
An Epigenetics Odyssey @ PyCon Taiwan 2013
40
-
Upload
wen-wei-liao -
Category
Technology
-
view
632 -
download
2
description
PyCon Taiwan 2013 "An Epigenetics Odyssey" by Wen-Wei Liao
Transcript of An Epigenetics Odyssey @ PyCon Taiwan 2013

AN EPIGENETICS ODYSSEYWen-Wei Liao

Wen-Wei Liao (廖玟崴)
Work in Pao-Yang Chen’s Lab, Academia Sinica (陳柏仰)
EducationM.S. in Dept. of Systems Neuroscience, NTHUB.S. in Dept. of Life Science, NTHU
Talks“Use the Matplotlib, Luke,” PyCon Taiwan 2012“Matplotlib for Python Programmers,” PyHUG
PyCon Taiwan
Our Lab

Who’s Paul Graham?

“So if you're a CS major and you want to start a startup, instead of taking a class on entrepreneurship you're better off taking a class on, say, genetics. Or better still, go work for a biotech company. CS majors normally get summer jobs at computer hardware or software companies. But if you want to find startup ideas, you might do better to get a summer job in some unrelated field.
ー How to Get Startup Ideas, Paul Graham
Human
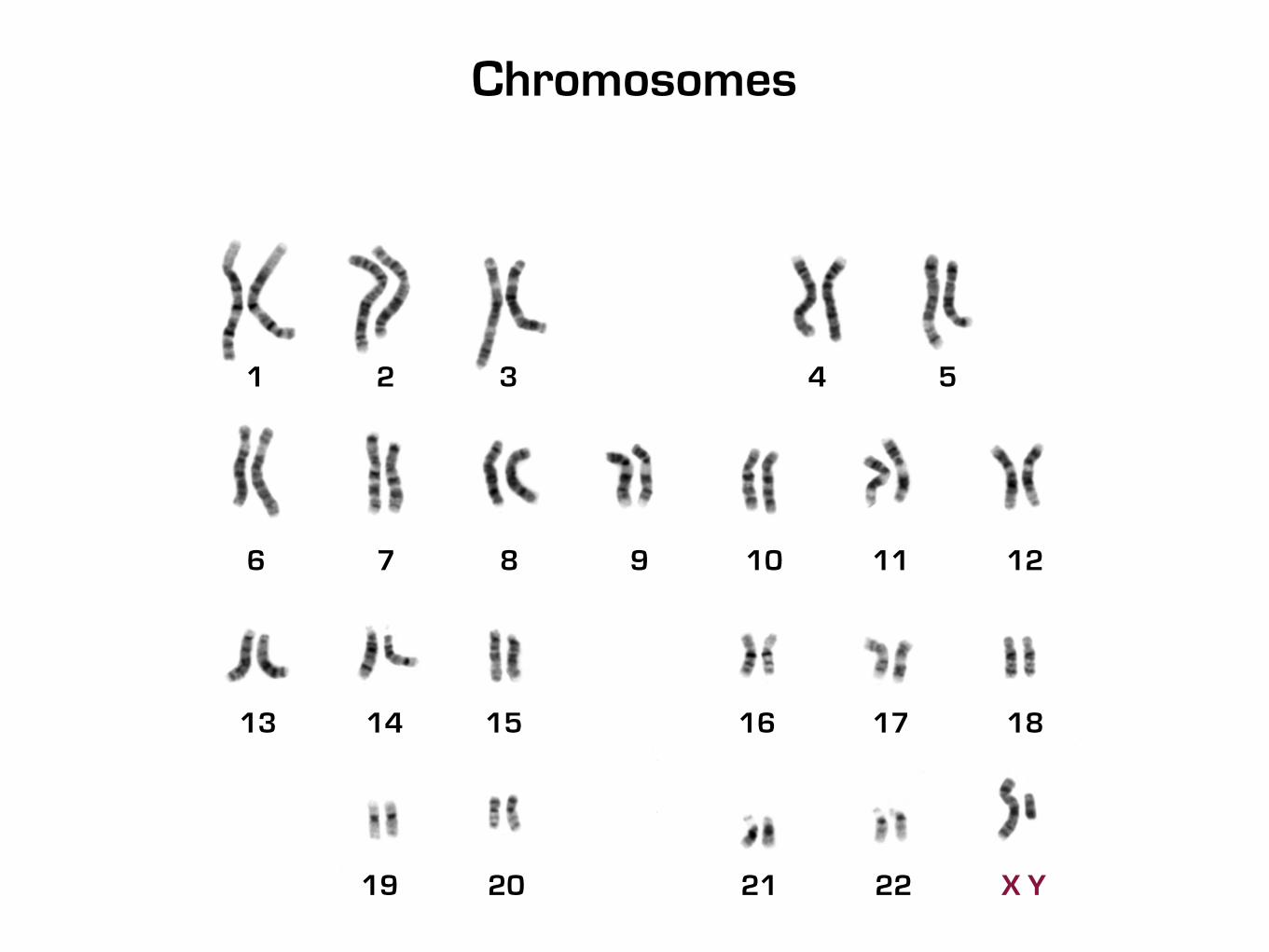
19 20 21 22 X Y
1 2 3 4 5
6 7 8 9 10 11 12
13 14 15 16 17 18
Chromosomes
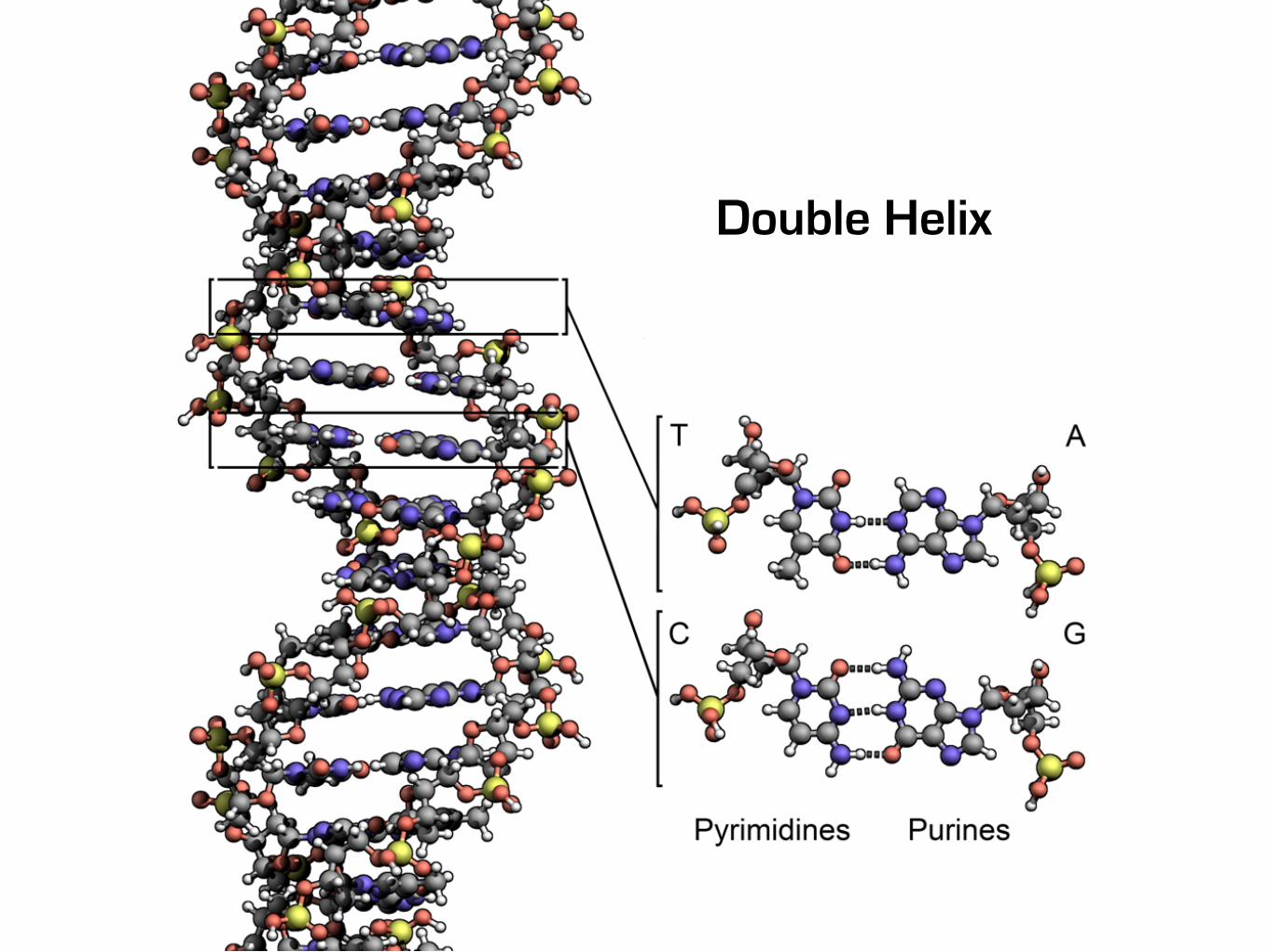
Double Helix

2003Human Genome Project
3 billion bases (Gb)30 億


Why are Identical Twins different?

...GATTACACCCATGTCAGTGCG...
...CTAATGTGGGTACAGTCACGC...
DNA Sequences

...GATTACACCCATGTCAGTGCG...
...CTAATGTGGGTACAGTCACGC...
DNA Sequences

...GATTACACCCATGTCAGTGCG...
...CTAATGTGGGTACAGTCACGC...
m m
DNA Sequences

...GATTACACCCATGTCAGTGCG...
...CTAATGTGGGTACAGTCACGC...
m m
DNA Sequences
m = methylation (甲基化)
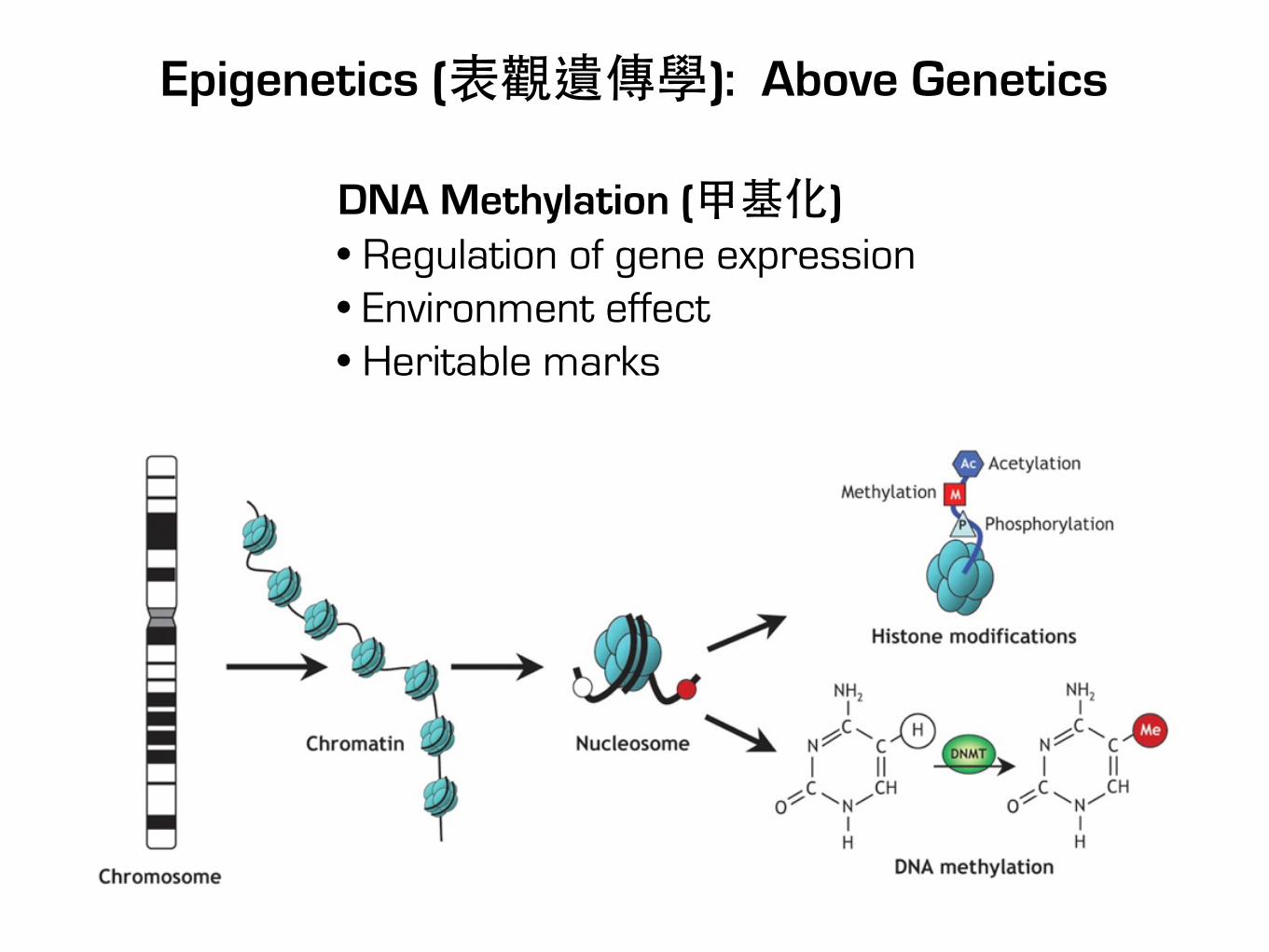
Epigenetics (表觀遺傳學): Above Genetics
DNA Methylation (甲基化)• Regulation of gene expression• Environment effect• Heritable marks

“The new science of epigenetics reveals how the choices you make can change your genes and those of your kids.”
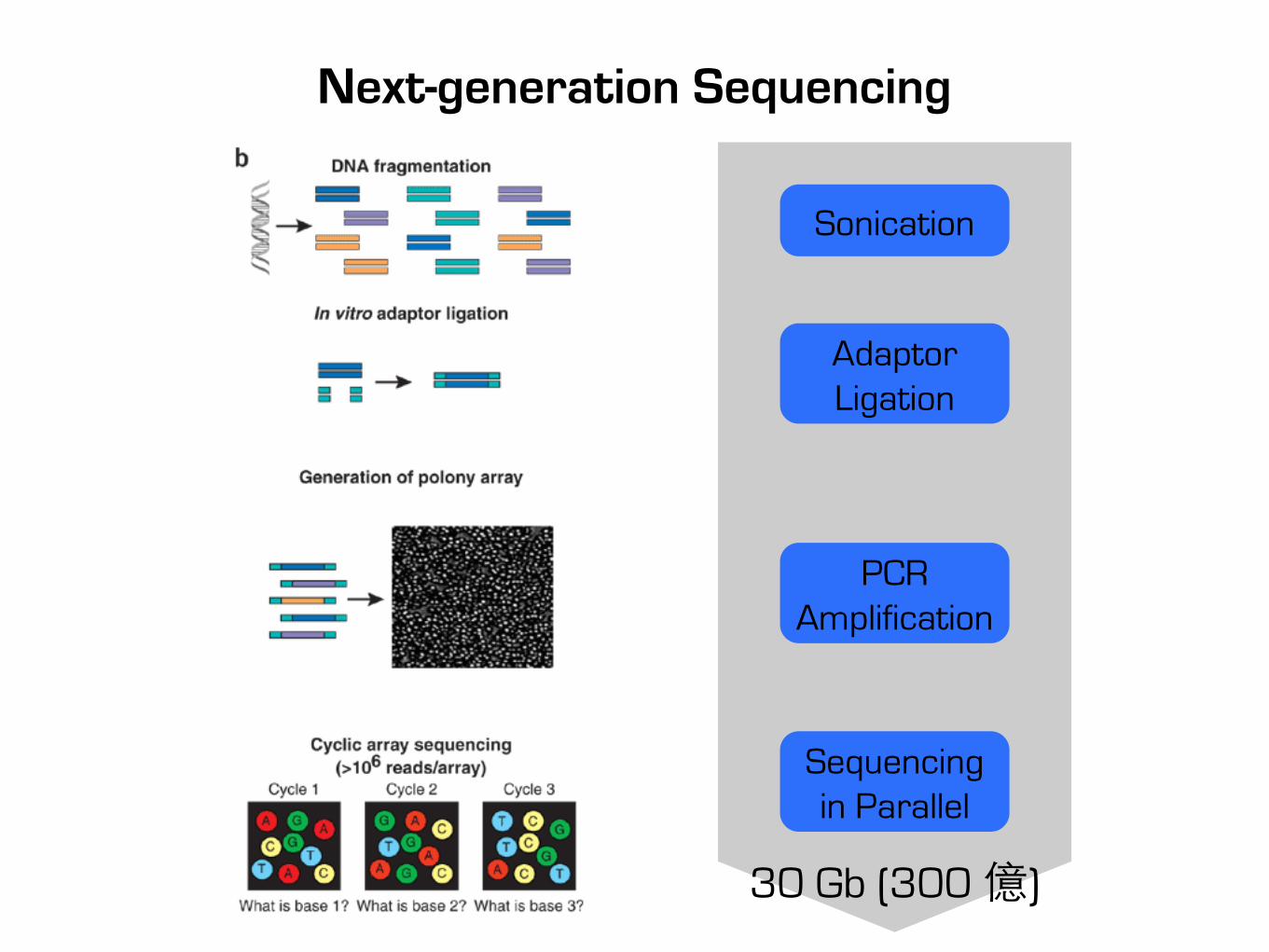
Next-generation Sequencing
30 Gb (300 億)
Sonication
Adaptor Ligation
PCR Amplification
Sequencing in Parallel
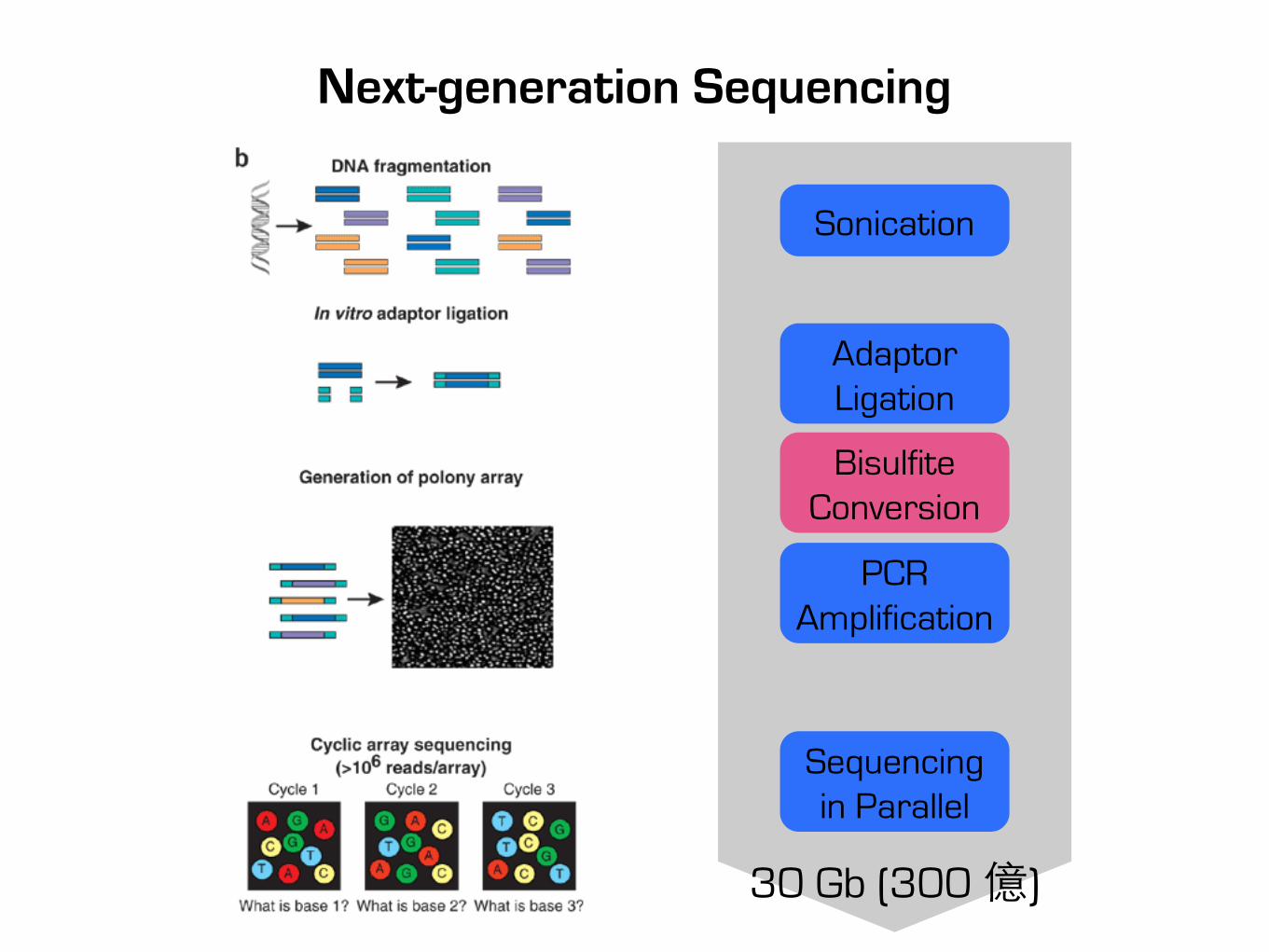
Next-generation Sequencing
30 Gb (300 億)
Sonication
Adaptor Ligation
PCR Amplification
Sequencing in Parallel
Bisulfite Conversion
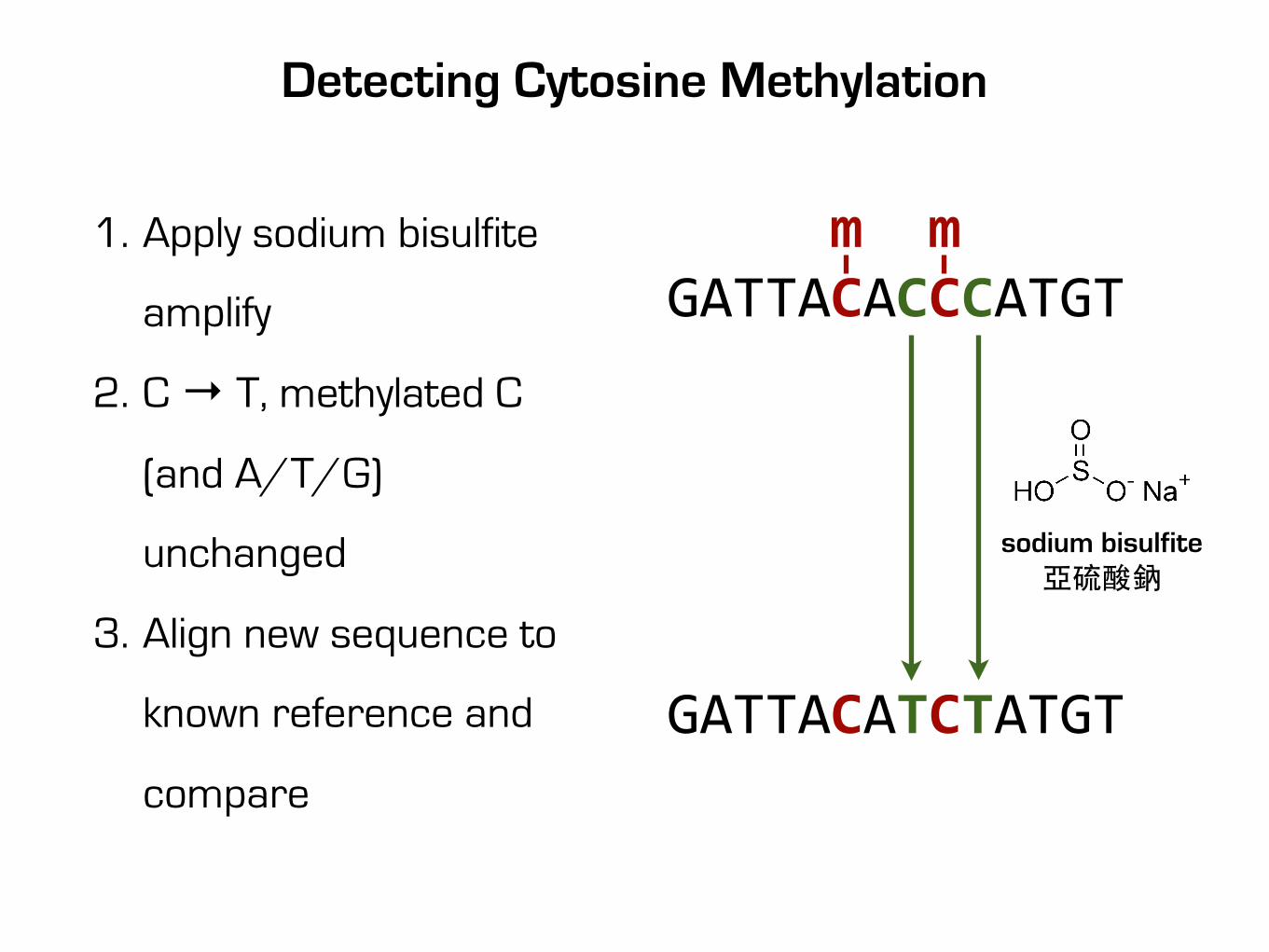
Detecting Cytosine Methylation
m mGATTACACCCATGT
GATTACATCTATGT
sodium bisulfite亞硫酸鈉
1. Apply sodium bisulfite
amplify
2. C → T, methylated C
(and A/T/G)
unchanged
3. Align new sequence to
known reference and
compare

Analysis Workflow
Sequence Mapping
MethylationCalling
Statistics&
Plotting
BS Seeker* mapping of bisulfite-treated reads
Biopython*parse bioinformatics files into Python utilizable data structures
PyTables*manage hierarchical datasets (HDF5) and design to efficiently cope with extremely large amounts of data
PysamPython wrapper of SAMtools C-APIread and manipulate SAM files
rePython built-in module for regular expression
NumPy&
SciPy
matrix operations, statistics, clustering, and more
Matplotlib data visualization
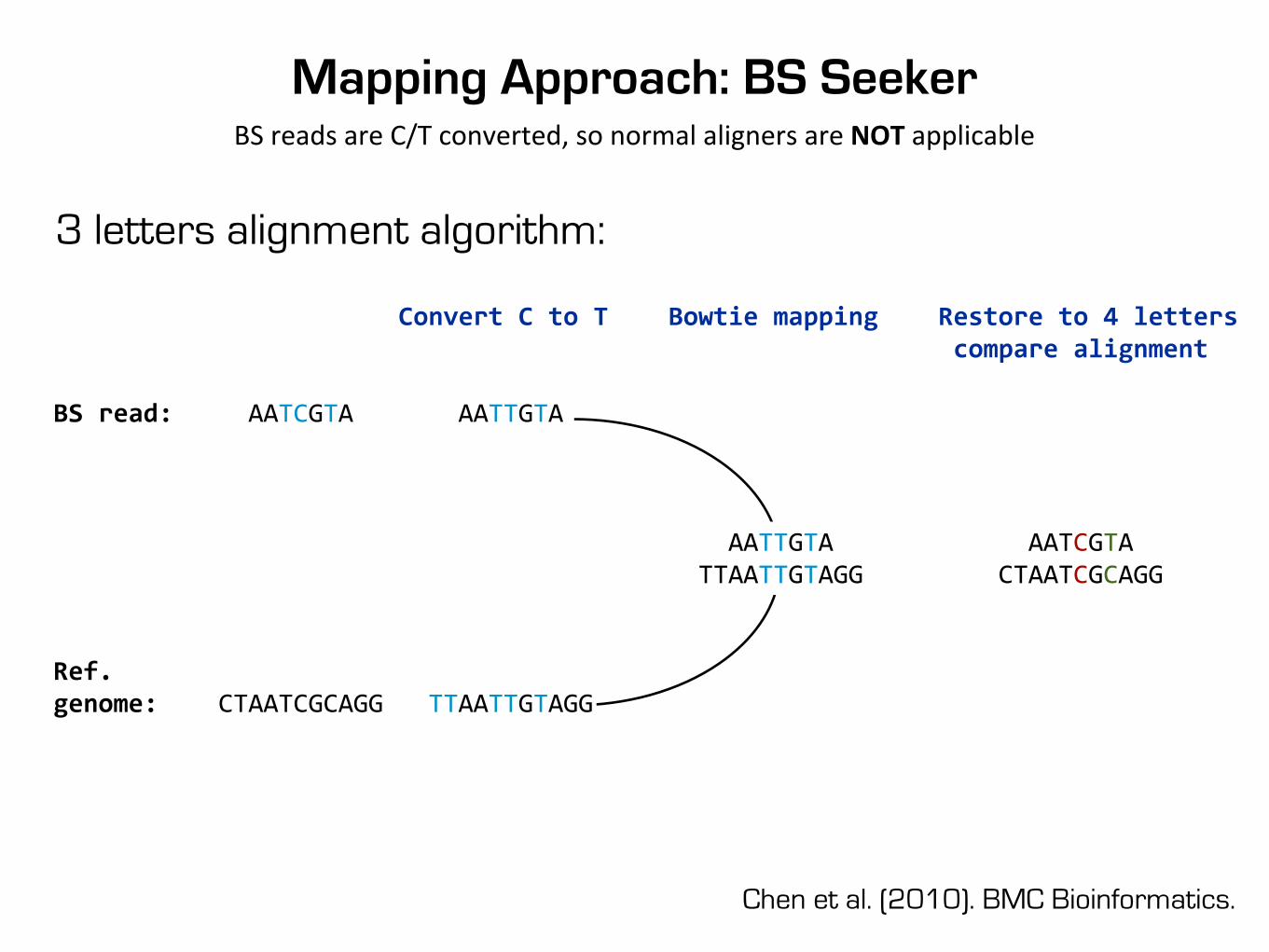
Mapping Approach: BS SeekerBS reads are C/T converted, so normal aligners are NOT applicable
3 letters alignment algorithm:
Chen et al. (2010). BMC Bioinformatics.
Convert C to T Bowtie mapping Restore to 4 letters compare alignment
BS read: AATCGTA AATTGTA
AATTGTA AATCGTA TTAATTGTAGG CTAATCGCAGG
Ref.genome: CTAATCGCAGG TTAATTGTAGG
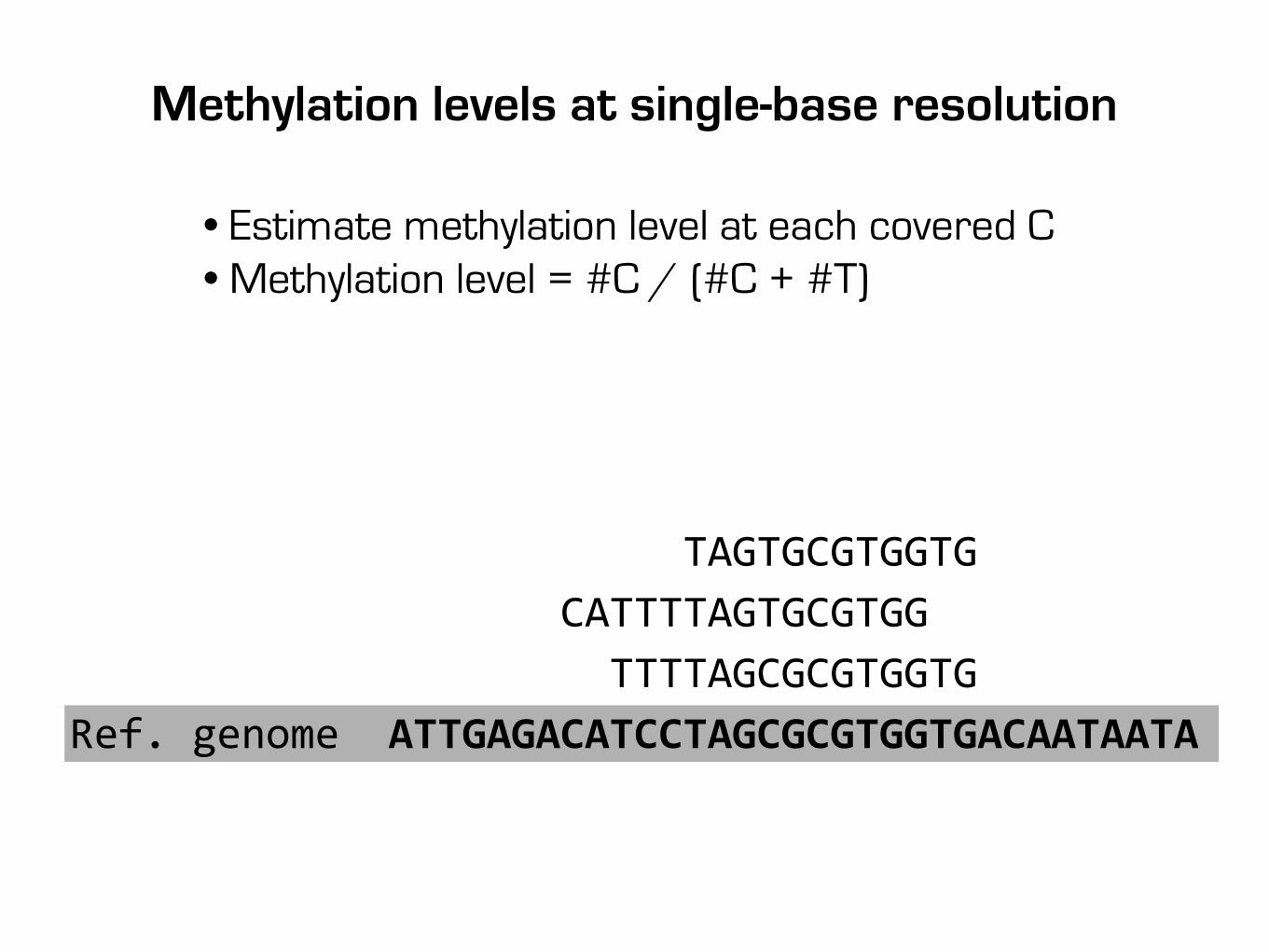
TAGTGCGTGGTG CATTTTAGTGCGTGG TTTTAGCGCGTGGTGRef. genome ATTGAGACATCCTAGCGCGTGGTGACAATAATA
Methylation levels at single-base resolution
• Estimate methylation level at each covered C• Methylation level = #C / (#C + #T)

TAGTGCGTGGTG CATTTTAGTGCGTGG TTTTAGCGCGTGGTGRef. genome ATTGAGACATCCTAGCGCGTGGTGACAATAATA
Methylation levels at single-base resolution
• Estimate methylation level at each covered C• Methylation level = #C / (#C + #T)

TAGTGCGTGGTG CATTTTAGTGCGTGG TTTTAGCGCGTGGTGRef. genome ATTGAGACATCCTAGCGCGTGGTGACAATAATA
Methylation levels at single-base resolution
• Estimate methylation level at each covered C• Methylation level = #C / (#C + #T)
1 / (1 + 2) = 33.3%

TAGTGCGTGGTG CATTTTAGTGCGTGG TTTTAGCGCGTGGTGRef. genome ATTGAGACATCCTAGCGCGTGGTGACAATAATA
Methylation levels at single-base resolution
• Estimate methylation level at each covered C• Methylation level = #C / (#C + #T)
1 / (1 + 2) = 33.3%

TAGTGCGTGGTG CATTTTAGTGCGTGG TTTTAGCGCGTGGTGRef. genome ATTGAGACATCCTAGCGCGTGGTGACAATAATA
Methylation levels at single-base resolution
• Estimate methylation level at each covered C• Methylation level = #C / (#C + #T)
1 / (1 + 2) = 33.3%
3 / (3 + 0) = 100%

Biopython
from Bio import SeqIO
# load all the records into memory at oncewith open('some.fasta') as infile: seq = SeqIO.to_dict(SeqIO.parse(infile, 'fasta'))
# indexing approach for very large file# provide dictionary-like access to any recordseq = SeqIO.index('some.fasta', 'fasta')
>chr1TTTAATTATCTCTGAAATTTAAACCCCCAAATCCAGGTAATAAAGCAAGGAAATGTCTTACAGCCCAACACTTGCCATCAATACTTTTTCGATGTTA...>chr2GGCTGCTCTATCCTTTTCTGCACATTTGAACTCCTCCGCTGTGGGCCATTCTCATTTGCTTTACTTCCTAGTCTGAATTCCATGGGAACTGCATTTA...
FASTA file
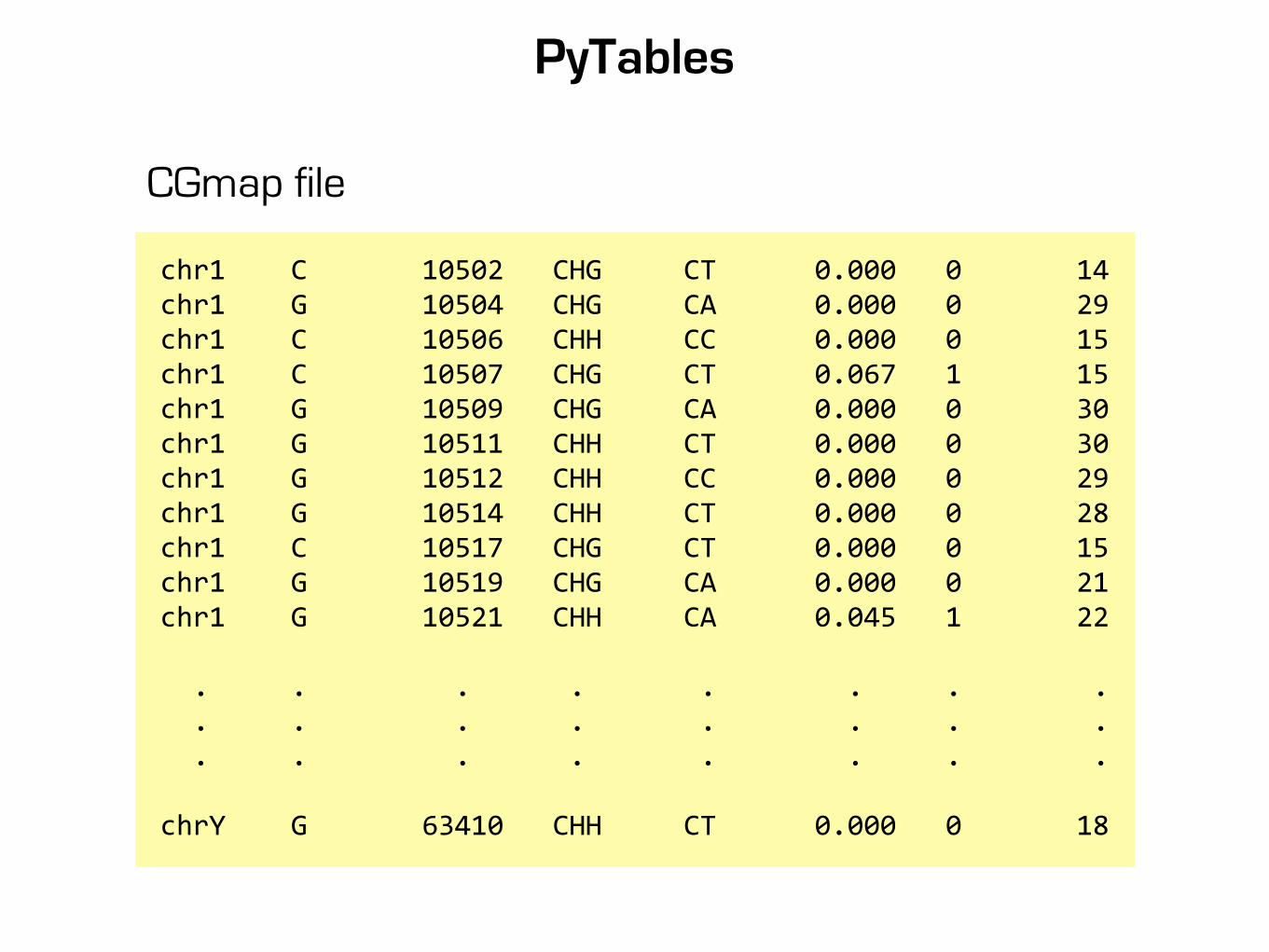
PyTables
chr1 C 10502 CHG CT 0.000 0 14chr1 G 10504 CHG CA 0.000 0 29chr1 C 10506 CHH CC 0.000 0 15chr1 C 10507 CHG CT 0.067 1 15chr1 G 10509 CHG CA 0.000 0 30chr1 G 10511 CHH CT 0.000 0 30chr1 G 10512 CHH CC 0.000 0 29chr1 G 10514 CHH CT 0.000 0 28chr1 C 10517 CHG CT 0.000 0 15chr1 G 10519 CHG CA 0.000 0 21chr1 G 10521 CHH CA 0.045 1 22
. . . . . . . . . . . . . . . . . . . . . . . .
chrY G 63410 CHH CT 0.000 0 18
CGmap file
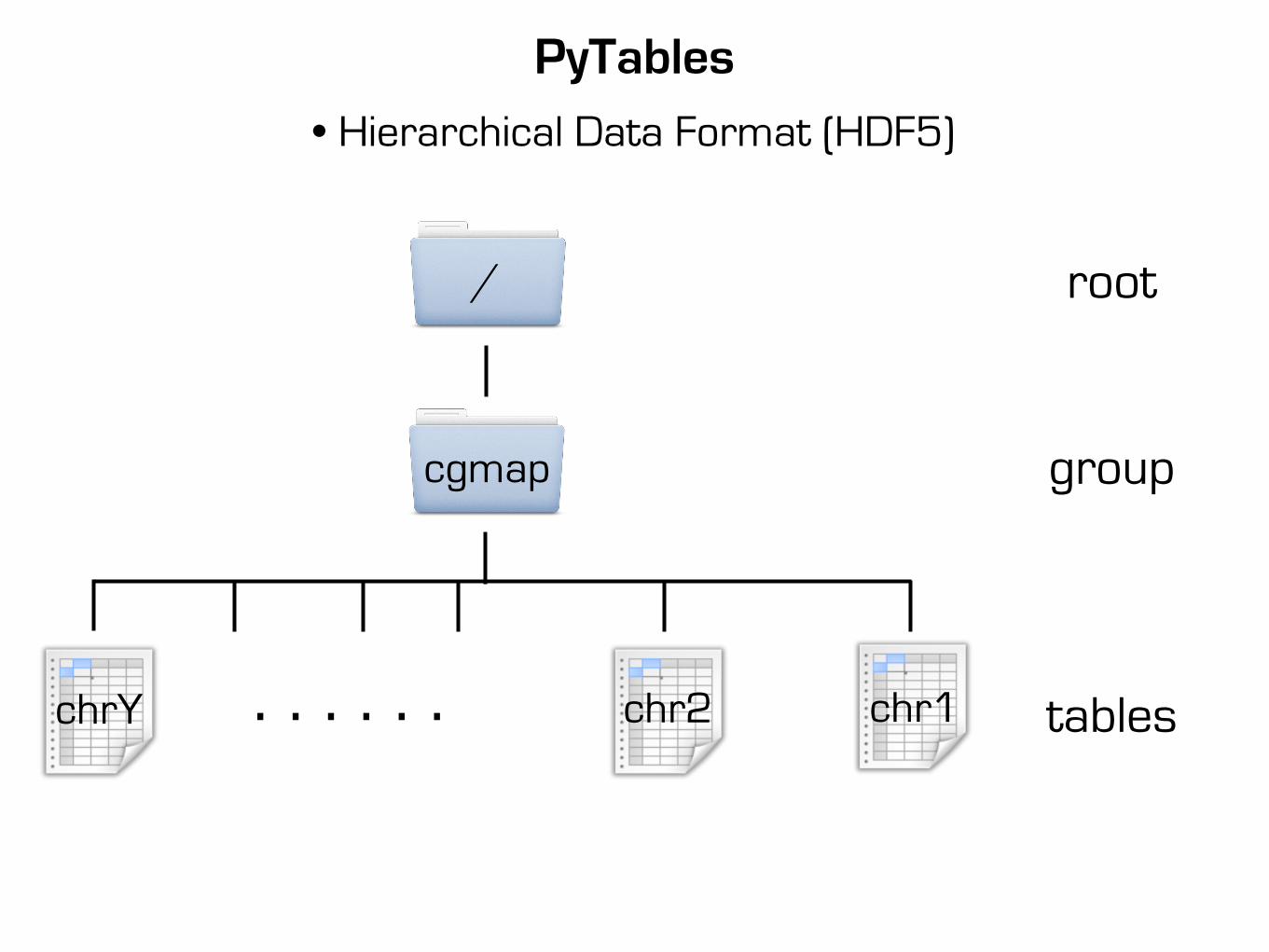
PyTables
root
group
tables
/
. . . . . .
cgmap
chr1chr2chrY
• Hierarchical Data Format (HDF5)

PyTables
import tables
class MethylSite(tables.IsDescription): strand = tables.StringCol(1) position = tables.Int64Col() context = tables.StringCol(3) dint = tables.StringCol(2) level = tables.Float32Col() mdepth = tables.Int32Col() depth = tables.Int32Col()

with open(args.cgmap) as cgmap: root = os.path.splitext(os.path.basename(args.cgmap))[0] with tables.openFile('{0}.h5'.format(root), 'w', root) as h5file: group = h5file.createGroup('/', 'cgmap') for line in cgmap: line = line.strip().split() try: table = group._f_getChild(line[0]) except tables.NoSuchNodeError: table = h5file.createTable(group, line[0], MethylSite) methylsite = table.row methylsite['strand'] = line[1] methylsite['position'] = int(line[2]) methylsite['context'] = line[3] methylsite['level'] = float(line[5]) methylsite['mdepth'] = int(line[6]) methylsite['depth'] = int(line[7]) methylsite.append() table.flush()
PyTables

with open(args.cgmap) as cgmap: root = os.path.splitext(os.path.basename(args.cgmap))[0] with tables.openFile('{0}.h5'.format(root), 'w', root) as h5file: group = h5file.createGroup('/', 'cgmap') for line in cgmap: line = line.strip().split() try: table = group._f_getChild(line[0]) except tables.NoSuchNodeError: table = h5file.createTable(group, line[0], MethylSite) methylsite = table.row methylsite['strand'] = line[1] methylsite['position'] = int(line[2]) methylsite['context'] = line[3] methylsite['level'] = float(line[5]) methylsite['mdepth'] = int(line[6]) methylsite['depth'] = int(line[7]) methylsite.append() table.flush()
PyTables

with open(args.cgmap) as cgmap: root = os.path.splitext(os.path.basename(args.cgmap))[0] with tables.openFile('{0}.h5'.format(root), 'w', root) as h5file: group = h5file.createGroup('/', 'cgmap') for line in cgmap: line = line.strip().split() try: table = group._f_getChild(line[0]) except tables.NoSuchNodeError: table = h5file.createTable(group, line[0], MethylSite) methylsite = table.row methylsite['strand'] = line[1] methylsite['position'] = int(line[2]) methylsite['context'] = line[3] methylsite['level'] = float(line[5]) methylsite['mdepth'] = int(line[6]) methylsite['depth'] = int(line[7]) methylsite.append() table.flush()
PyTables

with open(args.cgmap) as cgmap: root = os.path.splitext(os.path.basename(args.cgmap))[0] with tables.openFile('{0}.h5'.format(root), 'w', root) as h5file: group = h5file.createGroup('/', 'cgmap') for line in cgmap: line = line.strip().split() try: table = group._f_getChild(line[0]) except tables.NoSuchNodeError: table = h5file.createTable(group, line[0], MethylSite) methylsite = table.row methylsite['strand'] = line[1] methylsite['position'] = int(line[2]) methylsite['context'] = line[3] methylsite['level'] = float(line[5]) methylsite['mdepth'] = int(line[6]) methylsite['depth'] = int(line[7]) methylsite.append() table.flush()
PyTables

with open(args.cgmap) as cgmap: root = os.path.splitext(os.path.basename(args.cgmap))[0] with tables.openFile('{0}.h5'.format(root), 'w', root) as h5file: group = h5file.createGroup('/', 'cgmap') for line in cgmap: line = line.strip().split() try: table = group._f_getChild(line[0]) except tables.NoSuchNodeError: table = h5file.createTable(group, line[0], MethylSite) methylsite = table.row methylsite['strand'] = line[1] methylsite['position'] = int(line[2]) methylsite['context'] = line[3] methylsite['level'] = float(line[5]) methylsite['mdepth'] = int(line[6]) methylsite['depth'] = int(line[7]) methylsite.append() table.flush()
PyTables

with open(args.cgmap) as cgmap: root = os.path.splitext(os.path.basename(args.cgmap))[0] with tables.openFile('{0}.h5'.format(root), 'w', root) as h5file: group = h5file.createGroup('/', 'cgmap') for line in cgmap: line = line.strip().split() try: table = group._f_getChild(line[0]) except tables.NoSuchNodeError: table = h5file.createTable(group, line[0], MethylSite) methylsite = table.row methylsite['strand'] = line[1] methylsite['position'] = int(line[2]) methylsite['context'] = line[3] methylsite['level'] = float(line[5]) methylsite['mdepth'] = int(line[6]) methylsite['depth'] = int(line[7]) methylsite.append() table.flush()
PyTables

PyTables
with tables.openFile(args.h5file, 'a') as h5file: # indexed mode. Indexing is just a kind of sorting operation # over a column, so that searches along such a column will look # at this sorted information by using a binary search table = h5file.root.cgmap.chr1 table.cols.position.createIndex() table.cols.level.createIndex() table.cols.depth.createIndex()
# in-kernel mode, the condition is passed to the PyTables kernel, # written in C, and evaluated there at full C speed condition = "(depth >= 4) & (level >= 0.05)" res = [row['position'] for row in table.where(condition)]
condition = "(position > 1000) & (position < 5000)" res = [row['position'] for row in table.where(condition)]

“So if you're a CS major and you want to start a startup, instead of taking a class on entrepreneurship you're better off taking a class on, say, genetics. Or better still, go work for a biotech company. CS majors normally get summer jobs at computer hardware or software companies. But if you want to find startup ideas, you might do better to get a summer job in some unrelated field.
ー How to Get Startup Ideas, Paul Graham

join us :)


















