
BIOQUIMICA DE PROTEÍNAS - iib.unsam.edu.ar · Planificación inicial •Cómo vamos a ensayarla?...
98
BIOQUIMICA DE PROTEÍNAS
Transcript of BIOQUIMICA DE PROTEÍNAS - iib.unsam.edu.ar · Planificación inicial •Cómo vamos a ensayarla?...

BIOQUIMICA DE PROTEÍNAS

BIOTECNOLOGIA DE PROTEINAS
Es la producción y el aislamiento comerciales de proteínas específicas, de fuentes
animales, vegetales o microbianas, y/o su utilización ulterior para producir un
evento biológico pre-definido.
LA VIABILIDAD COMERCIAL ES ESENCIAL PARA EL
EXITO DE CUALQUIER EMPRESA BIOTECNOLOGICA
Biotecnología clásica: Procesos fermentativos (cerveza, vino, quesos) (al menos
4000 años).
Biotecnología moderna: Tecnologías de DNA recombinante y anticuerpos
monoclonales (desde mediados de la década del 1970).
Muchas proteínas tienen aplicación industrial: enzimas, anticuerpos, hormonas,
factores de coagulación sanguinea, factores de crecimiento. Se emplean como
agentes diagnósticos y terapéuticos, y en la fabricación de una gran variedad de
productos biológicos. Se las puede obtener de sus fuentes naturales, pero
actualmente es muy frecuente su obtención a partir de otros organismos, por
técnicas de DNA recombinante.

PROTEINAS DE USO FARMACEUTICO: Factores de
coagulación (hemofilias); eritropoietinas (anemias);
Factor de crecimiento de fibroblastos (ciertas
úlceras); Insulina (diabetes); Interferones,
interleukinas (cancer, SIDA); anticuerpos
monoclonales (diagnóstico); vacunas (hepatitis B).
Se producen en cantidades moderadas (gramos o
kilos) y requieren la máxima pureza. La mayoría se
produce por técnicas de DNA recombinante.

La producción de proteínas recombinantes para uso clínico es un
emprendimiento de alto riesgo y alta recompensa. La American
Pharmaceutical Manufacturers Association ha estimado el costo del
desarrollo de una nueva droga de aplicación farmacéutica en 200 – 250
millones de US$, y el tiempo requerido puede ser de unos 12 años,
desde su concepciòn en el laboratorio hasta su llegada a los anaqueles
de una farmacia.
La primera proteína recombinante empleada en clínica fué la insulina
humana, producida en Escherichia coli y aprobada por USA, UK,
Alemania Occidental y Holanda en 1982. La primera vacuna
recombinante administrada a seres humanos fué la vacuna contra
hepatitis B, producida en levadura (Saccharomyces cerevisiae). Se
producen actualmente proteínas recombinantes para uso clínico en
hongos filamentosos, plantas y animales transgénicos.

Muchas proteínas con APLICACION INDUSTRIAL, en general enzimas de
origen microbiano, se producen en grandes cantidades, a menudo cientos
de toneladas y con mucha menor pureza. Es una industria que moviliza
cientos de millones de US$ por año.
EJEMPLOS DE ENZIMAS CON APLICACIÓN INDUSTRIAL:
PROTEINASAS (preparados detergentes, fabricación de quesos, industrias de
la cerveza y el pan, de la carne y del cuero; digestivos de uso humano y
veterinario).
AMILASAS (procesado de almidones, industrias fermentativas).
PECTINASAS (industrias de jugos frutales y procesado de frutas).
LIPASAS (industria lechera, industria de aceites vegetales).
LACTASA (hidrólisis de lactosa en leche).
GLUCOSA ISOMERASA (producción de jarabes con alta concentración de
fructosa).
PENICILIN ACILASA (producción de penicilinas semisintéticas).
La mayoría de estas enzimas se obtiene de fuentes naturales, pero algunas son
recombinantes (quimosina, para la hidrólisis parcial de la caseína en la fabricación de
quesos).

PURIFICACIÓN DE PROTEÍNAS
MÉTODOS – PRINCIPIOS - EQUIPAMIENTO
Marzo 2017

Planificación inicial
• Cuál es el uso que se le va a dar a la proteína?El grado de pureza necesario dependerá del uso que se va a dar a la proteína.
Para investigación, la escala es reducida y es más importante la pureza que el rendimiento.
Para usos terapéuticos la escala es mayor y la pureza requerida es máxima.
Para usos industriales, frecuentemente se requiere gran cantidad, bajo costo y pureza menor.
Para act. biológica debe estar nativa; para determinar estr. 1° puede estar desnaturalizada.
• Cuál es la fuente más adecuada?Fuentes naturales donde la proteína de interés es más abundante y estable.
Fuentes recombinantes. Elección del organismo productor.
• Qué sabemos de la proteína?Características químicas, físicas, localización celular.
El saber si la proteína es extracelular, intracelular, soluble, insoluble, asociada a membrana o localizada en una organela particular, influirá en la elección del método de extracción.
Si se trata de una enzima o un receptor se puede explotar la afinidad por sustrato o ligando.
Tamaño y pI. Estabilidad, sensibilidad a la T, extremos de pH, proteasas, aire, metales.

Esquema general de unmétodo de purificación• Clarificado y extracción.
• Sucesivos pasos de fraccionamiento cromatográfico.
entre los que se pueden requerir pasos de concentración
y/o cambios de buffer.

Estrategia de purificación
• Definir los criterios aceptables de cantidad, calidad y costos.
• Preservar la actividad minimizando la desnaturalización, inactivación, proteólisis, etc.
• Planificar una estrategia de purificación considerando:
- Capacidad
- Resolución
- Rendimiento
- Costos

Planificación inicial
• Cómo vamos a ensayarla?Utilizar un método rápido, simple y específico que nos permita evaluar la proteína de interés.
Métodos basados en la act enzimática, propiedades inmunológicas o en la act biológica.
Gran ayuda para el seguimiento de la purificación:
- Nos permite identificar y poolear las fracciones que contienen la proteína de interés.
- Combinado con la determinación de proteínas totales nos da una idea del grado de purificación alcanzado.
EJEMPLOS: Glucosa 6P dh, proteasas

Proteínas totales
• espectrofotometría en el UV
(280 nm, Trp; 210-215 nm, unión peptídica)
• Métodos colorimétricos
- Reacción de Biuret (Cu2+ alcalino)
BCA (Cu2+ alcalino + ácido bicinconínico)
- Bradford (Coomassie Blue)

Espectrofotometría UV
• Abs 280 nm dada por los aminoácidos con anillos aromáticos (W,F,Y)
• Rápida y conveniente (no se requieren reactivos ni incubaciones adicionales, no requiere estándar, no se consume la proteína).
• Relación linear entre abs y cc de proteínas.
• Sujeta a error por presencia de interferencias que abs en la misma longitud de onda.
• La aplicación más frecuente es para monitorear fracciones de una cromatografía o para una estimación rápida (aunque no precisa).
Concentración (mg/ml) = (1.55 x A280) - 0.76 x A260)
• Abs 205 nm enlace peptídico
Concentración (mg/ml) = (A205)/31

Biuret
Biuret: Under alkaline conditions substances containing two or more peptide bonds form a purple complex with copper salts in the reagent. (Abs550 nm).

Lowry - BCA
• Lowry: Monovalent copper ion and the radical groups of tyrosine, tryptophan, and cysteine react with Folin reagent to produce an unstable product that becomes reduced to molybdenum/tungsten blue.
• BCA: BCA react with complexes between copper ions and peptide bonds to produce a purple end product.
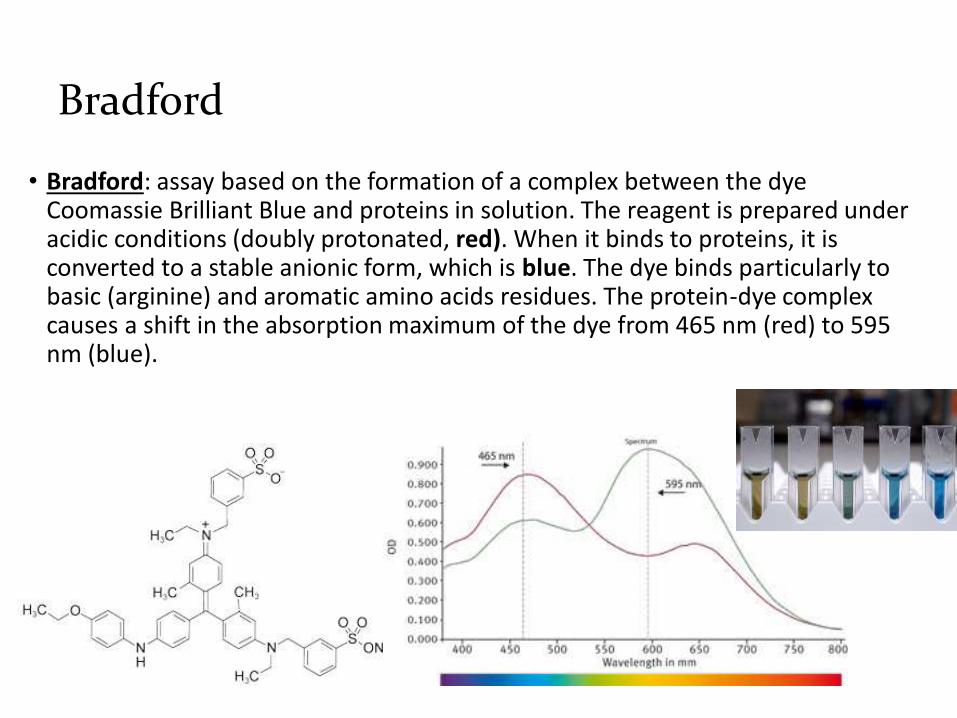
Bradford
• Bradford: assay based on the formation of a complex between the dye Coomassie Brilliant Blue and proteins in solution. The reagent is prepared under acidic conditions (doubly protonated, red). When it binds to proteins, it is converted to a stable anionic form, which is blue. The dye binds particularly to basic (arginine) and aromatic amino acids residues. The protein-dye complex causes a shift in the absorption maximum of the dye from 465 nm (red) to 595 nm (blue).

Actividad específica = Proteína de interés / Proteínas totales
Grado de purificación = Actividad específica 2/Actividad específica 1
Rendimiento = Proteína de interés 2/ Proteína de interés 1
DESPUÉS
Parámetros importantes!!
INICIAL
Purificacion step Total
Protein
Total
activity
Spec ific
Activity
Purification
factor
Yield
Cell-free extract
ConA- Sepharose
Mono Q
Mono P
mg
227.5
5.6
0.67
0.36
unitsa
56.00
43.75
25.95
14.70
unitsa/mg
0.246
7.81
38.73
40.83
Fold
1
31.75
157.44
165.97
%
100
78.1
46.34
26.25
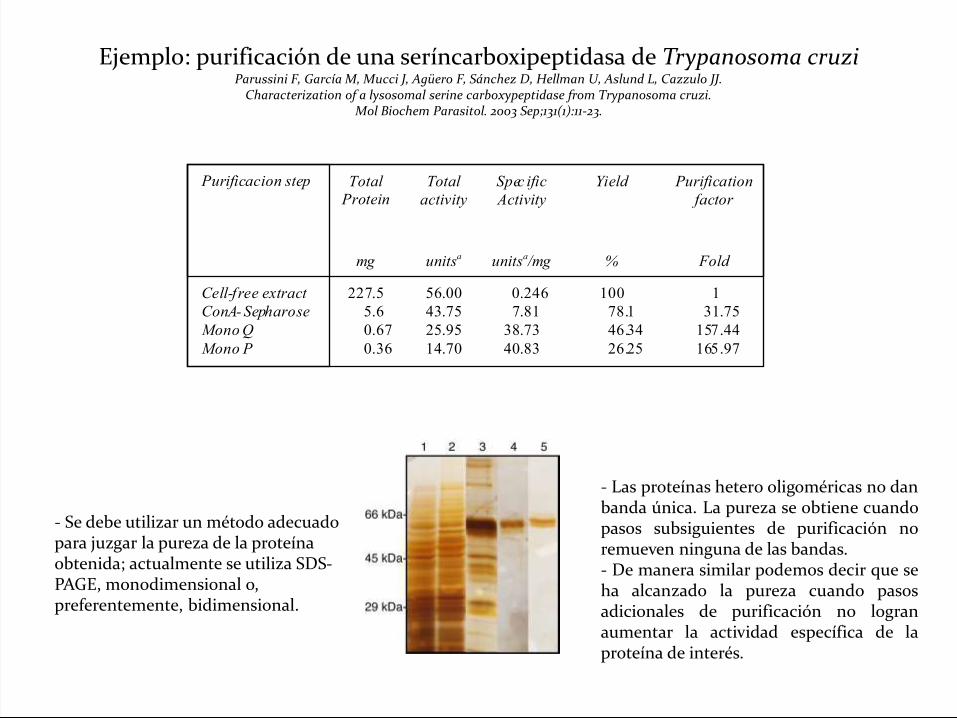
- Se debe utilizar un método adecuado para juzgar la pureza de la proteína obtenida; actualmente se utiliza SDS-PAGE, monodimensional o, preferentemente, bidimensional.
Ejemplo: purificación de una seríncarboxipeptidasa de Trypanosoma cruziParussini F, García M, Mucci J, Agüero F, Sánchez D, Hellman U, Aslund L, Cazzulo JJ.
Characterization of a lysosomal serine carboxypeptidase from Trypanosoma cruzi.Mol Biochem Parasitol. 2003 Sep;131(1):11-23.
- Las proteínas hetero oligoméricas no danbanda única. La pureza se obtiene cuandopasos subsiguientes de purificación noremueven ninguna de las bandas.- De manera similar podemos decir que seha alcanzado la pureza cuando pasosadicionales de purificación no logranaumentar la actividad específica de laproteína de interés.


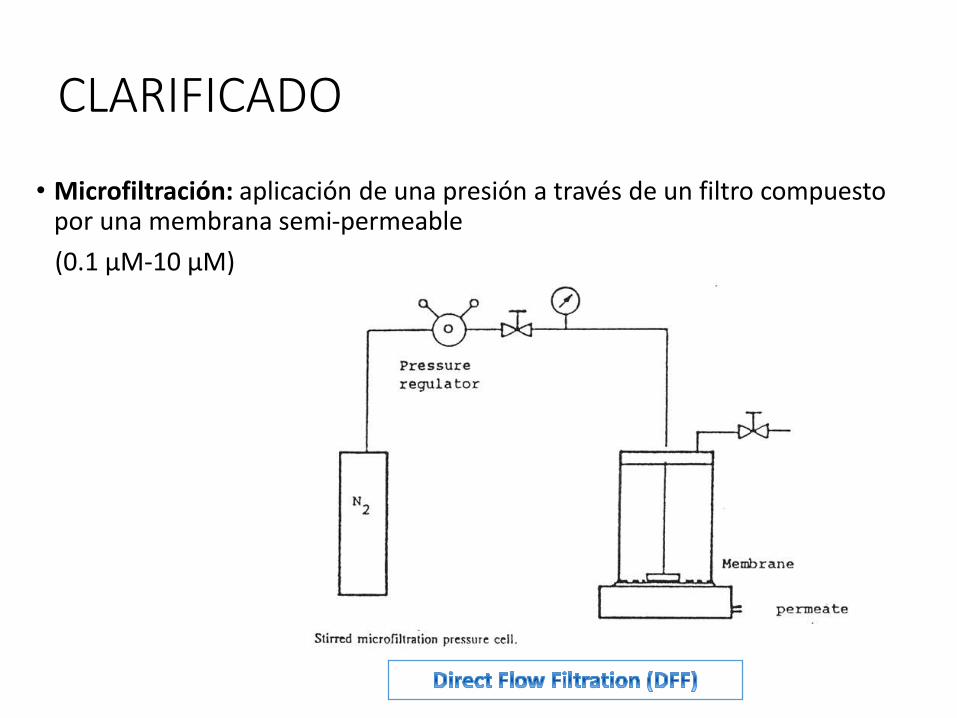
CLARIFICADO
• Centrifugación: aplicación de una aceleración radial a una suspensión de partículas por un movimiento rotacional.
Escala de lab
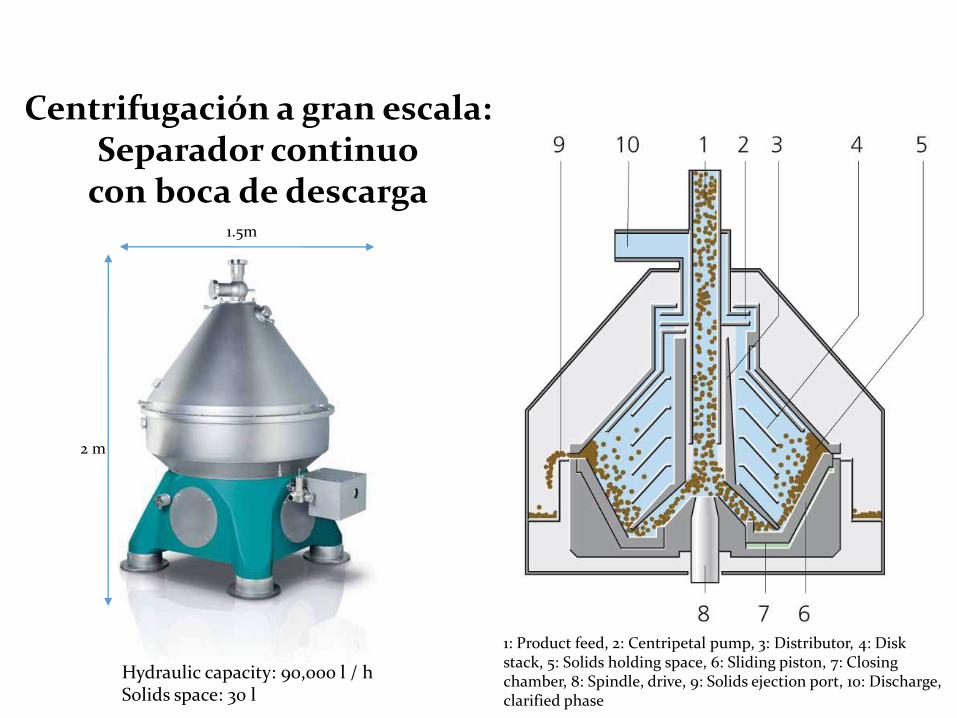
1: Product feed, 2: Centripetal pump, 3: Distributor, 4: Disk stack, 5: Solids holding space, 6: Sliding piston, 7: Closing chamber, 8: Spindle, drive, 9: Solids ejection port, 10: Discharge, clarified phase
Hydraulic capacity: 90,000 l / hSolids space: 30 l
2 m
1.5m
Centrifugación a gran escala: Separador continuo
con boca de descarga
CLARIFICADO
• Microfiltración: aplicación de una presión a través de un filtro compuesto por una membrana semi-permeable
(0.1 μM-10 μM)

Microfiltración: DFF
Escala industrialEscala de lab

Microfiltración: TFF
• In Tangential Flow Filtration (TFF) the feed stream passes parallel to the membrane face as one portion passes through the membrane (permeate) while the remainder (retentate) is recirculated back to the feed reservoir.
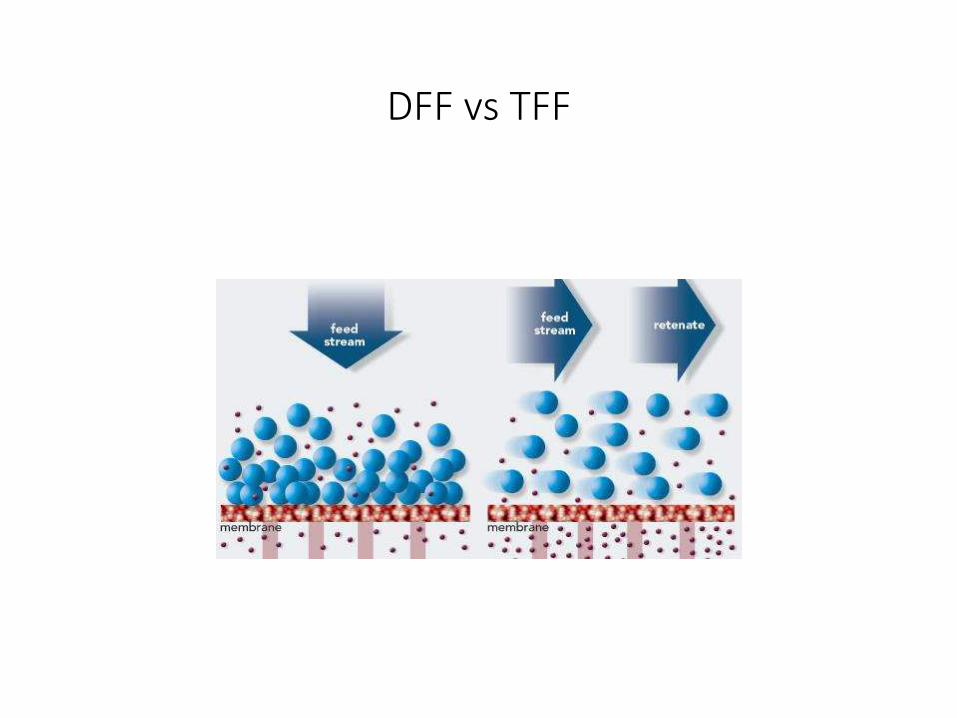
DFF vs TFF

EXTRACCIÓN
• Diferencias en la facilidad de la ruptura
- Las células animales son muy fáciles de romper, sólo están recubiertas por una membrana plasmática débilmente soportada por un citoesqueleto.
- Las vegetales también, pese a tener pared celular, su gran tamaño las hace vulnerables a la ruptura mecánica.
- Las células bacterianas son más resistentes, y esta propiedad depende de que sean Gram positivas o Gram negativas. El predominio del peptidoglicano en la pared de las primeras, las hace mucho más sensibles a métodos suaves, como la digestión con lisozima.
- Los hongos filamentosos y las levaduras son los más resistentes a la ruptura. Tienen paredes celulares que contienen hasta 80 - 90 % de polisacárido (quitina y glucanos en hongos, manano y glucano en levaduras), además de lípidos y proteínas.
• Consecuencias de la ruptura
- Calor, liberación de proteasas, liberación del ADN celular y aumento de la viscosidad del extracto

EXTRACCIÓN: MÉTODOS MECÁNICOS
• Homogeneizadores:
- - Mezcladoras y licuadoras para órganos y tejidos (cuchillas rotatorias)
- - French Press (extrusión de líquidos) “exprimir” las células a través de un tubo que es mucho menor que ellas. Utilizado para romper bacterias, levaduras.
• Agitación con abrasivos:
- Mortereado con alúmina, carburo de silicio, arena para pequeños vol (< 1ml)
- Bolitas de vidrio para volúmenes mayores (< 300 ml)
• Sonicado: >20kHz “cavitación gaseosa” pequeñas burbujas se forman y explotan produciendo una onda de shock local que produce cambios de presión y disrumpe las células.
• Congelado-Descongelado: formación de cristales que rompen la célula. Utilizado para materiales blandos.

EXTRACCIÓN: MÉTODOS NO MECÁNICOS
Involucran la adición de químicos o enzimas que degradan específicamente los componentes de la pared celular. Muchas veces se utilizan en combinación con métodos mecánicos para asegurar una ruptura completa.
La desventaja es que luego tienen que ser removidos de la muestra.
Ejemplos:
•Enzimas líticas
•Shock osmótico
•Detergentes y solventes

Uso de enzimas líticas• Bacterias: lisozima.
- from chicken egg white, from human milk, from human neutrophils
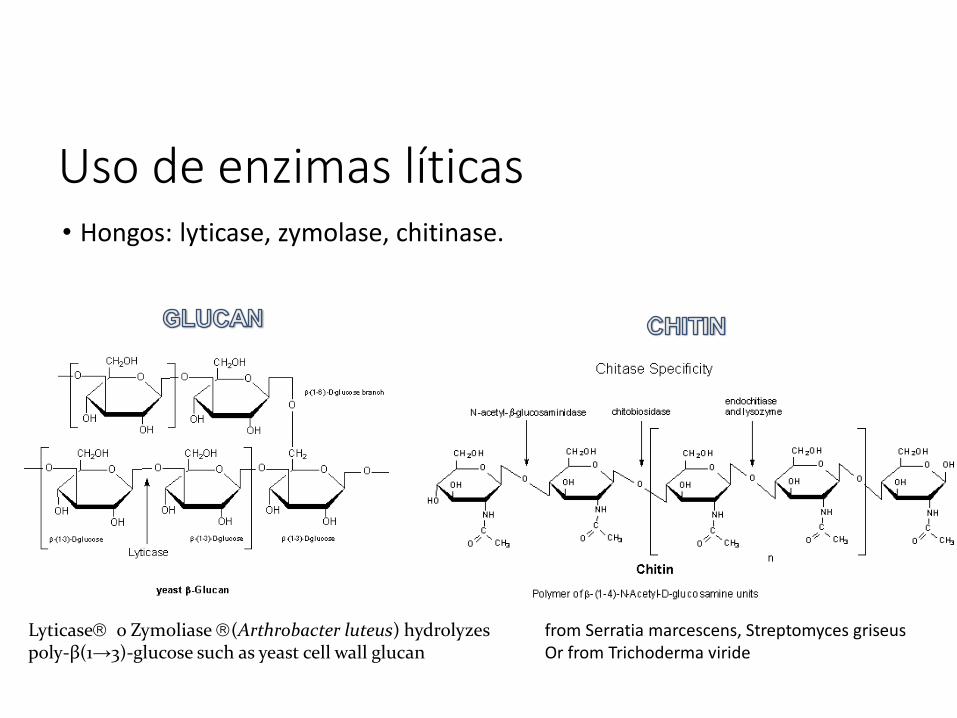
Uso de enzimas líticas• Hongos: lyticase, zymolase, chitinase.
Lyticase o Zymoliase (Arthrobacter luteus) hydrolyzes poly-β(1→3)-glucose such as yeast cell wall glucan
from Serratia marcescens, Streptomyces griseusOr from Trichoderma viride


MÉTODOS DE CONCENTRACIÓN
Remoción de agua y moléculas pequeñas por:
• Precipitación y resuspensión en un volumen menor.
• Remoción a través de una membrana semipermeable: Ultrafiltración.
• Remoción a través de una membrana semipermeable: Diálisis contra polietilenglicol.
• Agregado de un polímero seco con poros muy pequeños como para que la proteína penetre (Sephadex G-25).
• Remoción de agua en vacío: liofilización. Buffers volátiles, como el bicarbonato de amonio.

PRECIPITACIÓN
La precipitación fue utilizada como método de purificación.
Actualmente se utiliza como un paso muy grosero durante las primeras etapas de una purificación.
La precipitación se utiliza como método de concentración de proteínas.
• Efecto de las sales (salting-in, salting-out)
• Precipitación por alteración del pH (mínima solubilidad en el pI).
• Precipitación por solventes orgánicos (etanol, acetona), que disminuyen la constante dieléctrica de la solución y por ello su poder de solvatación.
• Desnaturalización de impurezas por altas temperaturas, extremos de pH, solventes orgánicos.
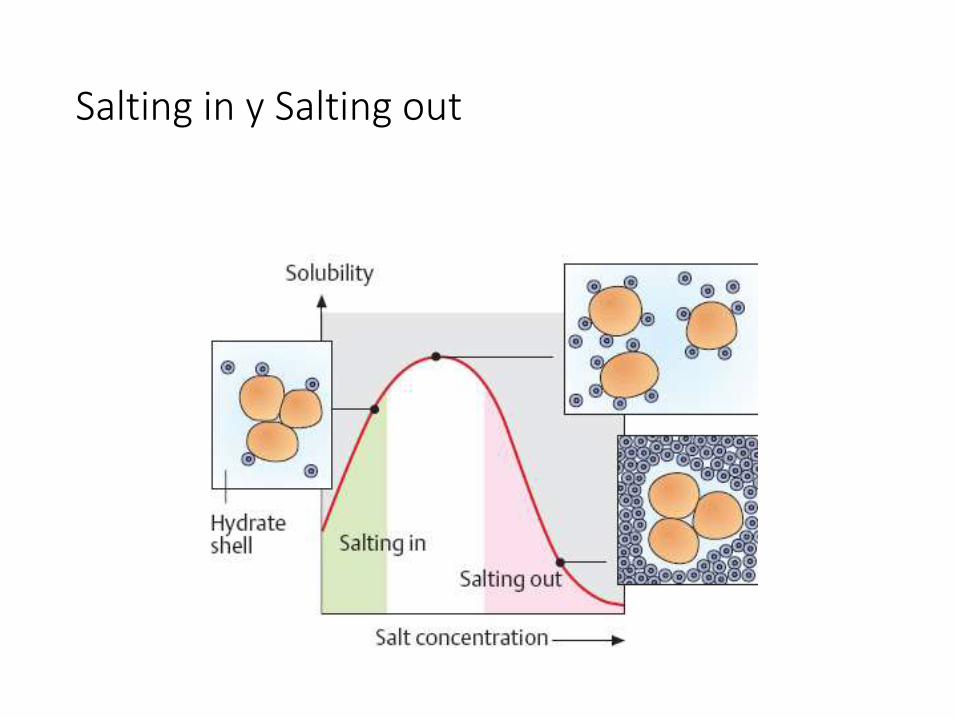
Salting in y Salting out
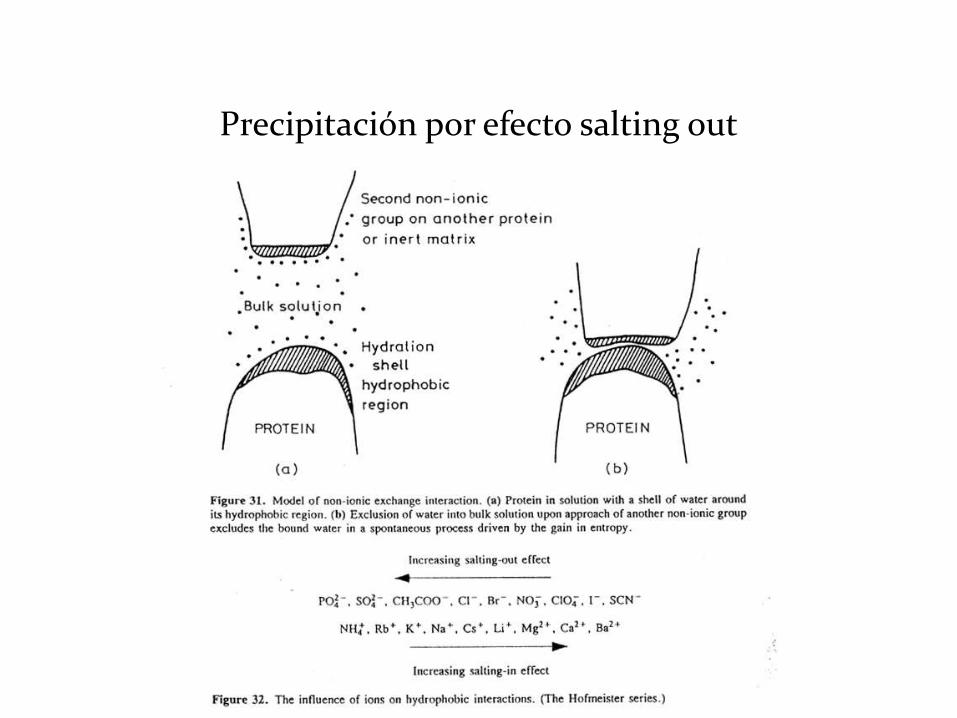
Precipitación por efecto salting out
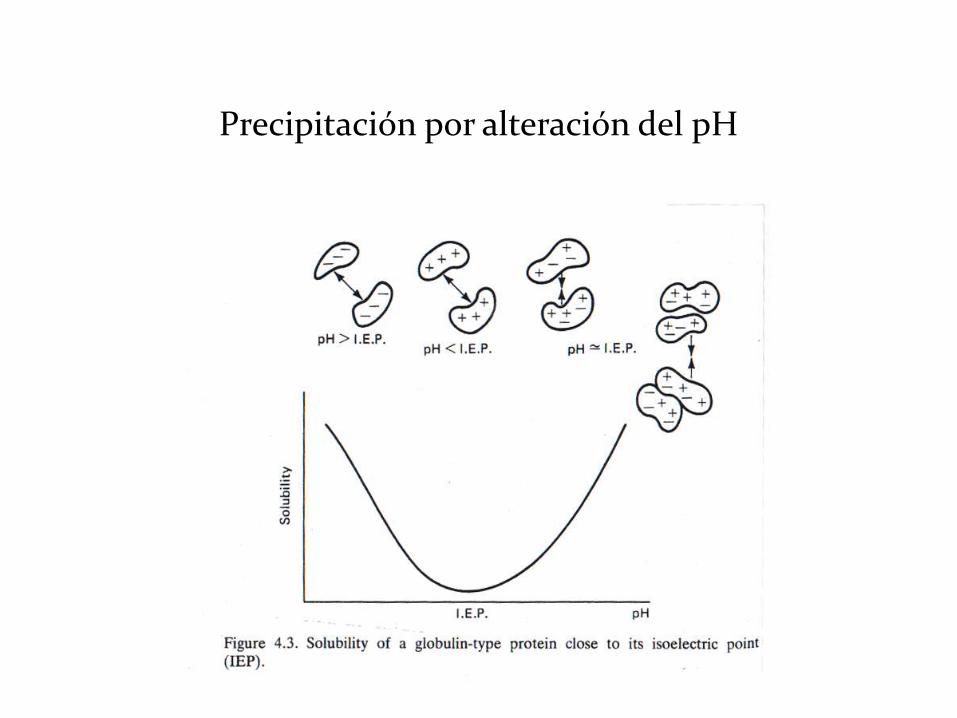
Precipitación por alteración del pH
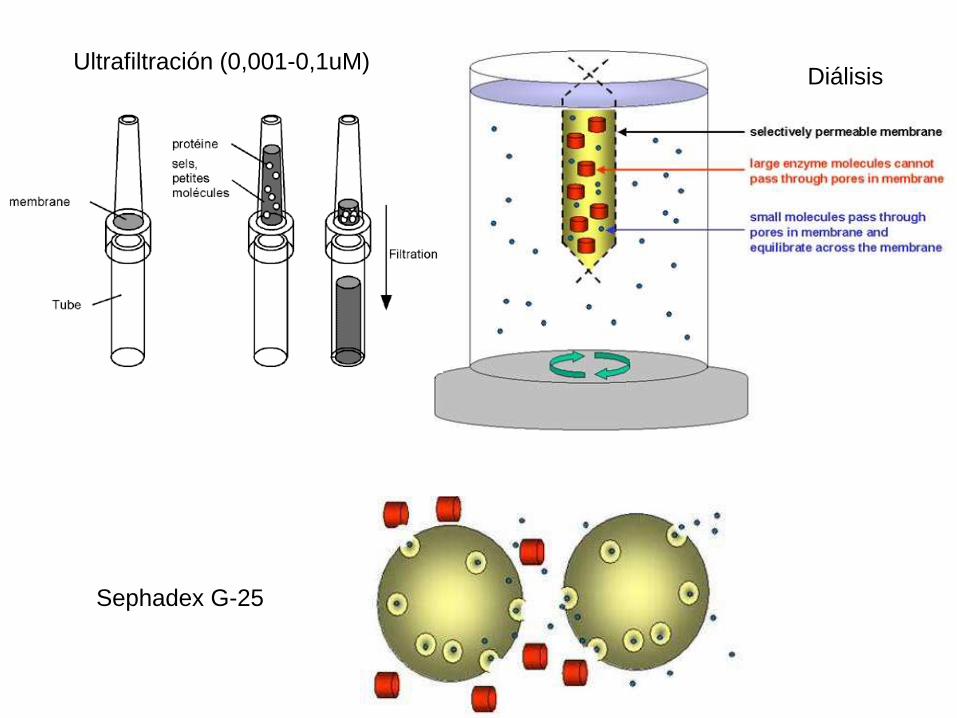
Sephadex G-25
Ultrafiltración (0,001-0,1uM)Diálisis

CROMATOGRAFÍA• Separación diferencial de los componentes de una muestra entre una fase
móvil y una fase estacionaria.
• La fase móvil es el solvente en que se introduce la muestra y con el que seeluyen las proteínas (puede ser el mismo o variar durante la corrida).
• La fase estacionaria comprende las partículas, en general esféricas, con quese llena la columna. Estas partículas están formadas por una matriz y, en lamayoría de los casos, moléculas de menor o mayor tamaño con las que se laderivatiza, para adaptarla a los diferentes procedimientos cromatográficos.
• La matriz es el sustrato sólido de la fase estacionaria. Las más comunes soncelulosa, dextrano, agarosa, poliacrilamida y silica.
• Deben tener buena estabilidad mecánica y química, alta capacidad, tamañoy forma adecuada de poros, superficie inerte para minimizar lasinteracciones no específicas, y tamaño de partícula adecuado al flujo desolvente deseado y a la presión operativa.

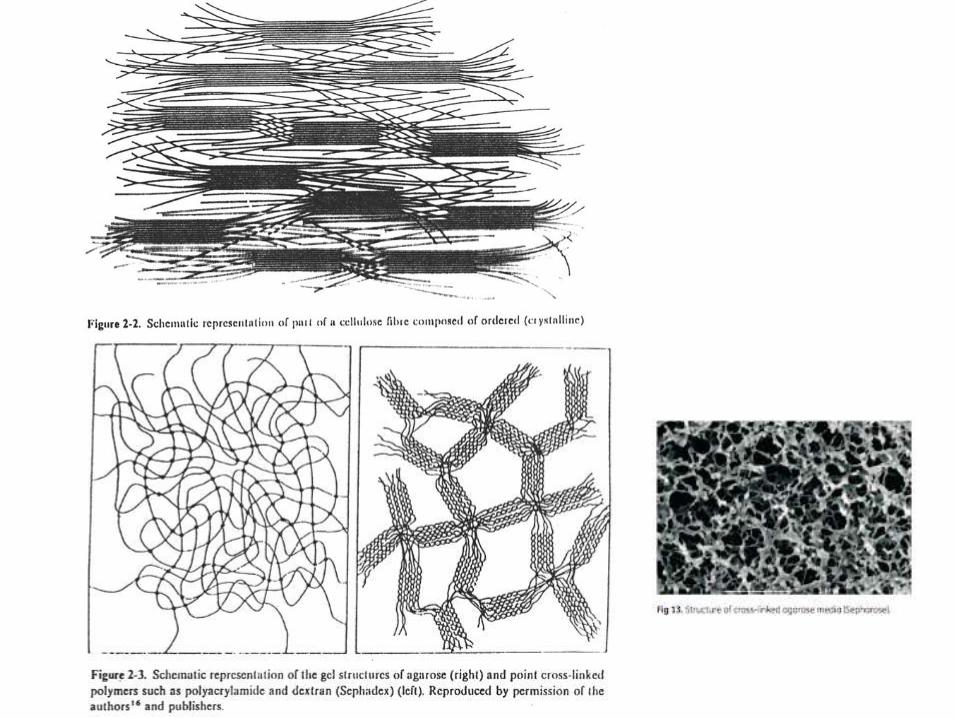
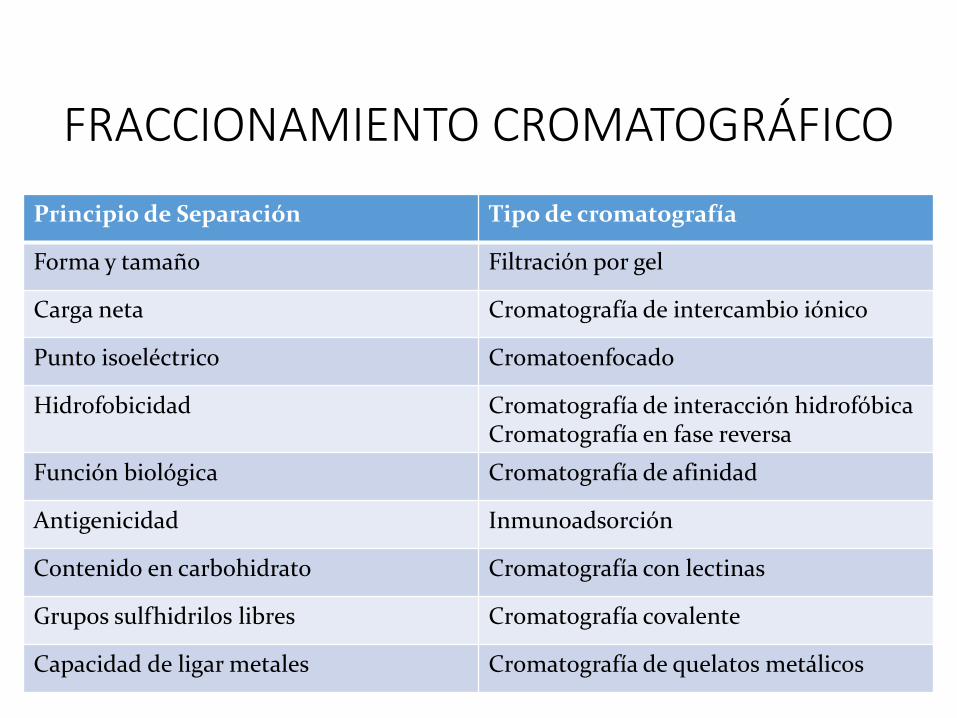
FRACCIONAMIENTO CROMATOGRÁFICO
Principio de Separación Tipo de cromatografía
Forma y tamaño Filtración por gel
Carga neta Cromatografía de intercambio iónico
Punto isoeléctrico Cromatoenfocado
Hidrofobicidad Cromatografía de interacción hidrofóbicaCromatografía en fase reversa
Función biológica Cromatografía de afinidad
Antigenicidad Inmunoadsorción
Contenido en carbohidrato Cromatografía con lectinas
Grupos sulfhidrilos libres Cromatografía covalente
Capacidad de ligar metales Cromatografía de quelatos metálicos

PASOS PRINCIPALES EN UNA CROMATOGRAFÍA• EMPAQUETADO DE LA RESINA (aunque puede venir la columna ya lista para usar). El
tamaño de la columna a utilizar dependerá del procedimiento cromatográfico aseguir y de la cantidad de muestra a purificar.
• EQUILIBRADO DE LA RESINA
• SIEMBRA DE LA MUESTRA. La muestra se encuentra resuspendida en un buffer decomposición similar al buffer de equilibrado de columna. La aplicación de la muestraa la columna se hace generalmente a través de un “loop”, imprescindible para FPLC oHPLC. En LPLC puede hacerse aplicación manual.
• LAVADO
• ELUCIÓN. La elución de las proteínas introducidas en la columna puede hacerse sincambiar la composición de la fase móvil (elución isocrática) o cambiándola, poretapas o continuamente (gradientes).
• REGENERACIÓN. Normalmente, luego de la corrida la columna debe ser regenerada,dejándola en condiciones de ser reutilizada. En general si las columnas no se utilizancontinuamente deben ser guardadas conteniendo un agente conservador, como eletanol al 20% o la azida sódica, para evitar el crecimiento de microorganismos.

TIPOS DE CROMATOGRAFÍA
• Según la presión a que se opera la corrida, se pueden considerar tres tipos:
1.Corridas a presión normal (LPLC, low-pressure liquid chromatography). Presión inferior a 5 bar. Pueden usarse matrices de escasa rigidez, como la agarosa o el dextrano.
2.Corridas a presión intermedia (MPLC, medium-pressure liquid chromatography, tambien llamada FPLC (Fast protein liquid chromatography). Presión entre 6 y 50 bar. Requiere matrices mas rígidas. Permite flujos mayores.
3.Corridas a alta presión (HPLC, high-pressure - o high-performance - liquid chromatography). Presiones mayores de 50 bar. Requiere matrices de rigidez alta, partículas pequeñas y columnas y tuberías de material altamente resistente a la presión, como el acero inoxidable o el titanio.
1 Atm corresponde a 1013.25 milibars

CARACTERÍSTICAS TÍPICAS DE LPLC Y HPLC
Característica LPLC HPLC
Tamaño de partículA (µm) 100 10
Flujo (ml.cm-2.h-1) 10-30 100-300
Presión de trabajo (bar) < 5 > 50
Tiempo de separación (hs) Hasta 24 1 – 3
Volumen de muestra ml – l µl – ml
Etapa de purificación Variable En general tardía
Resolución Buena Excelente
Dificultades Largo tiempo, cámara fría
Alto costo de equipo,posible desnaturalización
por presión
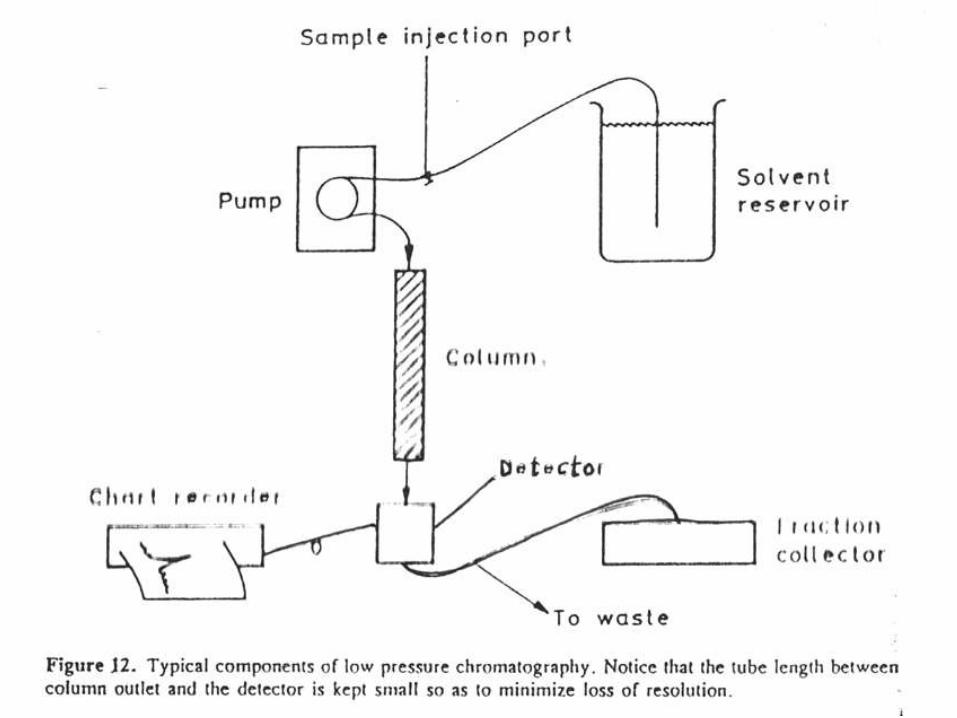

Generación del gradiente para la elución en LPLC


• El material cromatográfico es un gel en partículas esféricas, de porosidad controlada, sinderivatizaciones.
• La elución es isocrática. Se usa en general fuerza iónica alta, para minimizar lasinteracciones de las proteínas entre si, y de las proteínas con la matriz (en este último caso,no relacionadas con el tamaño de poro).
• La separación de las proteínas se hace en base a la forma y la masa de la proteína. Lasproteínas que no penetran en absoluto en los poros del gel son las que salen primero (en elvolumen de exclusión o volumen vacío, Vo). Las demás moléculas en la muestra se separan,saliendo últimas las que penetran por completo en las particulas de gel
• Los materiales mas comunes son geles de dextrano (Sephadex), de agarosa (Sepharosa), deagarosa y poliacrilamida (Biogel A), de poliacrilamida (Biogel P). Puede usarse en LPLC, ytambien en FPLC (Superosa, Superdex) y HPLC (silica porosa, como Lichrosorb).
• En la filtración por gel es crítica la relación entre el volumen de muestra y el volumen de lacolumna (1 a 5 % del volumen total (Vt) del gel), por lo cual es una técnica que no se puedeaumentar en escala mas que hasta un cierto punto. Para desalado de proteínas, se admitehasta un 25 - 30 % del Vt.
Aplicaciones: Purificación de proteínas - Desalado - Determinación de pesos moleculares deproteínas nativas - Determinación de constantes de equilibrio de unión (“binding”).
CROMATOGRAFÍA DE EXCLUSIÓN MOLECULAR


Gel filtration can be used as a purification technique. In most cases, gel filtration isused as a later step in the purification procedure, because it is a relatively lowresolution technique, and because it cannot tolerate large sample volumes.
However, gel filtration can be used as a desalting technique at any time during thepurification.
As mentioned above, gel filtration is used as an analytical technique to measurethe molecular weight of proteins in solution. In performing these experiments, it isworth examining the migration of different standard molecules
CROMATOGRAFÍA DE EXCLUSIÓN MOLECULARUSOS EXPERIMENTALES

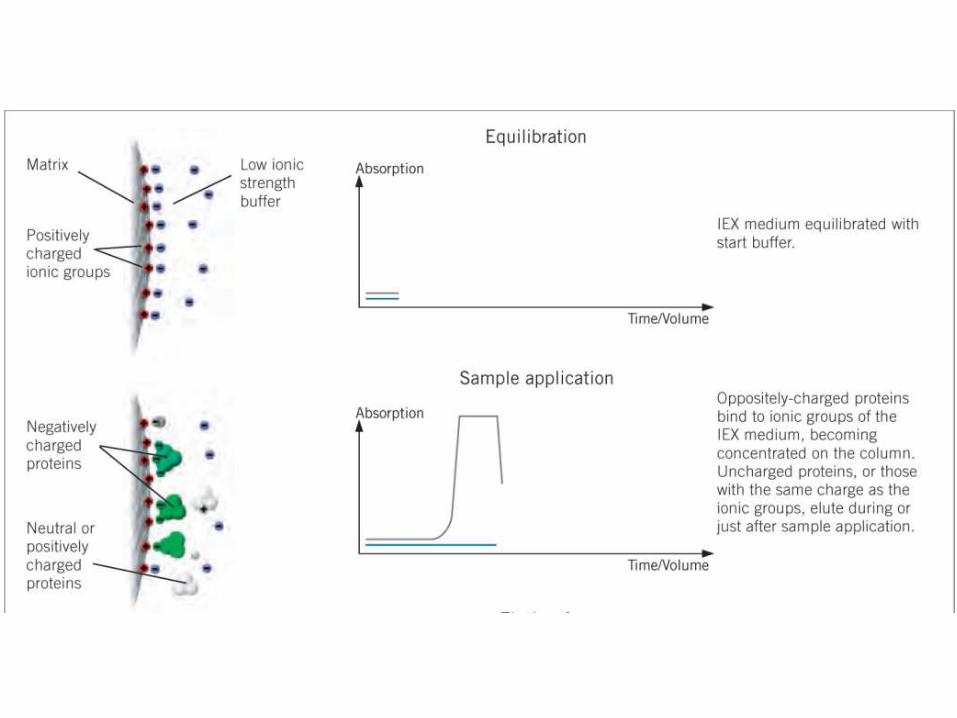
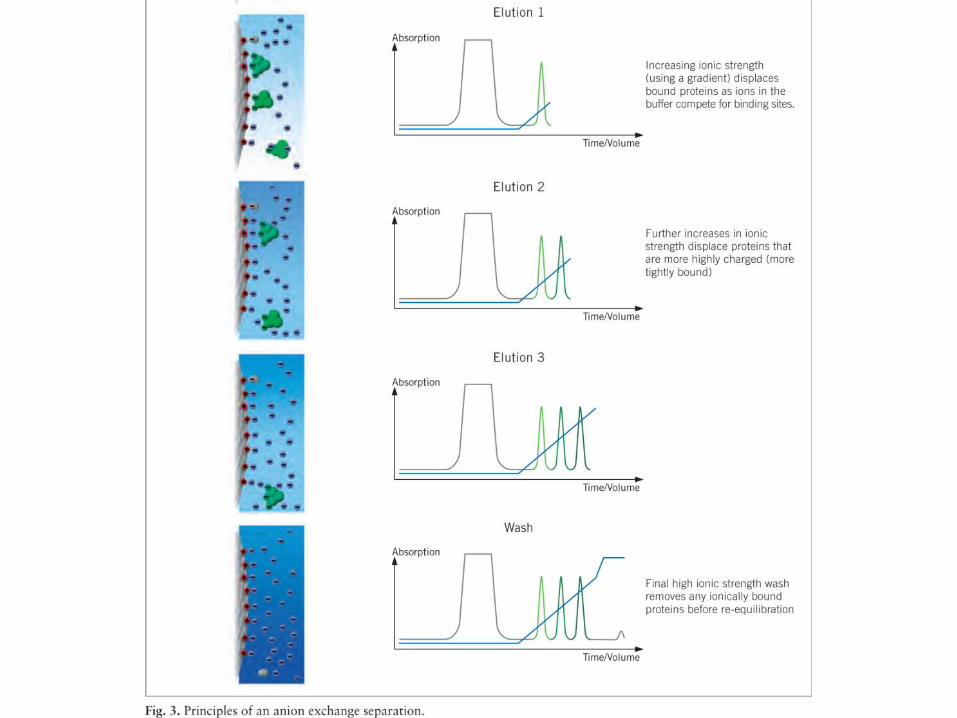
CROMATOGRAFÍADE INTERCAMBIO IÓNICO
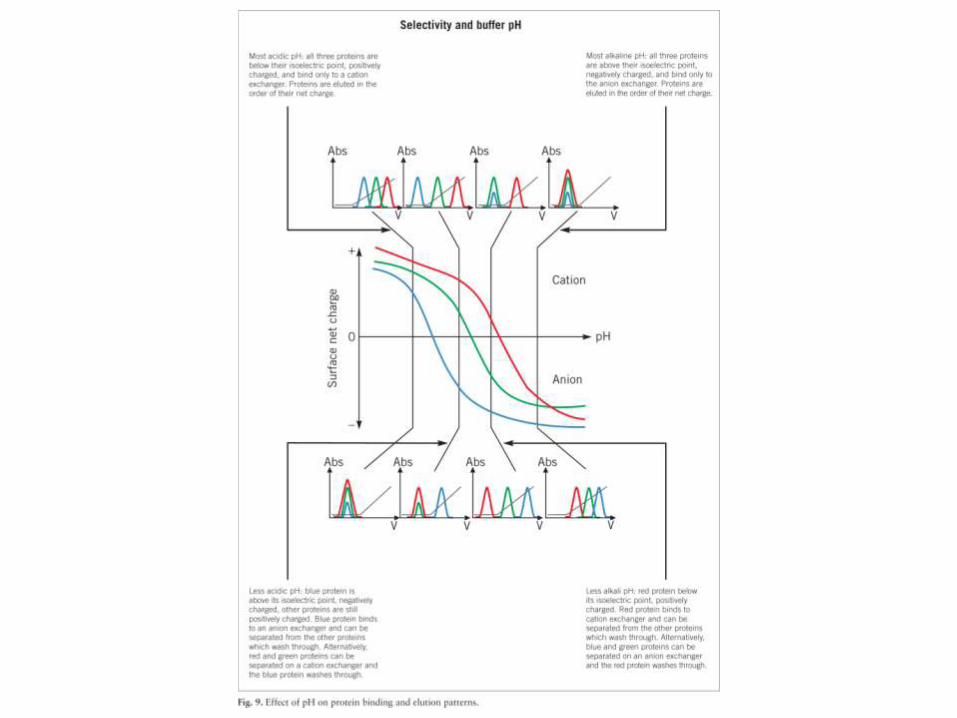
• Todas las proteínas contienen residuos cargados, positiva o negativamente, y su balance a undeterminado valor de pH da la carga neta de la molécula a ese pH. El valor de pH en el que las cargasse balancean, y en consecuencia la carga neta de la molécula es 0, es el punto isoeléctrico de laproteína.
• Los métodos cromatográficos basados en la carga son dos:
1) La cromatografía de intercambio iónico.
2) El cromatoenfocado.
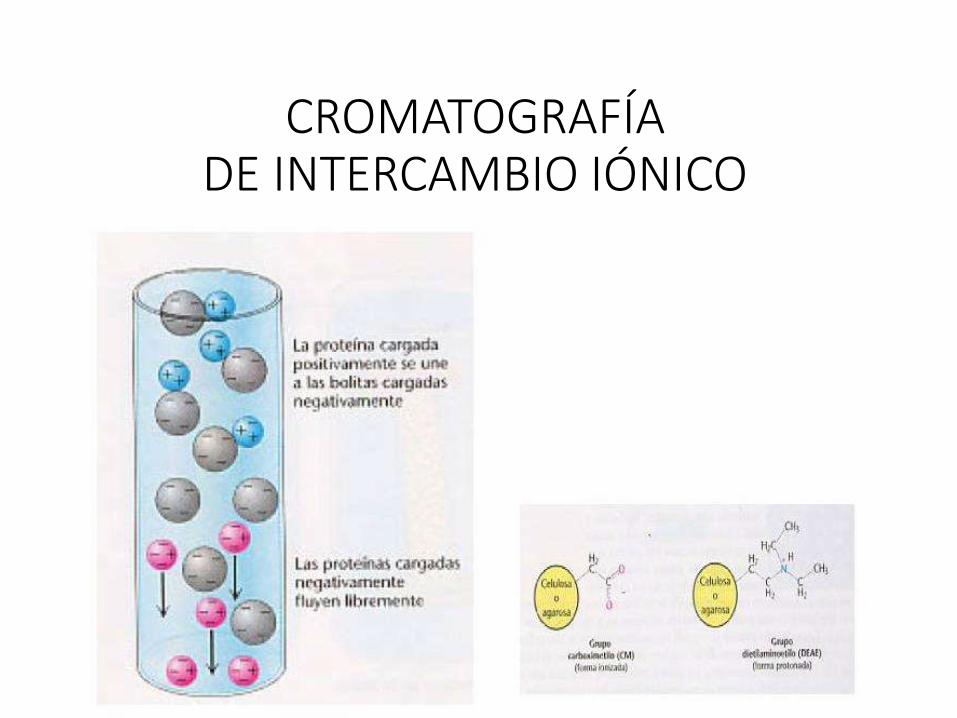
CROMATOGRAFÍADE INTERCAMBIO IÓNICO

MÉTODOS CROMATOGRÁFICOS BASADOS EN LA CARGA DE LA PROTEÍNA
• La mayoría de las proteínas tienen su pI entre pH 5 y 9. Por encima de su pI la carganeta de la proteína es negativa, y por debajo es positiva.
• El material utilizado es una matriz derivatizada con grupos cargados positivamente(que intercambian iones negativos, y se llaman por eso intercambiadores aniónicos,como DEAE-celulosa) o negativamente (intercambiadores catiónicos, como CM-celulosa).
• La adsorción puede hacerse en “batch” o en columna. Las proteínas migrarán enbase a su carga neta a ese determinado pH. La elución se hará aumentando la fuerzaiónica del buffer (eventualemente también cambiando el pH).
• Los intercambiadores pueden ser débiles (utilizables cuando la proteína tiene cargaelevada a ese pH) o fuertes (cuando la carga es baja). No conviene usarintercambiadores fuertes para proteínas muy cargadas a ese pH, pues la interacciónserá muy fuerte y la elución más dificil.
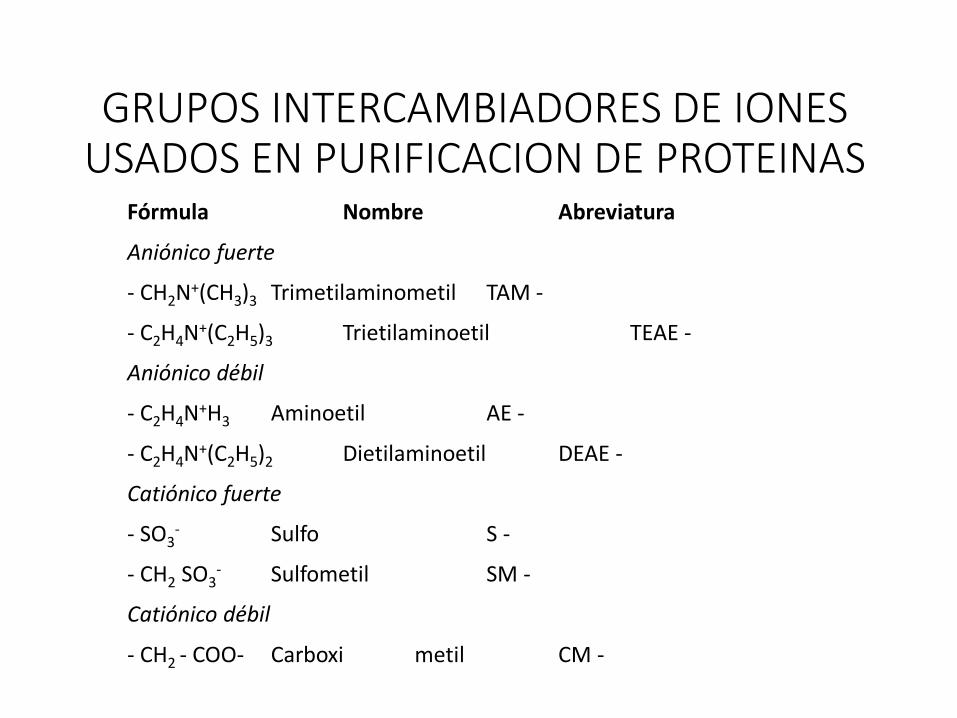
GRUPOS INTERCAMBIADORES DE IONES USADOS EN PURIFICACION DE PROTEINAS
Fórmula Nombre Abreviatura
Aniónico fuerte
- CH2N+(CH3)3 Trimetilaminometil TAM -
- C2H4N+(C2H5)3 Trietilaminoetil TEAE -
Aniónico débil
- C2H4N+H3 Aminoetil AE -
- C2H4N+(C2H5)2 Dietilaminoetil DEAE -
Catiónico fuerte
- SO3- Sulfo S -
- CH2 SO3- Sulfometil SM -
Catiónico débil
- CH2 - COO- Carboxi metil CM -

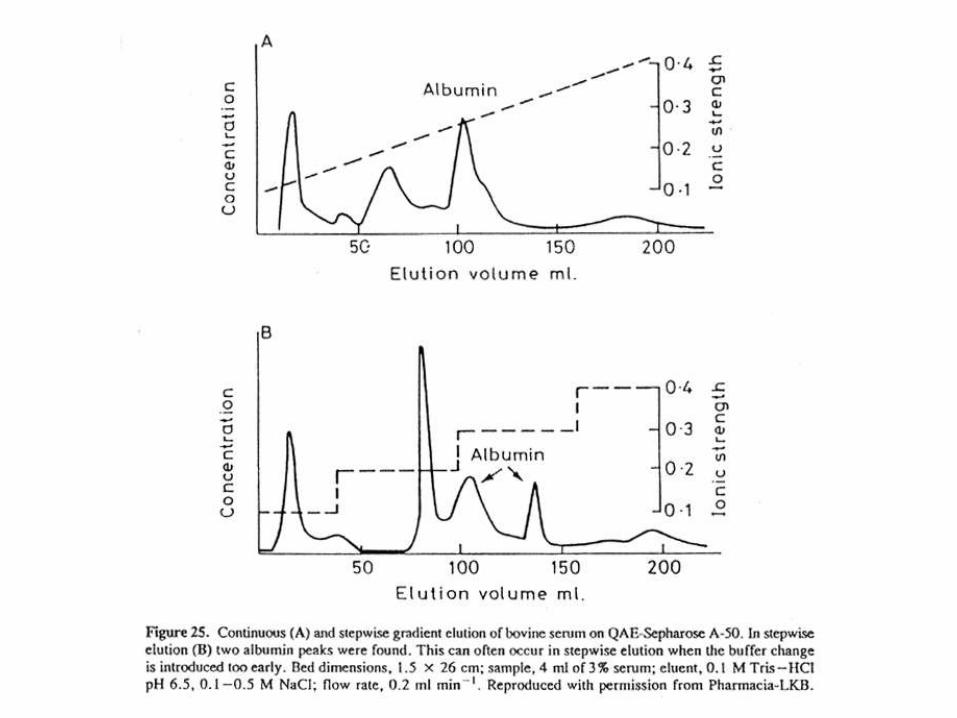

PURIFICACION DE LA SERINA CARBOXIPEPTIDASA DE TRYPANOSOMA CRUZI POR FPLC EN COLUMNA DE MONO Q (INTERCAMBIADOR ANIÓNICO)

Elución por pH
• Since the net charge on a protein is pH dependent, samples can also be eluted from an IEX medium by altering the pH of the elution buffer. As there is no salt gradient, samples are simply retained on the column at one pH and eluted by increasing or decreasing the pH.
• The various charged groups in the sample or on the column are titrated until they are neutral or of opposite charge to the medium and the sample elutes.
• Proteins bound to an anion exchanger will elute as pH is decreased.
• Proteins bound to a cation exchanger will elute as pH is increased.
• Since pH elution will involve working at pH values close to the isoelectric point of a protein and since many proteins show minimum solubility close to their isoelectric points, precautions must be taken to avoid precipitation on the column.

CROMATOENFOCADO
• En cromatoenfocado se forma un gradiente dentro de la columna,mezclando una matriz de intercambio aniónico pre-ajustada a unvalor de pH, con un buffer de pH más bajo.
• La proteína al descender en la columna encontrará valorescrecientes de pH; cuando llegue a su pI se volverá electronegativa,y se unirá a la matriz positivamente cargada. Como el buffer siguecorriendo, la proteína se separará y unirá sucesivamente, hastaeluirse en una banda muy definida (“enfocada”) al pHcorrespondiente a su pI.
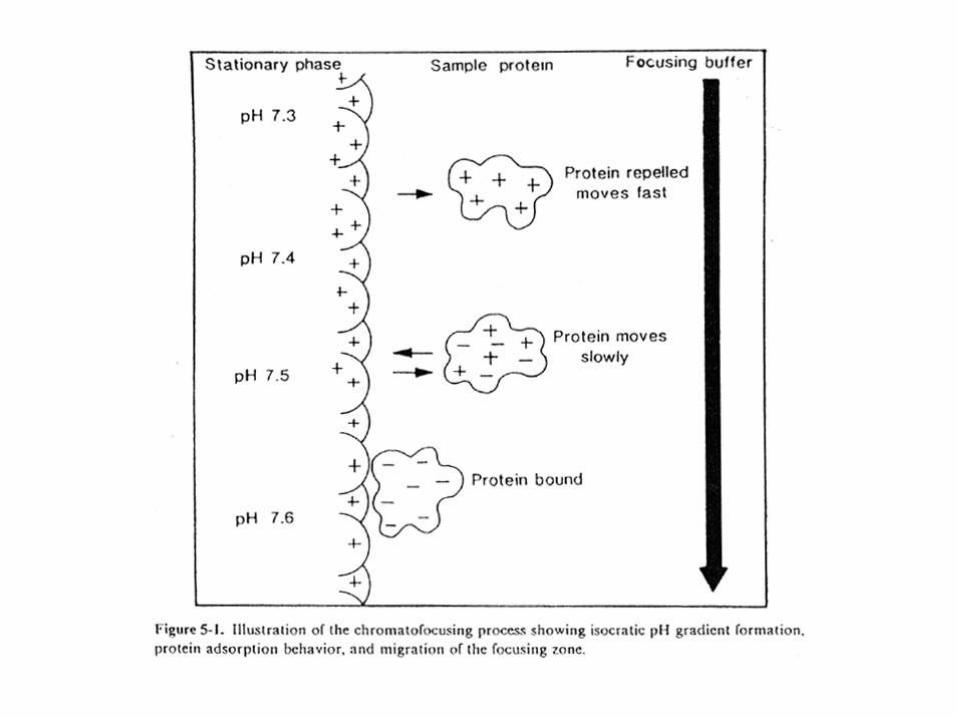


PURIFICACION DE LA SERINA CARBOXIPEPTIDASA DE TRYPANOSOMA CRUZI POR FPLC EN COLUMNA DE MONO P (CROMATOENFOCADO)

CROMATOGRAFÍA DE INTERACCIÓN HIDROFÓBICA
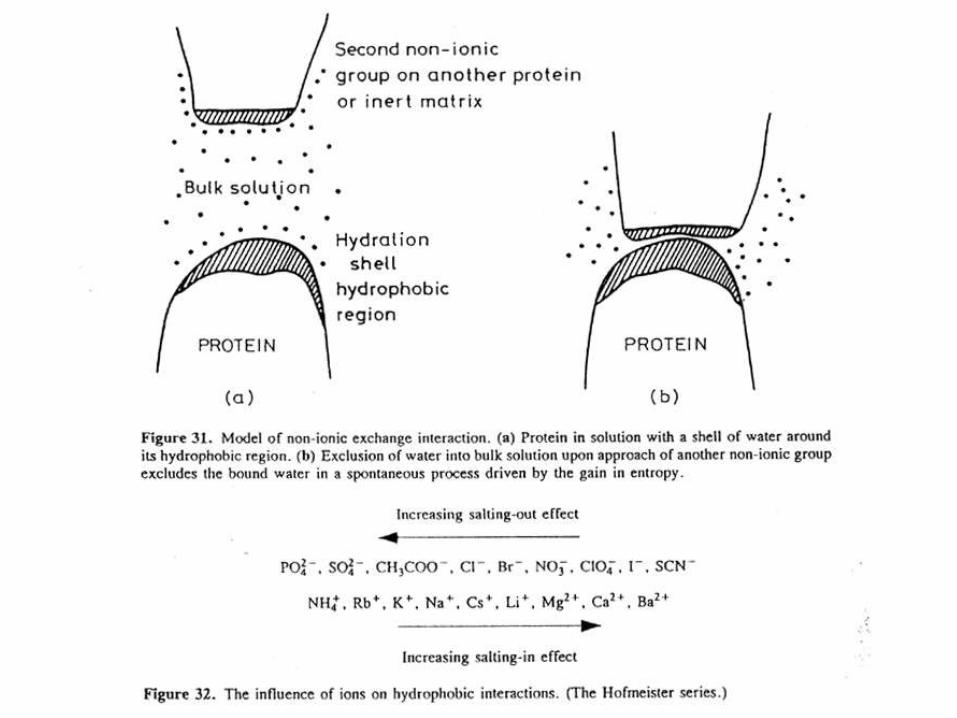
La mayoría de las proteínas tienen regiones hidrofóbicas en su superficie. Estos “parches”hidrofóbicos se deben a la presencia de cadenas laterales de aminoácidos hidrofóbicos o no polares como Phe, Trp, Ala, Met. Estas regiones hidrofóbicas están dispersas entre regiones más hidrofílicas o polares y su número, tamaño y distribución son una característica específica de cada proteína.
La cromatografía de interacción hidrofóbica separa proteínas de acuerdo a sus diferencias en la hidrofobicidad de superficie utilizando una interacción reversible entre las proteínas y la superficie hidrofóbica presente en la matriz..
La interacción entre las proteínas y la matriz se ve significativamente influenciada por la presencia de ciertas sales en el buffer de corrida.
Las sales provocan la eliminación de las moléculas de agua que se encuentran ordenadas cubriendo las regiones hidrofóbicas de las proteínas. Así las sales promueven la exposición de las porciones hidrofóbicas facilitando la interacción con la resina. A medida que la fuerza iónica disminuye, la interacción se revierte y las proteínas con el menor grado de hidrofobicidad se eluyen primero. Las proteínas más hidrofóbicas eluyen después requiriendo una mayor reducción en la concentración de sales para revertir la interacción.
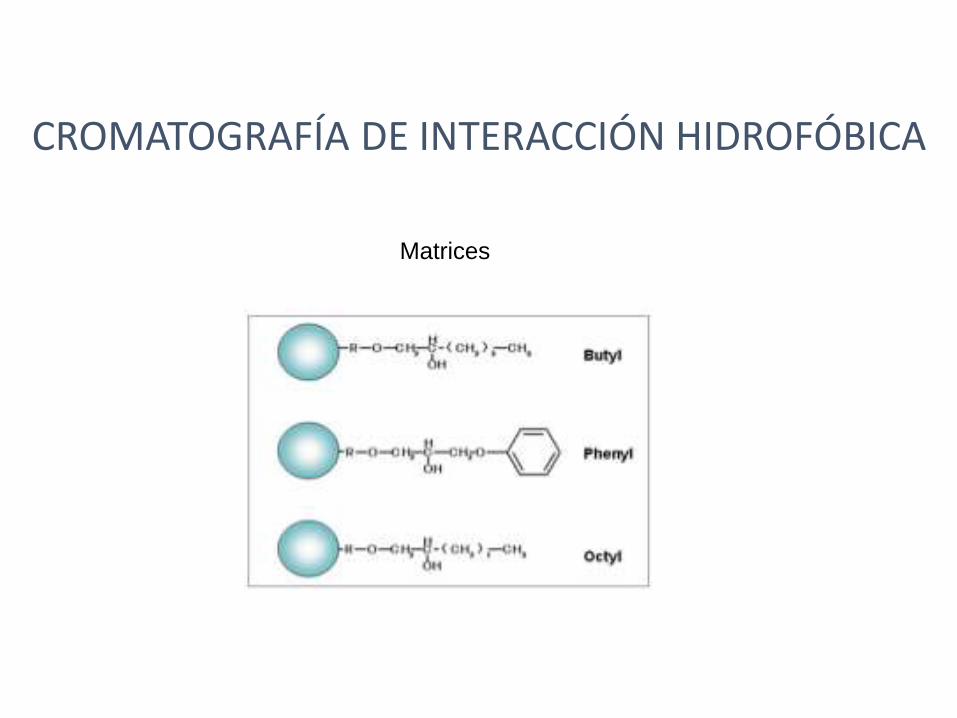
Matrices
CROMATOGRAFÍA DE INTERACCIÓN HIDROFÓBICA

CROMATOGRAFÍA DE INTERACCIÓN HIDROFÓBICA

La cromatografía en fase reversa se denomina de ese modo porque utiliza una faseestacionaria no polar y una fase móvil polar. La fase estacionaria está en generalformada por cadenas alifáticas de hasta C18 unidas a una matriz de silica. La fasemóvil es un solvente polar como metanol, propanol, etanol o acetonitrilo. Estascondiciones pueden causar la desnaturalización de muchas proteínas . Por ello selas utiliza en general en HPLC en fase reversa (RP-HPLC), para purificar proteínas ypéptidos para su secuenciación. Se usan contraiones como el ácido trifluoroacéticopara asociarse con grupos cargados en la proteína y aumentar su hidrofobicidad.
CROMATOGRAFÍA EN FASE REVERSA

• Principio: existencia de una interacción reversible entre una proteína y un ligando específico
acoplado a una matriz cromatográfica.
• Algunas interacciones biológicas típicas son:
Enzimaanálogo de sustrato, inhibidor, cofactor.
Anticuerpoantígeno, virus, célula.
Lectinapolisacárido, glicoproteina, receptor de sup celular, célula.
Acidos Nucleicossecuencias complementarias, histonas, polimerasa de ácidos
nucleicos, proteínas de unión a ácidos nucleicos.
Hormonas, vitaminasreceptores, proteínas transportadoras.
Glutationglutation-S-transferasa o proteínas de fusión a GST.
Iones metálicosproteínas de fusión a poli (His) fusion, proteínas nativas con residuos de
histidina, cisteína y/o triptofano en su superficie.
• La interacción biológica entre el ligando y el target puede ser resultado de interacciones
electrostáticas, hidrofóbicas, fuerzas de van der Waals’ y/o enlaces de hidrógeno. Para eluir
la interacción puede ser revertida específicamente utilizando un ligando competitivo, o de
manera no específica cambiando el pH, la fueza iónica o la polaridad.
• La afinidad de la proteína por el ligando debe ser moderada (KL entre 10-4 y 10-8 M) para
permitir una buena unión y una disociación ulterior del complejo en condiciones no
desnaturalizantes.
CROMATOGRAFÍA DE AFINIDAD

Pasos de purificación

Coupling through the primary amine of a ligand:NHS-activated Sepharose
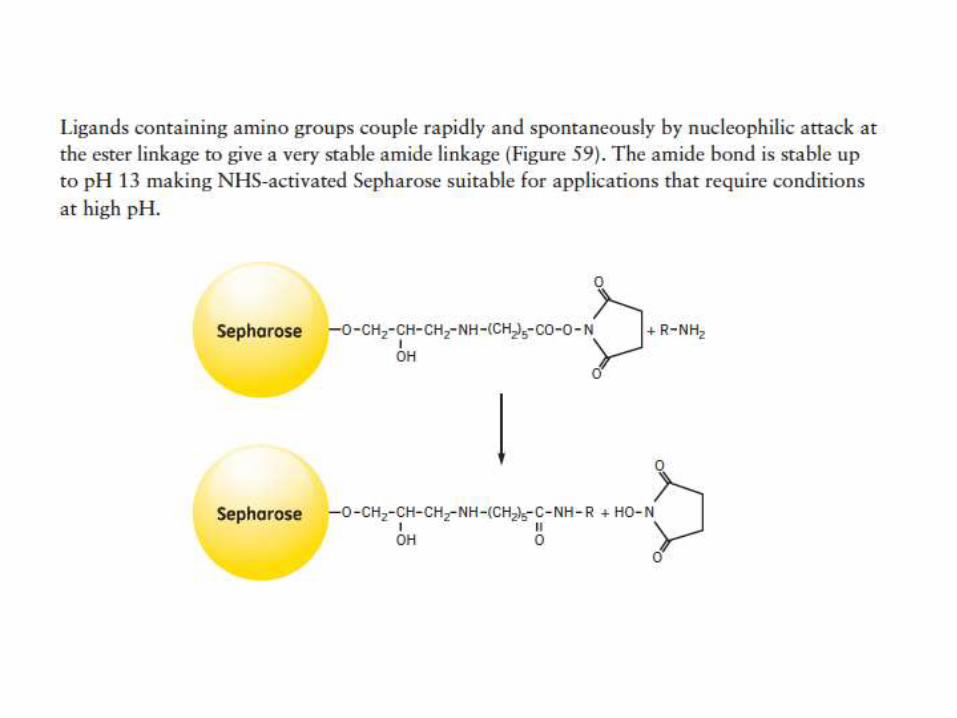

Dissolve the ligand in the coupling buffer (0.2 M NaHCO3, 0.5 M NaCl, pH
8.3) to a final concentration of 0.5–10 mg/ml (for protein ligands)


Coupling through the primary amine of a ligand: CNBr-activated Sepharose

Coupling through hydroxy, amino or thiol group: Epoxy-activated Sepharose 6B
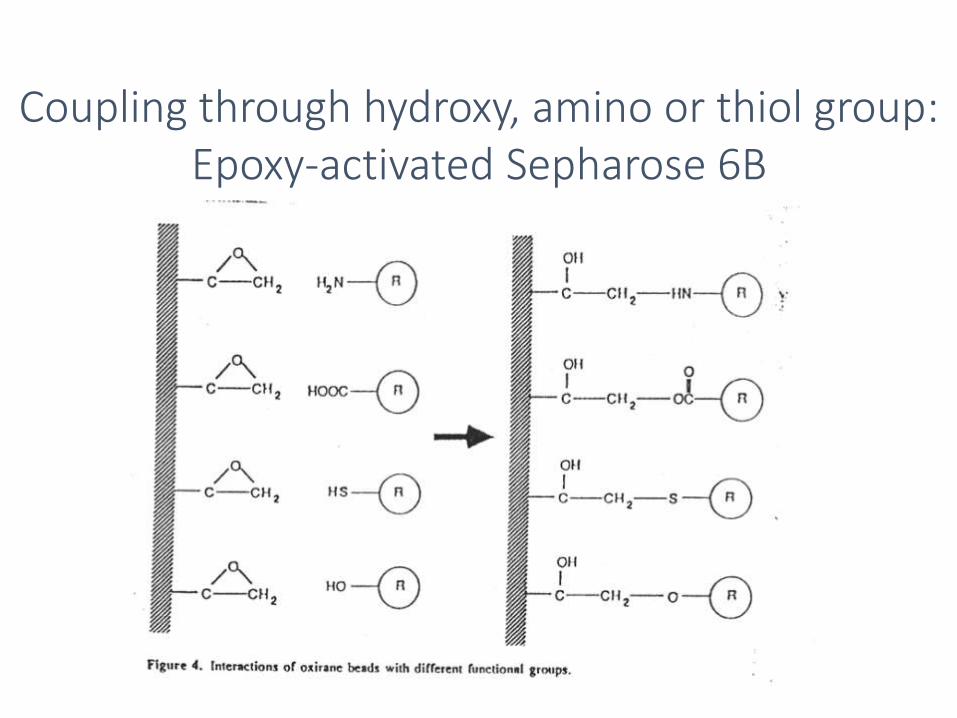
Coupling through hydroxy, amino or thiol group: Epoxy-activated Sepharose 6B

Coupling through a thiol groupThiopropyl Sepharose 6B
• Thiol-containing substances can be isolated selectively by covalent binding to an activated thiolated matrix via thiol-disulphide exchange to form a mixed disulphide bond. After washing away unbound material, the thiol-containing substance is eluted by reducing the disulphide bond.
• If the proteins to be purified contain disulphide bonds, the disulphide bridges must be reduced, for example with 2-mercaptoethanol (5 mM).
• The coupling reaction can be monitored and, in some cases, quantified by following the appearance of 2-thiopyridone in the eluent at 343 nm during the purification.
• Resolve different thiol proteins by sequential elution: 5–25 mM L-cysteine < 0.05 M glutathione
< 0.02–0.05 M 2-mercaptoethanol < and 0.02–0.05 M dithiothreitol.
• Reactivation: Pass one to two column volumes of a saturated solution (approximately 1.5 mM) of
2,2’-dipyridyl disulphide, pH 8.0 through the medium.
Principio de la cromatografía covalente basada en el 2-piridil disulfuro


ConA-Sepharose
• Lectinas: proteínas que interaccionan específicamente y de manera reversible con ciertos residuos de azúcares. Se utilizan para aislar y separar glicoproteínas, glicolípidos, polisacáridos.
• Con A es una metalloproteina tetramérica aislada de Canavalia ensiformis (jack bean). Con A se une a moléculas que contienen α-d-mannopyranosyl, α-d-glucopyranosyl, y residuos relacionados estéricamente.
• Para mantener las características de unión de la Con A Sepharose, es esencial la presencia de Mn2+ y Ca2+ (1 mM).
• La elución de las sustancias unidas se puede realizar con un gradiente creciente (linear o en pasos) de α-D-metilmanósido or α-D-metilglucósido. Estos azúcares actúan como eluyentes fuertes. Muchas sustancias eluyen a 0.1-0.2 M pero para sustancias unidas fuertemente se pueden requerir concentraciones más elevadas. Glucosa y manosa también se pueden usar pero son eluyentes más débiles.
• Las sustancias unidas fuertemente también pueden eluirse [por disminución del pH, pero no por debajo de pH 4. El Borato se sabe que forma complejos con los cis-diols presentes en los residuos de azúcares y así actúa como un eluyente competitivo (0.1 M buffer borato, pH 6.5).

ConA-Sepharose
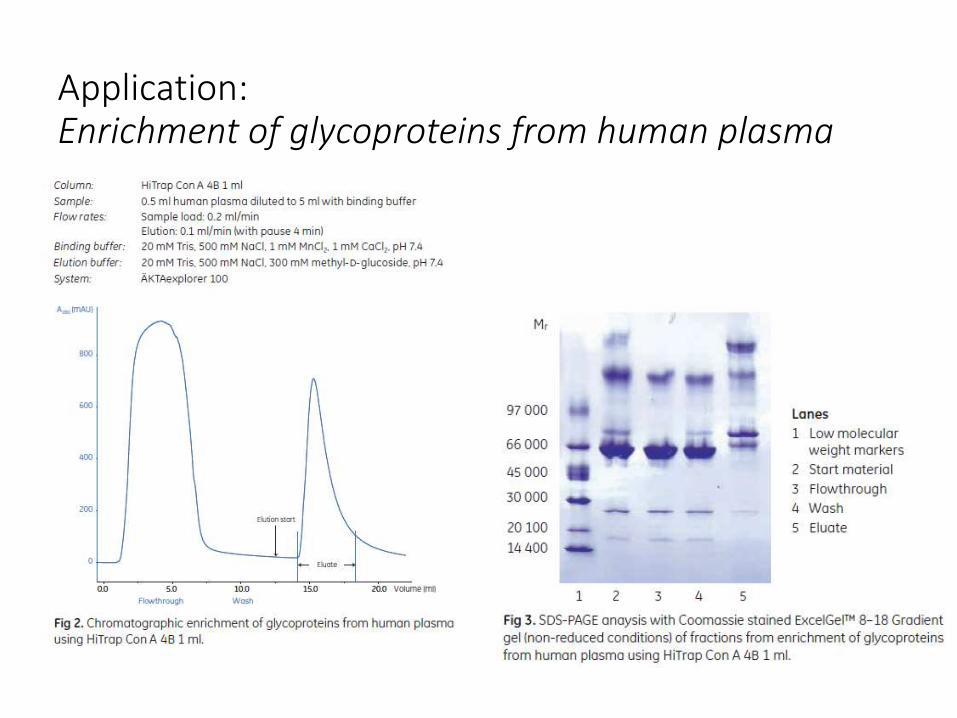
Application:Enrichment of glycoproteins from human plasma

Cromatografía de afinidad: proteínas recombinantes
•The purification of recombinant proteins can often be simplified by incorporating a tag of known size into the protein. As well as providing a marker for expression and facilitating detection of the recombinant protein, an important role for the tag is to enable a simple purification by affinity chromatography. The two most commonly used tags are glutathione-S-transferase (GST) and 6 x histidine residues (His)6.

Cromatografía de quelatos metálicos inmovilizados (IMAC)
• La matriz tiene unido covalentemente un quelante de metales, como el ácidoiminodiacético.
• Se carga la columna con un ión adecuado, que puede ser Ni2+, Co2+, Zn2+, Cu2+. Laproteína se adsorbe a la columna si tiene residuos agrupados de His, Cys o Trp. Sepuede eluir compitiendo con imidazol, con quelantes (EDTA) o bajando el pH a 4 - 6,o.
• Esta técnica es muy usada actualmente para purificar proteínas recombinantes,expresandolas como fusiones con un “tag” de poly-His (en general 6 residuos), quepueden agregarse en el N o en el C-terminal.
• Si todo funciona bien, se puede tener proteína recombinante pura en un solo pasode purificación. Debe tenerse en cuenta los metales pueden catalizar la oxidaciónde grupos en la proteína, en especial de –SH.

Cromatografía de Afinidad a Glutation Sepharose
REMOCIÓN DE SALES YCAMBIO DE BUFFER
• Diálisis
• Diafiltración (ultrafiltración y cambio de buffer)
• Filtración por gel (G-25, PD-10)

PURIFICACIÓN DEPROTEÍNAS DE MEMBRANA
1) Es fundamental determinar el modo de asociación de la proteína de interés con la membrana ya que de ello dependerán los métodos adoptados para su purificación.
• Las proteínas periféricas (o extrínsecas) están asociadas a la membrana a través de interacciones con otras proteínas o con las regiones expuestas de los fosfolípidos. En estos casos, la proteína puede a menudo ser disociada de la membrana alterando las condiciones iónicas del buffer o, en situaciones más resistentes, induciendo un cierto grado de desnaturalización. Aunque los detergentes pueden ayudar a liberar este tipo de proteínas, no suelen ser requeridos en los pasos subsiguientes.
• La liberación de las proteínas integrales de membrana (o intrínsecas) en una forma soluble requerirá la disrupción de la bicapa fosfolipídica o el clivado de su anclaje a membrana. Si una parte sustancial de la proteína se encuentra dentro de la bicapa lipoídica, durante todas los procedimientos de purificación posteriores se requerirá “proteger” las regiones hidrofóbicas de su exposición al medio acuoso, por lo que normalmente se usan detergentes.
2) El paso inicial más valioso consiste en tener una preparación de la membrana de interés tan pura como sea posible.
• Inducción de shedding, fraccionamiento de membranas, centrifugación en gradiente de densidad, etc

Six ways in which membrane proteins associate with the lipid bilayer. Most trans-membrane proteins are thought to extend across the bilayer as a
single a helix (1) or as multiple a helices (2); some of these "single-pass" and "multipass" proteins have a covalently attached fatty acid chain
inserted in the cytoplasmic monolayer (1). Other membrane proteins are attached to the bilayer solely by a covalently attached lipid - either a fatty
acid chain or prenyl group - in the cytoplasmic monolayer (3) or, less often, via an oligosaccharide, to a minor phospholipid, phosphatidylinositol,
in the noncytoplasmic monolayer (4). Finally, many proteins are attached to the membrane only by noncovalent interactions with other membrane
proteins (5) and (6).

SOLUBILIZACIÓN DEPROTEÍNAS DE MEMBRANA
• Proteínas de membrana periféricas
- Se utilizan tratamientos suaves (buffers de baja fuerza iónica conteniendo 0.1-1 mM EDTA o bufferes de alta fuerza iónica conteniendo hasta 1M NaCl o KCl. Para prevenir la desnaturalización irreversible de proteínas, el pH debería estar en el rango 6-8 y las incubaciones deben hacerse en hielo. Incluir inhibidores de proteasas.
- Condiciones más drásticas (6M clorhidrato de guanidina, 8M urea, ácidos pH 2-3 o bases pH 9.5-11 diluidas) usualmente conducen a la desnaturalización de las proteínas, a menudo irreversiblemente.

SOLUBILIZACIÓN DEPROTEÍNAS DE MEMBRANA
• Proteínas de membrana integrales
- Las que tienen una parte soluble sustancial, pueden ser solubilizadas cortando la parte que las ancla en la membrana con fosfolipasas o proteasas, según de qué ancla se trate. Las integrales de membrana que poseen una parte sustancial de la proteína inmersa en la bicapa lipídica se extraen por delipidación con solventes orgánicos, agentes caotrópicos o detergentes.
• Detergentes: Moléculas anfifílicas, relativamente pequeñas. Por encima de la concentración micelar crítica las moléculas coalescen a través de su parte hidrofóbica para formar micelas, en equilibrio con el monómero.


Non ionic detergents have a
hydrophilic head group that is
uncharged and are preferred for
their ability to break lipid-lipid
and lipid-protein interactions.
They have limited ability to break
protein-protein interactions and
are often referred to as non-
denaturing detergents and are
used to isolate biologically active
membrane proteins.
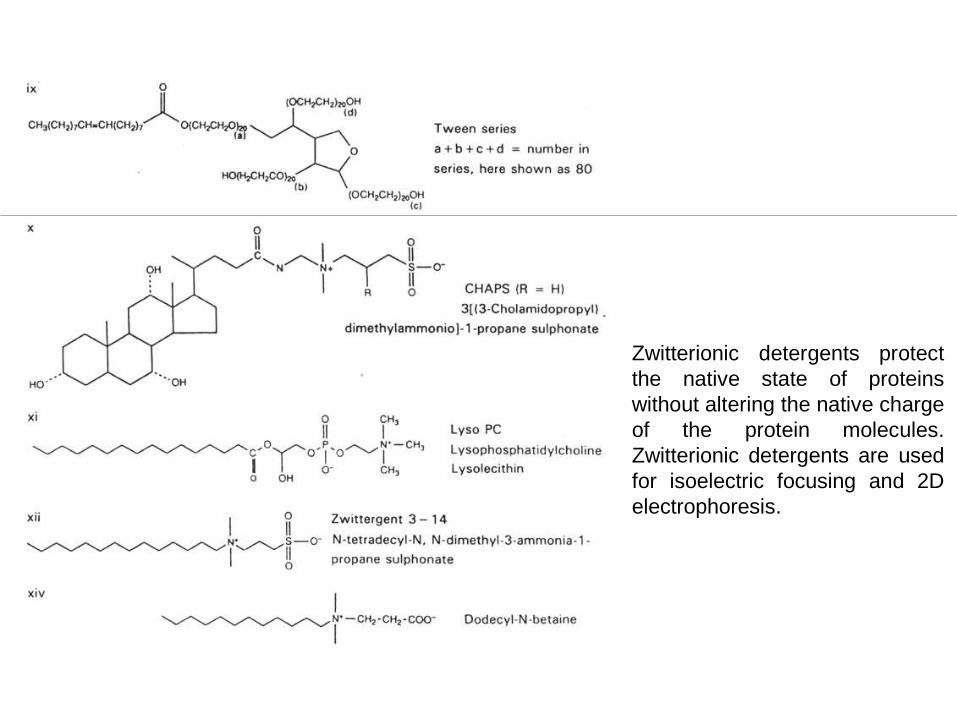
Zwitterionic detergents protect
the native state of proteins
without altering the native charge
of the protein molecules.
Zwitterionic detergents are used
for isoelectric focusing and 2D
electrophoresis.
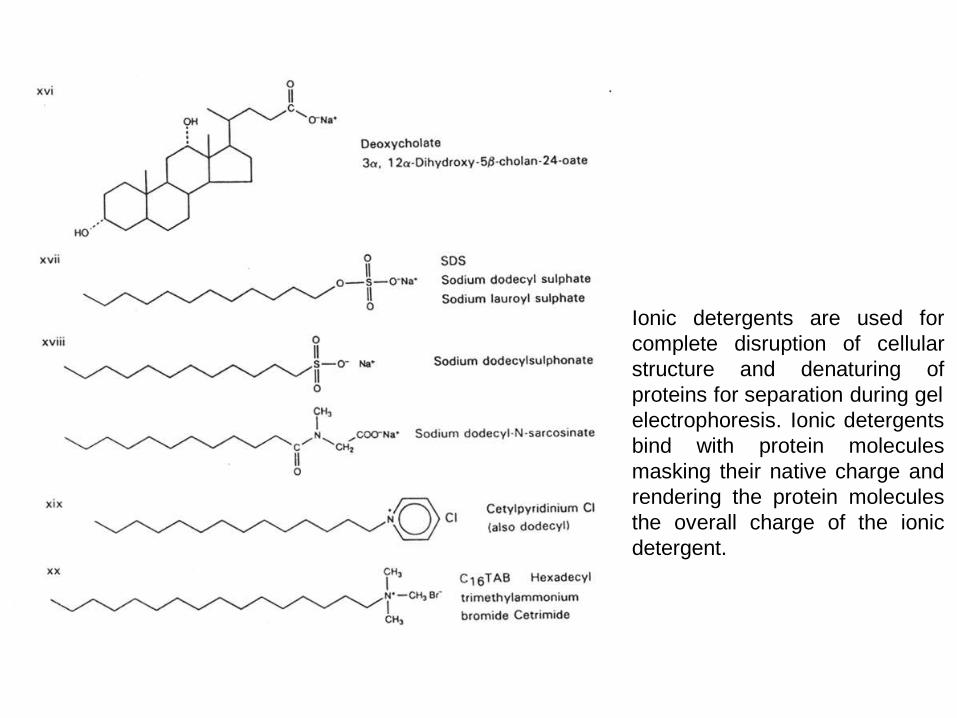
Ionic detergents are used for
complete disruption of cellular
structure and denaturing of
proteins for separation during gel
electrophoresis. Ionic detergents
bind with protein molecules
masking their native charge and
rendering the protein molecules
the overall charge of the ionic
detergent.

Actualmente se usa mucho el Triton X-114, que tiene la propiedad de ser miscible con el agua a 0 - 4oC, pero se separa en dos fases a 20 - 25oC. Las proteínas integrales, solubilizadas a baja temperatura, particionan en la fase de detergente al llevarlas a 20 - 25oC.

PURIFICACIÓN DE LASPROTEÍNAS SOLUBILIZADAS
• Las proteínas periféricas (o los fragmentos hidrofílicos de proteínas integrales) en general se purifican por los mismos métodos que los usados para las solubles. Las proteínas integrales presentan problemas especiales, debido a que requieren la presencia continua de detergentes o solventes orgánicos.
• La filtración por gel puede aplicarse; prácticamente todos los materiales para esta técnica pueden usarse en presencia de detergentes o desnaturalizantes, y existen algunas resinas (poliéteres hidroxilados, dextranos hidroxipropilados) admiten solventes orgánicos. La cromatografía de intercambio iónico y el cromatoenfocado pueden utilizarse, en presencia de detergentes no iónicos. La cromatografía en fase reversa en algunos casos puede aplicarse, con la muestra solubilizada en ácido fórmico al 50 - 70 % en agua. La cromatografía de afinidad es aplicable en general, requiriéndose a veces resinas de altas porosidad y brazos espaciadores largos.


















