
10 Smc
26
Síndrome Síndrome Mieloproliferativo Mieloproliferativo Crónico Crónico Blanca Acosta - UABC 1
Transcript of 10 Smc

Síndrome Síndrome Mieloproliferativo Mieloproliferativo CrónicoCrónico
Blanca Acosta - UABC1

SMPSMP Padecimientos caracterizados colectivamente
por mieloproliferación monoclonal que:
◦ Involucra linajes múltiples◦ Conserva un grado de maduración celular◦ Tiene el potencial para seguir una evolución clonal
2
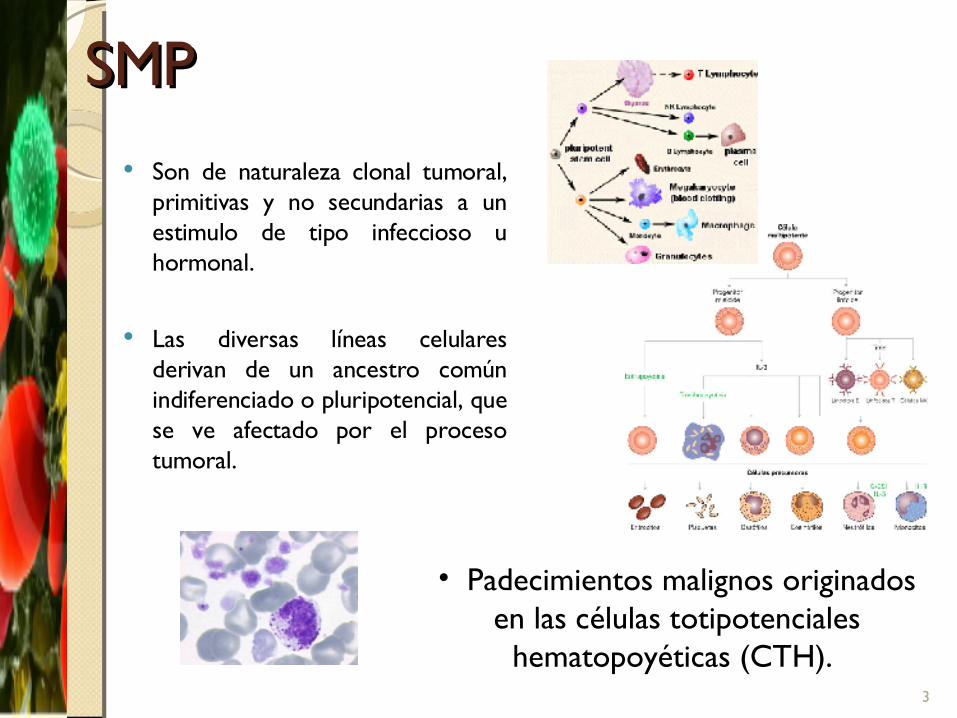
SMPSMP Son de naturaleza clonal tumoral,
primitivas y no secundarias a un estimulo de tipo infeccioso u hormonal.
Las diversas líneas celulares derivan de un ancestro común indiferenciado o pluripotencial, que se ve afectado por el proceso tumoral.
• Padecimientos malignos originados en las células totipotenciales
hematopoyéticas (CTH). 3
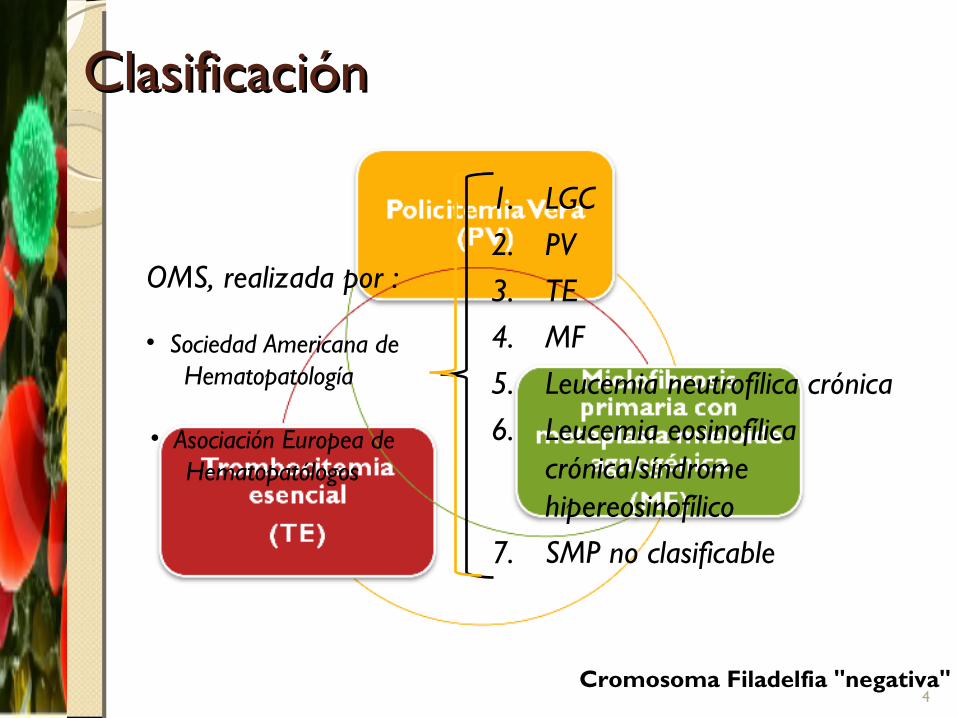
ClasificaciónClasificación
1. LGC2. PV3. TE4. MF5. Leucemia neutrofílica crónica6. Leucemia eosinofílica
crónica/síndrome hipereosinofílico
7. SMP no clasificable
OMS, realizada por :
• Sociedad Americana de Hematopatología
• Asociación Europea de Hematopatólogos
Cromosoma Filadelfia "negativa"4
PolicitemiaPolicitemia
VeraVera5

Policitemia VeraPolicitemia Vera Enfermedad clonal originada en CTH
Inicio insidioso
Curso crónico
Causa desconocida
MO hiperplásica, no secundaria a ningún estimulo medular.
25% con anormalidad cromosómica reproducible.
Anormalidad en las células de las líneas hematopoyéticas mayores, pero no en los tejidos no hematopoyéticos.
Proliferación excesiva en MO
de células:
Eritroides :
Aumento de volumen eritrocitario (VE)
Megacariocíticas
Granulocítica
6
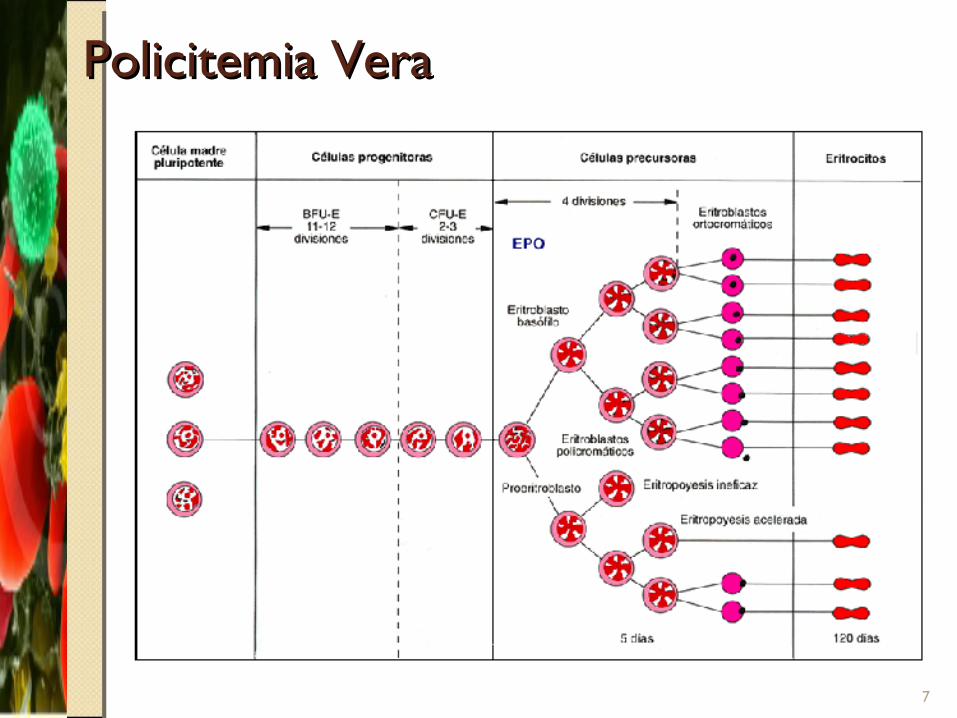
Policitemia VeraPolicitemia Vera
7

PatogénesisPatogénesis
Hiperplasia de precursores de glóbulos rojos, granulocitos y plaquetas
Producción excesiva
Aumento en numero en sangre periférica
Volumen eritrocítico aumentado (VE)
Mecanismos:◦ Proliferación neoplásica sin control◦ Presencia de factor proliferativo anormal que actúa sobre las células tronco normales◦ Sensibilidad aumentada de las células tronco a eritropoyetina.
80.037% P en M

Datos ClínicosDatos Clínicos
Atribuibles a:◦ Aumento del espacio vascular y
volumen sanguíneo (hipervolemia)
◦ Lentitud del flujo sanguíneo
◦ Hiperviscosidad
◦ Hipoxia tisular
Edad: 40-70 años 50 Hombres Hematocrito elevado Iniciados por procesos trombóticos o hemorragias
9

Datos ClínicosDatos Clínicos Comunes◦ Síntomas cerebrales◦ Síntomas cardiovasculares◦ Cara y ojos enrojecidos◦ Debilidad, laxitud y cansancio◦ Hallazgo accidental de un examen de rutina◦ Síntomas gastrointestinales◦ Alteraciones visuales◦ Complicaciones trombóticas◦ Manifestaciones hemorrágicas◦ Enfermedad vascular periférica◦ Esplenomegalia
Ocasionales◦ Prurito◦ Gota◦ Manifestaciones psiquiátricas
Cefalea: Leve o moderada, frecuente u ocasional, frontal u occipital, es posible que se intensifique al acostarse o levantarse
Mareos Sincope Pérdida de memoria Incapacidad para
concentrarse Irritabilidad Procesos vasculares
cerebrales
Plenitud Sed Meteorismo Estreñimiento Ulcera péptica Hemorragia Trombosis Ulcera duodenal (16%) Ulcera gástrica (7%)
10

Datos de laboratorioDatos de laboratorio Aumento del VE
Leucocitosis 12,000-20,000
Metamielocitos y mielocitos
Fosfatasa alcalina aumentada
Plaquetas 3000-6000/109
Anormalidades cromosómicas: (Supresión del brazo largo del cromosoma 5 (5q-),
ganancia de un 8 o 9 y supresión del brazo largo del 20)
Vitamina B12 y la capacidad de fijación de la misma están aumentadas no controlados
En MO: las tres series están hiperplasias y reemplazan la grasa medular
11
Mayores Menores
Volumen Eritrocitario
Varón: mas de 36mL/kg Trombocitosis: mas de 400/103
Mujer: mas de 32mL/kg Leucocitosis: mas de 12/103
Saturación arterial O2 Fosfatasa alcalina de los
leucocitos >100
Igual o mayor de 92% B12 sérica mayor de lo normal y
Esplenomegalia CLFB12 mayor de lo normal

TratamientoTratamiento Flebotomía◦ 465ml cada tercer día, hasta obtener hematocrito normal
Radiación Quimioterapia◦ Agentes mielosupresores Hidroxiurea
12

13

14
TrombocitemiaTrombocitemia EsencialEsencial

Trombocitemia esencialTrombocitemia esencial
Trombocitemia primaria Hemorrágica Idiopática.
Alteración de tipo Clonal
Aumento de Megacariocitos ◦ Volumen total y volumen medio
Aumento de plaquetas hasta 15 veces mayor de lo normal:◦ Hemorragia Anormalidad cuantitativa intrínseca de la función plaquetaria.◦ Trombosis Incremento del volumen plaquetario, junto con anormalidad
cualitativa (hiperagregables).
150.17% P en M

Datos ClínicosDatos Clínicos Edad : 50-70 años 61
Mujer
Hemorragia de intensidad variable
(espontanea o repetida).
Síntomas de isquemia del SNC y de la
vascularidad periférica.
Debilidad, mareo, cefalea.
Alteraciones visuales
Prurito
Eritromelalgia: Eritromelalgia:
◦ Dolor localizado tipo quemadura, enrojecimiento y aumento de temperatura en las porciones distales de las extremidades cianosis necrosis de dedos de manos y pies.
16

DiagnósticoDiagnóstico
17
1. Recuento plaquetario mayor de 600 000
2. Hemoglobina normal o VE normal
3. Presencia de hemosiderina en MO, o falta de respuesta a
tratamiento con hierro.
4. Ausencia de cromosoma Filadelfia5. Fibrosis de la colágena de la medula:
a. Ausente
b. Si existe, ha de ser menor a un tercio del área de biopsia y no deben existir esplenomegalia o reacción
leucoeritroblástica.
6. Sin causa de trombosis reactiva

TratamientoTratamiento
Pacientes con:◦ Bajo riesgo Aspirina
◦ Alto riesgo Agentes mielosupresores
Anagrelide
Esplenectomía contraindicada
18

19

20
Mielofibrosis primaria Mielofibrosis primaria con metaplasia con metaplasia
agnogénicaagnogénica

Mielofibrosis primaria Mielofibrosis primaria Hemopoyesis (células mieloides) extramedular
Asociada a fibrosis de la MO
Proliferación clonal de una célula tronco
mieloide multipotente anormal, pero restringida
Fibrosis reactiva a la proliferación clonal de
precursores granulocíticos y trombocíticos PDGF.
Mielodisplasia granulocitopenia y trombocitopenia
Anemia eritropoyesis ineficaz, supervivencia acortada y
efecto de la esplenomegalia (hemolisis).
21

Datos ClínicosDatos Clínicos Raza blanca Edad: 60 años En niñez, mas en mujeres.
Asintomática◦ Esplenomegalia
Sintomática◦ Fatiga, disnea, debilidad y palpitaciones◦ Perdida de peso, anorexia◦ Diaforesis nocturna◦ Fácil Equimosis ◦ Dolor óseo intenso, más en MI◦ Palidez
22

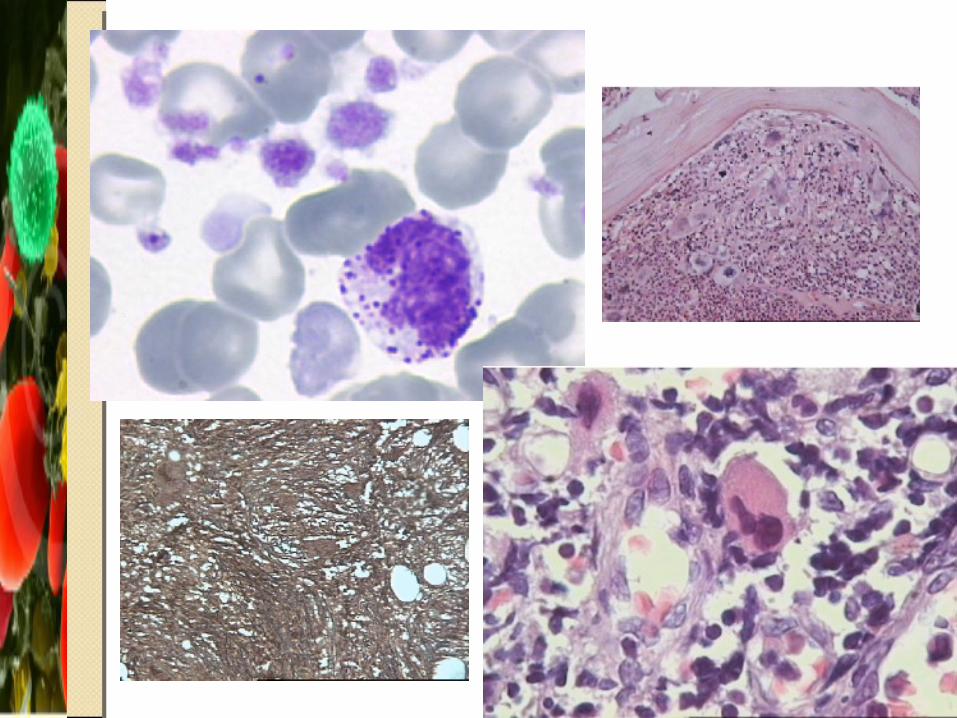
DiagnósticoDiagnóstico Eritrocitos en forma de lagrima y de normoblastos Células en diana Basofília difusa Reticulocitos aumentados Bilirrubina indirecta aumentada Leucocitos > 50,000 Anomalía adquirida de Pelger-Huet Plaquetas aumentadas, gigantes y granulación anormal Biopsia de MO Fibrosis Anormalidad Cromosómica
23
Criterios
1. Esplenomegalia
1. Leucoeritroblastosis2. Presencia acentuada de poiquilocitos en forma de lagrima o gota
en el extendido de la sangre periférica
4. Fibrosis en la biopsia de MO
TratamientoTratamiento Esplenectomía Clorhidrato de anagrelide◦ Disminuye cuentas de plaquetas
Anemia◦ Eritropoyetina humana
recombinante◦ Hemolítica prednisona
Hidroxiurea Trasplante de MO
24
25
26
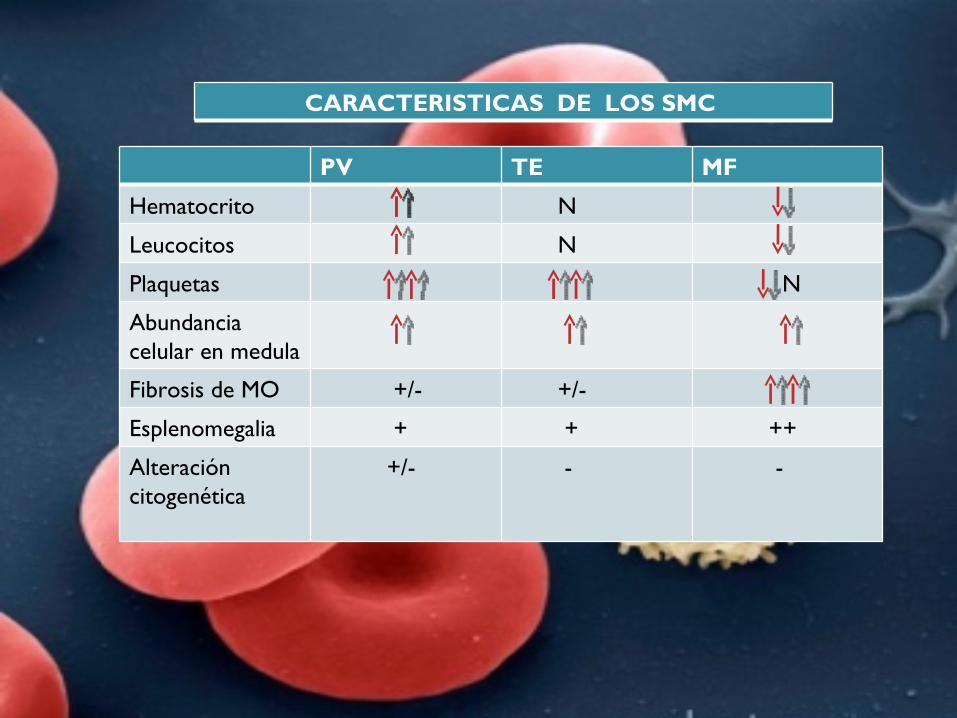
PV TE MF
Hematocrito N
Leucocitos N
Plaquetas N
Abundancia celular en medula
Fibrosis de MO +/- +/-
Esplenomegalia + + ++
Alteración citogenética
+/- - -
CARACTERISTICAS DE LOS SMC


















