
Регистрация медицинских изделий в ЕАЭС
11
Обзор особенностей регистрации медицинских изделий по правилам ЕАЭС Алексей Степанов 4-й международный форум для специалистов здравоохранения и холодовой цепи в России и ЕАЭС 4 декабря 2015 Москва
-
Upload
alexey-stepanov -
Category
Healthcare
-
view
4.519 -
download
30
Transcript of Регистрация медицинских изделий в ЕАЭС

Обзор особенностей
регистрации медицинских
изделий по правилам
ЕАЭС
Алексей Степанов
4-й международный форум для специалистов
здравоохранения и холодовой цепи
в России и ЕАЭС
4 декабря 2015
Москва
На 03.12.2015

Договор о Евразийском экономическом союзе - 29.05.2014
(статьи №№ 31, 100)
Соглашение о единых принципах и правилах обращения медицинских
изделий в рамках Евразийского экономического союза – 23.12.2014
Общие требования безопасности и эффективности
медицинских изделий
Правила
регистрации
Запрет
применения
23.11.2015:
…Рабочая группа
отмечает, что проект
решения нуждается в
доработке с учетом
замечаний… Заключение об оценке регулирующего
воздействия №64
“
Модель регулирования обращения
медицинских изделий в ЕАЭС
Мониторинг
безопасности
Знак
обращения
Классиф-ия
по классам
риска
Средства
измерения
Менеджмент
качества
Номенклатурн
.классиф-ия
Тех.исп.
Инф.
система
Био исп.
Клин. исп

Переходный период ?
…документы, подтверждающие факт государственной регистрации медицинских изделий выданные […] до вступления Соглашения в силу, действуют […] до окончания срока их действия, но не позднее 31 декабря 2021 г. “ Соглашение о единых принципах и правилах обращения медицинских изделий в рамках Евразийского экономического союза , п.11
В переходный период по выбору заявителя
экспертиза и регистрация медицинских изделий
осуществляется в соответствии с национальным
законодательством государства – члена Союза, либо
в соответствии с […] Правилами [Союза]… Проект правил регистрации и экспертизы безопасности, качества и
эффективности медицинских изделий, п.2
Сегодня Переходный
период
Законодательство
ЕЭК
Регистрация
Действие документов
? Регистрация
Действие документов
■ Регистрация
■Действие документов
■ Регистрация
■ Действие документов
? Регистрация
Действие документов
Регистрация
Действие документов
“ “
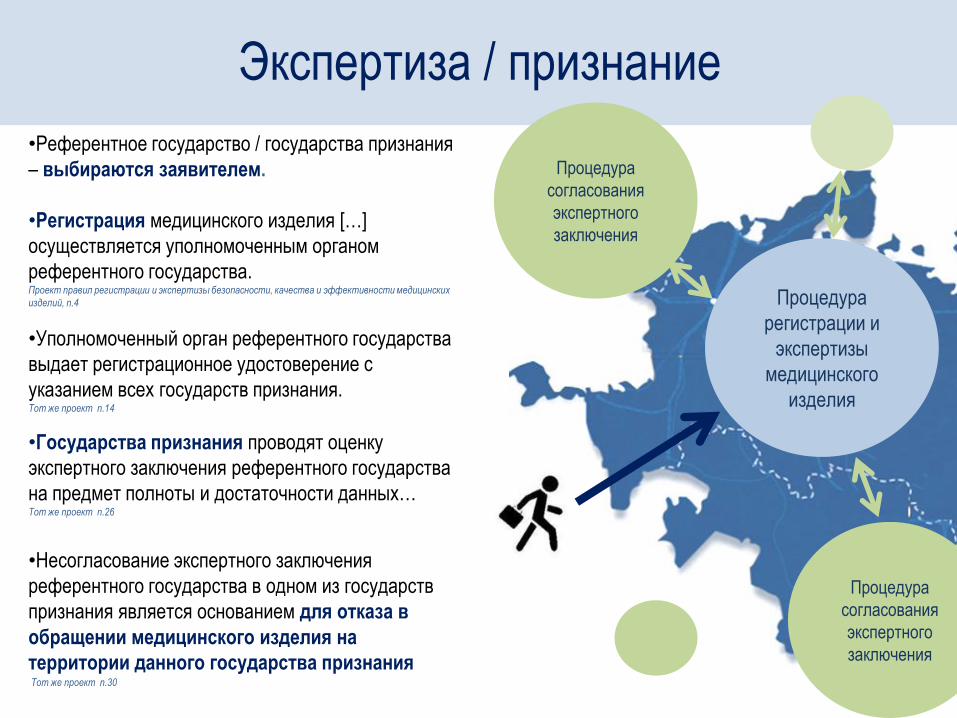
Процедура
регистрации и
экспертизы
медицинского
изделия
Процедура
согласования
экспертного
заключения
Процедура
согласования
экспертного
заключения
•Референтное государство / государства признания
– выбираются заявителем.
•Регистрация медицинского изделия […]
осуществляется уполномоченным органом
референтного государства. Проект правил регистрации и экспертизы безопасности, качества и эффективности медицинских
изделий, п.4
•Уполномоченный орган референтного государства
выдает регистрационное удостоверение с
указанием всех государств признания. Тот же проект п.14
•Государства признания проводят оценку
экспертного заключения референтного государства
на предмет полноты и достаточности данных… Тот же проект п.26
•Несогласование экспертного заключения
референтного государства в одном из государств
признания является основанием для отказа в
обращении медицинского изделия на
территории данного государства признания Тот же проект п.30
Экспертиза / признание
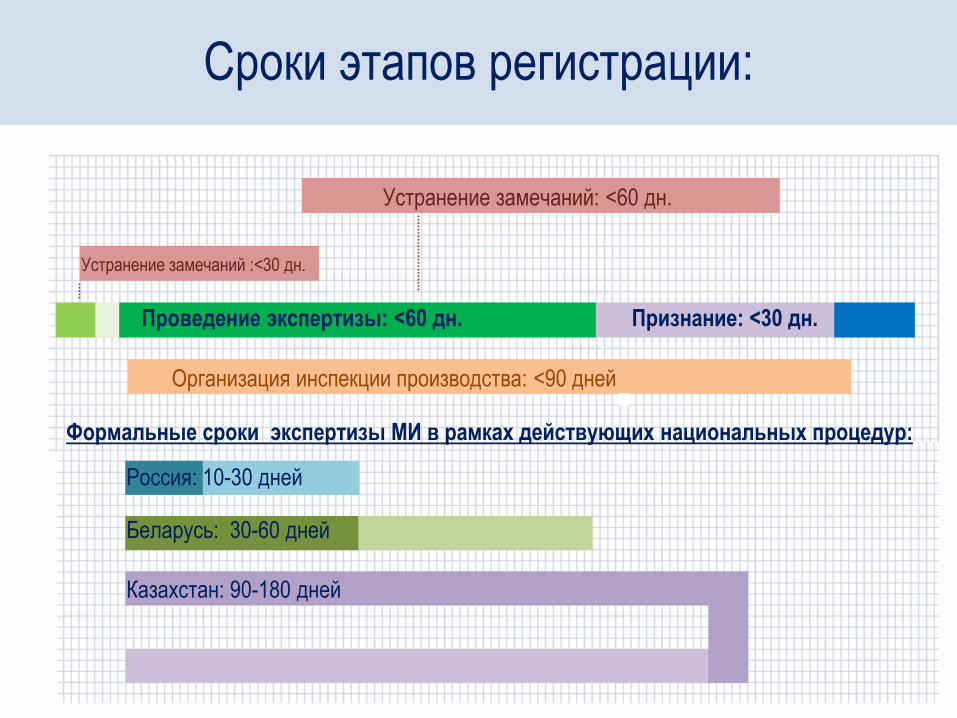
Устранение замечаний: <60 дн.
Устранение замечаний :<30 дн.
Проведение экспертизы: <60 дн. Признание: <30 дн.
Организация инспекции производства: <90 дней
Формальные сроки экспертизы МИ в рамках действующих национальных процедур:
Россия: 10-30 дней
Беларусь: 30-60 дней
Казахстан: 90-180 дней
Сроки этапов регистрации:

• Уполномоченный орган […] проводит инспекцию производства МИ в
соответствии с требованиями, установленными Комиссией […]Результаты
проведенной инспекции производства распространяются на выпускаемую
продукцию на момент инспектирования. Проект правил регистрации и экспертизы безопасности, качества и эффективности медицинских изделий, п.5
• «Инспектирование системы менеджмента качества» – аудит производителя МИ
с целью подтверждения соответствия системы менеджмента качества и
МИ регуляторным требованиям
Инспектирование производства
Для МИ класса потенциального риска
применения 2а наличие действующего
сертификата […] ISO 13485-2011
является достаточным
доказательством …[наличия в
актуальном состоянии системы
менеджмента качества]. Проект требований к внедрению, поддержанию и оценке системы менеджмента, п.7
Производители МИ классов
потенциального риска применения 3 и 2б,
а также МИ, выпускаемых в стерильном
виде класса потенциального риска
применения 2а, должны […] проходить
предрегистрационную и
пострегистрационную инспекции
Проект требований к внедрению, поддержанию и оценке системы менеджмента, п.8
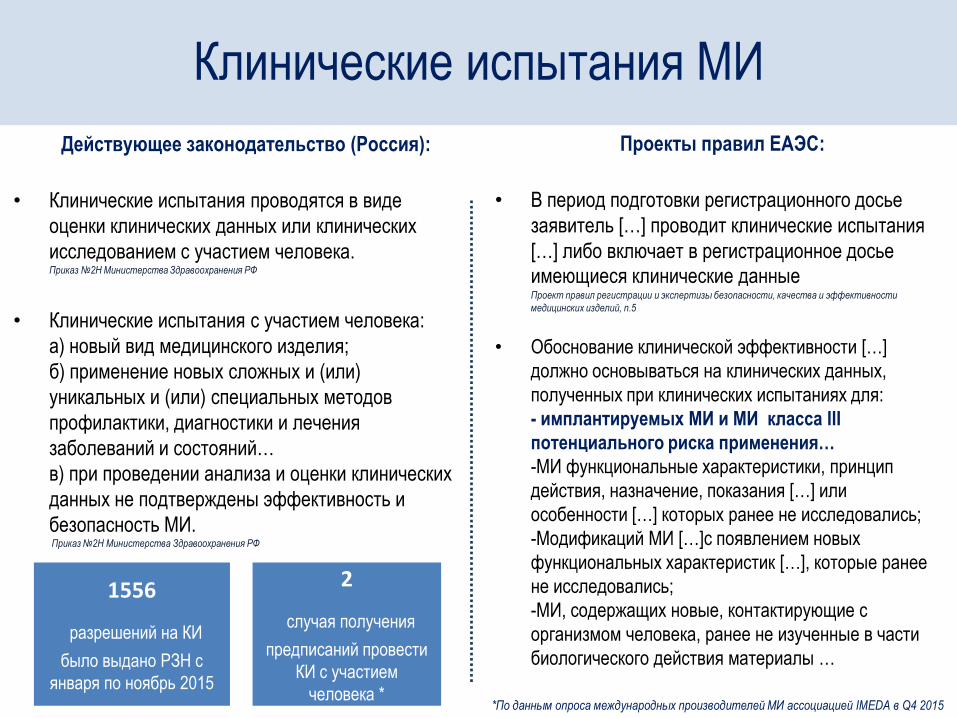
Проекты правил ЕАЭС:
• В период подготовки регистрационного досье
заявитель […] проводит клинические испытания
[…] либо включает в регистрационное досье
имеющиеся клинические данные Проект правил регистрации и экспертизы безопасности, качества и эффективности
медицинских изделий, п.5
• Обоснование клинической эффективности […]
должно основываться на клинических данных,
полученных при клинических испытаниях для:
- имплантируемых МИ и МИ класса III
потенциального риска применения…
-МИ функциональные характеристики, принцип
действия, назначение, показания […] или
особенности […] которых ранее не исследовались;
-Модификаций МИ […]с появлением новых
функциональных характеристик […], которые ранее
не исследовались;
-МИ, содержащих новые, контактирующие с
организмом человека, ранее не изученные в части
биологического действия материалы …
Действующее законодательство (Россия):
• Клинические испытания проводятся в виде
оценки клинических данных или клинических
исследованием с участием человека. Приказ №2Н Министерства Здравоохранения РФ
• Клинические испытания с участием человека:
а) новый вид медицинского изделия;
б) применение новых сложных и (или)
уникальных и (или) специальных методов
профилактики, диагностики и лечения
заболеваний и состояний…
в) при проведении анализа и оценки клинических
данных не подтверждены эффективность и
безопасность МИ. Приказ №2Н Министерства Здравоохранения РФ
1556
разрешений на КИ
было выдано РЗН с
января по ноябрь 2015
Клинические испытания МИ
2
случая получения
предписаний провести
КИ с участием
человека * *По данным опроса международных производителей МИ ассоциацией IMEDA в Q4 2015

В период подготовки регистрационного досье заявитель:
– получает предварительные консультации (при необходимости и по желанию заявителя)
по всем вопросам регистрации и экспертизы от экспертной организации… Проект правил регистрации и экспертизы безопасности, качества и эффективности медицинских изделий, п.5
“
Производитель имплантируемых медицинских изделий […] ежегодно в течение трех
лет со дня регистрации МИ должен представлять в уполномоченный орган отчет по
анализу безопасности и эффективности медицинского изделия на постпродажном
этапе в соответствии с Правилами мониторинга безопасности, качества и
эффективности медицинских изделий. Проект требований к внедрению, поддержанию и оценке системы менеджмента, п.6
“
Некоторые другие особенности:
Производитель в течение двух месяцев с момента внесения изменений обязан
инициировать процедуру внесения изменений в регистрационное досье….
Определен Перечень изменений, в отношении которых осуществляется
экспертиза при внесении изменений в регистрационное досье. Проект правил регистрации и экспертизы безопасности, качества и эффективности медицинских изделий, п.31
“

•На 04.12.2015 документы «второго уровня», регламентирующие обращение
МИ в ЕАЭС, официально не приняты, а наиболее актуальным вопросом
остается стратегия регулирования во время переходного периода.
• Основными этапами новой процедуры регистрации МИ является экспертиза
в референтном государстве и признание ее результатов другими странами
ЕАЭС; предполагаемые сроки новой процедуры соизмеримы с действующими
сегодня.
•Опубликованные проекты документов позволяют выделить принципиальные
особенности регистрации по новым правилам: введение инспекции
менеджмента качества, клинических испытаний с участием человека и
усиление требований к мониторингу безопасности для изделий высокого
класса риска, а также закрепление четких критериев внесения изменений в
регистрационные документы.
Регистрация МИ в ЕАЭС за 30 секунд….

СПАСИБО!
Возможно, Вам будет интересен мой блог о регулировании медицинских
изделий в России и странах Евразийского Союза:
www.MedicalDevicesInRussia.com


















