
Interstitial Spaces: Micro-Interaction Settings and the Genesis of ...
Upload
alexia-westCategory
view
224download
0
ιδιοπαθής πνευμονική ίνωση
Interstitial Pulmonary Fibrosis / IPF
ευρήματα από την μοριακή ανάλυση και συσχετισμοί με τον παθογενετικό μηχανισμό της νόσου
γιωτάννα δαλαβάγκα
αναπληρώτριακαθηγήτρια
ανατομίας-ιστολογίας-εμβρυολογίας ιατρική σχολή
πανεπιστήμιο ιωαννίνων μάρτιος
2014yo

Difuse Inderstitial Fibrosisδιάχυτες ινώσεις του διαμέσαου ιστού
του πνεύμονα
*γνωστής αιτιολογίας πνευμονοκονιώσεις, αμιάντωση,
τελικο στέδιο χρονίων πνευμονοπαθειών...
**αγνώστου αιτιολογίαςιδιοπαθείς διάμεσες πνευμονιτιδες
Idiopathic Inderstitial Pneumonias
από αυτές η συχνότερη:
Ιδιοπαθής Πνευμονική Ίνωση
Idiopathic Pulmonary Fibrosis / IPF
yo

IPF / Idiopathic Pulmonary Fibrosis
a chronic, relentlessly progressive fibrotic disorder
of the lower respiratory tract that
typically affects adults over the age of 40
Idiopathic Pulmonary Fibrosis (IPF), also referred to as cryptogenic
fibrosing alveolitis, is a progressive interstitial lung
disease of unknown aetiology
yo

Idiopathic Pulmonary Fibrosis / IPF is the most common form of the idiopathic interstitial pneumonias (IIP) and is characterized by * progressive dyspnoea, * interstitial infiltrates on radiologic imaging, * restriction on pulmonary function testing. a disease with a dire prognosis, the outcome in most patients with IPF is gradual progression with respiratory failure in 3-5 years unfortunately, short of lung transplantation, no clearly effective therapy exists.
perhaps, one of the main reasons for the continued lack of an effective therapy is that the cause of IPF and many aspects of its pathogenesis remain unknown.
[Am J Med Sci. 2011 June ; 341(6): 439]
yo

* about 5 million people are affected worldwide ** insidence : 4.6 - 7.4 cases per 100.000 persons
***the incidence continues to rise andIPF is now an important cause of respiratory
mortality
the median survival time is only 2.5 to 3.5 years, but
individual survival can vary from a few months to 10 years
in 0.5–19% of cases IPF is familial and is most likely caused by a single genetic mutation
yo
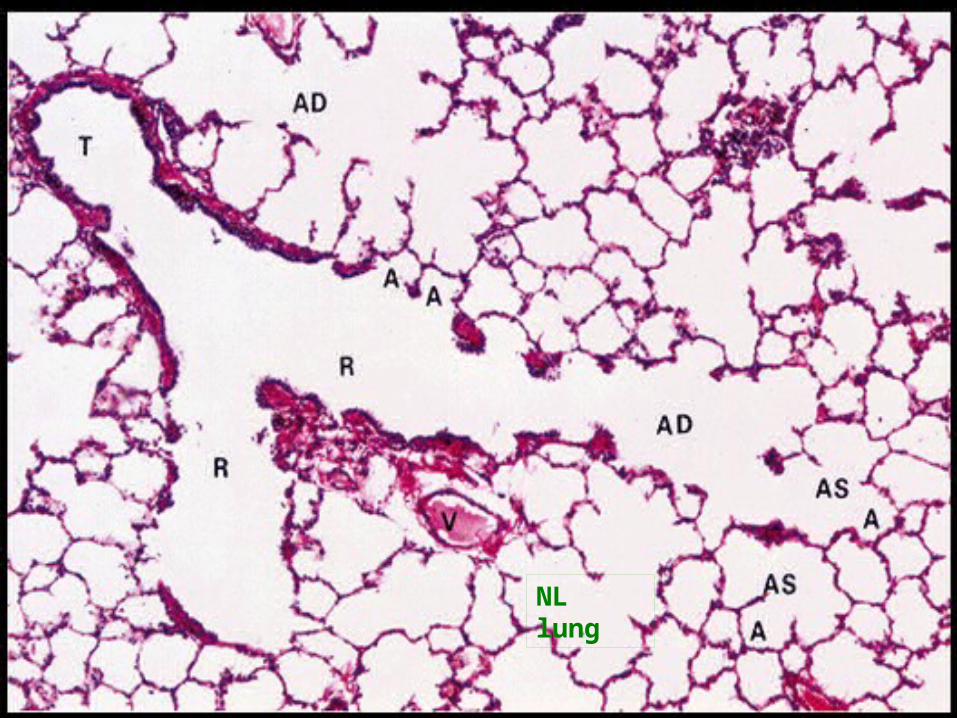
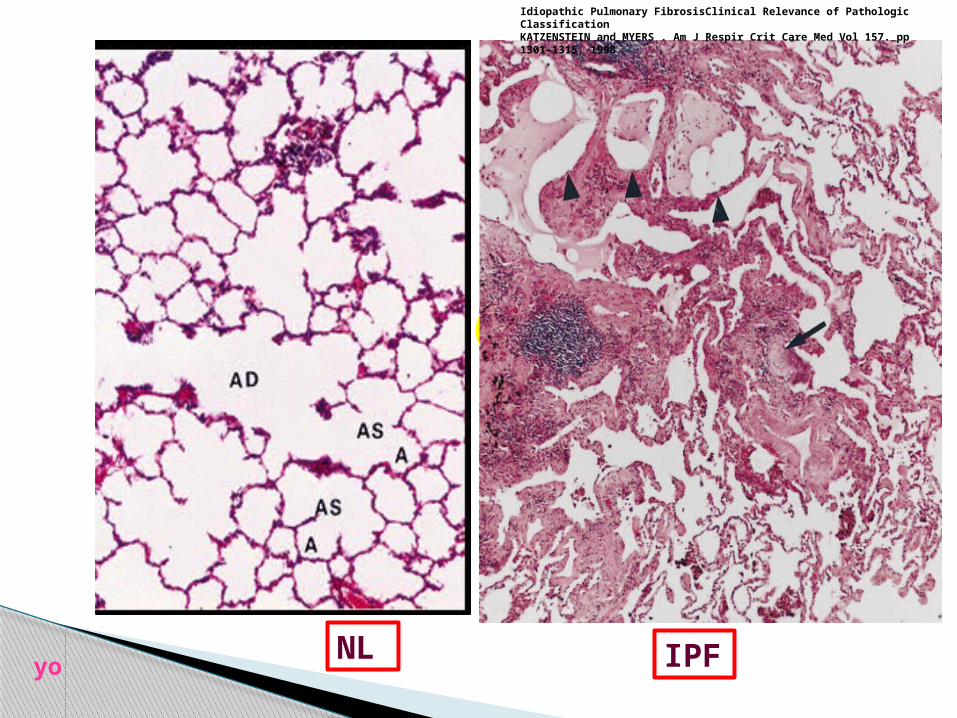
NL lung
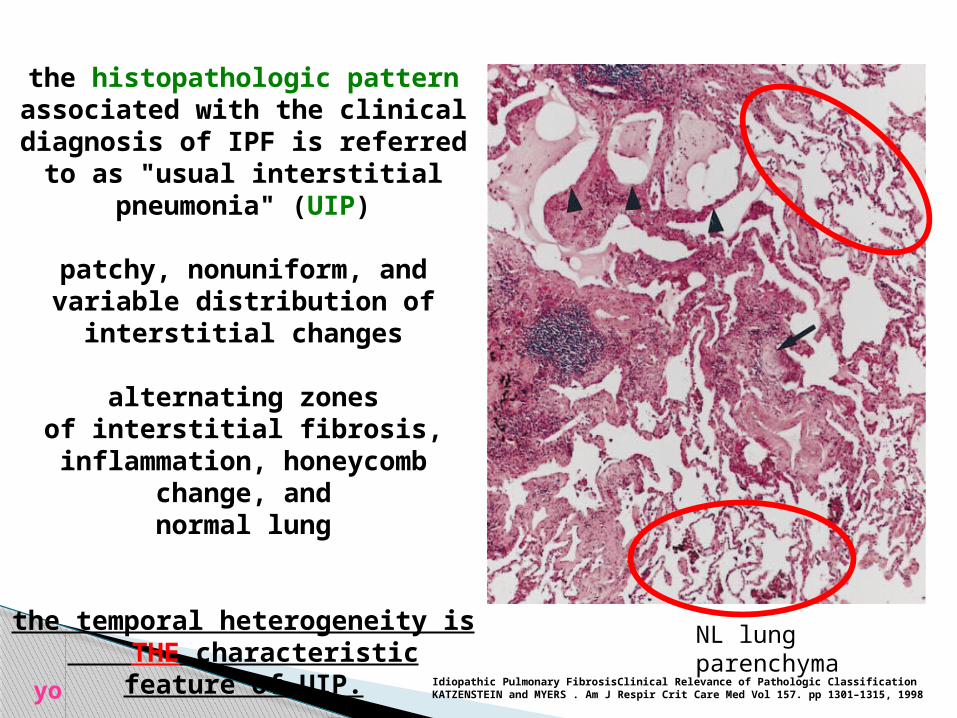
the histopathologic pattern associated with the clinical
diagnosis of IPF is referred to as "usual interstitial
pneumonia" (UIP)
patchy, nonuniform, and variable distribution of
interstitial changes
alternating zonesof interstitial fibrosis,
inflammation, honeycomb change, andnormal lung
the temporal heterogeneity is THE characteristic feature of
UIP.
NL lung parenchyma
Idiopathic Pulmonary FibrosisClinical Relevance of Pathologic ClassificationKATZENSTEIN and MYERS . Am J Respir Crit Care Med Vol 157. pp 1301–1315, 1998yo
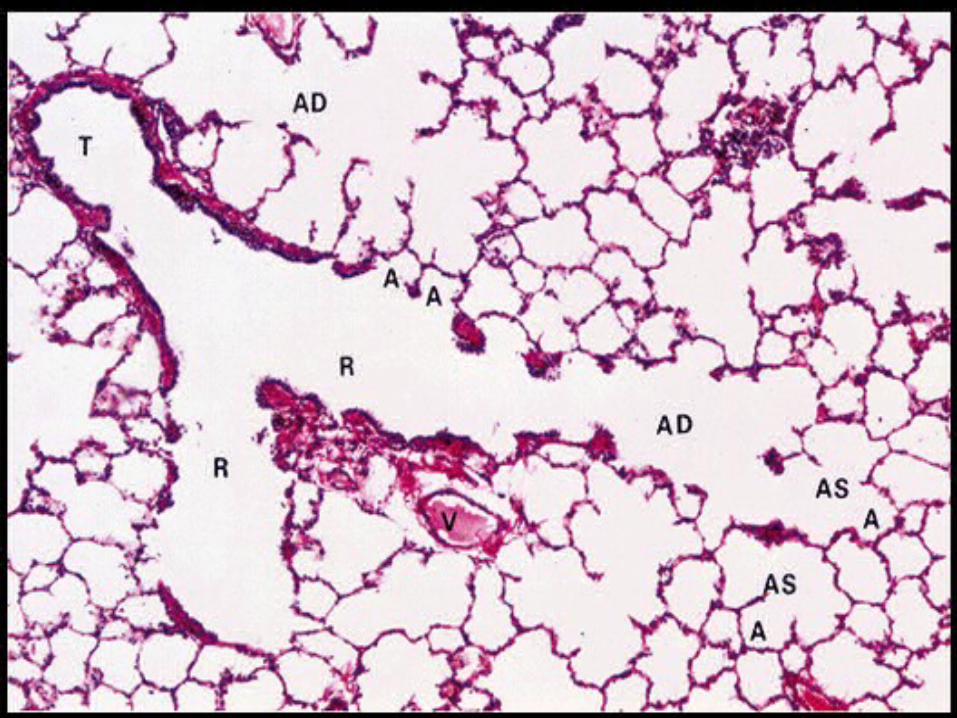

yo NL lung parenchyma / NL pleura
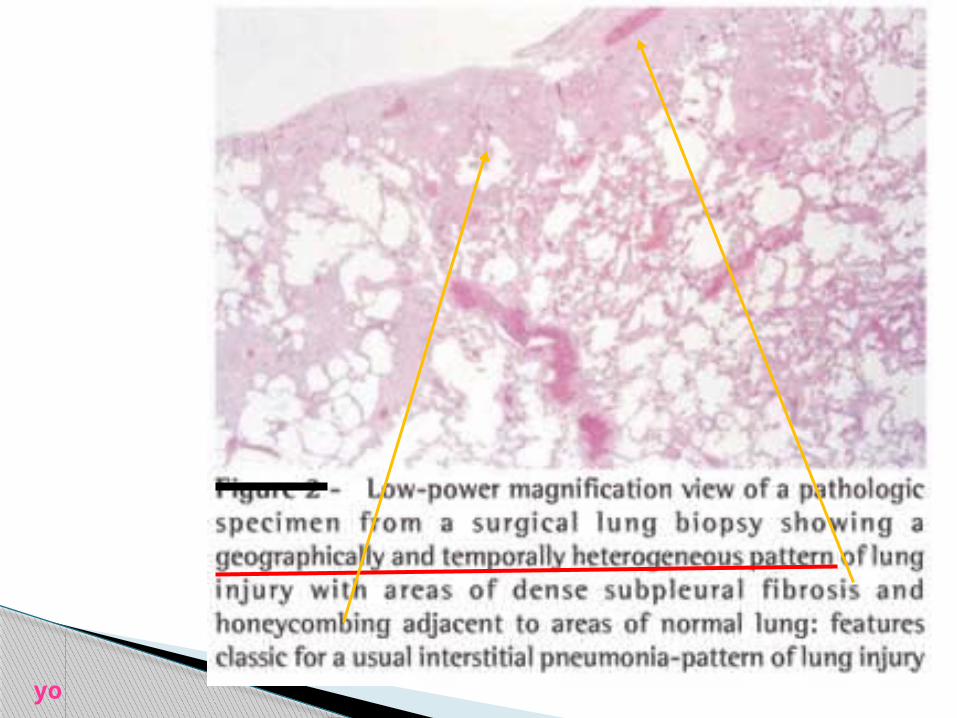
yo
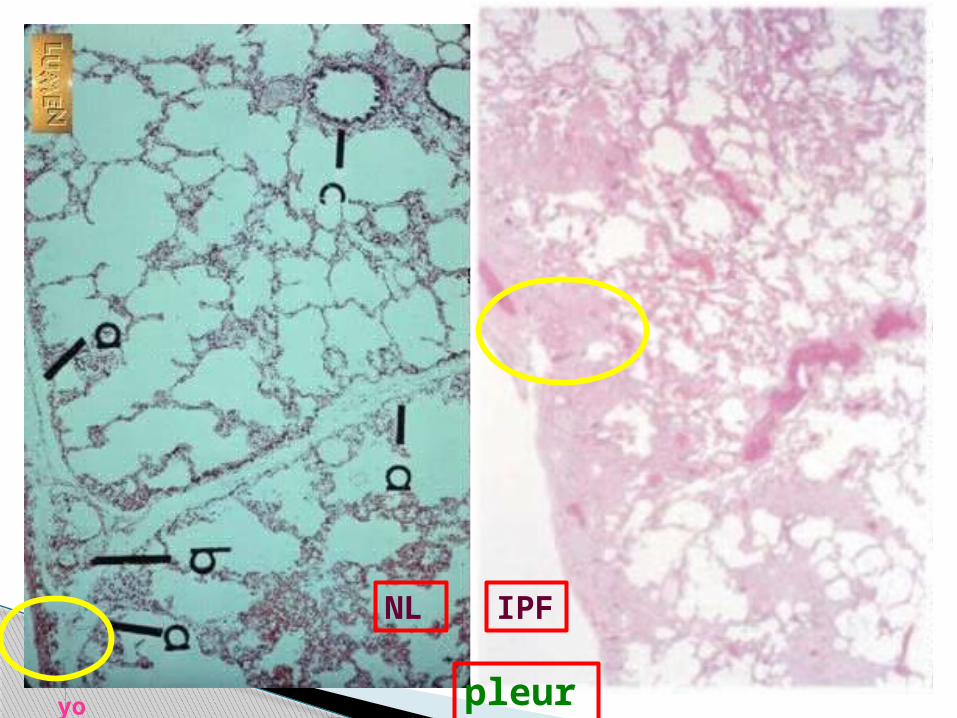
NL IPF
yo pleura
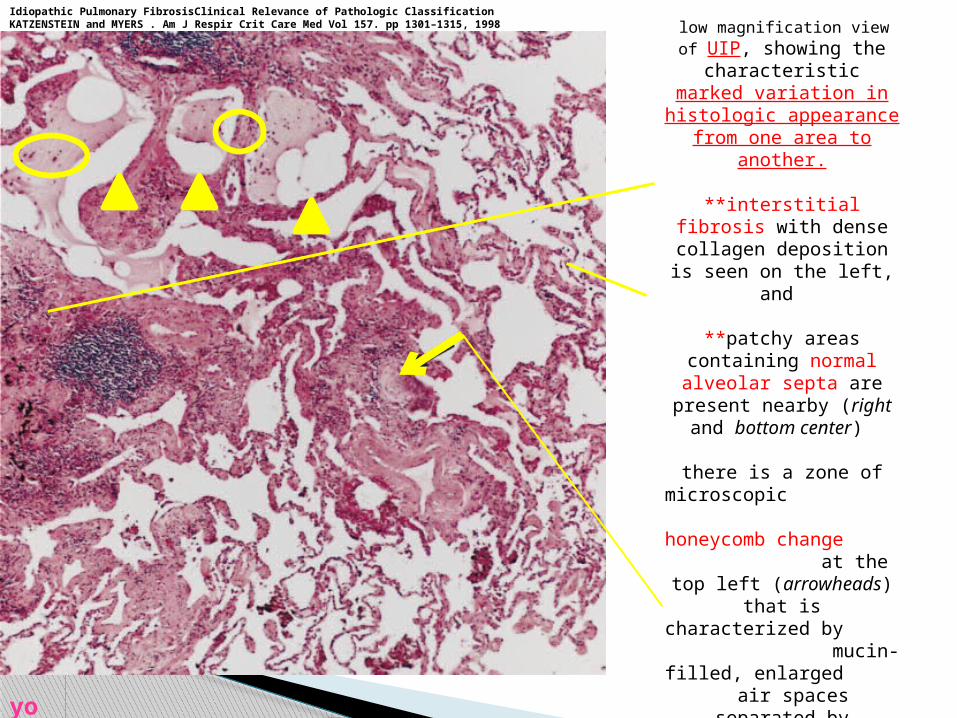
low magnification view of UIP, showing the
characteristicmarked variation in
histologic appearance from one area to
another.
**interstitial fibrosis with dense collagen
deposition is seen on the left, and
**patchy areas containing normal alveolar septa are
present nearby (right and bottom center)
there is a zone of microscopic honeycomb change at the top left
(arrowheads) that ischaracterized by
mucin-filled, enlarged air spaces separated by fibrosis.
**a fibroblast focus (arrow) is seen in an area of fibrosis and
inflammation in the center of the field.
yo
Idiopathic Pulmonary FibrosisClinical Relevance of Pathologic ClassificationKATZENSTEIN and MYERS . Am J Respir Crit Care Med Vol 157. pp 1301–1315, 1998
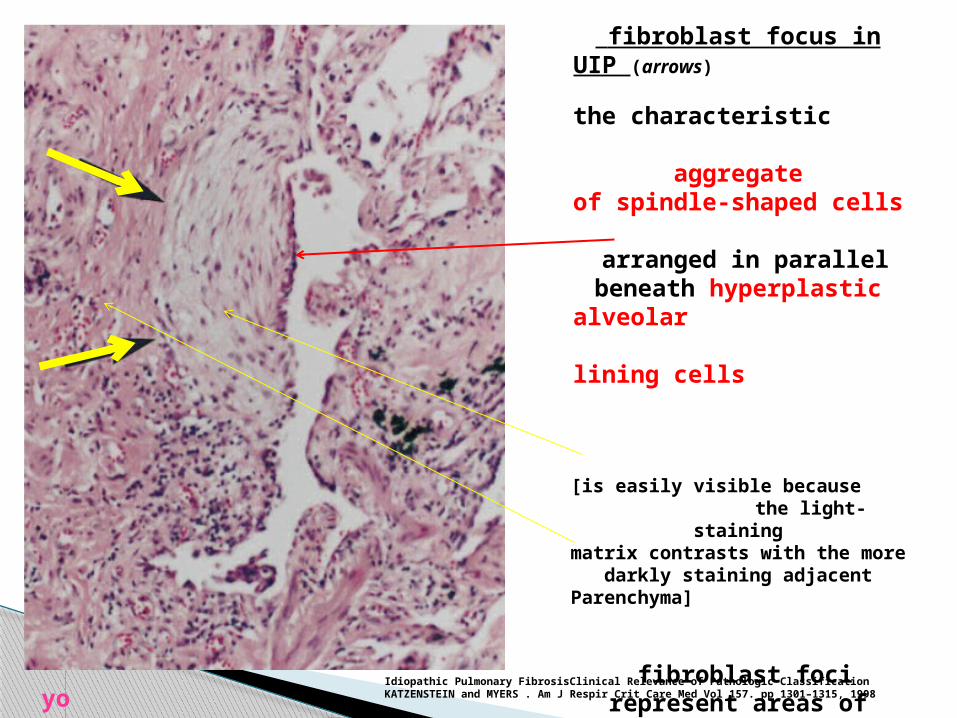
fibroblast focus in UIP (arrows)
the characteristic aggregateof spindle-shaped cells
arranged in parallel beneath hyperplastic
alveolar lining cells
[is easily visible because the light-staining
matrix contrasts with the more darkly staining adjacent
Parenchyma]
fibroblast foci represent areas of active fibrosis
thatcontrast with adjacent
areas of inactive collagen-type fibrosis
Idiopathic Pulmonary FibrosisClinical Relevance of Pathologic ClassificationKATZENSTEIN and MYERS . Am J Respir Crit Care Med Vol 157. pp 1301–1315, 1998yo
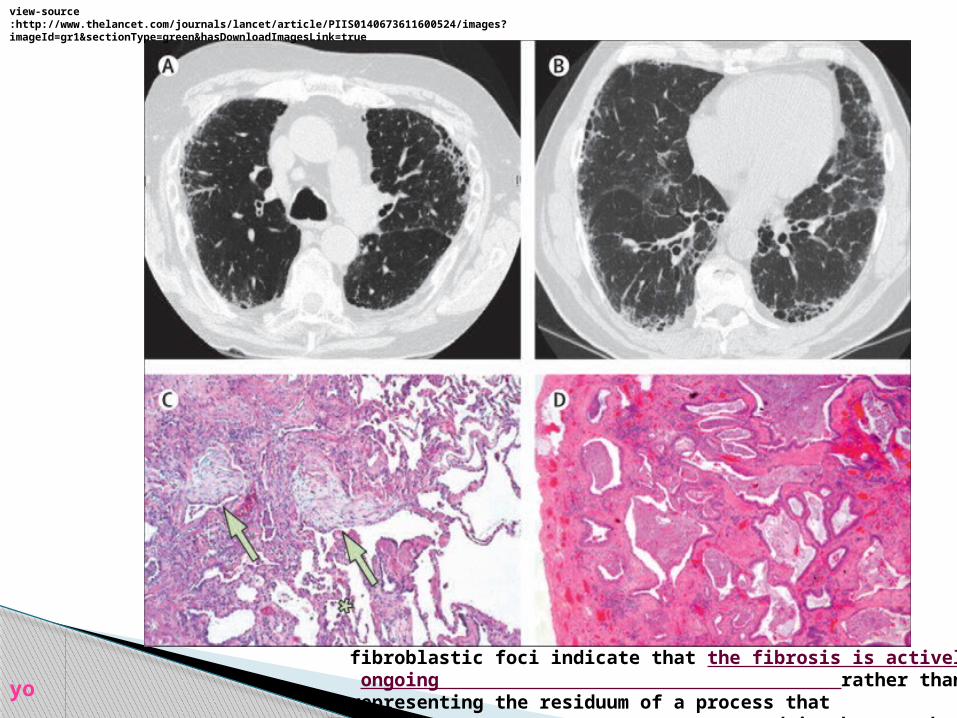
fibroblastic foci indicate that the fibrosis is actively ongoing rather than representing the
residuum of a process that occurred in the past but is now inactive
view-source:http://www.thelancet.com/journals/lancet/article/PIIS0140673611600524/images?imageId=gr1§ionType=green&hasDownloadImagesLink=true
yo

the characteristic histopathologic features of UIP in IPF -abnormal proliferation of mesenchymal cells, [fibroblasts/myofibroblasts] -varying degrees of fibrosis, -overproduction and disorganized deposition of *collagen and *extracellular matrix, -distortion of pulmonary architecture and -subpleural cystic airspaces (d=3-10 mm) called honeycomb cysts
fibroblast foci are clusters of fibroblasts and myofibroblasts that lie in continuity
with the established fibrosis and are a characteristic histologic feature of UIP
these abnormalities are typically associated with a mild, patchy, chronic inflammatory cell filtrate
yo
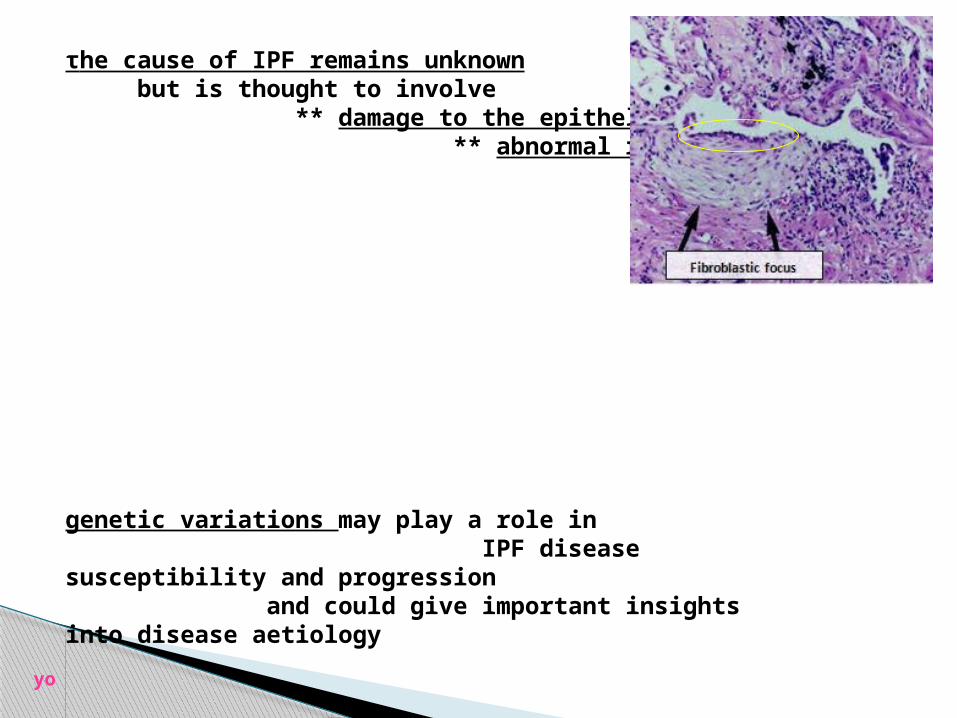
τhe cause of IPF remains unknown but is thought to involve ** damage to the epithelium and ** abnormal repair
genetic variations may play a role in IPF disease susceptibility and progression and could give important insights into disease aetiology
yo

facts (?) about IPF after ¬30 years of research
-cause ???-initiation ???? -extrinsic causative agent ????? [90 % of patients with IPF have physiologically detectable gastroesophageal reflux]
-genetic predisposition ----very probable -perpetuation unremitting fibrotic response why???-progression the fibrotic process is progressive why???? once initiated at the epithelial barrier of the alveoli, the injury to epithelial cells and basement membrane results in complex cell and cytokine interactions that extend the fibrotic process to involve the alveolar walls, alveolar lumen, and then adjacent areas of lung parenchyma.
-mechanism of fibrosis the mechanism of fibrosis in IPF remains elusive !!!!!/????-prognosis BAD !!!-treatment so far has been largely ineffective
yo

but
yo

IDEAS & BIOTECHNOLOGY HELP
yo

InitiationIt is possible that nonspecific injury to the epithelial barrier and pulmonary parenchyma, such as cigarette smoking, viral infection, environmental pollutants, chronic aspiration, genetic predisposition, and drugs, initiates the disease process of IPF in susceptible individuals.
?the stimulus that promotes pulmonary fibrosis instead of repair is unknown
yo Idiopathic Pulmonary Fibrosis: From Epithelial Injury to Biomarkers – Insights from the Bench SideKeren Borensztajn , Bruno Crestani , Martin Kolb. Respiration 2013;86:441–452
a

Genetic predisposition** Mutations in surfactant proteins A2 (SP-A2) and C (SFTPC)have been identified in several families with IPF **Mutations in the adenosine triphosphate binding cassette A3 (ABCA3) gene may affect the course of SFTPC-related IPF **A genome-wide linkage scan identified a risk locus for IPF on chromosome 11, and further fine mapping of that locus found a common variant in the promoter of the gene encoding mucin 5B (MUC5B) This variant was present in 34 % of subjects with familial pulmonary fibrosis, 38 % of those with IPF, and 9 % of controls. In a separate study, a MUC5B promotor single nucleotide polymorphism was significantly associated with IPF, but not lung fibrosis in sarcoidosis or systemic sclerosis MUC5B expression in lung tissue is 14 times higher in subjects with IPF, compared with unaffected subjects.
** Telomerase gene mutations, which cause shortened telomeres, have been identified in about 25 % of sporadic IPF and in about 15 % of families with familial pulmonary fibrosis.
yo

the “dogma” that lung inflammation is necessary and sufficient to produce fibrosis in IPF [ ‘80s ]
has been questioned for several reasons:
*inflammation is generally minimal in patients with IPF, and there is no evidence that patients with early disease have more prominent inflammatory changes *fibrosis can be induced in laboratory animals in the absence of inflammation
*anti-inflammatory therapy with systemic glucocorticoids has failed to alter the natural history of IPF.
need for effective therapy [pathogenetic mechanism] MORE RESEARCH
yo
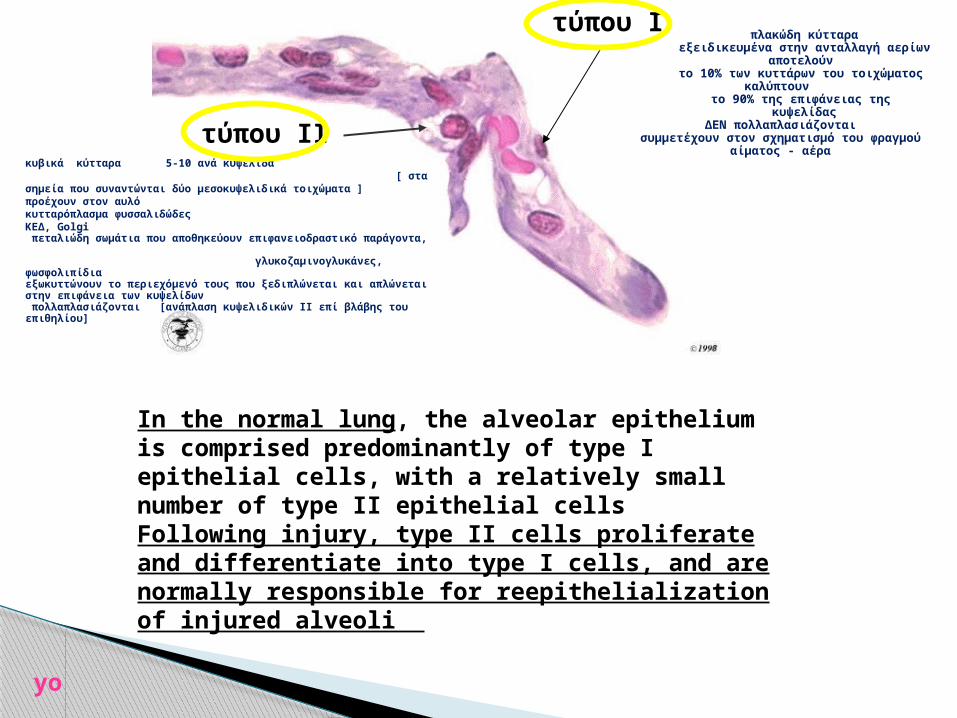
τύπου ΙΙ
τύπου Ι
πλακώδη κύτταραεξειδικευμένα στην ανταλλαγή αερίων
αποτελούν το 10% των κυττάρων του τοιχώματος
καλύπτουν το 90% της επιφάνειας της
κυψελίδαςΔΕΝ πολλαπλασιάζονται
συμμετέχουν στον σχηματισμό του φραγμού αίματος - αέρα
In the normal lung, the alveolar epithelium is comprised predominantly of type I epithelial cells, with a relatively small number of type II epithelial cells Following injury, type II cells proliferate and differentiate into type I cells, and are normally responsible for reepithelialization of injured alveoli
yo
κυβικά κύτταρα 5-10 ανά κυψελίδα [ στα σημεία που συναντώνται δύο μεσοκυψελιδικά τοιχώματα ] προέχουν στον αυλόκυτταρόπλασμα φυσσαλιδώδεςΚΕΔ, Golgi πεταλιώδη σωμάτια που αποθηκεύουν επιφανειοδραστικό παράγοντα, γλυκοζαμινογλυκάνες, φωσφολιπίδιαεξωκυττώνουν το περιεχόμενό τους που ξεδιπλώνεται και απλώνεται στην επιφάνεια των κυψελίδων πολλαπλασιάζονται [ανάπλαση κυψελιδικών ΙΙ επί βλάβης του επιθηλίου]

in IPF, loss of epithelial type I cells and marked proliferation of epithelial type II cells are noted; however, type II cells do not appear to reepithelialise the alveolar space this may be due to continued abnormalities of the basement membrane duplication and fragmentation which permit the migration of mesenchymal cells from the interstitium to the alveolar regions excessive deposition of collagen by mesenchymal cells the pattern of these changes suggests that chronic or recurrent injury to the alveolar epithelium may result in the progressive nature of intraalveolar exudates, fibrosis, and remodeling.
yo
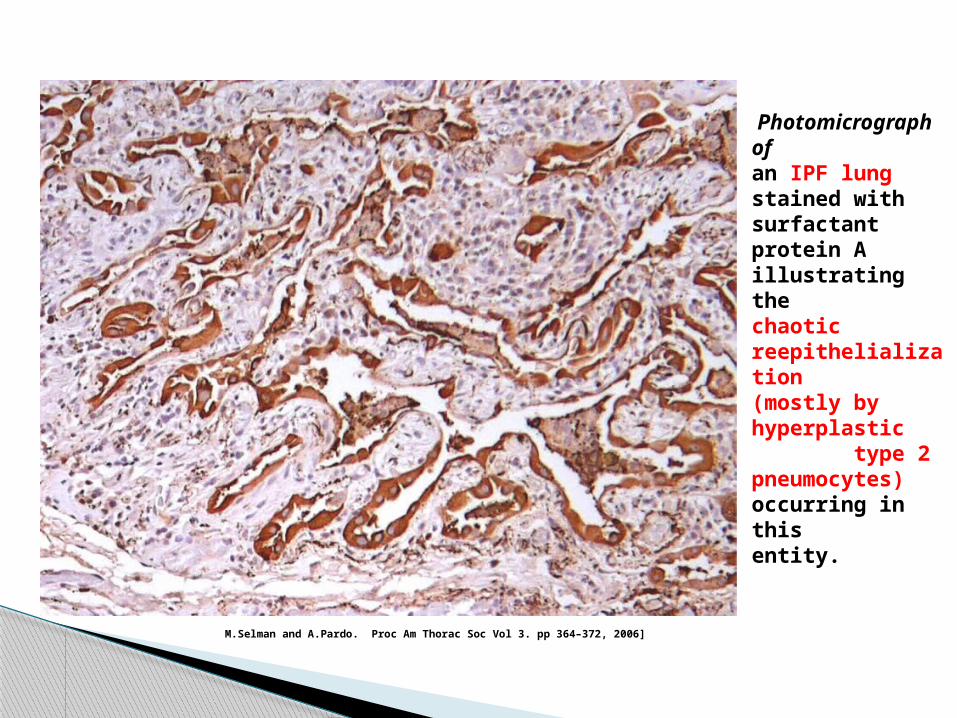
Photomicrograph ofan IPF lung stained withsurfactant protein A illustratingthe chaotic reepithelialization(mostly by hyperplastic type 2pneumocytes) occurring in thisentity.
M.Selman and A.Pardo. Proc Am Thorac Soc Vol 3. pp 364–372, 2006]
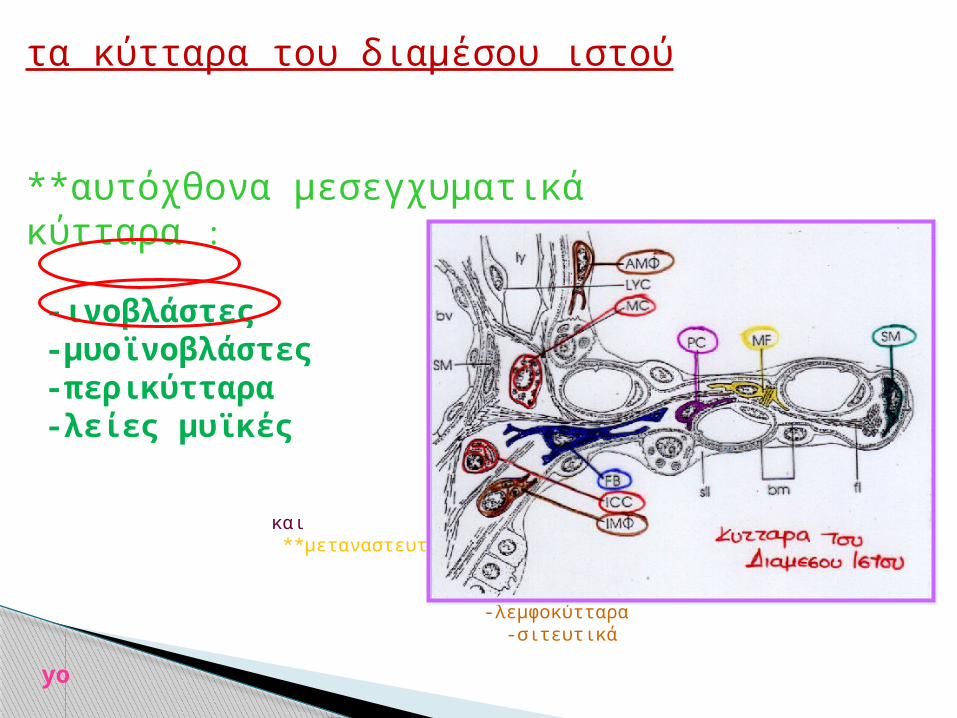
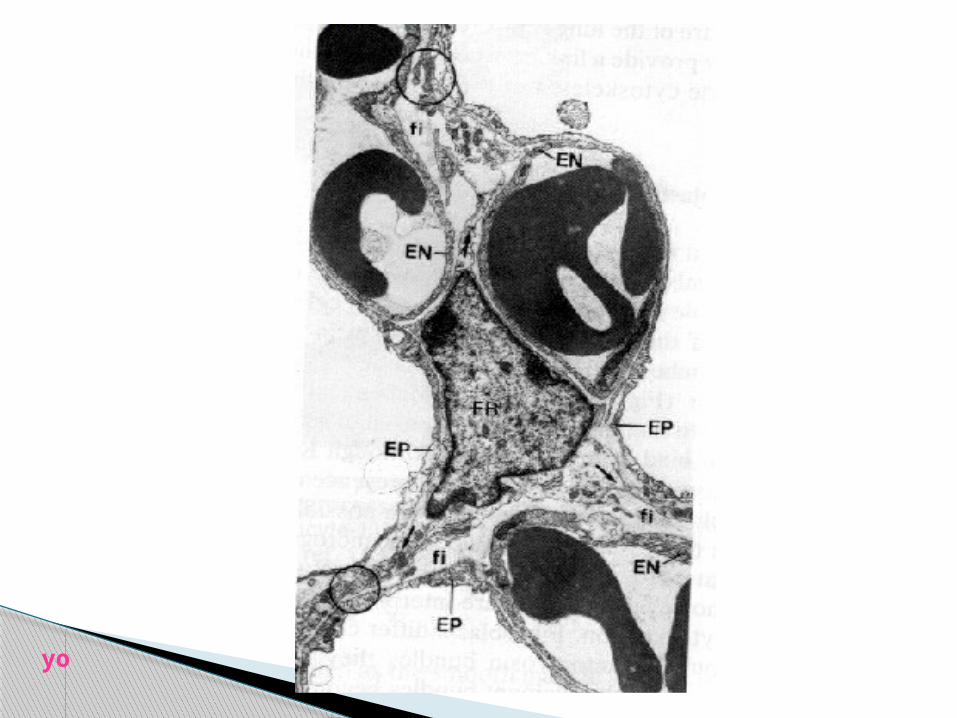
τα κύτταρα του διαμέσου ιστού
**αυτόχθονα μεσεγχυματικά κύτταρα : -ινοβλάστες -μυοϊνοβλάστες -περικύτταρα -λείες μυϊκές
και **μεταναστευτικά κύτταρα :
-ιστικά ΜΦ -λεμφοκύτταρα -σιτευτικά yo
yo
NL
IPF
IPFyo
Idiopathic Pulmonary FibrosisClinical Relevance of Pathologic ClassificationKATZENSTEIN and MYERS . Am J Respir Crit Care Med Vol 157. pp 1301–1315, 1998

the mechanism of fibrosis in IPF remains elusive however, a number of likely contributors have been identified so it is possible that
-multiple microinjuries to alveolar epithelial cells induce a fibrotic environment, and that -growth factors secreted by the injured epithelial cells recruit fibroblasts that differentiate into myofibroblasts.
-myofibroblasts secrete collagen, which accumulates
yo

— a considerable body of evidence suggests a critical role for alveolar epithelial cells (AEC) in the pathogenesis of IPF
there is loss of type I epithelial cells and proliferation of type II cells
orderly reepithelialization followed by differentiation of the type II cells to type I AECs does not occur this lack of normal reepithelialization may, in part, be due to an aberrant activation of the Wnt signalling pathway following to lung injury the Wnt proteins inhibit phosphorylation of beta-catenin by glycogen synthase kinase 3b (GSK3b), and prevent its translocation to nucleus and activation of the lymphoid enhancing factor/T-cell factor (LEF/TCF) transcription factors;
this is believed to activate proliferation of type II calls and
inhibit their differentiation,
yo

the interaction of growth factors, cytokines, and other mediators with cells resident in the lung is important to the evolution of the fibrotic response in IPF
following epithelial injury, fibrosis is believed to progress due to an imbalance between many groups of molecules related to fibrosis, that include pro-inflammatory and anti-inflammatory cytokines, fibrogenic and antifibrogenic polypeptides, oxidants-antioxidants, and angiogenic and angiostatic molecules
yo

it is possible that an initial injury and release of cytokines leads to long lasting effects on fibroblasts.
in an experimental model of IPF the release of transforming growth factor-beta1 (TGF-β1) from alveolar epithelial cells for 4 days led to induction of fibroblast growth factor-2, FGF-2, which was sequestered in the fibroblast matrix and led to an altered fibroblast phenotype with continued proliferation long after the exposure to TGF-β1
yo

BAL [BronchoAlveolar Lavage] from patients with IPF contains increased amounts of transforming growth factor-beta (TGF-β), including the active form TGF-β1.
TGF-β1 is one of the most potent regulators of connective tissue synthesis due to its ability to * increase connective tissue synthesis, * down-regulate connective tissue proteases, and * increase the inhibitors of connective tissue proteases
TGF-beta1 can also induce a number of growth factors and cytokines that participate in fibrosis, including -connective tissue growth factor (CTGF), -fibroblast growth factor (FGF-2), -platelet derived growth factor (PDGF), -insulin-like growth factor (IGF), and -interleukins (ILs).
the role of TGF-β in IPF is supported by inhibition of fibrosis in experimental animals by reducing its expression, signalling, and activity.
yo

TNF-alpha expression is increased in IPF
but its specific role is not clear
TNF-α has the ability to increase production of TGF-β1 and other peptide mediators, stimulate fibroblast proliferation, and induce collagen synthesis
ιn contrast, expression of IFN-γ, an inhibitor of fibroblast proliferation and connective tissue synthesis, appears to be deficient in lungs from patients with IPF.
yo

in IPF, there is also an overexpression of fibrotic cytokines
*monocyte chemoattractant protein 1 (MCP-1) its role in the recruitment of IPF cells is becoming increasingly recognized
*TGF-β, CTGF, IL-4, IL-13, FGF-2, IGF-1, PDGF and GM-CSF are fibrogenic and
*IFN-γ, IL-1, IL-10, IL-12, IL-17 are anti-fibrogenic molecules.
evolution of fibrosis appears to involve the activator protein (AP)-1 transcription factor and the fos-related protein Fra-2 yo

αdditional proteins that may contribute to pathogenesis or reflect disease activity of IPF have been identified using gene expression profiles
αs an example, genes overexpressed in lung tissue and blood from patients with IPF include matrix metalloprotein (MMP)-7, MMP-1, surfactant protein A1, cyclin A2 (CCNA2), and alpha-defensins
yo

wound healing involves a coordinated series of tissue movementsthat bears a striking resemblance to distinct stages inlung embryonic morphogenesis
comparing gene expression profiles from IPF lungs with profiles of genes associated with lung development may provide insight into the pathogenic mechanisms
yo

— fibrotic foci are now recognized as the characteristic histological feature of UIP
following the induction of fibroblast activity by epithelial injury, fibroblasts and myofibroblasts appear to organize into fibrotic foci that precede
the appearance of end-stage fibrosis
these fibroblast foci are situated adjacent to sites of epithelial cell and basement membrane damage, and consist of aggregates of actively proliferating fibroblasts and myofibroblasts.
the persistence of fibroblasts and formation of fibroblast foci may be regulated by inhibition of apoptosis of
fibroblasts [IL-4 ? ]. .
yo

soluble mediators secreted by alveolar epithelial cells and other cells in the surrounding milieu are believed to induce leukocyte influx, stimulate fibroblast activity, and promote fibrosis.
important among these are TGF-β, TNF-α, and IL-8
TGF-β is produced by activated epithelial cells, macrophages, and endothelial cells, and is the most crucial mediator in the development of pulmonary fibrosis. TGF-β is elevated both in patients with IPF and following intratracheal instillation of bleomycin in the murine model of IPF. fibrosis is attenuated by the inhibition of TGF-beta
specific polymorphisms of the gene encoding TGF-beta appear to increase the risk of disease progression
yo

IPF is characterized by fibroblast subpopulations that differ in their behaviour
when compare to fibroblasts isolated from normal lungs.
the fibroblast-like cell is the key target-effector cell that appears to determine the fibrotic response
in IPF
yo

myofibroblasts
produce interstitial collagens in significantly greater amounts than fibroblasts
two main sources of myofibroblasts in IPF have been identified
**it is believed that fibroblasts differentiate into myofibroblasts under the influence of mediators, such as TGF-β **in addition, a portion of fibroblasts may arise from mesenchymal transformation of local epithelial cells
***Growth factors promoting EMT [epithelial-mesenchymal transformation/transition] include TGF-β, epidermal growth factor (EGF), hepatocyte growth factor (HGF) and fibroblast growth factor (FGF) transcription factors Smads, Slug, Snail, Scatter, lymphoid enhancing factor-1, and β-catenin
yo

myofibroblast apoptosis
is a normal event during resolution of wound-repair, and failure of their apoptosis may also lead to accumulation of myofibroblasts and persistent production of extracellular matrix (ECM).
several studies in animal models
yo

using microRNA arrays on IPF lungs some interesting observations have been made.
microRNAs (miRNAs) are short RNA molecules that contain approximately 22 nucleotides and function to inhibit or promote the degradation of specific target mRNAs.
aberrant expression of the microRNA family miR-21 has been demonstrated in myofibroblasts from IPF lungs, but not from normal controls.
the miR-21 has been shown to function like an oncogene and in this manner prevents myofibroblast senescence and promotes proliferation
and differentiation In
contrast to miR-21, there is diminished expression of miR-29,
which inhibits fibrogenic proteins the
diminished expression of miR-29 in IPF lung fibroblasts may result in excessive connective tissue synthesis
yo

taken together these observations suggest that aberrant post-transcriptional gene regulation in IPF fibroblasts may result in a unique phenotype of fibroblasts that proliferate excessively and synthesize connective tissue proteins without control
AND excess collagen is produced by myofibroblasts and deposited in a disorganized manner within the extracellular matrix.
yo
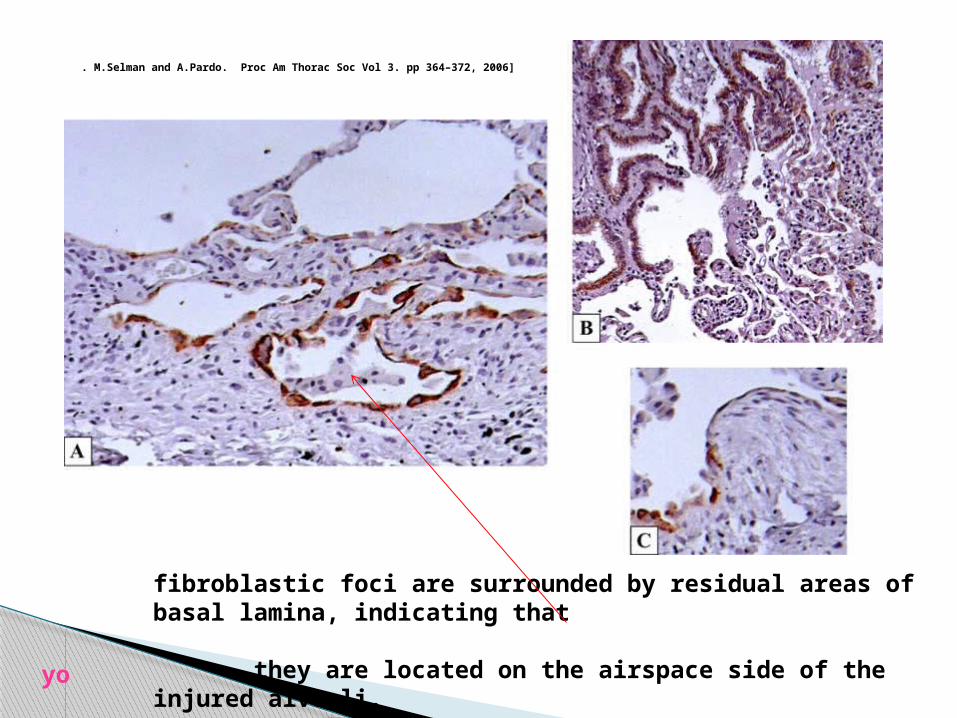
fibroblastic foci are surrounded by residual areas of basal lamina, indicating that they are located on the airspace side of the injured alveoli.
. M.Selman and A.Pardo. Proc Am Thorac Soc Vol 3. pp 364–372, 2006]
yo

increased numbers of collagen-synthesizing cells and increased collagen production have been noted in lung biopsies from patients with IPF
however, fibroblasts isolated from the lungs of patients with IPF and systemic sclerosis were found to synthesize a similar amount of collagen compared with control cell lines, [although the rate of collagen degradation was less]
yo

observations evaluating the expression of collagen type I and procollagen type I mRNA using in situ hybridization
have helped to reconcile these observations, demonstrating increased expression of type I collagen within fibroblast foci,
without evidence of increased type I procollagen mRNA expression in other
areas
therefore, it appears that the widely variable collagen secretion observed in IPF could be due to a focal, rather than a generalized, increase in fibroblast
activation.
yo

with increasing recognition of the pathogenic role of fibroblast proliferation and fibrotic foci, treatment strategies have shifted towards antifibrotic agents the rationale for the use of these agents rests upon the large amount of basic science data demonstrating an imbalance of cytokines, increased fibroblast proliferation, and excessive extracellular matrix deposition in the lungs of IPF patients
yo

τέλος
λεμε τωρα...
ακόμα στην αρχή είμαστε
yo

yo
lecture mainly based on :Pathogenesis of idiopathic pulmonary fibrosisAuthorsGanesh Raghu, MD A S Narayanan, PhD Nasreen Khalil, MDSection EditorsTalmadge E King, Jr, MDAndrew Nicholson, MDDeputy EditorHelen Hollingsworth, MDDisclosuresAll topics are updated as new evidence becomes available and our peer review process is complete.Literature review current through: Oct 2013. |This topic last updated:Ιουλ 8, 2013.UpToDate
articleshighly suggested :1. Integrating mechanisms of pulmonary fibrosisThomas A. Wynn, J. Exp. Med. Vol. 208 No. 7 1339-1350 review www.jem.org/cgi/doi/10.1084/jem.20110551 Supplemental Material can be found at: http://jem.rupress.org/content/suppl/2011/06/16/jem.20110551.DC1.html
2. Role of Epithelial Cells in Idiopathic Pulmonary Fibrosis. From Innocent Targets to Serial KillersMoise´s Selman and Annie Pardo. Proc Am Thorac Soc Vol 3. pp 364–372, 2006 review
3. Parallels between tissue repair and embryo morphogenesis Paul Martin and Susan M. Parkhurst; Development 131, 3021-3034. 2004 review


















