF1-Komplexes und Wechselwirkung mit Interaktionspartnern. (A) Die Primärstruktur von Ctr9 ist schematisch wiedergegeben, mögliche TPR-Motive sind als graduelle Füllung dargestellt. TPR-Motive mit signifikantem TPR-score sind fortlaufend nummeriert. Doppelpfeile symbolisieren eine direkte Protein-Protein-Interaktion. Ein stabilisierender Einfluss wird durch punktierte Doppelpfeile verdeutlicht. PBD: Paf1-bindende Domäne, CBD: Cdc73-bindende Domäne. (B) Schematisches Modell der Konstitution des PAF1-Komplexes. Protein-Protein Interaktionen zwischen Komponenten des PAF1-Komplexes werden durch die Überlappung der sie symbolisierenden Ellipsen dargestellt. Die Rtf1-Bindung wird erst durch die Interaktion zwischen Paf1 und Ctr9 ermöglicht, die ggf. eine gemeinsame Bindedomäne für Rtf1 ausbilden. Die räumliche Nähe zwischen Cdc73 und Rtf1 symbolisiert die Stabilisierung der Rtf1-Bindung durch Cdc73. Doppelpfeile kennzeichnen Wechselwirkungen zu den Interaktionspartnern RNA-Polymerase II (RNAPII), yFACT (Spt16) und Casein-Kinase II (CKII).

Diskussion
89
sprechen allerdings die allgemein relativ milden Phänotypen von cdc73Δ und rtf1Δ Mutanten, die im Vergleich zu paf1Δ oder ctr9Δ Mutanten eine weniger starke Vergrößerung des Zellvolumens oder Verlängerung der Generationszeit zeigen (Abb. 2.11). Interessanterweise hatte die Entfernung der vermuteten Bindungsdomäne für Cdc73 in Ctr9 einen geringeren Effekt auf die Assoziation mit der RNA-Polymerase II als die Deletion von CDC73 (Abb. 2.18). Daher besteht die Möglichkeit, dass der Befund aus den Studien mittels Chromatin-Immunpräzipitation zu einer Überbewertung des einzelnen Einflusses von Cdc73 oder Rtf1 auf die Assoziation des PAF1-Komplexes führt und die Bindung in vitro zumindest teilweise auch alleine durch Cdc73 bzw. Rtf1 vermittelt werden kann. Dies könnte jedoch auch darauf hinweisen, dass die Bindung von Cdc73 in vivo nicht vollständig durch die Entfernung dieser Ctr9-Domäne verloren geht, so dass die Assoziation des PAF1-Komplexes mit der RNA-Polymerase II zwar beeinträchtigt, aber nicht vollständig unterbunden wird.
Der Aufbau des Elongationsfaktors PAF1: ein Modell
Auf der Basis dieser Daten lässt sich ein vereinfachtes Modell des Aufbaus und der Interaktionen des PAF1-Komplexes erstellen (Abb. 3.1). Ctr9 ist vermutlich über seine N-terminalen TPR-Motive 1 & 2 mit Paf1 assoziiert. Die Ausbildung dieser Bindung ermöglicht die Anlagerung von Rtf1. Cdc73 ist über die C-terminalen TPR-Motive 9 & 10 ebenfalls mit Ctr9 verbunden. Durch die Struktur von Ctr9 wird Cdc73 dabei in räumliche Nähe zu Rtf1 gebracht, so dass ggf. ein stabilisierender Effekt auf die Rtf1-Bindung entsteht und eine Kooperativität zwischen Cdc73 und Rtf1 bei der Vermittlung der Assoziation mit der RNA-Polymerase II ermöglicht wird. Die Assoziation von Leo1 könnte, wie im Falle von Rtf1, die Bindung zwischen Paf1 und Ctr9 zur Voraussetzung haben. Die Interaktion mit Casein-Kinase II wird durch Paf1 vermittelt. Für die Interaktion mit yFACT ist wiederum die Bindung zwischen Paf1 und Ctr9 erforderlich. 3.2 Die Silencingdefekte in Mutanten des PAF1-Komplexes können
nicht alleine auf den Ausfall der Dot1-abhängigen Methylierung von Histon H3Lys79 zurückgeführt werden
Wie die in Kapitel 2.2.5 beschriebenen Untersuchungen zeigen, verursacht die Deletion
der Histon-Methyltransferase Dot1 gravierende Silencingdefekte in der betroffenen Mutante (Abb. 2.14). Dieser Effekt ist bereits gut dokumentiert (Singer et al., 1998) und kann vermutlich auf die Ausdünnung des begrenzten zellulären Pools, der für das Silencing notwendigen Sir-Proteine (Grunstein, 1997; Shore et al., 1984) zurückgeführt werden. Durch den Ausfall der bei 90% aller Nukleosomen vorkommenden Dot1-abhängigen Methylierung von Histon H3Lys79 (van Leeuwen et al., 2002) können die Sir-Proteine vermutlich an nahezu alle Nukleosomen binden und sind nicht mehr auf die heterochromatischen Regionen beschränkt, was zur Beeinträchtigung des Silencings an anderen heterochromatischen Loci führt (Maillet et al., 1996; Smith et al., 1998; van Leeuwen et al., 2002).

Diskussion
90
Kapitel 2.2.5 zeigt auch, dass die Deletion von PAF1 oder CTR9 ebenfalls eine ausgeprägte Beeinträchtigung des telomeren Silencings verursacht (vgl. Übersicht in Tabelle 3.1). Auch die Deletion von RTF1 führte zu dem Auftreten deutlicher aber weniger starker Silencingdefekte, die in cdc73Δ Mutanten dagegen kaum und in leo1Δ Mutanten gar nicht mehr beobachtet werden konnten (Abb. 2.14).
Die Untersuchung der Dot1-abhängigen Di- und Tri-Methylierung von Histon H3 in Kapitel 2.2.6 zeigt, dass Cdc73 und Leo1 keinen nachweisbaren Einfluss auf diese Histon-Methylierung besitzen (Abb. 2.16; vgl. Übersicht in Tabelle 3.1), was mit den geringen bzw. nicht erkennbaren Silencingdefekten der cdc73Δ bzw. leo1Δ Mutante übereinstimmt. Die Deletion von RTF1 führte dagegen eindeutig zu einer Beeinträchtigung der Di- und Tri-Methylierung von Histon H3, während im Rahmen dieser Studie in paf1Δ und ctr9Δ Mutanten nur ein Ausfall der Tri-Methylierung, nicht aber der Di-Methylierung an H3Lys79 nachgewiesen werden konnte (Abb. 2.16).
Da in der rtf1Δ Mutante jedoch im Vergleich zu paf1Δ und ctr9Δ Mutanten weniger starke Silencingdefekte auftreten, können diese nicht alleine auf die Beeinträchtigung der Dot1-abhängigen Histon-Methylierung zurückgeführt werden. Auch der Ausfall der Set1-abhängigen Methylierung von Histon H3Lys4 verursacht eine Störung des Telomersilencings (Krogan et al., 2002a). Da sowohl Set1 als auch Dot1 aufgrund ihrer Abhängigkeit von der Mono-Ubiquitinierung an Histon H2B ebenfalls von Rtf1 abhängig sind (Ng et al., 2003a), kann auch der Ausfall beider Histon-Methyltransferasen den unterschiedlichen Einfluss von Rtf1 bzw. Paf1 und Ctr9 auf das telomere Silencing nicht eindeutig erklären. Dies könnte daher auf eine zusätzliche, von der Methylierung an Histon H3 unabhängige Beeinflussung des telomeren Silencings durch den PAF1-Komplex hinweisen, beispielsweise durch Destabilisierung der Boundary-Elemente, die normalerweise die Ausbreitung des Heterochromatins einschränken (Rusche et al., 2003). Die Ausdehnung des heterochromatischen Bereichs an einem Locus würde dann wiederum zur Beeinträchtigung des Silencings an einem anderen Locus führen (siehe oben). Dieses Modell könnte auch die beobachteten Silencingdefekte der bdf1Δ Mutante erklären (vgl. Kapitel 2.3.3, Abb. 2.29), da Bdf1 als Bestandteil des SWR1-Komplexes für den Einbau der Histon-Variante H2A.Z in die Nukleosomen der Heterochromatingrenzen benötigt wird (Kobor et al., 2004; Krogan et al., 2003b; Ladurner et al., 2003). Die Acetylierung von H2A.Z durch die Histon-Acetyltransferase NuA4 verhindert dann die weitere Ausbreitung des Heterochromatíns über diese Boundary-Elemente hinweg (Babiarz et al., 2006; Keogh et al., 2006). Die Hypothese, dass der PAF1-Komplex unabhängig von seinem Einfluss auf Set1 und Dot1 die Stabilität der Heterochromatingrenzen beeinflusst, wird daher auch durch das gehäufte Auftreten von synthetisch letalen Interaktionen zwischen den Komponenten des PAF1-Komplexes und den Untereinheiten des SWR1-Komplexes, der Histon-Acetyltransferase NuA4 und auch H2A.Z unterstützt (vgl. Kapitel 2.2.3, Tabelle 2.3 und Kapitel 3.5).
Die Histon-Methylierungsdefekte der paf1Δ Mutante weichen teilweise von früheren Beobachtungen ab
Die Beobachtung, dass Rtf1 sowohl die Di- als auch die Tri-Methylierung von
Histon H3Lys79 beeinflusst, wird durch mehrere unabhängige Studien bestätigt (Krogan et al.,
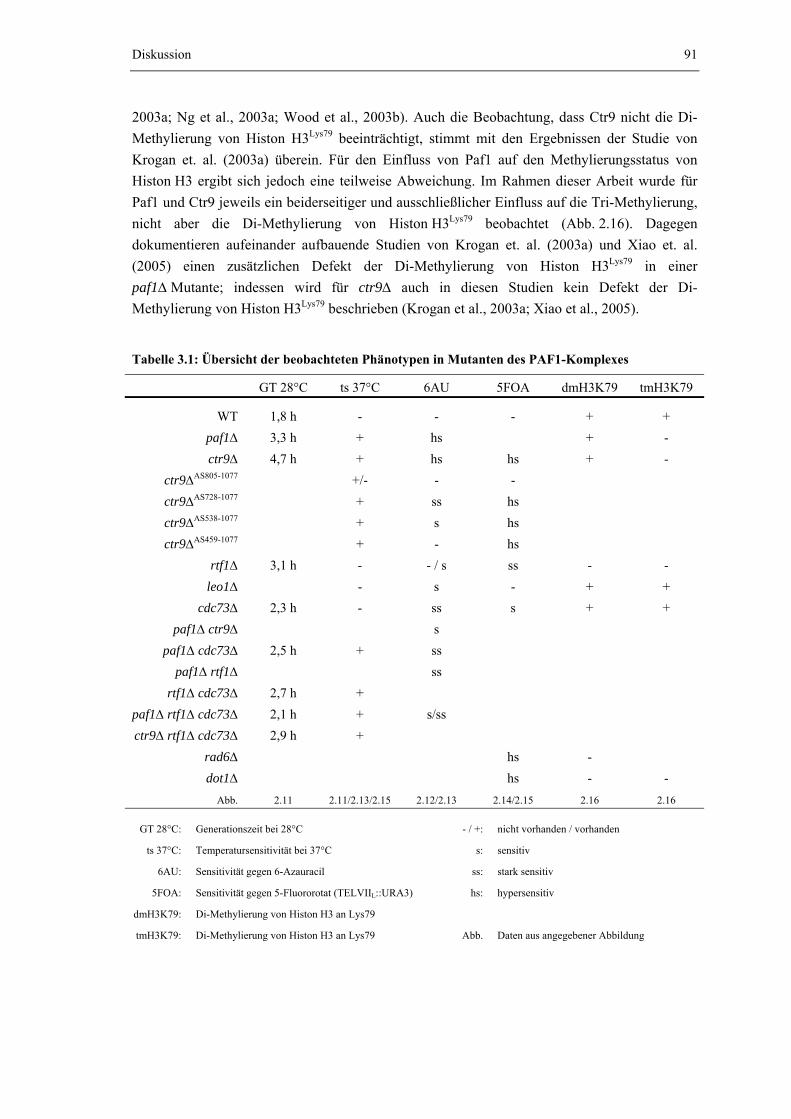
Diskussion
91
2003a; Ng et al., 2003a; Wood et al., 2003b). Auch die Beobachtung, dass Ctr9 nicht die Di-Methylierung von Histon H3Lys79 beeinträchtigt, stimmt mit den Ergebnissen der Studie von Krogan et. al. (2003a) überein. Für den Einfluss von Paf1 auf den Methylierungsstatus von Histon H3 ergibt sich jedoch eine teilweise Abweichung. Im Rahmen dieser Arbeit wurde für Paf1 und Ctr9 jeweils ein beiderseitiger und ausschließlicher Einfluss auf die Tri-Methylierung, nicht aber die Di-Methylierung von Histon H3Lys79 beobachtet (Abb. 2.16). Dagegen dokumentieren aufeinander aufbauende Studien von Krogan et. al. (2003a) und Xiao et. al. (2005) einen zusätzlichen Defekt der Di-Methylierung von Histon H3Lys79 in einer paf1Δ Mutante; indessen wird für ctr9Δ auch in diesen Studien kein Defekt der Di-Methylierung von Histon H3Lys79 beschrieben (Krogan et al., 2003a; Xiao et al., 2005).
Tabelle 3.1: Übersicht der beobachteten Phänotypen in Mutanten des PAF1-Komplexes
GT 28°C ts 37°C 6AU 5FOA dmH3K79 tmH3K79
WT 1,8 h - - - + + paf1Δ 3,3 h + hs + - ctr9Δ 4,7 h + hs hs + -
ctr9ΔAS805-1077 +/- - - ctr9ΔAS728-1077 + ss hs ctr9ΔAS538-1077 + s hs ctr9ΔAS459-1077 + - hs
rtf1Δ 3,1 h - - / s ss - - leo1Δ - s - + +
cdc73Δ 2,3 h - ss s + + paf1Δ ctr9Δ s
paf1Δ cdc73Δ 2,5 h + ss paf1Δ rtf1Δ ss
rtf1Δ cdc73Δ 2,7 h + paf1Δ rtf1Δ cdc73Δ 2,1 h + s/ss ctr9Δ rtf1Δ cdc73Δ 2,9 h +
rad6Δ hs - dot1Δ hs - -
Abb. 2.11 2.11/2.13/2.15 2.12/2.13 2.14/2.15 2.16 2.16
GT 28°C: Generationszeit bei 28°C - / +: nicht vorhanden / vorhanden
ts 37°C: Temperatursensitivität bei 37°C s: sensitiv
6AU: Sensitivität gegen 6-Azauracil ss: stark sensitiv
5FOA: Sensitivität gegen 5-Fluororotat (TELVIIL::URA3) hs: hypersensitiv
dmH3K79: Di-Methylierung von Histon H3 an Lys79
tmH3K79: Di-Methylierung von Histon H3 an Lys79 Abb. Daten aus angegebener Abbildung

Diskussion
92
Eine mögliche Erklärung für diese abweichenden Beobachtungen könnte die jeweils verwendete Antikörper-Charge sein. Da sich Paf1 und Ctr9 gleichermaßen auf die Stabilität des PAF1-Komplexes auswirken (Kapitel 2.1.3), ist eine unterschiedliche Beeinflussung der Histonmethylierung in paf1Δ und ctr9Δ Mutanten jedoch überraschend. Allerdings besitzen nur Rtf1 und Paf1 einen zu Rad6 vergleichbaren Einfluss auf die mono-Ubiquitinierung von Histon H2B (Ng et al., 2003a; Wood et al., 2003b; Xiao et al., 2005). Die Studie von Xiao et. al. (2005) dokumentiert aber dennoch, dass auch eine ctr9Δ Mutante zu einer, wenn auch nicht vollständigen, so doch zu einer signifikanten Beeinträchtigung der mono-Ubiquitinierung von Histon H2B führt. Dies stützt das Modell, dass Ctr9 und Paf1 eine funktionelle Einheit darstellen und steht im Einklang mit der in Kapitel 2.2.6 dokumentierten Beobachtung, dass die Methylierung von Histon H3Lys79 durch die Deletion von PAF1 und CTR9 in gleichem Maß beeinflusst wird.
3.3 Hinweise für eine mögliche regulatorische Selbstinhibierung des
PAF1-Komplexes
Die Bindung zwischen Paf1 und Ctr9 ist von entscheidender Bedeutung für den Zusammenhalt des PAF1-Komplexes und ein Verlust dieser Interaktion verursacht den Zerfall des Elongationsfaktors. Dieser Befund spiegelt sich unmittelbar in den auftretenden Phänotypen der einzelnen Deletionsmutanten wider, die in paf1Δ und ctr9Δ Mutanten generell wesentlich schwerwiegender in Erscheinung treten als in rtf1Δ, leo1Δ und cdc73Δ Mutanten (vgl. Übersicht in Tabelle 3.1). Beispiele hierfür sind das stark vergrößerte Zellvolumen, die verlängerte Generationszeit und die Temperatursensitivität (Kapitel 2.2.2, Abb. 2.11) sowie die anhand der Sensitivität gegen 6-Azauracil beobachteten Elongationsdefekte (Kapitel 2.2.4, Abb. 2.12). Ein möglicher Erklärungsansatz hierfür liegt in der Dissoziation des Komplexes in paf1Δ und ctr9Δ Mutanten. Während der Zerfall des Komplexes und die Degradation der dann frei vorliegenden Komponenten einen vollständigen Funktionsverlust des Elongationsfaktors PAF1 in diesen Mutanten bewirkt, bleibt bei Deletion von RTF1, LEO1 oder CDC73 jeweils der Komplex aus den verbleibenden Untereinheiten erhalten. Auch das in Kapitel 3.1 vorgeschlagene Modell für den Aufbau des PAF1-Komplexes würde mit diesen Befunden übereinstimmen.
Eine Reihe der beobachteten Phänotypen in Doppelmutanten können jedoch nicht durch dieses einfache Modell erklärt werden (vgl. Übersicht in Tabelle 3.1). Das stark vergrößerte Zellvolumen und die verlängerte Generationszeit der paf1Δ Mutante lässt sich beispielsweise durch die Deletion von CDC73 oder RTF1 teilweise unterdrücken (Mueller and Jaehning, 2002; eigene Beobachtung, Kapitel 2.2.2). Die gleichzeitige Deletion von CDC73 und RTF1 verursacht dagegen einen schwerwiegenden Wachstumsdefekt, der in seinem Ausmaß dem Phänotyp der paf1Δ Mutante entspricht bzw. diesen sogar noch übertrifft. Die gleichzeitige Deletion aller drei Gene (PAF1, RTF1 und CDC73) führt jedoch zur nahezu vollständigen Kompensierung dieses Phänotyps und kann in verringertem Ausmaß auch für die ctr9Δ rtf1Δ cdc73Δ Tripelmutante beobachtet werden (Kapitel 2.2.2, Abb. 2.11). Ein ähnliches Phänomen wurde auch für die Sensitivität der paf1Δ Mutante gegen 6-Azauracil beobachtet, die

Diskussion
93
bei gleichzeitiger Deletion mindestens einer der weiteren Komponenten des PAF1-Komplexes – CTR9 eingeschlossen - zumindest teilweise unterdrückt wird (Kapitel 2.2.4, Abb. 2.12). Paf1 und Ctr9 scheinen daher ohne die Bindung der anderen drei Untereinheiten eine für die Zelle schädliche Funktion zu besitzen, die im "Normalfall" einer Regulation durch Cdc73, Rtf1 und ggf. auch Leo1 unterliegt und daher als regulatorische Selbstinhibierung des PAF1-Komplexes aufgefasst werden könnte. Der Wegfall dieser Regulation verursacht dann möglicherweise den Abbruch der Elongation oder ein Pausieren der RNA-Polymerase II. Ein Pausieren der Polymerase könnte zudem mit der Beteiligung des PAF1-Komplexes am Überschreiten eines regulatorischen Kontrollpunkts während der Promotorfreigabe (z. B. dem Capping-Kontrollpunkt, vgl. Kapitel 1.3) erklärt werden. Allerdings könnte die Bindung anderer Elongationsfaktoren, wie z. B. yFACT, an den von der Polymerase losgelösten PAF1-Komplex auch deren Rekrutierung zur RNA-Polymerase II verhindern und dadurch ebenfalls ein Pausieren bzw. eine Blockade der Polymerase verursachen. Das Modell der regulatorischen Selbstinhibierung des PAF1-Komplexes würde den milderen Phänotyp von cdc73Δ bzw. rtf1Δ Mutanten im Vergleich zur cdc73Δ rtf1Δ Doppelmutante erklären, da bei Ausfall von Cdc73 oder Rtf1 noch immer eine regulatorische Untereinheit erhalten bleibt, während in der Doppelmutante aufgrund der Deletion beider Gene keine Regulation von Paf1/Ctr9 durch Cdc73 oder Rtf1 mehr erfolgen kann. Unklar bleibt jedoch, warum auch die Deletion von PAF1 oder CTR9 zum Auftreten schwerwiegender Phänotypen führt und warum diese durch die gleichzeitige Deletion von CDC73 und RTF1 kompensiert werden können. In das Modell lassen sich teilweise auch die beobachteten Phänotypen der Mutanten mit C-terminal verkürztem Ctr9-Protein integrieren. Wie in Kapitel 2.2.4 gezeigt, tritt bei sukzessiver Verkürzung des C-Terminus von Ctr9 zunächst ein Bereich auf, dessen Entfernung zur Sensitivität gegen 6-Azauracil führt. Dieser Bereich beinhaltet auch die Cdc73-bindenden TPR-Motive 9 & 10 und das Ausmaß der Sensitivität dieser Mutante gegen 6-Azauracil ist mit der 6AU-Sensitivität einer cdc73Δ Mutante vergleichbar. Die weitere Verkürzung von Ctr9 führt dagegen zu einer Minderung der Wachstumssensitivität gegen 6-Azauracil und kompensiert daher die auftretenden Elongationsdefekte (Abb. 2.13). In diesem Bereich von Ctr9, zwischen Aminosäure 459 und 538, könnte sich daher eine Domäne befinden, die in unbekannter Weise an der Ausbildung der 6AU-Sensitivität beteiligt ist, was im "Normalfall" durch die Bindung von Cdc73 verhindert würde. Die Entfernung dieser Domäne könnte daher die Ausbildung der 6AU-Sensitivität genauso verhindern, wie zuvor die Bindung von Cdc73. Unklar bleibt hingegen wiederum, warum auch die vollständige Deletion von CTR9 eine Hypersensitivität gegen 6-Azauracil verursacht. Aufgrund ähnlicher Beobachtungen wurde bereits an anderer Stelle eine mögliche Blockierung der Elongation durch die Deletion von PAF1 diskutiert, die durch den Ausfall von Rtf1 oder Leo1 aufgehoben werden kann (Mueller and Jaehning, 2002). Zur Klärung dieser Widersprüche und für das genaue mechanistische Verständnis des PAF1-Komplexes sind daher weitere Untersuchungen zwingend notwendig. Insbesondere zur Interpretation des Phänotyps der paf1Δ rtf1Δ cdc73Δ Tripelmutante müsste geklärt werden, welche Funktionen Cdc73 und Rtf1 in paf1Δ und ctr9Δ Mutante und ggf. allgemein außerhalb des PAF1-Komplexes besitzen.

Diskussion
94
3.4 Die Interaktion zwischen den Elongationsfaktoren PAF1 und yFACT im Kontext der Promotorfreigabe und des Capping-Kontrollpunkts
Die physikalische Interaktion zwischen den Elongationsfaktoren yFACT und PAF1 wird
nicht durch die Deletion von CDC73 oder RTF1 unterbunden (Abb. 2.20). Aber gerade diese beiden Komponenten erweisen sich während der Chromatin-Immunpräzipitation als erforderlich für die Assoziation des PAF1-Komplexes mit der RNA-Polymerase II (Kapitel 2.2.7, Abb. 2.18). Dies erscheint auf den ersten Blick widersprüchlich, da yFACT als Elongationsfaktor ebenfalls mit der RNA-Polymerase II assoziiert ist. Folgende Modelle bieten jedoch Erklärungsmöglichkeiten für diese Beobachtungen. i) Der PAF1-Komplex ist sowohl über Rtf1/Cdc73 als auch über yFACT mit der RNA-Polymerase II assoziiert. Die Stärke der Wechselwirkung zwischen yFACT und PAF1-Komplex wird in cdc73Δ oder rtf1Δ Mutanten jedoch geschwächt, so dass die Assoziation zwischen den beiden Elongationsfaktoren während der Chromatin-Immunpräzipitation verloren geht. Übereinstimmend mit dieser Interpretation wäre der beobachtete stabilisierende Einfluss von Cdc73 und Rtf1 auf die Assoziation der Elongationsfaktoren PAF1 und yFACT (Abb. 2.20). ii) Paf1-Komplex und yFACT sind unabhängig von ihrer Bindung an die RNA-Polymerase II miteinander assoziiert und haben eine weitere Funktion neben der transkriptionellen Elongation. Es wurde bereits dokumentiert, dass die Funktion von yFACT nicht ausschließlich an die Assoziation mit der RNA-Polymerase II geknüpft ist, da der Komplex auch im Kontext der Replikation als Chromatinremodulator wirkt (O'Donnell et al., 2004; Wittmeyer and Formosa, 1997; Zhou and Wang, 2004). Auch der PAF1-Komplex könnte unabhängig von seiner Assoziation mit der RNA-Polymerase II eine Funktion bei der Replikation oder der DNA-Schadenskontrolle besitzen, da die PAF1-abhängige Histonmethylierung und Mono-Ubiquitinierung auch bei diesen Prozessen eine Signalwirkung besitzt, die unter anderem zur Rad9-abhängigen Aktivierung der Rad53 Kinase und damit zum Zellzyklusarrest aufgrund von DNA-Schäden führt (Game et al., 2006b; Giannattasio et al., 2005a; Toh et al., 2006; Wysocki et al., 2005). Diese Vorstellung wird zudem durch das Auftreten synthetisch letaler Phänotypen zwischen Komponenten des PAF1-Komplexes und den Komponenten der DNA-Reparaturmaschinerie unterstützt (Kapitel 2.2.3, Tabelle 2.3). iii) Die Bindung von yFACT an die RNA-Polymerase II könnte über seine Interaktion mit dem PAF1-Komplex vermittelt werden. Die Hypothesen i und iii stehen in gewissem Widerspruch zueinander, da im ersten Fall yFACT PAF1-unabhängig zur RNA-Polymerase II rekrutiert wird oder sogar die Rekrutierung des PAF1-Komplexes vermitteln könnte. Obwohl ein Modell der Rekrutierung und Bindung des PAF1-Komplexes durch yFACT nicht befriedigend erklären kann, warum die Assoziation zwischen PAF1-Komplex und RNA-Polymerase II in cdc73Δ und rtf1Δ Mutanten unterbunden wird, während die Interaktion zwischen den beiden Elongationsfaktoren aufrecht erhalten bleibt, liefern Untersuchungen mittels eines in vitro nachgebildeten Transkriptionssystems jedoch stichhaltige Anhaltspunkte zugunsten der Hypothese, dass yFACT unabhängig von dem PAF1-Komplex zur RNA-Polymerase II rekrutiert wird (Pavri et al., 2006). Auch Untersuchungen anhand von Chromatin-Immunpräzipitationen über Spt16 aus paf1Δ Mutanten liefern keinen Hinweis auf eine

Diskussion
95
Abhängigkeit der Rekrutierung von yFACT durch den PAF1-Komplex (Mueller et al., 2004), so dass die Situation in vivo vermutlich am besten durch die Hypothesen i und ii beschrieben wird.
Die Bedeutung der Elongationsfaktoren PAF1 und yFACT für die Promotorfreigabe und den Capping-Kontrollpunkt
Unabhängig von ihrer Rekrutierungsreihenfolge werden die beiden Elongationsfaktoren
yFACT und PAF1 im Rahmen der Promotorfreigabe bzw. genauer des Capping-Kontrollpunkts (vgl. Kapitel 1.3 und Modell in Abb. 3.2) zur RNA-Polymerase II rekrutiert (Laribee et al., 2005; Pavri et al., 2006; Qiu et al., 2006). Auch eine regulatorische Beteiligung des PAF1-Komplexes an dem transkriptionellen Capping-Kontrollpunkt wäre daher denkbar (vgl. Modell in Abb. 3.2). Für yFACT ist bereits gezeigt worden, dass der Komplex unmittelbar an der Aufhebung des DSIF- und NELF-abhängigen Pausierens der RNA-Polymerase II beteiligt ist (Wada et al., 2000) und dass dies im Zusammenspiel mit den Elongationsfaktoren p-TEFb und Spt6 erfolgt (Endoh et al., 2004; Wada et al., 2000; Wada et al., 1998b). Die Rekrutierung des FACT-Komplexes zur RNA-Polymerase II wird in Säugerzellen durch DRB, einen Inhibitor der mRNA-Synthese, unterbunden (Gomes et al., 2006). Die transkriptionsinhibierende Wirkung beruht dabei auf der Beeinträchtigung des Elongationsfaktors DSIF (Wada et al., 1998a; DRB sensitivity inducing factor). DSIF wirkt in Hefe zusammen mit der zu p-TEFb äquivalenten BUR-Kinase (Wood and Shilatifard, 2006) als Rekrutierungsfaktor für den PAF1-Komplex (Laribee et al., 2005; Qiu et al., 2006). Daher besteht die Möglichkeit, dass der Elongationsfaktor DSIF sowohl für die Rekrutierung des PAF1-Komplexes als auch für die Rekrutierung des Elongationsfaktors yFACT zur RNA-Polymerase II notwendig ist. Dies schließt auch eine gegenseitige Abhängigkeit der Rekrutierung von yFACT und PAF1-Komplex nicht aus und unterstützt die Vermutung, dass beide Elongationsfaktoren am Durchlaufen des Capping-Kontrollpunkts während der Promotorfreigabe beteiligt sind.
Zusätzliche Hinweise hierfür liefern die funktionellen Überlappungen und wechselseitigen Abhängigkeiten zwischen PAF1-Komplex, yFACT und der BUR-Kinase. Beispielsweise führt sowohl die Deletion von BUR2 als auch einige mutierte Varianten von Spt16 (yFACT) zu einem veränderten Status der Set2-abhängigen Methylierung von Histon H3 an Lysin 36 (Chu et al., 2006). Die Rekrutierung von Set2 zur RNA-Polymerase II ist jedoch wiederum von dem PAF1-Komplex abhängig und dessen Funktionalität zudem eine zwingende Voraussetzung für die Aktivität der Histon-Methyltransferase Set2 (Krogan et al., 2003c). Auch die Untersuchungen der homologen Komplexe in Drosophila melanogaster (dm) zeigten, dass die Promotor-Rekrutierung sowohl von dmSpt6 als auch von dmFACT durch die RNAi-vermittelte Hemmung der Expression von dmPAF1 signifikant reduziert wird (Adelman et al., 2006).
Diese funktionellen Überlappungen sprechen zusammen mit der DSIF-abhängigen Rekrutierung daher für die Hypothese, dass sich PAF1 und yFACT in ihrer Rekrutierung zur RNA-Polymerase II gegenseitig beeinflussen und daher ggf. beide am Durchlaufen des Capping-Kontrollpunkts beteiligt sind.

Diskussion
96
3.5 Die Interaktion zwischen PAF1-Komplex, Bdf1 und Casein-Kinase II
Im Rahmen dieser Arbeit konnte erstmals eine physikalische Interaktion zwischen dem
Bromodomainfactor Bdf1 und dem Elongationsfaktor PAF1 nachgewiesen werden. Bdf1 ist, wie der PAF1-Komplex und yFACT, mit der Casein-Kinase II assoziiert und seine Aktivität ist in vivo von der reversiblen Phosphorylierung durch Casein-Kinase II abhängig (Sawa et al., 2004). Bdf1 ist, wie der PAF1-Komplex und yFACT, an dem Prozess der Transkriptionsinitiation beteiligt (Martinez-Campa et al., 2004; Matangkasombut et al., 2000). Wie in Kapitel 2.3.3 beschrieben, konnte übereinstimmend mit diesem Befund eine um ca. 50% verringerte Assoziation der RNA-Polymerase II mit dem Promotor und dem Leserahmen des CLN3-Gens beobachtet werden (Abb. 2.28). Die Chromatin-Immunpräzipitation von Bdf1 zeigt jedoch, dass seine Assoziation mit der RNA-Polymerase II, im Gegensatz zu den beiden Elongationsfaktoren PAF1 und yFACT, auf die Promotorregion und damit auf die Initiation beschränkt bleibt (Abb. 2.27). Der Bromodomainfactor 1 wirkt daher nicht als Elongationsfaktor. Zudem konnten keine offensichtlichen Elongationsdefekte der bdf1Δ Mutante festgestellt werden, da sie sich als relativ unempfindlich gegen 6-Azauracil erwiesen hat (Abb. 2.29).
Bdf1 ist als Bestandteil des Chromatinremodulators SWR1 entscheidend an dem Einbau der Histonvariante H2A.Z (Htz1) in Nukleosomen beteiligt (Kobor et al., 2004; Krogan et al., 2004). Auch zwischen dem SWR1-Komplex und dem Elongationsfaktor PAF1 besteht eine funktionelle Verbindung, wie das gehäufte Auftreten synthetisch letaler Phänotypen zwischen den Komponenten bzw. den entsprechenden Genen der beiden Komplexe und auch zu HTZ1 (H2A.Z) zeigt (Kapitel 2.2.3, Tabelle 2.3; siehe auch Kapitel 3.2). Die Anwesenheit von H2A.Z in Nukleosomen bewirkt eine Inhibierung der PAF1-abhängigen Histonmethyltransferasen Set2 und Dot1 sowie der Histon-Acetyltransferase NuA4 (Li et al., 2005). Die Analyse der genomweit auftretenden synthetisch letalen Phänotypen zeigt zudem, dass auch zwischen den Komponenten von NuA4 und dem PAF1-Komplex auffallend viele synthetisch letale Interaktionen auftreten (Kapitel 2.2.3, Tabelle 2.3), was auf eine überlappende Beteiligung dieser Faktoren an den gleichen zellulären Prozessen hinweist und auch bereits dokumentiert werden konnte. Beispielsweise setzt die Rekrutierung von NuA4 bzw. dessen katalytischer Komponente Esa1 zu aktiven Promotoren die Funktionalität von Set1 und Set2 voraus und fördert durch die Acetylierung von Histon H4 den Eintritt der RNA-Polymerase II in die aktive Elongationsphase (Morillon et al., 2005). Eine funktionale Verbindung zwischen PAF1-Komplex, Set2, NuA4 und auch zu Bdf1 bzw. dem SWR1-Komplex ist daher evident.
Diesbezüglich wurde im vorigen Kapitel die Hypothese aufgestellt, dass der Elongationsfaktor PAF1 zusammen mit yFACT während des Capping-Kontrollpunkts zur RNA-Polymerase II rekrutiert wird und auch funktionell daran beteiligt ist. Die beobachtete Assoziation zwischen dem Paf1-Komplex und Bdf1 könnte darauf hinweisen, dass auch Bdf1 an diesem Kontrollpunkt beteiligt ist. Diese Arbeit zeigt jedoch, dass Bdf1 kaum Einfluss auf die transkriptionelle Elongation besitzt, da die untersuchte bdf1Δ Mutante nur schwach sensitiv auf 6-Azaurazil reagierte und zudem der Dot1-abhängige Methylierungsstatus von Histon H3 an Lysin 79 weder durch die Deletion von BDF1 noch durch die Deletion von HTZ1 beeinflusst

Diskussion
97
RNAPII
S2 S5
PP
TFIIH
RNAPII
S2 S5
STOPP PPRNAPII
S2 S5
STOPP
Bdf1CKII
DSIF/NELF
Bdf1CKII
DSIF/NELF
Bdf1CKII
PPRNAPII
S2 S5
STOPP
pTEFb(Bur1/2)
Bdf1CKII
PPRNAPII
S2 S5
STOPP
pTEFb(Bur1/2)
CKII
FACT
PAF1
PP
CKIIFACT
PAF1
FACT
PAF1 Rad6/Bre1
Set1
PRNAPII
S2 S5P PP
RNAPII
S2 S5PP
PP
PP
I II III
IV V VI
Abb. 3.2 Hypothetische Rekrutierungskaskade während der Promotorfreigabe bzw. des Capping- Kontrollpunkts (verändert und ergänzt nach Sims et al., 2004)
(I → II) Während der Promotorfreigabe wird die CTD der RNA-Polymerase II (RNAPII) durch TFIIH an Serin 5 phosphoryliert. Casein-Kinase II (CKII) und Bdf1 sind vermutlich bereits mit der RNAPII assoziiert (II → III) Die Bindung von DSIF an die CTD führt zur Rekrutierung von NELF, der das Pausieren der RNA-Polymerase II verursacht. Das 5'-Ende der naszierenden mRNA wird mit dem 5'-Methylguanosin-Cap versehen (nicht dargestellt, vgl. Abb. 1.1). Bdf1 wird durch CKII phosphoryliert. (III → IV) Die Phosphorylierung von Bdf1 führt zur Rekrutierung von p-TEFb bzw. der BUR-Kinase (Bur1/2) in Hefe. (IV → V) p-TEFb rekrutiert die Elongationsfaktoren PAF1 und FACT und phosphoryliert die CTD an Serin 2. (IV → V) Der Eintritt in die prozessive Elongationsphase erfolgt nach Rekrutierung von Rad6/Bre1, COMPASS (Set1) und der Histon-Methyltransferase Set2 (nicht dargestellt).
wurde (Kapitel 2.3.3, Abb.2.29 bzw. 2.30). Auch die Rekrutierung des PAF1-Komplexes wird durch die Deletion von Bdf1 vermutlich nicht wesentlich beeinträchtigt, da die Assoziation des PAF1-Komplexes mit dem Promotor und Leserahmen des CLN3-Gens in einer bdf1Δ Mutante im vergleichbaren Ausmaß wie die Assoziation der RNA-Polymerase II selbst reduziert wird (Abb. 2.28). Diese Befunde sprechen zunächst gegen eine Bdf1-abhängige Rekrutierung des PAF1-Komplexes im Rahmen des Capping-Kontrollpunkts. Aber gerade der erhebliche Einfluss von Bdf1 auf die generelle Transkriptions- bzw. Initiationsrate der RNA-Polymerase II (Kapitel 2.3.3, Abb. 2.28) könnte dazu führen, dass ein spezifischer Einfluss von Bdf1 auf die Rekrutierung des PAF1-Komplexes durch diesen generellen Effekt überlagert und damit verschleiert wird. So wurde in der humanen HeLa-Zelllinie gezeigt, dass das zum Hefeprotein Bdf1 orthologe, humane Protein Brd4 an dem transkriptionellen Capping-Kontrollpunkt beteiligt ist und zudem für die Promotorrekrutierung

Diskussion
98
von p-TEFb zwingend erforderlich ist (Yang et al., 2005; Zhou and Yik, 2006). Die Funktion von p-TEFb wird in Hefe durch die BUR-Kinase und die Kinase CTDK-1 übernommen (Wood and Shilatifard, 2006), die beide einen entscheidenden Einfluss auf die Rekrutierung des PAF1-Komplexes bzw. auf dessen Funktion besitzen (Chu et al., 2006; Laribee et al., 2005; Wood et al., 2005; Wood et al., 2007). Auch Bdf1 ließe sich daher als der BUR-Kinase vorgeschalteter Faktor in ein Rekrutierungsmodell für die Elongationsfaktoren PAF1 und yFACT integrieren (vgl. Modell in Abb. 3.2). Zur Verifizierung dieser Hypothese sind jedoch zwingend weitere Untersuchungen erforderlich. Insbesondere müsste überprüft werden, ob auch in Hefezellen eine Interaktion zwischen Bdf1 und der BUR-Kinase auftritt und ob die Rekrutierung der BUR-Kinase, ähnlich wie in Säugerzellen, durch Bdf1 vermittelt wird.
Die Funktionen von Casein-Kinase II und PAF1-Komplex überlappen zu Beginn und am Ende des Transkriptionszyklus
Die räumliche Verbindung zwischen dem Bromodomainfactor 1 und dem
Elongationsfaktor PAF1 wird durch ihre beiderseitige physikalische Interaktion mit der Casein-Kinase II deutlich (Sawa et al., 2004; eigene Beobachtung, Abb. 2.28). Wie Bdf1 selbst, zählt auch die Spt5-Komponente des Elongationsfaktors DSIF zu den Substraten der Casein-Kinase II (Krogan et al., 2002b; Sawa et al., 2004). Damit besteht die Möglichkeit, dass die reversible Phosphorylierung durch Casein-Kinase II das Durchlaufen des Capping-Kontrollpunkts bzw. die Promotorfreigabe beeinflusst (vgl. Modell in Abb. 2.3), zumal auch die CTD der RNA-Polymerase II selbst zu den Substraten der Kinase zählt (Payne et al., 1989). Der Nachweis der in vitro Phosphorylierbarkeit des PAF1-Komplexes durch Casein-Kinase II (Abb. 2.23) schließt nicht aus, dass die Funktion des PAF1-Komplexes einer Regulation durch CKII-abhängige reversible Phosphorylierung unterliegt (siehe auch Kapitel 3.1). Die in Kapitel 2.3.2 beschriebene Beobachtung, dass die Casein-Kinase II auch während der transkriptionellen Elongation mit der RNA-Polymerase II assoziiert bleibt (Abb. 2.25), steht dabei im Einklang mit ihren bereits bekannten Funktionen bei der Initiation der Transkription und der Prozessierung des 3’-Endes der mRNA. Beispielsweise wird die Reinitiation der Transkription maßgeblich durch die CKII-abhängige Phosphorylierung des generellen Transkriptionsfaktors TFIIF und seiner assoziierten CTD-Phosphatase FCP1 reguliert, die ihrerseits zur Phosphorylierung bzw. Dephosphorylierung der C-terminalen Domäne der RNA-Polymerase II führen und dadurch den erneuten Eintritt bzw. das Ablaufen des Transkriptionszyklus ermöglichen (Kong et al., 2005; Sims et al., 2004).
Auch am 3’-Ende des transkribierten Genes bzw. bei der Polyadenylierung des 3’-Endes der naszierenden mRNA tritt eine räumliche bzw. funktionelle Überlappung zwischen PAF1-Komplex und Casein-Kinase II auf. Der Elongationsfaktor PAF1 beeinflusst dabei sowohl die Rekrutierung von Terminationsfaktoren als auch die Erkennung und Nutzung des Polyadenylierungssignals und die Casein-Kinase II ist durch die Phosphorylierung der Fip1-Untereinheit des CPA-Komplexes (CPA für „Cleavage and Polyadenylation“) unmittelbar an der Regulation der Poly(A)-Polymerase beteiligt (He and Moore, 2005; Penheiter et al., 2005; Sheldon et al., 2005; Zielinski et al., 2006). Daher könnte die Assoziation zwischen dem Elongationsfaktor PAF1 und der Casein-Kinase II auch auf die Funktionen der beiden Komplexe bei der Prozessierung des 3’-Endes der mRNA zurückzuführen sein.

Diskussion
99
3.6 Der PAF1-Komplex in höheren Eukaryoten Mit der Aufklärung der Funktion des PAF1-Komplexes als zentraler Regulator der Histonmethylierung in Hefe und dem fortschreitenden Verständnis der Zusammenhänge zwischen Histon-Code, Modulation der Chromatinstruktur und Regulation der Transkription, wurde auch die evolutionäre Konservierung und die Bedeutung des PAF1-Komplexes in höheren Eukaryoten immer offenkundiger. Vor allem seine Rolle bei der Mono-Ubiquitinierung von Histon H2B und der Methylierung von Histon H3 bleibt in allen untersuchten Organismen, von Pilzen, Pflanzen, Insekten bis hin zu Säugern, erhalten (Adelman et al., 2006; He et al., 2004; Moniaux et al., 2006; Oh et al., 2004; Tenney et al., 2006; Woodard et al., 2005). Entsprechend funktionell konserviert erweisen sich die Proteine Paf1 und Ctr9, deren Orthologe an den zum Elongationsfaktor PAF1 homologen Komplexen in A. thaliana, D. melanogaster und H. sapiens beteiligt sind. Auch die zu Leo1 orthologen Proteine wurden bislang speziesunabhängig in allen untersuchten Homologen des PAF1-Komplexes gefunden (Adelman et al., 2006; He et al., 2004; Oh et al., 2004; Rozenblatt-Rosen et al., 2005). Cdc73 bleibt als Bestandteil des humanen PAF1-Komplexes und des entsprechenden Komplexes der Fruchtfliege (D. melanogaster) erhalten. Die Assoziation und damit ggf. auch die Funktion von Rtf1 scheint sich dagegen evolutionär verändert zu haben, da das Protein nur noch lose mit dem jeweiligen Komplex verbunden ist (Adelman et al., 2006; Rozenblatt-Rosen et al., 2005). Im Gegensatz dazu wurde in A. thaliana zwar ebenfalls die Assoziation der zu Paf1, Ctr9, Leo1 und Rtf1 homologen Proteine in einem Proteinkomplex bestätigt, die Beteiligung des zu Cdc73 orthologen Proteins konnte dagegen nicht beobachtet werden und die Funktion dieses Proteins ist in A. thaliana bislang unbekannt (He et al., 2004; Oh et al., 2004). Obwohl die Funktion des PAF1-Komplexes als Regulator der Mono-Ubiquitinierung von Histon H2B und der Methylierung von Histon H3 speziesunabhängig konserviert ist, scheint der Komplex evolutionär betrachtet daher dennoch einer Reihe struktureller Veränderungen unterworfen worden zu sein, die jedoch die zentrale Rolle von Paf1 und Ctr9 innerhalb des PAF1-Komplexes unterstreicht.
Auch die Bedeutung und der Einfluss des PAF1-Komplexes auf die Entstehung einer Vielzahl menschlicher Tumore und Leukämien wird aufgrund der Entschlüsselung der Funktionen des PAF1-Komplexes zunehmend klarer (Moniaux et al., 2006; Okada et al., 2005; Tenney and Shilatifard, 2005; Woodard et al., 2005; Yokoyama et al., 2004). In diesem Zusammenhang könnte auch die immer offenkundigere Beteiligung von PAF1-Komplex, Rad6, Dot1 aber auch SWR1-Komplex, FACT und Casein-Kinase II an der DNA-Schadenskontrolle und der Reparatur von DNA-Schäden stehen (Ahmed et al., 2002; Giannattasio et al., 2005a; Keller et al., 2001; Pan et al., 2006; Papamichos-Chronakis et al., 2006). Die weitere eingehende Untersuchung des PAF1-Komplexes ist daher notwendig, um seine genaue Funktionsweise bei der Regulation der Transkription und auch der DNA-Schadenskontrolle zu entschlüsseln.

Material & Methoden
100
4. Material & Methoden Die prinzipielle Handhabung und Kultivierung der verwendeten Organismen (S. cerevisiae, E coli), sowie molekularbiologische und proteinbiochemische Standardmethoden wurden, soweit nicht extra aufgeführt oder abweichend angegeben, entsprechend der Protokollsammlungen des Cold Spring Harbour Laboratory Verlags bzw. des Verlags John Wiley and Sons, Inc. durchgeführt (Ausubel, 1998; Burke et al., 2000; Sambrook et al., 2001). 4.1 Organismen und Reagenzien 4.1.1 Saccharomyces cerevisiae Stämme
Alle eingesetzten Hefestämme sind congene Derivate der Saccharomyces cerevisiae Laborstämme W303 (K699) oder BY4741/BY4742. Kreuzungen aus W303 und BY4741/BY4742 Derivaten sind entsprechend gekennzeichnet. Zur Bestimmung des Paarungstyps wurden die Teststämme DC14a bzw. DC17α eingesetzt. Die Genotypen der Labor- und Teststämme sind in Tabelle 4.1, die der übrigen eingesetzten Hefestämme in Tabelle 4.2 aufgeführt. Angegeben sind jeweils nur die für die jeweiligen Experimente relevanten Genotypen. Die Transformation (Trf.) von Hefestämmen erfolgte jeweils durch homologe Rekombination von PCR-amplifizierten DNA-Modulen. Die dazu als Template verwendeten Plasmide und die eingesetzten Primer sind in Kapitel 4.1.3 aufgeführt.
Tabelle 4.1: Genotypen der Labor- und Teststämme
Stamm Genotyp MAT gen. Hintergrund Herkunft
K699 ade2-1, his3-11, 15, leu2-3, 112, trp1-1, ura3-1, can1-100, GAL, psi+
a W303
K. Nasmyth
K700 ade2-1, his3-11, 15, leu2-3, 112, trp1-1, ura3-1, can1-100, GAL, psi+
α W303
K. Nasmyth
BY4741 his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 a BY4741 Open Biosystems
BY4742 (CY4196)
his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0 α BY4742 Open Biosystems
CY274 his1, HIS3 a DC14a K. Nasmyth
CY275 his1, HIS3 α DC17α K. Nasmyth
Tabelle 4.2: Genotypen der verwendeten Hefestämme
Stamm Genotyp MAT congen zu Herkunft
CY1078 trp1-Δ63 α W303 AG Koch
CY1546 ctr9::CTR9-MYC6::LEU2 a W303 AG Koch
CY1615 pep4::URA3 α W303 AG Koch
CY1616 ctr9::CTR9-MYC6::LEU2, pep4::URA3 α W303 AG Koch
Material & Methoden
101
Tabelle 4.2: Genotypen der verwendeten Hefestämme (Fortsetzung)
Stamm Genotyp MAT congen zu Herkunft
CY1617 pep4::URA3 a W303 AG Koch
CY1994 paf1::URA3 a W303 AG Koch
CY1995 paf1::URA3 a W303 AG Koch
CY2002 cdc73::HIS3 α W303 AG Koch
CY2003 cdc73::HIS3 a W303 AG Koch
CY2038 paf1::PAF1-HA3::klTRP1 a W303 AG Koch
CY2239 ctr9::LEU2 α W303 AG Koch
CY2807 ctr9::CTR9-TAP::klTRP1, pep4::URA3 α W303 AG Koch
CY2976 ctr9::CTR9-TAP::klTRP1, pep4::URA3 α W303 AG Koch
CY2977 ctr9::CTR9-TAP::klTRP1, pep4::URA3 a W303 AG Koch
CY3099 paf1::PAF1-HA3::klTPR1, pep4::URA3 α W303 CY2038 × CY2976
CY3100 ctr9::CTR9-TAP::klTRP1, pep4::URA3 α W303 CY2038 × CY2976
CY3354 paf1::PAF1-TAP::klTRP1 α W303 Trf. CY1078
CY3364 ctr9::CTR9-MYC6::LEU2, pep4::URA3 a W303 AG Koch
CY3390 cdc73::CDC73-HA6::klTRP1 α W303 Trf. CY1078
CY3408 paf1::PAF1-TAP::klTRP1, pep4::URA3 a W303 CY3354 × CY3364
CY3444 ctr9::CTR9-TAP::klTRP1, pep4::URA3, cdc73::CDC73-HA6::klTRP1
a W303 CY3390 × CY2977
CY3445 cdc73::CDC73-HA6::klTRP1, pep4::URA3 a W303 CY3390 × CY2977
CY3466 cdc73::CDC73-HA6::klTRP1 α W303 Trf. CY1078
CY3468 rtf1::rtf1-HA6::klTRP1 α W303 Trf. CY1078
CY3469 rtf1::rtf1-HA6::klTRP1 α W303 Trf. CY1078
CY3475 cdc73::CDC73-TAP::klTRP1, pep4::URA3 a W303 CY3364 × CY3466
CY3482 ctr9::CTR9-TAP::klTRP1, pep4::URA , RTF1/rtf1::rtf1-HA6::klTRP1
a/α W303 CY2977 × CY3468
CY3483 rtf1::rtf1-HA6::klTRP1, pep4::URA3 a W303 CY2977 × CY3469,
CY3711 spt16::SPT16-HA6::klTRP1 α W303 Trf. CY1078
CY3472 spt16::SPT16-HA6::klTRP1 α W303 Trf. CY1078
CY3715 cka1::CKA1-HA6::klTRP1 α W303 Trf. CY1078
CY3718 paf1::PAF1-HA3::klTRP1, pep4::URA3 a W303 AG Koch
CY3725 paf1::PAF1-TAP::klTRP1, pep4::URA3, spt16::SPT16-HA6::klTRP1
a W303 CY3408 × CY3711
CY3728 spt16::SPT16-HA6::klTRP1, pep4::URA3 a W303 CY3408 × CY3711
CY3736 cka1::CKA1-TAP::klTRP1 α W303 Trf. CY1078
CY3769 cka1::CKA1-TAP::klTRP1 α W303 Trf. CY1078
CY3770 cka1::CKA1-TAP::klTRP1, pep4::URA3, paf1::PAF1-HA3::klTRP1
α W303 CY3718 × CY3736
CY3784 cka1::CKA1-TAP::klTRP1, pep4::URA3, spt16::SPT16-HA6::klTRP1
a W303 CY3728 × CY3769:
CY3785 cka1::CKA1-TAP::klTRP1, pep4::URA3 a W303 CY3728 × CY3769
CY3796 cka1::CKA1-HA6::klTRP1, pep4::URA3 a W303 CY3408 × CY3715
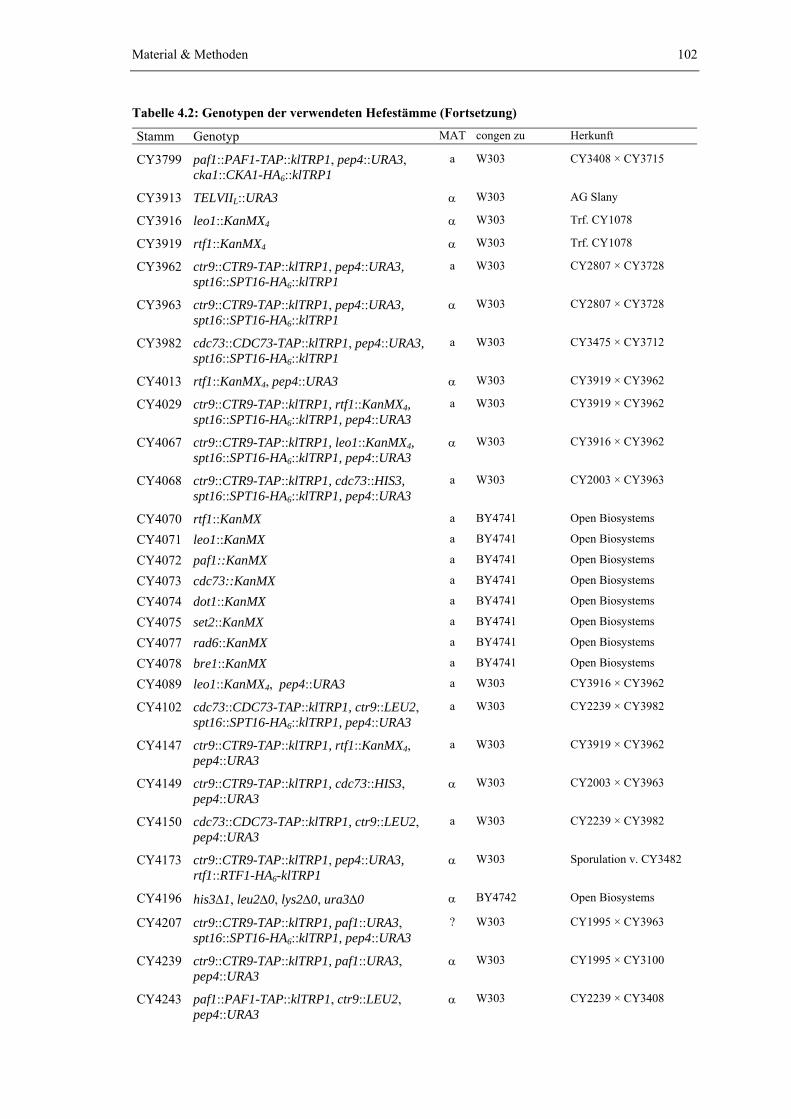
Material & Methoden
102
Tabelle 4.2: Genotypen der verwendeten Hefestämme (Fortsetzung)
Stamm Genotyp MAT congen zu Herkunft
CY3799 paf1::PAF1-TAP::klTRP1, pep4::URA3, cka1::CKA1-HA6::klTRP1
a W303 CY3408 × CY3715
CY3913 TELVIIL::URA3 α W303 AG Slany
CY3916 leo1::KanMX4 α W303 Trf. CY1078
CY3919 rtf1::KanMX4 α W303 Trf. CY1078
CY3962 ctr9::CTR9-TAP::klTRP1, pep4::URA3, spt16::SPT16-HA6::klTRP1
a W303 CY2807 × CY3728
CY3963 ctr9::CTR9-TAP::klTRP1, pep4::URA3, spt16::SPT16-HA6::klTRP1
α W303 CY2807 × CY3728
CY3982 cdc73::CDC73-TAP::klTRP1, pep4::URA3, spt16::SPT16-HA6::klTRP1
a W303 CY3475 × CY3712
CY4013 rtf1::KanMX4, pep4::URA3 α W303 CY3919 × CY3962
CY4029 ctr9::CTR9-TAP::klTRP1, rtf1::KanMX4, spt16::SPT16-HA6::klTRP1, pep4::URA3
a W303 CY3919 × CY3962
CY4067 ctr9::CTR9-TAP::klTRP1, leo1::KanMX4, spt16::SPT16-HA6::klTRP1, pep4::URA3
α W303 CY3916 × CY3962
CY4068 ctr9::CTR9-TAP::klTRP1, cdc73::HIS3, spt16::SPT16-HA6::klTRP1, pep4::URA3
a W303 CY2003 × CY3963
CY4070 rtf1::KanMX a BY4741 Open Biosystems
CY4071 leo1::KanMX a BY4741 Open Biosystems
CY4072 paf1::KanMX a BY4741 Open Biosystems
CY4073 cdc73::KanMX a BY4741 Open Biosystems
CY4074 dot1::KanMX a BY4741 Open Biosystems
CY4075 set2::KanMX a BY4741 Open Biosystems
CY4077 rad6::KanMX a BY4741 Open Biosystems
CY4078 bre1::KanMX a BY4741 Open Biosystems
CY4089 leo1::KanMX4, pep4::URA3 a W303 CY3916 × CY3962
CY4102 cdc73::CDC73-TAP::klTRP1, ctr9::LEU2, spt16::SPT16-HA6::klTRP1, pep4::URA3
a W303 CY2239 × CY3982
CY4147 ctr9::CTR9-TAP::klTRP1, rtf1::KanMX4, pep4::URA3
a W303 CY3919 × CY3962
CY4149 ctr9::CTR9-TAP::klTRP1, cdc73::HIS3, pep4::URA3
α W303 CY2003 × CY3963
CY4150 cdc73::CDC73-TAP::klTRP1, ctr9::LEU2, pep4::URA3
a W303 CY2239 × CY3982
CY4173 ctr9::CTR9-TAP::klTRP1, pep4::URA3, rtf1::RTF1-HA6-klTRP1
α W303 Sporulation v. CY3482
CY4196 his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0 α BY4742 Open Biosystems
CY4207 ctr9::CTR9-TAP::klTRP1, paf1::URA3, spt16::SPT16-HA6::klTRP1, pep4::URA3
? W303 CY1995 × CY3963
CY4239 ctr9::CTR9-TAP::klTRP1, paf1::URA3, pep4::URA3
α W303 CY1995 × CY3100
CY4243 paf1::PAF1-TAP::klTRP1, ctr9::LEU2, pep4::URA3
α W303 CY2239 × CY3408
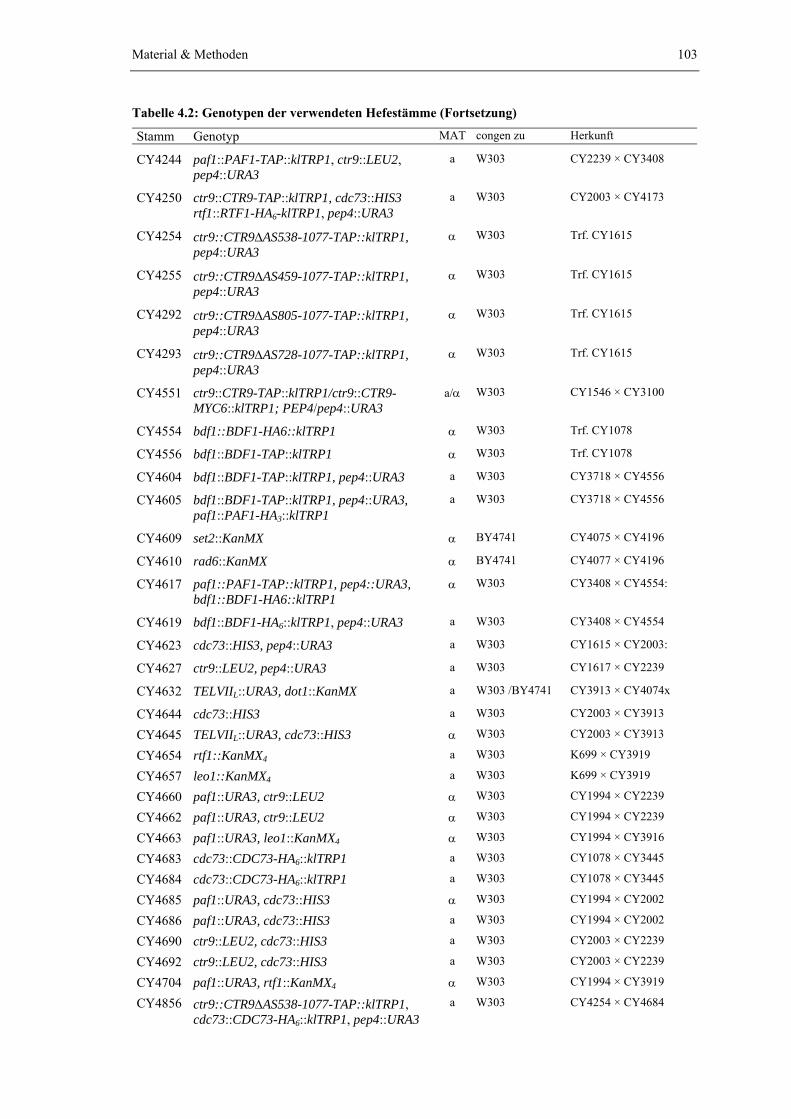
Material & Methoden
103
Tabelle 4.2: Genotypen der verwendeten Hefestämme (Fortsetzung)
Stamm Genotyp MAT congen zu Herkunft
CY4244 paf1::PAF1-TAP::klTRP1, ctr9::LEU2, pep4::URA3
a W303 CY2239 × CY3408
CY4250 ctr9::CTR9-TAP::klTRP1, cdc73::HIS3 rtf1::RTF1-HA6-klTRP1, pep4::URA3
a W303 CY2003 × CY4173
CY4254 ctr9::CTR9ΔAS538-1077-TAP::klTRP1, pep4::URA3
α W303 Trf. CY1615
CY4255 ctr9::CTR9ΔAS459-1077-TAP::klTRP1, pep4::URA3
α W303 Trf. CY1615
CY4292 ctr9::CTR9ΔAS805-1077-TAP::klTRP1, pep4::URA3
α W303 Trf. CY1615
CY4293 ctr9::CTR9ΔAS728-1077-TAP::klTRP1, pep4::URA3
α W303 Trf. CY1615
CY4551 ctr9::CTR9-TAP::klTRP1/ctr9::CTR9- MYC6::klTRP1; PEP4/pep4::URA3
a/α W303 CY1546 × CY3100
CY4554 bdf1::BDF1-HA6::klTRP1 α W303 Trf. CY1078
CY4556 bdf1::BDF1-TAP::klTRP1 α W303 Trf. CY1078
CY4604 bdf1::BDF1-TAP::klTRP1, pep4::URA3 a W303 CY3718 × CY4556
CY4605 bdf1::BDF1-TAP::klTRP1, pep4::URA3, paf1::PAF1-HA3::klTRP1
a W303 CY3718 × CY4556
CY4609 set2::KanMX α BY4741 CY4075 × CY4196
CY4610 rad6::KanMX α BY4741 CY4077 × CY4196
CY4617 paf1::PAF1-TAP::klTRP1, pep4::URA3, bdf1::BDF1-HA6::klTRP1
α W303 CY3408 × CY4554:
CY4619 bdf1::BDF1-HA6::klTRP1, pep4::URA3 a W303 CY3408 × CY4554
CY4623 cdc73::HIS3, pep4::URA3 a W303 CY1615 × CY2003:
CY4627 ctr9::LEU2, pep4::URA3 a W303 CY1617 × CY2239
CY4632 TELVIIL::URA3, dot1::KanMX a W303 /BY4741 CY3913 × CY4074x
CY4644 cdc73::HIS3 a W303 CY2003 × CY3913
CY4645 TELVIIL::URA3, cdc73::HIS3 α W303 CY2003 × CY3913
CY4654 rtf1::KanMX4 a W303 K699 × CY3919
CY4657 leo1::KanMX4 a W303 K699 × CY3919
CY4660 paf1::URA3, ctr9::LEU2 α W303 CY1994 × CY2239
CY4662 paf1::URA3, ctr9::LEU2 α W303 CY1994 × CY2239
CY4663 paf1::URA3, leo1::KanMX4 α W303 CY1994 × CY3916
CY4683 cdc73::CDC73-HA6::klTRP1 a W303 CY1078 × CY3445
CY4684 cdc73::CDC73-HA6::klTRP1 a W303 CY1078 × CY3445
CY4685 paf1::URA3, cdc73::HIS3 α W303 CY1994 × CY2002
CY4686 paf1::URA3, cdc73::HIS3 a W303 CY1994 × CY2002
CY4690 ctr9::LEU2, cdc73::HIS3 a W303 CY2003 × CY2239
CY4692 ctr9::LEU2, cdc73::HIS3 a W303 CY2003 × CY2239
CY4704 paf1::URA3, rtf1::KanMX4 α W303 CY1994 × CY3919
CY4856 ctr9::CTR9ΔAS538-1077-TAP::klTRP1, cdc73::CDC73-HA6::klTRP1, pep4::URA3
a W303 CY4254 × CY4684
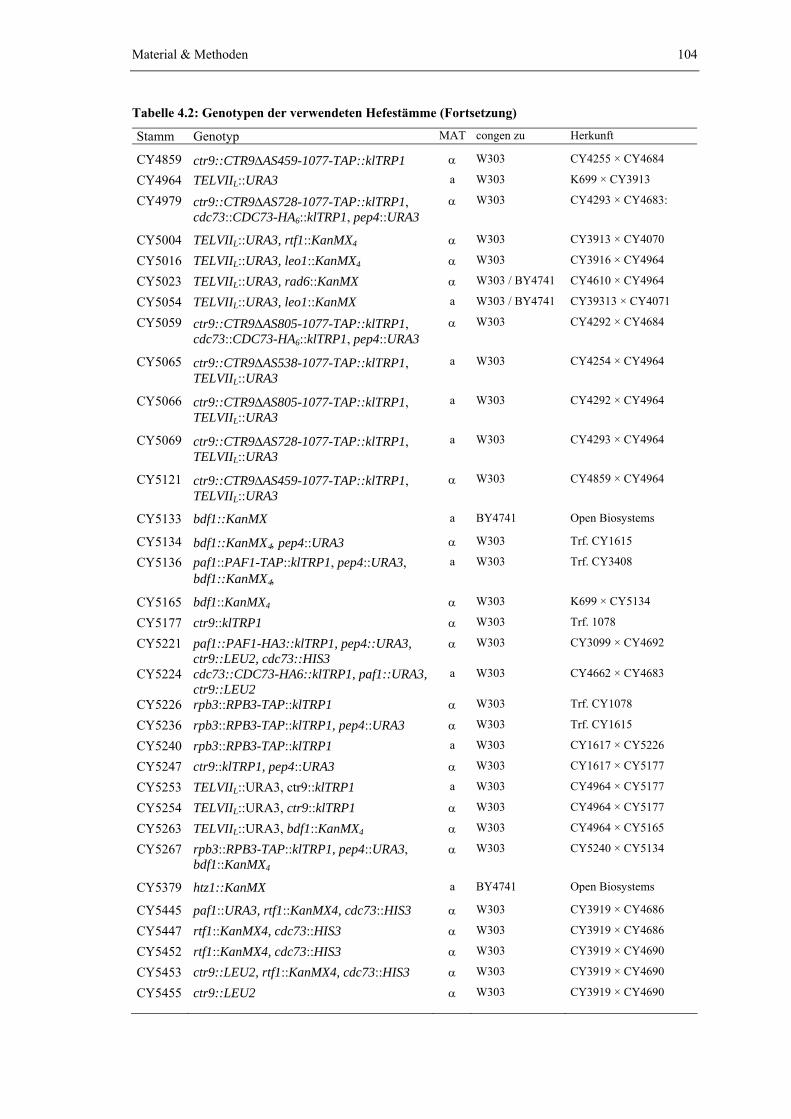
Material & Methoden
104
Tabelle 4.2: Genotypen der verwendeten Hefestämme (Fortsetzung)
Stamm Genotyp MAT congen zu Herkunft
CY4859 ctr9::CTR9ΔAS459-1077-TAP::klTRP1 α W303 CY4255 × CY4684
CY4964 TELVIIL::URA3 a W303 K699 × CY3913
CY4979 ctr9::CTR9ΔAS728-1077-TAP::klTRP1, cdc73::CDC73-HA6::klTRP1, pep4::URA3
α W303 CY4293 × CY4683:
CY5004 TELVIIL::URA3, rtf1::KanMX4 α W303 CY3913 × CY4070
CY5016 TELVIIL::URA3, leo1::KanMX4 α W303 CY3916 × CY4964
CY5023 TELVIIL::URA3, rad6::KanMX α W303 / BY4741 CY4610 × CY4964
CY5054 TELVIIL::URA3, leo1::KanMX a W303 / BY4741 CY39313 × CY4071
CY5059 ctr9::CTR9ΔAS805-1077-TAP::klTRP1, cdc73::CDC73-HA6::klTRP1, pep4::URA3
α W303 CY4292 × CY4684
CY5065 ctr9::CTR9ΔAS538-1077-TAP::klTRP1, TELVIIL::URA3
a W303 CY4254 × CY4964
CY5066 ctr9::CTR9ΔAS805-1077-TAP::klTRP1, TELVIIL::URA3
a W303 CY4292 × CY4964
CY5069 ctr9::CTR9ΔAS728-1077-TAP::klTRP1, TELVIIL::URA3
a W303 CY4293 × CY4964
CY5121 ctr9::CTR9ΔAS459-1077-TAP::klTRP1, TELVIIL::URA3
α W303 CY4859 × CY4964
CY5133 bdf1::KanMX a BY4741 Open Biosystems
CY5134 bdf1::KanMX4, pep4::URA3 α W303 Trf. CY1615
CY5136 paf1::PAF1-TAP::klTRP1, pep4::URA3, bdf1::KanMX4,
a W303 Trf. CY3408
CY5165 bdf1::KanMX4 α W303 K699 × CY5134
CY5177 ctr9::klTRP1 α W303 Trf. 1078
CY5221 paf1::PAF1-HA3::klTRP1, pep4::URA3, ctr9::LEU2, cdc73::HIS3
α W303 CY3099 × CY4692
CY5224 cdc73::CDC73-HA6::klTRP1, paf1::URA3, ctr9::LEU2
a W303 CY4662 × CY4683
CY5226 rpb3::RPB3-TAP::klTRP1 α W303 Trf. CY1078
CY5236 rpb3::RPB3-TAP::klTRP1, pep4::URA3 α W303 Trf. CY1615
CY5240 rpb3::RPB3-TAP::klTRP1 a W303 CY1617 × CY5226
CY5247 ctr9::klTRP1, pep4::URA3 α W303 CY1617 × CY5177
CY5253 TELVIIL::URA3, ctr9::klTRP1 a W303 CY4964 × CY5177
CY5254 TELVIIL::URA3, ctr9::klTRP1 α W303 CY4964 × CY5177
CY5263 TELVIIL::URA3, bdf1::KanMX4 α W303 CY4964 × CY5165
CY5267 rpb3::RPB3-TAP::klTRP1, pep4::URA3, bdf1::KanMX4
α W303 CY5240 × CY5134
CY5379 htz1::KanMX a BY4741 Open Biosystems
CY5445 paf1::URA3, rtf1::KanMX4, cdc73::HIS3 α W303 CY3919 × CY4686
CY5447 rtf1::KanMX4, cdc73::HIS3 α W303 CY3919 × CY4686
CY5452 rtf1::KanMX4, cdc73::HIS3 α W303 CY3919 × CY4690
CY5453 ctr9::LEU2, rtf1::KanMX4, cdc73::HIS3 α W303 CY3919 × CY4690
CY5455 ctr9::LEU2 α W303 CY3919 × CY4690
Material & Methoden
105
4.1.2 Escherichia coli Stämme
Zur Amplifikation von Plasmiden wurde der Stamm E. coli DH5α eingesetzt. Die Expression von GST-Ctr9-Fragmenten erfolgte in E. coli Rosetta(DE3). Die Genotypen dieser beiden Stämme sind in Tabelle 4.3 wiedergegeben. Die durch Transformation von E. coli Rosetta(DE3) mit dem jeweiligen Expressionsplasmid (pGEX-4T-3 Vektor-Rückgrat) erhaltenen Expressionsstämme sind in Tabelle 4.4 angegeben.
Tabelle 4.3: Genotypen der E. coli Stämme DH5a und Rosetta(DE3)
Stamm-Nr. E. coli Genotyp Quelle
CE0 DH5α supE44 ΔlacU169 (φ80 lacZ ΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1
(Sambrook et al., 2001)
CE58 Rosetta(DE3) F- ompT hsdSB(rB- mB
-) gal dcm lacY1 (DE3) pRARE (CmR)
(Novagen, TB055 pET Manual 10th Ed. 0702)
Tabelle 4.4: E. coli Rosetta(DE3) Expressionsstämme
Stamm-Nr. Plasmid exprimiertes Protein Herkunft
CE76 pCK1893 GST-Ctr9[AS550-816] A. Schwahn
CE77 pCK1894 GST-Ctr9[AS716-816] A. Schwahn
CE79 pCK1896 GST-Ctr9[AS716-1077] A. Schwahn
CE83 pCK1889 GST A. Alemay Schmidt, A. Schwahn
CE89 pCK2023 GST-Ctr9[AS368-549] A. Alemay Schmidt, A. Schwahn
CE90 pCK2024 GST-Ctr9[AS2-549] A. Alemay Schmidt, A. Schwahn
4.1.3 Vektoren, Plasmide und Oligonukleotide
Tabelle 4.5: Verwendete Vektoren Nr. Vektor Beschreibung Quelle
CK4 YEplac112 Hefe Expressionsplasmid für TRP1; AmpR (Gietz and Sugino, 1988)
CK1091 pYM3 HA6::klTRP1 tag-Modul; AmpR (Knop et al., 1999)
CK1279 pBS1479 TAP::klTRP1 tag-Modul; AmpR (Puig et al., 2001)
CK1889
pGEX-4T-3 E. coli Expressionsvektor (T7) für GST-Protein Fusionen; AmpR
(Smith and Johnson, 1988; GeneBank Acc. No. U13855)
CK2082 pFA6KanMX KanMX tag-Modul; AmpR (Wach et al., 1994)
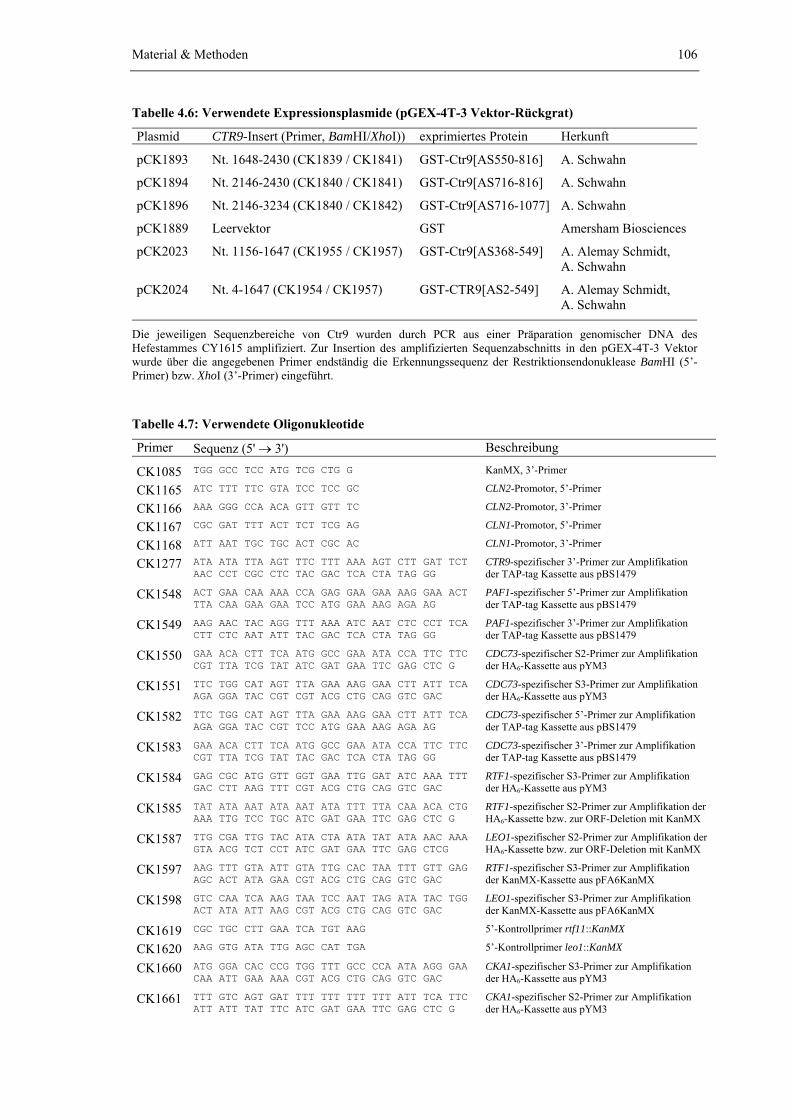
Material & Methoden
106
Tabelle 4.6: Verwendete Expressionsplasmide (pGEX-4T-3 Vektor-Rückgrat) Plasmid CTR9-Insert (Primer, BamHI/XhoI)) exprimiertes Protein Herkunft
pCK1893 Nt. 1648-2430 (CK1839 / CK1841) GST-Ctr9[AS550-816] A. Schwahn
pCK1894 Nt. 2146-2430 (CK1840 / CK1841) GST-Ctr9[AS716-816] A. Schwahn
pCK1896 Nt. 2146-3234 (CK1840 / CK1842) GST-Ctr9[AS716-1077] A. Schwahn
pCK1889 Leervektor GST Amersham Biosciences
pCK2023 Nt. 1156-1647 (CK1955 / CK1957) GST-Ctr9[AS368-549] A. Alemay Schmidt, A. Schwahn
pCK2024 Nt. 4-1647 (CK1954 / CK1957) GST-CTR9[AS2-549] A. Alemay Schmidt, A. Schwahn
Die jeweiligen Sequenzbereiche von Ctr9 wurden durch PCR aus einer Präparation genomischer DNA des Hefestammes CY1615 amplifiziert. Zur Insertion des amplifizierten Sequenzabschnitts in den pGEX-4T-3 Vektor wurde über die angegebenen Primer endständig die Erkennungssequenz der Restriktionsendonuklease BamHI (5’-Primer) bzw. XhoI (3’-Primer) eingeführt.
Tabelle 4.7: Verwendete Oligonukleotide Primer Sequenz (5' → 3') Beschreibung
CK1085 TGG GCC TCC ATG TCG CTG G KanMX, 3’-Primer
CK1165 ATC TTT TTC GTA TCC TCC GC CLN2-Promotor, 5’-Primer
CK1166 AAA GGG CCA ACA GTT GTT TC CLN2-Promotor, 3’-Primer
CK1167 CGC GAT TTT ACT TCT TCG AG CLN1-Promotor, 5’-Primer
CK1168 ATT AAT TGC TGC ACT CGC AC CLN1-Promotor, 3’-Primer
CK1277 ATA ATA TTA AGT TTC TTT AAA AGT CTT GAT TCT AAC CCT CGC CTC TAC GAC TCA CTA TAG GG
CTR9-spezifischer 3’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1548 ACT GAA CAA AAA CCA GAG GAA GAA AAG GAA ACT TTA CAA GAA GAA TCC ATG GAA AAG AGA AG
PAF1-spezifischer 5’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1549 AAG AAC TAC AGG TTT AAA ATC AAT CTC CCT TCA CTT CTC AAT ATT TAC GAC TCA CTA TAG GG
PAF1-spezifischer 3’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1550 GAA ACA CTT TCA ATG GCC GAA ATA CCA TTC TTC CGT TTA TCG TAT ATC GAT GAA TTC GAG CTC G
CDC73-spezifischer S2-Primer zur Amplifikation der HA6-Kassette aus pYM3
CK1551 TTC TGG CAT AGT TTA GAA AAG GAA CTT ATT TCA AGA GGA TAC CGT CGT ACG CTG CAG GTC GAC
CDC73-spezifischer S3-Primer zur Amplifikation der HA6-Kassette aus pYM3
CK1582 TTC TGG CAT AGT TTA GAA AAG GAA CTT ATT TCA AGA GGA TAC CGT TCC ATG GAA AAG AGA AG
CDC73-spezifischer 5’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1583 GAA ACA CTT TCA ATG GCC GAA ATA CCA TTC TTC CGT TTA TCG TAT TAC GAC TCA CTA TAG GG
CDC73-spezifischer 3’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1584 GAG CGC ATG GTT GGT GAA TTG GAT ATC AAA TTT GAC CTT AAG TTT CGT ACG CTG CAG GTC GAC
RTF1-spezifischer S3-Primer zur Amplifikation der HA6-Kassette aus pYM3
CK1585 TAT ATA AAT ATA AAT ATA TTT TTA CAA ACA CTG AAA TTG TCC TGC ATC GAT GAA TTC GAG CTC G
RTF1-spezifischer S2-Primer zur Amplifikation der HA6-Kassette bzw. zur ORF-Deletion mit KanMX
CK1587 TTG CGA TTG TAC ATA CTA ATA TAT ATA AAC AAA GTA ACG TCT CCT ATC GAT GAA TTC GAG CTCG
LEO1-spezifischer S2-Primer zur Amplifikation der HA6-Kassette bzw. zur ORF-Deletion mit KanMX
CK1597 AAG TTT GTA ATT GTA TTG CAC TAA TTT GTT GAG AGC ACT ATA GAA CGT ACG CTG CAG GTC GAC
RTF1-spezifischer S3-Primer zur Amplifikation der KanMX-Kassette aus pFA6KanMX
CK1598 GTC CAA TCA AAG TAA TCC AAT TAG ATA TAC TGG ACT ATA ATT AAG CGT ACG CTG CAG GTC GAC
LEO1-spezifischer S3-Primer zur Amplifikation der KanMX-Kassette aus pFA6KanMX
CK1619 CGC TGC CTT GAA TCA TGT AAG 5’-Kontrollprimer rtf11::KanMX
CK1620 AAG GTG ATA TTG AGC CAT TGA 5’-Kontrollprimer leo1::KanMX
CK1660 ATG GGA CAC CCG TGG TTT GCC CCA ATA AGG GAA CAA ATT GAA AAA CGT ACG CTG CAG GTC GAC
CKA1-spezifischer S3-Primer zur Amplifikation der HA6-Kassette aus pYM3
CK1661 TTT GTC AGT GAT TTT TTT TTT TTT ATT TCA TTC ATT ATT TAT TTC ATC GAT GAA TTC GAG CTC G
CKA1-spezifischer S2-Primer zur Amplifikation der HA6-Kassette aus pYM3

Material & Methoden
107
Tabelle 4.7: Verwendete Oligonukleotide (Fortsetzung) Primer Sequenz (5' → 3') Beschreibung
CK1662 ATG GGA CAC CCG TGG TTT GCC CCA ATA AGG GAA CAA ATT GAA AAA TCC ATG GAA AAG AGA AG
CKA1-spezifischer 5’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1663 TTT GTC AGT GAT TTT TTT TTT TTT ATT TCA TTC ATT ATT TAT TTC TAC GAC TCA CTA TAG GG
CKA1-spezifischer 3’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1666 GAG AAA AAG GCT GCT AGG GCT GAT AGG GGT GCA AAC TTT AGA GAT CGT ACG CTG CAG GTC GAC
SPT16-spezifischer S3-Primer zur Amplifikation der HA6-Kassette aus pYM3
CK1667 TCA GAT CAA GGT CTT GCT GGT GAA ACC CAG TAA GTG TTA TAA AGT ATC GAT GAA TTC GAG CTC G
SPT16-spezifischer S2-Primer zur Amplifikation der HA6-Kassette aus pYM3
CK1724 ATA GTG ATG CCA CTG TAG AC CLN2-ORF, 5’-Primer
CK1725 CAT GAT GGG GTT GAT ATG GT CLN2-ORF, 3’-Primer
CK1738 TTC TTT GCT GAT GGC ACT CA CLN2-3’-UTR, 5’-Primer
CK1739 GAC GTC TTG CTT TTT TCG GT CLN2-3’-UTR, 3’-Primer
CK1740 AAA TAA CGC TGC TGG TAC TG CLN3-ORF, 5’-Primer
CK1741 TAG GTC TTG GCA GTT TTC CT CLN3-ORF, 3’-Primer
CK1742 CCA AGA CAG CCA AAA ACA CT CTR9-ORF, 5’-Primer
CK1743 CAC TTT TGC CTT TGC TTG CT CTR9-ORF, 3’-Primer
CK1744 CAG TTT AAC CCG AAG TTC TG ORF-freie Region, Chr. V (10562), 5’-Primer
CK1745 AAC AAC GCA GCT GCT TTA AC ORF-freie Region, Chr. V (10699), 3’-Primer
CK1819 TAT TTG GAG AGA TAC CTA AAA TTA ACG CTT GCC ACA AAG AAC CAA TCC ATG GAA AAG AGA AG
CTR9-spez. 5’-Primer zur Amplifikation der TAP-tag Kassette, C-terminale Deletion ab AS 459
CK1820 CTC TTT AAG CAA GCA AAG GCA AAA GTG AGT GAT AAA GAT GAA AGT TCC ATG GAA AAG AGA AG
CTR9-spez. 5’-Primer zur Amplifikation der TAP-tag Kassette, C-terminale Deletion ab AS 538
CK1821 GGA CCA GCT CTA GAA ATT TTA AGG AAG GTA AGA GAT TCT TCC ATG GAA AAG AGA AG
CTR9-spez. 5’-Primer zur Amplifikation der TAP-tag Kassette, C-terminale Deletion ab AS 728
CK1822 TTT TAT CAA AAA GCC TTG GAA AAT GCC AAA ACT GCT TTG GAT CTT TCC ATG GAA AAG AGA AG
CTR9-spez. 5’-Primer zur Amplifikation der TAP-tag Kassette, C-terminale Deletion ab AS 805
CK1839 CGG GAT CCA ATG AGA AAA ACG ACT GT CTR9-Subklonierung (Nt. 1648), 5’-Primer
CK1840 CGG GAT CCC CAG CTC TAG AAA TTT TA CTR9-Subklonierung (Nt. 2146), 3’-Primer
CK1841 CCG CTC GAG ATG AAT GAA CTT GCT CTT CTR9-Subklonierung (Nt. 2430), 5’-Primer
CK1842 CCG CTC GAG GAA CAA TCC GTC GTT ATC CTR9-Subklonierung (Nt. 3234), 3’-Primer
CK1856 TGA AGA AGA CGA TGA AGA CG PAF1-ORF, 5’-Primer
CK1857 GTT TCC TTT TCT TCC TCT GG PAF1-ORF, 3’-Primer
CK1858 AAA ACT AGG CGA GTT GAC CT RTF1-ORF, 5’-Primer
CK1859 GAA TTG TGC CAC AAG TAG CT RTF1-ORF, 3’-Primer
CK1860 CTT GAA GCA GTG GTT AAC AG SPT16-ORF, 5’-Primer
CK1861 CTA ATT CAT CCC AGT CTT CA SPT16-ORF, 3’-Primer
CK1862 GAC CAG CTT GTC AAG ATC GT CKA1-ORF, 5’-Primer
CK1863 TCC CTT ATT GGG GCA AAC CA CKA1-ORF, 3’-Primer
CK1864 TTC AAG CAG AAG GCA TCT CA BRE1-ORF, 5’-Primer
CK1865 GCA TGT AGG GCA TTT TCT CA BRE1-ORF, 3’-Primer
CK1866 CAA CTG GGG TTT ATA TGC TC SET1-ORF, 5’-Primer
CK1867 GAT ATC ACG CAG TGC ATA GA SET1-ORF, 3’-Primer
CK1868 AAG AAA AAC GGT CTC CCA GT SET2-ORF, 5’-Primer
CK1869 TCC CAA CGC CTT TTT TTG CT SET2-ORF, 3’-Primer
CK1870 AAT AAC AGG GTC GCT GAA CT DOT1-ORF, 5’-Primer
CK1871 CAC TAT GCG TCC ATG AAA CA DOT1-ORF, 3’-Primer
CK1930 TCC TCA TCT TCA GAT GAC GAT GTT AGC AGC GAA AGT GAA GAA GAG TCC ATG GAA AAG AGA AG
BDF1-spezifischer 5’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1931 GCT CAT TCT TCT CAG TCG TTG AAG ATA ATC AAA TTC AAA ATT CAG TAC GAC TCA CTA TAG GG
BDF1-spezifischer 3’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK1932 TCC TCA TCT TCA GAT GAC GAT GTT AGC AGC GAA AGT GAA GAA GAG CGT ACG CTG CAG GTC GAC
BDF1-spezifischer S3-Primer zur Amplifikation der HA6-Kassette aus pYM3

Material & Methoden
108
Tabelle 4.7: Verwendete Oligonukleotide (Fortsetzung) Primer Sequenz (5' → 3') Beschreibung
CK1933 GCT CAT TCT TCT CAG TCG TTG AAG ATA ATC AAA TTC AAA ATT CAG ATC GAT GAA TTC GAG CTC G
BDF1-spezifischer S2-Primer zur Amplifikation der HA6-Kassette bzw. zur ORF-Deletion mit KanMX
CK1954 CGG GAT CCA CAA ACG CAA TGA AGG TG CTR9-Subklonierung (Nt. 4), 5’-Primer
CK1955 CGG GAT CCA ACT TGT TGG AGG AAA GT CTR9-Subklonierung (Nt. 1156), 5’-Primer
CK1957 CCG CTC GAG AGT TCT TGC TAT GTT ATA CTR9-Subklonierung (Nt. 1647), 3’-Primer
CK2065 ACA AGC TAA AAG GCG GTC GAA TCT CAA CGG CTC TGA TAA ACG TAC CGT ACG CTG CAG GTC GAC
BDF1-spezifischer S3-Primer zur Amplifikation der KanMX-Kassette aus pFA6KanMX
CK2099 CTT AGC ACG TGG ATC AAA GA BDF1-ORF, 5’-Primer
CK2100 CTC TTG GCT TAA TGC CTT AG BDF1-ORF, 3’-Primer
CK2103 TTC TAA TTG TCT GGT CCA TTT GTG TTG AGA GCA AGA AAA AAA AAT ATC GAA TTC CTG CAG CCC
CTR9-spezifischer 5’-Primer zur Amplifikation der klTRP1 Kassette aus pBS1479 (ORF-Deletion)
CK2104 ATA TTA AGT TTC TTT AAA AGT CTT GAT TCT AAC CCT CGC CTC TTC GGG CCC ATG TTT GAT ACA
PAF1-spezifischer 3’-Primer zur Amplifikation der klTRP1 Kassette aus pBS1479 (ORF-Deletion)
CK2105 TCT CAA ATG GGT AAT ACT GGA TCA GGA GGG TAT GAT AAT GCT TGG TCC ATG GAA AAG AGA AG
RPB3-spezifischer 5’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
CK2113 TAG CTT GGC AGC AAC AGGA 5’-Kontrollprimer TELVIL::URA3
CK2114 GGC ACA CGC ATA ATT GACG 3’-Kontrollprimer TELVIL::URA3
CK2129 ATT TTC GGT TCG TTC ACT TGT TTT TTT TCC TCT ATT ACG CCC ACT TAC GAC TCA CTA TAG GG
RPB3-spezifischer 3’-Primer zur Amplifikation der TAP-tag Kassette aus pBS1479
Zur Verifizierung der ORF-Deletion in Stämmen aus der Open Biosystems YKO-Kollektion wurden Primerpaare (A primer/kanB primer bzw. kanC primer/D primer) entsprechend der Angaben des „Saccharomyces Genome Deletion Project“ eingesetzt (www-sequence.stanford.edu/group/yeast_deletion _project/deletions3.html). 4.1.4 Enzyme, Substrate und spezielle Feinchemikalien
Die folgenden Tabellen geben einen Überblick über die verwendeten Enzyme (Tabelle 4.8) sowie die eingesetzten Substrate und speziellen Feinchemikalien (Tabelle 4.9)
Tabelle 4.8: Verwendete Enzyme
Enzym Aktivität Hersteller
BamHI 10 U·µL-1 New England Biolabs
Casein-Kinase II 10 U·µL-1 Promega
Tabelle 4.8: Verwendete Enzyme (Fortsetzung)
Enzym Aktivität Hersteller
Glusulase --- Du Pont
Lysozym (Hühnereiweiß) 185000 U·mg-1 Serva
Proteinase K 20 mg·mL-1 Promega
PstI 20 U⋅µL-1 New England Biolabs
T4-DNA-Ligase 400 U⋅µL-1 New England Biolabs
Taq DNA-Polymerase 3,5 U⋅µL-1 Roche, Expand High Fidelity

Material & Methoden
109
Tabelle 4.8: Verwendete Enzyme (Fortsetzung)
Enzym Aktivität / Reinheit Hersteller
Taq DNA-Polymerase ca. 2,5 U⋅µL-1 Laborpräparat, AG Koch
TEV-Protease 10 U⋅µL-1 Invitrogen XhoI 10 U⋅µL-1 New England Biolabs Zymolyase T20 20000 U⋅g-1 Seikugaku Zymolyase T100 100000 U⋅g-1 Seikugaku
Tabelle 4.9: Substrate und spezielle Feinchemikalien
Bezeichnung Konzentration / Reinheit
N-acetyl-D,L-phenylalanin-ß-naphthylester ≥ 98% Sigma-Aldrich Ampicillin > 99,8% Sigma-Aldrich Aprotinin --- Roche ATP, GTP ≥ 99% Sigma-Aldrich γ-32P-ATP, γ-32P-GTP 3 kCi·mM-1;10 mCi·mL-1 Perikin Elmer dATP, dCTP, dGTP, dTTP 100 mmol⋅L-1 Promega 6-Azauracil ≥ 98% Sigma-Aldrich Benzamidin ≥ 95% Sigma-Aldrich BSA 2,0 g⋅L-1 Bio-Rad BSA (Fraktion V) ≥ 92% Sigama-Aldrich Chloramphenikol ≥ 98,5% Roth CKII Peptidsubstrat 10 mg·mL-1 Promega Complete™ Tabletten Roche EGTA ≥ 92% Sigma-Aldrich Ethidiumbromid 10 mg·mL-1 Roth Fast Garnet GBR ≥ 97% Sigma-Aldrich 5-Flourorotsäure ≥ 98% Fermentas G418-Sulfat --- Serva Glutathion, reduziert ≥ 98% Serva Glykogen 10 mg·mL-1 Roche Histon H1 (Kalbsthymus) --- Roche Leupeptin --- Roche Pepstatin --- Roche Phenylmethylsulfonylfluorid ≥ 99% Roth DNA Na-Salz (salmon testes) 10 g·L-1 Sigma-Aldrich
4.1.5 Antikörper und Affinitätsmatrizes Tabelle 4.10: Verwendete Primär- und Sekundär-Antikörper
Bezeichnung Spezifität WB Quelle Herkunft
12CA5 HA-Epitop (monoclonal) 1:1000 Maus Laborpräparat sc-789 Myc-Epitop (polyclonal) 1:2000 Kaninchen Santa Cruz Chemicals 9E11 Myc-Epitop (monoclonal) --- Maus NeoMarkers H3K79-dm (NL79) di-Methyl-H3Lys79 (monoclonal) 1:20000 Kaninchen Upstate

Material & Methoden
110
Tabelle 4.10: Verwendete Primär- und Sekundär-Antikörper (Fortsetzung)
Bezeichnung Spezifität WB Quelle Herkunft
H3K79-tm tri-Methyl-H3Lys79 (polyclonal) 1:500 Kaninchen Abcam anti-GST GST (monoclonal) 1:100000 Maus Sigma-Aldrich anti-CBP CBP des TAP-tags 1:2000 Kaninchen Sigma-Aldrich PAP (ProtA)# 1:20000 Kaninchen Sigma-Aldrich anti-Mouse-HRP IgG (Maus) 1:15000 Ziege Promega anti-Rabbit-HRP IgG (Kaninchen) 1:15000 Ziege Promega
WB eingesetzte Antikörperverdünnung im Westernblot # Der PAP-Komplex wurde zum Nachweis des TAP-tags im Westernblot unter Ausnutzung der ProtA-IgG- Bindung eingesetzt
Tabelle 4.11: Verwendete Affinitätsmatrizes
Matrix Herkunft
Calmodulin-Affinitätsmatrix Stratagene Dynabeads™ (Pan-Mouse IgG) Dynal Glutathion-Sepharose 4B Sigma-Aldrich IgG-Agarose (Kaninchen) Sigma-Aldrich
4.1.6 Protein- und DNA-Größenstandards DNA-Größenstandards 1 kbp DNA Ladder, New England Biolabs: 0,5; 1,0; 1,5; 2,0; 3,0; 4,0; 5,0; 6,0; 8,0; 10,0 kbp 100 bp DNA Ladder, Fermentas (O’GeneRuler): 80; 100; 200, 300, 400, 500, 600, 700, 800, 900, 1031 bp
Protein-Größenstandards High Range Protein Standard, Bio-Rad: peqGOLD Prestained Protein-Marker, Peqlab: 200; 116,3; 97,4; 66,2; 45,0 kDa 122, 79, 47, 33, 24, 20 kDa Low Range Protein Standard, Bio-Rad: 97,4; 66,2; 45,0; 31,0; 21,5 kDa 4.1.7 Verwendete Kits QIAquick Gel Extraction Kit, Qiagen NucleoSpin Plasmid Isolation Kit, Macherey-Nagel
QIAquick PCR Purification Kit, Qiagen Protein Assay Kit (Bradford), Bio-Rad
QIAprep Spin Miniprep Kit, Qiagen Silver Stain Plus, Bio-Rad
QIAGEN Plasmid Midi Kit, Qiagen
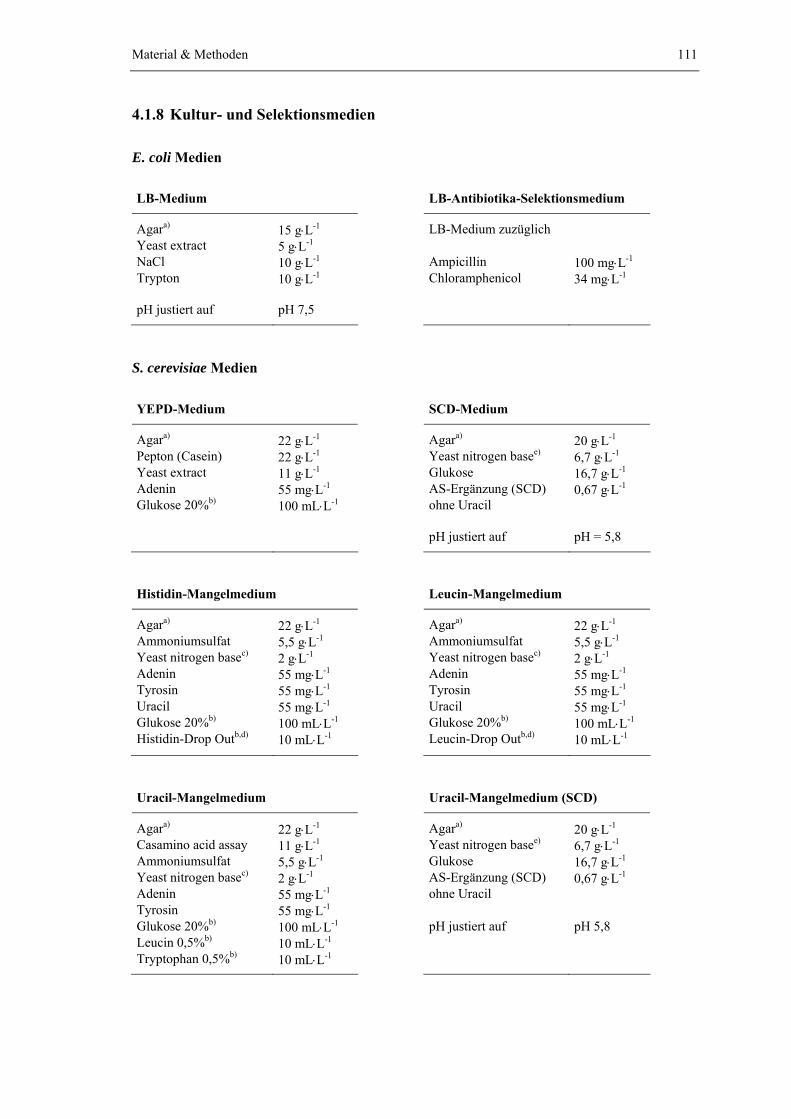
Material & Methoden
111
4.1.8 Kultur- und Selektionsmedien E. coli Medien LB-Medium LB-Antibiotika-Selektionsmedium
Agara) 15 g⋅L-1 LB-Medium zuzüglich Yeast extract 5 g⋅L-1 NaCl 10 g⋅L-1 Ampicillin 100 mg⋅L-1 Trypton 10 g⋅L-1 Chloramphenicol 34 mg⋅L-1 pH justiert auf pH 7,5
S. cerevisiae Medien
YEPD-Medium SCD-Medium
Agara) 22 g⋅L-1 Agara) 20 g⋅L-1 Pepton (Casein) 22 g⋅L-1 Yeast nitrogen basee) 6,7 g⋅L-1 Yeast extract 11 g⋅L-1 Glukose 16,7 g⋅L-1 Adenin 55 mg⋅L-1 AS-Ergänzung (SCD) 0,67 g⋅L-1 Glukose 20%b) 100 mL⋅L-1 ohne Uracil pH justiert auf pH = 5,8
Histidin-Mangelmedium Leucin-Mangelmedium
Agara) 22 g⋅L-1 Agara) 22 g⋅L-1 Ammoniumsulfat 5,5 g⋅L-1 Ammoniumsulfat 5,5 g⋅L-1 Yeast nitrogen basec) 2 g⋅L-1 Yeast nitrogen basec) 2 g⋅L-1 Adenin 55 mg⋅L-1 Adenin 55 mg⋅L-1 Tyrosin 55 mg⋅L-1 Tyrosin 55 mg⋅L-1 Uracil 55 mg⋅L-1 Uracil 55 mg⋅L-1 Glukose 20%b) 100 mL⋅L-1 Glukose 20%b) 100 mL⋅L-1 Histidin-Drop Outb,d) 10 mL⋅L-1 Leucin-Drop Outb,d) 10 mL⋅L-1
Uracil-Mangelmedium Uracil-Mangelmedium (SCD)
Agara) 22 g⋅L-1 Agara) 20 g⋅L-1 Casamino acid assay 11 g⋅L-1 Yeast nitrogen basee) 6,7 g⋅L-1 Ammoniumsulfat 5,5 g⋅L-1 Glukose 16,7 g⋅L-1 Yeast nitrogen basec) 2 g⋅L-1 AS-Ergänzung (SCD) 0,67 g⋅L-1 Adenin 55 mg⋅L-1 ohne Uracil Tyrosin 55 mg⋅L-1 Glukose 20%b) 100 mL⋅L-1 pH justiert auf pH 5,8 Leucin 0,5%b) 10 mL⋅L-1 Tryptophan 0,5%b) 10 mL⋅L-1

Material & Methoden
112
Tryptophan-Mangelmedium Aminosäure-Drop Out Lösung
Agara) 22 g⋅L-1 Arginin 2 g⋅L-1 Casamino acid assay 11 g⋅L-1 Histidin 1 g⋅L-1 Ammoniumsulfat 5,5 g⋅L-1 Leucin 6 g⋅L-1 Yeast nitrogen basec) 2 g⋅L-1 Iso-Leucin 6 g⋅L-1 Adenin 55 mg⋅L-1 Lysin 4 g⋅L-1 Tyrosin 55 mg⋅L-1 Methionin 1 g⋅L-1 Uracil 55 mg⋅L-1 Phenylalanin 6 g⋅L-1 Glukose 20%b) 100 mL⋅L-1 Threonin 5 g⋅L-1 Leucin 0,5%b) 10 mL⋅L-1 Tryptophan 4 g⋅L-1
5FOA Selektionsmedium AS-Ergänzung (SCD)
Lösung Af): Adenin-Hemisulfat 2,0 g Yeast nitrogen basec) 4 g⋅L-1 p-Aminobenzoesäure 0,2 g Ammoniumsulfat 11 g⋅L-1 Argenin HCl 2,0 g 5-Fluororotsäure 1 g⋅L-1 Histidin HCl 2,0 g Uracil 100 mg⋅L-1 Myo-Inositol 2,0 g Adenin 100 mg⋅L-1 Leucin 4,0 g Tryptophan 0,8 g⋅L-1 Iso-Leucin 2,0 g Leucin 1,2 g⋅L-1 Lysin HCl 2,0 g Histidin 0,2 g⋅L-1 Methionin 2,0 g Methionin 0,2g⋅L-1 Phenylalanin 3,0 g Glukose 40 g⋅L-1 Serin 2,0 g Threonin 2,0 g Lösung B: Tryptophan 3,0 g Agar 40 g⋅L-1 Tyrosin 2,0 g Uracil 1,2 g Valin 9,0 g
Lösung A wird mit der autoklavierten und Verwendung als Pulver, homogenisiert auf ca. 50°C abgekühlten Agar-Lösung (Lösung B) im Verhältnis 1:1 gemischt.
Minimalmedium Sporulationsmedium
Agara) 22 g⋅L-1 Agara) 15 g⋅L-1 Ammoniumsulfat 5,5 g⋅L-1 Natriumacetat 8,2 g⋅L-1 Yeast nitrogen basec) 2 g⋅L-1 Kaliumchlorid 1,9 g⋅L-1 Glukose 20%b) 100 mL⋅L-1 Natriumchlorid 1,2 g⋅L-1 Magnesiumsulfat 0,35 g⋅L-1
a) Zusatz nur bei Festmedien d) Drop Out-Lösung ohne die angegebene Aminosäure
b) Zusatz nach dem Autoklavieren e) mit Ammoniumsulfat, ohne Aminosäuren
c) ohne Aminosäuren und Ammoniumsulfat f) filtersterilisiert
Prä-Sporulations und Sporulationsmedium für Kreuzungen mit congenen BY4741 Derivaten
Zur Prä-Sporulation und Sporulation der Kreuzungen mit Stämmen aus der Open Biosystems YKO-Kollektion wurden Medien entsprechend den Protokollen des „Saccharomyces Genome Deletion Project“ eingesetzt (www-sequence.stanford.edu/group/yeast_deletion _project/deletions3.html).

Material & Methoden
113
6AU Selektionsmedium
Zusammensetzung entsprechend Uracil-Mangelmedium bzw. Uracil-Mangelmedium (SCD) mit zuzüglich 50, 75 oder 100 mg⋅L-1 6-Azauracilb,f) (Endkonzentration).
G418-Selektionsmedien
Grundmedium wie oben angegeben zuzüglich G418-Sulfatb,f) 200 mg⋅L-1 (Endkonzentration).
4.2 Mikro- und Molekularbiologische Methoden
4.2.1 Kultivierung von S. cerevisiae und E. coli
S. cerevisiae Flüssigkulturen wurden in YEPD-Medium bei 30°C, Deletionsmutanten bei 25°C oder 28°C, unter ständiger Durchmischung des Mediums kultiviert. Die Zelldichte im Flüssigmedium wurde optisch bei einer Wellenlänge von 600 nm verfolgt (OD600). Hefekolonien wurden auf den entsprechenden Festmedien ebenfalls bei 30°C bzw. 25°C inkubiert und anschließend bei 4°C gelagert. Die Anzucht von E. coli erfolgte in gleicher Weise unter Verwendung von LB-Medium mit Zusatz des jeweiligen Antibiotikums (Amp und/oder Cm) bei 37°C. 4.2.2 Amplifikation von DNA-Sequenzen durch Polymerase-Kettenreaktion (PCR)
Amplifikation aus Plasmiden und genomischer DNA
Gendisruptionskassetten, Module für Affinitäts- und Epitoptags sowie genomische DNA-Sequenzen wurden aus den entsprechenden Plasmiden (Tabelle 4.5) bzw. aus genomischer Hefe-DNA durch Polymerase-Kettenreaktion amplifiziert. Der dazu verwendete Reaktionsansatz und das zugehörige PCR-Programm ist in Tabelle 4.12 angegeben. Die Isolierung und Konzentrierung der entstandenen PCR-Produkte erfolgte jeweils durch Phenol/Chloroform-Extraktion und ethanolische Fällung der DNA. Die gefällte DNA wurde anschließend in TE-Puffer oder deionisiertem Wasser gelöst. Die isolierten PCR-Produkte wurden direkt zur Transformation von Hefezellen eingesetzt oder für die Insertion in einen Vektor verwendet.
Tabelle 4.12: DNA-Amplifikation aus Plasmiden und genomischer Hefe-DNA durch PCR
PCR-Ansatz PCR-Programm
5’-Primer (100 µM) 1,2 µL 1 94°C, 5min 3’-Primer(100 µM) 1,2 µL 2 94°C, 30 sec HiFi Taq- Puffer (10x) 20 µL 3 48°C (56°C)*, 30 sec dNTP-Mischung (jeweils 10 mM) 3 µL 4 72 C, 60 sec.kbp-1 HiFi Taq-Polymerase (Roche)* 2 µL 5 35 Zyklen Template (Plasmid oder gen. Hefe-DNA) ca. 100 ng 6 72°C, 5 min H20MQ ad. 200 µL Reaktionsvolumen 4-mal 50 µL
* Die Amplifikation der HA6::klTRP1-Kassette aus pYM3 wurde im Hot-Start Verfahren (0,5 µL HiFi Taq-Polymerase pro Reaktionsansatz) mit einer Hybridisierungstemperatur von 56°C durchgeführt.
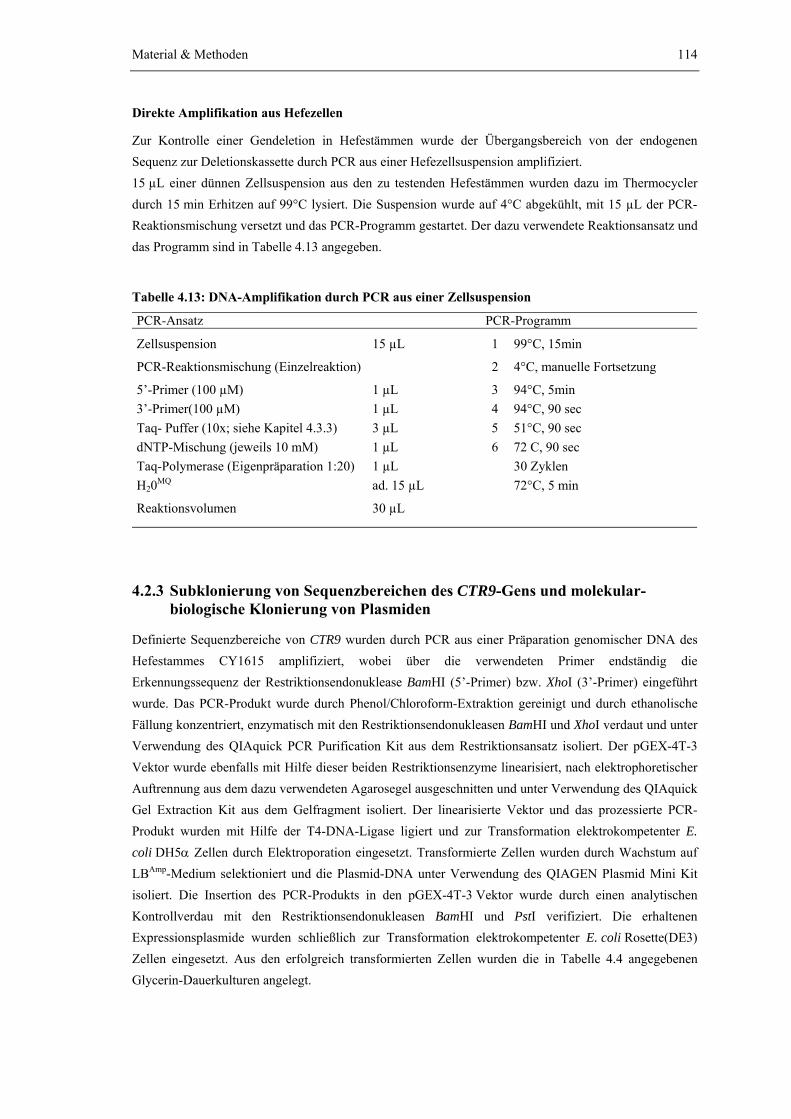
Material & Methoden
114
Direkte Amplifikation aus Hefezellen
Zur Kontrolle einer Gendeletion in Hefestämmen wurde der Übergangsbereich von der endogenen Sequenz zur Deletionskassette durch PCR aus einer Hefezellsuspension amplifiziert. 15 µL einer dünnen Zellsuspension aus den zu testenden Hefestämmen wurden dazu im Thermocycler durch 15 min Erhitzen auf 99°C lysiert. Die Suspension wurde auf 4°C abgekühlt, mit 15 µL der PCR-Reaktionsmischung versetzt und das PCR-Programm gestartet. Der dazu verwendete Reaktionsansatz und das Programm sind in Tabelle 4.13 angegeben.
Tabelle 4.13: DNA-Amplifikation durch PCR aus einer Zellsuspension
PCR-Ansatz PCR-Programm
Zellsuspension 15 µL 1 99°C, 15min
PCR-Reaktionsmischung (Einzelreaktion) 2 4°C, manuelle Fortsetzung
5’-Primer (100 µM) 1 µL 3 94°C, 5min 3’-Primer(100 µM) 1 µL 4 94°C, 90 sec Taq- Puffer (10x; siehe Kapitel 4.3.3) 3 µL 5 51°C, 90 sec dNTP-Mischung (jeweils 10 mM) 1 µL 6 72 C, 90 sec Taq-Polymerase (Eigenpräparation 1:20) 1 µL 30 Zyklen H20MQ ad. 15 µL 72°C, 5 min
Reaktionsvolumen 30 µL
4.2.3 Subklonierung von Sequenzbereichen des CTR9-Gens und molekular- biologische Klonierung von Plasmiden
Definierte Sequenzbereiche von CTR9 wurden durch PCR aus einer Präparation genomischer DNA des Hefestammes CY1615 amplifiziert, wobei über die verwendeten Primer endständig die Erkennungssequenz der Restriktionsendonuklease BamHI (5’-Primer) bzw. XhoI (3’-Primer) eingeführt wurde. Das PCR-Produkt wurde durch Phenol/Chloroform-Extraktion gereinigt und durch ethanolische Fällung konzentriert, enzymatisch mit den Restriktionsendonukleasen BamHI und XhoI verdaut und unter Verwendung des QIAquick PCR Purification Kit aus dem Restriktionsansatz isoliert. Der pGEX-4T-3 Vektor wurde ebenfalls mit Hilfe dieser beiden Restriktionsenzyme linearisiert, nach elektrophoretischer Auftrennung aus dem dazu verwendeten Agarosegel ausgeschnitten und unter Verwendung des QIAquick Gel Extraction Kit aus dem Gelfragment isoliert. Der linearisierte Vektor und das prozessierte PCR-Produkt wurden mit Hilfe der T4-DNA-Ligase ligiert und zur Transformation elektrokompetenter E. coli DH5α Zellen durch Elektroporation eingesetzt. Transformierte Zellen wurden durch Wachstum auf LBAmp-Medium selektioniert und die Plasmid-DNA unter Verwendung des QIAGEN Plasmid Mini Kit isoliert. Die Insertion des PCR-Produkts in den pGEX-4T-3 Vektor wurde durch einen analytischen Kontrollverdau mit den Restriktionsendonukleasen BamHI und PstI verifiziert. Die erhaltenen Expressionsplasmide wurden schließlich zur Transformation elektrokompetenter E. coli Rosette(DE3) Zellen eingesetzt. Aus den erfolgreich transformierten Zellen wurden die in Tabelle 4.4 angegebenen Glycerin-Dauerkulturen angelegt.

Material & Methoden
115
Zur Amplifikation der zusätzlich verwendeten Plasmide und Vektoren wurden ebenfalls E. coli DH5α Zellen eingesetzt. Die Transformation erfolgte wiederum durch Elektroporation. Zur Plasmidisolierung im Midi-Maßstab wurden Präparationskits von Qiagen oder Machery-Nagel verwendet.
4.2.4 Transformation von S. cerevisiae mit Lithium-Acetat (Ito et al., 1983) Zur Transformation wurden die Hefestämme CY1078 oder CY1615 verwendet. Die Anzucht der Hefezellen erfolgte in YEPD-Medium bis zu einer optischen Zelldichte von OD600 = 1 - 2. Pro Transformationsansatz wurden ca. 10 mL bis 15 mL Hefekultur mit einer Zelldichte von OD600 = 1 eingesetzt. Alle folgenden Angaben beziehen sich auf ein Kulturvolumen von 50 - 100 mL. Die Zellen wurden durch Zentrifugation (3000 rpm, 3 min) geerntet. Das Zellpellet wurde in 10 mL sterilem, deionisierten Wasser resuspendiert und erneut zentrifugiert (3000 rpm, 3 min). Der Waschschritt wurde mit 10 mL TE/Li-Acetat-Puffer wiederholt und die durch Zentrifugation sedimentierten Zellen abschließend in ca. 200 µL TE/Li-Acetat-Puffer resuspendiert. Die Suspension wurde bis zur Transformation auf Eis gelagert. Zur Transformation wurden 50 µL der Zellsuspension mit bis zu 5 µL des gereinigten PCR-Produktes und 5 µL der Träger-DNA (10 g⋅L-1, sonifiziert) gemischt und 300 µL PEG/Li-Acetat-Puffer zugesetzt. Der Transformationsansatz wurde nach erneutem Durchmischen 30 min bei 30°C und direkt im Anschluss 15 min bei 45°C inkubiert. Zur Selektion auf Tryptophanprototrophie wurde der gesamte Transformationsansatz auf Tryptophan-Mangelmedium ausplattiert und bei 25°C inkubiert. Transformierte Hefekolonien wurden vor der weiteren Verwendung auf Tryptophan-Mangelmedium vereinzelt und erneut 1-2 Tage bei 25°C inkubiert. Zur Kontrolle des Transformationserfolges wurde der eingesetzte Hefestamm zusätzlich mit dem TRP1-Expressions-plasmid YEplac112, bzw. in einer Negativkontrolle nur mit der Träger-DNA transformiert. Zur Selektion auf Geneticinresistenz wurde der gesamte Transformationsansatz zunächst mit 3 mL YEDP-Medium gemischt und für 3-4 h bei 25°C inkubiert. Die durch Zentrifugation sedimentierten Zellen wurden in 250 µL YEPD-Medium resuspendiert, auf YEPDG418-Selektionsmedium ausplattiert und mehrere Tage bei 25°C inkubiert. Transformierte Hefekolonien wurden vor der weiteren Verwendung auf YEPDG418-Selektionsmedium vereinzelt und erneut 1-2 Tage bei 25°C inkubiert. Spezielle Puffer
TE/Li-Acetat-Puffer: PEG/Li-Acetat-Puffer:
Tris-Cl pH 7,5 10 mM Tris-Cl pH 7,5 10 mM EDTA 1 mM EDTA 1 mM Lithium-Acetat 100 mM Lithium-Acetat 100 mM Polyethylenglycol 40 %
4.2.5 Kreuzung von Hefestämmen und Tetradenanalyse
Kreuzung und Sporulation von congenen W303 Derivaten
Eine etwa streichholzkopfgroße Menge Zellen aus Kolonien der zu kreuzenden Hefestämme wurde in 50 µL YEPD-Medium vereinigt und suspendiert. 10 -20 µL der Zellsuspension wurden auf YEPD-Agarplatten 24 - 48 h bei 30°C inkubiert. Der Kreuzungsansatz wurde entweder direkt auf Sporulationsmedium übertragen und 1 - 3 weitere Tage bei 25°C inkubiert oder zur Selektion auf diploide Hefezellen auf ein jeweils geeignetes Selektionsmedium übertragen und bei 25°C inkubiert. Die

Material & Methoden
116
Diploiden Zellen wurden auf YEPD-Medium vermehrt (25°C), auf Sporulationsmedium übertragen und 1-3 weitere Tage bei 25°C inkubiert. Die Sporulation wurde mikroskopisch verfolgt.
Kreuzung, Präsporulation und Sporulation von congenen BY4741 Derivaten
Die Kreuzung von congenen BY4741 Derivaten der Open Biosystems YKO-Kollektion erfolgte wie für die Stämme mit genetischem W303 Hintergurnd beschrieben. Die Prä-Sporulation und Sporulation wurde im Anschluss entsprechend den Protokollen des „Saccharomyces Genome Deletion Project“ durchgeführt (www-sequence.stanford.edu/group/yeast_deletion _project/deletions3.html).
Tetradenanalyse
Nach erfolgreicher Tetradenbildung wurde ein Teil der sporulierten Zellen in 180 µL sterilem, deionisiertem Wasser und 20 µL Zymolyase T20 (1 mg⋅mL-1) resuspendiert und ca. 45 min bei 37°C inkubiert. Die Suspension wurde je nach Zelldichte ggf. weiter verdünnt. Etwa 5 - 10 µL der Zellsuspension wurden am Rand einer YEPD-Agarplatte ausgestrichen. Einzelne Tetraden wurden mit Hilfe der Rastermatrix eines Mikromanipulators (Singer Instruments) auf definierten Positionen der Agarplatte abgelegt und die Ascosporen entlang diskreter Reihen vereinzelt. Die Ascosporen wurden 2-3 Tage bei 25°C inkubiert, bis die resultierenden Kolonien zu einer für die Replikaplattierung ausreichenden Größe angewachsen waren. Zur Replikaplattierung wurden Zellen jeder Kolonie durch Andrücken der "Tetradenplatte" auf sterilen Stempelsamt übertragen. Die am Samt anhaftenden Zellen wurden anschließend in gleicher Weise auf eine Agarplatte des entsprechenden Selektionsmediums übertragen, so dass das Koloniemuster der "Tetradenplatte" eindeutig erhalten blieb. Die Selektionsplatten wurden mehrere Tage bei 25°C inkubiert und die Wachstumsfähigkeit der einzelnen Kolonien überprüft. Der Paarungstyp der meiotischen Segreganten wurde durch die Komplementationsfähigkeit einer Testkreuzung in ähnlicher Weise ermittelt. Dazu wurden Zellen der haploiden Teststämme CY274 (MATa, his1), bzw. CY275 (MATα, his1) über die gesamte Fläche einer Minimalmedium-Agarplatte ausgestrichen und über das Stempelsamt mit Zellen der zu testenden Kolonien vereinigt. Alle zur Kreuzung eingesetzten Parentalstämme sind Träger eines HIS1-Allels. Die meiotischen Segreganten sind also prinzipiell dazu in der Lage die His-Auxotrophie der Teststämme zu komplementieren, allerdings nur dann, wenn sie aufgrund des entgegengesetzten Paarungstyps mit den Zellen der Teststämme fusionieren können. Die Bildung diploider Kolonien, nach ein- bis zweitägiger Inkubation der Testkreuzungen bei 30°C, erlaubt daher einen Rückschluss auf den Paarungstyp der meiotischen Segreganten.
4.2.6 Kolonie-Färbetest zur Ermittlung der Carboxypeptidase-Aktivität
Das Verfahren beruht auf der enzymatischen Spaltung von N-acetyl-DL-phenylalanin-ß-naphthylester durch die Proteinase C. Das entstehende 2-Naphthol kann dann mit dem Diazoniumsalz Fast Garnet GBR zu einem roten Azofarbstoff reagieren, der die Aktivität der Proteinase C anzeigt. Deletion von PRC1 oder PEP4 führt zum Ausfall der Proteinase C, so dass keine Farbstoffbildung eintritt (Jones, 1991). Die Methode kann direkt zur Kontrolle der Peptidaseaktivität auf YEPD-Agarplatten wachsender Kolonien eingesetzt werden. Das Substrat (3 g⋅L-1 N-acetyl-DL-phenyl-alanin ß-naphthyl-ester in Dimethylformamid) wurde im Verhältnis 2:3 mit einer ca. 50°C warmen 0,6% Agaroselösung gemischt

Material & Methoden
117
und die zu testenden Kolonien damit überschichtet. Nach dem Erstarren der Agarose wurde eine Lösung des Diazoniumsalzes Fast Garnet GBR (1 g⋅L-1 Fast Garnet GBR in 0,1 M Tris-Cl pH 7,3) zugegeben, die bereits nach kurzer Zeit zu einer Färbung der Kolonien mit aktiver Proteinase C führt. Carboxypeptidasedefiziente Kolonien (pep4Δ) bleiben dagegen weiß.
4.2.7 Untersuchung von Hefestämmen hinsichtlich einer auftretenden Temperatursensitivität sowie Elongations- und Silencingdefekten
Frisch auf YEPD-Medium gewachsene Zellen der zu testenden Hefestämme wurden in sterilem, deionisiertem Wasser resuspendiert und die Zelldichte der Suspension auf einen Wert von OD600 = 4,0 eingestellt. Aus dieser Suspension wurden durch serielle Verdünnung zusätzliche Suspensionen mit Zelldichten von OD600 = 1,0; 0,40; 0,040; 0,0040 und 0,00040 entsprechend 100.000, 25.000, 10.000, 1.000, 100 und 10 Zellen·µL-1 hergestellt. Ca. 2 µL dieser Suspensionen wurden mit Hilfe eines Stempelrechens auf die jeweiligen Selektionsmedien übertragen und nach dem Eintrocknen bis zu zehn Tage bei 25°C, 30°C oder 37°C (nur Temperatursensitivitätstest) inkubiert. Das Zellwachstum wurde im Abstand von zwei Tagen fotografisch dokumentiert. Zur Ermittlung von Elongationsdefekten in Uracil-prototrophen Stämmen wurden 6AU Selektionsmedien mit 50, 75 und 100 mg.L-1 6-Azauracil eingesetzt. Die Inkubation der Zellen erfolgte bei 30°C. Die Ermittlung von Silencingdefekten in TELVIIL::URA3 Stämmen erfolgte durch Test der Wachstumsfähigkeit in Gegenwart von 1 g·L-1 Fluororotsäure bei 30°C.
4.3 Spezielle biochemische Methoden
4.3.1 Tandem-Affinitätsreinigung (Puig et al., 2001) Zellernte und Aufschluss
YEPD-Kulturen der entsprechenden Hefestämme mit einem Kulturvolumen von 600 mL bzw. 1000 mL wurden bis zu einer Zelldichte zwischen OD600 = 1,5 und OD600 = 3,0 kultiviert. Die Zellen wurden durch Zentrifugation geerntet (Heraeus-Kühlzentrifuge, 4400 rpm, 20 min, 4°C) und nach Resuspendierung in 25 mL deionisiertem Wasser (H2OMQ, 4°C) erneut durch Zentrifugation sedimentiert (4400 rpm, 15 min, 4°C). Die Zellen wurde im Anschluss in 5 mL HEPES-Puffer resuspendiert und in ein 50 mL Schraubdeckelgefäß überführt. Nach erneuter Zentrifugation wurden die Zellen noch einmal durch Resuspendieren in 2,5 mL HEPES-Aufschlusspuffer gewaschen, abschließend durch Zentrifugation (4400 rpm, 5 min, 4°C) erneut pelletiert und nach vollständiger Entfernung des Puffers durch Inkubation des geschlossenen Schraubdeckelgefäßes in flüssigem Stickstoff schockgefroren (Inkubation min. 20 min). Das gefrorene Zellpellet wurde bis zum Zellaufschluss bei -80°C gelagert. Für den Zellaufschluss wurde das noch gefrorene Zellpellet bei Raumtemperatur in einer etwa dem Pelletvolumen entsprechenden Menge HEPES-Aufschlusspuffer (4°C) resuspendiert. Alle weiteren Arbeitsschritte wurden im Kühlraum bei 4°C durchgeführt. Die Zellsuspension wurde in Portionen zu 750 µL auf 2 mL Eppendorfgefäße aufgeteilt und jede Fraktion mit ca. 750 µL Glasperlen (∅ 0,5 mm, mit HCl gewaschen) versetzt. Der Zellaufschluss erfolgte mechanisch, durch heftiges Schütteln der Zellsuspension auf dem Vibrax-Schüttler (maximale Frequenz), über einen Zeitraum von 20 min.

Material & Methoden
118
Glasperlen und Zelltrümmer wurden durch Zentrifugation in einer -Kühlzentrifuge (13 krpm, 10 min, 4°C) abgetrennt und die Zellextrakte eines Ansatzes vereinigt.
Ultrazentrifugation
Die Rohextrakte wurden in Ultrazentrifugationsgläser überführt und mit 3 M KCl auf eine Konzentration von 0,1 mol⋅L-1 KCl eingestellt. Der Rohextrakt wurde 70 min bei 4°C einer Ultrazentrifugation (Beckman L8-60M, 60Ti-Rotor) mit 35000 rpm unterzogen.
Dialyse
Der Dialyseschlauch (Servapor, ∅ 16 mm, MWCO 12 kDa - 14 kDa) wurde 30 min in deionisiertem Wasser konditioniert und mehrfach durchspült. Die ultrazentrifugierten Proteinextrakte wurden in den an einem Ende durch Verknoten geschlossenen Dialyseschlauch pipettiert und das offene Ende des Dialyseschlauches durch eine dichte Klemme verschlossen. Die Dialyse erfolgte gegen HEPES-Dialysepuffer über Nacht in 500 mL Zentrifugengefäßen. Der Dialysepuffer wurde dabei permanent durch Überkopfschwenken des Zentrifugengefäßes auf einem Rotor durchmischt.
Tandem-Affinitätsreinigung
Der dialysierte Proteinextrakt wurde in ein 12,5 mL Probenröhrchen mit Schraubdeckel überführt und auf 10 mM Tris-Cl pH 8,0 (mit 1 M Tris-Puffer pH 8,0), 100 mM NaCl (mit 4 M NaCl) und 0,1% NP40 (mit 10% NP40-Lösung) eingestellt, wobei in den Dialysaten eine Chlorid-Konzentration von 50 mmol⋅L-1 angenommen wurde. Zur Adsorption der Protein-TAP-tag Fusion an die IgG-Agarosematrix (Sigma) wurden 125 µL der auf 4°C gekühlten Matrixsuspension eingesetzt. Die IgG-Matrix wurde zuvor durch mehrmaliges Resuspendieren und Zentrifugation (Eppendorf-Kühlzentrifuge, 4000 rpm, 1 min, 4°C) mit Glycin-Puffer pH 2,4 gewaschen und anschließend mit IPP100-Puffer äquilibriert. Der dialysierte Proteinextrakt wurde 2 h mit der IgG-Agarose inkubiert, wobei die Sedimentation der IgG-Matrix durch permanente Durchmischung des Dialysates auf einem Rotor unterbunden wurde. Die IgG-Agarose wurde durch Filtration über eine 2,5 mL Säule (Mobitec) mit Bodenfilter (Porengröße: 35 µm) vom Überstand abgetrennt, viermal mit 2,5 mL IPP100-Puffer und zweimal mit 2,5 mL TEV-Puffer gewaschen. Der Auslauf der Säule wurde verschlossen und die IgG-Agarosematrix in 250 µL TEV-Puffer resuspendiert. Nach Zugabe von 30 U TEV-Protease wurde gebundenes Protein 2 h bei 16°C proteolytisch eluiert. Die Matrixsuspension wurde dabei konstant bei 650 rpm mit einem Thermomixer durchmischt. Das Eluat (TEV-Eluat) wurde in einem 12,5 mL Probenröhrchen aufgefangen und die Matrix zur Elution des Säulentotvolumens mit 100 µL TEV-Puffer gewaschen. Waschfraktion und Eluat wurden vereinigt und mit dem dreifachen Volumen (1050 µL) Calmodulin-Adsorptionspuffer versetzt. Das im TEV-Puffer vorhandene EDTA wurde durch Zugabe von 21 µL 50 mM CaCl2 komplexiert. Zur Adsorption an die Calmodulin (CaM)-Affinitätsmatrix (Stratagene) wurden 125 µL der Matrixsuspension eingesetzt, die zuvor durch mehrfaches Resuspendieren in Calmodulin-Adsorptionspuffer äquilibriert wurde (Kühlzentrifuge: 4000 rpm, 1 min, 4°C). Das TEV-Eluat wurde 1 h bis 1,5 h mit der Calmodulin-Affinitätsmatrix inkubiert. Die Sedimentation der Matrix wurde dabei wiederum durch die Verwendung des Rotors verhindert. Nach der Inkubation wurde die Calmodulin-Matrix durch Filtration über eine 2,5 mL Säule mit Bodenfilter (Porengröße: 35 µm) abgetrennt und viermal mit 2,5 mL CaM-Adsorptionspuffer gewaschen. Die Elution des an die Calmodulin-Affinitätsmatrix gebundenen Proteins
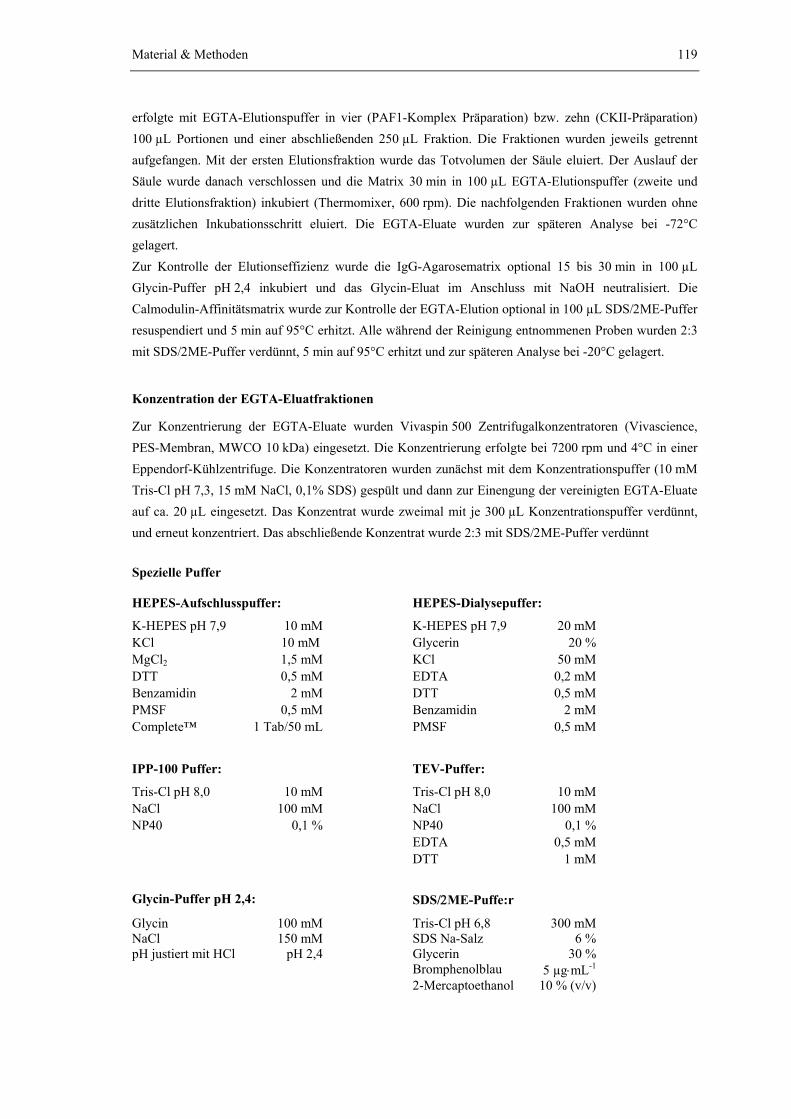
Material & Methoden
119
erfolgte mit EGTA-Elutionspuffer in vier (PAF1-Komplex Präparation) bzw. zehn (CKII-Präparation) 100 µL Portionen und einer abschließenden 250 µL Fraktion. Die Fraktionen wurden jeweils getrennt aufgefangen. Mit der ersten Elutionsfraktion wurde das Totvolumen der Säule eluiert. Der Auslauf der Säule wurde danach verschlossen und die Matrix 30 min in 100 µL EGTA-Elutionspuffer (zweite und dritte Elutionsfraktion) inkubiert (Thermomixer, 600 rpm). Die nachfolgenden Fraktionen wurden ohne zusätzlichen Inkubationsschritt eluiert. Die EGTA-Eluate wurden zur späteren Analyse bei -72°C gelagert. Zur Kontrolle der Elutionseffizienz wurde die IgG-Agarosematrix optional 15 bis 30 min in 100 µL Glycin-Puffer pH 2,4 inkubiert und das Glycin-Eluat im Anschluss mit NaOH neutralisiert. Die Calmodulin-Affinitätsmatrix wurde zur Kontrolle der EGTA-Elution optional in 100 µL SDS/2ME-Puffer resuspendiert und 5 min auf 95°C erhitzt. Alle während der Reinigung entnommenen Proben wurden 2:3 mit SDS/2ME-Puffer verdünnt, 5 min auf 95°C erhitzt und zur späteren Analyse bei -20°C gelagert.
Konzentration der EGTA-Eluatfraktionen
Zur Konzentrierung der EGTA-Eluate wurden Vivaspin 500 Zentrifugalkonzentratoren (Vivascience, PES-Membran, MWCO 10 kDa) eingesetzt. Die Konzentrierung erfolgte bei 7200 rpm und 4°C in einer Eppendorf-Kühlzentrifuge. Die Konzentratoren wurden zunächst mit dem Konzentrationspuffer (10 mM Tris-Cl pH 7,3, 15 mM NaCl, 0,1% SDS) gespült und dann zur Einengung der vereinigten EGTA-Eluate auf ca. 20 µL eingesetzt. Das Konzentrat wurde zweimal mit je 300 µL Konzentrationspuffer verdünnt, und erneut konzentriert. Das abschließende Konzentrat wurde 2:3 mit SDS/2ME-Puffer verdünnt
Spezielle Puffer
HEPES-Aufschlusspuffer: HEPES-Dialysepuffer:
K-HEPES pH 7,9 10 mM K-HEPES pH 7,9 20 mM KCl 10 mM Glycerin 20 % MgCl2 1,5 mM KCl 50 mM DTT 0,5 mM EDTA 0,2 mM Benzamidin 2 mM DTT 0,5 mM PMSF 0,5 mM Benzamidin 2 mM Complete™ 1 Tab/50 mL PMSF 0,5 mM
IPP-100 Puffer: TEV-Puffer:
Tris-Cl pH 8,0 10 mM Tris-Cl pH 8,0 10 mM NaCl 100 mM NaCl 100 mM NP40 0,1 % NP40 0,1 % EDTA 0,5 mM DTT 1 mM
Glycin-Puffer pH 2,4: SDS/2ME-Puffe:r
Glycin 100 mM Tris-Cl pH 6,8 300 mM NaCl 150 mM SDS Na-Salz 6 % pH justiert mit HCl pH 2,4 Glycerin 30 % Bromphenolblau 5 µg⋅mL-1 2-Mercaptoethanol 10 % (v/v)
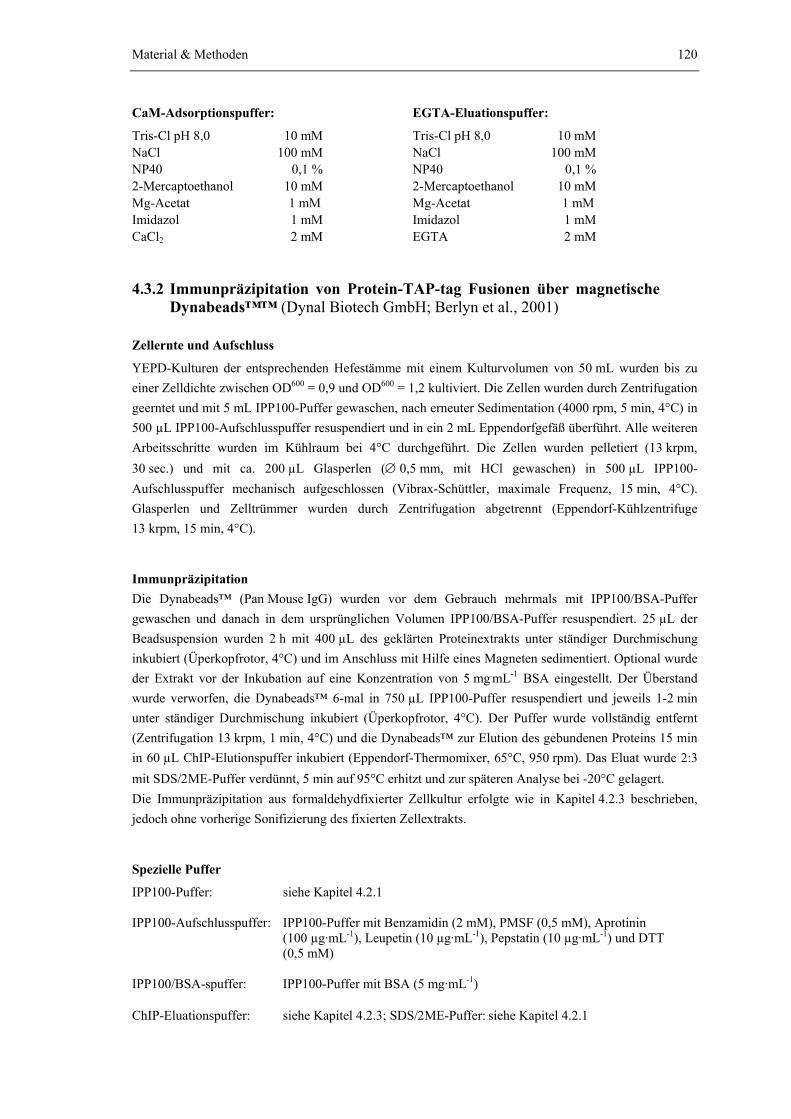
Material & Methoden
120
CaM-Adsorptionspuffer: EGTA-Eluationspuffer:
Tris-Cl pH 8,0 10 mM Tris-Cl pH 8,0 10 mM NaCl 100 mM NaCl 100 mM NP40 0,1 % NP40 0,1 % 2-Mercaptoethanol 10 mM 2-Mercaptoethanol 10 mM Mg-Acetat 1 mM Mg-Acetat 1 mM Imidazol 1 mM Imidazol 1 mM CaCl2 2 mM EGTA 2 mM
4.3.2 Immunpräzipitation von Protein-TAP-tag Fusionen über magnetische Dynabeads™™ (Dynal Biotech GmbH; Berlyn et al., 2001)
Zellernte und Aufschluss
YEPD-Kulturen der entsprechenden Hefestämme mit einem Kulturvolumen von 50 mL wurden bis zu einer Zelldichte zwischen OD600 = 0,9 und OD600 = 1,2 kultiviert. Die Zellen wurden durch Zentrifugation geerntet und mit 5 mL IPP100-Puffer gewaschen, nach erneuter Sedimentation (4000 rpm, 5 min, 4°C) in 500 µL IPP100-Aufschlusspuffer resuspendiert und in ein 2 mL Eppendorfgefäß überführt. Alle weiteren Arbeitsschritte wurden im Kühlraum bei 4°C durchgeführt. Die Zellen wurden pelletiert (13 krpm, 30 sec.) und mit ca. 200 µL Glasperlen (∅ 0,5 mm, mit HCl gewaschen) in 500 µL IPP100-Aufschlusspuffer mechanisch aufgeschlossen (Vibrax-Schüttler, maximale Frequenz, 15 min, 4°C). Glasperlen und Zelltrümmer wurden durch Zentrifugation abgetrennt (Eppendorf-Kühlzentrifuge 13 krpm, 15 min, 4°C).
Immunpräzipitation Die Dynabeads™ (Pan Mouse IgG) wurden vor dem Gebrauch mehrmals mit IPP100/BSA-Puffer gewaschen und danach in dem ursprünglichen Volumen IPP100/BSA-Puffer resuspendiert. 25 µL der Beadsuspension wurden 2 h mit 400 µL des geklärten Proteinextrakts unter ständiger Durchmischung inkubiert (Üperkopfrotor, 4°C) und im Anschluss mit Hilfe eines Magneten sedimentiert. Optional wurde der Extrakt vor der Inkubation auf eine Konzentration von 5 mg.mL-1 BSA eingestellt. Der Überstand wurde verworfen, die Dynabeads™ 6-mal in 750 µL IPP100-Puffer resuspendiert und jeweils 1-2 min unter ständiger Durchmischung inkubiert (Üperkopfrotor, 4°C). Der Puffer wurde vollständig entfernt (Zentrifugation 13 krpm, 1 min, 4°C) und die Dynabeads™ zur Elution des gebundenen Proteins 15 min in 60 µL ChIP-Elutionspuffer inkubiert (Eppendorf-Thermomixer, 65°C, 950 rpm). Das Eluat wurde 2:3 mit SDS/2ME-Puffer verdünnt, 5 min auf 95°C erhitzt und zur späteren Analyse bei -20°C gelagert. Die Immunpräzipitation aus formaldehydfixierter Zellkultur erfolgte wie in Kapitel 4.2.3 beschrieben, jedoch ohne vorherige Sonifizierung des fixierten Zellextrakts.
Spezielle Puffer
IPP100-Puffer: siehe Kapitel 4.2.1 IPP100-Aufschlusspuffer: IPP100-Puffer mit Benzamidin (2 mM), PMSF (0,5 mM), Aprotinin
(100 µg·mL-1), Leupetin (10 µg·mL-1), Pepstatin (10 µg·mL-1) und DTT (0,5 mM)
IPP100/BSA-spuffer: IPP100-Puffer mit BSA (5 mg·mL-1) ChIP-Eluationspuffer: siehe Kapitel 4.2.3; SDS/2ME-Puffer: siehe Kapitel 4.2.1

Material & Methoden
121
4.3.3 Chromatin-Immunpräzipitation (modifiziert nach Tanaka et al., 1997)
Formaldehydfixierung und Zellernte
YEPD-Kulturen der entsprechenden Hefestämme mit einem Kulturvolumen von 50 mL pro Präzipitationsansatz wurden bei 28°C bis zu einer optischen Zelldichte von OD600 = 1,0 - 1,2 kultiviert. Parallele Ansätze wurden vor der Fixierung mit Formaldehyd auf eine optische Zelldichte von OD600 = 1,0 eingestellt. Bezogen auf ein Kulturvolumen von 50 mL wurden 1,4 mL Formaldehyd 37% zur Kultur gegeben (Endkonzentration 1% (v/v)) und die Zellen unter stetiger Durchmischung 15 min bei Raumtemperatur fixiert. Die Fixierungsreaktion wurde durch Zugabe von 3 mL 2,3 M Glycin-Lösung (in 10 mM Tris-Puffer, pH 8,0; Endkonzentration 125 mM) gestoppt und überschüssiges Formaldehyd durch eine weitere Inkubation über 5 min gebunden. Mehrfachansätze wurden vor der Ernte in getrennte 50 mL Portionen aufgeteilt. Die Zellen wurden durch Zentrifugation (10 min, 4000 rpm, 4°C) geerntet, einmal mit 5 mL TBS-Puffer gewaschen und nach erneuter Zentrifugation (5 min, 4000 rpm, 4°C) in 1 mL TBS-Puffer resuspendiert. Die Zellen wurden nach Überführung der Suspension in ein 2 mL Eppendorfgefäß noch weitere 4-mal durch Resuspendieren in 1,5 mL TBS-Puffer und abschließend einmal in 750 µL Lysepuffer/PI gewaschen (Sedimentation durch Zentrifugation, 20 sec. 13 krpm). Nach erneuter Zentrifugation wurde der Puffer komplett abgezogen, das Zellpellet durch Inkubation des geschlossenen Eppendorfgefäßes in flüssigem Stickstoff schockgefroren (Inkubationszeit min. 15 min) und bis zum Zellaufschluss bei -80°C gelagert.
Zellaufschluss und Sonifizierung
Das gefrorene Zellpellet wurde mit ca. 200 µL Glasperlen (∅ 0,5 mm, mit HCl gewaschen) versetzt, in 500 µL Lysepuffer/PI resuspendiert und mechanisch aufgeschlossen (Vibrax-Schüttler, maximale Frequenz, 20 min, 4°C). Alle weiteren Arbeitsschritte wurden im Kühlraum bei 4°C durchgeführt. Der Rohextrakt wurde zur Sedimentation der Glasperlen 5 min auf Eis inkubiert. 400 µL des ungeklärten Rohextraktes wurden in ein 1,5 mL Eppendorfgefäß überführt und zur Erhöhung des Volumens mit 150 µL Lysepuffer verdünnt. Zur Scherung des Chromatins vor der Immunpräzipitation wurde der Zellextrakt mit Hilfe einer Ultraschallsonde sonifiziert (Ultraschallsonde Sonoplus UW2070, 5-mal 30 sec, 25-35% Leistung). Zwischen den einzelnen Ultraschallpulsen wurde der Rohextrakt mindestens 30 sec auf Eis gelagert. Der sonifizierte Rohextrakt wurde 10 min bei 10 krpm zentrifugiert (4°C) und der geklärte Extrakt in ein neues Eppendorfgefäß überführt. Von dem geklärten Extrakt wurden ca. 20 µL abgetrennt und für die spätere Analyse bei -80°C gelagert. Zusätzlich wurde eine weitere Probe des sonifizierten und geklärten Extrakts 2:3 mit SDS/2ME-Puffer verdünnt, 5 min auf 95°C erhitzt und zur späteren Analyse im Westernblot bei -20°C gelagert.
Chromatin-Immunpräzipitation
Die Chromatin-Immunpräzipitation über Protein-TAP-tag Fusionen erfolgte entweder durch Adsorption an die IgG-Agarosematrix oder die Bindung an magnetische Dynabeads™™ (Pan Mouse IgG). Zur Chromatin-Immunpräzipitation über Ctr9-Myc6 Fusionen wurden ebenfalls magnetische Dynabeads™ eingesetzt, die zuvor mit dem Antikörper 9E11 beladen wurden (Inkubation mit 0,05 µg·µL-1 9E11 Antikörper über Nacht in IPP100/BSA-Puffer). Die IgG-Agarose wurde vor der Chromatin-Immunpräzipitation mehrmals mit Glycin-Puffer pH 2,4 und anschließend mit IPP100/BSA-Puffer

Material & Methoden
122
gewaschen. Zum Waschen der unbehandelten bzw. mit 9E11 Antikörpern beladenen Dynabeads™ wurde Lyse/BSA-Puffer verwendet. Pro Präzipitationsansatz wurden 20 µL der IgG-Agarosematrix bzw. 25 µL Dynabead-Suspension eingesetzt (Massenanteil der Matrix in der Suspension entsprechend Ausgangssuspension). 400 µL des sonifizierten und geklärten Zellextrakts wurden mit der entsprechenden Matrix 3 h unter ständiger Durchmischung inkubiert (Überkopfrotor, 4°C) und im Anschluss durch Zentrifugation (IgG-Agarose, 800 rpm, 60 sec) oder mit Hilfe eines Magneten (Dynabeads™) sedimentiert. Der Überstand wurde verworfen, die jeweilige Affinitätsmatrix wurde jeweils 3-mal mit 750 µL Lysepuffer, Lyse/NaCl-Puffer, Waschpuffer und abschließend noch zweimal mit 750 µL TE-Puffer durch Resuspendierung gewaschen und jeweils 1-2 min unter ständiger Durchmischung inkubiert (Üperkopfrotor, 4°C). Der Puffer wurde vollständig entfernt (Zentrifugation 13 krpm, 1 min, 4°C) und die Dynabeads™ zur Elution des gebundenen Proteins 15 min in 60 µL ChIP-Elutionspuffer inkubiert (Eppendorf-Thermomixer, 65°C, 950 rpm). 30 µL des Eluats wurden zur Isolierung der gefällten DNA-Fragmente eingesetzt, der Rest wurde 2:3 mit SDS/2ME-Puffer verdünnt, 5 min auf 95°C erhitzt und zur späteren Analyse im Westernblot bei -20°C gelagert.
Revertierung der Quervernetzung, Proteinase K Verdau und DNA-Isolierung
Zum Lösen der kovalenten Verknüpfungen zwischen Proteinen und DNA wurden 30 µL des Eluats mit 120 µL SDS/TE-Puffer (1% (w/v) SDS in 1x TE) und 5 µL des sonifizierten und geklärten Zellextrakts mit 95 µL SDS/TE-Puffer gemischt und bei 65°C über Nacht inkubiert. Die Eluat-Probe wurden danach mit 140 µL TE-Puffer verdünnt und mit 7,5 µL Proteinase K (20 mg·mL-1
.) sowie 3 µL Glykogen (10 mg·mL-1
.) versetzt. Entsprechend wurden zu der Probe des Zellextrakts 93 µL TE-Puffer, 5 µL Proteinase K und 2 µL Glykogen gegeben. Die Lösungen wurden kurz gevortext und 2 h bei 37 °C inkubiert. Nach dem Proteinase K Verdau wurde die DNA durch zweimalige Extraktion mit Phenol/Chloroform gereinigt. Dazu wurden die Proben jeweils mit dem gleichen Volumen Phenol/Chloroform versetzt und gründlich gemischt (Vortex). Die Phasentrennung wurde durch Zentrifugation beschleunigt (Biofuge I3, Heraeus; 12 krpm, 2 min). Die gewonnenen Extrakte wurden mit 3 M Natriumacetat-Lösung (pH 5.5; 1/10 des Extraktvolumens) und Ethanol (2,5-faches Extraktvolumen) gemischt und für 30 min bei –20°C inkubiert. Nach Zentrifugation (13 krpm, 20 min, 4 °C) wurden die erhaltenen Pellets zweimal mit Ethanol 70% gewaschen und in 30 µL TE-Puffer gelöst. Die so gewonnenen DNA-Proben wurden über Polymerasekettenreaktion (PCR) analysiert. Die aus den Eluaten der Chromatin-Immunpräzipitation isolierten DNA-Proben werden im Folgenden und in Kapitel 2 zur sprachlichen Vereinfachung im Kontext der PCR-Analyse ebenfalls als ChIP-Eluate bezeichnet.
Analyse der Chromatin-Immunpräzipitate durch PCR
Der Nachweis definierter genspezifischer Sequenzen in den gefällten DNA-Fragmenten (ChIP-Eluate) erfolgte durch Amplifikation des jeweiligen Sequenzbereichs durch Polymerasekettenreaktion (PCR) mit den in Tabelle 4.7 angegebenen Primern. Alle verwendeten Primer wurden für eine Hybridisierungstemperatur von 56°C optimiert. Die ChIP-Eluate wurden in unterschiedlicher Menge und ggf. zuvor verdünnt als Template in den PCR-Reaktionsansätzen eingesetzt. Als DNA-Polymerase wurde eine Eigenpräparation der Taq DNA-Polymerase, je nach Anzahl der Reaktionsansätze in einer 1:2 oder 1:20 Verdünnung verwendet. Ein standardisierter Reaktionsansatz und das eingesetzte PCR-Programm sind in Tabelle 4.14 aufgeführt. Die

Material & Methoden
123
Reaktionsansätze wurden nach der Amplifikation mit 5 µL 6x DNA-Auftragspuffer verdünnt. 25 µL dieser Verdünnung wurden elektrophoretisch im 2% Agarosegel (0,5 mg.mL-1 Ethidiumbromid im Gel) getrennt. Das Agarosegel wurde vor der Photodokumentation 10 min in einer Ethidiumbromid-Lösung (10 mg·mL-1) inkubiert und im Anschluss zweimal 20 min in H2OMQ gewaschen.
Tabelle 4.14: Standardansatz und Programm für die Analyse der ChIP-Eluate durch PCR
PCR-Ansatz für eine Einzelreaktion PCR-Programm
5’-Primer (100 µM) 0,125 µL 1 94°C, 5min
3’-Primer(100 µM) 0,125 µL 2 94°C, 60 sec
Taq Puffer (10x) 2,5 µL 3 56°C, 60 sec
dNTP-Mischung (jeweils 10 mM) 0,5 µL 4 72 C, 2 min
Taq-Polymerase 1:20 (ca. 2,5 U.µL-1) 0,5 µL 5 30 Zyklen
Template (ChIP-Eluat, verdünnt) 5,0 µL 6 72°C, 7 min
H20MQ 16,25 µL
Reaktionsvolumen, gesamt 25 µL
Spezielle Puffer
Lysepuffer: Waschpuffer:
K-HEPES pH 7.5 50 mM TrisHCl pH 8.0 10 mM NaCl 140 mM LiCl 250 mM EDTA 1 mM EDTA 1 mM Triton X-100 1% (v/v) NP40 0,5% (v/v) Deoxycholsäure Na-Salz 0,1% (w/v) Deoxycholsäure Na-Salz 0,5% (w/v)
ChIP-Elutionspuffer: Taq Puffer (10x):
TrisHCl pH 8.0 50 mM Tris-Cl pH 8,55 200 mM EDTA 10 mM Ammoniumsulfat 100 mM SDS 1% (w/v) MgCl2 20 mM Tween 20 0,1% Lysepuffer/PI: Lysepuffer mit Benzamidin (2 mM), PMSF (0,5 mM), Aprotinin (100 µg·mL-1), Leupetin (10 µg·mL-1), Pepstatin (10 µg·mL-1) undDTT (0,5 mM)
Lyse/NaCl-Puffer: Lysepuffer mit 500 mM NaCl
SDS/2ME-Puffer: siehe Kapitel 4.3.1
IPP100/BSA-Puffer: siehe Kapitel 4.3.2

Material & Methoden
124
4.3.4 In vitro Bindestudien von heterolog in E. coli exprimierten GST-Ctr9 Fragmenten mit Cdc73-HA6 bzw. Paf1-HA3 aus Hefe
Induktion, Zellernte und Zellaufschluss der E. coli Zellen
25 mL LBAmp/Cm-Kultur der entsprechenden E. coli Stämme wurden bis zu einer optischen Dichte von OD600 = 0,6 bei 37°C im inkubiert. Die Expression des Zielproteins wurde mit 25 µL IPTG (1 M) induziert (Endkonzentration 1 mM). Die Kultur wurde nach der Induktion weitere 2,5 h bei 37°C inkubiert und die Zellen anschließend durch Zentrifugation (6000 rpm, 15 min, 4°C) sedimentiert. Die Zellen wurden durch Resuspendieren in 10 mL H2OMQ gewaschen und erneut durch Zentrifugation pelletiert (6000 rpm, 15 min, 4°C). Das Zellpellet wurde in flüssigem Stickstoff schockgefroren und bei -80°C gelagert. Zum Zellaufschluss wurden die Zellen auf Eis aufgetaut und in 1 mL E. coli-Aufschlusspuffer resuspendiert. Die Zellsuspension wurde 30 min auf Eis gehalten und die Zellen daraufhin durch Sonifizieren (3 x 20 sec, 30%, 4°C) aufgeschlossen. Das Lysat wurde durch Zentrifugation (13 krpm, 20 min, 4°C) geklärt.
Anzucht, Zellernte und Zellaufschluss der Hefezellen
YEPD-Kulturen der entsprechenden Hefestämme mit einem Kulturvolumen von 50 mL wurden bis zu einer Zelldichte von OD600 = ~ 1,5 bei 28°C inkubiert. Die Zellen wurden durch Zentrifugation (4400 rpm, 15 min, 4°C) sedimentiert, zweimal in 2 mL IPP100-Puffer (pH 7,3) durch Resuspendierung gewaschen und anschließend erneut pelletiert. Das Zellpellet wurde entweder in flüssigem Stickstoff schockgefroren und bis zu seiner Verwendung bei -80°C gelagert oder sofort aufgeschlossen. Das Zellpellet wurde in etwa dem gleichen Volumen IPP100-Aufschlusspuffer (pH 7,3; 4°C) resuspendiert, mit ca. 1/3 des Puffervolumens an Glasperlen versetzt (Ø = 0,5 mm) und mechanisch Aufgeschlossen (Vibrax-Schüttler, maximale Frequenz, 20 min, 4°C). Der Rohextrakt wurden durch Zentrifugation in einer Kühlzentrifuge (13200 rpm, 20 min, 4°C) geklärt.
Beladung der Glutathion-Sepharosematrix mit GST-Ctr9-Fragmenten und in vitro Bindung von Cdc73-HA6 bzw. Paf1-HA3
Zur Adsorption der GST-Ctr9-Fragmente an die GSH-Sepharosematrix wurden pro Ansatz 15 µL der Matrixsuspension eingesetzt. Die GSH-Matrix wurde zuvor durch dreimaliges Resuspendieren und Zentrifugation (2400 rpm, 3 min, 4°C) in 150 µL IPP100-Puffer (pH 7,3) äquilibriert und anschließend in 10 µL IPP100-Puffer (pH 7,3) resuspendiert. 1 mL des geklärten E. coli Zellextrakts wurde zur vorbereiteten Matrix gegeben. Zur Bindung der GST-Fusionsproteine an die Matrix wurde der Ansatz unter ständiger Durchmischung (Überkopfrotor) 1,5 h bei 4°C inkubiert. Die Sepharosematrix wurde anschließend durch Zentrifugation sedimentiert (2400 rpm, 2 min, 4°C) und der Überstand abgezogen. Die sedimentierte Matrix wurde in 1 mL IPP100-Puffer (pH 7,3) resuspendiert und 5 min bei 4°C inkubiert (Überkopfrotor). Nach erneuter Zentrifugation (2400 rpm, 2 min, 4°C) wurde die GSH-Matrix in 1 mL IPP100-Puffer (pH 7,3) aufgenommen und wiederum durch Zentrifugation sedimentiert. Dieser Waschschritt wurde viermal wiederholt. Der Heferohextrakt aus 50 mL Kultur wurde zur sedimentierten Sepharosematrix gegeben. Die Suspension aus Matrix und Heferohextrakt wurde 1,5 h bei 4°C unter ständiger Durchmischung (Überkopfrotor) inkubiert. Die GSH-Matrix wurde durch Zentrifugation (2400 rpm, 2 min, 4°C) sedimentiert und der Überstand verworfen. Die Matrix wurde sechsmal wie nach der

Material & Methoden
125
ersten Inkubation mit dem E. coli-Zellextrakt gewaschen. Die Elution gebundenen Proteine erfolgte durch Inkubation der GSH-Matrix in 75 µL 3x SDS/2ME-Puffer (5 min, 95°C).
Spezielle Puffer
IPP-100 Puffer (pH 7,3):
Tris-Cl pH 7,3 10 mM NaCl 100 mM NP40 0,1 %
IPP100-Aufschlusspuffer (pH 7,3):
IPP100-Puffer mit RNAse 5 µg.mL-1 DNAse 5 µg.mL-1 Lysozym 1 mg.mL-1 Benzamidin 2 mM PMSF 0,5 mM DTT 0,5 mM Complete™ 1 Tab./50mL
SDS/2ME-Puffer: siehe Kapitel 4.3.1
4.3.5 In vitro Phosphorylierung und Kinaseaktivitätstest
In vitro Phosphorylierungsreaktion
Der durch Tandem-Affinitätsreinigung isolierte PAF1-Komplex bzw. die aus der Hefe isolierte Casein-Kinase II oder die kommerzielle CKII-Präparation aus der Rattenleber wurden entsprechend des in Tabelle 4.15 angegebenen Reaktionsansatzes mit γ-32P-markiertem ATP bzw. GTP mit oder ohne Zusatz des jeweiligen Testsubstrats bzw. mit oder ohne Zugabe von Heparin 30 min bei 30°C inkubiert.
Autoradiographischer Phosphorylierungsnachweis
Für den autoradiographischen Phosphorylierungsnachweis wurde die Phosphorylierungsreaktion durch Zugabe von 15 µL 3x SDS/2ME-Puffer und 5 min Inkubation bei 95°C gestoppt. 15 µL der gestoppten Reaktionslösung wurden elektrophoretisch im Polyacrylamidgel (10%) getrennt. Das Gel wurde im Anschluss 20 min in Silbergel-Fixierlösung (Bio-Rad Silver Stain Plus Kit) inkubiert, danach 3-mal 20 min in deionisiertem Wasser gewaschen und mit Hilfe eines Vakuumgeltrockners getrocknet. Das getrocknete Gel wurde schließlich zur Exposition eines Röntgenfilms für den autoradiographischen Phosphorylierungsnachweis eingesetzt.
Kinaseaktivitätstest nach der Filtermethode (P81-Filtertest)
29 µL des Phosphorylierungsansatzes wurden auf ein trockenes P81-Phosphocellulose Filterpapier (Whatman P81-Rundfilter; Ø 2,1 cm) pipettiert und die Phosphorylierungsreaktion durch Inkubation des

Material & Methoden
126
P-81 Filters in 300 mL Phosphorsäurelösung 0,5% gestoppt. Das Filter wurde insgesamt 5-mal 10 min in 300 mL Phosphorsäurelösung 0,5% gewaschen und im Anschluss kurz in Ethanol 95% inkubiert. Das getrocknete Filter wurde mit 3 mL Szintillationslösung versetzt und in einem Szintillationszähler vermessen.
Tabelle 4.15: Zusammensetzung des Standard-Reaktionsansatzes für die in vitro Phosphorylierung
Zusammensetzung einer Einzelreaktiona) Autoradiographie P81-Filtertest
Kinase
PAF1-Komplex 5 µL 2,5 µL Casein-Kinase II (Hefepräparation) 5 µL 2,5 µL Casein-Kinase II (Rattenleber; 10·U.µL-1) 10 U 2,5 U
phosphorylierbare Substrate
Histon H1 (10 mg·mL-1) 6 µL 6 µL
beladene GSH-Matrixb) 10 µL ---
CKII Peptidsubstrat (10 mg·mL-1) --- 0,4 µL
Inhibitor
Heparin (100 µg·mL-1) 3 µL 3 µL
Puffer und Phosphatdonorsubstrat
BSA (10 mg·mL-1) --- 0,3 µL ATP bzw. GTP (10 mM) 0,3 µL 0,3 µL γ32P-ATP bzw. GTP (3 kCi·mM-1;10 mCi·mL-1) 1 µCi 1 µCi Kinase-Puffer (10x) 3 µL 3 µL H2OMQ ad. 35µL ad. 30 µL
a) Pro Reaktionsansatz wurde jeweils nur eine Kinase und ein Substrat unter optionalem Zusatz von Heparin eingesetzt.
b) Die Beladung der GSH-Matrix mit den heterolog exprimieren GST-Ctr9-Fragmenten erfolgte wie in Kapitel 4.3.4 beschrieben. Die beladene Matrix wurde vor dem Einsatz als Phosphorylierungssubstrat in 1x Kinase-Puffer äquilibriert.
Spezielle Puffer
Kinase-Puffer (10x):
Tris-Cl pH 7,4 250 mM NaCl 1000 mM MgCl2 100 mM
4.3.6 TCA-Extraktion und Westerntransfer von Histonen Zellernte und TCA-Extraktion (Wright et al., 1989)
25 mL YEPD-Kultur der entsprechenden Hefestämme wurden bei 28°C bis zu einer optischen Dichte zwischen OD600 = 1,5 und 2,0 inkubiert. Parallele Ansätze unterschiedlicher Hefestämme wurden vor der Ernte auf die gleiche Zelldichte eingestellt. Die Zellen wurden durch Zentrifugation (5 min, 4000 rpm, 4°C) sedimentiert und einmal in 5 mL H2OMQ gewaschen. Das Zellpellet wurde nach erneuter

Material & Methoden
127
Zentrifugation in 750 µL Trichloressigsäure 20% (TCA) resuspendiert, in ein 2 mL Eppendorfgefäß überführt und erneut sedimentiert (13 krpm, 20 sec). Die Zellen wurden mit 100 µL Glasperlen in 350 µL TCA 20% mechanisch aufgeschlossen (Vibrax-Schüttler, 20 min, 4°C). 300 µL des ungeklärten Extrakts wurden nach dem Absetzen der Glasperlen in ein 1,5 mL Eppendorfgefäß überführt und 30 min zentrifugiert (13 krpm, 4°C). Der Überstand wurde verworfen und das Pellet in 100 µL Tris-Base 1 M, 200 µL H2OMQ und 150 µL 3x SDS/2ME-Puffer resuspendiert und 5 min bei 95°C inkubiert. Die abgekühlte Suspension wurde erneut zentrifugiert (1 min, 13 krpm, 4°C) und der Überstand bis zur Analyse im Westernblot bei -20°C gelagert.
Westerntransfer von Histonen (Thiriet and Albert, 1995)
Die TCA-Extrakte wurden elektrophoretisch im 16,6% High-Tris-Polyacrylamidgel (Fling and Gregerson, 1986; Vernetzungsgrad 5,1%) getrennt und das Gel nach der Elektrophorese 20 min in Tris-Glycin-Elektrophoresepuffer mit 20% Methanol inkubiert. Der Westerntransfer auf eine PVDF-Membran (0,2 µm Porenweite) erfolgte bei 4°C, entweder im Tankblot-Verfahren (2 h, UStart = 175 V, Imax = 300 mA ) oder nach der Semi-dry Methode (100 min, I = 65 mA, konstant), jeweils unter Verwendung von Tris-Glycin-Elektrophoresepuffer mit 20% Methanol als Transferpuffer.
Spezielle Puffer
High-Tris Polyacrylamidgel: 4x High-Tris Trenngelpuffer:
4x High-Tris Trenngelpuffer 25% (v/v) Tris-Cl 2,6 M 2% Bisacrylamid 21% (v/v) SDS Na-Salz 0,2% (m/v) Rotiphorese Gel30 54% (v/v) pH 10,5 Sammelgel und Tris-Glycin-Elektrophoresepuffer: siehe Kapitel 4.4.1
4.4 Bioanalytische Verfahren 4.4.1 Elektrophoretische Trennung von DNA-Fragmenten im Agarosegel
Die Trennung von DNA-Fragmenten erfolgte in einem Agarosegel (1-2%, je nach DNA-Fragmentgröße) mit 0,5 µg⋅mL-1 Ethidiumbromid in der Gelmatrix unter Verwendung von TAE-Puffer für die Elektrophorese. Die Proben wurden zum Auftrag mit 6x DNA-Auftragspuffer verdünnt und bei einer Spannung von ca. 100 V elektrophoretisch in TAE-Puffer getrennt. Die Position der DNA-Fragmente wurde durch Anregung der Ethidiumbromid-Fluoreszenz im UV-Licht visualisiert und fotografisch dokumentiert.
4.4.2 Proteinbestimmung nach Bradford
Zur Bestimmung der Gesamtprotein-Konzentration wurde die Bradford-Methode eingesetzt. Für die Proteinbestimmung wurden die Reagenzien des Bio-Rad Protein-Assay verwendet. Die Kalibrierung

Material & Methoden
128
erfolgte mit BSA im Konzentrationsbereich zwischen 10 µg⋅mL-1 und 250 µg⋅mL-1 (bezogen auf die eingesetzte Standardlösung), durch polynomische Regression. Zur Messung wurden jeweils 50 µL der Probe, bzw. Standardlösung mit 1 mL des 1:5 verdünnten Farbstoffkonzentrates gemischt und nach 5 min die Absorption bei 595 nm registriert. Als Leerwert wurde anstelle der Probe 50 µL deionisiertes Wasser in gleicher Weise behandelt. 4.4.3 Trennung von Proteinen durch SDS-Polyacrylamidgelelektrophorese Die elektrophoretische Trennung von Proteinen erfolgte in einer vertikalen, SDS-haltigen und diskontinuierlich gepufferten Polyacrylamid-Gelmatrix, bestehend aus einem 10%-Trenngel pH 8,8, das zur Fokussierung der Proteinbanden mit einem 5% Sammelgel pH 6,8 überschichtet wurde. Die Zusammensetzung der jeweiligen Polyacrylamidgele ist in Tabelle 4.16 wiedergegeben. Die Elektrophorese erfolgte mit Tris-Glycin-Elektrophoresepuffer bei einer Stromstärke von 15 mA - 20 mA pro Gel.
Tabelle 4.16: Zusammensetzung der Polyacrylamidgele zur Trennung von Proteinen
Trenngel 10 %; Sammelgel 5 %
Trenngelpuffer 25 % (v/v) 50 % (v/v)
Acrylamid 30 %1 33 % (v/v) 17 % (v/v)
Radikaldonor:
TEMED 100 µL pro 10 mL Gellösung 100 µL pro 10 mL Gellösung
APS 10 % 5 µL pro 10 mL Gellösung 25 µL pro 10 mL Gellösung
Maße (b×h×t)2 ca. 8 cm × 6 cm × 0,1 cm ca. 8 cm × 2 cm × 0,1 cm
1) enthält 0,8 % Bisacrylamid
2) Breite × Höhe × Tiefe
Spezielle Puffer
Trenngelpuffer: Sammelgelpuffer:
Tris-Cl pH 8,8 1,5 M Tris-Cl pH 6,8 0,25 M SDS 0,4% (m/v) SDS 0,2% (m/v)
Tris-Glycin-Elektrophoresepuffer:
Tris-Base 0,25 M Glycin 1,92 M SDS 1% (m/v)

Material & Methoden
129
4.4.4 Färbung von Polyacrylamidgelen zum Nachweis von Proteinen
Die im Polyacrylamidgel elektrophoretisch getrennten Präparationen von Proteinkomplexen wurden entweder durch Reduktion von Silbernitrat (Silberfärbung) oder durch colloidales Coomassie Brillantblau gefärbt. Exprimierte Proteine wurden durch herkömmliche Coomassie Brillantblau-Lösung gefärbt. Zur Silberfärbung wurde das Bio-Rad Silver Stain Plus Kit eingesetzt. Die Färbung erfolgte nach dem entsprechenden Bio-Rad Protokoll. Die Inkubationszeit der Polyacrylamidgele in der Fixativ Enhancer-Lösung wurde dabei zum Teil von 20 min auf bis zu 12 h (über Nacht) ausgedehnt. Die entwickelten Gele wurden zur Dokumentation und weiteren Aufbewahrung in deionisiertem Wasser gelagert. Zur Färbung mit colloidalem Coomassie Brillantblau wurden die im Polyacrylamidgel getrennten Proteine 1 h oder über Nacht in 40% (v/v) Methanol, 7% (v/v) Essigsäure fixiert. Die Gele wurden vor der Färbung dreimal 10 min in deionisiertem Wasser gewaschen. Zur Färbung wurde die Brillantblau G-Colloidlösung (Sigma) 4:1 mit Methanol gemischt und intensiv geschüttelt. Die Polyacrylamidgele wurden ca. 12 h bis 24 h in der methanolischen Färbelösung inkubiert, wobei ca. 100 mL der Lösung pro Gel eingesetzt wurden. Überschüssiges Colloid wurde nach dem Färben durch kurzes Waschen (maximal 30 sec) der Gele mit 25% (v/v) Methanol, 10% (v/v) Essigsäure gelöst. Zur Dokumentation und weiteren Aufbewahrung wurden die Gele in deionisiertem Wasser gelagert. Die Dokumentation erfolgte jeweils fotografisch über eine Digitalkamera.
4.4.5 Immunologischer Nachweis von Proteinen durch Westernblot-Analyse
Elektrotransfer von Proteinen (Westernblot)
Vor dem Westernblot wurden die Proteinproben elektrophoretisch im SDS-Polyacrylamidgel getrennt. Das Sammelgel wurde entfernt und die im Trenngel vorhandenen Proteine im Semi-dry Verfahren mit diskontinuierlichem Puffersystem auf eine PVDF-Membran (0,45 µm Porenweite) übertragen. Das Polyacrylamidgel und die PVDF-Membran (6 cm × 9 cm) wurden dabei, flankiert von mehreren Lagen, in unterschiedlichen Puffern getränkter Filterpapiere (Whatman), zwischen zwei Plattenelektroden platziert. Ausgehend von der Anode wurden dazu zwei Lagen in 300 mM Tris-Base getränktes Whatman-Papier und eine Lage in 25 mM Tris-Base getränktes Whatman-Papier gestapelt. Auf die oberste Lage dieses Filterpapierstapels wurde die mit Methanol benetzte PVDF-Membran ausgebreitet und das Polyacrylamidgel luftblasenfrei aufgelegt, gefolgt von drei weitere Lagen in 40 mM DL-Norleucin, 25 mM Tris-Base getränkter Whatman-Papiere und schließlich der Kathode. Der Westernblot wurde 60 min bei einer konstanten Stromstärke von 32 mA pro Polyacrylamidgel durchgeführt.
Immunologische Detektion mit Antikörpern
Die PVDF-Membran wurde über Nacht in PTM5-Lösung inkubiert. Zur immunologischen Detektion von Proteinen wurde die PVDF-Membran 1-2 h in einer PTM5-Lösung des entsprechenden primären Antikörpers inkubiert (vgl. Tabelle 4.5). Der nicht-adsorbierte Antikörperüberschuss wurde durch dreimaliges Waschen der Membran (jeweils 15 min) mit PTM5-Lösung entfernt. Die PVDF-Membran wurde danach mit einer ebenfalls in PTM5-Lösung angesetzten Verdünnung des sekundären Antikörper-HRP Konjugats (vgl. Tabelle 4.5) über einen Zeitraum von min. 45 min inkubiert. Der PAP-Komplex wurde dabei wie ein sekundärer Antikörper behandelt und, bei gleichzeitigem Nachweis von Protein-

Material & Methoden
130
TAP-tag und Protein-HA Fusionen, zusammen mit dem anti-Maus-HRP Konjugat inkubiert. Nach der Inkubation mit dem sekundären Antikörper wurde die Membran dreimal 15 min in PBS/Tween-Puffer gewaschen und die Peroxidaseaktivität durch eine "Enhanced-Chemoluminescence"-Reaktion (ECL) nachgewiesen. Jeweils 10 mL ECL-Puffer (100 mM Tris-Cl pH 8,5) wurden dazu mit 145 µL Cumarin-Luminol-Substrat, bzw. mit 7 µL Wasserstoffperoxid 30% versetzt und anschließend vereinigt. Die PVDF-Membran wurde ca. 60 sec in dieser Reaktionslösung inkubiert, wobei die mit dem sekundären Antikörper konjugierte Meerrettichperoxidase (HRP) die Oxidation von Luminol katalysiert, die von einer Chemolumineszenz begleitet wird. Die Sensitivität der Methode wird dabei durch die anwesende Cumarinsäure verstärkt. Die Chemolumineszenz wurde, durch die Schwärzung eines Röntgenfilmes, zum Nachweis der untersuchten Proteine eingesetzt, wobei, je nach Intensität der Lumineszenzstrahlung, Expositionszeiten zwischen 15 sec und 15 min gewählt wurden.
Spezielle Puffer
PBS/Tween-Puffer: Cumarin-Luminol Substrat:
K2HPO4 40 mM Cumarinsäure 4,52 mg⋅mL-1 KH2PO4 10 mM 3-Aminophtalhydrazid 30,8 mg⋅mL-1 NaCl 150 mM in DMSO Tween 20 1% Thimerosal 1:40000 PTM5-Lösung: PBS/Tween-Puffer mit 5% (m/v) gelöstem Magermilchpulver

Danksagungen
131
Danksagungen Mein herzlicher Dank gilt all jenen, die mich bei der Erstellung dieser Arbeit begleitet und unterstützt haben und so durch ihre aktive Mitwirkung, Beratung und konstruktive Kritik sowie durch ihren Rückhalt und ihre Ermutigungen zu dieser Arbeit beitrugen.
Mein besonderer Dank gilt:
Prof. Dr. Christian Koch für die Überlassung des Themas und das große Engagement bei der Betreuung des Projekts und dem Erstellen dieser Arbeit ebenso wie für das entgegengebrachte Vertrauen und die gewährten Freiheiten
Prof. Dr. Georg Kreimer für die kurzfristige Übernahme des Zweitgutachtens für diese Arbeit sowie die wertvollen Tipps, v. a. bezüglich der Optimierung der Trennleistung im Polyacrylamidgel
Harald-Peter (Harry) Tartler, Alexandra Alemany Schmidt und Sabrina Haßler für ihr aktives Mitwirken an meinem Dissertationsprojekt im Rahmen ihrer Diplomarbeiten Eva Düll und Sandra Alt für ihre engagierte technische Unterstützung und Assistenz Dr. Guido Sauer für die Durchführung und Interpretation der massenspektrometrischen Analysen Prof. Dr. Heinrich Sticht für die freundliche Unterstützung bei der Strukturvorhersage von TPR-Motiven aus Proteinsequenzen Martin Stiebritz für die Durchführung und freundliche Überlassung der Homologiemodellierung von Ctr9 Dr. Robert Slany für die wertvollen Tipps bzgl. des Westerntransfers von Histonen und die Bereitstellung der Hefestämme mit telomergekoppeltem URA3-Reportergen Sabine Meixner und Isabel Jungkunz für das Korrekturlesen der Arbeit und das Austreiben des Tippfehlerteufels sowie
Octavian Stephan, Dr. Eva Gottwald, Dr. Marlis Dahl, Dr. Christoph Rieck, Dr. Thomas Delong und „last but not least“ Dr. Jörg Hoffmann für den konstruktiven fachlichen und methodischen Austausch

Abkürzungsverzeichnis
132
Häufig verwendete Abkürzungen 2ME 2-Mercaptoethanol
3'/5'-UTR 3’- bzw. 5’-nichttranslatierte Region
5FOA 5-Fluororotsäure
6AU 6-Azauracil
Amp Ampizillin
CaM Calmodulin
CAS Casaminoacid
CBP Calmodulin-binding Peptide
CC Coiled Coil
ChIP Chromatin-Immunpräzipititation
CKII Casein-Kinase II
Cm Chloramphenikol
COMPASS Complex Proteins Associatede with Set1
CTD C-terminal Domain
DRB 5,6-Dichloro-1-β -D-ribofuranosylbenzimidazol
DSIF DRB sensitivity inducing factor
EGTA Ethylenglykol-bis-(2-aminoethylethyl)-tetraessigsäure
EGTA-Eluat Eluat der zweiten Affinitätsreinigungsstufe bei einer Tandem-Affinitätsreinigung
FACT Facilitates Chromatin Transcription
GSH Glutathion, reduziert
GST Glutathione S-transferase
H2OMQ deionisiertes Wasser
HA Hämagglutinin
INT ORF-freie Region auf Chromosom V (internal region)
IP Immunpräzipitation
MAT Mating type
MW Molecular weight
MWCO Molecular weight cut off
NELF Negative Elongation Factor
NuA4 Nucleosomal acetyltransferase of H4
ORF Leserahmen
PAF1 RNA-Polymerase II Associated Factor 1
PAP Peroxidase-Antiperoxidase Komplex
PCR Polymerase Kettenreaktion
PIC Prä-Inititationskomplex der RNA-Polymease II
p-TEFb Positive Transcription Elongation Factor b
RNAPII RNA-Polymerase II
rpm Rounds per minute

Abkürzungsverzeichnis
133
SCD Synthetic complete dextrose
SWR1 Swr1 containing complex
TAP Tandem-Affinitätsreinigung
TCA Tri-Chloressigsäure
TEC Transcription elongation complex
TELVIIL Telomer des linken Arms von Chromosom VII
TEV Tobacco etch virus protease
TEV-Eluat Eluat der ersten Affinitätsreinigungsstufe bei einer Tandem-Affinitätsreinigung
TPR Tetratricopeptide Repeat
Vi Realtives Bandenvolumen
WT Wildtyp

Literaturverzeichnis
134
Literaturverzeichnis
Abbott, K.L., Renfrow, M.B., Chalmers, M.J., Nguyen, B.D., Marshall, A.G., Legault, P. and Omichinski, J.G. (2005) Enhanced binding of RNAP II CTD phosphatase FCP1 to RAP74 following CK2 phosphorylation. Biochemistry, 44, 2732-2745.
Ackermann, K., Waxmann, A., Glover, C.V. and Pyerin, W. (2001) Genes targeted by protein kinase CK2: a genome-wide expression array analysis in yeast. Mol Cell Biochem, 227, 59-66.
Adam, M., Robert, F., Larochelle, M. and Gaudreau, L. (2001) H2A.Z is required for global chromatin integrity and for recruitment of RNA polymerase II under specific conditions. Mol Cell Biol, 21, 6270-6279.
Adelman, K., Wei, W., Ardehali, M.B., Werner, J., Zhu, B., Reinberg, D. and Lis, J.T. (2006) Drosophila Paf1 modulates chromatin structure at actively transcribed genes. Mol Cell Biol, 26, 250-260.
Ahmed, K., Gerber, D.A. and Cochet, C. (2002) Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol, 12, 226-230.
Allard, S., Utley, R.T., Savard, J., Clarke, A., Grant, P., Brandl, C.J., Pillus, L., Workman, J.L. and Cote, J. (1999) NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. Embo J, 18, 5108-5119.
Allen, J.B., Zhou, Z., Siede, W., Friedberg, E.C. and Elledge, S.J. (1994) The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev, 8, 2401-2415.
Allison, L.A., Wong, J.K., Fitzpatrick, V.D., Moyle, M. and Ingles, C.J. (1988) The C-terminal domain of the largest subunit of RNA polymerase II of Saccharomyces cerevisiae, Drosophila melanogaster, and mammals: a conserved structure with an essential function. Mol Cell Biol, 8, 321-329.
Asturias, F.J., Meredith, G.D., Poglitsch, C.L. and Kornberg, R.D. (1997) Two conformations of RNA polymerase II revealed by electron crystallography. J Mol Biol, 272, 536-540.
Ausubel, F.M. (1998) Current protocols in molecular biology. John Wiley & Sons, New York.
Babiarz, J.E., Halley, J.E. and Rine, J. (2006) Telomeric heterochromatin boundaries require NuA4-dependent acetylation of histone variant H2A.Z in Saccharomyces cerevisiae. Genes Dev, 20, 700-710.
Bates, P.A., Kelley, L.A., MacCallum, R.M. and Sternberg, M.J. (2001) Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins, Suppl 5, 39-46.
Belotserkovskaya, R., Oh, S., Bondarenko, V.A., Orphanides, G., Studitsky, V.M. and Reinberg, D. (2003) FACT facilitates transcription-
dependent nucleosome alteration. Science, 301, 1090-1093.
Belotserkovskaya, R. and Reinberg, D. (2004) Facts about FACT and transcript elongation through chromatin. Curr Opin Genet Dev, 14, 139-146.
Berger, B., Wilson, D.B., Wolf, E., Tonchev, T., Milla, M. and Kim, P.S. (1995) Predicting coiled coils by use of pairwise residue correlations. Proc Natl Acad Sci U S A, 92, 8259-8263.
Berkey, C.D. and Carlson, M. (2006) A specific catalytic subunit isoform of protein kinase CK2 is required for phosphorylation of the repressor Nrg1 in Saccharomyces cerevisiae. Curr Genet, 50, 1-10.
Berlyn, K.A., Schultes, B., Leveugle, B., Noujaim, A.A., Alexander, R.B. and Mann, D.L. (2001) Generation of CD4(+) and CD8(+) T lymphocyte responses by dendritic cells armed with PSA/anti-PSA (antigen/antibody) complexes. Clin Immunol, 101, 276-283.
Bidwai, A.P., Reed, J.C. and Glover, C.V. (1994) Casein kinase II of Saccharomyces cerevisiae contains two distinct regulatory subunits, beta and beta'. Arch Biochem Biophys, 309, 348-355.
Biswas, D., Imbalzano, A.N., Eriksson, P., Yu, Y. and Stillman, D.J. (2004) Role for Nhp6, Gcn5, and the Swi/Snf complex in stimulating formation of the TATA-binding protein-TFIIA-DNA complex. Mol Cell Biol, 24, 8312-8321.
Biswas, D., Yu, Y., Prall, M., Formosa, T. and Stillman, D.J. (2005a) The Yeast FACT Complex Has a Role in Transcriptional Initiation. Mol. Cell. Biol., 25, 5812-5822.
Biswas, D., Yu, Y., Prall, M., Formosa, T. and Stillman, D.J. (2005b) The yeast FACT complex has a role in transcriptional initiation. Mol Cell Biol, 25, 5812-5822.
Blatch, G.L. and Lassle, M. (1999) The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays, 21, 932-939.
Boeke, J.D., LaCroute, F. and Fink, G.R. (1984) A positive selection for mutants lacking orotidine-5'-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet, 197, 345-346.
Borggrefe, T., Davis, R., Bareket-Samish, A. and Kornberg, R.D. (2001) Quantitation of the RNA polymerase II transcription machinery in yeast. J Biol Chem, 276, 47150-47153.
Bressan, D.A., Baxter, B.K. and Petrini, J.H. (1999) The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol, 19, 7681-7687.
Briggs, S.D., Bryk, M., Strahl, B.D., Cheung, W.L., Davie, J.K., Dent, S.Y., Winston, F. and Allis, C.D. (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev, 15, 3286-3295.
Broomfield, S., Hryciw, T. and Xiao, W. (2001) DNA postreplication repair and mutagenesis in

Literaturverzeichnis
135
Saccharomyces cerevisiae. Mutat Res, 486, 167-184.
Burke, D., Dawson, D., Stearns, T. and Cold Spring Harbor Laboratory. (2000) Methods in yeast genetics : a Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
Cardenas, M.E., Walter, R., Hanna, D. and Gasser, S.M. (1993) Casein kinase II copurifies with yeast DNA topoisomerase II and re-activates the dephosphorylated enzyme. J Cell Sci, 104 ( Pt 2), 533-543.
Carrozza, M.J., Li, B., Florens, L., Suganuma, T., Swanson, S.K., Lee, K.K., Shia, W.J., Anderson, S., Yates, J., Washburn, M.P. and Workman, J.L. (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell, 123, 581-592.
Chen-Wu, J.L., Padmanabha, R. and Glover, C.V. (1988) Isolation, sequencing, and disruption of the CKA1 gene encoding the alpha subunit of yeast casein kinase II. Mol Cell Biol, 8, 4981-4990.
Cho, E.J., Kobor, M.S., Kim, M., Greenblatt, J. and Buratowski, S. (2001) Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev, 15, 3319-3329.
Chu, Y., Sutton, A., Sternglanz, R. and Prelich, G. (2006) The BUR1 cyclin-dependent protein kinase is required for the normal pattern of histone methylation by SET2. Mol Cell Biol, 26, 3029-3038.
Cliff, M.J., Williams, M.A., Brooke-Smith, J., Barford, D. and Ladbury, J.E. (2005) Molecular recognition via coupled folding and binding in a TPR domain. J Mol Biol, 346, 717-732.
Copenhaver, G.P., Keith, K.C. and Preuss, D. (2000) Tetrad analysis in higher plants. A budding technology. Plant Physiol, 124, 7-16.
Cortajarena, A.L. and Regan, L. (2006) Ligand binding by TPR domains. Protein Sci, 15, 1193-1198.
Cosma, M.P., Tanaka, T. and Nasmyth, K. (1999) Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell, 97, 299-311.
Craven, R.J. and Petes, T.D. (2000) Involvement of the checkpoint protein Mec1p in silencing of gene expression at telomeres in Saccharomyces cerevisiae. Mol Cell Biol, 20, 2378-2384.
D'Andrea, L.D. and Regan, L. (2003) TPR proteins: the versatile helix. Trends Biochem Sci, 28, 655-662.
Dedon, P.C., Soults, J.A., Allis, C.D. and Gorovsky, M.A. (1991) A simplified formaldehyde fixation and immunoprecipitation technique for studying protein-DNA interactions. Anal Biochem, 197, 83-90.
Desmoucelles, C., Pinson, B., Saint-Marc, C. and Daignan-Fornier, B. (2002) Screening the yeast "disruptome" for mutants affecting resistance to the immunosuppressive drug, mycophenolic acid. J Biol Chem, 277, 27036-27044.
Dhillon, N. and Kamakaka, R.T. (2000) A histone variant, Htz1p, and a Sir1p-like protein, Esc2p, mediate silencing at HMR. Mol Cell, 6, 769-780.
Dhillon, N. and Kamakaka, R.T. (2002) Breaking through to the other side: silencers and barriers. Curr Opin Genet Dev, 12, 188-192.
Di Como, C.J., Chang, H. and Arndt, K.T. (1995) Activation of CLN1 and CLN2 G1 cyclin gene expression by BCK2. Mol Cell Biol, 15, 1835-1846.
Durant, M. and Pugh, B.F. (2006) Genome-wide relationships between TAF1 and histone acetyltransferases in Saccharomyces cerevisiae. Mol Cell Biol, 26, 2791-2802.
Endoh, M., Zhu, W., Hasegawa, J., Watanabe, H., Kim, D.K., Aida, M., Inukai, N., Narita, T., Yamada, T., Furuya, A., Sato, H., Yamaguchi, Y., Mandal, S.S., Reinberg, D., Wada, T. and Handa, H. (2004) Human Spt6 stimulates transcription elongation by RNA polymerase II in vitro. Mol Cell Biol, 24, 3324-3336.
Exinger, F. and Lacroute, F. (1992) 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Curr Genet, 22, 9-11.
Feng, Q., Wang, H., Ng, H.H., Erdjument-Bromage, H., Tempst, P., Struhl, K. and Zhang, Y. (2002) Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol, 12, 1052-1058.
Fingerman, I.M., Wu, C.L., Wilson, B.D. and Briggs, S.D. (2005) Global loss of Set1-mediated H3 Lys4 trimethylation is associated with silencing defects in Saccharomyces cerevisiae. J Biol Chem, 280, 28761-28765.
Finn, R.D., Mistry, J., Schuster-Bockler, B., Griffiths-Jones, S., Hollich, V., Lassmann, T., Moxon, S., Marshall, M., Khanna, A., Durbin, R., Eddy, S.R., Sonnhammer, E.L. and Bateman, A. (2006) Pfam: clans, web tools and services. Nucleic Acids Res, 34, D247-251.
Fling, S.P. and Gregerson, D.S. (1986) Peptide and protein molecular weight determination by electrophoresis using a high-molarity tris buffer system without urea. Anal Biochem, 155, 83-88.
Formosa, T., Ruone, S., Adams, M.D., Olsen, A.E., Eriksson, P., Yu, Y., Rhoades, A.R., Kaufman, P.D. and Stillman, D.J. (2002) Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics, 162, 1557-1571.
Friedl, E.M., Lane, W.S., Erdjument-Bromage, H., Tempst, P. and Reinberg, D. (2003) The C-terminal domain phosphatase and transcription elongation activities of FCP1 are regulated by phosphorylation. Proc Natl Acad Sci U S A, 100, 2328-2333.
Game, J.C., Williamson, M.S., Spicakova, T. and Brown, J.M. (2006a) The RAD6/BRE1 histone modification pathway in Saccharomyces confers radiation resistance through a RAD51-dependent process that is independent of RAD18. Genetics, 173, 1951-1968.
Game, J.C., Williamson, M.S., Spicakova, T. and Brown, M. (2006b) The RAD6/BRE1 histone modification pathway in Saccharomyces confers radiation resistance through a RAD51-dependent process that is independent of RAD18. Genetics.

Literaturverzeichnis
136
Gattiker, A., Gasteiger, E. and Bairoch, A. (2002) ScanProsite: a reference implementation of a PROSITE scanning tool. Appl Bioinformatics, 1, 107-108.
Giaever, G., Chu, A.M., Ni, L., Connelly, C., Riles, L., Veronneau, S., Dow, S., Lucau-Danila, A., Anderson, K., Andre, B., Arkin, A.P., Astromoff, A., El-Bakkoury, M., Bangham, R., Benito, R., Brachat, S., Campanaro, S., Curtiss, M., Davis, K., Deutschbauer, A., Entian, K.D., Flaherty, P., Foury, F., Garfinkel, D.J., Gerstein, M., Gotte, D., Guldener, U., Hegemann, J.H., Hempel, S., Herman, Z., Jaramillo, D.F., Kelly, D.E., Kelly, S.L., Kotter, P., LaBonte, D., Lamb, D.C., Lan, N., Liang, H., Liao, H., Liu, L., Luo, C., Lussier, M., Mao, R., Menard, P., Ooi, S.L., Revuelta, J.L., Roberts, C.J., Rose, M., Ross-Macdonald, P., Scherens, B., Schimmack, G., Shafer, B., Shoemaker, D.D., Sookhai-Mahadeo, S., Storms, R.K., Strathern, J.N., Valle, G., Voet, M., Volckaert, G., Wang, C.Y., Ward, T.R., Wilhelmy, J., Winzeler, E.A., Yang, Y., Yen, G., Youngman, E., Yu, K., Bussey, H., Boeke, J.D., Snyder, M., Philippsen, P., Davis, R.W. and Johnston, M. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature, 418, 387-391.
Giannattasio, M., Lazzaro, F., Plevani, P. and Muzi-Falconi, M. (2005a) The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem.
Giannattasio, M., Lazzaro, F., Plevani, P. and Muzi-Falconi, M. (2005b) The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem, 280, 9879-9886.
Gietz, R.D. and Sugino, A. (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene, 74, 527-534.
Goffeau, A., Barrell, B.G., Bussey, H., Davis, R.W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J.D., Jacq, C., Johnston, M., Louis, E.J., Mewes, H.W., Murakami, Y., Philippsen, P., Tettelin, H. and Oliver, S.G. (1996) Life with 6000 genes. Science, 274, 546, 563-547.
Goldberg, A.L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature, 426, 895-899.
Goldberg, A.L. and Dice, J.F. (1974) Intracellular protein degradation in mammalian and bacterial cells. Annu Rev Biochem, 43, 835-869.
Gomes, N.P., Bjerke, G., Llorente, B., Szostek, S.A., Emerson, B.M. and Espinosa, J.M. (2006) Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev, 20, 601-612.
Goodrich, J.A. and Tjian, R. (1994) Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II. Cell, 77, 145-156.
Gottschling, D.E., Aparicio, O.M., Billington, B.L. and Zakian, V.A. (1990) Position effect at S.
cerevisiae telomeres: reversible repression of Pol II transcription. Cell, 63, 751-762.
Grant, P.A., Duggan, L., Cote, J., Roberts, S.M., Brownell, J.E., Candau, R., Ohba, R., Owen-Hughes, T., Allis, C.D., Winston, F., Berger, S.L. and Workman, J.L. (1997) Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev, 11, 1640-1650.
Grunstein, M. (1997) Molecular model for telomeric heterochromatin in yeast. Curr Opin Cell Biol, 9, 383-387.
Guarente, L. (1984) Yeast promoters: positive and negative elements. Cell, 36, 799-800.
Guillemette, B., Bataille, A.R., Gevry, N., Adam, M., Blanchette, M., Robert, F. and Gaudreau, L. (2005) Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol, 3, e384.
Hadwiger, J.A., Wittenberg, C., Richardson, H.E., de Barros Lopes, M. and Reed, S.I. (1989) A family of cyclin homologs that control the G1 phase in yeast. Proc Natl Acad Sci U S A, 86, 6255-6259.
Hanna, J.S., Kroll, E.S., Lundblad, V. and Spencer, F.A. (2001) Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol, 21, 3144-3158.
Hathaway, G.M., Lubben, T.H. and Traugh, J.A. (1980) Inhibition of casein kinase II by heparin. J Biol Chem, 255, 8038-8041.
He, X. and Moore, C. (2005) Regulation of yeast mRNA 3' end processing by phosphorylation. Mol Cell, 19, 619-629.
He, Y., Doyle, M.R. and Amasino, R.M. (2004) PAF1-complex-mediated histone methylation of FLOWERING LOCUS C chromatin is required for the vernalization-responsive, winter-annual habit in Arabidopsis. Genes Dev, 18, 2774-2784.
Helsens, K., Martens, L., Vandekerckhove, J. and Gevaert, K. (2007) MascotDatfile: An open-source library to fully parse and analyse MASCOT MS/MS search results. Proteomics.
Hulo, N., Bairoch, A., Bulliard, V., Cerutti, L., De Castro, E., Langendijk-Genevaux, P.S., Pagni, M. and Sigrist, C.J. (2006) The PROSITE database. Nucleic Acids Res, 34, D227-230.
Ito, H., Fukuda, Y., Murata, K. and Kimura, A. (1983) Transformation of intact yeast cells treated with alkali cations. J Bacteriol, 153, 163-168.
Johnson, C.N., Adkins, N.L. and Georgel, P. (2005) Chromatin remodeling complexes: ATP-dependent machines in action. Biochem Cell Biol, 83, 405-417.
Jona, G., Wittschieben, B.O., Svejstrup, J.Q. and Gileadi, O. (2001) Involvement of yeast carboxy-terminal domain kinase I (CTDK-I) in transcription elongation in vivo. Gene, 267, 31-36.
Jones, E.W. (1991) Tackling the protease problem in Saccharomyces cerevisiae. Methods Enzymol, 194, 428-453.
Joshi, A.A. and Struhl, K. (2005) Eaf3 chromodomain interaction with methylated H3-K36 links

Literaturverzeichnis
137
histone deacetylation to Pol II elongation. Mol Cell, 20, 971-978.
Keller, D.M., Zeng, X., Wang, Y., Zhang, Q.H., Kapoor, M., Shu, H., Goodman, R., Lozano, G., Zhao, Y. and Lu, H. (2001) A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol Cell, 7, 283-292.
Keogh, M.C., Kurdistani, S.K., Morris, S.A., Ahn, S.H., Podolny, V., Collins, S.R., Schuldiner, M., Chin, K., Punna, T., Thompson, N.J., Boone, C., Emili, A., Weissman, J.S., Hughes, T.R., Strahl, B.D., Grunstein, M., Greenblatt, J.F., Buratowski, S. and Krogan, N.J. (2005) Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell, 123, 593-605.
Keogh, M.C., Mennella, T.A., Sawa, C., Berthelet, S., Krogan, N.J., Wolek, A., Podolny, V., Carpenter, L.R., Greenblatt, J.F., Baetz, K. and Buratowski, S. (2006) The Saccharomyces cerevisiae histone H2A variant Htz1 is acetylated by NuA4. Genes Dev, 20, 660-665.
Kim, M., Ahn, S.H., Krogan, N.J., Greenblatt, J.F. and Buratowski, S. (2004) Transitions in RNA polymerase II elongation complexes at the 3' ends of genes. Embo J, 23, 354-364.
Kim, T.K., Ebright, R.H. and Reinberg, D. (2000) Mechanism of ATP-dependent promoter melting by transcription factor IIH. Science, 288, 1418-1422.
Kim, Y.J., Bjorklund, S., Li, Y., Sayre, M.H. and Kornberg, R.D. (1994) A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell, 77, 599-608.
Knop, M., Siegers, K., Pereira, G., Zachariae, W., Winsor, B., Nasmyth, K. and Schiebel, E. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast, 15, 963-972.
Kobor, M.S., Venkatasubrahmanyam, S., Meneghini, M.D., Gin, J.W., Jennings, J.L., Link, A.J., Madhani, H.D. and Rine, J. (2004) A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol, 2, E131.
Koch, C., Moll, T., Neuberg, M., Ahorn, H. and Nasmyth, K. (1993) A role for the transcription factors Mbp1 and Swi4 in progression from G1 to S phase. Science, 261, 1551-1557.
Koch, C., Schleiffer, A., Ammerer, G. and Nasmyth, K. (1996) Switching transcription on and off during the yeast cell cycle: Cln/Cdc28 kinases activate bound transcription factor SBF (Swi4/Swi6) at start, whereas Clb/Cdc28 kinases displace it from the promoter in G2. Genes Dev, 10, 129-141.
Koch, C., Wollmann, P., Dahl, M. and Lottspeich, F. (1999) A role for Ctr9p and Paf1p in the regulation G1 cyclin expression in yeast. Nucleic Acids Res, 27, 2126-2134.
Komarnitsky, P., Cho, E.J. and Buratowski, S. (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev, 14, 2452-2460.
Kong, S.E., Kobor, M.S., Krogan, N.J., Somesh, B.P., Sogaard, T.M., Greenblatt, J.F. and Svejstrup,
J.Q. (2005) Interaction of Fcp1 phosphatase with elongating RNA polymerase II holoenzyme, enzymatic mechanism of action, and genetic interaction with elongator. J Biol Chem, 280, 4299-4306.
Kornberg, R.D. (1996) RNA polymerase II transcription control. Trends Biochem Sci, 21, 325-326.
Krogan, N.J., Baetz, K., Keogh, M.C., Datta, N., Sawa, C., Kwok, T.C., Thompson, N.J., Davey, M.G., Pootoolal, J., Hughes, T.R., Emili, A., Buratowski, S., Hieter, P. and Greenblatt, J.F. (2004) Regulation of chromosome stability by the histone H2A variant Htz1, the Swr1 chromatin remodeling complex, and the histone acetyltransferase NuA4. Proc Natl Acad Sci U S A, 101, 13513-13518.
Krogan, N.J., Dover, J., Khorrami, S., Greenblatt, J.F., Schneider, J., Johnston, M. and Shilatifard, A. (2002a) COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem, 277, 10753-10755.
Krogan, N.J., Dover, J., Wood, A., Schneider, J., Heidt, J., Boateng, M.A., Dean, K., Ryan, O.W., Golshani, A., Johnston, M., Greenblatt, J.F. and Shilatifard, A. (2003a) The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell, 11, 721-729.
Krogan, N.J., Keogh, M.C., Datta, N., Sawa, C., Ryan, O.W., Ding, H., Haw, R.A., Pootoolal, J., Tong, A., Canadien, V., Richards, D.P., Wu, X., Emili, A., Hughes, T.R., Buratowski, S. and Greenblatt, J.F. (2003b) A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell, 12, 1565-1576.
Krogan, N.J., Kim, M., Ahn, S.H., Zhong, G., Kobor, M.S., Cagney, G., Emili, A., Shilatifard, A., Buratowski, S. and Greenblatt, J.F. (2002b) RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol, 22, 6979-6992.
Krogan, N.J., Kim, M., Tong, A., Golshani, A., Cagney, G., Canadien, V., Richards, D.P., Beattie, B.K., Emili, A., Boone, C., Shilatifard, A., Buratowski, S. and Greenblatt, J. (2003c) Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol, 23, 4207-4218.
Kubinski, K., Domanska, K., Sajnaga, E., Mazur, E., Zielinski, R. and Szyszka, R. (2006) Yeast holoenzyme of protein kinase CK2 requires both beta and beta' regulatory subunits for its activity. Mol Cell Biochem.
Kuo, M.H. and Allis, C.D. (1999) In vivo cross-linking and immunoprecipitation for studying dynamic Protein:DNA associations in a chromatin environment. Methods, 19, 425-433.
Lacoste, N., Utley, R.T., Hunter, J.M., Poirier, G.G. and Cote, J. (2002) Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem, 277, 30421-30424.
Ladurner, A.G., Inouye, C., Jain, R. and Tjian, R. (2003) Bromodomains mediate an acetyl-histone

Literaturverzeichnis
138
encoded antisilencing function at heterochromatin boundaries. Mol Cell, 11, 365-376.
Laribee, R.N., Krogan, N.J., Xiao, T., Shibata, Y., Hughes, T.R., Greenblatt, J.F. and Strahl, B.D. (2005) BUR kinase selectively regulates H3 K4 trimethylation and H2B ubiquitylation through recruitment of the PAF elongation complex. Curr Biol, 15, 1487-1493.
Laybourn, P.J. and Dahmus, M.E. (1989) Transcription-dependent structural changes in the C-terminal domain of mammalian RNA polymerase subunit IIa/o. J Biol Chem, 264, 6693-6698.
Lee, T.I. and Young, R.A. (2000) Transcription of eukaryotic protein-coding genes. Annu Rev Genet, 34, 77-137.
Letunic, I., Copley, R.R., Pils, B., Pinkert, S., Schultz, J. and Bork, P. (2006) SMART 5: domains in the context of genomes and networks. Nucleic Acids Res, 34, D257-260.
Li, B., Howe, L., Anderson, S., Yates, J.R., 3rd and Workman, J.L. (2003) The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J Biol Chem, 278, 8897-8903.
Li, B., Pattenden, S.G., Lee, D., Gutierrez, J., Chen, J., Seidel, C., Gerton, J. and Workman, J.L. (2005) Preferential occupancy of histone variant H2AZ at inactive promoters influences local histone modifications and chromatin remodeling. Proc Natl Acad Sci U S A, 102, 18385-18390.
Li, J., Moazed, D. and Gygi, S.P. (2002) Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J Biol Chem, 277, 49383-49388.
Lindstrom, D.L., Squazzo, S.L., Muster, N., Burckin, T.A., Wachter, K.C., Emigh, C.A., McCleery, J.A., Yates, J.R., 3rd and Hartzog, G.A. (2003) Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol, 23, 1368-1378.
Magdolen, V., Lang, P., Mages, G., Hermann, H. and Bandlow, W. (1994) The gene LEO1 on yeast chromosome XV encodes a non-essential, extremely hydrophilic protein. Biochim Biophys Acta, 1218, 205-209.
Maillet, L., Boscheron, C., Gotta, M., Marcand, S., Gilson, E. and Gasser, S.M. (1996) Evidence for silencing compartments within the yeast nucleus: a role for telomere proximity and Sir protein concentration in silencer-mediated repression. Genes Dev, 10, 1796-1811.
Main, E.R., Xiong, Y., Cocco, M.J., D'Andrea, L. and Regan, L. (2003) Design of stable alpha-helical arrays from an idealized TPR motif. Structure, 11, 497-508.
Mandal, S.S., Cho, H., Kim, S., Cabane, K. and Reinberg, D. (2002) FCP1, a phosphatase specific for the heptapeptide repeat of the largest subunit of RNA polymerase II, stimulates transcription elongation. Mol Cell Biol, 22, 7543-7552.
Martinez-Campa, C., Politis, P., Moreau, J.L., Kent, N., Goodall, J., Mellor, J. and Goding, C.R. (2004) Precise nucleosome positioning and the TATA box dictate requirements for the histone H4 tail
and the bromodomain factor Bdf1. Mol Cell, 15, 69-81.
Mason, P.B. and Struhl, K. (2005) Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell, 17, 831-840.
Matangkasombut, O., Buratowski, R.M., Swilling, N.W. and Buratowski, S. (2000) Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev, 14, 951-962.
Maxon, M.E., Goodrich, J.A. and Tjian, R. (1994) Transcription factor IIE binds preferentially to RNA polymerase IIa and recruits TFIIH: a model for promoter clearance. Genes Dev, 8, 515-524.
Mayer, M.L., Gygi, S.P., Aebersold, R. and Hieter, P. (2001) Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol Cell, 7, 959-970.
Meneghini, M.D., Wu, M. and Madhani, H.D. (2003) Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell, 112, 725-736.
Moniaux, N., Nemos, C., Schmied, B.M., Chauhan, S.C., Deb, S., Morikane, K., Choudhury, A., Vanlith, M., Sutherlin, M., Sikela, J.M., Hollingsworth, M.A. and Batra, S.K. (2006) The human homologue of the RNA polymerase II-associated factor 1 (hPaf1), localized on the 19q13 amplicon, is associated with tumorigenesis. Oncogene, 25, 3247-3257.
Morillon, A., Karabetsou, N., Nair, A. and Mellor, J. (2005) Dynamic lysine methylation on histone H3 defines the regulatory phase of gene transcription. Mol Cell, 18, 723-734.
Mortensen, U.H., Bendixen, C., Sunjevaric, I. and Rothstein, R. (1996) DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci U S A, 93, 10729-10734.
Mueller, C.L. and Jaehning, J.A. (2002) Ctr9, Rtf1, and Leo1 are components of the Paf1/RNA polymerase II complex. Mol Cell Biol, 22, 1971-1980.
Mueller, C.L., Porter, S.E., Hoffman, M.G. and Jaehning, J.A. (2004) The Paf1 complex has functions independent of actively transcribing RNA polymerase II. Mol Cell, 14, 447-456.
Myer, V.E. and Young, R.A. (1998) RNA polymerase II holoenzymes and subcomplexes. J Biol Chem, 273, 27757-27760.
Nasmyth, K. and Dirick, L. (1991) The role of SWI4 and SWI6 in the activity of G1 cyclins in yeast. Cell, 66, 995-1013.
Ng, H.H., Dole, S. and Struhl, K. (2003a) The Rtf1 component of the Paf1 transcriptional elongation complex is required for ubiquitination of histone H2B. J Biol Chem, 278, 33625-33628.
Ng, H.H., Robert, F., Young, R.A. and Struhl, K. (2003b) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell, 11, 709-719.
Ng, H.H., Xu, R.M., Zhang, Y. and Struhl, K. (2002) Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated

Literaturverzeichnis
139
methylation of histone H3 lysine 79. J Biol Chem, 277, 34655-34657.
O'Brien, T., Hardin, S., Greenleaf, A. and Lis, J.T. (1994) Phosphorylation of RNA polymerase II C-terminal domain and transcriptional elongation. Nature, 370, 75-77.
O'Donnell, A.F., Brewster, N.K., Kurniawan, J., Minard, L.V., Johnston, G.C. and Singer, R.A. (2004) Domain organization of the yeast histone chaperone FACT: the conserved N-terminal domain of FACT subunit Spt16 mediates recovery from replication stress. Nucleic Acids Res, 32, 5894-5906.
Oh, S., Zhang, H., Ludwig, P. and van Nocker, S. (2004) A mechanism related to the yeast transcriptional regulator Paf1c is required for expression of the Arabidopsis FLC/MAF MADS box gene family. Plant Cell, 16, 2940-2953.
Okada, Y., Feng, Q., Lin, Y., Jiang, Q., Li, Y., Coffield, V.M., Su, L., Xu, G. and Zhang, Y. (2005) hDOT1L links histone methylation to leukemogenesis. Cell, 121, 167-178.
Orphanides, G., Lagrange, T. and Reinberg, D. (1996) The general transcription factors of RNA polymerase II. Genes Dev, 10, 2657-2683.
Orphanides, G. and Reinberg, D. (2000) RNA polymerase II elongation through chromatin. Nature, 407, 471-475.
Orphanides, G. and Reinberg, D. (2002) A unified theory of gene expression. Cell, 108, 439-451.
Orphanides, G., Wu, W.H., Lane, W.S., Hampsey, M. and Reinberg, D. (1999) The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature, 400, 284-288.
Osborn, A.J. and Elledge, S.J. (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev, 17, 1755-1767.
Padmanabha, R., Chen-Wu, J.L., Hanna, D.E. and Glover, C.V. (1990) Isolation, sequencing, and disruption of the yeast CKA2 gene: casein kinase II is essential for viability in Saccharomyces cerevisiae. Mol Cell Biol, 10, 4089-4099.
Palancade, B., Dubois, M.F. and Bensaude, O. (2002) FCP1 phosphorylation by casein kinase 2 enhances binding to TFIIF and RNA polymerase II carboxyl-terminal domain phosphatase activity. J Biol Chem, 277, 36061-36067.
Pan, X., Ye, P., Yuan, D.S., Wang, X., Bader, J.S. and Boeke, J.D. (2006) A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell, 124, 1069-1081.
Papamichos-Chronakis, M., Krebs, J.E. and Peterson, C.L. (2006) Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev, 20, 2437-2449.
Pavri, R., Zhu, B., Li, G., Trojer, P., Mandal, S., Shilatifard, A. and Reinberg, D. (2006) Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell, 125, 703-717.
Payne, J.M., Laybourn, P.J. and Dahmus, M.E. (1989) The transition of RNA polymerase II from initiation to elongation is associated with phosphorylation of the carboxyl-terminal domain of subunit IIa. J Biol Chem, 264, 19621-19629.
Penheiter, K.L., Washburn, T.M., Porter, S.E., Hoffman, M.G. and Jaehning, J.A. (2005) A posttranscriptional role for the yeast Paf1-RNA polymerase II complex is revealed by identification of primary targets. Mol Cell, 20, 213-223.
Pokholok, D.K., Hannett, N.M. and Young, R.A. (2002) Exchange of RNA polymerase II initiation and elongation factors during gene expression in vivo. Mol Cell, 9, 799-809.
Proudfoot, N.J., Furger, A. and Dye, M.J. (2002) Integrating mRNA processing with transcription. Cell, 108, 501-512.
Puig, O., Caspary, F., Rigaut, G., Rutz, B., Bouveret, E., Bragado-Nilsson, E., Wilm, M. and Seraphin, B. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods, 24, 218-229.
Qiu, H., Hu, C., Wong, C.M. and Hinnebusch, A.G. (2006) The Spt4p subunit of yeast DSIF stimulates association of the Paf1 complex with elongating RNA polymerase II. Mol Cell Biol, 26, 3135-3148.
Queimado, L., Rao, M., Schultz, R.A., Koonin, E.V., Aravind, L., Nardo, T., Stefanini, M. and Friedberg, E.C. (2001) Cloning the human and mouse MMS19 genes and functional complementation of a yeast mms19 deletion mutant. Nucleic Acids Res, 29, 1884-1891.
Raisner, R.M., Hartley, P.D., Meneghini, M.D., Bao, M.Z., Liu, C.L., Schreiber, S.L., Rando, O.J. and Madhani, H.D. (2005) Histone variant H2A.Z marks the 5' ends of both active and inactive genes in euchromatin. Cell, 123, 233-248.
Reguly, T., Breitkreutz, A., Boucher, L., Breitkreutz, B.J., Hon, G.C., Myers, C.L., Parsons, A., Friesen, H., Oughtred, R., Tong, A., Stark, C., Ho, Y., Botstein, D., Andrews, B., Boone, C., Troyanskya, O.G., Ideker, T., Dolinski, K., Batada, N.N. and Tyers, M. (2006) Comprehensive curation and analysis of global interaction networks in Saccharomyces cerevisiae. J Biol, 5, 11.
Rhoades, A.R., Ruone, S. and Formosa, T. (2004) Structural features of nucleosomes reorganized by yeast FACT and its HMG box component, Nhp6. Mol Cell Biol, 24, 3907-3917.
Richards, E.J. and Elgin, S.C. (2002) Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell, 108, 489-500.
Richardson, H.E., Wittenberg, C., Cross, F. and Reed, S.I. (1989) An essential G1 function for cyclin-like proteins in yeast. Cell, 59, 1127-1133.
Riles, L., Shaw, R.J., Johnston, M. and Reines, D. (2004) Large-scale screening of yeast mutants for sensitivity to the IMP dehydrogenase inhibitor 6-azauracil. Yeast, 21, 241-248.
Robzyk, K., Recht, J. and Osley, M.A. (2000) Rad6-dependent ubiquitination of histone H2B in yeast. Science, 287, 501-504.

Literaturverzeichnis
140
Rodriguez-Navarro, S., Fischer, T., Luo, M.J., Antunez, O., Brettschneider, S., Lechner, J., Perez-Ortin, J.E., Reed, R. and Hurt, E. (2004) Sus1, a functional component of the SAGA histone acetylase complex and the nuclear pore-associated mRNA export machinery. Cell, 116, 75-86.
Rodriguez, C.R., Cho, E.J., Keogh, M.C., Moore, C.L., Greenleaf, A.L. and Buratowski, S. (2000) Kin28, the TFIIH-associated carboxy-terminal domain kinase, facilitates the recruitment of mRNA processing machinery to RNA polymerase II. Mol Cell Biol, 20, 104-112.
Rosenberg, A.H., Lade, B.N., Chui, D.S., Lin, S.W., Dunn, J.J. and Studier, F.W. (1987) Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene, 56, 125-135.
Rosonina, E. and Manley, J.L. (2005) From transcription to mRNA: PAF provides a new path. Mol Cell, 20, 167-168.
Rozenblatt-Rosen, O., Hughes, C.M., Nannepaga, S.J., Shanmugam, K.S., Copeland, T.D., Guszczynski, T., Resau, J.H. and Meyerson, M. (2005) The parafibromin tumor suppressor protein is part of a human paf1 complex. Mol Cell Biol, 25, 612-620.
Rusche, L.N., Kirchmaier, A.L. and Rine, J. (2003) The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem, 72, 481-516.
Sambrook, J., Russell, D.W. and Cold Spring Harbor Laboratory. (2001) Molecular cloning : a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
Sawa, C., Nedea, E., Krogan, N., Wada, T., Handa, H., Greenblatt, J. and Buratowski, S. (2004) Bromodomain factor 1 (Bdf1) is phosphorylated by protein kinase CK2. Mol Cell Biol, 24, 4734-4742.
Seol, J.H., Kim, H.J., Yang, Y.J., Kim, S.T., Youn, H.D., Han, J.W., Lee, H.W. and Cho, E.J. (2006) Different roles of histone H3 lysine 4 methylation in chromatin maintenance. Biochem Biophys Res Commun, 349, 463-470.
Shahbazian, M.D., Zhang, K. and Grunstein, M. (2005) Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol Cell, 19, 271-277.
Sheldon, K.E., Mauger, D.M. and Arndt, K.M. (2005) A Requirement for the Saccharomyces cerevisiae Paf1 complex in snoRNA 3' end formation. Mol Cell, 20, 225-236.
Shi, X., Chang, M., Wolf, A.J., Chang, C.H., Frazer-Abel, A.A., Wade, P.A., Burton, Z.F. and Jaehning, J.A. (1997) Cdc73p and Paf1p are found in a novel RNA polymerase II-containing complex distinct from the Srbp-containing holoenzyme. Mol Cell Biol, 17, 1160-1169.
Shi, X., Finkelstein, A., Wolf, A.J., Wade, P.A., Burton, Z.F. and Jaehning, J.A. (1996) Paf1p, an RNA polymerase II-associated factor in Saccharomyces cerevisiae, may have both positive and negative roles in transcription. Mol Cell Biol, 16, 669-676.
Shilatifard, A., Conaway, R.C. and Conaway, J.W. (2003) The RNA polymerase II elongation complex. Annu Rev Biochem, 72, 693-715.
Shore, D., Squire, M. and Nasmyth, K.A. (1984) Characterization of two genes required for the position-effect control of yeast mating-type genes. Embo J, 3, 2817-2823.
Sikorski, R.S., Boguski, M.S., Goebl, M. and Hieter, P. (1990) A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis. Cell, 60, 307-317.
Simic, R., Lindstrom, D.L., Tran, H.G., Roinick, K.L., Costa, P.J., Johnson, A.D., Hartzog, G.A. and Arndt, K.M. (2003) Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. Embo J, 22, 1846-1856.
Sims, R.J., 3rd, Belotserkovskaya, R. and Reinberg, D. (2004) Elongation by RNA polymerase II: the short and long of it. Genes Dev, 18, 2437-2468.
Singer, M.S., Kahana, A., Wolf, A.J., Meisinger, L.L., Peterson, S.E., Goggin, C., Mahowald, M. and Gottschling, D.E. (1998) Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics, 150, 613-632.
Singer, R.A. and Johnston, G.C. (2004) The FACT chromatin modulator: genetic and structure/function relationships. Biochem Cell Biol, 82, 419-427.
Smith, D.B. and Johnson, K.S. (1988) Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene, 67, 31-40.
Smith, J.S., Brachmann, C.B., Pillus, L. and Boeke, J.D. (1998) Distribution of a limited Sir2 protein pool regulates the strength of yeast rDNA silencing and is modulated by Sir4p. Genetics, 149, 1205-1219.
Solomon, M.J. and Varshavsky, A. (1985) Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc Natl Acad Sci U S A, 82, 6470-6474.
Spector, M.S., Raff, A., DeSilva, H., Lee, K. and Osley, M.A. (1997) Hir1p and Hir2p function as transcriptional corepressors to regulate histone gene transcription in the Saccharomyces cerevisiae cell cycle. Mol Cell Biol, 17, 545-552.
Squazzo, S.L., Costa, P.J., Lindstrom, D.L., Kumer, K.E., Simic, R., Jennings, J.L., Link, A.J., Arndt, K.M. and Hartzog, G.A. (2002) The Paf1 complex physically and functionally associates with transcription elongation factors in vivo. Embo J, 21, 1764-1774.
Stolinski, L.A., Eisenmann, D.M. and Arndt, K.M. (1997) Identification of RTF1, a novel gene important for TATA site selection by TATA box-binding protein in Saccharomyces cerevisiae. Mol Cell Biol, 17, 4490-4500.
Strahl, B.D. and Allis, C.D. (2000) The language of covalent histone modifications. Nature, 403, 41-45.
Strahl, B.D., Grant, P.A., Briggs, S.D., Sun, Z.W., Bone, J.R., Caldwell, J.A., Mollah, S., Cook, R.G., Shabanowitz, J., Hunt, D.F. and Allis, C.D. (2002) Set2 is a nucleosomal histone H3-selective methyltransferase that mediates

Literaturverzeichnis
141
transcriptional repression. Mol Cell Biol, 22, 1298-1306.
Strasser, K., Masuda, S., Mason, P., Pfannstiel, J., Oppizzi, M., Rodriguez-Navarro, S., Rondon, A.G., Aguilera, A., Struhl, K., Reed, R. and Hurt, E. (2002) TREX is a conserved complex coupling transcription with messenger RNA export. Nature, 417, 304-308.
Struhl, K. (1987) Promoters, activator proteins, and the mechanism of transcriptional initiation in yeast. Cell, 49, 295-297.
Sun, Z.W. and Allis, C.D. (2002) Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature, 418, 104-108.
Tanaka, T., Knapp, D. and Nasmyth, K. (1997) Loading of an Mcm protein onto DNA replication origins is regulated by Cdc6p and CDKs. Cell, 90, 649-660.
Tenney, K., Gerber, M., Ilvarsonn, A., Schneider, J., Gause, M., Dorsett, D., Eissenberg, J.C. and Shilatifard, A. (2006) Drosophila Rtf1 functions in histone methylation, gene expression, and Notch signaling. Proc Natl Acad Sci U S A, 103, 11970-11974.
Tenney, K. and Shilatifard, A. (2005) A COMPASS in the voyage of defining the role of trithorax/MLL-containing complexes: linking leukemogensis to covalent modifications of chromatin. J Cell Biochem, 95, 429-436.
Thiriet, C. and Albert, P. (1995) Rapid and effective western blotting of histones from acid-urea-Triton and sodium dodecyl sulfate polyacrylamide gels: two different approaches depending on the subsequent qualitative or quantitative analysis. Electrophoresis, 16, 357-361.
Timmers, H.T. and Tora, L. (2005) SAGA unveiled. Trends Biochem Sci, 30, 7-10.
Toh, G.W., O'Shaughnessy, A.M., Jimeno, S., Dobbie, I.M., Grenon, M., Maffini, S., O'Rorke, A. and Lowndes, N.F. (2006) Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair (Amst), 5, 693-703.
Tong, A.H., Evangelista, M., Parsons, A.B., Xu, H., Bader, G.D., Page, N., Robinson, M., Raghibizadeh, S., Hogue, C.W., Bussey, H., Andrews, B., Tyers, M. and Boone, C. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science, 294, 2364-2368.
Tong, A.H., Lesage, G., Bader, G.D., Ding, H., Xu, H., Xin, X., Young, J., Berriz, G.F., Brost, R.L., Chang, M., Chen, Y., Cheng, X., Chua, G., Friesen, H., Goldberg, D.S., Haynes, J., Humphries, C., He, G., Hussein, S., Ke, L., Krogan, N., Li, Z., Levinson, J.N., Lu, H., Menard, P., Munyana, C., Parsons, A.B., Ryan, O., Tonikian, R., Roberts, T., Sdicu, A.M., Shapiro, J., Sheikh, B., Suter, B., Wong, S.L., Zhang, L.V., Zhu, H., Burd, C.G., Munro, S., Sander, C., Rine, J., Greenblatt, J., Peter, M., Bretscher, A., Bell, G., Roth, F.P., Brown, G.W., Andrews, B., Bussey, H. and Boone, C. (2004) Global mapping of the yeast genetic interaction network. Science, 303, 808-813.
Tuazon, P.T. and Traugh, J.A. (1991) Casein kinase I and II--multipotential serine protein kinases: structure, function, and regulation. Adv Second Messenger Phosphoprotein Res, 23, 123-164.
van Leeuwen, F., Gafken, P.R. and Gottschling, D.E. (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell, 109, 745-756.
VanDemark, A.P., Blanksma, M., Ferris, E., Heroux, A., Hill, C.P. and Formosa, T. (2006) The structure of the yFACT Pob3-M domain, its interaction with the DNA replication factor RPA, and a potential role in nucleosome deposition. Mol Cell, 22, 363-374.
Wach, A., Brachat, A., Pohlmann, R. and Philippsen, P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 10, 1793-1808.
Wada, T., Orphanides, G., Hasegawa, J., Kim, D.K., Shima, D., Yamaguchi, Y., Fukuda, A., Hisatake, K., Oh, S., Reinberg, D. and Handa, H. (2000) FACT relieves DSIF/NELF-mediated inhibition of transcriptional elongation and reveals functional differences between P-TEFb and TFIIH. Mol Cell, 5, 1067-1072.
Wada, T., Takagi, T., Yamaguchi, Y., Ferdous, A., Imai, T., Hirose, S., Sugimoto, S., Yano, K., Hartzog, G.A., Winston, F., Buratowski, S. and Handa, H. (1998a) DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev, 12, 343-356.
Wada, T., Takagi, T., Yamaguchi, Y., Watanabe, D. and Handa, H. (1998b) Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. Embo J, 17, 7395-7403.
Wade, P.A., Werel, W., Fentzke, R.C., Thompson, N.E., Leykam, J.F., Burgess, R.R., Jaehning, J.A. and Burton, Z.F. (1996) A novel collection of accessory factors associated with yeast RNA polymerase II. Protein Expr Purif, 8, 85-90.
Watson, J.D. and Crick, F.H. (1953) Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature, 171, 737-738.
Weinmann, R. and Roeder, R.G. (1974) Role of DNA-dependent RNA polymerase 3 in the transcription of the tRNA and 5S RNA genes. Proc Natl Acad Sci U S A, 71, 1790-1794.
Winkler, G.S., Kristjuhan, A., Erdjument-Bromage, H., Tempst, P. and Svejstrup, J.Q. (2002) Elongator is a histone H3 and H4 acetyltransferase important for normal histone acetylation levels in vivo. Proc Natl Acad Sci U S A, 99, 3517-3522.
Wittekind, M., Dodd, J., Vu, L., Kolb, J.M., Buhler, J.M., Sentenac, A. and Nomura, M. (1988) Isolation and characterization of temperature-sensitive mutations in RPA190, the gene encoding the largest subunit of RNA polymerase I from Saccharomyces cerevisiae. Mol Cell Biol, 8, 3997-4008.
Wittmeyer, J. and Formosa, T. (1997) The Saccharomyces cerevisiae DNA polymerase alpha catalytic subunit interacts with

Literaturverzeichnis
142
Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol Cell Biol, 17, 4178-4190.
Wood, A., Krogan, N.J., Dover, J., Schneider, J., Heidt, J., Boateng, M.A., Dean, K., Golshani, A., Zhang, Y., Greenblatt, J.F., Johnston, M. and Shilatifard, A. (2003a) Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell, 11, 267-274.
Wood, A., Schneider, J., Dover, J., Johnston, M. and Shilatifard, A. (2003b) The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J Biol Chem, 278, 34739-34742.
Wood, A., Schneider, J., Dover, J., Johnston, M. and Shilatifard, A. (2005) The Bur1/Bur2 complex is required for histone H2B monoubiquitination by Rad6/Bre1 and histone methylation by COMPASS. Mol Cell, 20, 589-599.
Wood, A. and Shilatifard, A. (2006) Bur1/Bur2 and the Ctk complex in yeast: the split personality of mammalian P-TEFb. Cell Cycle, 5, 1066-1068.
Wood, A., Shukla, A., Schneider, J., Lee, J.S., Stanton, J.D., Dzuiba, T., Swanson, S.K., Florens, L., Washburn, M.P., Wyrick, J., Bhaumik, S.R. and Shilatifard, A. (2007) Ctk complex-mediated regulation of histone methylation by COMPASS. Mol Cell Biol, 27, 709-720.
Woodard, G.E., Lin, L., Zhang, J.H., Agarwal, S.K., Marx, S.J. and Simonds, W.F. (2005) Parafibromin, product of the hyperparathyroidism-jaw tumor syndrome gene HRPT2, regulates cyclin D1/PRAD1 expression. Oncogene, 24, 1272-1276.
Wright, A.P., Bruns, M. and Hartley, B.S. (1989) Extraction and rapid inactivation of proteins from Saccharomyces cerevisiae by trichloroacetic acid precipitation. Yeast, 5, 51-53.
Wysocki, R., Javaheri, A., Allard, S., Sha, F., Cote, J. and Kron, S.J. (2005) Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol, 25, 8430-8443.
Xiao, T., Hall, H., Kizer, K.O., Shibata, Y., Hall, M.C., Borchers, C.H. and Strahl, B.D. (2003) Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev, 17, 654-663.
Xiao, T., Kao, C.F., Krogan, N.J., Sun, Z.W., Greenblatt, J.F., Osley, M.A. and Strahl, B.D. (2005) Histone H2B Ubiquitylation Is Associated with Elongating RNA Polymerase II. Mol Cell Biol, 25, 637-651.
Xu, H., Kim, U.J., Schuster, T. and Grunstein, M. (1992) Identification of a new set of cell cycle-regulatory genes that regulate S-phase transcription of histone genes in Saccharomyces cerevisiae. Mol Cell Biol, 12, 5249-5259.
Yang, Z., Yik, J.H., Chen, R., He, N., Jang, M.K., Ozato, K. and Zhou, Q. (2005) Recruitment of P-TEFb for stimulation of transcriptional elongation by
the bromodomain protein Brd4. Mol Cell, 19, 535-545.
Ye, P., Peyser, B.D., Pan, X., Boeke, J.D., Spencer, F.A. and Bader, J.S. (2005) Gene function prediction from congruent synthetic lethal interactions in yeast. Mol Syst Biol, 1, 2005 0026.
Yokoyama, A., Wang, Z., Wysocka, J., Sanyal, M., Aufiero, D.J., Kitabayashi, I., Herr, W. and Cleary, M.L. (2004) Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol, 24, 5639-5649.
Zhang, H., Roberts, D.N. and Cairns, B.R. (2005) Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell, 123, 219-231.
Zhou, Q. and Yik, J.H. (2006) The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev, 70, 646-659.
Zhou, Y. and Wang, T.S. (2004) A coordinated temporal interplay of nucleosome reorganization factor, sister chromatin cohesion factor, and DNA polymerase alpha facilitates DNA replication. Mol Cell Biol, 24, 9568-9579.
Zielinski, R., Hellman, U., Kubinski, K. and Szyszka, R. (2006) Fip1--an essential component of the Saccharomyces cerevisiae polyadenylation machinery is phosophorylated by protein kinase CK2. Mol Cell Biochem, 286, 191-197.
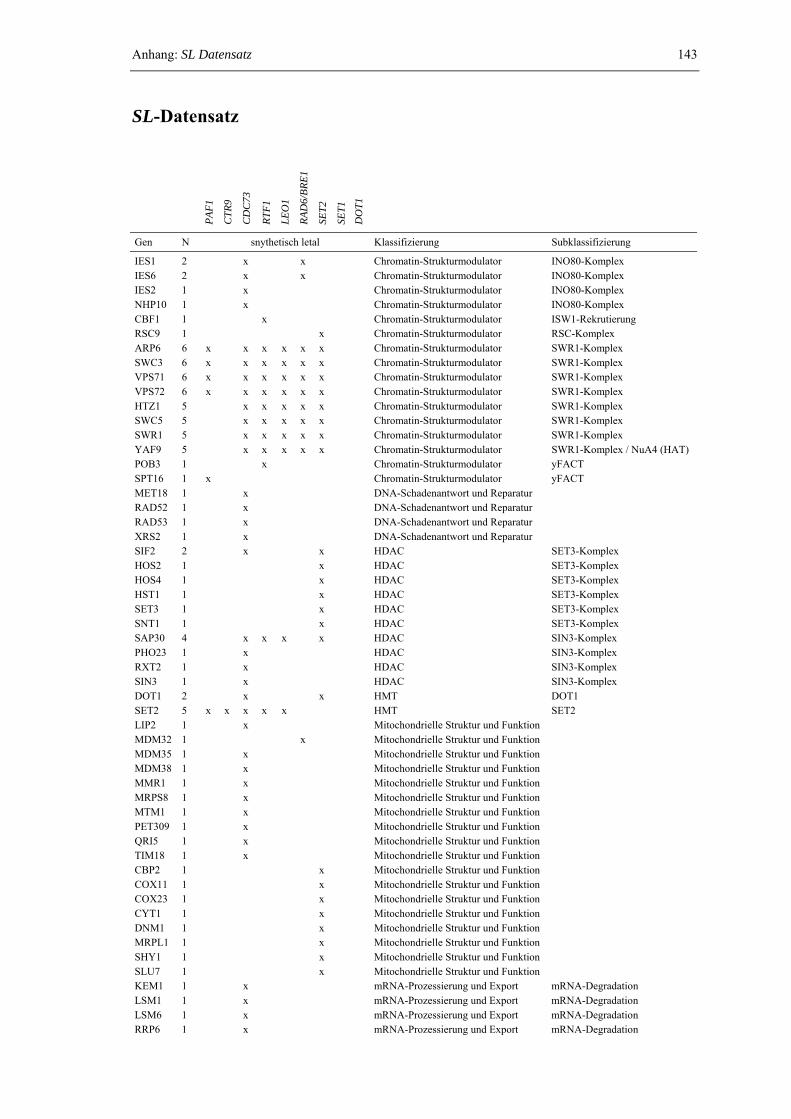
Anhang: SL Datensatz
143
SL-Datensatz
PA
F1
CTR
9
CD
C73
RTF1
LEO
1
RAD
6/BR
E1
SET2
SET1
DO
T1
Gen N snythetisch letal Klassifizierung Subklassifizierung
IES1 2 x x Chromatin-Strukturmodulator INO80-Komplex IES6 2 x x Chromatin-Strukturmodulator INO80-Komplex IES2 1 x Chromatin-Strukturmodulator INO80-Komplex NHP10 1 x Chromatin-Strukturmodulator INO80-Komplex CBF1 1 x Chromatin-Strukturmodulator ISW1-Rekrutierung RSC9 1 x Chromatin-Strukturmodulator RSC-Komplex ARP6 6 x x x x x x Chromatin-Strukturmodulator SWR1-Komplex SWC3 6 x x x x x x Chromatin-Strukturmodulator SWR1-Komplex VPS71 6 x x x x x x Chromatin-Strukturmodulator SWR1-Komplex VPS72 6 x x x x x x Chromatin-Strukturmodulator SWR1-Komplex HTZ1 5 x x x x x Chromatin-Strukturmodulator SWR1-Komplex SWC5 5 x x x x x Chromatin-Strukturmodulator SWR1-Komplex SWR1 5 x x x x x Chromatin-Strukturmodulator SWR1-Komplex YAF9 5 x x x x x Chromatin-Strukturmodulator SWR1-Komplex / NuA4 (HAT) POB3 1 x Chromatin-Strukturmodulator yFACT SPT16 1 x Chromatin-Strukturmodulator yFACT MET18 1 x DNA-Schadenantwort und Reparatur RAD52 1 x DNA-Schadenantwort und Reparatur RAD53 1 x DNA-Schadenantwort und Reparatur XRS2 1 x DNA-Schadenantwort und Reparatur SIF2 2 x x HDAC SET3-Komplex HOS2 1 x HDAC SET3-Komplex HOS4 1 x HDAC SET3-Komplex HST1 1 x HDAC SET3-Komplex SET3 1 x HDAC SET3-Komplex SNT1 1 x HDAC SET3-Komplex SAP30 4 x x x x HDAC SIN3-Komplex PHO23 1 x HDAC SIN3-Komplex RXT2 1 x HDAC SIN3-Komplex SIN3 1 x HDAC SIN3-Komplex DOT1 2 x x HMT DOT1 SET2 5 x x x x x HMT SET2 LIP2 1 x Mitochondrielle Struktur und Funktion MDM32 1 x Mitochondrielle Struktur und Funktion MDM35 1 x Mitochondrielle Struktur und Funktion MDM38 1 x Mitochondrielle Struktur und Funktion MMR1 1 x Mitochondrielle Struktur und Funktion MRPS8 1 x Mitochondrielle Struktur und Funktion MTM1 1 x Mitochondrielle Struktur und Funktion PET309 1 x Mitochondrielle Struktur und Funktion QRI5 1 x Mitochondrielle Struktur und Funktion TIM18 1 x Mitochondrielle Struktur und Funktion CBP2 1 x Mitochondrielle Struktur und Funktion COX11 1 x Mitochondrielle Struktur und Funktion COX23 1 x Mitochondrielle Struktur und Funktion CYT1 1 x Mitochondrielle Struktur und Funktion DNM1 1 x Mitochondrielle Struktur und Funktion MRPL1 1 x Mitochondrielle Struktur und Funktion SHY1 1 x Mitochondrielle Struktur und Funktion SLU7 1 x Mitochondrielle Struktur und Funktion KEM1 1 x mRNA-Prozessierung und Export mRNA-Degradation LSM1 1 x mRNA-Prozessierung und Export mRNA-Degradation LSM6 1 x mRNA-Prozessierung und Export mRNA-Degradation RRP6 1 x mRNA-Prozessierung und Export mRNA-Degradation
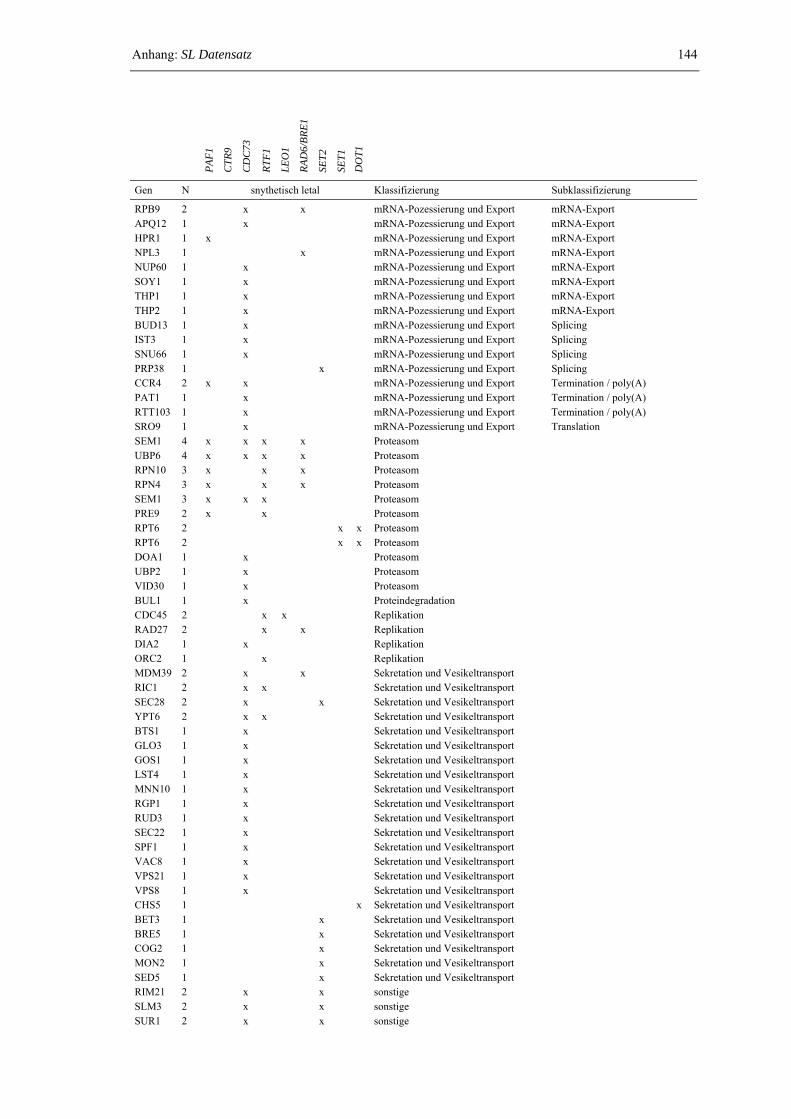
Anhang: SL Datensatz
144
PAF1
CTR
9
CD
C73
RTF1
LEO
1
RAD
6/BR
E1
SET2
SET1
DO
T1
Gen N snythetisch letal Klassifizierung Subklassifizierung
RPB9 2 x x mRNA-Pozessierung und Export mRNA-Export APQ12 1 x mRNA-Pozessierung und Export mRNA-Export HPR1 1 x mRNA-Pozessierung und Export mRNA-Export NPL3 1 x mRNA-Pozessierung und Export mRNA-Export NUP60 1 x mRNA-Pozessierung und Export mRNA-Export SOY1 1 x mRNA-Pozessierung und Export mRNA-Export THP1 1 x mRNA-Pozessierung und Export mRNA-Export THP2 1 x mRNA-Pozessierung und Export mRNA-Export BUD13 1 x mRNA-Pozessierung und Export Splicing IST3 1 x mRNA-Pozessierung und Export Splicing SNU66 1 x mRNA-Pozessierung und Export Splicing PRP38 1 x mRNA-Pozessierung und Export Splicing CCR4 2 x x mRNA-Pozessierung und Export Termination / poly(A) PAT1 1 x mRNA-Pozessierung und Export Termination / poly(A) RTT103 1 x mRNA-Pozessierung und Export Termination / poly(A) SRO9 1 x mRNA-Pozessierung und Export Translation SEM1 4 x x x x Proteasom UBP6 4 x x x x Proteasom RPN10 3 x x x Proteasom RPN4 3 x x x Proteasom SEM1 3 x x x Proteasom PRE9 2 x x Proteasom RPT6 2 x x Proteasom RPT6 2 x x Proteasom DOA1 1 x Proteasom UBP2 1 x Proteasom VID30 1 x Proteasom BUL1 1 x Proteindegradation CDC45 2 x x Replikation RAD27 2 x x Replikation DIA2 1 x Replikation ORC2 1 x Replikation MDM39 2 x x Sekretation und Vesikeltransport RIC1 2 x x Sekretation und Vesikeltransport SEC28 2 x x Sekretation und Vesikeltransport YPT6 2 x x Sekretation und Vesikeltransport BTS1 1 x Sekretation und Vesikeltransport GLO3 1 x Sekretation und Vesikeltransport GOS1 1 x Sekretation und Vesikeltransport LST4 1 x Sekretation und Vesikeltransport MNN10 1 x Sekretation und Vesikeltransport RGP1 1 x Sekretation und Vesikeltransport RUD3 1 x Sekretation und Vesikeltransport SEC22 1 x Sekretation und Vesikeltransport SPF1 1 x Sekretation und Vesikeltransport VAC8 1 x Sekretation und Vesikeltransport VPS21 1 x Sekretation und Vesikeltransport VPS8 1 x Sekretation und Vesikeltransport CHS5 1 x Sekretation und Vesikeltransport BET3 1 x Sekretation und Vesikeltransport BRE5 1 x Sekretation und Vesikeltransport COG2 1 x Sekretation und Vesikeltransport MON2 1 x Sekretation und Vesikeltransport SED5 1 x Sekretation und Vesikeltransport RIM21 2 x x sonstige SLM3 2 x x sonstige SUR1 2 x x sonstige
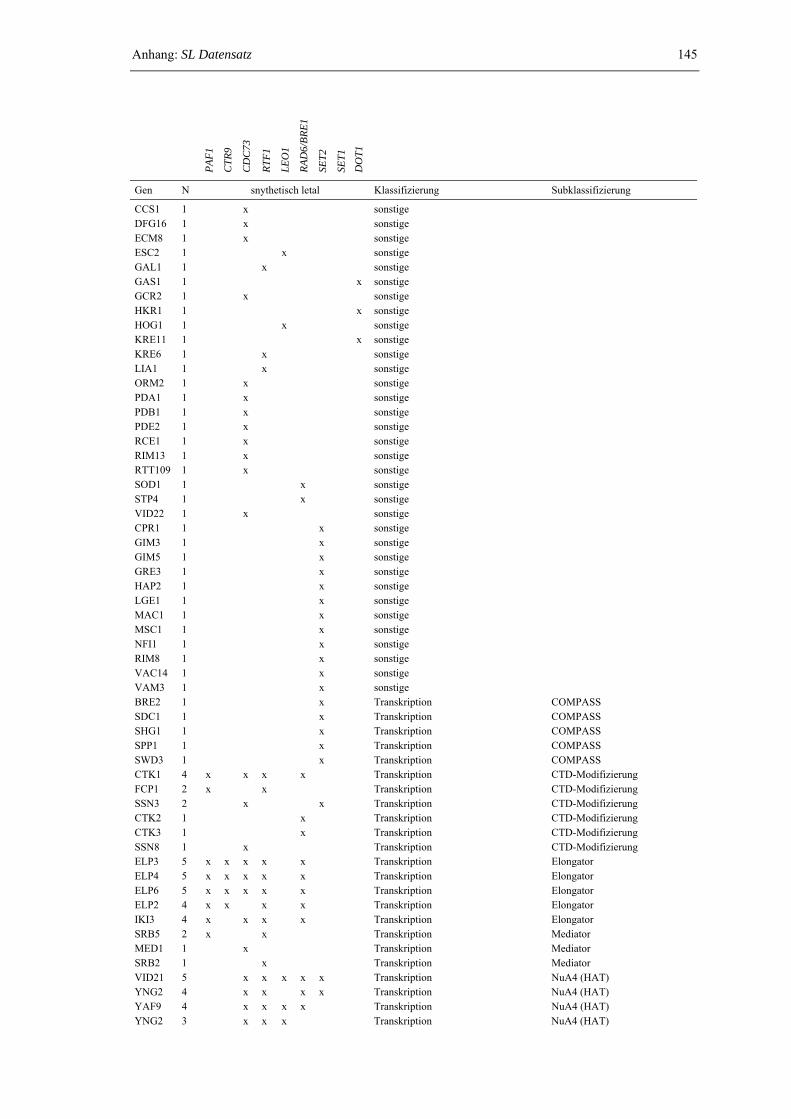
Anhang: SL Datensatz
145
PAF1
CTR
9
CD
C73
RTF1
LEO
1
RAD
6/BR
E1
SET2
SET1
DO
T1
Gen N snythetisch letal Klassifizierung Subklassifizierung
CCS1 1 x sonstige DFG16 1 x sonstige ECM8 1 x sonstige ESC2 1 x sonstige GAL1 1 x sonstige GAS1 1 x sonstige GCR2 1 x sonstige HKR1 1 x sonstige HOG1 1 x sonstige KRE11 1 x sonstige KRE6 1 x sonstige LIA1 1 x sonstige ORM2 1 x sonstige PDA1 1 x sonstige PDB1 1 x sonstige PDE2 1 x sonstige RCE1 1 x sonstige RIM13 1 x sonstige RTT109 1 x sonstige SOD1 1 x sonstige STP4 1 x sonstige VID22 1 x sonstige CPR1 1 x sonstige GIM3 1 x sonstige GIM5 1 x sonstige GRE3 1 x sonstige HAP2 1 x sonstige LGE1 1 x sonstige MAC1 1 x sonstige MSC1 1 x sonstige NFI1 1 x sonstige RIM8 1 x sonstige VAC14 1 x sonstige VAM3 1 x sonstige BRE2 1 x Transkription COMPASS SDC1 1 x Transkription COMPASS SHG1 1 x Transkription COMPASS SPP1 1 x Transkription COMPASS SWD3 1 x Transkription COMPASS CTK1 4 x x x x Transkription CTD-Modifizierung FCP1 2 x x Transkription CTD-Modifizierung SSN3 2 x x Transkription CTD-Modifizierung CTK2 1 x Transkription CTD-Modifizierung CTK3 1 x Transkription CTD-Modifizierung SSN8 1 x Transkription CTD-Modifizierung ELP3 5 x x x x x Transkription Elongator ELP4 5 x x x x x Transkription Elongator ELP6 5 x x x x x Transkription Elongator ELP2 4 x x x x Transkription Elongator IKI3 4 x x x x Transkription Elongator SRB5 2 x x Transkription Mediator MED1 1 x Transkription Mediator SRB2 1 x Transkription Mediator VID21 5 x x x x x Transkription NuA4 (HAT) YNG2 4 x x x x Transkription NuA4 (HAT) YAF9 4 x x x x Transkription NuA4 (HAT) YNG2 3 x x x Transkription NuA4 (HAT)
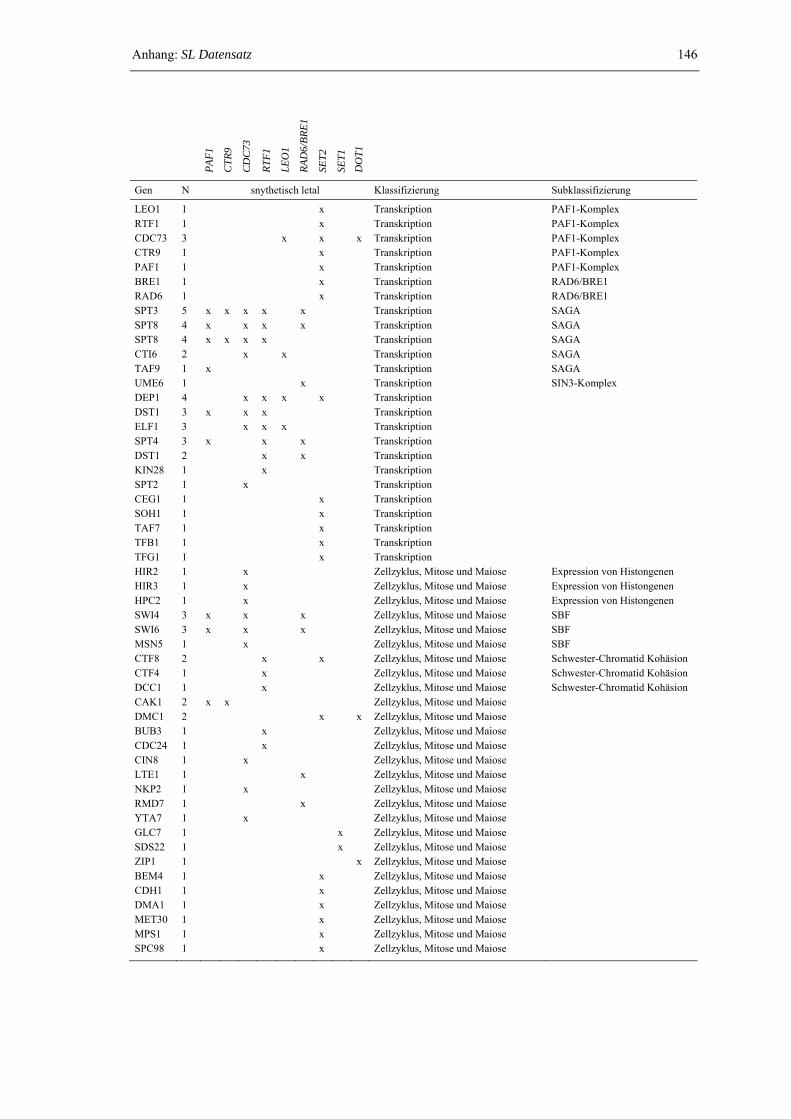
Anhang: SL Datensatz
146
PAF1
CTR
9
CD
C73
RTF1
LEO
1
RAD
6/BR
E1
SET2
SET1
DO
T1
Gen N snythetisch letal Klassifizierung Subklassifizierung
LEO1 1 x Transkription PAF1-Komplex RTF1 1 x Transkription PAF1-Komplex CDC73 3 x x x Transkription PAF1-Komplex CTR9 1 x Transkription PAF1-Komplex PAF1 1 x Transkription PAF1-Komplex BRE1 1 x Transkription RAD6/BRE1 RAD6 1 x Transkription RAD6/BRE1 SPT3 5 x x x x x Transkription SAGA SPT8 4 x x x x Transkription SAGA SPT8 4 x x x x Transkription SAGA CTI6 2 x x Transkription SAGA TAF9 1 x Transkription SAGA UME6 1 x Transkription SIN3-Komplex DEP1 4 x x x x Transkription DST1 3 x x x Transkription ELF1 3 x x x Transkription SPT4 3 x x x Transkription DST1 2 x x Transkription KIN28 1 x Transkription SPT2 1 x Transkription CEG1 1 x Transkription SOH1 1 x Transkription TAF7 1 x Transkription TFB1 1 x Transkription TFG1 1 x Transkription HIR2 1 x Zellzyklus, Mitose und Maiose Expression von Histongenen HIR3 1 x Zellzyklus, Mitose und Maiose Expression von Histongenen HPC2 1 x Zellzyklus, Mitose und Maiose Expression von Histongenen SWI4 3 x x x Zellzyklus, Mitose und Maiose SBF SWI6 3 x x x Zellzyklus, Mitose und Maiose SBF MSN5 1 x Zellzyklus, Mitose und Maiose SBF CTF8 2 x x Zellzyklus, Mitose und Maiose Schwester-Chromatid Kohäsion CTF4 1 x Zellzyklus, Mitose und Maiose Schwester-Chromatid Kohäsion DCC1 1 x Zellzyklus, Mitose und Maiose Schwester-Chromatid Kohäsion CAK1 2 x x Zellzyklus, Mitose und Maiose DMC1 2 x x Zellzyklus, Mitose und Maiose BUB3 1 x Zellzyklus, Mitose und Maiose CDC24 1 x Zellzyklus, Mitose und Maiose CIN8 1 x Zellzyklus, Mitose und Maiose LTE1 1 x Zellzyklus, Mitose und Maiose NKP2 1 x Zellzyklus, Mitose und Maiose RMD7 1 x Zellzyklus, Mitose und Maiose YTA7 1 x Zellzyklus, Mitose und Maiose GLC7 1 x Zellzyklus, Mitose und Maiose SDS22 1 x Zellzyklus, Mitose und Maiose ZIP1 1 x Zellzyklus, Mitose und Maiose BEM4 1 x Zellzyklus, Mitose und Maiose CDH1 1 x Zellzyklus, Mitose und Maiose DMA1 1 x Zellzyklus, Mitose und Maiose MET30 1 x Zellzyklus, Mitose und Maiose MPS1 1 x Zellzyklus, Mitose und Maiose SPC98 1 x Zellzyklus, Mitose und Maiose



















