Charakterisierung Styrol exponierter Arbeitnehmer mittels ...
139
Charakterisierung Styrol exponierter Arbeitnehmer mittels biochemischem Effekt- und Dosismonitoring anhand aromatischer Carbonsäuren, Mercaptursäuren und Hämoglobinaddukten Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der RWTH Aachen University zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigte Dissertation vorgelegt von Diplom-Biologe Marcus Reska aus Geilenkirchen Berichter: Universitätsprofessor Dr. med. Thomas Kraus Universitätsprofessor Dr. rer. nat. Andreas Schäffer Tag der mündlichen Prüfung: 26.09.2011 Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.
Transcript of Charakterisierung Styrol exponierter Arbeitnehmer mittels ...

Charakterisierung Styrol exponierter Arbeitnehmer mittels
biochemischem Effekt- und Dosismonitoring anhand
aromatischer Carbonsäuren,
Mercaptursäuren und Hämoglobinaddukten
Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der
RWTH Aachen University zur Erlangung des
akademischen Grades eines Doktors der Naturwissenschaften genehmigte Dissertation
vorgelegt von
Diplom-Biologe
Marcus Reska
aus Geilenkirchen
Berichter:
Universitätsprofessor Dr. med. Thomas Kraus
Universitätsprofessor Dr. rer. nat. Andreas Schäffer
Tag der mündlichen Prüfung: 26.09.2011
Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.


Widmung
Meinen Eltern


Danksagung
Herrn Prof. Dr. med. Thomas Kraus gilt mein besonderer Dank. Nicht nur für seine
wissenschaftliche Unterstützung, aufmunternden Worte und Betreuung dieser Arbeit,
sondern auch dafür, deren Durchführung am Institut für Arbeitsmedizin und Sozialmedizin
Aachen ermöglicht zu haben.
Ich bedanke mich herzlich bei Herrn Prof. Dr. rer. nat. Andreas Schäffer von der Fakultät
für Mathematik, Informatik und Naturwissenschaften für die Betreuung und die
Begutachtung dieser Arbeit.
Ebenso möchte ich mich bei Herrn Prof. Dr. rer. nat. Hans Toni Ratte bedanken, der sich
bereit erklärt hat, sich als weiterer Prüfer seitens der Fakultät für Mathematik,
Informatik und Naturwissenschaften zur Verfügung zu stellen.
Besonderer Dank gilt auch Herrn Dr. rer. nat. Thomas Schettgen für die thematische
Betreuung dieser Arbeit und der Beratung in allen chemischen und analytischen
Fragestellungen.
Ich möchte mich ebenfalls sehr herzlich bei meinen Kollegen und Freunden Dr. Elke
Ochsmann und Dr. Jessica Lang für die immer willkommenen Ratschläge, Disskussionen
und erheiternden Worte bedanken.
Für ihr immer offenes Ohr, ihre Unterstützung und der Versorgung mit guter Laune im
Labor möchte ich mich bei meinen Kollegen Dr. Angela Tings, Jens Bertram, Petra Dewes,
Kerstin Gerards und Anne Alt bedanken.
Rosi Kohl möchte ich danken für die immer wiederkehrende Versorgung mit Keksen und
allen erdenklichen Dingen um nicht nur im Labor sondern auch im Büro ein „zweites zu
Hause“ vorzufinden.
Ich möchte mich bei allen Mitgliedern des Institutes für Arbeitsmedizin und Sozialmedizin
für die gute und erfahrungsreiche interdisziplinäre Zusammenarbeit bedanken.

Natürlich obliegt ein grosser Dank den kooperierenden Firmen und deren Geschäftsleitung,
die nach längerer Suche gefunden werden konnten und mit der Bereitstellung von
Räumlichkeiten und dem Zugang zu ihren Mitarbeitern unterstützend zur Seite standen.
Ebenfalls grosser Dank an die freiwilligen Probanden, für die Teilnahme an den
Untersuchungen.
Mein Dank auch an meine Freunde am Universitätsklinikum Dr. Marcel Esser und Agnes
Dreier für den institutsübergreifenden fachlichen als auch privaten Austausch.
Ich möchte mich sehr bei meiner guten Freundin Dr. Kristin Michael für private
Unterstützung mittels amerikanischer Leckereien, tollen redsamen Abenden und
motivierenden Worten bedanken.
Besonders danke ich meinen Eltern und dem Rest meiner Familie.
Mein herzlichster Dank an meine Frau Anna. Danke, dass Du da warst und bist.

Die grössten Ereignisse - das sind nicht unsre lautesten, sondern unsre stillsten Stunden
Friedrich Nietzsche


Inhaltsverzeichnis
1 Einleitung ............................................................................................................. 1
2 Grundlagen .......................................................................................................... 3
2.1 Styrol ..................................................................................................................... 3
2.1.1 Stoffeigenschaften ................................................................................................. 3
2.1.2 Entdeckung ............................................................................................................ 4
2.1.3 Belastung und Vorkommen ................................................................................... 4
2.1.4 Metabolismus und Toxikologie ............................................................................. 6
2.1.5 Suszeptibilität ........................................................................................................ 9
2.2 Schadstoffexposition und ihre Erfassung am Arbeitsplatz.................................. 10
2.2.1 Biomarker der Exposition.................................................................................... 10
2.2.2 Monitoring von Gefahrstoffexpositionen ............................................................ 10
2.2.3 Ambient Monitoring ............................................................................................ 11
2.2.4 Dosismonitoring .................................................................................................. 12
2.2.5 Biochemisches Effektmonitoring ........................................................................ 14
2.2.6 Biologisches Effektmonitoring............................................................................ 15
2.2.6.1 Mercaptursäuren als Dosismarker der inneren Exposition .............................. 15
2.2.6.2 Addukte als biochemische Effektmarker ......................................................... 16
Hämglobinaddukte als Langzeitmarker ........................................................... 17
Humanes Hämoglobin ..................................................................................... 18
Entstehung eines Hämoglobinaddukts ............................................................. 19
2.2.7 Cotinin als Marker des Raucherstatus ................................................................. 22
3 Zielsetzung der Arbeit....................................................................................... 23
4 Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin ...................... 25
4.1 Grundlage des Verfahrens ................................................................................... 25
4.2 Geräte, Chemikalien, Lösungen .......................................................................... 25
4.2.1 Geräte................................................................................................................... 25
4.2.2 Chemikalien......................................................................................................... 26
4.2.3 Lösungen ............................................................................................................. 27
4.3 Probennahme und Probenaufbereitung................................................................ 29
4.3.1 Probenaufbereitung des Urins ............................................................................. 29
4.4 Instrumentelle Arbeitsbedingungen..................................................................... 30
4.4.1 Hochleistungschromatographische Arbeitsbedingungen .................................... 30
4.4.2 Identifizierung der Analyte.................................................................................. 32

Inhaltsverzeichnis
4.4.3 Massenspektrometrische Arbeitsbedingungen .................................................... 33
4.5 Kalibrierung und Ermittlung der Analyseergebnisse .......................................... 34
4.6 Standardisierung und Qualitätssicherung ............................................................ 35
4.7 Beurteilung des Verfahrens ................................................................................. 35
4.7.1 Präzision in Serie und von Tag zu Tag................................................................ 35
4.7.2 Richtigkeit ........................................................................................................... 36
4.7.3 Frier-Auftau-Stabilität der Phenylhydroxyethyl Mercaptursäuren...................... 38
4.7.4 Methodenspezifische Störeinflüsse ..................................................................... 39
4.8 Applikabilität der Methode.................................................................................. 40
4.9 Diskussion der Methode ...................................................................................... 42
5 Analytik der Hämoglobinaddukte im Blut...................................................... 43
5.1 Grundlage des Verfahrens............................................................................................... 43
5.2 Geräte, Chemikalien und Lösungen................................................................................ 44
5.2.1 Geräte.............................................................................................................................. 44
5.2.2 Chemikalien .................................................................................................................... 45
5.2.3 Lösungen......................................................................................................................... 45
5.3 Probennahme und Probenaufbereitung ........................................................................... 48
5.3.1 Gewinnung des Erythrozythämolysats ........................................................................... 48
5.3.2 Globinisolierung ............................................................................................................. 48
5.3.3 Herstellung der Kalibrierstandards ................................................................................. 49
5.3.4 Derivatisierung des Globins............................................................................................ 50
5.3.5 Acetylierung des Pentafluorphenylthiohydantoin-Derivates .......................................... 50
5.4 Gaschromatographische Arbeitsbedingungen................................................................. 52
5.5 Analytische Bestimmung................................................................................................ 53
5.6 Kalibrierung .................................................................................................................... 57
5.7 Berechnung der Analyseergebnisse ................................................................................ 59
5.8 Standardisierung und Qualitätssicherung ....................................................................... 59
5.9 Beurteilung des Verfahrens............................................................................................. 59
5.9.1 Präzision in Serie und von Tag zu Tag ........................................................................... 59
5.9.2 Richtigkeit....................................................................................................................... 61
5.9.3 Methodenspezifische Störeinflüsse................................................................................. 62
5.10 Diskussion der Methode ................................................................................................. 64

Inhaltsverzeichnis
6 Analytik der aromatischen Carbonsäuren...................................................... 65
6.1 Grundlage des Verfahrens............................................................................................... 65
6.2 Geräte, Chemikalien und Lösungen................................................................................ 65
6.3 Geräte.............................................................................................................................. 65
6.4 Chemikalien .................................................................................................................... 66
6.5 Lösungen......................................................................................................................... 67
6.6 Probennahme und Probenaufarbeitung ........................................................................... 68
6.6.1 Probenaufbereitung......................................................................................................... 68
6.7 Gaschromatographische Arbeitsbedingungen................................................................. 69
6.8 Analytische Betimmung ................................................................................................. 70
6.9 Kalibrierung und Berechnung der Analyseergebnisse.................................................... 70
7 Humanbiomonitoring belasteter Personengruppen und der
Allgemeinbevölkerung....................................................................................... 73
7.1 Kollektivbeschreibung .................................................................................................... 73
7.1.1 Beruflich belastete Personengruppen.............................................................................. 73
7.1.2 Kontrollkollektiv der Allgemeinbevölkerung................................................................. 74
7.1.3 Auswertung..................................................................................................................... 74
7.2 Ergebnisse des Humanbiomonitorings ........................................................................... 76
8 Diskussion des Humanbiomonitorings ............................................................ 94
9 Zusammenfassung ........................................................................................... 101
10 Abbildungsverzeichnis .................................................................................... 105
11 Tabellenverzeichnis ......................................................................................... 109
12 Literaturverzeichnis ........................................................................................ 111
13 Anhang.............................................................................................................. 121
13.1 Information und Einverständniserklärung für Probanden............................................. 121
13.2 Fragebogen für Probanden............................................................................................ 123
13.3 Veröffentlichungen ....................................................................................................... 124

Abkürzungsverzeichnis
Abkürzungsverzeichnis 4-ViP 4-Vinylphenol
3,4 SO Styrol 3,4-oxid
% Prozent
µg Mikrogramm
µl Mikroliter
Abb. Abbildung
AGW Arbeitsplatzgrenzwert
AUC area under curve
BAR Biologischer Arbeitsstoff-Referenzwert
BAT Biologischer Arbeitsstoff-Toleranzwert
BER Basenexcisionsreparatur
BGW Biologischer Grenzwert
BLW Biologischer Leitwert
BMAS Bundesministerium für Arbeit und Soziales
bspw. beispielsweise
bzw. beziehungsweise
ChemG Chemikaliengesetz
cm Zentimeter
CYP P450 Cytochrom P450 Monooxygenasen
DE Deutschland
DFG Deutsche Forschungsgesellschaft
DGAUM Deutschen Gesellschaft für Arbeitsmedizin und Umweltmedizin
DNA Desoxyribonucleinsäure (engl.: deoxyribonucleicacid)
DNA-ssb DNA Einzelstrangbrüche (engl. DNA single strand breaks)
EI electron impact
EKA Expositionsäquivalente für krebserzeugende Arbeitsstoffe
EPHX1 Epoxidhydrolase 1
ERCC Gen des DNA Reparatursystems
(engl. excision repair cross-complementing)
Fa Firma
FDA Food and Drug Administration
FPB(-G) 4-Fluoryphenetylbromid (-Globin)

Abkürzungsverzeichnis
g Gramm
GC Gaschromatographie
GefStoffV Gefahrstoffverordnung
GfPI Glasfaserverstärkende Plastikindustrie
Glu Glutamat
Gly Glycin
GSH Glutathion
GST Glutathion-S-Transferase
Hb Hämoglobin
HbA Hämoglobinaddukt
Hb-Val Valin Addukt des Hämoglobins
HPB(-G) 4-Hydroxyphenetylbromid (-Globin)
HPV 2-Hydrox-2-Phenylethyl-Valin
HPLC Hochleistungsflüssigkeitschromatographie
(engl.: high performance liquid chromatography
HPRT Hypoxanthin-Guanin-Phosphoribosyltransferase
i.d.R. in der Regel
IARC International Agency for Research on Cancer
IFCC International Federation of Clinical Chemistry
ISTD interner Standard
kDa Kilodalton
kg Kilogramm
Kon Kontrollen
l liquid
L Liter
LC/MS/MS Tandem-Massenspektrometrie
m/z Masse-zu-Ladung-Verhältnis
MA Mandelsäure (engl.: mandelic acid)
MAK maximale Arbeitsplatzkonzentration
mg Milligramm
min Minute(n)
ml Milliliter
MSD massenselektiver Detektor
NATs N-Acethyltransferasen

Abkürzungsverzeichnis
NER Nukleotidexcisionsreparatur
nm Nanometer
NS Nachschicht
OSHA Occupational Safety and Health Administration
PE Phenoxyethanol
PFPITC Pentafluorphenylisothiocyanat
PFPTH Pentafluorphenylthiohydantoin
PGA Phenylglyoxylsäure (engl.: phenylglyoxylic acid)
PHEMA Phenylhydroxyethylmercaptursäuren
(engl.: phenylhydroxyethylmercapturic acids)
pmol Picomol
ppm parts per million
RT Raumtemperatur
R(t) Retentionszeit
RAM Restricted Access Material
RT Raumtemperatur
S Styrol
s solid
s. siehe
s.u. siehe unten
SIM single ion monitoring
SG Styrolglykol
SO Styrol 7, 8-Oxid
t Zeit
Tab. Tabelle
TRGS Technische Richtlinien für Gefahrstoffe
TRK technische Richtkonzentration
vgl. vergleichend
Vol% Volumenprozent
vs versus
VS Vorschicht
WFR Wiederfindungsrate
XRCC Gen des DNA Reparatursystems
(engl. X-ray repair cross-complementing)

Einleitung 1
1 Einleitung
Die meiste Zeit des Tages verbringt der Mensch an seinem Arbeitsplatz. Hier sollte er
Bedingungen vorfinden, in denen auf ihn bezogene Gesundheits- und Sicherheitsrisiken
kontrolliert und bestmöglich eliminiert werden. Die praktische Umsetzung stellt, in diesem
Fall an den Arbeitgeber, höchste Anforderungen, denn die Gestaltung des Arbeitsplatzes,
ist vielschichtig. Komplex sind zahlreiche Gesetzestexte, Richtlinien und Verordnungen
und zu klein sind oft die finanziellen Mittel bspw. für neuere Gerätschaften oder
Umbaumassnahmen. Notwendiges medizinisches, biochemisches und technisches
Fachwissen ist darüber hinaus beim Arbeitgeber meist nicht vorhanden. Diesen Aufgaben
unter Berücksichtigung aller Gesundheits- und Sicherheitsaspekte gerecht zu werden, ist
daher Ziel der Arbeitsmedizin. Sie ist Schnittstelle zwischen Betrieb und Individuum mit
Ihrem zentralen Schwerpunkt der Verhältnis- und Verhaltensprävention.
Gerade wenn im Betrieb die Exposition und die Gesundheitsgefährdung überprüft werden
soll, stellt die Arbeitsmedizin eine wichtige Beratungsstelle für Arbeitgeber und
Arbeitnehmer dar.
Gefahrstoffe sind allgegenwärtig. Wir nehmen sie in Form des kanzerogenen Acrylamids
mit der Nahrung oder über den Zigarettenrauch auf (Schettgen et al, 2002), sind u.a. in
vielen alten Gebäuden einer kontinuierlichen Belastung mit polychlorierten Biphenylen
ausgesetzt und müssen uns Gedanken machen, ob die Nanotechnologie revolutionierenden
carbon-nanotubes mit ihrem asbestähnlichen Pathomechanismus die Mesotheliom-
inzidenzen des 21. Jahrhundert bestimmen werden (Poland et al, 2008). Für etwa 15% der
europäischen Arbeitnehmer ist der direkte Umgang mit Gefahrstoffen Teil ihrer täglichen
Arbeit. In der EU kommt es jährlich zu geschätzten 167.000 berufsbedingten Todesfällen,
von denen ~ 44% auf den Gebrauch und die Nutzung von Gefahrstoffen zurückzuführen
sind (OSHA, 2009). Hier besteht grosses Potential für Prävention.
Im Teilgebiet der klinisch orientierten arbeitsmedizinischen Toxikologie sind
Präventionsmassnahmen und Forschungsansätze vereint. Wissenschaftlich bedient sie sich
biochemischen, toxikologischen und molekularbiologischen Methoden. Zur Ermittlung und
Risikoabschätzung von Gefahrstoffexpositionen am Arbeitsplatz dienen das Instrument des
Biological- und Ambientmonitorings sowie das gesetzliche Regelwerk des
Chemikaliengesetzes (ChemG), der Gefahrstoffverordnung (GefstoffV) und der
technischen Regeln für Gefahrstoffe (TRGS), sowie der Verordnung zur

Einleitung 2
arbeitsmedizinischen Vorsorge (ArbMedVV) Die grösste Bedeutung, in Hinblick auf
Gesundheitsgefährdungen am Arbeitsplatz, kommt den Substanzen zu, die mit dem Erbgut
in Wechselwirkung treten und somit als potentielle Karzinogene eine erhebliche Gefahr
darstellen können. Der Nachweis verschiedener Metabolite, die unterschiedlichen
Abbauwegen eines Gefahrstoffes repräsentieren, zur Bestimmung der inneren individuellen
Dosis liefert für die Risikobewertung beim Umgang mit bestimmten Substanzen ein
umfassenderes Bild belasteter Personen an ihrem Arbeitsplatz und erlaubt z.T. eine
spezifischere Risikoabschätzung.
Das Ziel der aktuellen Forschung im Bereich des Biomonitorings muss es daher sein, durch
ein kontinuierliches Voranschreiten der biomedizinischen Anwendungs- und
Grundlagenforschung (bspw. die Identifizierung neuer/alternativer Abbauwege) eine
stetige Verbesserung des präventiven Arbeitsschutzes zu gewährleisten.
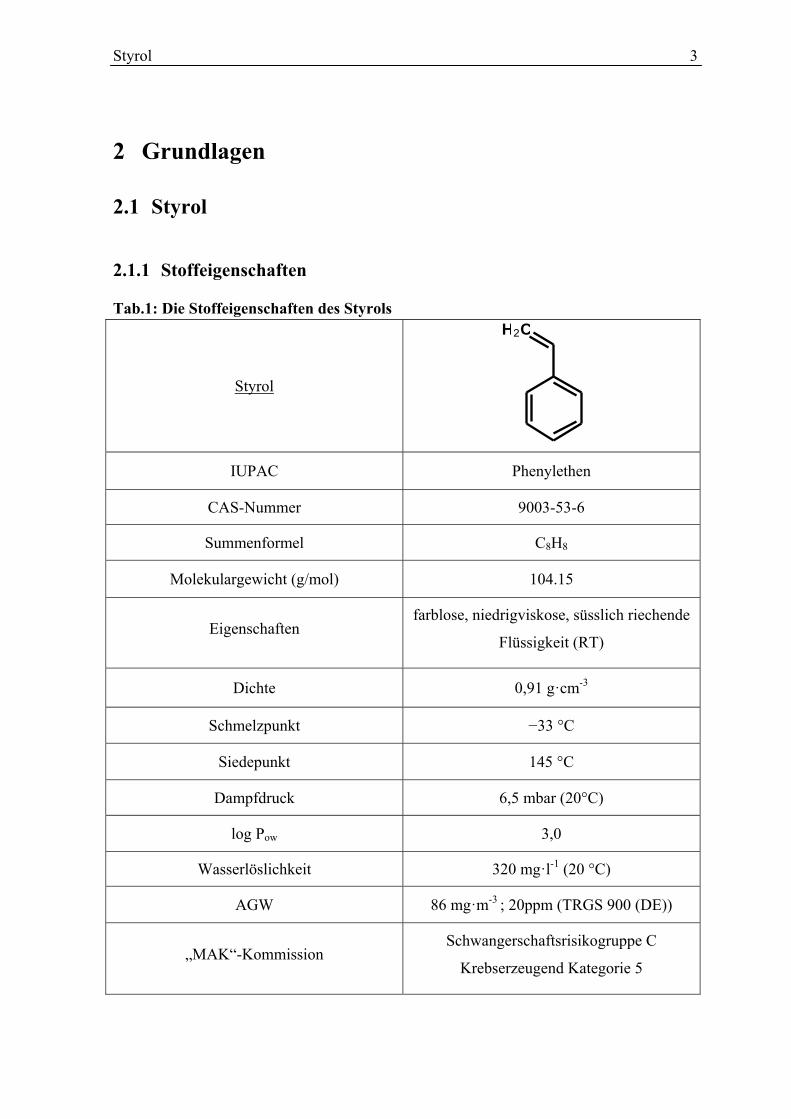
Styrol 3
2 Grundlagen
2.1 Styrol
2.1.1 Stoffeigenschaften
Tab.1: Die Stoffeigenschaften des Styrols
Styrol
IUPAC Phenylethen
CAS-Nummer 9003-53-6
Summenformel C8H8
Molekulargewicht (g/mol) 104.15
Eigenschaften farblose, niedrigviskose, süsslich riechende
Flüssigkeit (RT)
Dichte 0,91 g·cm-3
Schmelzpunkt −33 °C
Siedepunkt 145 °C
Dampfdruck 6,5 mbar (20°C)
log Pow 3,0
Wasserlöslichkeit 320 mg·l-1 (20 °C)
AGW 86 mg·m-3 ; 20ppm (TRGS 900 (DE))
„MAK“-Kommission Schwangerschaftsrisikogruppe C
Krebserzeugend Kategorie 5
CH2

Styrol 4
2.1.2 Entdeckung
Styrol (s. Tab.1) wurde 1839 durch den Berliner Pharmazeuten Eduard Simon zufällig
entdeckt, als er aus einem Pflanzenextrakt des orientalischen Amberbaums durch
Wasserdampfdestillation eine klare, ölige und farblose Flüssigkeit gewann. Diese von ihm
als Styrol benannte Flüssigkeit entwickelte nach Erhitzen einige Tage später eine gelartige
Konsistenz (Scheirs, 2003) – eine Reaktion, die viele Jahre später die Basis für die
Kunststoffauskleidung in Autos dienen, Bootrümpfe eine hohe Wiederstandsfähigkeit
verleihen und als Hauptbestandteil in Spielzeug (bspw. Playmobil®) Generationen von
Kindern erfreuen sollte. 1922 beschrieb Hermann Staudinger erstmals Simons Entdeckung
als Polymerisationsprozess zu Polystyrol und erhielt 1953 für seine Arbeit zu Polymeren
den Nobelpreis (Staudinger, 1953). Staudingers Entdeckung revolutionierte die gesamte
Kunststoffindustrie.
2.1.3 Belastung und Vorkommen
Der umweltbedingte Kontakt der Allgemeinbevölkerung mit Styrol kommt meist durch
Inhalation und über die Nahrung zustande. Schätzungen ergaben, dass hierbei über die
Nahrung Mengen zwischen 0,3-0,8 µg/kg Körpergewicht aufgenommen werden. Bei einer
Studie der Food and Drug Administration (FDA) konnten bei 49 von 320 untersuchten
Lebensmitteln Grössenordnungen von Styrol zwischen 10 µg/kg (bei Eiern) bis hin zu 274
µg/kg (bei Erdbeeren) bestimmt werden (IARC, 2002). Steele et al. konnten die grössten
Mengen Styrol in indonesischem Zimt finden (39,2 mg/kg). Die Gründe für Styrol in der
Nahrung können speziell bei Zimt auf metabolische Effekte (Steele, 1994) oder
Mikroorganismen (Lafeuille et al, 2009) zurückgeführt werden. Gerade bei kleinen
Mengen kann jedoch auch eine Migration von Styrol aus polymerhaltigen
Lebensmittelverpackungen als Grund erachtet werden (Tang et al, 2000).
Die inhalative Aufnahme von Styrol geschieht meist durch Emissionen industrieller
Styrolverarbeitungen, Autoabgasen oder Zigarettenrauch (Adam et al, 2009; IARC, 2002).
Es konnte gezeigt werden, dass Tabakrauch schätzungsweise 0,2-7,2 µg Styrol pro
Zigarette enthält (Darrall et al, 1998). In einer US amerikanischen Befragung zur
Exposition von Kindern durch flüchtige organische Substanzen wurden
Innenraumvorkommen von Styrol mit Durchschnittswerten von 1,2 µg/m³ und
Aussenbereichvorkommen von 0,5 µg/m³ angegeben (Adgate et al, 2004).

Styrol 5
Die industrielle Hauptanwendung von Styrol liegt in der Polystyrolherstellung,
Plastikverarbeitung, Fiberglas-Produktion als auch in der Gummiproduktion sowie bei
Laminierarbeiten und der Herstellung von Isolierstoffen (Rybak, 1992). Zu den durch
Styrol erzeugten Produkten zählen Verpackungsmaterialien (Schaumstoff, Füllmittel),
Konstruktionselemente (Rohre, Formstücke, korrosionsgeschützte Materialien),
Fahrzeugbestandteile (Autoreifen) und Haushaltswaren (Fussbodenbeläge,
Kunststoffschalen hitzebeständiger Küchengeräte). Styrol wird meist zu Polystyrolgranulat
verarbeitet. Dies ist kompatibel mit einer Vielzahl von Farbstoffen und stellt den Rohling
für die weitere Verarbeitung dar. Darüber hinaus werden auch Granulate hergestellt, in
denen Styrol als Copolymer mit anderen Polymeren vorliegt (Acrylnitril-Butadien-Styrol,
Styrol-Acrylnitril) und dem Material je nach Anwendung zusätzliche Eigenschaften
verleiht (Transparenz, Hitzebeständigkeit, Stossfestigkeit). Die vielfältige Verwendung
erhöht gleichfalls die Wahrscheinlichkeit von hohen Belastungen und gesundheitlichen
Gefährdungen beim Menschen am Arbeitsplatz. Dabei treten die grössten Expositionen in
der Produktion von glasfaserverstärktem Plastiks (GfP) und bei Styrol basierten Laminier-
und Lackierarbeiten auf. Allein im Jahr 1994 wurden weltweit 5,1 Millionen Tonnen
Styrol produziert (WHO, 2000).

Styrol 6
2.1.4 Metabolismus und Toxikologie
Die Aufnahme von Styrol in den Körper geschieht oral, inhalativ und dermal. Die Mengen
des dermal aufgenommenen Styrols können jedoch als nicht signifikant angesehen werden
(Brooks et al, 1980). Eine Schlüsselfunktion und gleichzeitig der initiale Schritt der
Biotransformation kommt der Proteinsuperfamilie der ubiquitären Cytochrom P450
Monooxygenasen (CYP P450) zu, die sich in der Phospholipidmatrix des
endoplasmatischen Retikulums eukaryotischer Zellen befinden. Diese Enzyme gehören zu
den Hämproteinen, die im menschlichen Genom von mindestens 50 P450-Genen codiert
werden und sich in 10 Familien aufspalten (Laffon et al, 2003). Bedeutend für den
humanen Styrolmetabolismus sind CYP2B6 und CYP2E1 in der Leber sowie CYP1A1,
CYP2F1, CYP2B1 und CYP2B6 in der Lunge (Nakajima et al, 1994). Die Epoxidierung
des Styrols durch Cytochrom P450 führt in geringen Mengen zu Styrol 3,4-oxid und weiter
zu 4-Vinylphenol, dem kürzlich entdeckten 2-,3- Venylphenol (Linhart et al, 2010) und
hauptsächlich zum sehr reaktiven Styrol 7,8-oxid (SO) welches in der Folge, überwiegend
über die mikrosomale Epoxidhydrolase EPHX1, zu Styrolglykol (SG) hydrolysiert wird.
Abschliessend entstehen Mandelsäure (MA) und Phenylglyoxylsäure (PGA) durch
Oxidation mittels Aldehyd- und Alkoholdehydrogenase und repräsentieren dabei 95% der
sich im Urin befindlichen Styrolmetabolite. Ein weiterer Stoffwechselweg ist die
Konjugierung von SO am reaktiven Epoxidring mit Glutathion (GSH), katalysiert durch
die Gluthation-S-Transferase (GST) zu den Glutathionkonjugaten GSH Konjugat 1 und
GSH Konjungat 2. Die Phenylhydroxyethylmercaptursäuren (PHEMA) N-Acetyl-S-(2-
Hydroxy-1-Phenylethyl)-Cystein (PHEMA1) und N-Acetyl-S-(2-Hydroxy-2-Phenylethyl)-
Cystein (PHEMA2) stellen die Endmetaboliten der zuvor durch γ-Glutamyltranspeptidase,
Cystein-Glycin-Dipeptidase und der N-Acetyl-S-Transferase katalysierten Reaktionen am
Glutathionkojugat dar (Sumner & Fennell, 1994). Ein weiterer reaktiver Schritt zwischen
SO und Hämoglobin führt zum Hämoglobinaddukt N-(2-Hydroxy-2-Phenylethyl)-Valin
(HPV) und N-(2-Hydroxy-1-Phenylethyl)-Valin (Abb.1).
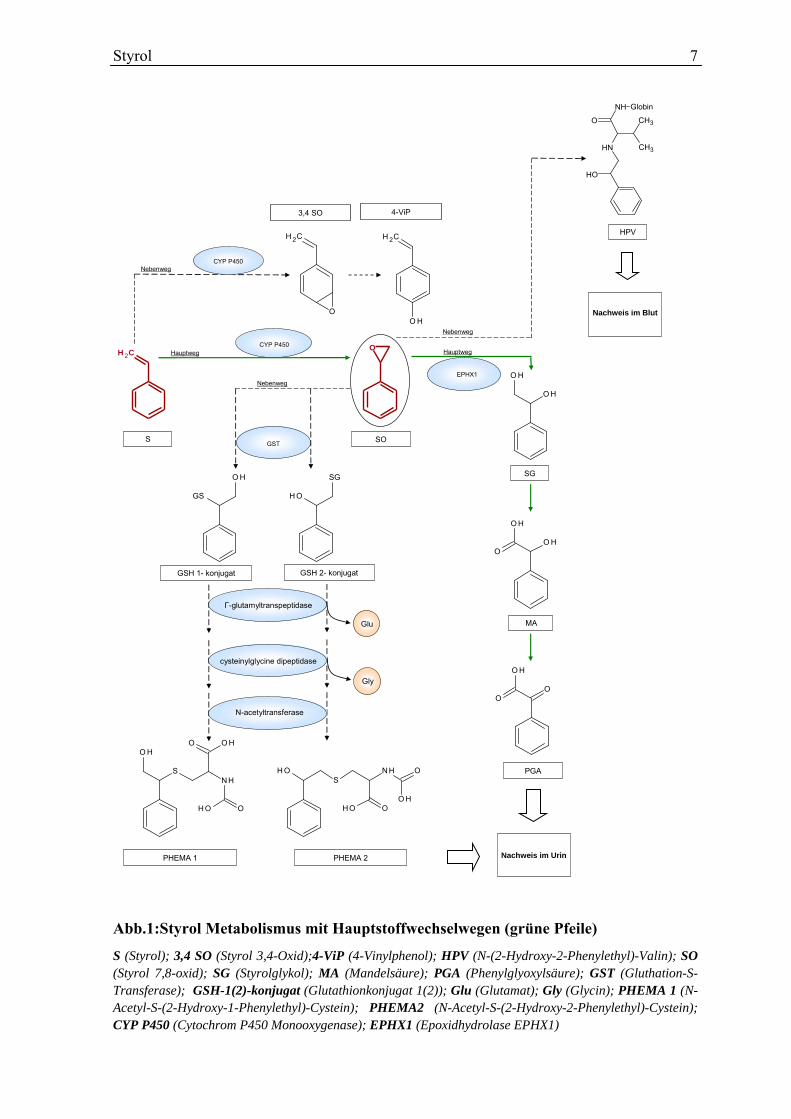
Styrol 7
CH 2
S
O
SO
SG
MA
O H
O H
O
O H
O H
O
O H
O
PGA
GSH 1- konjugat GSH 2- konjugat
OH
SG
GS
O H
OH O
N H
O O H
S
O H
PHEMA 1 PHEMA 2
O H
ON H
OOH
SOH
Γ-glutamyltranspeptidase
cysteinylglycine dipeptidase
N-acetyltransferase
GST
CYP P450Hauptweg
Nebenweg
Glu
CH 2
OO H
CH 2
3,4 SO 4-ViP
CYP P450Nebenweg
Gly
Nachweis im Urin
Hauptweg
NH
O CH3
CH3NH
OH
Globin
HPV
Nebenweg
Nachweis im Blut
EPHX1
Abb.1:Styrol Metabolismus mit Hauptstoffwechselwegen (grüne Pfeile)
S (Styrol); 3,4 SO (Styrol 3,4-Oxid);4-ViP (4-Vinylphenol); HPV (N-(2-Hydroxy-2-Phenylethyl)-Valin); SO (Styrol 7,8-oxid); SG (Styrolglykol); MA (Mandelsäure); PGA (Phenylglyoxylsäure); GST (Gluthation-S-Transferase); GSH-1(2)-konjugat (Glutathionkonjugat 1(2)); Glu (Glutamat); Gly (Glycin); PHEMA 1 (N-Acetyl-S-(2-Hydroxy-1-Phenylethyl)-Cystein); PHEMA2 (N-Acetyl-S-(2-Hydroxy-2-Phenylethyl)-Cystein); CYP P450 (Cytochrom P450 Monooxygenase); EPHX1 (Epoxidhydrolase EPHX1)

Styrol 8
Die Auswirkungen von Styrolexpositionen beinhalten mannigfaltige, toxikologische
Effekte. So kann eine Belastung zu Augen- Rachen- und Atemwegsreizungen führen.
Beeinträchtigende Effekte des zentralen und peripheren Nervensystems in Form
dopaminerger, psychischer und funktionaler Anomalien konnten bei Konzentrationen ab
100 parts per million (ppm) beobachtet werden (IARC, 2002). Störungen des Farbsehens
wurden ebenfalls auf styrolbedingten neuronalen (Campagna et al, 1996; Gobba, 2000)
aber reversiblen (Triebig et al, 2001) Funktionsverlust zurückgeführt. Unterschiedliche
Autoren untersuchten die Ototoxizität von Styrol (Morioka et al, 1999; Sliwinska-
Kowalska et al, 2003) und beschreiben im Rattenversuch einen grösseren Verlust der
Cochleafunktion und der Haarsinneszellen bei kombinierter Exposition von Lärm und
Styrol als bei Einzelexposition gegenüber einem der beiden Faktoren (Chen & Henderson,
2009; Lataye et al, 2000).
In Vitro (Bastlova et al, 1995; Dypbukt et al, 1992; Fracasso et al, 2009) als auch in vivo
Versuche (Maki-Paakkanen et al, 1991) zeigten, dass Styrol und sein reaktiver Metabolit
Styrol 7,8-Oxid (SO) DNA-Einzelstrangbrüche und Doppelstrangbrüche hervorrufen kann
und somit Einfluss auf die Differenzierungsfähigkeit von Zellen hat. Durch die
Behandlung mit SO konnte eine signifikante Erhöhung der Mutationsrate der
Hypoxanthin-Guanin-Phosphoribosyltransferase (HPRT) humaner T-Zellen herbeigeführt
werden (Bastlova et al, 1995). In Mäusestudien wurde ein erhöhtes Auftreten von
Lungenadenomen und Karzinomen beobachtet: Weiblichen, trächtigen O20-Mäusen wurde
vom 17. Trächtigkeitstag an wöchentlich 1,35 g/kg Styrol zugeführt. Die Jungtiere
erhielten diese Dosis ab dem Geburtszeitpunkt weiterhin für über 100 Wochen
(Ponomarkov & Tomatis, 1978). Vermehrtes Auftreten von Lungenkarzinomen und
bronchoalveolären Adenomen wurde auch bei Styrol exponierten CD-10 Mäusen
beobachtet (Cruzan et al, 2001). Epidemiologische Studien zum Einfluss von Styrol
bedingten Krebsentstehungen bei Arbeitern aus Firmen, in denen glasfaserverstärktes
Plastik verarbeitet und produziert wird, konnten bisher keinen klaren Beweis für einen
kausalen Zusammenhang zwischen Styrol und dem Auftreten von Krebs erbringen. So
konnten Kogevinas et al. in einer Studie an 40.000 Arbeitern aus 8 Ländern Europas keine
Korrelation zwischen Exposition und erhöhtem Neoplasien-bedingtem Sterblichkeitsrisiko
finden. Zu ähnlichen Ergebnissen kamen Wong et al., bei denen bei einer Untersuchung
von 15.826 Arbeitern keine Korrelation zwischen Styrolexposition und Sterberate zu
finden war. Vielmehr gab es die grössten Sterblichkeiten bei den niedrig exponierten

Styrol 9
Arbeitern, was in der Folge nicht mit Styrol in Verbindung gebracht werden konnte
(Kogevinas et al, 1994; Wong et al, 1994).
Trotz des Mangels eindeutiger Belege für einen direkten Zusammenhang zwischen einer
Styrolexposition und der Inzidenz von Krebs beim Menschen, stufte die International
Agency for Research on Cancer (IARC) auf Basis des (1) Auftretens von Styrol bedingten
malignen Lungenerkrankungen bei Mäusen, (2) dem kanzerogenen Potential des SO im
Tierversuch und (3) dem Auftreten von DNA Addukten nach Exposition beim Menschen
Styrol als möglicherweise (Gruppe 2B) humankanzerogen und seinen Metaboliten SO als
wahrscheinlich (Gruppe 2A) humankanzerogen ein (IARC, 1994; IARC, 2002).
2.1.5 Suszeptibilität
Die Gesundheitsrisiken hängen nicht nur von der Häufigkeit und Konzentration einer
Umwelt- bzw. Arbeitsplatzexposition ab, sondern auch von den Charakteristika eines
Gefahrstoffes individuell genotoxisch zu wirken. Genetische Prädispositionen, die sich in
Form unterschiedlicher Polymorphismen zeigen, können Auwirkung auf die spätere
Funktion eines Proteins oder Enzyms haben und deren Funktion nachteilig beeinflussen
(z.B. Enzyme, die verantwortlich für die Aktivierung und Detoxifizierung von
Schadstoffen oder der DNA Reparatur sind) (Godderis et al, 2006; Laffon et al, 2003).
Relevante Polymorphismen sind bspw. Gene der Gluthation-S-Transferasen (GSTs), N-
Acetyltransferasen (NATs), Cytochrom P450 Monooxygenasen (CYPs) (Raunio et al,
1995) als auch Gene der Basenexcisions- (BER) und Nukleotid-Exzisions-Reparatur
(NER) XRCC (X-ray repair cross-complementing) und ERCC (Excision repair cross-
complementing) (Duell et al, 2000).

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 10
2.2 Schadstoffexposition und ihre Erfassung am Arbeitsplatz
2.2.1 Biomarker der Exposition
Messungen von Änderungen spezifischer Parameter (Biomarker) im biologischen System
eines Organismus als Indikator einer bestimmten Krankheit (bspw. Kreatinkinasen oder
Troponine bei einem Herzinfarkt) haben eine lange Vergangenheit. Im Gegensatz dazu ist
die Messung und Bestimmung von biologischen Markern einer Gefahrstoffexposition oder
die durch aufgenommene Chemikalien verursachte Ermittlung toxikologischer Effekte ein
relativ neuer Ansatz in der Medizin. Diese, als „toxikologische Biomarker“ bezeichneten
Parameter, haben ihren Ursprung in der arbeitsmedizinischen Forschung und der
forensischen Toxikologie. Innerhalb der toxikologischen Biomarker wird zwischen den
Expositionsbiomarkern, den Effektbiomarkern und den Prädispositionsbiomarkern (auch
Suszeptibilitätbiomarkern) unterschieden. Als Expositionsbiomarker gelten Chemikalien,
ihre Metabolite oder das Produkt aus einer Reaktion zwischen Molekül oder
Makromolekül und einer Chemikalie. Der Effektbiomarker beschreibt hingegen eine
strukturelle oder funktionale Veränderung im Organismus, die mit einer spezifischen
gesundheitlichen Beeinträchtigung bzw. Krankheit in Verbindung gebracht werden kann.
Die Bezeichnung des Prädispositionsbiomarkers beinhaltet alle genetischen Faktoren, die
für eine individuelle Suszeptibilität verantwortlich sind und bestimmt werden können.
Trotz des Versuchs verschiedene Biomarker zu definieren, ist es nicht immer möglich sie
einer bestimmten Kategorie zuzuordnen. Für eine Definition ist vielmehr der gesamte
Kontext, in dem der zu untersuchende Marker steht, zu beurteilen. So stellen bspw.
Benzol-Addukte der DNA Expositionsmarker dar. Benzol als genotoxisches kanzerogen
kann jedoch auch als Effektbiomarker gesehen werden.
2.2.2 Monitoring von Gefahrstoffexpositionen
Der Schutz vor Gesundheitsgefahren, insbesondere auch der Schadstoffexpositionen am
Arbeitsplatz ist eines der Hauptziele der Arbeitsmedizin. Als analytische Instrumente um
diese Gefahren frühzeitig aufzudecken und somit präventiv handeln zu können, stellen das
Biological Monitoring und das Ambientmonitoring die Methoden der Wahl dar. Das
Biological Monitoring kann als ein Instrument der individuellen Prävention angesehen
werden. Durch seine Anwendung ist es möglich die Reichweite einer Schadstoffbelastung

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 11
beim Menschen und der daraus folgenden Gesundheitlichen Effekte abzuschätzen (Angerer
& Gundel, 1996; Kommission „Human-Biomonitoring“des Umweltbundesamtes, 1996;
Schaller & Angerer, 1998; Zielhuis, 1980). Unterteilt wird das Biological Monitoring in
das Dosismonitoring, das biochemische Effektmonitoring sowie das biologische
Effektmonitoring. Die prädiktive Bedeutung im Hinblick auf eine gesundheitliche
Risikoabschätzung steigt dabei vom Ambientmonitoring bis zur arbeitsmedizinischen
Vorsorgeuntersuchung stetig an (Abb.2).
Abb.2: Monitoring von Gefahrstoffen in der Arbeits- und Umweltmedizin (Angerer,
2001)
2.2.3 Ambient Monitoring
Das Ambientmonitoring hat die Aufgabe Gefahrstoffe in der Umwelt (Luft, Boden,
Wasser, Lebensmitteln, Staub) zu ermitteln. Dabei gibt es stationäre Messungen, die an
beliebiger Stelle im Raum stattfinden, als auch personengebundene Messungen, die direkt
an der Expositionsquelle bzw. dem direkten Aufnahmeort des Schadstoffes (bspw. orale
Aufnahme) stattfinden. Zur Bestimmung des Schweregrades werden im Ausschuss für
Gefahrstoffe des Bundesministerium für Arbeit und Soziales (BMAS) so genannte
Arbeitsplatzgrenzwerte (AGWs) festgelegt, die anschliessend in den technischen Regeln
für Gefahrstoffe 900 (TRGS 900) veröffentlicht werden. Der AGW definiert sich als „Der
Grenzwert für die zeitlich gewichtete durchschnittliche Konzentration eines Stoffes (mg/m³
und ml/m³ (ppm)) in der Luft am Arbeitsplatz in Bezug auf einen gegebenen
Referenzzeitraum. Er gibt an bei welcher Konzentration eines Stoffes akute oder
chronische schädliche Auswirkungen auf die Gesundheit im Allgemeinen nicht zu erwarten
sind (§ 3 Abs. 6 GefStoffV).“. Die Reformierung der Gefahrstoffverordnung (GefStoffV)

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 12
und die Einführung der AGWs traten am 1. Januar 2005 in Kraft und ersetzten damit die
bis zu diesem Zeitpunkt gültige maximale Arbeitsplatzkonzentration (MAK) und die
technische Richtkonzentration (TRK). Diese beiden Parameter wurden von der
„Senatskommission zur Prüfung gesundheitsschädlicher Arbeitsstoffe“ der Deutschen
Forschungsgesellschaft (DFG) vorgegeben (DFG, 2009).
Innerhalb der TRGS 900 werden Grenzwerte auf Einzelsubstanzen bezogen. Bei
Produktionsarbeiten kommt es allerdings häufiger zu Expositionen gegenüber
Stoffgemischen. Die Richtlinien zum Schutz des Arbeitnehmers und zur
Risikoabschätzung von Stoffgemischen werden den TRGS 403 entnommen. Bei
Substanzen, die noch nicht in die TRGS 900 übertragen wurden, ist zur Bewertung
weiterhin der MAK bzw. der entsprechende TRK-Wert zu verwenden. Diese haben
allerdings keine rechtliche Grundlage mehr. Beim Ambientmonitoring ist es nicht möglich
die eigentliche pathologische, molekulare Wirkung eines Schadstoffes zu analysieren und
zu quantifizieren, denn das bspw. kanzerogene Potential einer Chemikalie kann nicht durch
ihr Vorhandensein in der Raumluft bestimmt werden. Ebenso ist die Ermittlung der
tatsächlichen Stoffmengenkonzentrationen im Körper, die dermal oder inhalativ
aufgenommen wurde, nicht möglich.
2.2.4 Dosismonitoring
Das Dosismonitoring dient dem Nachweis von Schadstoffen oder ihrer Metaboliten in
Körperflüssigkeiten. Als Matrices dienen Vollblut, Plasma, Serum oder Urin. Hierbei wird
die so genannte „innere Dosis“ bestimmt, deren ermittelte Werte bspw. mit Referenzwerten
des Umweltbundesamtes oder dem 2008 von der DFG vorgestellten Biologischen
Arbeitsstoff-Referenzwert (BAR) zu vergleichen sind (DFG, 2009). Beide beruhen auf
Hintergrundbelastung bei Menschen der Allgemeinbevölkerung, die beruflich nicht mit
einer Exposition mit dem jeweiligen zu vergleichenden Stoff in Verbindung stehen. Im
Vergleich mit diesen Referenzwerten für die Allgemeinbevölkerung (definiert über das 95.
Perzentil), können überdurchschnittliche Belastungen und gesundheitliche Risiken durch
zwischenzeitliche Belastungen am Arbeitsplatz mit den zu vergleichenden Noxen
aufgedeckt werden. Folglich wird der Referenzwert für die Allgemeinbevölkerung zur
toxikologischen Begründung abgelehnt. Hintergrundbelastungen treten hier meist durch die
ubiquitären und unvermeidbaren Belastungen mit jeweiligen Noxen der Umwelt auf.
Darüber hinaus können Hintergrundbelastungen durch das Alter, Gewicht oder individuelle

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 13
Lebensgewohnheiten bspw. Rauchen und Alkohol beeinflusst werden. Hier kann der
Referenzwert Hinweise bieten, ob eine eventuelle zusätzliche Belastung gegenüber dem
Schadstoff am Arbeitsplatz oder in der Umwelt vorliegt (Lewalter & Neumann, 1998).
Als Richtlinie zur Bestimmung arbeitsplatzbezogener Konzentrationsüberschreitungen
diente bis zum 1. Januar 2005 der Biologische Arbeitsplatztoleranzwert (BAT) aus dem
sich der seit dem rechtlich verbindliche Biologischen Grenzwert (BGW) (veröffentlicht in
der TRGS 903) ableitet. Er stellt die toxikologisch-arbeitsmedizinisch abgeleitete
Konzentration eines Stoffes, seines Metaboliten oder eines Beanspruchungsindikators im
entsprechenden biologischen Material dar, bei dem im Allgemeinen die Gesundheit eines
Beschäftigten nicht beeinträchtigt wird (§ 3 Abs. 7 GefStoffV). Da die im Bereich der
BAT (BGW)-Werte liegenden, beobachteten Effekte voll reversibel sein müssen, wurden
und werden für kanzerogene Substanzen der Kategorien 1, 2 und 3 keine BAT (BGW)-
Werte erstellt. Eine toxikologisch unbedenkliche Dosis kann hier nicht festgelegt werden.
Anstelle des BAT (BGW) wurden für diese Gruppe und ihren Konzentrationen so genannte
EKAs (Expositionsäquivalente für krebserzeugende Arbeitsstoffe) definiert. Diese wurden
aus zahlreichen Daten zu Luftanalysen und Biomonitoringergebnissen gewonnen, die
gegeneinander korreliert und anschliessend zu einem EKA abgeleitet wurden. Dieser
definierte sich darüber, welche innere Belastung bei ausschliesslich inhalativer
Stoffaufnahme vorliegen würde (Bender, 2008). Als relevanter EKA wurde der Wert
bestimmt, der am ehesten dem jeweiligen TRK entspricht. So gibt es bspw. für einige
kanzerogene Expositionsäquivalente auf Basis von Hämoglobinaddukt-Analysen
(Acrylnitril (7 mg/m3 ≙ EKA 420 µg/L Blut, Ethylenoxid 1,83 mg/m3
≙ EKA 90 µg/L Blut,
Dimethylsulfat 0,20 mg/m3 ≙ EKA 40 µg/L Blut)). Durch die Gefahrstoffverordnung 2005
wurde den TRK Werten (und sich daraus ableitenden EKA-Werten) die gesetzliche
Grundlage entzogen, da sich die TRK Werte auf den Stand der Technik beziehen, nicht
gesundheitsbasiert sind und somit ein Schutz vor gesundheitlichem Schaden nicht
auszuschliessen war. In der Bewertung bei der arbeitsmedizinischen Praxis können sie
jedoch weiterhin als Anhaltspunkte angesehen werden. Ist es für bestimmte Stoffe nicht
möglich einen BAT (BGW)-Wert zu definieren und die Datenlage zu gering um EKA
Korrelationen aufzustellen, kann für die praktische Arbeitsmedizin der sog. Biologische
Leitwert (BLW) für eine Beurteilung hinzugezogen werden. Er definiert sich auf Basis von
toxikologischem Wissen sowie arbeitsmedizinischen und umwelthygenischen Erfahrungen,
kann jedoch auch bei gemessenen Werten unterhalb des BLW eine Gesundheitsgefährdung
nicht ausschliessen (DFG, 2009) .

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 14
Zur Evaluation oder Aufstellung bspw. neuer AGWs, BGWs oder EKA Korrelationen
müssen grosse Mengen von Messdaten aus Expositionen mit Schadstoffen verschiedener
Kollektive und Firmen ermittelt und zusammengetragen werden. Darüber hinaus müssen
die dazu benötigten analytischen Methoden etabliert/entwickelt und in regelmässigen
Abständen extern verifiziert werden. Zur Einhaltung und Prüfung der Entwicklung
einheitlicher analytischer und valider Messmethoden, nahm die Senatskommission zur
Prüfung gesundheitsschädlicher Arbeitsstoffe der DFG bereits 1975 eine Vorreiterrolle ein.
Mit dem Ziel der Sicherung der Qualität des Biomonitorings führt die Deutsche
Gesellschaft für Arbeitsmedizin und Umweltmedizin (DGAUM) seit 1984 jährliche
Ringversuche für arbeitsmedizinisch-toxikologische Laboratorien durch. Im Hinblick auf
Styrol nimmt das Institut für Arbeitsmedizin und Sozialmedizin ebenfalls jedes Jahr
erfolgreich an der Bestimmung der Hauptmetaboliten MA und PGA teil und bestätigt
damit die Validität dieser Methode.
2.2.5 Biochemisches Effektmonitoring
Angerer definiert das biochemische Effektmonitoring als die Quantifizierung von
Reaktionsprodukten aus mutagenen Substanzen mit der Erbsubstanz: Erst ein direkter oder
indirekter Nachweis dieser Reaktion kann einen validen Schluss über das tatsächliche
Risiko für die Gesundheit des Beschäftigten zulassen. Das biochemische Effektmonitoring
gibt, wie auch das Dosismonitoring, eine Auskunft über die innere Dosis, allerdings
spezifisch über die innere effektive Dosis. Diese Nachweismethode zeigt demnach nicht
nur das Vorhandensein des Schadstoffes im Körper an, sondern liefert gleichzeitig den
Beweis seiner potentiell toxikologischen Wirksamkeit. Geeignete Analysekandidaten sind
Metabolite, Aminosäuren, Enzyme, Protein und DNA-Addukte (Angerer, 2001).
Reagieren genotoxische Substanzen mit nukleophilen Bereichen der DNA-Nukleotide
bilden sie ein sogenanntes Addukt. Diese Addukte liefern nicht nur einen „Fingerabdruck
der Exposition“, sondern sind auch Indikator eines prokarzinogenen DNA-Schadens
(Perera, 1997). Die Bestimmung von DNA-Addukten birgt allerdings einige
Schwierigkeiten. So wirkt sich das essentielle DNA-Reparatursystem modulierend auf das
Messergebnis aus. Betroffene DNA Nukleotide werden entfernt und ersetzt, gehen nicht
mehr in die Konzentrationsbestimmung mit ein, verfälschen somit die Aussage des
Messergebnisses und machen besonders retrospektive Risikoabschätzungen oft schwierig.
Zielgewebe eines toxikologischen oder kanzerogenen Effektes ist beim lebenden

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 15
Menschen in der Regel nicht zugänglich und verhindert die Durchführung routinemäßiger
Screenings betroffener Personengruppen (Angerer, 2001).
2.2.6 Biologisches Effektmonitoring
Das Biologische Effektmonitoring beschreibt Funktionsstörungen als Reaktion auf
Schadstoffbelastungen. Zu den standardmäßigen Untersuchungen zählt der Nachweis von
DNA Einzel- und Doppelstrangbrüchen, chromosomalen Aberrationen (bspw.
Schwesterchromatidaustausch) und veränderten Enzymaktivitäten. Der größte Nachteil der
alleinigen Anwendung des Biologischen Monitorings ist die mangelnde Substanzspezifität,
da sich auftretende Mutationen oder Funktionsstörungen nicht eindeutig auf eine Substanz
zurückführen lassen und daher als Marker nur begrenzt eingesetzt werden können.
Interindividuelle Schwankungen erschweren zudem die Aussagekraft, weshalb eine
Anwendung des Biologischen Effektmonitorings nur bei sehr großen Studien und
mindestens mit einem ergänzenden Dosismonitoring zu empfehlen ist.
2.2.6.1 Mercaptursäuren als Dosismarker der inneren Exposition
Elektrophile Substanzen oder Intermediate gelten als potentiell toxisch. Glutathion (GSH)
stellt die Komponente dar, die als nukleophiles Tripeptid zusammen mit dem Elektrophil
zu einem reaktionsträgen und wasserlöslichen Molekül reagiert, dadurch detoxifiziert wird
und vom Körper über den Urin ausgeschieden werden kann. Diese durch Glutathion-S-
transferasen katalysierten Glutathionkonjugationsreaktionen dienen daher dem natürlichen
Schutz vor Elektrophilen und Radikalen.
Mercaptursäuren leiten sich aus weiteren Umwandlungsreaktionen des zuvor mit GSH
konjugierten Elektrophils ab (Abb.1) und weisen damit direkt auf eine „toxikologisch
relevante absorbierte Dosis“ hin, während andere Metabolite (MA, PGA) gewöhnlich nur
auf die „absorbierte Dosis“ hinweisen. Aus diesem Grund stellen die Mercaptursäuren
wichtige Biomarker der inneren Exposition durch am Arbeitsplatz und in der Umwelt
vorkommende, relevante Substanzen dar (Boettcher & Angerer, 2005; Hecht et al, 2008;
Kellert et al, 2006; Kopp et al, 2008). Zur quantitativen Bestimmung der Mercaptursäuren
von Styrol beim Menschen, gibt es derzeit nur wenige Arbeiten. Hallier et al. untersuchten
20 Arbeiter der Polyesterindustrie, konnten jedoch, durch die vergleichsweise wenig
sensitive Dünnschichtchromatographie, nur bei einer Person das Vorhandensein von Styrol

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 16
spezifischen Mercaptursäuren detektieren (Hallier et al, 1995). Eine nicht ausreichende
Sensitivität lag auch bei Verwendung der Hochleistungsflüssigkeitschromatographie mit
gekoppelter elektronischer Detektion vor. Hier konnten bei sechs mit Styrol exponierten
Probanden keine Mercaptursäuren detektiert werden. (Norström et al, 1992). Methoden der
Hochleistungsflüssigkeitschromatographie basierend auf Fluoreszenzdetektion (Ghittori et
al, 1997) oder Flüssigkeitschromatographie gekoppelte Tandem-Massenspektrometrie
(LC/MS/MS) (Fustinoni et al, 2008; Manini et al, 2000) schienen vielversprechender zur
Analyse Styrol bedingter PHEMAs zu sein. Problematisch war hier jedoch der intensive
Zeitaufwand der Probenaufarbeitung, was Belastungsuntersuchungen in der Routine nicht
möglich macht (Maestri et al, 1997) oder die fehlende Hinzunahme interner Standards
(Fustinoni et al, 2008; Manini et al, 2000), was Quenching Effekte kompensieren und die
Richtigkeit der Ergebnisse sichern würde.
2.2.6.2 Addukte als biochemische Effektmarker
Als Addukt wird in der arbeitsmedizinischen Toxikologie die Reaktion zwischen einem
Schadstoff oder seinem Metaboliten mit Makromolekülen (Proteine, DNA) des Körpers
bezeichnet. Dieses zusammengesetzte Molekül repräsentiert in der Analytik einen Effekt
des Schadstoffes auf den Organismus und dient als Parameter der Genotoxizität und damit
der gesundheitlichen Risikoabschätzung. Indikatoren können demnach ein nach
Schadstoffexposition vermehrtes Auftreten von Protein oder DNA Addukten sein.
Im Hinblick auf eine kanzerogene Wirkung kann nur von Auftrittswahrscheinlichkeiten
gesprochen werden, da DNA-Addukte mittels Reparaturmechanismen entfernt werden
können. Dabei ist die Persistenz eines Adduktes stark von der Bindungsstelle abhängig. So
finden Adduktbildungen, hervorgerufen durch SO, in vitro bevorzugt am N7, N2–Guanin
als auch am N3-Adenin statt (Koskinen & Plna, 2000; Vodicka & Hemminki, 1988) Beim
Menschen wird die stärkste Bindung am O6 –Guanin vermutet (Vodicka et al, 1994). In der
Vergangenheit haben verschiedenste Humanstudien bspw. durch den Nachweis
styrolbedingter N2- und O6-Guaninaddukt Konzentrationen (Horvath et al, 1994; Vodicka
et al, 1995; Vodicka et al, 1999; Vodicka et al, 1994) sowie von Addukten des 1-Adenins
(Koskinen et al, 2001) in Lymphozyten von Laminierern den Nachweis eines
genotoxischen Potentials von Styrol erbracht.
Die von Törnqvist et al. entwickelte Methode zur Bestimmung von Addukten des
Hämoglobins (Törnqvist et al, 1986), wurde bereits mehrfach eingesetzt, um styrolbedingte

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 17
Expositionen nachzuweisen (Christakopoulos et al, 1993; Teixeira et al, 2008; Vodicka et
al, 1999; Yeowell-O'Connell et al, 1996). Speziell Untersuchungen der Addukte am N-
terminalen Valin des Hämoglobins (Hb-Val) im Tierversuch (Osterman-Golkar et al, 1995;
Pauwels et al, 1997) als auch beim Menschen (Christakopoulos et al, 1993; Teixeira et al,
2008) konnten diese Addukte als geeignete Biomarker der internen Exposition bestätigen.
Gleichwohl gab es in der Vergangenheit unterschiedlichste Meinungen zur Wahl des
Proteinaddukts, da sie sich in ihrem Detektionslimit als auch in ihrem
Korrelationsverhalten von anderen Metaboliten des Styrols unterscheiden. Christakopoulos
et al. konnten signifikante Erhöhungen von N-terminalen Valin Addukten des
Hämoglobins bei hoch exponierten Arbeitern der glasfaserverstärkenden Plastikindustrie
(GfPI) nachweisen (Christakopoulos et al, 1993), während Brenner et al. keine
signifikanten Unterscheide zwischen exponierten Arbeitern und Kontrollen feststellen
konnte (Brenner et al, 1991). Severi et al. konnten bei 52 Arbeitern mit Styrolexposition
keine Erhöhungen der Adduktkonzentration von N-terminalen Valin Addukten des
Hämoglobins nachweisen, was einerseits auf die geringen Expositionsbedingungen
andererseits auf das Detektionslimit von 10 pmol/g Globin zurückzuführen war (Severi et
al, 1994). Kein Beweis für eine Korrelation zwischen Exposition und einem erhöhten
Auftreten von Hämoglobinaddukten an der Cystein Seitenkette und an Carbonsäuren
konnte durch die Studie von Yeowell-O’Connell et al. erbracht werden. Sie analysierten
Addukte von 48 Bootsbauern, die mit Styrol und Styroloxid exponiert waren.
Konzentrationen von Cystein Addukten des Albumins stiegen allerdings signifikant mit der
Exposition an (Yeowell-O'Connell et al, 1996).
Hämoglobinaddukte als Langzeitmarker
1974 führte Lars Ehrenberg Versuche an Ethylenoxid exponierten Mäusen durch und
untersuchte das Bindungsverhalten an DNA und Hämoglobin (Hb) mit dem Ziel ein neues
Messinstrument für die Bewertung des Risikos der Schadstoffbelastung zu generieren. Sein
begründetes Konzept der „Tissue-Dose“ war der Beginn des Effektmonitoring, bei dem die
effektive innere Dosis direkt oder indirekt durch das Addukt beschrieben wird (Ehrenberg
et al, 1974). Die Bestimmung der inneren Belastung anhand von Hämoglobinaddukten
(HbA) bietet Vorteile gegenüber der von DNA-Addukten.
Bereits 1953 konnten Jackson und Thompson zeigen, dass die Bindung einer elektrophilen
Substanz an Hb sehr stark ist und nur durch die Degradierung des Erythrozyten eliminiert

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 18
werden kann (Jackson, 1953). Die stabile Bindung von Hb innerhalb des Erythrozyten wird
durch die Bildung des Addukts insbesondere am endständigen Hb-Valins nicht beeinflusst
(Osterman-Golkar et al, 1995). Der Abbau von Hb wird demnach nur durch die eigene
Lebensdauer (ca. 120 Tage beim Menschen, ca. 40 Tage bei der Maus) bestimmt und
verläuft nach einer Reaktion nullter Ordnung. Durch ein nicht vorhandenes
Reparatursystem bleibt die Adduktbildung bestehen und kann somit über den maximalen
Lebenszeitraum nachgewiesen werden. Durch den langen Akkumulationszeitraum können
Expositionen im Niedrig-Dosis-Bereich als auch weiter zurück liegende
Expositionsereignisse detektiert werden. Für die Bestimmung der Alkylierung wird eine
wesentlich größere Menge DNA im Blut benötigt; im Gegensatz zur hohen Konzentration
des Hb (Ehrenberg & Osterman-Golkar, 1980). In der praktischen Anwendung bietet das
Blut als Matrix somit nicht nur Vorteile der Verfügbarkeit, sondern in Hinblick auf
belastete Personen auch eine weniger invasive Form der Probenbeschaffung, als bspw. eine
spezifische Gewebebiopsie. Diese würde sicherlich den Mehrwert an Information
quantitativ steigern, ist jedoch in der Anwendungsforschung nicht praktikabel.
Humanes Hämoglobin
Physiologisch hat Hämoglobin die Funktion des Sauerstofftransportes sowie in geringen
Mengen des Kohlendioxid- und Protonentransportes in den Erythrozyten des Blutes. Es
besitzt eine Gesamtmolekülmasse von ungefähr 64,5 Kilodalton (kDa) und besteht als
Proteintetramer aus 4 Polypeptidketten (2 alpha-Ketten mit je 141 Aminosäuren und 2
beta-Ketten mit je 146 Aminosäuren), die primär durch schwache nicht kovalente
Bindungen (bspw. Van der Waals Kräfte oder Wasserstoffbrückenbindungen)
zusammengehalten werden. Die letzte Aminosäure der 4 Ketten ist Valin. Innerhalb der
vier Globinketten befinden sich ebenfalls vier prosthetische Gruppen (Häm), die über das
proximale Histidin mit der entsprechenden Aminosäurekette verbunden sind. Hier ist der
eigentliche Bindungsort des Sauerstoffs lokalisiert (Schechter, 2008).
Die kovalente Bindung eines Gefahrstoffes oder eines seiner Metaboliten an das
Hämoglobin (Abb. 3) setzt ganz allgemein die Verfügbarkeit elektrophiler und
nukleophiler Gruppen voraus.

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 19
Entstehung eines Hämoglobinaddukts
Die meisten Karzinogene gehören zu den elektrophilen, alkylierenden Substanzen. Ihr
gesundheitsgefährdendes, toxisches Potential wird primär bestimmt durch ihre direkte oder
indirekte (exogen oder endogen induzierte) Reaktivität und die daraus resultierende
Fähigkeit eine kovalente Verbindung mit nukleophilen Bereichen in Makromolekülen
bspw. Proteinen oder der DNA einzugehen. Die Nukleophilie einzelner Aminosäuren ist
abhängig vom Grad ihrer Protonierung, denn nur im unprotoniertem Zustand kommt die
Adduktbildung zustande. Die verschiedenen Seitenketten des Hämoglobins eignen sich
daher unterschiedlich gut für einen nukleophilen Angriff. Die stark basische Aminogruppe
von Lysin oder Arginin besitzt pKa-Werte im Bereich zwischen 9.5-12.5. Bei einem
physiologischen pH-Wert von 7.4 liegt diese Aminogruppe fast vollständig protoniert vor.
Bedeutender für die Bindungsreaktion sind dagegen die Aminosäuren, deren pKa sich im
naheliegenden Bereich des Blut-pH-Wertes befinden. Dazu zählen die Aminogruppen des
N-terminalen Valins, Histidins und die Thiol-Gruppe des Cysteins (Törnqvist et al, 2002),
deren durch verschiedene Alkylantien (bspw. Styrol-7,8-oxid) entstandenen Addukte als
Marker biochemischer Effekte verwendet werden können.
Abb. 3: Mechanismus einer Hämoglobinadduktbildung an das N-terminale Valin
(skizziert) am Beispiel des Epoxids Styrol-7,8-oxid
nukleophiler Angriff +
-
Styrol - 7,8 - oxid
Addukttragendes
Globin
2 - Hydroxy-2-Phenyl-Ethyl-
Globin
UmlagerungenN-terminales Valin der
Globin Aminos ä ure Globin
O
C H 3 C H 3
N H OH
HH
Globin
O
CH3
CH3
H
H
N+
O C-
+ Globin
C H 3 C H 3
N H 2 O
O

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 20
Zur Bestimmung von Addukten des N-terminalen Valins wird meistens ein modifizierter
Edman-Abbau verwendet. Anschließend erfolgt eine chromatographische Trennung mittels
Gaschromatographie und massenspektrometrischer Detektion. Pehr Edman entwickelte
1949 eine Methode zur Bestimmung der Aminosäuresequenz eines Proteins. Diese gehört
seither zum Standardrepertoire vieler molekularbiologischer und biochemischer
Untersuchungsmethoden (Edman, 1949). Durch eine Abänderung des Verfahrens konnte
Törnqvist et al. im Jahr 1986 erstmals ein praktikable Bestimmung von Addukten des N-
terminalen Valins zeigen. Anstelle des ursprünglich eingesetzten Sequenzierungsreagenz
Phenylisothiocyanat verwendeten sie Pentafluorphenylisothiocyanat (PFPITC). Während
beim klassischen Edman-Abbau die elektrophile Addition des Phenylisothiocyanats im
wässrigen Milieu bei einem pH-Wert von etwa 9-10 abläuft, findet die Reaktion an das N-
terminale Valin bei modifiziertem Edman-Abbau unter neutralen, nicht-wässrigen
Bedingungen statt. Die Bildung des Thiocarbamoylderivat wird durch die veränderten pH
Bedingungen nicht gestört, da das endständige Valin deprotoniert vorliegt (pks = 6,8-7,8).
SN
F
F
F
F
F
++
NH
O
CH3
CH3
N
H
R
Globin
PFPITC Endständige Aminosäure Valin mit gebundenem Addukt „R“
R
S
NH
O
CH3
CH3
N
NHF
F
F F
F
Globin
Thiocarbamoylderivat
Abb. 4: Bildung des Thiocarbamoylderivats durch die Reaktion von PFPITC mit der
Aminogruppe des N-terminalen Valins
Nachdem sich das Thiocarbamoylderivat gebildet hat (Abb. 4), kommt es zwischen dem
Schwefel- und dem Sauerstoffatom zu einer Zyklisierungsreaktion, die bevorzugt an dem
addukt-tragendem Valin (im Gegensatz zum unsubstituierten) stattfindet. Dies liegt an der
alkylierten Aminofunktion, die bedingt durch den elektronenliefernden Substituenten die
auftretende negative Ladung am Schwefelatom stabilisieren kann und zu einer selektiven
Abspaltung des addukt-tragenden Valins führt. Diese Gegebenheit führt gleichzeitig zu
einer verbesserten Analytik, da das unsubstituierte N-terminale Valin mehrere
Größenordnungen oberhalb des addkut-tragenden Valins liegt und so das
Hintergrundrauschen vermindert wird. Das entstandene Thiazolinonderivat erfährt durch
Temperaturerhöhung eine Umlagerung in das stabilere Pentafluorphenylthiohydantoin
(PFPTH)-Derivat, welches den Analyten zur Identifizierung der Hämoglobinaddukte
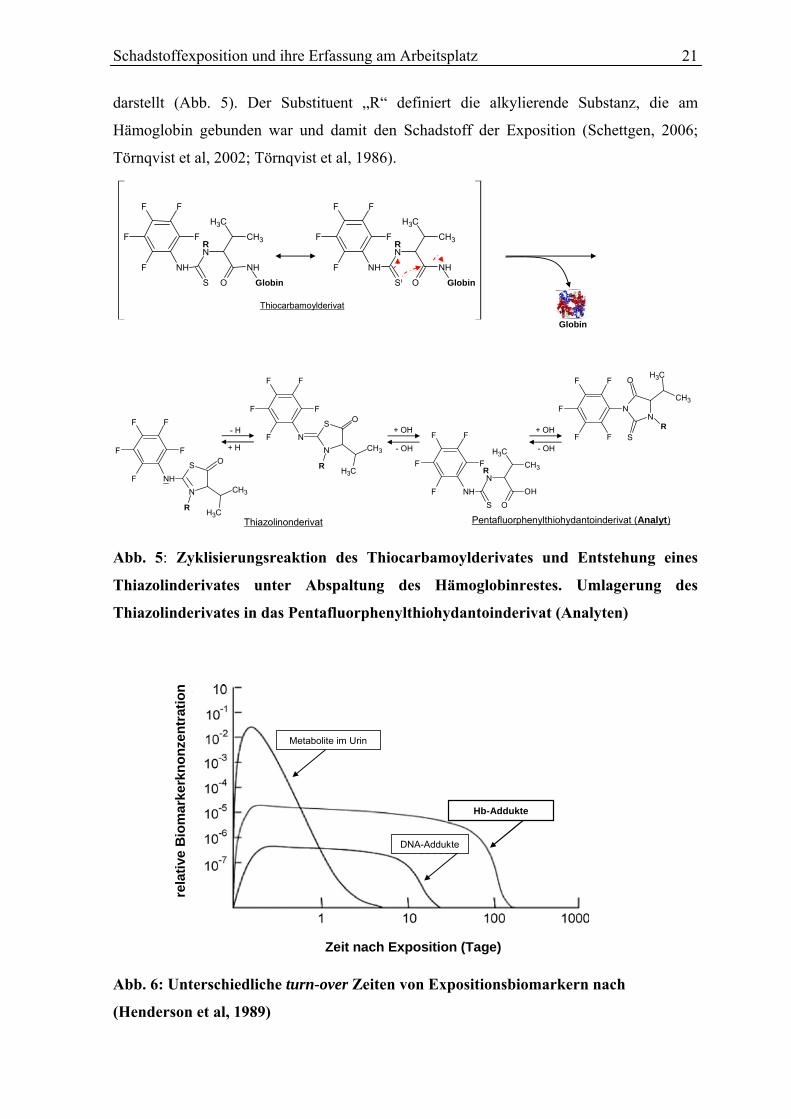
Schadstoffexposition und ihre Erfassung am Arbeitsplatz 21
Metabolite im Urin
DNA-Addukte
Hb-Addukte
rela
tive
Bio
mar
kerk
no
nze
ntr
atio
n
Zeit nach Exposition (Tage)
darstellt (Abb. 5). Der Substituent „R“ definiert die alkylierende Substanz, die am
Hämoglobin gebunden war und damit den Schadstoff der Exposition (Schettgen, 2006;
Törnqvist et al, 2002; Törnqvist et al, 1986).
R
S O
CH3
CH3N
NHF
F
F F
F
R
SO
CH3
CH3N
NF
F
F F
F
- H
+ H - OH
+ OH
CH3
CH3
R
OH
O
N
S
NHF
F
F F
F
- OH
+ OH
O
CH3
CH3
RS
NN
FF
F
F F
Thiazolinonderivat Pentafluorphenylthiohydantoinderivat (Analyt)
Abb. 5: Zyklisierungsreaktion des Thiocarbamoylderivates und Entstehung eines
Thiazolinderivates unter Abspaltung des Hämoglobinrestes. Umlagerung des
Thiazolinderivates in das Pentafluorphenylthiohydantoinderivat (Analyten)
Abb. 6: Unterschiedliche turn-over Zeiten von Expositionsbiomarkern nach
(Henderson et al, 1989)
R
S
NH
O
CH3
CH3
N
NHF
F
F F
F
Globin
R
S
NH
O
CH3
CH3
N
NHF
F
F F
F
Globin
Globin
Thiocarbamoylderivat

Schadstoffexposition und ihre Erfassung am Arbeitsplatz 22
Untersuchungen zur Bindung kanzerogener Substanzen an Proteine, insbesondere
Hämoglobin, konnten im Tierversuch (Li et al, 2003; Osterman-Golkar et al, 2003; Wu,
1997) als auch beim Menschen (Czene et al, 2002) zeigen, dass diese proportional zur
DNA-Bindung geschieht. Hämoglobinaddukte können daher als geeignete DNA-Addukt-
Surrogate angesehen (Angerer, 2001; van Sittert et al, 2000a) und als Risikoparameter
eines makromolekularen Schadens verwendet werden.
2.2.7 Cotinin als Marker des Raucherstatus
Tabakrauch kann Einfluss haben auf die Ergebnisse von Expositionsanalysen. Oft sind
bereits im Tabak verschiedenste Gefahrstoffe enthalten oder entstehen während der
Pyrolyse beim Rauchvorgang selbst. Speziell für Styrol konnte bereits nachgewiesen
werden, dass es auch während des Rauchvorgangs gebildet wird (Adam et al, 2009;
Wallace et al, 1987). Der Beitrag dieser Stoffe zur Gesamtbelastung kann marginal sein
oder aber auch weitreichende Bedeutung haben (Hagmar et al, 2001; Schettgen et al,
2003). Beim Biomonitoring ist es deshalb essentiell den individuellen Raucherstatus über
den Nikotin Metaboliten Cotinin zu erfassen und den Einfluss des Rauchens auf bestimmte
Expositionsäquivalente hin zu überprüfen.

Zielsetzung der Arbeit
23
3 Zielsetzung der Arbeit
Um den Arbeitnehmer an seinem Arbeitsplatz vor einer Belastung mit Gefahrstoffen zu
schüzten oder etwaige Expositionen zu ermitteln, wird am Arbeitsplatz klassischerweise
ein konventionelles Luftmonitoring durchgeführt. Hierbei erfolgt die Abschätzung der
potentiellen Gefahrstoffbelastung nur über Messungen der jeweiligen Substanz in der
Raumluft. Diese Messungen geben aber keinen Aufschluss über das tatsächliche
Vorkommen des Schadstoffes im Körper des Betroffenen und sind zudem stark anfällig für
Störeffekte.
Im Gegensatz dazu liefern das Dosis- und biochemische Effektmonitoring Informationen
zu den tatsächlich aufgenommenen Mengen eines Gefahrstoffes im Körper. Erfassungen
von Styrol bedingten Belastungen, bei denen alle genannten Parameter (MA_PGA, Hb-
Val, PHEMAs) zur gleichen Zeit beim Menschen erfasst wurden, um eine möglichst
umfassende Risikoabschätzung durchführen zu können, lagen bislang nicht vor.
Vor diesem Hintergrund war die Zielsetzung der vorliegenden Arbeit ein
Humanbiomonitoring an einem Styrolexponierten Kollektiv und einem Referenzkollektiv
durchzuführen, bei denen sowohl die als Standardparameter des Dosismonitorings
beschriebenen aromatischen Carbonsäuren (Mandelsäure und Phenylglyoxylsäure)
bestimmt wurden sowie die Addukte am endständigen Valin der Hämoglobinseitenkette als
Langzeitmarker. Als spezifischer Marker eines durch Glutathion vermittelten Schutzes vor
körperständigen elektrophilen Substanzen wurde zudem die Bestimmung styrolbedingter
Mercaptursäuren durchgeführt. Diese Parameter repräsentieren unterschiedliche
Abbauwege im Metabolismus des Styrols. Weiterhin galt es den potentiellen Einfluss des
Rauchens auf die Metabolitkonzentrationen zu berücksichtigen und das Verhalten der
Metabolite innerhalb eines Vorschicht-Nachschichtsystems zu untersuchen.
Um eine Expositionserfassung durchzuführen war ein weiteres Ziel dieser Arbeit zunächst
die Etablierung und Validierung einer Methode zur Bestimmung des Hämoglobinaddukts
N-(2-Hydroxy-2-Phenylethyl)-Valin des Styrols und weiterhin aufbauend auf Methoden
zur Bestimmung von Mercaptursäuren des 1,3-butadiens und des Acrylonitrils eine
Methode zur Bestimmung der Mercaptursäuren N-Acetyl-S-(2-Hydroxy-1-Phenylethyl)-
Cystein und N-Acetyl-S-(2-Hydroxy-2-Phenylethyl)-Cystein zu etablieren und validieren.

Zielsetzung der Arbeit 24
Nach abschließender Validierung beider Methoden war ein Firmenkollektiv mit
produktionsbedingten Styrolexpositionen zu akquirieren und die dortigen Belastungen
mittels des oben genannten Humanbiomonitorings zu erfassen.

Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin
25
4 Analytik der Phenylhydroxyethyl Mercaptursäuren
im Urin
4.1 Grundlage des Verfahrens
Dieses Verfahren diente der Messung und der daraus resultierenden
Konzentrationsbestimmung der Styrol spezifischen Mercaptursäuren N-Acetyl-S-(2-
Hydroxy-1-Phenylethyl)-Cystein (PHEMA1) und N-Acetyl-S-(2-Hydroxy-2-Phenylethyl)-
Cystein (PHEMA2). Die Mercaptursäuren wurden mittels HPLC online selektiv an einer
RAM-Phase (Restricted Access Material) konzentriert und von restlichen
Matrixkomponenten isoliert. Durch das Prinzip des Größenausschlusses können kleinste
Moleküle durch Poren in das Innere des Säulenmaterials gelangen, während
Makromoleküle (bspw. Proteine) eluieren und die Säule verlassen. Das Prinzip der
Adsorptionschromatographie hält kleine Moleküle (bspw. Analyt) im inneren, alkylierten
Bereich des Säulenmaterials zurück. Im so genannten Backflush-Verfahren werden die
Analyte auf die analytische Säule übertragen und nach erfolgter Trennung tandem-
massenspektrometrisch detektiert. Die Kalibrierung erfolgte mittles Vergleichsstandards,
die in Poolurin angesetzt und in der gleichen Weise behandelt wurden wie die zu
analysierenden Proben. Als interner Standard wurde den Urinproben eine 1:1 Mischung
aus N-Acetyl-S-(2-Hydroxy-1-Phenylethyl)-Cystein-13C6 und N-Acetyl-S-(2-Hydroxy-2-
Phenylethyl)-Cystein-13C6 zugefügt.
4.2 Geräte, Chemikalien, Lösungen
4.2.1 Geräte
HPLC-Apperatur (Agilent 1100 Series):
binäre Gradientenpumpe (G 1312A)
Isokratische Pumpe mit Vorrichtung zum Entgasen der jeweiligen Eluenten
Säulenthermostat
Injektionsventil
Probenschleife 100 µl, Pumpenkopf 100 µl

Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 26
Automatisierter Probengeber (G 1313A)
Sixport-Ventil (Valco Systems)
Tandem-massenspektrometrischer Detektor (Applied Biosystems API 3000)
Computersystem zur Datenauswertung
HPLC-Säule (Phenomenex): Luna C8, 150 mm; Ø innen 4,6 mm; Teilchendurchmesser 3
µm
Vorsäulenfilter (Supelco) (0,5 µm)
Vorsäule (Phenomenex): C8 3 mm, Ø innen 4 mm, Teilchendurchmesser 3 µm
RAM-Phase (Merck): LiChroSpher RP-8 ADS; 25 mm; Ø innen 4 mm;
Teilchendurchmesser 25 µm
Magnetrührer (IKA Labortechnik)
1,8 ml Schraubgläser mit dazugehörigen PTFE-kaschierten Septen und Schraubkappen
(Macherey-Nagel)
Verschlusszange (Macherey-Nagel)
pH-Meter mit Einstabmesskette (Mettler-Toledo)
Mikroliterpipetten, verstellbar 10-100 µl (100- 1000) µl (Eppendorf)
Messkolben 10- und 25-ml (Brand)
Zentrifuge Rotina 420R (Hettich)
Erlenmeyer Kolben 10 ml (Brand)
Labor „Blaukopfflaschen“ 1 Liter (Schott)
4.2.2 Chemikalien
Sofern keine weiteren Angaben gemacht werden, sind alle benannten Chemikalien in der
Reinheit von mindestens p.a.-Qualität verwendet worden.
Standards: N-Acetyl-S-(2-Hydroxy-1-Phenylethyl)-Cystein und N-Acetyl-S-(2-Hydroxy-2-
Phenylethyl)-Cystein (Verhältnis 1:1) (Toronto Research Chemicals Inc)
Interne Standards: N-Acetyl-S-(2-Hydroxy-1-Phenylethyl)-Cystein-13C6 und N-Acetyl-S-
(2-Hydroxy-2-Phenylethyl)-Cystein-13C6 (Verhältnis 1:1) (Toronto Research Chemicals
Inc)

Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 27
Ameisensäure; 100% (Merck)
Chromatographie-Wasser (LiChrosolv®) (Merck)
Acetonitril; HPLC grade (J.T. Baker)
aqua bidest (Millipore®)
4.2.3 Lösungen
1% Ameisensäure
50 ml aqua bidest wurden in einem 100-ml Messkolben vorgelegt. 1 ml 100%
Ameisensäure wurde hinzugegeben und der Kolben mit aqua bidest bis zur Marke
aufgefüllt. Die Lösung war bei 4°C ca. 1 Monat haltbar.
Eluent A (0,1% Ameisensäure, pH 2.5)
500 ml aqua bidest wurde in einem 1000-ml Messkolben vorgelegt. 100 ml der 1%
Ameisensäure (siehe oben) wurden mit Hilfe eines Standzylinders abgemessen und in den
Messkolben hinzugegeben. Anschließend wurde der Messkolben mit ca. weiteren 400 ml
aqua bidest aufgefüllt und der pH-Wert der Lösung unter Rühren mittels Magnetrührer
unter ständiger Kontrolle des pH-Meters tropfenweise mit Ameisensäure (100%) auf
pH 2.5 eingestellt. Der Messkolben wurde anschließend mit aqua bidest Wasser bis zur
Marke aufgefüllt.
Eluent B (Acetonitril)
Eluent C
(isokratische Pumpe, 95:5 0,1% Ameisensäure pH 2.5 / Acetonitril):
475 ml des hergestellten Eluenten A wurden mit Hilfe eines Standzylinders in einen
500 ml-Messkolben gefüllt. Anschließend wurde der Messkolben mit Acetonitril bis zur
Marke aufgefüllt.
Alle Eluenten wurden vor der Benutzung zur Vermeidung von Luftblasen für 10 min in ein
Ultraschallbad gestellt.

Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 28
Lösungen der Standards
Stammlösung:
10 mg der PHEMA Standards wurden in 10 ml Methanol gelöst (1 g/L). 100 µl dieser
Stammlösung wurden anschließend in einen 10-ml Messkolben mit 5 ml vorgelegter 0,1%
Ameisensäure gegeben und bis zur Marke weiter aufgefüllt Dotierlösung 1 (10 mg/L)
500 µl der Dotierlösung 1 wurden anschließend in einen 10-ml Messkolben mit 5 ml
vorgelegter 0,1% Ameisensäure gegeben und bis zur Marke weiter aufgefüllt
Dotierlösung 2 (500 µg/L).
Lösungen der internen Standards
Stammlösung:
1 mg der 13C6 PHEMAs (komplette erworbene Menge) wurde in 1 ml Methanol gelöst
(1 g/L). 10 µl dieser Stammlösung wurden anschließend in einen 10-ml Messkolben mit 5
ml vorgelegter 0,1% Ameisensäure gegeben und bis zur Marke weiter aufgefüllt
Gebrauchslösung (1 mg/L)
Alle Lösungen wurden bei –20°C in braunen Schraubgläschen gelagert. Die
Vergleichsstandardlösungen wurden in angesäuertem, filtriertem Mischharn von vier
unbelasteten Personen der Allgemeinbevölkerung angesetzt. Um die Hintergrundbelastung
der Mercaptursäuren im verwendeten Poolurin (Kreatinin 0,56 g/L) möglichst niedrig zu
halten, sollte hier lediglich Urin von Nichtrauchern verwendet werden. Zur Herstellung des
Mischharns wurden Harnproben der Probanden in einem geeigneten Gefäß gesammelt, gut
durchmischt und bis zur Herstellung der Standards und des Kontrollmaterials bei -20°C
gelagert. Nach dem Auftauen wurde der Mischharn durch einen Faltenfilter filtriert, um
ausgefallene Proteine abzutrennen, mit 1 % Ameisensäure (100%) angesäuert und dann
zum Ansetzen der Standards benutzt.
Aus den Dotierlösungen wurden durch Verdünnen mit dem filtrierten Mischharn (bzw.
aqua bidest für den Matrixeffektvergleich) Vergleichsstandards in einem
Konzentrationsbereich von 0,5 – 250 µg/L nach dem Pipettierschema in Tab.2 hergestellt.
Als Leerwert wurden der filtrierte Mischharn und ein Wasserblindwert in jeder Serie
mitgeführt.
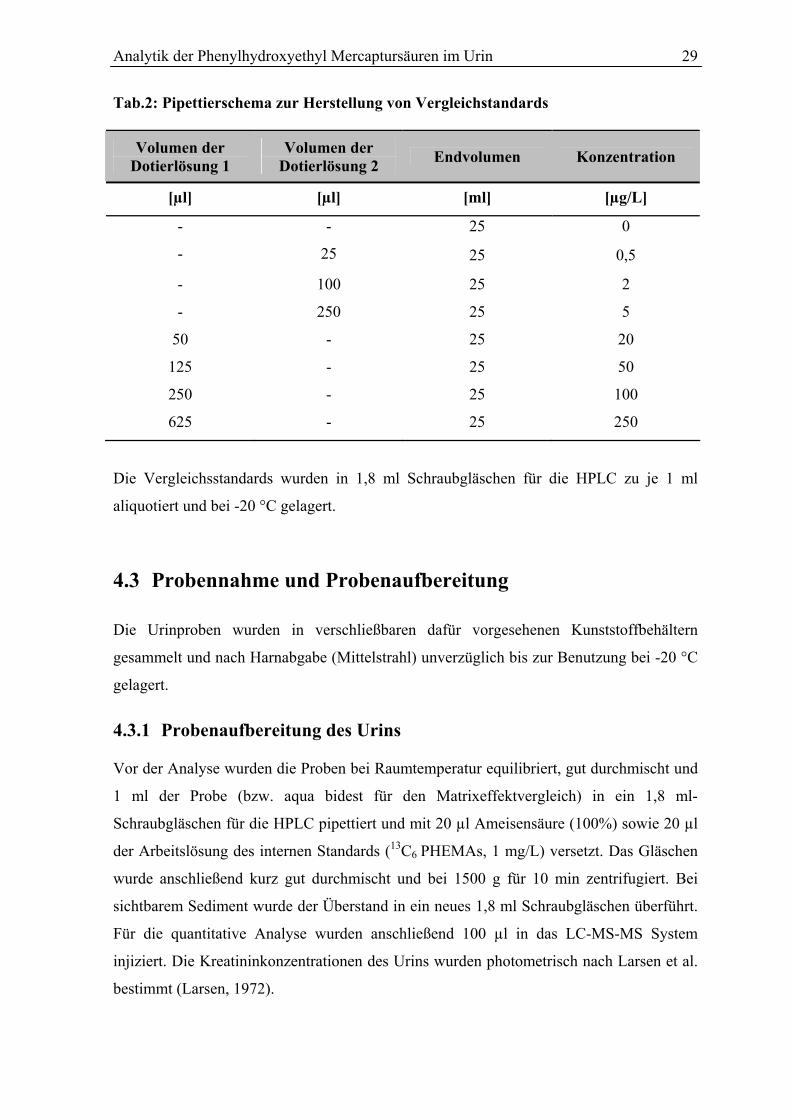
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 29
Tab.2: Pipettierschema zur Herstellung von Vergleichstandards
Volumen der Dotierlösung 1
Volumen der Dotierlösung 2
Endvolumen Konzentration
[µl] [µl] [ml] [µg/L]
- - 25 0
- 25 25 0,5
- 100 25 2
- 250 25 5
50 - 25 20
125 - 25 50
250 - 25 100
625 - 25 250
Die Vergleichsstandards wurden in 1,8 ml Schraubgläschen für die HPLC zu je 1 ml
aliquotiert und bei -20 °C gelagert.
4.3 Probennahme und Probenaufbereitung
Die Urinproben wurden in verschließbaren dafür vorgesehenen Kunststoffbehältern
gesammelt und nach Harnabgabe (Mittelstrahl) unverzüglich bis zur Benutzung bei -20 °C
gelagert.
4.3.1 Probenaufbereitung des Urins
Vor der Analyse wurden die Proben bei Raumtemperatur equilibriert, gut durchmischt und
1 ml der Probe (bzw. aqua bidest für den Matrixeffektvergleich) in ein 1,8 ml-
Schraubgläschen für die HPLC pipettiert und mit 20 µl Ameisensäure (100%) sowie 20 µl
der Arbeitslösung des internen Standards (13C6 PHEMAs, 1 mg/L) versetzt. Das Gläschen
wurde anschließend kurz gut durchmischt und bei 1500 g für 10 min zentrifugiert. Bei
sichtbarem Sediment wurde der Überstand in ein neues 1,8 ml Schraubgläschen überführt.
Für die quantitative Analyse wurden anschließend 100 µl in das LC-MS-MS System
injiziert. Die Kreatininkonzentrationen des Urins wurden photometrisch nach Larsen et al.
bestimmt (Larsen, 1972).
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 30
4.4 Instrumentelle Arbeitsbedingungen
Die analytischen Messungen erfolgten an einer Gerätekopplung, bestehend aus einer
HPLC-Gradientenpumpe Agilent 1100 Series und einem API-3000 LC-MS-MS-System
der Firma Applied Biosystems. Die
100 µl Probe wurden mittels
isokratischer Pumpe zur
Anreicherung auf eine RAM Phase
befördert. Anschließend wurde der
Analyt im Rückflussmodus mittels
der Eluenten der Gradientenpumpe
über ein zeitgesteuertes 6-fach
Ventil (Tab.3) auf die analytische
Umkehrphasensäule transferiert
(Abb.7). Um die Lebensdauer der
analytischen Säule zu steigern
waren hier ein Vorfilter und eine
„Wächter“-Säule vorgeschaltet.
Abb.7: Anreicherungsschaltung (A) und Transferschaltung (B) des 6-fach Ventils
4.4.1 Hochleistungschromatographische Arbeitsbedingungen
Anreicherungssäule: Material: Stahl Länge: 25 mm Innerer Durchmesser: 4 mm Säulenfüllung: LiChroSpher RP-8 ADS Trennsäule: Material: Stahl Länge: 150 mm innerer Durchmesser: 4,6 mm Säulenfüllung: Phenomenex Luna C 8 (2), 3 µm
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 31
Trennprinzip: Reversed phase Temperatur: 35°C Detektion: Tandem-Massenspektrometrischer Detektor Mobile Phase: Eluent A: 0,1 % Ameisensäure pH 2,5 Eluent B: 100 % Acetonitril Eluent C: 95 % Eluent A, 5 % Acetonitril (Die Eluenten wurden vor ihrem Einsatz entgast) Methode: PHEMA (Projekt: Mercaptursäuren) Isokratische Pumpe: Eluent C Flussrate: 0,3 ml/min Gradientenpumpe: Eluent A bzw. Eluent B Gradient: s. Tabelle Tab.3: Programm der Gradientenpumpe
Zeit (min) Eluent A (vol.%) Eluent B (vol. %) Ventilposition
0 70 30 A
1,95 70 30 B
3,15 70 30 A
3,8 70 30 A
7,8 0 100 A
13,8 0 100 A
17,8 70 30 A
21,0 80 20 A
Stopzeit für das MS: 17 Minuten Flussrate: 0,3 ml/min Injektionsvolumen: 100 µl
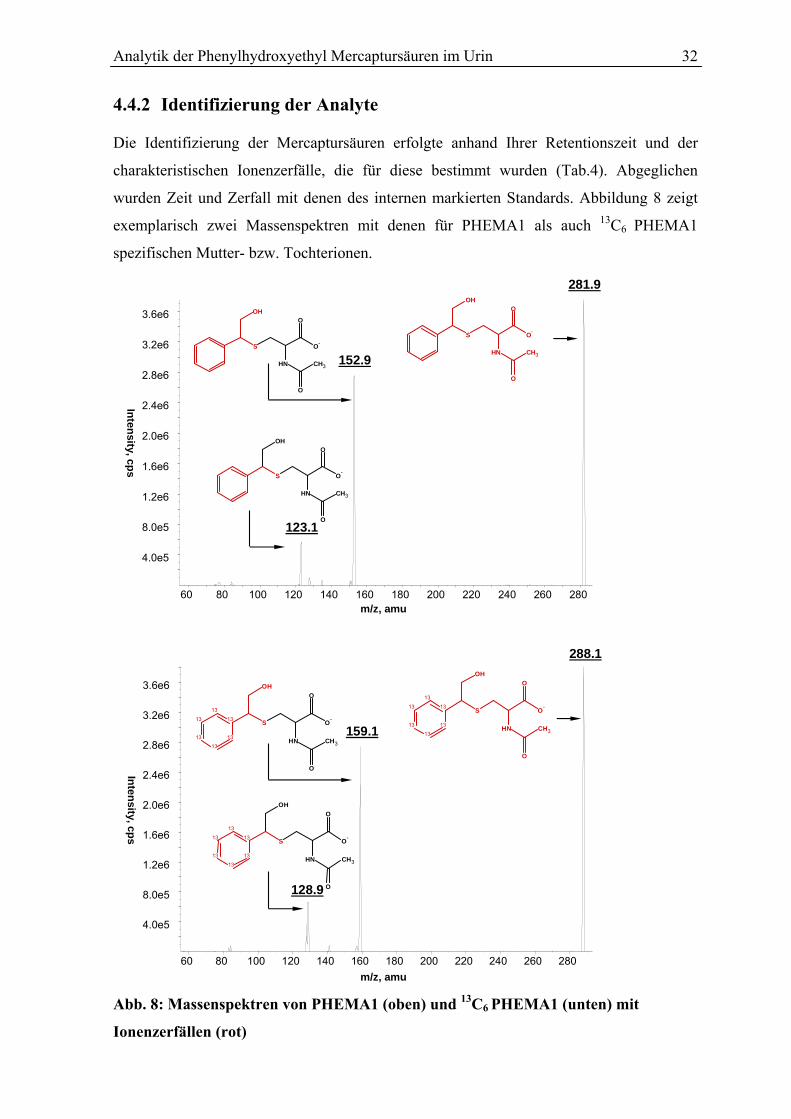
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 32
4.4.2 Identifizierung der Analyte
Die Identifizierung der Mercaptursäuren erfolgte anhand Ihrer Retentionszeit und der
charakteristischen Ionenzerfälle, die für diese bestimmt wurden (Tab.4). Abgeglichen
wurden Zeit und Zerfall mit denen des internen markierten Standards. Abbildung 8 zeigt
exemplarisch zwei Massenspektren mit denen für PHEMA1 als auch 13C6 PHEMA1
spezifischen Mutter- bzw. Tochterionen.
60 80 100 120 140 160 180 200 220 240 260 280m/z, amu
4.0e5
8.0e5
1.2e6
1.6e6
2.0e6
2.4e6
2.8e6
3.2e6
3.6e6
Inten
sity, cps
281.9
152.9
123.1
60 80 100 120 140 160 180 200 220 240 260 280
m/z, amu
4.0e5
8.0e5
1.2e6
1.6e6
2.0e6
2.4e6
2.8e6
3.2e6
3.6e6
Inten
sity, cps
288.1
159.1
128.9
O
OH
S
O
O-
NH CH3
OH
S
O
O-
NH CH3
O
OH
S
O
O-
NH CH3
O
OH
S
O
O-
NH CH3
O
13
13
13
13
13
13
OH
S
O
O-
NH CH3
O
13
13
13
13
13
13
OH
S
O
O-
NH CH3
O
13
13
13
13
13
13
Abb. 8: Massenspektren von PHEMA1 (oben) und 13C6 PHEMA1 (unten) mit
Ionenzerfällen (rot)
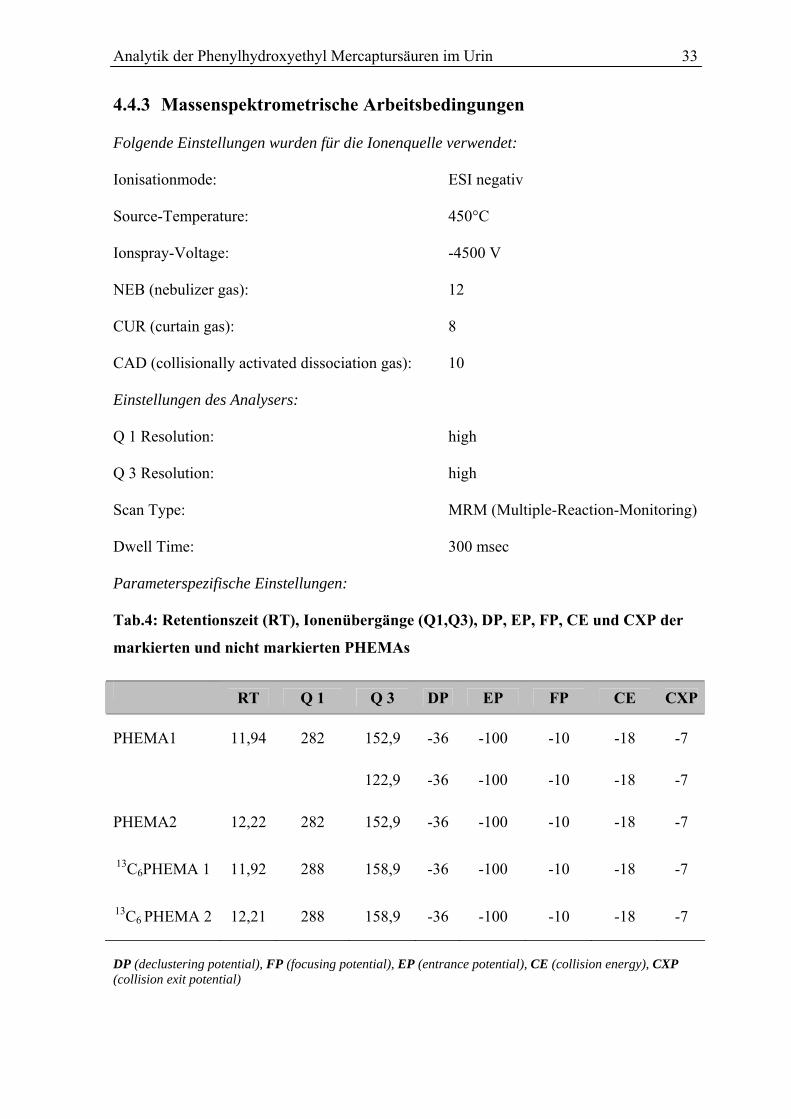
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 33
4.4.3 Massenspektrometrische Arbeitsbedingungen
Folgende Einstellungen wurden für die Ionenquelle verwendet: Ionisationmode: ESI negativ Source-Temperature: 450°C Ionspray-Voltage: -4500 V NEB (nebulizer gas): 12 CUR (curtain gas): 8 CAD (collisionally activated dissociation gas): 10 Einstellungen des Analysers: Q 1 Resolution: high Q 3 Resolution: high Scan Type: MRM (Multiple-Reaction-Monitoring) Dwell Time: 300 msec Parameterspezifische Einstellungen: Tab.4: Retentionszeit (RT), Ionenübergänge (Q1,Q3), DP, EP, FP, CE und CXP der
markierten und nicht markierten PHEMAs
RT Q 1 Q 3 DP EP FP CE CXP
PHEMA1 11,94 282 152,9 -36 -100 -10 -18 -7
122,9 -36 -100 -10 -18 -7
PHEMA2 12,22 282 152,9 -36 -100 -10 -18 -7
13C6PHEMA 1 11,92 288 158,9 -36 -100 -10 -18 -7
13C6 PHEMA 2 12,21 288 158,9 -36 -100 -10 -18 -7
DP (declustering potential), FP (focusing potential), EP (entrance potential), CE (collision energy), CXP (collision exit potential)
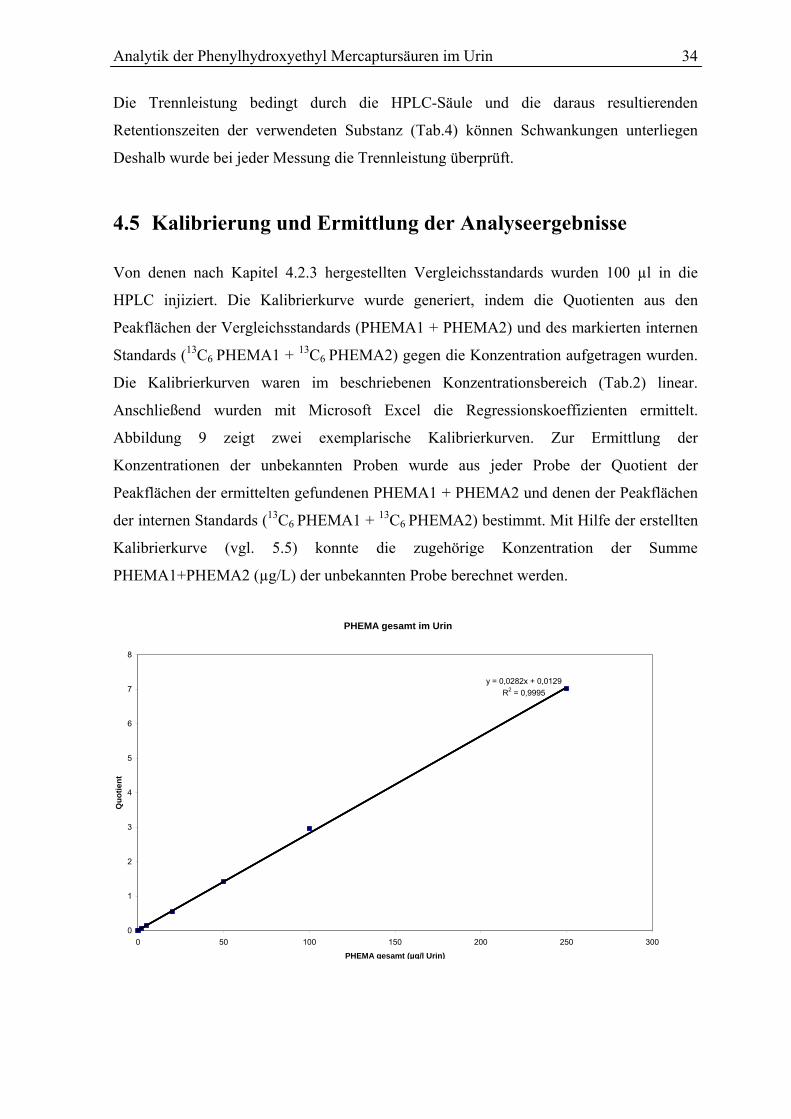
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 34
Die Trennleistung bedingt durch die HPLC-Säule und die daraus resultierenden
Retentionszeiten der verwendeten Substanz (Tab.4) können Schwankungen unterliegen
Deshalb wurde bei jeder Messung die Trennleistung überprüft.
4.5 Kalibrierung und Ermittlung der Analyseergebnisse
Von denen nach Kapitel 4.2.3 hergestellten Vergleichsstandards wurden 100 µl in die
HPLC injiziert. Die Kalibrierkurve wurde generiert, indem die Quotienten aus den
Peakflächen der Vergleichsstandards (PHEMA1 + PHEMA2) und des markierten internen
Standards (13C6 PHEMA1 + 13C6 PHEMA2) gegen die Konzentration aufgetragen wurden.
Die Kalibrierkurven waren im beschriebenen Konzentrationsbereich (Tab.2) linear.
Anschließend wurden mit Microsoft Excel die Regressionskoeffizienten ermittelt.
Abbildung 9 zeigt zwei exemplarische Kalibrierkurven. Zur Ermittlung der
Konzentrationen der unbekannten Proben wurde aus jeder Probe der Quotient der
Peakflächen der ermittelten gefundenen PHEMA1 + PHEMA2 und denen der Peakflächen
der internen Standards (13C6 PHEMA1 + 13C6 PHEMA2) bestimmt. Mit Hilfe der erstellten
Kalibrierkurve (vgl. 5.5) konnte die zugehörige Konzentration der Summe
PHEMA1+PHEMA2 (µg/L) der unbekannten Probe berechnet werden.
PHEMA gesamt im Urin
y = 0,0282x + 0,0129
R2 = 0,9995
0
1
2
3
4
5
6
7
8
0 50 100 150 200 250 300
PHEMA gesamt (µg/l Urin)
Qu
oti
ent
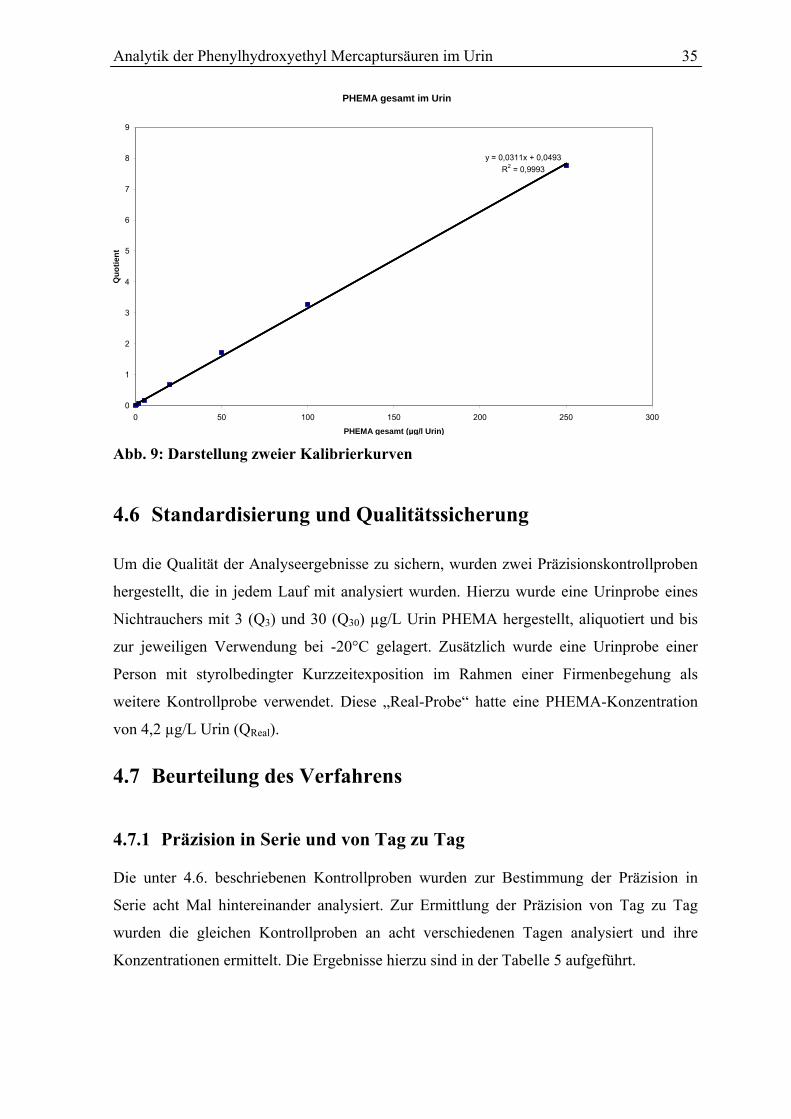
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 35
PHEMA gesamt im Urin
y = 0,0311x + 0,0493
R2 = 0,9993
0
1
2
3
4
5
6
7
8
9
0 50 100 150 200 250 300
PHEMA gesamt (µg/l Urin)
Qu
oti
ent
Abb. 9: Darstellung zweier Kalibrierkurven
4.6 Standardisierung und Qualitätssicherung
Um die Qualität der Analyseergebnisse zu sichern, wurden zwei Präzisionskontrollproben
hergestellt, die in jedem Lauf mit analysiert wurden. Hierzu wurde eine Urinprobe eines
Nichtrauchers mit 3 (Q3) und 30 (Q30) µg/L Urin PHEMA hergestellt, aliquotiert und bis
zur jeweiligen Verwendung bei -20°C gelagert. Zusätzlich wurde eine Urinprobe einer
Person mit styrolbedingter Kurzzeitexposition im Rahmen einer Firmenbegehung als
weitere Kontrollprobe verwendet. Diese „Real-Probe“ hatte eine PHEMA-Konzentration
von 4,2 µg/L Urin (QReal).
4.7 Beurteilung des Verfahrens
4.7.1 Präzision in Serie und von Tag zu Tag
Die unter 4.6. beschriebenen Kontrollproben wurden zur Bestimmung der Präzision in
Serie acht Mal hintereinander analysiert. Zur Ermittlung der Präzision von Tag zu Tag
wurden die gleichen Kontrollproben an acht verschiedenen Tagen analysiert und ihre
Konzentrationen ermittelt. Die Ergebnisse hierzu sind in der Tabelle 5 aufgeführt.
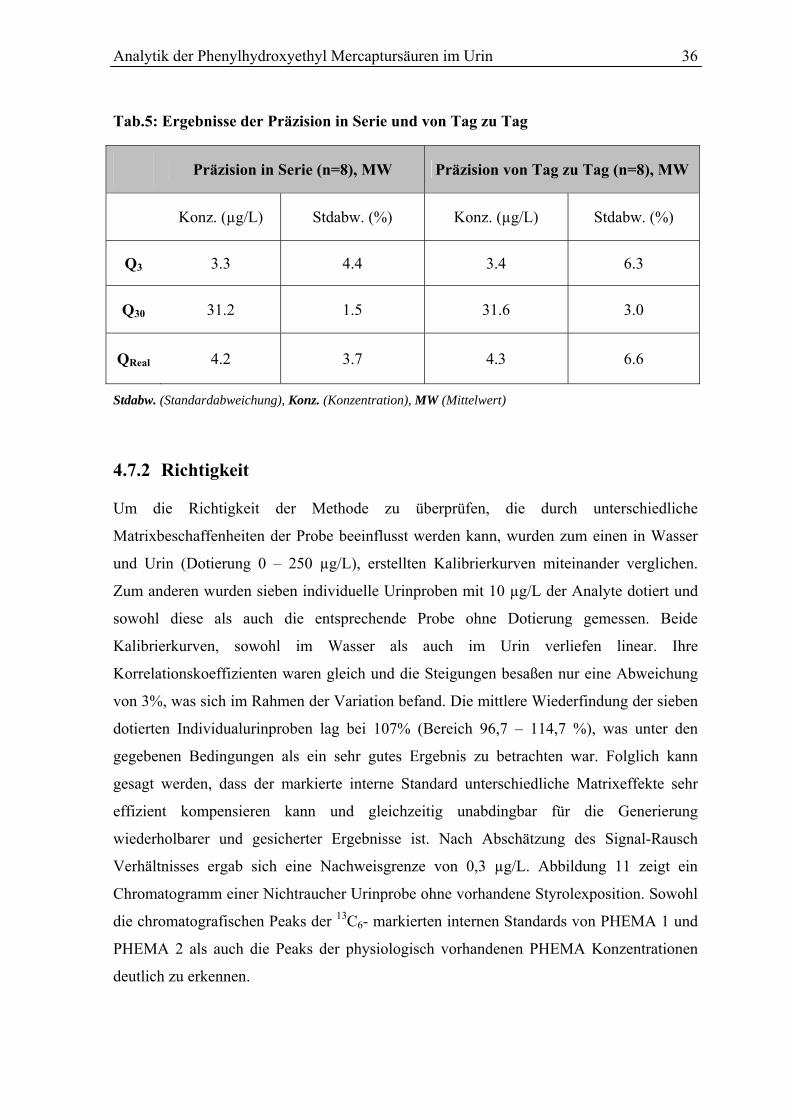
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 36
Tab.5: Ergebnisse der Präzision in Serie und von Tag zu Tag
Präzision in Serie (n=8), MW Präzision von Tag zu Tag (n=8), MW
Konz. (µg/L) Stdabw. (%) Konz. (µg/L) Stdabw. (%)
Q3 3.3 4.4 3.4 6.3
Q30 31.2 1.5 31.6 3.0
QReal 4.2 3.7 4.3 6.6
Stdabw. (Standardabweichung), Konz. (Konzentration), MW (Mittelwert)
4.7.2 Richtigkeit
Um die Richtigkeit der Methode zu überprüfen, die durch unterschiedliche
Matrixbeschaffenheiten der Probe beeinflusst werden kann, wurden zum einen in Wasser
und Urin (Dotierung 0 – 250 µg/L), erstellten Kalibrierkurven miteinander verglichen.
Zum anderen wurden sieben individuelle Urinproben mit 10 µg/L der Analyte dotiert und
sowohl diese als auch die entsprechende Probe ohne Dotierung gemessen. Beide
Kalibrierkurven, sowohl im Wasser als auch im Urin verliefen linear. Ihre
Korrelationskoeffizienten waren gleich und die Steigungen besaßen nur eine Abweichung
von 3%, was sich im Rahmen der Variation befand. Die mittlere Wiederfindung der sieben
dotierten Individualurinproben lag bei 107% (Bereich 96,7 – 114,7 %), was unter den
gegebenen Bedingungen als ein sehr gutes Ergebnis zu betrachten war. Folglich kann
gesagt werden, dass der markierte interne Standard unterschiedliche Matrixeffekte sehr
effizient kompensieren kann und gleichzeitig unabdingbar für die Generierung
wiederholbarer und gesicherter Ergebnisse ist. Nach Abschätzung des Signal-Rausch
Verhältnisses ergab sich eine Nachweisgrenze von 0,3 µg/L. Abbildung 11 zeigt ein
Chromatogramm einer Nichtraucher Urinprobe ohne vorhandene Styrolexposition. Sowohl
die chromatografischen Peaks der 13C6- markierten internen Standards von PHEMA 1 und
PHEMA 2 als auch die Peaks der physiologisch vorhandenen PHEMA Konzentrationen
deutlich zu erkennen.
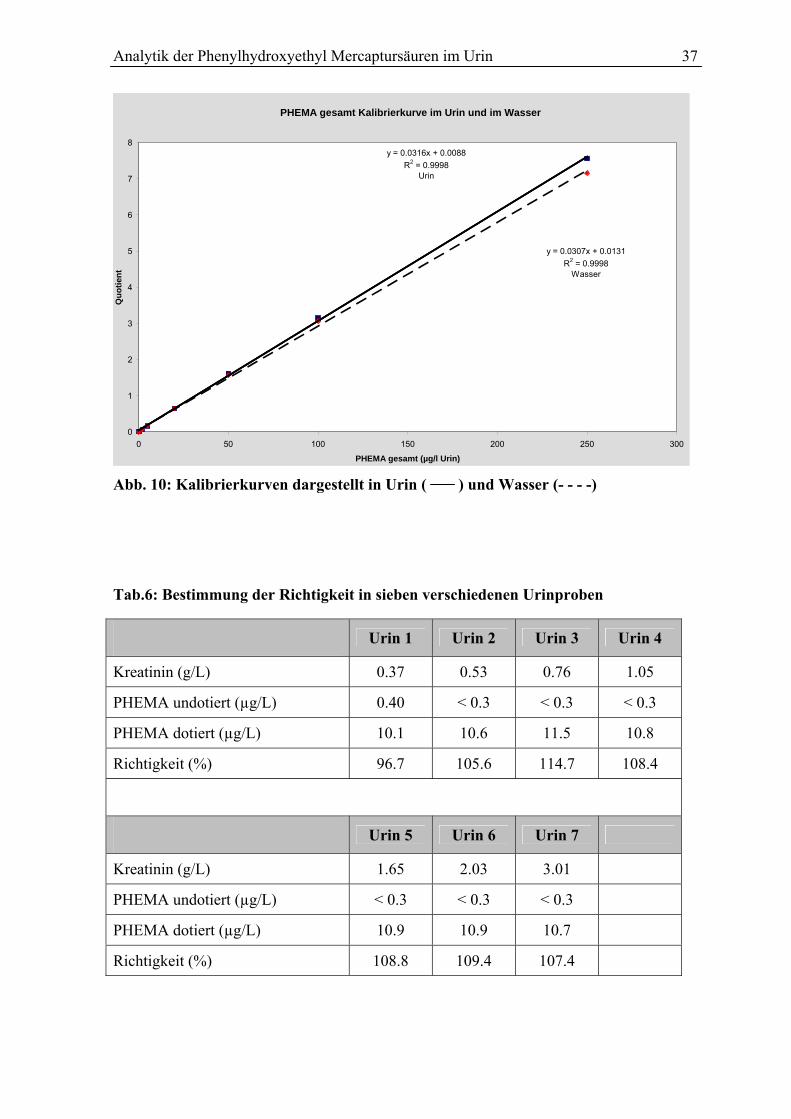
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 37
PHEMA gesamt Kalibrierkurve im Urin und im Wasser
y = 0.0316x + 0.0088
R2 = 0.9998Urin
y = 0.0307x + 0.0131
R2 = 0.9998Wasser
0
1
2
3
4
5
6
7
8
0 50 100 150 200 250 300
PHEMA gesamt (µg/l Urin)
Qu
oti
ent
Abb. 10: Kalibrierkurven dargestellt in Urin ( ) und Wasser (- - - -)
Tab.6: Bestimmung der Richtigkeit in sieben verschiedenen Urinproben
Urin 1 Urin 2 Urin 3 Urin 4
Kreatinin (g/L) 0.37 0.53 0.76 1.05
PHEMA undotiert (µg/L) 0.40 < 0.3 < 0.3 < 0.3
PHEMA dotiert (µg/L) 10.1 10.6 11.5 10.8
Richtigkeit (%) 96.7 105.6 114.7 108.4
Urin 5 Urin 6 Urin 7
Kreatinin (g/L) 1.65 2.03 3.01
PHEMA undotiert (µg/L) < 0.3 < 0.3 < 0.3
PHEMA dotiert (µg/L) 10.9 10.9 10.7
Richtigkeit (%) 108.8 109.4 107.4
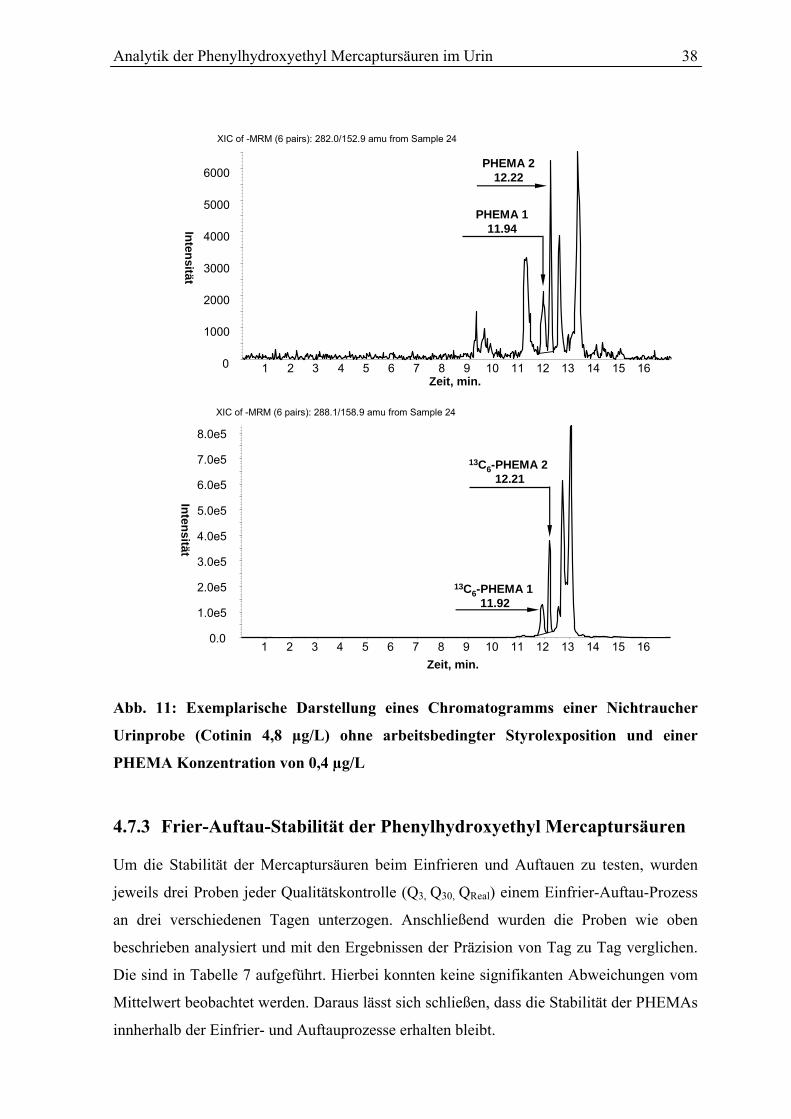
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 38
XIC of -MRM (6 pairs): 288.1/158.9 amu from Sample 24
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
Zeit, min.
0.0
1.0e5
2.0e5
3.0e5
4.0e5
5.0e5
6.0e5
7.0e5
8.0e5
Inten
sität
13C6-PHEMA 212.21
XIC of -MRM (6 pairs): 282.0/152.9 amu from Sample 24
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16Zeit, min.
0
1000
2000
3000
4000
5000
6000
Inten
sität
PHEMA 212.22
PHEMA 111.94
13C6-PHEMA 111.92
Abb. 11: Exemplarische Darstellung eines Chromatogramms einer Nichtraucher
Urinprobe (Cotinin 4,8 µg/L) ohne arbeitsbedingter Styrolexposition und einer
PHEMA Konzentration von 0,4 µg/L
4.7.3 Frier-Auftau-Stabilität der Phenylhydroxyethyl Mercaptursäuren
Um die Stabilität der Mercaptursäuren beim Einfrieren und Auftauen zu testen, wurden
jeweils drei Proben jeder Qualitätskontrolle (Q3, Q30, QReal) einem Einfrier-Auftau-Prozess
an drei verschiedenen Tagen unterzogen. Anschließend wurden die Proben wie oben
beschrieben analysiert und mit den Ergebnissen der Präzision von Tag zu Tag verglichen.
Die sind in Tabelle 7 aufgeführt. Hierbei konnten keine signifikanten Abweichungen vom
Mittelwert beobachtet werden. Daraus lässt sich schließen, dass die Stabilität der PHEMAs
innherhalb der Einfrier- und Auftauprozesse erhalten bleibt.
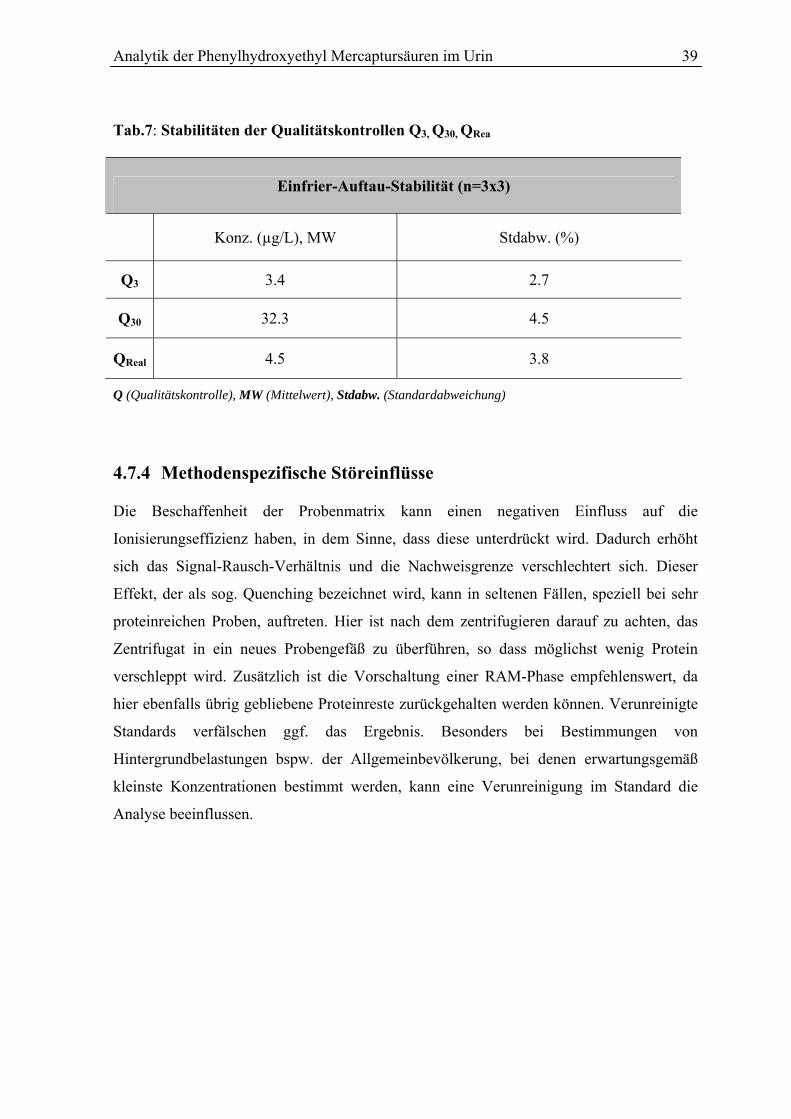
Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 39
Tab.7: Stabilitäten der Qualitätskontrollen Q3, Q30, QRea
Einfrier-Auftau-Stabilität (n=3x3)
Konz. (µg/L), MW Stdabw. (%)
Q3 3.4 2.7
Q30 32.3 4.5
QReal 4.5 3.8
Q (Qualitätskontrolle), MW (Mittelwert), Stdabw. (Standardabweichung)
4.7.4 Methodenspezifische Störeinflüsse
Die Beschaffenheit der Probenmatrix kann einen negativen Einfluss auf die
Ionisierungseffizienz haben, in dem Sinne, dass diese unterdrückt wird. Dadurch erhöht
sich das Signal-Rausch-Verhältnis und die Nachweisgrenze verschlechtert sich. Dieser
Effekt, der als sog. Quenching bezeichnet wird, kann in seltenen Fällen, speziell bei sehr
proteinreichen Proben, auftreten. Hier ist nach dem zentrifugieren darauf zu achten, das
Zentrifugat in ein neues Probengefäß zu überführen, so dass möglichst wenig Protein
verschleppt wird. Zusätzlich ist die Vorschaltung einer RAM-Phase empfehlenswert, da
hier ebenfalls übrig gebliebene Proteinreste zurückgehalten werden können. Verunreinigte
Standards verfälschen ggf. das Ergebnis. Besonders bei Bestimmungen von
Hintergrundbelastungen bspw. der Allgemeinbevölkerung, bei denen erwartungsgemäß
kleinste Konzentrationen bestimmt werden, kann eine Verunreinigung im Standard die
Analyse beeinflussen.

Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 40
4.8 Applikabilität der Methode
Um die Applikabilität der Methode zu testen, wurden Urinproben von 40 Personen der
Allgemeinbevölkerung ohne berufliche Belastung mit Styrol auf das Vorhandensein
styrolspezifischer Mercaptursäuren untersucht. Bis zur Analyse waren alle Proben bei
-20°C gelagert.
Cotinin
Zusätzlich zu den von den Probanden angegebenen Informationen zum Raucherstatus,
wurde dieser mittels urinständigem Cotinin bestimmt. Die zu messenden Proben wurden
bei Raumtemperatur equilibriert, durchmischt und in ein 1,8 ml Schraubglas pipettiert.
Hinzugegeben wurden ein deuterierter interner Standard (d-3-Cotinin),
Ammoniumacetatpuffer und Acetonitril. Es folgte ein mehrmaliges resuspendieren mit
anschließender Zentrifugation der Proben. Der Überstand wurde bei Auftreten eines
sichtbaren Sediments vorsichtig in ein neues Probenglas pipettiert. Für die Analyse wurden
10 µl der Probe flüssigkeitschromatographisch aufgetrennt und anschließend tandem-
massenselektiv quantifiziert (Schettgen, 2009). Bei Messungen von Cotinin in Harnproben
wurde zur Bestimmung von Rauchern / Nicht-Rauchern ein Proben cut-off von 100 µg/L
definiert (Haufroid & Lison, 1998).
Alle Personen mussten im Vorfeld für die weitere Analyse ihrer Urinproben ihr
Einverständnis erklären. Zum direkten Vergleich mit einer akzidentellen Belastung
konnten noch Urinproben von zwei Personen, durch die im Rahmen einer
Betriebsbesichtigung einer styrolverarbeitenden Firma aufgekommenen Kurzzeitexposition
(~ 1,5 h) mit Styrol, hinzugezogen werden. Die Ergebnisse sind in Tab.8 zusammengefasst.
Die Urinproben der exponierten Personen zeigten deutlich erhöhte PHEMA
Konzentrationen im Vergleich zum Median der nicht exponierten. Auch im Vergleich der
Raucher zu den Nichtrauchern zeigte sich ein signifikanter Unterschied (p< 0,05) in der
PHEMA Konzentration.

Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 41
Tab.8: Ergebnisse des Biomonitorings von Personen der Allgemeinbevölkerung
PHEMA
(µg/g Kreatinin)
Cotinin
(µg/L Urin)
Nichtraucher (n=22) Median
Spannbreite
< 0.3
< 0.3 – 1.4
4.0
2.9 – 7.7
Raucher (n=18) Median
Spannbreite
0.35
< 0.3 – 1.4
1398
283 - 4943
Exp.Person 1
Exp.Person 2
3.0
2.7
4.4
3.0

Analytik der Phenylhydroxyethyl Mercaptursäuren im Urin 42
4.9 Diskussion der Methode
In der hier etablierten Methode konnte gezeigt werden, dass diese zuverlässige Messungen
der urinständigen Mercaptursäuren des Styrols erlaubt. Durch den Nutzen eines
markierten, internen Standards war es möglich potentielle Matrixeffekte zu kompensieren
und quantitativ sichere Ergebnisse zu generieren. Die Ergebnisse der Präzisionen als auch
der Richtigkeit mit sehr geringen Standardabweichungen zeigten die sehr gute
Wiederholbarkeit und Robustheit der Methode. Andere Autoren berichteten über die
chromatographische Auftrennung der beiden PHEMA Isomere in ihre Diastereomere, was
als Resultat 4 Peaks ergab (Manini et al, 2000). Unter den beschriebenen
chromatographischen Bedingungen konnte diese Auftrennung nicht nachvollzogen werden.
Definierte Standards der Diastereomere waren zudem nicht käuflich zu erwerben. Darüber
hinaus konnte der Zulieferer keine Angaben zum vorliegenden Verhältnis der einzelnen
Diastereomere zueinander machen, so dass hier kein weiterer Informationsgewinn zu
erwarten war. Hier wurde der Schwerpunkt auf die Optimierung der Methode für eine
maximale Sensitivität gelegt, durch die sogar PHEMA-Konzentrationen in der
Normalbevölkerung (Raucher und Nichtraucher) nachgewiesen werden konnten. Eine
signifikante Korrelation zwischen der Subgruppe der Raucher und dem spezifischen
Marker Cotinin, konnte nicht beobachtet werden (r=0,36; p=0,13), was auch auf andere
Expositionsquellen als durch Rauchen oder interindividuelle Aktivitäten von Styrol
metabolisierenden Enzymen (bspw. Glutathion S-transferasen) zurückgeführt werden
könnte. Bisher wurden Hintergrundbelastungen im Bereich der Nachweisgrenze von 0,7-
1.0 µg/L bei nicht-exponierten Personengruppen der Allgemeinbevölkerung nachgewiesen.
Dies allerdings ausschließlich bei dem rauchenden Anteil des untersuchten Kollektivs
(Manini et al, 2000). Auch Fustinoni et al. wiesen PHEMAs in ihrem Kontrollkollektiv
nach (<1,6-8 µg/L), allerdings ohne eine explizite Auftrennung von Rauchern und
Nichtrauchern (Fustinoni et al, 2008). Im Kollektiv, das innerhalb dieser Arbeit untersucht
wurde, konnte neben den Konzentrationen der Subgruppe der Raucher auch das
Vorkommen von Mercaptursäuren bei den Nichtrauchern ermittelt und definiert werden
(s.Tab.8) Beide Gruppen unterschieden sich signifikant voneinander (p<0.05). Die im
Vergleich zu den Werten der nicht exponierten Personen deutlich höheren Konzentrationen
der Kurzzeitexponierten zeigt die hohe Sensitivität der Methode.

Analytik der Hämoglobinaddukte im Blut
43
5 Analytik der Hämoglobinaddukte im Blut
5.1 Grundlage des Verfahrens
Das Verfahren welches hier Anwendung fand, diente zur Bestimmung der
Hämoglobinaddukte des Styrols bei Personengruppen, die beruflich oder umweltbedingt
mit dieser Substanz in Kontakt kamen. Um die Addukte am N-terminalen Valin bestimmen
zu können, mussten die Erythrozyten aus gewonnenem Vollblut isoliert und von den
Plasmabestandteilen abgetrennt werden. Anschließend erfolgte die Lyse der Zellen. Über
Ethylacetat wurde in der Folge aus dem durch die Lyse gewonnenen Erythrozythämolysat
die Proteinfraktion, das Globin, ausgefällt und durch organisches Lösemittel getrocknet.
Der modifizierte Edmann-Abbau diente der Abspaltung des an die Proteinkette alkylierten
Valins, wodurch das sog. Pentafluorphenylthiohydantoin-Derivat entstand. Durch eine
flüssig-flüssig Extraktion konnte dieses mittels Diethylether aus der Proteinmatrix
extrahiert werden. Weitere Waschschritte dienten der Eliminierung von Störsubstanzen.
Abschließend entstand der Analyt durch die Acetylierung des 2-Hydrox-2-Phenylethyl-
Valin-Pentafluorphenylthiohydantoin. Mittels kapillarer Gaschromatographie wurden
weitere Störsubstanzen vom Analyten abgetrennt. Die weitere Detektion und
Quantifizierung erfolgte massenspektrometrisch im electron-impact (EI) Modus.
Zur Quantifizierung wurden Kalibrierkurven vom Poolglobin von Nichtrauchern der
Allgemeinbevölkerung mit keiner bekannten arbeitsspezifischen Styrolbelastung erstellt
und verwendet. Dieses Poolglobin wurde mit unterschiedlichen Konzentrationen eines
Dipeptidstandards versetzt, der die letzten beiden N-terminalen Aminosäuren der
Hämoglobinkette simuliert, an denen sich das Addukt bildet. Diese Kalibrierstandards
wurden analog mit den zu untersuchenden Proben aufgearbeitet. Als interner Standard
wurde den Proben eine Substanz zugegeben, welche ein Valin-Addukt bildet, das sowohl
arbeitsmedizinisch als auch umweltmedizinisch nicht vorkommt (4-Hydroxyphenethyl-
bromid, 4-Fluorphenethylbromid).

Analytik der Hämoglobinaddukte im Blut
44
5.2 Geräte, Chemikalien und Lösungen
5.2.1 Geräte
Gaschromatograph-System (Hewlett Packard 6890 Series):
mit Split/Splitless-Injektor (7683 Series)
massenselektiver Detektor (MSD 5973)
Autosampler
Kapillargaschromatographische Säule:
DB-17HT; Länge 30 m; Ø innen 0,25 mm; Filmdicke 0,15 µm (Wicom)
Vortex Mixer (Heidolph)
Zentrifuge Rotina 420R (Hettich)
Magnetrührer (IKA Labortechnik)
Probenwippe (Ortho Diagnostic Systems)
EDTA Monovetten (Sarstedt)
Schraubgläschen mit teflonkaschierten Gummisepten und Schraubdeckel (Grössen 5 ml,
20 ml) (Macherey-Nagel)
Bördelkappen für 1,5 ml Rollrandampullen (Macherey-Nagel)
Rollrandampullen 1,5 ml und Mikrovials (1 ml) (Macherey-Nagel)
Bördelzange (Macherey-Nagel)
Wasserbad (Memmert)
Reacti-Vap III Evaporatorstation (Pierce)
Erlenmeyer Kolben 10 ml (Brand)
Mikroliterpipetten, verstellbar 10-100 (100- 1000) µl (Eppendorf)
Messkolben 10- und 25-ml (Brand)
Falcon Tubes; 50 ml (BD)

Analytik der Hämoglobinaddukte im Blut
45
5.2.2 Chemikalien
Sofern keine weiteren Angaben gemacht werden, wurden alle benannten Chemikalien in
der Reinheit von mindestens p.a.-Qualität verwendet.
Natriumchlorid (Merck)
aqua bidest (Millipore®)
Salzsäure konzentriert 37 % (Merck)
2-Propanol (Merck)
Ethylacetat (Merck)
n-Hexan (Gaschromatographie) (Merck)
Formamid ultrapure (United States Biochemicals)
Natriumhydroxid (Merck)
Pentafluorphenylisothiocyanat (Fluka)
Diethylether (Merck)
Toluol (Gaschromatographie) (Merck)
Natriumhydrogencarbonat (Merck)
Acetonitril; HPLC grade (J.T. Baker)
Essigsäureanhydrid (Merck)
N-(2-Hydroxy-2-Phenylethyl)-Val-Leu-anilid (Bachem)
4-Hydroxyphenetylbromid (Sigma-Aldrich)
4-Fluorphenethylbromid (Sigma-Aldrich)
Aceton (Gaschromatographie) (Merck)
5.2.3 Lösungen
50 mM HCl in 2-Propanol
In einen 1000 ml Messkolben wurden ca. 500 ml 2-Propanol vorgelegt. Es wurden 4,1 ml
konzentrierte Salzsäure (37%) zugegeben und der Messkolben mit 2-Propanol bis zur
Messmarke aufgefüllt.
0,9 %ige NaCl-Lösung
9 g NaCl wurden in einen 1000 ml Messkolben eingewogen und der Kolben mit aqua
bidest bis zur Messmarke aufgefüllt.

Analytik der Hämoglobinaddukte im Blut
46
1 M NaOH-Lösung
2 g NaOH wurden in einem Becherglas eingewogen und mit ca. 30 ml aqua bidest
aufgelöst. Die Lösung wurde unter mehrmaligem Nachspülen mit aqua bidest in einen
50-ml Messkolben überführt und der Kolben mit Wasser bis zur Messmarke aufgefüllt.
Diese Lösung war im Kühlschrank ca. 3 Tage haltbar und sollte nach diesem Zeitraum
frisch angesetzt werden.
0,1 M Na2CO3-Lösung
530 mg Na2CO3 (wasserfrei) wurden in einem Becherglas eingewogen und in ca. 30 ml
aqua bidest aufgelöst. Die Lösung unter mehrmaligem Nachspülen mit aqua bidest in einen
50-ml Messkolben überführt und der Kolben mit aqua bidest bis zur Messmarke aufgefüllt.
Lösung des Standards
Lösung N-(2-Hydroxy-2-Phenylethyl)-Val-Leu-anilid (1mM):
NH
O
CH3
CH3
CH3CH3
NH
OOH
NH
NH
O
CH3
CH3
CH3CH3
NH
OOH
NH
Abb.12: Molekülstruktur des N-(2-Hydroxy-2-Phenylethyl)-Val-Leu-anilid
Stammlösung
10 ml Ethanol wurden in einem 20 ml Rundkolben vorgelegt, 8,51 mg des
Dipeptidstandards anschließend eingewogen und in den Rundkolben überführt.
Anschließend wurde dieser bis zur Messmarke mit Ethanol aufgefüllt, bis zur Lösung auf
dem Magnetrührer vermischt und bei -20° gelagert.
10 µl dieser Stammlösung wurden in einen mit bereits 5 ml Ethanol vorgelegten 10-ml
Rundkolben pipettiert und bis zur Messmarke mit Ethanol aufgefüllt Dotierlösung 1
(1 µM/L)

Analytik der Hämoglobinaddukte im Blut
47
Zur Herstellung der Dotierlösung 2 (0,1 µM/L) wurden 5 ml Ethanol in einen 10 ml-
Rundkolben vorgelegt und 1 ml der Dotierlösung 1 hinzupipettiert. Anschließend wurde
der Rundkolben bis zur Messmarke mit Ethanol aufgefüllt.
Herstellung der internen Standards (ISTDs):
4-Hydroxyphenetylbromid (s) und 4-Fluorphenethylbromid (l)
10 mg bzw. 10 µl wurden in 100 µl Ethanol gelöst. Anschließend wurden diese zu 2,5 ml
Erythrozythämolysat und 100 µl 0,1 M Na2CO3 hinzugegeben und für 20 Stunden
durchmischt. Aus der Bindung an das N-terminale Valin des Globins entsteht das Addukt
(interner Standard) des 4-Hydroxyphenetylbromids (4-Hydroxyphenetylbromid-Globin,
HBP-G) bzw. des Fluorphenethylbromids (4-Fluorphenethylbromid-Globin, FBP-G). Im
Anschluss wurden die Globine isoliert (s.u. Globinisolierung). 100 mg des isolierten
Globins wurden in 3 ml Formamid gelöst und aus dieser Stammlösung eine Verdünnung
von 1:5 mit Formamid hergestellt.
Abb.13: Molekülstruktur des 4-Hydroxyphenetylbromid (HPB) (a), 4-Fluor-
phenethylbromid (FPB) (b) und der daraus hergestellten internen Standards 4-
Hydroxyphenetylbromid-Globin sowie 4-Hydroxyphenetylbromid-Globin
Br
OH+ Globin
CH3 CH3
NH2
O
OH Globin
NH
O
Addukt tragendes Globin4-Hydroxyphenetylbromid -
Globin
CH3
CH3
N-terminales Valin der
Globinseitenkette
(A) 4-Hydroxyphenetylbromid (HPB)
Br
F+ Globin
CH3 CH3
NH2
O
F Globin
NH
O
CH3
CH3
Addukt tragendes Globin4-Hydroxyphenetylbromid -
Globin
N-terminales Valin der
Globinseitenkette
(B) 4-Fluorphenetylbromid (FBP)

Analytik der Hämoglobinaddukte im Blut
48
5.3 Probennahme und Probenaufbereitung
5.3.1 Gewinnung des Erythrozythämolysats
5 ml Vollblut wurden dem Probanden mittels einer EDTA Monovette entnommen.
Anschließend wurde die Monovette 10 min bei 800 g zentrifugiert, danach der obere
Flüssigkeitsstand an der Monovette markiert und die Plasamafraktion entnommen und
verworfen. Mit 0,9% NaCl wurde die Fraktion der Erythrozyten wieder auf das
Ursprungsvolumen aufgefüllt, kurz mit dem Vortexer durchmischt für 10 min bei 800 g
zentrifugiert. Erneut wurde der Überstand bis zur Erythrozytfraktion verworfen. Der
Vorgang wurde wiederholt, bis der Überstand der NaCl-Lösung klar und farblos war
(i.d.R. 2-3 Mal). Die Lyse der Erythrozyten wurde durch Auffüllen mit Wasser auf das
Ursprungsvolumen durchgeführt. Die erhaltenen Erythrozythämolysate konnten
abschließend bis zur weiteren Nutzung bis zu 12 Monate bei -20° C gelagert werden.
5.3.2 Globinisolierung
2 ml Erythrozythämolysat pro Probe wurden in ein 50 ml Plastikreaktionsgefäß pipettiert
und 12 ml 50 mM HCl - 2-Propanol hinzugegeben. Anschließend erfolgte ein 1-minütiges
vortexen der Proben. Um Zellreste zu sedimentieren erfolgte im Anschluss ein
Zentrifugationsschritt von 10 min bei 2773 g. Der klare, braune Überstand wurde in ein 20
ml Schraubglas dekantiert. Zur Ausfällung des Hämoglobins wurden langsam mittels
Tropftrichter 8 ml Ethylacetat zu jeder Probe hinzugegeben und diese anschließend für 60
Minuten bei 4°C gelagert. Es folgte ein 5-minütiges zentrifugieren bei 800 g, um das
ausgefällte Globin zu sedimentieren. Der Überstand wurde verworfen und das
sedimentierte Globin in 10 ml Ethylacetat resuspendiert, 2 min mit dem Vortexer
vermischt, erneut bei 800 g für 5 min zentrifugiert und der Überstand verworfen. Dieser
Vorgang wurde wiederholt und das Globin abschließend in 5 ml n-Hexan aufgenommen
und für 2 min durchmischt. Nachdem die Proben nochmal für 5 min bei 800 g zentrifugiert
wurden, lag das gereinigte Globin als weißlich-graues Pulver vor, welches über Nacht im
Vakuumexsikkator getrocknet wurde.
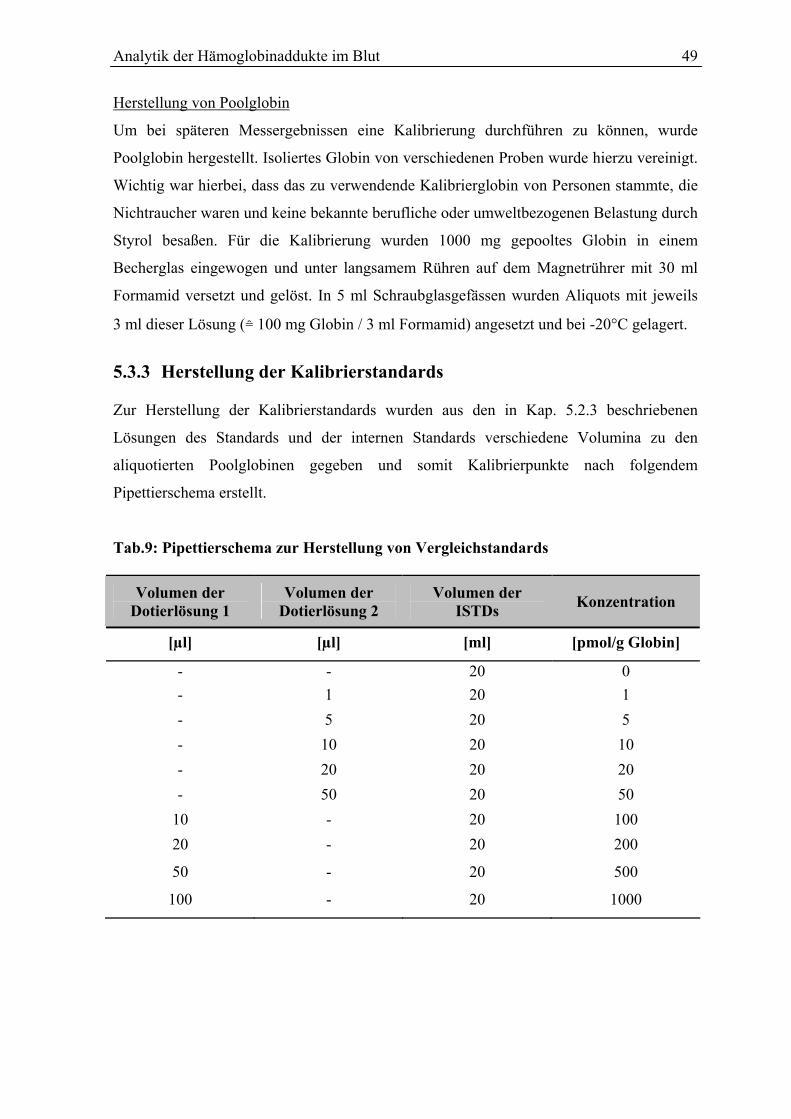
Analytik der Hämoglobinaddukte im Blut
49
Herstellung von Poolglobin
Um bei späteren Messergebnissen eine Kalibrierung durchführen zu können, wurde
Poolglobin hergestellt. Isoliertes Globin von verschiedenen Proben wurde hierzu vereinigt.
Wichtig war hierbei, dass das zu verwendende Kalibrierglobin von Personen stammte, die
Nichtraucher waren und keine bekannte berufliche oder umweltbezogenen Belastung durch
Styrol besaßen. Für die Kalibrierung wurden 1000 mg gepooltes Globin in einem
Becherglas eingewogen und unter langsamem Rühren auf dem Magnetrührer mit 30 ml
Formamid versetzt und gelöst. In 5 ml Schraubglasgefässen wurden Aliquots mit jeweils
3 ml dieser Lösung (≙ 100 mg Globin / 3 ml Formamid) angesetzt und bei -20°C gelagert.
5.3.3 Herstellung der Kalibrierstandards
Zur Herstellung der Kalibrierstandards wurden aus den in Kap. 5.2.3 beschriebenen
Lösungen des Standards und der internen Standards verschiedene Volumina zu den
aliquotierten Poolglobinen gegeben und somit Kalibrierpunkte nach folgendem
Pipettierschema erstellt.
Tab.9: Pipettierschema zur Herstellung von Vergleichstandards
Volumen der Dotierlösung 1
Volumen der Dotierlösung 2
Volumen der ISTDs
Konzentration
[µl] [µl] [ml] [pmol/g Globin]
- - 20 0
- 1 20 1
- 5 20 5
- 10 20 10
- 20 20 20
- 50 20 50
10 - 20 100
20
50
100
-
-
-
20
20
20
200
500
1000

Analytik der Hämoglobinaddukte im Blut
50
5.3.4 Derivatisierung des Globins
100 mg des zuvor isolierten Globins der jeweiligen Probe wurden in ein 5 ml Schraubglas
abgewogen und in 3 ml Formamid bis zur Lösung auf dem Wippschüttler durchmischt.
Nach Lösung des Globins wurden 40 µl 1 M NaOH, 20 µl der Lösung der internen
Standards und 15 µl Pentafluorphenylisothiocyanat hinzugegeben und die Probe bis zum
nächsten Tag auf dem Wippschüttler durchmischt. Anschließend wurde die Probe für 90
Minuten im Wasserbad bei 45°C inkubiert. Nachdem die Proben abgekühlt waren, wurden
weitere 75 µl NaOH hinzugegeben und die Formamidphase zwei Mal mit jeweils 3 ml
Diethylether extrahiert. Dafür wurden die Proben zwischen jedem Extraktionsschritt
jeweils für eine Minute mit dem Vortex-Mixer durchmischt bevor sie anschließend bei
2773 g zentrifugiert wurden. Die aus jedem Extraktionsschritt gewonnen Etherphasen
wurden in einem neuen 5 ml Schraubglas gesammelt und unter einem leichten
Stickstoffstrom bis zur Trockenheit eingedampft. Die trockenen Rückstände der Proben
wurde anschließend in 1,5 ml Toluol und 2 ml aqua bidest aufgenommen, 1 Minute auf
dem Vortex-Mixer durchmischt und 10 min bei 1083 g zentrifugiert. Die Wasserphase
wurde verworfen, der Vorgang ein weiteres Mal mit 2 ml aqua bidest und ein
abschließendes Mal mit 2 ml 0,1 M Na2CO3-Lösung wiederholt. Nach diesem letzten
Schritt wurde die Toluolphase entnommen und in ein neues 5 ml Schraubglas überführt.
Bei geringer Wärmezufuhr wurde das Toluol unter leichtem Stickstoffstrom bis zur
Trockene eingeengt.
5.3.5 Acetylierung des Pentafluorphenylthiohydantoin-Derivates
Der unter 5.3.4 erhaltene trockene Rückstand wurde in 150 µl Acetonitril, 20 µl Pyridin
und 20 µl Essigsäureanhydrid gegeben und über Nacht auf dem Wippschüttler
durchmischt. Anschließend wurden 1,5 ml Toluol und 2 ml aqua bidest hinzugegeben, 1
Minute auf dem Vortex-Mixer durchmischt und 10 min bei 1083 g zentrifugiert. Die
Wasserphase wurde verworfen der Vorgang ein weiteres Mal wiederholt. Danach wurde
die Toluolphase abgenommen, in eine 2 ml Rollrandampulle überführt und unter geringer
Wärmezufuhr bei leichtem Stickstoffstrom bis zur Trockene eingeengt. Der Rückstand
wurde in 50 µl Toluol resuspendiert und in ein Mikrovial überführt, dieses in die
Rollrandampulle gestellt und mit einer teflonkaschierten Aluminiumverschlusskappe
verschlossen. 1 µl dieser Lösung wurde in den Gaschromatographen injiziert.
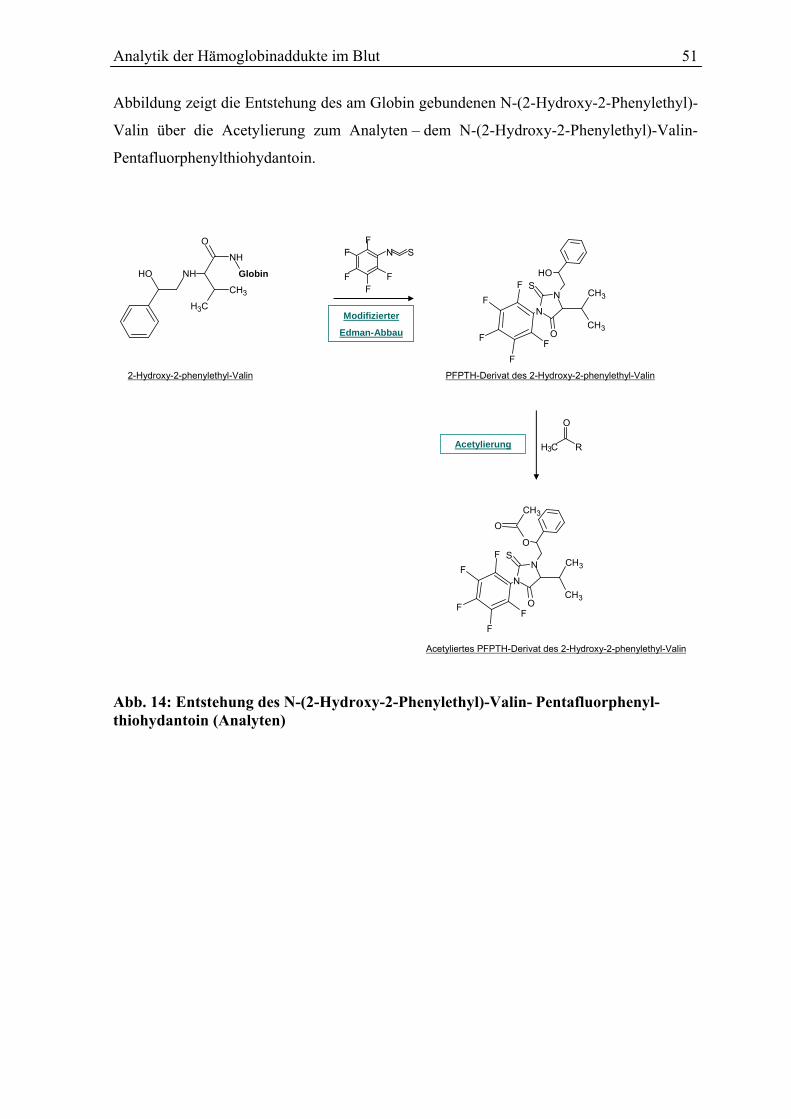
Analytik der Hämoglobinaddukte im Blut
51
Abbildung zeigt die Entstehung des am Globin gebundenen N-(2-Hydroxy-2-Phenylethyl)-
Valin über die Acetylierung zum Analyten – dem N-(2-Hydroxy-2-Phenylethyl)-Valin-
Pentafluorphenylthiohydantoin.
NH
O
CH3
CH3
NHOH Globin
SN
FF
F
FF
O
CH3
OS
NF
F
F
F
FN
O
CH3
CH3
OHS
NF
F
F
F
FN
O
CH3
CH3
R
O
CH3
Modifizierter
Edman-Abbau
Acetylierung
2-Hydroxy-2-phenylethyl-Valin PFPTH-Derivat des 2-Hydroxy-2-phenylethyl-Valin
Acetyliertes PFPTH-Derivat des 2-Hydroxy-2-phenylethyl-Valin
Abb. 14: Entstehung des N-(2-Hydroxy-2-Phenylethyl)-Valin- Pentafluorphenyl-thiohydantoin (Analyten)

Analytik der Hämoglobinaddukte im Blut
52
5.4 Gaschromatographische Arbeitsbedingungen
Kapillarsäule DB 17-HT
30 m x 0,25 mm, 0,15 µm Filmdicke (Wicom)
Trägergas Helium 5.0
Flussgeschwindigkeit 1,20 ml/min (constant flow)
Injektion 1 µl, splitless
Inlet purge off time 1 min
Gerätetemperaturen
Injektor 300°C
Transferline 300°C
Säulenofen 90°C, 1 min halten
25 °C/min auf 120°C
10°C/min auf 255 °C
5°C/min auf 290°C, 20 min halten
25°C/min auf 310°C, 5 min halten
Ionisierung Negative chemische Ionisation(NCI)
Ionisierungsenergie 100,5 eV
Detektion Selected Ion Monitoring (SIM)
Messzeit pro Ion (dwell time) 100 ms
Elektronenmultiplier 306 V rel (~2106 V)
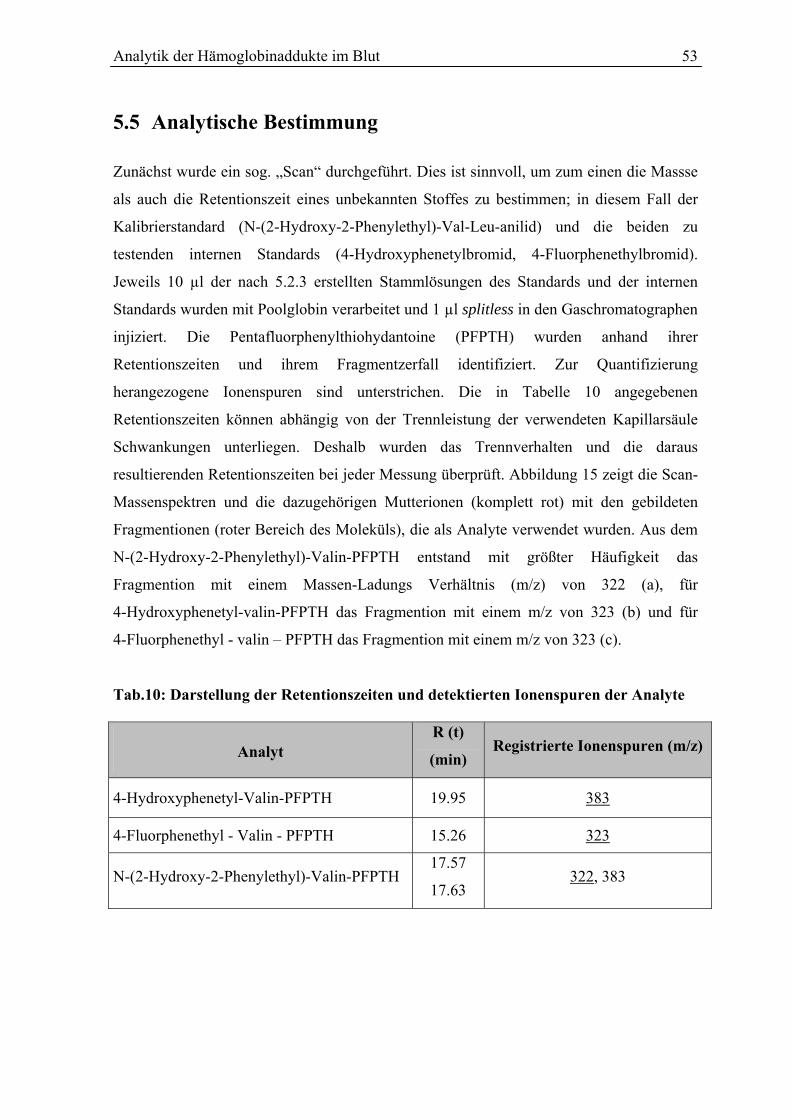
Analytik der Hämoglobinaddukte im Blut
53
5.5 Analytische Bestimmung
Zunächst wurde ein sog. „Scan“ durchgeführt. Dies ist sinnvoll, um zum einen die Massse
als auch die Retentionszeit eines unbekannten Stoffes zu bestimmen; in diesem Fall der
Kalibrierstandard (N-(2-Hydroxy-2-Phenylethyl)-Val-Leu-anilid) und die beiden zu
testenden internen Standards (4-Hydroxyphenetylbromid, 4-Fluorphenethylbromid).
Jeweils 10 µl der nach 5.2.3 erstellten Stammlösungen des Standards und der internen
Standards wurden mit Poolglobin verarbeitet und 1 µl splitless in den Gaschromatographen
injiziert. Die Pentafluorphenylthiohydantoine (PFPTH) wurden anhand ihrer
Retentionszeiten und ihrem Fragmentzerfall identifiziert. Zur Quantifizierung
herangezogene Ionenspuren sind unterstrichen. Die in Tabelle 10 angegebenen
Retentionszeiten können abhängig von der Trennleistung der verwendeten Kapillarsäule
Schwankungen unterliegen. Deshalb wurden das Trennverhalten und die daraus
resultierenden Retentionszeiten bei jeder Messung überprüft. Abbildung 15 zeigt die Scan-
Massenspektren und die dazugehörigen Mutterionen (komplett rot) mit den gebildeten
Fragmentionen (roter Bereich des Moleküls), die als Analyte verwendet wurden. Aus dem
N-(2-Hydroxy-2-Phenylethyl)-Valin-PFPTH entstand mit größter Häufigkeit das
Fragmention mit einem Massen-Ladungs Verhältnis (m/z) von 322 (a), für
4-Hydroxyphenetyl-valin-PFPTH das Fragmention mit einem m/z von 323 (b) und für
4-Fluorphenethyl - valin – PFPTH das Fragmention mit einem m/z von 323 (c).
Tab.10: Darstellung der Retentionszeiten und detektierten Ionenspuren der Analyte
Analyt R (t)
(min) Registrierte Ionenspuren (m/z)
4-Hydroxyphenetyl-Valin-PFPTH 19.95 383
4-Fluorphenethyl - Valin - PFPTH 15.26 323
N-(2-Hydroxy-2-Phenylethyl)-Valin-PFPTH 17.57
17.63 322, 383
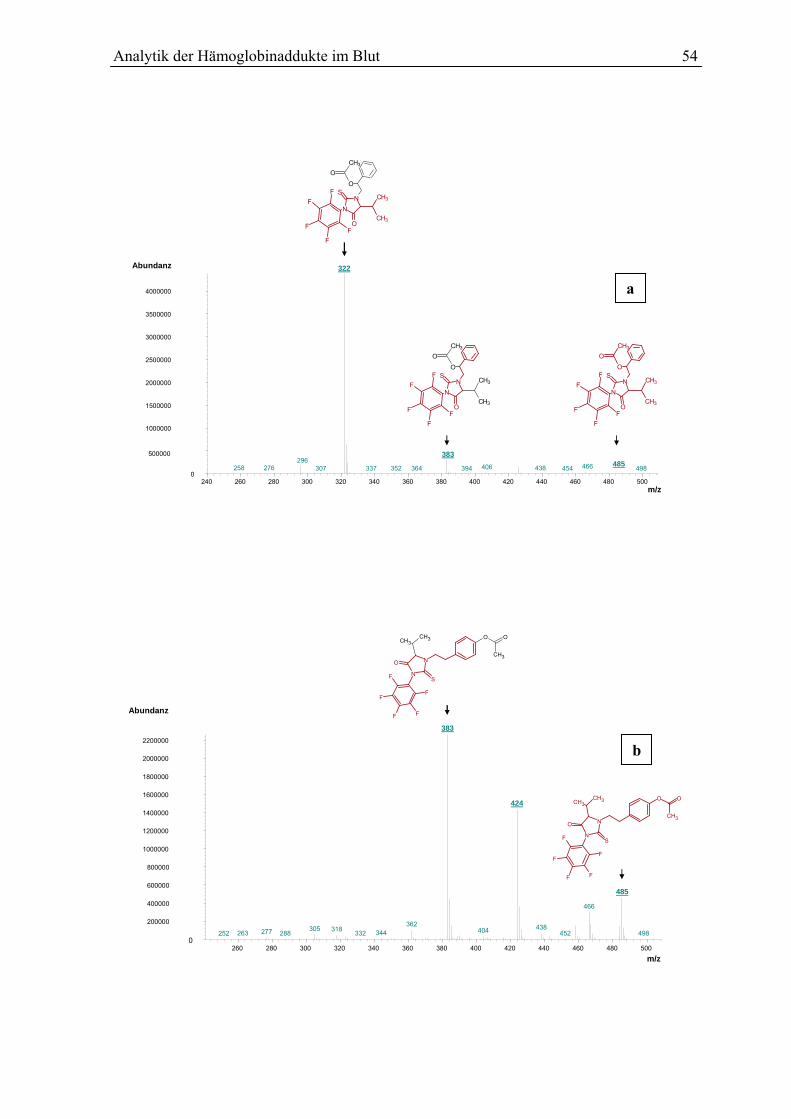
Analytik der Hämoglobinaddukte im Blut
54
240 260 280 300 320 340 360 380 400 420 440 460 480 5000
500000
1000000
1500000
2000000
2500000
3000000
3500000
4000000
m/z
Abundanz 322
383296
466406 485276258 438352 364337307 454 498394
O
CH3
OS
NF
F
F
F
FN
O
CH3
CH3
O
CH3
OS
NF
F
F
F
FN
O
CH3
CH3
O
CH3
OS
NF
F
F
F
FN
O
CH3
CH3
260 280 300 320 340 360 380 400 420 440 460 480 5000
200000
400000
600000
800000
1000000
1200000
1400000
1600000
1800000
2000000
2200000
383
424
485
466
362 438305 318 404277 344263 332288 452 498252
Abundanz
m/z
O
SN
F
F F
F
F
NO
CH3CH3 O
CH3
O
SN
F
F F
F
F
NO
CH3CH3 O
CH3
a
b
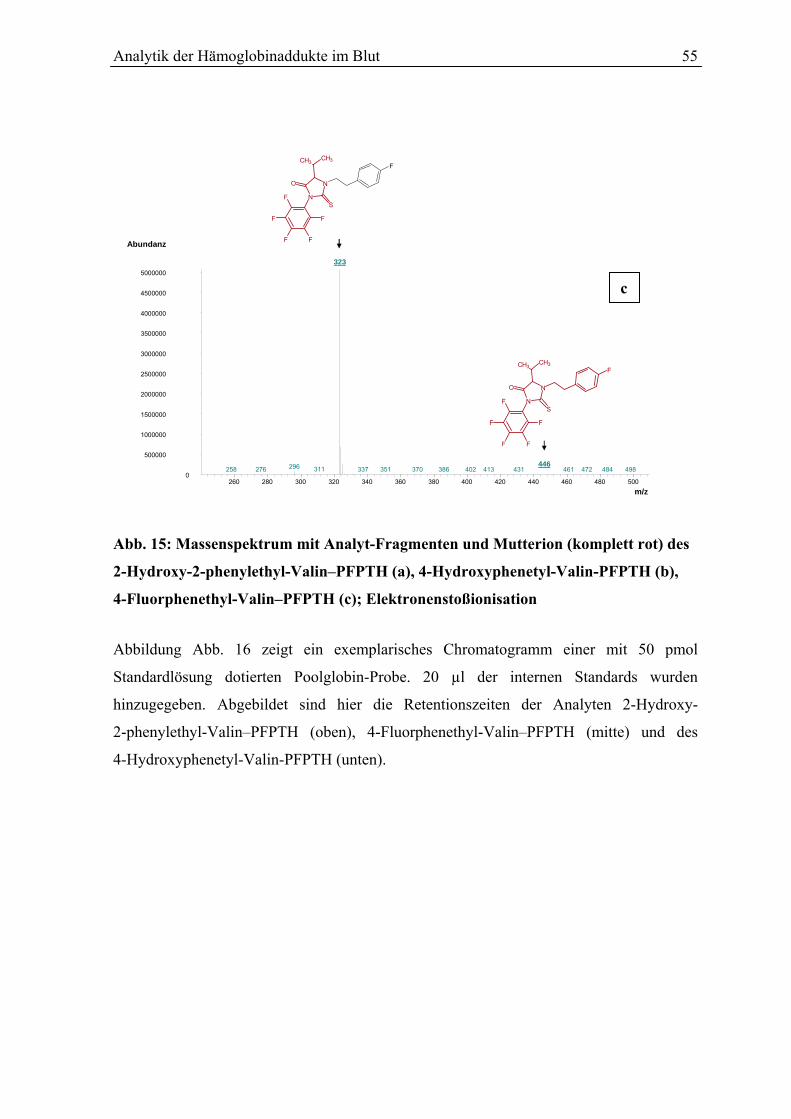
Analytik der Hämoglobinaddukte im Blut
55
260 280 300 320 340 360 380 400 420 440 460 480 5000
500000
1000000
1500000
2000000
2500000
3000000
3500000
4000000
4500000
5000000
323
296 311 351 370 386276 413446
258 402337 484472461431 498
Abundanz
m/z
F
SN
F
F F
F
F
NO
CH3CH3
F
SN
F
F F
F
F
NO
CH3CH3
Abb. 15: Massenspektrum mit Analyt-Fragmenten und Mutterion (komplett rot) des
2-Hydroxy-2-phenylethyl-Valin–PFPTH (a), 4-Hydroxyphenetyl-Valin-PFPTH (b),
4-Fluorphenethyl-Valin–PFPTH (c); Elektronenstoßionisation
Abbildung Abb. 16 zeigt ein exemplarisches Chromatogramm einer mit 50 pmol
Standardlösung dotierten Poolglobin-Probe. 20 µl der internen Standards wurden
hinzugegeben. Abgebildet sind hier die Retentionszeiten der Analyten 2-Hydroxy-
2-phenylethyl-Valin–PFPTH (oben), 4-Fluorphenethyl-Valin–PFPTH (mitte) und des
4-Hydroxyphenetyl-Valin-PFPTH (unten).
c

Ana
lyti
k de
r H
ämog
lobi
nadd
ukte
im B
lut
56
15.0
015
.50
16.0
016
.50
17.0
017
.50
18.0
018
.50
19.0
019
.50
20.0
020
.50
0
500
00
100
000
150
000
200
000
Ab
un
dan
zIo
n 32
2.0
0 (3
21.7
0 to
322
.70)
: 50
.D 17.
5717
.63
15.0
015
.50
16.0
016
.50
17.0
017
.50
18.0
018
.50
19.0
019
.50
20.0
020
.50
0
500
00
100
000
150
000
200
000
Ion
323
.00
(322
.70
to 3
23.7
0): 5
0.D
15.2
6
15.0
015
.50
16.0
016
.50
17.0
017
.50
18.0
018
.50
19.0
019
.50
20.0
020
.50
0
500
00
100
000
150
000
200
000
Ion
383
.00
(382
.70
to 3
83.7
0): 5
0.D
19.9
5
Zei
t
Zei
t
Zei
t
Pe
nta
fluor
phen
ylth
ioh
ydan
toin
-De
rivat
des
2-H
ydro
xy-
2-P
he
nyl
eth
ylva
lin
Pe
nta
fluor
phen
ylth
ioh
ydan
toin
-De
rivat
des
4-F
luo
rph
en
eth
ylb
rom
id
Pe
nta
fluor
phen
ylth
ioh
ydan
toin
-De
rivat
des
4-H
ydro
xyp
he
ne
tylb
rom
idA
bu
nd
anz
Ab
un
dan
z
Ab
b. 1
6: C
hro
mat
ogra
mm
ein
er d
otie
rten
Poo
lglo
bin
pro
be
(50
pm
ol)
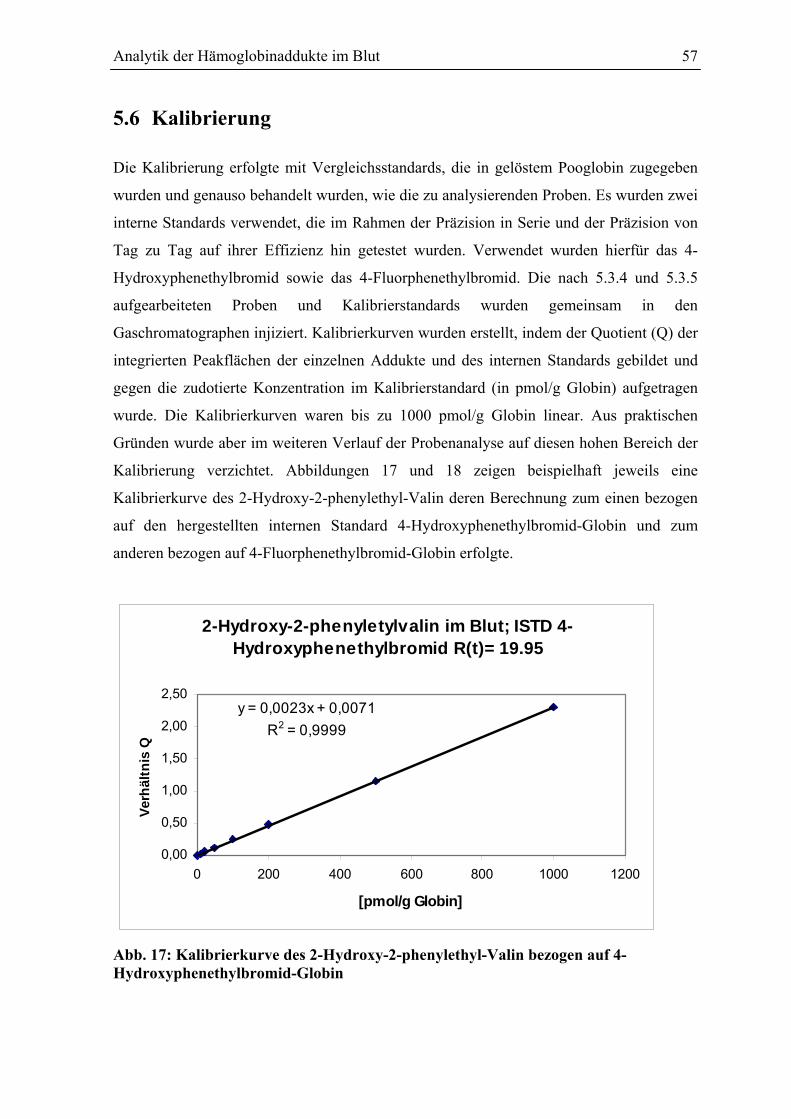
Analytik der Hämoglobinaddukte im Blut
57
5.6 Kalibrierung
Die Kalibrierung erfolgte mit Vergleichsstandards, die in gelöstem Pooglobin zugegeben
wurden und genauso behandelt wurden, wie die zu analysierenden Proben. Es wurden zwei
interne Standards verwendet, die im Rahmen der Präzision in Serie und der Präzision von
Tag zu Tag auf ihrer Effizienz hin getestet wurden. Verwendet wurden hierfür das 4-
Hydroxyphenethylbromid sowie das 4-Fluorphenethylbromid. Die nach 5.3.4 und 5.3.5
aufgearbeiteten Proben und Kalibrierstandards wurden gemeinsam in den
Gaschromatographen injiziert. Kalibrierkurven wurden erstellt, indem der Quotient (Q) der
integrierten Peakflächen der einzelnen Addukte und des internen Standards gebildet und
gegen die zudotierte Konzentration im Kalibrierstandard (in pmol/g Globin) aufgetragen
wurde. Die Kalibrierkurven waren bis zu 1000 pmol/g Globin linear. Aus praktischen
Gründen wurde aber im weiteren Verlauf der Probenanalyse auf diesen hohen Bereich der
Kalibrierung verzichtet. Abbildungen 17 und 18 zeigen beispielhaft jeweils eine
Kalibrierkurve des 2-Hydroxy-2-phenylethyl-Valin deren Berechnung zum einen bezogen
auf den hergestellten internen Standard 4-Hydroxyphenethylbromid-Globin und zum
anderen bezogen auf 4-Fluorphenethylbromid-Globin erfolgte.
2-Hydroxy-2-phenyletylvalin im Blut; ISTD 4-Hydroxyphenethylbromid R(t)= 19.95
y = 0,0023x + 0,0071
R2 = 0,9999
0,00
0,50
1,00
1,50
2,00
2,50
0 200 400 600 800 1000 1200
[pmol/g Globin]
Ver
häl
tnis
Q
Abb. 17: Kalibrierkurve des 2-Hydroxy-2-phenylethyl-Valin bezogen auf 4-Hydroxyphenethylbromid-Globin
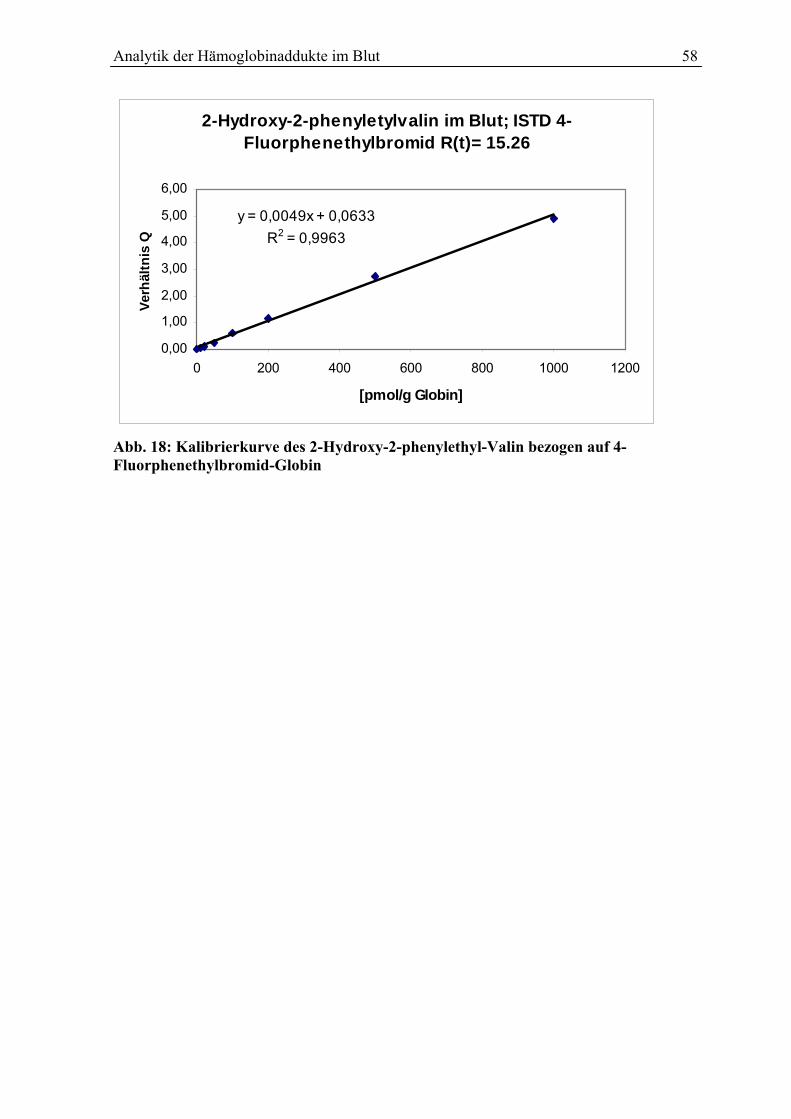
Analytik der Hämoglobinaddukte im Blut
58
2-Hydroxy-2-phenyletylvalin im Blut; ISTD 4-Fluorphenethylbromid R(t)= 15.26
y = 0,0049x + 0,0633
R2 = 0,9963
0,00
1,00
2,00
3,00
4,00
5,00
6,00
0 200 400 600 800 1000 1200
[pmol/g Globin]
Ver
häl
tnis
Q
Abb. 18: Kalibrierkurve des 2-Hydroxy-2-phenylethyl-Valin bezogen auf 4-Fluorphenethylbromid-Globin

Analytik der Hämoglobinaddukte im Blut
59
5.7 Berechnung der Analyseergebnisse
Die Konzentration des ermittelten Hämoglobinadduktes wurde durch die Bildung eines
Quotienten (Q) der jeweiligen Probe bestimmt. Dieser setzt sich zusammen aus der
integrierten Fläche des Analytpeaks und der integrierten Fläche des verwendeten internen
Standards. Anhand der Steigung der erstellten Kalibrierkurve (K) konnte anschließend die
Konzentration (C) der Probe berechnet werden. (Formel C = Q/K)
Der Achsenabschnitt ist begründet durch den physiologischen Adduktgehalts des
Poolglobins, welches für die Kalibrierung eingesetzt wurde und musste nicht für die
Berechnung berücksichtigt werden. Der internationale und nationale Standard gibt vor,
dass der Gehalt des Globinadduktes auf ein Gramm Globin bezogen und als pmol/g Globin
angegeben wird.
5.8 Standardisierung und Qualitätssicherung
Um die Qualität der Analyseergebnisse zu sichern, wurden zwei Präzisionskontrollproben
hergestellt, die in jedem Lauf mit analysiert wurden. Hierzu wurden Poolglobinproben
hergestellt, diese mit jeweils mit 40 pmol (Q40) und 250 pmol (Q250) des Standards dotiert.
Die so hergestellten Poolglobine wurden anschließend bis zur jeweiligen Verwendung bei
-20°C eingefroren.
5.9 Beurteilung des Verfahrens
5.9.1 Präzision in Serie und von Tag zu Tag
Die nach 5.3.2 hergestellten Poolglobinproben wurden zur Bestimmung der Präzision in
Serie verwendet und an sechs, mit 40 und 250 pmol dotierten Proben durchgeführt. Zur
Ermittlung der Präzision von Tag zu Tag wurden die gleichen Kontrollproben, dotiert mit
40 und 250 pmol, an sechs unterschiedlichen Tagen aufgearbeitet, analysiert und ihre
Konzentrationen ermittelt. Die Ergebnisse hierzu sind in der Tab.11 und 12 aufgeführt. Bei
den Ergebnissen der Präzision in Serie (Q40 ) zeigt sich eine höhere Wiederfindungsrate bei
Verwendung des internen Standards HPB im Vergleich zu FPB. Umgekehrt bei den
Ergebnissen zu Q250. Deutlicher zeigt sich dieser Unterschied beim Vergleich der
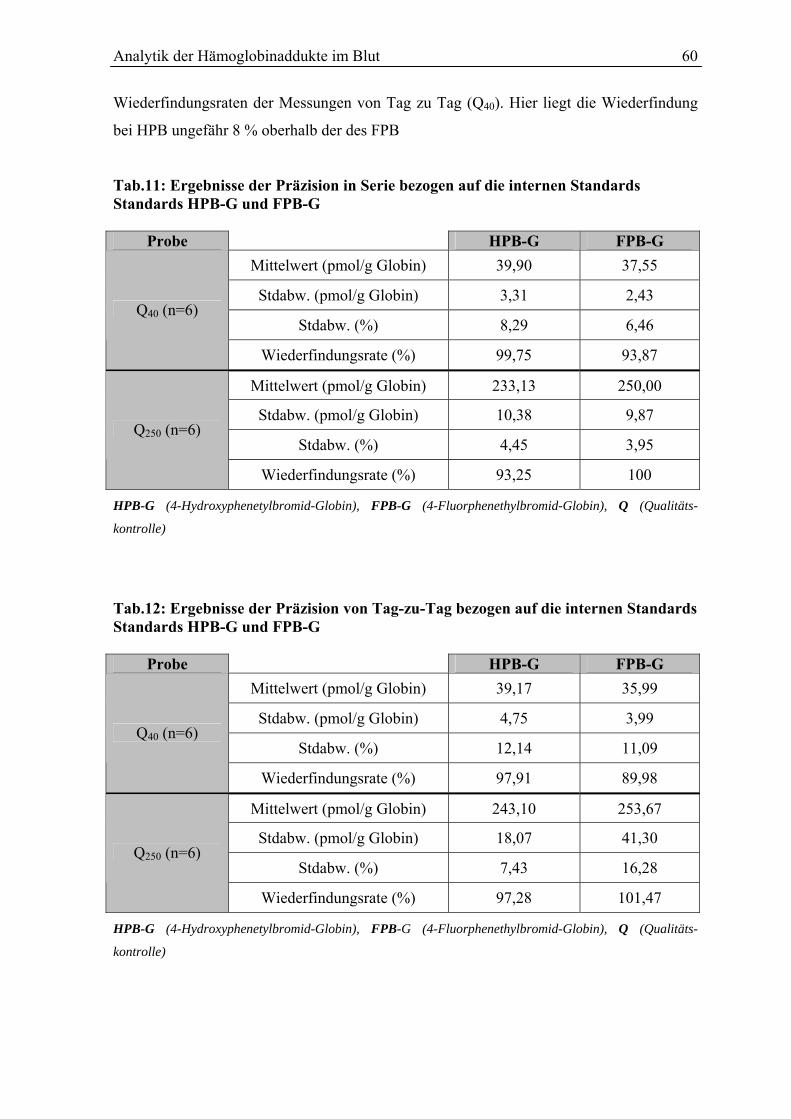
Analytik der Hämoglobinaddukte im Blut
60
Wiederfindungsraten der Messungen von Tag zu Tag (Q40). Hier liegt die Wiederfindung
bei HPB ungefähr 8 % oberhalb der des FPB
Tab.11: Ergebnisse der Präzision in Serie bezogen auf die internen Standards Standards HPB-G und FPB-G
Probe HPB-G FPB-G
Mittelwert (pmol/g Globin) 39,90 37,55
Stdabw. (pmol/g Globin) 3,31 2,43
Stdabw. (%) 8,29 6,46 Q40 (n=6)
Wiederfindungsrate (%) 99,75 93,87
Mittelwert (pmol/g Globin) 233,13 250,00
Stdabw. (pmol/g Globin) 10,38 9,87
Stdabw. (%) 4,45 3,95 Q250 (n=6)
Wiederfindungsrate (%) 93,25 100
HPB-G (4-Hydroxyphenetylbromid-Globin), FPB-G (4-Fluorphenethylbromid-Globin), Q (Qualitäts-
kontrolle)
Tab.12: Ergebnisse der Präzision von Tag-zu-Tag bezogen auf die internen Standards Standards HPB-G und FPB-G
Probe HPB-G FPB-G
Mittelwert (pmol/g Globin) 39,17 35,99
Stdabw. (pmol/g Globin) 4,75 3,99
Stdabw. (%) 12,14 11,09 Q40 (n=6)
Wiederfindungsrate (%) 97,91 89,98
Mittelwert (pmol/g Globin) 243,10 253,67
Stdabw. (pmol/g Globin) 18,07 41,30
Stdabw. (%) 7,43 16,28 Q250 (n=6)
Wiederfindungsrate (%) 97,28 101,47
HPB-G (4-Hydroxyphenetylbromid-Globin), FPB-G (4-Fluorphenethylbromid-Globin), Q (Qualitäts-
kontrolle)
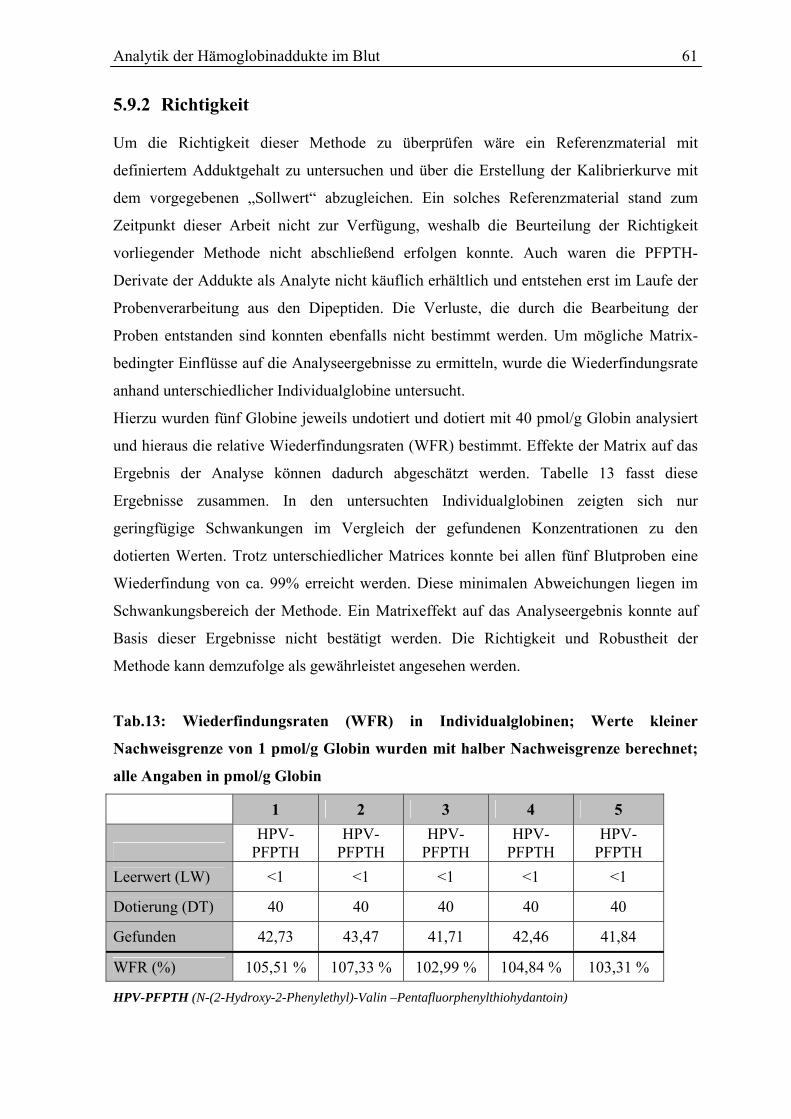
Analytik der Hämoglobinaddukte im Blut
61
5.9.2 Richtigkeit
Um die Richtigkeit dieser Methode zu überprüfen wäre ein Referenzmaterial mit
definiertem Adduktgehalt zu untersuchen und über die Erstellung der Kalibrierkurve mit
dem vorgegebenen „Sollwert“ abzugleichen. Ein solches Referenzmaterial stand zum
Zeitpunkt dieser Arbeit nicht zur Verfügung, weshalb die Beurteilung der Richtigkeit
vorliegender Methode nicht abschließend erfolgen konnte. Auch waren die PFPTH-
Derivate der Addukte als Analyte nicht käuflich erhältlich und entstehen erst im Laufe der
Probenverarbeitung aus den Dipeptiden. Die Verluste, die durch die Bearbeitung der
Proben entstanden sind konnten ebenfalls nicht bestimmt werden. Um mögliche Matrix-
bedingter Einflüsse auf die Analyseergebnisse zu ermitteln, wurde die Wiederfindungsrate
anhand unterschiedlicher Individualglobine untersucht.
Hierzu wurden fünf Globine jeweils undotiert und dotiert mit 40 pmol/g Globin analysiert
und hieraus die relative Wiederfindungsraten (WFR) bestimmt. Effekte der Matrix auf das
Ergebnis der Analyse können dadurch abgeschätzt werden. Tabelle 13 fasst diese
Ergebnisse zusammen. In den untersuchten Individualglobinen zeigten sich nur
geringfügige Schwankungen im Vergleich der gefundenen Konzentrationen zu den
dotierten Werten. Trotz unterschiedlicher Matrices konnte bei allen fünf Blutproben eine
Wiederfindung von ca. 99% erreicht werden. Diese minimalen Abweichungen liegen im
Schwankungsbereich der Methode. Ein Matrixeffekt auf das Analyseergebnis konnte auf
Basis dieser Ergebnisse nicht bestätigt werden. Die Richtigkeit und Robustheit der
Methode kann demzufolge als gewährleistet angesehen werden.
Tab.13: Wiederfindungsraten (WFR) in Individualglobinen; Werte kleiner
Nachweisgrenze von 1 pmol/g Globin wurden mit halber Nachweisgrenze berechnet;
alle Angaben in pmol/g Globin
1 2 3 4 5
HPV-
PFPTH HPV-
PFPTH HPV-
PFPTH HPV-
PFPTH HPV-
PFPTH
Leerwert (LW) <1 <1 <1 <1 <1
Dotierung (DT) 40 40 40 40 40
Gefunden 42,73 43,47 41,71 42,46 41,84
WFR (%) 105,51 % 107,33 % 102,99 % 104,84 % 103,31 %
HPV-PFPTH (N-(2-Hydroxy-2-Phenylethyl)-Valin –Pentafluorphenylthiohydantoin)
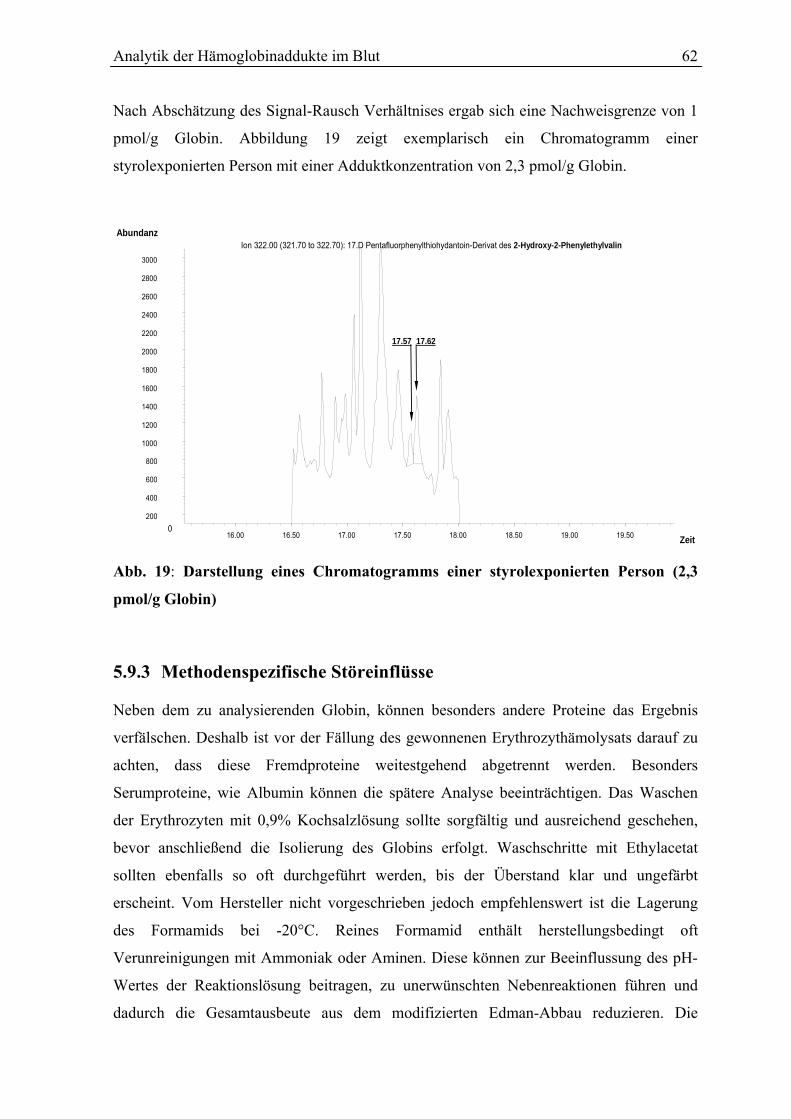
Analytik der Hämoglobinaddukte im Blut
62
Nach Abschätzung des Signal-Rausch Verhältnises ergab sich eine Nachweisgrenze von 1
pmol/g Globin. Abbildung 19 zeigt exemplarisch ein Chromatogramm einer
styrolexponierten Person mit einer Adduktkonzentration von 2,3 pmol/g Globin.
16.00 16.50 17.00 17.50 18.00 18.50 19.00 19.50
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
AbundanzIon 322.00 (321.70 to 322.70): 17.D Pentafluorphenylthiohydantoin-Derivat des 2-Hydroxy-2-Phenylethylvalin
Zeit
17.57 17.62
0
Abb. 19: Darstellung eines Chromatogramms einer styrolexponierten Person (2,3
pmol/g Globin)
5.9.3 Methodenspezifische Störeinflüsse
Neben dem zu analysierenden Globin, können besonders andere Proteine das Ergebnis
verfälschen. Deshalb ist vor der Fällung des gewonnenen Erythrozythämolysats darauf zu
achten, dass diese Fremdproteine weitestgehend abgetrennt werden. Besonders
Serumproteine, wie Albumin können die spätere Analyse beeinträchtigen. Das Waschen
der Erythrozyten mit 0,9% Kochsalzlösung sollte sorgfältig und ausreichend geschehen,
bevor anschließend die Isolierung des Globins erfolgt. Waschschritte mit Ethylacetat
sollten ebenfalls so oft durchgeführt werden, bis der Überstand klar und ungefärbt
erscheint. Vom Hersteller nicht vorgeschrieben jedoch empfehlenswert ist die Lagerung
des Formamids bei -20°C. Reines Formamid enthält herstellungsbedingt oft
Verunreinigungen mit Ammoniak oder Aminen. Diese können zur Beeinflussung des pH-
Wertes der Reaktionslösung beitragen, zu unerwünschten Nebenreaktionen führen und
dadurch die Gesamtausbeute aus dem modifizierten Edman-Abbau reduzieren. Die

Analytik der Hämoglobinaddukte im Blut
63
Lagerung bei -20°C hilft weitere, lagerungsbedingte Verunreinigungen zu vermeiden. Mit
dem Gebrauch des hier verwendeten Formamids der Firma USB Biochemicals konnten
bisher gute Erfahrungen gemacht werden. Wie bereits erwähnt sollte die Kalibrierkurve in
gepoolter Globinlösung von Nichtrauchern angesetzt werden. Dies erhöht die Präzision
durch die Reduzierung der Variabilität innerhalb des Versuchsmaterials (Matrix). Die
Dotierung des Dipeptid-Standards in reiner Formamidlösung ohne die vorhandene
Proteinmatrix des Globins führt nicht nur zu großen Störungen in der Chromatographie
sondern auch zu sehr geringen Ausbeuten im Edman-Abbau. Bei der praktischen
Durchführung kann dies unter Umständen das „Auffinden“ des PFPTH-Analyten
erschweren, da diese erst mit Fortschreiten der Aufarbeitung der Blutproben aus den
Dipeptiden gebildet werden. Wie unter 5.5 bereits erwähnt, kann es hilfreich sein dafür
eine undotierte und eine hoch dotierte Probe im Scan-Modus zu analysieren und beide
Chromatogramme miteinander zu vergleichen. Erklärungen für dieses Phänomen sind nicht
bekannt, allerdings könnte ein Erklärungsversuch in der Tatsache liegen, dass das
Vorhandensein von vielen kationischen und anionischen Aminosäureresten einen pH-
puffernden Effekt haben kann. So wäre es vorstellbar, dass die Proteinmatrix des Globins
einen puffernden Effekt auf die Lösung während des Edman-Abbaus hat, für dessen
Reaktion ein neutrales Milieu erforderlich ist.
Störeinflüsse können außerdem hervorgerufen werden durch die Bildung einer gelartigen
Konsistenz des Formamid-Diethylether-Gemisches. Die Proteinmatrix könnte hier als eine
Art Emulgator wirken wodurch keine Phasentrennung mehr möglich ist. Mittels Zugabe
von 75 µl 1 molarer NaOH vor der Extraktion mit Diethylether kann dieses Gelieren
reduziert werden.
„Fronting“-Erscheinungen im Chromatogramm lassen Rückschlüsse auf Verschmutzungen
im Bereich des Injektionssystems oder am Beginn der analytischen Säule zu. Mit dem
Wechsel des Liners bzw. dem Kürzen der Säule können diese Erscheinungen oftmals
behoben werden (Schettgen, 2006). Um Reinigungsrückstände in den zu verwendenen
Probengläschen und damit potentielle Störsubstanzen in den Proben zu vermeiden,
empfiehlt es sich diese nur als einmal-Gläser zu verwenden oder vor Gebrauch mit Aceton
mehrfach durchzuspülen, zu trocknen und anschließend in aqua bidest für 60 min
auszukochen.

Analytik der Hämoglobinaddukte im Blut
64
5.10 Diskussion der Methode
Mit der in dieser Arbeit beschriebenen Methode ließ sich das Addukt des Styrols N-(2-
Hydroxy-2-Phenylethyl)-Valin am N-terminalen Valin des Hämoglobins empfindlich und
reproduzierbar nachweisen. Durch den käuflich erwerbbaren Dipeptidstandard liegt eine
Standardsubstanz vor, die nicht nur eine hohe Reinheit aufweist, sondern auch den
kompletten Aufarbeitungsprozess mit durchläuft. Ähnliches gilt auch für die hergestellten
hier verwendeten internen Standards. Diese durchlaufen ebenfalls den gesamten
Aufarbeitungsprozess. Verluste von Probenmaterial während der Probenaufarbeitung und
Analytik selbst (bspw. durch Verdünnungseffekte, unvollständige Extraktionen oder
instrumentelle Unterschiede) können durch den internen Standard ausgeglichen werden. Im
Zuge der Präzision wurden in dieser Arbeit zwei interne Standards (HPB-G, FPB-G)
getestet. Bezüglich der sehr aufwendigen Probenaufarbeitung können die Ergebnisse der
Präzision (5.9.1) als sehr gut bezeichnet werden. Die Wiederfindungsraten von HPB-G Q40
waren in der Präzision in Serie als auch von Tag zu Tag oberhalb derer von FPB-G. Bei
Q250 wurden demgegenüber mit FPB-G bessere Werte erreicht (Tab.12). Da jedoch eher
von kleinen Expositionskonzentrationen ausgegangen werden musste, wurde bei weiteren
Messungen innerhalb dieser Arbeit HPB-G als interner Standard verwendet. Ein weiterer
Vorteil von HPB-G ist das Vorhandensein der Hydroxygruppe am Phenylring. Dadurch ist
gewährleistet, dass dieses Molekül auch den abschließenden Acetylierungsschritt bei der
Probenaufarbeitung mit durchlaufen kann. Aufarbeitungsbedingte Verluste oder
Reaktionsträgheiten von Probenmaterial speziell in diesem Schritt können durch diesen
internen Standard wesentlich besser kompensiert werden als über FPB-G, welches nur den
potentiellen Probenverlust bis zum Zeitpunkt vor der Acetylierung wiedergeben kann.
Um die Richtigkeit der Methode zu prüfen, wurden Wiederfindungsversuche durchgeführt.
Dazu wurden Dotierungen von Globin unterschiedlicher Matrices hergestellt und
gemeinsam mit der jeweiligen undotierten Probe aufgearbeitet. Die relativen
Wiederfindungsraten zwischen 102,99 % und 107,33 % konnte als sehr gut und damit ein
Matrixeffekt als marginal bezeichnet werden. Die verwendete Methode stellte bezogen auf
die Präzisions- und Wiederfindungsdaten als sehr robust dar. Im weiteren Verlauf der
Arbeit wurde anhand dieser Methode ein Biomonitoring von belasteten Personengruppen
durchgeführt (s. Kap.7).

Analytik der aromatischen Carbonsäuren
65
6 Analytik der aromatischen Carbonsäuren
Die hier beschriebene Analytik wird seit mehreren Jahren am Institut für Arbeitsmedizin
und Sozialmedizin der RWTH Aachen angewendet. Das Prinzip dieser Methode basiert
ursprünglich auf dem in der DFG Methodensammlung zu „Analysen in biologischem
Material“ aufgenommenen Verfahren von Lewalter und Schucht (Lewalter, 1984). Im
Rahmen von erfolgreichen Teilnahmen an Ringversuchen wird diese Methode qualitativ
und quantitativ jährlich bestätigt. Zu diesem Verfahren werden im Folgenden Ergänzungen
bzw. Abwandlungen beschrieben.
6.1 Grundlage des Verfahrens
Die sich im (mit Essigsäure angesäuerten) Harn befindlichen aromatischen Carbonsäuren
Mandelsäure (MA) und Phenylglyoxylsäure (PGA) wurden mit Diethylether extrahiert. Als
interner Standard wurde die 3-Chlor-4-hydroxybenzoesäure verwendet. Der aus der
eingeengten Etherphase gewonnene Rückstand wurde in einem Milliliter der mobilen
Phase gelöst, die beiden Metabolite durch HPLC getrennt und mittels UV detektiert. Die
zur Kalibrierung benötigten wässrigen Standards wurden parallel mit den zu bestimmenden
Proben aufgearbeitet und analysiert. Deren Peakflächen wurden anschließend auf die des
internen Standards bezogen und der erhaltene Quotient als Funktion der dotierten
aromatischen Carbonsäuren zur Kalibrierkurve aufgetragen.
6.2 Geräte, Chemikalien und Lösungen
6.3 Geräte
HPLC-System mit UV-Detektor (L-7400), Autosampler (L-7200) und Pumpe (L-7100),
Interface (D-7000) (Merck-Hitachi)
Chromatographische Säule: HyPurity C18; 250 x 4,6 mm; Teilchendurchmesser 5 µm
(Thermo Electron Corporation)
Magnetrührer (IKA Labortechnik)

Analytik der aromatischen Carbonsäuren
66
1,8 ml Schraubgläser mit dazugehörigen PTFE-kaschierten Septen und Schraubkappen
(Macherey-Nagel)
Schraubgläschen mit teflonkaschierten Gummisepten und Schraubdeckel (5 und 20 ml)
(Macherey-Nagel)
Verschlusszange (Macherey-Nagel)
pH-Meter mit Einstabmesskette (Mettler-Toledo)
Mikroliterpipetten, verstellbar 10-100 (100- 1000) µl (Eppendorf)
Erlenmeyerkolben 10, 25, 50 1000, 2000-ml (Brand)
Zentrifuge Rotina 420R (Hettich)
Erlenmeyer Kolben 10 ml (Brand)
Labor „Blaukopfflaschen“ 1 und 2 Liter (Schott)
Ultraschallgerät (Bandelin Sonnorex RK510)
Wasserbad (Memmert)
Probenschüttler (Kottermann 4020)
Reacti-Vap III Evaporatorstation (Pierce)
Urin-Kunsstoffbehälter (Sarstedt)
6.4 Chemikalien
Sofern keine weiteren Angaben gemacht werden, wurden alle benannten Chemikalien in
der Reinheit von mindestens p.a.-Qualität verwendet.
Essigsäure (100%) (Merck)
Methanol Suprasolv (Merck)
Trikaliumphosphattrihydrat (K3PO4 x 3H2O) (Merck)
Salzsäure, 37% (HCL) (Merck)
Diethylether (Merck)
Phenylglyoxylsäure (PGA) (Fluka)
DL-Mandelsäure (MA) (Merck)
3-Chlor-4-hydroxybenzoesäure (Sigma-Aldrich)
aqua bidest (Millipore®)
Phosphorsäure konz. (H3PO4) (Merck)
Methanol (Merck)

Analytik der aromatischen Carbonsäuren
67
6.5 Lösungen
Phosphatpuffer 0,01 M
5,33 g K3PO4 x 3H2O wurden in 2 Liter aqua bidest gelöst und anschließend 3,25 ml
H3PO4 hinzugegeben und der pH-Wert auf 3.0 eingestellt. Zur Vermeidung von Blasen im
Puffer, wurde dieser in der Folge für 30 Minuten in das Ultraschallbad gestellt.
Laufmittel A
Phosphatpuffer 0,01 M und Methanol wurden im Verhältnis 4:1 gemischt
Laufmittel B
Methanol/H2O im Verhältnis 1:1
Spülphase
Methanol/Wasser im Verhältnis 1:1
Interner Standard
100 mg 3-Chlor-4-hydroxybenzoesäure wurden in 5 ml Ethanol gelöst und mit aqua bidest
auf 50 ml aufgefüllt (2g/L). Bei schlechter Löslichkeit, konnte der Standard durch leichtes
Erwärmen im Ultraschallbad zur Lösung gebracht werden.
Stammlösung für Kalibrierstandards
100 mg Mandelsäure und 100 mg Phenylglyoxylsäure wurden in 2,5 ml Methanol
vollständig gelöst (erwärmen u. Ultraschall) und mit aqua bidest auf 10 ml aufgefüllt
(10g/L).

Analytik der aromatischen Carbonsäuren
68
Kalibrierstandards
Die Kalibrierstandards wurden in aqua bidest nach folgendem Pipettierschema angesetzt:
Tab.14: Pipettierschema für die Kalibrierkurve MA und PGA
Stammlösung Endvolumen Konzentration
Standard
[µl] [ml] [mg/L]
0 50 0
25 50 5
50 50 10
125 50 25
250 50 50
375 50 75
500 50 100
1000 50 200
Zusätzlich wurde noch eine Qualitätskontrolle (35 mg/L) (Q35) in gepooltem Urin von vier
Personen der Allgemeinbevölkerung angesetzt und parallel mit den Proben und den
Kalibrierpunkten verarbeitet.
6.6 Probennahme und Probenaufarbeitung
Die Urinproben (Mittelstrahl) wurden in verschließbaren dafür vorgesehenen
Kunststoffbehältern gesammelt, mit 1 ml Essigsäure/100 ml Harn angesäuert und
unverzüglich bis zur Benutzung bei -20 °C gelagert.
6.6.1 Probenaufbereitung
1 ml des gewonnenen Harns bzw. Wassers (Probe, Standard oder Leerwert) wurden in ein
20-ml Schraubglas vorgelegt und dazu 100 µl der hergestellten Lösung des internen
Standards pipettiert. Anschließend wurden 50 µl 37 % HCL und 5 ml Diethylether
zugegeben. Das Schraubglas wurde verschlossen und für 10 Minuten auf dem
Probenschüttler gelegt. Es folgte ein Zentrifugationsschritt für ebenfalls 10 Minuten bei
1083 g. Die obere Etherphase wurde anschließend entnommen und in ein 5 ml Schraubglas
überführt.

Analytik der aromatischen Carbonsäuren
69
Im Folgenden wurde der Ether unter leichtem Stickstoffstrom bis zur Trockene eingeengt.
Der Rückstand wurde abschließend in 1 ml Laufmittel A aufgenommen, in 1,8 ml
Schraubgläschen überführt und verschlossen. 10 µl wurden in die HPLC injiziert.
6.7 Gaschromatographische Arbeitsbedingungen
Säule: HyPurity C18; 250 x 4,6 mm, Teilchendurchmesser 5 µm
(Thermo Electron Corporation)
Säulentemperatur: 30°C
Trennprinzip: Reversed phase
Detektor: UV-Detektor (L-7400); 252 nm (PGA); 220 nm (MA)
Flussgeschwindigkeit: 0,9 ml/min (constant flow)
Laufmittel A: 0,01 M Phosphatpuffer:MeOH 4:1
Laufmittel B: MeOH/H2O (1:1)
Druck: ~ 163 bar

Analytik der aromatischen Carbonsäuren
70
6.8 Analytische Betimmung
10 µl des in 1 ml Rückstand aufgenommenen Analyten wurden in den
Hochdruckflüssigkeitschromatographen injiziert.
Tab.15: Gradientenprogramm zur Bestimmung der aromatischen Carbonsäuren
Zeit (min) Laufmittel A (%) Laufmittel B (%) Flussgeschwindigkeit
(ml/min)
0,0
24,5
100,0
100,0
0,0
0,0
0,9
0,9
30,0 20,0 80,0 0,9
38,0 20,0 80,0 0,9
39,0 0,0 100,0 0,9
44,0 0,0 100,0 0,9
45,0 100,0 0,0 0,9
48,0 100,0 0,0 0,9
6.9 Kalibrierung und Berechnung der Analyseergebnisse
Die hergestellten Vergleichsstandards als auch die Qualitätskontrolle (Q35) wurden parallel
mit den zu untersuchenden Proben aufgearbeitet und im selben HPLC-Lauf mit analysiert.
Die jeweiligen Retentionszeiten lagen für PGA bei R (t) ~7,13 min und für MA bei R (t)
~9,57 min Diese Retentionszeiten können nur als Anhaltspunkt dienen und abhängig von
der Trennleistung der verwendeten Kapillarsäule Schwankungen unterliegen. Die
Berechnung der Analyseergebnisse erfolgte analog zu Kapitel 5.7.
Abbildung 20 zeigt zwei exemplarische Kalibrierkurven zur Berechnung der
Konzentrationen von Phenylglyoxylsäure (oben) und Mandelsäure (unten), die für jede
Probenaufarbeitungsreihe parallel mit erstellt wurde. Abbildung 21 zeigt zwei
Chromatogramme der aromatischen Carbonsäuren. Chromatogramm „A“ zeigt die
jewiligen Peaks von MA und PGA der Kalibrierdotierung von 25 mg/L und dem internen
Standard mit ihren jeweiligen Retentionszeiten. Im unteren Chromatogramm „B“ sind die
gleichen Peaks in einer Realprobe einer nicht beruflich belasteten Person gekennzeichnet.

Analytik der aromatischen Carbonsäuren
71
Abb. 20: Kalibrierkurve für Phenylglyoxylsäure (oben) und Mandelsäure (unten),
“area under curve” (AUC), interner Standard (IS), Mandelsäure (MA),
Phenylglyoxylsäure (PGA)
Kalibrierkurve PGA
y = 0,0046x
R2 = 0,9976
0,000000,100000,200000,300000,400000,500000,600000,700000,800000,900001,00000
0 50 100 150 200 250
[PGA] mg/L
AU
C P
GA
/IS
Kalibrierkurve MA
y = 0,0015x + 0,0003
R2 = 0,999
0,00000
0,05000
0,10000
0,15000
0,20000
0,25000
0,30000
0,35000
0 50 100 150 200 250
[MA] mg/L
AU
C M
A/IS
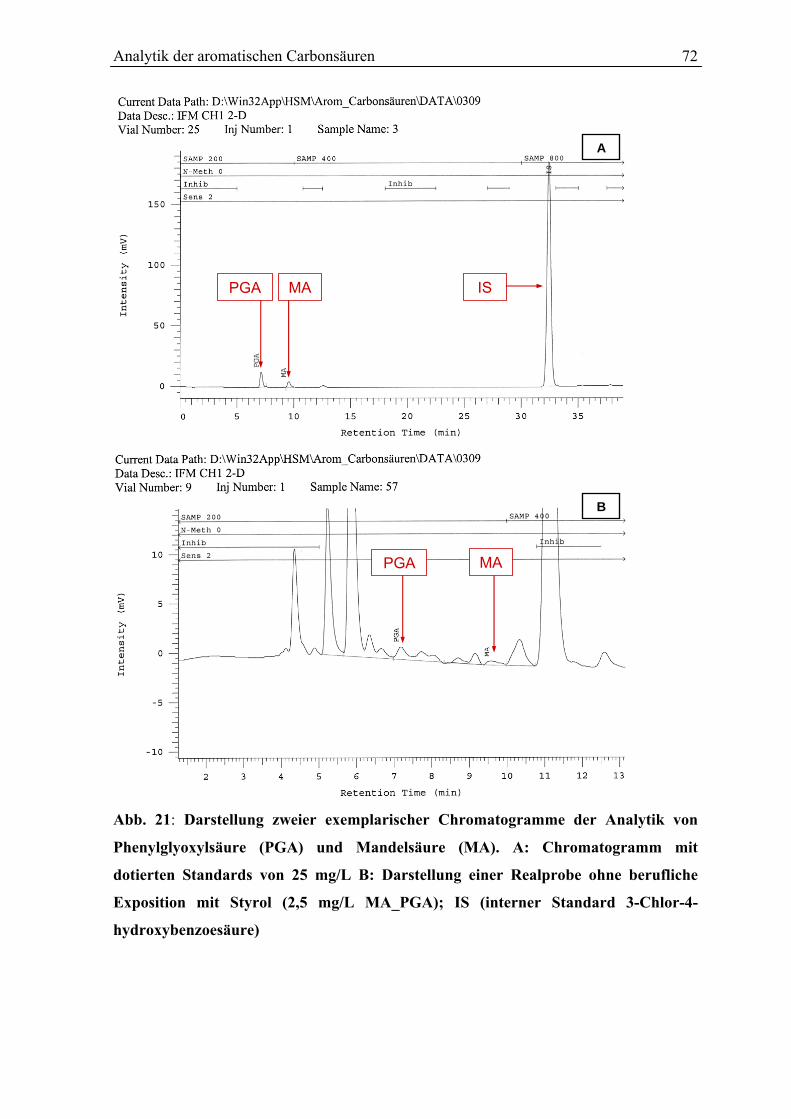
Analytik der aromatischen Carbonsäuren
72
MAPGA IS
MAPGA
A
B
Abb. 21: Darstellung zweier exemplarischer Chromatogramme der Analytik von
Phenylglyoxylsäure (PGA) und Mandelsäure (MA). A: Chromatogramm mit
dotierten Standards von 25 mg/L B: Darstellung einer Realprobe ohne berufliche
Exposition mit Styrol (2,5 mg/L MA_PGA); IS (interner Standard 3-Chlor-4-
hydroxybenzoesäure)

Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
73
7 Humanbiomonitoring belasteter Personengruppen
und der Allgemeinbevölkerung
7.1 Kollektivbeschreibung
7.1.1 Beruflich belastete Personengruppen
Für die Durchführung des biochemischen Effektmonitorings anhand der in unserem Institut
angepassten und etablierten Methoden, erklärten sich zwei styrolverarbeitende Firmen
(GfPI ) dazu bereit ein Biomonitoring ihrer Mitarbeiter durchführen zu lassen und die
gewonnenen Ergebnisse für die vorliegende Arbeit in anonymer Form weiterzuverwenden.
Während in der ersten Firma (Fa.1) die Nutzung von Styrol-basierten Harzen
kontinuierlich durchgeführt wurde, ist in der zweiten Firma (Fa.2) Styrol zum Zeitpunkt
der Probennahme nach längerem wirtschaftlich bedingtem Produktionsstop erst seit 6
Wochen verarbeitet worden (Probennahme nach 6 Wochen Exposition). Durch
produktionsspezifische Unterschiede ergaben sich dennoch in Firma 1 potentiell weniger
hohe Expositionen als in Firma 2. Zur analytischen Bestimmung der Konzentrationen der
Mercaptursäuren als auch der aromatischen Carbonsäuren und Cotinin im Urin und der
Hämoglobinaddukte im Blut, wurden von den Mitarbeitern vor Ort jeweils vor ihrer
Schicht (VS) als auch nach ihrer Schicht (NS; Δt~8h) Urin sowie zu Beginn ihrer Tätigkeit
eine Vollblutprobe gewonnen. Die Teilnahme am Biomonitoring setzte eine vorab vom
Mitarbeiter zu unterschreibende Probandeninformation „Biomonitoring-Styrol“ als auch
eine Einwilligungserklärung voraus (s. Anlage), wodurch vor Ort einige Probanden nicht
mehr mit in das Humanbiomonitoring mit eingeschlossen werden konnten. Insgesamt
konnten in Fa.1 sieben und in Fa.2 29 Probanden für die Teilnahme gewonnen werden.
Alle Probanden waren Männer im Alter zwischen 26 und 60 Jahren mit einem
Altersmedian von 50 (Fa.1) und im Alter zwischen 21 und 61 mit einem Altersmedian von
46 (Fa.2). Die Tätigkeitsjahre lagen im Median bei 9 Jahren (Fa.1) und 10 Jahren (Fa.2).
Bei allen 35 Personen bestand der Verdacht einer potentiellen Belastung mit Styrol als
Copolymer durch firmenspezifische Arbeiten mit Vinylesterharzen. Die Bestimmungen der
einzelnen Metabolite erfolgten analog beschriebener Methoden.

Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
74
7.1.2 Kontrollkollektiv der Allgemeinbevölkerung
Über die berufliche Belastung mit Styrol hinaus stellte sich die Frage nach
umweltbedingten Styrolbelastungen in einem Kontrollkollektiv (Kon) bestehend aus
Probanden der Allgemeinbevölkerung. Es wurden hierzu 18 Personen untersucht, bei
denen eine beruflich bedingte Styrolexposition nicht zu erwarten war. Eine unterschriebene
Information als auch eine Einverständniserklärung war auch hier die Voraussetzung um an
dem Humanbiomonitoring teilzunehmen. Pro Person wurden sowohl eine Urinprobe als
auch eine Vollblutprobe akquiriert und diese anschließend sofort bis zur Lagerung bei
-20°C aufgearbeitet. Da bei diesen Personengruppen kein Schichtsystem bestand, wurde
hier jeweils ein Wert für den jeweiligen Metaboliten erhoben. Alle Probanden waren
Männer im Alter zwischen 21 und 79 Jahren mit einem Altersmedian von 44. Unter ihnen
befanden sich acht Raucher und zehn Nichtraucher.
7.1.3 Auswertung
Die statistische Auswertung und Berechnung der Ergebnisse wurde mit Microsoft Excel
Office 2003 (Microsoft, 1985-2003) und Polar Engineering PASW Statistics 18 (IBM,
1993-2007) durchgeführt. Der Vergleich der Metabolitkonzentrationen von Vorschicht zu
Nachschicht innerhalb einer Firma wurde mittels Wilcoxon Rangsummentest für
abhängige und analog die Vergleiche zum Kontrollkollektiv der Allgemeinbevölkerung
mittels Wilcoxon Rangsummentest für unabhängige Stichproben durchgeführt. Als
signifikant wurde p < 0,05 (*) und p < 0,01 (**) angenommen. Die Beziehungen der
einzelnen VS/NS Metabolite zueinander wurden mittels zweiseitiger Spearman Korrelation
untersucht. Da in der Gruppe der Unbelasteten keine Schichtsysteme vorhanden waren,
wurden die Korrelationen im Kontrollkollektiv unabhängig von dem Kollektiv der zuvor
zusammengefassten Firmenmitarbeiter bestimmt. Die Bewertung des
Korrelationskoeffizienten (r) wurde anhand folgender Einstufung vorgenommen (Sachs &
Hedderich, 2006).

Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
75
0,0 ≤ r ≤ 0,2: kein bis geringer Zusammenhang
0,2 ≤ r ≤ 0,5: schwacher bis mäßiger Zusammenhang
0,5 ≤ r ≤ 0,8: deutlicher Zusammenhang
0,8 ≤ r ≤ 1,0: hoher bis perfekter Zusammenhang
Für die Definition von Ausreißerwerten (○) wurde ein Interquartilsabstand (englisch IQR
interquartile range) von 1,5 – 3 IQR’s ab Boxende angenommen. Extremwerte (◊) lagen
per Definition oberhalb von 3 IQR’s ab Boxende.
Mittels hierarchisch linearer Regression wurde überprüft, ob die Konzentrationen der
einzelnen Metabolite (VS/NS Mercaptursäuren, VS/NS aromatische Carbonsäuren und
Hämoglobinaddukte) mit der Zugehörigkeit zu der exponierten Gruppe (Fa.1, Fa.2) vs. des
Kontrollkollektivs zu erklären sind. Hierfür wurde die Gruppenzugehörigkeit mittels
Dummy-Codierung in die Variable „Gruppe“ (0 = Kontrollkollektiv, 1 = Firmenkollektiv)
vorgenommen. Für die Durchführung der Regression wurden im ersten Schritt die
Kontrollvariablen Alter und Cotinin in das Regressionsmodell aufgenommen. Im zweiten
Schritt wurde die Gruppenvariable hinzugefügt.
Die Einstufung des Raucherstatus erfolgte anhand eigener Angaben der Probanden als auch
durch die Bestimmung von Cotinin, wobei der Cotininwert auschlaggebend war. Bei einem
cut-off von 100 µg/L wurden Nichtraucher von Rauchern unterschieden. Ein Auszug der
Fragen aus dem Fragebogen zur Ermittlung des Raucherstatus und des Alters ist im
Anhang dargestellt.

Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
76
7.2 Ergebnisse des Humanbiomonitorings
Die Ergebnisse des biochemischen Effektmonitorings der untersuchten Kollektive für alle
untersuchten Metabolite sind in Tabelle 16 zusammengefasst. Hier wird deutlich, dass bei
allen drei Gruppen (Fa.1, Fa.2 und Allgemeinbevölkerung) Styrol-assoziierte Biomarker
nachgewiesen werden konnten.
Bei den Konzentrationen der aromatischen Carbonsäuren konnte zwischen den
Vorschichtwerten (VS-MA_PGA) und Nachschichtwerten (NS-MA_PGA) ein deutlicher
Anstieg beobachtet werden. Die MA_PGA Werte der Vorschicht lagen im Median für Fa.1
bei 39,98 mg/g und für Fa.2 bei 54,48 mg/g und damit fast um eine Zehnerpotenz oberhalb
des Medians des Kontrollkollektivs (6,63 mg/g). Der biologische Arbeitsplatztoleranzwert
für MA_PGA (die Summe der Werte der Mandelsäure und der Phenylglyoxylsäure); (600
mg/g Kreatinin) (DFG, 2009) wurde in Fa.1 in den Vor- und Nachschichtproben
unterschritten, während in der Nachschicht der Fa.2 dieser Wert in Einzelproben
überschritten wurde.
Die Konzentrationen der Hämoglobinaddukte lagen im Bereich von <1 – 2,76 pmol/g
Globin bei Fa.1 und zwischen <1 – 3,45 pmol/g Globin bei Fa.2. Werte die unterhalb der
Nachweisgrenze von 1 pmol/g Globin lagen, wurden für die weiteren Berechnungen auf
die halbe Nachweisgrenze (0,5 pmol/g Globin) gesetzt. Alle Probanden des
Kontrollkollektivs lagen unterhalb von 1 pmol. Der Maximalwert in der Fa.1 betrug das
6-fache, der in Fa.2 das 7-fache des Wertes der unbelasteten Personengruppe.
Bei der Beobachtung der Vorschicht- und Nachschichtwerte der PHEMAs konnte ebenfalls
ein Anstieg der Konzentrationen von VS zu NS im Median beobachtet werden (Tab.16).
Die Medianwerte der Fa.1 betrug dabei im Vergleich zum Kontrollkollektiv das 60-fache
(VS) bzw. das 101-fache (NS) und die der Fa.2 das 83-fache (VS) bzw. 251-fache (NS) des
Medianwertes der Kontrollen von 0,39 µg/g Kreatinin. Beim Vergleich der
Schichtmediane der Firmen lag der Vorschichtmedian der Fa.2 bei dem 1,4-fachen Wert
der Fa.1 und im Nachschichtmedian 2,5-fach oberhalb des Medians der Fa.1.
Die Cotininwerte, die zur Einstufung des Raucherstatus verwendet wurden, lagen zwischen
2,75 – 1945,74 µg/L (Fa.1), 2,98 – 3102,01 µg/L (Fa.2) und 3,10 – 3026,80 µg/L
(Kontrollkollektiv) (Tab.16).
Insgesamt zeigten sich die höchsten Konzentrationen bei den aromatischen Carbonsäuren
(Bereich: mg/L). Weniger hohe Konzentrationen konnten bei den Mercaptursäuren

Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
77
(Bereich: µg/L) und die kleinsten Konzentrationen bei den Hämoglobinaddukten
nachgewiesen werden (Bereich: pmol/g Globin ≙ ng/L).
Da sich in der linearen Regression (Tab.19-25) keine signifikante Ausprägung von Cotinin
auf die Konzentration der Mercaptursäuren und aromatischen Carbonsäuren beim beruflich
exponierten Kollektiv zeigte, wurde für dieses Kollektiv auf eine tabellarische Darstellung
von Rauchern und Nichtraucher verzichtet. Tabelle 17 zeigt das Kontrollkollektiv der
Allgemeinbevölkerung unterteilt in Raucher und Nichtraucher. Raucher zeigten im Median
mehr als ein Vierfaches der Konzentrationen der aromatischen Carbonsäuren im Vergleich
zu den Nichtrauchern. Die Konzentrationen der Mercaptursäuren lagen bei den
Nichtrauchern im Gegensatz zu denen der Raucher (0,74 µg/g Kreatinin) im Median
unterhalb der Nachweisgrenze. Hämoglobin Adduktkonzentrationen lagen bei beiden
Gruppen unterhalb der Nachweisgrenze.
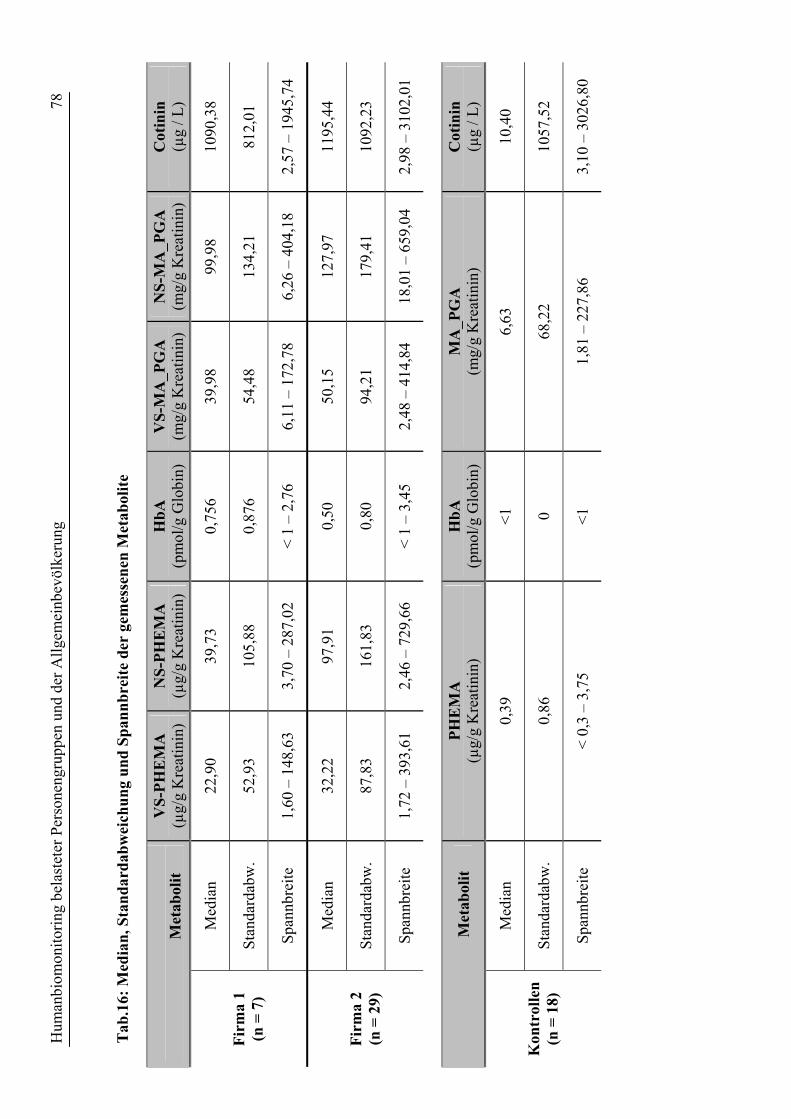
Hum
anbi
omon
itor
ing
bela
stet
er P
erso
neng
rupp
en u
nd d
er A
llge
mei
nbev
ölke
rung
78
Tab
.16:
Med
ian
, Sta
nd
ard
abw
eich
un
g u
nd
Sp
ann
bre
ite
der
gem
esse
nen
Met
abol
ite
M
etab
olit
V
S-P
HE
MA
(µ
g/g
Kre
atin
in)
NS
-PH
EM
A
(µg/
g K
reat
inin
) H
bA
(p
mol
/g G
lobi
n)
VS
-MA
_PG
A
(mg/
g K
reat
inin
) N
S-M
A_P
GA
(m
g/g
Kre
atin
in)
Cot
inin
(µ
g / L
)
Med
ian
22,9
0 39
,73
0,75
6 39
,98
99,9
8 10
90,3
8
Sta
ndar
dabw
. 52
,93
105,
88
0,87
6 54
,48
134,
21
812,
01
Fir
ma
1 (n
= 7
)
Spa
nnbr
eite
1,
60 –
148
,63
3,70
– 2
87,0
2 <
1 –
2,7
6 6,
11 –
172
,78
6,26
– 4
04,1
8 2,
57 –
194
5,74
Med
ian
32,2
2 97
,91
0,50
50
,15
127,
97
1195
,44
Sta
ndar
dabw
. 87
,83
161,
83
0,80
94
,21
179,
41
1092
,23
Fir
ma
2 (n
= 2
9)
Spa
nnbr
eite
1,
72 –
393
,61
2,46
– 7
29,6
6 <
1 –
3,4
5 2,
48 –
414
,84
18,0
1 –
659,
04
2,98
– 3
102,
01
Met
abol
it
PH
EM
A
(µg/
g K
reat
inin
) H
bA
(p
mol
/g G
lobi
n)
MA
_PG
A
(mg/
g K
reat
inin
) C
otin
in
(µg
/ L)
Med
ian
0,39
<
1 6,
63
10,4
0
Sta
ndar
dabw
. 0,
86
0 68
,22
1057
,52
Kon
trol
len
(n =
18)
Spa
nnbr
eite
<
0,3
– 3
,75
<1
1,81
– 2
27,8
6 3,
10 –
302
6,80
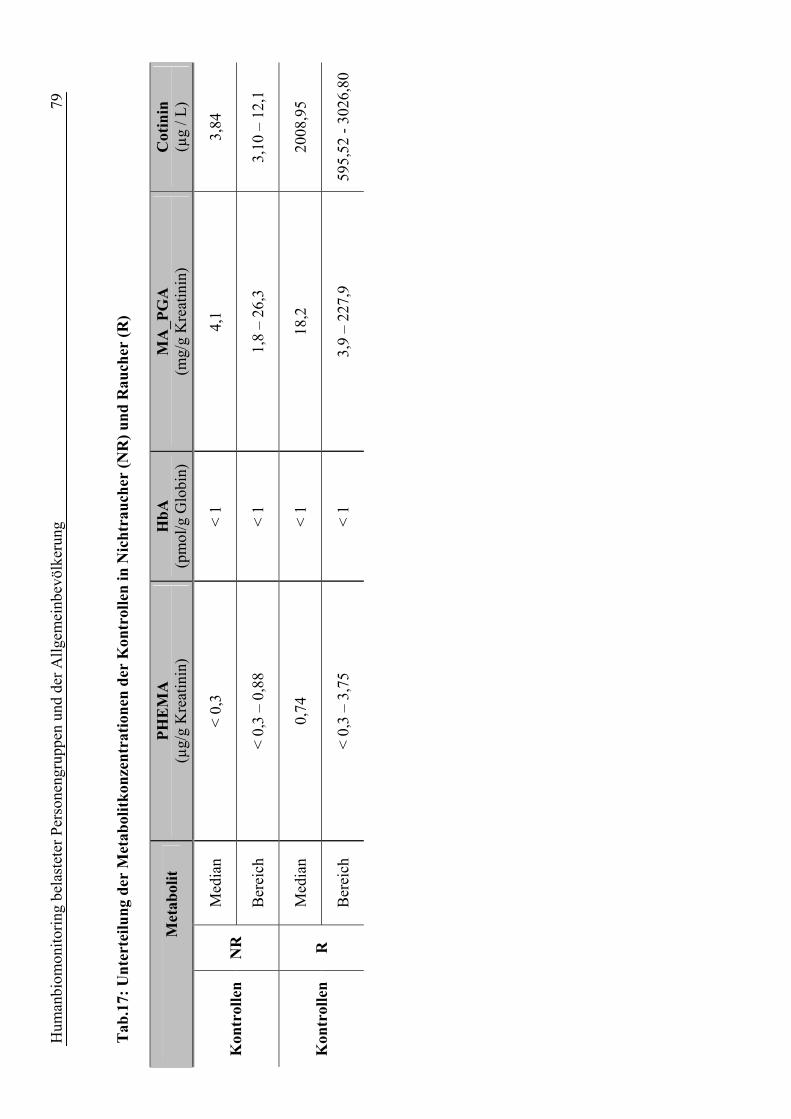
Hum
anbi
omon
itor
ing
bela
stet
er P
erso
neng
rupp
en u
nd d
er A
llge
mei
nbev
ölke
rung
79
Tab
.17:
Un
tert
eilu
ng
der
Met
abol
itk
onze
ntr
atio
nen
der
Kon
trol
len
in N
ich
trau
cher
(N
R)
un
d R
auch
er (
R)
Met
abol
it
PH
EM
A
(µg/
g K
reat
inin
) H
bA
(p
mol
/g G
lobi
n)
MA
_PG
A
(mg/
g K
reat
inin
) C
otin
in
(µg
/ L)
Med
ian
< 0
,3
< 1
4,
1 3,
84
Kon
trol
len
NR
Ber
eich
<
0,3
– 0
,88
< 1
1,
8 –
26,3
3,
10 –
12,
1
Med
ian
0,74
<
1
18,2
20
08,9
5 K
ontr
olle
n R
B
erei
ch
< 0
,3 –
3,7
5 <
1
3,9
– 22
7,9
595,
52 -
302
6,80

Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
80
Abbildungen 22 bis 26 zeigen Boxplot-Diagramme der Vergleiche der jeweiligen
Metaboliten in der Vor- und Nachschicht innerhalb einer Firma als auch im Vergleich zum
entsprechenden Metaboliten des Kontrollkollektivs.
Es fanden sich signifikante Unterschiede (s. Kap. 7.1.3.) der PHEMA Konzentrationen
beim innerbetrieblichen Vergleich von VS zu NS (*) als auch im Vergleich der VS und der
NS-Werte zu den Werten der Kontrollen (**) (Abb.22). Bei der Gegenüberstellung der
Mercaptursäurenkonzentrationen in der VS und der NS zeigten sich ebenfalls signifikante
(**) Unterschiede bei Fa.2. Auch gegenüber den Konzentrationen der Referenzgruppe
unterschieden sich die Werte aus VS und NS der Firmen jeweils signifikant (**) (Abb.23).
Die VS und NS Werte der aromatischen Carbonsäuren der Fa.1 zeigten wesentliche
Unterschiede (*) zu den Werten der Kontrollen. Im innerbetrieblichen Vergleich von VS
zu NS konnten die Werte ebenfalls als signifikant angesehen werden (*). Im Gegensatz zu
den PHEMA Werten der Kontrollen, wurden hier drei Extremwerte beobachtet (Abb.24).
Beide Schichtwerte (VS, NS) der Carbonsäuren zeigten einen deutlichen Unterschied (**)
zur Gruppe der nicht-Exponierten und auch die Schichwerte untereinander konnten
signifikant (**) voneinander unterschieden werden (Abb.25).
In beiden Fällen (Fa.1, Fa.2) ergaben sich signifikante Unterschiede der
Hämoglobinadduktkonzentrationen beim Vergleich der exponierten Firmenarbeiter mit den
unbelasteten Probanden (Abb.26).
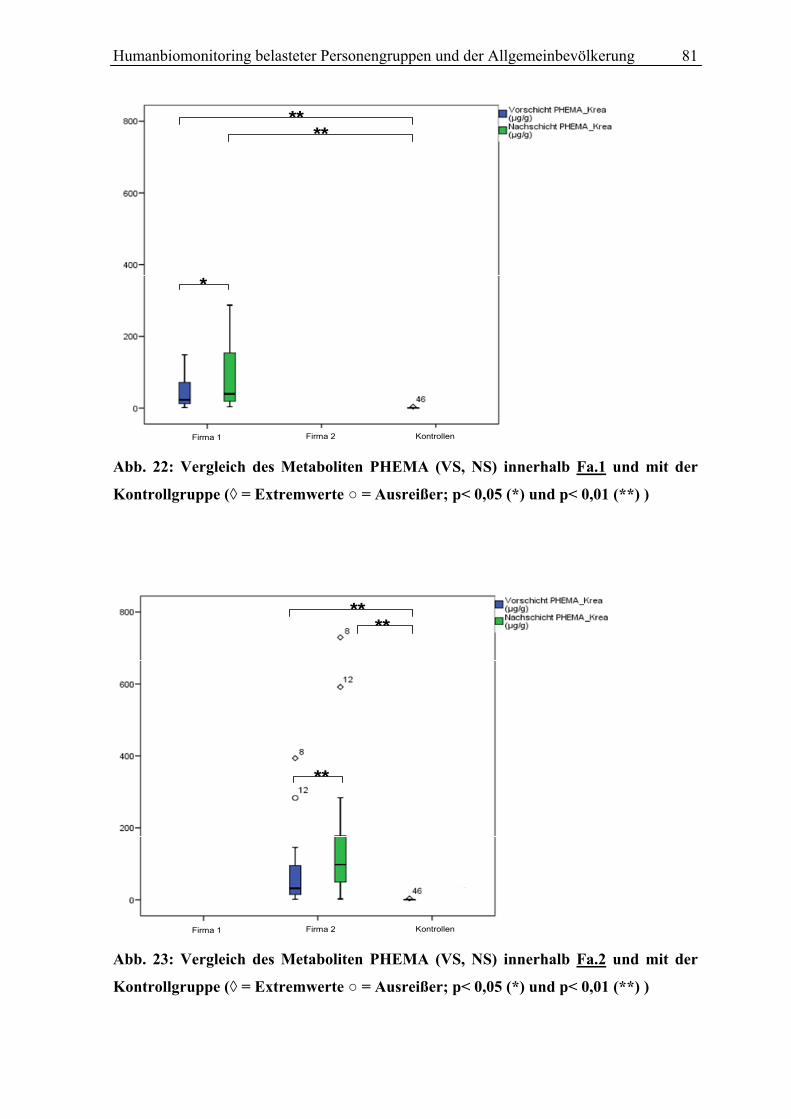
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
81
Abb. 22: Vergleich des Metaboliten PHEMA (VS, NS) innerhalb Fa.1 und mit der
Kontrollgruppe (◊ = Extremwerte ○ = Ausreißer; p< 0,05 (*) und p< 0,01 (**) )
Abb. 23: Vergleich des Metaboliten PHEMA (VS, NS) innerhalb Fa.2 und mit der
Kontrollgruppe (◊ = Extremwerte ○ = Ausreißer; p< 0,05 (*) und p< 0,01 (**) )
Firma 1 KontrollenFirma 2
****
*
Firma 1 KontrollenFirma 2
****
**
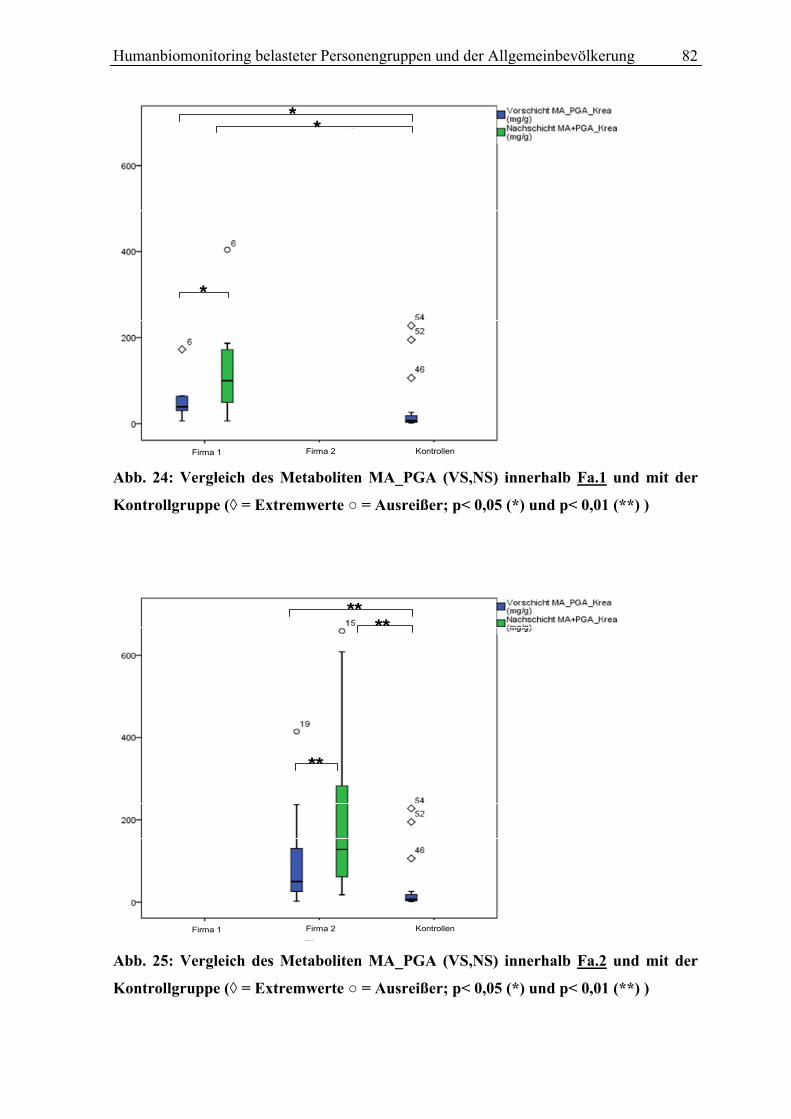
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
82
Abb. 24: Vergleich des Metaboliten MA_PGA (VS,NS) innerhalb Fa.1 und mit der
Kontrollgruppe (◊ = Extremwerte ○ = Ausreißer; p< 0,05 (*) und p< 0,01 (**) )
Abb. 25: Vergleich des Metaboliten MA_PGA (VS,NS) innerhalb Fa.2 und mit der
Kontrollgruppe (◊ = Extremwerte ○ = Ausreißer; p< 0,05 (*) und p< 0,01 (**) )
Firma 1 KontrollenFirma 2
**
*
Firma 1 KontrollenFirma 2
****
**
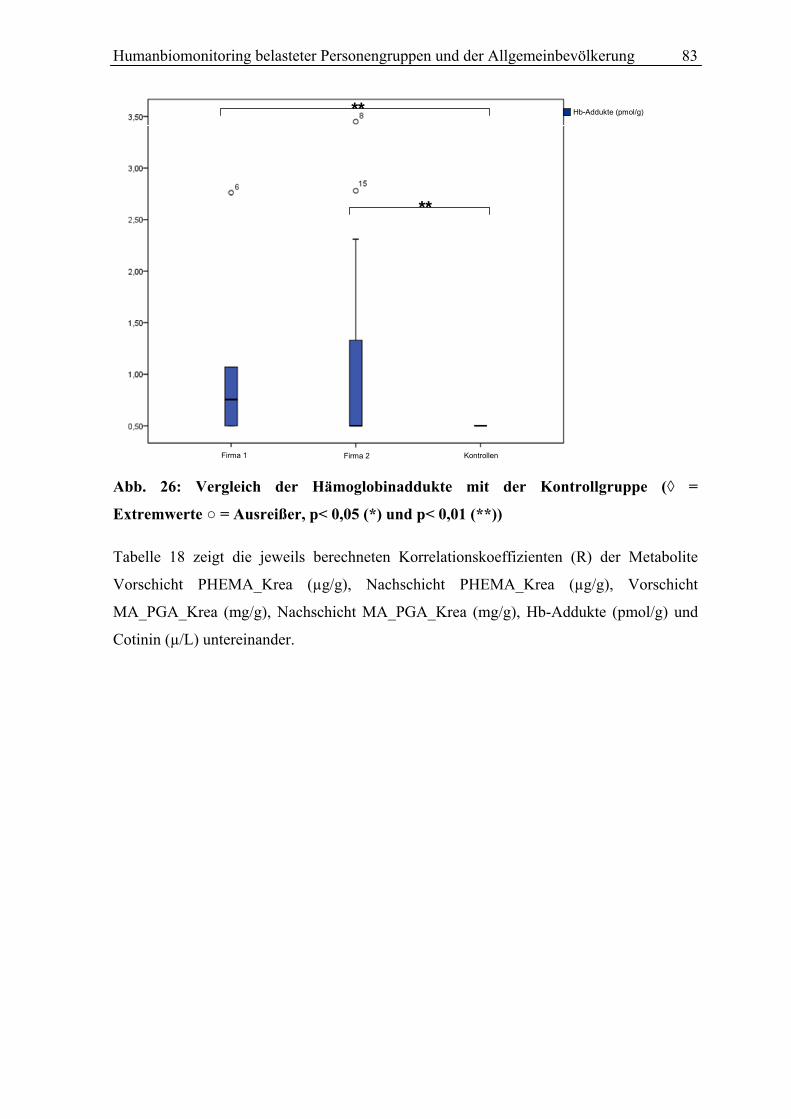
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
83
Abb. 26: Vergleich der Hämoglobinaddukte mit der Kontrollgruppe (◊ =
Extremwerte ○ = Ausreißer, p< 0,05 (*) und p< 0,01 (**))
Tabelle 18 zeigt die jeweils berechneten Korrelationskoeffizienten (R) der Metabolite
Vorschicht PHEMA_Krea (µg/g), Nachschicht PHEMA_Krea (µg/g), Vorschicht
MA_PGA_Krea (mg/g), Nachschicht MA_PGA_Krea (mg/g), Hb-Addukte (pmol/g) und
Cotinin (µ/L) untereinander.
Hb-Addukte (pmol/g)
Firma 1 Firma 2 Kontrollen
**
**
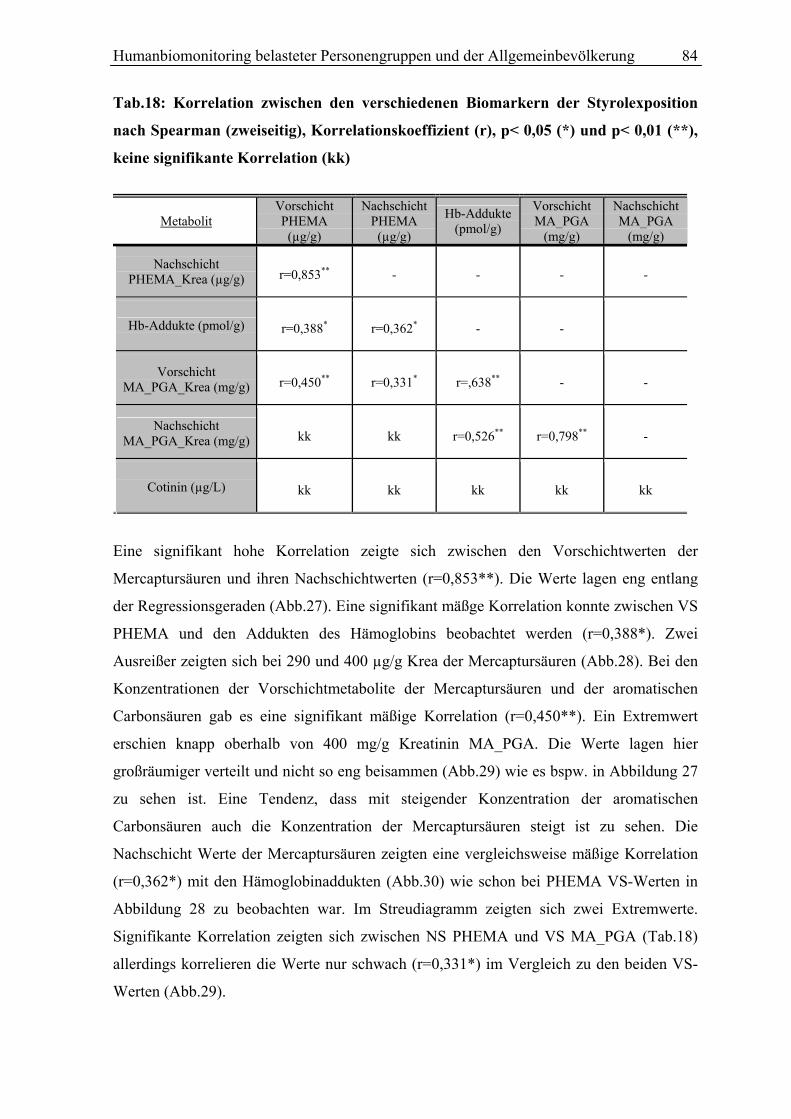
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
84
Tab.18: Korrelation zwischen den verschiedenen Biomarkern der Styrolexposition
nach Spearman (zweiseitig), Korrelationskoeffizient (r), p< 0,05 (*) und p< 0,01 (**),
keine signifikante Korrelation (kk)
Metabolit Vorschicht PHEMA (µg/g)
Nachschicht PHEMA (µg/g)
Hb-Addukte (pmol/g)
Vorschicht MA_PGA
(mg/g)
Nachschicht MA_PGA
(mg/g)
Nachschicht PHEMA_Krea (µg/g) r=0,853** - - - -
Hb-Addukte (pmol/g) r=0,388* r=0,362* - -
Vorschicht MA_PGA_Krea (mg/g) r=0,450** r=0,331* r=,638** - -
Nachschicht MA_PGA_Krea (mg/g) kk kk r=0,526** r=0,798** -
Cotinin (µg/L) kk kk kk kk kk
Eine signifikant hohe Korrelation zeigte sich zwischen den Vorschichtwerten der
Mercaptursäuren und ihren Nachschichtwerten (r=0,853**). Die Werte lagen eng entlang
der Regressionsgeraden (Abb.27). Eine signifikant mäßge Korrelation konnte zwischen VS
PHEMA und den Addukten des Hämoglobins beobachtet werden (r=0,388*). Zwei
Ausreißer zeigten sich bei 290 und 400 µg/g Krea der Mercaptursäuren (Abb.28). Bei den
Konzentrationen der Vorschichtmetabolite der Mercaptursäuren und der aromatischen
Carbonsäuren gab es eine signifikant mäßige Korrelation (r=0,450**). Ein Extremwert
erschien knapp oberhalb von 400 mg/g Kreatinin MA_PGA. Die Werte lagen hier
großräumiger verteilt und nicht so eng beisammen (Abb.29) wie es bspw. in Abbildung 27
zu sehen ist. Eine Tendenz, dass mit steigender Konzentration der aromatischen
Carbonsäuren auch die Konzentration der Mercaptursäuren steigt ist zu sehen. Die
Nachschicht Werte der Mercaptursäuren zeigten eine vergleichsweise mäßige Korrelation
(r=0,362*) mit den Hämoglobinaddukten (Abb.30) wie schon bei PHEMA VS-Werten in
Abbildung 28 zu beobachten war. Im Streudiagramm zeigten sich zwei Extremwerte.
Signifikante Korrelation zeigten sich zwischen NS PHEMA und VS MA_PGA (Tab.18)
allerdings korrelieren die Werte nur schwach (r=0,331*) im Vergleich zu den beiden VS-
Werten (Abb.29).

Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
85
Eine Signifikanz mit deutlicher Korrelation war beim Vergleich der aromatischen
Carbonsäuren in der Vorschicht mit den Addukten des Hämoglobins zu beobachten
(r=0,638**) (Abb.31), ebenso im Vergleich mit den Nachschichtwerten der Carbonsäuren
(Abb.32). Beide Korrelationen weisen auf einen linearen Zusammenhang hin, hier
allerdings mit einem gerinfügig niedrigeren Korrelationskoeffizienten bei den
Nachschichtwerten (r=0,526**). Wird die Relation ohne die Wertepaare, für die eine
Adduktkonzentration von 0,5 pmol/g Globin vorliegt, durchgeführt, ergab sich aus VS
MA_PGA zu HbA ein Korrelationskoeffizient von r = 0,631* und für NS MA_PGA zu
HbA ein Korrelationskoefizient von r= 0,883**.
Die gleichen Tendenzen konnten auch für die PHEMA VS- und NS-Korrelation mit HbA
beobachtet werden (VS:HbA: r = 0,311; NS:HbA: r = 0,420).
Eine hochsignifikante Korrelation fand sich zwischen den Werten der MA_PGA
Vorschicht und Nachschicht (r=0,798**). Ein deutlich linearer Zusammenhang ist gut
erkennbar (Abb.33).
Keine signifikanten Korrelationen wurden zwischen den Nachschichtwerten der
Carbonsäuren und denen der Vor- und Nachschicht der Mercaptursäuren nachgewiesen.
Außerdem bestanden keine signifikanten Korrelationen zwischen Cotinin und allen
anderen Metaboliten (Tab.18).
Zwischen den Styrol- Metabolitkonzentrationen des Kontrollkollektivs konnten keine
signifikanten Korrelationen beobachtet werden, jedoch ein mäßiger Zusammenhang
zwischen MA_PGA und PHEMA (r= 0,406). Zwischen dem Metaboliten Cotinin, der beim
Rauchen entsteht, konnte allerdings ein signifikanter, deutlicher Zusammenhang mit den
Konzentrationen der aromatischen Carbonsäuren festgestellt werden (r=0,513*) (Abb.34).
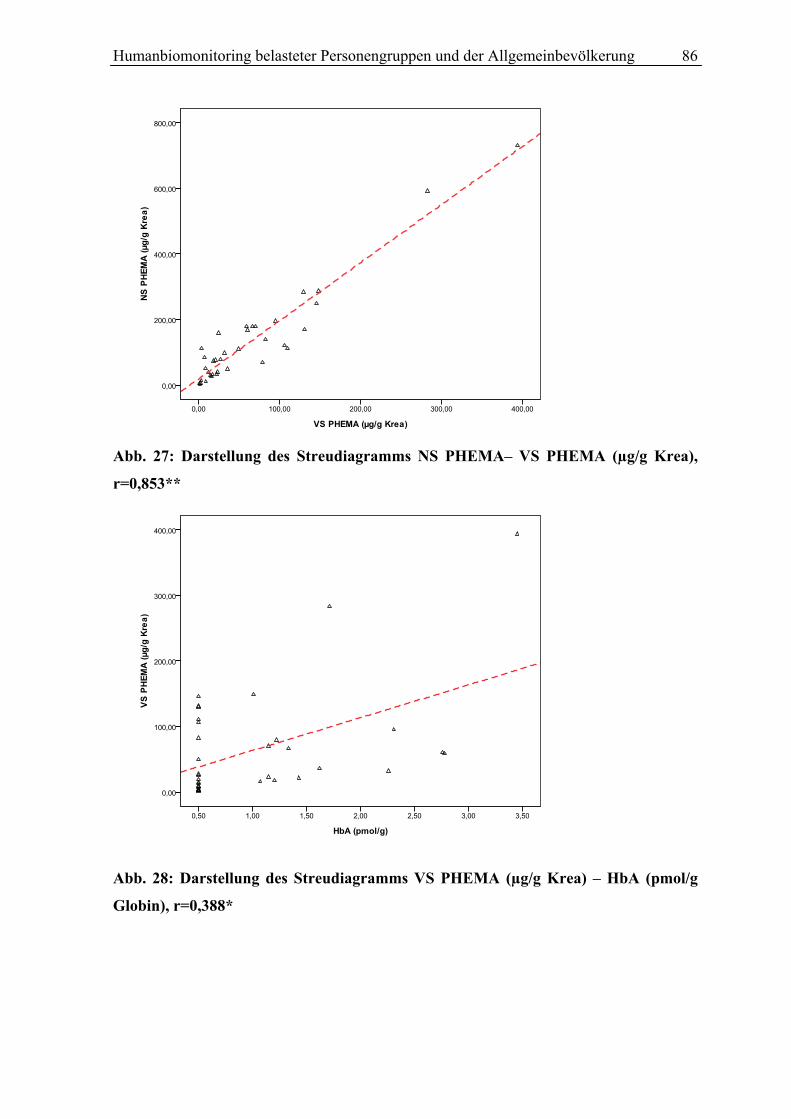
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
86
Abb. 27: Darstellung des Streudiagramms NS PHEMA– VS PHEMA (µg/g Krea),
r=0,853**
Abb. 28: Darstellung des Streudiagramms VS PHEMA (µg/g Krea) – HbA (pmol/g
Globin), r=0,388*
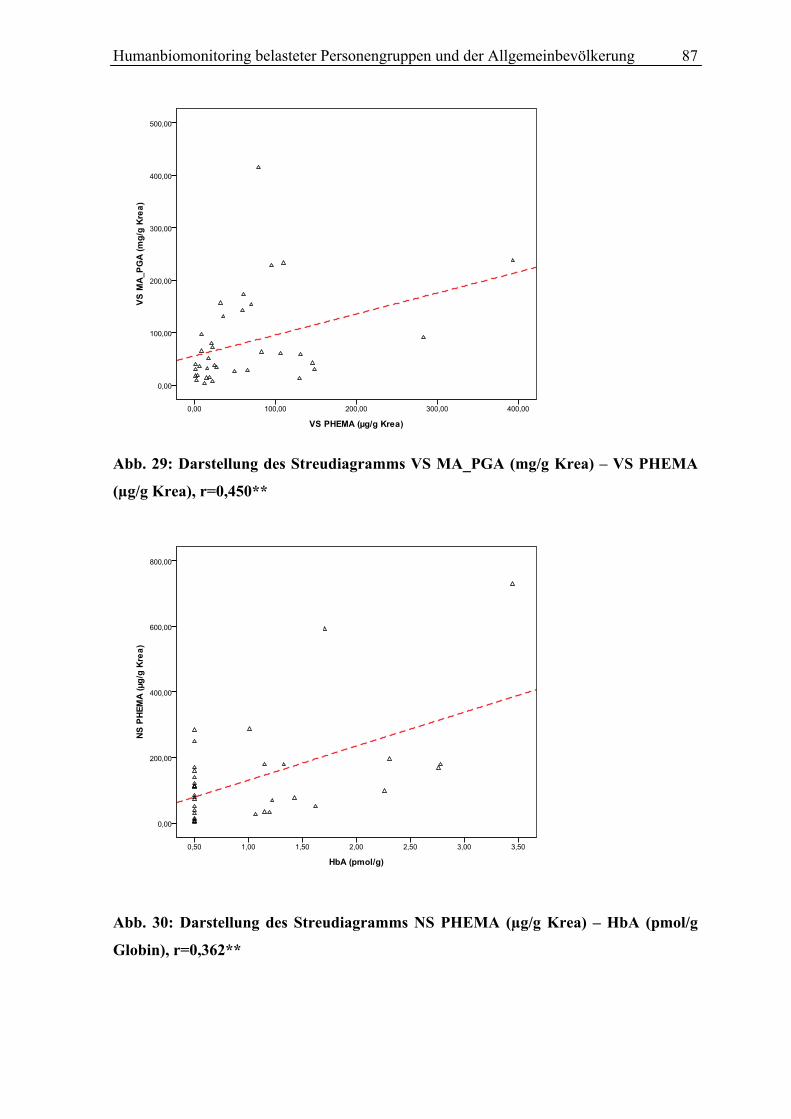
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
87
Abb. 29: Darstellung des Streudiagramms VS MA_PGA (mg/g Krea) – VS PHEMA
(µg/g Krea), r=0,450**
Abb. 30: Darstellung des Streudiagramms NS PHEMA (µg/g Krea) – HbA (pmol/g
Globin), r=0,362**
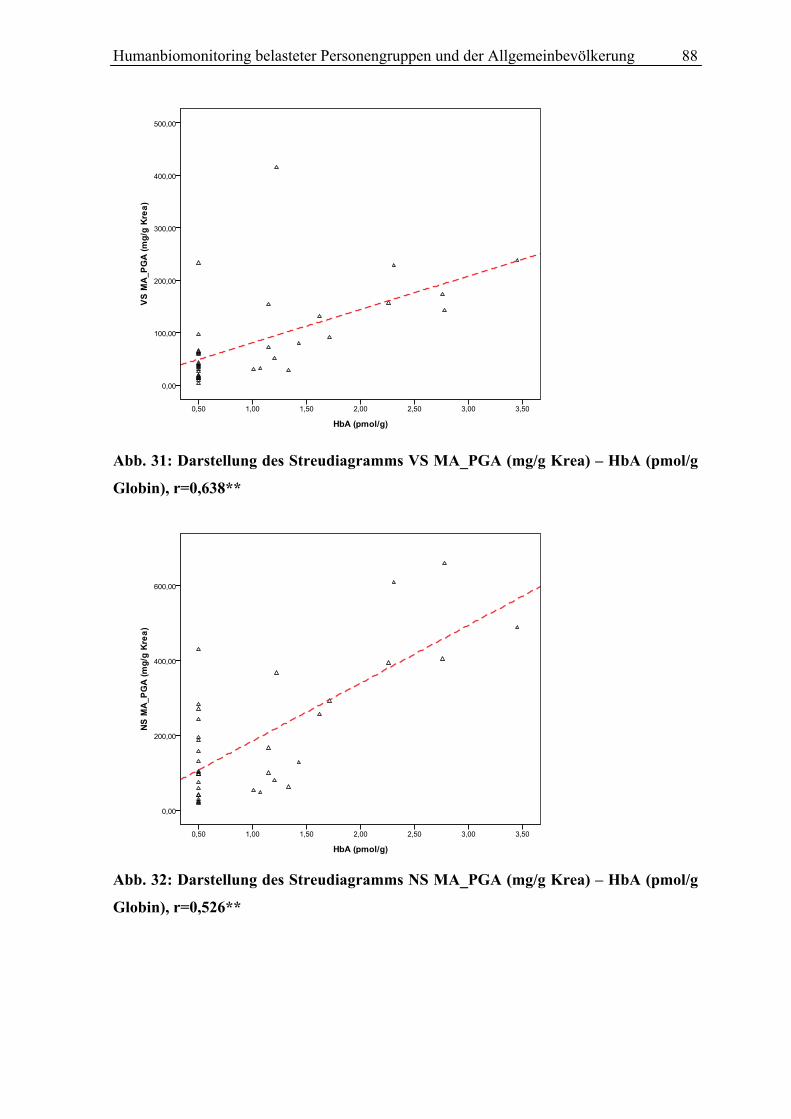
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
88
Abb. 31: Darstellung des Streudiagramms VS MA_PGA (mg/g Krea) – HbA (pmol/g
Globin), r=0,638**
Abb. 32: Darstellung des Streudiagramms NS MA_PGA (mg/g Krea) – HbA (pmol/g
Globin), r=0,526**
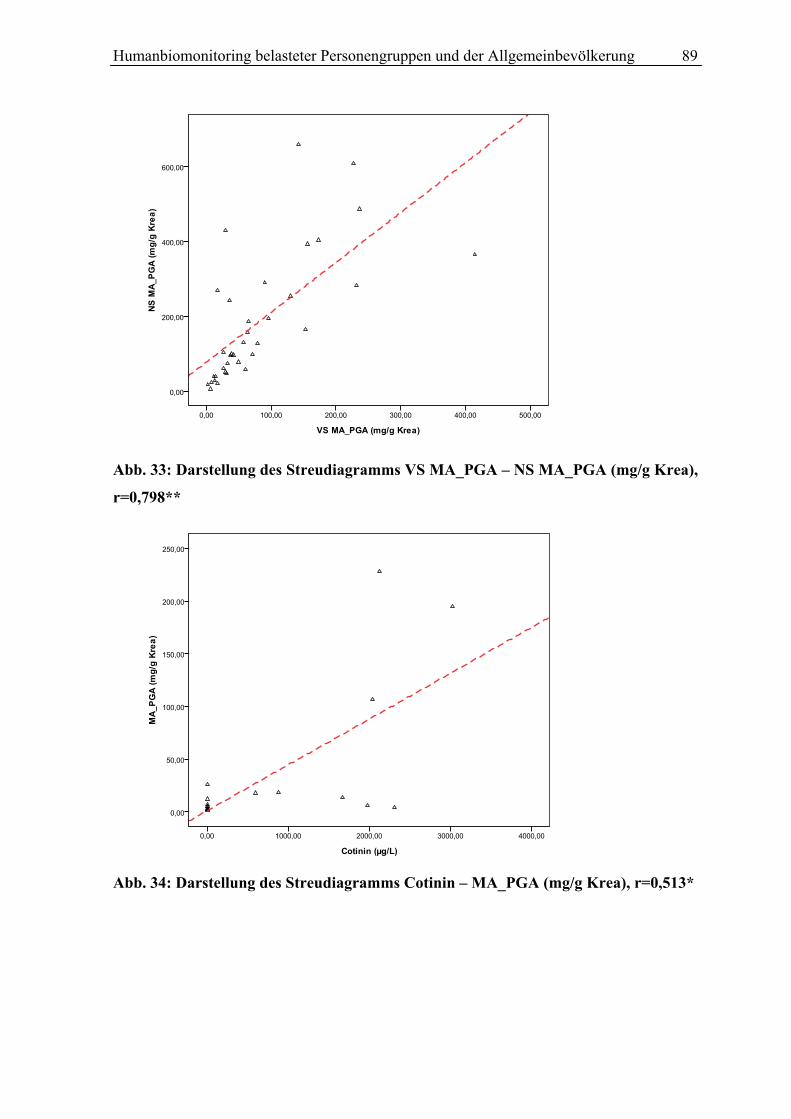
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
89
Abb. 33: Darstellung des Streudiagramms VS MA_PGA – NS MA_PGA (mg/g Krea),
r=0,798**
Abb. 34: Darstellung des Streudiagramms Cotinin – MA_PGA (mg/g Krea), r=0,513*

Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
90
Inferenzstatistische Auswertung
Die Tabellen 19-23 zeigen die Ergebnisse der Regressionen für das gesamte Kollektiv.
Folgende Parameter wurden über die Regressionen berechnet: ß (standardisiertes
Regressionsgewicht), SE (Standardfehler), t (T-Wert), F (F-Wert), ΔR2 (inkrementelle
Varianzaufklärung), R2 (Determinationskoeffizient).
Die Gruppenzugehörigkeit als Maß für die beruflich bedingte Exposition mit Styrol hatte
bis auf die Variable der VS MA_PGA (Tab.22) durchweg einen signifikanten positiven
Einfluss auf die Ausprägung der Konzentrationen der einzelnen Metaboliten (PHEMA VS:
Beta = 0,37, t = 2,74** PHEMA NS: Beta = 0,43, t = 3,26** ; HbA: Beta = 0,38 ,
t =2,75**; MA_PGA VS: Beta = 0,14; t = 0,98; MA_PGA NS: Beta = 0,39, t = 3,01**).
Die Kontrollvariablen Alter und Cotinin lieferten keinen signifikanten Erklärungsbeitrag
für die Ausprägung der Metaboliten.
Da Styrol erwiesenermaßen in kleinen Mengen im Tabakrauch vorkommt, wurde hier das
Kollektiv der Kontrollgruppen zusätzlich alleine einer linearen Regression unterzogen.
Hier zeigte sich eine signifikante Ausprägung der Variablen „Cotinin“ (PHEMA: Beta =
0,56; t = 2,63* ; MA_PGA: Beta = 0,70, t = 3,75**) gegenüber den Metaboliten, jedoch
nicht bei der Variable „Alter“ (Tab.24, 25). Da bei den Kontrollen die Hämoglobinaddukt-
Konzentrationen alle <1 pmol/g Globin waren, wurde die lineare Korrelation für das
Kontrollkollektiv nicht durchgeführt.
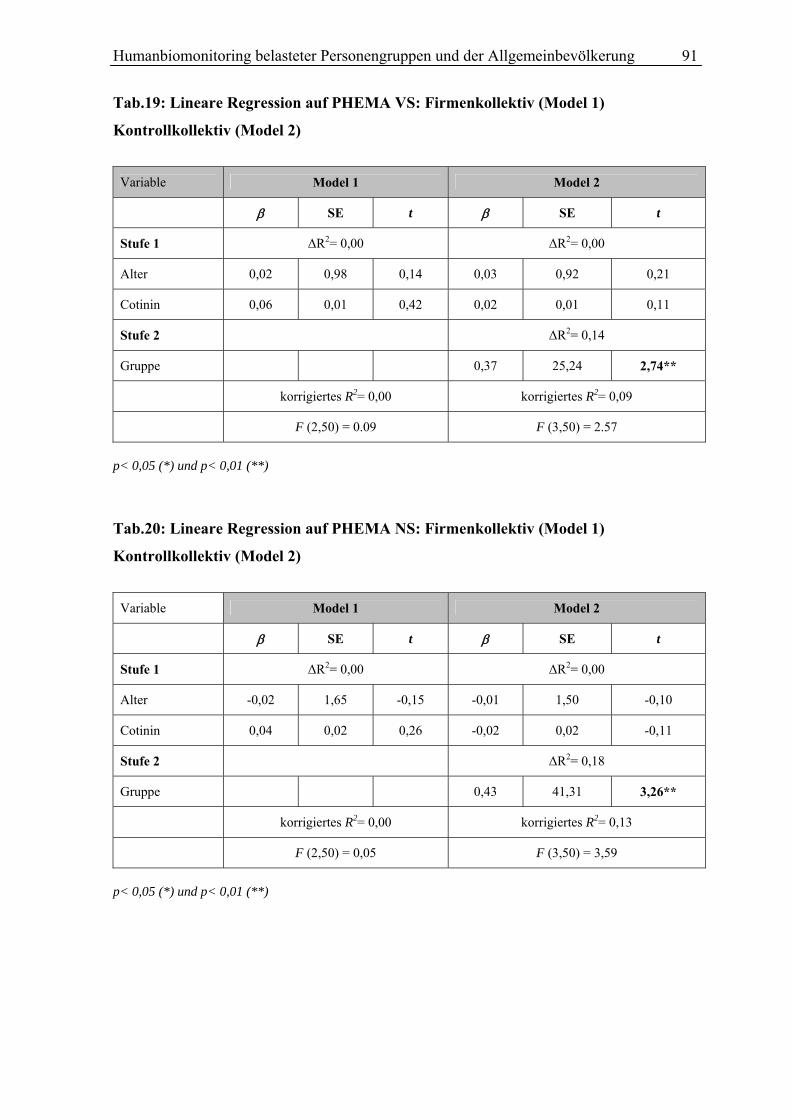
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
91
Tab.19: Lineare Regression auf PHEMA VS: Firmenkollektiv (Model 1)
Kontrollkollektiv (Model 2)
Variable Model 1 Model 2
SE t SE t
Stufe 1 ΔR2= 0,00 ΔR2= 0,00
Alter 0,02 0,98 0,14 0,03 0,92 0,21
Cotinin 0,06 0,01 0,42 0,02 0,01 0,11
Stufe 2 ΔR2= 0,14
Gruppe 0,37 25,24 2,74**
korrigiertes R2= 0,00 korrigiertes R2= 0,09
F (2,50) = 0.09 F (3,50) = 2.57
p< 0,05 (*) und p< 0,01 (**) Tab.20: Lineare Regression auf PHEMA NS: Firmenkollektiv (Model 1)
Kontrollkollektiv (Model 2)
Variable Model 1 Model 2
SE t SE t
Stufe 1 ΔR2= 0,00 ΔR2= 0,00
Alter -0,02 1,65 -0,15 -0,01 1,50 -0,10
Cotinin 0,04 0,02 0,26 -0,02 0,02 -0,11
Stufe 2 ΔR2= 0,18
Gruppe 0,43 41,31 3,26**
korrigiertes R2= 0,00 korrigiertes R2= 0,13
F (2,50) = 0,05 F (3,50) = 3,59
p< 0,05 (*) und p< 0,01 (**)
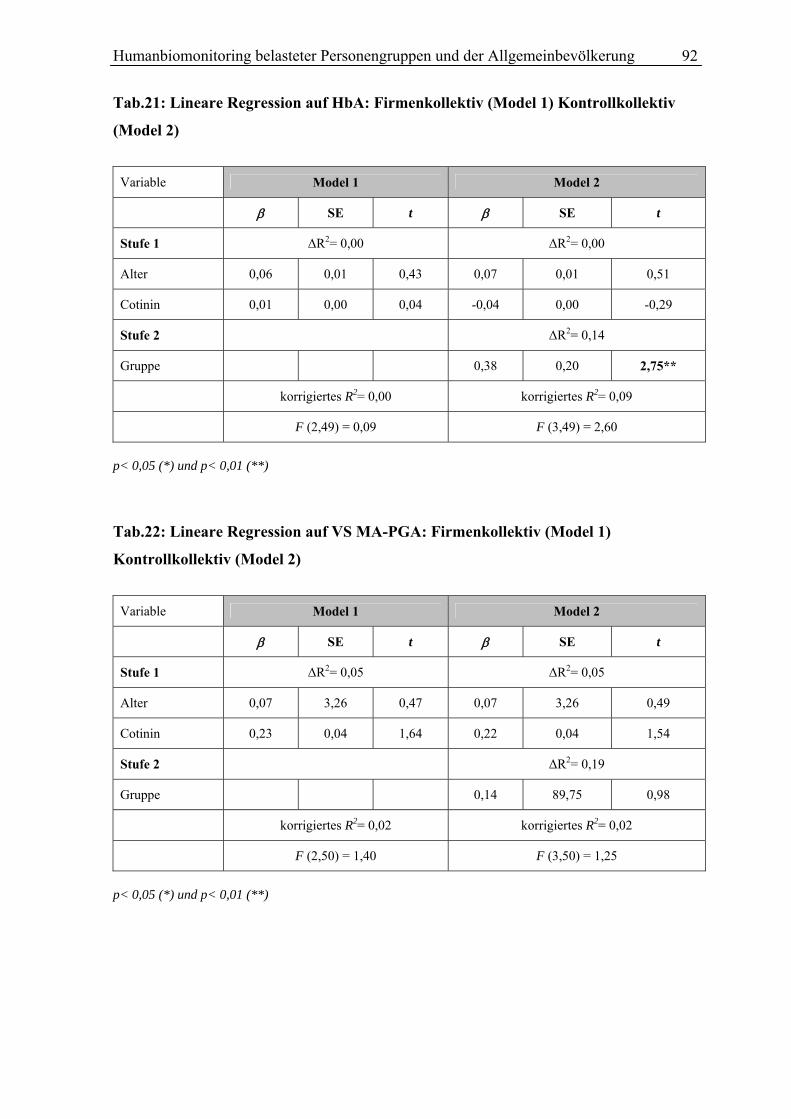
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
92
Tab.21: Lineare Regression auf HbA: Firmenkollektiv (Model 1) Kontrollkollektiv
(Model 2)
Variable Model 1 Model 2
SE t SE t
Stufe 1 ΔR2= 0,00 ΔR2= 0,00
Alter 0,06 0,01 0,43 0,07 0,01 0,51
Cotinin 0,01 0,00 0,04 -0,04 0,00 -0,29
Stufe 2 ΔR2= 0,14
Gruppe 0,38 0,20 2,75**
korrigiertes R2= 0,00 korrigiertes R2= 0,09
F (2,49) = 0,09 F (3,49) = 2,60
p< 0,05 (*) und p< 0,01 (**) Tab.22: Lineare Regression auf VS MA-PGA: Firmenkollektiv (Model 1)
Kontrollkollektiv (Model 2)
Variable Model 1 Model 2
SE t SE t
Stufe 1 ΔR2= 0,05 ΔR2= 0,05
Alter 0,07 3,26 0,47 0,07 3,26 0,49
Cotinin 0,23 0,04 1,64 0,22 0,04 1,54
Stufe 2 ΔR2= 0,19
Gruppe 0,14 89,75 0,98
korrigiertes R2= 0,02 korrigiertes R2= 0,02
F (2,50) = 1,40 F (3,50) = 1,25
p< 0,05 (*) und p< 0,01 (**)
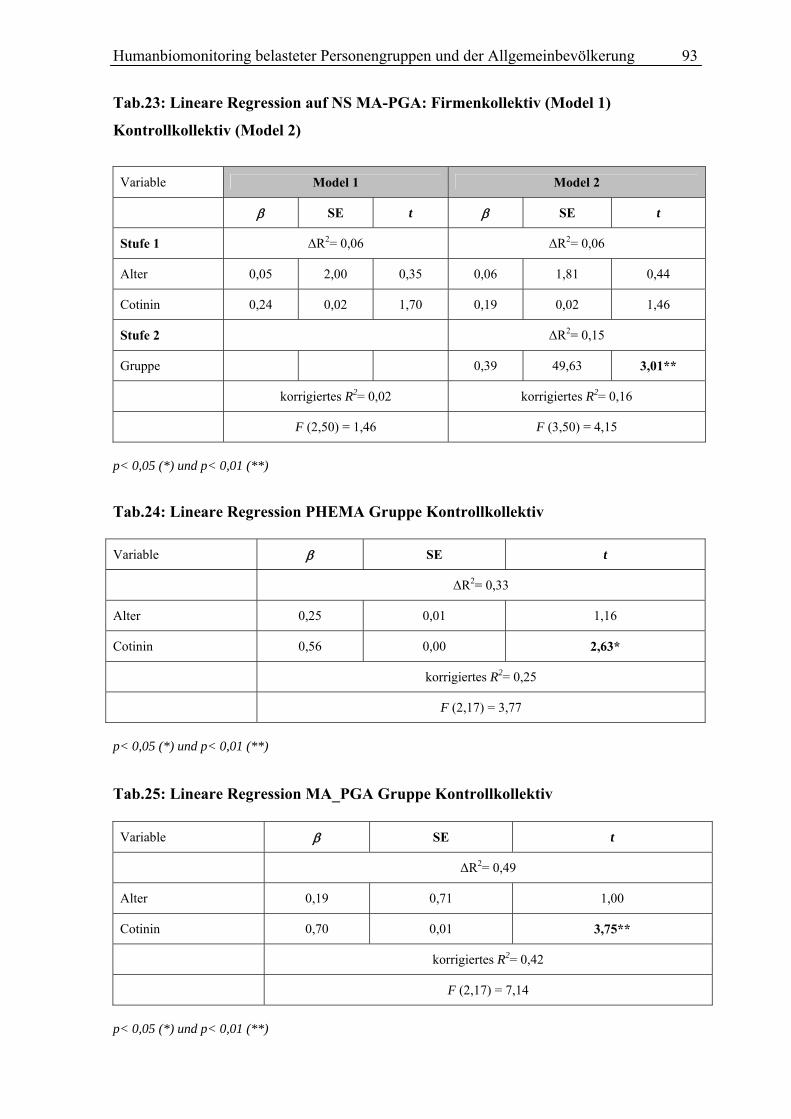
Humanbiomonitoring belasteter Personengruppen und der Allgemeinbevölkerung
93
Tab.23: Lineare Regression auf NS MA-PGA: Firmenkollektiv (Model 1)
Kontrollkollektiv (Model 2)
Variable Model 1 Model 2
SE t SE t
Stufe 1 ΔR2= 0,06 ΔR2= 0,06
Alter 0,05 2,00 0,35 0,06 1,81 0,44
Cotinin 0,24 0,02 1,70 0,19 0,02 1,46
Stufe 2 ΔR2= 0,15
Gruppe 0,39 49,63 3,01**
korrigiertes R2= 0,02 korrigiertes R2= 0,16
F (2,50) = 1,46 F (3,50) = 4,15
p< 0,05 (*) und p< 0,01 (**) Tab.24: Lineare Regression PHEMA Gruppe Kontrollkollektiv
Variable SE t
ΔR2= 0,33
Alter 0,25 0,01 1,16
Cotinin 0,56 0,00 2,63*
korrigiertes R2= 0,25
F (2,17) = 3,77
p< 0,05 (*) und p< 0,01 (**) Tab.25: Lineare Regression MA_PGA Gruppe Kontrollkollektiv
p< 0,05 (*) und p< 0,01 (**)
Variable SE t
ΔR2= 0,49
Alter 0,19 0,71 1,00
Cotinin 0,70 0,01 3,75**
korrigiertes R2= 0,42
F (2,17) = 7,14

Diskussion des Humanbiomonitorings
94
8 Diskussion des Humanbiomonitorings
Mittels der vorgestellten validierten Methoden war es möglich ein Humanbiomonitoring
styrolexponierter Personengruppen durchzuführen. Zur Untersuchung der zwei
Firmenkollektive als auch des Kontrollkollektivs dienten drei Marker-Metabolite des
Styrolstoffwechsels. Neben den in der Routine des Dosismonitorings verwendeten
aromatischen Carbonsäuren, Mandelsäure und Phenylglyoxylsäure, dienten darüber hinaus
die Bestimmung der Mercaptursäuren als auch die der Hämoglobinaddukte einer
Charakterisierung der Exposition. Für die aromatischen Carbonsäuren wurden von allen
Metaboliten die höchsten gemessenen Werte bestimmt. Während hier Mengen im Bereich
von mg/g Kreatinin vorlagen, befanden sich Werte der Mercaptursäuren um den Faktor
1000 unterhalb dieser Mengen (µg/g Kreatinin). Die kleinsten Mengen eines Metaboliten
wurden bei den Hämaglobinaddukten gemessen (Tab.16). Diese Ergebnisse unterstreichen
die bisherige Erkenntnis, dass der Weg des Styroloxids bevorzugt über die Umwandlung
von Styrolglykol hin zu den aromatischen Carbonsäuren führt und diese 95% (Bardodej &
Bardodejova, 1970) der gesamten aufgenommenen Styrolmenge repräsentieren.
Messungen der aromatischen Carbonsäurenkonzentrationen mittels HPLC im Urin gehören
daher zu den Standardmessungen des Styrol-bedingten Dosismonitorings (Kivisto et al,
1993; Laffon et al, 2001; Teixeira et al, 2010).
Der biologische Grenzwert des Styrols (abgeleitet aus dem BAT Wert) liegt bei 600 mg
Mandelsäure plus Phenylglyoxylsäure pro g Kreatinin (DFG, 2009). Dieser Wert wurde
innerhalb des Kollektivs der Firma 2 im Nachschichturin überschritten. Die Datenlage zu
Konzentrationen der aromatischen Carbonsäuren bei Menschen der Allgemeinbevölkerung
mit keiner beruflich bedingten Styrolexposition ist nicht sehr groß. Manini et al. gaben eine
obere Spannbreite von 2,56 – 3,38 mg/g Kreatinin für die Summe aus MA und PGA an
(Manini et al, 2004). Die Spannbreite bei den Konzentrationen des hier untersuchten
Kontrollkollektivs liegt zum Teil oberhalb dieser Konzentrationen. Dieser Unterscheid
könnte durch private, nicht-beruflich bedingte Exposition erklärt werden. Confounder
(Störfaktor) kann hierbei Ethylbenzol sein. Diese farblose Flüssigkeit wird häufig als
Antiklopfmittel in Motorenkraftstoffen zugefügt. Zudem findet es grosse Verwendung als
Lösemittelersatz des Benzols für Farben und Anstrichmittel und ist in einer Vielzahl von
Bodenbelägen enthalten (Knecht et al, 2000). Sie wird wie Styrol innerhalb ihres
Metabolismus zu Mandelsäure und Phenylglyoxylsäure umgewandelt und könnte dadurch
die Spezifität mindern (Schaller, 1976).

Diskussion des Humanbiomonitorings
95
Trotz dieses zusätzlichen Bias konnten sowohl die Vorschichtwerte von Fa.1 (p< 0,05) als
auch die der Fa.2 (p<0,01) signifikant von den Werten des unbelasteten Kontrollkollektivs
unterschieden werden (s. Abb. 24, 25). Dies zeigten die deutlichen durch Styrol bedingten
Belastungen auf Seiten der Firmenmitarbeiter, die auch im Vorschichturin messbare
Konzentrationen von MA und PGA aufwiesen. Die erhöhten Konzentrationen im
Vorschichturin können durch ihre Halbwertszeit von ca. 4 – 9 h erklärt werden (Triebig et
al, 1989; Wigaeus et al, 1984). Im Gegensatz zu den aromatischen Carbonsäuren MA und
PGA wird angenommen, dass die Phenylhydroxyethyl-Mercaptursäuren des Styrols nur zu
weniger als 1% über den Urin ausgeschieden werden (Norström et al, 1992). Trotz dieser
vergleichbar kleinen Mengen und der daraus resultierenden Anforderungen an eine
sensitive Methodik gelten sie nach derzeitigem Stand als substanzspezifischer (Manini et
al, 2003) als die Carbonsäuren. Daher wäre eine routinemäßige Mitbestimmung der
Mercaptursäuren im Humanbiomonitoring, sowie eine Verbesserung und Entwicklung
neuer Analysemethoden gefordert (Ghittori et al, 1997). Zur Konzentrationsbestimmung
wurde in dieser Untersuchung die Summe der PHEMAs (PHEMA1 und 2) berechnet. Dies
kann, analog zu der Summierung der aromatischen Carbonsäuren (MA+PGA) (Laffon et
al, 2001) zur Reduzierung potentieller individueller Unterschiede in der
Umbaugeschwindigkeit von Mandelsäure zu Phenylglyoxylsäure, Abweichungen in der
PHEMA Zusammensetzung reduzieren.
In allen Kollektiven konnten PHEMAs zu jedem Zeitpunkt nachgewiesen werden. Ähnlich
wie bei den aromatischen Carbonsäuren wurden auch bei der Analyse der Mercaptursäuren
diese bereits in den Vorschichturinen gefunden. Generell waren die Konzentrationen der
Mercaptursäuren im Vorschichturin ebenfalls signifikant höher als bei den Kontrollen
(Fa.1 p<0,05 , Fa.2 p<0,01). Das kann ein Indiz dafür sein, dass der Abbau des durch die
Exposition vorhandenen Styrols im Körper zum Zeitpunkt des erneuten Arbeitsbeginns
noch nicht vollständig geschehen ist (van Sittert et al, 2000b). Nach einer Unterbrechung
der Arbeit von mindestens 10 Stunden konnten noch Mercaptursäuren im Urin festgestellt
werden. Hintergrundkonzentrationen einer Kurzzeitexposition bei Nichtrauchern konnten
bereits 2-4 Stunden nach Exposition nachgewiesen werden.
Die Kontrollwerte der nicht exponierten Personengruppen sind im Median vergleichbar mit
anderen, unbelasteten Personengruppen aus der Literatur (Reska et al, 2010). Die
Extremwerte des Kontrollkollektivs bei MA_PGA und PHEMA (s. Spannbreite, Tab.16)
resultieren von drei Personen und lassen auf einen möglichen starken Einfluss von

Diskussion des Humanbiomonitorings
96
Konfoundern schließen. Einer der Probanden hatte den höchsten Wert bei den
Mercaptursäuren allerdings nur den drittgrößten bei den Carbonsäuren. Dies könnte
interpretiert werden als eine durch Konfounder verursachte Konzentration auf Seiten der
Carbonsäuren, jedoch ebenfalls eine Exposition gegenüber Styrol selbst, bedingt durch den
hohen Wert der sehr spezifischen Mercaptursäuren. In der Tat stellte sich bei dieser Person
erst nach Metabolitanalyse heraus, dass sie beruflich als Maler/Lackierer arbeitet, was
durchaus zu Styrolexpositionen einerseits und Ethylbenzolbelastungen andererseits führen
kann. Die beiden weiteren Probanden zeigten Werte, die im Bereich der bereits
publizierten Spannbreite nicht beruflich exponierter Personen lagen (Reska et al, 2010).
Beide lagen um das 3,7-fache bzw. 2,5-fache unterhalb des Höchstwertes (3,75 µg/g
Kreatinin) (Tab.16). Die parallel hohen Werte von MA_PGA könnten demzufolge eine
Konfounder vermittelte Ursache haben. Maestri et al. berichteten über urinständige mittlere
NS gesamt-PHEMA Konzentrationen von 1880 µg/g Kreatinin. Untersucht wurden hier 25
Arbeiter der glasfaserverstärkenden Plastikindustrie (Maestri et al, 1997). NS
Gesamtkonzentration von 474 µg/g Kreatinin im Mittelwert (VS=255,7) fanden Manini et
al. in einem Kollektiv von 56 Arbeitern der GfPI (Manini et al, 2000). In einer weiteren
Arbeit untersuchten Fustinoni et al. 13 Mitarbeiter der GfPI und stellten NS gesamt-
PHEMA Konzentrationen zwischen 39,4 – 3464 µg/g Kreatinin (VS= 2,9 – 653) mit einem
Median von 262 µg/g Kreatinin (VS= 105) fest. Die selbe Arbeitsgruppe untersuchte auch
ein Kollektiv aus der Lackproduktion und stellten dort NS Konzentrationen zwischen 3,2 –
559 (VS= <1,6 – 387,8) µg/g Kreatinin mit einem Median von 206 µg/g Kreatinin (VS =
93) fest (Fustinoni et al, 2008).
Die Spannbreiten der in der vorliegenden Arbeit ermittelten Konzentrationen, sowohl der
gesamt Vorschicht- als auch der Nachschicht (Tab.16), kongruieren sehr gut mit den oben
beschriebenen Werten der internationalen Literatur.
In beiden untersuchten Firmenkollektiven konnte das Vorkommen von
Hämoglobinaddukten nachgewiesen werden. Im Kollektiv unbelasteter Personengruppen
lagen demgegenüber alle Werte unterhalb der Nachweisgrenze von 1 pmol/g Globin
(Tab.16). Hier konnte ein signifikanter Unterschied (p<0,01) im Vergleich der
Adduktkonzentrationen zwischen den Kontrollen mit denen beider Firmen festgestellt
werden (Abb. 26). Die Ergebnisse können so interpretiert werden, dass es bei bestimmten
Arbeitern eine längerfristige, kontinuierliche Belastung gegeben haben muss, die jenseits
einer akuten, kurzfristigen Exposition lag und mittels der Kurzzeitparameter MA_PGA
sowie PHEMAs nachgewiesen werden kann. Der Median der Messwerte (MA_PGA und

Diskussion des Humanbiomonitorings
97
PHEMA) von Fa.1 lag unterhalb des Medians der Messwerte in Fa.2, obwohl die Werte
von Fa.2 nur eine Exposition von 6 Wochen repräsentieren. Die ebenfalls höheren NS-
Werte von Mercaptursäuren und aromatischen Carbonsäuren in Fa.2 gegenüber den
Werten der Fa.1, lassen die Vermutung zu, dass die Belastung in Fa.2 generell höher war
und eher nicht als ein Hauptresultat einer Suszeptibilität zu verstehen ist.
Um die innere Belastung einer Styrolexposition zu ermitteln wurden von anderen Gruppen
Hämoglobinaddukte des N-terminalen Valins bestimmt. So wiesen Christakopoulos et al.
Adduktwerte von 15-52 pmol/g Globin bei 17 Arbeitern der GfPI nach (Christakopoulos et
al, 1993). Hier waren allerdings extrem hohe Belastungen mit 300 mg/m³ Styrol gegeben
(AGW=86 mg/m³). Bei einem Detektionslimit von 10 pmol/g Globin untersuchten Severi
et al. 52 Arbeiter mit geringen Styrolexpositionen (31mg/m³) und konnten hier keine
Addukte nachweisen (Severi et al, 1994). Teixeira et al. berichteten von einer medianen
Konzentration von 5,78 pmol/g Globin in einem Kollektiv von 57 Arbeitern der GfPI mit
einer 2,5-fach niedrigeren Belastung (~ 120 mg/m³) im Vergleich zur Studie von
Christakopoulos et al. (Teixeira et al, 2008). Für die untersuchten Kollektive lagen keine
Luftmesswerte vor. Vergleicht man jedoch die Mediane der HbA-Val der neuesten Arbeit
auf diesem Gebiet, mit den hier gemessenen, ergäbe sich ein Verhältnis, welches auf eine
durchschnittliche Belastung mit 15 mg/m³ (Fa.1) und 10 mg/m³ (Fa.2) schließen lässt
(Teixeira et al, 2008). Dies sind allerdings rein geschätzte Werte und erlauben keine
Rückschlüsse auf akute Spitzenbelastungswerte.
Die Werte des hier untersuchten Kontrollkollektivs der Allgemeinbevölkerung lagen
erwartungsgemäß alle unterhalb 1 pmol/g Globin.
Der generelle Trend einer stärkeren Exposition in Fa.2 gegenüber Fa.1 zeigt sich auch in
den höheren Werten der Adduktkonzentrationen. Die Adduktwerte der Mitarbeiter in Fa.2
wären mit großer Wahrscheinlichkeit höher ausgefallen, wenn anstelle einer 6-wöchigen
eine 12-wöchige Exposition möglich gewesen und damit ein Expositionszeitraum
abgedeckt wäre, der die Lebenszeit des Hämoglobins widerspiegelt. Ab einer Exposition
von mehr als 120 Tagen wird ein steady state der HbA Konzentration erreicht und es
herrscht ein Gleichgewicht zwischen dem turn over der Erythrozyten und ihrer
Neubildung. Die individuelle maximale Konzentration ist demnach erst nach Ablauf von
120 Tagen ermittelbar.
Andere Autoren führten Messungen von Addukten der Cystein-Seitenketten und der
Carbonsäurereste des Albumins durch (Fustinoni et al, 1998; Yeowell-O'Connell et al,
1996). Etwa 5,2 % der Hb-Addukte in vitro bilden Addukte am endständigen Valin

Diskussion des Humanbiomonitorings
98
(Phillips & Farmer, 1994). Dennoch stellen Hämoglobinaddukte repräsentativere Surrogate
einer Adduktbildung auf Ebene der DNA dar als die Bildung von Albumin Addukten. Das
in den Blutgefäßen zirkulierende Albumin ist frei zugänglich und damit angreifbar. Somit
kann viel leichter mit Albumin ein Addukt gebildet werden, als es mit Hämoglobin
möglich ist, welches zusätzlich von der Erythrozytenmembran geschützt wird und dadurch
den Zugang seitens des Epoxides erschwert. Dies kann dazu führen, dass sich beim
direkten Vergleich mehr Albuminaddukte als Hämoglobinaddukte nachweisen lassen und
somit die Anforderungen an die Sensitivität der Messgeräte bzgl. der Detektierbarkeit eines
Albuminaddukts sinken. Der Weg vom Epoxid zum Hämoglobinaddukt hingegen ist, wie
oben beschrieben, komplexer, da das Styroloxid zunächst die Erythrozytmembran
passieren muss. Im Inneren des Erythrozyten triff das Epoxid auf hohe Konzentrationen
von Glutathion, so dass eine Konkurrenzreaktion zwischen einer Bindung des Epoxids an
das Hämoglobin und Glutathion entsteht. Da die DNA Adduktbildung ebenfalls erst mit
der Fähigkeit des Passierens einer Membran (Bader et al, 1994), der Kernmembran,
verbunden ist, ähneln Hämoglobinaddukte in ihrem Entstehungsmechanismus eher den
DNA-Addukten und können dadurch wesentlich mehr zu einer Risikobeurteilung und der
Einschätzung eines kanzerogenen Potentials beisteuern, als es bei den Albumin-Addukten
der Fall ist. Darüber hinaus besitzt das Hämoglobinaddukt eine Lebensdauer von 120
Tagen. Das Albumin dagegen besitzt nur eine Lebensdauer von ca. 60 Tagen und wird im
Gegensatz zum Hämoglobin durch eine Alkylierung in seiner Lebensdauer beeinträchtigt
(Ehrenberg & Osterman-Golkar, 1980; Granath et al, 1992).
Die hochsignifikanten Korrelationen zwischen den Vor- und Nachschichtwerten der
aromatischen Carbonsäuren sowie der Mercaptursäuren lassen für den Nachweiszeitraum
beider Metabolite auf eine individuelle gleichbleibende Kurzzeitbelastung schließen
(Abb.27, 33). Mäßige bis keine Signifikanz zeigten alle Korrelationen mit den
Mercaptursäuren. Hier könnten GSTM1- Polymorphismen eine beeinflussende Wirkung
auf die urinständigen Konzentrationen der Mercaptursäuren gehabt haben (De Palma et al,
2001; Haufroid et al, 2001), was folglich die Effektstärke einer Korrelation mindern kann
(Tab.18). Eine Genotypisierung der GSTM1 wurde im Rahmen dieser Arbeit nicht
durchgeführt. Bei den geplanten fortführenden Untersuchungen am beschriebenen
Kollektiv werden diese Typisierungen mit eingeschlossen.
Hier stand zunächst die Etablierung einer effizienten Methode zur Messung der
Mercaptursäuren im Vordergrund deren Anwendung es ermöglichte, styrolspezifische

Diskussion des Humanbiomonitorings
99
Metabolitkonzentrationen bei exponierten sowie nicht exponierten Personen zu
identifizieren (Tab.16 und 17).
Es konnten signifikante Korrelationen der aromatischen Carbonsäuren in Vor- und
Nachschicht mit den Hb-Addukten festgestellt werden. Hier ist folglich von weitgehend
individuell gleichbleibenden Expositionsbedingungen der vorhergegangenen 6 bzw. 12
Wochen im jeweiligen Betrieb auszugehen. Somit kann das Hämoglobinaddukt als
korrelierender Langzeitmarker zu den Kurzzeitmarkern der aromatischen Carbonsäuren
angesehen werden.
Mit Hilfe der hierarchisch linearen Regression konnte gezeigt werden, dass die
Konzentrationen der einzelnen gemessenen Metabolite signifikant von der Zugehörigkeit
zur Gruppe „Firmenkollektiv“ bzw. „Kontrollkollektiv“ abhängig sind (Tab.19-23).
Demnach hat die berufsnahe Styrolexposition einen eindeutigen Einfluss auf die
Metabolitkonzentration. Lediglich bei den Vorschichtwerten der aromatischen
Carbonsäuren ließ sich die Metabolitkonzentration nicht über die Zugehörigkeit zur
Gruppe definieren. Die in diesem Zusammenhang lineare Regression mit den potentiellen
Einflussvariablen „Alter“ und „Cotinin“ , bei der nur das Kontrollkollektiv einbezogen
wurde, zeigte für die aromatischen Carbonsäuren einen hochsignifikanten Einfluss des
Rauchens auf die Konzentration dieser Metabolite (Tab.25). Dieser Einfluss konnte
insgesamt nur bei nicht-berufsbedingter Exposition (Tab.25) bzw. bei den
Vorschichtwerten (Tab.22) gefunden werden. Dies könnte damit erklärt werden, dass die
Carbonsäuren in der arbeitsfreien Zeit bereits soweit abgebaut wurden, dass der Effekt des
Rauchens nun einen signifikanten Beitrag zur Gesamtkonzentration liefert. Die Vermutung
von Rappaport et al., dass Zigarettenrauchen einen Beitrag zur Hintergrundbelastung mit
Styroloxid liefert, kann dazu eine weitere Erklärung bieten (Rappaport et al, 1996). Im
Kontrollkollektiv konnte ebenfalls ein hoch signifikanter Einfluss der Variable Cotinin auf
die Konzentration der Mercaptursäuren beobachtet werden (Tab.24). In der Regression mit
beiden Gruppen kam dieser Einfluss nicht zum tragen (Tab.19).
Der Einfluss des Rauchens auf das Biomarkerlevel beider Metabolite wurde auch in
anderen Arbeiten untersucht (Fustinoni et al, 2008; Reska et al, 2010). Fustinoni et al.
klärten den Raucherstatus jedoch nicht über die Bestimmung des Metaboliten Cotinin
sondern mittels Fragebogen. (Fustinoni et al, 2008). Weiterhin wurde in dieser Arbeit keine
Aufspaltung der linearen Korrelation auf das Kollektiv der Kontrollgruppe gemacht,
sondern nur das gesamte Kollektiv untersucht. Es empfiehlt sich nicht nur eine Befragung
durchzuführen, sondern zusätzlich die analytische Cotininbestimmung. Eine Befragung

Diskussion des Humanbiomonitorings
100
unterliegt den subjektiven Angaben und Tendenzen zu sozial erwünschten Antworten
durch die Probanden. Mit Hilfe der analytischen Cotininbestimmung können diese
Fehlerquellen objektiv ausgeschlossen und Fehlinterpretationen der Ergebnisse vermieden
werden. Teixeira et al. konnte keinen durch das Rauchen verursachten Effekt auf die
Carbonsäuren beobachten, jedoch einen Effekt auf die Konzentration der
Hämoglobinaddukte. Dies kann darauf zurückzuführen sein, das die Nachweisgrenze zur
Detektion der Carbonsäuren sehr hoch war (14 mg/g Kreatinin); knapp unter dem in dieser
Arbeit ermittelten Median der Raucher im Kollektiv der Allgemeinbevölkerung (18,2 mg/g
Kreatinin) (s. Tab 17). Weiterhin wurde der Raucherstatus ebenfalls nur über einen
Fragebogen abgeklärt (Teixeira et al, 2008). Erwartungsgemäß müsste sich ein Effekt auf
Seiten der Hämoglobinaddukte als Langzeitmarker auch auf Seiten der Carbonsäuren
zeigen, es sei denn, die Probanden haben zur Zeit der Probennahme und innerhalb des
Nachweiszeitraums von Cotinin nicht geraucht. Signifikante altersbedingte Effekte
konnten in dieser Arbeit nicht beobachtet werden. Zwar zeigen die beiden -Werte des
Alters im Kontrollkollektiv höhere Ausprägungen als mit Hinzunahme der Gruppe des
exponierten Kollektivs (Tab.24 und 25) jedoch ohne eine Signifikanz. Es könnte einen
marginalen Einfluss der Variablen „Alter“ bei Nichtexponierten geben. Dieser wurde aber
in der Literatur im Zusammenhang mit Styrol bisher nicht als signifikant beschrieben.

Zusammenfassung
101
9 Zusammenfassung
Schadstoffbelastungen am Arbeitsplatz zu detektieren und somit frühzeitig und präventiv
Gesundheitsrisiken vorzubeugen ist von immenser Bedeutung. Insbesondere Gefahrstoffe
mit neurotoxischem oder kanzerogenem Potential bedürfen dabei größter Aufmerksamkeit.
Letztere zeichnen sich durch ihre hohe Reaktivität aus. Diese ist bereits vor der Aufnahme
in den Körper vorhanden oder wird durch metabolische Aktivierung hervorgerufen. In
Folge einer kovalenten Bindung dieser hochreaktiven Moleküle an die Nukleotide der
DNA können erste Schritte einer Kanzerogenese eingeleitet werden.
Im Rahmen einer Expositionsprüfung wird heutzutage oft ein Ambientmonitoring
durchgeführt, bei dem die Schadstoffkonzentration in der Luft am Arbeitsplatz bestimmt
wird. Diese Messungen liefern jedoch keine Informationen über die tatsächliche
Konzentration des Stoffes im Körper und obliegen häufig Störeffekten. In diesem
Zusammenhang kommt dem Biologischen Monitoring eine besondere Bedeutung zu.
Hierbei können Marker der inneren Dosis ebenso wie Marker eines Effektes ermittelt
werden und liefern dadurch Informationen, die in ihrer Bedeutung näher am Auftreten
einer potentiellen Gesundheitsbeeinträchtigung liegen, als die der herkömmlichen
Luftmessung. Die Messung verschiedener Endmetabolite innerhalb eines
Stoffmetabolismus liefert dabei ein vollständigeres Abbild der inneren Belastung und
bildet die Basis, das individuelle Risiko besser einschätzen zu können.
Styrol gehört zu einem der wichtigsten Monomere der Plastikindustrie. Expositionen sind
assoziiert mit unterschiedlichsten Gesundheitsgefährdungen. Von der International Agency
for Research on Cancer wurde Styrol als möglich kanzerogen (Gruppe 2B) und sein
direkter Metabolit das Styrol 7-8 Oxid als wahrscheinlich kanzerogen (Gruppe 2A)
eingestuft. Die DFG stufte Styrol in die Kategorie 5 krebsauslösender Stoffe ein.
Vor diesem Hintergrund bestand ein Ziel dieser Arbeit in der Durchführung eines Human
Biomonitorings, bei dem verschiedene Endmetabolite des Styrols im Blut und im Urin
bestimmt werden sollten. Es galt ferner zu untersuchen, ob und in wie weit die einzelnen
Metabolite in ihrer Gemeinsamkeit als Marker der Exposition fungieren können. Zu den
untersuchten Parametern zählten die klassischen Kurzzeit-Parameter des Dosismonitorings
Mandelsäure und Phenyglyoxylsäure. Sie repräsentieren den mittels Epoxidhydrolase
katalysierten Weg des Styrols. Weitere Metabolite waren die Styrol spezifischen
Mercaptursäuren (PHEMA) N-Acetyl-S-(2-Hydroxy-1-Phenylethyl)-Cystein (PHEMA1)
und N-Acetyl-S-(2-Hydroxy-2-Phenylethyl)-Cystein (PHEMA2). Diese Kurzzeitmarker

Zusammenfassung
102
resultieren aus einem weiteren Stoffwechselweg der die Detoxifizierung von Styrol mittels
Gluthation-S-transferase einleitet. Als Langzeit-Parameter wurde das Hämoglobinaddukt
N-(2-Hydroxy-2-Phenylethyl)-Valin bestimmt. Aus der Reaktion des Styrol Epoxids
(Styrol 7-8 Oxid) mit dem endständigen Valin der Hämoglobinseitenkette, entsteht ein
Addukt welches als Surrogat möglicher DNA-Addukte angesehen werden kann. Durch
Reparaturmechanismen bedingte Eliminationen des DNA-Adduktes, machen eine genaue
Quantifizierung hierbei oft nicht möglich. Im Gegensatz dazu unterliegen
Hämoglobinaddukte keinen Reparaturvorgängen und bleiben trotz Alkylierung über ihren
gesamten Lebenszeitraum von ca. 120 Tagen stabil.
Die Vorraussetzung zur Durchführung eines Biomonitorings ist das Vorhandensein von
geeigneten analytischen Methoden. Zwei dieser Methoden galt es im Rahmen dieser Arbeit
zunächst zu erarbeiten und zu validieren.
Die etablierte Methode zur Messung des Hämoglobinadduktes N-(2-Hydroxy-2-
Phenylethyl)-Valin lieferte sehr gute Ergebnisse für die Präzisionen in Serie als auch für
die Präzisionen von Tag zu Tag sowohl bei den niedrig konzentrierten als auch bei den
höher konzentrierten Qualitätskontrollen. Die Wiederfindungsversuche zur Ermittlung des
Einflusses unterschiedlicher Blutmatrices auf das Analyseergebnis, lieferten sehr gute
Wiederfindungsraten, was nicht zuletzt auf den hier verwendeten internen Standard und
des zur Dotierung verwendeten käuflichen Dipeptidstandards zurückzuführen war
(s. Kap.5).
Im Rahmen dieser Arbeit wurde weiterhin eine Methode zur Messung von Styrol
spezifischen Mercaptursäuren erarbeitet und etabliert (Kap.4). Nach derzeitigem
Wissenstand des Autors dieser Arbeit wurde hierbei erstmals mittels Backflushverfahren
gearbeitet und ein isotopenmarkierter interner Standard verwendet - eine Voraussetzung
zur Generierung zuverlässiger quantitativer Ergebnisse. Die Präzisionsversuche sowie die
Wiederfindungsversuche und die der Richtigkeit lieferten sehr gute Ergebnisse.
Die Validierungsergebnisse zeigten, dass beide Methoden als robust angesehen werden
können und geeignet sind, im Rahmen von zukünftigen humanen Biomonitoring Studien
von Styrol Anwendung zu finden.
Im Anschluss an die Erarbeitung der Analysemethodik wurde ein Dosis- und
Effektbiomonitoring an Kollektiven zweier Firmen der styrolverarbeitenden Industrie
durchgeführt. Ein weiteres Kollektiv nicht beruflich exponierter Personen der
Allgemeinbevölkerung fungierte als Kontrolle (Kap. 7).

Zusammenfassung
103
Die höchsten Konzentrationen der hier bestimmten Marker wurden bei den aromatischen
Carbonsäuren Mandelsäure und Phenylglyoxylsäure in allen Kollektiven gemessen. Die
Expositionsbiomarker Mercaptursäuren und die Hämoglobinaddukte konnten ebenfalls
nachgewiesen werden – letztere allerdings nicht im Kollektiv der Allgemeinbevölkerung.
Insgesamt konnten mittels aller gemessenen Parameter die Firmenkollektive signifikant
von dem Kontrollkollektiv unterschieden werden. Signifikante Korrelationen der
Metabolitkonzentrationen untereinander (auch Vor- und Nachschicht) konnten bei beiden
Firmenkollektiven gezeigt werden. Ohne Signifikanz aber dennoch mit einem schwach bis
mäßigen Zusammenhang wurden die Korrelationen zwischen Nachschicht MA_PGA mit
Vor- und Nachschicht PHEMA abgebildet.
Ein signifikanter Einfluss des Rauchens auf die Konzentrationen der aromatischen
Carbonsäuren als auch der Mercaptursäuren konnte bei den Firmenkollektiven nicht, sehr
wohl aber bei den Probanden der Allgemeinbevölkerung beobachtet werden. Ein alters-
bedingter Einfluss sowohl bei den beruflich Exponierten als auch bei Nicht-Exponierten
zeigte sich nicht.
Zusammenfassend kann gesagt werden, dass in dieser Arbeit zwei valide Methoden
etabliert werden konnten und diese im Rahmen eines Dosis- und Effektmonitorings an
zwei Firmenkollektiven Anwendung fanden. Erstmals wurden dabei parallel die beiden
Parameter HbA-Val und PHEMA des Styrols am gleichen Kollektiv bestimmt.
PHEMA Korrelationen mit den übrigen Metaboliten (HbA-Val; MA_PGA) zeigten trotz
eines möglichen GSTM1-Polymorphismus schwach bis mäßige Zusammenhänge. Der
grosse Nutzen der Mercaptursäuren liegt nicht zuletzt in ihrer Spezifität, die gerade bei
Hintergrundbelastungen beruflich nicht belasteter Personen mit Mischexpositionen einen
Nutzen erbringen können (bspw. Styrol vs. Ethylbenzol). Generell liefern sie
Informationen über die metabolische Aktivität der Glutathion S-transferase. Somit können
sie als eine Ergänzung zur Geno- und Phänotypisierung der GSTM1 angesehen werden, die
in weiteren follow-up Studien der untersuchten Firmen implementiert werden und die hier
beobachteten Korrelationseffekte verbessern sollen. Hier können die Mercaptursäuren
eindeutig als ergänzende Marker zu den aromatischen Carbonsäuren angesehen werden.
Die Erfassung der Langzeitparameter der Hämoglobinaddukte stellt ebenfalls einen
weiteren wichtigen Parameter dar, der Hinweise erster genotoxischer Effekte liefern kann.
Konzentrationen von Hämoglobinaddukten konnten bei einzelnen Probanden innerhalb der
Firmenkollektive gemessen werden, was über die Validierung der Methode hinaus deren
Applikabilität im Biomonitoring bestätigen konnte. Hier werden geplante fortführende

Zusammenfassung
104
Untersuchungen, insbesondere beim Kollektiv der Firma 2 nach einer mindestens 12-
wöchigen Dauerexposition erfolgen. Zusätzlich zur relativ aufwendigen
Probenaufarbeitung, ist die Bestimmung der HbA als Routineparameter oft nur in
speziellen Laboren möglich. Dennoch sollten diese Effektmarker möglichst immer
Bestandteil von Expositionserfassungen sein und zumindest zum Standard initialer
betrieblicher Vorsorgeuntersuchungen werden. Nur so können frühzeitig mögliche
genotoxische Effekte bei beruflich bedingten Styrolbelastungen erkannt und präventive
Maßnahmen auf betrieblicher und individueller Ebene eingeleitet werden.
Die etablierten Methoden werden zukünftig in externe Qualitätskontrollen durch
Ringversuche eingebunden und im Human Biomonitoring bei follow-up Studien
untersuchter Kollektive sowie bei weiteren neuen Firmen Anwendung finden.

Abbildungsverzeichnis
105
10 Abbildungsverzeichnis
Abb.1: Styrol Metabolismus mit Hauptstoffwechselwegen
Abb.2: Monitoring von Schadstoffen in der Arbeits- und Umweltmedizin
Abb.3: Mechanismus einer Hämoglobinadduktbildung an das N-terminale
Valin(skizziert) am Beispiel des Epoxids Styrol-7,8-oxid (Quelle:
http://commons.wikimedia.org/wiki/File:1GZX_Haemoglobin.png?uselang
=de)
Abb.4: Bildung des Thiocarbamoylderivats durch die Reaktion von PFPITC mit der
Aminogruppe des N-terminalen Valins
Abb.5: Zyklisierungsreaktion des Thiocarbamoylderivates und Entstehung eines
Thiazolinderivates unter Abspaltung des Hämoglobinrestes. Umlagerung
des Thiazolinderivates in das Pentafluorphenylthiohydantoinderivat
(Analyten)
Abb.6: Unterschiedliche turn-over Zeiten von Expositionsbiomarkern
Abb.7: Anreicherungsschaltung und Transferschaltung des 6-fach Ventils
Abb.8: Massenspektren von PHEMA1 (oben) und 13C6 PHEMA1 (unten) mit
Ionenzerfällen (rot)
Abb.9: Darstellung zweier Kalibrierkurven
Abb.10: Kalibrierkurven dargestellt in Urin ( ) und Wasser (- - - -)
Abb.11: Exemplarische Darstellung eines Chromatogramms einer Nichtraucher
Urinprobe (Cotinin 4,8µg/L) ohne arbeitsbedingter Styrolexposition und
einer PHEMA Konzentration von 0,4 µg/L
Abb.12: Molekülstruktur des N-(2-Hydroxy-2-Phenylethyl)-Val-Leu-anilid

Abbildungsverzeichnis
106
Abb.13: Molekülstruktur des 4-Hydroxyphenetylbromid (HPB) (a), 4-Fluor-
phenethylbromid (FPB) (b) und der daraus hergestellten internen Standards
4-Hydroxyphenetylbromid-Globin sowie 4-Hydroxyphenetylbromid-Globin
Abb.14: Entstehung des N-(2-Hydroxy-2-Phenylethyl)-Valin- PFPTH (Analyten)
Abb.15: Massenspektrum mit Analyt-Fragmenten (rot) des N-(2-Hydroxy-2-
Phenylethyl)-Valin–PFPTH (a), 4-Hydroxyphenetyl-Valin-PFPTH (b), 4-
Fluorphenethyl-Valin–PFPTH (c); Elektronenstossionisation
Abb.16: Chromatogramm einer dotierten Poolglobinprobe (50 pmol)
Abb.17: Kalibrierkurve des 2-Hydroxy-2-phenylethyl-Valin bezogen auf 4-
Hydroxyphenethylbromid-Globin
Abb.18: Kalibrierkurve des 2-Hydroxy-2-phenylethyl-Valin bezogen auf 4-
Fluorphenethylbromid-Globin
Abb.19: Darstellung eines Chromatogramms einer styrolexponierten Person (2,3
pmol/g Globin)
Abb.20: Kalibrierkurve für Phenylglyoxylsäure (oben) und Mandelsäure (unten),
“area under curve” (AUC), interner Standard (IS), Mandelsäure (MA),
Phenylglyoxylsäure (PGA)
Abb.21: Darstellung zweier exemplarischer Chromatogramme der Analytik von
Phenylglyoxylsäure (PGA) und Mandelsäure (MA). A: Chromatogramm mit
dotierten Standards von 25 mg/L B: Darstellung einer Realprobe ohne
berufliche Exposition mit Styrol (2,5 mg/L MA_PGA); IS (interner Standard
3-Chlor-4-hydroxybenzoesäure)
Abb.22: Vergleich des Metaboliten PHEMA (VS,NS) innerhalb Fa.1 und mit der
Kontrollgruppe (◊ = Extremwerte ○ = Ausreißer, * Signifikanz auf dem 0,05
Niveau ** Signifikanz auf dem 0,01 Niveau )

Abbildungsverzeichnis
107
Abb.23: Vergleich des Metaboliten PHEMA (VS,NS) innerhalb Fa.2 und mit der
Kontrollgruppe Kontrollgruppe (◊ = Extremwerte ○ = Ausreißer, *
Signifikanz auf dem 0,05 Niveau ** Signifikanz auf dem 0,01 Niveau)
Abb.24: Vergleich des Metaboliten MA_PGA (VS,NS) innerhalb Fa.1 und mit der
Kontrollgruppe (◊ = Extremwerte ○ = Ausreißer, * Signifikanz auf dem 0,05
Niveau ** Signifikanz auf dem 0,01 Niveau)
Abb.25: Vergleich des Metaboliten MA_PGA (VS,NS) innerhalb Fa.2 und mit der
Kontrollgruppe (◊ = Extremwerte ○ = Ausreißer, * Signifikanz auf dem 0,05
Niveau ** Signifikanz auf dem 0,01 Niveau)
Abb.26: Vergleich der Hämoglobinaddukte mit der Kontrollgruppe Kontrollgruppe
(◊ = Extremwerte ○ = Ausreißer, * Signifikanz auf dem 0,05 Niveau **
Signifikanz auf dem 0,01 Niveau)
Abb.27: Darstellung des Streudiagramms NS PHEMA– VS PHEMA (µg/g Krea),
r=0,853**
Abb.28: Darstellung des Streudiagramms VS PHEMA (µg/g Krea) – HbA (pmol/g
Globin), r=0,388*
Abb.29: Darstellung des Streudiagramms VS MA_PGA (mg/g Krea) – VS PHEMA
(µg/g Krea), r=0,450**
Abb.30: Darstellung des Streudiagramms NS PHEMA (µg/g Krea) – HbA (pmol/g
Globin), r=0,362**
Abb.31: Darstellung des Streudiagramms VS MA_PGA (mg/g Krea) – HbA (pmol/g
Globin), r=0,638**
Abb.32: Darstellung des Streudiagramms NS MA_PGA (mg/g Krea) – HbA (pmol/g
Globin), r=0,526**
Abb.33: Darstellung des Streudiagramms VS MA_PGA – NS MA_PGA (mg/g
Krea), r=0,798**

Abbildungsverzeichnis
108
Abb.34: Darstellung des Streudiagramms Cotinin – MA_PGA (mg/g Krea), r=0,513*

Tabellenverzeichnis
109
11 Tabellenverzeichnis
Tab.1: Die Stoffeigenschaften des Styrols
Tab.2: Pipettierschema zur Herstellung von Vergleichstandards
Tab.3: Programm der Gradientenpumpe
Tab.4: RT, Ionenübergänge (Q1,Q3), DP, EP, FP, CE und CXP der markierten und
nicht markierten PHEMAs
Tab.5: Ergebnisse der Präzision in Serie und von Tag zu Tag
Tab.6: Bestimmung der Richtigkeit in sieben verschiedenen Urinproben
Tab.7: Stabilitäten der Qualitätskontrollen Q3, Q30, QRea
Tab.8: Ergebnisse des Biomonitorings von Personen der Allgemeinbevölkerung
Tab.9: Pipettierschema zur Herstellung von Vergleichstandards
Tab.10: Darstellung der Retentionszeiten und detektierten Ionenspuren der Analyte
Tab.11: Ergebnisse der Präzision in Serie der internen Standards HPB und FPB
Tab.12: Ergebnisse der Präzision von Tag-zu-Tag der internen Standards HPB und
FPB
Tab.13: Wiederfindungsraten (WFR) in Individualglobinen; Werte kleiner
Nachweisgrenze von 1 pmol/g Globin werden mit halber Nachweisgrenze
berechnet; alle Angaben in pmol/g Globin
Tab.14: Pipettierschema für die Kalibrierkurve MA und PGA
Tab.15: Gradientenprogramm zur Bestimmung der aromatischen Carbonsäuren
Tab.16: Median, Standardabweichung und Spannbreite der gemessenen Metabolite
Tab.17: Unterteilung der Metabolitkonzentrationen der Kontrollen in Nichtraucher
(NR) und Raucher (R)

Tabellenverzeichnis
110
Tab.18: Korrelation zwischen den verschiedenen Biomarkern der Styrolexposition
nach Spearman (zweiseitig), Korrelationskoeffizient (r), p< 0,05 (*) und
p<0,01 (**), keine signifikante Korrelation (kk)
Tab.19: Lineare Regression auf PHEMA VS: Firmenkollektiv Kontrollkollektiv
Tab.20: Lineare Regression auf PHEMA NS Firmenkollektiv Kontrollkollektiv
Tab.21: Lineare Regression auf HbA: Firmenkollektiv Kontrollkollektiv
Tab.22: Lineare Regression auf VS MA-PGA: Firmenkollektiv Kontrollkollektiv
Tab.23: Lineare Regression auf NS MA-PGA: Firmenkollektiv Kontrollkollektiv
Tab.24: Lineare Regression PHEMA Gruppe 0
Tab.25: Lineare Regression MA_PGA Gruppe 0

Literaturverzeichnis
111
12 Literaturverzeichnis
Adam T, McAughey J, McGrath C, Mocker C, Zimmermann R (2009) Simultaneous on‐line size and chemical analysis of gas phase and particulate phase of cigarette mainstream smoke. Anal Bioanal Chem 394(4): 1193‐1203
Adgate JL, Eberly LE, Stroebel C, Pellizzari ED, Sexton K (2004) Personal, indoor, and outdoor VOC exposures in a probability sample of children. J Expo Anal Environ Epidemiol 14 Suppl 1: S4‐S13
Angerer J (2001) Biological Monitoring ‐ Heutige und künftige Möglichkeiten in der Arbeits‐ und Umweltmedizin, Weinheim: Wiley ‐ VCH Verlag GmbH.
Angerer J, Gundel J (1996) Biomonitoring and occupational medicine. Possibilities and limitations. Ann Ist Super Sanita 32(2): 199‐206
Bader M, Lehnert G, Angerer J (1994) GC/MS determination of N‐phenylvaline, a possible biomarker for benzene exposure in human hemoglobin by the "N‐alkyl Edman method". Int Arch Occup Environ Health 65(6): 411‐414
Bardodej Z, Bardodejova E (1970) Biotransformation of ethyl benzene, styrene, and alpha‐methylstyrene in man. Am Ind Hyg Assoc J 31(2): 206‐209
Bastlova T, Vodicka P, Peterkova K, Hemminki K, Lambert B (1995) Styrene oxide‐induced HPRT mutations, DNA adducts and DNA strand breaks in cultured human lymphocytes. Carcinogenesis 16(10): 2357‐2362
Bender HF (2008) Das Gefahrstoffbuch: Sicherer Umgang mit Gefahrstoffen nach REACH und GHS, Vol. 3: Wiley‐VCH Verlag GmbH & Co. KGaA.
Boettcher MI, Angerer J (2005) Determination of the major mercapturic acids of acrylamide and glycidamide in human urine by LC‐ESI‐MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 824(1‐2): 283‐294
Brenner DD, Jeffrey AM, Latriano L, Wazneh L, Warburton D, Toor M, Pero RW, Andrews LR, Walles S, Perera FP (1991) Biomarkers in styrene‐exposed boatbuilders. Mutat Res 261(3): 225‐236
Brooks SM, Anderson L, Emmett E, Carson A, Tsay JY, Elia V, Buncher R, Karbowsky R (1980) The effects of protective equipment on styrene exposure in workers in the reinforced plastics industry. Arch Environ Health 35(5): 287‐294

Literaturverzeichnis
112
Campagna D, Gobba F, Mergler D, Moreau T, Galassi C, Cavalleri A, Huel G (1996) Color vision loss among styrene‐exposed workers neurotoxicological threshold assessment. Neurotoxicology 17(2): 367‐373
Chen GD, Henderson D (2009) Cochlear injuries induced by the combined exposure to noise and styrene. Hear Res
Christakopoulos A, Bergmark E, Zorcec V, Norppa H, Maki‐Paakkanen J, Osterman‐Golkar S (1993) Monitoring occupational exposure to styrene from hemoglobin adducts and metabolites in blood. Scand J Work Environ Health 19(4): 255‐263
Cruzan G, Cushman JR, Andrews LS, Granville GC, Johnson KA, Bevan C, Hardy CJ, Coombs DW, Mullins PA, Brown WR (2001) Chronic toxicity/oncogenicity study of styrene in CD‐1 mice by inhalation exposure for 104 weeks. J Appl Toxicol 21(3): 185‐198
Czene K, Osterman‐Golkar S, Yun X, Li G, Zhao F, Perez HL, Li M, Natarajan AT, Segerback D (2002) Analysis of DNA and hemoglobin adducts and sister chromatid exchanges in a human population occupationally exposed to propylene oxide: a pilot study. Cancer Epidemiol Biomarkers Prev 11(3): 315‐318
Darrall KG, Figgins JA, Brown RD, Phillips GF (1998) Determination of benzene and associated volatile compounds in mainstream cigarette smoke. Analyst 123(5): 1095‐1101
De Palma G, Manini P, Mozzoni P, Andreoli R, Bergamaschi E, Cavazzini S, Franchini I, Mutti A (2001) Polymorphism of xenobiotic‐metabolizing enzymes and excretion of styrene‐specific mercapturic acids. Chem Res Toxicol 14(10): 1393‐1400
DFG (2009) MAK‐ und BAT‐Werte‐Liste 2009: Maximale Arbeitsplatzkonzentrationen und Biologische Arbeitsstofftoleranzwerte. Mitteilung 45: Wiley‐VCH Verlag GmbH & Co. KGaA.
Duell EJ, Wiencke JK, Cheng TJ, Varkonyi A, Zuo ZF, Ashok TD, Mark EJ, Wain JC, Christiani DC, Kelsey KT (2000) Polymorphisms in the DNA repair genes XRCC1 and ERCC2 and biomarkers of DNA damage in human blood mononuclear cells. Carcinogenesis 21(5): 965‐971
Dypbukt JM, Costa LG, Manzo L, Orrenius S, Nicotera P (1992) Cytotoxic and genotoxic effects of styrene‐7,8‐oxide in neuroadrenergic Pc 12 cells. Carcinogenesis 13(3): 417‐424
Edman P (1949) A method for the determination of amino acid sequence in peptides. Arch Biochem 22(3): 475
Ehrenberg L, Hiesche KD, Osterman‐Golkar S, Wenneberg I (1974) Evaluation of genetic risks of alkylating agents: tissue doses in the mouse from air contaminated with ethylene oxide. Mutat Res 24(2): 83‐103

Literaturverzeichnis
113
Ehrenberg L, Osterman‐Golkar S (1980) Alkylation of macromolecules for detecting mutagenic agents. Teratog Carcinog Mutagen 1(1): 105‐127
Fracasso ME, Doria D, Carrieri M, Bartolucci GB, Quintavalle S, De Rosa E (2009) DNA single‐ and double‐strand breaks by alkaline‐ and immuno‐comet assay in lymphocytes of workers exposed to styrene. Toxicol Lett 185(1): 9‐15
Fustinoni S, Campo L, Manini P, Buratti M, Waidyanatha S, De Palma G, Mutti A, Foa V, Colombi A, Rappaport SM (2008) An integrated approach to biomonitoring exposure to styrene and styrene‐(7,8)‐oxide using a repeated measurements sampling design. Biomarkers 13(6): 560‐578
Fustinoni S, Colosio C, Colombi A, Lastrucci L, Yeowell‐O'Connell K, Rappaport SM (1998) Albumin and hemoglobin adducts as biomarkers of exposure to styrene in fiberglass‐reinforced‐plastics workers. Int Arch Occup Environ Health 71(1): 35‐41
Ghittori S, Maestri L, Imbriani M, Capodaglio E, Cavalleri A (1997) Urinary excretion of specific mercapturic acids in workers exposed to styrene. Am J Ind Med 31(5): 636‐644
Gobba F (2000) Color vision: a sensitive indicator of exposure to neurotoxins. Neurotoxicology 21(5): 857‐862
Godderis L, Aka P, Mateuca R, Kirsch‐Volders M, Lison D, Veulemans H (2006) Dose‐dependent influence of genetic polymorphisms on DNA damage induced by styrene oxide, ethylene oxide and gamma‐radiation. Toxicology 219(1‐3): 220‐229
Granath F, Ehrenberg L, Tornqvist M (1992) Degree of alkylation of macromolecules in vivo from variable exposure. Mutat Res 284(2): 297‐306
Hagmar L, Tornqvist M, Nordander C, Rosen I, Bruze M, Kautiainen A, Magnusson AL, Malmberg B, Aprea P, Granath F, Axmon A (2001) Health effects of occupational exposure to acrylamide using hemoglobin adducts as biomarkers of internal dose. Scand J Work Environ Health 27(4): 219‐226
Hallier E, Goergens HW, Karels H, Golka K (1995) A note on individual differences in the urinary excretion of optical enantiomers of styrene metabolites and of styrene‐derived mercapturic acids in humans. Arch Toxicol 69(5): 300‐305
Haufroid V, Lison D (1998) Urinary cotinine as a tobacco‐smoke exposure index: a minireview. Int Arch Occup Environ Health 71(3): 162‐168

Literaturverzeichnis
114
Haufroid V, Toubeau F, Clippe A, Buysschaert M, Gala JL, Lison D (2001) Real‐time quantification of cytochrome P4502E1 mRNA in human peripheral blood lymphocytes by reverse transcription‐PCR: method and practical application. Clin Chem 47(6): 1126‐1129
Hecht SS, Villalta PW, Hochalter JB (2008) Analysis of phenanthrene diol epoxide mercapturic acid detoxification products in human urine: relevance to molecular epidemiology studies of glutathione S‐transferase polymorphisms. Carcinogenesis 29(5): 937‐943
Henderson RF, Bechtold WE, Bond JA, Sun JD (1989) The use of biological markers in toxicology. Crit Rev Toxicol 20(2): 65‐82
Horvath E, Pongracz K, Rappaport S, Bodell WJ (1994) 32P‐post‐labeling detection of DNA adducts in mononuclear cells of workers occupationally exposed to styrene. Carcinogenesis 15(7): 1309‐1315
IARC. (1994) Some Industrial Chemicals. Vol. 60.
IARC. (2002) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. IARC Press Lyon France, Vol. 82.
IBM. (1993‐2007) PASW Statistics 18. SPSS Inc.
Jackson H (1953) Studies with erythrocytes labelled with radioactive p‐lodophenylhydroxylamine. Nature 172(4367): 80‐81
Kellert M, Scholz K, Wagner S, Dekant W, Volkel W (2006) Quantitation of mercapturic acids from acrylamide and glycidamide in human urine using a column switching tool with two trap columns and electrospray tandem mass spectrometry. J Chromatogr A 1131(1‐2): 58‐66
Kivisto H, Pekari K, Aitio A (1993) Analysis and stability of phenylglyoxylic and mandelic acids in the urine of styrene‐exposed people. Int Arch Occup Environ Health 64(6): 399‐403
Knecht U, Reske A, Woitowitz HJ (2000) Biological monitoring of standardized exposure to ethylbenzene: evaluation of a biological tolerance (BAT) value. Arch Toxicol 73(12): 632‐640
Kogevinas M, Ferro G, Andersen A, Bellander T, Biocca M, Coggon D, Gennaro V, Hutchings S, Kolstad H, Lundberg I, et al. (1994) Cancer mortality in a historical cohort study of workers exposed to styrene. Scand J Work Environ Health 20(4): 251‐261
Kommission „Human‐Biomonitoring“des Umweltbundesamtes (1996) Human‐Biomonitoring: Definitionen, Möglichkeiten und Voraussetzungen sowie Qualitätssicherung und Konzept der

Literaturverzeichnis
115
Referenz‐ und Human‐Biomonitoring‐Werte in der Umweltmedizin. Bundesgesundheitsbl 6: 205–244
Kopp EK, Sieber M, Kellert M, Dekant W (2008) Rapid and sensitive HILIC‐ESI‐MS/MS quantitation of polar metabolites of acrylamide in human urine using column switching with an online trap column. J Agric Food Chem 56(21): 9828‐9834
Koskinen M, Plna K (2000) Specific DNA adducts induced by some mono‐substituted epoxides in vitro and in vivo. Chem Biol Interact 129(3): 209‐229
Koskinen M, Vodicka P, Hemminki K (2001) Identification of 1‐adenine DNA adducts in workers occupationally exposed to styrene. J Occup Environ Med 43(8): 694‐700
Lafeuille JL, Buniak ML, Vioujas MC, Lefevre S (2009) Natural formation of styrene by cinnamon mold flora. J Food Sci 74(6): M276‐283
Laffon B, Lema M, Mendez J (2001) Simultaneous high‐performance liquid chromatographic determination of urinary mandelic and phenylglyoxylic acids as indirect evaluation of styrene exposure. J Chromatogr B Biomed Sci Appl 753(2): 385‐393
Laffon B, Perez‐Cadahia B, Pasaro E, Mendez J (2003) Individual sensitivity to DNA damage induced by styrene in vitro: influence of cytochrome p450, epoxide hydrolase and glutathione S‐transferase genotypes. Toxicology 186(1‐2): 131‐141
Larsen K (1972) Creatinine assay by a reaction‐kinetic principle. Clin Chim Acta 41: 209‐217
Lataye R, Campo P, Loquet G (2000) Combined effects of noise and styrene exposure on hearing function in the rat. Hear Res 139(1‐2): 86‐96
Lewalter J, Neumann H‐G (1998) Biologische Arbeitsstoff‐Toleranzwerte (Biomonitoring). Arbeitsmedizin Sozialmedizin Umweltmedizin 33(8): 352‐364
Lewalter JS, Th. (1984) Analytische Methoden zur Prüfung gesundheitsschädlicher Arbeitsstoffe ‐ Analysen in biologischem Material (2007): DFG ‐ Deutsche Forschungsgemeinschaft.
Li H, Wang H, Sun H, Liu Y, Liu K, Peng S (2003) Binding of nitrobenzene to hepatic DNA and hemoglobin at low doses in mice. Toxicol Lett 139(1): 25‐32
Linhart I, Mraz J, Scharff J, Krouzelka J, Duskova S, Nohova H, Vodickova L (2010) New urinary metabolites formed from ring‐oxidized metabolic intermediates of styrene. Chem Res Toxicol 23(1): 251‐257

Literaturverzeichnis
116
Maestri L, Imbriani M, Ghittori S, Capodaglio E, Gobba F, Cavalleri A (1997) Excretion of N‐acetyl‐S‐(1‐phenyl‐2‐hydroxyethyl)‐cysteine and N‐acetyl‐S‐(2‐phenyl‐2‐hydroxyethyl)‐cysteine in workers exposed to styrene. Sci Total Environ 199(1‐2): 13‐22
Maki‐Paakkanen J, Walles S, Osterman‐Golkar S, Norppa H (1991) Single‐strand breaks, chromosome aberrations, sister‐chromatid exchanges, and micronuclei in blood lymphocytes of workers exposed to styrene during the production of reinforced plastics. Environ Mol Mutagen 17(1): 27‐31
Manini P, Andreoli R, Bergamaschi E, De Palma G, Mutti A, Niessen WM (2000) A new method for the analysis of styrene mercapturic acids by liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom 14(21): 2055‐2060
Manini P, Buzio L, Andreoli R, Goldoni M, Bergamaschi E, Jakubowski M, Vodicka P, Hirvonen A, Mutti A (2003) Assessment of biotransformation of the arene moiety of styrene in volunteers and occupationally exposed workers. Toxicol Appl Pharmacol 189(3): 160‐169
Manini P, De Palma G, Andreoli R, Goldoni M, Mutti A (2004) Determination of urinary styrene metabolites in the general Italian population by liquid chromatography‐tandem mass spectrometry. Int Arch Occup Environ Health 77(6): 433‐436
Microsoft. (1985‐2003) Microsoft Office Excel 2003.
Morioka I, Kuroda M, Miyashita K, Takeda S (1999) Evaluation of organic solvent ototoxicity by the upper limit of hearing. Arch Environ Health 54(5): 341‐346
Nakajima T, Elovaara E, Gonzalez FJ, Gelboin HV, Raunio H, Pelkonen O, Vainio H, Aoyama T (1994) Styrene metabolism by cDNA‐expressed human hepatic and pulmonary cytochromes P450. Chem Res Toxicol 7(6): 891‐896
Norström Å, Löf A, Aringer L, Samuelsson R, Andersson B, Levin J‐O, Näslund P (1992) Determination of N‐acetyl‐S‐(2‐phenyl‐2‐hydroxyethyl)cysteine in human urine after experimental exposure to styrene Chemosphere 24(11): 1561
OSHA. (2009) OSHA ‐ Factsheet 84 ‐ Expert forecast on emerging chemical risks related to occupational safety and health. European Agency for Safety and Health at Work.
Osterman‐Golkar S, Christakopoulos A, Zorcec V, Svensson K (1995) Dosimetry of styrene 7,8‐oxide in styrene‐ and styrene oxide‐exposed mice and rats by quantification of haemoglobin adducts. Chem Biol Interact 95(1‐2): 79‐87

Literaturverzeichnis
117
Osterman‐Golkar S, Czene K, Lee MS, Faller TH, Csanady GA, Kessler W, Perez HL, Filser JG, Segerback D (2003) Dosimetry by means of DNA and hemoglobin adducts in propylene oxide‐exposed rats. Toxicol Appl Pharmacol 191(3): 245‐254
Pauwels W, Farmer PB, Osterman‐Golkar S, Severi M, Cordero R, Bailey E, Veulemans H (1997) Ring test for the determination of N‐terminal valine adducts of styrene 7,8‐oxide with haemoglobin by the modified Edman degradation technique. J Chromatogr B Biomed Sci Appl 702(1‐2): 77‐83
Perera FP (1997) Environment and cancer: who are susceptible? Science 278(5340): 1068‐1073
Phillips DH, Farmer PB (1994) Evidence for DNA and protein binding by styrene and styrene oxide. Crit Rev Toxicol 24 Suppl: S35‐46
Poland CA, Duffin R, Kinloch I, Maynard A, Wallace WA, Seaton A, Stone V, Brown S, Macnee W, Donaldson K (2008) Carbon nanotubes introduced into the abdominal cavity of mice show asbestos‐like pathogenicity in a pilot study. Nat Nanotechnol 3(7): 423‐428
Ponomarkov V, Tomatis L (1978) Effects of long‐term oral administration of styrene to mice and rats. Scand J Work Environ Health 4 Suppl 2: 127‐135
Rappaport SM, Yeowell‐O'Connell K, Bodell W, Yager JW, Symanski E (1996) An investigation of multiple biomarkers among workers exposed to styrene and styrene‐7,8‐oxide. Cancer Res 56(23): 5410‐5416
Raunio H, Husgafvel‐Pursiainen K, Anttila S, Hietanen E, Hirvonen A, Pelkonen O (1995) Diagnosis of polymorphisms in carcinogen‐activating and inactivating enzymes and cancer susceptibility‐‐a review. Gene 159(1): 113‐121
Reska M, Ochsmann E, Kraus T, Schettgen T (2010) Accurate quantification of mercapturic acids of styrene (PHEMAs) in human urine with direct sample injection using automated column‐switching high‐performance liquid chromatography coupled with tandem mass spectrometry. Anal Bioanal Chem 397(8): 3563‐3574
Rybak LP (1992) Hearing: the effects of chemicals. Otolaryngol Head Neck Surg 106(6): 677‐686
Sachs L, Hedderich J (2006) Angewandte Statistik, Vol. 12, Berlin: Springer Verlag.
Schaller KH (1976) Gaschromatographische Methoden zur Bestimmung von Styrol im Blut sowie von Mandelsäure und Phenylglyoxylsäure im Urin. Arbeitsmed Sozialmed Praventivmed 11: 24‐26

Literaturverzeichnis
118
Schaller KH, Angerer J (1998) Biomonitoring in der Umweltmedizin. Umweltmed Forsch Prax 3(3): 168–175
Schechter AN (2008) Hemoglobin research and the origins of molecular medicine. Blood 112(10): 3927‐3938
Scheirs J (2003) Modern Styrenic Polymers: Polystyrenes and Styrenic Copolymers, Vol. 1: Wiley.
Schettgen (2006) Biochemisches Effekt‐Monitoring in der Umweltmedizin : Hämoglobin‐Addukte von Acrylamid, Glycidamid und Acrylnitril im Blut der Allgemeinbevölkerung: Deutsche Nationalbibliothek.
Schettgen T. (2009) Methodenvorschriften zur Bestimmung von Cotinin im Harn (personal communication). Institut für Arbeitsmedizin und Sozialmedizin Aachen (IASA), RWTH Aachen.
Schettgen T, Broding HC, Angerer J, Drexler H (2002) Hemoglobin adducts of ethylene oxide, propylene oxide, acrylonitrile and acrylamide‐biomarkers in occupational and environmental medicine. Toxicol Lett 134(1‐3): 65‐70
Schettgen T, Weiss T, Drexler H, Angerer J (2003) A first approach to estimate the internal exposure to acrylamide in smoking and non‐smoking adults from Germany. Int J Hyg Environ Health 206(1): 9‐14
Severi M, Pauwels W, Van Hummelen P, Roosels D, Kirsch‐Volders M, Veulemans H (1994) Urinary mandelic acid and hemoglobin adducts in fiberglass‐reinforced plastics workers exposed to styrene. Scand J Work Environ Health 20(6): 451‐458
Sliwinska‐Kowalska M, Zamyslowska‐Szmytke E, Szymczak W, Kotylo P, Fiszer M, Wesolowski W, Pawlaczyk‐Luszczynska M (2003) Ototoxic effects of occupational exposure to styrene and co‐exposure to styrene and noise. J Occup Environ Med 45(1): 15‐24
Staudinger H. (1953) Macromolecular chemistry.
Steele DH (1994) Determination of styrene in selected foods. J Agric Food Chem 42 (8): 1661–1665
Sumner SJ, Fennell TR (1994) Review of the metabolic fate of styrene. Crit Rev Toxicol 24 Suppl: S11‐33
Tang W, Hemm I, Eisenbrand G (2000) Estimation of human exposure to styrene and ethylbenzene. Toxicology 144(1‐3): 39‐50

Literaturverzeichnis
119
Teixeira JP, Gaspar J, Coelho P, Costa C, Pinho‐Silva S, Costa S, Da Silva S, Laffon B, Pasaro E, Rueff J, Farmer P (2010) Cytogenetic and DNA damage on workers exposed to styrene. Mutagenesis
Teixeira JP, Silva S, Torres J, Gaspar J, Roach J, Farmer PB, Rueff J, Mayan O (2008) Styrene‐oxide N‐terminal valine haemoglobin adducts as biomarkers of occupational exposure to styrene. Int J Hyg Environ Health 211(1‐2): 59‐62
Törnqvist M, Fred C, Haglund J, Helleberg H, Paulsson B, Rydberg P (2002) Protein adducts: quantitative and qualitative aspects of their formation, analysis and applications. J Chromatogr B Analyt Technol Biomed Life Sci 778(1‐2): 279‐308
Törnqvist M, Mowrer J, Jensen S, Ehrenberg L (1986) Monitoring of environmental cancer initiators through hemoglobin adducts by a modified Edman degradation method. Anal Biochem 154(1): 255‐266
Triebig G, Lehrl S, Weltle D, Schaller KH, Valentin H (1989) Clinical and neurobehavioural study of the acute and chronic neurotoxicity of styrene. Br J Ind Med 46(11): 799‐804
Triebig G, Stark T, Ihrig A, Dietz MC (2001) Intervention study on acquired color vision deficiencies in styrene‐exposed workers. J Occup Environ Med 43(5): 494‐500
van Sittert NJ, Boogaard PJ, Natarajan AT, Tates AD, Ehrenberg LG, Tornqvist MA (2000a) Formation of DNA adducts and induction of mutagenic effects in rats following 4 weeks inhalation exposure to ethylene oxide as a basis for cancer risk assessment. Mutat Res 447(1): 27‐48
van Sittert NJ, Megens HJ, Watson WP, Boogaard PJ (2000b) Biomarkers of exposure to 1,3‐butadiene as a basis for cancer risk assessment. Toxicol Sci 56(1): 189‐202
Vodicka P, Bastlova T, Vodickova L, Peterkova K, Lambert B, Hemminki K (1995) Biomarkers of styrene exposure in lamination workers: levels of O6‐guanine DNA adducts, DNA strand breaks and mutant frequencies in the hypoxanthine guanine phosphoribosyltransferase gene in T‐lymphocytes. Carcinogenesis 16(7): 1473‐1481
Vodicka P, Hemminki K (1988) Identification of alkylation products of styrene oxide in single‐ and double‐stranded DNA. Carcinogenesis 9(9): 1657‐1660
Vodicka P, Tvrdik T, Osterman‐Golkar S, Vodickova L, Peterkova K, Soucek P, Sarmanova J, Farmer PB, Granath F, Lambert B, Hemminki K (1999) An evaluation of styrene genotoxicity using several biomarkers in a 3‐year follow‐up study of hand‐lamination workers. Mutat Res 445(2): 205‐224
Vodicka P, Vodickova L, Trejbalova K, Sram RJ, Hemminki K (1994) Persistence of O6‐guanine DNA adducts in styrene‐exposed lamination workers determined by 32P‐postlabelling. Carcinogenesis 15(9): 1949‐1953

Literaturverzeichnis
120
Wallace L, Pellizzari E, Hartwell TD, Perritt R, Ziegenfus R (1987) Exposures to benzene and other volatile compounds from active and passive smoking. Arch Environ Health 42(5): 272‐279
WHO. (2000) WHO air quality guidelines for Europe. World Health Organization.
Wigaeus E, Lof A, Nordqvist MB (1984) Uptake, distribution, metabolism, and elimination of styrene in man. A comparison between single exposure and co‐exposure with acetone. Br J Ind Med 41(4): 539‐546
Wong O, Trent LS, Whorton MD (1994) An updated cohort mortality study of workers exposed to styrene in the reinforced plastics and composites industry. Occup Environ Med 51(6): 386‐396
Wu XHW, H.F.;Liu, Y.F.; Lux,Y.;Wang, J.J.;Li, K. (1997) Histone adduction with nicotine : A bio‐AMS study. Radiocarbon 39(3): 293‐297
Yeowell‐O'Connell K, Jin Z, Rappaport SM (1996) Determination of albumin and hemoglobin adducts in workers exposed to styrene and styrene oxide. Cancer Epidemiol Biomarkers Prev 5(3): 205‐215
Zielhuis RL. (1980) Recent and potential advances applicable to the protection of workers’ health: Biological Monitoring. Presented at the international seminar Assessment of toxic agents at the workplace – Roles of ambient and biological monitoring. (Workshop, December, 8th‐12th). Luxembourg.

Anhang
121
13 Anhang
13.1 Information und Einverständniserklärung für Probanden
UNIVERSITÄTSKLINIKUM - RHEINISCH-WESTFÄLISCHE TECHNISCHE HOCHSCHULE AACHEN
Institut für Arbeitsmedizin und Sozialmedizin
Ambulanz Leiter: Univ.-Prof. Dr. med. T. Kraus
Pauwelsstraße 30 52074 Aachen Tel. +49-(0)241-80 80410 Fax: +49-(0)241-80 82086
Information für Teilnehmer „Biomonitoring“
Sehr geehrte Teilnehmerin, sehr geehrter Teilnehmer, innerhalb Ihrer beruflichen Tätigkeit haben Sie häufig Kontakt zu Styrol. Die Chemikalie Styrol ist eine farblose, süßlich riechende Flüssigkeit und findet besonders in der kunststoffherstellenden und kunststoffverarbeitenden Industrie große Verwendung. Durch Einatmen kann Styrol aus der Luft in den Körper gelangen. Dort wird der Großteil normalerweise in eine für den Körper besser weiterzuverarbeitende Form umgewandelt und verlässt diesen zusammen mit dem Urin. Ein kleinerer Teil verbleibt im Blut und kann über einen wesentlich längeren Zeitraum nachgewiesen werden als im Urin. Styrol im Blut dient daher als Langzeitparameter und kann uns im Gegensatz zu Raumluftmessungen einen viel größeren Einblick über das tatsächliche Vorkommen von Styrol im Körper geben. Ziel der vom Institut für Arbeitsmedizin und Sozialmedizin der RWTH Aachen (IASA) durchgeführten Vorsorgeuntersuchung ist die Ermittlung der Styrol‐Stoffwechselprodukte im Urin (Mandelsäure, Phenylglyoxylsäure ggf. Mercaptursäuren des Styrols) sowie von Styrol im Blut zur Ermittlung der tatsächlich

Anhang
122
an Ihrem Arbeitsplatz aufgenommenen Menge an Styrol‐Blut‐Verbindungen. Zusätzlich werden ggf. noch Phänotypisierungen der am Styrolmetabolismus beteiligten Enzyme durchgeführt. Bei der Teilnahme an dieser Untersuchung benötigen wir Ihre Unterstützung. Für die Konzentrationsbestimmungen im Blut sowie im Urin benötigen wir von Ihnen sowohl eine Blutprobe als auch eine Urinprobe. Weiterhin erheben wir einige personenbezogene Daten in Form eines Fragebogens. Die Beantwortung der Fragen wird nicht mehr als 10 Minuten Ihrer Zeit in Anspruch nehmen und erfolgt im Anschluss an die Blutabnahme durch eine(n) unserer Ärzte/Arzthelfer(innen).
Die Teilnahme an der Beantwortung sowohl der Fragebögen als auch die Abgabe von Blut – und Urinproben ist ganz und gar freiwillig. Sie können jederzeit und ohne Angabe von Gründen Ihr Einverständnis zur Teilnahme zurücknehmen, ohne dass Ihnen hieraus irgendwelche Nachteile entstehen. Datenerfassung und Datenverarbeitung: Das Institut für Arbeitsmedizin und Sozialmedizin der RWTH Aachen (IASA) führt als durchführende Stelle die Ermittlung Ihrer personenbezogenen Probandenddaten im Sinne des Datenschutzgesetzes NRW durch. Sie ist somit einzige datenhaltende Stelle und führt eine Anonymisierung Ihrer personenbezogenen Daten mit Hilfe einer Identifikationsnummer (ID)) durch. Das IASA trägt dafür Sorge, dass alle Mitglieder innerhalb des Institutes, die personenidentifizierende Daten einsehen können, zur Einhaltung der Datenschutzbestimmungen verpflichtet werden. Die Datenübermittlung an Dritte, die statistische Auswertung sowie die spätere Veröffentlichung der Ergebnisse erfolgt/erfolgen in anonymisierter Form. Wir bedanken uns sehr für Ihre Mitarbeit! Die mündliche Aufklärung wurde am (Datum) ________________ von (Unterschrift des aufklärenden Arztes) (Name Arzt in Druckschrift)

Anhang
123
13.2 Fragebogen für Probanden
UNIVERSITÄTSKLINIKUM - RHEINISCH-WESTFÄLISCHE TECHNISCHE HOCHSCHULE AACHEN
Institut für Arbeitsmedizin und Sozialmedizin
Ambulanz Leiter: Univ.-Prof. Dr. med. T. Kraus
Pauwelsstraße 30 52074 Aachen Tel. +49-(0)241-80 80410 Fax: +49-(0)241-80 82086
Fragebogen für Teilnehmer „Biomonitoring“
Vorname: _____________ Nachname: _____________ Geburtsdatum: _______________ Gewicht: _______________ Größe (cm): _______________ Teilnehmer ID: Datum: _________ Firma: ______________
Ihre Antworten, die Sie in diesem Fragebogen geben, fallen unter die ärztliche Schweigepflicht! Ihre Identität wird nicht veröffentlicht oder an Ihren Arbeitgeber weitergegeben. Sie wird anonymisiert und verbleibt alleine beim Institut für Arbeitsmedizin und Sozialmedizin der RWTH Aachen.
Geschlecht [ ]A männlich [ ]B weiblich
Seit wann arbeiten Sie an Ihrem jetzigen Arbeitsplatz? [ ]a Seit: Monat: _______ Jahr: ________
Haben Sie früher Zigaretten geraucht oder rauchen Sie zurzeit? [ ]A Ich habe noch nie geraucht (bis auf ganz seltenes Probieren) [ ]B Ja, ich habe früher geraucht (seit dem Jahr 19___ ___ bis 19/20___ ___) rauche aber jetzt nicht
mehr; damals durchschnittlich ____ Zigaretten pro Tag [ ]C Ja, ich rauche zur Zeit (seit dem Jahr 19/20___ ___ bis heute) durchschnittlich ____ Zigaretten pro
Tag und habe meine letzte Zigarette vor ____ Stunden geraucht

Anhang
124
13.3 Veröffentlichungen
Paper
Reska M, Ochsmann E, Kraus T, Schettgen T (2010) Accurate quantification of
mercapturic acids of styrene (PHEMAs) in human urine with direct sample injection using
automated column-switching high-performance liquid chromatography coupled with
tandem mass spectrometry. Anal Bioanal Chem 397(8): 3563-3574
Posterbeitrag
Reska, M., Ochsmann, E., Kraus, T., Schettgen, T., Dosis- und biochemisches
Effektmonitoring styrolexponierter Mitarbeiter der Polyesterharz verarbeitenden Industrie.
Posterbeitrag auf der: 51. Jahrestagung der Deutschen Gesellschaft für Arbeitsmedizin und
Umweltmedizin (DGAUM); März, 9-12, 2011; Heidelberg.

Anhang
125
Lebenslauf:
Persönliche Daten
Name: Marcus Reska
Geburt: 12.07.1977 in Geilenkirchen / Deutschland Nationalität: deutsch
Schulausbildung
1984- 1988 Grundschule in Geilenkirchen 1989- 1991 Bischöfliches Gymnasium St. Ursula in Geilenkirchen 1991- 1995 Städtische Realschule Geilenkirchen (Fachoberschulreife) 1995- 1998 Städtisches Gymnasium Übach – Palenberg (Abitur) 1998- 1999 Zivildienst im St.-Antonius-Hospital in Eschweiler Universitätsausbildung
1999- 2006 Biologiestudium an der RWTH Aachen (Hauptfach: Molekularbiologie/Zellbiologie; Nebenfächer:
Molekulare Medizin, Humanbiologie, Entwicklungsbiologie) Promotion
2007- 2011 Institut für Arbeitsmedizin und Sozialmedizin Aachen (Gruppe„Biomarker“), Prof. Dr. med. T. Kraus